
INFORME TÉCNICO FINAL DE PROYECTO DE INVESTIGACIÓN...
21
1 INFORME TÉCNICO FINAL DE PROYECTO DE INVESTIGACIÓN (ENERO 2007-DICIEMBRE 2007) CARACTERIZACIÓN DE LAS PROTEÍNAS QUE SE UNEN A REGIONES ACTIVADORAS DEL PROMOTOR DEL GEN EhPgp1 DE Entamoeba histolytica . RESUMEN En este trabajo se estudiaron las interacciones DNA-proteína que corren en la región comprendida entre –234 y –196 pb y específicamente en el repetido R9(2) del promotor del gen EhPgp1, ya que como lo habíamos reportado anteriormente esta secuencia de 9 pb, en la posición -226 a -218 pb, regula positivamente la actividad del promotor. En este trabajo encontramos que los EN de trofozoítos de la clona C2 forman tres complejos DNA-proteína con la secuencia de -234 a -196 pb y las proteínas que se unen a esta región, reconocen DNA de cadena doble y sencilla; los EN de trofozoítos de la clona C2 forman tres complejos DNA-proteína con el elemento R9(2). Además, se purificaron tres proteínas, de 64.4, 56.7 y 27.4 kDa, que se unen a la región de -234 a-196 pb, sin embargo el repetido R9(2) es reconocido específicamente por tres proteínas de 122, 61 y 42 kDa. Finalmente, se purificaron dos proteínas, de 63.9 y de 31 a 29 kDa, que se unen al repetido R9(2). INTRODUCCIÓN. Desde hace algunos años se ha documentado que Entamoeba histolytica, el protozoario causante de la amibiasis, posee el fenotipo MDR que es la capacidad para desarrollar resistencia a una droga que se usa como agente selectivo, y además presentar resistencia cruzada a otras drogas. Este fenotipo está asociado con la sobreexpresión de una glicoproteína de membrana llamada PGP, la cual es un transportador que expulsa las drogas quimioterapeúticas fuera de la célula, evitando su acción. Uno de los genes que codifican para estas PGPs es el gen EhPgp1, cuyo promotor minimo está siendo caracterizado. Hasta el momento se han identificado dos secuencias C/EBPs que participan activamente en la actividad del promotor, sin embargo recientemente hemos identificado secuencias repetidas de 9 y 7 pb cada una, localizadas en la región de -234 a -196 pb de este promotor, que también son importantes para la actividad del mismo. Particularmente un repetido de 9 pb localizado en la posición de -226 a-218 es fundamental para la actividad del promotor, ya que la incorporación de 4 o màs mutaciones en el mismo, produjo una reducción del 70% en la actividad. Es muy probable que en este sitio esté
Transcript of INFORME TÉCNICO FINAL DE PROYECTO DE INVESTIGACIÓN...

1
INFORME TÉCNICO FINAL DE PROYECTO DE INVESTIGACIÓN (ENERO 2007-DICIEMBRE 2007)
CARACTERIZACIÓN DE LAS PROTEÍNAS QUE SE UNEN A REGIONES ACTIVADORAS DEL PROMOTOR DEL GEN EhPgp1 DE Entamoeba histolytica . RESUMEN
En este trabajo se estudiaron las interacciones DNA-proteína que corren en la región
comprendida entre –234 y –196 pb y específicamente en el repetido R9(2) del promotor del
gen EhPgp1, ya que como lo habíamos reportado anteriormente esta secuencia de 9 pb, en
la posición -226 a -218 pb, regula positivamente la actividad del promotor. En este trabajo
encontramos que los EN de trofozoítos de la clona C2 forman tres complejos DNA-proteína
con la secuencia de -234 a -196 pb y las proteínas que se unen a esta región, reconocen
DNA de cadena doble y sencilla; los EN de trofozoítos de la clona C2 forman tres
complejos DNA-proteína con el elemento R9(2). Además, se purificaron tres proteínas, de
64.4, 56.7 y 27.4 kDa, que se unen a la región de -234 a-196 pb, sin embargo el repetido
R9(2) es reconocido específicamente por tres proteínas de 122, 61 y 42 kDa. Finalmente, se
purificaron dos proteínas, de 63.9 y de 31 a 29 kDa, que se unen al repetido R9(2).
