PROTOCOLO CENTROAMERICANO PARA EL … · Web viewPROTOCOLO CENTROAMERICANO PARA EL TRATAMIENTO DE...
47
PROTOCOLO CENTROAMERICANO PARA EL TRATAMIENTO DE LA ENFERMEDAD DE HODGKIN EN NIÑOS Y ADOLESCENTES EH CA 3-2012 DR. MAURICIO CASTELLANOS ALQUIJAY (COORDINADOR DEL ESTUDIO) UNIDAD NACIONAL DE ONCOLOGIA PEDIATRICA, GUATEMALA E MAIL: [email protected] DR. PEDRO DE ALARCON (CONSULTANTE INTERNACIONAL) DRA. MONIKA METZGER( CONSULTANTE INTERNACIONAL) PAISES PARTICIPANTES: Costa Rica Dra. Yessika Gamboa E mail: El Salvador: Dra. Soad Alevi Honduras: Dr. Armando Peña Nicaragua: Dr. Fulgencio Báez Dra. Mercedes Arguello Panamá: Dra. María Sabina Ah-Chu Dra. Rosario Batista Dra. Hilze
Transcript of PROTOCOLO CENTROAMERICANO PARA EL … · Web viewPROTOCOLO CENTROAMERICANO PARA EL TRATAMIENTO DE...

PROTOCOLO CENTROAMERICANO PARA EL TRATAMIENTODE LA ENFERMEDAD DE HODGKIN EN NIÑOS Y ADOLESCENTES
EH CA 3-2012
DR. MAURICIO CASTELLANOS ALQUIJAY (COORDINADOR DEL ESTUDIO)UNIDAD NACIONAL DE ONCOLOGIA PEDIATRICA, GUATEMALAE MAIL: [email protected]
DR. PEDRO DE ALARCON (CONSULTANTE INTERNACIONAL)DRA. MONIKA METZGER( CONSULTANTE INTERNACIONAL)
PAISES PARTICIPANTES:
Costa Rica Dra. Yessika GamboaE mail:
El Salvador: Dra. Soad Alevi
Honduras: Dr. Armando Peña
Nicaragua: Dr. Fulgencio BáezDra. Mercedes Arguello
Panamá:Dra. María Sabina Ah-ChuDra. Rosario BatistaDra. Hilze
Republica DominicanaDra Rosa NievesDra Wendy Gomez Dra. Margarita Montero ArgentinaDr David Veron

INDICE
1- OBJETIVOS GENERALES Y SECUNDARIOS2- INTRODUCCIÓN3- DISEÑO DEL PROTOCOLO4- ELEGIBILIDAD DE PACIENTES5 FACTORES PRONÓSTICOS6- DIAGNÓSTICO7- ESTADIFICACIÓN8- PATOLOGÍA9- MEDICAMENTOS
10- PLAN DE TRATAMIENTO10.1 Quimioterapia para Riesgo Favorable Dosis y Esquema 10.2 Quimioterapia para Riesgo Intermedio Dosis y Esquema10.3 Quimioterapia para Riesgo Desfavorable Dosis y Esquema10.4 Guía para la Quimioterapia y terapia de soporte10.5 Definición de respuesta al tratamiento10.6 Directrices Radioterapeutitas10.7 Inicio de Radioterapia10.8 Directrices Quirúrgicas10.9 Evaluación de la rrespuesta al tratamiento
11- BIBLIOGRAFÍA 12- APENDICES
I ESTUDIOS PRE, TRANS Y POST TRATAMIENTOII DEFINICIONES DE CRITERIOS DE RESPUESTAIII REPORTE DE CASOS Y SEGUIMIENTOIV HOJA DE CONSENTIMIENTO INFORMADOV ESCALA DE TOXICIDAD

1- OBJETIVOS
GENERALESa. Estudiar y comparar las características clínicas de los niños con Enfermedad de
Hodgkin (EH) con histología esclerosis nodular, celularidad mixta, deplesion linfocitaria y predominio linfocitario y rica en linfocitos en los diferentes países centroamericanos.
b. Documentar la tasa de curación de los pacientes con esta enfermedad según los diferentes estadios y a la vez disminuir la morbi-mortalidad secundaria a tratamientos.
SECUNDARIOSa. Documentar la toxicidad producida a causa del tratamiento.
b. Disminuir la intensidad del tratamiento, mediante una adecuada estadificación en base de los grupos pronósticos y la experiencia de protocolos anteriores.
c. Determinar la tasa de recaída.
d. Estudiar las diferencias en las presentaciones de la enfermedad entre niños menores de 5 años y aquellos mayores.

2- INTRODUCCION
En los años 60, la radioterapia era el tratamiento estandard para los pacientes pediátricos con linfoma de Hodgkin, eran dosis de 35 a 44 Gy, sin embargo, la radioterapia producia muchos efectos tardíos como malignidades secundarias, retardo y asimetrías en el crecimiento, problemas hormonales y funcionales. Al reconocer como un problema grande estos efectos secundarios, se inicio una nueva experiencia, con el uso de quimioterapia. El primer reporte usando solo quimioterapia con MOPP viene de Uganda, donde no había posibilidades de radioterapia. El grupo de Australia-Alemania dieron 6 a 12 ciclos MOPP de quimioterapia para los pacientes favorables sin enfermedad de Bulky, y radioterapia adicional para los que tenían Bulky. En este estudio tuvieron un relativo pequeño número de pacientes pero revelaban supervivencia a 5 años en un promedio del 90% en estadios tempranos, sin envargo tuvieron una supervivencia libre de enfermedad a 5 años para pacientes con estadio avanzado del 40-55%. Estas experiencias dieron las directrices para la necesidad de conducir estudios prospectivos randomizados y valorar el tratamiento de modalidad combinada. El uso de MOPP y radiación a dosis menos tóxicas y a campos definidos empezó a dar mejores resultados y a la vez a disminuir un poco mas los efectos tardíos, por lo que comparado a los resultados anteriores iniciaron una nueva era para el tratamiento de la enfermedad de Hodgkin. La experiencia de Stanford reveló una sobrevida libre de recaída a 20 años del 85% después de dar radioterapia en dosis de 15, 20, 25 Gy, y quimioterapia con seis ciclos de MOPP en niños estadiados quirúrgicamente. Después, el grupo de Toronto extendió este tipo de tratamiento a los pacientes estadiados clínicamente y reportaron una supervivencia libre de recaída a 10 años de 80% con 6 ciclos de MOPP y 20 a 30 Gy. Pronto la modalidad combinada fue avanzando en aceptación en el Intergroup Hodgkin´s disease Study los cuales redujeron la radioterapia a campos involucrados y MOPP, lo cual dio una supervivencia libre de enfermedad de un 90% para estadios quirúrgicos I-II. Este mismo entusiasmo llevó al Grupo Francés e Italiano a usar quimioterapia y radioterapia a dosis de 20- 25 Gy, el grupo Alemán uso 35, 30, o 25 Gy y dos, cuatro y seis ciclos de quimioterapia en pacientes con manejo según el riesgo. Todos reportaron una supervivencia libre de enfermedad entre el 80% y 95%. Cuando los reportes de infertilidad masculina y leucemias mielógenas se atribuyeron a los agentes alcalinizantes, el grupo italiano hizo una substitución del MOPP por Doxorubicina, Bleomicina, Dacarbacina y Vinblastina, como una terapia de no resistencia cruzada para usar con bajas dosis de radioterapia a campos involucrados por la enfermedad.El grupo de estudio Nacional Italiano usó tres ciclos de ABVD y 20-25 Gy para pacientes con estadio I-IIA sin enfermedad Bulky, y el grupo pediátrico de Milán investigó la necesidad de tres versus seis ciclos de ABVD con 25 Gy para niños con estadios I-III. Resultados optimistas fueron reportados en EFS y OS.

