Medicalum*Revista*de*Casos*Clínicos*para*Estudiantes*de...
52
Diagnóstico genético preimplantacional en pareja portadora de fibrosis quística. Brucelosis. Manejo clínico de la leucemia aguda mieloblástica y su diagnóstico diferencial. Insuficiencia renal crónica en Atención Primaria. Síndrome coronario agudo con elevación del segmento ST. Síndrome de Alport autosómico dominante. Ictus en el contexto de una fibrilación auricular anticoagulada. Pseudomixona peritoneal. página 5 9 14 20 24 30 37 46
Transcript of Medicalum*Revista*de*Casos*Clínicos*para*Estudiantes*de...

Diagnóstico genético preimplantacional en pareja portadora de fibrosis quística.
Brucelosis.
Manejo cl ínico de la leucemia aguda mieloblástica y su diagnóstico diferencial.
Insuficiencia renal crónica en Atención Primaria.
Síndrome coronario agudo con elevación del segmento ST.
Síndrome de Alport autosómico dominante.
Ictus en el contexto de una fibrilación auricular anticoagulada.
Pseudomixona peritoneal.
página
5
9
14
20
24
30
37
46

!!Medicalum*Revista*de*Casos*Clínicos*para*Estudiantes*de*Medicina*
Volumen(1,(número(1(
(
Medicalum,*2018*
(
Editores*
Dr.(Pedro(A.(Cascales(Campos(
D.(Juan(Manuel(Quiñonero(Rubio(
(
Diseño*y*maquetación:(Medicalum(
(
Proyecto*Medicalum*
San(Nicolás(7,(1º.(
Murcia,(España.(
Página(Web:(www.medicalum.com(
EHmail:([email protected](
(
ISSN:(en(tramitación.(
(
(
(
Esta( obra( está( editada( bajo( una( Licencia( de(
Creative( Commons( ReconocimientoHNoComercialH
SinObraDerivada(4.0(Internacional(
!!!
!

!!Dirección(Editorial(
Editor'Jefe'Dr.$Pedro$A.$Cascales$Campos$
$
Coordinación'Editorial'D.$Juan$Manuel$Quiñonero$Rubio
$Editores(Adjuntos(
Dr.$Joaquín$García.Estañ$López$Área$de$Educación$Médica.$Centro$de$Estudios$universitario$de$Educación$Médica,$Universidad$de$Murcia.$
Dr.$Pablo$Ramírez$Romero$Cirugía$General$y$del$Aparato$Digestivo,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
Dr.$Pedro$A.$Cascales$Campos$Cirugía$General$y$del$Aparato$Digestivo,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
Dra.$Beatriz$Febrero$Sánchez$Cirugía$General$y$del$Aparato$Digestivo,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
Dr.$Antonio$López$López.Guerrero$Neurocirugía,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
Dra.$Elisa$García$Vázquez$MI$–$Infecciosas,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
Dr.$Sergio$Manzano$Fernández$Cardiología,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
Dr.$Manuel$Moreno$Ramos$Reumatología,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
Dra.$María$José$Moreno$Martínez$Reumatología,$Hospital$Rafael$Méndez$(Lorca,$Murcia).$
Dr.$Juan$Antonio$Ortega$García$Pediatría,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
Dr.$Oscar$Girón$Vallejo$Cirugía$Pediátrica,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
Dr.$Anibal$Nieto$Díaz$Ginecología$y$Obstetricia,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
Dra.$Pilar$Marín$Sánchez$Ginecología$y$Obstetricia,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
Dr.$Guillermo$Carbonell$López$del$Castillo$Radiodiagnóstico,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
Editores(Asociados(
Dra.$Alida$González$Gil$Cirugía$General$y$del$Aparato$Digestivo,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
Dr.$Felipe$Alcolchel$Gago$Cirugía$General$y$del$Aparato$Digestivo,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
Dr.$Antonio$García$López$Neurocirugía,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
Dr.$Sergio$Alemán$Belando$Medicina$Interna,$Hospital$Universitario$Morales$Meseguer$(Murcia).$
Dr.$Antonio$Ortega$Sabater$Digestivo,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
Dr.$Ginés$Elvira$Ruiz$Cardiología,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
Dra.$Esther$Tobarra$Sánchez$Pediatría,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
Dra.$Encarnación$García$Campaña$Ginecología$y$Obstetricia,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
Dr.$Javier$Sánchez$Romero$Ginecología$y$Obstetricia,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
Dr.$José$Antonio$Villa$Carpes$Oftalmología,$Hospital$Clínico$Universitario$Virgen$de$la$Arrixaca$(Murcia).$
$'
'
'
$
$
!

! ! ! ! ! ! ! ! ! Editorial
¡Bienvenidos!
Es un auténtico placer para nosotros el poder presentaros este primer número de la Revista de Casos Clínicos para Estudiantes de Medicina, proyecto original de Medicalum.
Somos conscientes de que determinadas tendencias se ponen de moda con el paso del tiempo porque en realidad, dan respuesta a necesidades reales de una población. Esto es precisamente lo que ha ocurrido con la Educación Médica, que ha resurgido
Dr. Pedro A. Cascales Campos
Editor!Jefe!de!Medicalum!Revista!de!Casos!Clínicos.!Servicio!de!Cirugía!General!y!del!Ap.!Digestivo,!Hospital!
Clínico!Universitario!Virgen!de!la!Arrixaca!(Murcia).!Profesor!Asociado!en!la!Faculdad!de!Medicina!de!la!
Universidad!de!Murcia.
con fuerza especialmente en la última década, para enfocar los viejos problemas relacionados con la formación del estudiante de Medicina (que no de “MIRicina”). El ser “estudiante de Medicina” no debe ser visto como un mal que termina con un último suspiro antes de pasar a mejor vida, que puede interpretarse como la residencia y cuyo último suspiro puede verse como el examen MIR. Si la capacidad de trabajo y la determinación de un residente puede marcar su futuro profesional, malo será el no darnos cuenta que estos atributos ya pueden cultivarse durante la etapa de estudiante.
El presente número es el primero de una larga serie, que esperamos pueda contribuir a la formación de aquellas personas que en el futuro se encargarán de los cuidados de nuestra salud y la de nuestros familiares. Solamente desde este punto de vista, aun siendo un punto de vista legítimamente egoísta, esto es, el de que nos traten bien, merece la pena el esfuerzo.
La Revista de Casos Clínicos para Estudiantes de Medicina se complementa con otros proyectos que van encaminados a la mejora de la formación del estudiante de Medicina. La revista es realmente solo uno de los pilares del proyecto, que puede consultarse íntegramente en www.medicalum.com, y que se completa con un blog, un apartado amplio de material audiovisual (Aula Medicalum) y una biblioteca de casos clínicos, en continuo crecimiento, que esperamos satisfaga las necesidades teóricas básicas de muchos.
Somos conscientes de que la Medicina no se aprende solamente en los libros. No queremos virtualizar la realidad ni pretendemos sustituir la formación existente, aunque nos gustaría, sino complementarla. Nuestra visión va más allá, y deseamos que los estudiantes de Medicina vuelvan a poblar los hospitales de nuevo. No es esta una idea novedosa pero es pertinente actualmente ya que el sistema educativo, enfocado exclusivamente a superar ese último suspiro del estudiante, que es el MIR, ha convertido a éste en una especie en extinción dentro del ecosistema hospitalario. En cierto modo, el futuro de esta revista depende de vuestra presencia en los hospitales.
Murcia, 26 de octubre de 2018

!
www.medicalum.com
!
A PROPÓSITO DE UN CASO: DIAGNÓSTICO GENÉTICO PREIMPLANTACIONAL EN PAREJA PORTADORA DE FIBROSIS QUÍSTICA
García García, A. Mª.1; Martínez Morata, I.1; Sánchez Hernández, A.1; Tomás Martínez, G.1; Galvez Pradillo, J.2
(1): Facultad de Medicina, Universidad de Murcia (2): Servicio de Ginecología y Obstetricia, Hospital Clínico Universitario Virgen de la Arrixaca (Murcia).
VALORACIÓN Y JUSTIFICACIÓN El gran avance tecnológico en el campo de la reproducción asistida y en lo que respecta al estudio genético de embriones, nos lleva a la posibilidad de estudiar y prevenir enfermedades de las que conocemos su causa a nivel genómico, como la fibrosis quística. Distintos argumentos apoyan la realización de un diagnóstico genético preimplantacional (DGP) cuando los progenitores son portadores, como el hecho de ser una enfermedad sin cura y con gran afectación de la calidad de vida. Con esta técnica evitamos la transmisión de la enfermedad a la descendencia, las repercusiones psicológicas y el tratamiento crónico de elevado coste.
RESUMEN El diagnóstico genético preimplantacional (DGP) se basa en el análisis genético de embriones obtenidos por fecundación in vitro (FIV) o por inyección intracitoplasmática de esperma (ICSI). Está dirigido a parejas con antecedentes de enfermedad genética. En nuestro caso, se le realizó a una pareja portadora de la mutación de la Fibrosis quística que fue descubierto como hallazgo casual durante el estudio de esterilidad primaria en la Unidad de Disfunción Reproductiva del hospital donde se desarrolla el caso clínico. La fibrosis quística es una enfermedad genética autosómica recesiva de alta prevalencia en la raza caucásica (1/25 portadores) y que supone una esperanza de vida menor que en población sana, un elevado coste sanitario y una calidad de vida disminuida del paciente. Por estos motivos se justifica la realización de un cribado prenatal a todas aquellas parejas que se someten a un tratamiento de fecundación in vitro.
PALABRAS CLAVE Esterilidad, cribado preimplantacional, diagnóstico genético preimplantacional, fibrosis quística y coste efectividad.
ABSTRACT Preimplantation genetic diagnosis (PGD) is based on the genetic analysis of embryos obtained by in vitro fertilization (IVF) or by intracytoplasmic sperm injection (ICSI). It is aimed at couples with a history of genetic disease. In our case, a couple carrying the cystic fibrosis mutation was discovered as a casual finding during the primary sterility study in the Reproductive Dysfunction Unit. Cystic fibrosis is an autosomal recessive genetic disease of high prevalence in the Caucasian race (1/25 carriers) and that supposes a life expectancy lower than in a healthy population, a high health cost and a diminished quality of life of the patient. For these reasons, it is justified to carry out a prenatal screening to all those couples that undergo in vitro fertilization treatment.
KEYWORDS
Sterility, preimplantation screening, preimplantation genetic diagnosis, cystic fibrosis and cost effectiveness.
5

!
www.medicalum.com
!
INTRODUCCIÓN En la Unidad de Reproducción Asistida del Hospital Universitario Virgen de la Arrixaca se realiza por defecto en todas las parejas que acuden para someterse a un tratamiento de Fecundación in Vitro (FIV) un cribado de portadores de Fibrosis Quística. Debido a que no encontramos ninguna publicación científica que justificara la aplicación de este cribado en nuestro medio decidimos contactar con los responsables de la Unidad de Reproducción Asistida y de Genética Molecular, para que nos informaran de la evidencia científica en la que se habían basado para implantar esta estrategia preventivo-terapéutica en sus respectivos servicios. Ambos refirieron que la decisión se basó en la alta prevalencia de portadores en nuestro medio, así como, las graves consecuencias de desarrollar la enfermedad y el elevado coste, tanto en términos económicos como en calidad de vida, del tratamiento de estos enfermos. Por tanto, consideraban sostenible el coste de la realización del cribado de portadores a todas las parejas que se sometían a FIV, incluso si no existían antecedentes familiares de Fibrosis Quística en sus familias. Numerosos artículos científicos han demostrado la eficiencia del screening de portadores frente a los costes del tratamiento de un hijo nacido enfermo1.
CASO CLÍNICO Presentamos el caso de una pareja que fue remitida a la Unidad de Reproducción Asistida del Hospital Universitario Virgen de la Arrixaca por no conseguir embarazo tras dos años y medio de búsqueda.
La paciente era una mujer de 32 años sin antecedentes heredofamiliares de interés. Con relación a sus antecedentes personales presentaba hipotiroidismo en tratamiento con Levotiroxina sódica 25 mg. Era alérgica a Azitromicina. En lo relativo a hábitos tóxicos la paciente fumaba 2,5 paquetes/año. Presentaba un índice de masa corporal (IMC) de 21.
Antecedentes Ginecológicos:
-Menarquia a los 13 años con ciclo menstrual regular cada 28 días y de 3 días de duración.
-Análisis hormonal del 3º día de ciclo: LH de 7,1 mUl/ml; FSH de 5,8 mUl/ml ; estradiol de 49,4 pg/ml; hormona anti-mülleriana de 1,3 ng/ml ; TSH de 2,6 uUl/ml ; niveles de T4L de 1,6 ng/dl ; prolactina de 36 ng/ml .
-Ecografía vaginal: recuento de folículos antrales (RFA) de 12 (buena reserva ovárica entre 6-10 folículos antrales).
-Cariotipo: 46 XX. En las 20 metafases estudiadas no se observaron anomalías numéricas ni estructurales. No se detectan mosaicismos. Este estudio no excluye cuadros de origen monogénico o multifactorial.
-Análisis directo de 50 mutaciones frecuentes en el gen CFTR y la secuencia poliT en IVS8 mediante PCR-ARMS y análisis de fragmentos mediante electroforesis capilar. Se identificó la mutación N1303K en gen CFTR en heterocigosis. Se le diagnosticó como portadora de mutación asociada a Fibrosis Quística. La sensibilidad del estudio en la población española y en la población caucásica europea se sitúa en torno al 83%. El riesgo de ser portador de otras mutaciones no analizadas es inferior al 1%.
-No antecedentes obstétricos.
El paciente era un varón de 33 años sin antecedentes heredofamiliares de interés. En relación a sus antecedentes personales informó de criptorquidia intervenida a los 6 meses de edad. No presentaba alergias medicamentosas conocidas. En lo relativo a hábitos tóxicos el paciente fumaba 1 paquetes/año.
Seminograma:
-Volumen: 2,6 ml (valor de normalidad >1,5 ml)
-Recuento/concentración: 19,6 millones/ml (valor de normalidad >15 millones/ml).
-Movilidad: 5% móviles progresivos (valor de normalidad >32%). Espermatozoides móviles no progresivos del 35%. Espermatozoides inmóviles del 60%.
-REM (recuento de espermatozoides móviles): 0,18 millones (valor de normalidad 3 millones)
DIAGNÓSTICO: Astenozoospermia severa.
6

!
www.medicalum.com
!
-Cariotipo: 46XY. El cariotipo del paciente era normal, aunque no puede excluir anomalías cromosómicas estructurales por debajo del poder de resolución del microscopio óptico, patologías de origen monogénico o mosaicismo de baja frecuencia o presentes en otros tejidos.
-Análisis directo de las mutaciones más frecuentes en el gen CFTR mediante el kit Elucigene CF-EU2 (gen-probe) que identificó la mutación F508del del gen CFTR en heterocigosis. Este resultado es compatible con el diagnóstico de portador sano de Fibrosis Quística.
DIAGNÓSTICO La esterilidad primaria es la ausencia de fertilidad desde el inicio de las relaciones sexuales. Cuando una pareja lleva más de un año intentando tener un hijo sin conseguir gestación, está indicado comenzar el estudio de esterilidad.
El estudio de esterilidad se debe apoyar en tres pilares básicos: análisis de datos que confirman la existencia de ovulación, pruebas que valoran la permeabilidad tubárica y seminograma.
En la primera visita a la Unidad de Reproducción Asistida se realiza:
-ANAMNESIS COMPLETA.
-HISTORIAL DETALLADO DE ESTERILIDAD.
-EXPLORACIÓN FÍSICA ADECUADA.
-SEMINOGRAMA. Se estudia el volumen, número, movilidad, vitalidad y morfología de los espermatozoides. Un parámetro muy importante es el REM, ya que cifras bajas en el REM desaconsejan la inseminación artificial 2.
-ESTUDIO DE LA OVULACIÓN: La determinación hormonal del tercer día del ciclo puede orientarnos sobre la reserva ovárica de la mujer (FSH y estradiol basales) 3. Otro marcador es el recuento de los folículos antrales (RFA) (adecuado si >6) 4. Actualmente, la determinación de HAM se considera el mejor test para valorar la reserva ovárica de la mujer y para orientar el tratamiento y el protocolo de
estimulación de una forma más acorde a la situación basal del ovario 5.
-ECOGRAFÍA TRANSVAGINAL. Puede orientar sobre la presencia de patología cervical y uterina y permite valorar posibles alteraciones ováricas.
-CARIOTIPO: está indicado realizarlo ante varios seminogramas patológicos ya que la causa puede deberse a espermatozoides con alteraciones genéticas. También se ha de realizar antes del comienzo del primer ciclo de FIV.
TRATAMIENTO Nos encontramos ante un caso de esterilidad primaria en el que el varón presenta una astenozoospermia severa con un REM muy inferior a 3 millones de espermatozoides/ml (0.18 millones/ml). Por este motivo se descarta la inseminación artificial como técnica de reproducción asistida, ya que se requiere un recuento de espermatozoides móviles (REM) superior a 3 millones. Además, ambos progenitores son portadores de la mutación del gen de la fibrosis quística, por lo que se les ofrece y aceptan la posibilidad de realizar una fecundación in vitro asociada a diagnóstico genético preimplantacional 6.
La fecundación in vitro (FIV) es una técnica de reproducción asistida (TRA) que consiste en la fecundación del ovocito en condiciones de cultivo in vitro, previa obtención y preparación de los gametos, para posteriormente transferir los embriones a la cavidad uterina. La tasa de gestación por ciclo iniciado de FIV/ ICSI en España, según los últimos datos del registro de la Sociedad Española de Fertilidad (SEF), es del 27,5%.
El DGP se basa en el análisis genético de embriones obtenidos por fecundación in vitro (FIV) o por inyección intracitoplasmática de esperma (ICSI) para seleccionar los no afectados, transferirlos al útero, y tratar de conseguir un embarazo viable 7.
El número de patologías genéticas que pueden ser detectadas por DGP está en constante expansión. Actualmente, las principales situaciones en las que está indicado realizar un DGP son:
7

