
4919612 Monografia de Aines
77
U U NIVERSIDAD NIVERSIDAD P P ERUANA ERUANA C C AYETANO AYETANO H H EREDIA EREDIA F F ACULTAD ACULTAD D DE E E STOMATOLOGÍA STOMATOLOGÍA D D EPARTAMENTO EPARTAMENTO DE DE C C IRUGÍA IRUGÍA O O RAL RAL Y M M AXILOFACIAL AXILOFACIAL “ANALGÉSICOS ANTIPIRÉTICOS Y ANTINFLAMATORIOS NO ESTEROIDEOS (AINEs)” Alumno : Dr .Ludeña Manco, Marco LIMA – PERÚ 2003
-
Upload
leydi-heredia-espinoza -
Category
Documents
-
view
61 -
download
0
Transcript of 4919612 Monografia de Aines

UUNIVERSIDADNIVERSIDAD P PERUANAERUANA C CAYETANOAYETANO H HEREDIAEREDIA
FFACULTADACULTAD D DEE E ESTOMATOLOGÍASTOMATOLOGÍA
DDEPARTAMENTOEPARTAMENTO DEDE C CIRUGÍAIRUGÍA O ORALRAL YY M MAXILOFACIALAXILOFACIAL
“ANALGÉSICOS ANTIPIRÉTICOS Y ANTINFLAMATORIOS NO ESTEROIDEOS
(AINEs)”
Alumno : Dr .Ludeña Manco, Marco
LIMA – PERÚ
2003

INTRODUCCIÓN
Las drogas analgésicas antipiréticas antiinflamatorias no esteroides (AINEs)
son un grupo de agentes de estructura química diferente que tienen como efecto
primario inhibir la síntesis de prostaglandinas, a través de la inhibición de la
enzima cicloxigenasa.
Estas drogas comparten acciones farmacológicas y efectos indeseables
semejantes.
La aspirina es el prototipo del grupo y es la droga con la cual los distintos
agentes son comparados. Debido a esto también son llamadas drogas ”tipo
aspirina”; otra denominación común para este grupo de agentes es el de “AINEs”
(antiinflamatorios no esteroideos) o drogas “anticicloxigenasa” debido a que
inhiben esta enzima, responsable de la síntesis de prostaglandinas, las cuales son
mediadoras de la producción de fiebre, dolor e inflamación.
En farmacología existen dos grupos importantes de agentes
antiinflamatorios:
a) Los antiinflamatorios esteroides o glucocorticoides, que son los más potentes
antinflamatorios
b) Los analgésicos, antipiréticos, antinflamatorios no esteroides (AINEs) o drogas
tipo aspirina.
Las drogas tipo aspirina son los agentes más vendidos en el mundo, son muy
comúnmente utilizadas por prescripción o automedicación.
2

Se expenden toneladas por año. Existe una alta prevalencia de
enfermedades reumáticas en el mundo. Aproximadamente un 8% de la población
tiene un síndrome reumático alguna vez. Sin embargo se sabe poco sobre cuales
AINEs son realmente necesarios para un óptimo tratamiento de estas afecciones.
Hasta la fecha se sigue buscando el analgésico ideal, es decir que posee
gran potencia y mínimos efectos indeseables.
Esta familia de drogas está compuesta por innumerables agentes, cuya
síntesis e incorporación al mercado farmacológico se realiza permanentemente.
Existe una gran variación interindividual en la respuesta a estos agentes a los
efectos adversos y tóxicos que aparecen en un porcentaje de pacientes. La
potencia analgésica, antiinflamatoria, antitérmica y antiagregante plaquetaria, es
vari ables con los distintos agentes.
El nuevo conocimiento de que existen 2 isoenzimas ciclooxigenasas y que
se han desarrollado inhibidores selectivos de la ciclooxigenasa 2 (COX2) como el
meloxicam, salicilato y nimesulida, abre un camino para la terapéutica más segura
en pacientes con riesgos de hemorragia gastrointestinal o deterioro de la función
renal. Además serían de utilidad en pacientes con trastornos de la coagulación,
que reciben anticoagulantes o están programados para cirugía, es decir
cuando se necesita la función plaquetaria intacta.
La aspirina, indometacina, piroxicam, diclofenac e ibuprofeno, son
inhibidores no selectivos de COX1 y COX2, aunque algunas como el piroxicam y
la indometacina poseen afinidad alta in vitro por COX1, pudiendo se más tóxicas a
nivel GI y renal.
El dolor de origen dentario es la urgencia odontoestomatológica de más
frecuente consulta, tanto en servicios de urgencia hospitalarios como
3

extrahospitalarios. Además constituye la mayor demanda de consulta en una
clínica dental fuera de las citas programadas.
El dolor en la región orocervical es relativamente frecuente y posee una
serie de connotaciones especiales debidas a las peculiaridades de la región
anatómica.
La inervación sensitiva orofacial procede fundamentalmente del V par
craneal o nervio trigémino, aunque también coadyuvan el VII par o nervio facial
(áreas profundas de la cara), el IX par o nervio glosofaríngeo ( orofaringe, paladar
blando, epiglotis, fauces y amígdalas, y pared faríngea hasta su unión con el
esófago, tercio posterior de la lengua, oído medio), el X par o nervio vago (cara
inferior de la epiglotis, laringe, conducto auditivo externo) y el plexo cervical (C1-
C4) superficial y profundo (piel cervical y parte del cuero cabelludo).
La inervación simpática muy importante por su participación en los
fenómenos de vasodilatación/vasoconstricción asociados al dolor procede del
ganglio estrellado para toda la cabeza. En el caso del nervio trigémino, el
componente sensitivo emerge del troncocerebral en la protuberancia y se dirige
hasta el ganglio de Gasser. Este, está situado en la cara anterolateral del
peñasco, en la confluencia del suelo de la fosa craneal y el seno cavernoso,
rodeado de un repliegue de la duramadre y bañado en líquido cefalorraquídeo. Del
ganglio de Gasser salen las tres ramas, los nervios oftálmico, maxilar y
mandibular.
El nervio oftálmico, el más craneal de los tres, asciende hasta la hendidura
esfenoidal y se divide en sus tres ramas terminales: nervios lacrimal, frontal y
nasal, que recogen la sensibilidad de la parte superior de la cara. El nervio maxilar
sale del cráneo por el agujero redondo mayor a la fosa pterigomaxilar y entra en la
órbita por la hendidura esfenopalatina. Ramas suyas son los nervios
esfenopalatino, temporomalar, y den-tales o alveolares posteriores, medios y
anteriores. Recogen la sensibilidad de la mejilla, párpado inferior, labio y dientes
superiores, paladar, amígdalas y techo de la boca. El nervio mandibular sale del
4

cráneo por el agujero oval. Entre sus ramas se encuentran los ner-vios pterigoideo
externo e interno, maseterino, temporal superficial, dental o alveolar inferior y
lingual.
Recoge sensibilidad de la porción inferior de la cara (mandíbula, labio y
dientes inferiores, glándulas sali-vales), conducto auditivo externo, articulación
tempo-romandibular y región temporal
El origen del mayor número de dolores orofaciales son las piezas dentarias.
Las ramas terminales de los nervios maxilar (alveolares anterior, medio y poste-
rior, palatino posterior y nasopalatino) y mandibular (nervios bucal largo o
buccinador, lingual, dentario inferior y mentoniano) penetran en los dientes a
través de sus raíces (foramen apical) y viajan, dividiéndose, a través de la pulpa
dentaria en dirección a la corona. Parte de ellas llegan a la porción interna de la
dentina. Tanto los estímulos térmicos como los eléctricos, quí-micos y los cambios
osmóticos pueden producir dolor dental. La forma en que estos estímulos
producen dolor no está clara y, de hecho, se han postulado dife-rentes teorías
para su explicación. La más aceptada, la hidrodinámica, establece la existencia de
túbulos capi-lares en la dentina, en contacto con las terminaciones nerviosas, y
llenos de líquido extracelular. Los cam-bios de este líquido (por presión,
temperatura, pH, etc.) estimularían a su vez la terminación nerviosa.
El dolor bucodental se produce por un exceso de afluencia nociceptiva
procedente de la periferia. Este hecho es un fenómeno físico y psíquico que
resulta de la suma de varios factores, como es el caso del estado psíquico y
personal del paciente y el equilibrio de las áreas nerviosas centrales. Desde el
punto de vista etio-lógico, el dolor de esta localización puede producirse por:
pulpitis, alveolitis, pericoronaritis, periodontitis, abscesos periodontales y celulitis,
entre los procesos más importantes
5

La Asociación Internacional para el Estudio y Tra-tamiento del Dolor establece
cinco categorías de dolor dental:
1) Los relacionados con defectos de esmalte y la dentina.
2) Los originados en la pulpa dental (pulpitis).
3) Los originados en áreas vecinas al diente (perio-dontitis).
4) El síndrome del diente roto.
5) Aquellos dolores dentales no asociados a lesión dental objetivable alguna.
Las características de una odontalgia son plurales: desde un dolor agudo y
sostenido, irradiado a la cara, occipucio y cráneo, hasta otro difuso, sordo,
localiza-do en mandíbula o maxilar.
6