Si bien los grupos americanos han realizado estudios randomizados donde se busca el demostrar que a los pacientes con RC al final del tratamiento, dependiendo el estadio, deben ser observados, continúan apoyando el valor de la terapia combinada usando bajas dosis de radioterapia y quimioterapia con multiagentes para dar una mejor sobrevida.
El comité multi-institucional, multi-nacional para la terapia combinada en niños con Enfermedad de Hodgkin apoya nuevos estudios basados en el estadiaje no quirurgico solo que adaptados al riesgo clinico y eso separa a los pacientes en estadio I y II con características favorables de los pacientes con enfermedad avanzada y de características no favorables, estableciendo a la terapia adaptada al riesgo del paciente.
El grupo de Stanford/Dana Farber/St jude redefinieron sus protocolos adaptados al riesgo en tres grupos de riesgo, para los grupos de riesgo favorables requerian un estado clinico estadio I-IIA con masa mediastinal <1/3 y <3 regiones nodales involucradas, sin bulky nodal. Grupo intermedio incluye a estadios I-IIA con masa mediastinal ≥ 1/3, bulky nodal y≥3 areas nodales involucradas, estadios IIIA. Alto riesgo es reservado para pacientes con estadio IIB, IIIB, y IV.
En 1975, Bonadonna y sus colegas introdujieron el regimen ABVD( ) para pacientes que recurrian después del uso de MOPP. Ademas el grupo de milan empezo a compara MOPP y ABVD usando 3 ciclos para una de la combinación de estas drogas, seguida de un campo extenso de radioterapia y nuevamente otros 3 ciclos adicionales de la misma quimioterapia . Esta comparación demostro una significante superioridad para ABVD con FFP de 63% para MOPP y 81% para ABVD. Sabiendo que ambos regimes son altamente activos para esta enfermedad y no tienen toxicidad sobrelapadas, fue importante empezar a trabajar en los regimes hibridos.
Investigadores de Vancouver y Milan independientemente designaron 2 hibridos de MOPP y ABVD in orden de probar la hipótesis prospectiva de Goldie-coldman. El grupo NCI-C comparo el MOPP-ABVD alternando MOPP/ABVD en pacientes con estadio avanzado IIIB y IV de EH. A los 5 años no hubo una diferencia significativa en la sobrevida global entre los dos brazos del estudio; pero es importante mencional que el brazo hibrido tuvo mas toxicidad hematologica y no hematologica. Como consecuencia de estos estudios secuenciales de comparación primero el CALGB, como fue reportado por Canellos y luego mas tarde por North American Intergroup concluyeron que ABVD solo es igualmente efectivo como MOPP/ABVD hibrido pero menos toxico y en todas la combinaciones son mas efectivos que MOPP solo. Algo mas ABVD solo tiene menos toxicidad aguda, especialmente no esterilidad y menos o no leucemia mieloide aguda secundaria a tratamiento, pero se debe tener en mente la cardiotoxicidad por doxorubicina y efectos secundarios pulmonares devido a bleomicina si se dan mas de 6 ciclos y se consolida con radioterapia. Importante mencionar que es internacionalmente aceptado que ABVD sea el regimen estandar contra cualquier combinación de drugas experimentales para EH ( ). Hay nuevos regimenes que se estan usando en estadios avanzados de EH (IIB, IIIB, y IV) uno de ellos es el Stanford V , una combinación de 7 drogas, fue desarrollado para una corta duracion, reducir el grado de toxicidad incluyendo doxorubicina, vinblastina, mostaza, bleomicina, vincristina, etoposido, y prednisona. El regimen se da en 12 semanas,

y se consolida con radioterapia a areas de bulky inicial y de respuesta parcial al final del tratamiento( ). Como fue en el regimen MOPP donde la Mostaza fue substituida por ciclofosfamida COPP, han habido sugerencias para esta substitución en el Stanford V. La Freedom from progresión estimada a 5 años fue de 89% y el overall survival fue de 96% con una mediana de observación de 5.4 años. En Guatemala se ha usado el Stanfor v desde el año 2002 y se tienen 7 pacientes fuera de tratamiento con un EFS de 100% a 2 años de seguimiento( ). Ademas en 1992, el GHSG designo el BEACOPP que usa drogas similares a COPP/ABVD, excluyendo el velban y la dacarbazina y agregando etoposido, tratando de aumentar la eficacia de dos formas: dosis-densidad y dosis-intensidad. Luego el GHSG designo un estudio de 3 ramas comparando COPP/ABVD, un BEACOPP Standard y un BEACOPP escalado en pacientes con EH avanzada. Radioterapia se dio de consolidación a las areas donde habia bulky al diagnostico (30Gy) y a areas residuales (40Gy) después de 8 ciclos de quimioterapia. En el informe final en Junio 2001 donde OS para COPP/ABVD fue de 83%, para BEACOPP basico fue de 88% y para BEACOPP escalado 91 %. Es importante mencionar que son estudios hechos en pacientes adultos y que a pesar de su gran efectividad tiene tambien un alto incremento en la toxicidad.
Este progreso que se esta teniendo en Enfermedad de Hodgkin pediátrico, ha traído un 19% de incremento en la cura desde 1969 para el presente. El control adecuado, la epidemiología, y los resultados finales reportan un 94% de relativa survival rate a 5 años en niños con Hodgkin en USA. La enfermedad de Hodgkin tiene la sobrevida más alta en niños con cancer.
La Enfermedad de Hodgkin (EH) sigue siendo una enfermedad controversial, especialmente en cuanto a la mejor manera de hacer la estadificación y tratamiento (1). En países como los nuestros en los cuales los recursos económicos son limitados y hay un alto índice de abandono del tratamiento (dato suministrado en el 2do curso de la MISPHO), sobre todo si esta terapia es intensa y conlleva una morbi-mortalidad aumentada, es importante tener un plan de tratamiento adaptado lo mejor posible a nuestra realidad. Por esto, se inició el primer protocolo Centro Americano de Linfoma de Hodgkin (EH CA 1-99), en el cual se utilizó quimioterapia con la combinación COPP para estadios tempranos y COPP/AVB para tardíos. La radioterapia se dejó sólo para los casos de enfermedad residual o recaída. Este estudio se basó en la experiencia previa reportada por Costa Rica y Nicaragua (2,3)
En el EH CA 1-99 se trataron 198 pacientes de cuatro países Centroamericanos, observamos datos importantes como la edad al diagnostico , el 60% fueron pacientes menores de 10 años, con una predominacia en el sexo masculino, ademas la histología predominante fue la celularidad mixta, estos datos son muy diferentes a los reportados por paises desarrollanos; se presentaron 22 abandonos, 25 recaídas y 7 fallecidos. Durante este periodo fue claro que se requería una mejor categorización de la clasificación de los casos en A o B y una mejor estratificación de los pacientes en grupos de riesgo. Además fue necesario no eliminar completamente la radioterapia como parte del tratamiento primario, ya que en algunos casos es imposible evitarla si se pretende que los niños no recaigan. El EH CA 1-99 fue detenido en el año 2003, la principal razón para la suspensión del estudio fue la no disponibilidad de Procarbazina por algunos de los países participantes, y este

medicamento fue considerado esencial en el esquema usado. Para el nuevo estudio se han tomado en consideración todos estos puntos.
3- DISEÑO DEL ESTUDIO
Este es un estudio prospectivo no randomizado sobre la epidemiología, estadio, evaluación temprana y resultado del tratamiento de la Enfermedad de Hodgkin en niños. Participarán los países de Guatemala, Honduras, El Salvador, Nicaragua, Costa Rica , Panamá , Republica Dominicana y Argentina. Todos los pacientes serán tratados con un protocolo común luego de comprobación histológica del diagnóstico. El análisis histopatológico será hecho en cada país por el grupo correspondiente, aunque se tratará de implementar una revisión centralizada, con un arbitraje por parte del St. Jude Children´s Hospital en los casos de desacuerdo o duda.
El estudio está diseñado para tratar a los pacientes de acuerdo a tres grupos de riesgo con quimioterapia solamente, o quimioterapia más radioterapia de acuerdo a la situación que presenten. Además se valorará la respuesta completa en forma temprana con la terapia inicial (ver sección de plan de tratamiento).
4- ELEGIBILIDAD DE PACIENTES
Todos los niños entre 0 y 17 años 364 días con EH comprobada histológicamente y previamente no tratados, podrán entrar al estudio. El estadío de los pacientes se hará de acuerdo a la clasificación de Ann Arbor (Apéndice III y sección de estadío). El diagnóstico se deberá hacer siguiendo los lineamientos establecidos en el protocolo, esto es especialmente importante para aquellos niños que fueron inicialmente investigados en otro hospital, pero en los que no se inició tratamiento y fueron referidos al centro participante. Al paciente también se le pudo haber realizado la biopsia en otro hospital diferente al centro donde recibirá el tratamiento, pero la histología debe ser confirmada por el equipo tratante. El intervalo entre la cirugía y el inicio de la quimioterapia debe de ser tan corto como sea posible. No serán elegibles aquellos pacientes que hayan tenido tratamiento previo o que no tengan diagnóstico histológico de EH o que se utilice una estadificación quirúrgico patológica en lugar de clínica. Los pacientes que abandonen tratamiento podrán ser tomados en cuenta en la evaluación de la epidemiología de la región, así como la toxicidad del tratamiento hasta el momento en que lo recibieron.
5- FACTORES PRONÓSTICOS
Los principales factores pronósticos están relacionados con el estadío y la presencia o no de síntomas agregados. Los niños que no presentan sintomatología se clasificarán como A y aquellos que tengan uno o más de los síntomas presentes se clasificarán como B.

