Protein=Aminoacidos
115
METABOLISMO DE AMINOACIDOS Dr. Iván Patricio Jácome A.
-
Upload
gaby-ortega -
Category
Documents
-
view
97 -
download
7
Transcript of Protein=Aminoacidos

METABOLISMO DE AMINOACIDOS
Dr. Iván Patricio Jácome A.

Proteínas
• Comprenden un grupo de estructuras químicas formadas por polímeros de AA, que uniendose por enlaces peptídicos, forman cadenas polipeptídicas.
• Son moléculas estructrurales básicas de los organismos vivos
• Una cadena polipeptídica promedio de una proteína tiene alrededor de 500 residuos de AA, y unas pocas tienen más de 2000 residuos.
• Estructuralmente están conformadas por C (50-55%), O2 (20-40%), H (15 -20%), N (6-7%).
• Son solubles en agua excepto las proteínas fibrosas: colágeno, elastina, las de pelos y uñas.

Proteínas
• Generalmente funcionan a un pH óptimo de 6.8 a 7.2 con ciertas excepciones
• Su pH hace que unas proteínas sean ácidas, otras alcalinas y otras neutras.
• Esto depende básicamente de la presencia en su estructura de grupos amino, o grupos carboxilo.
• Su oxidación metabólica genera 4,1 Kcal/mol de proteína oxidada.

AMINOACIDOS
• Químicamente se los considera como derivados de los ácidos orgánicos su fórmula general es:
• H• R- C- COOH• NH3• El C tiene sus 4 valencias saturadas por
elementos diferentes.• Si así ocurre excepto con la Glicina, este C se
denomina C asimétrico o C quiral

Aminoácidos
• De acuerdo a la nomenclatura del alfabeto griego al C que sigue al grupo carboxilo se lo denomina como Alfa, al siguiente Beta y así sucesivamente.
• Por esta denominación de sus carbonos se habla de alfa aminoácidos.etc…
• El esqueleto de los AA se los numera desde el C del carboxilo como 1, 2 etc

AMINOACIDOS
• Constituyen los componentes estructurales de las proteínas.
• Proteínas son moléculas estructurales y funcionales básicas de los organismos vivos.
• Su metabolismo se da principalmente en el hígado.
• También participan en el metabolismo de compuestos no proteínicos.

AMINOACIDOS
• Se derivan de la descomposición de las proteínas de la dieta y del organismo.
• El 85% de los AA que derivan de la hidrólisis de las proteínas endógenas son reutilizados para la síntesis de nuevas proteínas.
• Oxidación del esqueleto de C de los AA significa el 12 al 15% de la E metabólica.
• C/AA proporciona una pequeña cantidad del total de la E metabólica humana.

Digestión de proteínasConsta de 2 fases1.-Fragmentación endopentidasas péptidos + cortos(Protein.menores)2.- Péptidos exopeptidasas liberación aa de las proteinas.Mecanismo: Proteínas dieta.
1) Estomago inicio digestión. Favorecidas x ph ácido ph 3.7 * Pepsinogeno activa a zimogenos ph 1.5 Pepsina proteínas C. act: aspartato rompe las degradada a enlaces peptid peptidos grandes 600- 3.000 kd. Pepsina activa + lentamente si aa que sigue a un aa aromático es
Leu .*Zimogeno: Precursores enzimàticos inactivos. (dar lugar a las enzimas)
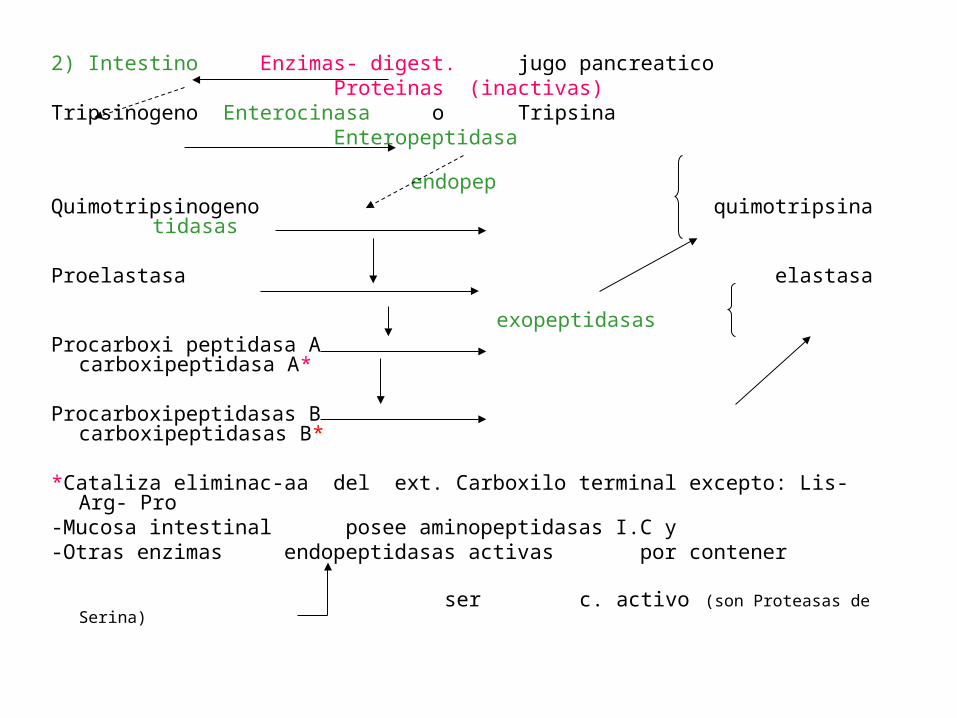
2) Intestino Enzimas- digest. jugo pancreatico Proteinas (inactivas)Tripsinogeno Enterocinasa o Tripsina Enteropeptidasa endopepQuimotripsinogeno quimotripsina tidasas
Proelastasa elastasa exopeptidasasProcarboxi peptidasa A carboxipeptidasa A*
Procarboxipeptidasas B carboxipeptidasas B*
*Cataliza eliminac-aa del ext. Carboxilo terminal excepto: Lis-Arg- Pro-Mucosa intestinal posee aminopeptidasas I.C y-Otras enzimas endopeptidasas activas por contener ser c. activo (son Proteasas de Serina)
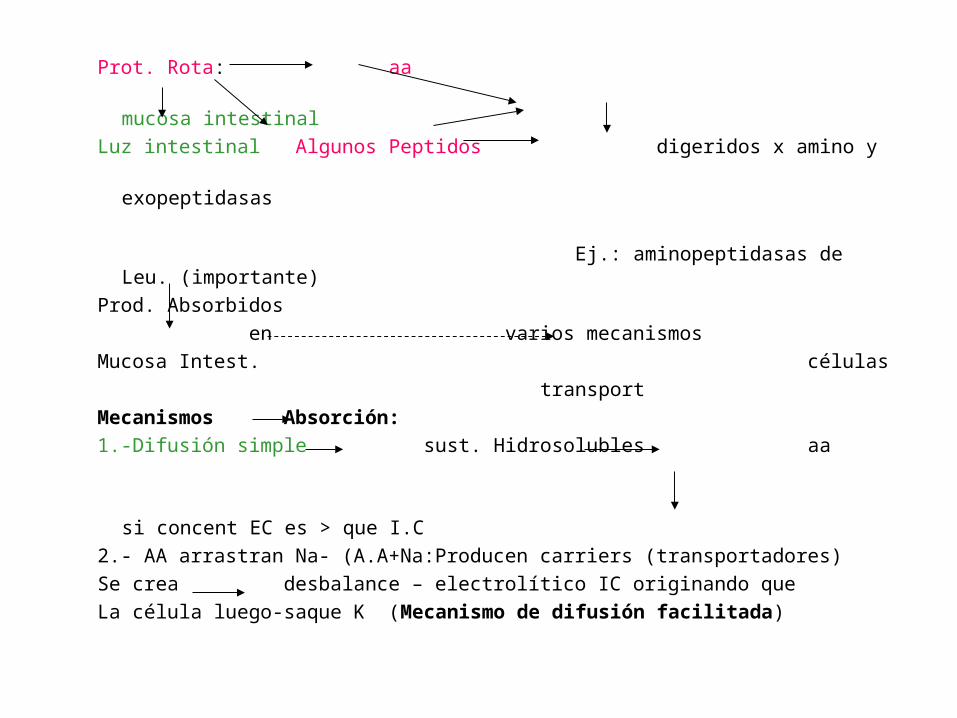
Prot. Rota: aa
mucosa intestinal
Luz intestinal Algunos Peptidos digeridos x amino y
exopeptidasas
Ej.: aminopeptidasas de Leu. (importante)
Prod. Absorbidos
en varios mecanismos
Mucosa Intest. células
transport
Mecanismos Absorción:
1.-Difusión simple sust. Hidrosolubles aa
si concent EC es > que I.C
2.- AA arrastran Na- (A.A+Na:Producen carriers (transportadores)
Se crea desbalance – electrolítico IC originando que
La célula luego-saque K (Mecanismo de difusión facilitada)

-Transporte activo: (contragradiente) de concentracion
X hidrólisis ATP (hay consumo E)
Todos Peptidos liberad Intestino
hidrolizados
aa
*Algunas Prot. Pueden absorberse sin previa digest
Ej. Ig A. (calostro) Inmunidad temprana RN
Propiedad dura 1-2 dias
Posteriormente: aa trasporta x circ. Portal hacia hígado excepto: los de cadena ramificada como Val-Leu-Isoleu.
A pesar de esto la cantidad de péptidos absorbidos es muy pequeña y al parecer muchos de ellos son de orígen endógeno en la circulación portal.

Absorcion Proteìnas• Hay muchos dipeptidos y tripeptidos que no sufren
hidrólisis enzimática, y van desde la luz hacia el interior celular más rápidamente.
• Ya que tienen un transporte electrogénico dirigido por un gradiente de pH, mantenido por intercambio Na/H, y en la membrana apical por intercambio basolateral Na/K.
• Los péptidos que así se transportan suelen ser portadores de grán cantidad de Prolina e Hidroxiprolina

Absorción Proteínas.
• Proteínas lacteas: que son una mezcla de factores de crecimiento, hormonas, Igs, métaloproteínas se absorben en intestino por permeabilización temporal.
• En calostro a más de IGA secretora hay IGG.
• Se ha demostrado en RN de bajo peso la absorción de lactoferrina.

Aminoácidos• Clasificación:1.- Por Predominio de : Ácidos o Bases
Ácidos: Ac. Glutamico Ac. Aspartico Bases: Lisina- Arginina- HistidinaNeutros: Resto de a.a.
2.- Polares y no Polares. Depende Existencia o no de grupos
Polares en la cadena lateral. R. Polares: Iónicos: con carga eléctrica (-) pueden ser ácidos o básicos.
-Ac. Aspartico- Glutamico- Lis-Arg-His

• Poco Iónicos: sin carga eléctrica apreciable pero con grupos , OH, SH, amidas o
Indol. con OH:serina- treonina-tirosina-hidroxiprolina con SH: Cisteína, Metionina. con Función amida: Aspargina- Glutamina con Anillo Indol: Triptófano.• No polares: Glicina- Alanina- Valina- Leucina
Isoleucina-Metionina- Cisteina- Fenilalanina - Prolina

3.- De acuerdo a su origen
• Esenciales: Arg- Lis- Trip- His- Met- Val- Leu- Fenil- Isoleu- Treo.
• No Esenciales: Gli,- Ala,- Ser,- Tir,- Prol,- Asparg- Ac. Aspart- Glutamina-Ac.Glutamico.
4.-Por Composición de Cadena Lateral(R)
a:Cadena Lateral Alifática: (Hidrocarburos)
Glicina(Gli), Alanina(ala), Valina(val), Leucina(leu), Isoleucina(ILeu)

B: AA Cadenas Laterales con radical-OH(Hidroxilo).
• Serina(Ser), Treonina(Tre).
• C: AA. Cadenas Laterales con Azufre.
• Cisteína(cis), Metionina(Met)
• D: AA con grupos ácidos o sus amidas en cadena lateral.
• Ac.Aspartico(Asp) Ac.Glutamico(Glu)
• Aspargina(Asn), Glutamina (Gin)

E : AA Con grupo básico en su Cadena Lateral
• Arginina(Arg), Lisina(Lis), Histidina(His).
• F: AA. con anillo aromático en su Cadena Lateral.
• Fenilalanina(Fen), Tirosina(Tir), Triptófano(Trp).
• G: AA. con anillo pirrol (Iminoácidos)
• Prolina(Pro)

Consideraciones generales
• Algunos AA, como la Gli, y otros que tienen grupos ácidos, participan en reacciones de transaminación, y cumplen un rol en la exitación y neurotransmisión
• Ac.Glutàmico: se transforma en Ac.alfacetoglu tarico por transaminación mediada por GABA , en las
terminaciónes nerviosas y en las células gliales, y por acción de la glutaminasa sobre la glutamina en las terminaciones neuronales.
Es el principal neurotransmisor en la coclea, retina, bulbo olfatorio y células piramidales del cerebro.

Consideraciones Generales
• El Oxalacetato se conviete en Ac.Aspartico por transaminación mediada por la aminotransferasa , o también el Piruvato se convierte en Ac.Aspartico por descarboxilación en las células gliales, así como en las terminaciones neuronales.
En la liberación de éstos AA, intervienen mecanismos de exocitosis dependientes del Calcio iónico, así como transporte activo para la estimulación nerviosa transmiti-
da desde Corteza cerebral hasta Médula espinal.