!
www.medicalum.com
!
1. Enfermedades monogénicas (autonómicas dominantes o recesivas y ligadas al X):
2. Anomalías cromosómicas numéricas y estructurales (trisomías, translocaciones, etc.)
EVOLUCIÓN Y PRONÓSTICO La fibrosis quística (FQ) es la enfermedad genética letal más frecuente en la raza caucásica. Se hereda de forma autosómica recesiva y tiene su origen en la mutación del gen de la FQ (CFTR) situado en el brazo largo del cromosoma 7 8. Desde el descubrimiento del gen CFTR en 1989, se han descrito más de 1900 mutaciones y variantes en el gen. La prueba sistemática de todas las posibles mutaciones no es ni viable ni rentable, por lo que el test de screening que se emplea se limita al análisis de las mutaciones más comunes. El CF-EU2v1 es un kit de prueba para la Fibrosis Quística diseñado específicamente para el análisis de las mutaciones más comunes encontradas en poblaciones de origen europeo. Identifica 50 mutaciones en total 9.
DISCUSIÓN Y CONCLUSIONES Como método no natural de selección de embriones en función de su carga genética, el DGP ha planteado un amplio debate sobre cuestiones éticas relacionadas con la vida humana. Las múltiples metodologías disponibles actualmente para la selección prenatal de embriones han originado discrepancias en cuanto al nivel de “gravedad” de algunas enfermedades genéticas, así como el riesgo de favorecer la discriminación de mujeres y de personas con discapacidad. Sin embargo, las encuestas de parejas con alto riesgo de transmitir un trastorno genético informan que del 30 al 74 por ciento preferirían el DGP. Entre todas estas controversias, parece que hoy día se ha llegado a un cierto consenso en relación a dos aspectos del DGP: 1) el apoyo incondicional de su uso como servicio médico preventivo (malformaciones o enfermedades graves), y 2) el rechazo a su uso como técnica de selección de seres humanos “de diseño”.
El comité de ética de la Sociedad Estadounidense de Medicina Reproductiva (ASRM) ha declarado que el
DGP para enfermedades de inicio en adultos es "éticamente” justificable cuando las condiciones son graves y cuando no se conocen intervenciones o las que hay disponibles son inadecuadamente efectivas o con consecuencias graves 10.
REFERENCIAS 1. Davis LB, Champion SJ, Fair SO, Baker VL, Garber AM. A cost-benefit analysis of preimplantation genetic diagnosis for carrier couples of cystic fibrosis. Fertil Steril. 2010; 93 (6): 1793-804.
2. Kruger TF, Acosta AA, Simmons KF et al. Predictive value of abnormal sperm morphology in in vitro fertilization. Fertil Steril. 1988; 49:112-117.
3. Muasher SJ, Oehninger S, Simonetti S, et al. The value of basal and/or stimulated serum gonadotropin levels in prediction of stimulation response and in vitro fertilization outcome. Fertil Steril. 1988; 50: 298-307.
4. Tarlatzis BC, Zepiridis L, Grimbizis G, Bontis J. Clinical management of low ovarian response to stimulation for IVF: a systematic review. Hum Reprod Update. 2003; 9(1): 61-76.
5. Dolleman M, Verschuren WM, Eijkemans MJ, et al. Reproductive and lifestyle determinants of anti-Müllerian hormone in a large population-based study. J Clin Endocrinol Metab. 2013; 98 (5): 2106-15.
6. Remohí J. Manual práctico de Esterilidad y Reproducción Humana. 5º ed. Madrid: Editorial Médica Panamericana; 2017.
7. Ramos Fuentes FJ, Ribate Molina MP. Diagnóstico genético preimplantacional. Rev Esp Pediatr. 2007; 63 (6): 443-499.
8. Santoro Biazotti MC, Walter Pinto J, Romano Maciel de Albuquerque MC. Preimplantation genetic diagnosis for cystic fibrosis: a case report. Einstein (Sao Paulo). 2015; 13 (1): 110-113.
9. Óscar Fielbaum C. Avances en fibrosis quística. Rev Médica Clínica Las Condes. 2011; 22(2):150-9.
10. Schattman, Glenn. 2016. Preimplantation genetic diagnosis. Post Tw,ed.UpToDate. Wilkins-Haug, Louise: UpToDate Inc. Disponible en: http://www.uptodate.com/contents/preimplantation-genetic-diagnosis.
8

!
www.medicalum.com
!
BRUCELOSIS
Ortiz Castro, J.1; García Vázquez, E.2 (1): Facultad de Medicina, Universidad de Murcia
(2): Servicio de MI-Infecciosas, Hospital Clínico Universitario Virgen de la Arrixaca (Murcia).
VALORACIÓN Y JUSTIFICACIÓN La brucelosis es una enfermedad de declaración obligatoria, cuya incidencia en nuestro medio ha ido disminuyendo significativamente con los controles actuales del ganado. Sin embargo, aún existen zonas rurales en las que esta patología continúa siendo endémica, por lo que debe de ser tenida en cuenta como diagnostico diferencial en procesos febriles fluctuantes acompañados por el antecedente epidemiológico de trato con ganado (vacuno y ovino fundamentalmente)1. Siendo como es, una enfermedad que responde muy bien al tratamiento y cuyo pronóstico es excelente en el caso de que sea detectada y tratada rápidamente, siendo las secuelas el resultado del retraso en su diagnóstico. RESUMEN Nuestro caso clínico trata de un paciente infectado por brucella, donde vamos a valorar las principales características clínicas que presentan este tipo de pacientes, como los episodios tan característicos de fiebre fluctuante y sudoración, los cuales, asociados a los antecedentes epidemiológicos del contacto con el ganado, son sugestivos de enfermedad brucelosa. Veremos además una de las complicaciones potencialmente más graves de esta infección, la invasión del sistema nervioso central o neurobrucelosis, por parte de la brucella, la cual se encuadra en el capítulo de las meningitis crónicas, prestando especial interés, en el manejo diagnóstico y terapéutico de la brucelosis, así como el de sus posibles complicaciones. Además, revisaremos los principales métodos diagnósticos empleados en nuestro medio para el diagnóstico de esta infección, así como una breve explicación de las diferentes pautas de tratamiento que se pueden emplear en función de la clínica del paciente y de las posibles complicaciones que pueden ir surgiendo durante el transcurso de la enfermedad. Terminando con unas pinceladas sobre el pronóstico y la evolución del cuadro, así como la importancia de la rápida instauración del diagnóstico y tratamiento, y la influencia de estos en las posibles secuelas. PALABRAS CLAVE Brucella, brucella mellitensis,brucelosis.
ABSTRACT Our clinical case is about a patient infected by brucella, where we will evaluate the main clinical characteristics of this type of patient, such as the characteristic episodes of fluctuating fever and sweating, which, associated with the epidemiological background of contact with cattle , are suggestive of brucellosis disease. We will also see one of the potentially more serious complications of this infection, the invasion of the central nervous system or neurobrucellosis, by the brucella, which falls within the chapter of chronic meningitis, paying special attention in the diagnostic and therapeutic management of brucellosis, as well as its possible complications. In addition, we will review the main diagnostic methods used in our environment for the diagnosis of this infection, as well as a brief explanation of the different treatment guidelines that can be used depending on the patient's clinic and the possible complications that may arise during the course of the disease. Finishing with some brushstrokes on the prognosis and the evolution of the picture, as well as the importance of the rapid establishment of the diagnosis and treatment, and the influence of these in the possible sequels.
KEYWORDS Brucella, brucella mellitensis,brucellosis.
9

!
www.medicalum.com
!
INTRODUCCIÓN La brucelosis, aunque cada vez menos frecuente, sigue siendo endémica en determinadas zonas rurales de España, concretamente en Murcia. Debemos estar atentos a los antecedentes epidemiológicos y al patrón tan característico de la fiebre ondulante. Con estos dos sencillos datos podemos hacer una aproximación diagnostica precoz y bastante acertada en el estudio del paciente con fiebre prolongada.
El diagnóstico se basa en la sospecha clínica y en el aislamiento de la bacteria en muestras biológicas, así como en test serológicos2,3.
CASO CLÍNICO Varón de 53 años que acude al servicio de urgencias aquejado de malestar general, con episodios fluctuantes de fiebre de hasta 39ºC, acompañado de episodios de sudoración y debilidad de varias semanas de evolución.
Entre los antecedentes del paciente destaca que trabaja como ganadero en la zona de la Vega Baja en la Región de Murcia.
No presenta alergias conocidas ni hábitos tóxicos.
El paciente es traído por su familia; estos refieren que el enfermo ha perdido algún kilo en las últimas semanas y que ha estado en contacto con secreciones de sus ovejas, que estuvieron en época de parir en los últimos meses. Además, han percibido episodios de desorientación temporo-espacial en las últimas 24 horas y signos inflamatorios en rodilla izquierda con impotencia funcional secundaria a dolor, esto de una semana de evolución.
Exploración física:
Regular estado general. Temperatura 38ºC, 100 lpm, Tensión arterial 145/89 mmHg. Frecuencia respiratoria 18 por minuto. Peso 90 kg. Talla 175 cm. IMC 29,38. Consciente, con tendencia al sueño y desorientación témporo-espacial, con signos de irritación meníngea y rigidez de nuca (Kernig y Bruzinski positivos) sin otra focalidad neurológica. No
se palpan adenopatías en los territorios accesibles a la palpación.
Auscultación pulmonar: murmullo vesicular conservado.
AC: rítmico, sin soplos, carótidas rítmicas, pulsos temporales palpables.
ABD: se palpa hepatomegalia a 3 cm. del reborde costal, ligeramente dolorosa. No se palpa bazo.
Extremidades: normotónicas, normotróficas, extremidad derecha fuerza 5/5; extremidad izquierda fuerza 2/5 (limitada por dolor en rodilla). Dolor a la flexión y extensión de la rodilla en extremidad inferior izquierda con signos inflamatorios (tumefacción, eritema y aumento de temperatura local con datos que sugieren la presencia de derrame articular).
Laboratorio:
Hemograma: leucopenia con linfocitosis, resto de parámetros normales.
Bioquímica: función renal conservada y en el perfil hepático lo único destacable fue una gammaglutamiltranspeptidasa (GGT) de 79 U/l, con bilirrubina normal.
Reactantes de fase aguda: aumento de procalcitonina (PCT), proteína C reactiva (PCR) Y VSG.
Punción Lumbar (previo fondo de ojo y TC craneal que descartan signos de hipertensión intracraneal): líquido opalescente, con aumento de células de predominio linfocitario, elevación de proteínas y leve disminución de la glucorraquia.
Radiología:
Rx tórax, sin hallazgos patológicos.
Rx de columna lumbo-sacra: alteraciones en columna lumbar sugestivas espondilitis con aspecto en “caña de bambú”.
Rx de rodilla izquierda: signos de osteoporosis yuxtaarticular.
10

!
www.medicalum.com
!
Microbiología:
- Se procesan 2 parejas de hemocultivos
- Gram de extensión de LCR, con tinción Zielh-Neelsen negativa. Se procesa la muestra para cultivo habitual con sospecha de brucelosis y en medio de cultivo para micobacterias.
- Se realiza serología con Rosa de Bengala, obteniéndose un resultado positivo con títulos de IgM >1/80.
Se decide ingresar al paciente con el diagnóstico de brucelosis con afectación meníngea (Neurobrucelosis) y probable sacroileitis asociada, bajo tratamiento con la pauta de Doxiciclina 100mg/12h vía oral, ceftriaxona 2 g cada 12 horas y estreptomicina 1g/24h intramuscular3.
Tras su valoración en planta de hospitalización a cargo del Servicio de Enfermedades Infecciosas se completa el estudio con pruebas de seroaglutinación en la que obtenemos un título de IgM > 1/160 (considerando positivos valores de > 1/80 en zona no endémica y > 1/160 en zona endémica) y test Brucellacapt, prueba de inmunocaptura-aglutinación con un resultado de 1/320.
A las 72 horas del ingreso informan desde microbiología de positividad de los hemocultivos (2/2) para Brucella spp. En nuestro centro se utilizan frascos de hemocultivos Bactec®, que sustituyen al clásico medio de cultivo de Ruiz Castañeda, en el que se obtiene crecimiento de forma más tardía, frente a los 3-4 días de los actuales medios de cultivo.
También se informa a los 4 días del crecimiento en LCR de Brucella spp., siendo negativo el cultivo para micobacterias.
Otras pruebas complementarias:
Se procedió a realizar artrocentesis de la rodilla izquierda, obteniendo liquido articular de aspecto turbio con 2500 leucocitos/mm3, de predominio linfocitario y una glucosa articular de 50 mg/dl. La tinción de Gram no mostró microorganismos y
posteriormente el cultivo se informó como positivo para Brucella spp.
Dado que una de las complicaciones más frecuentes y graves de la brucelosis es la endocarditis, se solicitó ecocardiograma transtorácico y luego transesofágico que no objetivaron lesiones valvulares.
DIAGNÓSTICO
El antecedente epidemiológico, la clínica de fiebre prolongada con sudoración, la afectación articular, lumbo-sacra y el cuadro de meningitis son manifestaciones todas propias de la brucelosis, siendo la neurobrucelosis una de las complicaciones más graves4.
Para el diagnóstico de brucelosis, las pruebas serológicas y los cultivos de sangre, líquido articular y LCR son claves.
Leucocitos 6,7%
Neutrófilos 4,7%
Linfocitos 1,5%
Monocitos 0,5%
Eosinófilos 0,02%
Basófilos 0,02%
Hemoglobina 14 g/dL
Hematocrito 42,3%
VCM 81,1 micromm3
HCM 28,7 pg
Plaquetas 227 000/mm3
PCR 95 mg/dl
VSG 123 mm
PCT 6 ng/dl
Tabla 1. Valores analíticos en Urgencias.
11

!
www.medicalum.com
!
!Aspecto del líquido Opalescente
Células 100 cel/mm3(89% linfocitos) Proteínas 200 mg/dl Glucosa 30 mg/dl
Tabla 2. Valores de punción lumbar.
TRATAMIENTO El tratamiento de la infección aguda sin clínica focal consiste en la combianción e doxiciclina (6 semanas) con gentamicina durante las 1-2 primeras semanas, pudiendo ser esta sustituida por estreptomicina5.
El tratamiento de la neurobrucelosis y de la endocarditis puede realizarse con la triple asociación de doxiciclina, rifampicina y cotrimoxazol, durante un mínimo de 6 meses. En caso de endocarditis puede asociarse gentamicina durante las 3 primeras semanas6 y en la neurobrucelosis ceftriaxona. Otras alternativas menos eficaces son las asociaciones menos eficaces incluyen levofloxacino.
En casos de enfermedad complicada se suele asociar al tratamiento con antimicrobianos la cirugía valvular (endocarditis) o la administración de esteroides en la neurobrucelosis complicada por iritis, papiledema, mielopatía, polineuropatía y / o parálisis de pares craneales.
Nuestro paciente recibió una pauta combinada de doxiciclina, rifampicina y ceftriaxona.
EVOLUCIÓN Y PRONÓSTICO Tras la instauración del tratamiento antibiótico el paciente mejora, por lo que tras 14 días de ingreso se decide remitir a domicilio para completar tratamiento antibiótico de forma ambulatoria durante 6 meses.
En el seguimiento se repitió ecocardiograma transtorácico a los 2 meses y al final del tratamiento, que no mostró lesiones sugestivas de endocarditis
Se hizo seguimiento serológico con BRUCELLACAPT (aglutinación - inmunocaptura), que mostró un descenso paulatino de los niveles siendo de 1/160 a los 6 meses.
DISCUSIÓN Y CONCLUSIONES Para el diagnostico de brucelosis debemos emplear métodos directos que se basan en la detección de la bacteria. En nuestro medio clásicamente se ha empleado el Ruiz Castañeda, que se basa en la inoculación de sangre en frascos cerrados que contienen un líquido rico en triptosa y un medio de agar triptosa, manteniendo el cultivo en incubación unos 30 días, debido al lento crecimiento de la bacteria. Más recientemente se han desarrollado métodos automáticos como el de Bactec® que nos permite detectar en 7 días hasta el 95% de los cultivos positivos. Conforme la enfermedad avanza, la concentración de microorganismo en sangre baja, por lo que habrá que ir a ganglios, hígado o bazo a obtener las muestras. También se pueden estudiar muestras de LCR o líquido articular, según la clínica focal que presente el paciente.
Para la detección de los antígenos en sangre podemos ayudarnos de las técnicas de ELISA, inmunofluorescencia directa o hemaglutinación reversa.
Con respecto a los métodos indirectos existen numerosas pruebas de detección siendo las más usadas el Rosa de Bengala, prueba de aglutinación con 2 mercaptopurinol, seroaglutinación y Coombs.
Una vez que tenemos el diagnóstico la evolución suele ser favorable en la mayoría de los pacientes, a excepción de los casos de endocarditis infecciosa, que suelen precisar cirugía de reemplazo valvular, y los casos de neurobrucelosis, que pueden curar con secuelas neurológicas.
12

!
www.medicalum.com
!
REFERENCIAS 1. Álvarez-Hernández, N., Díaz-Flores, M. and Ortiz-
Reynoso, M. (2015). Brucelosis, una zoonosis frecuente. Medigraphic [Internet], 3(2), pp.129-133.
2. Cardenal XA. Brucelosis. En: C Rozman, F Cardellach, JM Ribera, el al, editores. Medicina interna. Vol 2. 17ª ed. Barcelona: Elsevier; 2012. p. 2045-2047.
3. Corbel MJ, Beeching NJ. Brucelosis. En: Barnes PJ. Longo DL, Fauci AS, et al, editores. Harrison principios de medicina interna. Vol 1. 18a ed. México: McGraw-Hill; 2012. p. 1296-1300.
4. M.E. Jiménez Mejías, J. Pachón-Diaz. Síndrome meníngeo agudo, encefalitis y meningoencefalitis. En: Gómez Gómez J, Gobernado M. En: Enfoque clínico de los grandes síndromes infecciosos. 5ª ed. Madrid: Novartis; 2013. P 157-194.
5. Gómez Gómez, J. and García Vázquez, E. (2017). Los antimicrobianos en la clínica práctica. Murcia: Servicio de Publicaciones de la Universidad de Murcia.
6. Hernández Torres A. Brucella una causa infrecuente de endocarditis infecciosa. En: Gómez Gómez, J, García Vázquez, Hernández Torres A. En. Casos de Patología Infecciosa. Una aproximación desde la clínica. 1ª ed. Murcia: Editum; Servicio de Publicaciones de la Universidad de Murcia; 2016. P55-59.
13

!
www.medicalum.com
!
MANEJO CLÍNICO DE LA LEUCEMIA AGUDA MIELOBLÁSTICA Y SU DIAGNÓSTICO DIFERENCIAL: A PROPÓSITO DE UN CASO
Iniesta González, S.1 ; Casado Prieto, A. Mª.2
(1): Facultad de Medicina, Universidad de Murcia (2): Servicio de Hematología y Hemoterapia, Hospital Universitario Morales Meseguer.
VALORACIÓN Y JUSTIFICACIÓN La leucemia aguda mieloblástica es una patología maligna, de curso rápido y agresivo, pero potencialmente curable. El retraso en su diagnóstico y tratamiento puede contribuir a un desenlace fatal de la enfermedad o de sus complicaciones. El interés de este caso clínico radica en la importancia de su conocimiento por el médico general, que en cualquier escenario clínico debería ser capaz de sospecharla, lo que constituye el primer paso para un manejo diagnóstico-terapéutico eficaz. Además, se trata de un tema de actualidad, cada vez más presente en los medios de comunicación y en las redes sociales.
RESUMEN La leucemia aguda mieloblástica es el tipo de leucemia aguda más frecuente en adultos en el mundo occidental (80%), con una mediana de edad al diagnóstico de 65 años. Se trata de una enfermedad grave y rápidamente mortal sin tratamiento específico. Presentamos el caso de un varón de 56 años sin patología previa que debutó con anemia y diátesis hemorrágica leve y ha sido recientemente diagnosticado de una leucemia aguda mieloblástica con cambios displásicos. Haremos una breve revisión del diagnóstico, el tratamiento y el pronóstico de esta enfermedad, con el objetivo de un mejor conocimiento de la misma por el médico general.
PALABRAS CLAVE Leucemia aguda mieloblástica, aspirado de médula ósea, quimioterapia, trasplante de progenitores hematopoyéticos.
ABSTRACT Acute myeloblastic leukaemia is the most common type of acute leukaemia in adults in the Western world (80%), with a median age at diagnosis of 64 years. It is considered a severe and rapidly life-threatening condition when not specifically treated. We describe the case of a 56 year old male with no previous pathology that presented with anaemia and mild haemorrhagic diathesis and was recently diagnosed with an acute myeloblastic leukaemia with myelodysplasia related changes. Diagnosis, treatment and prognosis of this disorder will be briefly reviewed with the aim of a better knowledge of it by the general practitioner.
KEYWORDS
Acute myeloblastic leukaemia, bone marrow examination, chemotherapy, stem cell transplantation.
INTRODUCCIÓN Las leucemias agudas son enfermedades malignas caracterizadas por una proliferación clonal de células hematopoyéticas inmaduras (blastos) en médula
ósea, alterando la producción de células sanguíneas normales, con o sin infiltración de otros órganos. A su vez, se pueden clasificar en dos grandes grupos en
14

