
mclworld.netmclworld.net/equiposMedicinas/docs/fileInformeTecnico... · Web viewHaga girar el...
52
RELACION DE PRODUCTOS A INSCRIBIR Y FORMA DE PRESENTACION
Transcript of mclworld.netmclworld.net/equiposMedicinas/docs/fileInformeTecnico... · Web viewHaga girar el...

RELACION DE PRODUCTOS A INSCRIBIR Y FORMA DE
PRESENTACION
RELACION DE PRODUCTOS A INSCRIBIR Y FORMA DE PRESENTACION

Nombre del producto
Códigos Forma de presentación
Marinr EP Diagnostic Catheters
072322M072302072402
Caja de cartón conteniendo 01 unidad estéril del producto de la referencia el cual a su vez viene
sellado en un blíster de papel grado médico y polietileno de alta densidad
preformado.
Mariner CS Coronary Sinus Diagnostic
Catheters
043302M043325M043328M
RF Marinr MC Ablation Catheters
075302075305075312075402075405
5F RF Marinr Ablation Catheters
0765083076584076585076586076514076515

DESCRIPCION DEL DISPOSITIVO
DESCRIPCION DEL DISPOSITIVOMARINR EP DIAGNOSTIC CATHETERS
072322M, 072302, 072402

MARINR CS CORONARY SINUS DIAGNOSTIC CATHETERS043302M, 043325M, 043328M
RF MARINR MC ABLATION CATHETERS075302, 075305, 075312, 075402, 075405
5F RF MARINR ABLATION CATHETERS076583, 076584, 076585, 076586, 076514, 076515
El catéter orientable de ablación por radiofrecuencia Marinr de Medtronic, es un catéter radio-opaco flexible fabricado con polímero extruido sobre un cordoncillo de acero inoxidable. Este catéter está concebido para ablación intracardiaca por radiofrecuencia mediante el electrodo de la punta y un electrodo dispensor independiente que se conectan al generador de RF Atakr de Medtronic. El catéter de RF Marinr puede utilizarse también para registro o estimulación intracardiaca. El mango del catéter Marinr permite una exacta colocación de la punta dentro del corazón.
IndicacionesLos catéteres orientables de ablación por radiofrecuencia Marinr de Medtronic, están concebidos para ser utilizados con el generador de RF Atakr que produce energía de radiofrecuencia que puede utilizarse para la ablación intracardiaca de los trayectos de conducción aurícula ventriculares (AV) asociados con una taquicardia, para el tratamiento de taquicardias reentrantes del nodo AV y para causar bloqueos aurícula ventriculares completos en pacientes en los que es difícil controlar la respuesta ventricular a una arritmia auricular.
Contraindicaciones
El uso de este catéter está contraindicado en pacientes con infecciones sistémicas activas.La aproximación transeptal está contraindicada en pacientes con trombos o mixomas en la aurícula izquierda o con parches o deflectores intrauriculares.La aproximación retrógrada trans aórtica está contraindicada en pacientes en los que se haya sustituido la válvula aórtica.
Advertencias
Con los catéteres orientables de ablación por radiofrecuencia Marinr de Medtronic, la ablación efectuada desde el interior del seno coronario y sus ramas venosas puede

presentar riesgos adicionales debido a una mayor probabilidad de lesionar las estructuras adyacentes de las arterias coronarias.
Los procedimientos de ablación por catéter conllevan una exposición potencial significativa a rayos X, lo que puede causar un daño por radiación aguda así como un aumento del riesgo de efectos somáticos y genéticos tanto para los pacientes como para el personal de laboratorio, debido a la intensidad del haz de rayos X y la duración de la imagen de fluoroscopía. La ablación por catéter sólo debe realizarse una vez que se hayan considerado adecuadamente la posible exposición a la radiación asociada con el procedimiento y se hayan tomado medidas para reducir al mínimo esta exposición. El uso de este dispositivo con mujeres embarazadas debe considerarse cuidadosamente.
Durante una aproximación trans aórtica, es necesario utilizar una visualización fluoroscópica adecuada para evitar colocar el catéter en el interior de los vasos coronarios. La colocación de catéteres y aplicación de radiofrecuencia dentro de una arteria coronaria están asociados con infartos de miocardio y muerte.
Los materiales con que está fabricado este catéter no son compatibles con las imágenes de resonancia magnética (IMR).
La perforación de los vasos sanguíneos es un riesgo inherente en la colocación de cualquier catéter. Se ha documentado cierto número de otros acontecimientos adversos graves para los procedimientos de ablación mediante catéter, incluyendo embolias pulmonares, infartos de miocardio, ataques cardíacos, taponamiento cardíaco y muerte.
En los pacientes sometidos a procedimientos de ablación por el lado izquierdo es necesario supervisar cuidadosamente la aparición de signos clínicos de infarto durante el periodo posterior a la ablación.
No deben utilizarse catéteres con una separación de los electrodos del par distal superior a 2 mm para la ablación de trayectos accesorios septales o para el tratamiento de la taquicardia reentrante del nodo aurícula ventricular (AV) debido al peligro de causar un bloqueo AV completo no intencionado.
Los marcapasos y cardioversores/desfibriladores implantables (ICD) pueden ser afectados adversamente por la corriente de radiofrencuencia (RF), por lo que es importante disponer de fuentes externas provisionales de estimulación y desfribrilación durante el procedimiento de ablación, tener sumo cuidado si se realiza la ablación en la proximidad de cables permanentes auriculares o ventriculares y realizar un análisis completo de los dispositivos implantables en todos los pacientes después de la ablación.
Conviene desactivar los cardioversores/desfibriladores implantados antes de aplicar energía de radiofrecuencia.
Los pacientes sometidos a una modificación de nodo auriculoventricular o a una ablación de trayecto accesorio septal presentan el riesgo de un bloqueo AV completo. Un 1,6% (2/128) de los pacientes sometidos a ablación del trayecto accesorio septal, un 3,1% (1/32) de los tratados en los trayectos posteriores izquierdos y un 1,7% (4/238) de los sometidos a modificación del nodo AV, que experimentaron un bloqueo auriculoventricular parcial o completo no intencionado durante el estudio, requirieron una estimulación permanente. Durante la aplicación de energía RF es necesario supervisar cuidadosamente la conductividad auriculoventricular. Interrumpa inmediatamente la aplicación de energía si detecta un bloqueo AV parcial o completo.
El generador de RF Atakr puede producir una importante cantidad de energía de radiofrecuencia. No toque la punta distal del catéter de RF CardioRhythm y el electrodo dispersor al mismo tiempo (especialmente si está encendido el generador de RF Atakr) ya que puede lesionarse el operario.

