Resumen FARMACO
57
Farmacología General Generalidades: Farmacología: ciencia que estudia a un grupo de sustancias (drogas, fármacos o medicamentos) y los cambios que producen en el organismo. Para ello se divide en varias ramas, las 3 más importantes son: Fármacoterapia: estudia el uso de los medicamentos útiles en la prevención y tratamiento de las enfermedades de los seres vivos. Dentro de esta rama se encuentra la quimioterapia (estudia agentes químicos específicos en contra de patologías definidas, actualmente sólo se asocia al tratamiento de neoplasias). Farmacodinamia: estudia el efecto de los fármacos, sus mecanismos de acción sobre el organismo desde los puntos bioquímicos y fisiológicos, y establece las relaciones químicas existentes entre la estructura química de los fármacos y su actividad farmacológica. Para entenderla bien hay que conocer bien al fármaco y su receptor. Farmacocinética: estudio de las transformaciones de los fármacos en el organismo (desde la aplicación a la salida del fármaco). Estudia cómo es capaz de moverse el medicamento para lograr un efecto. Para entenderla se subdivide en el sistema LADME (liberación, absorción, distribución, metabolización y eliminación). Este sistema puede ser expresado en parámetros cinéticos. Fármaco principio activo (PA). La mayoría son ácidos o bases débiles presentes en soluciones como especies ionizadas y no ionizadas. Medicamento fármaco integrado en una forma farmacéutica y listo para su utilización. Se refiere a la forma de presentación (FP). Droga origen natural Formas de presentación: - FP PA + vehículo (líquido, gaseoso, semi sólido o semi líquido) o excipiente (sólido) - Biofarmacia: estudia las formas de presentación, las cuales básicamente pueden ser sólido, líquido o gaseoso. - El vehículo o excipiente acompaña al fármaco para facilitar la liberación, absorción, mejorar la palatabilidad y proteger los tejidos. Además, ayudan a que el ingreso del fármaco sea rápido o lento según necesidad, y seguro. Ej: agua, saborizantes, celulosa, gelatinas, polímeros sintéticos, azúcares, sustancias oleosas. - Al mismo tiempo, la forma de presentación define la vía de administración. Farmacocinética: El paso del fármaco por el organismo puede ser descrito por el sistema LADME: 1. Liberación: involucra liberar un principio activo desde una forma de presentación. Actualmente existen 4 formas de presentación en el mercado: a) Sólidos: i.- Tabletas: muy aglutinados y difíciles de tragar.
-
Upload
catita2389 -
Category
Documents
-
view
3.582 -
download
1
Transcript of Resumen FARMACO

Farmacología General
Generalidades:Farmacología: ciencia que estudia a un grupo de sustancias (drogas, fármacos o medicamentos) y los cambios que producen en el organismo. Para ello se divide en varias ramas, las 3 más importantes son:
Fármacoterapia: estudia el uso de los medicamentos útiles en la prevención y tratamiento de las enfermedades de los seres vivos. Dentro de esta rama se encuentra la quimioterapia (estudia agentes químicos específicos en contra de patologías definidas, actualmente sólo se asocia al tratamiento de neoplasias).
Farmacodinamia: estudia el efecto de los fármacos, sus mecanismos de acción sobre el organismo desde los puntos bioquímicos y fisiológicos, y establece las relaciones químicas existentes entre la estructura química de los fármacos y su actividad farmacológica. Para entenderla bien hay que conocer bien al fármaco y su receptor.
Farmacocinética: estudio de las transformaciones de los fármacos en el organismo (desde la aplicación a la
salida del fármaco). Estudia cómo es capaz de moverse el medicamento para lograr un efecto. Para entenderla se subdivide en el sistema LADME (liberación, absorción, distribución, metabolización y eliminación). Este sistema puede ser expresado en parámetros cinéticos.
Fármaco principio activo (PA). La mayoría son ácidos o bases débiles presentes en soluciones como especies ionizadas y no ionizadas. Medicamento fármaco integrado en una forma farmacéutica y listo para su utilización. Se refiere a la forma de presentación (FP). Droga origen natural
Formas de presentación:- FP PA + vehículo (líquido, gaseoso, semi sólido o semi líquido) o excipiente (sólido) - Biofarmacia: estudia las formas de presentación, las cuales básicamente pueden ser sólido, líquido o gaseoso. - El vehículo o excipiente acompaña al fármaco para facilitar la liberación, absorción, mejorar la palatabilidad y
proteger los tejidos. Además, ayudan a que el ingreso del fármaco sea rápido o lento según necesidad, y seguro. Ej: agua, saborizantes, celulosa, gelatinas, polímeros sintéticos, azúcares, sustancias oleosas.
- Al mismo tiempo, la forma de presentación define la vía de administración.
Farmacocinética:El paso del fármaco por el organismo puede ser descrito por el sistema LADME:1. Liberación: involucra liberar un principio activo desde una forma de presentación. Actualmente existen 4 formas de presentación en el mercado:a) Sólidos:
i.- Tabletas: muy aglutinados y difíciles de tragar.ii.- Pastillas: más suaves en la partición. Tienen menor aglutinación por lo tanto se absorben más rápido que las tabletas.iii.- Comprimidos: la incorporación de forma facilita su administración, se les mejora la aglutinación, son recubiertos de un baño de azúcar por lo tanto se les mejora la palatabilidad, son divisibles. Los comprimidos con recubierto entérico tienen una gruesa capa de azúcar que los hace indivisibles y que protegen al estómago del PA permitiendo así su liberación en intestino. iv.- Cápsulas: permite 2 administraciones, blanda (celulosa que en su interior contiene líquido por lo tanto se salta el proceso de disolución) y perla. v.- Grageas: son lo mismo que los comprimidos pero con la diferencia de que tienen forma. Al ser de forma alargada permiten entregar mayor volumen y por lo tanto mayor concentración. vi.- Polvos: se les puede cambiar la forma a líquido.vii.- Pellet: permite administración a lo largo del tiempo (depósito), por lo tanto es de acción larga (LA).
b) Líquidos: Oral:
i.- Suspensión: soluto + solvente (inestables)ii.- Solución: si tiene precipitado está mala.

iii.- Jarabe: solución saturada de azúcar, altamente palatable.iv.- Cápsulas blandas: contienen una solución en su interior.
Inyectable:- Vía EV se pueden aplicar reconstituciones, soluciones y suspensiones (si son muy viscosas o no son
tranparentes no se pueden aplicar por esta vía).- Vía IM se pueden aplicar suspensiones viscosas de LA.
Local:- Pueden ser suspensiones o soluciones. - Poseen características determinadas para que permanezcan en el lugar de acción.- Ej: gotas óticas, depósito subconjuntiva ocular, suspensión intramamaria.
c) Gaseosa:- Vía muy eficiente ya que los pulmones son muy irrigados.- Tiene las restricciones que es la más cara y los pesos moleculares deben ser lo más pequeños posibles.
d) Semi-líquida o semi-sólida:- SL cremas, lociones, ungüentos (difieren en la cantidad de partículas)- SS supositorios- Útiles en patologías dérmicas, sin embargo tienen la desventaja que al estar al alcance del animal este se lame.
Absorción:- Involucra el paso del fármaco a través de las membranas biológicas a la sangre. - Su objetivo es alcanzar una concentración eficiente del PA en el torrente sanguíneo.- Biodisponibilidad biológica (F): cantidad de fármaco en el torrente que está disponible para llegar al sitio de
acción.
Factores que influyen en la velocidad de absorción: i. Vía de administración: determina principalmente la superficie absorbente. Oral (PO):
- Vía más simple.- Rápida, segura y económica.- No tiene restricciones de temperatura y no genera dolor.- Biodisponibilidad y velocidad variable, depende de varios factores que la van disminuyendo:
pH: la acidez del estómago destruye algunos compuestos. Tasa de vaciamiento gástrico (cuánto de demora en llegar a duodeno): cualquier factor que acelera el
vaciamiento gástrico probablemente incrementará la velocidad de absorción, mientras que cualquier factor que lo retrase tendrá un efecto contrario.
Ambiente gástrico Presencia de mucus Presencia de alimento en el estómago Vómitos: como consecuencia a irritación de la mucosa gastrointestinal. Efecto de 1er paso o 1era pasada: parte del fármaco pasa a la vena porta y llega al hígado donde es
metabolizado, por lo tanto disminuye la cantidad disponible para ser absorbido. Además del hígado, los fármacos pueden ser metabolizados por las enzimas digestivas o la flora
intestinal.
Parenteral: en general la disponibilidad es más rápida que si el agente se administra vía oral, y la dosis efectiva puede ser determinada con mayor exactitud.
- Intramuscular (IM): La absorción es rápida en soluciones acuosas (con dependencia de la velocidad del flujo sanguíneo en el
sitio de inyección), y lenta y sostenida en preparados con depósitos. La velocidad de absorción también depende en parte de la cantidad y distribución de grasa subcutánea.

Vía propia de la presentación LA La biodisponibilidad y la velocidad dependen del vehículo y principalmente del pH del músculo. Tiene limitaciones de volumen (en animales menores no más de 5 ml por zona de aplicación en mayores
10 ml). Produce dolor al distender el músculo. Pinchar solamente en masa muscular (nunca fascia) y generalmente en extremidades. El producto debe estar formulado para esta vía (nunca puede ser necrosante y ni irritante).
- Endovenosa (EV): Vía de urgencia. Requiere de limpieza obligatoria de la zona de aplicación. Restringe muy poco el volumen. Requiere de control de velocidad. Dada para fármacos transparentes, de baja viscosidad e irritantes. Los agentes en un vehículo oleoso o aquellos que forman precipitados con los compuestos sanguíneos o
hemolizan los eritrocitos no deben ser administrados por esta vía. Permite un control de concentración-efecto 100% biodisponibilidad.
- Subcutáneo (SC): Rápido para soluciones acuosas, lento y sostenido para preparados con depósitos. La incorporación de un agente vasoconstrictor en una solución inyectable por esta vía retarda la
absorción. Conveniente para algunas suspensiones insolubles y para la implantación de pellets sólidos. No es conveniente para grandes volúmenes.
Locales:- Son una modificación de la parenteral.- Intraosea: única vía para aplicar líquido en un animal con deshidratación extrema (más de un 10%) ya que en
estas condiciones en cualquier otro lugar el líquido se encapsula. - Intraarterial: los fármacos administrados por esta vía evitan los efectos del primer paso y depuración pulmonar.- Articular: vía de depósito, tiene distintas restricciones como volumen, no debe generar precipitado ni ser irritante.- Intratecal: se puede usar como vía de urgencia, o también cuando se necesita un efecto rápido y puntual (Ej:
anestesia epidural).- Rectal: aproximadamente un 50% del compuesto que es absorbido en recto evade el pasaje por hígado, por lo
tanto tiene efecto primer paso menor que en la vía oral, sin embargo, la absorción rectal a menudo es irregular e incompleta y varios agentes irritan la mucosa rectal.
- Intraperitoneal: gran superficie de absorción por lo que otorga rapidez, principalmente a través de la vena porta, en consecuencia, las pérdidas de primer paso son posibles. Alto peligro de causar infecciones y adherencias.
- Pulmonar: absorción casi instantánea, puede haber pérdida por efecto de primer paso, también puede ser muy irritante y causar molestias.
- Ocular: la absorción sistémica que se debe al drenaje a través del conducto nasolagrimal en general no es deseada, además el compuesto absorbido luego de este drenaje no está sometido a la eliminación por primer paso hepático, por esta razón se pueden producir efectos farmacológicos indeseados.
- Otras: ótica, dérmica, sublingual, etc.
ii. Características físico-químicas del fármaco: - Peso molecular: determina el mecanismo de paso que usa el fármaco para ingresar al torrente. Fármacos que
tienen un peso muy alto (sobre 40.000 Dalton) se quedan restringidos al lugar de aplicación ya que no pueden atravesar la membrana (Ej: soluciones coloidales, manitol).
- Grado de disociación: Dependiendo del pH del medio y del pKa (constante de disociación) del fármaco, éste se puede
encontrar asociado o disociado.