INTRODUCCIÓN.
Desde hace algunos años se ha documentado que Entamoeba histolytica, el protozoario
causante de la amibiasis, posee el fenotipo MDR que es la capacidad para desarrollar
resistencia a una droga que se usa como agente selectivo, y además presentar resistencia
cruzada a otras drogas. Este fenotipo está asociado con la sobreexpresión de una
glicoproteína de membrana llamada PGP, la cual es un transportador que expulsa las
drogas quimioterapeúticas fuera de la célula, evitando su acción. Uno de los genes que
codifican para estas PGPs es el gen EhPgp1, cuyo promotor minimo está siendo
caracterizado. Hasta el momento se han identificado dos secuencias C/EBPs que participan
activamente en la actividad del promotor, sin embargo recientemente hemos identificado
secuencias repetidas de 9 y 7 pb cada una, localizadas en la región de -234 a -196 pb de
este promotor, que también son importantes para la actividad del mismo. Particularmente
un repetido de 9 pb localizado en la posición de -226 a-218 es fundamental para la
actividad del promotor, ya que la incorporación de 4 o màs mutaciones en el mismo,
produjo una reducción del 70% en la actividad. Es muy probable que en este sitio esté

2
interactuando algun factor proteico involucrado en la regulación transcripcional del gen
Ehpgp1. El conocimiento de las moleculas involucradas en la expresíon de los genes de
amiba nos permitirá contar con las herramientas indispendables para poder combatir el
problema de la amibiasis.
MÉTODOS Y MATERIALES
Cultivo de los trofozoítos de E. histolytica
Los trofozoítos de la clona C2 (resistente a emetina) se cultivaron axénicamente en
medio TYI-S-33 a 37º C.
Obtención de Extractos Nucleares
Se obtuvieron extractos nucleares (EN) de trofozoítos de la clona C2 usando el método de
Schreiber modificado (Gómez y col., 1998). Se cosecharon los trofozoítos de 6 cajas de
cultivo 225 cm2 (Corning) y se lavaron dos veces con PBS pH 6.8, centrifugando a 450 x
g por 10 min. El paquete se resuspendió en 8 volúmenes de buffer A (Hepes 10 mM pH
7.9, MgCl2 1.5 mM, KCl 10 mM, DTT 0.5 mM y PMSF 0.5 mM) y se incubaron
aproximadamente 20 min a 4º C, hasta que la mayoría de las células se mostraban
redondas. Se centrifugó a 2,809 x g por 10 min en un rotor Sorvall SS 34, el paquete se
resuspendió en 5 volúmenes de buffer A con 41 μg/ml de mezcla de inhibidores de
proteasas (PMSF 0.5 mM, benzamidina 2 mM, aprotinina 5μg/ml, pepstatina A 5 μg/ml y
leupeptina 5 μg/ml). Las células se lisaron usando un homogeneizador con pistilo, dando
25 golpes y observando al microscopio el grado de lisis. Los núcleos se recuperaron
centrifugando a 5,637 x g por 10 min. El paquete se resuspendió en 200 μl de buffer C
(Hepes 200 mM pH 7.9, NaCl 0.42 M, EDTA 1 mM, EGTA 1 mM y DTT 0.5 mM)
adicionado con mezcla de inhibidores de proteasas, se incubaron 50 min a 4º C con
agitación rotatoria y se centrifugaron a 20,800 x g por 10 min a 4º C. El sobrenadante se
distribuyó en fracciones y se almacenó a -70º C.
La integridad de los extractos se visualizó mediante el corrimiento electroforético de
estos en geles de poliacrilamida-SDS al 12%.
