Leucemias
41
HOSPITAL NACIONAL ARZOBISPO LOAYZA VIII- CICLO FIORELLA PALACIOS PINILLOS LEUCEMIAS ASOCIACIÓN UNIVERSIDAD PRIVADA SAN JUAN BAUTISTA
-
Upload
upsjb2014ii -
Category
Health & Medicine
-
view
108 -
download
5
Transcript of Leucemias

HOSPITAL NACIONAL ARZOBISPO LOAYZA
VIII- CICLO
FIORELLA PALACIOS PINILLOS
LEUCEMIAS
ASOCIACIÓN UNIVERSIDAD PRIVADA SAN JUAN BAUTISTA

LEUCEMIAS-EPIDEMIOLOGÍAPARA EL AÑO 2014, LOS
CÁLCULOS DE LA SOCIEDAD AMERICANA
CONTRA EL CÁNCER PARA ESTE CÁNCER EN LOS
ESTADOS UNIDOS SON:
ALREDEDOR DE 52,380 NUEVOS CASOS DE
LEUCEMIA (TODOS LOS TIPOS) Y 24,090 MUERTES
A CAUSA DE LEUCEMIA (TODOS LOS TIPOS).
ALREDEDOR DE 18,860 NUEVOS CASOS DE LEUCEMIA MIELOIDE
AGUDA (AML). LA MAYORÍA SE REPORTARÁ EN
ADULTOS.
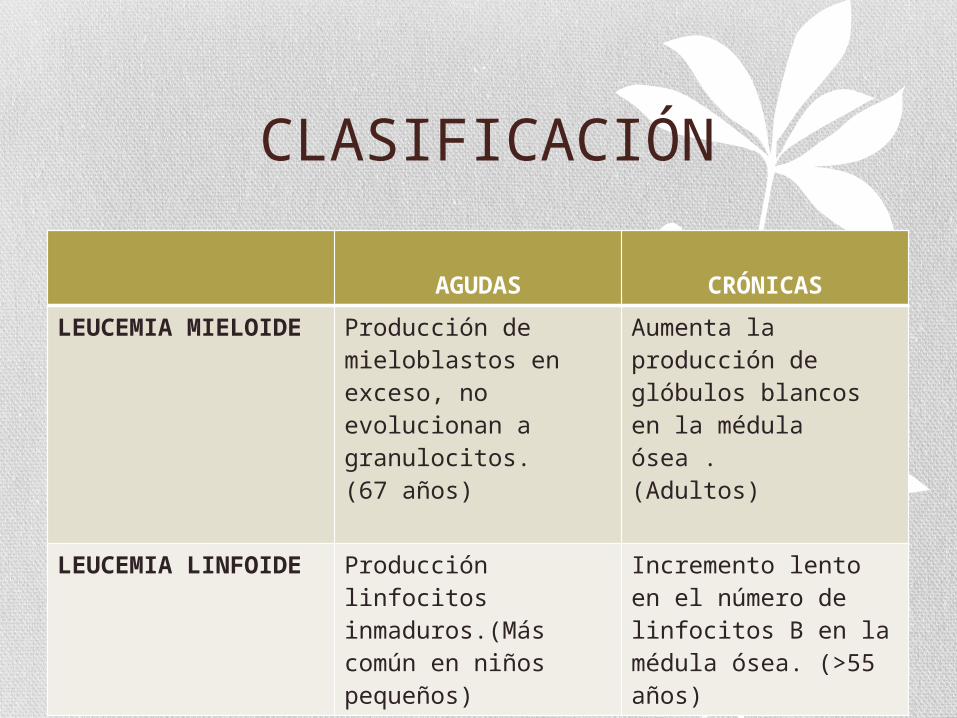
CLASIFICACIÓN
AGUDAS CRÓNICAS
LEUCEMIA MIELOIDE
Producción de mieloblastos en exceso, no evolucionan a granulocitos.(67 años)
Aumenta la producción de glóbulos blancos en la médula ósea .(Adultos)
LEUCEMIA LINFOIDE
Producción linfocitos inmaduros.(Más común en niños pequeños)
Incremento lento en el número de linfocitos B en la médula ósea. (>55 años)

LEUCEMIA MIELOIDE
LEUCEMIA
MIELOIDE AGUDA
Incidencia: 3.5 / 100000
hab. año
hombres:mujeres 5:3
Edad media de
presentación:
60 – 65 años
LEUCEMIA
MIELOIDE CRÓNICA
Incidencia: 1.5 /100000
hab. año
Predominio en sexo
masculino(15-20% de
todas las leucemias)
Edad de presentación
: 40-60 años