Utilice solamente amplificadores, equipo de estimulación y electrógrafos aislados. En caso contrario, el paciente puede sufrir daños o morir. Las corrientes de fuga de cualquier dispositivo conectado al paciente no deben exceder los 10 microamperios (µA) en ningún caso.
Los catéteres de radiofrecuencia son desechables y de un solo uso. No vuelva a esterilizar o utilizar el catéter, ya que ello podría causar la pérdida de su adecuada función eléctrica y mecánica.
Precauciones
Debido al menor diámetro del catéter de RF Marinr, las dimensiones de la lesión pueden ser menores que las logradas con un sistema de ablación de 7 u 8 French y potencia semejante.
Durante las pruebas in vitro con un catéter de RF Marinr en condiciones de ausencia de flujo, hubo una mayor frecuencia de interrupciones por temperatura.
No intente utilizar el sistema de ablación Atakr o conectar el catéter al generador de RF Atakr antes de haber leído completamente y entendido el manual técnico del sistema de ablación Atakr y las instrucciones de empleo del catéter RF Marinr.
Los procedimientos de ablación cardíaca sólo deben ser realizados por personal con formación adecuada en electrofisiología en un laboratorio de electrofisiología completamente equipado.
Los catéteres sólo deben ser utilizados por médicos o bajo la supervisión de médicos con una formación adecuada en electrofisiología, incluyendo la colocación y uso de catéteres de electrodos intracardiacos y con experiencia en los procedimientos de ablación con un catéter de radiofrecuencia.
No se han establecido los riesgos a largo plazo de la fluoroscopia de protracción, por lo que debe considerarse cuidadosamente la utilización de este dispositivo en niños antes de la adolescencia.
No se han establecido todavía los riesgos a largo plazo de la ablación por radiofrecuencia. En concreto, no se conocen los efectos a largo plazo de las lesiones causadas en la proximidad del sistema de conducción especializado o de los vasos coronarios. Tampoco se ha estudiado la relación riesgo/beneficio en pacientes asomáticos.
Los catéteres de radiofrecuencia están concebidos para utilizarse solamente con el generador de RF Atakr. Su uso con otros generadores de radiofrecuencia no ha sido comprobado. Utilice solamente los cables Medtronic que se suministran con el sistema de ablación Atakr.
Un doblado o retorcido excesivo del catéter puede dañar sus cables de electrodos internos o reducir capacidad de conformación de la punta distal.
No permita que entre agua o fluidos en los conectores del catéter de RF, del generador de RF Atakr o de los cables. El sistema puede no funcionar correctamente si los conectores están húmedos.
Inspeccione el paquete estéril y el catéter antes de utilizarlo. Si han sufrido daños, no los utilice y consulte a su representante local de Medtronic.
El catéter debe manipularse con cuidado para evitar daños, perforación o taponamiento cardiaco. El catéter debe introducirse bajo orientación fluoroscopica. Nunca introduzca o retire a la fuerza el catéter si encuentra resistencia.
Vea el manual técnico del sistema de ablación Atakr sobre la información correspondiente a la retirada del catéter después de una detención de seguridad.

No aplique corriente continua a través del sistema de ablación Atakr o del catéter de RF. Ni el sistema de ablación Atakr ni el catéter de RF han sido concebidos para aplicar corriente continua y no se han realizado pruebas en este sentido.
Compruebe que la punta del catéter de radiofrecuencia no esté en contacto con los electrodos de otros catéteres que puedan existir en el corazón para evitar el recalentamiento de los restantes electrodos.
Es necesario supervisar la pantalla de impedancia del catéter durante la aplicación de energía. Si se observa un aumento repentino de la impedancia durante el procedimiento de ablación, interrumpa la aplicación de energía, examine la punta del catéter por si hay algún coágulo y retírelo en tal caso.
No inicie un procedimiento de ablación si se enciende la indicación LOW BATERY (batería descargada) en el sistema generador de RF Atakr, Sustituya la pila.
Para mantener una óptima seguridad del paciente y la integridad de los electrodos del catéter no limpie este catéter con disolventes orgánicos, tales como alcohol.
Los ensayos realizados en el banco de pruebas de los catéteres de RF han demostrado que pueden resistir veinticinco aplicaciones de energía de radiofrecuencia sin merma de sus características funcionales. Durante las pruebas clínicas se realizaron hasta 73 aplicaciones de energía con un mismo catéter, aunque el número medio fue de siete aplicaciones.
Especificaciones técnicas
Colocación Rápida del Catéter:
Curvas diseñadas para fácil avance y colocación precisa de la punta, a fin de adaptarse a las diversas anatomías cardíacas.
La composición del catéter proporciona una respuesta de torque 1:1, permitiendo una colocación rápida y precisa de la punta y minimizando el fenómeno de “whipping”.
Construcción del Eje:
El eje de cable trenzado mejora el torque y mantiene la forma del catéter. El eje de 7F está diseñado con un eje distal blando para minimizar el trauma intracardiaco,
mientras que el eje proximal es más rígido, lo cual mejora el empuje. El eje de 5F está diseñado para reducir el riesgo de complicaciones, así como para
minimizar el posible trauma en el tejido intracardiaco.Rendimiento Fiable:
Reposicionamiento mínimo del catéter. Lecturas intracardiacas fiables.
MARINR EP DIAGNOSTIC CATHETERS
Nº de Modelo
Tamaño French
(Fr.)
Alcance de la Curva en
90º (mm)
Electrodo Distal (mm)
Nº de Electrodos
Espacio entre
Electrodos (mm)
Longitud Usable (cm.)
072302 7 55-75 4 4 2/5/2 110