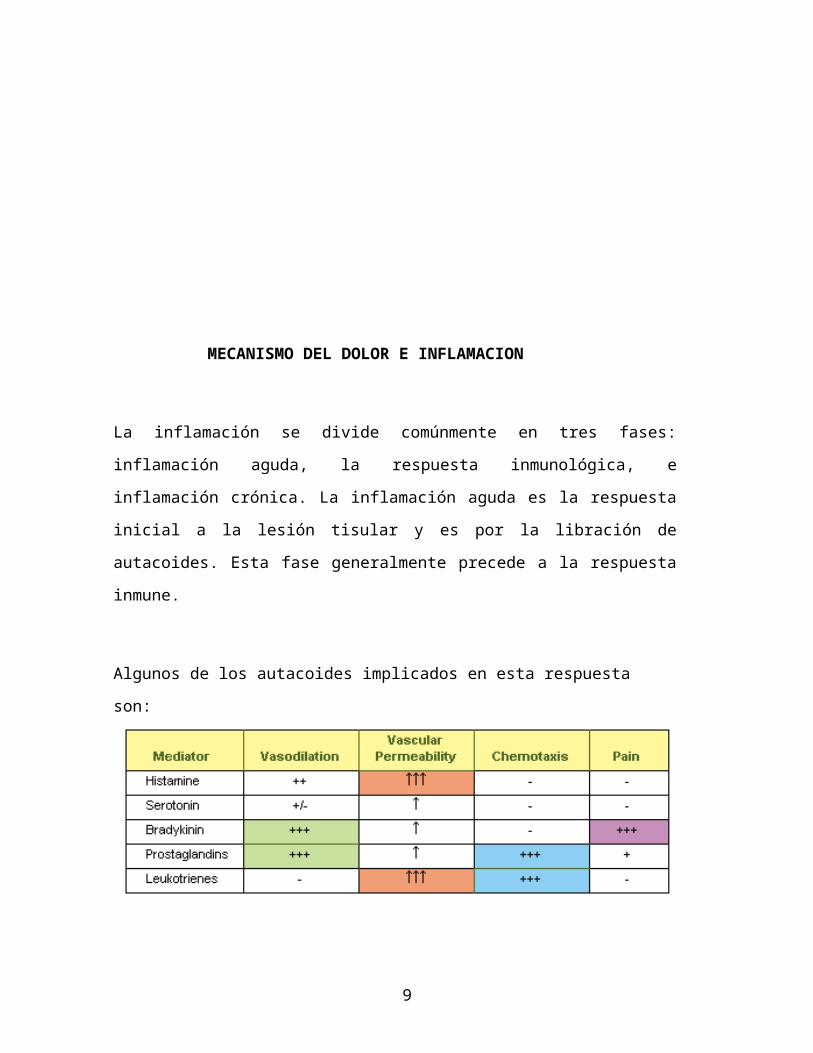
MECANISMO DEL DOLOR E INFLAMACION
La inflamación se divide comúnmente en tres fases: inflamación aguda, la
respuesta inmunológica, e inflamación crónica. La inflamación aguda es la
respuesta inicial a la lesión tisular y es por la libración de autacoides. Esta fase
generalmente precede a la respuesta inmune.
Algunos de los autacoides implicados en esta respuesta son:
La respuesta inmune ocurre cuando las células inmunocompetentes se activan
ante organismos extraños o sustancias antigenicas liberadas durante la
respuesta inflamatoria aguda o crónica. El resultado de esta inmunorespuesta
es benéfico para el huesped ya que los organismos invasores son fagocitados
o neutralizados.
7

El resultado de este proceso puede ser deletéreo si conduce una inflamación
crónica sin resolución del proceso perjudicial subyacente.
La inflamación crónica implica la liberación de un número de mediadores que
no son prominentes en la respuesta aguda tales como: Interleuquinas (1, 2 y
3), factor de necrosis tumoral alfa2, Inferferones y factores de crecimiento
derivados de las plaquetas.
La artritis reumatoidea es un claro ejemplo clínico de este fenómeno, ya que en
esta entidad, la inflamación crónica produce dolor y lleva a la destrucción del
hueso y el cartílago que pueden conducir a la inhabilidad severa y a cambios
sistemicos que pueden acortar la vida.
El daño celular asociado a la inflamación actúa en las membranas celulares
produciendo liberación de enzimas lisosómicas en los leucocitos.
8
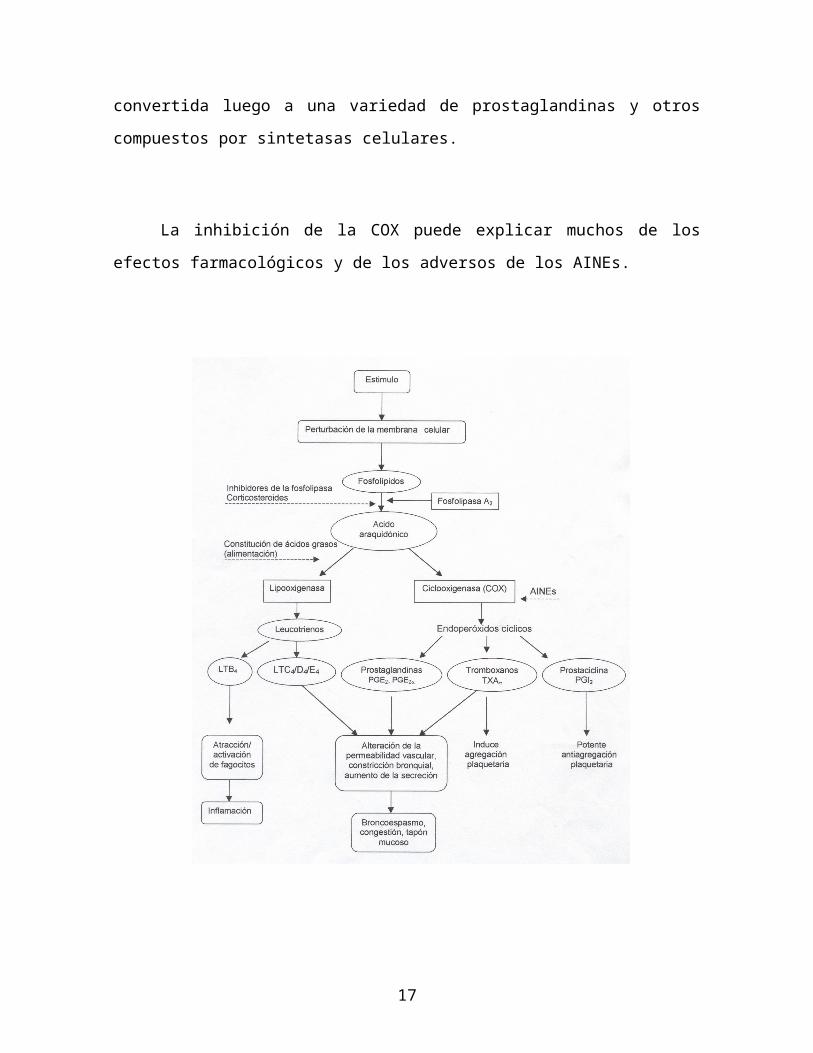
Posteriormente se produce la liberación del ácido araquidónico de los fosfolípidos
de membrana mediante las lipasas del tipo Fosfolipasa A2. Estas son inhibidas por
los antiinflatorios esteroideos (como los corticoesteroides).
Este ácido araquidónico es fundamental para la síntesis de varios eicosanoides
que participan directamente en el proceso inflamatorio.
Productos de la Lipoxigenasa
El metabolismo del ácido araquidónico por las lipooxigenasas (principalmente
la 5-Lipooxigenasa) da como resultado la producción de ácidos
hidroperoxieicosatetraenoicos (HPETES), los cuales rápidamente se convierten
en derivados hidróxidos (HETES) y Leucotrienos.
La 5-Lipooxigenasa está presente en las células inflamatorias (PMN,
mastocitos, basófilos, macrófagos y eosinófilos) y está involucrada en la
fisiopatología del asma y el shock anafiláctico. Esta enzima produce el
Leucotrieno A4 que se convierte en Leucotrieno B4 o Leucotrieno C4 (si se
conjuga con glutatión). Este a su vez puede convertirse en LTD4 y LTE4,
mediante peptidasas.
Los LTC4 y LTD4 son potentes broncoconstrictores y hoy en día se reconocen
como los componentes primarios de la sustancia de reacción lenta en la
anafilaxia (SRS-A), que se secretan en el asma y en la anafilaxia. Por esta
razón las últimas investigaciones farmacológicas han ido dirigidas a buscar
inhibidores de los leucotrienos en estas patologías.
9