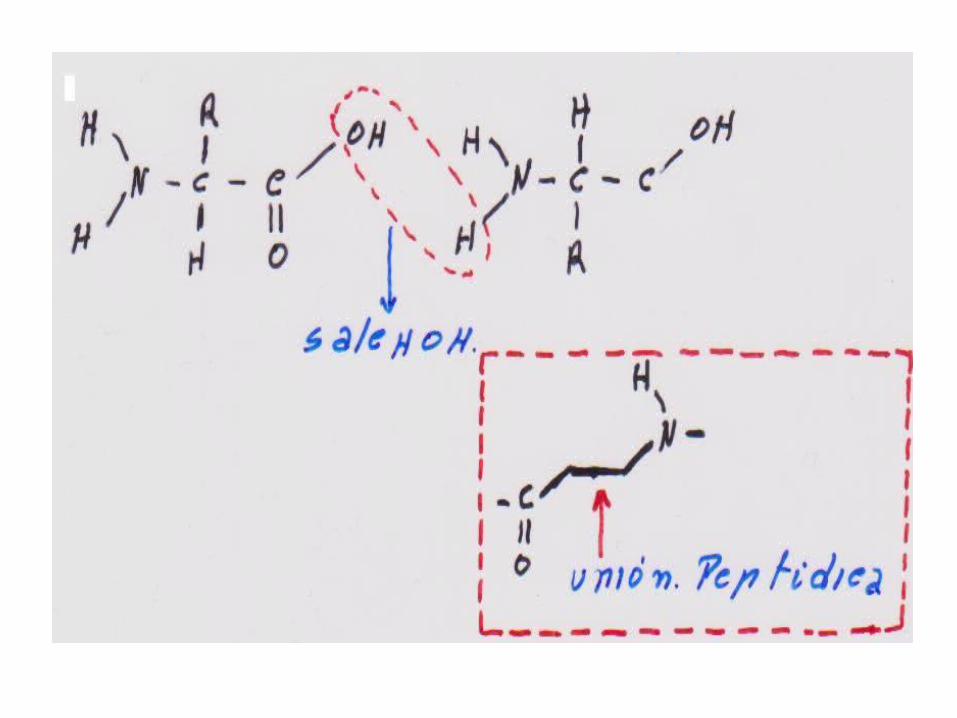
• El Enlace Peptidico:
• Reacción entre grupo amino de un a.a. y el carboxilo de otro a.a.:forman un dipéptido.
• Si son 3 Tripeptido, etc. Oligopeptido:8-9 aa.
• Siempre se elimina H2O al formarse el enlace.
• Mas de 10 a.a. Polipéptido
• Más de 100 aa es ya una Proteína.

Generalidades de los AA
• Residuo Aminoacilo: son los AA cuyo grupo COO participa en la formación del enlace peptídico.
• Usualmente el grupo NH3 en las formulas de los AA va a la izquierda y se denomina N-terminal.
• Y el grupo COO se dispone a la derecha denominàndose C terminal

METABOLISMO Grupo Amino
1. Transaminación
2. Desaminación ox. de Glutamato3. Transporte de NH3
4. Formación Urea(ciclo)

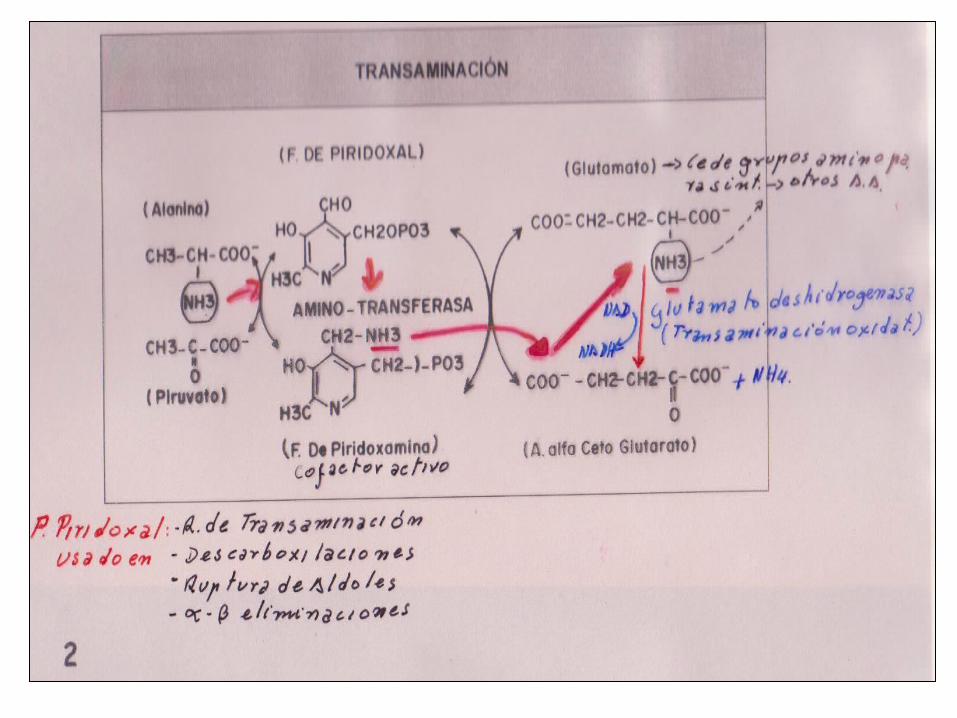
1. TRANSAMINACIÓN• Remoción e intercambio de nitrógeno• Todos los AA excepto Lis-Tir• Catalizadas por transaminasas, P de piridoxal cofactor.• Permiten inter conversión: AA cetoácido• Reacciones reversibles• Transferencia NH2 α Cetoglutarato: forma el
*Glutamato:donador comùn de grupos NH2 para los AA.• Liberación post N - como NH4 catalizadas por Glutamato deshidrogenasa usa NAD – NADP (oxidantes) Regulada por ATP – GTP – NADHGlutamato o alfacetoglutarato son sustratos clave en estas
reacciónes.

PRODUCCION DEL ION AMONIO
• La vía principal constituye la desaminación oxidativa del glutamato catalizada por la glutamato deshidrogenasa
• A más de amonio, se generan alfacetoglutarato, NAD(P)H; el NAD, y el NADP sirven como oxidantes a la enzima.
• La reacción es de oxidoreducción simple y es reversible.
• La direccion de la reaccion depende de las necesidades fisiològicas.
• El glutamato tiene un papel central en el metabolismo de los aminoácidos.

PRODUCCION DE AMONIO
• A más de la glutamato deshidrogenasa hay otras 2 enzimas que oxidan los aminoácidos a cetoácidos.
• Son la L-amino oxidasa con FMN como cofactor. • La D-aminooxidasa con FAD como cofactor.
• Se localizan en los peroxisomas y generan peróxido de hidrógeno el que es degradado por la catalasa a O2 molecular y H2O.
• Estas enzimas son cuantitativamente menos importantes en la producción del ión Amonio que la glutamato deshidrogenasa.

PRODUCCION DE AMONIO• Por reacciones de hidrólisis por acción de la glutaminasa
transforma la glutamina a glutamato y ión amonio.• Es importante en riñón donde genera la > parte de amonio
excretado por orina.• La asparginasa cataliza la hidrólisis de la aspargina y
produce aspartato y amonio.
• La asparginasa puede usarse con fines terapèuticos en trastornos causados por Escherichia Coli, y en algunas leucemias agudas que no responden a otros agentes. (es tòxica para cèlulas de higado, riñòn, y pancreas)
• Los Cs del glutamato y aspartato pueden convertise en intermediarios del C. de Krebs por transaminación.

Glutamina
*Glutamato Glutamina concent Séricas Sintetaza +que otros AA ATP ADP+Pi
• Hígado• Músculo• El principal transportador de iones NH3 es la glutamina.
• Glutamato por desaminación Ox.se convierte en CO2, NH3, o NH4 este va al ciclo de la urea .
• La glutaminsintetasa es muy activa en riñòn en casos de acidosis para producir NH4 que es excretado en forma de Cloruro de amonio.

UREA
• Metabolito nitrogenado más importante
• Ingesta 100gr proteína
• Excreta 16,5 gr. N/día 5% por heces
Urea 80-90% N excretados
Síntesis: HIGADO Otros productos finales
* Amonio - Urato
CICLO DE LA UREACooperación Mitocondria y Citoplasma. Los grupos amino de los AA generados en otros tejidosson transportados al higado principalmente como alanina y glutamina.
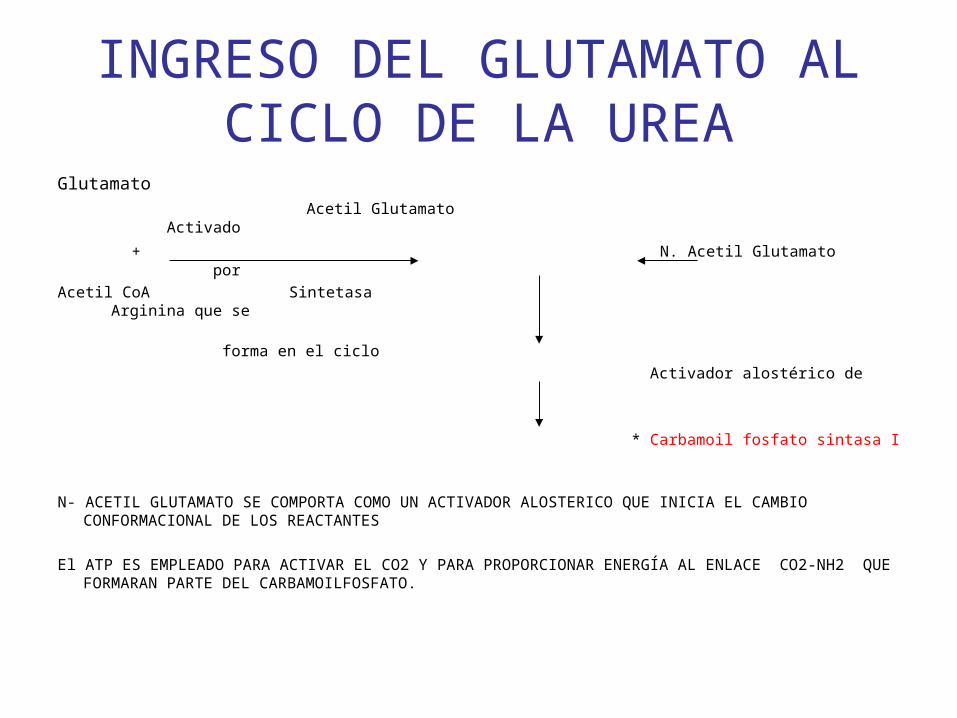
INGRESO DEL GLUTAMATO AL CICLO DE LA UREA
Glutamato
Acetil Glutamato Activado
+ N. Acetil Glutamato por
Acetil CoA Sintetasa Arginina que se
forma en el ciclo
Activador alostérico de
* Carbamoil fosfato sintasa I
N- ACETIL GLUTAMATO SE COMPORTA COMO UN ACTIVADOR ALOSTERICO QUE INICIA EL CAMBIO CONFORMACIONAL DE LOS REACTANTES
El ATP ES EMPLEADO PARA ACTIVAR EL CO2 Y PARA PROPORCIONAR ENERGÍA AL ENLACE CO2-NH2 QUE FORMARAN PARTE DEL CARBAMOILFOSFATO.

CICLO DE LA UREA
• Es Cooperación mitocondria y citoplasma • *Enzima participa primer paso del ciclo
intramitocondrialmenteCO2 proviene de combustión celular y del bicarbonato. *carbamoil fosfato+ carbamoil fosfatoNH4 sintetasa I Mg 2ATP 2ADP+Pi N – acetil Glutamato (activador) (Se usan 2 ATP, 1º es fuente de
fosfato, 2ºimpulsa formación del enlace amida CO2-NH4)
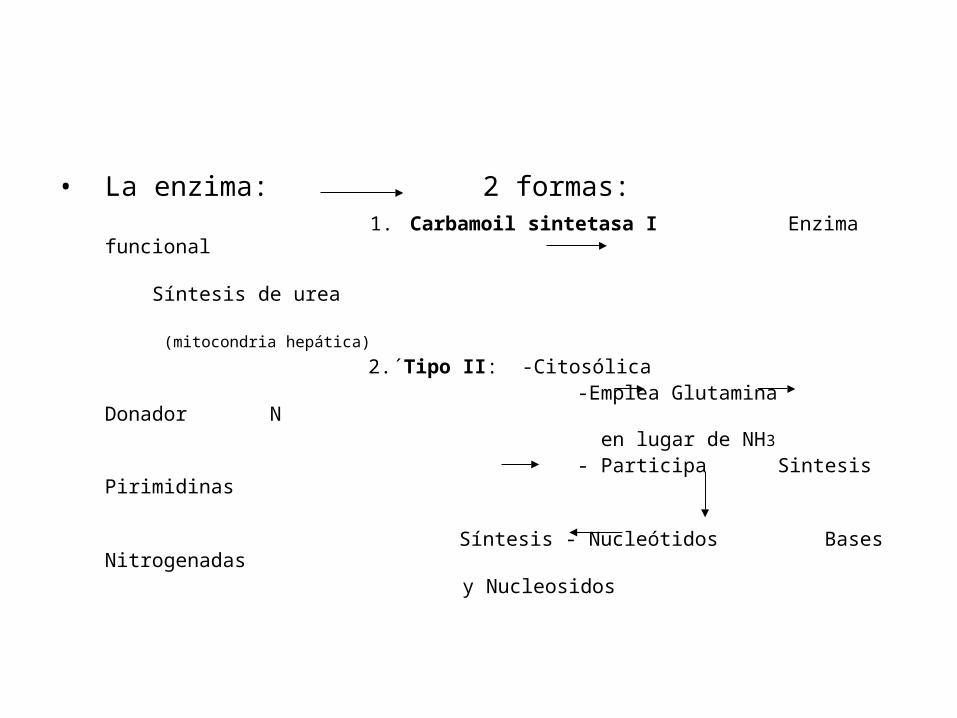
• La enzima: 2 formas: 1. Carbamoil sintetasa I Enzima funcional
Síntesis de urea (mitocondria hepática)
2.´Tipo II: -Citosólica -Emplea Glutamina Donador N en lugar de NH3
- Participa Sintesis Pirimidinas
Síntesis - Nucleótidos Bases Nitrogenadas y Nucleosidos