A las moléculas cargadas les cuesta más pasar porque deben recurrir a un carrier (se vuelven hidrosolubles), por lo tanto uno busca que el fármaco esté asociado ya que así no tiene carga, es más liposoluble y puede atravesar fácilmente la bicapalipídica.
Fármacos ácidos prefieren ambientes ácidos y fármacos básicos ambientes básicos. - Liposolubilidad: a mayor liposolubilidad con mayor facilidad se atraviesan los tejidos. - Concentración: a mayor concentración, mayor velocidad de absorción.
iii. Membranas biológicas (barreras):- Influye el número de capas, permeabilidad (n° de poros, diámetro), presencia de carriers.- Los fármacos pueden atravesar la membrana a través de procesos pasivos (poro, difusión) o activos (carrier)
dependiendo de sus características físico-químicas. - En general la difusión pasiva es en mecanismo dominante en la absorción y la distribución.- El transporte activo de algunos fármacos se produce a través de las membranas neuronales, el plexo coroideo,
las células tubulares renales y los hepatocitos.
iv. Sangre:- A mayor irrigación (estimulado mediante un masaje o aplicación de calor local) mayor absorción.- Un menor flujo sanguíneo, producido por agentes vasoconstrictores, shock u otros factores patológicos puede
retardar la absorción.
3. Distribución: - Proceso por el cual el principio activo alcanza el sitio de acción.- Parámetro cinético: volumen de distribución (Vd) medida del espacio aparente del organismo para contener el
fármaco (volumen de líquido que se requeriría para contener todo el fármaco presente en el organismo con la misma concentración que en sangre o plasma). Relaciona la cantidad de fármaco en el organismo con su concentración en sangre o plasma.
- El fármaco sólo alcanza el sitio de acción una vez que alcanza un equilibrio sistémico, primero llega a los órganos de gran irrigación (corazón, pulmón, hígado, riñón, cerebro, etc) y luego a los otros tejidos (músculos, vísceras, piel y grasa).
- El Vd puede variar ampliamente con dependencia del pKa del compuesto, del grado de fijación a proteínas plasmáticas, del la liposolubilidad del fármaco, del grado de fijación a otros tejidos, etc. Además, puede cambiar en función de la edad, sexo, patologías y composición orgánica del paciente.
Factores que influyen en la distribución:a) Unión a proteínas plasmáticas (UPP):
Principalmente a albumina ya que es la más abundante. Se determina como un porcentaje, a mayor afinidad mayor % y viceversa. La fracción de fármaco total presente en el plasma fijado a proteínas está determinado por la
concentración del agente, su afinidad por los sitios de unión y el número de estos últimos. A mayor afinidad más lenta es la distribución ya que al estar unido con una proteína plasmática el
fármaco se ve obligado a quedarse en el torrente y no podrá migrar a tejidos Permite ejercer un control en el paso de un fármaco. Finalmente el fármaco es liberado por diferencias de concentración entre el torrente y el tejido. UPP promedio= 90% La UPP pasa a ser importante dependiendo del tipo de fármaco, y puede ser tanto negativa como
positiva. Ej: de nada me sirve un anestésico con alto UPP porque requiero que llegue rápidamente al sistema nervioso.
Esta unión es poco específica, por eso muchos fármacos con características similares pueden competir entre sí y con sustancias endógenas por los sitios de unión. Ej: las sulfonamidas compiten con la bilirrubina no conjugada, aumentando el riesgo de encefalopatía bilirrubinémica en recién nacidos.
En el caso de fármacos que están extensamente unidos a las proteínas plasmáticas, pero que no están fijados a los componentes tisulares, el volumen de distribución se aproxima al volumen plasmático.
b) Irrigación:

Una irrigación normal viene dada por una adecuada hidratación. A peor irrigación peor distribución.
c) Tejido de reservorio: Cada paciente tiene distinto % de tejidos por lo tanto % distribución. Los fármacos que se han acumulado en un tejido dado pueden actuar como reservorio prolongando la
acción del agente en ese mismo tejido o en un sitio distante al cual llega mediante la circulación. Si el reservorio tiene gran capacidad y se ocupa rápidamente, la distribución del fármaco se altera de
modo que se requieren inicialmente cantidades mayores de éste para proporcionar una concentración terapéuticamente efectiva en el órgano blanco.
El principal tejido de reservorio es el adiposo, pero también lo son el muscular y el óseo. Para que el fármaco vaya a un determinado tejido debe tener afinidad con él.
Modelo de distribución de fármacos: Unicompartimental o de un compartimiento:
- El organismo actúa como una unidad homogénea en donde el fármaco se distribuye en un sólo lugar.- Esta teoría sólo sirve para el manitol y la tinción de fenol. - Ej: el manitol ingresa al torrente, pero al ser tan grande se queda ahí, de esta forma atrae agua y sirve para
disminuir edemas. Posteriormente es eliminado renalmente ya que el glomérulo permite su filtración. Actúa como diurético.
Multicompartimental:- En este modelo el fármaco ingresa primero a un compartimiento central (tejidos altamente irrigados), y una vez
que alcanza un equilibrio en este compartimiento pasa a un compartimiento periférico (tejidos menos irrigados). Una vez que disminuye la concentración en el Cc, el fármaco pasa desde el Cp al Cc para posteriormente ser eliminado.
Factores que influyen el grado de distribución:a) Velocidad de absorciónb) Capacidad de distribución tisularc) Constante de eliminación tisular y plasmática, realizada por procesos metabólicos y excretores.
4. Metabolización:
Vk12: cte de distribución de Cc a CpVk21: cte de distribución de Cp a CcVke10: cte de eliminación

- Su principal objetivo es la formación de metabolitos, estos son moléculas que toman carga para así hacerse más hidrosolubles y ser más fácilmente eliminadas.
- Esto se logra gracias al proceso de biotransformación transformación de la molécula original en una molécula con mayor capacidad de carga.
- El principal órgano metabolizador es el hígado (pero no el único), éste contiene sistemas de degradación como el citocromo P450 (CP450), los cuales son un grupo enzimático (encontrados en REL hepatocitos), específicamente hemoproteínas que tienen la capacidad de recibir u otorgar electrones gracias al hem que contienen. Son especie específicos.
- Estas enzimas también se encuentran en riñón, pulmón y epitelio gastrointestinal, aunque en cantidades menores.
- Existen una serie de factores genéticos, ambientales y fisiológicos que afectan el destino metabólico de un fármaco, como por ejemplo uso concomitante de otros fármacos inductores o inhibidores de las enzimas que participan en su metabolismo, o hepatopatías severas.
Fases de la metabolización:a) Oxido- reducción:
El CP450 actúa como oxidante y se acompaña de NADPH para la reducción. El fármaco se une al citocromo formando el complejo fármaco – citocromo, el cual es reducido por la
reductasa (NADPH) y se combina con oxígeno molecular. En esta fase adquieren la carga necesaria para poder ser eliminadas. La obtención de carga puede ocurrir en varias pasadas por el hígado, no siempre ocurre a la primera.
Mientras más liposoluble es la molécula más pasadas se requieren. Como producto se pueden obtener metabolitos inactivos (que no necesariamente se eliminan más
rápido), activos (que pueden tener una capacidad superior que el PA, en este caso se dice que el compuesto original es un profármaco) o tóxicos (como el acetominofeno en gatos).
b) Conjugación: En esta fase el metabolito se une a un ácido orgánico para poder ser eliminado. Los principales ácidos orgánicos son el ácido glucurónico, ácido acético y ácido sulfónico. Las concentraciones de estos ácidos son especie específico. El ácido le otorga más carga al metabolito para facilitar el paso al tejido de eliminación. Este proceso puede ser potenciado o limitado para favorecer la eliminación y evitar la saturación del
sistema de eliminación (gatos tienen bajas concentraciones de ácido glucurónico por lo tanto esa vía es altamente saturable).
5. Eliminación:- Las 2 vías más importantes son la renal y la biliar (hepática), pero también puede ser por saliva, sudor, leche,
etc.- Los parámetros cinéticos son Ke, vida media (T1/2), y clearence (CL).- Vida media: es el tiempo necesario para que la concentración plasmática o la cantidad de un fármaco presente
en el organismo se reduzca en un 50%. Es un parámetro derivado que varía en función de la depuración y del volumen de distribución.
- Clearence o depuración: cantidad de plasma que queda libre de fármaco por unidad de tiempo. Es una medida de la capacidad del organismo para eliminar el compuesto, por lo tanto también nos indica que tanta funcionalidad renal se necesita para eliminar al fármaco. Es el concepto más importante que debe tenerse en cuenta para la administración de una terapia durante un periodo prolongado.
i. Renal:- Cuantitativamente es la más importante.- Los fármacos pueden pasar por 3 procesos: filtración (pasivo), reabsorción o secreción (ambos activos). - La cantidad de un agente que llega al lumen tubular por filtración depende de la fracción que se fija a las
proteínas plasmáticas y de la velocidad de filtración glomerular. - El pH de la orina y el pKa del metabolito son fundamentales ya que determinan si el fármaco se disocia o se
asocia.

- Al estar asociado se reabsorbe, por lo tanto se prefiere la disociación porque al tener carga la molécula es eliminada.
- pKa ácido/básico en pH básico/ácido alta disociación, rápida eliminación. - pKa ácido/básico en pH ácido/ácido alta asociación.- En el tratamiento del envenenamiento por fármacos, la excreción de algunos compuestos puede ser acelerada
mediante la alcalinización o acidificación apropiada de la orina. Ej: La T1/2 de la aspirina en el gato es de 38,5 horas, en caso de intoxicación por este fármaco se puede basificar la orina (dando bicarbonato o ringer lactato) para así acelerar la eliminación.
- La ke puede dar la vida media del fármaco.
Factores que determinan la velocidad de la eliminación renal:a) pHb) Irrigación da la tasa de filtración glomerular.c) Funcionalidad renald) UPP cuando la albumina está unida al fármaco no se filtra.e) Vd alto a mayor Vd, mayor llegada del fármaco a tejidos, menor Ke (porque hay menos fármaco en sangre, se
filtra menos y se demora más en ser eliminado). A menor Vd, menor T1/2 (porque se elimina más rápido).
ii. Biliar o hepática:- Los metabolitos son vertidos a duodeno a través de las sales biliares, ahí pueden ser reabsorbidos y pasar a
circulación entero hepática o simplemente ser eliminados por fecas.- Son reabsorbidos cuando tienen una alta liposolubilidad, cuando esto ocurre pasan a la sangre y finalmente son
eliminados por la orina.- Solamente importan aquellos metabolitos activos que son reabsorbidos y siguen ejerciendo su función (los
inactivos dan lo mismo), por lo tanto cuando se dice que un fármaco tiene circulación entero hepática quiere decir que cumple con estas condiciones.
- Las sustancias excretadas con las heces son principalmente las que han sido administradas por vía oral o metabolitos excretados en la bilis y no reabsorbidos en el tracto gastrointestinal.
Farmacodinamia:- Estudia las modificaciones que genera el fármaco al llegar al receptor, o sea, el efecto.- Según su dinámica, los fármacos se pueden caracterizar de 2 formas:
i. Agonista: estimula o inhibe una funciónii. Antagonista: evita la acción del agonista, actúan bloqueando los receptores.
Antagonista competitivo: depende de la concentración del agonista porque tiene que competir.
Receptores:- Una de las características que más importan en un receptor es la especificidad.- Mientras más amplia es su distribución menos específico es.- Receptores poco específicos tiene un efecto muy difuso y por lo tanto menos controlable.
Tipos: Específicos: como los que se encuentran en la pared celular de las bacterias y permiten que los antibióticos ejerzan su función. Semi-específicos: quelantes de receptor catión bivalente (++), anestésicos. No específicos: agua (manitol), protones (drogas acidificantes), hidroxilos (drogas alcalinizantes).
Propiedades de los receptores auténticos:1. Saturabilidad:
- Existe un n° finito de receptores, los cuales al saturarse alcanzan un estado de equilibrio, lo cual a la curva de saturación se expresa como un plateu.

- Hay fármacos que alcanzan la saturabilidad antes que otros, esto se debe a 2 propiedades: Especificidad: el fármaco es más afín con el receptor (se unen con más fuerza) Potencia: se requiere menos cantidad de fármaco en comparación con otros para lograr el mismo
efecto. - Que se sature antes significa que ejerce su función más rápido.
2. Especificidad:- Existe una complementariedad estructural entre el receptor y la droga.
3. Reversibilidad:- Un fármaco debe ser capaz de librarse del receptor sin sufrir cambios.- Todo fármaco tarde o temprano se suelta del receptor, pero si dice que la unión es “irreversible” cuando una vez
que lo suelta éste pierde su función para siempre o muere, por lo tanto los fármacos que se unen “irreversiblemente” se consideran tóxicos. Ej: órganos fosforados y acetilcolinesterasa (receptor).
Medición de un efecto:Las 3 formas más comunes de la expresión de un efecto son:
1. Respuesta molecular: incluye efectos como modulación enzimática y movilización de iones a través de membranas.
2. Respuesta celular: modulación en la secreción de hormonas o neurotransmisores.3. Respuesta orgánica: incluye respuestas como la concentración de las fibras musculares tras la administración
de una droga.
Otra forma de clasificar es según el receptor:1. Ionotrópicos:
- Provoca apertura o cierre de canales iónicos (Na, K, Ca, Cl).- Muy abundantes en el SNC- La respuesta es muy rápida.- Ej: tiopental, fenorbital, diazepam son muy liposolubles, atraviesan fácilmente la barrera
hematoencefálica y se dirigen a la membrana neuronal donde se encuentra su receptor Gaba. Su acción consiste en bloquear el canal iónico generando hiperpolarización, al no haber estimulación disminuye la actividad cerebral.
2. Metabotrópicos:- Receptores ligados a cadenas metabólicas (estimulación de un evento metabólico).- Abundantes en el SNP.- La respuesta es más pausada.- Ej: el salbutanol (adrenalina), el cual es un agonista selectivo, se une a su receptor y lo estimula.
Esto provoca que se active la proteína G, la cual gatilla la estimulación de la enzima adeninciclasa (genera fosforliación de GMPc a AMPc), por lo tanto aumenta la concentración de AMPc. Esto estimula fosforilaciones, las cuales provocan secuestro de Ca y finalmente broncodilatación.
Cuantificación de un efecto:Esto se hace según:
- Efectividad- Potencia- Comparación

Terapias:- Involucra una dosis y un ritmo (ciencia que estudia esto posología)- El objetivo es obtener una determinada concentración en un determinado tiempo bajo márgenes de seguridad.
- Índice o rango terapéutico: relación entre la concentración máxima y mínima. Origina el margen de seguridad.- Mientras más pequeño es el índice menos seguro es el fármaco (el margen de seguridad es muy estrecho).- Existen las terapias únicas y las múltiples.
Múltiples:Para poder realizarlas hay que conocer las interacciones farmacológicas de los distintos compuestos ya que no todos se pueden mezclar:a) Cinético: interacciones en etapas de LADME. Dentro de estas las más importantes son las metabólicas y de eliminación, las otras en general no importan porque se pueden solucionar con la biofarmacia.b) Dinamia:i. Aditiva:
- Se suman efectos, por lo tanto el resultado en general es mejor (ambos efectos se expresan).- A + B A + B
ii. Sinérgica:- Al asociarse 2 fármacos, el efecto resultante está compuesto por la sumatoria de los efectos de ambos fármacos,
por lo tanto se obtiene un efecto superior.- Da como resultado final un efecto distinto al que cada fármaco por separado daría. - Es la interacción más común.- A + B C
iii. Potenciación:- Hay un potenciador (A) y un potenciado (B), ambos tienen un efecto, pero al juntarse, A aumenta el efecto de B.- Interacción común en antibióticos.
- A + B A + B- Ej: trimetroprim (potenciador) + sulfadizina (potenciado) = sulfan (pasa de tener efecto bacteriostático a uno
bactericida).
iv. Antagonismo: - Puede ser de función o receptor (antagonismo verdadero o también conocido como antídoto).- Muy importante en toxicología.