Productos de la Cicloxigenasa
Solo se han descubierto dos isoenzimas de la ciclooxigenasa que son capaces
de convertir el ácido araquidónico en prostaglandinas: La PGH Sintetasa 1
(llamada COX-I) y la PGH Sintetasa 2 (llamada COX-II). La primera se
encuentra constantemente en los tejidos mientras que la segunda puede ser
inducida.
La COX-I tiene funciones de "mantenimiento" principalmente (por ejemplo la
protección gástrica), mientras que la COX-II se produce tempranamente y de
forma masiva por las células inflamatorias e inmunes y su producción se
aumenta por factores de crecimiento, promotores tumorales, citoquinas y
especialmente por endotoxinas bacterianas.
El conocimiento de estas isoenzimas es importante para el manejo
farmacológico de la inflamación con agentes no esteroideos, ya que ellos
actúan precisamente inhibiendolas. Por ejemplo la Aspirina (Acido
AcetilSalicílico) inhibe ambas isoenzimas, mientras que la nabumetona inhibe
prefencialmente la COX-II.
Los medicamentos que inhiben exclusivamente la COX-II pueden ser útiles
para el tratamiento de las enfermedades inflamatorias crónicas, ya que no
comprometen las prostaglandinas que participan en la protección gástrica
(mediada por la COX-I).
Las prostaglandinas que se producen por esta vía tienen una gran variedad de
efectos en los vasos sanguíneos, las terminaciones nerviosas y en las células
involucradas en el proceso inflamatorio.
Las kininas, los neuropeptidos, y la histamina también se liberan en el sitio de
lesión tisular, al igual que los componentes del complemento, las citoquinas, y
10

otros productos leucocitarios y plaquetarios.
El estímulo de las membrana del neutrofilo produce radicales libres derivados
del oxígeno. La interacción de estas sustancias con el ácido araquidónico da
lugar a la generación de sustancias quimiotácticas, que perpetuan el proceso
inflamatorio.
En esta sesión estudiaremos los fármacos anti-inflamatorios no esteroideos.
Como veremos mas adelante este grupo de medicamentos actúan
precisamente en este punto de la inflamación es decir de las ciclooxigenasas.
11

MECANISMO DE ACCIÓN DE LOS AINES
Mecanismo de acción: Inhibición de la enzima cicloxigenasa
La COX, también conocida como H2-sintetasa, cataliza el primero
de los dos pasos en la síntesis de prostaglandinas, tromboxanos y
prostaciclinas (colectivamente conocidos como prostanoides) a partir del
ácido araquidónico.
Prostanoides
Prostaglandinas
Prostaciclinas
Tromboxanos
Las prostaglandinas están implicadas en muchos procesos hemostáticos y
son mediadores importantes de la inflamación. La COX es una enzima bifuncional
la cual cataliza la oxidación del ácido araquidónico a endoperóxido cíclico
PGG2 y la reducción peroxidativa de la PGG2 a PGH2. La PGH2 es convertida
luego a una variedad de prostaglandinas y otros compuestos por sintetasas
celulares.
La inhibición de la COX puede explicar muchos de los efectos
farmacológicos y de los adversos de los AINEs.
12

Se muestran los sitios donde los AINEs inhiben a la COX
La variabilidad en las propiedades inhibitorias de los AINEs en los
diferentes tejidos del cuerpo, y las aparentes discrepancias entre la eficacia clínica
de ciertos AINEs y su potencia inhibitoria sobre la COX, sugirió que podrían existir
diferentes formas de la isoenzima. Los cambios en la producción de
prostaglandinas no siempre se asociaron, por ejemplo, con cambios similares en
13

la síntesis de prostaglandina ARNm. Este punto se aclaró cuando en 1991 se
describieron dos tipos distintos de la enzima COX: la COX-1 y la COX-2.
La COX-1 es la forma constitutiva responsable de la producción de
prostaglandinas comprometidas en las señales de célula a célula y en las
funciones celulares de mantenimiento, tales como la regulación de la homeostasis
vascular y la coordinación de las acciones de las hormonas circulantes.
La COX-2 es inducida en células activadas por la exposición a
mediadores de la inflamación tales como citokinas y endotoxinas, y es
responsable de la producción de prostanoides que median la inflamación, el
dolor y la fiebre. La COX-2 se expresa también constitutivamente en los riñones y
el partes del sistema nervioso central (SNC).
COX
COX-1 Prostaglandinas Funciones de
mantenimiento celular
COX-2 ProstanoidesMedian en inflamación,
dolor y fiebre
Luego de la lesión o inflamación de los tejidos periféricos, las
prostaglandinas sintetizadas por los COX-2 sensibilizan receptores periféricos por
medio de activación de los nociceptores localizados en la terminaciones
nerviosas periféricas. Esto da como resultado un comportamiento exagerado
frente al dolor que incluye: hiperalgesia y alodinia.
14

Hiperalgesia Respuesta aumentada al estímulo doloroso
Alodinia Dolor en respuesta a un estímulo que
normalmente es inocuo
Esto contribuye a elevar la sensibilidad en el sitio donde los tejidos han sido
dañados, hiperalgesia primaria, a través de la proteinkinasa A que media la
fosforilación de los canales sódicos en las terminales nociceptivas.
Durante mucho tiempo se ha pensado que los efectos analgésicos de los
AINEs son mediados primariamente a través de la inhibición de las
prostaglandinas en la periferia. Evidencias recientes sugieren que acciones en el
SNC pueden también contribuir a su actividad analgésica.
Aunque los niveles de COX-2 en el SNC son normalmente bajos, la
regulación de la enzima es estimulada en la médula espinal por la inflamación
periférica, conduciendo a la producción de prostaglandina E2 (PGE2).
La PGE2 puede facilitar la transmisión nociceptiva a través de la alteración
de la excitabilidad neuronal del asta dorsal. Se ha visto que la administración
intratecal de AINEs, en animales, estimula el aumento de PGE2 espinal y la
hiperalgesia inducida por la inflamación periférica.
Muchos de los AINEs poseen otros efectos bioquímicos no del todo
aclarados, sin embargo la inhibición de la cicloxigenasa o prostaglandin sintetasa,
parece ser el principal mecanismo de ac-ción de estos agentes y por lo tanto la
15

inhibi-ción de la síntesis de prostaglandinas. El orden de potencia como
inhibidores de la síntesis de prostaglandinas in vitro refleja su poder
antiinflamatorio in vivo.
La mayoría de los AINEs son inhibidores reversibles y competitivos de la
cicloxigena-sa, mientras que el ácido acetil salicílico es un inhibidor irreversible,
acetila la enzima en el sitio activo, por ello es uno de los agentes más útiles como
antiagregante plaquetario ya que inhibe la enzima cicloxigenasa pla-quetaria
(COX1) por toda la vida de la pl a-queta (7-11 días), como las plaquetas son
fragmentos celulares son incapaces de sinte-tizar nueva enzima.
Algunos estudios sugieren que existen otros mecanismos de acción , sobre
todo para sus acciones antiinflamatorias. De acuerdo a estas teorías se vio que
algunos AINEs in-hiben la enzima lipoxigenasa in vitro y en algunos modelos
animales utilizando diclofe-nac e indometacina, estos 2 agentes dismi-nuyen los
leucotrienes y prostaglandinas de leucocitos y células sinoviales por estimular la
reincorporación de ácido araquidónico libre en los triglicéridos de las membranas.
16
FARMACOLOGIA MOLECULAR
Las isoformas COX-1 y COX-2 derivan de genes diferentes, los que en el
ser humano se localizan en el cromosoma 9 y 1, respectivamente. El orden de
las porciones intron/exon es idéntico, excepto que los exones 1 y 2 de la COX-1
se condensan en un exon simple en la COX-2.
Ambas enzimas tienen en común entre el 60 y 80% de su secuencia de
aminoácidos, dependiendo de la especie que se trate, pero los sitios residuales
activos se encuentran altamente conservados y difieren en sólo dos
localizaciones. El sitio de unión activo para el ácido araquidónico se localiza en
la mitad superior de un canal largo, estrecho e hidrófobo que se extiende desde
la superficie externa de la membrana, lo cual permite que el ácido araquidónico
tenga acceso a los sitios catalíticos directamente desde la membrana.
Figura . Secuencia de aminoácidos en canal COX
Diagrama que representa el extremo de un canal COX, demostrando la
localización de los aminoácidos que están comprometidos en el proceso
catalítico, lo mismo que el sitio del canal que participa en la actividad de la COX-
2. El aminoácido en posición 523 es la isoleucina para la COX-1 y la valina para
la COX-2
17

El ácido araquidónico, liberado desde las membranas dañadas, es "chupado"
hacia dentro del canal, se le insertan dos átomos de oxígeno y se le extraen
radicales libres, lo que da por resultado un anillo de 5 átomos de carbono,
característico de las prostaglandinas.
Los AINEs COX-1 bloquean éste canal a la mitad de su recorrido. Este bloqueo
ocurre por el hidrógeno ligado a la arginina polar en posición 120. Sin embargo,
es el aminoácido vecino, en posición 523, el que resulta crítico para la
selectividad. En los COX-1 es el aminoácido isoleucina el que está en posición
523, mientras que en los COX-2 es la valina (más pequeña por poseer
simplemente un grupo metilo).
Isoleucina ◄ COX-1
Aminoácido 523
Valina ◄ COX-2
La pequeña molécula de valina en los COX-2, deja en la pared del canal
una brecha, dando acceso a un bolsillo lateral el que se piensa que es el sitio de
unión para los fármacos selectivos COX-2.
La abultada isoleucina, en posición 523 en la COX-1 es lo suficientemente
grande para bloquear el acceso a este bolsillo lateral. Simplemente la sustitución
del aminoácido isoleucina por la valina permite que la enzima COX-1 sea inhibida
por los agentes selectivos COX-2.
18