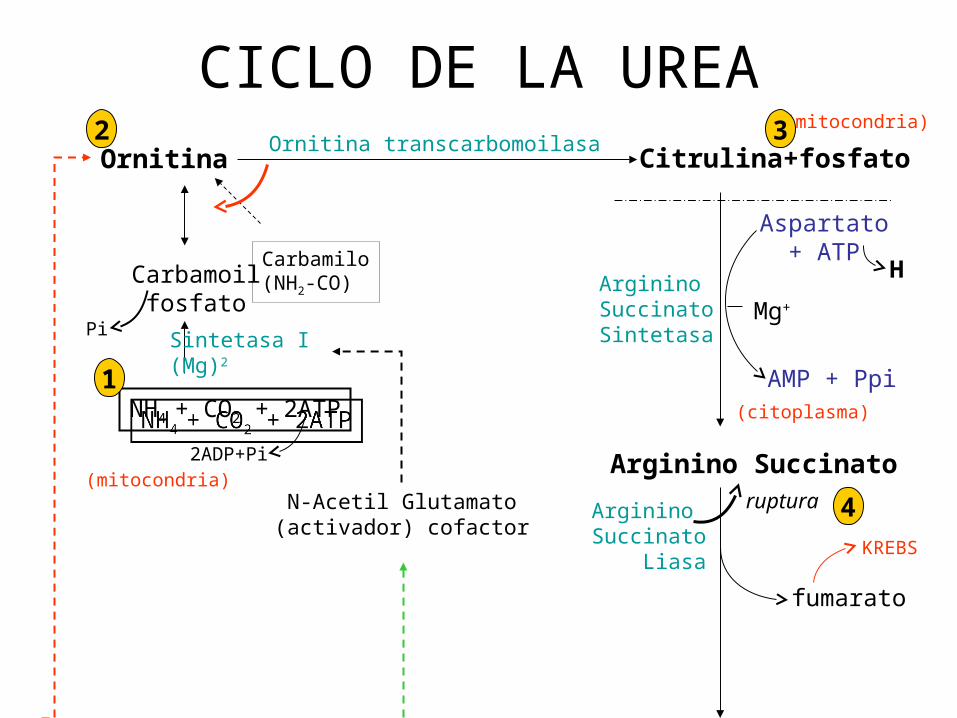
CICLO DE LA UREA
• Sustrato regenerante: Ornitina.• La secuencia se inicia con Ornitina y se añaden
componentes a la molécula progenitora para formar Arginina.
• El Carbono y el O2 de la Urea se derivan del CO2 el cual proviene del bicarbonato.
• El CO2 se combina con H2O para producir acido carbónico catalizado por la anhidrasa carbónica.
• Un grupo amino se deriva del NH4, y otro del aspartato.
• El ciclo es exergónico implica el gasto de 3 molèculas de ATP.
CICLO DE LA UREA
NH4 + CO2 + 2ATPNH4 + CO2 + 2ATP
Carbamoilfosfato
Ornitina Citrulina+fosfato
Arginino Succinato
fumarato
Ornitina transcarbomoilasa(mitocondria)
Sintetasa I (Mg)2
Carbamilo(NH2-CO)
Pi
Aspartato+ ATP
AMP + Ppi
H
Mg+
(citoplasma)
2ADP+Pi
KREBS
rupturaArginino Succinato
Liasa
Arginino SuccinatoSintetasa
(mitocondria)
1
2 3
4N-Acetil Glutamato(activador) cofactor
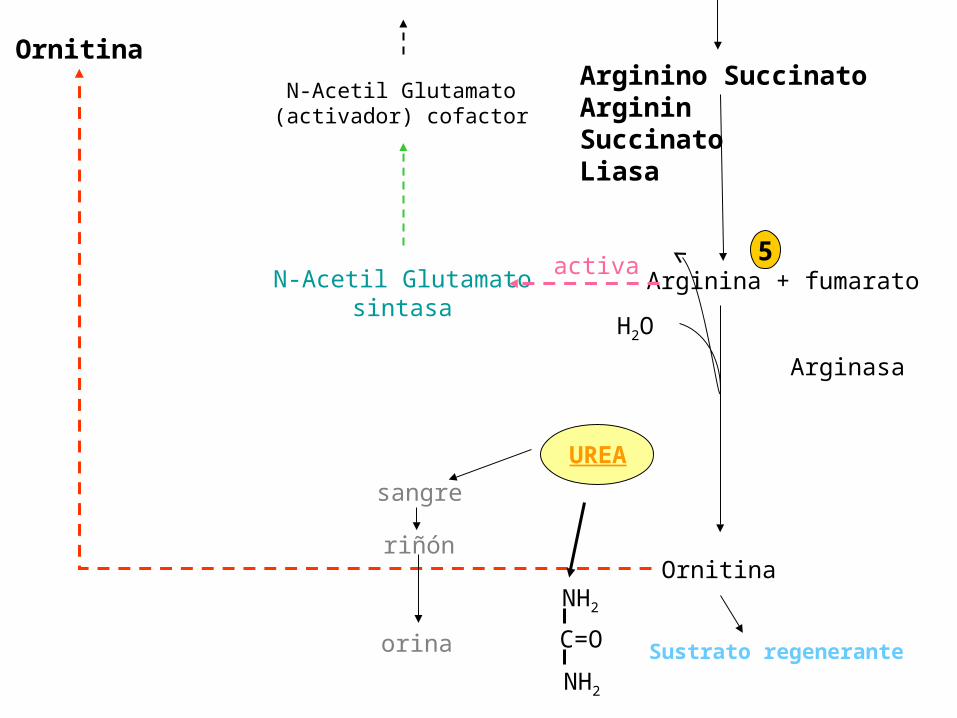
5Arginina + fumarato
Arginasa
N-Acetil Glutamatosintasa
activa
Ornitina
Arginino SuccinatoArgininSuccinatoLiasa
N-Acetil Glutamato(activador) cofactor
Sustrato regenerante
H2O
UREA
sangre
riñón
orina
NH2
C=O
NH2
Ornitina
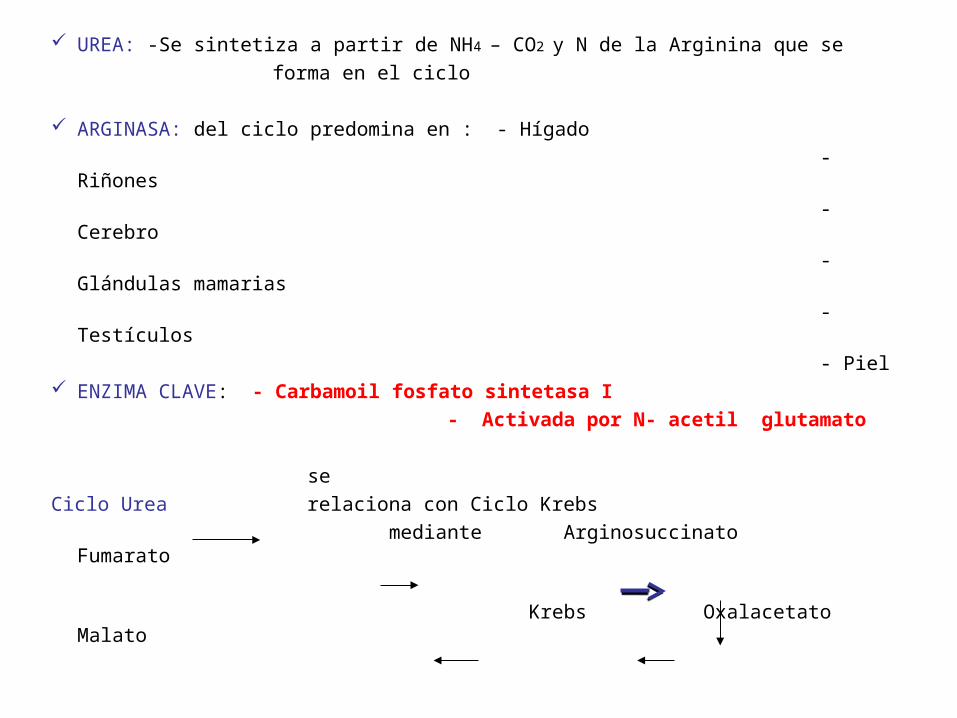
UREA: -Se sintetiza a partir de NH4 – CO2 y N de la Arginina que se
forma en el ciclo
ARGINASA: del ciclo predomina en : - Hígado
- Riñones
- Cerebro
- Glándulas mamarias
- Testículos
- Piel ENZIMA CLAVE: - Carbamoil fosfato sintetasa I
- Activada por N- acetil glutamato
se
Ciclo Urea relaciona con Ciclo Krebs
mediante Arginosuccinato Fumarato
Krebs Oxalacetato Malato
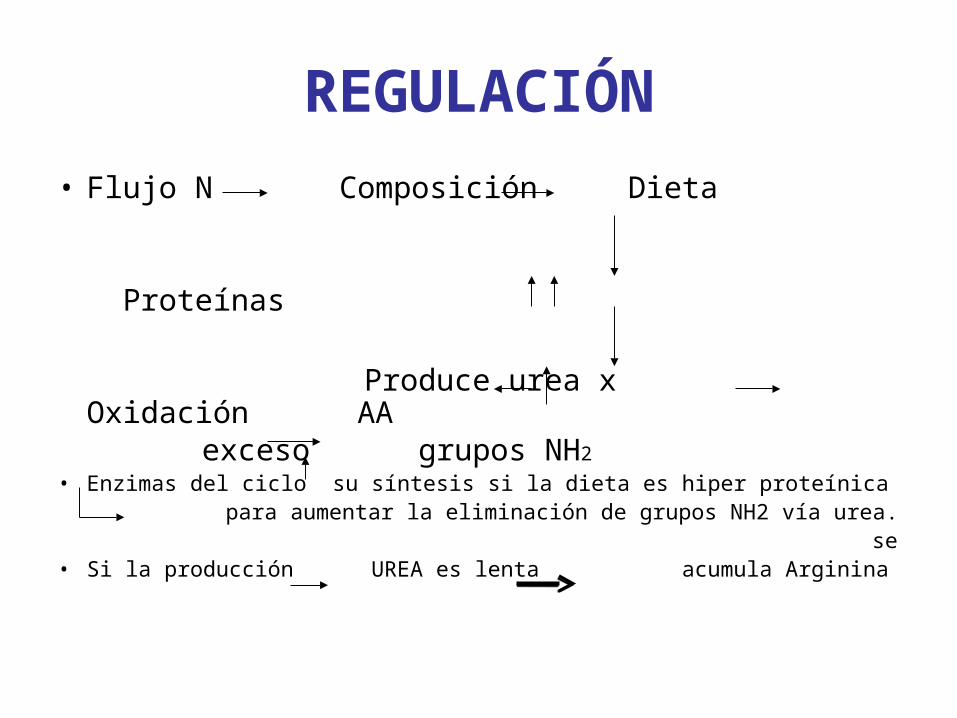
REGULACIÓN
• Flujo N Composición Dieta Proteínas Produce urea x Oxidación AA exceso grupos NH2
• Enzimas del ciclo su síntesis si la dieta es hiper proteínica para aumentar la eliminación de grupos NH2 vía urea. se• Si la producción UREA es lenta acumula Arginina
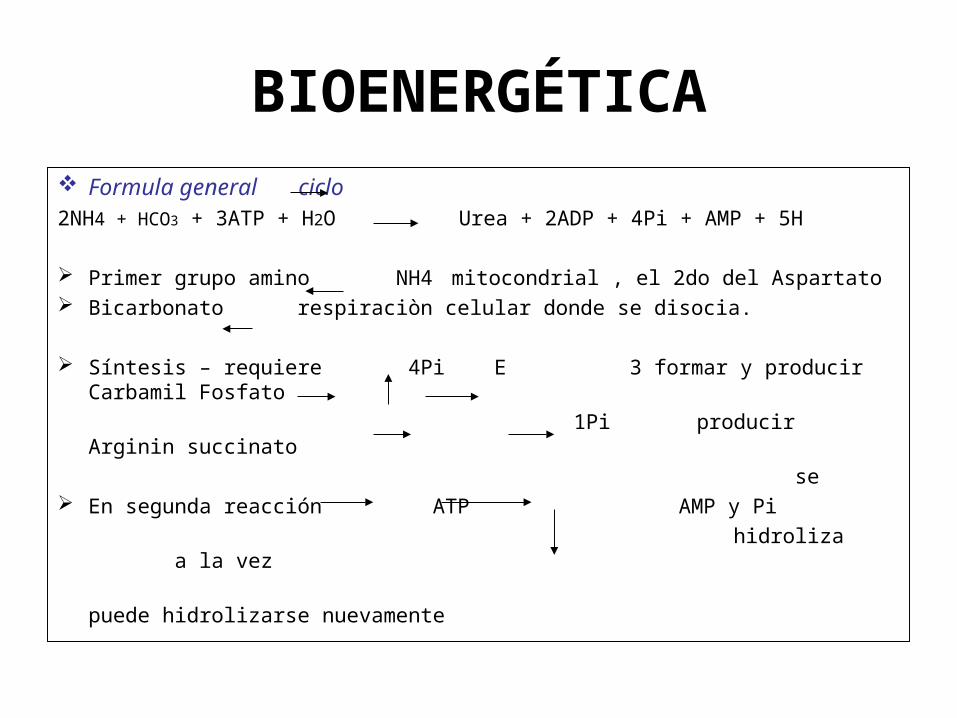
BIOENERGÉTICA
Formula general ciclo2NH4 + HCO3 + 3ATP + H2O Urea + 2ADP + 4Pi + AMP + 5H
Primer grupo amino NH4 mitocondrial , el 2do del Aspartato Bicarbonato respiraciòn celular donde se disocia.
Síntesis – requiere 4Pi E 3 formar y producir Carbamil Fosfato
1Pi producir Arginin succinato
se En segunda reacción ATP AMP y Pi
hidroliza a la vez
puede hidrolizarse nuevamente

Org. → Ureotélicos → pierden + - 15% de Energía ↓
de los AA → que forman urea.
Urea → representa → 80-90% → N → excretado por orina
El porcentaje restante está ocupado por iones NH4 y uratos La urea se filtra en glomérulo, y el 40% se reabsorbe en túbulos Sus valores de depuración son iguales a los índices de filtración. Por eso el N ureico urinario es considerado como prueba de filtración
glomerular, cuando la filtración disminuye la urea en sangre aumenta.

TRASTORNOS DEL CICLO
• Enfermedades renales se asocian con elevación del Nitrógeno Ureico en sangre.(NUS)
• La Urea es poco tóxica, la elevación del NUS sugiere enfermedad renal acompañada de desequilibrio hidroelectrolítico y ácido base.
• En enfermedades hepáticas graves aumenta la concentración de NH4 en sangre.
• La toxicidad del NH4 conduce a encefalopatía hepática que se acompaña de confusión, estupor convulsiones, coma y aún muerte.