LEUCEMIA MIELOIDE AGUDA
HERENCIA
SÍNDROMES ( DOWN, KOSTMANN, BLOOM)
ANEMIA DE FANCONI, ATAXIA-TELANGIECTASICA
SINDROMES MIELOPROLIFERATIVOS
MUTACIONES ( p53, RUNX1, CCAAT)
EXPOSICIÓN QUÍMICAS Y A RADIACIÓN
AL BENCENO, TABAQUISMO, PRODUCTOS DEL PETRÓLEO, PINTURA, LÍQUIDOS, ÓXIDO DE ETILENO, HERBICIDAS, PESTICIDAS,.
RADIACIÓN A DOSIS ALTAS (5-7 AÑOS DESPUÉS DE LA EXPOSICÍÓN)
FÁRMACOS
ALQUILANTES (5-7 AÑOS DESPUÉS DE EXPOSICIÓN) (ABERRACIÓN CR 5 Y 7)INHIBIDOR DE TOPOISOMERASA II (1-3 AÑOS DESPUÉS locus 11q23CLORANFENICOL, FENILBUTAZONA, CLOROQUINA, METOXIPSORALENO)

CLASIFICACIÓN LMA• OMS / FAB
Blastos para el diagnóstico: OMS 20%, FAB 30%
OMS: Considera rasgos genéticos
con características morfológicas; se basa en
inmunofenotipos, caracerísticas clínicas,
moleculares y morfológicasDiferencia LAM de LLA, identifica tipos de LAM
FAB:Más antiguo
base de varios estudios actualesSe fiaba de estudios citoquímicos
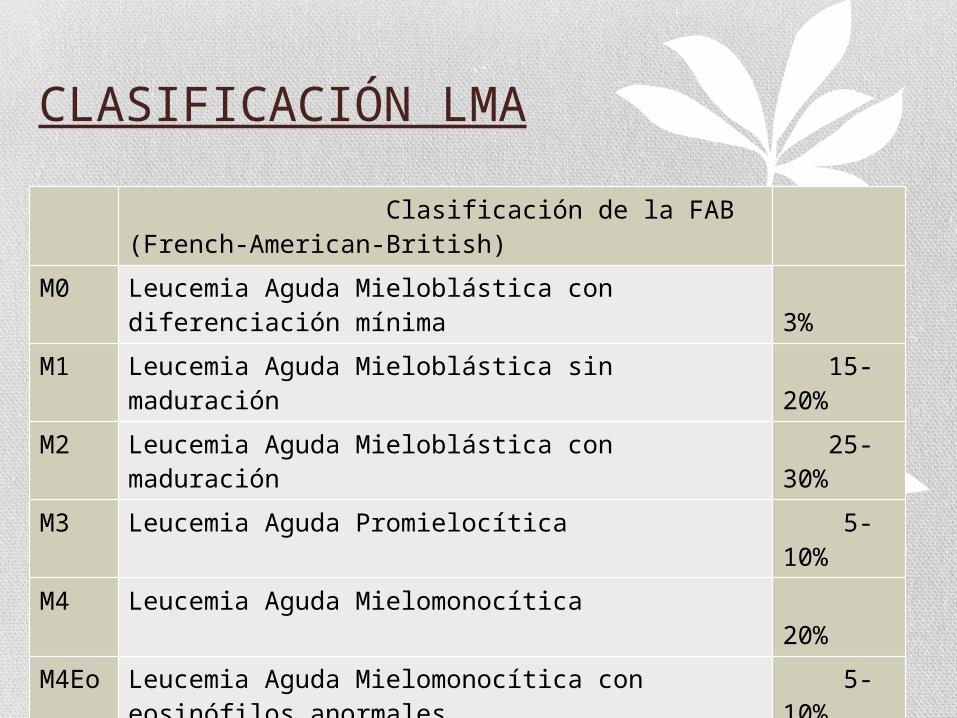
CLASIFICACIÓN LMA
Clasificación de la FAB (French-American-British)
M0 Leucemia Aguda Mieloblástica con diferenciación mínima
3%
M1 Leucemia Aguda Mieloblástica sin maduración 15-20%
M2 Leucemia Aguda Mieloblástica con maduración 25-30%
M3 Leucemia Aguda Promielocítica 5-10%
M4 Leucemia Aguda Mielomonocítica 20%
M4Eo Leucemia Aguda Mielomonocítica con eosinófilos anormales
5-10%
M5 Leucemia Aguda Monocítica 2-9%
M6 Leucemia AgudaEritroleucemia 3-5%
M7 Leucemia Aguda Megacariocítica 3-12%

-DECITABINA
-5-AZACITIDIN
A

CUADRO CLÍNICO-LMA
• Múltiples e Inespecíficas• Periodo de 3 meses
PRIMERAS MANIFESTACIONE
S
• Fatiga (1er síntoma en 50%) • Infecciones / fiebre (10%)• Hemorragia (5%) • Dolores óseos; adenopatías; tos; cefalalgias o
sudores• Palidez, disnea de esfuerzo
PRESENTACIÓN
• Hígado hepatomegalia• Bazo esplenomegalia• Piel leucemia cutis• Ganglios linfadenopatía• Huesos dolor • Gingiva, SNC, etc• Sarcoma granulocítico o cloromas: masa aislada de blastos ( t 8:21).
INFILTRACIÓN