La punta del canal contiene una tirosina en posición 385, la que se piensa
que está comprometida en la actividad de la COX.
Los sitios donde el ácido araquidónico se une a la enzima son la serina en
posición 530 y la arginina en posición 120.
Acido Araquidónico
La aspirina inhibe de manera irreversible a la COX por acetilación de la
serina 530, la que se sitúa por debajo de la tirosina 385, previniendo el contacto
del ácido araquidónico con la tirosina 385.Todos los inhibidores de la COX-2 que
se disponen en la actualidad carecen del grupo ácido carboxílico que se encuentra
presente en los inhibidores de la COX-1, pero poseen una extensión específica
sulfuro que contiene una cadena lateral, ya sea un grupo sulfonamida, sulfonilo o
sulfona.
Esta extensión lateral rígida puede acceder a la cadena lateral de la COX-2.
Mientras ambas isoformas de la COX usan el mismo sustrato, el ácido
araquidónico, y forman los mismos productos por igual reacción catalítica, difieren
de manera marcada en sus funciones fisiológicas.
Mientras que la actividad de la COX-1 está restringida al ácido
araquidónico, la COX-2 es capaz de metabolizar un rango amplio de sustratos,
incluyendo varios ácidos grasos.
19

La COX-1 se expresa continuamente en casi todos los tejidos, mientras
que la COX-2 es una enzima inducible que es casi indetectable en la mayoría de
los tejidos en condiciones fisiológicas. Sin embargo, se expresa constitutivamente
en pequeñas cantidades en la mucosa gástrica, en las neuronas y el los riñones.
La COX-2 se localiza en la vasculatura renal, en la mácula densa cortical, y el las
células intersticiales medulares, y aparece ligada a la producción de renina. Por
el contrario, la COX-1 se encuentra en la vasculatura, en los túbulos colectores y
en la asa fina de Henle. En muchos tejidos humanos ambas isoformas se
expresan en una extensión similar.
Mientras que la expresión de la COX-1 puede aumentar 2 y 3 veces por
encima de su valor basal durante la estimulación, la de la COX-2 puede aumentar
de 20 a 80 veces cuando es inducida por productos bacterianos como las
endotoxinas, las citokinas (ejemplo: IL-1 y TNF) el factor de crecimiento,
importante en el proceso de curación.
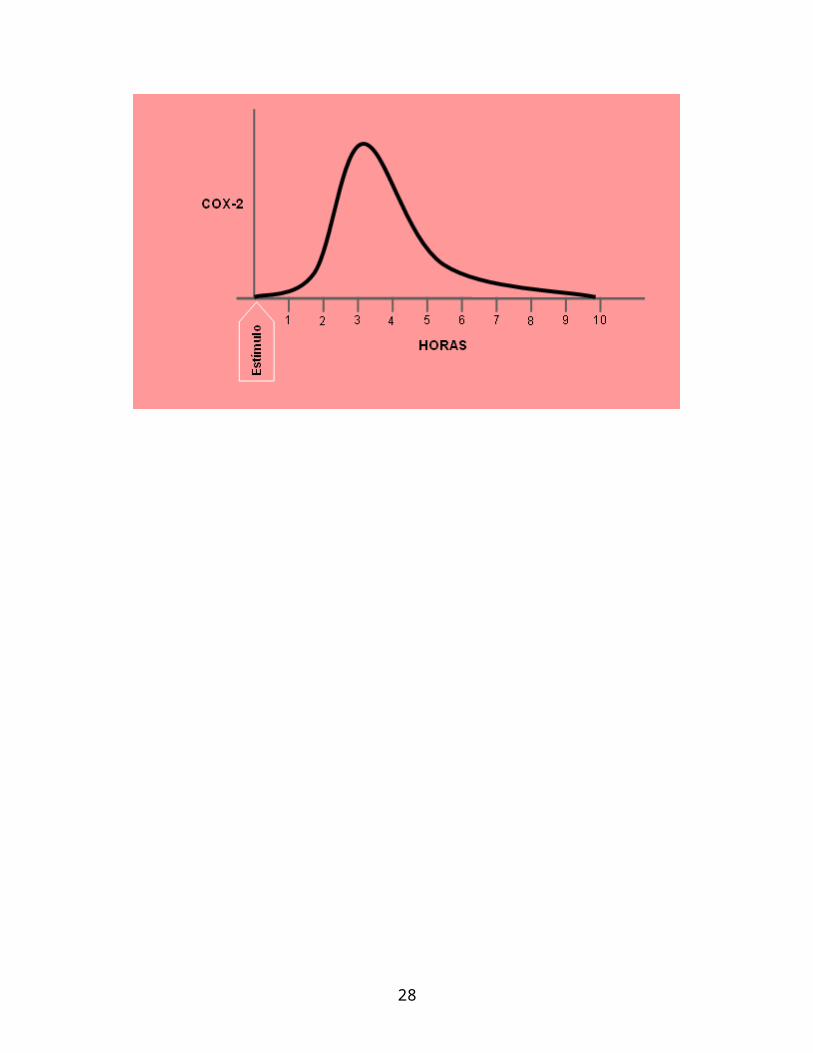
La inducción de la COX-2 se caracteriza por la producción súbita de una
gran cantidad de prostaglandinas en un período relativamente corto de tiempo
La inducción de COX-2 tarda en iniciarse 1 hora, alcanza su pico a las 3 horas y
amaina a las 6 horas. La inducción de COX-2 es suprimida por los antinflamatorios
esteroides, y por aquellas citokinas que inhiben la inflamación.
20

21

CLASIFICACION DE LOS AINES
1. Salicilatos:
Ácido acetil-salicílico (AAS)
2. Derivados Pirazolónicos
Dipirona o Metamizol
Fenilbutazona
Azapropazona
3. Derivados del Paraminofenol
Acetaminofén o Paracetamol
4. Derivados del Ácido Propiónico
Ibuprofeno
Ketoprofeno
Flubiprofeno
Naproxeno
Ácido Tiaprofénico
5. Derivados del Ácido N-Acetil Antranílico o Fenamatos
Ac. Mefenámico
Ac. Niflúmico
Meclofenámico
Clonixiato de lisina
22

6. Derivados del Ácido Fenilácetico
Diclofenac sóido y potásico
7. Derivados del Ácido Indolacético
Indometacina
Sulindaco
Glucametacina
8. Derivados del Ácido Pirrolacético
Ketoralaco
9. Derivados Enólicos
Peroxicam
Tenoxicam
10. Inhibidores específicos de la Ciclooxigenasa 2 (COX2)
Nimesulide
Celecoxib
Rofecoxid
11.Derivados del ácido Fenil Propiónico
Loxoprofén sódico
23

Existen AINEs que bloquean, tanto la vía de la Ciclooxigenasa como la de
la Lipooxigenasa como el Diclofenac y el Ketoprofeno, los cuales pueden tener
una ventaja por lo menos en términos de seguridad y eficacia, frente a los que
solamente bloquean la vía de la ciclooxigenasa.
24

INHIBIDORES COX-2 SELECTIVOS
Hay vastas diferencias en la selectividad de los AINEs convencionales para
las dos isoformas (Tabla) . Algunos, tales como la aspirina, la indometacina y el
ibuprofeno son inhibidores más potentes para la COX-1 que para la COX-2.
El diclofenac, el paracetamol, y el naproxeno son inhibidores equipotentes
para ambos tipos.
La nimesulida y el meloxicam son inhibidores selectivos de la COX-2.
La nueva generación de COX-2 selectivos, los coxibs, tienen muy baja o
ninguna actividad contra la COX-1.
25

La selectividad para la COX-2 ha sido medida usando una variedad de
ensayos, los que frecuentemente dan valores muy diferentes en la relación de
selectividad COX-2:COX-1.
Los ensayos bioquímicos usando enzimas recombinantes dan relaciones
mucho más altas, pero ellas no reflejan necesariamente la complejidad de las
interacciones droga-enzima in vivo. Los ensayos que se han realizado con sangre
entera es más probable que reflejen de manera más cierta la selectividad en los
pacientes. Estos se basan en la producción de tromboxano B2 durante la
coagulación de la sangre (un índice de la actividad plaquetaria de la COX-1) y en
la producción de prostaglandina E2 por lipopolisacáridos bacterianos (LPS) en
sangre entera (un índice de la actividad de la COX-2 de los monocitos). Estos
procedimientos han demostrado que el rofecoxib y el celecoxib no tienen efectos
significativos sobre la actividad de la COX-1 a las dosis recomendadas para su
uso terapéutico. Sin embargo, el celecoxib, puede inhibir la COX-1 mediante la
26

producción de TXB2 cuando se le administra en dosis que exceden las
terapéuticas.
Los primeros inhibidores selectivos de la COX-2, los coxibs celecoxib,
rofecoxib y parecoxib se encuentran disponibles para su uso clínico. El valdecoxib
y el etoricoxib se encuentran en la actualidad sometidos a ensayos clínicos. El
extremadamente corto lapso de tiempo transcurrido (aproximadamente una
década) entre la identificación de las isoformas de COX-2 y el desarrollo de éstas
drogas es un logro remarcable
Figura
Estructura química del Celecoxib, Rofecoxib y Etoricoxib
Disponibles para uso clínico Celecoxib
Rofecoxib
Parecoxib
Valdecoxib
Etoricoxib Aún en etapa de ensayos clínicos
27
1.- CELECOXIB
Celecoxib, el primer inhibidor específico de la COX-2 aprobado para uso
clínico, es aproximadamente siete veces más específico para la COX-2 que para
la COX-1. Está disponible sólo para administración oral.
El celecoxib es altamente lipofílico con un volumen de distribución grande (>400
litros).
Es metabolizado en el hígado por la encima citocromo CYP 2C9 a metabolitos
inactivos. La Biodisponibilidad oral es del 22 al 40%. Su vida media de
eliminación es de 11 hs., aproximadamente. La unión a las proteínas
plasmáticas es >97%.
Celecoxib
Especificidad COX-2:COX-1 7:1
Vía de suministro Oral
Lipofilicidad / Hidrofilicidad Lipofílico
Volumen de Distribución >400 litros
Metabolización Hepática
Biodisponibilidad oral 22-40%
Vida Media de Distribución 11 horas
Unión Pts Plasmáticas >97%
28

