
CARDIOPATIAS CONGENITAS
21
INTRODUCION Las cardiopatías congénitas son defectos anatómicos del corazón y de los grandes vasos que se producen en las diferentes etapas del desarrollo fetal y el presente al nacer. Es toda anomalía estructural del corazón o de los grandes vasos que presentan o potencialmente tienen el riesgo de un compromiso funcional. Las anomalías tienen espectro clínico amplio, que van desde defectos que se desarrollan de forma asintomática a los que determinan los síntomas importantes y un alto índice de mortalidad. Algunas veces se debe a factores tales como la inclusión de los defectos que pueden pasar totalmente desapercibido en el examen físico, por ejemplo, la presencia de válvula aórtica bicúspide o la clasificación conducto arterioso ya sea como enfermedad o como persistencia todavía se considera fisiológica.
-
Upload
histeffany-mendonca -
Category
Documents
-
view
15 -
download
0
description
pediatria
Transcript of CARDIOPATIAS CONGENITAS

INTRODUCION
Las cardiopatías congénitas son defectos anatómicos del corazón y de los
grandes vasos que se producen en las diferentes etapas del desarrollo fetal y el presente
al nacer. Es toda anomalía estructural del corazón o de los grandes vasos que presentan
o potencialmente tienen el riesgo de un compromiso funcional.
Las anomalías tienen espectro clínico amplio, que van desde defectos que se
desarrollan de forma asintomática a los que determinan los síntomas importantes y un
alto índice de mortalidad.
Algunas veces se debe a factores tales como la inclusión de los defectos que
pueden pasar totalmente desapercibido en el examen físico, por ejemplo, la presencia de
válvula aórtica bicúspide o la clasificación conducto arterioso ya sea como enfermedad
o como persistencia todavía se considera fisiológica.

ETIOLOGÍA E INCIDENCIA
Deben a alteraciones en el desarrollo embrionario del corazón, entre la 3era y 10ma
semanas de la gestación.
En la mayoría de los casos es desconocida pero:
80-85% de origen genético o multifactorial: Podría suceder que no haya
ningún caso en la familia y se trate de una alteración espontánea, como ocurre
con las mutaciones. O puede ocurrir que la alteración sea aportada por la
información genética los padres, o sea hereditaria.
10 a 25% se asocian a anomalías cromosómicas: más frecuentes trisomías 13
o 18 que presentan anomalías más graves y la Trisomía 21, Síndrome de Turner
o Sindrome de Holt-Oram causan anomalías menos graves.
2-3% pueden ser causadas por factores ambientales (enfermedades maternas
o causadas por teratógenos) dentro de estos encontramos los siguientes:
FÁRMACOS/
DROGAS
AGENTES
INFECCIOSOSAGENTES MATERNOS
Trimetadiona Rubeola Diabetes
Ácido retinoico Enfermedades del Colágeno
Anfetaminas Fenilcetonuria
Sales de Litio AGENTES FÍSICOS OTROS
Alcohol Radiaciones Disolventes
Hidantoínas Hipoxia Pinturas
Hormonas sexuales Lacas y colorantes
Simpaticomiméticos Pesticidas
Las anomalías congénitas tienen una incidencia de alrededor del 5% en forma
global, siendo las anomalías congénitas mayores entre un 1,8 y 3%, presentándose en
uno de cada 30 recién nacidos vivos y en 0,1 a 1 de cada 10 mortinatos. Las anomalías
congénitas mayores representan por sí sola el 25% de la mortalidad perinatal.
En orden de frecuencia, el primer lugar lo comparten, con un 21% del total, las
malformaciones cardíacas y genitourinarias, siguiendo en frecuencia con un 16% las del
sistema nervioso central y luego las musculoesqueléticas, faciales y gastrointestinales
con un 5 a 7% cada una.