Ensayos de retardamiento
Oligonucleótidos complementarios de doble cadena con las secuencias de -234 a -196
pb (5’ TATCTGATAAAAATGTTATCTGAAAAAATGTTATCTGA 3’) y de -226 a –218 pb
repetida tres veces y separada por 6 pb (5’

3
AAAAATGTTCCGATCAAAAATGTTCCGATCAAAA ATGTT 3’) fueron marcados
con [γ-32P]- ATP 10 mCi/ml, 3000 mCi/mmol, usando 10 unidades de cinasa T4 en un
volumen de reacción de 15 μl (In vitrogen), la reacción se incubó 45 min a 37º C, se
adicionaron 85 μl de agua bidestilada y la marca no incorporada se limpió utilizando una
columna de Sephadex G-50. La actividad se determinó en el contador de centelleo. Las
interacciones DNA-proteína se hicieron mezclando 20 μg de extractos nucleares,
(Amersham Pharmacia Biotech), 5 μl de DNA mix (Espermidina 4 mM, MgCl2 4 mM, 1
μg de poli [d(I-C)]), 4 μl de buffer de unión de proteínas (Hepes 12 mM pH 7.9, KCl 60
mM, DTT 1 mM, EDTA 1mM, Tris HCl 4 mM pH 7.9, 10% de glicerol) y oligonucleótido
competidor. Se incubó 10 min a 4º C, posteriormente se agregó 1 ng de la sonda marcada
(20,000 cpm) y se incubó 10 min más a 4º C. Para los ensayos de competencia se utilizó
un exceso molar de 150 veces del mismo oligonucleótido sin marca u oligonucleótidos de
doble cadena conteniendo las secuencias consenso para los factores de transcripción
C/EBP (5’-CTGATGAATTGGAAAAGAAA GA-3’), HOX (5’-GTAAGAGTTATTATT
GAT-3’) y GATA1 (5’-GTTGCAGATAAA CATT-3’). Como competidor inespecífico se
utilizó un exceso molar de 350 veces de poli [d(I-C)]. Los complejos se separaron en geles
de poliacrilamida al 6 % en TBE 0.5 X (Tris-HCl 44.5 mM pH 7.9, ácido bórico 44.5 mM,
EDTA 1 mM) a 120 V por 3 h. Los geles se secaron con vació y se visualizaron por
autoradiografía utilizando un equipo Storage Phosphor Imaging System (Bio Rad). La
intensidad de las bandas se comparó mediante un análisis densitométrico.
Purificación de proteínas de unión al DNA
Se utilizó el kit de purificación DNA-binding Protein Purification Kit (Roche) y EN de
trofozoítos de la clona C2. Este método consiste en la purificación de proteínas que se
unen específicamente a una secuencia blanco de DNA conocida y se basa en el uso de
partículas magnéticas cubiertas con un oligonucleótido de 16 pb, al cual se puede ligar un
concatámero del oligonucleótido conteniendo la secuencia blanco. Cuando se agregan a
este sistema los EN conteniendo la proteína de unión al DNA deseada, la proteína es
capturada por el complejo concatámero-partícula, mientras que las proteínas inespecíficas
no se unen. Finalmente, las proteínas que se unieron específicamente al DNA, se eluyen
utilizando un amortiguador de fuerza iónica alta.
Como primer paso, se diseñaron oligonucleótidos complementarios con la secuencias: -
234 a -196 pb y -226 a -218 pb, la última repetida tres veces y separadas por un numero
igual de bases que las presentes en la secuencia original. De cada par de oligonucleótidos

4
se obtuvo un concatámero por la técnica de self-primed PCR en la cual se utilizó como
molde a los oligonucleótidos complementarios, las condiciones de la PCR se muestran en
la tabla 6.
Tabla 1. Reacción de PCR para la obtención de concatámeros. Se utilizaron 95º C como temperatura de desnaturalización, 55º C para alinear los oligonucleótidos y 72º C para amplificar.
Mezcla de dNTPs 20 mM 2 μl
Oligonucleótido sentido 20 pM
Oligonucleótido antisentido 20 pM
Thermo Pol Buffer 10X 10 μl
Deep Vent Polimerasa 2 U
Agua para 100 μl
MgSO4 1 μl
BSA 1 μl
La formación de los concátameros y su variedad de tamaños se verificó corriendo 10 μl
del producto de la self-primed PCR en geles de agarosa al 1%, junto a los marcadores de
tamaño molecular lambda digerido con HindIII y PhiX digerido con HaeIII.