El pronóstico está relacionado con la extensión de la enfermedad: Estadíos I, II, III y IV. Cada uno de estos se dividirá en A o B para especificar la ausencia o presencia, respectivamente, : pérdida de peso mayor de 10% en los seis meses precedentes sin causa aparente, fiebre de 38°C o más sin explicación y sudoración nocturna profusa.
6- DIAGNÓSTICO
El diagnóstico de la EH se hará mediante la historia clínica completa, prestando especial interés a los siguientes aspectos:
1- Presencia o no de síntomas (B) 2- Si hay alguna malformación congénita.3- Antecedente de linfomas u otros tumores en la familia.
Examen físico completo, incluyendo medición de los ganglios linfáticos. El estado nutricional o grado de malnutrición es especialmente importante, por lo que todo paciente deberá ser seguido por el departamento de nutricion de cada hospital y tener peso, talla , área de superficie corporal, albumina , pliegue y circunfencia de brazo antes de iniciar la terapia.
Laboratorio: Hemograma Velocidad de Eritrosedimentación (VES) Función hepática ( BBSS total, directa, indirecta, TTSS, gglutamil transferasa) Función renal ( creatinina, BUN, uroanalisis, filtración glomerular –formula swartz-) Fosfatasa alcalina Rx de tórax
TAC de cuello ( con medidas recomendadas). Ver apéndice (importante seguir especificaciones)
TAC de tórax ( con medidas recomendadas). Ver apéndice (importante seguir especificaciones)
TAC de abdomen y pelvis ( con medidas recomendadas). Ver apéndice ( importante seguir especificaciones)
Ecocardiograma antes de iniciar la terapia y al final de la misma. Funcionalidad endocrinológica de base. Funcionalidad Respiratoria.

Biopsia de Médula ósea (MO) en una o más sedes ( espina iliaca postero –superior o cresta iliaca) en todo paciente con síntomas B no importando estadio.
Gammagrafía ósea ( en niños con sospecha de metástasis óseas.) Biopsia de ganglio linfático y/ o de otra sede donde se sospeche enfermedad.
No se hará en ningún paciente al inicio laparotomía exploradora ni linfangiografía.
7- ESTADIFICACIÓN
Se hará un estadío clínico en todos los casos y se usará la clasificación de Ann Arbor (4), además se realizará una clasificación en A o B de acuerdo a la presencia de síntomas o no al diagnóstico.
Estadío I: Compromiso de 1 región linfática o de 1 órgano o sitio extralinfático.
Estadío II: Compromiso de 2 o más regiones linfáticas en el mismo lado del diafragma o compromiso localizado de 1 órgano o sitio extralinfático y 1 o más regiones linfáticas del mismo lado del diafragma.
Estadío III: Compromiso de regiones linfáticas en ambos lados del diafragma, los cuales pueden acompañarse de afección del bazo o compromiso localizado de 1 órgano o sitio extralinfático o ambos (bazo + órgano extralinfático).
Estadío IV: Enfermedad difusa o diseminada de 1 o más órganos o tejidos extralinfáticos con o sin compromiso de ganglios linfáticos.(medula osea, pulmon, higado,etc)
CRITERIOS PARA LA MEDIDA DE LA MASA MEDIASTINICA
Masa Mediastínica positiva se entiende como masa anterior la cual en su largues máxima es superior a 1/3 del diámetro torácico medido a nivel de D5 (M/T > 0.33). Esta medida se realiza mediante la radiografía de tórax antero-posterior.
8- PATOLOGÍA
El esquema de clasificación morfológica más ampliamente aceptado es el sistema de clasificación de Rye (5), el cual define 5 subtipos histológicos:
A- Predominio linfocítico (PL): Se observa en 10 a 15% de los casos y es más común en varones jóvenes. También se ve más en los casos de enfermedad localizada. La

arquitectura del ganglio puede estar parcial o completamente destruida y podría ser difícil su diferenciación con una hiperplasia reactiva, debido a su característica proliferación de linfocitos de apariencia benigna, es importante demostrar con CD 20 positivo.
B- Celularidad mixta (CM): Las células de Reed-Sternberg son más comunes en la CM. Este subtipo se observa en cerca de un 30% de los casos y es más común en niños de 10 años o menos y en aquellos con infección por el virus de inmunodeficiencia humana (HIV). Además se observa en enfermedad avanzada y con extensión extranodal.
C- Esclerosis nodular (EN): Este es el subtipo más común de todos, afectando cerca de un 40% de los niños y 70% de los adolescentes. Es caracterizado por ganglios con cápsula gruesa y bandas de colágeno que dividen el tejido. En la EN se afecta más los ganglios linfáticos cervicales bajos, supraclaviculares y mediastinales.
D- Depleción linfocítica (DL): Es muy rara en niños y muchos casos reportados en el pasado pueden en realidad haber sido linfomas difusos de células grandes (1). La presencia de numerosas células reticulares malignas bizarras, muchas células de Reed-Sternberg y pocos linfocitos, caracterizan este subtipo. Pacientes con DL frecuentemente se presentan con enfermedad avanzada y compromiso de MO y hueso.
9-MEDICAMENTOS
9.1 VINBLASTINA
Fuente y farmacología:La Vinblastina es un alcaloide extraído de la Vinca Rosea. Se une en forma irreversible a los microtúbulos y proteínas causando un arresto en metafase. Puede también bloquear la utilización celular de ácido glutámico, produciendo una inhibición de la síntesis de purina y la formación de urea a través del ciclo del ácido úrico. Es un medicamento ligado a las proteínas y penetra poco al sistema nervioso central. El metabolismo en el hígado es muy importante, produciendo un metabolito, la deacetil vinblastina, la cual es más activa que el medicamento original. La eliminación principal es a través de la bilis, por ende, las dosis deben ser ajustadas a pacientes con compromiso hepático (bilirrubina > a 3 mg/dl).
Presentación y estabilidad:La Vinblastina se encuentra en frascos de 10 ml que contienen 1mg/ml de Vinblastina en solución. También se puede adquirir la Vinblastina como polvo blanco liofilizado en frascos de 10 mg del medicamento. Los frascos que no hayan sido abiertos y el polvo

liofilizado deben mantenerse bajo refrigeración. Al medicamento en polvo se les agrega 10 ml de solución de cloruro de sodio para dar una concentración final de 1 mg/ml, y ya disueltos, el medicamento puede refrigerarse durante 28 días sin pérdida de su potencia. Las dosis pueden ser disueltas posteriormente en solución con dextrosa al 5% o solución salina al 0,9%.
ToxicidadLa dosis límite por toxicidad es la mielosupresión. Otras toxicidades reportadas incluyen la alopecia, náuseas y vómitos, anorexia, diarrea, o constipación. Vinblastina puede producir importante daño tisular si ocurre extravasación. Raramente produce neurotoxicidad, pero si ésta ocurre puede dar neuropatía periférica, ausencia de reflejos osteotendinosos, debilidad, y dolor mandibular. Aun así, la toxicidad es mucho menor si se compara con la Vincristina. Otras toxicidades reportadas incluyen crisis convulsivas, infarto del miocardio, estomatitis, y fotosensiblidad.
Dosis y vía de administración
La Vinblastina es administrada en forma intravenosa a 6 mg/m2 por dosis en los días 1, semana 1, 3, 5, 7, y 9 de esquema Stanford V modificado.
9.2 DOXORRUBICINA (Adriamicina®)
Fuente y farmacología:La Doxorrubicina es un antibiótico antracíclico producido por el Streptomyces peucetius.Tiene su efecto antitumoral a través de diferentes vías. Primeramente la Doxorrubicina se intercala entre los pares de base del ADN causando una disrupción de la función del ADN e inhibición de la síntesis del ARN. Adicionalmente produce una inhibición de la topoisomerasa II, la enzima responsable de separar ambas cadenas de ADN en la replicación. Finalmente, la Doxorrubicina lleva a una reducción enzimática que genera moléculas altamente reactivas como por ejemplo radicales libres tipo hidroxilos, que se cree son responsables de la toxicidad cardíaca del medicamento, además de tener acción antitumoral. La Doxorrubicina no tiene especificidad de fase en el ciclo celular. El medicamento se distribuye ampliamente en los tejidos y el plasma, pero no cruza la barrera hematoencefálica en forma importante. La Doxorrubicina es metabolizada a doxorubicinol, que se piensa que es el metabolito activo más importante, y aglicones. Aproximadamente el 80% de la Doxorrubicina y sus metabolitos son excretados por las bilis y las heces, el restante es excretado por la orina. Por esto, las dosis en pacientes con alteraciones en la función hepática (bilirrubina > 1,2 mg/dl) o en la función renal (creatinina > 3 mg/dl) deben ser reducidas.
Presentación y estabilidadLa Doxorrubicina viene en presentaciones de 10 mg, 20 mg, 50 mg y 200 mg así como en una solución rojo-anaranjada a 2 mg/ml. Estos frascos deben ser guardados bajo refrigeración. También puede venir en un polvo liofilizado rojo-anaranjado de 10 mg, 20 mg, 50 mg y 150 mg los cuales deben ser guardados bajo temperatura ambiente. Ambos productos deben protegerse de la luz. La Doxorrubicina liofilizada puede ser reconstituida