• Anemia normocítica normocrómica no proliferativa
• Leucostasis disfunción ocular y cerebrovascular o sangrado
• Metabólicas: hiperuricemia e hipocalcemia (raro)
• Frotis: 5 % sin células leucémicas
LABORATORIO
• LAM M3: hemorragia GI, intrapulmonar o intracraneal• LAM monocítica: hemorragia (retiniana 15%) /
coagulopatía • Leucemias monocíticas / 11q23: infiltración de encias,
piel, tejidos blandos o meningitis
LABORATORIO
• Leucocitosis : > 15 000/mcL • 25 – 40 % < 5 000 /mcL• 20 % > 100 000 /mcL
• Plaquetas grandes y abigarradas, granulaciones anormales y disfuncionales
VARIANTES
Plaquetas:75% < 100,000 / mcL25% < 25,000 / mcL
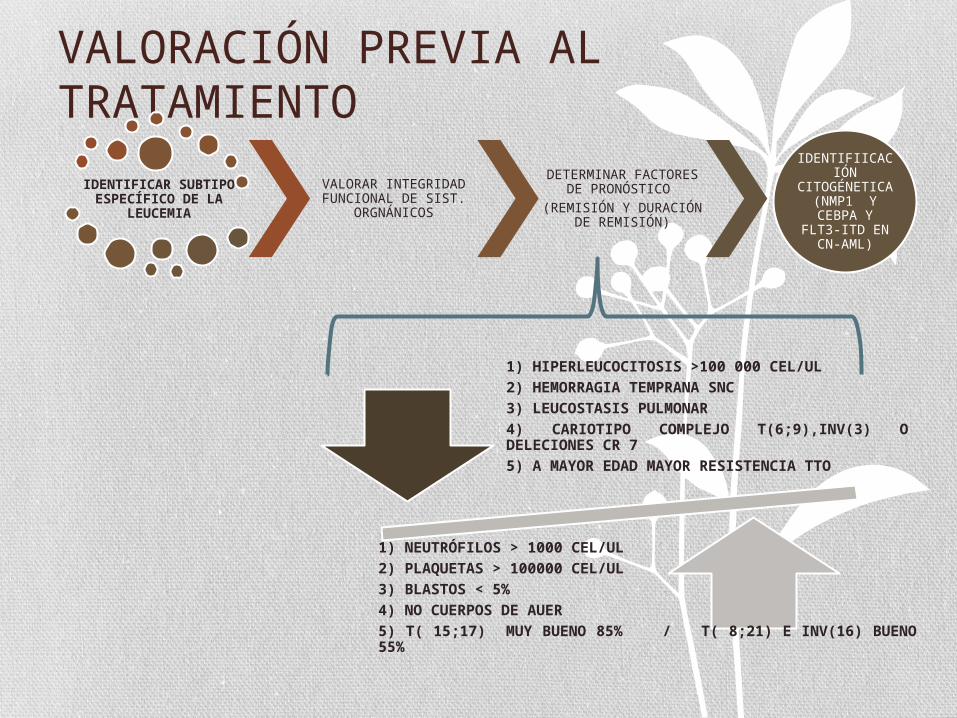
VALORACIÓN PREVIA AL TRATAMIENTO
IDENTIFICAR SUBTIPO ESPECÍFICO DE LA
LEUCEMIA
VALORAR INTEGRIDAD FUNCIONAL DE SIST.
ORGNÁNICOS
DETERMINAR FACTORES DE PRONÓSTICO
(REMISIÓN Y DURACIÓN DE REMISIÓN)
IDENTIFIICACIÓN
CITOGÉNETICA (NMP1 Y
CEBPA Y FLT3-ITD EN CN-
AML)
1) HIPERLEUCOCITOSIS >100 000 CEL/UL2) HEMORRAGIA TEMPRANA SNC3) LEUCOSTASIS PULMONAR4) CARIOTIPO COMPLEJO T(6;9),INV(3) O DELECIONES CR 75) A MAYOR EDAD MAYOR RESISTENCIA TTO
1) NEUTRÓFILOS > 1000 CEL/UL 2) PLAQUETAS > 100000 CEL/UL 3) BLASTOS < 5%4) NO CUERPOS DE AUER5) T( 15;17) MUY BUENO 85% / T( 8;21) E INV(16) BUENO 55%

Dosis: 100-200 mg/m2/día
Dosis: (60 mg/m2/día)O Idarrubucina (12-13 mg/m2/día)
TRATAMIENTO-LMA

PRONÓSTICO Remisión completa
LLA: 90% LMA: 60% a 70%
Sobrevida libre de enfermedad a 5 años LLA: 60% LMA: 20%