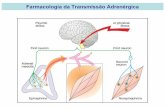
Neurofarmacología
SISTEMA NERVIOSO PERIFÉRICO:a) Involuntario o autónomo:
i. Simpáticoii. Parasimpático
b) Somático:NT Receptor Efecto
Acetilcolina Nicotínicos Contracción
Simpático : NT: adrenalina, noradrenalina
Sinapsis:
- Al ingresar sodio, ingresa calcio.- El calcio estimula el avance de las vesículas (a través del estimulo de las sinapsinas que están unidas a la
noradrenalina) hasta el espacio presináptico.- Las vesículas se fusionan con la membrana y se liberan por exocitosis.- La noradrenalina se dirige a su receptor.- Mecanismos de regulación:
Enzimas: COMT (encontrada en el espacio sináptico), y MAO (encontrada en el terminal). Ambas degradan a la noradrenalina.
Recapturación: cuando la concentración de noradrenalina aumenta mucho en el espacio es recapturada y degradada por MAO.
Autorregulación: los receptores (encontrados en la membrana presináptica) al ser estimulados por la noradrenalina inhiben la liberación de las vesículas.
Efectos:Órgano Receptor EfectoCorazón Aumento frecuencia cardiaca
Aumenta inotropismoRespiratorio BroncodilataciónDigestivo Disminuye peristaltismoOjo Midriasis
Disminuye drenajeNo altera o puede disminuir la presión intraocular.
Vasos Constricción

Útero Relajación de la musculatura (tocolítico)* A nivel digestivo el efecto es muy pobre porque generalmente predomina el sistema parasimpático, por eso lo que se busca es obtener un efecto parasimpáticolitico para lograr una acción simpáticomimetica. * La adrenalina en bajas concentraciones actúa sobre los receptores generando una vasodilatación. * Los receptores son dependientes de concentración metabotrópicos
Fármacos agonistas adrenérgicos:1. Respiratorio: fármacos acción 2.
- Efecto broncodilatador de tipo periférico con una acción antitusígena.- Se usan en cuadros respiratorios obstructivos.- Administrando a bajas concentraciones y por poco tiempo la administración constante en el tiempo genera un
acostumbramiento, lo que hace al fármaco menos eficiente (taquifilaxis), es común a todos los fármacos de la familia adrenérgica.
- En patologías crónicas se deben usar terapias alternas como: corticoides intranasal, AINES, metilxantina. - En bovinos no se tratan cuadros respiratorios con adrenérgicos por ser animales de consumo y su eventual
retención del fármaco en hígado. Tampoco se usan corticoides por su potencial efecto abortivo, por lo que el tratamiento es por otras vías.
a) Clembuterol:- Broncodilatador de elección en equinos, no está permitido en animales de consumo (porque induce a problemas
hepáticos, pulmonares o cardiacos en el hombre y además porque es un anabólico prohibido) y no hay presentaciones para menores. Prohibido en humanos en muchos países.
- Eficiente en cuadros de COPD (Enfermedad Pulmonar Crónica Obstructiva) y asma.- Muy similar a catecolaminas.- Tiene una vida media muy larga (aprox. 35 horas).- Metabolización hepática con formación de algunos metabolitos de noradrenalina tóxicos.- Eliminación renal, con una cinética lineal.- Generalmente se da por vía oral, pero también se puede dar EV bajo monitoreo.- En tratamientos prolongados se tiende a acumular en hígado, riñón, músculos, retina, folículo piloso (en dosis muy
altas) y piel.- Consideraciones:
Controlar acción sobre receptores 1 y 1. Lo que se manifiesta con aumento de frecuencia cardiaca y aumento de la presión sanguínea, ya sea por vasoconstricción o por el aumento del gasto cardiaco.
Controlar efectos secundarios por acción simpaticomimético, que se manifiesta con la presencia de temblores musculares.
- RAM: cefaleas, vómitos, nauseas, taquicardias, alteración de la presión sanguínea, temblores musculares.
b) Salbutamol o Albuterol:- Se utiliza en clínica menor.- Tiene una mayor selectividad sobre 2.- Menor incidencia de RAM.- Efectividad dependiente de la sensibilidad del paciente.- Se usa para tratar asmas, COPD y cuando se asocia con antibióticos sirve para tratar neumonías.- Disminuye contracciones y dilata útero sirve en casos de partos distócicos para detener el parto y darle tiempo al
MV para que llegue y asista al parto.- En animales geriátricos no se recomienda ya que necesita una alta tasa de metabolización, en estos casos mejor
usar mucolíticos.- Estimula a los receptores 2 que se encuentran en gran número en el músculo liso bronquial; esta estimulación activa
las proteínas Gs y aumenta el AMPc, lo que causa disminución del tono muscular (broncodilatación). Además, también aumenta la conductancia de calcio y potasio en las células musculares bronquiales causando hiperpolarización de la membrana y relajación. El fármaco también actúa sobre las células inflamatorias estimulando los receptores 2 presentes en ellas y evitando así la liberación de mediadores y citocinas inflamatorias.
- Las dosis para lograr un efecto tocolítico son 2 a 3 veces superiores a las que se usan cuadros respiratorios.- Vía de administración: IM, EV, SC, PO, inhalatorio.

2. Vascular: agonistas a) Fenilefrina:- Catecolamina sintética- Se comporta ligeramente como un agonista adrenérgico indirecto, estimulando la liberación de la noradrenalina de
sus vesículas, lo que potencia los efectos adrenérgicos.- En humanos se usa como descongestionante nasal.
b) Epinefrina:- Catecolamina sintética de mayor uso.- También actúa sobre receptores - Se administra EV para tratar crisis cardiológicas, colapso circulatorio agudo, resucitación cardiopulmonar,
broncoespasmo, reacciones anafilácticas, shock, hipotensión, hemorragias abundantes. La vía SC o IM se emplea en reacciones anafilácticas agudas y broncoespasmo.
- Aumenta el inotropismo al actuar sobre 1.- En ojo su aplicación en gotas logra un efecto midriático sin provocar un aumento de la presión intraocular.- Forma de presentación: generalmente viene en ampollas de 1 ml con 1 mg de clorhidrato de Epinefrina.
3. Cardiaco: agonista Dobutamina:
- Catecolamina sintética de acción directa que aumenta el gasto cardíaco, la distribución de oxígeno y el consumo de oxígeno con más garantías que la dopamina, principalmente porque no causa vasoconstricción.
- Tiene una potente acción 1 (estimulación cardíaca) y una modesta acción (vasodilatación), y poca acción 1 agonista.
- En pacientes con insuficiencia cardíaca reduce la resistencia vascular periférica y aumenta el gasto cardíaco mientras la presión arterial permanece constante.
- También puede mejorar el cronotropismo ventricular y aumentar la conducción AV. - Forma de administración: EV a goteo lento- Eliminación renal- No puede ser usada en conjunto con halotano ya que aumenta el riesgo de arritmias ventriculares.- La dosis varía entre 2.5 y 15 mg/kg. En los gatos, dosis de más de 5mg/kg pueden inducir ataques.
Fármacos antagonistas adrenérgicos:1. Antagonistas - Tienen una mala selectividad.- Fármacos de vida media corta.- Se busca un efecto hipotensor.- Entre los mecanismos alternativos que se tienen para conseguir el mismo efecto están:
Diuréticos: aumentan la diuresis, aumentando el volumen de orina, disminuyendo el flujo sanguíneo. Acción tanto osmótica como diurética por su efecto sobre la Aldosterona.
Bloqueadores de Angiotensina. Inhibidores ECA: bloquean el eje Renina - Angiotensina - Aldosterona. Vasodilatadores directos: hay un secuestro del Ca presente específicamente en la musculatura lisa vascular,
evitando que se produzca la contracción.
a) Fenoxibenzamina:- Es un antagonista no selectivo ya que no solamente actúa sobre 1 también lo hace sobre 2.- Es un antihipertensivo y se usa en el tratamiento de la enfermedad vascular periférica (hipertonía vascular) y en la
insuficiencia vascular periférica para mejorar la perfusión deteriorada por la vasoconstricción intensa.- Los efectos principales son el resultado del bloqueo de los receptores alfa adrenérgicos en el músculo liso vascular.- Produce disminución progresiva de las resistencias periféricas vasculares y aumento del gasto cardiaco, que se debe
en parte a la estimulación nerviosa simpática refleja.- Inhibe la captación de catecolaminas, tanto en las terminaciones nerviosas adrenérgicas como en los tejidos
extraneuronales puede acentuar taquicardia por bloqueo 2.- Se le reconoce un efecto irreversible al tener una potente unión a receptor cuando es dado en dosis altas.- En la actualidad no se ocupa en MV.

b) Fentolamina:- Es un vasodilatador (antihipertensivo).- Administración IV o IM- También bloquea receptores 2.- Similar a fenoxibenzamina pero con mayor selectividad y es reversible por lo que se puede controlar sus efectos
con la dosis.- Metabolización hepática.
c) Prazosin.- Vasodilatador.- Su mayor efecto es que bloquea los receptores de arteriolas y venas, por lo tanto disminuye la resistencia
periférica y el retorno venoso.- Casi no tiene efecto sobre receptores 2.- Tiene buena absorción tras su administración oral y tiene alta UPP.- Metabolización hepática.- Es la cuarta alternativa disponible para tratar tumores hipersecretores de catecolaminas,
2. Antagonistas - Se busca selectividad sobre el receptor 1.- Fármacos de vida media corta.- Efectos a nivel cardiaco:
Inotropismo de tipo negativo. Cronotropismo de tipo negativo: se controla la conductancia a este nivel para evitar la generación de
arritmias, disminuyendo la fuerza de contracción como forma de compensación en un insuficiente cardiaco.- Los ß-bloqueantes se contraindican en presencia de: asma, neumopatía obstructiva crónica, diabetes mellitus,
falla cardíaca congestiva, tirotoxicosis, vasculopatías periféricas y problemas conductivos AV.
a) Propanolol:- Es el más antiguo.- Su selectividad por 1 es moderada, por lo que dependiendo de dosis podría llegar a actuar sobre 2.- Tiene alta potencia a bajas dosis.- Es muy liposoluble y su absorción es casi completa luego de ser administrado oralmente, sin embargo tiene un gran
efecto de primera pasada, por lo tanto aproximadamente sólo un 25% llega a circulación.- Su biodisponibilidad aumenta al ser ingerido en conjunto con alimentos, y en terapias a largo plazo.- UPP aproximadamente 90%.- Eliminación renal.- Se usa para tratar hipertensión.- Buena respuesta en insuficiencia cardiaca obstructiva.- Generalmente se administra vía oral (jarabe), sin embargo vía EV sirve para tratar arritmias.- También se usa en terapias de agresión, fobia a ruidos y síntomas somáticos de la ansiedad en perros y gatos.
b) Atenolol:- Tiene mayor selectividad.- Muy hidrosoluble- No tiene buena absorción pero si buena biodisponibilidad.- Eliminación renal.- Se usa para tratar hipertensión.- Tiene una vida media más extensa, por lo que requiere de una dosificación menos frecuente.- Generalmente administrado vía oral.
c) Metoprolol:- Es selectivo.- Muy similar al atenolol.

- Se usa en dosis baja terapéutica para tratar arritmias e hipotensión sin embargo no es muy utilizado en perros.
d) Esmolol:- Su acción dura muy poco y se usa principalmente en pacientes críticos (cuando se requiere una solución rápida a
la taquicardia, falla cardiaca o hipotensión).- Eliminación renal.- Se administra EV.- En caninos se usa como manejo médico para la cardiomiopatía dilatada.
e) Acebutolol:- Tiene muy buena absorción.- Su metabolito activo (diacetolol) es el responsable de gran parte su actividad.- Eliminación renal.- En equinos se usa como broncodilatador.
Fármacos que aumentan la liberación de catecolaminas: a) Anfetaminas:
- Es un adrenérgico sintético, potente estimulante del sistema nervioso central.- Tiene acción simpáticomimetica broncodilatador, vasoconstricción.- También se usa para tratar narcolepsia, obesidad (disminuyen sensación de hambre al actuar sobre el centro del
apetito), depresión, mejorar rendimiento deportivo y cognitivo.- Administración oral, buena absorción.- Mecanismo de acción: es un agonista directo de los receptores presinápticos para noradrenalina y dopamina a
nivel del sistema nervioso central. La anfetamina se une a estos receptores y los activa, induciendo la liberación de los neurotransmisores de reserva alojados en las vesículas de los terminales nerviosos, convirtiendo los respectivos transportadores moleculares en canales abiertos. También tiene una acción agonista serotoninérgica, aunque relativamente más débil.
- Al ser un estupefaciente puede generar dependencia.- No se usan en animales.
b) Metanfetaminas:- Similar a la anfetamina pero sus efectos son más pronunciados ya que atraviesa con mayor facilidad la barrera
hematoencefálica, lo cual le permite alcanzar concentraciones en el cerebro mucho mayores, logrando ejercer su acción casi exclusivamente sobre el SNC.
c) Efedrina:- Es una amina simpaticomimética de origen vegetal, principio activo aislado originalmente de Ephedra vulgaris- Fue originalmente el precursor químico para la síntesis de la anfetamina- Es un agonista adrenérgico (directo e indirecto), muy activo sobre los receptores del sistema nervioso simpático,
pero relativamente poco potente como estimulante del sistema nervioso central. Esto se debe a la limitada destreza de la molécula para atravesar la barrera hematoencefálica, en relación con otros compuestos similares como la anfetamina.
- Es un broncodilatador adrenérgico, vasopresor. Estimula los receptores beta-2 adrenérgicos en los pulmones para relajar el músculo liso bronquial; alivia el broncoespasmo, aumenta la capacidad respiratoria, disminuye el volumen residual y reduce la resistencia de las vías aéreas.
- Puede también inhibir la liberación de histamina inducida por antígenos. - Como vasopresor actúa en los receptores beta-1 adrenérgicos en el corazón y aumenta la fuerza de contracción
mediante un efecto inotrópico positivo en el miocardio. - Actúa sobre los receptores alfa adrenérgicos de los vasos sanguíneos de la mucosa nasal; produce
vasoconstricción, lo que origina descongestión nasal. - Estimula la corteza cerebral y los centros subcorticales, y muestra sus efectos en la narcolepsia y estados
depresivos, aunque estos usos están acotados por la excesiva activación simpaticomimética periférica que produce la efedrina, respecto de otros estimulantes más selectivos sobre el sistema nervioso central.
- Se absorbe en forma rápida luego de su administración oral, intramuscular o subcutánea.