adicionando 5, 10, 25, 50, o 75 ml de solución salina al 0,9% respectivamente a los frascos de 10 mg, 20 mg, 50 mg y 150 mg para producir una concentración final de 2 mg/ml. Diluyentes bacteriostáticos no son recomendables. Después de la agregación de la solución salina, la solución restante debe ser protegida de la luz y es estable durante 7 días si es almacenada a temperatura ambiente, o durante 15 días si es refrigerada.
ToxicidadToxicidades dosis limitantes incluyen mielosupresión y cardiotoxicidad. Dos formas de cardiotoxicidad pueden ocurrir: aguda o crónica. La toxicidad aguda puede verse con arritmias, bloqueos cardíacos o pericarditis, y pueden ser fatales. La forma crónica está relacionada a la dosis acumulada y es caracterizada por fallo cardiaco. Radioterapia mediastinal o el uso de otras drogas cardiotóxicas pueden aumentar el riesgo de la cardiotoxicidad. En general, no deben excederse dosis acumuladas de 450-550 mg/m2. Otras toxicidades incluyen náuseas, vómitos, mucositis, alopecia, diarrea, y coloración rojiza de la orina y otros fluidos. Daño severo de tejido y necrosis pueden ocurrir si ocurre extravasación. Raramente reacciones alérgicas pueden ocurrir con la Doxorrubicina.
Dosis y vía de administración Se administra intravenosa a 25 mg/ m2 por dosis en día 1 de las semanas 1, 3,5,7, y 9 del Stanford V. 9.3 PREDNISONA
Fuente y farmacología La Prednisona es un análogo de la hidrocortisona, la hormona adrenal corporal que se presenta como un polvo blanco o amarillento. La droga se une a los receptores de esteroides de las membranas nucleares, comprometiendo la mitosis celular e inhibiendo la síntesis de proteínas. La Prednisona también tiene un importante componente antinflamatorio y suprime el sistema inmune. La Prednisona es bien absorbida oralmente, y es convertida en prednisona, el metabolito activo, en el hígado. Posteriormente, la prednisona es metabolizada a otros compuestos en el hígado. Los metabolitos son excretados principalmente en la orina.
Presentación y estabilidadLa Prednisona viene en tabletas de 2.5, 5, 10, 20, 25, y 50 mg. Adicionalmente viene en una solución de 1 mg/ml. Todas las presentaciones pueden almacenarse a temperatura ambiente.
ToxicidadLos efectos adversos de la Prednisona varían dependiendo de la duración del tratamiento. Los efectos adversos que pueden ocurrir a corto plazo incluyen retención de sodio y agua asociando hipertensión, úlcera péptica con una posible perforación o hemorragia, alteraciones en la cicatrización, aumento en la susceptibilidad de infecciones, inestabilidad emocional, insomnio, aumento del apetito, incremento de peso, náuseas, acné, e hiperglicemia. Otros efectos adversos que pueden ocurrir incluyen pancreatitis, reacciones de hipersensibilidad, y necrosis aséptica. Los efectos adversos asociados al uso prolongado incluyen cataratas, aumento de la presión intraocular y glaucoma asociado, desarrollo de un

estado cushinoide, fracturas, irregularidades menstruales, supresión del crecimiento en niños, inhibición adrenal o pituitaria secundaria particularmente en tiempos de estrés (trauma, enfermedad o cirugías), osteoporosis, y gasto muscular.
Dosis y vía de administraciónSe administra oralmente a 40 mg/m2 por dosis (20 mg VO TID máxima dosis) cada otro día en las semanas 1-10 del protocolo Stanford V y disminuyendo la dosis a 10 mg cada otro día en las semanas 11-12.
9.4 CICLOFOSFAMIDA (Cytoxan®)
Fuente y farmacologíaLa Ciclofosfamida es un derivado de la mostaza nitrogenada. Actúa como un agente alcalinizante que causa bloqueo cruzado de cadenas de ADN al unirse con los ácidos nucleicos y otras estructuras intracelulares, interfiriendo así con la función normal del ADN. La droga no es fase específica en el ciclo celular. La Ciclofosfamida es bien absorbida vía oral con una biodisponibilidad de más del 75%, pero es una pro droga que requiere activación. Ésta es metabolizada por oxidasas en el hígado para producir hidroxiciclofosfamida, encontrándose en equilibrio con aldofosfamida. Aldofosfamida se rompe espontáneamente dando mostaza de Ciclofosfamida, que es considerado el metabolito más activo, y acroleína. Adicionalmente, la 4-hidroxiciclofosfamida puede metabolizarse a 4-ketociclofosfamida, y aldofosfamida a carboxifosfamida que son considerados metabolitos inactivos. La Ciclofosfamida y sus metabolitos son excretados principalmente en la orina. Ajustes de dosis deben hacerse en pacientes con aclaramiento de creatinina menores de 50 ml/min.
Presentación y estabilidadLa Ciclofosfamida está disponible en tabletas de 25 y 50 mg. También se encuentra en frascos de 100, 200, 500, 1000, y 2000 mg de la droga liofilizada, y 75 mg de manitol por cada 100 mg de Ciclofosfamida. Ambas presentaciones pueden almacenarse a temperatura ambiente. La droga liofilizada puede ser reconstituida con 5, 10, 25, 50, o 100 ml de agua estéril respectivamente para obtener una concentración final de 20 mg/ml. Las soluciones reconstituidas pueden ser disueltas posteriormente ya sea en solución glucosada al 5% o solución salina al 0,9%. Las soluciones disueltas son estables durante 24 horas a temperatura ambiente o durante 6 días si son refrigeradas, pero es recomendable que sean utilizadas en las primeras 24 horas posteriores a la preparación.
ToxicidadLas toxicidades dosis limitantes de la Ciclofosfamida son supresión de la médula ósea y toxicidad cardiaca. Toxicidad cardiaca es generalmente manifestada como insuficiencia cardiaca congestiva, necrosis cardiaca, o miocarditis hemorrágica, y puede ser fatal. Cistitis hemorrágica puede ocurrir y es necesario terapia adyuvante. La incidencia de cistitis hemorrágica se relaciona con la dosis utilizada y la duración del tratamiento. Aumento en la ingesta de líquidos y la administración de Mesna disminuye la incidencia y severidad de la misma. Otras toxicidades reportadas incluyen náusea, vómitos, diarrea, anorexia, alopecia, inmunosupresión, y esterilidad. Fibrosis pulmonar, secreción

inadecuada de hormona antidiurética, anafilaxis y neoplasias secundarias han sido reportadas.
Dosis y vía de administración Se administra en forma intravenosa a 600 mg/m2 por dosis en los días 1 y 8 de esquema COP.
9.5 VINCRISTINA (Oncovin®)
Fuente y farmacologíaLa Vincristina es un alcaloide obtenido de una planta (Vinca rosea). Se une a las proteínas de los microtúbulos causando una inhibición de la metafase. Este medicamento tiene una pobre penetrancia en el líquido cefalorraquídeo ya que aproximadamente un 75% de la droga está unida a las proteínas. Un metabolismo extenso ocurre en el hígado. La excreción es principalmente a través de la bilis y las heces. Se recomienda una disminución de la dosis en pacientes con bilirrubinas de más de 3 mg/dl.
Presentación y estabilidadLa Vincristina viene en frascos de 1 ml, 2 ml, y 5 ml. Cada ml de solución contiene 1 mg de Vincristina y 100 mg de manitol. Adicionalmente, ácido acético y acetato de sodio son agregados para el control del pH. Los frascos deben ser refrigerados y protegidos de la luz. También se puede diluir la droga con solución glucosada al 5% o solución salina al 0,9%.
ToxicidadLa toxicidad dosis limitante es la neurotoxicidad que se caracteriza por constipación, y/o íleo paralítico, ptosis, parálisis de cuerdas vocales, debilidad, dolor mandibular, dolor abdominal, neuropatías periféricas, y pérdida de los reflejos osteotendinosos. La neuropatía periférica es generalmente la primera manifestación de neurotoxicidad y es inicialmente reversible. Otros efectos adversos comúnmente reportados son la alopecia, náuseas, y vómitos. La Vincristina puede causar daño tisular severo si ocurre extravasación. Otros efectos adversos menos frecuentes incluyen el síndrome de secreción inadecuada de hormona antidiurética, mielosupresión, estomatitis, crisis convulsivas, alteración del estado mental, anafilaxis, cefalea, y atrofia óptica. Es importante conocer que una disminución de la dosis puede ser necesaria en pacientes menores de 1 año de edad (dosis por kg en lugar de por metro cuadrado se han recomendado en infantes para así disminuir la toxicidad).
Dosis y vía de administración Se administra a 1,4 mg/m2 (2 mg como dosis máxima) en día 1, semanas 2,4,6, 8, y 12 del esquema Stanford V.
9.6 BLEOMICINA (Blenoxane®)
Fuente y farmacologíaLa Bleomicina es un antibiótico antitumoral producido por la fermentación de Streptomyces verticillus. Se piensa que actúa inhibiendo la incorporación de timidina en el ADN, inhibiendo así la síntesis de ADN. También se ha observado que produce una inestabilidad y rompimiento de las cadenas de ADN. Actúa principalmente en las fases G2