072402 7 55-75 4 4 2/5/2 110
MARINR CS CORONARY SINUS CATHETERS
Nº de Modelo
Tamaño French
(Fr.)
Alcance de la Curva en
90º (mm)
Electrodo Distal (mm)
Nº de Electrodos
Espacio entre
Electrodos (mm)
Longitud Usable (cm.)
043302M 7 55 2 10 2/2/2 90043325M 7 55 2 10 2/5/2 90043328M 7 55 2 10 2/8/2 90
RF MARINR MC ABLATION CATHETERS
Nº de Modelo
Tamaño French
(Fr.)
Alcance de la Curva en
90º (mm)
Electrodo Distal (mm)
Nº de Electrodos
Espacio entre
Electrodos (mm)
Longitud Usable (cm.)
075302 7 40-60 4 4 2/5/2 110075305 7 40-60 4 4 5/5/5 110075312 7 40-60 4 4 2/5/2 110075402 7 40-60 4 4 2/5/2 110
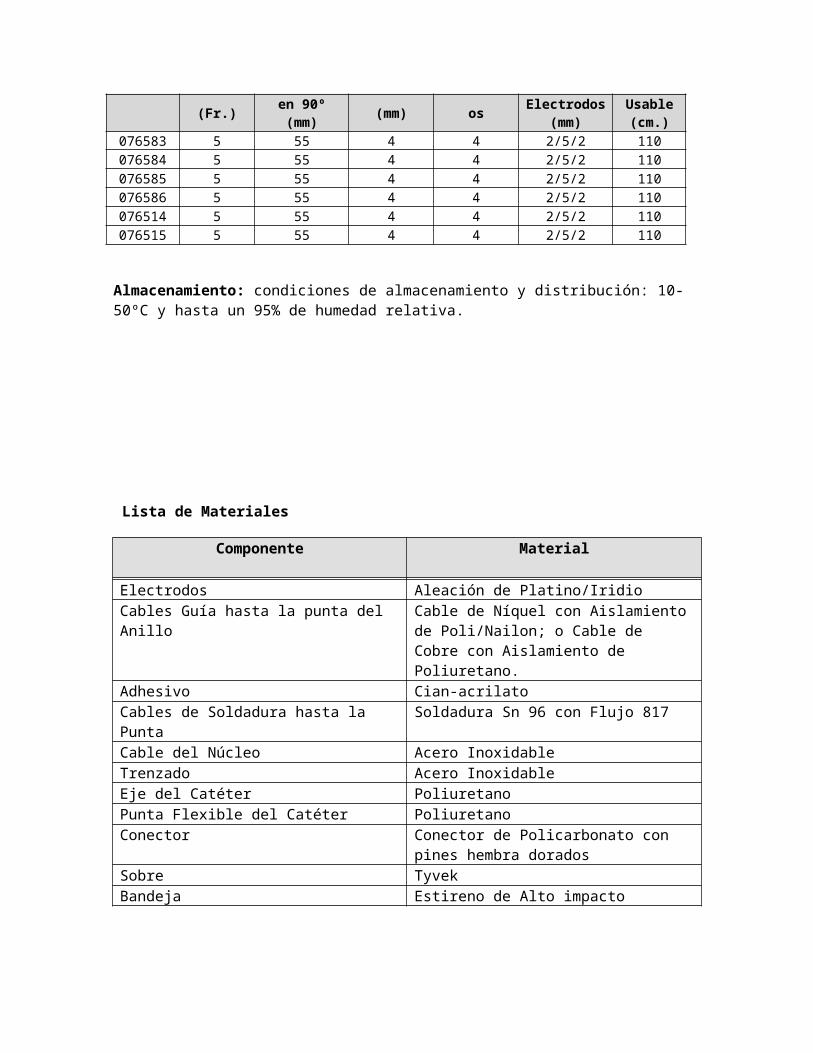
5F RF MARINR ABLATION CATHETERS
Nº de Modelo
Tamaño French
(Fr.)
Alcance de la Curva en
90º (mm)
Electrodo Distal (mm)
Nº de Electrodos
Espacio entre
Electrodos (mm)
Longitud Usable (cm.)
076583 5 55 4 4 2/5/2 110076584 5 55 4 4 2/5/2 110076585 5 55 4 4 2/5/2 110076586 5 55 4 4 2/5/2 110076514 5 55 4 4 2/5/2 110076515 5 55 4 4 2/5/2 110
Almacenamiento: condiciones de almacenamiento y distribución: 10-50ºC y hasta un 95% de humedad relativa.
Lista de Materiales
Componente Material
Electrodos Aleación de Platino/Iridio

Cables Guía hasta la punta del Anillo Cable de Níquel con Aislamiento de Poli/Nailon; o Cable de Cobre con Aislamiento de Poliuretano.
Adhesivo Cian-acrilatoCables de Soldadura hasta la Punta Soldadura Sn 96 con Flujo 817Cable del Núcleo Acero InoxidableTrenzado Acero InoxidableEje del Catéter PoliuretanoPunta Flexible del Catéter PoliuretanoConector Conector de Policarbonato con pines hembra
doradosSobre TyvekBandeja Estireno de Alto impacto

INSTRUCCIONES DE USO
INSTRUCCIONES DE USOMARINR EP DIAGNOSTIC CATHETERS
072322M, 072302, 072402
MARINR CS CORONARY SINUS DIAGNOSTIC CATHETERS043302M, 043325M, 043328M
RF MARINR MC ABLATION CATHETERS075302, 075305, 075312, 075402, 075405
5F RF MARINR ABLATION CATHETERS076583, 076584, 076585, 076586, 076514, 076515
El catéter de electrodos de punta orientable/flexionable Marinr, de Medtronic ® CardioRhythm®, es un catéter radio-opaco y flexible fabricado con un polímero extruido sobre un cordoncillo de acero inoxidable. Este catéter está concebido para registro o estimulación intracardíaca. El mango del catéter Marinr permite una exacta colocación de la punta dentro del corazón.
INSTRUCCIONES DE EMPLEO
GENERAL
1. Revise el paquete del catéter antes de abrirlo. El contenido de este paquete se ha esterilizado antes de su distribución. No lo use si el paquete está abierto o dañado.
2. Use técnica aséptica para sacar el catéter del paquete y colóquelo en un lugar de trabajo estéril (Figura 1).
3. Revise el catéter cuidadosamente para comprobar la integridad del electrodo y sus condiciones generales. No use el catéter si los electrodos o la punta están flojos, deformados o visiblemente dañados.
4. Cree un acceso vascular con técnica estéril. El catéter puede usarse desde emplazamientos de acceso femoral, braquial, subclávico o yugular.
5. Haga coincidir los colores de los conectores para efectuar correctamente la conexión cable a catéter. Conectar el cable (Figura 2): Coloque las flechas del conector de cables alineadas con las ranuras de anclaje del conector del catéter. Inserte el cable dentro del catéter hasta que quede bloqueado. Desconectar el cable (Figura 3): Tire del anillo de sujeción grande en el conector del cable hasta liberar el mecanismo de bloqueo.