Alteración → Ciclo → Urea
↓ Se presenta → uno de cada 30000 Recién Nacidos
↓ Se afecta → cualquiera de 5 enzimas → del ciclo
Transtorno ligado → cromosomas → X
Causa: Mutación → Brazo corto
↓
Cromosoma 2
↓regula
Síntesis y actividad
↓
Carbamil fosfato sintetasa I

SÍNTOMAS Y SIGNOS
Inicio → asintomático Luego: - Anorexia
- Vómito
- Letargia
- Irritabilidad
Retardo mental
- Convulsiones → Coma
- Hiperamonemia (no se forma urea)
- Muerte por hemorragia cerebral o pulmonar
Roskosky, pag233, Tabla 13-1

ENCEFALOPATÍA POR ↑ AMONIO
Mecanismos de detoxificación: - C→ Urea
- Creatinina
- Transporte por glutamina
- Ciclo de Krebs
Amonio Tóxico (no metabolizado) → Encefalopatía ↓produce
Depresión → Flujo cerebral Estados exitatorios
↓
Depresión → Metabolismo → Glucosa Convulsiones

AA. GLUCOGENICOS
• Son convertidos a glucosa.• Catabolizados a piruvato o intermediarios del Krebs.• Gli, Ala, Ser, cisteina• Aspartato, Aspargina.• Glutamato, glutamina.• Pro, His, Arg.• Metionina.• Treonina.• Valina.

AA.Gluco y Cetogènicos
• Pueden ser convertidos a glucosa y cuerpos cetònicos.• Isoleucina,Lis + AA aromàticos como: Fen, Tir, Tript.
• AA. Cetogènicos.Pueden ser convertidos a cuerpos cetònicos.
Son catabolizados a Acetil CoA, Acetoacetil CoA, o ambos, desviandose del piruvato o de los intermediarios del ciclo de Krebs.
Leucina.

Aminoácidos Esenciales y Balance Nitrogenado
• Si un AA esencial existe en cantidades inadecuadas la síntesis de proteínas disminuye en forma correspondiente.
• Esto conduce a un balance nitrogenado negativo (cuando la excreción de N excede a la ingesta).También en infecciones, quemaduras, estrés post quirurgico.
• Se relaciona también con secreción aumentada de glucocorticoides por corteza suprarrenal.
• En adultos existe un balance nitrogenado en equilibrio.• En niños en crecimiento o en embarazo existe un
Balance + donde la ingesta excede a la excreta

AA. Esenciales
• 8 de los 20 AA son esenciales, y deben proporcionarse con la dieta.
• FVT—TIM—HALL.• Fenilalanina, Valina, Treonina-• Triptofano, Isoleusina, Metionina-• Histidina, Arginina, Leucina, Lisina.
• H,A: AA adicionales requeridos por lactantes y niños.

Síntesis de AA no esenciales
• Su síntesis es simple no así la de los esenciales• De 12 AA no esenciales, 9 se forman a partir de AA
anfibólicos.• Cis,Tir, His,se forman a partir de AA esenciales.• Síntesis de proteínas citoplasmáticas se da en
ribosomas libres. • Sìntesis de proteínas de membrana se hace en retículo
endoplásmico rugoso.• Tres enzimas claves en la síntesis: glutamato
deshidrogenasa, glutamina sintetasa, y transaminasas.
(Tabla 13-3, Pag 235 Roskoski, familias de AA)

1.- Glutamato(C5)
COO - COO-
H2N-C-H glutamato deshidrogenasa O = C H-C-H+ NADP NADPH H-C-H+ NH4
H-C-H R de transaminacion H-C-H
COO - R- Exergónica COO-
Glutamato a ceto glutarato
Oxidación +NH4
Alfa cetoglutarato se convierte en malato y oxalacetato x el ciclo de Krebs lo que explica la naturaleza glucogénica del Glutamato.
Tambièn pueden convertirse en piruvato y acetil CoA, antes de su oxidacion completa por el ciclo de Krebs.
BIOSINTESIS y DEGRADACION.
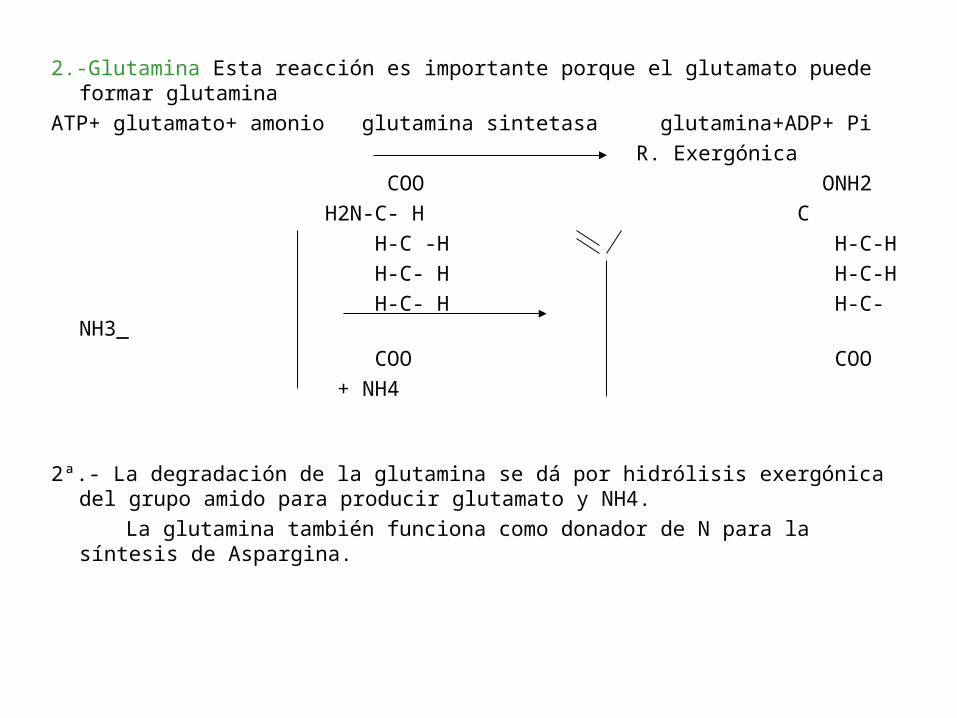
2.-Glutamina Esta reacción es importante porque el glutamato puede formar glutamina
ATP+ glutamato+ amonio glutamina sintetasa glutamina+ADP+ Pi
R. Exergónica
COO ONH2
H2N-C- H C
H-C -H H-C-H
H-C- H H-C-H
H-C- H H-C-NH3_
COO COO
+ NH4
2ª.- La degradación de la glutamina se dá por hidrólisis exergónica del grupo amido para producir glutamato y NH4.
La glutamina también funciona como donador de N para la síntesis de Aspargina.

O OO O O O - P-O C C O H-C-H asparagina sintetasa H-C-H H-C-NH3 ATP Mg+ ADP H-C-NH3 COO COO Asparto (C4) B Aspargil adenilato +NH2 H2O aspargina sintetasa
ONH2 C H-C-H H-C-NH3 OXALACETATO GLUTAMATO COO ASPARGINA(C4)
Su degradación implica la producción de Aspartato y amonio por hidrólisis exergónica.Este por hidròlisis produce oxalacetato y glutam.

Alanina(C2) y Serina(C3)
• Ala: por transaminación isoergónica y en una reacciòn bidireccional con alfa cetoglutarato forma piruvato y glutamato.
• Ser: sufre deshidratación + hidrólisis catalizada por la deshidratasa de Ser, con fosfato de piridoxal como cofactor, forma piruvato.
• Tambièn por transaminación produce 3-hidroxipiruvato seguida por reducciòn NADH dependiente a D-glicerato , seguidas por fosforilación ATP dependiente catalizada por la gliceratocinasa produciendo 3-fosfoglicerato.
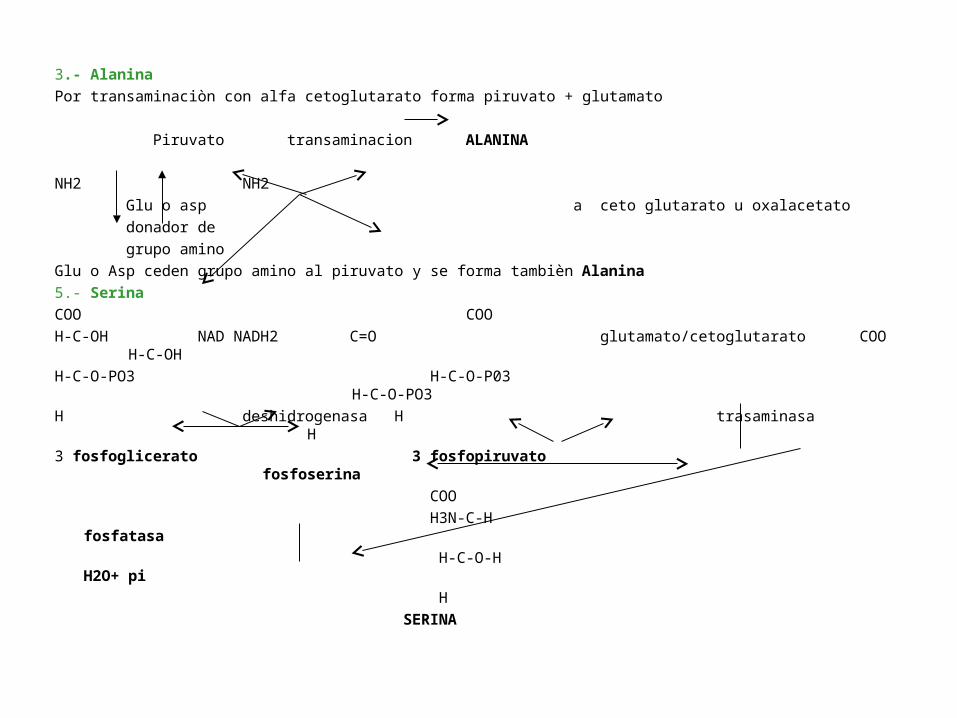
3.- Alanina
Por transaminaciòn con alfa cetoglutarato forma piruvato + glutamato
Piruvato transaminacion ALANINA
NH2 NH2
Glu o asp a ceto glutarato u oxalacetato
donador de
grupo amino
Glu o Asp ceden grupo amino al piruvato y se forma tambièn Alanina
5.- Serina
COO COO
H-C-OH NAD NADH2 C=O glutamato/cetoglutarato COO H-C-OH
H-C-O-PO3 H-C-O-P03 H-C-O-PO3
H deshidrogenasa H trasaminasa H
3 fosfoglicerato 3 fosfopiruvato fosfoserina
COO
H3N-C-H fosfatasa
H-C-O-H H2O+ pi
H
SERINA

Glicina
• Su metabolismo se asocia al tetrahidrofolato.
• Este AA reacciona con metilentetrahidrofolato para producir Ser.
• La Ser puede luego convertirse en piruvato o fosfoglicerato y estos luego en glucosa.
• Su síntesis puede provenir del glutamato, Ala, y del glioxilato.
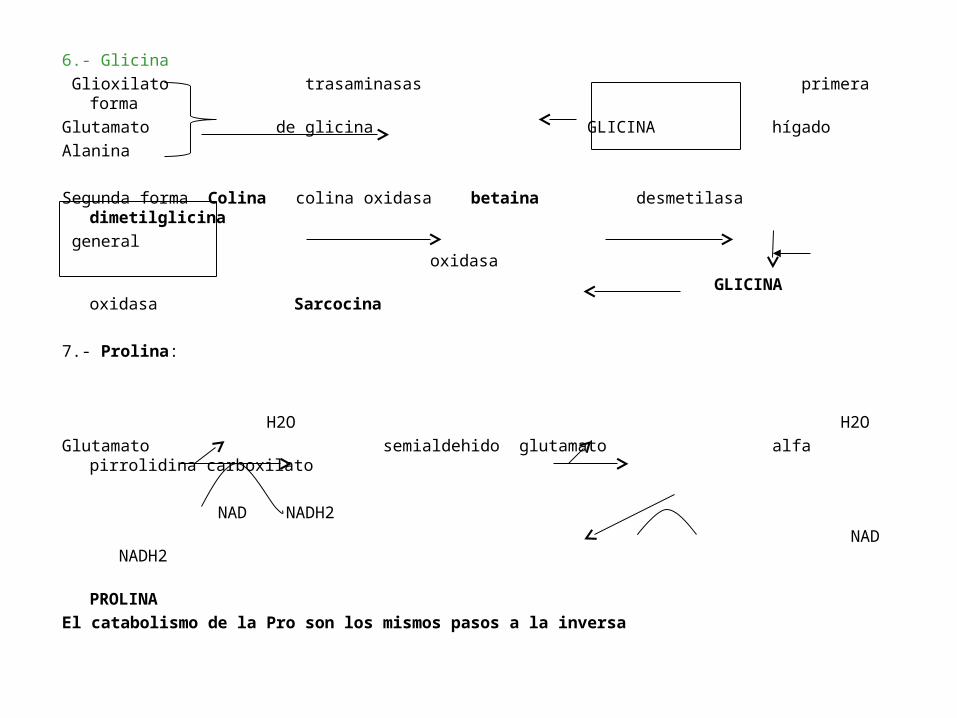
6.- Glicina
Glioxilato trasaminasas primera forma
Glutamato de glicina GLICINA hígado
Alanina
Segunda forma Colina colina oxidasa betaina desmetilasa dimetilglicina
general oxidasa
GLICINA oxidasa Sarcocina
7.- Prolina:
H2O H2O
Glutamato semialdehido glutamato alfa pirrolidina carboxilato
NAD NADH2
NAD NADH2
PROLINA
El catabolismo de la Pro son los mismos pasos a la inversa

Prolina e Hidroxiprolina
• Hidroxiprolina,es su forma hidroxilada,= que hidroxilisina son AA más abundantes en fibras colagenas .
• Así se evita la digestión de la proteína por proteasas del Tejido Conectivo.
• La hidroxilacion es activa en piel, pulmones, corazón, músculos y tejido conectivo articular.

prolil - hi
. - Hidroxiprolina: Prolina
droxililasa
-Prolil- Hidroxilasa: + -Piel pulmones
-hígado- Corazón-Músculo,tej.conectivo articular
*Formación Colágeno Tej Conjuntivo
1/3 Hidroxilisina Estabilizan- Triple
Colágeno
1/3 Hidroxi Prolina hélice Colágeno
_AA dietéticos no se incorporan Colágeno
Excepto Pro precursor hidroprolina Colágeno.