LEUCEMIA MIELOIDE CRÓNICA
Translocación
cromosómica
(Cr 9 y 22)
Fusión cabeza-cola en el BCR( zona de conglomerados del sitio de rotura) l (22q11) con el gen ABL1 l(9q34)
1) La proteína Abl activación constitutiva como TK y activa cinasas distales ( Impide apoptosis).2) Se atenúa la actividad de unión a proteínas del ADN de Abl.3) Incrementa unión a los microfilamentoos de actina
Proliferación maligna de células mieloides
(Sin tto fase crónica acelerada (crisis blástica)

CUADRO CLÍNICO-LMC• Sistémicos: diaforesis, fatiga, malestar general.
• Por esplenomegalia: malestar abdominal, saciedad temprana.
• Síntomas por hiperviscosidad por cifras leucocitarias altas (raro): ACV, IMA, TVP, insuficiencia respiratoria ,priapismo.
• La px de histamina en respuesta a la basofilia (fases posteriores) prurito, diarrea y fenómenos vasomotores.
SÍNTOMAS
• Esplenomegalia mínima o moderada• Hepatomegalia leve (raro)• Linfadenopatías (inusual)• Sarcoma mieloide (inusual)
HALLAZGOS FÍSICO
• Leucocitosis: <5% blastos circulantes, <10% blastos y promielocitos
• Incremento de mielocitos , metamielocitos y en banda en sangre periférica y médula ósea.
• Plaquetas normales o elevadas, eosinofilia, basofilia, monocitosis (MO y SP)
• Anemia n normocítica normocrómica• DHL y ácido úrico incrementado• Pacientes con CML positiva a p230 BCR-ABL1 tienen una
evolución más gradual
LABORATORIO
Usual fase tardía (mal prnóstico)

CRITERIOS DE DIAGNÓSTICO
• 10%-19% blastos• Basófilos > 20%• Plaquetas <100 x 10^9/L• Plaquetas >100 x 10^9/L• Esplenomegalia• Leucocitosis • Alteraciones citogenéticas
FASE ACELERADA
(1 o + CRITERIOS)
• Blastos >20%• Proliferación blástica extramedular• Cantidad aumentada de blastos en biopsia de médula ósea.
FASE BLÁSTICA
(1 o + CRITERIOS)

ACELERACIÓN DE LA ENFERMEDAD
ANEMIA PROGRESIVA ( SIN HEMORRAGIA Y NO POR TTO)
EVOLUCIÓN CITOGÉNETICA DE CLONAS
10-20% DE BLASTOS EN MO O SP
PLAQUETAS < 100000 /ul
CRISIS BLÁSTICALEUCEMIA AGUDA CON 20% O MÁS BLASTOS EN SP O MO
NEUTRÓFILOS CON HIPERSEGMENTACIÓN (Anomalías de Pelger-Hüet)
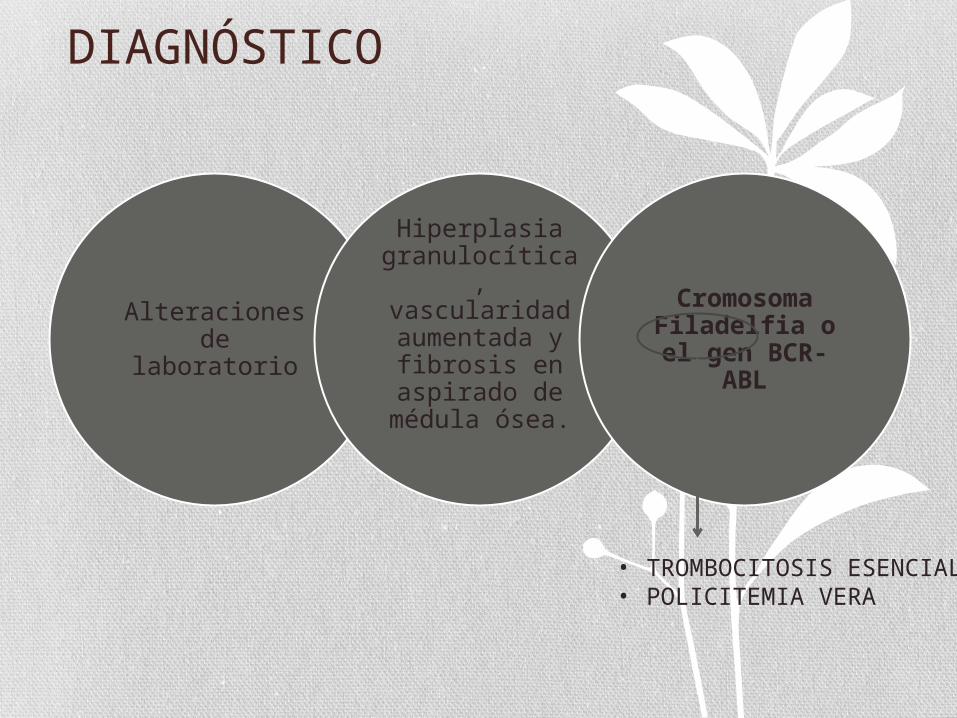
DIAGNÓSTICO
Alteraciones de laboratorio
Hiperplasia granulocítica, vascularidad aumentada y
fibrosis en aspirado de
médula ósea.
Cromosoma Filadelfia o el gen BCR-ABL
• TROMBOCITOSIS ESENCIAL• POLICITEMIA VERA