6. Conecte las espigas del cable de conexión de catéteres al equipo electrónico de registro y/o estimulación correspondiente. La espiga “D” corresponde al electrodo de la punta distal y las demás espigas (2, 3 y 4) corresponden a la ubicación de las bandas de electrodos desde la punta distal. Para más información, consulte las instrucciones para el uso del cable de conexión de catéteres.
7. Haga avanzar el catéter hasta el lugar deseado en el corazón, mediante orientación fluoroscópica y ECG.
MANEJO DEL MANGO CONFORMABLE
1. Tire hacia atrás el Control de flexión de la punta (Figura 4) para flexionar la punta del catéter. La punta se puede flexionar al menos 180
Para enderezar la punta, empuje el Control de flexión de la punta hacia adelante (Figura 5).
2. Haga girar el Control de flexión de la punta en el sentido de las manecillas del reloj para aumentar la fricción, y en sentido contrario para disminuir la fricción (Figura 6); así se ajusta la fuerza necesaria para mantener la posición de la punta.

3. Tire hacia atrás del Control del radio de la curva (Figura 7) para aumentar el radio de la curva flexionada. Para disminuir el radio de la curva flexionada, empuje el Control del radio de la curva hacia adelante.
4. Haga girar el Control del radio de la curva en el sentido de las manecillas del reloj para aumentar la fricción y en sentido contrario para disminuir la fricción (Figura 6) a fin de ajustar la fuerza necesaria para mantener el radio de la curva.
5. Haga rotar el Control de flexión lateral en el sentido de las manecillas del reloj o en sentido contrario para girar la punta lateralmente hasta un máximo de 45° en cada dirección (Figura 8).
6. Antes de retirar el catéter, vuelva a alinear las marcas en el Control de flexión lateral, empuje el Control de flexión de la punta hacia adelante y verifique fluoroscópicamente que la punta se encuentre en una posición neutral.
Almacenamiento
Condiciones de almacenamiento y distribución: 10-50 °C y hasta un 95% de humedad relativa.

PRUEBAS FUNCIONALES
PRUEBAS FUNCIONALESMARINR EP DIAGNOSTIC CATHETERS
072322M, 072302, 072402

MARINR CS CORONARY SINUS DIAGNOSTIC CATHETERS043302M, 043325M, 043328M
RF MARINR MC ABLATION CATHETERS075302, 075305, 075312, 075402, 075405
5F RF MARINR ABLATION CATHETERS076583, 076584, 076585, 076586, 076514, 076515
La Inspección del Control de Calidad en el Proceso se Realiza en las Siguientes Etapas de Manufactura
Inspección de los Componentes Moldeados. Inspección en el proceso interno. Inspección del Empaque Blister.
Inspección de los Componentes MoldeadosEl componente moldeado se verifica por lo menos una vez en el turno por defectos de moldeado, dimensiones y encaje. La evaluación de fuga se realiza si se requiere. Se realiza la inspección adicional cuando llega una nueva carga de producción.
Inspección en el Proceso InternoLos productos semi-ensamblados de las líneas de ensamblaje se inspeccionan por lo menos una vez durante el turno. Se verifican diversos encajes funcionales.
Inspección del Empaque BlisterLos productos empacados en blisters se verifican una vez en el turno sobre una base de muestreo. El blister se comprueba visualmente si existe algún defecto y por la fuerza del sellado.
Inspección del Producto TerminadoLos productos terminados (esterilizados) son retirados sobre una base de muestreo desde la inspección final y sección de empaque. Los productos son inspeccionados/evaluados por sus propiedades físicas de acuerdo a los estándares de las empresas. Todo y cada uno de los productos terminados se evalúan con evaluaciones extractivas acuosas. El lote esterilizado del producto terminado se verifica por esterilización de acuerdo a los requerimientos de transfusión & ensamblajes de acuerdo a USP XXX. Además todos estos productos son evaluados según su tipo para cumplir los requerimientos de la evaluación de Compatibilidad Biológica según ISO 10993. Periódicamente, se realizan evaluaciones de reacción biológica, se lleva a cabo la citotoxicidad in-vitro para verificar la continuidad del cumplimiento de la compatibilidad biológica.Los sellos /marcas en los empaque unitarios, empaques multi-unitarios y empaques de cajas principales se inspeccionan de acuerdo los estándares de la empresa por impresos/estampados e.g. número de lote, fecha de manufactura, fecha de vencimiento.
Normas y Directivas Consideradas en la Prueba:

Se emprendió la realización de actividades de verificación de diseño a fin de confirmar las especificaciones funcionales del dispositivo. Se llevaron a cabo pruebas selectivas para cumplir o sobrepasar los requisitos de las siguientes normas y directivas:
ISO 594-1, Guarniciones Cónicas con un 6% (Luer) de conificación. ISO 10555-1, Catéteres Electrodos, estériles, de uso único. ISO 10993-1, Evaluación Biológica para Dispositivos Médicos. ASTM-F640-79, Método de Prueba Standard para Radiopacidad de los Plásticos para Uso
Médico. Documento de la Directiva de FDA para Catéteres Electrodos de Corto Plazo y Largo Plazo. USP XXX Monografía 788.
Análisis Dimensional:Medición de la conformidad del catéter terminado con las especificaciones dimensionales.
Prueba de integridad del Catéter:Medición de la resistencia a la tracción del catéter, resistencia al retorcimiento, fatigas y frenado de la punta.