Aunque varios estudios han sugerido que los coxibs pueden ser bien
tolerados en pacientes que padecen asma inducido por aspirina, se han reportado
reacciones alérgicas al grupo sulfonamida. En un ensayo clínico grande se
investigó la seguridad gastrointestinal del celecoxib (estudio CLASS), la incidencia
de rash dérmico fue más del doble que con los AINEs comparados. Por lo tanto,
basado en las evidencias al presente, el celocoxib debería se considerado como
contraindicado en pacientes que sean portadores de alergia a la sulfonamida.
29

2.- ROFECOXIB
Rofecoxib, el segundo coxib en ser aprobado para uso clínico, es un
inhibidor selectivo de la COX-2 cuyos estudios in vivo han demostrado que hasta
en dosis de 1000 mg/día, no posee efectos sobre la COX-1. Tanto en estudios in
vivo como in vitro ha tenido una selectividad COX-2 más alta que el celecoxib y el
meloxicam.
Al igual que el celecoxib sólo se dispone de la presentación oral.
Su Biodisponibilidad oral es de aproximadamente el 90%, y su
concentración plasmática máxima se alcanza a las 2-3 horas.
El rofecoxib es metabolizado en hígado por reducción citosólica, por medio
de la flavoproteína reductasa, una vía independiente de la citocromo P450.
La vida media de eliminación es de 10-17 horas.
En los pacientes que toman warfarina el rofecoxib puede dar como
resultado un aumento en el Porcentaje Normalizado Internacional (INR). Esto
puede reflejar un aumento de la concentración plasmática de una R(+)warfarina,
biológicamente menos activa.En muchos pacientes que toman warfarina, un
pequeño aumento en el INR en estado de equilibrio es probable que no tenga
significación clínica importante.
Sin embargo, se debe tener la conducta de monitoreo estándar del INR en
los pacientes que están medicados con warfarina y que inician tratamiento con
rofecoxib o cambian el mismo, particularmente durante los primeros días.
30

Lo mismo que otros AINEs, el rofecoxib puede reducir la efectividad de la
terapéutica antihipertensiva.
La coadministración de rofecoxib con el inductor enzimático de la citocromo
P-450, rifampicina, puede producir una disminución del 50% de la concentración
plasmática de rofecoxib, por lo que debe considerarse aumentar la dosis inicial
en los pacientes que están tomando inductores enzimáticos.
En ensayos clínicos controlados el rofecoxib fue más efectivo que el
celecoxib, y similar a los AINEs convencionales, para el tratamiento de la
osteoartritis y la artritis reumatoidea. También se ha asociado con una
significativamente menor incidencia, endoscópicamente confirmada, de úlcera
duodenal y de eventos adversos gastroduodenales, en comparación con los
AINEs convencionales.
Rofecoxib
Especificidad COX-2:COX-1 35:1 [53]
Vía de suministro Oral
Concentración Plasmática
Máxima2-3 hs
Metabolización Hepática
Biodisponibilidad oral 90%
Vida Media de Eliminación 10-17 horas
31

3.- PARECOXIB
El parecoxib sódico es una pro-droga, soluble en agua, del valdecoxib un
inhibidor potente y selectivo de la COX-2.
Recientemente ha sido aprobado para su uso clínico en Europa
El parecoxib es el único inhibidor de la COX-2 disponible para ser usado
por vía endovenosa e intramuscular.
En los humanos y en las ratas el parecoxib es llevado rápida y
completamente a valdecoxib por hidrólisis amídica. Luego de la inyección
venosa a las 0,5 horas alcanza la máxima concentración plasmática; mientras
que si se suministra por vía intramuscular aquella se logra en 1,5 horas.
El valdecoxib, el metabolito activo del parecoxib, es 28.000 veces más
selectivo COX-2 que COX-1 en los ensayos en humanos usando enzimas
recombinante.
En los ensayos con sangre entera en humanos, los valores de la IC50 los
valores para la producción de LPS inducido por PGE2 (una función de la
actividad de COX-2) y la producción de TBX2 (una función de la actividad COX-1)
fueron de 0,89 ± 0,033 µM y 25,4 ± 1,2 µM, resultando en una selectividad COX-
2 de 28,5.
El valdecoxib es metabolizado primariamente por las enzimas hepáticas
citocromo P-450, CYP 2C9 y CYP 3A4. Tanto el parecoxib como el valdecoxib
son inhibidos por la CYP 2C9y por lo tanto, potencialmente, hay probabilidades
32

de que interactúen con otras drogas metabolizadas de manera similar como es el
caso del propofol.
Parecoxib
Especificidad COX-
2:COX-128,5:1
Vía de suministro EV - IM
Concentración
Plasmática Máxima
0,5 hs (EV)
1,5 hs (IM)
MetabolizaciónHepática
a Valdecoxib (metabolito activo)
Ibrahin y colaboradores han demostrado que el parecoxib, en las dosis que
se usa perioperatoriamente, no altera la disponibilidad, la magnitud, ni el tiempo
de acción de los efectos clínicos del propofol.
Cuando se suministran 20 a 80 mg por vía endovenosa el parecoxib , y
antes del inicio de la cirugía de boca, es efectivo como analgésico. No se
presentaron diferencias significativas entre las dosis de 40 y 80 mg, lo que ha
sugerido que se alcanza un efecto analgésico plateau a los 40 mg preoperatorios,
en éste modelo.
También se ha evaluado al parecoxib a dosis de hasta 100 mg, tanto en
estudios de dosis únicas como múltiples para el tratamiento del dolo
agudo postoperatorio en una variedad de cirugías. Tanto en el
comienzo de la acción, en el efecto pico como en la duración de la
analgesia, 40 mg parecoxib son comparables a 30 mg de ketorolac,
pero con menos efectos colaterales. 40 mg de parecoxib ha sido
superior a 4 mg de morfina.
33

Figura
Vía metabólica para la formación de valdecoxib desde su prodroga, parecoxib. El
metabolismo del valdecoxib ocurre vía de la glucuronidación del grupo
sulfonamida y la hidroxilacón, mediante la P-450, del grupo metilo, a una forma
menor de metabolito (SC-66905), el que también posee actividad inhibitoria COX-2
34

4.- ETORICOXIB
El etoricoxib es un coxib de segunda generación, potente y de acción
rápida que se encuentra actualmente bajo ensayos clínicos.
Es el más altamente COX-2 selectivo de todos los coxibs.
En los ensayos con sangre entera la razón de la selectividad de la COX-2
fue de 106, haciendo que el etoricoxib sea aproximadamente 3 veces más
selectivo que el rofecoxib y el valdecoxib, y 15 veces más selectivo que el
celecoxib.
35

¿Cuán Seguros son los Inhibidores Selectivos de la COX-2?
Los inhibidores selectivos de la COX-2 también han recibido la
denominación de "aspirinas mejoradas" y no están asociadas a los efectos
colaterales de los AINEs convencionales.
Esta afirmación se basó en la premisa de que los inhibidores selectivos de la
COX-2 no interferirían con las funciones fisiológicas relacionadas con la COX-1.
La toxicidad asociada con la terapéutica con AINEs se debe, principalmente, a la
inhibición de la COX-1, mientras que los efectos terapéuticos beneficiosos derivan
de la inhibición de la enzima inducible COX-2.
Los compuestos que inhiben de manera selectiva a la COX-2 son analgésicos y
anti-inflamatorios, y presentan menor toxicidad gástrica y renal, las que
normalmente se asocian al uso de los AINEs.
Existe considerable evidencia con respecto a que los inhibidores selectivos COX-
2 causan significativamente menos complicaciones gastrointestinales que los
AINEs no selectivos.Asimismo, debido a que la única isoforma presente en las
plaquetas es la COX-1, los inhibidores selectivos de la COX-2 no tendrían que
tener implicancias sobre la hemostasia.
36

Analgésicas
accionesAnti-
inflamatorias
AINEs COX-2
selectivos
menorToxicidad
gástrica
Toxicidad renal
Sin embargo, a pesar de que las evidencias sugirieron que la inhibición selectiva
de la COX-2 serían muy beneficiosas, van apareciendo nuevas evidencias que
llevan a pensar que la inhibición de la función fisiológica de las prostaglandinas
podría no ser tan beneficiosa como se creyó.
La idea de que existe una clara distinción entre los roles fisiológicos y patológicos
de las dos isoformas de la COX está comenzando a ser menos sustentable ya
que sus acciones se entremezclan considerablemente. De hecho, en algunas
circunstancias, las prostaglandinas podrían ser beneficiosas en la resolución de
la inflamación y del daño tisular.Así, mientras que los inhibidores selectivos de la
COX-2 son eficaces en la inflamación aguda, podrían exacerbar una fase tardía
en la que se producen prostaglandinas anti-inflamatorias por mediación de la
ciclo-oxigenasa
37