Una vez verificada la formación de los concatámeros, éstos se acoplaron a partículas
magnéticas cubiertas con un oligonucleótido de 16 pb, mediante la siguiente metodología:
se tomaron 50 μl de la suspensión de partículas magnéticas, se lavaron cuatro veces con 50
μl de buffer TEN 2000 (tris-HCl 10 mM pH 8, EDTA 1mM, NaCl 2 M) y dos veces con
50 μl de buffer de ligación 1X (tris-HCl 52.5 mM, DTT 10.5 mM, MgCl2 5.24 mM, ATP
0.104 mM), utilizando un separador magnético. Después de descartar el sobrenadante del
último lavado se agregaron 10 μl de buffer de ligación 5X (Tris-HCl 262.5 mM pH 7.5,
DTT 52.5 mM, MgCl2 26.2 mM, ATP 0.52 mM), 18.75 μl de PEG 6000 (40%), 4 μl de
KCl 2.5 M, 5 μl de la reacción de PCR para la obtención del concátamero y 9.75 μl de
agua. La mezcla se homogeneizó en un vortex y se adicionaron 2.5 μl de ligasa T4 (5
U/μl). Se incubó 30 min a temperatura ambiente, agitando el tubo ocasionalmente. Las
partículas se lavaron una vez en buffer de ligación IX, tres veces con buffer de unión de
proteínas (Hepes 20 mM pH 7.6, EDTA 1 mM, (NH4)2SO4 10 mM, DTT 1 mM, 0.2% de
tween 20, KCl 30 mM), tres veces con buffer de elusión (KCl 2 M, Hepes 20 mM pH 7.6,

5
EDTA 1 mM, (NH4)2SO4 10 mM, DTT 1 mM, 0.2% de tween 20, KCl 30 mM) y tres
veces más con buffer de unión de proteínas 1X.
Para la unión de las proteínas al complejo de perlas magnéticas-concatámero, se hizo la
siguiente reacción: el complejo se incubó con 50 μg de extractos nucleares (EN) de la
clona C2, 6.25 μg de poli [d(I-C)], 0.625 μg de poli L- lisina y 25 μl de buffer de unión de
proteínas 5X (Hepes 100 mM pH 7.6, EDTA 5 mM, (NH4)2SO4 50 mM, DTT 5 mM, 1%
de Tween 20 y KCl 150 mM) en un volumen final de 125 μl y se incubaron durante 1 h a
4º C. Las partículas se lavaron 6 veces con 50 μl de buffer de unión de proteínas 1X, para
eliminar las proteínas contaminantes. Las proteínas de unión al DNA se eluyeron dos veces
con 15 μl de buffer de elusión incubando 20 min a 4º C cada vez. Las fracciones colectadas
se analizaron por electroforesis en geles de poliacrilamida-SDS al 10% (Laemmli, 1970) y
tinción con plata.
Ensayos de retardamiento con proteínas parcialmente purificadas
Se utilizó como sonda el oligonucleótido de doble cadena conteniendo la secuencia de -
226 a -218 repetida tres veces y separada por un numero igual de bases a las presentes en
la secuencia original
(5’AAAAATGTTCCGATCAAAAATGTTCCGATCAAAAATGTT 3’), se marcó con
[γ-32P]- ATP 10 mCi/ml, 3000 mCi/mmol, de la forma ya descrita. Las interacciones
DNA-proteína se hicieron incubando 1 ng de la sonda marcada, con 20 μg de EN de la
clona C2 o con 45 μl de la proteína eluída y se pusieron a interactuar bajo las condiciones
ya descritas. Los complejos se separaron en geles de poliacrilamida al 6% en TBE 0.5 X
(Tris-HCl 44.5 mM pH 7.9, ácido bórico 44.5 mM, EDTA 1 mM) a 120 V por 3 h. Los
geles se secaron con vació y se visualizaron por autoradiografía utilizando un equipo
Storage Phosphor Imaging System (Bio Rad).