Propiedades Químicas, Físicas y Biológicas
1. Los dispositivos deben estar diseñados y manufacturados de manera que garanticen las características y rendimientos referidos en la Sección I de los “Requisitos Generales”. Se debe prestar especial atención en:La opción de materiales utilizados, particularmente de acuerdo a toxicidad y donde es apropiado, si es inflamable.La compatibilidad entre los materiales utilizados y los tejidos biológicos, células y fluidos corporales, se toman en cuenta de acuerdo al propósito del dispositivo.
2. Los dispositivos deben estar diseñados y manufacturados y empacadas de manera que se minimice el riesgo presentado por los contaminantes y residuos de las personas involucradas en el transporte, almacenamiento y uso de dispositivos y de los pacientes, tomando en cuenta del propósito del producto. Se debe tener especial atención a los tejidos expuestos y a la duración y la frecuencia de exposición.
ISO-10993-5:1992
ISO 10993-7
Prueba de Citotoxicidad
El producto está diseñado para el uso deseado & fabricado en una habitación limpia. Empacado individualmente en un blister & esterilizado con óxido de etileno.
Análisis de los residuos de Óxido de Etileno
Departamento de control de Calidad
3. Los dispositivos deben estar diseñados y manufacturados de manera que puedan ser utilizados en forma segura con los materiales sustancias y gases con los que ellos pueden entrar en contacto durante su uso normal o durante los procedimientos de rutina; si los dispositivos están dirigidos a administrar productos medicinales ellos deben estar diseñados y manufacturados de manera que sean
ISO-10555-1
CLAÚSULAS-4.2, 4.4
Pruebas del Material que Ingresa y Prueba de la AgujaObservación: Material de contacto; La Aguja está evaluada por resistencia a la corrosión
Departamento de control de Calidad

compatibles con los productos medicinales de acuerdo a las regulaciones y restricciones dictadas por dichos productos y que su rendimiento se mantiene de conformidad con el uso propuesto.
Infección y Contaminación Microbial
1. Los dispositivos deben estar diseñados y manufacturados de manera que se elimine o reduzca lo máximo posible el riesgo de infección al paciente, usuario y terceros. El diseño debe permitir el fácil manipuleo y donde sea necesario, minimizar la contaminación del dispositivo por el paciente o viceversa durante su uso.
FED-STD-209EEN-550
La manufactura de los productos se realiza en un área limpia y controlada de manufactura y se monitorea según el procedimiento HH-QSP-6.4-01&02.El producto se esteriliza según el procedimiento HH-QSP-7.5-08
Departamento de control de Calidad
3. Los dispositivos administrados en estado esterilizado deben estar diseñados y manufacturados y empacados en un empaque no reutilizable y/o de acuerdo a los procedimientos adecuados para asegurar que están esterilizados cuando se colocan en el mercado y permanecen esterilizados durante el almacenamiento y condiciones de transporte, hasta que el empaque protector se daña o se abre.
EN-550 i) Prueba de esterilizaciónii) Prueba de estabilidad
El producto está esterilizado según el procedimiento HH-QSP-7.5-08
Departamento de control de Calidad
4. El empaque y/o la etiqueta del dispositivo debe distinguirse entre los productos que se venden idénticos o similares en condiciones esterilizadas y no esterilizadas.


BIOCOMPATIBILIDAD

BIOCOMPATIBILIDADMARINR EP DIAGNOSTIC CATHETERS
072322M, 072302, 072402
MARINR CS CORONARY SINUS DIAGNOSTIC CATHETERS043302M, 043325M, 043328M
RF MARINR MC ABLATION CATHETERS075302, 075305, 075312, 075402, 075405
5F RF MARINR ABLATION CATHETERS076583, 076584, 076585, 076586, 076514, 076515
IntroducciónEl catéter orientable de ablación por radiofrecuencia Marinr de Medtronic, son catéteres radiopacos, flexibles y de un solo uso. El eje del catéter ha sido evaluado de conformidad con el estándar ISO 10993-1. Esta parte de ISO 10993 es todo el documento guía para la selección de pruebas que permiten la evaluación de las respuestas biológicas relacionadas a la seguridad de los dispositivos y materiales médicos.Contacto con el Material y el TejidoLos catéteres orientables de ablación por radiofrecuencia Marinr de Medtronic están compuestos de los siguientes materiales que tienen el potencial de tener contacto directo y/o indirecto con el tejido del cuerpo y los fluidos del paciente. Dichos materiales fueron evaluados en conjunto. Los materiales individuales que comprende los Ejes de los catéteres orientables de ablación por radiofrecuencia Marinr de Medtronic se identificaron con la siguiente información (ver el Cuadro 2.1).
Cuadro 2.1Material Número
de Parte del
Material
Proveedor del Material
Componente del
Dispositivo
Número de Parte del
Componente
Proveedor del
Componente
Tejido en Contacto
Duración del
Contacto
90% Platino/10% Iridio
163163-006
Noblemet Anillo de Electrodo (9)
163163-006 MedtronicSangre, Tejido
vascular< 24 horas
90% Platino/10% Iridio
163163-007
Noblemet Anillo de Electrodo (1)
163163-007 MedtronicSangre, Tejido
vascular< 24 horas
90% Platino/10% Iridio
120168-004
Pulse Technology
Punta de electrodo 8
mm120168-004 Medtronic
Sangre, Tejido
vascular< 24 horas
Polieter-eterketo
na (PEEK)
170343-004,
170447-001
Pulse Technology
Punta de Aislante 7F,
anclaje170343-004, 170447-001
MedtronicSangre, Tejido
vascular< 24 horas
Polieter Amida-Pebax 72D
177885-001
MECCTubería, Eje
externo trenzado
177885-001 MedtronicSangre, Tejido
vascular< 24 horas