Cisteina• Puede formarse a partir de la Met, y Ser.• Su catabolismo es complejo debido a las distintas vías en
las que entra por el metabolismo del Azufre.• Cisteina y Cistina son la fuente dietética + importante de
Azufre.• Cisteina no es escencial.• La Met AA. esencial, tambièn contiene azufre pero en muy
poca cantidad y el contenido de Met en las proteínas es escaso, en su catabolismo el Azufre de la Met, se convierte al de la Cisteina.
• El producto principal de la excreción del Azufre es el Sulfato.• Por último X transaminación la Cisteina se convierte en
piruvato.
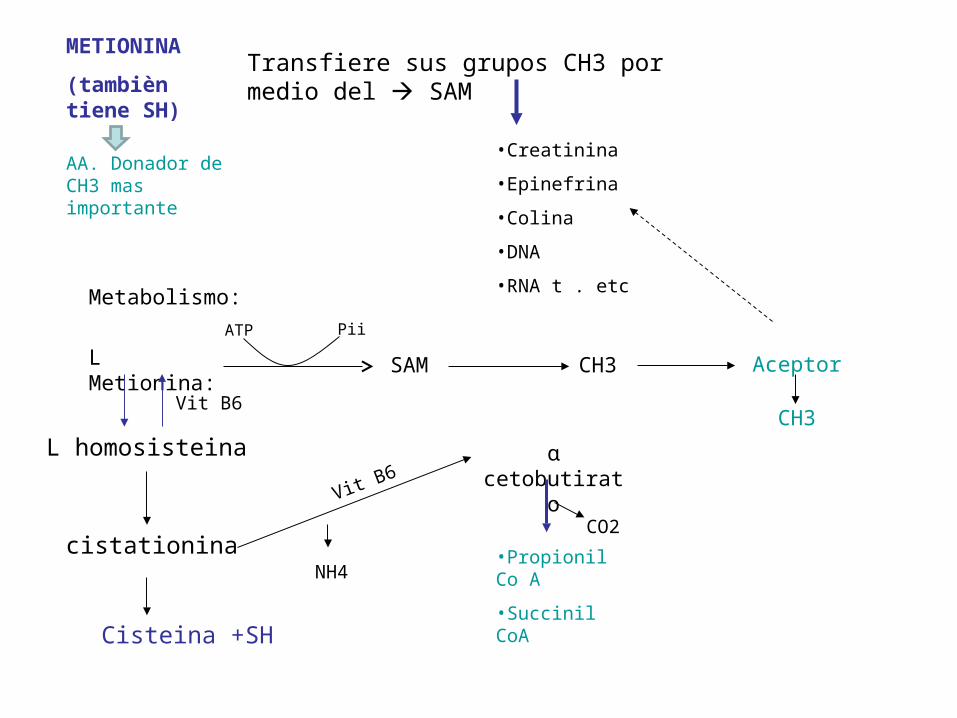
METIONINA
(tambièn tiene SH)
Transfiere sus grupos CH3 por medio del SAM
•Creatinina
•Epinefrina
•Colina
•DNA
•RNA t . etc
AA. Donador de CH3 mas importante
Metabolismo:
L Metionina: SAM CH3 Aceptor
L homosisteina
cistationina
ɑ cetobutirato
•Propionil Co A
•Succinil CoA
Cisteina +SH
CH3
ATP Pii
NH4
Vit B6
Vit B6
CO2
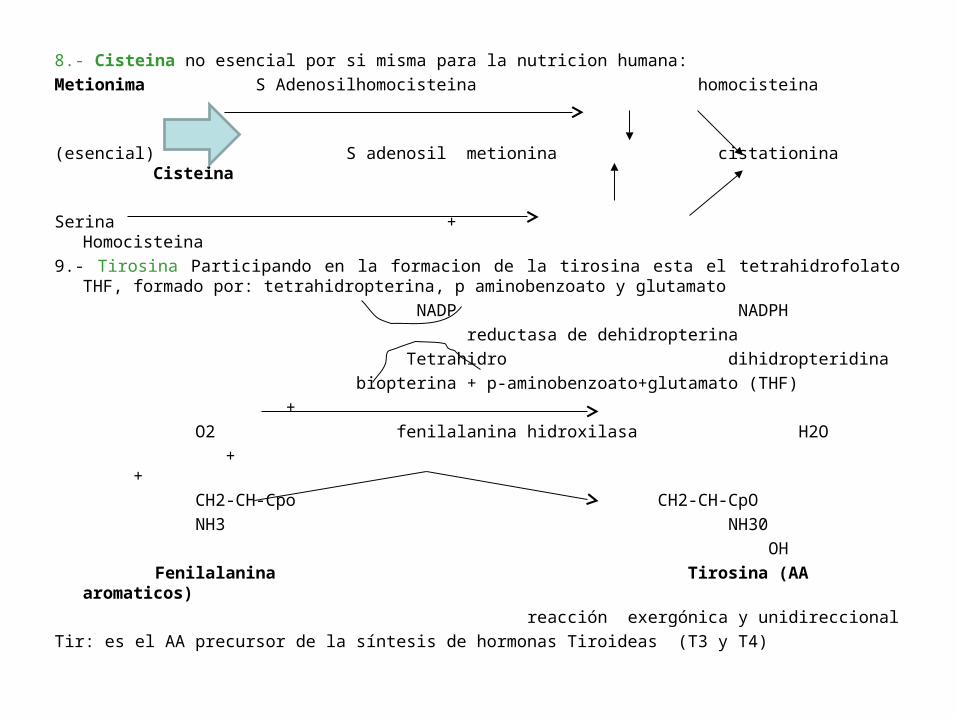
8.- Cisteina no esencial por si misma para la nutricion humana:
Metionima S Adenosilhomocisteina homocisteina
(esencial) S adenosil metionina cistationina Cisteina
Serina + Homocisteina
9.- Tirosina Participando en la formacion de la tirosina esta el tetrahidrofolato THF, formado por: tetrahidropterina, p aminobenzoato y glutamato
NADP NADPH
reductasa de dehidropterina
Tetrahidro dihidropteridina
biopterina + p-aminobenzoato+glutamato (THF)
+
O2 fenilalanina hidroxilasa H2O
+ +
CH2-CH-Cpo CH2-CH-CpO
NH3 NH30
OH
Fenilalanina Tirosina (AA aromaticos)
reacción exergónica y unidireccional
Tir: es el AA precursor de la síntesis de hormonas Tiroideas (T3 y T4)

Tirosina y H. Tiroideas• 115 res de tirosina conforman la proteína
tiroglobulina.
• El yodo se halla en un 70% en los precursores inactivos de las h. Tiroideas MIT, DIT también incluidos en la tiroglobulina, que es una proteina glucosilada en la que los CHO representan el 8 al 10% del peso de la proteína.
• El 30% restante del Yodo se halla en los residuos yodotironilo T3 y T4.

Síntesis de H. Tiroideas
• Tir por acción de una peroxidasa forma MIT + MIT = DIT .
• MIT + DIT = T3 (Triyodotirosina)
• DIT + DIT = T4 (tetrayodotirosina)

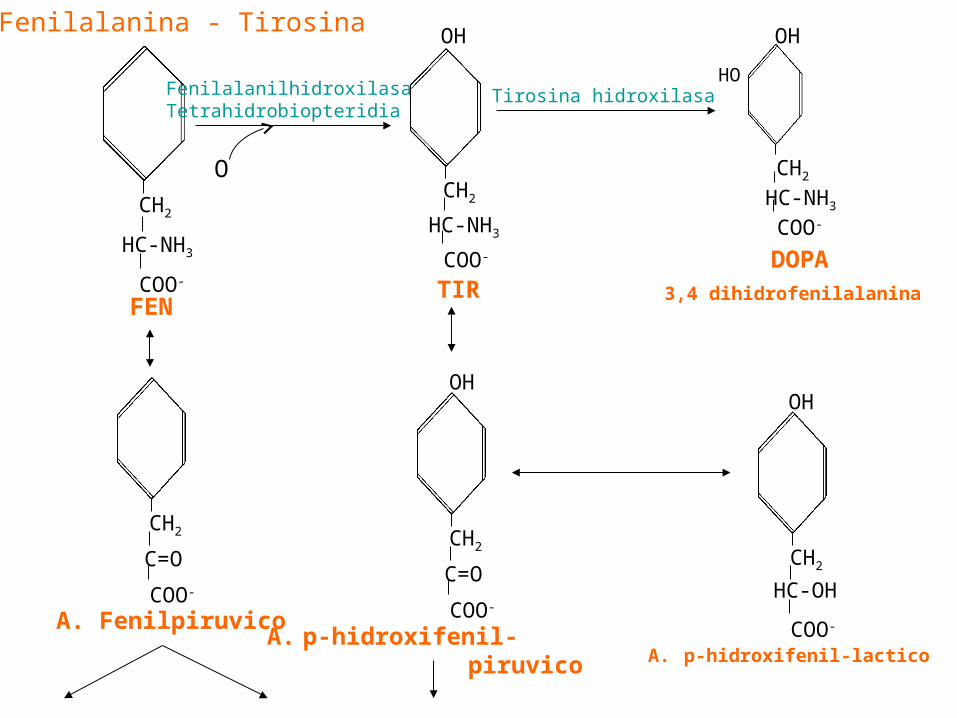
FENILALANINA
• Su metabolismo se inicia por su oxidación a Tir.• Es un AA esencial, la Tir no lo es.• Sin embargo la Tir dietètica disminuye los
requerimientos de Fen. • A este fenómeno se lo llama no esencial o de
ahorro.• La enzima que cataliza la oxidación es la
fenilalanina hidroxilasa hepática, en la reacción participan el O2, la tetrahidropterina los productos son la Tir, H2O, y dihidropterina.

-Deficit fenilalanima hidroxilasa (Ej de error congénito del Metabolismo)
fenilalanina Fenilcetonuria (PKU)
Carácter autosómico recesivo x alteración del
Brazo largo cromosoma 12q (MUTACIÓN GENÉTICA)
Sintomas: Neurologicos- hiperactividad reflejos
osteotendinosos profundos
retardo mental, los niños son normales al nacimiento.
Laboratorio: fenililalanina > a 1.2 mMol
Tirosina-normal
Fenil piruvato orina
Tratamiento: dieta sintética carente de fenilalanina.
-Fenilalanina: AA esencial
Tir no si dieta contiene suficiente Fen
- Reacciones irreversibles- Tir no remplaza
requerimientos Fen
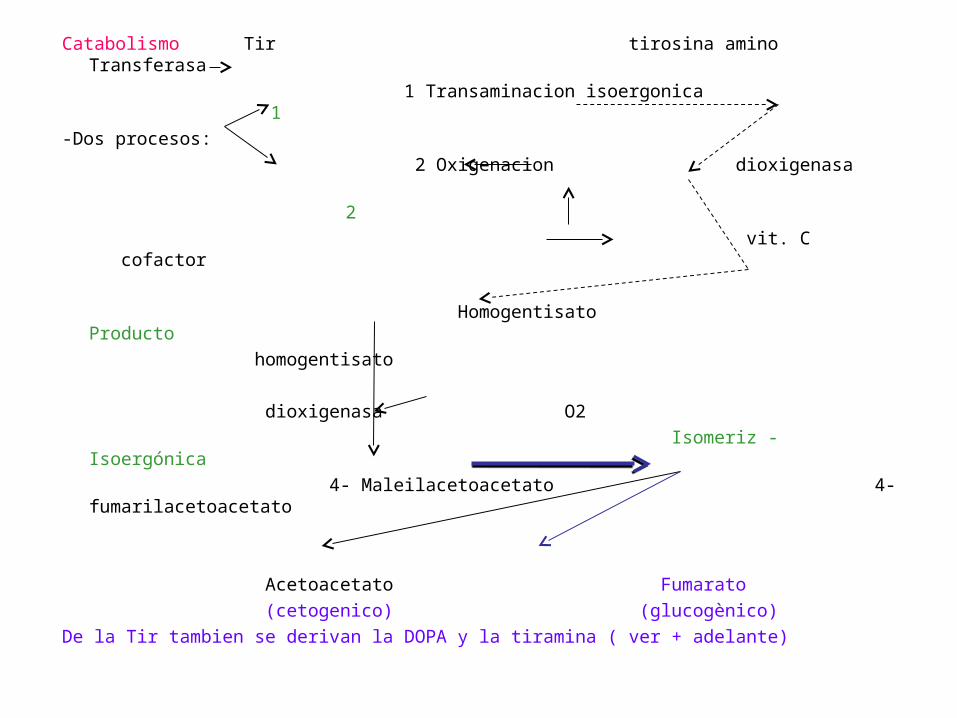
Catabolismo Tir tirosina amino Transferasa
1 Transaminacion isoergonica 1
-Dos procesos:
2 Oxigenacion dioxigenasa
2
vit. C cofactor
Homogentisato Producto
homogentisato
dioxigenasa O2
Isomeriz - Isoergónica
4- Maleilacetoacetato 4-fumarilacetoacetato
Acetoacetato Fumarato
(cetogenico) (glucogènico)
De la Tir tambien se derivan la DOPA y la tiramina ( ver + adelante)

-Deficit + Homogentisato dioxigenasa
Alcaptonuria (Error congenito Metabolismo)
-Homogentisato no se metaboliza
Excretado orina se hace obscura O2 ambiental. (luz)
+ ràpido si pH es alcalino
-pigmentación Tej conectivo (ocronosis)
-En largo tiempo Artritis
Triada clàsica de Alcaptonuria: orina oscura, ocronosis y artritis.