Ensayos de entrecruzamiento con luz UV
Se utilizó la misma sonda que en el ensayo anterior y se marco de la manera ya descrita.
Se mezclaron 70 μg de EN de la clona C2, 12 μl de buffer de unión de proteínas, 15 μl de
DNA mix y oligonucleótido competidor, en un volumen final de 60 μl y se incubaron 10
min a 4º C; se agregó la sonda (2 ng) y la reacción se incubó 10 min más a 4º C. Los tubos
se irradiaron con luz ultravioleta (UV) durante 5 min en un transiluminador Cole-Parmer
serie 97500, 115V, 60 Hz. Se agregó buffer de corrida de proteínas 6X a cada tubo (7 ml
de tris HCl 0.5M pH 6.8, 1g de SDS, 3 ml de glicerol, 1.2 mg de azul de bromofenol, agua

6
para 10 ml) con β-mercaptoetanol y se colocaron en ebullición por 5 min. Para comprobar
la naturaleza de los complejos observados, se incluyeron dos testigos conteniendo 1 U de
DNasa I (In Vitrogene) y 20 μg de Proteinasa K (In Vitrogene), respectivamente, los
cuales se incubaron durante 30 min a 37º C. Las muestras se cargaron en un gel de
poliacrilamida al 8 % y se corrieron a 120 V durante 4 h, se incluyó un pozo con 15 μl del
marcador de peso molecular preteñido (Precision Plus Protein Dual Color Standards,
BioRad). Las competencias se hicieron utilizando un exceso molar de 150 veces del
oligonucleótido frío o de poli [d(I-C)]. Los geles se secaron con vació y se visualizaron
por autoradiografía utilizando un equipo Storage Phosphor Imaging System (Bio Rad).
RESULTADOS
Reconocimiento de la región de -234 a -196 pb por proteínas nucleares de E.
histolytica.
A fin de conocer las interacciones DNA-proteína que pudieran estar ocurriendo en esta
región del promotor se llevaron a cabo ensayos de retardamiento usando oligonucleótidos
conteniendo la región de -234 a -196 pb, que incluye todos los elementos identificados, y
la región de -226 a -218 pb que incluye el repetido R9 repetido tres veces y separado por
seis pb. La integridad de los EN de la clona C2 se visualizó mediante su migración
electroforética en un gel de poliacrilamida-SDS al 12 % (figura 1). Los extractos nucleares
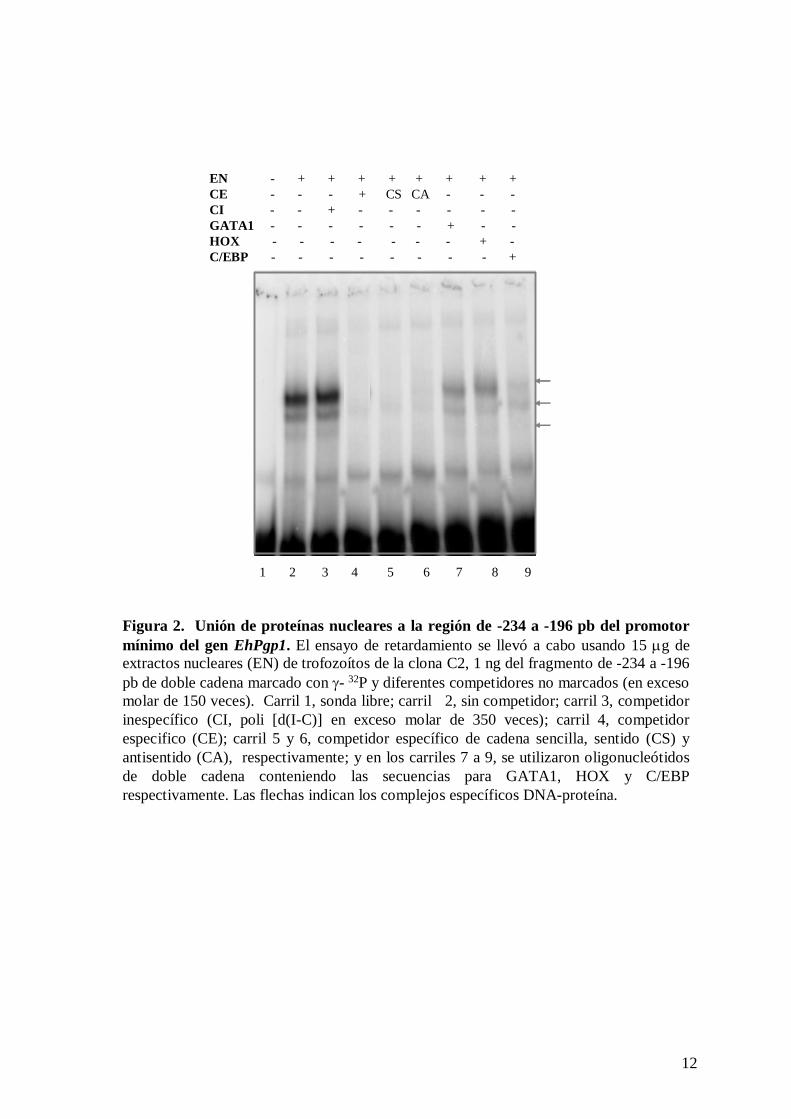
incubados con el fragmento de -234 a -196 pb formaron tres complejos DNA-proteína
(Figura 2, carril 2), los cuales fueron competidos específicamente por un exceso molar de
150 veces de sonda fría (carril 4), pero no por el competidor inespecífico poli [d(I-C)]