Polieter Amida-Pebax 55D
170410-003,
153575-007
MECC Tubería, eje 170410-003, 153575-007
MedtronicSangre, Tejido
vascular< 24 horas
Polieter Amida-Pebax 35D
170411-001
MECCTubería, extremo
distal del eje170411-001 Medtronic
Sangre, Tejido
vascular< 24 horas
El eje del catéter tendrá contacto directo con el tejido y la sangre a través del ingreso vascular en el paciente. Así, los catéteres orientables de ablación por radiofrecuencia Marinr de Medtronic, se clasifica como un dispositivo de comunicación externa, con contacto directo de tejido y sangre y se cataloga como un dispositivo de uso agudo, con una duración limitada menor de 24 horas. Se asevera una duración del uso clínico de un mínimo de 2 a un máximo de 8 horas. La selección de las pruebas se basó es esta asesoría de contacto con el tejido.
Evaluación del Cumplimiento de ISO 10993A continuación un debate de cómo la evaluación biológica del eje de los catéteres orientables de ablación por radiofrecuencia Marinr de Medtronic cumple con ISO 10993:
Cuadro 3.1
Designación del
Estándar ISO
Nombre Cumplimiento del
Estándar
Debate del CumplimientoNúmero de Referencia del Informe
de Medtronic
10993-1 Evaluación y prueba SiUna evaluación de la necesidad de desarrollar la evaluación y las pruebas que fueron elegidas, siguiendo las normas.
El presente documento y su Plan de Evaluación Biológico correspondiente P000008
10993-2 Requerimiento de bienestar de los animales
SiProcedimientos de prueba utilizando animales de acuerdo a las normas. Un Comité Institucional de Uso y Cuidado de los Animales revisó todas las pruebas que involucraban animales.
0077D0434
10993-3Pruebas de genotoxicidad, carcinogenicidad y toxicidad reproductiva
N/ADebido a la naturaleza de este dispositivo de exposición limitada, aguda y de un solo uso, las pruebas de genotoxicidad no son requeridas.
N/A
10993-4Sección de pruebas para la interacción con la sangre
Si Las pruebas in Vitro para coagulación, hematología y complementos han sido realizadas en este dispositivo.
0077D0434
10993-5Pruebas para la citotoxicidad: métodos in vitro
Si La prueba de Elusión MEM ha sido realizada en este dispositivo.
0077D0435
10993-6 Pruebas para efectos locales después del
N/AEste dispositivo no está hecho para implantes de más de 30 días. Debido a su naturaleza
N/A

implante de exposición limitada, aguda y de un solo uso las pruebas de implante no se requieren.
10993-7 Residuales de esterilización ETO
N/A Este dispositivo está esterilizado en gamma
N/A
10993-8Guía para materiales de referencia
Si La selección del material de referencia para la evaluación seleccionada de acuerdo a las normas.
0077D0434,0077D0435
10993-9 Marco para identificación y cuantificación de productos de degradación potencial
N/AEste dispositivo no está hecho para implantes de más de 30 días. Debido a su naturaleza de exposición limitada, aguda y de un solo uso las pruebas de degradación no se requieren.
N/A
10993-10Pruebas de irritación y sensibilización
Si Pruebas de Inyección intracutánea y Sensibilidad Máxima en Cuyes han sido realizados en este dispositivo
0077D0434
10993-11Pruebas de toxicidad sistémica
Si Pruebas de Inyección sistémica y pirogenicidad en conejos han sido realizados en este dispositivo
0077D0434
10993-12 Preparación de muestra y materiales de referencia
SiLa selección del material y Prueba junto con las condiciones de extracción para las pruebas recomendadas siguieron esta norma.
0077D0434,0077D0435
10993-13Identificación y cuantificación de productos de degradación de dispositivos médicos poliméricos
N/AEste dispositivo no está hecho para implantes de más de 30 días. Debido a su naturaleza de exposición limitada, aguda y de un solo uso las pruebas de degradación no se requieren.
N/A
10993-14Identificación y cuantificación de productos de degradación de cerámicos.
N/A Este material no es de cerámica
N/A
10993-15Identificación y cuantificación de productos de degradación de metales y aleaciones.
N/AEste dispositivo no está hecho para implantes de más de 30 días. Debido a su naturaleza de exposición limitada, aguda y de un solo uso las pruebas de degradación no se requieren.
N/A
10993-16Estudio de diseño toxico cinético para productos de degradación y filtrables
N/AEste dispositivo no está hecho para implantes de más de 30 días. Debido a su naturaleza de exposición limitada, aguda y de un solo uso las pruebas de degradación no se requieren.
N/A
Proceso de ManufacturaNo se efectuó una comparación en el proceso para esta evaluación biológica de los componentes, ya que es un producto para el que no existe información histórica. La información del proceso para este dispositivo se detalla en el archivo de Medtronic 03-CR-1130 Biomatrix Test Plan. Se afirma

que no existen condiciones en el proceso que hayan tenido impacto en la seguridad biológica de los materiales. Este dispositivo es un producto esterilizado en gamma.
ConclusiónLa evaluación biológica ha sido realizada en los ejes de los catéteres orientables de ablación por radiofrecuencia Marinr de Medtronic. Se ha documentado el raciocinio para la selección de pruebas y para las pruebas que han sido retiradas. La información en este informe de evaluación biológica demuestra el cumplimiento del eje de los catéteres orientables de ablación por radiofrecuencia Marinr de Medtronic con el estándar ISO 10993 para su uso en esta aplicación.Los catéteres orientables de ablación por radiofrecuencia Marinr de Medtronic, están concebidos para procedimientos electrofisiológicos de diagnóstico. Estos catéteres están destinados al registro de los electrogramas intracardiacos y la estimulación temporal utilizados durante los estudios electrofisiológicos.