CLASIFICACIÓN
Las cardiopatías congénitas pueden dividirse en dos grupos principales basados
en la presencia o ausencia de cianosis. Estos dos grupos se subdividir si la radiografía de
tórax muestra signos de un flujo pulmonar aumentado, disminuido o normal. El
electrocardiograma puede utilizarse para determinar si existe hipertrofia ventricular
izquierda, derecha o biventricular. Las características de los ruidos cardíacos y la
presencia y características de cualquier soplo permiten acotar aún más el diagnóstico
diferencial. La ecocardiografía, la TC o la RM, o el cateterismo confirman el
diagnóstico final.
Siendo así, de acuerdo al tipo de cardiopatías las podemos clasificar de la
siguiente manera:
CC I-D No cianóticas CC D-I Cianóticas CC Obstáculo VD-VI
Comunicación Interauricular Tetralogía de Fallot Estenosis Pulmonar
Comunicación InterventricularTransposición de
Grandes VasosEstenosis Aortica
Persistencia del Ductus
ArteriosoAtresia Tricuspídea Coartación de la Aorta
CARDIOPATÍAS CONGÉNITAS NO CIANÓTICAS
Las cardiopatías congénitas a cianóticas se clasifican en:
1. Lesiones de cortocircuito o shunt de izquierda a derecha: Son con sobrecarga
de volumen y la mayoría de ellas tienen en común la existencia de un cortocircuito
desde la circulación sistémica al circuito pulmonar, con recirculación pulmonar de
sangre ya oxigenada. La importancia de la cardiopatía viene condicionada por la
magnitud del shunt que se determina relacionando el flujo de sangre circulante a nivel
pulmonar (Qp) con el que circula a nivel sistémico (Qs), es decir: la relación Qp : Qs.
En situación normal ésta relación es uno (Qp/Qs = 1), representando su aumento un
indicio claro de shunt izquierda derecha.

Así, un Qp/Qs igual a 2 nos informa de que el volumen de sangre del circuito
pulmonar es el doble del sistémico. Por el contrario, un Qp/Qs inferior a la unidad nos
indicará un shunt de derecha a izquierda.
Los factores de los que depende la dirección y la magnitud del cortocircuito a través
de la comunicación son principalmente dos:
1. El tamaño del defecto.
2. La relación entre las presiones y resistencias vasculares existentes en los
circuitos sistémico y pulmonar.
Estos factores no son fijos si no dinámicos, cambiando a lo largo del tiempo. Los
defectos intracardíacos (CIV, CIA) pueden disminuir de tamaño, especialmente a lo
largo del primer año de vida.
En condiciones normales, las resistencias pulmonares elevadas del neonato
descienden a niveles adultos pocas semanas después del nacimiento.
La existencia de una excesiva cantidad de sangre circulante a nivel de los vasos
pulmonares (originando por ello una elevación de la presión a dicho nivel), puede
terminar produciendo una alteración anatómica de las arteriolas pulmonares y un
aumento progresivo e irreversible de las resistencias vasculares a su nivel.
En estas cardiopatías la magnitud del shunt varía a lo largo del tiempo.
Además de las comunicaciones mencionadas, hay otras cardiopatías que se
acompañan de sobrecarga de volumen. De ellas destacaremos tres situaciones:
1. Insuficiencias de las válvulas auriculoventriculares: La más importante es el
defecto de cojinetes endocárdicos (canal atrioventricular), el cual presenta –
además de un cortocircuito izquierda derecha una insuficiencia mitral, tricúspide
o de ambas válvulas. La Enfermedad de Ebstein, que presenta insuficiencia
tricuspídea importante.
2. Insuficiencia aórtica.
3. Las miocardiopatías (primarias o secundarias). Estas enfermedades se
acompañan de disminución de la función miocárdica, produciéndose un aumento de la
presión de llenado tanto a nivel cardíaco como de los capilares pulmonares. Ello
conduce a la aparición de edema pulmonar.
Las principales cardiopatias de lesiones com cortocircuito son:

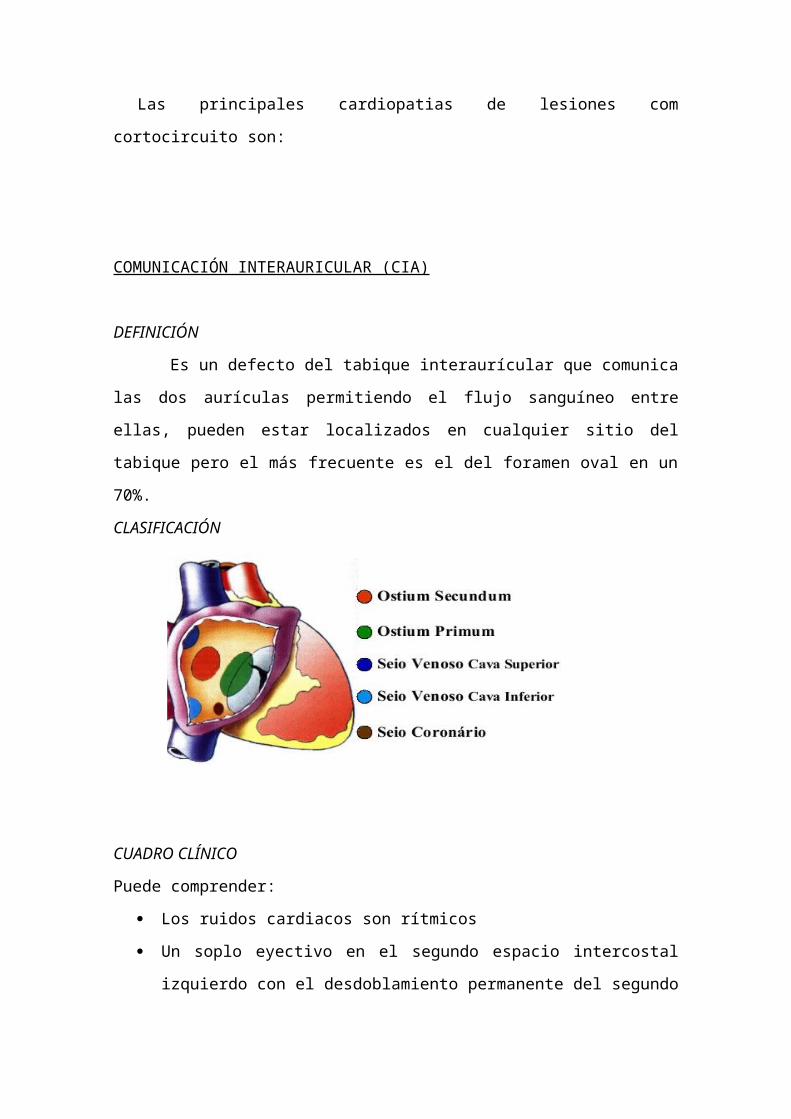
COMUNICACIÓN INTERAURICULAR (CIA)
DEFINICIÓN
Es un defecto del tabique interaurícular que comunica las dos aurículas
permitiendo el flujo sanguíneo entre ellas, pueden estar localizados en cualquier sitio
del tabique pero el más frecuente es el del foramen oval en un 70%.
CLASIFICACIÓN
CUADRO CLÍNICO
Puede comprender:
Los ruidos cardiacos son rítmicos
Un soplo eyectivo en el segundo espacio intercostal izquierdo con el
desdoblamiento permanente del segundo ruido debido a la sobrecarga del
ventrículo derecho y retraso del cierre de la válvula pulmonar.
Ocasionalmente, cuando hay una CIA grande puede producirse insuficiencia
cardíaca.
No hay cianosis
Antecedentes importante de problemas pulmonares a repetición
Hipodesarrollo
Hiperactividad del ventrículo derecho tanto en el tercio inferior como en el
superior.
Frémito en el segundo espacio intercostal izquierdo con línea paraesternal
Cuando la CIA es grande hay un QP/QS mayor de 2:1, un retumbo diastólico en el
tercio inferior del borde esternal izquierdo, si la evolución es prolongada, la CIA es
grande y ya hay hipertensión pulmonar se encuentra un reforzamiento importante del
segundo ruido y se pierde la característica del desdoblamiento del ruido.