y M1 del ciclo celular. Es poco absorbido a través del TGI, por lo que se debe administrar en forma parenteral. Aproximadamente un 50-70% de la dosis es excretada sin cambios por los riñones, y el restante es hidrolizada por una aminopeptidasa intracelular, encontrada en la mayoría de tejidos. La Bleomicina tiene una vida media de eliminación de aproximadamente 3 a 5 horas en pacientes con una función renal normal. Por esto, la dosis del medicamento debe ser reducida en pacientes con alguna disfunción renal.
Presentación y estabilidadLa Bleomicina se encuentra en frascos de 15 IU y 30 IU de sulfato de Bleomicina, un polvo liofilizado de color blanco o amarillento que deben ser guardados bajo refrigeración. El contenido de cada frasco debe ser diluido con 1-5 ml o 2-10 ml respectivamente de solución salina al 0,9% (no se debe diluir en solución glucosada porque puede haber disminución de la potencia). Las soluciones diluidas son estables por 24 horas a temperatura ambiente
Toxicidad
Efectos adversos pueden incluir náusea, vómitos, anorexia, alopecia, rash, hiperpigmentación y frialdad de la piel. Menos comúnmente asociados se encuentran la nefrotoxicidad, la hepatotoxicidad, el infarto de miocardio y la mielosupresión. Reacciones anafilácticas son raras pero potencialmente fatales y pueden caracterizarse por fiebre alta, escalofríos, hipotensión y obstrucción de la vía aérea. Neumonitis puede ocurrir y puede progresar a fibrosis pulmonar. Esto ocurre generalmente con dosis acumulativas de más de 400 U (200 U/ m2) en adultos. Utilización de oxígeno puede aumentar la toxicidad pulmonar y debe usarse con precaución. Es recomendable que se realicen pruebas de función pulmonar durante el curso del tratamiento y hasta un año después de la finalización de éste.
Dosis y vía de administraciónSe administra en forma intravenosa a 5 U/ m2 (no hay dosis máxima) por dosis en el día 1 de las semanas 2, 4, 6, 8, 10, y 12 del Stanford V.
9.7 DACARBAZINA (DTIC-Dome®)
Fuente y farmacologíaLa Dacarbazina es un agente alcalinizante que causa cross-linking y rompimiento de las cadenas de ADN, inhibiendo así las síntesis de ADN y ARN. También se ha postulado que la Dacarbazina no es específico en el ciclo celular. La Dacarbazina es metabolizada en el hígado con una excreción renal del 35-50% en 6 horas, teniendo una vida media bifásica, inicial de 35 min y terminal de 5 horas. Solamente un 5 % de la droga está unida a las proteínas.
Presentación y estabilidadLa Dacarbazina se encuentra como droga liofilizada en frascos de 100, 200, y 500 mg. La solución diluida es estable por 8 horas a temperatura ambiente y por 72 horas si es

refrigerada (a 4 grados Celsius). La solución es de color amarillenta, y es indicativo de descomposición si se vuelve de un color rosado. Se debe almacenar sin ser expuesta a la luz ya que pierde rápidamente su actividad.
ToxicidadLa toxicidad de la Dacarbazina puede dividirse en efectos inmediatos o efectos retardados. Entre los efectos inmediatos que se observan están las náuseas, vómitos (que generalmente inician de 1 a 3 horas posterior a la inyección y pueden durar hasta 12 horas después), un síndrome pseudogripal caracterizado por fatiga, mialgias, malestar general, y fiebre. Muy raramente pueden ocurrir calor, alergias, eosinofilia, parestesias, y fotosensibilidad. Si hay extravasación del medicamento puede ocurrir dolor y daño local. Los efectos adversos que se podrían observar son la leucopenia, trombocitopenia, y eosinofilia. Raramente ocurre toxicidad renal o hepática, y esterilidad.
Dosis y vía de administraciónSe da a 375 mg/m² IV en infusión de 30 minutos en 50 a 100 cc de SG5%, días 1 y 14. (si < de 10 Kg la dosis es 2 mg/Kg/dosis) para el protocolo de ABVD.
9.8 ETOPOSIDO (VP-16) (Vepesid®)
Fuente y farmacologíaEl Etopósido es una epipodofilotoxina derivada de la Podophyllum pelatatum que se piensa que actúa principalmente inhibiendo la topoisomerasa II, rompiendo las cadenas de ADN. Este medicamento es específico para las fases G2 y S del ciclo celular. La absorción del Etopósido es aproximadamente de un 30-40%, pero es muy variable y muchas veces dosis dependiente. Se une fuertemente a las proteínas y es metabolizado por el hígado, incluyendo el metabolismo por el citocromo P450 3A. El Etopósido y sus metabolitos son excretados principalmente en la orina y en menor cantidad por las heces. A los pacientes con disfunción hepática, hipoalbuminemia, o insuficiencia renal se les debe ajustar la dosis.
Presentación y estabilidadEl Etopósido puede encontrarse en frascos de 100 mg, 150 mg, 500 mg, y 1000 mg en una solución de 20 mg/ml y 30 % de alcohol que pueden ser almacenados a temperatura ambiente. También puede encontrarse en cápsulas de 50 mg que deben ser almacenadas bajo refrigeración. La solución de Etopósido debe ser diluida en solución con dextrosa al 5% o en solución salina al 0,9% previa a la administración. Estas soluciones finales con concentraciones de 0,2 y 0,4 mg/ml son estables a temperatura ambiente por 96 horas y 24 horas respectivamente.
ToxicidadLa toxicidad dosis limitante es la mielosupresión. Náuseas, vómitos (usualmente de baja o moderada severidad), diarrea, mucositis (generalmente con dosis altas), alopecia, y anorexia son relativamente comunes. Hipotensión puede ocurrir con infusiones rápidas. Otros efectos adversos reportados incluyen: hepatitis, fiebre, escalofríos, anafilaxis, y neuropatía periférica. También una leucemia secundaria ha sido reportada.

Dosis y vía de administraciónSe administra en forma intravenosa a 60 mg/m2 (sin dosis máxima) por dosis en los días 1 y 2 de las semanas 3, 7, y 11 del protocolo de Stanford V modificado.
10.- PLAN DE TRATAMIENTO
El tratamiento de la enfermedad va a depender del estadío y de la presencia de síntomas (A o B). Basado en esto, se establecerán tres grupos principales de tratamiento (I, II y III).
Grupo I o de Bajo Riesgo: enfermedad en estadíos IA , II A supradiafragmático sin compromiso mediastinito o con M/T<0.33, sin compromiso de nódulos pulmonares y con < 4 sedes nodales de enfermedad, o los pacientes con estadio IA, IIA infradiafragmático con <4 sedes nodales de enfermedad; ninguno debe de tener Bulky nodal (definido como mayor de 6 cm en su diámetro transverso mas largo. ) Estos pacientes recibirán el protocolo TERAPEUTICO 1
Grupo II o de Riesgo Intermedio: Los pacientes no incluidos en el grupo I y en el grupo III recibirán el protocolo TERAPEUTICO 2
Grupo III o de Riesgo Alto: enfermedad en estadíos II B, III B, IV A y IV B (estadíos avanzados) recibirán el protocolo TERAPEUTICO 3.
10.1 Esquema terapeutico 1 (Bajo Riesgo)
Todos aquellos pacientes en el grupo I y de bajo riesgo recibirán este protocolo de tratamiento.CADA CICLO Adriamicina (ADRIA) 25 mg/m² IV en bolo, días 1 y 14. (Si < 10 Kg la dosis es 0.67
mg/Kg/dosis) Bleomicina (BLEO) 10 U/m² IV en bolo, días 1 y 14. (Si < de 10 Kg la dosis es 0.33
U/Kg/dosis) Vinblastina (VINBLA) 6 mg/m² IV en bolo, días 1 y 14. (Si < de 10 Kg la dosis es 0.2
mg/Kg/dosis) Dacarbazina (DTIC) 375 mg/m² IV en infusión de 30 minutos en 50 a 100 cc de SG5%,
días 1 y 14. (Si < de 10 Kg la dosis es: ¿ ? )