LMC-ATÍPICA

DIAGNÓSTICO DIFERENCIAL
LEUCEMIA MIELOMONOCÍTIC
A JUVENIL
• Infancia y adolescencia
• Sobreproducción de células mieloides maduras
• Infiltración de órganos
• Muerte por falla orgánica o infección
LEUCEMIA MIELOMONOCÍTIC
A CRÓNICA
• Sobreproducción de células monocíticas maduras y displásicas y de neutrófilos.
LEUCEMIA EOSINOFÍLICA
CRÓNICA
• Sobreproducción de eosinófilos displásicos
• Hiperplasia médula ósea, con displasia mieloide y tendencia a progresión a leucemia mieloide aguda.
• Eosinófilos asociados a daño tisular por liberación de proteínas de gránulos, especialmente pulmonar y endocárdica

TRATAMIENTO• Suprimen hiperplasia mieloide, con reducción de
leucocitosHidroxiurea y
Busulfan
• (5 millones U/m2/día SC) produce respuesta hematológica en la mayoría de los pacientes con remisión citogenética en 13-27%.
Interferón alfa 2a
• Más respuesta citogenética (41%), con mejoría en supervivencia. Esta se consideraba previamente el tratamiento inicial estándar.
Interferón alfa 2a con
citarabina• inhibe selectivamente la actividad de BCR/ABL de
tirosina quinasa. • En estudios IRIS, compararon imatinib (400 mg/día)
con tratamiento convencional (IFN con arabinósido de citosina) tratamiento con imatinib tuvo mejor respuesta citogenética (87% con imatinib contra 35% en grupo combinado)
Imatinib (Gleevec)
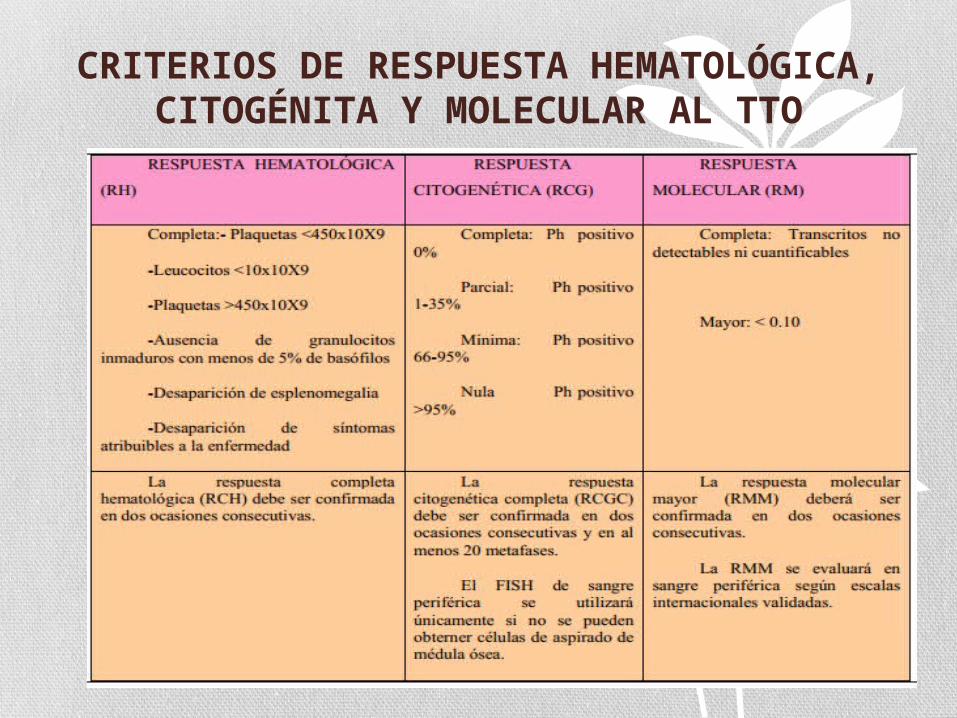
CRITERIOS DE RESPUESTA HEMATOLÓGICA, CITOGÉNITA Y
MOLECULAR AL TTO

CRITERIOS DE RESPUESTA AL TTO

Hurtado R, Vargas P, Cortés J. Chronic Myeloid Leukemia: Current Concepts in Physiopathology and Treatment. Cancerología 2 (2007), 137-147
OTROS TRATAMIENTOS
Inhibidores de ABL cinasa
• Tiazolcarboxamida: se une a dominio de Abl cinasa, inhibe familia de Src cinasas.
• Efectos adversos: citopenia, diarrea, náusea, eritema, sangrado gastrointestinal, derrame pleural.
• Aminopirimidina:• Derivado estructural de imatinib,se une a Abl cinasa.• Útil en casi todas las mutaciones que provocan
resistencia a Imatinib, excepto T3151.• Inhibe factor de crecimiento derivado de plaquetas y c-
Kit, pero no la familia de Scr cinasas.