DIAGNÓSTICO
Es muy difícil de diagnosticar en los primeros días de vida sobre todo si hay
retraso en la disminución de las resistencias pulmonares como ocurre al nacer a grandes
alturas sobre el nivel del mar o secundario a un problema pulmonar en el recién nacido
donde experimentará un aumento de las presiones pulmonares ya que se distiende el
tabique interatrial llevando a una distensión del foramen oval. Generalmente se la
descubre después de los dos años de vida.
Vamos a utilizar métodos como:
Electrocardiograma: Generalmente podemos apreciar una desviación del eje a la
derecha mientras más HT Pulmonar presente. Además el Bloqueo de rama
derecha es característico de CIA, la R única mellada en V1 se la aprecia en una
HT Pulmonar importante.
Ecocardiograma: Lo podemos utilizar para precisar la localización y el tamaño
del defecto; las anomalías asociadas, el cálculo de la magnitud del cortocircuito,
y la presión pulmonar
Rx de Tórax: Hay un crecimiento variable del ventrículo derecho y de aurícula
derecha dependiente del tamaño del defecto, un abombamiento del tronco de la
pulmonar, la aorta ascendente y cayado aórtico son poco aparentes y flujo
pulmonar se encuentra aumentado.
Estudio con Doppler: Nos proporciona beneficios para determinar la dirección
del cortocircuito, amplitud y tamaño además de la valoración de la presión
pulmonar.
Cateterismo cardiaco: Útil en caso de persistencia de alguna duda o en
cardiopatías asociadas.
TRATAMIENTO:
En el momento actual existen dos formas reconocidas de tratamiento de la CIA:
el cierre quirúrgico y el cierre percutáneo con dispositivos.
DEFECTO SEPTAL INTERVENTRICULAR (CIV)
DEFINICIÓN
Describe un orificio en el tabique interventricular, que puede encontrarse en
cualquier punto del mismo, ser único o múltiple, y con tamaño y forma variable. Las
comunicaciones interventriculares pueden presentarse aisladas o formando parte
integrante de otras cardiopatías más complejas como: tronco arterioso común, tetralogía
de Fallot, ventrículo derecho de doble salida, transposición de grandes arterias, canal
auriculo-ventricular común, etc).
CLASIFICACIÓN
Las CIV se clasifican, atendiendo a su situación en el tabique en:
CUADRO CLÍNICO
Al examen físico es común encontrar:
• cardiomegalia, frémito, soplo
• R2 puede estar reforzado o aumentado.
• La presencia de estertores, sibilancias por el compromiso de los alvéolos o
bronquios de pequeño calibre es común en lactantes.
• La obstrucción del bronquio izquierdo por la compresión.
En defectos del Septum Interventricular grandes con repercusión hemodinámica
podemos encontrar:
MEMBRANOSA
MUSCULAR
Porción de entrada
Porción trabeculada
Porción de salida
PERIMEMBRANOSA
Extensión entrada
Extensión trabeculada
Apical
Media
Extensión de salida subaórtica
Subpulmonar
Doblemente relacionada
POR MALA
ALINEACIÓN DE
LOS SEPTUM

• Diaforesis
• Fatiga para alimentarse en los neonatos y lactantes,
• Cansancio con el ejercicio en niños mayores,
• Poca ganancia de peso
• Procesos pulmonares repetidos
DIAGNÓSTICO
ELECTROCARDIOGRAMA: Desviación del eje a la izquierda por crecimiento
de cavidades izquierdas.
RX DE TORAX: Se evidencia un Índice Cardiotorácico aumentado para la edad
a expensas de cavidades izquierdas o biventriculares, aorta angosta y
abombamiento del tronco pulmonar.
ECOCARDIOGRAMA: Permite hacer el diagnóstico anatómico y su
clasificación, la evaluación funcional de la repercusión hemodinámica.
CATETERISMO CARDIACO: Con estudio hemodinámico y
angiocardiográfico, permite evaluar la magnitud del cortocircuito, medir la
presión arterial pulmonar y estimar las resistencias vasculares, además de
determinar el tamaño, el número y la localización de los defectos y de excluir
lesiones asociadas.
ECOCARDIOGRAFIA DOPPLER: Permite obviar la necesidad de cateterismo
en la mayoría de los pacientes.
TRATAMIENTO
Tratamiento farmacológico con IECA (captopril o enalapril) y diuréticos
(furosemida, espironolactona). La administración de digoxina está en discusión pero con
su uso se ha evidenciado una mejoría sintomática, y algunos protocolos experimentales
han mostrado un beneficio agudo en parámetros hemodinámicos. Es habitual asociarla a
los vasodilatadores y diuréticos en casos muy sintomáticos, en ocasiones se puede
requerir catecolaminas
El tratamiento inicial en el lactante sintomático debe incluir también un control
nutricional meticuloso que utilice fórmulas hipercalóricas concentradas o suplementos
cuando la lactancia materna normal sea insuficiente, ocasionalmente se requiere la
nutrición por sonda nasogástrica.
TRATAMIENTO QUIRURGICO
Son motivos de intervención la presencia de ICC no controlada, hipertensión
pulmonar, hipodesarrollo importante y/o infecciones respiratorias recurrentes.