- Se metaboliza en el hígado y se elimina por vía renal.
Parasimpático o Colinérgico:- Es un sistema depresor.- No es común que se busquen sus efectos directos, sino que desde el punto de vista farmacológico se busca un
efecto parasimpaticomimetico para obtener una acción simpaticolítica.- La acetilcolina es el NT de los:
Ganglios sistema nervioso vegetativo Terminación neuroefectoras Placa motora Algunas sinapsis del SNC
Sinapsis:- Las vesículas contienen acetilcolina.- El único método de regulación es la acetilcolinesterasa encontrada en el espacio.
Receptores colinérgicos:
Tipo Tejido Lugar AntagonistaNicotínico Nm Muscular. Unión Neuromuscular. Tubocuramina.
Nn Nervioso. Ganglios Autónomos. Trimetafan.Muscarínico M1 Ganglios Autónomos, SNC,
Mucosa Gástrica, Secreciones.Atropina.Pirenzepina.
M2 Corazón. Atropina.M3 Músculo liso,
Glándulas Secretoras.Atropina.
* M3 se considera el receptor más importante.
Efectos colinérgicos:Órgano Receptor EfectoCorazón m3 Disminuye frecuencia cardiacaRespiratorio m3 Bronconstricción
Aumenta secrecionesDigestivo m3 Aumenta peristaltismoUrinario m3 Favorece micción (disminuye la tensión del músculo
detrusor)Aumenta la motilidad musculatura lisa
Ojo m3 MiosisDisminuye la presión intraocular (por aumento de drenaje del humor acuoso)Acomodación del cristalino para visión de cerca
Vasos m3 Dilatación (por relajación de la musculatura lisa)Secreciones m3 Aumento en tubo digestivo, bronquios, glándulas
lagrimales, salivales y sudoríparas.
Fármacos colinérgicos: actúan como agonistas muscarínicos ya que estimulan los receptores muscarínicos1. Colinérgicos directos: se unen al receptor de la Ach estimulando intrínsecamente al receptor muscarínico.a) Naturales o colinesteres:
- Muscarina: no tiene uso actual porque no tiene especificidad- Pilocarpina:
Similar a la muscarina, pero se puede concentrar su efecto a través del manejo de la vía de administración.
Ejemplo: se usa a nivel de ojo en pacientes con presión ocular elevada ya que facilita el drene ocular por lo tanto disminuye la presión.
También se puede usar a nivel oral en casos de xerostomía (boca seca) para estimular las secreciones. Casi no se usa en MV

Su uso se restringe localmente ya que al ser tan poco específico, si se aplica EV se estimula a todo el parasimpático.
b) Sintéticos:- Carbacol:
Funciona 50% en digestivo y 50% en urinario. Sirve para vaciar la vejiga. Es muy poco específico, por eso sólo se usa en casos de urgencia y bajo monitoreo.
- Betanecol: Tiene mayor afinidad a los receptores encontrados en la musculatura lisa digestiva por lo tanto sirve para
aumentar el peristaltismo y las secreciones (mejora el avance gástrico). Puede generar hipersensibilidad. Se usan en MV pero han sido reemplazados por los procinéticos (actúan en receptores de serotonina en
el músculo liso digestivo). Vía de administración generalmente oral. Es parcialmente selectivo (por eso si es dado vía EV en dosis muy altas puede exacerbar todo el
parasimpático).
2. Colinérgicos indirectos: inhiben la enzima que degrada la acetilcolina.a) Reversibles:
- Principales representantes: fisostigmina, neostigmina (derivado de la fisostigmina), pirodostigmina.- Estos fármacos se unen lábilmente con la acetilcolinesterasa (Ache) provocando un aumento de Ach.- Al aumentar la Ach a nivel somático sirven para aumentar la funcionalidad de la placa motora aumentan la
contracción muscular a través de la estimulación de los receptores nicotínicos.- Los derivados de fisostigmina se usan para revertir los efectos de los relajantes musculares periféricos (como los
derivados del curare los cuales actúan como antagonista competitivo de receptores nicotínicos).
b) Irreversibles- Nacen con la finalidad de ser usados como insecticidas, acaricida, pesticida y antiparasitarios. Ej:
órganosfosforados- Otros: malathion, tetraetilerfosfato, parathion o nitrostigmina (prohibido en todo el mundo por ser altamente
cancerígeno).- Su acción es dependiente de su concentración.- Son muy eficientes pero tienen el defecto que se mantienen mucho tiempo en circulación generando serios
daños, y además al ser altamente liposolubles se absorben fácilmente por piel y mucosas pudiendo causar un cuadro de hipercolinergia.
*Tratamiento de un cuadro de hipercolinergia:- Oximas: libera el organofosforado de la Ache (al tener mayor afinidad hace que se suelte y se una a él, liberando
la enzima). Su efectividad depende del tiempo en que fue aplicado. - Atropina
Fármacos anticolinérgicos: antagonistas del parasimpático1. Atropina:
- Sustancia de origen natural (proviene de arbusto atropa belladora), pero hoy en día es sintético.- Mecanismo de acción: bloqueo de receptor muscarínico sin especificidad (acción muscarínica generalizada).- Muy eficiente y de bajo costo.- Usos:
Actúa como un potente midriático, por lo tanto sirve para realizar exámenes de fondo de ojo. Su desventaja es que aumenta la presión intraocular por lo tanto está contraindicado en pacientes con glaucoma (en estos casos se usa otro midriático alternativo como agonistas ya que estos no alteran las presiones a menos que su uso sea muy prolongado, su desventaja es que demoran más que la atropina).
Pre anestésico (por su control vagal). Es un soporte cardiovascular

Tratamiento a intoxicaciones por órganosfosforados. Disminuye motilidad intestinal. Disminuye secreciones. Broncodilatador
- Presentación: gotas, jarabes, comprimidos y todas las imaginables. Para administración EV o SC viene en ampollas de 1 ml de sulfato de atropina al 0,1%
- Se puede aplicar por cualquier vía pero generalmente se hace EV. Cuando se usa la vía IM se tiene una respuesta 15 minutos después de su aplicación.
- Dosis: 0,044 mg/Kg- Para comprobar si su aplicación ha hecho efecto se observan las pupilas (debería haber midriasis)- Metabolización hepática y eliminación renal.- Vida media no muy larga: 6 a 8 horas (pero tiende a concentrarse en ojo, y demorarse más en ser eliminado de
ahí).
2. Homotropina:- Derivado sintético de la atropina.- Tiene uso exclusivo en ojo.- Su T1/2 es más baja que la de la atropina 30 min a 1 hora.- Tiene menor potencia que la atropina pero a nivel práctico casi no se nota.- Alto costo por lo tanto no se usa mucho en MV.
3. Bromuro de Ipatropium:- Derivado de la atropina.- Es una sal que se administra principalmente vía oral.- Tiene mayor especificidad en el sistema respiratorio tiene un potente efecto broncodilatador.- Ayudan a complementar terapias con otros broncodilatadores como los agonistas 2 (disminuyen la dosis
requerida del antagonista para disminuir los efectos colaterales que produce como taquicardia).- Es caro, por eso se usa principalmente en equinos.- Efectos colaterales en equinos: disminuyen el peristaltismo.- En especies menores no se usan mucho a pesar que no producen efectos colaterales.
4. Escopolamina:- Es un derivado directo de la atropina (nace como alternativa sintética).- No tiene la misma potencia que la atropina.- Es constituyente de compuestos antiespasmódicos, anti diarreicos, o fármacos para disminuir la hipermotilidad.- Disminuye el peristaltismo.- Generalmente se administra vía PO, también se puede hacer EV pero bajo estricto monitoreo.
SISTEMA NERVIOSO CENTRAL:
Neurotransmisor (NT) Receptor

Gaba gA, gBAspartato glutamato NMDAAdrenalina AdrenérgicoNoradrenalina AdrenérgicoSerotonina 5HTDopamina D1, D2, D3
Anestesiología:- El protocolo anestésico consta de una combinación de fármacos que buscan controlar 4 factores fundamentales:
1. Motor: controlar movimientos.2. Mental: generar amnesia para evitar estrés operatorio.3. Sensitivo: controlar la sensación dolorosa a través de la analgesia o estado de profundidad cortical
(inconsciencia).4. Vagal: componente reflejo
- En el proceso anestésico lo que se va generando es una depresión cortical (disminución de la actividad), esto se logra a través de la potenciación de la actividad Gaba.
- La depresión cortical tiene distintas etapas:1. Fase de inducción o depresión de tipo involuntaria:
Hay conciencia, pero también hay analgesia. Es corta y de poca importancia porque no compromete la integridad del individuo.
2. Fase excitatoria: Estado de depresión, pero con excitación. Hay alucinaciones y exacerbación de sensaciones. Hay fármacos que solamente llegan hasta esta etapa, como por ejemplo la ketamina (se
mantiene en la fase “C” o catalectoide, donde hay amnesia, analgesia, pérdida de movimientos voluntarios, desconexión con el medio, las extremidades quedan rígidas y se mantienen algunos reflejos).
3. Fase de anestesia:a. Superficial: anestesia “ideal”, se busca llegar hasta esta etapa. Para saber si fue alcanzada se
hace un pequeño corte en la piel y si hay fasiculaciones aún hay sensibilidad por lo tanto no se ha logrado totalmente la anestesia. Hay ausencia de reflejo podal y deglutorio.
b. Profunda: hay una depresión mayor de la frecuencia respiratoria (límite con parálisis bulbar).4. Parálisis bulbar:
Los barbitúricos (tiopental) llegan fácilmente a esta etapa. Hay una disminución brusca de la frecuencia respiratoria. Los anestésicos inhalatorios permiten mayor control y salida de esta etapa.
- La anestesiología presenta 2 etapas: pre anestesia y anestesia general (se pueden considerar 3 si se agrega la inducción).
Preanestésicos:- Fármacos que tienen la finalidad de tranquilizar y de hacer la anestesia más segura ya que disminuyen la
cantidad de anestésico requerido.- Para lograr su objetivo se asocian más de un fármaco.- Hay 3 tipos:
Tranquilizantes Opióides Hipnóticos o sedantes
Tranquilizantes:- Ayudan a controlar el componente motor, mental y algunos también el sensitivo.- También tienen la finalidad de sujeción química.
a) Fentiacínicos:

- Tienen una amplia gama de representantes, en Chile los 2 más importantes son la acepromazina (viene en presentación de acepromazina maliato y comercialmente se conoce como pacifor o también acedam) y la clorpromacina (se usa menos porque tiene más efectos colaterales).
- Mecanismo de acción: Actúan a nivel de tronco cerebral. Controla acciones de noradrenalina y dopamina (compite con ellos) a nivel de SNC y SNP, por lo tanto
controla la vigilia. Cuando es dado en concentraciones muy altas puede producir una marcada hipotensión ya que al
competir con la noradrenalina inhibe su acción simpaticomimética a nivel de SNP. - Características:
Tiene un efecto ataráxico el animal pierde coordinación de movimientos pero aún es responsivo. Induce a un sueño ligero. Antiemético control secundario del vómito. Antihistamínico permite controlar mareos y vómito Protectores del miocardio al inhibir catecolaminas evita una sobreestimulación. Hipotensores gatillado por bloqueo de noradrenalina. Apneas poco comunes y relacionadas con aumento de prolactina (por bloqueo de dopamina). Aumentan la secreción de prolactina esto es de mucha relevancia en terapias prolongadas, en el caso
de los preanestésicos no tiene ninguna importancia porque se aplican 1 vez.- Cinética:
Administración: PO, IM, SC, EV Efecto: 15 a 30 minutos Lenta absorción Vida media corta Lenta eliminación renal 50% activa, 50% pasiva Metabolización hepática Presentación: ampollas o frascos (color ámbar para proteger de la luz) en concentración de 1 %.
- Contraindicado en pacientes agresivos porque puede exacerbar agresividad, o pacientes epilépticos porque puede exacerbar convulsiones.
- Ante un efecto hipotensor se debe tratar con un agonista .- Incompatibilidades
Cloranfenicol hay competencia por su vía de metabolización. Fenilbutazona idem Sulfonamidas idem Órganosfosforados puede generar convulsiones Epinefrina puede provocar una sobreestimulación adrenérgica.
- Dosificación: 0,022 – 0,44 mg/kg Depende de la especie ej: perro 0,5 mg/kg, conejo 1 mg/kg (lagomorfos requieren dosis más altas). Máximo de administración: 3 mg totales
- Usos: Transporte, cinetosis, pre anestesia. Se usa principalmente en perros y gatos donde tienen pocos efectos colaterales y son muy eficientes. En equinos se debe usar bajo estricto monitoreo y medidas de seguridad (al generar descoordinación las
patas pueden ser peligrosas). Sirve para inspeccionar pene ya que provoca relajación peneana (solamente se puede usar acepromazina porque otros derivados fenotiacinicos como la propinilpromazina o la clorpromacina producen protrusión peneana).
b) Butirofenonas:- Derivan de los fenotiacinicos (también tienen anillo) y tienen mucha semejanza en cuanto a cinética y función, sin
embargo son más seguros y nacen para evitar todos sus problemas.- Los más comunes son haloperidol, droperidol, azaperona (no se usan en veterinaria por su alto costo).