!
www.medicalum.com
!
función de la línea celular proliferante: mieloblásticas y linfoblásticas1.
En este trabajo nos centraremos en la leucemia aguda mieloblástica (LAM) por ser la más frecuente en adultos, con un discreto predominio en varones y una mediana de edad al diagnóstico de 64 años1,3. Su incidencia aumenta con la edad y en ocasiones puede considerarse secundaria a enfermedades o tratamientos previos, pero en la mayoría de los casos su etiología es desconocida. Su clasificación en subtipos es compleja y se basa en criterios clínicos, citológicos, citogenéticos y moleculares2 (tabla 1).
Presentamos un caso de nuevo diagnóstico de LAM, a partir del cual haremos una breve revisión del manejo de esta patología.
CASO CLÍNICO Se trata de un varón de 56 años, sin antecedentes de interés y con una situación basal adecuada a su edad, remitido a Urgencias por pancitopenia moderada. A su llegada, se encontraba estable y con buen estado general, no precisando soporte transfusional de forma urgente. A la anamnesis destacaba una clínica de un mes de evolución de astenia, hiporexia y pérdida no intencionada de 8 kilogramos de peso, sin fiebre ni clínica infecciosa. A la exploración física se objetivó una discreta palidez cutáneo-mucosa y múltiples equimosis no complicadas.
Con la sospecha de hemopatía maligna, se realizó una extensión de sangre periférica, que mostró un 43% de blastos indiferenciados, una serie granulocítica displásica, anemia macrocítica y una trombopenia confirmada (Figura 1). El paciente fue ingresado en Hematología, donde al día siguiente se le realizó una analítica completa, incluyendo serología y estudio transfusional, y un aspirado de médula ósea (Figura 2), que confirmó el diagnóstico de LAM con cambios displásicos. Se envió material a un centro externo para análisis citogenético y molecular. Además, se solicitó una radiografía de tórax, una ecografía abdominopélvica y un estudio de función cardíaca.
Incluso antes de conocer los resultados de todas las pruebas complementarias y previa obtención del consentimiento informado, se canalizó un catéter venoso yugular y se comenzó el tratamiento de inducción a la remisión, siguiendo el protocolo del Programa Español de Tratamientos en Hematología (PETHEMA) para el tratamiento de primera línea de la LAM en pacientes menores de 65 años candidatos a terapia intensiva4.
F igura 1. Frotis de sangre periférica. A la derecha, un blasto indiferenciado con una alta relación núcleo/citoplasma; a la izquierda, un polimorfonuclear neutrófilo con anomalías en la segmentación nuclear y en la granulación citoplasmática.
!Figura 2. Aspirado de médula ósea. Infiltración a expensas de mieloblastos tipo I y II
15

!
www.medicalum.com
!
Aunque la quimioterapia de inducción se completó en siete días, el ingreso se prolongó cinco semanas, ya que produce una aplasia medular transitoria que requiere una estrecha monitorización clínica y analítica, así como tratamiento de soporte transfusional y de las posibles complicaciones, hasta la recuperación del hemograma. Se realizó profilaxis del síndrome de lisis tumoral y de infecciones bacterianas, víricas y fúngicas, llegando a precisar tratamiento antibiótico empírico por fiebre neutropénica de probable foco pulmonar. Además presentó náuseas, vómitos, diarrea, alopecia y un exantema secundario a la citarabina, que requirieron tratamiento sintomático.
Tras la recuperación de la aplasia se realizó una reevaluación medular, que cumplía criterios de remisión completa. Dada la buena respuesta inicial al tratamiento, sumada a los datos citogenéticos y moleculares obtenidos al diagnóstico, se consideró al paciente dentro del grupo de pronóstico favorable, por lo que ha recibido dos ciclos de quimioterapia de consolidación y posteriormente será sometido a un trasplante autólogo de progenitores hematopoyéticos.
DIAGNÓSTICO
La sospecha de leucemia aguda se basa en datos clínicos y analíticos. Aunque no es infrecuente detectarla de forma casual, los signos y síntomas más frecuentes son los derivados de las citopenias –fiebre o infección, sangrado, síndrome anémico– y de la infiltración de órganos –adenopatías, visceromegalias, hipertrofia gingival, leucémides–, así como el síndrome constitucional1. Los hallazgos del hemograma pueden ser más o menos llamativos en
función del estado de avance de la enfermedad. La cifra de leucocitos puede estar aumentada a expensas de células inmaduras, ser normal (con una fórmula leucocitaria alterada o no) o estar disminuida. Ante cualquier duda se debe realizar un contaje celular manual al microscopio, ya que los contadores automáticos pueden interpretar los blastos como células normales o no. Es frecuente la alteración de parámetros bioquímicos relacionados con un alto recambio celular, pero esto es variable, pues la leucemia puede afectar a la función de cualquier órgano.
Para el diagnóstico definitivo es necesario realizar un aspirado de médula ósea a la mayor brevedad posible, con el fin de no retrasar su manejo terapéutico, que debe mostrar al menos un 20% de blastos según la definición de leucemia aguda de la Organización Mundial de la Salud1-4. Un porcentaje del 6 al 10% se puede considerar una “preleucemia”.
Es recomendable completar el análisis citomorfológico de la sangre medular con técnicas especiales: tinciones histoquímicas, inmunofenotipo por citometría de flujo, citogenética y estudio de mutaciones por biología molecular3. Esto ayuda a caracterizar mejor la clona de células malignas, definiendo su grado de maduración y diferenciación (subtipos de LAM, Tabla 1) y permitiendo su diagnóstico diferencial con otros tipos de leucemia, síndromes mielodisplásicos, linfomas con afectación medular, infiltración por células tumorales de origen no hematológico o aplasia medular. Estos datos también tienen implicaciones pronósticas y en muchos casos condicionan nuestra actitud terapéutica3.
16

!
www.medicalum.com
!
LAM con alteraciones genéticas recurrentes
! LAM con t(8;21)(q22;q22.1); RUNX-RUNX1T1
! LAM con inv(16)(p13;1q22) o t(16;16)(p13;1q22); CBFB-MYH11
! LAP con PML-RARA
! LAM con t(9;11)(p21.3;q23.3); MLLT3-KMT2A
! LAM con t(6;9)(p23;q34.1); DEK-NUP214
! LAM con inv(3)(q21.3;3q26.2) o t(3;3)(q21.3;q26.2); GATA2, MECOM
! Leucemia aguda megacarioblástica con t(1;22)(p13.3;q13.3), RBM15-MKL1
! Entidad provisional: LAM con BCR-ABL1
! LAM con mutaciones de NPM1
! LAM con mutaciones bialélicas de CEBPA
! Entidad provisional: LAM con mutaciones de RUNX1
LAM con cambios relacionados con mielodisplasia
Neoplasia mieloide relacionada con terapia previa
LAM, otras categorías
! LAM indiferenciada
! LAM con diferenciación mínima
! LAM con maduración
! Leucemia aguda mielomonocítica
! Leucemia aguda monoblástica/monocítica
! Leucemia aguda eritroide pura
! Leucemia aguda megacarioblástica
! Leucemia aguda basofílica
! Panmielosis aguda con mielofibrosis
Sarcoma mieloide
Proliferaciones mieloides relacionadas con el s índrome de Down
Neoplasia blástica plasmacitoide de células dendrít icas
Leucemias agudas de l inaje ambiguo
Tabla 1. Adaptación de la clasificación de las leucemias agudas según la Organización Mundial de la Salud (2016). LAM: Leucemia Aguda Mieloblástica. LAP: Leucemia Aguda Promielocítica.
17

!
www.medicalum.com
!
Grupo de riesgo favorable
Grupo de riesgo intermedio
Grupo de riesgo desfavorable
inv(16) o t(16,16) con EMR baja y remisión completa con
primer ciclo de inducción
Enfermedad mínima residual alta
t(8;21) con EMR baja y remisión completa con primer
ciclo de inducción
Alteraciones citogenéticas de alto riesgo
Mutación en NPM1 con cariotipo normal y ausencia
de mutación en FLT3
Duplicaciones en tándem de FLT3
Mutación en CEBPα con cariotipo normal y ausencia
de mutación FLT3
Pacientes sin factores de buen ni mal pronóstico
Remisión completa no alcanzada tras primer ciclo
de inducción
Tabla 2. Estratificación pronóstica en la leucemia aguda mieloblástica en base a criterios clínicos, citogenéticos y moleculares1, 3. EMR: Enfermedad Mínima Residual.
TRATAMIENTO Con la excepción de la leucemia aguda promielocítica (ver Tabla 1), que por su fisiopatología y su tratamiento específico constituye una entidad aparte, el tratamiento de inducción es estándar para todas las LAM y consiste en una combinación de idarrubicina y citarabina4. La tasa de remisiones completas con este esquema ronda el 80%.
El tratamiento post-remisión tiene por objetivo evitar las recaídas de la enfermedad. Su elección se basa en la estratificación de los pacientes en grupos de riesgo4 (ver Tabla 2):
• En los casos de riesgo favorable se realiza una consolidación con dos ciclos de citarabina, seguida de un trasplante autólogo de progenitores hematopoyéticos. El fundamento del trasplante autólogo es poder dar una quimioterapia tan intensiva (“acondicionamiento”) que acabe definitivamente con la enfermedad, pero que a su vez produce una mieloablación
irreversible que requiere un rescate con células madre para regenerar la función hematopoyética. Estas células se obtienen del propio paciente mediante aféresis de sangre periférica durante la fase de remisión, se criopreservan y tras el acondicionamiento se reinfunden en modo similar a una transfusión.
• En los casos de riesgo intermedio y alto, la consolidación se realiza con citarabina e idarrubicina y se opta por un trasplante autólogo o alogénico en función de la disponibilidad de donante. Dado que la enfermedad se considera de entrada más agresiva, es preferible el alotrasplante por el efecto de alorreactividad de las células del donante contra las de la leucemia, buscando ir más allá de la mera regeneración de la médula tras el acondicionamiento. Esta alorreactividad no es selectiva contra las células malignas, pudiendo afectar a múltiples órganos del receptor –enfermedad injerto contra huésped, ni es unilateral, de manera
18

!
www.medicalum.com
!
que el receptor también puede rechazar el injerto. Es preciso conseguir un nivel óptimo de inmunosupresión para evitar estas complicaciones y ajustarlo ante cualquier dato de recaída, enfermedad injerto contra huésped, fallo del injerto o infecciones. En definitiva, la morbimortalidad del alotrasplante no es despreciable y es por ello que se intenta reservar para pacientes con peor pronóstico.
En la mayoría de los casos el trasplante es el único tratamiento curativo, y lo ideal es realizarlo en situación de remisión completa.1,3,4 Para la leucemia refractaria o en recaída existe un creciente arsenal de opciones terapéuticas que incluye múltiples esquemas de quimioterapia de rescate, inmunoterapia contra dianas específicas e incluso segundos trasplantes. Es recomendable la participación en ensayos clínicos cuando no existe una opción claramente superior a otras, contribuyendo al avance científico sin que ello conlleve un perjuicio para los pacientes.
EVOLUCIÓN Y PRONÓSTICO La probabilidad de curación de la LAM en pacientes jóvenes se estima por debajo del 50%.
A pesar de la alta tasa de remisiones con el tratamiento de inducción, la LAM es una enfermedad con gran tendencia a la recaída. En los últimos años está cobrando mucha importancia la monitorización de la enfermedad mínima residual (EMR), un concepto surge de la aparición de nuevas técnicas de laboratorio capaces de detectar una mínima fracción de células malignas en pacientes que por morfología se consideran en remisión completa. Una EMR positiva se relaciona con una mayor probabilidad de recaída3.
El tratamiento de la LAM tampoco está exento de riesgos, incluyendo infecciones oportunistas, hemorragias graves, daño de órganos, complicaciones específicas del trasplante y neoplasias secundarias. Cada vez es más frecuente tratar e incluso trasplantar a pacientes mayores o con comorbilidades, recurriendo a esquemas de menor
intensidad o a fármacos nuevos, lo que hace aún más impredecible la evolución de la enfermedad.
CONCLUSIONES La leucemia aguda mieloblástica es una enfermedad grave y de manejo complejo que precisa atención urgente por parte de un servicio especializado en Hematología. Su mejor conocimiento por parte del clínico en cualquier ámbito sanitario puede ayudar a su detección y tratamiento de forma precoz.
REFERENCIAS 1. Figuera Álvarez A, Sierra Gil J. Leucemias. Concepto
y clasificación. Leucemias agudas. En: Moraleda Jiménez JM. Pregrado de Hematología. 4ª ed. Madrid: Luzán 5; 2017. p. 227-264.
2. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. American Society of Hematology. 2016 mayo; 127(20): [2391-2405].
3. Döhner H, Estey E, Grimwade D, Amadori S, Frederick R. Büchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017; 129(4): [424-447].
4. Montesinos P, Sanz MA. Recomendaciones para el tratamiento de primera línea adaptado al riesgo de la leucemia mieloblástica aguda en pacientes de edad menor o igual a 65 años candidatos a quimioterapia intensiva. Valencia: PETHEMA; 2010.
19

!
www.medicalum.com
!
INSUFICIENCIA RENAL CRÓNICA EN ATENCIÓN PRIMARIA. A PROPOSITO DE UN CASO.
Iniesta González, S.1 ; Martínez Vélez, Mª V.2
(1): Facultad de Medicina, Universidad de Murcia (2): Atención Primaria, Servicio Murciano de Salud.
VALORACIÓN Y JUSTIFICACIÓN La enfermedad renal crónica (ERC) es una patología frecuente, con una prevalencia cercana al 10% de la población española, y posiblemente infradiagnósticada1-2. Es común encontrar dicha alteración en edades avanzadas, a diferencia del caso que presentamos, un paciente de 38 años que acude a consulta de atención primaria por un motivo diferente. Debido a la discrepancia en la edad de presentación, es importante en atención primaria conocer los datos clínicos y analíticos que orienten hacia una alteración renal para detectarlo rápidamente y saber discernir entre la amplia gama de etiologías posibles gracias a la anamnesis y la exploración física.
RESUMEN La insuficiencia renal crónica es una patología infradiagnosticada que es vista cada vez con mayor frecuencia en el mundo occidental. Se trata de una enfermedad que lleva a un desenlace mortal si no es detectada y tratada a tiempo. Presentamos el caso de un varón de 38 años que es diagnosticado de una insuficiencia renal crónica tras acudir a Atención Primaria consultando por un motivo totalmente diferente. Haremos una breve revisión del diagnóstico diferencial de esta enfermedad, incidiendo en la importancia de la temprana edad del paciente a la hora de conducir las decisiones clínicas.
PALABRAS CLAVE Insuficiencia renal crónica, tasa de filtración glomerular, hipertensión arterial, monorreno.
ABSTRACT Chronic kidney disease is an underdiagnosed disease, whose prevalence is growing in the western world. This disease needs an early detection and treatment in order to prevent its fatal ending. We describe the case of a 38 year old male who is diagnosed of chronic kidney disease after looked for medical attention due to a different chief complaint. Differential diagnosis of this disorder will be briefly reviewed, and we will stress the early age of our patient at the moment of taking the clinical decisión.
KEYWORDS
Chronic kidney disease, glomerular filtration rate, hypertension, solitary kidney.
INTRODUCCIÓN La enfermedad renal crónica se define como la presencia de deterioro de la función renal (marcado por una tasa de filtrado glomerular [TFG] menor a 60 mL/min/1,7m2) o existencia de daño renal (detectado por una excreción urinaria de albúmina igual o mayor
a 30 mg/día) por un período mayor a tres meses, cualquiera que sea la causa de la misma2-4.
Es una enfermedad muy frecuente, con una prevalencia aproximada del 10% en la población adulta española, con un discreto predominio en mujeres y una mediana de edad al diagnóstico de 68
20

!
www.medicalum.com
!
años. Su incidencia aumenta con la edad y es considerada una enfermedad infradiagnosticada1-2. La clasificación de la enfermedad renal crónica aporta una guía para el tratamiento y las complicaciones de la misma, y se realiza mediante la tasa de filtración glomerular y el grado de albuminuria3-4 (Tabla 1).
A continuación se presenta un caso de nuevo diagnóstico de IRC, a partir del cual se hará una breve revisión del diagnóstico diferencial y manejo de esta patología.
CASO CLÍNICO Paciente varón de 38 años acude a Consulta de Atención Primaria por lumbalgia crónica.
Como antecedentes personales solo destaca haber presentado cefalea ocasional y lumbalgia tratada con AINE (Ibuprofeno) de manera ocasional. Sin antecedentes familiares ni psicosociales de interés. Tratamiento crónico: Ibuprofeno a demanda desde hace varios años.
La paciente acudió a consulta en el episodio actual por lumbalgia crónica sin irradiación. Localizaba el dolor en zona lumbar, ampliado en los últimos días a zona dorsal derecha. La exploración física resultó anodina salvo dolor a la palpación en zona lumbar, con puño-percusión negativa.
Se solicitó como pruebas complementarias una radiografía de columna dorso-lumbar y una analítica. Los hallazgos encontrados en la radiografía fueron semejantes a los encontrados en la radiografía realizada hace dos años al diagnóstico de la lumbalgia (estrechamiento espacio intervertebral L5-S1). En la analítica se halló creatinina de 2,1 mg/dL y urea de 85 mg/dL.
Ante el hallazgo de alteración de la función renal, se reevaluó al paciente, indagando en sintomatología
nefro-urinaria. El paciente refirió polidipsia y poliuria así como orina levemente espumosa en loso últimos días. Resto de aparatos y sistemas sin sintomatología. A la exploración física presentó una tensión arterial de 174/11 mmHg. Tras la alteración de función renal y la hipertensión, la actitud tomada fue derivar al paciente a Urgencias del Hospital Virgen de la Arrixaca para completar estudio.
En Urgencias se le realizó una nueva analítica confirmando la alteración renal, así como una ecografía renal donde no se detectó riñón izquierdo así como un riñón derecho con signos de nefropatía parenquimatosa. El paciente ingresó en planta de Nefrología donde se completó el estudio con una amplia batería de pruebas diagnósticas, destacando un filtrado glomerular de 27 ml/min, orina de 24 horas con proteinuria 1,8 gr/día y albuminuria 1,5 gr/día así como un renograma que confirmaba que el paciente era monorreno.
DIAGNÓSTICO
La alteración de la función renal con un filtrado glomerular de 27 ml/min y ausencia de riñón izquierdo llevó al diagnóstico de Enfermedad Renal Crónica estadio IV en paciente monorreno y abuso de AINEs por probable glomerulonefritis segmentaria y focal.
En Atención Primaria, al encontrar a un paciente con alteración de la función renal se nos presentó un amplio diagnóstico diferencial representado en la tabla 25.
Tras la valoración en el Hospital y detectando la cronicidad de la patología, el diagnóstico diferencial se centró en las causas crónicas de la misma, destacando como causas más frecuentes la Diabetes
21