FACTORES PRONÓSTICOSÍNDICE DE SOKAL:-Edad-Tamaño esplénico-Porcentaje blástico en sangre periférica.-Número de plaquetas al diagnóstico.-Evolución citogenética de las clonas
FASES:•Supervivencia media en fase crónica es de 4 a 6 años.
•En fase acelerada supervivencia media menor a un año.
•En fase blástica supervivencia media de 3 a 6 meses.
SISTEMA HASFORD(INF-alfa):• % blastos• Tamaño bazo• #plaquetas• Edad• % Eosinófilos, basófilos

LEUCEMIA LINFOIDE AGUDAINCIDENCIA
• 3000 a 4000 casos nuevos al año en EEUU, 2/3 de los cuales son en niños.
• Leve predominio en sexo masculino(2,9 : 1,9)
ETIOLOGÍA
• Respuesta inmunológica anormal a una infección común
• Hereditaria: • Gemelos idénticos
probabilidad 5 veces mayor que población general.
• Anemia de Fanconi, Sd. De Bloom, Sd. Ataxia-telangectasia, agamaglobulinemia congénita, Sd. De Down, Sd. De Kleinefelter
• Radiación:• Mayor número de casos
en población expuesta a radiación después de bombas nucleares, 5 a 7 años después.
• También RT, en altas dosis en períodos cortos y en personas jóvenes
ETIOLOGÍA
• Químicos:• Derivados del benceno,
como kerosene, tetracloruro de carbono, tabaco y pesticidas (LLAg)
• Drogas:• Agentes alquilantes:
Melfalán, 2/3 1° como mielodisplasia y luego de éstos 1/3 avanza a leucemia (crom.5 y 7)
• Inhibidores de la topoisomerasa II: Etopósido, Doxirrubicina (crom 11)
• Inmunosupresores y factores de crcimiento: Ciclosporina y G-CSF (crom. 7) . Fenilbutazonas, CAF y cloroquinas

VARIEDAD INMUNITARIA
CASOS (%) VARIEDAD FAB ALTERACIONES CITOGENÉTICA
S
ALL pre-B 75 L1,L2 T(9;22),t(4;11),t(1;19)
ALL de linfocitos T
20 L1,L2 14q11 o 7q34
ALL de linfocitos B
5 L3 (Leuc. Burkitt)
T(8;14),t(8;22),t(2;8)
CLASIFICACIÓN DE LA LEUCEMIA LINFOIDE AGUDA (LLA)
LEUCEMIA LINFOBLÁSTICA AGUDA CON CROMOSOMA FILADELFIA (+)Alteración de mal pronóstico
5% LLAg infantiles 25% de las LLAg en adultos
Se asocia a menor % y menor duración de la remisión completa (RC) y mayor compromiso del SNC

CLÍNICA
Reemplazo de MO y sangre
periférica por blastos
atípicos
FATIGA
DEBILIDAD
ANOREXIA
PÉRDIDAPESO
HEMORRA-GIAS
DOLOR ÓSEO
ADENPATÍAS
INFILTRACIÓN DE
TEJIDOS BLANDOS
HEPATO-ESPLENOMEGALIA

LABORATORIO
Anemia, normo-normo, eritopoyesis inefectiva, disminución de reticulocitos
GB. 15000, 25-40% menor a 5000, 20% mayores a 100000, 5% sin blastos en la periferia.
Plaquetas: 75% menor a 100.000,25% menor a 25.000.

TRATAMIENTO-LLA
PRE-TRATAMIENTO• ESTABLECER EL SUBTIPO DE LEUCEMIA• EVALUAR INTEGRIDAD Y FUNCIÓN DE SIST.
CARDIOVASCULAR, PULMONAR, HEPÁTICO Y RENAL
• INFECCIONES• TRANSFUSIONES GR, PLAQUETAS O PLASMA• 50% CON HIPERURICEMIA (PRECIPITACIÓN
CON QT) HIDRATAR, ALOPURINOL, ALCALINIZAR ORINA
• ESTUDIO DE LCR
TRATAMIENTO• INDUCCIÓN: CICLOS CORTOS DE
ASOCIACIÓN DE DROGAS EN QUE SIEMPRE SE INCLUYE UN CORTICOIDE; VINCRISTINA, ANTRACICLINA; TAMBIÉN ALTAS DOSIS DE CICLOFOSFAMIDA, METROTEXATO Y CITARABINA
• PROFILAXIS DEL SNC, 35% DE RECAÍDAS- METROTEXATE INTRATECAL.
• CONSOLIDACIÓN: METROTEXATE O 6-MERCAPTOPURINA
• NIÑOS 95-99% RC, 60-70% SOBREVIDA LIBRE DE ENFERMEDAD. ADULTOS 70-90% RC, 25-50% SLE
• BUEN PRONÓSTICO: MENOR DE 30 AÑOS, GB MENOS DE 30.000 Y AUSENCIA DE CROM. PH(+).
• TRASPLANTE DE MO, AUTÓLOGA O HALÓGENA EN MENORES DE 55 AÑOS