(Figura 2, carril 3).
Se ha reportado que algunos factores de transcripción que reconocen DNA de doble
cadena, y en particular secuencias repetidas, también pueden reconocer DNA de cadena
sencilla (Schaenman y col., 2001), por lo cual decidimos investigar si las proteínas que
están formando los complejos observados poseen esta característica.
En los carriles 5 y 6 de la figura 2 se utilizaron como competidores los oligonucleótidos
fríos de cadena sencilla, sentido y antisentido, que conforman la sonda marcada,
respectivamente. Los resultados mostraron que estos oligonucleótidos compiten
completamente la formación de los complejos DNA-proteína, lo cual indica que estas
proteínas también reconocen DNA de cadena sencilla.

7
Debido a que en diferentes organismos los promotores presentan características
estructurales similares, se han podido identificar secuencias conservadas de
reconocimiento para diferentes factores de transcripción. En E. histolytica se conoce muy
poco acerca de la presencia de dichas secuencias consenso y de las proteínas que pudieran
estar reconociéndolas. Para investigar la identidad de las proteínas que están formando
estos complejos se llevaron a cabo experimentos de competencia usando oligonucleótidos
de doble cadena conteniendo secuencias consenso para factores de transcripción,
previamente identificadas en esta región (C/EBP y GATA1), o de las que se ha sugerido
participan en la formación de un complejo multiproteíco (HOX) en está región (Gómez y
col., 1998). El análisis densitométrico de los complejos formados mostró que los
oligonucleótidos GATA1 y HOX compiten en un 49 y 26 % respectivamente en la
formación de estos complejos (figura 2, carriles 7 y 8 respectivamente), mientras que
C/EBP los compitió en un 68% (figura 2, carril 9). Estos resultados sugieren que los
complejos DNA-proteína obtenidos pueden estar formados por más de una proteína, y que
estas proteínas reconocen secuencias presentes en estos oligonucleótidos, teniendo mayor
afinidad por el oligonucleótido C/EBP. Aun cuando no se identificó alguna secuencia
consenso para el factor HOX en esta región, este oligonucleótido compitió parcialmente en
la formación de los tres complejos, lo cual sugiere que las proteínas que conforman el
complejo también pueden interactuar con éste.
Reconocimiento del repetido R9 por proteínas nucleares de E. histolytica.