ESTERILIZACION

ESTERILIZACIONMARINR EP DIAGNOSTIC CATHETERS
072322M, 072302, 072402
MARINR CS CORONARY SINUS DIAGNOSTIC CATHETERS043302M, 043325M, 043328M
RF MARINR MC ABLATION CATHETERS075302, 075305, 075312, 075402, 075405
5F RF MARINR ABLATION CATHETERS076583, 076584, 076585, 076586, 076514, 076515
1.0 PROPÓSITO/OBJETIVOS
1.1 El propósito de este plan de prueba es resaltar los pasos necesarios para
validar la dosis de esterilización para el catéter orientable de ablación por
radiofrecuencia Marinr de Medtronic por radiación gamma (Cobalt 60) en las
instalaciones de radiación gamma contratados. Este plan también resalta los
pasos necesarios para las auditorias de control trimestrales del producto.
2.0 ENTORNO
2.1 La última validación de la esterilización de los catéteres de ablación dirigible
por radiofrecuencia del catéter orientable de ablación por radiofrecuencia
Marinr de Medtronic, por medio de radiación gamma fue en Febrero de 2004.
2.2 Las auditorias trimestrales de las dosis han sido realizadas para liberar la
dosimetría desde que la validación se completó.
2.3 No ha habido cambios en el producto ni en el proceso desde la última
validación.
2.4 La prueba de Bacteriostasis y fungistasis se realizó en Julio de 1998 mediante
el procedimiento del Laboratorio Geneva ST1013 Rev. E. Los resultados de la
prueba se pueden encontrar en Estudio de Prueba, Prueba de Bacteriostasis y
Fungistasis TS-A0020-125, Rev. EOO.

3.0 DOCUMENTOS APLICABLES
3.1 29113-H, Documentos y Archivo de Planes de prueba, Informes de Pruebas y
Estudios de las pruebas.
3.2 AAMI/ANSI/ISO-11137, Esterilización de Productos para Cuidado de la Salud –
Requisitos para Va1idación y Control de Rutina – Esterilización de la
Radiación (Aprobado en Julio de 1994).
3.3 20925, Procedimiento de Auditoría para los Esterilizadores Contratados.
3.4 20932, Procedimiento, Validación de la Esterilización por radiación Gamma
del catéter orientable de ablación por radiofrecuencia Marinr de Medtronic.
3.5 Procedimiento, Prueba de Esterilización de los Laboratorios Geneva ST1010
Rev. D, Anexo MB-1ª.
3.6 Procedimiento, Prueba de Carga Biológica de los Laboratorios Geneva ST2003
Rev. C.
3.7 USP Revisión Actual.
4.0 RESPONSABILIDAD
4.1 Las instalaciones de radiación gamma contratada, SteriGenics, es
responsable por la calibración de los instrumentos, mantener registros de
la actividad de la fuente, inspección de todo el proceso relacionado a la
documentación, requisitos del proceso, requisitos del contrato GMP,
mantenimiento preventivo y de rutina del equipo, modelo de carga del
producto, mapeo de la distribución de las dosis y toda la documentación
necesaria para asegurar el cumplimiento del contrato.
4.2 El equipo de la bomba es responsable de ejecutar el plan de prueba. Las
instalaciones de prueba contratada, laboratorios Geneva, es responsable de
realizar la prueba de esterilización y la prueba de carga biológica y
proporcionar los resultados a Medtronic.
5.0 EQUIPO / INSTRUMENTOS / VÁLVULAS UTILIZADOS

5.1 El equipo necesario para realizar la esterilización por irradiación gamma se
determina por el esterilizador contratado.
5.2 El equipo necesario para realizar la prueba de esterilización y la prueba de
carga biológica se determina por la instalación de prueba contratada.
6.0 MATERIALES / MUESTRAS UTILIZADAS
6.1 Para determinar la carga biológica, una muestra de por lo menos 10
catéteres orientables de ablación por radiofrecuencia Marinr de
Medtronic se deben tomar al azar de cada tres lotes de producción
(por un total de 30 catéteres orientables de ablación por
radiofrecuencia Marinr de Medtronic por lo menos), inmediatamente
antes de la esterilización.
6.2 La prueba de verificación de esterilización se realiza irradiando 100
muestras de los catéteres orientables de ablación por radiofrecuencia
Marinr de Medtronic en el nivel de dosis de verificación del sub proceso.
6.3 Las auditorias trimestrales de la dosis se realizaran seleccionando al azar
110 muestras de los catéteres orientables de ablación por radiofrecuencia
Marinr de Medtronic de un lote de producción.
7.0 SOFTWARE
7.1 No es aplicable.
8.0 PROCEDIMIENTO
8.1 Configuración de la Dosis: Método 1 según AAMII ANSVISO-11137 se
utilizará para configurar la dosis de esterilización en base a la
información de la carga biológica (el promedio de la carga biológica por
equipo) como sigue:
8.1.1 Seleccionar una muestra de por lo menos 10 catéteres
orientables de ablación por radiofrecuencia Marinr de Medtronic
tomadas al azar de cada tres lotes de producción,
inmediatamente antes de la esterilización.

8.1.2 Se deberá calcular el promedio de la carga biológica por cada lote y
el promedio total. La prueba de carga biológica la debe realizar la
instalación de prueba contratada de conformidad con el protocolo
suscrito siguiendo el procedimiento de prueba descrito en la
revisión actual de USP. También se le puede solicitar a la
instalación de prueba el cálculo de los promedios y la
recomendación de la verificación y las dosis del SAL (Nivel de
Seguridad de Esterilización) por el personal de Medtronic.
NOTA: MIENTRAS SE CALCULA EL PROMEDIO DE LA CARGA
BIOLÓGICA DEL DISPOSITIVO, ONL Y CFU (CONTEO DE CARGA
BIOLÓGICA) NECESITAN SER CONSIDERADOS, NO LA CUENTA DE
ESPORAS. ADEMÁS, LA PRUEBA DE CARGA BIOLÓGICA NECESITA
REFLEJAR EL PORCENTAJE DE RECUPERACIÓN DE LA CARGA
BIOLÓGICA
8.1.3 El conteo de la carga biológica a utilizarse por el Método 1 es el
promedio total a menos que uno de los promedios del lote sea dos
o más veces mayor que el total del promedio, en cuyo caso se
deberá utilizar el promedio mayor del lote.
8.1.4 Ya que toda la bomba se utiliza como una muestra, SIP (Porción de
muestra) es 1.0. El método de inmersión completo se emplea para
el cálculo de la carga biológica y se considera un SIP de 1.0.
8.1.5 Elegir la dosis de verificación del sub proceso del Cuadro 7 (del
documento de requisitos AAMI/ISO) que está diseñado para la
prueba para la resistencia de la carga biológica hasta el proceso de
esterilización en un SAL de 10-2. Cuando se utiliza el cuadro 7 para
determinar la dosis de verificación del sub proceso, utilizar el
número de la carga biológica más cerca igual o mayor a los números
de la carga biológica actual. El número de la carga biológica
utilizado para obtener la dosis de verificación del sub proceso es la
carga biológica promedio del dispositivo multiplicada por la SIP. La