Indicaciones quirúrgica para CIA
DUCTUS ARTERIOSO PERSISTENTE (DAP)
DEFINICIÓN
Es una estructura vascular que comunica la porción
distal del arco aórtico con la región proximal de la arteria
pulmonar izquierda. 1
CLASIFICACIÓN
SILENTES: Pacientes que no presentan soplo ni
datos de hipertensión arterial pulmonar y son diagnosticados solo por
ecocardiografía.
PEQUEÑOS: Pacientes con soplo continuo audible, insignificantes cambios
hemodinámicos, sin sobrecarga en cavidades izquierdas ni hipertensión arterial
pulmonar.
MODERADOS: Pacientes con soplo continuo, pulsos amplios, sobrecarga de
volumen en cavidades izquierdas, hipertensión arterial pulmonar leve a
moderada. Con o sin datos de insuficiencia cardiaca leve (compensada).
1

GRANDES: Pacientes con soplo continuo, pulsos amplios, sobrecarga
importante de volumen en cavidades izquierdas, hipertensión arterial pulmonar
moderada o severa, con datos clínicos de insuficiencia cardiaca descompensada.
MANIFESTACIONES CLÍNICAS
Los síntomas van a depender del tamaño y las resistencias pulmonares manejadas
por el paciente.
Ductus pequeño menor de 1.5 mm: Generalmente no se presentan síntomas y el
único hallazgo es la presencia de un soplo sistólico eyectivo en el foco pulmonar
o en la región infraclavicular izquierda.
Ductus moderado de 2 mm en la lactancia hasta 3,5 mm en la edad escolar: Se
presentan disnea, infecciones respiratorias recurrentes y disminución en el
crecimiento. Se encuentra un soplo sisto-diastólico continuo o en maquinaria a
nivel del foco pulmonar o de la región infraclavicular izquierda; el R2 está
ligeramente reforzado y los pulsos son hiperdinámicos en forma difusa.
Ductus grande mayores de 4 mm en la infancia: Encontramos disnea, taquicardia
en reposo, historia de infecciones respiratorias recurrentes y desnutrición
crónica. Se observa hiperdinamia precordial a expensas del ventrículo izquierdo,
punto de máximo impulso hacia la línea axilar anterior izquierda, presión de
pulso amplia, soplo sistólico eyectivo en el foco pulmonar y soplo diastólico en
foco mitral con R2 reforzado. Cuando se presenta hipertensión pulmonar y se
invierte el cortocircuito se presenta cianosis.
DIAGNÓSTICO
Dentro de los principales medios de diagnóstico encontramos:
Electrocardiograma: En Ductus pequeños puede ser normal, en Ductus mayores
se observa crecimiento de cavidades izquierdas y signos de hipertensión
pulmonar como P picudas, y S profundas en V5 y V6.
Radiografía de tórax: Normal en Ductus pequeños, en Ductus mayores puede
observarse aumento en el flujo pulmonar, cardiomegalia, aumento del botón
aórtico y pulmonar.
Ecocardiograma: Determina la presencia del defecto, su tamaño, y si hay
repercusión hemodinámica.
Cateterismo cardiaco: Su realización solo se hará en casos donde este indicado el
cierre percutáneo o en pacientes con signos clínicos y ecocardiográficos de
hipertensión pulmonar importante.

TRATAMIENTO
Depende de: la edad del paciente y el diámetro del ductus.
RN pretérmino <28 semanas DAP > o igual a 1,6 mm de diámetro y en RN pretérmino
de 29-35 semanas DAP > o igual a 2 mm de diámetro:
Indometacina 0,2 mg/kg dosis inicial, seguida de 0,1 mg/kg cada 12 horas hasta
completar 3 dosis.
Ibuprofeno a dosis de 10 mg/kg/IV dosis inicial, seguido de dos dosis de 5
mg/kg/IV cada 24 horas.
Control ecocardiográfico al terminar el ciclo.
En reapertura se puede repetir y si es fallido el cierre farmacológico y hay
repercusión hemodinámica se debe llevar a cirugía.
RN a término con DAP > o igual a 3 mm:
medidas anticongestivas inicialmente y si no hay mejoría cierre quirúrgico.
RN a término DAP < 2 mm:
observación clínica
Si hay signos de hiperflujo pulmonar iniciar diuréticos e IECA
Con control de los síntomas vigilancia clínica por tendencia natural al cierre
espontáneo.
Lactante < 6 meses DAP >3 mm:
Cierre quirúrgico
En niños >6 meses DAP < 3 mm:
Cierre percutáneo con dispositivo tipo resorte metálico y cierre quirúrgico si no
hay esta posibilidad.
Escolares y adolescentes DAP >4 mm:
Cierre percutáneo con dispositivos tipo resorte metálico o Amplatzer vs. cierre
quirúrgico.
2. Cardiopatías obstructivas: Con sobrecarga de presión, se deben
principalmente a la obstrucción del tracto de salida de uno de los ventrículos
(estenosis de la válvula aórtica o pulmonar), o a un estrechamiento de la aorta
(coartación de aorta). Los principales son:
ESTENOSIS AORTICA