ERRORES CONGÉNITOS DEL METABOLISMO
Proteina
Transtornos → Alteración o
Enzima
Interrumpe → Ciclo Metabólico
↓
Acumulación → Productos previos al sitio bloqueado
Causas: Mutaciones → 1 gen (autosómica receciva)
Frecuencia: 1 en 800 a 5000 Recién Nacidos
Se afecta más frecuentemente SNC → 34%

ALTERACIONES CONGÉNITAS MÁS FRECUENTES:
Aminoácidos Ácidos orgánicosMetabolismo → Amonio Metabolismo Hidratos de CarbonoGluconeogénesisMetabolismo → Vitaminas y Minerales
Por almacenamiento y Peroxisomales
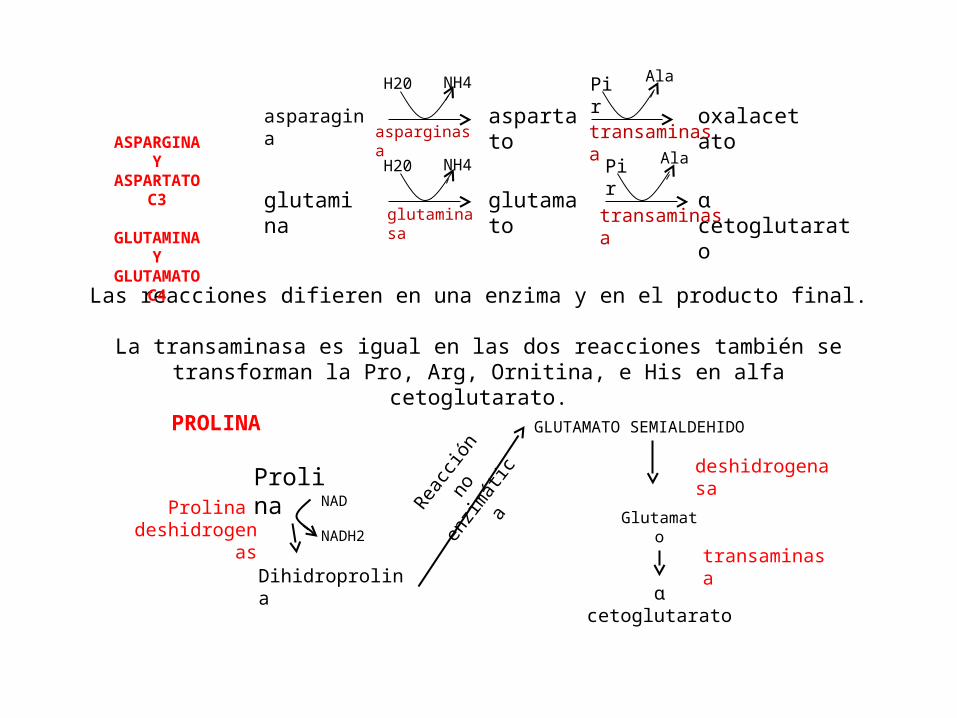
Las reacciones difieren en una enzima y en el producto final. La transaminasa es igual en las dos reacciones también se transforman
la Pro, Arg, Ornitina, e His en alfa cetoglutarato.
PROLINA
ProlinaProlina
deshidrogenas
Dihidroprolina
NAD
NADH2
GLUTAMATO SEMIALDEHIDO
Glutamato
ɑ cetoglutarato
deshidrogenasa
transaminasa
Reac
ción n
o
enzim
ática
ASPARGINA Y ASPARTATO
C3
GLUTAMINA Y GLUTAMATO
C4
asparagina
glutamina
aspartato
glutamato
oxalacetato
ɑ cetoglutarato
H20 NH4
H20 NH4
Pir Ala
Pir Ala
transaminasa
transaminasa
asparginasa
glutaminasa

Arginina-ornitina
Arg ɑ cetoglutarato OrnitinaTRANSAMINASA
Guanidino arginasa
Glutamato y semialdehido
ORNITINA TRANSAMINASA
ɑ cetoglutarato
transaminasa
Hiperornitonemia: DEFICIT DE ORNITINA TRANSAMINASA-Hereditario: atrofia retiniana en espiral
perdida progresiva de visión periférica
visión en túnel ceguera VISION EN TUNEL
Sd: Hiperornitonemia-Hiperamonemiadefecto del transporte de ornitina al interior de la mitocondria NH4 -> alteración por defecto en su metabolismo
esquema
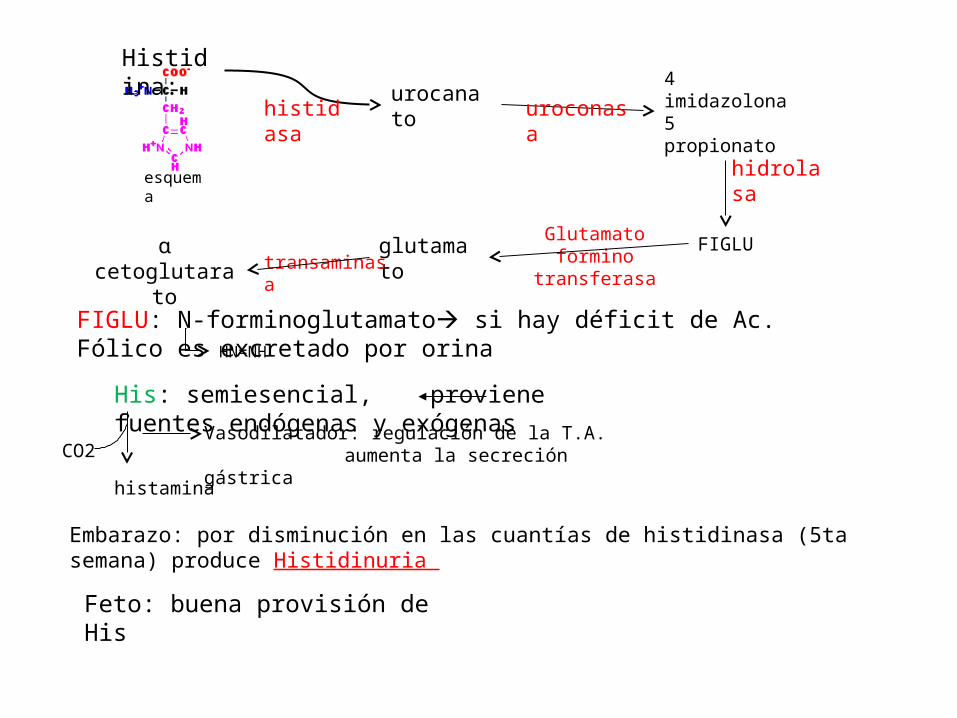
Histidina:urocanato
4 imidazolona 5 propionato
ɑ cetoglutarato glutamato FIGLUtransaminasa
histidasa uroconasa
Glutamato formino transferasa
hidrolasa
FIGLU: N-forminoglutamato si hay déficit de Ac. Fólico es excretado por orinaHN=NH
His: semiesencial, proviene fuentes endógenas y exógenas
histamina
CO2Vasodilatador: regulación de la T.A.
aumenta la secreción gástrica
Embarazo: por disminución en las cuantías de histidinasa (5ta semana) produce Histidinuria
Feto: buena provisión de His

En alergias la histamina es liberada en exceso
Ac. ɞimidazolacético orina
histaminasa
Histidinemia: defecto de Histidasa
Aciduria urocánica: déficit parcial de urocanasa (recesivo)

AA que forman Piruvato
• Gli, Ala,Cisteina (proviene de la Cistina por reducción), Ser (proviene de la Gli por transferencia de NH2): usan todos sus C para formar Piruvato.
• La Cisteina forma piruvato mediante oxidación directa o por transaminación.
• Cistina: 2 residuos de cisteina unidos por un enlace disulfuro, contiene 2 grupos NH2 que permiten el reconocimiento y transporte por translocasas
• Cistinuria: excreción urinaria de cistina hasta 30 veces lo normal, puede formar cálculos (litiasis) causada por defecto en el transporte de AA en intestino delgado y riñones.
• Cistinosis: por almacenamiento de cistina, se forman cristales en retículo endotelial, y riñón, puede causar muerte.
• Treonina usa solo 2 C para formar piruvato.

AA que forman Acetil Co A.
• Ala, Cis, Gli, Ser, Hidpro, Treo: por acción de la piruvato deshidrogenasa.
• Fen, Tir, Leu: sin previa formación de piruvato se convierten en Acetil CoA, y acetato.
• Con relación a los AA aromáticos se puede indicar lo siguiente:

TRIPTOFANO
• Es convertido a alanina (glucogénico) y a glutaril CoA (cetogénico).
• Menos del 2% se convierte en nicotinato que se utiliza en la formación de niacina.
• La cantidad de triptofano metabolizada diariamente es pequeña, y la energía derivada de su metabolismo es poca, contribuye con menos de 2 Kcal/d, por 500mg metabolizados.
• Del triptofano también se derivan la Serotonina, y la Melatonina.
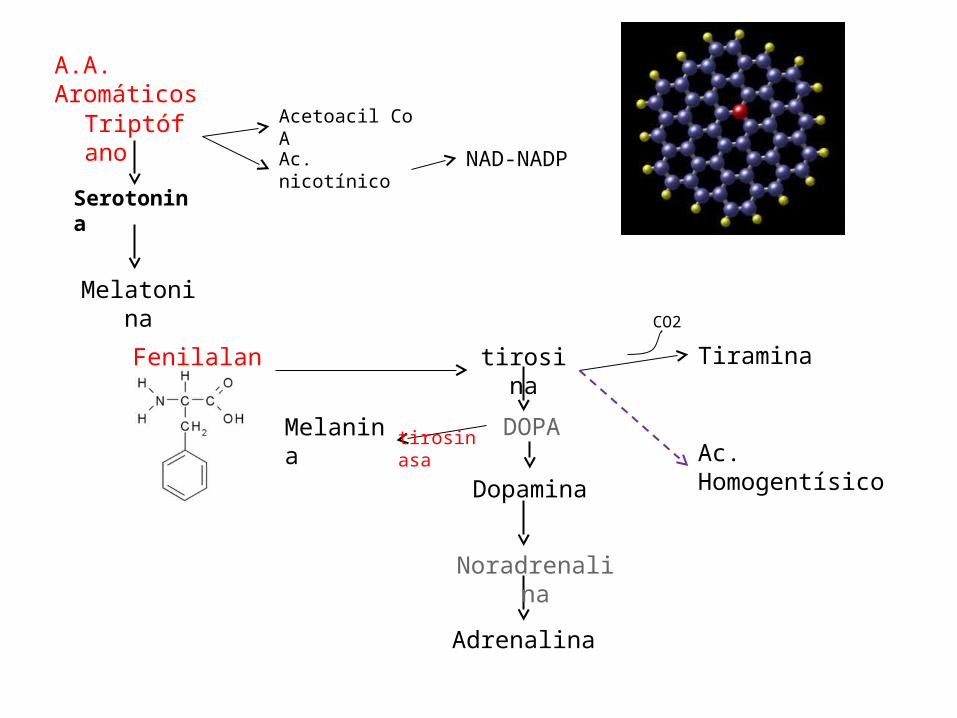
A.A. Aromáticos
Triptófano
Serotonina
Melatonina
Acetoacil Co A
Ac. nicotínico NAD-NADP
Fenilalanina
Melanina
tirosina
DOPA
Dopamina
Noradrenalina
Adrenalina
Tiramina
Ac. Homogentísico
CO2
tirosinasa
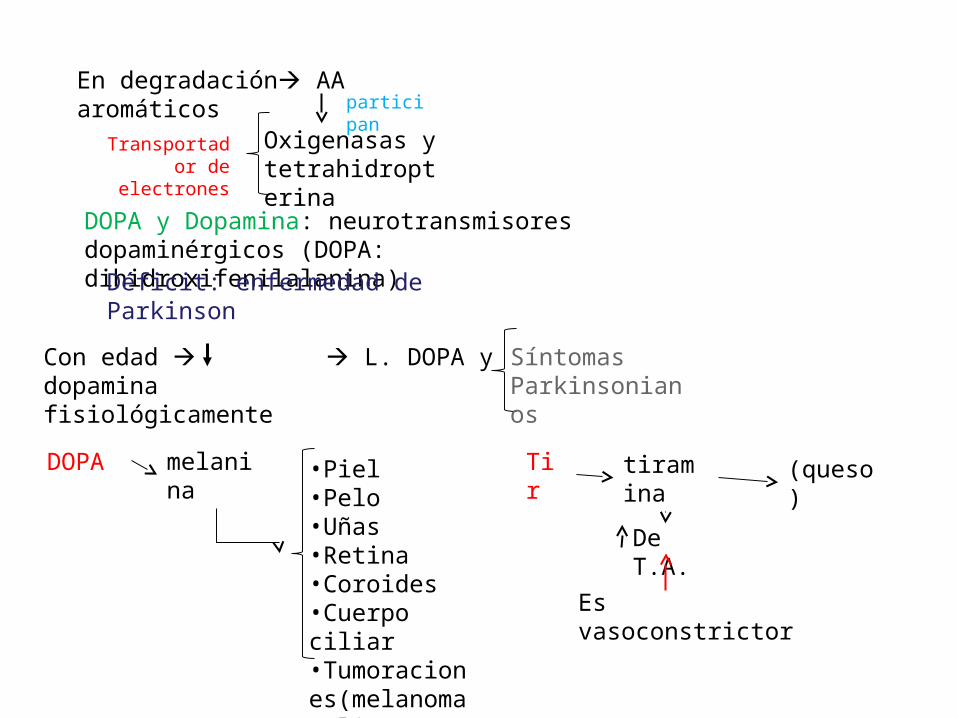
En degradación AA aromáticosparticipan
Oxigenasas y tetrahidropterina
Transportador de electrones
DOPA y Dopamina: neurotransmisores dopaminérgicos (DOPA: dihidroxifenilalanina)
Déficit: enfermedad de Parkinson
Con edad L. DOPA y dopaminafisiológicamente
Síntomas Parkinsonianos
DOPA melanina •Piel•Pelo•Uñas•Retina•Coroides•Cuerpo ciliar•Tumoraciones(melanoma maligno)
Tir tiramina (queso)
De T.A.
Es vasoconstrictor