Mediante estudios recientes se ha demostrado que la COX-2 se encuentra
expresada constitutivamente en las neuronas y en las células epiteliales
gástricas, y que podría ser importante en la transmisión neural de la protección
gástrica.
La COX-2 del endotelio vascular también podría ser importante para la
vasoconstricción. En estudio reciente ha sugerido que la COX-2 podría tener un
rol crucial en la protección contra el daño de la mucosa gástrica.
Las prostaglandinas desempeñan un papel importante en la cicatrización de las
úlceras estomacales y el RNAm que codifica la síntesis de la COX-2 es
intensamente inducido en la mucosa gástrica vecina a una úlcera.En la rata, la
inhibición de la COX-2 tiene una influencia negativa sobre la cicatrización de la
úlcera gástrica.La administración en las ratas de celecoxib y el rofecoxib durante
cinco días no provocó lesiones en las mucosas gastrointestinales sanas. Por el
38

contrario, cuando se les administró a ratas con mucosas gastrointestinales
dañadas, se agravaron y complicaron las úlceras gástricas y provocaron,
además, necrosis del intestino delgado.
La inhibición completa de la COX-2 en los humanos podría implicar riesgos
aún desconocidos.
Las prostaglandinas desempeñan un rol fisiológico importante en los riñones
manteniendo la perfusión renal, especialmente ante una disminución de la
presión de perfusión o del volumen plasmático.
En pacientes susceptibles, los AINEs pueden provocar trastornos de la
hemodinamia renal que terminen en insuficiencia renal aguda.
Las prostaglandinas también regulan la liberación de ADH e inhibirlas puede
resultar en retención de sal, edema y en una reducción del efecto antihipertensivo
de los diuréticos.
Mientras que los efectos renales adversos han sido principalmente atribuidos a la
inhibición de la COX-1, ahora se reconoce que la COX-2 desempeña un rol
fisiológico en la homeostasis renal.
La COX-2 es la única isoforma que ha sido detectada en la mácula densa y su
expresión se dispara ante la restricción de sal. Podría tener también un rol en el
desarrollo ya que los ratones privados de COX-2 (knock out) desarrollan una
nefropatía progresiva en la medida que van envejeciendo.
39

Hasta hoy no existe evidencia sólida en humanos sobre los efectos renales
adversos con los AINEs inhibidores selectivos de la COX-2 que se disponen
actualmente. Sin embargo, la experiencia limitada con éstas drogas en la
actualidad hace recomendable su uso con precaución en pacientes susceptibles.
Como se señaló antes, los AINEs pueden desestabilizar el control de la presión
en pacientes tratados por hipertensión, efecto en parte debido a sus acciones
sobre la función renal.
Los pacientes que reciben tratamiento antihipertensivo, especialmente
inhibidores de la enzima de conversión, e inhibidores selectivos de la COX-2,
deberían ser vigilados en cuanto al desarrollo de eventos cardiorrenales.
Los pacientes que recibieron celecoxib experimentaron menos edema y menos
desestabilización del control de su presión arterial que los que recibieron
rofecoxib.
Los ratones privados de COX-2 también desarrollaron fibrosis cardíaca y
murieron precozmente. Las hembras son infértiles.
Las prostaglandinas están involucradas en diversos estadios de preñez. Se cree
que tanto la COX-1 como la COX-2 son importantes para la implantación del
huevo y para la angiogénesis necesaria para el establecimiento de la placenta.
Inmediatamente antes del comienzo del parto la expresión de la COX-2 se
incrementa significativamente en el amnios y en la placenta y se cree que las
40

prostaglandinas derivadas de la COX-2 son necesarias para la iniciación del
trabajo de parto.
41

EFECTOS FARMACOLÓGICOS DE LOS AINES
Efecto analgésico:
Los AINEs son leves a moderados analgésicos. Como vimos, el efecto analgésico
parece depender la inhibición de la síntesis de las prostaglandinas. Las
prostaglandinas parecen sensibilizar los receptores del dolor a la estimulación
mecánica o a otros media-dores químicos (ej: bradiquinina, histamina).
Las prostaglandinas producen hiperalgesia, es decir se produce dolor con
maniobras como la estimulación mecánica que comunmente no lo produce. Los
analgésicos antipiréticos no modifican el umbral del dolor y no previenen el dolor
causado por prostaglandinas exógenas o ya formadas, estas drogas pueden
producir analgesia por prevenir la síntesis de prostaglandinas involucradas en el
dolor. Parecería que los efectos analgésicos son principalmente periféricos,
aunque estas drogas pueden tener actividad semejante u otro mecanismo de
acción similar en el SNC, posiblemente en el hipotálamo.
Además su mecanismo de acción antinflamatorio puede contribuir a sus efectos
analgésicos.
No existen evidencias que durante el uso crónico de los AINEs se desarrolle
dependencia psíquica o física a estos agentes.
Efectos antinflamatorios:
Debido a la complejidad de la respuesta inflamatoria, el mecanismo exacto de los
efectos antinflamatorios no está totalmente aclarado.
(Ver acciones no dependientes de prostaglandinas de los AINEs). Las
prostaglandinas parecen mediar muchos efectos infla-matorios y han mostrado
producir directa-mente muchos de los síntomas y signos de
42

la inflamación, los efectos antiinflamatorio pueden deberse en parte a la inhibición
de la síntesis y liberación de estos autacoides durante la inflamación. Sin embargo
parecerían existir, como vimos, otros mecanismos que contribuyen a este efecto.
La patología inflamatoria es atenuada por los AINEs, aunque en los procesos
reumáticos no se evitan las lesiones de los tejidos (articulares) ni se detiene el
progreso de la enfermedad.
Efectos antipiréticos:
La aspirina y los agentes AINEs reducen la temperatura elevada, mientras que la
temperatura corporal normal es solo suavemente
afectada. La disminución de la temperatur generalmente se relaciona por un
incremento en la disipación causado por vasodilatación
de vasos sanguíneos superficiales y puede acompañarse de sudoración profusa.
El mecanismo de acción antipirético es por inhibi-ción de síntesis y liberación de
prostaglandi-nas en el hipotálamo. Prostaglandinas y fiebre de este Volúmen).
Casi todas las prostaglandinas con excepción de la I2 son piretógenas.
Paradójicamente la intoxicación con salicilatos puede producir elevación de la
temperatura corporal, por aumento del consumo de óxigeno y de la tasa
metabólica, aparente-mente por desacople de la fosforilación oxidativa.
Efectos antiagregantes plaquetarios:
La aspirina y los demás agentes antiinflama-torios no esteroides inhiben la
agregación plaquetaria y prolongan el tiempo de sangría debido a una inhibición
de la síntesis de tromboxano A2 en las plaquetas. En general, el agente de
elección para este efecto es la aspirina por ser inhibidor irreversible de la
cicloxigenasa, es decir, que acetila la enzima. Como las plaquetas son fragmentos
celulares, la cicloxigenasa queda inhibida por el resto de la vida de esas plaquetas
(7-11 días) hasta que nuevas plaquetas son forma-das, sin embargo la PGI2 o
prostaciclina que se sintetiza en el endotelio vascular puede seguir liberandose y
43

produciendo su efecto antiagregante y vasodilatador, sobre todo cuando se utilizan
dosis bajas de aspirina (350 y hasta 100 mg/día). Numerosos ensayos clínicos han
aportado evidencias que la aspirina reduce la incidencia de trombosis arterial
coronaria; en un prolongado estudio multicéntrico fue observado que 325 mg de
aspirina por día redujeron en un 40% la incidencia de infarto en médicos varones.
Esta acción de los AINEs como antiagregantes, muchas veces puede ser un
efecto colateral sobre todo cuando los pacientes deben ser sometidos a cirugía.
Efectos a nivel vascular:
Los AINEs inhiben la síntesis de prostaciclina (PGI2) PGE2 que poseen
propiedades vasodilatadoras, pudiendo de este modo disminuir el efecto
hipotensor de bloqueado-res beta, inhibidores de la enzima de conversión de
angiotensina (IECA), diuréticos, entre otros.
44

AINES UTILZADOS EN ODONTOLOGIA
Existen similitudes y diferencias entre los AINEs, aunque en general estos
agentes se absorben completamente por vía oral. Tienen escasa dependencia del
aclaramiento hepá-tico y del metabolismo de primer paso hepático.
Se unen con alta afinidad a la albúmina (el de menor unión es el
paracetamol) y tienen volumen de distribución pequeño.
Fue observado que el naproxeno y el ibuprofeno tienen acciones
antiinflamatorias semejantes, dependiendo de la dosis y la concentración
plasmática, en la artritis reumatoidea, sin embargo, esto no explica la variación
individual en la respuesta a estos y otros agentes. No se puede demostrar aún una
diferencia farmacocinética entre los pacientes que responden a AINEs y los que
no responden.
Con respecto a la vida media de estos agentes, podemos dividirlos en dos
grupos de acuerdo a su vida media de eliminación. Los de vida media corta
(menos de 6 horas) y los de vida media larga (más de 10 horas)
45