PRONÓSTICO-LLA
REMISIÓN COMPLETA
LEUCOCITOS MAYOR O IGUAL A 1500, PLAQ. MAYOR A 100000 Y AUSENCIA
DE BLASTOS PERIFÉRICOS
POR UN PLAZO DE 4 SEMANAS
HACER PCR E INMUNOFENOTIPO PARA DETECTAR ENFERMEDAD RESIDUAL
FACTORES PRONÓSTICOS
-EDAD: A MAYOR, PEOR PRONÓSTICO + DE 60-STATUS FUNCIONAL Y PATOLOGÍAS ASOCIADAS-PATOLOGÍAS AGUDAS INTERCURRENTES-HALLAZGOS CROMOSÓMICOS AL DG.-HISTORIA DE SINT. PROLONGADOS, PANCITOPENIA MAYOR A UN MES-PATOLOGÍA HEMATOLÓGICA PREVIA-MÁS DE 100.000 LEUCOCITOS AL DG., Y MÁS BLASTOS PERIFÉRICOS-CLASIFICACIÓN FAB, INMUNOFENOTIPO

LEUCEMIA LINFOIDE CRÓNICA
DEFINICIÓN:
ENFERMEDAD LINFOPROLIFERATIVA CON ACUMULACIÓN DE LINFOCITOS B MADUROS, 5% LINFOCITOS T
INCIDENCIA
5 X 100,000 HAB. (35 - 59 AÑOS)30 X 100,000 HAB. (80 - 84 AÑOS)
FACTORES DE RIESGO
LA MÁS FRECUENTE ANORMALIDAD ES EN EL CROMOSOMA 12 O 11.TRISOMIAS CON EL CROMOSOMA (+12) Y LA PRESENCIA DE DELECCIONES 11, ESTÁ RELACIONADA CON AUMENTO DEL CONTEO LEUCOCITARIO Y POBRE RESPUESTA A LA TERAPIA.

Alteración citogenética (mutación) Incidencia Pronóstico
Sin alteraciones citogenéticas 18 % Buen pronóstico
Deleción* 13 (q14) 55 % Buen pronóstico
Trisomía 12 20 % Malo / Regular
Deleción* 11 (q22-23) 20 % Mal pronóstico
Gen TP53 10 % Mal pronóstico

DIAGNÓSTICO
CLÍNICATRIADA: LINFOCITOSIS (20%),
LINFODENOPATÍA 5% (CERVICAL Y AXILAR COMO 3%, MASA MEDIASTINAL 1% Y
MASAS RETROPERITONEALES 1%) ESPLENOMEGALIA (75%).
ANEMIA HB < 12 GR % (35%) HEMOLÍTICA AUTO INMUNE (15%).
SÍNTOMAS GENERALES: FATIGA, DISMINUCIÓN PONDERAL.
LA FIEBRE ES RARA.
REQUISITOS
LINFOCITOSIS DE MÁS DE 5 X 109/L POR MÁS DE 4 SEMANAS EN SANGRE
PERIFÉRICA.
CÉLULAS CON INMUNOGLOBULINAS DE SUPERFICIE CADENA LIGERA KAPPA O
LAMBDA.
INMUNOFENOTIPO TÍPICO ES: CD5, CD19, CD20, CD23.
M0 > 30% DE LINFOCITOS.
SOMBRAS DE GUMPRECHT EN MIELOGRAMA
Linfocitos pequeños con
núcleo de cromatina
madura y componiendo
grumos (grumelé), escaso
citoplasma y tendencia a
romperse al realizar la
extensión sanguínea, formando lo que se
denomina Sombras de Gumprecht
CLÍNICA+2
REQUISITOS

CLASIFICACIÓN DE ESTADIAJE
ESTADIO RIESGO # CASOS (%)
0Solo linfocitosis en SP (> 15 x 109)y en MO (>40%)
120 Bajo 30
I Linfocitosis + linfodenopatía 95 Intermedio 30
IILinfocitosis (+/- linfodenopatía)+ esplenomegalia
72 Intermedio 30
IIILinfocitosis (+/- linfodenopatía+/- esplenomegalia) + anemia 4b < 11gr/dl
30 Alto 5
IVLinfocitosis (+/- linfodenopatía +/- esplenomegalia +/- anemia) + plaquetas < 100 x 10/L
30 Alto 5