!
www.medicalum.com
!
Estadio TFG TFG (mL/min/1.73m2) Característ icas función renal
G1 >90 Normal o aumentada
G2 60-89 Ligeramente disminuida
G3a 45-59 Ligera a moderadamente disminuida
G3b 30-44 Moderada a severamente disminuida
G4 15-29 Severamente disminuida
G5 <15 Fallo renal terminal
Albuminuria Excreción albúmina (mg/día) Función renal
A1 <30 Normal o aumentada, función normal
A2 30-300 Aumentada
A3 >300 Muy aumentada, rango nefrótico
Tabla 1. Clasificación de la insuficiencia renal crónica.
!!!!!!!!!!!!!!!!!
!Tabla 2. Diagnóstico diferencial ante alteración de la función renal.
Insuficiencia prerrenal
- Hipovolemia: hemorragia, pérdida digestiva o renal, hipoalbuminemia, quemaduras
- Bajo gasto cardíaco: insuficiencia cardíaca, arritmias, infarto de miocardio
- Vasodilatación periférica: sepsis, fístulas arteriovenosas, cirrosis con ascitis
- Vasoconstricción renal: noradrenalina, dopamina, hipercalcemia, contrastes yodados, tacrolimus, ciclosporina, anfotericina
- Interferencia con autorregulación renal: AINE, IECA, ARAII
- Síndrome de hiperviscosidad: mieloma múltiple, macroglobulinemia, policitemia
Causa parenquimatosa
- Necrosis tubular aguda: fármacos, isquemia renal mantenida
- Vascular: trombosis arterial/venosa bilateral, ateroembolia, vasculitis
- Glomerular: glomerulonefritis agudas, radiación, esclerodermia, contrastes yodados, cocaína
- Túbulo-intersticial: nefritis intersticial aguda, infecciones, obstrucción tubular difusa, fármacos
Causa postrenal
- Lesión ureteral: litiasis, coágulos, fibrosis, tumor
- Lesión vesical: tumor, litiasis, cistitis, fármacos
- Lesión uretral: traumatismos, fimosis, válvulas congénitas, estenosis, tumor
22

!
www.medicalum.com
!
Mellitus y la Angionefroesclerosis, descartadas con la amplia batería de pruebas diagnósticas que se realizaron mientras el paciente estuvo ingresado.
TRATAMIENTO Durante el ingreso se pautó IECA a dosis bajas.
EVOLUCIÓN Y PRONÓSTICO Tras iniciar tratamiento con IECA a baja dosis se produjo un empeoramiento de la función renal, aunque sí se pudo controlar la hipertensión arterial hasta cifras normotensionales junto con dieta hiposódica. Finalmente se dio de alta al paciente con el siguiente tratamiento:
1. Dieta sin sal.
2. Evitar tomar fármacos nefrotóxicos (AINEs, fibratos…)
3. Control estricto de cifras tensionales.
4. Lisinopril 5 mg un comprimido cada 24 horas por la noche.
5. Si la tensión arterial aumenta por encima de 140/80 mmHg: Amlodipino 5 mg un comprimido cada 24 horas por la mañana.
DISCUSIÓN Y CONCLUSIONES La insuficiencia renal crónica es un hallazgo inusual en personas jóvenes, lo que se convierte en un auténtico reto desde el inicio. Debido a ello, una de las principales limitaciones ha sido la dificultad para buscar en la literatura científica otros casos de insuficiencia renal en pacientes de semejante edad.
Sin embargo, debido a que nuestro paciente presentaba una TFG menor a 30 mL/min/m2, la aparición de síntomas inespecíficos unidos a los datos hallados en el estudio analítico permitió la detección de la disfunción renal que padecía nuestro paciente 2. El dato más orientativo fue el consumo crónico de Ibuprofeno por parte del paciente, ya que
es conocido el efecto nefrotóxico de los AINE, así como otros fármacos como aminoglucósidos o fibratos 6.
Conclusiones:
1. La insuficiencia renal crónica es una patología común, siendo una de las causas el consumo de fármacos nefrotóxicos (AINEs).
2. Tener una visión global del paciente, puesto que a veces podemos encontrar hallazgos no relacionados con el motivo de consulta que nos llevan a un nuevo diagnóstico.
3. La HTA suele ser causa o estar asociada a la ERC. En pacientes con ERC tratar la HTA para mejorar la función renal.
REFERENCIAS 1. Martín de Francisco AL, Piñera C, Gago M, Ruiz J,
Robledo C, Arias M. Epidemiología de la enfermedad renal crónica en pacientes no nefrológicos. Nefrología. 2009; 29(5): 101-105
2. Lorenzo V. Enfermedad Renal Crónica. En: Lorenzo V, López Gómez JM. Nefrología al Día [monografía]. Tenerife: Sociedad Española de Nefrología; 2017.
3. Alcázar R, Egocheaga I, Orte L, Lobos M, González Parra E, Álvarez Guisasola F et al. Documento de consenso SEN-semFYC sobre la enfermedad renal crónica. Nefrología. 2008; 28(3): 273-282.
4. Rosenberg M. Overview of the management of chronic kidney disease in adults. [Monografía en Internet]. UpToDate: Curhan G (Ed); 2017 Diciembre [acceso 5 de febrero de 2018].
5. López de Briñas EP. Insuficiencia renal aguda. En: Rozman C, director. Medicina Interna. 17ª ed. Barcelona: Elsevier; 2012. p. 809-16.
6. Narváez Tamayo MA, Castañeda de la Lanza C, O Shea Cuevas GJ, Lozano Herrera J, Castañeda Martínez C. Paciente con enfermedad renal: manejo del dolor. Pain control in patient with kidney disease. Gaceta Mexicana de Oncología. 2015 diciembre; 14(6): 335-41.
23

!
www.medicalum.com
!
SÍNDROME CORONARIO AGUDO CON ELEVACIÓN DE SEGMENTO ST (SCACEST)
Buendía Santiago, F.1 ; Gil Rueda, B.2
(1): Facultad de Medicina, Universidad de Murcia (2): Hospital Universitario Morales Meseguer (Murcia).
VALORACIÓN Y JUSTIFICACIÓN El síndrome coronario agudo es un síndrome de una enorme importancia. Se trata de la patología más prevalente en cuanto a ingresos en unidades de cuidados intensivos (UCI) se refiere, y una de las más comunes en la práctica clínica general. Es indispensable para cualquier profesional o futuro profesional médico conocer y dominar el manejo de esta situación clínica.
RESUMEN Varón con de 43 años que llama a emergencias por dolor centrotorácico irradiado a región dorsal. A la llegada de los servicios de emergencias realizan un primer ECG que evidencia signos de elevación del segmento ST. A la llegada al servicio de urgencias el paciente no refiere dolor y las alteraciones del ECG se han normalizado. Se encuentran troponinas elevadas en analítica, y tras un segundo examen bioquímico se evidencia un nuevo aumento de estas, derivando al paciente al servicio de urgencias del hospital de referencia. El paciente se encuentra en el servicio de UCI, y tras una nueva exploración declara sufrir el mismo dolor centrotorácico anteriormente descrito, y refiere que éste no ha remitido desde el inicio de los síntomas. EL paciente es trasladado a la unidad de hemodinámica para la realización de una coronariografía, donde se evidencia una oclusión del 95% de la arteria descendente anterior (DA) en su segmento proximal. Se realiza una ecocardiografía en la que se detecta una fracción de eyección del ventrículo izquierdo (FEVI) conservada, y una discreta hipocinesia de la cara anterior del ventrículo izquierdo. Como tratamiento se coloca un stent farmacoactivo y el paciente es trasladado nuevamente a UCI.
PALABRAS CLAVE Infarto del miocardio, dolor torácico, elevación del ST, cateterismo cardíaco.
ABSTRACT A 43-year-old-man who call Emergencies due to a chest pain radiated to the dorsal región. At the arrival of the emergency services, an ECG is recorded, which shows evidence of elevation of the ST segment. The patient did not refer chest pain after arriving at the emergency service. The ECG disturbances disappeared. A high level of troponines are detected at the analytics, and after a second biochemist exam, a new increasement is shown, therefore the patient is transferred to the Intensive Care Unit at his referente hospital. The patient, after a new physical exploration, declares having the same chest pain he had at the beginning of the symtomps, and that it never disappeared. At that moment, we contact with the hemodynamics unit. There, a coronariography is performed and we detected a 95% occlusion of left anterior descending artery, in its proximal segment. An ecocardiography is performed. The left ventricle eyection fraction is conseved, and a minor hipokynesia is detected in the anterior wall of left ventricle. A pharmacoactive stent is set and the patient is sent back to the Intensive Care Unit.
KEYWORDS
Myocardial infarction, Cheste pain, ST elevation, cardiac catheterization.
24

!
www.medicalum.com
!
INTRODUCCIÓN La enfermedad coronaria es la causa más frecuente de muerte a nivel mundial, responsable del 20% del total de los fallecimientos en Europa.
Se considera infarto agudo de miocardio (IAM) cuando exista evidencia de daño miocárdico, definido por la elevación de las troponinas cardíacas a valores superiores al percentil 99 del límite superior de referencia, con presencia de necrosis en un contexto clínico compatible con isquemia miocárdica1, 2.
Se define a un paciente con síndrome coronario agudo con elevación del ST (SCACEST), a aquel que presenta un dolor torácico persistente (u otros síntomas indicativos de isquemia) y elevación del segmento ST en al menos 2 derivaciones contiguas. En estos pacientes se recomienda iniciar inmediatamente estrategias de tratamiento como la reperfusión, la cual favorece un gran descenso en la mortalidad aguda y a largo plazo tras el SCACEST1.
CASO CLÍNICO Antecedentes personales:
Varón de 43 años. No AMC, no HTA, no DM, no DLP. Ligero sobrepeso. Fumador, 20 cigarrillos diarios. No antecedentes familiares de cardiopatía. Sin limitación para la actividad física. Sin clínica cardinal de insuficiencia cardíaca. No tratamiento de base al ingreso.
Historia actual:
El paciente despierta a las 4:00h por intranquilidad, con comienzo brusco de dolor centrotorácico irradiado a región dorsal, molestia en brazo izquierdo. No se acompaña inicialmente de disnea, nauseas, vómitos, ni cortejo vegetativo. Niega cambios con los movimientos o con la respiración. Intenta descansar, pero el dolor le levanta de la cama de nuevo, avisando al 112 a las 5.30h. En ECG se evidencia elevación del ST en aVR, V1 y V2, como se aprecia en la figura 1; ondas T hipervoltadas-hiperagudas en V3 y V4 con descenso de ST en derivaciones de cara inferior y
lateral. Tras administración de doble antiagregación es trasladado al Hospital de Cieza, donde continúa con fluctuación en la intensidad del dolor a pesar de la normalización de las alteraciones ECG. Cuando se confirma aumento de Troponina I (0.006 a 0.4) se consulta con nuestra unidad (UCI HUMM) acordando traslado a Servicio de Urgencias donde continúa asintomático y con ECG sin signos de isquemia aguda. Nuevo incremento ligero de Troponinas (1,5) decidiéndose ingreso en nuestra unidad.
Exploración física:
- Glasgow 15. Normocoloreado y normohidratado. TAS 118 mmHg. TAD 60 mmHg. No IY. Temperatura 36,2 ºC. FC 70 lpm. FR 16.
- AC: ruidos cardíacos rítmicos y apagados, no soplos.
- AR: murmullo vesicular disminuido de forma generalizada, sin ruidos sobreañadidos.
- Abdomen: blando, depresible, no doloroso a la palpación
- Extremidades: no edemas ni signos de TVP. Pulsos presentes y simétricos en las cuatro extremidades
- Exploración neurológica: sin alteraciones.
Pruebas complementarias:
- Anatítica: glucemia 111,0 mg/L. Creatinina 0,73 mg/L. Na 139,0 mmol/L. Troponina 1,557 µg/L. Hb 14,1 g/L. Hematocrito 41,5 L/L. Plaquetas 176,0 109/L. Actividad de Protrombina 110,0%. PTTA 19,0. FiO2 0,21. pH 7,31. pCO2 51,0 mmHg. LDH 553,0 mg/dL, Lactato 1,0 u int/L.
- Rx tórax: ICT normal. Campos pulmonares sin infiltrados ni condensaciones
- ECG: ritmo sinusal, 60 lpm. Alguna extrasístole ventricular aislada. Bloqueo incompleto de
25

!
www.medicalum.com
!
rama derecha del haz de His. Normalización de las alteraciones del ST y onda T descritas.
- Puntuación Grace 130, TIMI 3, CRUSADE 5, Killip I
- Coronariografía (ver Figura 2): acceso radial derecho. No se observan lesiones significativas en el tronco de coronaria
izquierda. Lesión severa en la descendente anterior (DA) proximal que produce estenosis del 95% (flujo TIMI III) y una placa ligera en la DA media. Sin lesiones significativas en circunfleja y coronaria derecha. Se identifica la lesión de la DA como culpable del SCASEST.
Figura 1. !Electrocardiograma pre-hospitalario.
F igura 2. Hallazgos coronariografía.
26

!
www.medicalum.com
!
DIAGNÓSTICO Diagnóstico al ingreso de IAMCEST de alto riesgo.
Podemos afirmar que se trata de un IAM debido al valor de troponinas que se detecta al ingreso (1,557 µg/L), el cual evidencia que existe daño con afectación miocárdica.
Asimismo, se evidencia que es un SCACEST ya que el enfermo presenta dolor torácico persistente, así como elevación del ST en al menos dos derivaciones contiguas.
El paciente, pese a declarar encontrarse asintomático al ingreso de nuestra unidad, en exploraciones posteriores refiere dolor centrotorácico, persistente desde el inicio de los síntomas.
Se trata de un caso de IAM con elevación transitoria del ST, de localización anteroseptal, ya que en el ECG prehospitalario encontramos la elevación del ST en las derivaciones aVR, V1 y V2 (característico de la obstrucción del tronco común de la coronaria izquierda y/o de la DA proximal).
TRATAMIENTO El paciente no es reperfundido en un inicio porque declara encontrarse asintomático y por presentar un ECG normal a su llegada a urgencias. Por ello, no se describe como SCACEST en un primer momento.
Intervención farmacológica inicial:
- Antiagregantes plaquetarios: ácido acetil salicílico (AAS) y clopidogrel.
- Anticoagulantes: enoxaparina.
- Hipolipemiantes: atorvastatina.
- Beta-bloqueantes: bisoprolol.
- Inhibidor bomba de protones gástrica: pantoprazol.
- Ansiolíticos (si precisa): lorazepam.
- Laxantes (si precisa): lactulosa.
Los días posteriores el dolor centrotorácico no remite de forma completa, intensificándose en repetidas
ocasiones, no siendo comunicado por parte del paciente hasta pasadas 48 horas. Las troponinas sufren un pico máximo de 13.915 µg/L (ver tabla 1). Tras recoger esta información y saber de la persistencia del dolor del paciente, se realiza la petición de cateterismo cardíaco preferente. En dicho procedimiento se objetiva estenosis del 95% en DA proximal.
Intervención quirúrgica: implantación de un Stent farmacoactivo en la DA proximal. Tras ello, ecocardiografía que presenta discreta hipocinesia de cara anterior, con fracción de eyección del ventrículo izquierdo (FEVI) visual conservada.
Se mantiene misma intervención farmacológica tras intervencionismo coronario. Tras pasar un día en observación en nuestra unidad es dado de alta a planta.
Se recomienda abandonar hábito tabáquico, así como una dieta más saludable y compaginar estos hábitos con ejercicio físico de manera regular.
EVOLUCIÓN Y PRONÓSTICO En los días siguientes descienden las troponinas, teniendo 1,74 µg/L en el alta a planta. El dolor centrotorácico va remitiendo casi por completo.
FEVI mayormente conservada, no existe una gran afectación funcional del miocardio. Queda una notable hipocinesia en la cara afectada, pudiendo limitar la realización de esfuerzos que precisen de aumento del GC.
Si el paciente se ajusta a las indicaciones terapéuticas, tanto farmacológicas como los cambios en el estilo de vida, es altamente probable que se pueda evitar cualquier otro accidente cardiovascular similar en el futuro.
DISCUSIÓN Y CONCLUSIONES Ante cualquier paciente con sospecha de IAMCEST, debe realizarse el tratamiento de reperfusión lo antes posible mediante intervención coronaria percutánea (ICP)3,4,5,6. El objetivo es realizar el diagnóstico antes
27

!
www.medicalum.com
!
de 10 minutos tras el primer contacto médico (PCM), e inmediatamente después, remitir al paciente directamente al laboratorio de cateterismos si se dispone de uno, o trasladándolo lo más rápido posible a un hospital con dicho equipamiento.
La fibrinólisis es una importante estrategia de reperfusión cuando la ICP primaria no puede realizarse dentro de los plazos recomendados (antes de 120 minutos desde inicio de los síntomas). Está recomendada en las primeras 12 horas1, preferiblemente iniciarlo lo antes posible.
En nuestro caso, tras el PMC, o a la llegada al servicio de urgencias del Hospital de Cieza, debía haber sido diagnosticado de SCACEST y haber sido remitido directamente al laboratorio de cateterismos para reperfusión, derivándose temporalmente a un servicio de UCI para monitorización continua si no fuera posible su intervención directa.
Cuando valoramos al paciente en UCI, unas 48 horas tras los síntomas iniciales, se solicita cita para cateterismo para realizar ICP primaria (indicada siempre que existan síntomas compatibles con isquemia1).
Se implanta stent metálico farmacoactivo debido a sus mejores resultados en comparación a stents metálicos7 y a la angioplastia de balón. No se recomienda aplazar su implantación si está indicada1.
Se recomienda la administración de un tratamiento antiagregante plaquetario doble (combinación de AAS + un inhibidor del P2Y12) y un anticoagulante parenteral1. Se recomienda mantener el AAS indefinidamente8, así como el inhibidor del P2Y12
durante al menos 6 meses (se recomiendan prasugrel o ticagrelor, y clopidogrel si los anteriores no están disponibles o están contraindicados)9.
Debe considerarse la administración de beta-bloqueantes a todo paciente tras un IAMCEST, aunque no está claro si existe beneficio claro para pacientes con FEVI >40%, ni la duración del tratamiento. No pueden establecerse recomendaciones al respecto puesto que no hay ningún estudio que las avale1.
Hay que administrar estatinas a altas dosis independientemente de la concentración de colesterol en la fase aguda, ya que se relaciona con beneficios a corto y largo plazo10.
Según los criterios PAMI-II, se trata de un paciente de bajo riesgo, por lo que puede recibir un alta temprana. Debe permanecer en UCI las primeras 24h tras ICP primaria, y otras 24-48h en monitorizado en planta.
Se considera clave en la prevención el abandono del hábito tabáquico como medida más efectiva de la prevención secundaria, debido a su efecto protombótico11. Además, se recomienda un control óptimo de la presión arterial, mejorar hábitos alimenticios y control del peso y la actividad física10.
REFERENCIAS 1. Ibáñez B, James S, Agewall S, Antunes M,
Bucciarelli-Ducci C, Bueno H et al. Guía ESC 2017 sobre el tratamiento del infarto agudo de miocardio en pacientes con elevación del segmento ST. Revista Española de Cardiología. 2017;70(12):1082.e1-1082.e61.
2. Thygesen K, Alpert J, Jaffe A, Simoons M, Chaitman B, White H et al. Third Universal Definition of Myocardial Infarction. Journal of the American College of Cardiology. 2012;60(16):1581-1598.
3. Keeley E, Boura J, Grines C. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. The Lancet. 2003;361(9351):13-20.
4. Andersen H, Nielsen T, Rasmussen K. A comparison of coronary angioplasty with fibrinolytic therapy in acute myocardial infarction. ACC Current Journal Review. 2003;12(6):47.
5. D'Souza S, Mamas M, Fraser D, Fath-Ordoubadi F. Routine early coronary angioplasty versus ischaemia-guided angioplasty after thrombolysis in acute ST-elevation myocardial infarction: a meta-analysis. European Heart Journal. 2010;32(8):972-982.
28