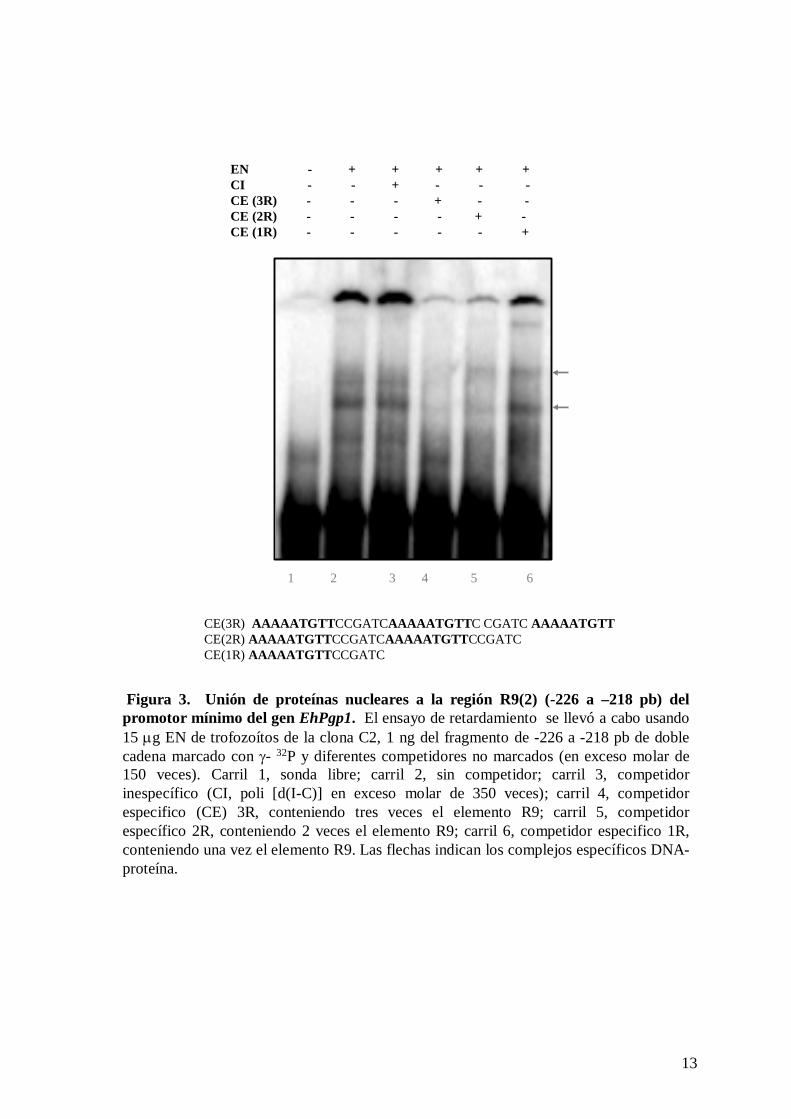
Cuando los EN se incubaron con el oligonucleótido conteniendo el elemento R9 (-226
a -218 pb) repetido tres veces, observamos la formación de tres complejos específicos
DNA-proteína (figura 3, carril 2), los cuales no fueron competidos por el competidor
inespecífico poli [d(I-C)] (figura 3, carril 3). Así mismo, debido a que existe un segundo
elemento con la misma secuencia, denominado R9(1) en la posición de -211 a -203 pb,
decidimos realizar ensayos de competencia para determinar si este segundo elemento R9
también participa en la formación de los complejos observados. Para lo cual se utilizaron
como competidores oligonucleótidos conteniendo la secuencia R9 repetida tres (3R), dos
(2R) y una (1R) vez. El análisis densitométrico mostró que el oligonucleótido 3R, compitió
un 65 % con la formación de los complejos (figura 3, carril 4), el oligonucleótido 2R,
compitió un 31% (figura 3, carril 5) y cuando se utilizó como competidor el
oligonucleótido 1R, hubo una competencia del 22 % (figura 3, carril 6). Los resultados
muestran que es necesaria la presencia de más de un elemento R9 para que ocurra la

8
interacción DNA-proteína, lo cual sugiere que el primer repetido R9 (R9(1)) también está
involucrado en la regulación transcripcional del gen EhPgp1.