- Excelente alternativa para generar neuroleptoanalgesia (anestesia para procedimientos cortos con buen manejo del dolor y amnesia, se logra combinando un tranquilizante con un opioide), ideal para castraciones, cortes de cola, corte de oreja. Ej: haloperidol + meperdina (opio) = innovan vet (muy caro por lo tanto ya salió del mercado)
- No son epileptoides y tiene bajísimas probabilidades de generar un cuadro hipotensor.- Tienen mayor margen de seguridad.- Su única desventaja es que provocan cambio de conducta por un tiempo prolongado.
c) Benzodiacepinas:- Potencian la actividad gabaérgica.- Según la dosis se puede lograr distinto grado de depresión cortical (como se puede manjar se considera más
“seguro”).- También tienen efecto sedante e hipnótico.- Influye en la liberación de serotonina.- Principal representante: diazepam- Otros: clonazepam, alprazolam, midazolam, dromazepam (no se usa en MV).- Antagonista flumazenil - Características:
Potente anticonvulsionante Miorrelajante Antiepiléptico y estimulante del apetito SOLO en gatos Acción paradojal es una de sus desventajas, no se puede prevenir y es individual. No provoca depresión miocardica ni respiratoria (su acción se concentra en SNC manifestando poca
acción en el SNP). Amplio margen de seguridad Es muy lipídico, por eso debe administrarse con un vehículo que lo haga más soluble como el
propinelglicol (mejora la estabilidad pero puede causar depresión miocardica, y generar dolor cuando es aplicado via IM, por eso se debe administrar EV a goteo lento o PO)
Es muy compatible con la ketamina, la cual produce convulsiones y rigidez, por eso se suelen dar en conjunto para que contrarreste los efectos de la ketamina.
Son considerados estupefacientes y en tratamientos prolongados puede generar dependencia. De difícil adquisición para MV.
- Cinética: Mala absorción oral Requiere de una alta tasa de metabolización hepática. En la primera pasada por hígado se genera un metabolito activo que tiene mayor potencia que el
diazepam, a la tercera se genera un metabolito inactivo. Viene en una amplia gama de presentaciones, en veterinaria la más común es de ampollas de 2 ml de 10
mg. Eliminación renal 50% filtración, 50% secreción,
Hipnóticos sedantes:Xilacina:
- Agonista 2 (disminuye concentración de noradrenalina)- Potente analgésico (aparte de controlar lo motor también controla lo sensitivo)- Potente relajante muscular- Su gran desventaja es que tiene un gran efecto a nivel de SNP depresor cardiaco, genera bloqueos
atrioventriculares, depresor respiratorio, hipotensor. Estos efectos pueden ser rápidamente revertidos al aplicar un antagonista (yohimbina).
- La yohimbina es muy potente y actúa casi de inmediato, por lo tanto frente a un cuadro de intoxicación por Xilacina se obtiene rápidamente una recuperación completa, además permite controlar el tiempo que el animal pasa postrado (evitando compresión de órganos) esto hace que sea muy seguro.
- Usos: Principalmente en equinos ya que provoca tranquilización sin que el animal se desplome. Procesos simples (castraciones, limpieza de abscesos, procedimientos dentales).

- Administración EV o IM.- En bovinos aumenta la motilidad intestinal pudiendo causar cuadros de torsión abomasal.- Sus desventajas dieron paso a la demetodina y medotomida las cuales no generan tantos efectos adversos, sin
embargo son muy caras y se usan principalmente en equinos.
Opiáceos:- Son narcóticos, sedantes, hipnóticos.- Prohibidos para los MV, si se permite el uso de derivados sintéticos de la morfina como el tramal o tramadol.- Características:
Analgésico nada más potente que morfina Antitusivo la codeína es un derivado de la morfina exclusivamente usado para controlar la tos. Ej;
codetos. Disminuyen motilidad intestinal antidiarreicos. Pueden generar excitación se mantienen en la fase II (alucinaciones, vocalizaciones, etc) No atraviesan barrera hematoencefálica
- Fármacos usados en MV: Oximorfona Butorfanol Buprenorfina Fentanilo Meperidina Etorfina muy potente y liposoluble, dosis bajas permiten sedar a animales de gran tamaño. Su
presentación más conocida es en dardos. Su antídoto es la dipenorfina (en humanos es el narcan).- Su principal desventaja es que son un depresor respiratorio (provoca desbalance informativo de concentración
de O2 y CO2), por eso un manejo común antes de su aplicación es pre oxigenar por 5 minutos.
Anestésicos:- Dependiendo de la concentración del fármaco se puede usar como inductor o como anestésico.
Inyectables disociativos:En general:
- Controlan dolor, inducen amnesia y generan catatonia.- Como no llegan a la fase 3 se mantiene el reflejo palpebral, laríngeo, corneal y faríngeo el animal queda con lo
ojos abiertos, lengua serpenteante y mastica.- Hay 2 efectos que permiten saber si se está bajo su efecto: midriasis y nistagmo.- Su mecanismo de acción los hace bastante seguros.
Ketamina:- De acción ultra corta.- Buen analgésico.- Anti arrítmico sostenedor cardiaco.- Provoca respuesta apneutica (genera alteración del ritmo pero sin producir depresión respiratoria)- En el gato no sensibiliza al laringoespasmo (disminuye la posibilidad que al colocar el traqueo tubo colapsen las
paredes de la laringe).- Se puede asociar en una misma jeringa (combinado externo) con otros tranquilizantes como diazepam,
acepromazina o xilazina.- Amplio margen de seguridad.- Desventajas:
No se puede determinar profundidad anestésica. Ausencia de relajación muscular. No apto para procedimientos prolongados máx. 30 minutos porque si no aumentan probabilidades de
riesgo. Convulsiones tónico crónicas.

En perros al actuar en la fase 2 excitatoria aumenta las probabilidades de convulsión (esto no ocurre en gatos, por eso es la única especie permitida por la FDA).
Administraciones sobre 10 mg/kg son dolorosas.- Sus deficiencias se mejoran con un buen protocolo previo se puede asociar en una misma jeringa (combinado
externo) con otros tranquilizantes como diazepam, acepromazina o xilazina.- Metabolización hepática y eliminación renal. - Administración: EV o IM- Dosis de inducción: 5 mg/kg- Dosis de anestesia: 10-20 mg/kg- Contraindicaciones:
Animales viejos, débiles, deshidratados. Animales destinados a consumo. Animales con afección renal. Animales con TEC o epilépticos. Intoxicados con organofosforados, estricina. Perros (en Chile igual se usa).
Inyectables barbitúricos:- Existen de acción corta (como el fenobarbital, el cual se usa como anticonvulsionante y no anestésico) y de
acción ultra corta (como el tiopental sódico).
Tiopental: - Mecanismo de acción: deprime la actividad a nivel de SNC a través del control de la actividad gabaérgica.- Desventajas:
Depresión SNC Depresión respiración genera apnea post inducción Depresión cardiovascular Hipotensor puede generar cuadro de hipotermia Cruzan barrera placentaria Aumentan la sensibilidad al laringoespasmo en el gato Mal analgésico Profundidad anestésica difícil de controlar.
- Ventaja: es muy liposoluble por lo tanto tiene un rápido efecto.- Indicaciones:
Procedimientos de corta duración Dosis de anestesia general: 20 – 30 mg/kg Dosis de inducción: 8-10 mg/kg
- Metabolización y eliminación lenta.- Administración: EV solamente porque es muy necrosante (a excepción de ratas)- Es muy inestable, a concentraciones mayores de 2,5% no se debe almacenar más de 15 días (a concentraciones
mayores aumenta su inestabilidad).
Inhalatorios:- Ventajas: permiten inducir fase 3 y operar.- Desventajas:
Pueden producir hipertermia maligna la temperatura sube por sobre los 37°C y no hay forma de hacerla bajar. Su origen no está muy determinado, se cree que es un cuadro alérgico, por eso sus probabilidades aumentan a partir de la segunda aplicación.
Requieren de maquinas inhalatorias que son caras. Pueden provocar coagulopatias.
Halotano:- No inflamable.

- Principal ventaja: 60 a 80% no se metaboliza y es eliminado por respiración.- Aumenta la presión intracraneana (porque aumenta circulación cerebral).- Disminuye circulación a hígado y riñones algunos pacientes pueden presentar hepatitis por halotano.- Depresor circulatorio y cardiaco.- Depresor respiratorio.- Broncodilatador.- Relajante muscular.- Actualmente es el anestésico inhalatorio menos usado porque ya casi no se hacen las maquinas para él.
Isofluorano:- Anticonvulsionante.- No aumenta la presión intracraneana.- No se metaboliza, es eliminado 100% por los pulmones altísima seguridad.- Depresor cardiovascular.- Mayor relajación y menor incidencia a efectos colaterales.
Antiepilépticos:Generación una convulsión:
1. Alteración de la membrana neuronal2. Disminución de neurotransmisores inhibitorios (GABA)3. Aumento de neurotransmisores excitatoria (glutamato)4. Alteración de las concentraciones de potasio y calcio extracelular.
Causas de convulsión:- Golpes- Descomposición hormonal- Intoxicaciones- Problemas metabólicos
Terapias:- Tipos:
1. De mantención: es crónico evita convulsiones2. De estatus para manejar crisis trata convulsiones
- Criterios:1. Frecuencia de las crisis2. Violencia de las crisis
- Varían mucho de animal en animal.- Una terapia que disminuya las crisis a la mitad es considerada como exitosa. - En general, no son caras, pero si tienen restricciones en cuanto al daño que pueden causar (como por ejemplo al
hígado por la metabolización).- Como regla una vez que se inicia el tratamiento por primera vez, debe durar como mínimo 6 meses, y a la vez
hay que ir evaluando constantemente la función hepática y los niveles plasmáticos.- A los 6 meses se debe hacer una evaluación de la terapia se va disminuyendo la dosis progresivamente hasta
llegar a 0, en cuyo caso se da por controlada la patología. Si mientras se disminuye se presenta al menos una crisis, se debe volver a subir la dosis y se debe mantener la terapia de por vida ya que la patología no se podrá curar nunca.
- Un paciente con terapia de por vida requerirá evaluación hepática siempre y se deberá siempre tener presente posibles interacciones con otros medicamentos.
- Las terapias alternativas como flores de Bach o dietas especiales generalmente tienen buena respuesta.
Fenobarbital: - Misma familia del tiopental.

- De acción corta.- Mecanismo de acción: estimula la actividad GABA, por lo tanto deprime el SNC (por eso también produce sueño)- Es un inductor enzimático puede alterar las enzimas hepáticas, pero para evitar esto se trabaja con
concentraciones plasmáticas entre 15 a 40 ug/ml. - Administración :
Oral con 100% de absorción permite terapias crónicas. EV terapia de estatus
- UPP: 45%- Peak de concentración: 4 a 6 horas.- Dosis: 3-5 mg/kg se aplica terapia de ensayo y error, se parte con la dosis máxima y se va disminuyendo de a
poco, y apenas se presenta una crisis se vuelve a subir y se mantiene esa dosis por 6 meses.- Concentraciones muy altas (sobre 40 mg/kg) producen hepatototoxicidad, para evitar esto se puede asociar con
otros fármacos, como por ejemplo el bromuro de potasio, y así disminuir la cantidad requerida de fenobarbital.- Los veterinarios no están autorizados para recetar este fármaco (a menos que estén inscritos en la liga contra la
epilepsia).
Primidona:- Es una prodroga se activa en el hígado donde se forman 2 metabolitos: fenobarbital y
feniletilmalondiaminapema.- Administración oral solamente sirve para terapias de mantención.- No tiene absorción 100% gástrica.- Dosis: 15-30 mg/kg su dosificación es más lenta.- Asociaciones: bromuro de potasio o ácido valproico y/o carbamazepina.- Su terapia también debe ser evaluada por hepatoxicidad.- Niveles plasmáticos: 15 a 40 ug/ml
Fenitoina:- Es una hidantoina.- Su gran desventaja es la cinética: baja vida media (6 a 8 horas) y mala absorción oral (36%).- Contraindicado en gatos porque su via de eliminación se satura, por lo tanto es altamente tóxico.- Darlo sólo se deja como última opción, pero no tiene restricciones para darlo asociado.
Bromuro de potasio:- Administración oral (en suspensión o cápsulas) se debe mandar a hacer (recetario magistral)- Dosis de carga: 120 mg/kg y de ahí bajar a 30 mg/kg- Concentraciones plasmáticas: 0.8 – 3 ug/kg- Antiepiléptico de terapia múltiple (nunca se da sólo porque su acción no es tan buena).- Primer antiepiléptico usado en animales.
Alprazolam y Diazepam:- Se usan en terapias de mantención en gatos.- El resto de las benzodiacepinas se usan para terapias de estatus.- Se usan para disminuir la dosis de otros antiepilépticos.
Lamotrigina:- Es el preferido para humanos porque no produce hepatotoxicidad y tiene excelente respuesta.- En animales su cinética y dinámica no ha sido estudiadas, pero al parecer no es tan efectiva como en humanos,
además es muy cara.
Antiinflamatorios

Antiinflamatorios de tipo esteroidal:Corticoides:
- Mecanismo de acción: Receptor: glucoreceptores (también tiene actividad con los mineraloreceptores). El receptor gluco activa un sistema de modificación de síntesis proteica. Al alterarse la síntesis proteica se forma lipocortina enzima que inhibe la síntesis de fosfolipasa 2 y
factor de agregación plaquetaria. Al inhibirse la fosfolipasa no se podrá dar la cadena de ácido araquidónico.
- Efectos fisiológicos: Mantienen la homeostasis Gluconeogénicos Mantienen la microcirculación Estabilizadores de membrana Supresión de la inflamación
- Cinética: Absorción oral, parenteral, tópica múltiples presentaciones UPP 90% Metabolización hepática y eliminación de metabolitos inactivos por la orina. Vida media alta
GlucocorticoideDosis
equivalentePotencia antiinfl.
Potencia mineralocor.
VM Plasma (min)
VM Bio (hrs)
Acción corta
Cortisona 25 0.8 2 30 8-12
Hidrocortisona 20 1 2 80-118 8-12Acción intermedia
Prednisona 5 4 1 60 18-36
Prednisolona 5 4 1 115-212 18-36
Triamcinolona 4 5 0 200 + 18-36
Metilprednisolona 4 5 0 78-188 18-36
Acción larga
Dexametasona 0.75 20-30 0 110-210 36-54
Betametasona 0.6-0.75 20-30 0 300 + 36-54
- Usos clínicos: Terapias de reemplazo Antiinflamatorio Antialérgico Shock Terapias inmunosupresoras
- RAM: Síndrome de Cushing iatrogénico Gastritis
Alergias:- Las más comunes son atopias, por picaduras de pulgas o alimentarias.- Se debe determinar la causa para determinar el tratamiento.- Los tratamientos sintomáticos en casos puntuales (como prurito por picadura de pulga) generalmente no duran
más de 5 días.- La mayoría de las atopias son por polen (alergia a la primavera) y suelen no durar más de 4 a 5 meses.