!
www.medicalum.com
!
6. Dalby M. Transfer for Primary Angioplasty Versus Immediate Thrombolysis in Acute Myocardial Infarction: A Meta-Analysis. Circulation. 2003;108(15):1809-1814.
7. Kastrati A, Dibra A, Spaulding C, Laarman G, Menichelli M, Valgimigli M et al. Meta-analysis of randomized trials on drug-eluting stents vs. bare-metal stents in patients with acute myocardial infarction. European Heart Journal. 2007;28(22):2706-2713.
8. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. The Lancet. 2009;373(9678):1849-1860.
9. Wallentin L, Becker R, Budaj A, Cannon C, Emanuelsson H, Held C et al. Ticagrelor versus Clopidogrel in Patients with Acute Coronary Syndromes. New England Journal of Medicine. 2009;361(11):1045-1057.
10. Piepoli M, Hoes A, Agewall S, Albus C, Brotons C, Catapano A et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice. European Heart Journal. 2016;37(29):2315-2381.
11. Chow C, Jolly S, Rao-Melacini P, Fox K, Anand S, Yusuf S. Association of Diet, Exercise, and Smoking Modification With Risk of Early Cardiovascular Events After Acute Coronary Syndromes. Circulation. 2010;121(6):750-758.
29

!
www.medicalum.com
!
A PROPOSITO DE UN CASO: SÍNDROME DE ALPORT AUTOSÓMICO DOMINANTE.
Martínez Segura, A.B.1 ; González Soriano, M. J.2 ; Martínez Jiménez, V.2 ; Morales Caravaca, F.2 (1): Facultad de Medicina, Universidad de Murcia
(2): Hospital Clínico Universitario Virgen de la Arrixaca (Murcia).
VALORACIÓN Y JUSTIFICACIÓN Lo consideramos un caso clínico de relevancia, puesto que para llegar al diagnóstico de esta nefropatía hereditaria fue importante estudiar los antecedentes personales y familiares, así como integrar la clínica con las exploraciones complementarias, teniendo que recurrir en última instancia al estudio genético. Además, nos recuerda que hay que tener presente que una enfermedad puede tener distintas variantes, con diferente expresión clínica, para poder pensar en ella y diagnosticarla, para así tratarla correctamente, intentado influir en el pronóstico de la enfermedad.
RESUMEN El síndrome de Alport (SA) es una enfermedad hereditaria que afecta a las membranas basales del glomérulo, aparato coclear y cristalino, causada por alteraciones en una de sus proteínas estructurales: el colágeno tipo IV. El SA se transmite mediante dos patrones de herencia: la ligada al cromosoma X y la autosómica recesiva. Clásicamente, se consideraba también la herencia autosómica dominante, pero actualmente dicha entidad queda englobada, junto a la hematuria familiar benigna y al estado de portador del SA autosómico recesivo, en la «nefropatía del colágeno IV, α3/α4»1,2. Presentamos a una paciente con SA con herencia autosómica dominante por mutación del gen COL4A3 en heterocigosis.
PALABRAS CLAVE Síndrome de Alport, colágeno tipo IV, hematuria, insuficiencia renal.
ABSTRACT Alport syndrome (AS) is a hereditary disease that affects the basal membranes of the glomerulus, the cochlear and crystalline apparatus, caused by alterations in one of its structural proteins: type IV collagen. The AS is transmitted by two inheritance patterns: linked to the X chromosome and autosomal recessive. Classically, autosomal dominant inheritance was also considered, but now this entity is included, together with the benign familial hematuria and the carrier state of the autosomal recessive AS, in the «nephropathy of collagen IV, α3/α4»1,2. We present a patient with AS with autosomal dominant inheritance due to mutation of the COL4A3 gene in heterozygosis.
KEYWORDS Alport syndrome, type IV collagen, hematuria; renal insufficiency.
INTRODUCCIÓN El SA es una nefropatía genéticamente heterogénea, que a menudo se asocia con alteraciones auditivas y oculares, y se caracteriza por alteraciones en las cadenas de colágeno α3, α4 y α5 de la membrana basal glomerular (MBG), codificadas por los genes COL4A3, COL4A4 y COL4A5, respectivamente. Mutaciones en estos genes del colágeno tipo IV afectan la estructura de la MBG, dando lugar a nefropatías cuyos síntomas oscilan desde la
hematuria aislada hasta la insuficiencia renal. Estas alteraciones renales han sido consideradas como distintas entidades: SA ligado al cromosoma X, SA autosómico recesivo, SA autosómico dominante, hematuria familiar benigna y portadores del SA autosómico recesivo. Pero el conocimiento molecular de estas enfermedades ha hecho posible agrupar las tres últimas en una entidad denominada «nefropatía del colágeno tipo IV». El modo de herencia del SA ha sido durante mucho tiempo controvertido, pero
30

!
www.medicalum.com
!
ahora se sabe que mutaciones del gen COL4A5 son responsables del SA ligado al cromosoma X, mutaciones en los genes COL4A3/COL4A4 son la base molecular el SA autosómico recesivo, y mutaciones en los genes COL4A3/COL4A4 en heterocigosis dan lugar a la «nefropatía del colágeno tipo IV, α3/α4». Estos descubrimientos tienen importantes consecuencias en el diagnóstico, pronóstico y asesoramiento genético de los pacientes1,2.
CASO CLÍNICO Motivo de consulta: mujer de 56 años que consulta por astenia.
Antecedentes personales:
-Alergia a metamizol.
-Factores de riesgo cardiovasculares:
Hipertensión arterial (HTA) conocida desde el año 2012 bien controlada con manidipino.
Hipercolesterolemia conocida desde el año 2012 bien controlada con atorvastatina.
No diabetes mellitus.
-Hábitos tóxicos:
Fumadora de 10 cigarrillos/día.
No hábito enólico.
-Antecedentes quirúrgicos:
Amigdalectomía a los 8 años de edad.
Cesárea en el año 1993.
-Antecedentes médicos:
Nefro-urológicos: microhematuria y proteinuria desde el año 2014 en seguimiento en consultas externas de Nefrología. En febrero de 2016, se solicitó una ecografía renal, que mostró unos riñones disminuidos de tamaño y una leve pérdida de la diferenciación corticomedular. En enero de 2017, la paciente rechazó la realización de biopsia renal y, ante sus antecedentes familiares nefropatológicos, se solicitó estudio genético por la sospecha de hematuria familiar, cuyo resultado estaba aún
pendiente de recibir. En marzo de 2017, aceptó realizarse la biopsia renal con resultado de: tres glomérulos con proliferación endocapilar y leve mesangial y focos de necrosis fibrinoide sin semilunas verdaderas; fibrosis e infiltrado crónico túbulo-intersticial; no rojo congo positivo; inmunofluorescencia (IF): vestigios de Inmunoglobulina M (IgM); y hallazgos sugestivos de vasculitis, descartada en base a la clínica y por tener anticuerpos anticitoplasma de neutrófilos (ANCA) negativos. Como tratamiento renal sustitutivo, la paciente había elegido la diálisis peritoneal, que aún no había comenzado a realizarse. Actualmente presentaba enfermedad renal crónica (ERC) en estadio 5.
Sin otros antecedentes médicos de interés.
-Situación basal: independiente para las actividades básicas de la vida diaria. Vive con su marido y un hijo. Actualmente jubilada; ha sido cocinera como única profesión.
-Tratamiento crónico: Manidipino 20mg (1-0-0), Atorvastatina 40mg (0-1-1) (3 veces/semana), Calcitriol 0,25mcg/48h y Bicarbonato sódico 1 sobre/día.
Antecedentes familiares:
-Padre fallecido en el año 1989 por infarto agudo de miocardio.
-Madre fallecida en el año 2000 por enfermedad renal en tratamiento con diálisis.
-Una hermana menor fallecida a los 36 años por enfermedad renal en tratamiento con diálisis. Además, padecía diabetes mellitus mal controlada.
-Un hermano diagnosticado a los 39 años de ERC (con proteinuria y microhematuria glomerular), el cual ha recibido dos trasplantes de riñón.
-Tiene otros seis hermanos (una mujer y cinco varones) sin patologías renales conocidas.
-Tiene cinco hijos y una hija sin patologías renales conocidas.
Enfermedad actual: paciente mujer de 56 años que acudió a urgencias por astenia progresiva asociada a
31
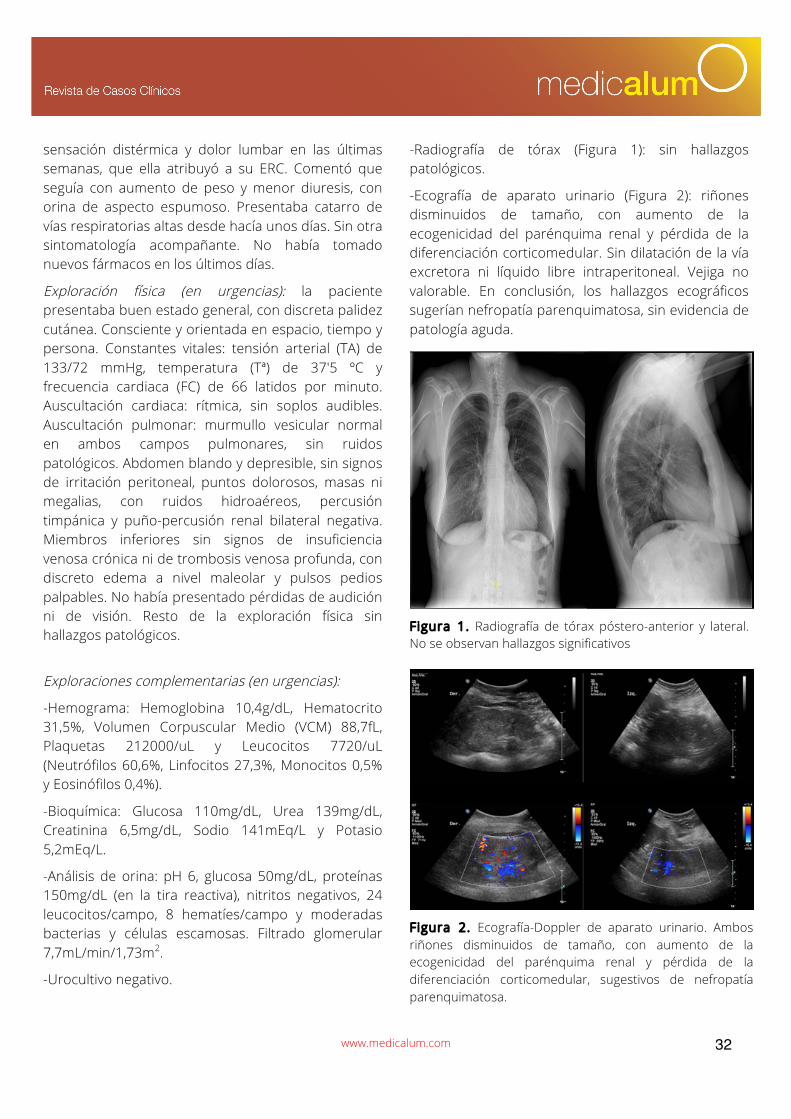
!
www.medicalum.com
!
sensación distérmica y dolor lumbar en las últimas semanas, que ella atribuyó a su ERC. Comentó que seguía con aumento de peso y menor diuresis, con orina de aspecto espumoso. Presentaba catarro de vías respiratorias altas desde hacía unos días. Sin otra sintomatología acompañante. No había tomado nuevos fármacos en los últimos días.
Exploración física (en urgencias): la paciente presentaba buen estado general, con discreta palidez cutánea. Consciente y orientada en espacio, tiempo y persona. Constantes vitales: tensión arterial (TA) de 133/72 mmHg, temperatura (Tª) de 37'5 ºC y frecuencia cardiaca (FC) de 66 latidos por minuto. Auscultación cardiaca: rítmica, sin soplos audibles. Auscultación pulmonar: murmullo vesicular normal en ambos campos pulmonares, sin ruidos patológicos. Abdomen blando y depresible, sin signos de irritación peritoneal, puntos dolorosos, masas ni megalias, con ruidos hidroaéreos, percusión timpánica y puño-percusión renal bilateral negativa. Miembros inferiores sin signos de insuficiencia venosa crónica ni de trombosis venosa profunda, con discreto edema a nivel maleolar y pulsos pedios palpables. No había presentado pérdidas de audición ni de visión. Resto de la exploración física sin hallazgos patológicos.
Exploraciones complementarias (en urgencias):
-Hemograma: Hemoglobina 10,4g/dL, Hematocrito 31,5%, Volumen Corpuscular Medio (VCM) 88,7fL, Plaquetas 212000/uL y Leucocitos 7720/uL (Neutrófilos 60,6%, Linfocitos 27,3%, Monocitos 0,5% y Eosinófilos 0,4%).
-Bioquímica: Glucosa 110mg/dL, Urea 139mg/dL, Creatinina 6,5mg/dL, Sodio 141mEq/L y Potasio 5,2mEq/L.
-Análisis de orina: pH 6, glucosa 50mg/dL, proteínas 150mg/dL (en la tira reactiva), nitritos negativos, 24 leucocitos/campo, 8 hematíes/campo y moderadas bacterias y células escamosas. Filtrado glomerular 7,7mL/min/1,73m2.
-Urocultivo negativo.
-Radiografía de tórax (Figura 1): sin hallazgos patológicos.
-Ecografía de aparato urinario (Figura 2): riñones disminuidos de tamaño, con aumento de la ecogenicidad del parénquima renal y pérdida de la diferenciación corticomedular. Sin dilatación de la vía excretora ni líquido libre intraperitoneal. Vejiga no valorable. En conclusión, los hallazgos ecográficos sugerían nefropatía parenquimatosa, sin evidencia de patología aguda.
Figura 1. Radiografía de tórax póstero-anterior y lateral. No se observan hallazgos significativos
Figura 2. Ecografía-Doppler de aparato urinario. Ambos riñones disminuidos de tamaño, con aumento de la ecogenicidad del parénquima renal y pérdida de la diferenciación corticomedular, sugestivos de nefropatía parenquimatosa.
32

!
www.medicalum.com
!
DIAGNÓSTICO Diagnóstico de sospecha: la paciente tenía una ERC, que evolucionó a terminal y precisaba iniciar diálisis, aun no filiada, con sospecha de una etiología hereditaria, que presentó un rápido deterioro de la función renal, por lo que nuestro diagnóstico de sospecha fue una ERC agudizada.
Diagnóstico diferencial3: hubo que descartar causas que hubieran podido acelerar ese deterioro de la función renal de la paciente:
Proceso infeccioso respiratorio: ante la clínica de catarro de vías respiratorias altas, se realizó una radiografía de tórax para descartar un foco pulmonar.
Proceso infeccioso urinario: la paciente no presentaba síndrome miccional ni fiebre, refería dolor lumbar, pero la puño-percusión renal bilateral fue negativa. En la analítica de sangre no había leucocitosis, y en la de orina había leucocituria, bacteriuria y microhematuria, pero los nitritos eran negativos y el urocultivo también negativo.
Uropatía obstructiva: la paciente no presentaba síntomas de un cólico nefrítico y en la ecografía no se halló ningún dato de patología obstructiva.
Síndrome urémico: la situación clínica de la paciente encuadraba dentro de la evolución de la ERC. La astenia y la palidez eran secundarias a la anemia (presente en este tipo de pacientes debido al déficit de eritropoyetina, principalmente).
Por otra parte, para intentar identificar la causa de la ERC, hubo que tener en cuenta que la paciente presentaba un síndrome nefrítico (hematuria, proteinuria en rango no nefrótico, HTA y fracaso renal), por lo que la patología estaba afectando al glomérulo. Las glomerulonefritis (GN) se clasifican en primarias y secundarias:
Las GN primarias que cursan con síndrome nefrítico son la endocapilar, membrano-proliferativa, rápidamente progresiva y mesangial IgA (enfermedad de Berger). De las cuales, la endocapilar, la
membrano-proliferativa y la rápidamente progresiva tipo II cursan con hipocomplementemia. Como esta paciente tenía unos niveles normales de complemento, nos quedamos como posibles candidatas la rápidamente progresiva tipo I y tipo III y la mesangial IgA. La rápidamente progresiva se descartó porque la paciente no tenía verdaderas semilunas en la biopsia (además, tampoco tenía anticuerpos contra la MBG ni depósitos lineales en la IF para ser la tipo I, ni ANCA para ser la tipo III). La mesangial IgA cursa con brotes de hematuria recidivantes coincidentes con infecciones, sobre todo respiratorias; pero en este caso la paciente ya presentaba hematuria mantenida desde hacía años sin relación con infecciones y en la biopsia solo se halló una leve proliferación mesangial con positividad vestigial para IgM, no para IgA, lo cual descarta esta GN.
Dentro de las GN secundarias encontramos como posibles candidatas en este caso las vasculitis, de las cuales, la poliangeítis microscópica y la granulomatosis de Wegener cursan con síndrome nefrítico, pero se descartaron por la clínica y por tener ANCA negativos, a pesar de que en la biopsia se hallaran signos sugestivos.
En las GN hereditarias se incluye el síndrome de Alport, que cursa con afectación renal (microhematuria y proteinuria, que la paciente sí presentaba), ocular y auditiva (que la paciente no presentaba). Se sospechaba que fuera ésta la causa de la ERC de la paciente por sus antecedentes familiares, por ello se había solicitado el estudio genético.
Diagnóstico definitivo: descartadas las causas de empeoramiento agudo de la función renal en urgencias y seguida la evolución de la paciente en planta, llegaron los resultados del estudio genético, con diagnóstico de síndrome de Alport con herencia autosómica dominante por mutación del gen COL4A3 en heterocigosis. Otros diagnósticos: hiperparatiroidismo secundario (por déficit de vitamina D), anemia multifactorial (por déficit de eritropoyetina y de ácido fólico) y acidosis metabólica.
33