Enfermedad de Hartnup• Rara, autosòmica recesiva.• Por defecto en el transporte de AA neutros en intestino
delgado y riñones. No es grave.• Dx: Hiperaminoaciduria (Val, Leu, Tir, Fen, Tript).• Tambièn absorciòn intestinal disminuida de estos AA.• Por la disminucion de absorber Triptofano conduce a las 3 D
de la Pelagra: dermatitis, diarrea, demencia.• La dermatìtis tipo pelagra y otros sìntomas responden a la
niacina(nicotinato): este es el tratamiento recomendado en la enfermedad de Hartnup.
• Pero la hiperaminoaciduria y el transporte intestinal no se corrigen con este règimen.
Fenilalanina - Tirosina
CH2
HC-NH3
COO-
CH2
HC-NH3
COO-
OH
FenilalanilhidroxilasaTetrahidrobiopteridia
O
Tirosina hidroxilasa
FENTIR
DOPA
CH2
C=O
COO-
A. FenilpiruvicoA. p-hidroxifenil- piruvico A. p-hidroxifenil-lactico
3,4 dihidrofenilalanina
CH2
HC-NH3
COO-
OH
HO
CH2
C=O
COO-
OH
CH2
HC-OH
COO-
OH
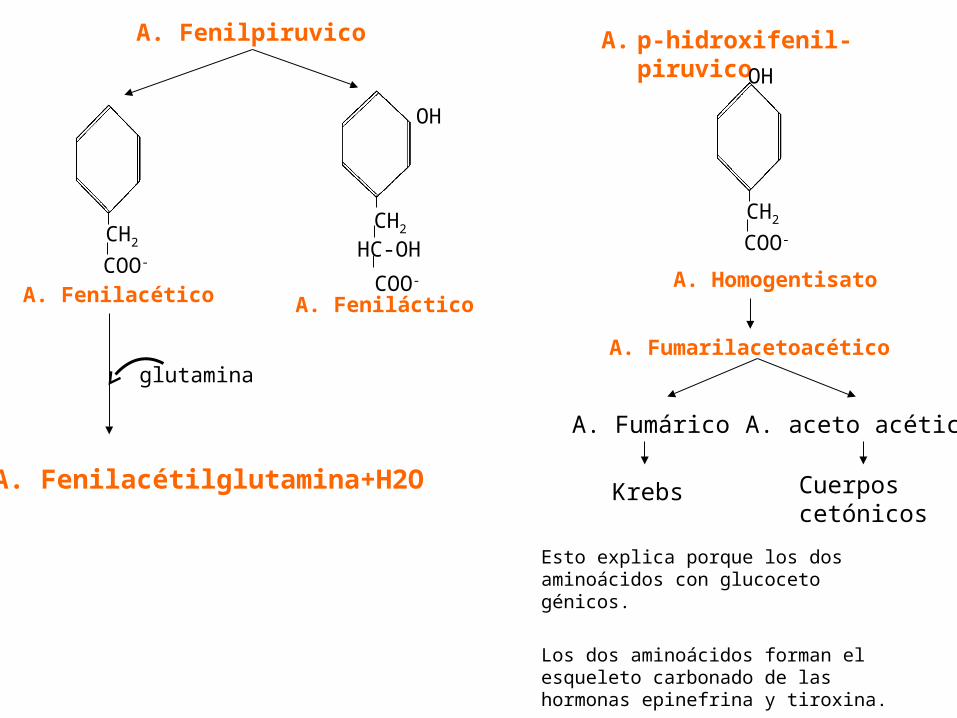
A. Fenilpiruvico A. p-hidroxifenil-piruvico
COO-
CH2
CH2
HC-OH
COO-
OH
A. Fenilacético A. Feniláctico
COO-
CH2
OH
A. Homogentisato
A. Fumarilacetoacéticoglutamina
A. Fenilacétilglutamina+H2O
A. Fumárico
Krebs
A. aceto acético
Cuerposcetónicos
Esto explica porque los dos aminoácidos con glucoceto génicos.
Los dos aminoácidos forman el esqueleto carbonado de las hormonas epinefrina y tiroxina.
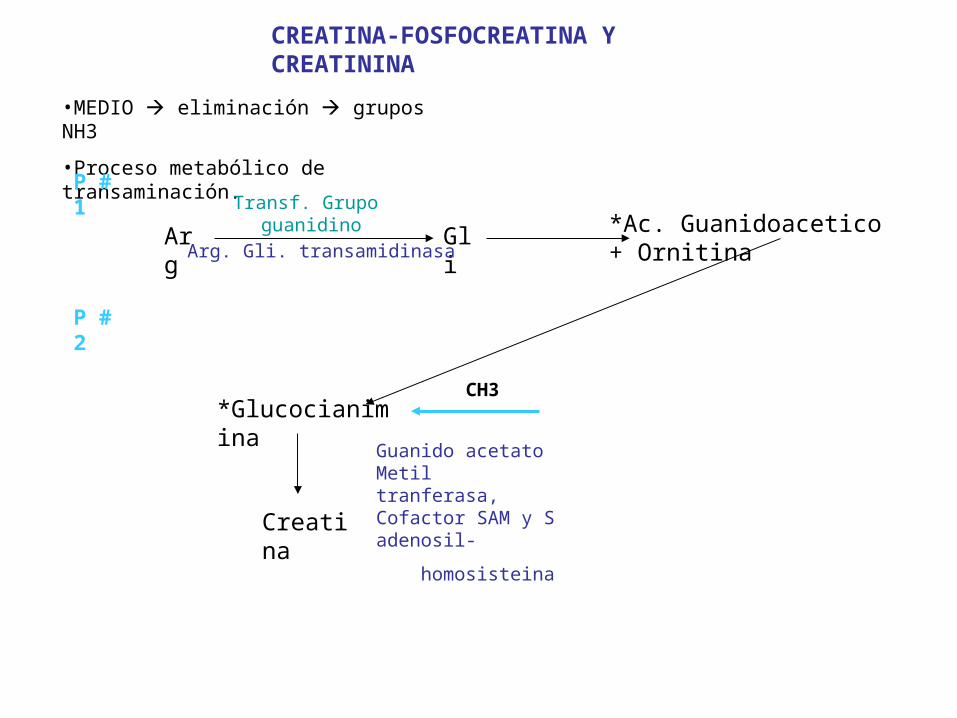
CREATINA-FOSFOCREATINA Y CREATININA
•MEDIO eliminación grupos NH3
•Proceso metabólico de transaminación.
P # 1
Arg Gli *Ac. Guanidoacetico+ Ornitina
Transf. Grupo guanidino
Arg. Gli. transamidinasa
P # 2
*Glucocianimina
Creatina
Guanido acetato Metil tranferasa, Cofactor SAM y S adenosil-
homosisteina
CH3

Síntesis de Creatina(S- Metilguanidoacetato)
• La formación del ácido guanidoacético+
ornitina es catalizada por la Arg-Gli trans-
aminidasa.
. La formación de la Creatina es catalizada por la Guanidoacetato Metiltransferasa, y tiene como cofactores a S adenosilmetioni
na y S Adenosilhomosisteina.
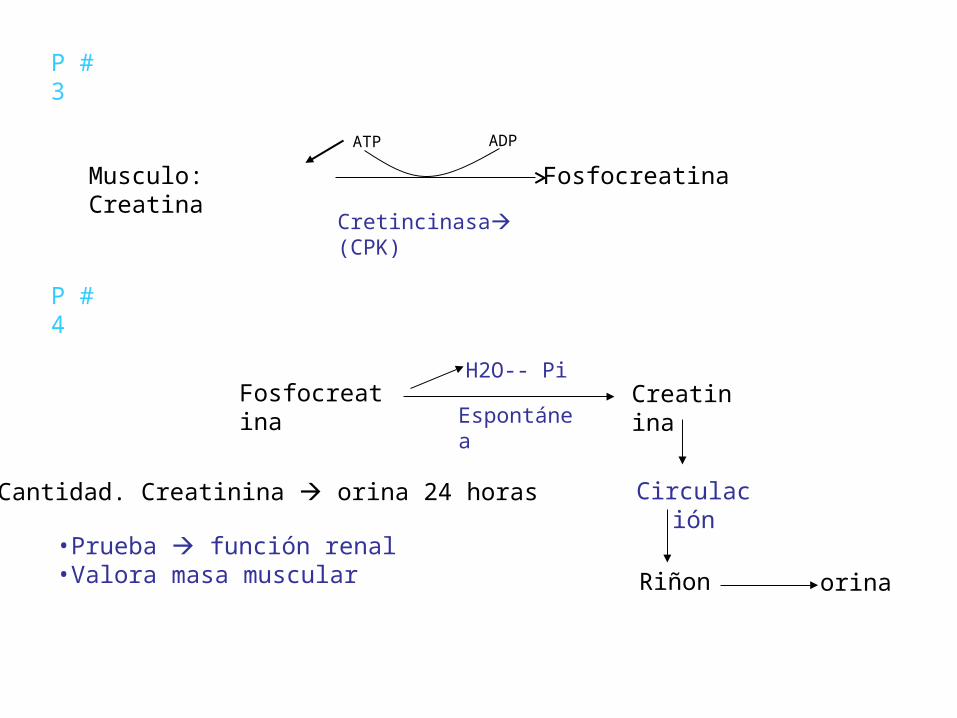
P # 3
Musculo: Creatina Fosfocreatina
Cretincinasa (CPK)
ATP ADP
Fosfocreatina Creatinina
Circulación
Riñon orina
P # 4
Espontánea
H2O-- Pi
Cantidad. Creatinina orina 24 horas
•Prueba función renal•Valora masa muscular
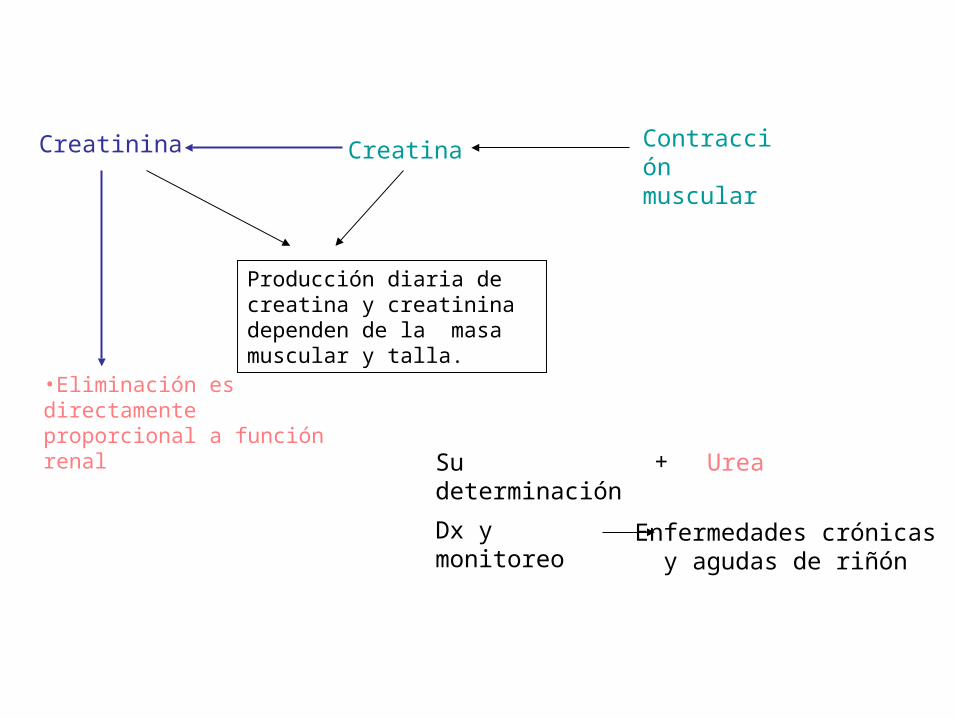
Creatinina Creatina Contracción muscular
•Eliminación es directamente proporcional a función renal
Producción diaria de creatina y creatinina dependen de la masa muscular y talla.
Su determinación + Urea
Dx y monitoreo Enfermedades crónicasy agudas de riñón

Creatinina.
• Es la menos variable de los nitrogenados no proteicos en sangre, no es influenciada por la dieta, se excreta en índices relativamente constantes, ya que no es reabsorbida por túbulos renales,
• Los valores pueden ser normales en algunos casos de uremia (ins.renal crón.) porque el valor plasmático de creatinina emplea de 7 a 10 días para estabilizarse si el índice de filtración glomerular disminuye, valores altos indican disfunción renal de larga evolución.(crónica)

ESPERMIDINA Y ESPERMINA
• Son poliaminas que forman complejos con el DNA.
• Se derivan de la ornitina y metionina.• La ornitina es descarboxilada y forma
Putrescina + CO2.• La S-adenosilmtionina se descarboxila y
forma S-adenosilmetipropilamina, esta dona propilamina a la putrescina y a la espermidina para formar Espermina.

Masa muscular existente: se valora indirectamente por su excreción de creatinina
P. de creatinina
Para esto se usa: ICT. (incide: creatinina/Talla)
ICT (indice creatinina totl=Creat. orina-24h (mg/día)x100
Creat. orina-24h (según talla, edad, sexo)
ICT: 60-80% déficit moderado Masa muscular
<60% déficit grave Masa muscular
Componentes: Colageno 3 Hidroxiprolina urinaria
Calcificados o no
IHC:Hidro. prolina (mg/24h)
Creatinina (mg/24h)

Balance. N: Cambios en proteínas corporales N Corporal incorporado a
Prot. Corporal: 16% N: Bal. Nit: bueno: Si ingestas adecuadas
para reemplazar perdidas en d N
BN= I – (u – ue) + (N – Fe) + D
I: Ingesta N (g de prot/6.25)
U: N urinario total
Ue: N. Urinario endogeno
N: N. eliminado por heces (prot no absorbida)
Fe: Perdidas de N fecal. Endogeno
D: Perdidas de N. dermico
BNE: Ingesta Pro. G – Nureico
Urinario eng + 41

Las proteínas se forman por unión de AA mediante uniones peptidicas
• Nomenclatura: nombre Aa del N terminal incompleto terminado en (il) seguido del Aa del C terminal completo y con su nombre propio.
• Así: varios AA:Ej: glicil, valil, treonil, Valina.
• Si es incierta la secuencia se los encierra en ().
• Ej.: (valíl, glicil, Valina)

Estructura de las Proteínas• Primaria:
• Es el numero, orden o secuencia especifica de los AA en una cadena polipeptidica y con la ubicación de enlaces disulfuro si los tiene.
• Tiene una determinación genética.
• Se determina por:
1.-Hidrólisis de la cadena se separan los a.a.

• 2.- los a.a. separados se someten a cromatografía.
• 3.- el numero y orden de sucesión se hace por el método de Edmanutiliza Isotiocianato.
• Secundaria: Es el plegamiento deCad.Polipepticas.
• A:Pleg en forma de Hélice:(): giros alrededor de un hélice estabilizado por uniones H.
• Existen 3-6 a.a. por giro

La formación de la helice es favorecida por presencia de: Ala, Met, Leu.
b.- Lamina(hoja) plegadaplegamientode a.a. Forman estructura Distendid.
Se favorece por presencia de: Val, Tir, Isoleu. Pueden ser paralelas y antiparalelas,estas son +estables.
• Terciaria : disposición tridimensional de cadenas polipeptidicas en el espacio.
• Ej.: Mioglobina.
• Cuaternaria: forma en que las distintas cadenas se articulan entre si.