- La triada fundamental en el tratamiento de alergia consisten en antihistamínicos, ácidos grasos y corticoides de baja potencia (por sí solos no son muy efectivos, pero combinados sí).
- Los corticoides más fuertes se usan cuando los otros tratamientos no sirven.
Inmunoterapias:- Causas: enfermedades autoinmunes.- Son terapias crónicas se administran oralmente para poder tener control dosis-efecto.- Los más comunes son los de acción intermedia como prednisona, prednisolona, metilprednisolona.- La metilprednisolona es el mejor, sin embargo es la más cara y generalmente se usa cuando los otros dos no
funcionan.- Se comienza el tratamiento con altas dosis, pero apenas aparecen las 3 P (polifagia-polidipsia-poliuria indican
Cushing es fase inicial) se disminuye un poco la dosis y se mantiene 2 semanas.- A la tercera semana se comienza a disminuir la dosis se puede bajar violentamente o progresivamente, pero
lo primordial es que se debe mantener una fluctuación en las concentraciones de cortisol para mantener al eje despierto.
- En la cuarta semana se comienza a dar día por medio (fundamental para mantener fluctuaciones).- El eje en los perros es muy sensible, por lo tanto terapias de más de 7 días se consideran prolongadas.- Los gatos son muy resistentes y pueden soportar terapias de 1 año sin que les pase nada, sin embargo
corticoides en gatos se usan muy poco.
Procesos inflamatorios:- Son una buena elección cuando el proceso inicial es muy violento. Ej. Dexametasona- Generalmente se usan más los AINES porque son más seguros.- Impiden el proceso inflamatorio al inhibir la formación de ácido araquidónico y las ciclooxigenasas.- En un trauman encefalocraneano se usan para disminuir la inflamación del cerebro con el fin de evitar la hipoxia
y la posterior lipoperoxidación (que trae como consecuencia la muerte celular). - No todos los corticoides son capaces de controlar la lipoperoxidación, sí lo pueden hacer la prednisolona y
prednisona pero en 30 veces la dosis convencional y no más de 24 horas.- El tratamiento y pronóstico depende del tiempo que pasa desde el trauma. Si fue reciente y sólo hay hipoxia se
aplica dexametasona. Se puede aplicar metilprednisolona antes de las 8 horas de ocurrido el daño. Si ha pasado mucho tiempo y el paciente presenta lipoperoxidación se aplica prednisona y prednisolona, en este caso el pronóstico no es bueno.
Antiinflamatorios no esteriodales (AINES):- Los 3 efectos fundamentales son:
Analgésico Antipirético Antiinflamatorio
- El efecto es dosis dependiente Ej: la aspirina en dosis bajas es analgésico y en altas es antiinflamatorio.- Función: bloquear la acción de las ciclooxigenasas para que no se formen prostaglandinas inhibe PG
fisiológicas (COX1) e inflamatorias (COX2) a menos que se le dé especificidad por una cierta ciclooxigenasa. - La gran mayoría tiene especificidad 50/50, además son los más estudiados.- Aquellos con selectividad COX1 causan serios efectos adversos.- A mayor especificidad por COX2 menores efectos secundarios (porque inhiben menos las PG fisiológicas).- No existe especificidad 100%.- Para que los con especificidad 50/50 no causen problemas (reacciones adversas) se debe respetar la dosis, el
tiempo de terapia (no más de 7 días) y asociarlo a otros fármacos cuando se dan por más de 15 días. Idealmente darlos con un protector gástrico.
- Clasificación: Derivados del ácido salicílico: poco uso en MV Derivados del para-aminofenol: paracetamol

Indol y ácido indenaceticos: indometacina, etodoloc Ácido eteroarilacetico: ketorolac Ácido propionico: ketoprofeno importante en MV Ácido enólico: oxicam gran potencia, precursores del meloxicam Fenamatos: ácido mefenamico no se usan en Chile Pirazalidindionas: fenilbutazona
- Cinética: Absorción casi completa. La metabolización depende de la familia: algunos tienen alta (como por ej. fenilbutazona), y otros muy
baja (como la aspirina). La eliminación renal, también depende de la familia: algunos son muy dependientes del ácido
glucurónico (como la aspirina) y otros se eliminan fácilmente. UPP 90% Volumen de distribución pequeño
- Efectos adversos: GI porque inhibe PGI2
Renales en terapias de larga duración (porque inhibe PGE2 que tiene función vasodilatadora) Hipertensivos Hematológicos Ginecológicos disminuye libido, produce fallas espermáticas, disminuye ovulación
Derivados del ácido propionico:- Son igual de eficientes en cuanto a analgesia y antiinflamatorios, en plano secundario queda su efecto
antipirético.- Controlan bien el dolor periférico y el dolor visceral en etapa inicial (ej: dolor post quirúrgico).
Ibuprofeno:- Excelente potencia antiinflamatoria (para analgesia hay que aumentar la dosis).- En veterinaria se usa principalmente en perros y gatos como no hay presentación para MV, y las disponibles
son fundamentalmente PO no se da mucho en animales mayores.- Dosis: 10mg/kg cada 24-48 horas (sólo en casos agudos cada 12) y no por más de 3 días.- Si es administrado por más de 3 días puede haber depresión, pérdida del apetito y hemorragia G.I (por erosiones
a nivel de estómago e intestinos). - Sólo se usa cuando no se tiene otra alternativa, y siempre bajo estricto monitoreo.
Ketoprofeno:- Alta potencia analgésica e antiinflamatoria.- Al ser más potente requiere menos dosis 1mg/kg cada 24 horas- Amplia gama de presentaciones: PO (comprimidos, gotas), EV, IM (solución de bajo dolor).- Es el más usado en MV, pero no el más seguro (FDA no permite su uso en perros).- Usos:
En animales menores: cualquier cuadro músculo-esquelético, control del dolor fundamentalmente pre y post operatorio (en ambos casos no se da por más de 5-7 días), y control de dolor crónico.
En animales mayores: en equinos se usa en el control del dolor agudo músculo-esquelético (no darlo por más de 5 a 7 días), y se caracteriza por tener bajos efectos colaterales, sin embargo algunos pueden presentar ulceraciones a nivel de la mucosa bucal (pero en baja frecuencia). También se usa en cerdos y rumiantes.
- Cuando es dado en terapias crónicas debe ir acompañado de un protector gástrico, darlo en la menor dosis posible y en días alternos (evaluando umbral del dolor).
- Tiene la ventaja que sus efectos pueden ser controlados por dosis.- Eliminación renal pasiva.- Baja incidencia de úlceras y problemas GI.
Carprofeno:

- Analgésico y antiinflamatorio (pero en menor potencia que ketoprofeno)- Alternativa para control de dolor crónico.- Se formuló para control de displasias uso exclusivo para MV, se usa más que nada en caninos.- Nombre comercial: Rymadyl- Forma de presentación: PO y EV (para casos agudos)- Muy similar al ketoprofeno, incluso algunos animales responden mejor que a él.
Pirazonólicos:Fenilbutazona:
- Potente antiinflamatorio, secundariamente analgésico.- Se usa en todas las especies, pero las presentaciones se centran más en equinos oral (pastas), EV, IM (debe
mencionar que fue formulado exclusivamente para esta vía porque es altamente irritante, si no se aplica EV).- Alta metabolización hepática (metabolitos activos) si no se usa controladamente puede causar hepatitis.- Eliminación renal saturable.- Dosis dependiente.- Algunos animales son muy susceptibles a úlceras bucales.- No usar por más de 5 días y solamente cuando es necesario.- Si se usa descontroladamente puede causar flebitis. - No tiene especificidad por COX2.- En perros es una alternativa secundaria porque generalmente reaccionan mal a ella (hemorragias GI y cuadros
hematológicos).- Dosis: 1-2 mg/kg cada 12 horas.
Dipirona: - Fundamentalmente analgésico.- Buena alternativa para control de dolor agudo en equinos (como por ej. cólicos).- Administración EV- Prohibido en muchos países porque puede causar CID cuando alcanza altas concentraciones, o también
anemias sin respuesta.- Muy económico.- Es el menos seguro, pero el más usado.
Flunixin Meglumin:- Extremo poder analgésico (secundariamente antiinflamatorio).- Se usa casi exclusivamente en equinos.- Administración PO-EV- Menos irritante que fenilbutazona- Eliminación de tipo renal no saturable (principal ventaja sobre fenilbutazona).- También puede presentar reacciones adversas, pero aparecen más lento.
Derivados enólicos o del oxicam:Meloxicam:
- Específico de COX2.- Sirve para manejo de dolor crónico.- Cinética similar a ketoprofeno.- Baja metabolización hepática y eliminación renal pasiva ideal para pacientes viejos o con enfermedades
hepatorenales.- Su gran desventaja es su alto costo.
Diclofenamico:- Muy popular en control de dolor en humanos.- En MV es una buena alternativa para tratar dolor en vez de ketoprofeno (que es más caro), sin embargo sus
cinética no ha sido estudiada y sobre los 7 días causa problemas gástricos.

Derivados anílicos Paracetamol: - Se diferencian del resto de los AINES porque controla las PG por una vía alterna las bloquea perno no inhibe
su formación (no inhibe COX)- No genera problemas gástricos.
Antihistamínicos
- Antagonistas competitivos H1 bloquean receptores de histamina por lo tanto antagonizan los efectos de la histamina endógena.

- Tienen un comportamiento errático.- Grupo de fármacos muy variables.- En animales tienen una respuesta de 3 en 10.- Su efectividad aumenta con la asociación de ácidos grasos.- También se pueden asociar con corticoides ya que disminuyen la dosis que se requiere de ellos.- Administración oral (nunca EV porque los que cruzan la barrera hematoencefálica pueden causar sedación).- En las terapias generalmente se parte con dosis altas, y se va disminuyendo a medida que disminuyen los
signos.- Pocos efectos secundarios (el principal es somnolencia)
Clasificación:A. De primera generación (cruzan la barrera):
- Clorfenamina- Difenidramina- Clemastina- Hidroxicina
B. De segunda generación (no cruzan la barrera)
Acciones farmacológicas:- SNP:
Anulan efectos nocivos de histamina por bloque HI síntomas cutáneos, cardiovasculares (hipotensión) y GI (fundamentalmente reflujo e hiperacidez).
Efecto anticolinérgico por bloqueo M todos los de 1° generación, y de la 2° generación solamente la azelastina.
- SNC: sólo los de 1° categoría Por bloqueo H1 acción depresora (sedación), antiemética, antivertiginosa, antitusígena y algunos
(como la ciprohepatidina) estimulan el apetito. Por bloqueo M acción anticolinérgica central
Efectos adversos:- 1° generación:
Somnolencia y sedación Efectos anticolinérgicos (sequedad de boca, retención urinaria, etc) Hipotensión (prometacina) Dermatitis alérgica o fotosensibilidad dérmica, Intoxicación aguda similar a por atropina.
- 2° generación: Taquicardia ventricular solamente astemizol y fenadina en caso de sobredosis o bloqueo del
metabolismo hepático.* Inhibidores del metabolismo hepático: eritromicina, ketokonazol, claritromicina.
Diuréticos
- Aumentan la eliminación de agua diuresis- 80% ejerce su efecto a través de la natriuresis eliminación de agua a través de la eliminación de sodio.- Otros los pueden hacer variando las presiones osmóticas y favoreciendo la eliminación de agua.

Acetozolamina:- Derivados de sulfonamidas- Es un inhibidor de anhidrasa carbónica bloquea la reabsorción de bicarbonato a nivel del túbulo proximal por
lo tanto aumenta su eliminación orina más básica - Absorción oral cada 12 horas- Excreción renal por túbulo colector- Potencia (techo) baja – moderada.- En veterinaria se usa poco porque generalmente los pacientes llegan en estados más críticos y requieren de
diuréticos más fuertes no útil en traumatismos- Usos patologías poco agudas:
Algunas patologías cardiacas Glaucoma Disminución de líquido céfalo raquídeo Alcalinización urinaria Alcalosis metabólica
- Toxicidad: Acidosis metabólica poco probable porque tienen potencia muy controlable. Cálculos renales por depósito de calcio Somnolencia y parestesia (disminución de capacidad de percepción) por aumento de CO2
Furosemida y Bumetanida:- Deriva de la familia de sulfonamidas- Provocan un aumento del flujo sanguíneo renal vasodilatadores, esto también los hace protectores renales.- Diurético (natriurético) de asa.- El más importante en MV es la furosemida.- Mecanismo de acción: inhiben el sistema de transporte acoplado Na/K/2Cl en la rama ascendente provocan
una disminución en la reabsorción de NaCl disminuyen el potencial positivo del lumen aumentan la excreción de Mg y Ca
- Alto techo su potencia se puede controlar según su administración y dosificación.- Administración: PO, EV- Absorción: aprox. 85%- Usos sirve para casos crónicos
ICC Hiperpotasemia IRA mejora el trabajo renal al aumentar el flujo sanguíneo En equinos su uso es muy común, especialmente antes de las carreras para disminuir la presión
sanguínea. También sirve para tratar patología del caballo sangrador Se suele dar en conjunto con inhibidores ECA como enalapril, captoril, lisinopril los cuales tienden a
causar concentración de potasio causando un cuadro de hipercalemia. - Toxicidad:
Alcalosis metabólica hipopotasémica Ototoxicidad en perros por depósito de furosemida a nivel coclear Hiperuricemia porque compite con el ácido úrico por la vía de eliminación. HipoMg afecta al sistema nervioso Deshidratación intensa
Tiacidas:- Techo moderado a alto (pero menos potente que furosemida).- Los más representativos son: cloroticida, clortalidona, indapamida, hidroclorotiacida.- Inhibe el transporte (reabsorción) de NaCl y cuando es aplicado de forma crónica inhibe la anhidrasa carbónica.- Es natriurético disminuye la reabsorción de sodio, de cloro y como consecuencia la reabsorción de calcio.- Usos:

ICC Hipertensión junto con inhibidores ECA y dieta adecuada Nefrolitiasis causadas por hipercalciuria
- Toxicidad más común que en otros diuréticos: Alcalosis metabólica hiperpotasémica e hiperuricemica Hiperlipidemia causado por hipersensibilidad Hiponatremia Debilidad y parestesia Reacciones alérgicas porque también proviene de la familia de las sulfonamidas
- La toxicidad puede ser controlada por la dosis, por eso normalmente se da cada 24 horas.
Ahorradores de potasio:- El representante más importante en MH es la espironolactona, en MV el triamtereno.- Actúan en la última porción distal del colector. - Es un antagonista directo de la aldosterona (bloquea sus receptores) por lo tanto inhiben la reabsorción de sodio.- Alto techo (pero menos que furosemida)- Menores efectos colaterales:- Usos:
ICC en fase media (no tan avanzada) Patologías mineralocorticoides Hiperaldosteronismo Cirrosis hepática Síndrome nefrótico
- Toxicidad: Hipercalemia
- Administración: PO, EV
Manitol:- Actúa en asa descendente- Alta potencia- No conviene usarlo en cuadro hemorrágicos porque aumenta la extravasación en politraumatisados se tiende
a usar más furosemida.
Antibióticos
Criterios para elegir un antibiótico adecuado:1. Espectro bacteriano2. Tejido a tratar3. Saber si la infección es reciente o crónica

Betalactámicos:- Familia que se caracteriza por poseer un anillo betalactámico.- Amplio espectro y afinidad por variados tejidos.- Muy usados en MV por precio y eficiencia.
Penicilinas:- Se le conoce estructuralmente como un ácido del aminopenicilanico- Es un antibiótico per se originado a partir de hongo Penicillium notatum (descubierto por Flehming en 1928).- Acción bactericida.- Mecanismo de acción: el anillo reconoce lo que se llama receptores de penicilinas en las bacterias (PBP), los
cuales corresponden a carboxipeptidasas, transpeptidasa y endopeptidasa (enzimas encontradas en la pared encargadas de su síntesis), y las inhibe (por lo tanto inhibe la síntesis de la pared).
- Espectro bacteriano: Fundamentalmente Gram (+) Streptococos y Staphylococos betalactamasa (-) Dependiendo de la clasificación se amplía el espectro.
- Resistencia: En general es una de las familias que presenta más resistencia Betalactamasa o penicilinasas (+) Incapacidad para unirse al receptor las PBP se modifican estructuralmente para que la penicilina
pierda afinidad. Resistencia intrínseca por variación de PBP los receptores se invaginan y en vez de presentarse
hacia afuera lo hacen hacia adentro.
Penicilinas naturales:- La más común es la penicilina G.- Mala solubilidad en líquidos (inestable, precipita) presentación en polvos para diluir- Se asocia a distintos vehículos (sódica, potásica, benzatina o procaína) para mejorar estabilidad y variar en
cuanto a su vida media. Se vehiculiza con sodio o potasio para mejorar la llegada a tejidos (entre ambas sales no hay
diferencia). Se puede administrar EV o IM cada 8 horas. Para disminuir tiempo de eliminación y aumentar vida media se puede unir a procaína (como es
vasoconstrictor no se puede aplicar EV sólo IM) o benzatina (macromolécula que genera depósito). El tiempo de administración de la procaína varía entre 24 a 48 horas (dependiendo si es oleosa u
acuosa) y el de la benzatina de 48 horas a 6 días.- Administración parenteral - Como no se puede dar vía oral (por su alta inestabilidad a pH ácido) se creó la variante penicilina V oral se
usa muy poco porque no es muy eficiente (hay que administrarla por más de 7 días cada 8 horas).- Vd alto en tejido y líquido sinovial, pericardio, bilis- Cuando es administrado EV el primer tejido que alcanza es el respiratorio.- Bajo paso por barrera hematoencefálica sólo la atraviesa cuando está dañada - Concentraciones sobre el 1% en líquido cefalorraquídeo generan convulsiones. - Metabolización hepática baja tasa- Eliminación renal por filtración y secreción.- Espectro bacteriano:
Cocos G(+) Muy sensible a Streptococos Staphylococos betalactamasa (-) Corynebacterium Bacillus anthracis Clostridium (anaerobios)
- RAM más importante hipersensibilidad.- Usos en perros y gatos:
Cuadros respiratorios altos Dermatitis en fase inicial (10 días)

Preventivo (post cirugías) Control de algunos cuadros reproductivos aplicación local (óvulos) o IM (10 días)
- Usos en equinos: Gurma o abscesos retrofaríngeos Cuadros respiratorios altos Se asocia con aminoglucocidos (gentamicina) para obtener amplio espectro Generan baja incidencia de disbacteriosis
- Usos en cerdos: Antes se usaban como prevención de cuadros respiratorios altos, hoy está prohibido se tiende a usar
más cefalosporinas. - Uso en bovinos:
Cuadros respiratorios iniciales Pericarditis Cuadros digestivos simples (como diarrea inicial) Asociación con gentamicina Administración local intramamaria (pomos) para tratamiento de mastitis contagiosas uso más
importante.- Triple penicilina sódica + procaína + benzatina administrada IM, asegura 7 días de protección, muy usada
en post operatorio y en casos de heridas por mordidas de perros.
Aminopenicilinas:- Amoxicilina, ampicilina - También tienen anillo betalactámico- Espectro bacteriano:
G (+) G (-) enterobacterias Cepas no productoras de penicilinasas
- Pueden actuar sobre bacterias simples G(-) porque pueden atravesar la capa de mucina y alcanzar los PBP.- Administración: oral (comprimidos o suspensión de reconstitución), parenteral (presentación LA), local
(intramamaria, ocular, ótica). - Absorción oral:
Amoxicilina: 85% Ampicilina: 75%
- La amoxicilina es más eficiente que la ampicilina sobre las E. coli- Dosis: 10 a 25 mg/kg cada 8-12 horas amplio margen de seguridad- Se pueden unir a antibióticos suicidas (sulbactan con ampicilina y ácido clavulanico con amoxicilina) los cuales
se unen por su gran afinidad a las betalactamasas permitiendo que las aminopenicilinas atraviesen y alcancen los PBP sin ser dañadas.
- Usos en animales menores: Cuadros respiratorios Urinario porque tienen eliminación 100% renal y con metabolitos activos Dérmico prevención a infecciones secundarias por corticoides, piodermas superficiales y profundas. Dientes gingivitis y periodontitis grado 1.
- RAM: muy bajos Hipersensibilidad Concentraciones altas y prolongadas pueden modificar la flora bacteriana generando cuadros como
diarreas leves o deshidratación leve. Pueden ser mortales en lagomorfos porque afectan mucho su flora bacteriana.
Penicilinas resistentes a penicilinasas:- Representantes: flucloxa, cloxacilina, dicloxacilina, oxacilina. - Presentación: PO, EV, IM SC- También se pueden asociar con benzatina, sodio o potasio.- Usos:

Tratamiento de mastitis mixtas Cuando se asocia con sodio o potasio sirve para tratar mastitis clínicas y subclínicas. Cuando se asocia con benzatina sirve para terapia de secado (45-60 días).
Penicilinas de última generación:- Representantes: carboxipenicilinas, carbenicilina, ticarcilina - Se administran principalmente vía oral- F oral: 90 %- Tienen la desventaja que son muy caras- Activas sobre pseudomonas.
Cefalosporinas:- Se originan a partir del hongo Cephalosporum acremonium- También tienen anillo 7-aminocefalosporámico- Propiedades:
Resistentes a penicilinasas Amplio espectro dependiendo de la generación Alta eliminación renal Alto Vd en tejido y líquido pericardico y sinovial. Cruzan barrera hematoencefálica y placenta Dependiendo de la generación requieren de más o menos pasadas por hígado y se eliminan metabolitos
activos o inactivos. Eliminación principalmente activa mayores riesgos de saturación de vía de eliminación.
- Espectro bacteriano: 1era generación: G(+) y moderada sobre G(-) 2da generación: mayor acción sobre G(-), pero menor que los de 3era 3era generación: G(+) y G(-) menor que la 1era pero mayor que la 2da.
- RAM: no son peligrosas pero si Daño renal no son nefrotóxicos pero si son mal usados causan problemas (si se satura vía de
eliminación pueden precipitar y potenciar algún grado de nefropatía). Hipersensibilidad. Hipoprotrombinemia: aumento tiempo de coagulación Trombocitopenia Disfunción plaquetaria Necesitan de funcionalidad renal
- No son de elección previo a cirugías por todos los problemas de coagulación que podrían causar.
1era generación:- Cefalotina:
Administración sólo EV. Metabolización hepática. No cruza barrera Baja resistencia a penicilinasas.
- Cefradina: En comprimidos sirve mucho para tratar dermatitis y mastitis en perros y gatos En pomos sirve para tratar mastitis en bovinos Buena cinética oral.
- Cefapirina: usada en control de mastitis contagiosa en bovinos- Cefazolina: administración parenteral- Cefalexina: administración PO- Cefadroxil: PO
2da generación:

- Su principal característica es su administración parenteral.- Representantes: cefoxitina, ceforoxima, cefotetan
3era generación: - Cefotaxima:
Muy eficientes en meningitis Administración parenteral.
- Cefoperazona: Uso exclusivo MV Usado contra pseudomonas
- Ceftifur: Uso exclusivo MV Parenteral Sin paso a leche Alta afinidad hacia tejido óseo y dérmico por eso se usa mucho en podopatologías Muy eficientes contra anaerobios como fusobacterium y bacterioides Altísima potencia en comparación con otras cefalosporinas (1 mg/kg cada 24 horas vs 25 mg/kg cada 8-
12 horas). Usado en bovinos, equinos (puede causar necrosis) y cerdos en cuadros respiratorios profundos.
- Cefoperazona: Se usa intramamaria para tratar mastitis contagiosas y ambientales Por el amplio uso que se les ha dado se ha generado gran resistencia por lo tanto su uso ha ido en
disminución.
Antibióticos suicidas: - Ácido clavulanico, sulbactan- No se dan solos porque su potencia es malísima. - Protegen la acción de las aminopenicilinas se unen por su gran afinidad a la betalactamasa permitiendo que la
aminopenicilina atraviese esa barrera y alcancen los receptores sin ser dañadas.
Aminoglicosidos:- Representantes: gentamicina, kanamicina, estreptomicina, amikacina, neomicina- Nacen en los 70 para controlar infecciones intrahospitalarias. - La principal característica de esta familia es su alta polaridad. - Se originan a partir de un MO- Cinética:
Administración: parenteral (LA), local (dérmica, ótica, ocular generan irritación pero eso se controla con la dosis, reactivan la reparación corneal), PO.
Baja absorción gástrica (podría aumentar si hay úlceras) Vd bajo, con buena llegada a tejido respiratorio y urinario No tienen metabolización hepática Eliminación 100% renal activa
- Espectro: Fundamentalmente G(-) y algunos G(+) Enterobacterias muy importante para control de E. coli Solamente actúan sobre aerobios. Gentamicina G(-) y algunos G(+) como ciertos Staphylococos Kanamicina, amikacina, neomicina G(-)
- Mecanismo de acción: bactericida interfieren con la replicación de la bacteria inhibiendo la síntesis proteica de subunidad 30 s.
- Efecto post antibiótico una vez que se termina el tratamiento se sigue teniendo una alta concentración en tejido respiratorio (hasta por 2 semanas).
- RAM muy comunes Toxicidades (ototoxicidad, nefrotoxicidad) depende de la polaridad (mientras mayor es, más tóxico)

- La toxicidad se debe principalmente a que el tejido otico y renal poseen un LPS ubicado a nivel de membrana conocido como receptor de aminoglucósidos, a mayor polaridad, mayor afinidad por el receptor, mayor gatillo de depósito la célula se comienza a llenar de antibiótico y pierde funcionalidad. También ocurre a nivel vestibular y coclear.
- Los que son muy polares (como neomicina) se restringen solamente a uso local.- La estreptomicina es el menos polar (por lo tanto el de menor toxicidad) pero tiene muy mala sensibilidad (por
eso generalmente se da asociado).- La gentamicina es el más popular en MV, como su polaridad es media se debe controlar su toxicidad a través de:
1. Dosis y tiempo de administración 3-5 mg/kg cada 8-12 horas por 7 días2. No aplicar en edades extremas3. No aplicar en nefrópatas4. No aplicarlo después de aplicar cefalosporinas (porque aumentan la predisposición a problemas renales por precipitación), dejar pasar 2 semanas.
- Principal desventaja elevada resistencia. - Usos en animales menores:
Administración IM cada 8-12 horas Patologías respiratorias Distemper y parvovirus iniciales Patologías digestivas simples (como intoxicaciones alimentarias) Otitis
- Usos en equinos: Cuadros respiratorios altos y bajos Cuadros digestivos simples alteran poco la flora MO Patologías reproductivas lavados uterinos
- Usos en bovinos: Cuadros respiratorios Cuadros mamarios Cuadros reproductivos principalmente post parto
Tetraciclinas:- Clasificación:
Acción corta: son lo de peor cinética Clortetracilina Oxitetracilcina Tetraciclina
Acción intermedia Demeclocilina poco importante, uso muy puntual
Acción larga: administración oral, cada 24 horas, muy liposolubles Minociclina Doxicilina es la más usada y más segura, cara
- Originados a partir de una bacteria (antibiótico per se)
- Espectro: G (-) y G(+) aerobios y anaerobios Rickettsia, mycoplasma, chlamydia, espiroquetas, protozoos Resistencias frecuentes.
- Mecanismo de acción: bacteriostático inhiben síntesis de la pared (subunidad 30 s)- Muy liposolubles.- Absorción:
Acción corta e intermedia: 30-70% depende de la presencia o ausencia de alimentos Acción larga: 95-100% Absorción en intestino superior Leche y sus derivados, Ca, Fe, Mg, AL, Bl reducen su absorción
- Distribución:

Amplia difusión en tejidos, especialmente los de acción larga Se acumulan en huesos y dientes en crecimiento Atraviesan barrera HE, placenta y llegan a leche materna
- Eliminación: Acción corta e intermedia:
Principalmente renal activa y metabolitos Vías fácilmente saturables Excreción parcial por bilis circulación enterohepática Una parte por heces disbacteriosis Insuficiencia renal o hepática aumentan la vida media.
Acción larga: Principalmente hepática por circulación enterohepática eliminación más eficiente y segura Insuficiencia renal no altera la vida media Excreción por heces disbacteriosis Excreción mínima activa renal
- RAM: generalmente dosis dependiente GI
Por irritación directa ardores, nauseas, vómitos, úlceras Por modificación de flora digestiva lengua negra (micosis), diarreas, colitis pseudomembranosa
(rara) Hepáticos: excepcionalmente necrosis grasa hepática, potencialmente mortal y descrito casi siempre en
niños y embarazadas. Toxicidad ósea:
Coloración irreversiblemente gris amarillenta Raramente hipoplasia dental o deformaciones óseas No administrar en gestantes o animales pequeños (hasta calcificación completa; 6 meses en perros y
1.5 año en equinos) Otras:
Demeclociclina: fototoxicidad Minociclina: afecta sistema vestibular (mareo, ataxia, vómito)
- Mucho ojo con la fecha de vencimiento porque pueden causar síndrome de falconi altísimo depósito, ruptura tubular, sangramiento e incluso muerte.
- Usos: De primera elección:
Brucelosis y cólera Infecciones por rickettsia, mycoplasma, chlamydia, borrelia
De reserva: Por resistencia o alergia a otros antibióticos
Animales menores: Doxicilina: administración cada 24 horas, patologías respiratorias extremas o muy avanzadas mixtas,
o cuadros digestivos, urinarios o reproductivos no iniciales. Oxitetraciclinas y tetraciclina: en patologías reproductivas y urinarias principalmente, también en
patologías mixtas y otitis. Bovinos:
Lo que más se usa es oxitetraciclina y tetraciclina (principalmente LA) para tratar podopatologías (por tener buena llegada a tejido óseo y acción sobre anaerobios), patologías mamarias (administración local y sistémica), óvulos uterinos y vaginales (uso descontinuado)
La doxicilina se usa principalmente en patologías respiratorio limitado por alto costo Cerdos:
Lo que más se usa es doxicilina en cuadros respiratorios, digestivos. Equinos:
NO se usa ninguna tetraciclina antigua (corta e intermedia) porque alteran la flora digestiva y al ser de lenta administración EV pueden causar problemas vasculares

La doxicilina generalmente se usa cuando el animal no responde a cefalosporinas de 3era generación o a las terapias convencionales.
Cloramfenicol:- Prohibido en animales de consumo, animales menores (excepto para aplicaciones locales como en spray), en
humanos sólo se permite administración local. - Alternativas: tiamfenicol (uso exclusivo humanos) y fluorfenicol (uso exclusivo MV) menor liposolubilidad y
eficiencia pero mayor seguridad. - Se prohíbe porque produce serias RAM hematológicas:
Reacciones dosis dependientes: el P-nitrofenol (grupo del cloramfenicol) afecta ribosomas mitocondriales causando anemia, leucopenia y trombocitopenia. Son previsibles (aparecen con niveles en sangre sobre 25 mg), frecuentes, benignas y reversibles.
Reacciones de idiosincrasia: se genera una anemia aplásica con panleucopenia, se gatilla por concentraciones mínimas de P-nitrofenol (mecanismo desconocido), ocurre sólo en humanos (1 de 200.000 casos), es imprevisible, irreversible y altamente mortal.
Otras: anemia hemolítica por déficit, síndrome gris del recién nacido (niveles sobre 40 mg). - Mecanismo de acción:
Bacteriostático dependiente de dosis (a altas concentraciones es bactericida pero no se usa a dosis altas).
Fijación subunidad 50s Impide la transpeptidasa
- Espectro: Amplio G (-) y G(+) anaerobios Clamidias, rickettsias, micoplasma y espiroquetas Frecuentes resistencias.
- Absorción oral rápida y completa.- Muy liposoluble- Distribución:
Muy buena distribución tisular Atraviesa BHE, placenta y llega a leche Niveles eficaces en líquido pleural, sinovial, peritoneal y prostático. Alcanza humor acuoso y útero
- Metabolismo: 90% hepático con circulación enterohepática Variable en prematuros, recién nacidos, enfermos hepáticos (cirrosis) y pacientes con terapias con
fármacos inductores o inhibidores hepáticos. Inhibe citocromo P450 Eliminación renal 10% activa y metabolitos Vida media condicionada por la tasa de metabolización
- Aplicaciones terapéuticas: Meningitis bacteriana en pacientes alérgicos a betalactámicos Infecciones por anaerobios de reserva Fiebre tifoidea grave o salmonelosis invasiva Alternativa a tetraciclinas Infecciones oculares
Florfenicol:- Se usa principalmente en cerdos y bovinos en presentación LA para cuadros respiratorios, digestivos, dérmicos. - En bovinos más que nada respiratorios.- Alto costo- Controlar límite máximo residual en cerdos.- Nace para estas 2 especies pero su uso se extrapola a equinos y animales menores.
Tiamfenicol:

- Actividad antibacteriana similar- Eliminación renal activa 90% - No presenta interacciones hepáticas- Depresión de médula ósea menos frecuente y grave- Experiencia clínica limitada.
Macrólidos:- Principal representante: eritromicina y espiromicina- Da origen a otras 2 familias:
Ketólidos: azitromicina, roxitromicina antibióticos de administración de 3 días como Trex Lincosamidas: lincomicina y clindamicina
- Familia muy segura.- Espectro:
G (+) aerobios estafilococos G (-)
- Mecanismo de acción: Bacteriostáticos dependiente de dosis (bactericidas) Penetración por difusión pasiva Concentración intracelular 100 veces mayor en G(+) Fijación a subunidad 50 s (contraindicado con cloramfenicol o sus derivados) Macrociclo de 14-15 átomos impiden translocación Macrociclo de 16 átomos impiden la transpeptidación
- Tienden a acumularse en árbol bronquial efecto post antibiótico variable- Alternativa a betalactámicos en cuadros de hipersensibilidad.- Muy eficiente para eliminar bacterias intracelulares.
Eritromicina:- Absorción:
Vía oral: es inactivada por los ácidos gástricos por eso se asocia con sales como esteres, estearato, propionato, estolato, etilsuccinato, las cuales mejoran la estabilidad en medios ácidos y permiten la liberación en duodeno. Los alimentos dificultan la absorción. A pesar de todo es la vía de preferencia.
Vía parenteral: genera dolor, por eso se vehiculiza con lactobionato. Para evitar tromboflebitis administrar lentamente y diluido.
- Distribución: Amplia No atraviesa BHE, sí la placenta y pasa a leche Llega a líquido ascítico, pleural, prostático, bronquial, humor acuoso. No llega a líquido sinovial- Alcanza altas concentraciones en macrófagos y leucocitos útil en bacterias intracelulares como
Legionella, Chlamydia, Brucella, etc.
- Eliminación: Metabolismo hepático 80% excreción biliar activa con circulación enterohepática 5% eliminación renal activa Vida media corta (1.5 horas) administración 4 veces al día cada 8 horas (dependiendo de la sal varía
entre 6-12)- Interacciones: inhibidor enzimático P-450 aumenta niveles de fármacos que son inactivados vía citocromo P-
450 como corticoides.- Usos animales menores (eritromicina y espiromicina):
Mastitis (vía sistémica) Cuadros reproductivos como brucelosis y prostatitis bacterianas. Cuadros respiratorios Patologías dentales, principalmente gingivitis
- Usos en bovinos:

Mastitis contagiosas (aplicación intramamaria) Alta tasa de resistencia Aplicación parenteral para manejo de cuadros respiratorios, digestivos y reproductivos.
- Usos en cerdos y equinos: se usan como segunda opción generalmente por su alta resistencia, por eso se dan asociados para patologías de baja intensidad.
Lincosanidos: - Clasificación:
Lincomicina Clindamicina Pilocina Tiamulina
- Mecanismo de acción: Subunidad 50 s (igual que espiromicina y cloranfenicol) Impiden la transpeptidación (acción bacteriostática) Activos frente a G(+) y anaerobios G(+) y G(-) Resistencias cruzadas con macrólidos
- Cinética: Administración oral principalmente, también parenteral Amplia distribución buena llegada a tejido GI, hueso y líquido sinovial También llega a bacterias intracelulares Efecto post antibiótico por concentración en tejido respiratorio. Eliminación biliar y heces 90%
- RAM: Diarreas (8%) Colitis pseudomembranosa (0.01-10%) exacerbación Clostridium difficile (tratar con vancomicina o
metronidazol o bacitracina) - Usos terapéuticos:
Infecciones por anaerobios resistentes a otros antibióticos En pacientes alérgicos a penicilina Alternativa a macrólidos Osteomielitis y artritis séptica
- No se usan en equinos.- Muy caros.
Clindamicina:- Mejor espectro- Mejor llegada a hueso insuperable en osteomielitis- Usos animales menores: osteomielitis, patologías dentales como periodontitis avanzada (grado III-IV), aplicación
tópica para ciertos cuadros dérmicos y aplicación local para tratar otitis crónicas o recurrentes.
Lincomicina:- Menor absorción oral- Alta tasa de colitis pseudomembranosa- Usos animales menores: principalmente para cuadros respiratorios y también reproductivos (como mastitis),
altísima resistencia.- Usos en bovinos: cuadros respiratorios, mastitis, alta resistencia.
Quinolonas:- Dan origen a las fluoroquinolonas.- Principal representante: ácido nalidixico - Alta potencia pero bajo espectro (sólo G - )- Muy seguro- Resistencia bacteriana casi nula

- Ya no se usan en la clínica (sólo en investigaciones)
Fluoroquinolonas:- Principales representantes:
Ciprofloxacino Norfloxacino Danofloxacino Enrofloxacino uso casi exclusivo veterinario
- Mecanismo de acción: Bactericida Inhibe la enzima ADN giraza mecanismo muy puntual, específico y eficiente.
- Administración: PO, local y parenteral con tasas de absorción sobre 80% en cualquier vía- Altos Vd buena llegada a respiratorio, digestivo, reproductivo, SNC; articular, pericardico, dérmico, urinario.- Espectro:
Depende del grupo Enrofloxacino G(-), G(+), estáfilos, Clostridios, rickettsias Malos para anaerobios
- Eliminación: Metabolización hepática y renal Sus vías non son saturables
- RAM: Afecciones digestivas muy bajas Depósito en cartílago articular en crecimiento
- Altísima seguridad amplio índice terapéutico- Muy usado en animales exóticos.- Usos animales menores:
Enrofloxacino: administración PO, parenteral. No se usa como antibiótico de elección (alto costo), alternativa en animales viejos, nefrópatas, hepatopatías o cuando no hay respuesta con un tratamiento convencional.
- Usos en cerdos: se usa como tercera opción (después de doxicilina y fluorfenicol), excelente alternativa para afecciones respiratorias.
- Usos en bovinos: pocos por alto costo, principalmente reproductores.- Dosis: 1-5 mg/kg cada 24 horas (alta potencia) - Alta resistencia.
Sulfonamidas:- Origen sintético
Antiguas:- Sulfas más comunes:
Sufadimexatona Sulfametazina Sulfadiazina Sulfaquinoxalina única sulfa que se sigue aplicando sola por su acción anticoccidial Sulfametotazol más usada
- Mecanismo de acción: Bacteriostáticos Inhibidores del metabolismo disminuyen la producción de ácido fólico al competir con Paba
- Espectro: Amplio, G(+), G(-) y protozoos Buena susceptibilidad: actinomices, bacillus, brucella, erisipelas, streptococcus, chlamydias Moderada susceptibilidad: Staphylococos, enterobacterias, proteus, Pasteurellas, pseudomonas.

Resistentes: mycoplasma, rickettsias - pH muy variable- Mala solubilidad en agua- Absorción:
Administración oral con rápida absorción excepto las sulfas entéricas (sulfasalazinas, sulfaguanadinas se usan como antiséptico intestinal)
Administración IM y SC con soluciones buffer para evitar reacción a la administración No se recomienda su uso tópico por competencia con Paba y reacciones alérgicas
- Distribución: Amplia en líquido cerebroespinal, sinovial UPP variable dependiendo de la sulfa 15 a 90% Bajo paso a leche, limitando su uso para mastitis por vías sistémicas
- Metabolización: Hepática acetilación es la más común formando cristales. Renales: filtración y secreción
- Principales cuadros: infecciones, SNC, tracto respiratorio, GI, urinario- RAM:
Cristaluria: baja incidencia en animales (aumenta en deshidratación y orina ácida) Queratoconjuntivitis seca: reacción alérgica en donde el lagrimal deja de producir lagrimas. Se ha
observado en perros tratados con sulfasalazina, sulfadiazina y sulfametaxol Hipoprotrombinemia: reportada en animales tratados con sulfaquinoxalina, la cual es un inhibidor de la
enzima reductasa de la vitamina K. Anemia aplásica y trombocitopenia: se produce en tratamientos prolongados donde se altera la flora
digestiva y se produce bloqueo del ácido fólico bacteriano. Modernas o potenciadas:
- Asociadas a trimetoprim (pertenece a familia de diaminopirimidinas) las potencia y hace que pasen de ser bacteriostáticas a bactericidas.
- El trimetoprim inhibe la enzima reductasa que convierte el ácido tetrahidrofónico en ácido fólico, por lo tanto se produce el ácido.
- Excelente solubilidad (por lo tanto no hay Cristaluria)- Metabolización en hígado y eliminación por ácido glucurónico en orina.- Baja incidencias de RAM- Dosis: 22 mg/kg cada 8-12 horas- Usos en animales menores:
Cuadros respiratorios como distemper inicial Urinario cistitis Prostatitis bacterianas buena respuesta frente a pH prostático