SIP en este caso es 0.67.
8.1.6 Se permite que la dosis administrada para verificación varíe desde
la dosis de verificación del sub proceso por + 10%kGy. La dosis de
verificación se proporciona mediante un proceso por lotes en el
esterilizador contratado.
8.2 Irradiar 100 muestras de los catéteres orientables de ablación por
radiofrecuencia Marinr de Medtronic en el nivel de dosis de verificación del
sub proceso.
8.3 Las muestras luego son enviadas a la instalación de prueba contratada de
acuerdo al protocolo suscrito (ST1010, Anexo II). La verificación estadística
se acepta si no existe más de dos pruebas esterilizadas positivas a partir de
cien muestras evaluadas.
8.4 La configuración de nuestra dosis establecerá una probabilidad de
presencia de un simple microorganismo viable en los catéteres orientables
de ablación por radiofrecuencia Marinr de Medtronic igual o menor a 10-6.
La dosis de esterilización elegida para los catéteres orientables de ablación
por radiofrecuencia Marinr de Medtronic será una correspondiente al
número más cerca de carga biológica en el cuadro que es igual o mayor al
total de la carga biológica promedio para este dispositivo.
8.5 Si la verificación falla, se debe utilizar un método alternativo para la
verificación de la dosis. El Método 1 no debe repetirse a menos que se
demuestre que las determinaciones de la carga biológica, la evaluación de
la esterilización o la dosis de verificación se realizaron en forma incorrecta.
8.6 Se requiere una auditoria periódica (se debe realizar cada tres meses) para
reafirmar que la dosis de esterilización elegida proporciona un apropiado
SAL. La auditoría se lleva a cabo para detectar cambios en la carga biológica
y/o la resistencia de la radiación de la población de microbios requiere un
incremento en la dosis de esterilización establecida. El procedimiento para
la auditoria periódica es así:

8.6.1 Seleccionar al azar 110 bombas de la producción de un lote.
8.6.2 Realizar una prueba de carga biológica en diez de las bombas de
muestra utilizando SIP de 1.0 para confirmar el promedio calculado
y la dosis SAL.
8.6.3 Irradiar cien (100) muestras con la dosis de verificación utilizada en
la validación (paso 8.2). La dosis administrada podría variar a partir
de la dosis meta por + 10%kGy.
8.6.4 Las bombas luego son enviadas a las instalaciones de prueba
contratadas para la evaluación de la esterilización de acuerdo al
protocolo suscrito.
8.6.5 Si se obtienen dos o menos positivos, la dosis actual SAL se
considera aceptable, y no se requiere ninguna acción.
NOTA: LA EVALUACIÓN DE LA CARGA BIOLÓGICA
NORMALMENTE SE REALIZA AL MISMO TIEMPO QUE
LAS MUESTRAS AUDITADAS SON IRRADIADAS. POR LO
TANTO, LOS RESULTADOS NO ESTARÁN DISPONIBLES HASTA QUE
LA AUDITORIA HAYA SIDO REALIZADA. LOS RESULTADOS DE LA
NUEVA CARGA BIOLÓGICA PODRIAN SER DE AYUDA PARA
INTERPRETAR LOS RESULTADOS ATIPICOS.
8.6.6 Una auditoria de los controles de manufactura y medio ambiente y
de la evaluación de la carga biológica se deberá revisar junto con la
evaluación de los resultados de la dosis auditada.
8.6.7 Las normas AAMI/ANSI/ISO-11137 se deberán consultar si existen
más de dos (2) positivos obtenidos en la prueba de esterilización.
La investigación de las causas podría requerir un equipo y análisis
de toda la información pertinente.
9.0 REQUISITOS Y CRITERIO DE ACEPTACIÓN

9.1 La verificación de la dosis se acepta si no existe más de dos pruebas de
esterilidad positivas a partir de las cien muestras evaluadas. La evaluación
de la carga biológica se monitorea continuamente y los valores deben
permanecer bajo los valores utilizados para determinar la dosis de
validación.
9.2 La documentación necesaria debe recibirse y encontrarse aceptable de la
instalación de irradiación gamma contratada, SteriGenics.
9.3 La documentación necesaria debe recibirse y encontrarse aceptable de la
instalación de prueba contratada, laboratorios Geneva.

ROTULADOS

ROTULADOSMARINR EP DIAGNOSTIC CATHETERS
072322M, 072302, 072402
MARINR CS CORONARY SINUS DIAGNOSTIC CATHETERS043302M, 043325M, 043328M
RF MARINR MC ABLATION CATHETERS075302, 075305, 075312, 075402, 075405
5F RF MARINR ABLATION CATHETERS076583, 076584, 076585, 076586, 076514, 076515
A continuación se presenta el proyecto de rotulado del envase mediato e inmediato del producto de la referencia.

ROTULADO DEL ENVASE MEDIATO E INMEDIATO


Nota. – Todos los dispositivos médicos Marinr EP Diagnostic Catheters tienen el mismo modelo de rotulado del envase mediato e inmediato cambiando solamente el modelo y

ROTULADO DEL ENVASE MEDIATO E INMEDIATO

Nota. – Todos los dispositivos médicos Marinr CS Coronary Sinus Diagnostic Catheters tienen el mismo modelo de rotulado del envase mediato e inmediato cambiando solamente el

ROTULADO DEL ENVASE MEDIATO E INMEDIATO

Nota. – Todos los dispositivos médicos RF Marinr MC Ablation Catheters tienen el mismo modelo de rotulado del envase mediato e inmediato cambiando solamente el modelo y

ROTULADO DEL ENVASE MEDIATO E INMEDIATO

Nota. – Todos los dispositivos médicos 5F RF Marinr Ablation Catheters tienen el mismo modelo de rotulado del envase mediato e inmediato cambiando solamente el modelo y