PARACETAMOL
El uso de paracetamol como antipirético incrementó cuando la fenacetina fue
sacada del mercado y más recientemente cuando se aceptó ampliamente a la
aspirina como agente etiológico del síndrome de Reye, lo cual hizo que este
agente (la aspirina) se restri n-ja en su utilización en niños sobre todo en fiebre y
enfermedades virales. Es un agente con pocos efectos colaterales, pero también
pobres efectos antiinflamatorios y efectos analgésicos y antipiréticos semejantes a
la aspirina.
Por ser bien tolerado y carecer de muchos de los efectos colaterales de la aspirina
ha ganado un lugar como analgésico casero, aunque las sobredosis agudas de
este agente pueden causar daño hepático fatal, en los últimos años ha aumentado
el número de intoxicaciones por automedicación. En general, se debe dejar bien
claro que el paracetamol posee débiles acciones antinflamatorias, por más que se
aumenten las dosis.
Farmacocinética
El paracetamol es un agente que se absorbe rápido y completamente en el tracto
gastrointestinal. Su vida media es de aproximada-mente 2 horas después de dosis
terapéuticas. La unión a proteínas plasmáticas es escasa. Cerca de un 60% de la
drogas se conjuga con ácido glucurónico, el 35% con ácido sulfúrico y cerca del
3% con cisteína, tambiémn se producen metabolitos hidroxilados y desacetilados.
Los niños poseen me-nor capacidad para la glucuronoconjugación.
46

Una pequeña proporción sufre N-hidroxilación por el citocromo P450 y forma un
metabolito intermedio de alta reactividad, en condiciones normales este metabolito
reacciona con los grupos sulfhifrilos del glutatión pero en sobredosis se agota el
glutatión hepático y los metabolitos reaccionas con los sulfhidrilos de otras
proteínas hepáticas y se puede producir necrosis hepáticas.
Usos Terapéuticos paracetamol
El paracetamol es un sustituto de la aspirina en las indicaciones como analgésico
o antipirético, o cuando la prolongación del tiempo de sangría que produce la
aspirina sería una desventaja.
47

IBUPROFENO
El ibuprofeno es el prototipo de los derivados del ácido propiónico, se
absorbe rápìdo por vía gastrointestinal, alcanzando picos máx imos en 1-2 hs. El
alimento afecta poco la biodisponibilidad de este agente. Se enlaza a proteínas
plasmáticas en un 99%, por lo que puede interaccionar con drogas que tienen alta
afinidad por estas proteínas. Se meta-boliza en el hígado, los metabolitos se
excretan por orina (cerca del 1% de droga libre). Se administra cada 6-8 horas.
Efectos colaterales: los efectos colaterales primarios son gastrointestinales
(nausea, dolor epigastrico precordialgia). Otros efectos colaterales incluyen
discrasias sanguineas, mareos, cefaleas, meningitis aséptica, edema, sangrado
GI, nefrotoxicidad, reacciones de piel y ambliopía tóxica.
Usos clínicos: se utiliza en el manejo de la artritis reumatoidea, osteoartritis,
dismenorrea primaria y síndromes dolorosos modera-dos.
Los pacientes que no responden a ibuprofeno pueden responder a otros AINEs.
Dosis: en artritis reumatoidea la dosis usual es de 1200 a 3200 mg/día, en
dolor modera-do 400 mg cada 4 a 6 hs, en dismenorrea primaria 400 mg cada 4
horas. La dosis máxima es de 3200 mg/día .
Dosis pediátrica: 20 mg/kg/día en dosis divididas, no exceder los 500 mg/día en
chi-cos con menos de 30 kg.
En chicos con fiebre de 6 meses a 12 años es de 5 mg/kg si la temperatura es
menor a 38..5 ºC o 10 mg/kg si la temperatura es
mayor. La dosis diaria máxima es de 40 mg/kg.
48

KETOROLACO
El ketorolac es un potente analgésico, antipi-rético y antiinflamatorio no esteroide,
(AINEs) comparable a los opiáceos, aparentemente sin mayores efectos adversos
que otros AINEs. Sin embargo, recientemente, la secretaría de Salud de Alemania
informó al fabri-cante de Ketorolac la intención de revocar la autorización para su
producción y venta, debido a comunicaciones de efectos adversos serios, como
insuficiencia renal aguda en pacientes tratados post-operatoriamente, úlceras,
hemorragias digestivas, perforación gástrica y duodenal. Y reacciones de
hipersensibilidad. El fabricante aunque no estuvo de acuerdo con esta opinión,
discontinuó la fabricación del producto en Alemania, lo que llama poderosamente
la atención.
El Comité de Medicamentos de la Comunidad Económica Europea en 1993, ha
pro-puesto que la forma inyectable (i.m. o i.v.) debe ser revisada precisando las
indicaciones, contraindicaciones y dosis máxima. Su indicación debe hacerse por
muy corto período de tiempo (2 días) en su administración i.m. o i.v. en el dolor
postoperatorio modera-do o severo.
A su vez contraindicó su utilización en: Asma, hipovolemia, deshidratación,
anteceden-tes de úlcera péptica o trastornos de la coagulación, hipersensibilidad a
los AINEs, en pacientes con pólipos nasales, antecedentes de angioedema,
broncoespasmos, tratamiento con litio, embarazo, lactancia, insuficien-cia renal
moderada o severa, pérdida de sangre en el tracto gastrointestinal , antecedentes
de ACV, diátesis hemorrágicas, riesgo de hemorragia postoperatoria,
administración de anticoagulantes.
No debe usarse junto con otros AINEs, con terapia anticoagulante (heparina,
anticoagu-lantes orales), ni con la pentoxifilina o el probenecid, tampoco en
menores de 12 años.
La dosis inicial (i.v./i.m.) debe ser de 10 mg seguida de 10-30 mg cada 4-6 hs,
según necesidad. Dosis diaria máxima en adultos: 90 mg, 60 mg en ancianos.
49

La duración máxima del tratamiento parenteral: (i.m./i.v.): dos días. En caso de ser
transferido el tratamiento a la vía oral el límite máximo de 90 y 60 mg debe ser
respetado y no debe administrarse crónicamente.
Indicación: Por vía oral: tratamiento a corto plazo del dolor agudo de moderado a
grave.
Por vía parenteral: tratamiento a corto plazo del dolor pos-operatorio agudo de
moderado a grave.
El Ketorolac no está indicado para el trata-miento de dolores crónicos.
Posología: La dosis diaria se adecuará a la intensidad del dolor aceptándose
dosis di aria máxima 90 mg.
Comprimidos: Dosis inicial 10 mg. Dosis de mantenimiento de 10 a 20 mg. cada
6 horas, no debiendo exceder la duración del trata-miento
los 5 días. Tratamientos más prolon-gados han sido asociados con un aumentos
de la incidencia de efectos adversos graves.
Ampollas: uso por vía intramuscular o endovenosa:
Dosis inicial: 10 mg. Dosis subsiguientes: 10 a 30 mg. cada 8 horas. Duración
máxima del tratamiento: 2 días. En los pacientes que han recibidos el Ketorolac
inyectable y que sean transferidos a compri midos de 10 ó 20 mg., la dosis diaria
combinada no deberá exceder los 90 mg.
Uso por venoclisis:
Se aconseja administrar Ketorolac por vía venoclisis utilizando una dilución de 60
mg. en 500 ml. de solución fisiológica o dextrosa al 5%. La dosis inicial deberá ser
de 10 mg. (equivalente a 83,33 ml. de la solución), respetando las dosis diarias
máximas especificadas anteriormente.
50

TOXICIDAD DE LOS AINEs
Toxicidad gástrica
La patogénesis de la gastropatía es multifactorial y depende de la
producción y metabolismo del ácido araquidónico, de los tipos de enzimas
existentes, de los cambios en el pH, de las propiedades estructurales, bioquímica
y funcionales del epitelio gástrico, del flujo sanguíneo, del vaciamiento gástrico y
de la circulación entero hepática. Otros estudios muestran que la pérdida de
sangre en materia fecal es rara. Los AINEs son los únicos culpables de las
lesiones gastrointestinales asociadas con su empleo, posiblemente hay factores
genéticos y ambientales que predisponen el daño. Los AINEs dañan el estómago
al privarlo del defecto citoprotector de las prostaglandinas pero también afectan la
mucosa gástrica localmente.
Toxicidad renal
Los pacientes con riesgo de sufrir toxicidad renal son aquellos que presentan: falla
cardiaca congestiva, cirrosis con ascitis, Síndrome nefrótico, estenoides de la
arteria renal, hipotensión, hipovolemia, deshidratación, pacientes que toman
inhibidores Eca (ej. Capotén). Los pacientes con función renal normal y que
reciban AINEs a dosis adecuadas no presentan riesgos de toxicidad.
Toxicidad hepática
El efecto puede ser hepatocelular (niveles de transaminasas elevadas),
colestáticos o de ambas categorías. Se presentan más a menudo con salicilatos,
derivados del ácido propiónico y derivados del ácido fenilacético; con salicilatos,
los niveles de transaminasas se aumentan de 3 a 4 veces y están implicadas en la
génesis del Síndrome de reyes. Los derivados del ácido fenilacético producen
elevaciones asintomáticas de las aminotransferasas séricas en cerca del 15% de
51