!
www.medicalum.com
!
TRATAMIENTO Se ingresó a la paciente en planta para conseguir una mejoría clínica, ajustando el tratamiento farmacológico, y para valorar el inicio de la terapia renal sustitutiva.
Se inició pauta de ceftriaxona 1g intravenosa para cubrir una posible infección. Para el control de la tensión arterial se pautó amlodipino 5mg y nifedipino 10mg. Se mantuvo su tratamiento crónico con atorvastatina (se le bajó la dosis a 10mg), calcitriol 0,25mcg y bicarbonato sódico 500mg. Para tratar la anemia, se pautó hierro intravenoso y epoetina beta 3000UI. Ante unos niveles altos de fósforo y bajos de calcio, se pautó calcio carbonato 500mg. Durante su estancia, la paciente llevó una dieta baja en potasio y en sal. Todos los días se tomaban las constantes vitales (TA, FC y Tª), se controlaba la diuresis y se iban realizando analíticas de control. A los dos días de su ingreso, se le realizó un ecocardiograma para descartar derrame pericárdico urémico (pues sería una indicación urgente de iniciar diálisis), con resultado de función sistólica biventricular normal y mínima separación de hojas pericárdicas sin signos de compromiso hemodinámico asociado. También se le realizó una serología con resultados negativos para los virus de la hepatitis B y C y retrovirus.
EVOLUCIÓN Y PRONÓSTICO Cada día la paciente iba mejorando clínicamente (con menos astenia y sin edemas en miembros inferiores), hasta que fue dada de alta a los siete días con la siguiente analítica: glucosa 113mg/dL, urea 158mg/dL, creatinina 5,3mg/dL, ácido úrico 8,9mg/dL, sodio 142mEq/L, potasio 4,6mEq/L, filtrado glomerular 10,9mL/min/1,73m2, calcio 9,1mg/dL, fósforo 6,7mg/dL, 25-hidroxi vitamina D 8,8ng/mL, folato 2,7ng/mL, hemoglobina 10,2g/dL, hematocrito 30,9%, VCM 93,1fL, leucocitos 9030/uL, plaquetas 202000/uL, pH venoso 7,3, bicarbonato 19,2mmol/L y pCO2 40mmHg. Su tratamiento al alta fue el siguiente: dieta baja en sal, abandonar el hábito tabáquico, manidipino 20mg (1-0-0), atorvastatina 10mg (0-0-1), calcitriol 0,25mcg (0-0-1), bicarbonato 500mg (1-2-2), calcio carbonato 500mg (0-1-0), ácido fólico 5mg (1-0-0) y epoetina beta 3000UI (martes y
viernes). Al día siguiente del alta se citó para valorar la técnica de diálisis peritoneal y el día 13/10/2017 se le colocó el catéter.
DISCUSIÓN Y CONCLUSIONES La enfermedad renal crónica se define por la presencia de alteraciones en la estructura o función renal, expresadas por marcadores de daño renal o descenso del filtrado glomerular <60mL/min/1,73m2 durante más de tres meses4. Una de sus causas son las glomerulonefritis. El síndrome de Alport es una enfermedad hereditaria que afecta a las membranas basales del glomérulo, aparato coclear y cristalino, causada por alteraciones en una de sus proteínas estructurales: el colágeno tipo IV, el cual está formado por una triple hélice constituida por las cadenas α3, α4 y α5, codificadas por los genes COL4A3, COL4A4 y COL4A5, respectivamente. Cuando se altera una de las tres, la MBG se hace más susceptible al ataque proteolítico1.
Se estima que la prevalencia del SA en la población general es de 1:50.000 nacidos vivos1. Se transmite mediante dos patrones de herencia1,5:
-La ligada al cromosoma X (80%): causada por mutaciones en el gen COL4A5. Los hombres son enfermos y las mujeres portadoras, las cuales suelen tener pocos síntomas atribuibles a la enfermedad, pero en ocasiones pueden presentar una forma florida.
-La autosómica recesiva (15%): causada por mutaciones en los genes COL4A3 y COL4A4. Afecta por igual a hombres y mujeres.
Mutaciones en los genes COL4A3 o COL4A4 en heterocigosis dan lugar a la «nefropatía del colágeno tipo IV, α3/α4», que engloba a la hematuria familiar benigna, al SA autosómico dominante (5%) y a los portadores del SA autosómico recesivo2,5,6,7.
También se han descrito familias que presentan herencia digénica debido a la transmisión de mutaciones en dos de los tres genes (COL4A3, COL4A4, COL4A5)2.
34

!
www.medicalum.com
!
La tríada clínica característica del SA se caracteriza por afectación renal, auditiva y ocular. Ésta es la presentación clásica del SA, basada en las manifestaciones clínicas de los hombres afectados con SA ligado al cromosoma X. A nivel renal cursan con hematuria (signo guía), proteinuria significativa que puede llegar a ser de rango nefrótico (pero es excepcional que los afectados desarrollen un síndrome nefrótico), HTA y progresión hacia ERC terminal, con necesidad de diálisis a una edad media de 25 años. La afectación auditiva se caracteriza por hipoacusia bilateral neurosensorial para frecuencias altas y suele aparecer antes del desarrollo de la insuficiencia renal. A nivel ocular, se han descrito lesiones de córnea, cristalino (el lenticono anterior es el único signo patognomónico del SA) y retina en pacientes con SA, que parecen exclusivas de familias que también presenten sordera neurosensorial. Otras manifestaciones clínicas del SA incluyen la leiomiomatosis y la enfermedad arterial (aneurismas). La presentación clínica y el curso en pacientes con SA autosómico recesivo son similares a aquellos con SA ligado al cromosoma X. Los pacientes con SA autosómico dominante suelen tener sólo microhematuria, pero pueden desarrollar proteinuria e incluso insuficiencia renal a partir de la quinta década de la vida, siendo poco comunes la hipoacusia neurosensorial y las anomalías oculares1,2.
El diagnóstico de SA generalmente se sospecha a partir de los antecedentes familiares y se confirma mediante biopsia de piel o riñón o pruebas genéticas moleculares. La biopsia renal apoya el diagnóstico del SA, tras la sospecha clínica. Los hallazgos en el microscopio óptico son inespecíficos, se pueden observar células espumosas, aumento de la matriz mesangial y un patrón compatible con lesiones de glomerulonefritis segmentaria y focal. La IF suele ser negativa, pero pueden detectarse depósitos inespecíficos de IgM o C3. La microscopía electrónica es más específica: engrosamiento y adelgazamiento variable de la MBG y su lamelación. Existen dos tipos de diagnóstico genético: análisis de ligamiento (diagnóstico indirecto, apoyándose en la historia clínica) y análisis mutacional (diagnóstico directo, detectando la mutación específica)1,2,8.
No existe un tratamiento específico para el SA. Los inhibidores de la enzima convertidora de angiotensina (IECA) y los antagonistas de los receptores de la angiotensina II (ARA-II) han sido los únicos fármacos que han demostrado efectividad y seguridad. Su acción disminuye la proteinuria y frena la progresión de la ERC. El tratamiento renal sustitutivo de elección es el trasplante renal. El SA no recidiva en el trasplante, pero sí puede aparecer una enfermedad de Goodpasture mediada por autoanticuerpos contra la cadena de colágeno de la MBG que previamente el paciente no poseía o no expresaba y, por lo tanto, se comporta como un nuevo epítopo antigénico1,2,5,8.
Como conclusiones, las nefropatías del colágeno tipo IV están causadas por mutaciones en los genes COL4A3 y COL4A4, que codifican las cadenas α3 y α4 del colágeno tipo IV, de manera que se altera la MBG, produciendo síntomas que varían desde la hematuria aislada hasta la insuficiencia renal. Forman un amplio espectro clínico, que incluye el SA con herencia autosómica dominante, producido por mutaciones de dichos genes en heterocigosis, y cursa con una pérdida gradual de la función renal, siendo inusuales la afectación auditiva y ocular. El diagnóstico se sospecha con la clínica y antecedentes familiares del paciente, confirmándose mediante biopsia o estudio genético, y no tiene tratamiento específico.
REFERENCIAS 1. Torra R. Síndrome de Alport y nefropatía del colágeno IV (α3/α4). Revista de Nefrología Suplemento Extraordinario. 2011;2(1):29-37. 2. Kashtan CE. Clinical manifestations, diagnosis, and treatment of Alport syndrome (hereditary nephritis) [Internet]. Walthman (MA): Uptodate; 2017 [actualizada en marzo de 2018; citado el 30 de abril de 2018]. Disponible en: https://www.uptodate.com 3. Botella J. Manual de Nefrología Clínica. Barcelona: Masson; 2002.
35

!
www.medicalum.com
!
4. Grupo de trabajo de la Guía de Práctica Clínica sobre la Detección y el Manejo de la Enfermedad Renal Crónica. Guía de Práctica Clínica sobre la Detección y el Manejo de la Enfermedad Renal Crónica. Ministerio de Sanidad, Servicios Sociales e Igualdad. Instituto Aragonés de Ciencias de la Salud; 2016. Guías de Práctica Clínica en el SNS. 5. Kashtan CE, Ding J, Gregory M, Gross O, Heidet L, Knebelmann B et al. Clinical practice recommendations for the treatment of Alport syndrome: a statement of the Alport Syndrome Research Collaborative. Pediatr Nephrol 2013; 28:5-11. 6. Longo I, Porcedda P, Mari F, Giachino D, Meloni I, Deplano C et al. COL4A3/COL4A4 mutations: From familial hematuria to autosomal-dominant or recessive Alport síndrome. Kidney International. 2002; 61(6):1947-56. 7. Van der Loop F, Heidet L, Timmer E, Van den Bosch B, Leinonen A, Antignac C et al. Autosomal dominant Alport syndrome caused by a COL4A3 splice site mutation. Kidney International. 2000;58:1870-75. 8. Asociación para la información y la investigación de las enfermedades renales genéticas. Síndrome de Alport. Nefrogen. 2014;10:20-6.
36

!
www.medicalum.com
!
A PROPOSITO DE UN CASO: ICTUS EN EL CONTEXTO DE UNA FIBRILACIÓN AURICULAR ANTICOAGULADA.
Martínez Segura, A.B.1 ; Albert Lacal, L.2
(1): Facultad de Medicina, Universidad de Murcia (2): Hospital Clínico Universitario Virgen de la Arrixaca (Murcia).
VALORACIÓN Y JUSTIFICACIÓN Consideramos que este caso clínico es de relevancia, pues el ictus es uno de los motivos más frecuentes de asistencia neurológica urgente. Este caso clínico nos recuerda que hay que tener en cuenta los ictus de otros territorios vasculares e indagar en la causa de los mismos, guiándonos inicialmente por la historia clínica y, posteriormente, con las pruebas de neuroimagen, para un adecuado diagnóstico y tratamiento de la patología del paciente, condicionando así su pronóstico. Destacar también la importancia de valorar la anticoagulación oral en la prevención primaria y secundaria del ictus en un paciente con fibrilación auricular.
RESUMEN El ictus se define como una focalidad neurológica brusca espontánea y supone una urgencia neurológica. Los más frecuentes son los isquémicos, los de territorio carotídeo y los de origen aterotrombótico. Los de territorio vertebrobasilar suponen el 20% del total. La mayoría de infartos de territorio de la arteria cerebral posterior son de origen cardioembólico (sobre todo, por fibrilación auricular) y su clínica más frecuente es la hemianopsia homónima contralateral con respeto macular. En los pacientes con fibrilación auricular es importante valorar el riesgo de sufrir un ictus, con escalas como la de CHA2DS2-VASC, para instaurar la anticoagulación oral como prevención primaria. Como prevención secundaria, en los ictus de origen cardioembólico, se debe iniciar la anticoagulación entre uno y doce días tras sufrir un ictus isquémico. En caso de que un paciente en tratamiento anticoagulante sufra un ictus, se debe considerar el cambio a otro anticoagulante oral, teniendo indicación los de acción directa.
PALABRAS CLAVE Ictus, hemianopsia homónima, fibrilación auricular, anticoagulantes.
ABSTRACT Ictus is defined as an abrupt and spontaneous neurological focal and it supposes a neurological urgency. The most frequent ictus are: the ischemic ictus, the carotid territory ictus and the ictus with an aterotrombic origin. The vertebrobasilar territory ictus suppose the 20% of the total. Most of the territory strokes of the rear cerebral artery have a cardioembolic origin (mainly, due to the atrial fibrillation) and its most frequent clinical presentation is the contralateral homonymous hemianopsia with macular sparing. In the patients with atrial fibrillation it is important to value the risk of suffering an ictus with scales like the CHA2DS2-VASC to beggin with the oral anticoagulant. As a secondary prevention, in the ictus with a cardioembolic origin the anticoagulant must be initiated between one and twelve days after the ischemic ictus. In case that a patient with an anticoagulant treatment suffers an ictus, the change to another oral anticoagulant must be considered, having instruction the oral anticoagulant of direct action.
KEYWORDS
Stroke, homonymous hemianopsia, atrial fibrillation, anticoagulants.
37

!
www.medicalum.com
!
INTRODUCCIÓN La fibrilación auricular (FA) es la cardiopatía más importante como factor de riesgo de ictus y causa aproximadamente la mitad de los infartos cerebrales de origen cardioembólico1. El tratamiento anticoagulante puede prevenir la mayoría de los ictus isquémicos en pacientes con FA. El tratamiento con fármacos antagonistas de la vitamina K, como el acenocumarol, reduce el riesgo de ictus y la mortalidad, aunque su uso se ve limitado por el estrecho intervalo terapéutico, lo que requiere una monitorización frecuente, mediante el International Normalized Ratio (INR), que debe situarse entre 2 y 3, pero en rango terapéutico son efectivos para la prevención del ictus en pacientes con FA. Un INR inferior a 2 aumenta el riesgo de sufrir ictus isquémicos. Se debe tener en cuenta que un paciente que sufra un ictus isquémico de origen cardioembólico, éste tiene más riesgo de sufrir una transformación hemorrágica, y esto influirá en el inicio de la anticoagulación oral como prevención secundaria del ictus, en la que tendrán indicación los anticoagulantes orales de acción directa2.
CASO CLÍNICO Motivo de consulta: varón de 85 años que consulta por cefalea y alteración de la visión.
Antecedentes personales:
-Alergia a pirazolonas, oxicams y sulfamidas.
-Factores de riesgo cardiovasculares: Hipertensión Arterial (HTA) conocida desde el año 1985, en tratamiento farmacológico. Obesidad, con índice de masa corporal de 32,3 kg/m2, en tratamiento nutricional. No diabetes mellitus (DM) ni dislipemia (DLP).
-Hábitos tóxicos: no hábito tabáquico, enólico ni otros hábitos tóxicos.
-Antecedentes quirúrgicos: hernia discal en el año 1975, cataratas de ambos ojos en el año 2012 y colecistectomía en el año 2017.
-Antecedentes médicos: pericarditis vírica en el año 1980, hiperplasia benigna de próstata diagnosticada en el año 1999, enfermedad pulmonar obstructiva crónica diagnosticada en el año 2005, FA crónica diagnosticada en el año 2007, epididimitis izquierda en el año 2012, aplastamiento de cuerpo vertebral L1 en el año 2012, epicondilitis derecha en el año 2013, lumbociática en el año 2015 y gota diagnosticada en el año 2015.
-Situación basal: independiente para las actividades básicas de la vida diaria. Camina con ayuda de un bastón. Vive con su mujer. Actualmente jubilado (trabajó en una industria de productos químicos).
-Tratamiento crónico: Irbesartán e hidroclorotiazida 300/12,5mg 1 cada 24 horas, verapamilo hidrocloruro 120mg 1/24h, acenocuramol 4mg, terazosina hidrocloruro 5mg 1/24h, salmeterol xinafoato y fluticasona propionato 500/50mcg 1/12h, bromuro de tiotropio 18mcg 1/24h, salbutamol sulfato 100mcg en caso de disnea, pantoprazol sódico 20mg 1/24h y paracetamol 650mg 1/12h.
Antecedentes familiares: episodios de trombosis por la rama materna, sin saber especificar la localización, y DM por la rama paterna. Tiene dos hijas con HTA y DLP, pero sin patología cerebrovascular conocida. Sin otros antecedentes familiares de interés.
Enfermedad actual: paciente varón de 85 años que acudió a urgencias por presentar cefalea hemicraneal derecha de carácter opresivo a las 6h de la mañana, refiriendo que, al querer saber la hora que era, advirtió una alteración visual, consistente en pérdida de visión por el hemicampo visual izquierdo (veía solo la mitad derecha de la esfera del reloj, tanto con ambos ojos abiertos como tapándose uno y otro ojo), sin otra sintomatología acompañante. Se había acostado a dormir sobre las 22:30h asintomático. El paciente comentó que había recibido ajustes en la dosificación del acenocumarol recientemente, sin otros cambios en la medicación.
Exploración física (en urgencias): el paciente presentaba buen estado general, normocoloreado y normohidratado. Constantes vitales: tensión arterial
38

!
www.medicalum.com
!
(TA) de 169/77mmHg, temperatura (Tª) de 37,2ºC, frecuencia cardiaca de 75 latidos por minuto (lpm), frecuencia respiratoria de 12 respiraciones por minuto y saturación de oxígeno (Sat.O2) al 93%. Auscultación cardiaca: arrítmica, sin soplos audibles. Auscultación pulmonar: murmullo vesicular normal en ambos campos pulmonares, sin ruidos patológicos. Ambas carótidas sin soplos. Abdomen blando y depresible, sin signos de irritación peritoneal, puntos dolorosos, masas ni megalias, con ruidos hidroaéreos y percusión timpánica. Miembros inferiores con signos de insuficiencia venosa crónica, sin edemas ni signos de trombosis venosa profunda y pulsos distales bilaterales presentes. Resto de la exploración física sin hallazgos patológicos.
Exploración neurológica (en urgencias): alerta, consciente y orientado en espacio, tiempo y persona. Obedecía órdenes sencillas y complejas. No presentaba alteraciones de la memoria. Lenguaje fluente, sin disartria ni elementos afásicos, nominaba y repetía. No presentaba negligencias. Pupilas isocóricas y normorreactivas. Movimientos oculares extrínsecos sin restricciones ni nistagmos. Campimetría por confrontación: hemianopsia homónima izquierda. No presentaba diplopía. Reflejo nauseoso abolido. Resto de pares craneales normales. Balance muscular conservado en las cuatro extremidades, sin claudicación en maniobras antigravitatorias. Reflejos osteotendinosos presentes, bilaterales y simétricos. Reflejo cutáneo plantar flexor bilateral. No presentaba déficits sensitivos. No presentaba dismetrías con las maniobras dedo-nariz ni talón-rodilla, ni ataxia (Romberg negativo), ni alteraciones de la marcha. No presentaba signos meníngeos. National Institute of Health Stroke Scale (NIHSS) 2.
Exploraciones complementarias (en urgencias):
-Bioquímica: Glucosa 179mg/dL, Urea 30mg/dL, Creatinina 0,72mg/dL, Sodio 137mEq/L, Potasio 4,1mEq/L y Cloro 96 mEq/L.
-Hemograma y coagulación: Hemoglobina 15,3g/dL, Hematocrito 44,8%, Volumen Corpuscular Medio (VCM) 95,1fL, Plaquetas 220000/uL, Leucocitos
8150/uL (Neutrófilos 60,6%, Linfocitos 28,2%, Monocitos 10,2%, Eosinófilos 0,6% y Basófilos 0,4%) e INR 1,35.
-Gases en sangre venosa: pH 7,37, pCO2 55mmHg, pO2 37mmHg, Bicarbonato 30,8mmol/L, Anion Gap 1,3mmol/L, Sat.O2 65,8%, Hematocrito 49%, Ion sodio 136mmol/L, Ion potasio 3,8mmol/L, Ion cloruro 103mmol/L, Calcio iónico 1,15mmol/L, Glucosa 179mg/dL e Ion lactato 3,5mmol/L.
-Electrocardiograma (ECG) (Figura 1): arritmia cardiaca por FA a 70 lpm, QRS estrecho con eje -30º, sin alteraciones del ST ni de la repolarización.
-Radiografía de tórax: índice cardiotorácico normal, senos costodiafragmáticos libres y sin signos de redistribución vascular, masas ni infiltrados. Sin hallazgos patológicos.
-Tomografía axial computarizada (TAC) craneal simple (Figura 2): tenue hipodensidad córtico-subcortical occipital derecha en territorio de arteria cerebral posterior (ACP) y cerebelosa derecha en territorio de la arteria cerebelosa postero-inferior (PICA), sugerentes de proceso isquémico agudo-subagudo. Hipodensidad córtico-subcortical parietal izquierda en territorio de arteria cerebral media izquierda, probablemente relacionada con lesión isquémica crónica. Área de encefalomalacia córtico-subcortical izquierda en territorio de PICA, atribuible a lesión isquémica crónica. Infartos lacunares en ganglios basales y en región frontal derecha. Discreta ampliación del sistema ventricular y surcos de la convexidad en relación con la edad. Estructuras de la línea media centradas. Afaquia bilateral. En conclusión, los hallazgos sugerían lesión occipital derecha y cerebelosa derecha establecida (aguda/subaguda).
Exploraciones complementarias (en planta de hospitalización):
-TAC craneal simple de control (Figura 3): transformación hemorrágica del infarto isquémico subagudo del territorio de la ACP derecha.
39