Agrupamiento de cadenas unidas por fuerzas, diferentes a los enlaces covalentes.Si una proteina esta compuesta por 2 o 4 subunidades se llama dimérica o tetramérica.
• Hay fuerzas estabilizadorasEnlaces de H. y Fuerzas Electroestáticas.
• Ej.: HEMOGLOBINA

Desnaturalización
• Inducida por:calor, disolventes orgánicos, pH,fuerzas iónicas etc.
• Desaparece total o parcialmente la envoltura acuosa, esto provoca neutralización de cargas elèctricas, y roptura de los puentes de H.
• Si recupera su estructura nativa la proteína se renaturaliza.

Desnaturalización.
• Provoca: a.- Pérdida de sus propiedades biológicas.
• b.- Disminución de su solubilidad por pérdida de los residuos hidrofílicos de la superficie y exposición a los aa hidrófobos
• c.-Cambios en las propiedades como aumento de la viscosidad y disminución de la velocidad de difusión.

Papel de AAComponente Proteínas.Metabolitos Energéticos.Precursores Comp.. Nitrogenados.Parte Hemo-Aminas-Nucleótidos-
GlutatiónParte: Enzimas y Coenzimas.Precursores: Ac.Grasos-glucosa-
C.cetonicosExesootros metabolitosAc.
OxalaceticoKrebs

Nitrógeno Molecular (N2)
• Org. Vivos N reducido
1.- Debe pasar d N2 o NO3 NH3- NH4.
Ion amonio base debil:en AA y proteínas.
esta en forma no protonada
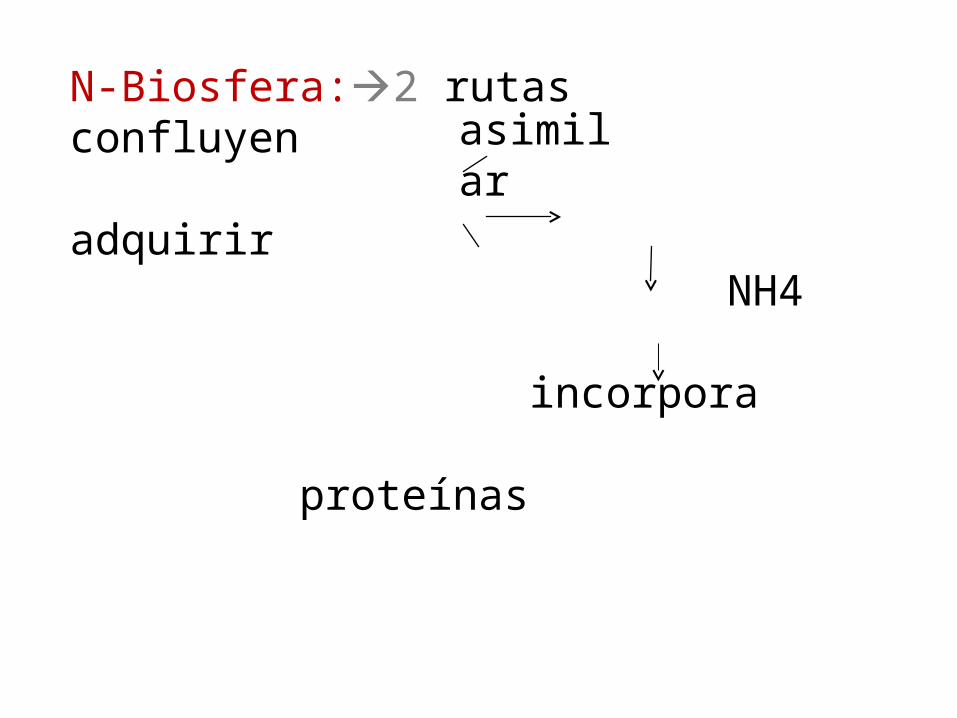
N-Biosfera:2 rutas confluyen adquirir
NH4 incorpora proteínas
asimilar

ASIMILACION
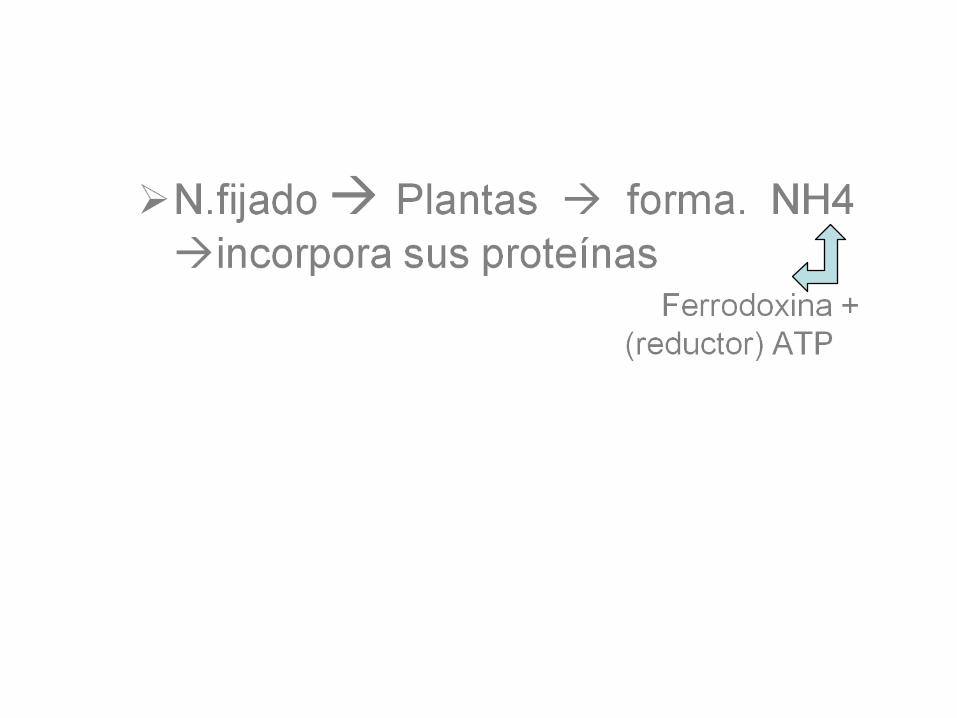

Regulación- Fijación N.• Relación ATP/ADP regula:
Actividad Nitrogenasa.• NH4regula exp.genetica.• Amonio: ingresacompuestos
orgánicos x 3 vías catalizadas:• Carbamoil sintasa I.• Glutamato Deshidrogenasa.• Glutamina sintasa.• El N de AAse incorpora al
organismo ingresando x dieta.

• 1.-Por recambio proteico
2.-Por transaminaciòn
a.a. esenciales a.a.no esenciales
unidireccional bidireccionalAa sea exógenos o endógenos experimentan
transaminación siendo su efecto la remoción e intercambio del N de un alfa Aa el cual se convierte en su cetoácido correspondiente.
Aa +Cetoácido = Nuevo Aa+Nuevo cetoácido.
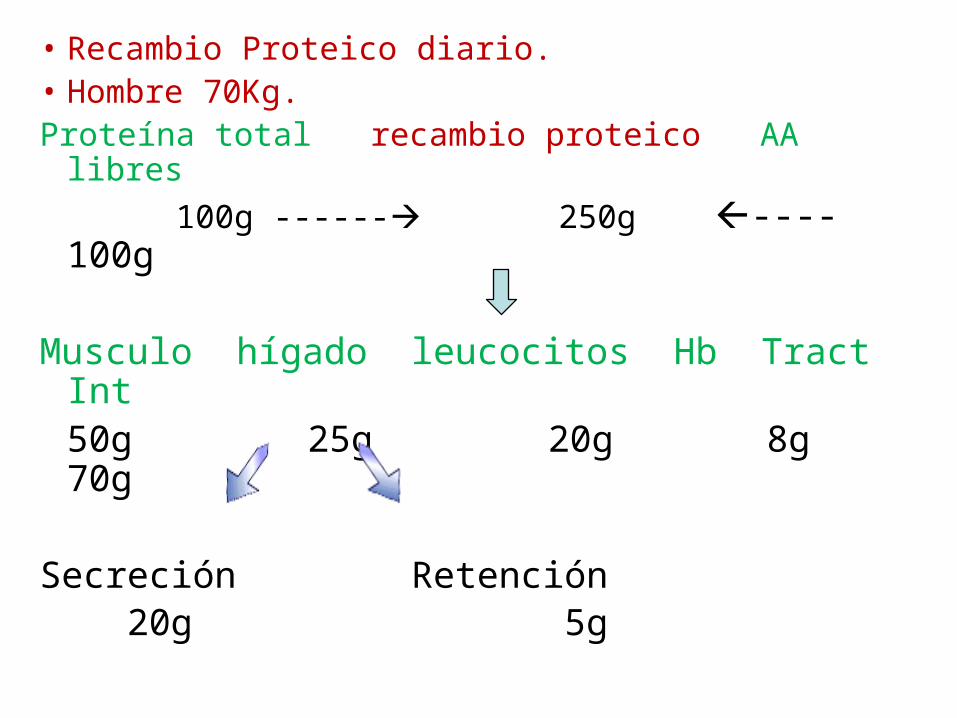
• Recambio Proteico diario.• Hombre 70Kg. Proteína total recambio proteico AA libres
100g ------ 250g ---- 100g
Musculo hígado leucocitos Hb Tract Int50g 25g 20g 8g 70g
Secreción Retención 20g 5g
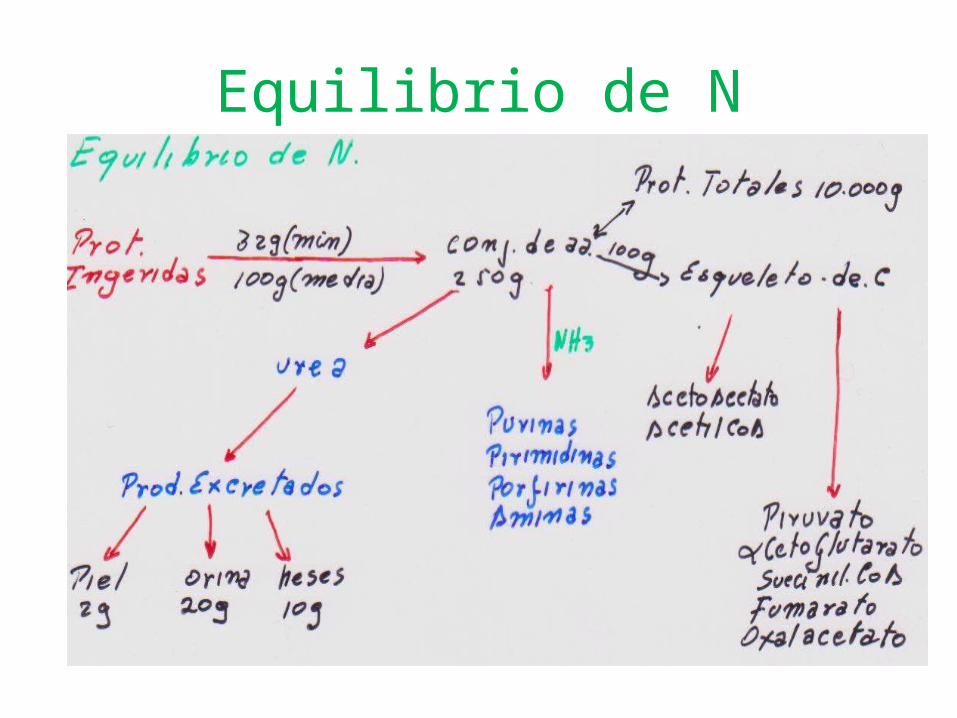
Equilibrio de N

Incorporación de N.Reacciones de transaminaciòn transf
NH2 aaotro a.a. a.a. convierte en cetoacido correspond.Participan todos a.a.- excepto Lis- Tir.Utilizadas: - Biosíntesis. a.a.
CatabolismoEnzimas: STGO (AST)-STGP(ALT).Cofactor: P de
Piridoxal(B6) U.Covalentemente a
NH2Lisina- Ala.

Complejo:transaminasa+cofactor se unen.
x Interacciones: Iónicas e Hidrofobicas.Si a.a. u. al complejo
Desplaza NH2 de Lisina
Cesa covalencias y se forma piridoxamina

FUENTES PROTEINAS• Animales: - Carnes
- pescados - huevos
- víceras - leche derivados.Vegetales:- Legumbres
- soja - frutos secos y otros - hortalizas

Calidad proteínas: se mide por capacidad de satisfacer necesidades orgánicas.
• Se determina por:• Valor biológico: % de N proteico
absorbido y retenidovalor máximo=100%
• Digestibilidad:proporc.Ningerido que se absorbe.valor max.=100%
• Score de AA: CantAA esenciales de una proteína comparada con una proteina de referencia internacional.

• Si 1 AA esencial falta: se llama a.a. Limitante.
• Prot.calidad: la que tiene: valor biolog digestibilidad score de A.A
"Recomendable dieta combine prot. Animales y vegetales"
Recomendación: 60g/diaadulto sano 40-80g/día 0.8g/Kg peso/díavaria: crecimiento - embarazo- lactancia.

Balance N= N ingerido - *N eliminadoorina(urea)
4g/día eliminheces y piel.
1g N ingerido procede de 6,25g de Prot. Ingerida.
*N.ureico: Cant. urea en orina/24h x 0,467.

Calculo requerimiento proteicos
Requerimientos:
Recomendados por peso en Kg.
0,8g X Kg.
Ej. peso 60kg- adulto
0,8 x 60= 48g proteínas/d.(recomendación)

Consumo calorías/ día.
Recomendado:
2000kcal_ adulto sano
Proteínas aportan: 12-15 %- cal.(promedio 13%)
Así: 100%------2000
13%------ x = 260kcal.aportan proteínas de las 2000 recomendadas.

Por otra parte: 1g proteína oxidado genera 4k cal.Entonces: 260 kcal / 4= 65g proteína se requieren / díaPara generar 260 kcal es el aporte calórico que deben dar las proteínas como parte de las 2000 kcal/día recomendadas para adulto (60-70kg de peso)Así :Consumo total Prot./día oscila: 48-65g promedio Ningún alimento aporta 100% proteínasSi se elige carne, pescado cubrir-48-65g Prot.Se requerirán: aproximadamente 325mg carne o pescado


