los casos, en general, no superan 3 veces el límite superior de lo normal y es
reversible.
Toxicidad hematológica
Las prostaglandinas desempeñan un papel importante en la función
plaquetaria y en el tono muscular. Los efectos hematológicos de los AINEs son
relativamente raros. Los mecanismos de reacción dependen de las propiedades
farmacológicas de los fármacos y de las reacciones dependientes de mecanismos
inmunes. De las reacciones hematológicas las más comunes son: Anemia
aplástica: El riesgo de esta enfermedad aumenta con la edad y con el sexo (mayor
en las mujeres). El mecanismo es por daño directo a la célula madre del sistema
hematopoyético por el medicamento o sus metabolitos o bien por mecanismo auto
inmune. La anemia puede ser debida a las lesiones gastrointestinales y es de tipo
hipercrómica o microlítica, se ve más con el ácido acetil-salicilíco, otra forma de
anemia inducida por ácido acetil-salicílico es medida por complejo inmune,
influyen además factores como la polifarmacia, el tiempo de uso, el empleo de
alcohol, etc. agranulacitosis: Hay tres tipos de causas por fármacos: TIPO I:
Mediado por anticuerpos y complejos inmunes dirigidos contra el medicamento
que se une a la superficie del granulocito, es la forma clásica y está asociada a
Aminopirina, pirazolonas, ácido acetilsalicílico, fenacetina, acetaminofén, con una
incidencia de 0,05%-1%. TIPO II: Está relacionado con la toxicidad directa sobre la
médula ósea dependiendo de la dosis, en estos casos hay: sensibilidad individual,
se han asociado pirazolonas y diclofenac. TIPO III: Son mixtas, están implicadas
indometacina, sulindaco, tolmetín, naproxeno.
NOTA: De todos modos son eventos raros; se considera que se producen 6,3
casos por millón por año.
Efecto sobre las plaquetas pueden ser: Cuantitativas: Trombocitopenia:
producidos por un mecanismo inmune análogo a la anemia hemolítica auto
inmune, se ha visto con indometacina, tolmetin, ácido acetil-salicílico, piroxicam,
52

fenilbutazona y diclofenac. Cualitativas: Trombocitopatía: se debe a la inhibición
de la síntesis de tromboxano. El ácido acetilsalicilico inhibe de forma irreversible la
actividad de la ciclooxigenasa, los otros AINEs la inhiben de forma reversible. Para
estudiar la función plaquetaria se utiliza el tiempo de sangría y agregación
plaquetaria. El efecto antiplaquetario persiste mientras el medicamento es
depurado, con el ácido acetilación es irreversible, tanto en el megacariocito como
en la plaqueta.
Par ala mayoría de los pacientes, este efecto antiplaquetario no tiene importancia
clínica o se usa para prevenir enfermedades cardiovasculares. En pacientes con
defectos hematológicos previos, debe tenerse cuidado por ejemplo en pacientes
con trombocitopenia con recuento plaquetario inferior a 50.000 por ml3, pacientes
hemofílicos por defecto de factor VIII y IX y, en pacientes intervenidos
quirúrgicamente que tengan grandes superficies sangrantes.
Hipoprotrombinemia: Se ven en casos de intoxicación aguda por fenilbutazona,
piroxicam, naproxeno y ácido mefenámico. En conclusión, la frecuencia de efectos
adversos de tipo hematológico es escasa.
53

REACCIONES ALÉRGICAS Y SEUDOALÉRGICAS ASOCIADAS CON AINEs
Hay dos tipos de reacciones farmacológicas:
TIPO A: Directamente producidas por el medicamento.
TIPO B: Indirectamente, como alergías, idiosinerasias e intolerancia.
Clasificación
Las reacciones alérgicas se basan en mecanismos inmunes en los cuales
anticuerpos o linfocitos sensibilizados inducen la reacción.
Reacción Tipo I: Son mediadas por IgE: urticaria, angioedema, bronco espasmos,
shock anafiláctico, rinitis alérgica, conjuntivitis.
Reacción Tipo II: Citotoxicidad inducida inmunológicamente con aglutinación o lisis
de eritrocitos, trombocitos o leucocitos.
Reacción tipo III: Por complejos inmunes, por ejemplo fiebre por medicamentos,
enfermedad del suero, vasculitis alérgica, fenómeno de Arthus.
Reacción Tipo IV: Reacciones de hipersensibilidad retardada por ejemplo:
Dermatitis por contacto, a veces se superponen dos o tres mecanismos diferentes.
Estos pacientes deben evitar los AINEs que inhiben fuertemente de ciclooxigenasa
y los ácidos acetil-salicílicos. Otro tipo de reacciones muy raras son las neumonitis
y el edema pulmonar descritos con fenilbutazona, oxifenbutazona, sulindac,
naproxeno.
54

COMPLICACIONES NEUROLÓGICAS DE LOS AINEs
La mayoría son leves, exceptuando las encefalopatías por salicilatos que pueden
ser fatales. Los mecanismos son: reacciones alérgicas por idiosincrasia: como la
meningitis aséptica producia por ibuprofeno, relacionadas con algunos
medicamentos como: litio e indometacina, por acción sobre el metabolismo de los
ácidos grasos, como la enfermedad de Rey, otros son propios de los fármacos por
acción sobre serotoninas como la cefalea. Los salicilatos producen: delirio,
demencia, sordera, diplopia, miopía, síndrome de Reye, convulsiones.
El Diclofenac produce retención de litio y precipita la porfiria.
El Ibuprofeno: Puede producir delirio, meningitis aséptica y pérdida de la audición.
La Indometacina: Puede causar cefalea, delirio, tención de litio, pérdida de la
audición, neuropatía periférica, depresión, entre otros.
El Naproxeno: Puede producir delirio, interacción con fenotiazinas.
El ácido Mefenámico: Puede desencadenar convulsiones, disquinesia, torticolis.
Se puede producir convulsiones por inhibición de la síntesis de Gaba.
55

REACCIONES DERMATOLÓGICAS DE LOS AINEs
Los productos de la lipooxigenasa y de las prostaglandinas pueden estar
comprometidos en enfermedades como la psoriasis, dermatitis por contacto,
mastocitosis urticaria al frío y eritema ultravioleta. En general, son comunes pero
raramente peligrosos, la incidencia de necrólisis epidérmica tóxica con diclofenac
es de 0,03 casos por millón de dosis diarias, también la producen el acetil-
salicílico, pirazolonas, indometacina, sulindac, tolmetin, naproxeno y oxianes.
Eritema Fijo: Eritema multiforme y Stevens y Johson con ácido acetil-salicílico,
sulindac, tolmetin, ibuprofeno, naproxeno, piroxicam, diclofenac.
Fotosensibilidad: La puede causar la indometacina, fenilbutazona, piroxicam,
ketoprofeno y naproxeno.
Pancreatitis: Se puede producir con ácido acetil-salicílico y con ibuprofeno.
EMBARAZO Y LACTANCIA
Según algunos investigadores, el uso de AINEs en la lactancia no tiene mayores
riesgos, excepto con la indometacina, cuando los niveles del fármaco en la leche
materna pueden exceder los niveles séricos y el medicamento está
contraindicado. Sin embargo, el uso de los mismos debe hacerse con precaución.
El ácido acetil-salicítico y otros antiinflamatorios no esteroideos, tomados de dos a
cuatro semanas antes del parto, pueden producir cierre prematuro del ductus
arterioso, provocando efectos adversos sobre la circulación pulmonar.
56

CONCLUSIONES
El dolor (algesia) es un problema común a todas las áreas de la odontología,
por lo tanto, es importante entenderlo, reducirlo al mínimo o prevenirlo
(analgesia).
El dolor clínico es más fácil de controlar si el analgésico se toma antes de la
aparición del dolor (no está claro si es un mecanismo psicológico o
farmacológico).
Si el dolor post-operativo es tan severo que los analgésicos orales son
incapaces de hacerlo tolerable al paciente, será necesario un mayor
tratamiento clínico y no una mayor analgesia.
Una de las herramientas de la terapéutica moderna en odontología es el uso
de los AINEs como mejor elección.
Todos los AINEs presentan efectos adversos que deben ser considerados y se
estima que, entre 1 y 3% de la población desarrolla efectos secundarios
graves, incluso con dosis mínimas.
Se destaca el uso de los AINEs en el dolor agudo de tipo leve a moderado y,
en casos de dolor moderadamente intenso a severo es útil añadir un opioide
ligeramente potente a una dosis total de un no opioide.
La importancia terapéutica de los inhibidores selectivos de la COX2 es que no
ocasionan tantas reacciones adversas como los AINEs clásicos. Sin embargo,
deben de medicarse con precaución y evitarlos en pacientes con problemas
renales, cardiovasculares y hepáticos.
57

BIBLIOGRAFÍA
Canalda, Carlos y Brau Esteban (2001). Endodoncia. Técnicas Clínicas y
Bases Científicas. España MASSON, S.A.
Clínicas Odontológicas de Norteamérica (1984). Endodoncia. 4 Interamericaba
MacGraw Hill.
Clínicas Odontológicas de Norteamericana (1984). Fármaco Odontología. 4.
Editorial Interamericana MacGrawq Hill
Cohen, Stephen y Bums Richard (1999). Vías de la Pulpa (7,2 ed) España:
Harcourt.
Díaz de León, Melva. Fármaco terapéutica (1989) 1 Venezuela, Universidad de
los Andes.
GAGE, Tommy y Pickett Frieda (1994). Dental Drug Reference Editorial
Mosby-Year Book, Inc.
Laboratorio Ciba (1995). Entrenamiento al día, 5. Venezuela Autor.
Páyele, Carlos y Bilbeny, Norberto (1997). El Dolor, Aspectos Básicos y
Clínicos (2à ed.) Chile: Mediterráneo.
América Dental Association (1985). Terapéutica Odontológica aceptada de la
América Dental Association (3911 ed). Argentina: Médica Panamericana, S.A.
58


