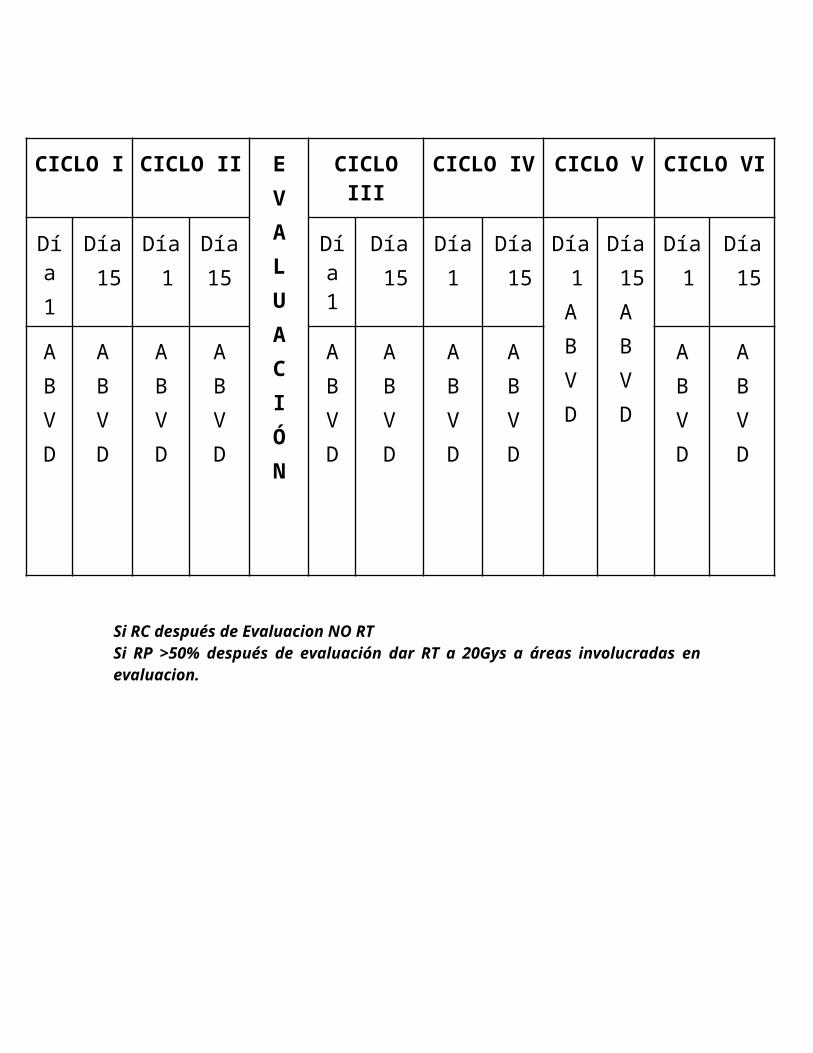
Este ciclo se repite cada 4 semanas por un total de 4 ciclos. La remisión se comprobará al final del mismo. En los pacientes con RP>50% al final de 4 ciclos se dará radioterapia a sedes Residuales a una dosis de 20 Gy cuatro semanas después de terminado el tratamiento con quimioterapia, pero previo se hara una discusión con el grupo de trabajo . Si después de 4 ciclos hay RP< 50% debe salir de este protocolo, y si hay RC se da por finalizado el tratamiento sin Radioterapia.
CICLO I CICLO II CICLO III CICLO IV EVALUACIÓN
Día 1
Día 15
Día 1
Día 15
Día 1
Día 15
Día 1
Día 15
ABVD
ABVD
ABVD
ABVD
ABVD
ABVD
ABVD
ABVD
RC después de evaluación NO RTPR>50% 20Gys áreas involucradas a la evaluación final.
Ciclo:Día 1 Día 14↑ ↑ABVD ABVD
El uso de antieméticos previo a la aplicación de la quimioterapia queda a criterio de cada médico tratante y a la disponibilidad de productos en cada centro.

10.2 Esquema terapeutico 2 (Riesgo intermedio)
Todos aquellos pacientes en el Grupo 2 o riesgo Intermedio, serán tratados con el siguiente protocolo:CADA CICLO Adriamicina (ADRIA) 25 mg/m² IV en bolo, días 1 y 14. (Si < 10 Kg la dosis es 0.67
mg/Kg/dosis) Bleomicina (BLEO) 10 U/m² IV en bolo, días 1 y 14. (Si < de 10 Kg la dosis es 0.33
U/Kg/dosis) Vinblastina (VINBLA) 6 mg/m² IV en bolo, días 1 y 14. (Si < de 10 Kg la dosis es 0.2
mg/Kg/dosis) Dacarbazina (DTIC) 375 mg/m² IV en infusión de 30 minutos en 50 a 100 cc de SG5%,
días 1 y 14. (si < de 10 Kg la dosis es: ¿ ? )
Este ciclo se repite cada 4 semanas por un total de 6 ciclos, se deben hacer estudios para valorar respuesta después de 2 ciclos, si RC (dar cuatro ciclos mas) y 4 semanas de terminada la quimioterapia (termina el tratamiento); si RP>50% después de 2 ciclos, dar 4 ciclos mas y 4 semanas después de terminada quimioterapia dar RT a sedes residuales a la evaluación temprana y sedes Bulky a 20 Gy y hacer estudios de fin de tratamiento 4 semanas después de terminada radioterapia, si persiste en RP después de RT hacer comprobación histológica de enfermedad por BIOPSIA. Para la radioterapia se usará Unidades de Cobalto 60 o Acelerador lineal de electrones, de acuerdo a disponibilidad del centro. En los pacientes que no responden al final del tratamiento, la conducta quedara a discreción de cada grupo tratante para escoger la terapia de salvamento.Es importante la discusión de cada caso, con el grupo de trabajo para definir la RC o RP después de 2 ciclos de ABVD y para definir la zona a irradiar.

CICLO I CICLO II EVALUACIÓN
CICLO III CICLO IV CICLO V CICLO VI
Día1
Día 15
Día 1
Día 15
Día 1
Día 15
Día 1
Día 15
Día 1ABVD
Día 15ABVD
Día 1
Día 15
ABVD
ABVD
ABVD
ABVD
ABVD
ABVD
ABVD
ABVD
ABVD
ABVD
Si RC después de Evaluacion NO RTSi RP >50% después de evaluación dar RT a 20Gys a áreas involucradas en evaluacion.

10.3 Esquema terapéutico 3 ( Alto Riesgo)
Este grupo corresponde a los pacientes de Grupo III o de Riesgo Alto. En estos niños se usará el esquema OEPA/COPDAC.
Tabla 1 OE*PA esquema
Prednisona/prednisolona60 mg/m2/day p.o. dividido en 3 dosis
día 1 – 15
Vincristina1.5 mg/m2 i.v., max. SD 2 mg
día 1 + 8 + 15
Doxorubicina40 mg/m2 as 1-6 horas infusión
día 1 + 15
Etopósido125 mg/m² as 1-2 horas infusión
día 1 – 5
Día 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15

Tabla 2 COPDAC esquema
Prednisona/Prednisolona40 mg/m2/day p.o. dividido en 3 dosis
día 1 – 15
Dacarbazina250 mg/m2 as 15 - 30-min. inf.
Día 1 – 3
Vincristina1.5 mg/m2 i.v. max. SD 2 mg
día 1 + 8
Ciclofosfamida500 mg/m2, 60-min. inf.
day 1 + 8 hidratacion endovenosa con solución glucosa/ salina en un rango de 3 l / m2/ 24 horas
Día 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15Se Hara una evaluación con imágenes a áreas involucradas a diagnostico después
del Segundo OEPA si RC dar RT a 20Gys a áreas involucradas a Diagnostico, y si RP>50% después de segundo OEPA dar 25 Gys a áreas involucradas al Diagnotico .
10.4 Guía para la Quimioterapia y terapia de soporte
Los ciclos se repiten cada mes en los grupos 1 y 2.
Si pacientes en estos grupo 1 y 2 tienen un atraso de administración de quimioterapia por cuentas bajas dos veces consecutivas se debe disminuir la dosis de los quimioterapéuticos a un 80% para el siguiente ciclo.
Se debe administrar la quimioterapia cuando los neutrófilos absolutos (ANC) sean > 1000/mm3, y las plaquetas > 100,000/mm3. Si estos están por debajo de estas cifras, se debe atrasar la administración 1 semana y evaluar la disminución de la dosis para evitar retrasos y mantener la intensidad..
Disfunción pulmonar sintomática: la presencia de disfunción pulmonar sintomática y un test de funcionalidad respiratoria alterado (en ausencia de infección) respecto al realizado al diagnóstico, llevará a la suspensión de la administración de BLEOMICINA.
Mucositis severa: en presencia de mucositis severa ( grado 3-4) el suministro de ADRIAMICINA será reducida a un 33% de la dosis inicial. Si después de la dosis