TRATAMIENTO
•NO TRATAMIENTO ,AL MENOS QUE DESARROLLEN MANIFESTACIONES TALES COMO:
•FIEBRE, SUDORACIÓN NOCTURNA, PESO (> 10%), ANEMIA Y/O TROMBOCITOPENIA, ESPLENOMEGALIA MASIVA, ENF. BULKY EN GANGLIOS, LINFOCITOSIS ABSOLUTA > 500,000/MM3, ANOMALÍAS CROMOSOMIALES, DOBLAJE EN < 12 MESES
BAJO RIESGO:ESTADIO 0:
•NO TRATAMIENTO, AL MENOS QUE DESARROLLEN MANIFESTACIONES ANTERIORMENTE MENCIONADAS.
•CLORAMBUCIL:DOSIS DIARIA 0.1 - 0.2 MG/KG PO POR DÍA•DOSIS INTERMITENTE 0.4-0.7 MG/KG 4 DÍAS PO/3-4SEM
+/- PREDNISONA (SI HAY SOLO FENÓMENOS AUTOINMUNES)
RIESGO INTERMEDIO:ESTADIO I-II
•CLORAMBUCIL: 6MG/D X 6 SEMANAS X 2 CURSOS•SI NO HAY RESPUESTA: CAP, CHOP, FLUDARA, PENTOSTATIN, VAD. (SEGUNDA LÍNEA)
RIESGO ALTO:ESTADIO III-IV
Radioterapia:
Si se asocia a
lesiones
locales.
Esplenectomía:
bazo con dolor
y/ofenómenos
autoinmunes.

LEUCEMIA PROLINFOCÍTICA
• Edad: 70 - 80 años• Es una variante más agresiva
de LLC, esplenomegalia masiva, linfocitosis > 100,000 pero adenopatías es raro
• Sobrevida: 24 meses (5 - 84 meses)
• Linfocitos más grandes, núcleo no completo
• La cel. B de las cel. T por tinción especial naphthyl acetato esterasa (ANAE)
• Tratamiento: Clorambutil + PD y/o CHOP y/o esplenectomía pero las respuestas son cortas.
LEUCEMIA PROLINFOCÍTICAS CRÓNICAS A CÉLULAS T
• 2% de casos de LLC.• 2 fenotipos CD4 - CD8
• CD4 : Curso maligno,se da en jovenes, adenopatías,infiltración a la piel, hiperlinfocitos e infiltración medular difusa sobrevivencia es < 2 años quimioresistente.
• CD8 : Curso benigno, SD linfocitosis granular por linfocitos con abundante citoplasma y gránulos azurofilos tienen neutropenia y menos frecuentemente aplasia de la serie roja.
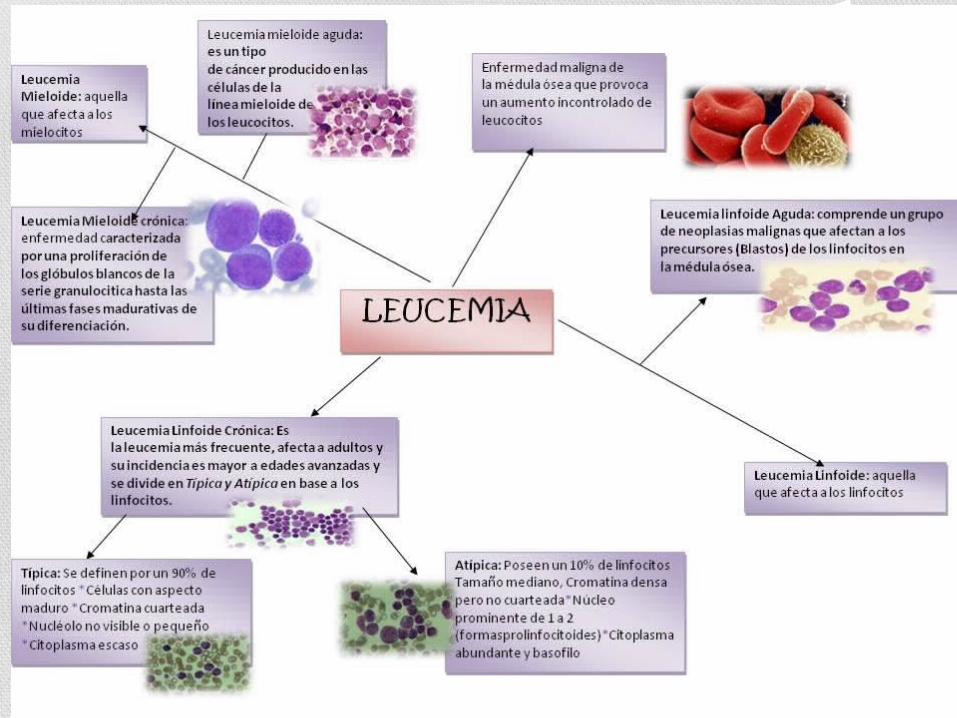
CLASIFICACIÓN

LEUCEMIA MIELOIDE AGUDA
LEUCEMIA MIELOIDE CRÓNICA
LEUCEMIA LINFOIDE AGUDALEUCEMIA LINFOIDE
CRÓNICA

CUERPOS DE AUER
CLASIFICACIÓN