Se produce cuando el diámetro de la válvula aórtica es reducido –tiene dos
valvas o velos en vez de tres- por lo que el flujo sanguíneo a través de la válvula se
reduce y el ventrículo izquierdo del corazón tiene que aumentar la presión para bombear
la cantidad de sangre necesaria (5 litros por minuto) por una apertura disminuida.
Debido a este esfuerzo extra, las paredes del ventrículo se engruesan. Los niños con
estenosis aórtica pueden sufrir otras alteraciones congénitas.
COARTACIÓ D'AORTA
Se trata de una estenosis de la parte inicial de la
aorta descendente que se localiza generalmente
después de la salida de la arteria subclavia izquierda.
Esta estenosis se asocia a menudo a una válvula aórtica
bicúspide.
Distinguimos dos grupos anatómicos (y
clínicos) de coartación de aorta:
1. F orma preductal o infantil , que como dice el
nombre la coartación se halla por encima de la embocadura del ductus en la aorta, se
asocia a hipoplasia del arco aórtico y a otras anomalías cardíacas. En estos casos existe
siempre al nacer un conducto arterioso persistente con flujo de derecha a izquierda.
Mientras que el ductus permitía en la vida fetal una circulación prácticamente
normal, cuando a poco de nacer ésta estructura
se cierra se origina una situación hemodinámica
de extrema gravedad al ser incapaz el corazón
de bombear sangre al terreno de la aorta
descendente. Por ello, pocos días después del
parto el recién nacido empeora bruscamente y
entra en una situación de insuficiencia cardíaca
que – por médios no quirúrgicos ‐ resulta
intratable.
2.Forma postductal o tipo adulto, no presenta anomalías cardíacas asociadas y en
el momento del diagnóstico el ductus se halla cerrado o – si persiste‐ es pequeño. Antes
de nacer estos niños ya han desarrollado una circulación colateral suficiente para

compensar parcialmente la interrupción aórtica, evitando así que la sintomatología se
inicie nada más nacer.
CLINICA
Mientras las formas preductales originan habitualmente un cuadro de
insuficiencia cardíaca aguda grave en las primeras semanas del periodo neonatal,
representando una urgencia vital, en las postductales la malformación
cardiovascular se manifiesta más tarde por la presencia de un soplo cardíaco de
eyección a nivel de los focos de la base (pulmonar y aórtico), y en la espalda, a
nivel interescapular.
La clave del diagnóstico la da la ausencia o disminución y retardo de los
pulsos femorales respecto de los braquiales, y la existencia de hipertensión
arterial a nivel de extremidades superiores. Más allá de la edad neonatal, los
pacientes (habitualmente 7 formas postductales) pueden aquejar cansancio en las
piernas, cefalea o epistaxis, siendo sin embargo generalmente escasas las
manifestaciones clínicas.
EVOLUCIÓN
Todos los pacientes, también los casos de diagnóstico más tardío, son
tributarios de tratamiento quirúrgico a la mayor brevedad posible. Con ello se
intenta evitar las posibles complicaciones tardías secundarias a una hipertensión
arterial sistémica mantenida (accidentes cerebrales), o que la hipertensión pueda
acabar haciéndose permanente.
El tratamiento quirúrgico consiste en la resección de la zona vascular
coartada, efectuándose una anastómosis término‐terminal. En las coartaciones
preductales a menudo se deberá ampliar el arco aórtico y la intervención
comporta un riesgo importante, siendo en cambio mínima la mortalidad
operatoria en las formas postductales. En caso de recidiva (recoartación) se evita
tener que recurrir de nuevo a la cirugía y se procede a la dilatación de la zona
recoartada por medio de un catéter dotado de un pequeño balón inflable en su
extremo (angioplastia aórtica). Éste procedimento sólo se halla indicado en las
recoartaciones, pero no en coartaciones nativas (originarias).
En las coartaciones se deberá tener presente la necesidad de efectuar
prevención de la endocarditis bacteriana en situaciones de riesgo, administrando
entonces el antibiótico oportuno.


