!
www.medicalum.com
!
-Ecografía-Doppler de troncos supraaórticos y transcraneal: abundantes placas ateromatosas calcificadas en ambas bifurcaciones carotídeas, de predominio en bulbo carotídeo, sin presentar estenosis
hemodinámicamente significativas en el sistema carotideo ni en el sistema vertebrobasilar, ni shunt intracardiaco derecha-izquierda.
Figura 1. Electrocardiograma. Arritmia cardiaca por fibrilación auricular crónica a 70 lpm, QRS estrecho con eje -30º, sin alteraciones del ST ni de la repolarización.
Figura 2. Tomografía axial computarizada craneal simple. Lesión isquémica aguda-subaguda occipital derecha.
Figura 3. Tomografía axial computarizada craneal simple de control. Transformación hemorrágica del infarto isquémico occipital derecho.
40

!
www.medicalum.com
!
DIAGNÓSTICO Diagnóstico de sospecha: ICTUS ISQUÉMICO OCCIPITAL DERECHO.
Diagnóstico diferencial: ante la clínica presentada por el paciente, nos preguntamos si lo que había sufrido había sido un ictus u otra entidad clínica. Debido a la presencia de focalidad neurológica brusca espontánea, primero pensamos en un ictus, cuyo origen lo indica la TAC craneal simple, que en el caso de este paciente se trataba de un ictus isquémico. Para diferenciar si es un accidente isquémico transitorio (AIT) o un ictus establecido, nos basamos en la duración de la clínica y en los resultados de las pruebas de neuroimagen. Para localizar el ictus nos basamos en la clínica, corroborada por la TAC, que en el caso de nuestro paciente era occipital derecho. Por último, había que investigar la etiopatogenia del ictus mediante pruebas complementarias, que en este caso era cardioembólica por FA. Así pues, las distintas patologías con las que hacer el diagnóstico diferencial se comentan a continuación:
-Hemorragia intraparenquimatosa: el paciente no presentaba vómitos ni deterioro progresivo del nivel de conciencia. Además, en la TAC realizada en urgencias no se evidenció hemorragia.
-Hemorragia subaracnoidea: el paciente presentaba alteración visual y cefalea, pero ésta no fue de intensidad grave, y tampoco presentó náuseas, vómitos, meningismo ni alteración del nivel de conciencia. Además, en la TAC realizada en urgencias no se evidenció hemorragia.
-Tumor cerebral: el paciente no presentaba síndrome constitucional ni antecedentes de tumor primario en otra localización. Además, en la TAC no se evidenció masa tumoral en el parénquima cerebral.
-Infección (encefalitis, absceso): el paciente no presentaba fiebre, disminución del nivel de conciencia ni otras alteraciones analíticas que orientaran hacia esta etiología. Además, en la TAC no se evidenció ningún absceso cerebral ni edema.
-AIT: se descartó porque la clínica duró más de 24 horas, aunque en intensidad decreciente y hubo evidencia de infarto en las técnicas de neuroimagen (TAC).
-Ictus de la arteria cerebral media derecha: las radiaciones ópticas atraviesan el lóbulo parietal y temporal, por lo que un infarto de la arteria cerebral media derecha puede provocar hemianopsia homónima izquierda, pero el paciente no presentaba hemiparesia izquierda, hemihipoestesia izquierda, afasia, desviación de la mirada hacia la derecha ni anosognosia. Además, en la TAC no se observó infarto de este territorio.
-Ictus de la ACP derecha: las radiaciones ópticas alcanzan la corteza visual a nivel occipital, pudiendo su lesión ocasionar hemianopsia homónima izquierda. Por ser ésta la clínica del paciente y objetivar hipodensidad en dicho territorio en la TAC, se llegó a este diagnóstico.
Diagnóstico definitivo: ICTUS ISQUÉMICO SUBAGUDO EN TERRITORIO DE ACP DERECHA DE ORIGEN CARDIOEMBÓLICO, CON TRANSFORMACIÓN HEMORRÁGICA.
TRATAMIENTO Se le suspendió el acenocumarol y se le administró enoxaparina sódica 40mg y ácido acetilsalicílico 100mg como prevención secundaria del ictus, manteniendo el resto de su medicación habitual. Por los hallazgos en la TAC de control, se decidió demorar el inicio de la anticoagulación, sustituyendo acenocuramol por rivaroxabán. El tratamiento al alta fue el siguiente: dieta baja en sal, irbesartán e hidroclorotiazida 300/12,5mg 1 cada 24 horas, verapamilo hidrocloruro 120mg 1/24h, terazosina hidrocloruro 5mg 1/24h, salmeterol xinafoato y fluticasona propionato 500/50mcg 1/12h, bromuro de tiotropio 18mcg 1/24h, salbutamol sulfato 100mcg en caso de disnea, pantoprazol sódico 20mg 1/24h, paracetamol 650mg 1/8h hasta el cese del dolor, metilprednisolona 4mg 1/24h durante 5 días,
41

!
www.medicalum.com
!
suspender acenocuramol 4mg e iniciar rivaroxabán 15mg en una semana.
EVOLUCIÓN Y PRONÓSTICO El paciente fue trasladado a la unidad de ictus, donde permaneció clínica y hemodinámicamente estable. A las 24h de observación y monitorización en la unidad de ictus, debido a una mejoría de la alteración visual, quedó ingresado en la planta de hospitalización de Neurología. Quedó asintomático a las 72h del inicio de la clínica. En ningún momento durante su estancia hospitalaria presentó otra sintomatología ni focalidad neurológica. Como único incidente, al cuarto día, el paciente refirió gonalgia izquierda, por lo que se le exploró, se solicitó una radiografía simple de rodilla izquierda en proyección anteroposterior y lateral, en la que se observaron signos de artrosis severa sin patología ósea aguda, y se realizó interconsulta a Reumatología, quienes refirieron tendinitis del bíceps crural, por lo que se añadió al tratamiento metilprednisolona 4mg. Al sexto día fue dado de alta.
DISCUSIÓN Y CONCLUSIONES El ictus está causado por un trastorno en la circulación cerebral, ocasionando una focalidad neurológica brusca espontánea, debido a una alteración transitoria o definitiva del funcionamiento de una o varias regiones del encéfalo. Según su naturaleza, puede ser isquémico (85%) o hemorrágico (15%). El ictus isquémico está ocasionado por la alteración del aporte circulatorio a un territorio encefálico, lo cual produce un déficit neurológico durante más de 24 horas e indica la presencia de una necrosis tisular, con su correspondiente evidencia en las pruebas de neuroimagen. Su principal factor de riesgo es la HTA. Su etiología más frecuente es la aterotrombótica, seguida de la cardioembólica (15-20%). El infarto de origen cardioembólico suele ser de tamaño medio o grande, de topografía habitualmente cortical, siendo imprescindible la presencia de una cardiopatía embolígena demostrada (siendo la más frecuente la FA) y la ausencia de oclusión o estenosis arterial significativa de forma concomitante1,3.
Según la topografía vascular, el infarto puede ser de territorio carotídeo o anterior, de territorio vertebrobasilar o posterior, de territorio frontera, venoso, de vaso arterial grande o de vaso arterial pequeño. Según la topografía parenquimatosa, puede ser infarto de la circulación anterior, infarto de la circulación posterior o infarto lacunar. El 20% de los eventos isquémicos en el cerebro involucran estructuras de la circulación vertebrobasilar o posterior1,4.
Como recordatorio de la circulación vertebrobasilar, de las arterias vertebrales surgen la arteria espinal anterior y las arterias cerebelosas posteroinferiores. Las dos arterias vertebrales se unen para formar la arteria basilar, de la cual nacen las arterias cerebelosas anteroinferiores, las arterias cerebelosas superiores, ramas para protuberancia y mesencéfalo, y las ACP. Las causas más comunes de isquemia del territorio de la circulación posterior son la aterosclerosis, la embolia y la disección. La mayoría de los infartos en el territorio de la ACP se deben a una embolia procedente del corazón, aorta o arterias vertebrales. Según el territorio irrigado por las ACP, se pueden producir los siguientes hallazgos1,4:
-Lóbulos occipitales: la clínica más frecuente de infarto de ACP es la hemianopsia homónima aislada contralateral con preservación macular5.
-Lóbulos temporales mediales: trastornos transitorios de la memoria, consistentes en amnesia anterógrada.
-Tálamo: anomalías somatosensitivas, con parestesias en cara, extremidades y tronco.
Ante un paciente con un cuadro clínico sugestivo de un ictus, el proceso debe ir dirigido a confirmar el diagnóstico de ictus y descartar otras entidades clínicas, determinar el tipo de ictus, establecer la topografía y extensión de la lesión encefálica, conocer la situación del sistema vascular y saber cuál es su etiopatogenia. El proceso diagnóstico incluye1:
-Historia clínica: se debe prestar una especial atención a los antecedentes vasculares, tanto personales como familiares, y la detección de otros factores de riesgo vascular.
42

!
www.medicalum.com
!
-Exploración general y neurológica: confirma la sospecha de una focalidad neurológica y orienta hacia la topografía del ictus. Los defectos en el campo visual, como la hemianopsia, pueden explorarse mediante pruebas manuales de confrontación o mediante campímetros automatizados. La NIHSS, con 11 ítems, es la escala más empleada para la valoración de las funciones neurológicas básicas (funciones corticales, pares craneales superiores, función motora, sensibilidad, coordinación y lenguaje) en la fase aguda del ictus isquémico, tanto al inicio como durante su evolución, teniendo valor pronóstico.
-Exploraciones complementarias:
• Analítica: pruebas hematológicas y bioquímicas de forma sistemática, ampliando el estudio según los datos de la historia clínica.
• Radiografía de tórax: a todos los pacientes, para la valoración de la silueta cardiaca, aportando indicios de posibles cardiopatías embolígenas, e información sobre posibles complicaciones del ictus.
• Neuroimagen:
-TAC craneal simple: de manera urgente, para excluir lesiones de origen no vascular y diferenciar los ictus isquémicos de los hemorrágicos. Además, permite identificar infartos crónicos, cuyas características topográficas pueden ayudar a establecer el diagnóstico etiopatogénico del ictus actual. También permite observar la existencia de una transformación hemorrágica del infarto cerebral durante los primeros días siguientes a éste, circunstancia que se produce de forma espontánea hasta en el 65% de los infartos cerebrales y hasta en el 90% de los de origen cardioembólico.
-La resonancia magnética (RM) craneal resulta útil en el tratamiento del ictus, pues ayuda a confirmar y localizar topográficamente los infartos. Con las secuencias de difusión y perfusión se puede identificar el tejido en
penumbra o mismatch (potencialmente recuperable con tratamientos recanalizadores).
• Estudio neurovascular por ultrasonidos: se debe realizar un estudio doppler de troncos supraaórticos y transcraneal en todos los pacientes que han experimentado un ictus isquémico.
• Estudios angiográficos craneocervicales: incrementa la especificidad diagnóstica y mejora la selección de pacientes candidatos a terapia trombolítica. En la actualidad, la técnica de referencia es la angiografía por sustracción digital. Sin embargo, con la progresiva utilización de técnicas no invasivas, como las angiografías por TAC y RM, que alcanzan aceptables niveles de precisión diagnóstica, se han reducido los procedimientos angiográficos invasivos con fines puramente diagnósticos.
• Evaluación cardiaca: ECG a todos los pacientes al ingreso y a las 24h, pudiendo ampliar el estudio con Holter y ecocardiografía. La valoración de cardiopatías potencialmente embolígenas se puede realizar en base a la anamnesis, la exploración física, la radiografía de tórax y el ECG, para así poder instaurar un tratamiento preventivo anticoagulante.
Respecto al tratamiento de la fase aguda del ictus isquémico, ya sea de territorio anterior o posterior, las medidas terapéuticas orientadas a la repermeabilización del vaso ocluido y a incrementar la resistencia del cerebro frente a la isquemia (neuroprotección) solo serán eficaces si se aplican durante las primeras horas desde el inicio de los síntomas. En cuanto a la repermeabilización del vaso, hay dos posibles opciones: trombólisis con activador tisular del plasminógeno recombinante (rtPA) intravenoso y/o trombectomía mecánica. La trombólisis con rtPA está indicada en pacientes con menos de 4,5h de evolución y está contraindicada en pacientes tratados con fármacos antivitamina K (AVK), salvo si el INR está por debajo de 1,7. Tras la repermeabilización del vaso, o si ésta no se ha podido realizar, se ingresará al paciente en la unidad de ictus1,3.
43

!
www.medicalum.com
!
Sobre los cuidados generales, el control intensivo de la TA, Tª y glucemias en las primeras horas es lo que ha demostrado mayor beneficio en el tratamiento del ictus (disminuye la mortalidad y las secuelas)1,3.
Respecto al tratamiento preventivo de la isquemia cerebral, la prevención primaria está orientada a la actuación sobre los factores de riesgo vascular modificables o potencialmente modificables: tratamiento antihipertensivo con diuréticos tiazídicos, inhibidores de la enzima convertidora de angiotensina (IECA) o antagonistas del receptor de angiotensina II (ARA II); tratamiento hipolipemiante con estatinas en pacientes que tengan además otros factores de riesgo vascular, según su nivel de riesgo; y tratamiento anticoagulante en caso de cardiopatías embolígenas como la FA1,3,6.
El tratamiento anticoagulante puede prevenir la mayoría de los ictus isquémicos en pacientes con FA, siendo superior a no tratar o a tratar con ácido acetilsalicílico. La decisión de iniciar anticoagulación en un paciente con FA se lleva a cabo determinando el riesgo tromboembólico y hemorrágico mediante escalas. Para la evaluación del riesgo tromboembólico se dispone de la escala CHA2DS2-VASc, que son las siglas en inglés correspondientes a: Congestive Heart Failure/Left ventricular dysfunction, Hypertension, Age ≥75 years, Diabetes Mellitus, previous Stroke/transient ischaemic attack/thromboembolism, Vascular disease, Age 65-74 years y Sex category (female). Para valorar el riesgo de hemorragia se recomienda la escala HAS-BLED, cuyas siglas en inglés corresponden a: Hypertension, Abnormal kidney and/or liver function, Stroke, Bleeding, Labile INR, Elderly y Drugs and/or alcohol, indicando alto riesgo de sangrado una puntuación ≥3. En general, los pacientes sin factores de riesgo de ictus no necesitan terapia antitrombótica, mientras que en los pacientes con factores de riesgo (es decir, CHA2DS2-VASc de 1 o más en hombres, y 2 o más en mujeres) se debe considerar la anticoagulación oral. El tratamiento con anticoagulantes orales (también denominados AVK o cumarínicos, como el acenocumarol o la warfarina) reducen el riesgo de ictus y la mortalidad, aunque su uso se ve limitado por el estrecho intervalo
terapéutico, lo que requiere una monitorización frecuente (mediante el INR) y ajustes de dosis, pero en rango terapéutico son efectivos para la prevención del ictus en pacientes con FA. El intervalo normal del INR se sitúa entre 0,9 y 1,2, pero en una persona en tratamiento con AVK debe estar entre 2 y 3. Un INR inferior a 2 conlleva un tiempo de protrombina corto y una mayor coagulabilidad de la sangre, aumentando el riesgo de trombosis e ictus isquémicos. Mientras que un INR superior a 3 traduce un tiempo de protombina largo y una mayor dificultad para la coagulación, que puede conducir a hemorragias e ictus hemorrágicos. Los nuevos anticoagulantes orales (también denominados anticoagulantes orales de acción directa –ACOD-), que incluyen el inhibidor directo de la trombina (dabigatrán) e inhibidores del factor X activado (apixabán, edoxabán y rivaroxabán) son alternativas adecuadas a los AVK en la prevención del ictus, con la ventaja de tener un efecto predecible sin necesidad de monitorización. Tanto los AVK como los ACOD son efectivos para la prevención del ictus en la FA, pero los ACOD son alternativas terapéuticas válidas, con un perfil beneficio/riesgo favorable, en FA no valvular y con al menos un factor de riesgo adicional de complicaciones tromboembólicas2,6.
El mecanismo patogénico y la presentación clínica de cada tipo de isquemia cerebral condicionan diferentes opciones de prevención secundaria. En general, están indicados los antiagregantes plaquetarios, salvo en los ictus de origen cardioembólico, que se optará por los anticoagulantes. Se propone iniciar la anticoagulación en pacientes con FA entre 1 y 12 días después de un ictus isquémico, dependiendo de la gravedad del ictus. Se sugiere repetir una prueba de neuroimagen para determinar el inicio óptimo de la anticoagulación en pacientes con un ictus en riesgo de transformación hemorrágica3,6.
La anticoagulación a largo plazo con un AVK o un ACOD aporta beneficios en pacientes con FA que sobreviven a un ictus. Los ACOD parecen aportar resultados ligeramente superiores, principalmente por menos hemorragias intracraneales e ictus hemorrágicos. Si un paciente en tratamiento
44