sucesiva no viene reportada toxicidad, la dosis deberá ser incrementada progresivamente del 15% hasta llegar a la dosis inicial.
Cardiotoxicidad: en presencia de evidencia clínica de cardiotoxicidad ( grado 3-4) será necesario suspender definitivamente la administración de ADRIAMICINA.
Neuropatía periférica e Ilio paralítico severo: en presencia de neuropatía periférica e ileo paralítico severo ( grado 3-4) la suministración de Vinblastina será reducida en un 33% respecto a la dosis inicial. Si la administración sucesiva no viene reportada de ulterior toxicidad, la dosis deberá ser progresivamente aumentada de un 15% hasta llegar a la dosis inicial. Si aun en los ciclos sucesivos se presenta una neurotoxicidad de grado severo, la suministración de Vinblastina tiene que ser suspendida definitivamente, y se deberá hablar con el coordinador del estudio.
Hipertensión grave: en presencia de hipertensión grave ( aumento ≥ 20 % de la diastólica y/o ≥40% de la sistólica) la dosis de Prednisona deberá ser reducida en un 33%. Si es necesario se debe reducir ulteriormente la dosis y recurrir a restricción de sodio y a los medicamentos antihipertensivos.
Factor estimulante de colonias se debe descontinuar si los neutrófilos absolutos son > 10,000/mm3 o > 1000/mm3 dos días seguidos 7 días después de administrar quimioterapia potencialmente mielosupresiva.
Todas las toxicidades deben reportarse, incluyendo el grado de la misma, usando la escala de toxicidad del NCI (Apéndice VI).
10. 5 Definición de respuesta al tratamiento
RESPUESTA COMPLETA (RC):Desaparición completa o > al 85% de todos los síntomas, signos clínicos e instrumentales (demostrables con examen físico o TC, Rx de tórax, biopsia ósea, etc.) de la enfermedad al diagnóstico.
RESPUESTA PARCIAL >50% (RP> 50%):Reducción > 50 % de la suma del producto de los diámetros de las lesiones medibles relevantes al momento del diagnóstico mediante examen físico e instrumental.
RESPUESTA PARCIAL ≤ 50% ( RP≤ 50%):Reducción ≤ 50% de la suma del producto de los diámetros de las lesiones medibles relevantes al diagnóstico mediante examen físico y instrumental.
FALTA DE RESPUESTA (FR):Permanencia de los signos y síntomas clínicos e instrumentales relevantes al momento del diagnóstico.

PROGRESIÓN DE LA ENFERMEDAD (PE):Progresión de los síntomas y signos clínicos e instrumentales relevantes al diagnóstico.
RECAÍDA:Presencia de enfermedad en sedes y/o extranodales (encontrada mediante examen físico o instrumental), no presente al diagnóstico, después de haber conseguido la remisión completa.
RECIDIVA:Presencia de enfermedad en sedes nodales y/o extranodales (encontrada mediante examen físico o instrumental) presente al diagnóstico, después de conseguir la remisión completa.
10.6- Directrices radioterapéuticas
La radioterapia (RT) será dada a las zonas afectadas en todos aquellos pacientes que no respondan a la quimioterapia o que presenten recaída. Cuando el área a irradiar esté en un lado del cuello, la RT debe comprender ambos lados del cuello. Cuidado particular se debe tener con la protección de la mandíbula, clavícula y también se debe proteger la zona de la columna en los casos de campos posteriores. Otro sitio importante de proteger es la zona genital: los testículos y ovarios en los casos que ameriten RT a las áreas cercanas (RT en Y invertida), la forma de proteger estos órganos, especialmente los ovarios, quedará a discreción de cada grupo (si el grupo prefiere hacer una laparotomía para movilizar los ovarios y protegerlos con el útero).
10.7 Inicio de radioterapia
La radioterapia se iniciará aproximadamente 3 a 4 semanas después de haber completado la última dosis de quimioterapia o posteriormente tan pronto se obtenga un recuento de neutrófilos absolutos > 1000/mm3, y plaquetas > 100,000/mm3.
Dosis
Para los pacientes del Grupo 1 que después de 4 ciclos de quimioterapia no alcancen la RC, pero tengan una RP≥50% RT a 20Gy a enfermedad residual, en fracciones de 150 cGy.
Para los pacientes del Grupo 2 que no alcanzaron una respuesta completa después de haber recibido 2 ciclos de quimioterapia deben completar la misma y luego 20 Gy en fracciones de 150 cGy cinco veces a la semana a áreas residuales al momento de la evaluación temprana después de 2 ABVD y Bulky; si alcanzaron la RC después de dos ciclos de quimioterapia, se completa la quimioterapia y no se da RT.
Definición de los campos

A- Anillo de Waldeyer: Cuando la enfermedad se localiza a nivel de los ganglios periauriculares, anillo de Waldeyer, o en los casos en donde se encuentren comprometidos ganglios linfáticos cervicales altos que no puedan ser cubiertos por un campo AP/PA de cuello.
B- Manto: Enfermedad supradiafragmática que involucra a nivel mediastinal, cervical, supraclavicular, infraclavicular, y axilar.
C- Manto modificado: Cuando se omite alguno de los nódulos linfáticos que se encuentran en el campo de manto (por ejemplo la zona axilar y/o infraclavicular).
D- Mini-manto: Enfermedad supramediastinal bilateral que involucra axilas, ganglios supraclaviculares, infraclaviculares, o cervicales, pero sin involucrar mediastino.
E- Mini-manto modificado: Enfermedad supramediastinal bilateral que no incluye alguno de las regiones que normalmente incluye el mini-manto.
F- Hemimini-manto: Enfermedad supramediastinal unilateral que involucra los ganglios axilares, supraclaviculares, infraclaviculares, o cervicales ipsilaterales con un mediastino normal.
G- Para-aórtico/esplénico: Enfermedad subdiafragmática a nivel esplénico o para-aórtico. El bazo se incluirá siempre en la enfermedad para-aórtica/peri-cava. Se incluirá al hígado si se comprueba que se involucra en la enfermedad.
H- Espada: Enfermedad que involucra ganglios para-aórticos y de la iliaca común y se extiende caudal de la articulación sacro-iliaca. El bazo será incluido en este campo, pero sin comprometer los ovarios y eliminar la necesidad de una ooforopexia. No se da ninguna radiación pélvica si los estudios de imágenes abdomino-pélvicos son negativos caudal al nivel S1.
I- Pelvis: Enfermedad a nivel de las áreas iliacas e inguino-femorales bilaterales.
J- Hemi-pelvis: Enfermedad a nivel del área iliaca e inguino-femoral unilateral si se comprueba que no hay compromiso del otro lado por estudios de imágenes abdomino-pélvicas.
K- Y-invertida: Enfermedad subdiafragmática en el bazo, hilio esplénico, áreas para-aórtica, pélvica y femoral.
Cabe recalcar que las zonas comprometidas no necesariamente serán irradiadas en forma simétrica:
Si solamente hay compromiso supraclavicular derecha, el tratamiento incluiría a nivel del cuello inferior derecho (bajo el hueso hioides) y axila superior (por encima del pliegue axilar para prevenir exceso de radiación de la mama cuando la axila es clínicamente negativa).

Para compromiso cervical por encima de la región supraclavicular puede limitarse la radiación a la zona supraclavicular.
El borde inferior del mediastino se limitará a nivel de T8 para los casos sin comprometer por debajo de la carina, o 2 cuerpos vertebrales o 5 cm por debajo del borde caudal de la enfermedad.
Cuando la enfermedad residual al evaluar la respuesta está limitada a la región por encima de la carina, el mediastino inferior se bloqueará al llegar a 15 Gy cuando está indicada una cobertura mediastinal total.
Un manto modificado puede usarse cuando se determina clínicamente que no hay compromiso ganglionar de alguna zona. La axila puede bloquearse para no dar exceso de radiación y evitar compromiso de la glándula mamaria.
En estadíos I-II de enfermedad mediastinal, el hilio bilateral debe ser tratado. En pacientes en estadío III, el área de tratamiento es una irradiación total
supradiafragmática seguida de irradiación del bazo y las zonas para-aórticas. Si hay compromiso de los ganglios iliacos comunes a nivel L4-S1, entonces el borde inferior del campo para-aórtico se coloca inferior de la articulación SI. Si hay compromiso de las zonas iliacas e inguinofemorales bilaterales, se irradiará toda la pelvis. Se tratará la hemi-pelvis si este compromiso es unilateral.
Radioterapia inmediata al diagnóstico o antes de la quimioterapia se dará a aquellos pacientes que se presenten con condiciones que ponen en riesgo su vida como por ejemplo: compresión de la vía aérea, compresión de la médula espinal, o enfermedad que interfiera con un tratamiento y seguimiento óptimo para el paciente.
Pacientes con enfermedad parenquimatosa pulmonar residual periférica al tiempo de evaluación de la respuesta recibirán 8 Gy en el pulmón comprometido bloqueando el otro pulmón para limitar la radiación a 100 cGy por fracción. Pacientes con evidente compromiso óseo en estudios por imágenes recibirán 25,5 Gy al sitio comprometido.
Se debe ofrecer ooforopexia previo a la irradiación pélvica a todas las mujeres que requieren esta radioterapia.
10.8- Directrices quirúrgicas
Todos los pacientes tendrán comprobación patológica de la enfermedad. El rol principal de la cirugía va dirigido a obtener el material necesario para hacer el diagnóstico histológico. En la mayoría de los casos esto se restringirá a una biopsia de uno o varios ganglios periféricos, dándole prioridad a los que clínicamente impresionen más representativos.
En aquellos pacientes con masas mediastinales o abdominales sin la presencia de adenopatías periféricas, será necesario hacer una toracotomía o una laparotomía para obtener la biopsia. La extensión de la resección en estos casos quedará a criterio del cirujano, tomando en consideración que sea lo suficientemente extensa para obtener una buena muestra para el diagnóstico.
La toma de muestra a través de punciones percutáneas, laparoscopía, toracoscopía será usada a discreción de cada grupo, siempre y cuando consigan el resultado deseado, el

diagnóstico. Cabe señalar que la toma de muestra con aguja fina no es útil en la EH y las muestras con el tipo de aguja conocido como “Tru - cut” van a representar una dificultad importante para la interpretación adecuada por parte del patólogo, por lo que no deben efectuarse estos procedimientos.
10.9- Evaluación de la respuesta al tratamiento
La evaluación durante el tratamiento se hará de acuerdo al esquema que se estableció en el rubro de evaluación de la respuesta.
La evaluación al finalizar el tratamiento se hará mediante un control en clínica de seguimiento cada mes durante el primer año, cada 3 meses por el segundo y tercer año, luego cada 6 meses durante el cuarto año y posteriormente visitas anuales.En cada visita el paciente tendrá: Examen físico completo, con control de peso, talla y desarrollo puberal. Además se debe hacer conciencia en las adolescentes que hayan recibido RT a mediastino sobre el riesgo de cáncer de mama en el futuro y la necesidad de valoraciones más frecuentes, incluyendo ultrasonido de mama y coordinación con hospitales de adultos donde seguirá control luego de salir del centro pediátrico.
Se harán en la visita mensual mínimo un examen físico, hemograma con V/S, funcionalidad renal y hepática, además se deben hacer estudios de tomografías de los lugares involucrados al diagnóstico cada tres meses durante el primer año, y luego cada seis meses durante el segundo y tercer año y una vez al año hasta completar cinco años.
III- REPORTE DE CASOS Y EVALUACION DEL PROTOCOLO
Nombre:__________________________________________________________________
Expediente_________ Sexo_______
Fecha de nacimiento__________ Edad__________
País___________________ Provincia______________________
Fecha inicio de Tx ____/____/____ Tiempo desde inicio de padecimiento_______________

Síntomas/Signos (A o B)
___________________________________________________________________________
Peso______Kg Talla______cm Superficie corporal______m²
Sitio tumor primario___________________________________________________________
Metástasis_______________________________________________________________
Estadío__________
Fecha cirugía inicial ____/____/____
Tipo de cirugía: Biopsia abierta ______ Laparotomía______ Toracotomía______
Histología__________________________________________________________________
Rx tórax____________________________________________________________________
TAC_______________________________________________________________________
Gamma_____________________________________________________________________
Otro_______________________________________________________________________
Seguimiento:
Remisión total._________ Remisión parcial________ Enfermedad progresiva__________
Fecha de remisión ___/_ ___/_ ___/ Fecha término de Tx ___ _/_ ___/_ ___
Fecha de recaída ____/__ __/__ _ Fecha de muerte ___ _/__ __/___ _
Causa de muerte _____________________________________________________________
Sobrevida____________ Condición actual_____________________________________

Tiempo de seguimiento________________________________________________________
Complicaciones______________________________________________________________
___________________________________________________________________________
Observaciones_______________________________________________________________
___________________________________________________________________________
IV- HOJA DE CONSENTIMIENTO
Estoy en pleno conocimiento de que mi hijo(a) _____________________________, tiene un tumor maligno llamado Linfoma de Hodgkin y se me ha indicado que debe ser tratado de acuerdo a un plan establecido, el cual puede incluir cirugía, quimioterapia y radioterapia, con el objetivo de curar la enfermedad.
Los riesgos de este tratamiento de acuerdo a los medicamentos a utilizarse incluyen: náuseas, vómitos, disminución de las defensas y plaquetas, anemia, mayor posibilidad de infecciones, pérdida de apetito, riesgo de esterilidad, principalmente en varones, posible retraso en el crecimiento, riesgo de problemas cardiacos y pulmonares y de una segunda malignidad. Todos estos riesgos del tratamiento serán observados por el equipo de trabajo muy de cerca con el fin de prevenirlos, cuando sea posible y tratarlos en caso de que ocurran. Ningún niño experimenta todas las complicaciones que se han escrito aquí, pero es frecuente que se presenten 2 o 3 de ellas. Efectos inesperados o desconocidos pueden ocurrir.
El beneficio de este tratamiento es el que puede aumentar la oportunidad de que el niño(a) se cure, sin embargo esto no se puede garantizar en todos los casos. Estoy consciente que de no recibir un tratamiento adecuado, la muerte de mi hijo sería inevitable. Entiendo que el equipo de salud que participa en este tratamiento ha escogido el más apropiado de acuerdo al nivel actual del conocimiento científico, para esta enfermedad.
Al firmar este documento, comprendo que la respuesta de mi hijo al tratamiento puede ser utilizada para reportar los resultados en publicaciones científicas, pero que él no será identificado en forma personal.
En caso que no quiera participar en este tratamiento puedo cambiar de idea y salir inmediatamente de él, sin que por esto pierda la relación con los médicos tratantes o el Hospital, quienes harán todo lo posible por tratar al paciente de la mejor manera. He leído toda la descripción del plan de tratamiento; cualquier cosa que yo no entendí me fue explicada por _________________________ y todas mis preguntas fueron contestadas a mi entera satisfacción.
Yo deseo que mi hijo(a) sea tratado de la manera que se me ha explicado.
Nombre__________________________________
Firma____________________________________
Dr.______________________________________
Firma____________________________________Fecha:

APENDICE DE ESPECIFICACIONES PARA LAS TOMOGRAFIAS
REQUERIMIENTOS PARA REALIZACION DE TOMOGRAFIAS
PLEASE CIRCLECT EXAM
SLICETHICKNESS
mmSPECIAL INSTRUCTIONS OR COMMENTS
IVCONTRAST
ml/kgORAL
CONTRAST
CHEST without 5 3.75mm for 6kg-9.5kg None None
CHEST with 5 3.75mm for 6kg-9.5kg 1.5ml/kg up to 100ml None
CHEST HI-RES 1.25 at 10 space Insp/Exp/Both None None
NECK NECK 5 3.75mm for 6kg-14.4kg 1.5ml/kg up to 100ml None
NECK & CHEST 5 3.75mm for 6kg-14.4kg 1.5ml/kg up to 55kg/ 2.0ml/kg above 55kg None
NCAP 5/5/5/5 3.75mm on neck and chest for 6kg-14.4kg 2.0ml/kg up to 150ml 3hr. 2hr. 30min.
CHEST & ABD 5/5 3.75mm for 6kg-14.4kg 2.0ml/kg up to 150ml 2hr 30min
ABD without 5 None None
ABD with 5 2.0ml/kg up to 150ml 2hr 30min
ABD/PEL with 5/5 2.0ml/kg up to 150ml 3hr 2hr 30min
CHEST / ABD / PEL with 5/5/5 3.75mm for 6kg-14.4kg 2.0ml/kg up to 150ml 3hr 2 hr 30 min
CHEST / ABD for Fungus 5/5 3.75mm for 6kg-14.4kg 2.0ml/kg up to 150ml None
CAF with PELVIS 5/5/5 3.75mm for 6kg-14.4kg 2.0ml/kg up to 150ml None
PELVIS without 5 None None
PELVIS with 5 2.0ml/kg up to 150ml None/ 3hr
RENAL STONE PROTOCOL 5 Low mAs None None
EXTREMITY 5 Check with Radiologist None/ 2.0ml/kg up to 150ml None
CT ANGIOGRAPHY Check with Radiologist 2.0ml/kg up to 150ml None
Three phase liver 5 30sec 70sec Delayed_______ 2.0ml/kg up to 150mL None
P.E. Protocol 1.25/1.25 Dual IV 2.0ml/kg up to 150ml None