!
www.medicalum.com
!
anticoagulante sufre un ictus o AIT, se debe considerar el cambio a otro anticoagulante. Los pacientes con antecedentes de hemorragia intracraneal, o ictus isquémico que presenten criterios clínicos y de neuroimagen de alto riesgo de hemorragia intracraneal, o en los que no es posible mantener un control del INR dentro de rango a pesar de un buen cumplimiento terapéutico, tienen indicación de ACOD2,6.
También como prevención secundaria, se realizará endarterectomía carotídea en las estenosis importantes, o angioplastia carotídea con colocación de stent en casos de alto riesgo quirúrgico. Se recomienda tratamiento con IECA combinado con un diurético o ARA II, con el objetivo terapéutico de TA inferior a 130/80mmHg, y tratamiento con estatinas, con el objetivo terapéutico de unos valores de lipoproteínas de baja densidad (LDL) inferiores a 100mg/dL1,3.
En cuanto al pronóstico, la evolución del ictus depende de la gravedad de la sintomatología neurológica, la presencia o ausencia de lesiones arteriales, la localización y extensión del infarto y el mecanismo de la isquemia. La tasa de mortalidad inmediatamente después del ictus de la circulación posterior es aproximadamente del 3-4%. El embolismo cardíaco y el compromiso de la arteria basilar y de territorios intracraneales múltiples aumentan el riesgo de una peor evolución, independientemente de la edad y los factores de riesgo del paciente. En lo referente a la hemianopsia homónima, muchos pacientes se recuperan espontáneamente, más de la mitad en el primer mes. La recuperación después de seis meses es poco probable, a menos que haya una mejoría de la enfermedad subyacente. Las opciones de tratamiento son limitadas e incluyen técnicas de rehabilitación1,3,5.
Como conclusiones, el ictus es una urgencia neurológica. Los ictus de territorio de ACP suelen ser de origen cardioembólico y su clínica más frecuente es la hemianopsia homónima contralateral con respeto macular. Ante un paciente con una hemianopsia homónima, hay que considerar como causa más frecuente una lesión en el lóbulo occipital,
que con mayor frecuencia tendrá un origen vascular. Como la FA es una arritmia con potencial embolígeno, es muy importante valorar la prevención primaria y secundaria de sufrir un ictus en estos pacientes mediante tratamiento anticoagulante.
REFERENCIAS 1. Sociedad española de Neurología. Guía oficial para el diagnóstico y tratamiento del ictus. 3ª ed. Barcelona: Prous Science; 2004. 2. Agencia Española de Medicamentos y Productos Sanitarios. Criterios y recomendaciones generales para el uso de los anticoagulantes orales directos (ACOD) en la prevención del ictus y la embolia sistémica en pacientes con fibrilación auricular no valvular. Noviembre 2016. Disponible en: https://www.aemps.gob.es/medicamentosUsoHumano/informesPublicos/docs/criterios-anticoagulantes-orales.pdf 3. Ustrell-Roig X, Serena-Leal J. Ictus: Diagnóstico y tratamiento de las enfermedades cerebrovasculares. Rev Esp Cardiol 2007; 60 (7): 753-69. 4. Caplan LR. Posterior circulation cerebrovascular syndromes [Internet]. Walthman (MA): Uptodate; 2017 [actualizado en marzo de 2018; citado el 14 de abril de 2018]. Disponible en: https://www.uptodate.com 5. Biousse V, Kedar S, Newman NJ. Homonymous hemianopia [Internet]. Walthman (MA): Uptodate; 2017 [actualizado en marzo de 2018; citado el 14 de abril de 2018]. Disponible en: https://www.uptodate.com 6. Kirchhof P, Benussi S, Kotecha D, Ahlsson A, Atar D, Casadei B et al. Guidelines for the management of atrial fibrillation developed in collaboration with EACTS. European Heart Journal. 2016; 37: 2921-23.
45

!
www.medicalum.com
!
PSEUDOMIXOMA PERITONEAL. UNA ENTIDAD INFRECUENTE CON UN TRATAMIENTO POCO CONOCIDO.
Simón Frapolli, V.1 ; González Gil, A.2; Gil Martínez, J.2
(1): Facultad de Medicina, Universidad de Murcia (2): Hospital Clínico Universitario Virgen de la Arrixaca (Murcia).
VALORACIÓN Y JUSTIFICACIÓN
El Pseudomixoma peritoneal es una entidad que puede considerarse por estadística como una “enfermedad rara” y que precisa de un tratamiento súperespecializado y poco conocido por la comunidad de estudiantes de Medicina. Su tratamiento actual permite ofrecer opciones reales de curación que con el manejo clásico no existían.
RESUMEN El Pseudomixoma peritoneal es una rara entidad clínica caracterizada por la diseminación de abundante moco en el interior de la cavidad peritoneal. En aproximadamente el 80% de los casos tiene su origen en una neoplasia mucinosa localizada en el apéndice cecal, que se rompe a la cavidad peritoneal. Con menor frecuencia, también puede deberse a tumores mucinosos localizados en el ovario, colon o estómago. Se trata de una entidad de lenta progresión, cuyo diagnóstico es fundamentalmente clínico.
El tratamiento es quirúrgico siempre que sea posible, y existe un abordaje terapéutico clásico, practicado hasta recientes fechas. Este ha consistido en la realización de laparotomías repetitivas con el objetivo de evacuar las ingentes cantidades de moco que invaden y ocupan progresivamente la cavidad peritoneal, siendo normalmente un tratamiento no curativo a largo plazo, y los pacientes fallecen generalmente por un cuadro de obstrucción intestinal intratable. El abordaje actual, que consiste en la realización de una citorreducción quirúrgica agresiva combinada con la administración de quimioterapia intraperitoneal intraoperatoria hipertérmica (HIPEC), ha supuesto una gran mejoría en el pronóstico de la enfermedad con respecto al abordaje clásico. Además, es interesante la determinación de los valores de algunos marcadores tumorales (CEA, CA 19.9 y CA 125), cuya elevación se ha relacionado de forma negativa con la supervivencia de los pacientes, constituyendo una herramienta excelente para el seguimiento de esta enfermedad.
PALABRAS CLAVE Pseudomixoma, peritoneo, pseudomixoma peritoneal, cirugía, quimioterapia hipertérmica intraperitoneal.
ABSTRACT Peritoneal Pseudomyxoma is a rare clinical entity characterized by the dissemination of abundant mucous in the interior of the peritoneal cavity. In approximately 80% of the cases it has its origin in a mucinous neoplasia located in the cecal appendix, which breaks into the peritoneal cavity. Less frequently, it can also be due to mucinous tumors located in the ovary, colon or stomach. It is an entity of slow progression, whose diagnosis is fundamentally clinical.
The treatment is surgical whenever possible, and there is a classic therapeutic approach, practiced until recent dates. This has consisted in performing repetitive laparotomies in order to evacuate the huge amounts of mucous material that invades and progressively occupies the peritoneal cavity, usually being a non-curative treatment in the long term, and patients usually die due to an intractable intestinal obstruction. . The current approach, which consists of performing an aggressive surgical debulking combined with the administration of hyperthermic intraoperative intraperitoneal chemotherapy (HIPEC), has led to a great improvement in the prognosis of the disease with respect to the classic approach. In addition, it is interesting to determine the values of some tumor markers (CEA, CA 19.9 and CA 125), whose elevation has been negatively related to the survival of patients, constituting an excellent tool for the follow-up of this disease.
KEYWORDS Pseudomyxoma, peritoneum, pseudomyxoma peritonei, surgery, hyperthermic intraperitoneal chemotherapy.
46

!
www.medicalum.com
!
INTRODUCCIÓN El pseudomixoma peritoneal es una entidad clínica caracterizada por la presencia de ascitis mucinosa en la cavidad peritoneal que, en la mayoría de las ocasiones, se produce de forma secundaria a la rotura de una neoplasia mucinosa apendicular1,2. El diagnóstico de la enfermedad se puede establecer en diferentes escenarios clínicos: como hallazgo incidental durante una cirugía de urgencias, frecuentemente por apendicitis aguda; en el transcurso del estudio de una neoplasia de ovario; durante el estudio de un paciente con ascitis y distensión abdominal; e incluso de formas más peregrinas como la cirugía de una hernia de la pared abdominal o como hallazgo casual durante otra prueba diagnóstica realizada por otros motivos.
El abordaje actual, basado en la realización de una citorreducción quirúrgica agresiva para la eliminación de la enfermedad macroscópica, acompañado de la administración de HIPEC para el tratamiento de la enfermedad microscópica, ofrece opciones reales de supervivencia a largo plazo y curación de la enfermedad3. En el presente trabajo, presentamos un caso clínico típico de Pseudomixoma peritoneal y discutimos algunos aspectos importantes de su manejo.
CASO CLÍNICO Mujer de 36 años, sin antecedentes personales de interés, que consulta al servicio de Urgencias por dolor abdominal. La paciente no refiere alergias conocidas a fármacos y está diagnosticada de ovarios poliquísticos en seguimiento por Ginecología. Intervenida de un sinus pilonidal hacía ya 10 años y de una cesárea de su tercer hijo de 7 años (G3A0P2C1).
El día de la consulta a Urgencias, la paciente refirió un dolor abdominal localizado en la Fosa Ilíaca Derecha (FID), de unas 24 horas de evolución, sin otra sintomatología acompañante. El dolor, continuo, no se modificaba con la postura corporal. No presentaba fiebre ni alteraciones del hábito intestinal ni urinario.
En la anamnesis, la paciente venía refiriendo cuadros similares desde hacía 6 meses, pero de menor intensidad y de carácter autolimitado. La exploración abdominal demostraba la presencia de una cicatriz de cesárea previa sin complicar y la palpación de una masa en FID, algo dolorosa, pero sin signos de irritación peritoneal.
La analítica practicada en Urgencias no mostró ningún hallazgo de interés y la ECOGRAFÍA ABDOMINAL hizo visible una masa quística en FID de unos 85x65x45 milímetros, por lo que se solicitó un TAC que informó de la presencia de una lesión quística del tamaño descrito y que parecía proceder de la base del ciego, con calcificaciones en la periferia y de contenido liquido denso, compatible con un posible mucocele de origen apendicular. Asociaba además, la presencia de ascitis densa de distribución perihepática y en pelvis menor.
La paciente, tras ser dada de alta fue remitida a la Unidad de Cirugía Oncológica Peritoneal para su evaluación y tratamiento, donde se obtuvieron unos marcadores tumorales que fueron normales (CEA, Ca 19.9 y Ca 125).
DIAGNÓSTICO PSEUDOMIXOMA PERITONEAL secundario a la rotura de una NEOPLASIA MUCINOSA APENDICULAR.
El pseudomixoma peritoneal es un concepto clínico que está basado en el hallazgo de ascitis mucinosa libre en la cavidad abdominal. El origen más frecuente es la rotura de una neoplasia mucinosa apendicular, en el 80% de los casos, pero puede tener otros orígenes. En el DIAGNÓSTICO DIFERENCIAL, debe tenerse en cuenta fundamentalmente al CARCINOMA MUCINOSO DE ORIGEN OVÁRICO. De hecho, en el protocolo quirúrgico de este último la apendicectomía es obligatoria, ya que en ocasiones el ovario es un invitado y no el actor principal (que puede ser el apéndice) del cuadro clínico.
47
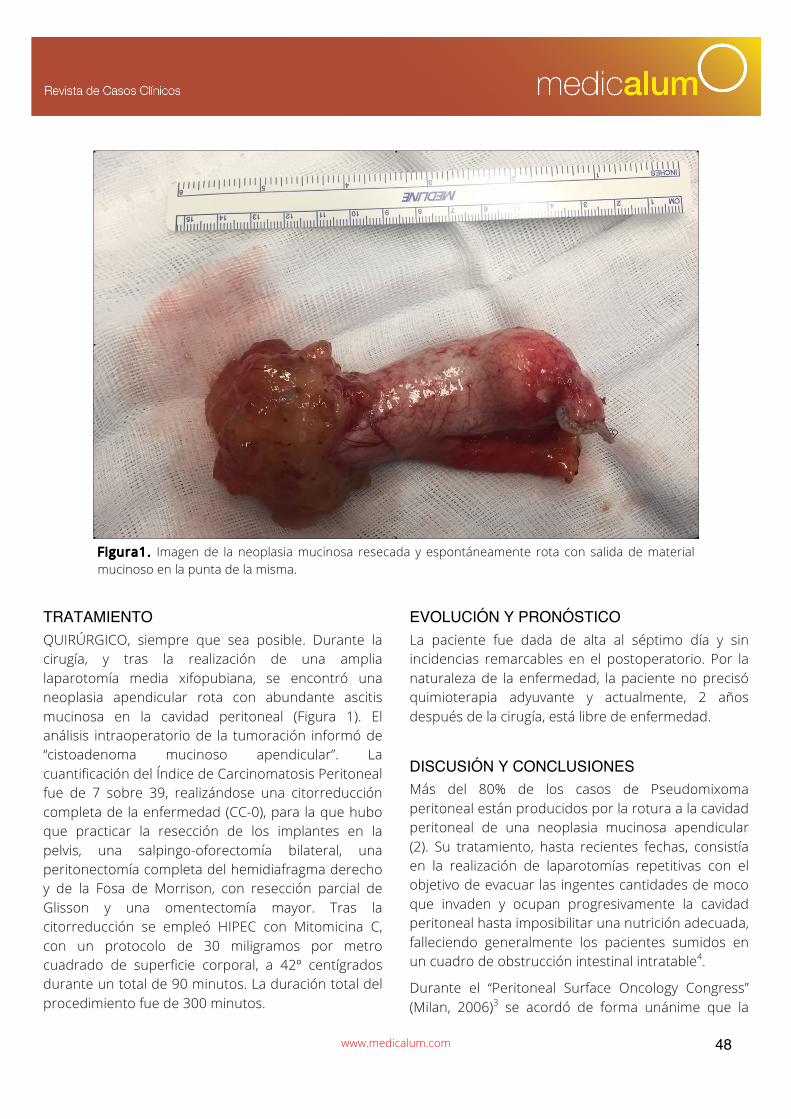
!
www.medicalum.com
!
Figura1. Imagen de la neoplasia mucinosa resecada y espontáneamente rota con salida de material mucinoso en la punta de la misma.
TRATAMIENTO QUIRÚRGICO, siempre que sea posible. Durante la cirugía, y tras la realización de una amplia laparotomía media xifopubiana, se encontró una neoplasia apendicular rota con abundante ascitis mucinosa en la cavidad peritoneal (Figura 1). El análisis intraoperatorio de la tumoración informó de “cistoadenoma mucinoso apendicular”. La cuantificación del Índice de Carcinomatosis Peritoneal fue de 7 sobre 39, realizándose una citorreducción completa de la enfermedad (CC-0), para la que hubo que practicar la resección de los implantes en la pelvis, una salpingo-oforectomía bilateral, una peritonectomía completa del hemidiafragma derecho y de la Fosa de Morrison, con resección parcial de Glisson y una omentectomía mayor. Tras la citorreducción se empleó HIPEC con Mitomicina C, con un protocolo de 30 miligramos por metro cuadrado de superficie corporal, a 42º centígrados durante un total de 90 minutos. La duración total del procedimiento fue de 300 minutos.
EVOLUCIÓN Y PRONÓSTICO La paciente fue dada de alta al séptimo día y sin incidencias remarcables en el postoperatorio. Por la naturaleza de la enfermedad, la paciente no precisó quimioterapia adyuvante y actualmente, 2 años después de la cirugía, está libre de enfermedad.
DISCUSIÓN Y CONCLUSIONES Más del 80% de los casos de Pseudomixoma peritoneal están producidos por la rotura a la cavidad peritoneal de una neoplasia mucinosa apendicular (2). Su tratamiento, hasta recientes fechas, consistía en la realización de laparotomías repetitivas con el objetivo de evacuar las ingentes cantidades de moco que invaden y ocupan progresivamente la cavidad peritoneal hasta imposibilitar una nutrición adecuada, falleciendo generalmente los pacientes sumidos en un cuadro de obstrucción intestinal intratable4.
Durante el “Peritoneal Surface Oncology Congress” (Milan, 2006)3 se acordó de forma unánime que la
48

!
www.medicalum.com
!
cirugía citorreductora con HIPEC era el tratamiento estándar para los tumores mucinosos asociados a esta diseminación peritoneal. La administración de HIPEC persigue la erradicación completa de la enfermedad peritoneal microscópica y se basa en la perfusión de la cavidad peritoneal mediante una solución de citostático que además se calienta para mantener una temperatura constante entre 41-43 grados centígrados, ya que parece ser que la hipertermia potencia los efectos citostáticos del fármaco. No hay estudios que demuestren la superioridad de ningún fármaco citostático en el pseudomixoma peritoneal, y los protocolos más utilizados incluyen el oxaliplatino o la Mitomicina C.
Respecto a los detalles operatorios, existe un consenso actual en pacientes con tumores mucinosos apendiculares de bajo grado, como es el caso de nuestra paciente, a favor de evitar la realización sistemática de una hemicolectomía derecha5. Incluso en pacientes con afectación del margen de resección del apéndice, esta puede evitarse con la realización de una extirpación amplia y con márgenes del ciego en la base de implantación del mismo. Por el contrario, en presencia de un adenocarcinoma de apéndice o de tumores mucinosos de alto grado o con células en anillo de sello, el tratamiento sí que debería completarse con hemicolectomía derecha.
En general, además del Pseudomixoma peritoneal, otras patologías malignas con diseminación peritoneal (Carcinomatosis peritoneal) pueden ser tratadas con este abordaje terapéutico, como por ejemplo ocurre en las metástasis peritoneales del cáncer de colon, gástrico, ovárico y en tumores primarios del peritoneo como el mesotelioma maligno peritoneal.
Algunos conceptos son pertinentes en este apartado al haberse nombrado, y son:
1. El volumen y la extensión de la enfermedad peritoneal se miden mediante el Índice de Carcinomatosis Peritoneal (PCI, Peritoneal Cancer Index), que es una estimación cuantitativa de la carga tumoral presente en la cavidad peritoneal. Se evalúa tanto al inicio de la intervención
quirúrgica como una vez finalizada esta y su valor oscila entre 0 y 39. Brevemente, el cálculo del PCI se realiza dividiendo la cavidad abdominal en 9 regiones a las que se suman 4 regiones más que corresponden al intestino delgado. En cada una de ellas se cuantifica el tamaño de las metástasis peritoneales y se otorga una puntuación. El valor del PCI se establece como la suma de todas las regiones descritas6.
2. El alcance de la citorreducción (CCS, completeness of cytoreduction score) determina el grado de citorreducción alcanzado tras la intervención quirúrgica. El resultado puede ser: CC-0 (citorreducción completa, sin residuo tumoral evidente al final de la cirugía), CC-1 (residuo menor de 2,5 milímetros), CC-2 (residuo entre 2,5 milímetros y 2,5 centímetros) y CC-3 (residuo superior a los 2,5 centímetros)6.
En el Pseudomixoma peritoneal, la elevación de algunos marcadores tumorales (CEA, CA 19.9 y CA 125) se ha relacionado de forma negativa con la supervivencia de los pacientes, siendo hasta 8 veces superior el riesgo de recaída en los pacientes con elevación de dichos marcadores en relación a los que no presentan cifras elevadas7, como por ejemplo nuestra paciente.
REFERENCIAS 1. González-Moreno S. Pseudomyxoma Peritonei: A
rare disease and a disease model in peritoneal surface oncology. Clin Transl Oncol. 2011; 13:211-12.
2. Ronnett BM, Zahn CM, Kurman RJ, Kass ME, Sugarbaker PH, Shmookler BM. Disseminated peritoneal denomucinosis and peritoneal mucinous carcinomatosis. A clinicopathologic analysis of 109 cases with emphasis on distinguishing pathologic features, site of origin, prognosis and relationship to “pseudomyxoma peritonei”. Am J Surg Pathol. 1995; 19: 1390-408.
3. Moran Bj, Baratti D, Yan Td, Kusamura S, Deraco M. Consensus statement on the loco-regional treatment of appendiceal mucinous neoplasms
49

!
www.medicalum.com
!
with peritoneal dissemination (pseudomyxoma peritonei). J Surg Oncol. 2008; 98:277-82.
4. Delhorme Jb, Elias D, Varatharajah S, Benhaim L, Dumont F, Honoré C, Et al. Can a benefit be expected from surgical debulking of unresectable pseudomyxoma peritonei? Ann Surg Oncol. 2016; 23:1618-24.
5. González-Moreno S, Sugarbaker PH. Right hemicolectomy does not confer a survival advantage in patients with mucinous carcinoma of the appendix and peritoneal seeding. Br J Surg. 2004; 91:304-11
6. Sugarbaker PH. Peritonectomy procedures. Ann Surg. 1995; 221:29-42.
7. Taflampas P, Dayal S, Chandrakumaran K, Mohamed F, Cecil T, Moran Bj. Preoperative tumor marker status predicts recurrence and survival after complete cytoreduction and hyperthermic intraperitoneal chemotherapy for appendiceal pseudomyxoma peritonei: analysis of 519 patients. Eur J Surg Oncol. 2014; 40:515-20.
50


!!
!!!
www.medicalum.com