Purificación de las proteínas nucleares que se unen a la región de -234 a -196 pb y a la
región R9 (-226 a -218 pb) del promotor del gen EhPgp1
Con el fin de identificar y caracterizar las proteínas que están formando estos complejos
se procedió a su purificación parcial. Los factores nucleares que se unen a las regiones -
234 a -196 pb y al oligonucleótido 3R, se purificaron parcialmente por cromatografía de
afinidad al DNA, usando el kit DNA binding protein purification (Roche), EN de la clona
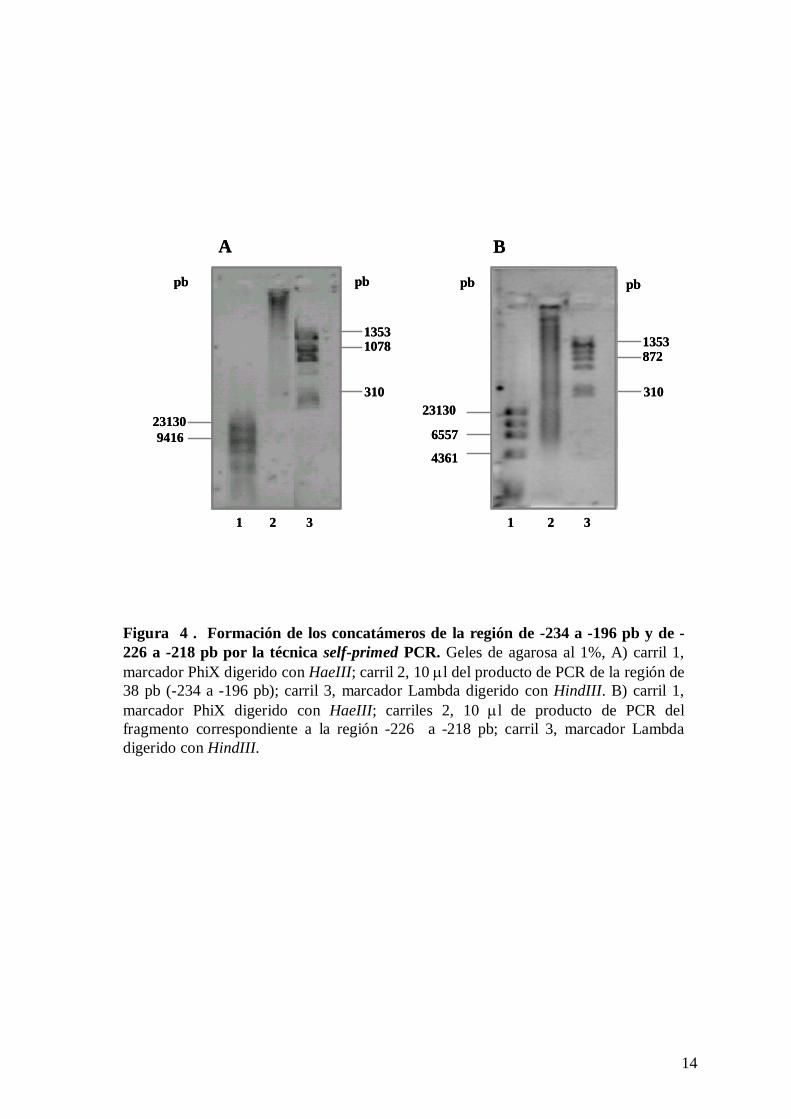
C2 y los oligonucleótidos concatamerizados. Estos concatámeros se obtuvieron por la
técnica de self-primed PCR, los productos se muestran en la figura 4. El barrido observado
indica la variedad de los amplificados obtenidos de diferente tamaño molecular (figura 4,
A y B, carril 2).
Los eluídos obtenidos de la purificación se corrieron en geles de poliacrilamida al 10% y
éstos se tiñeron con plata. Cuando se utilizó como sonda el concatámero de 38 pb (-234 a -
196), conteniendo todos los repetidos que estamos estudiando, se observó el
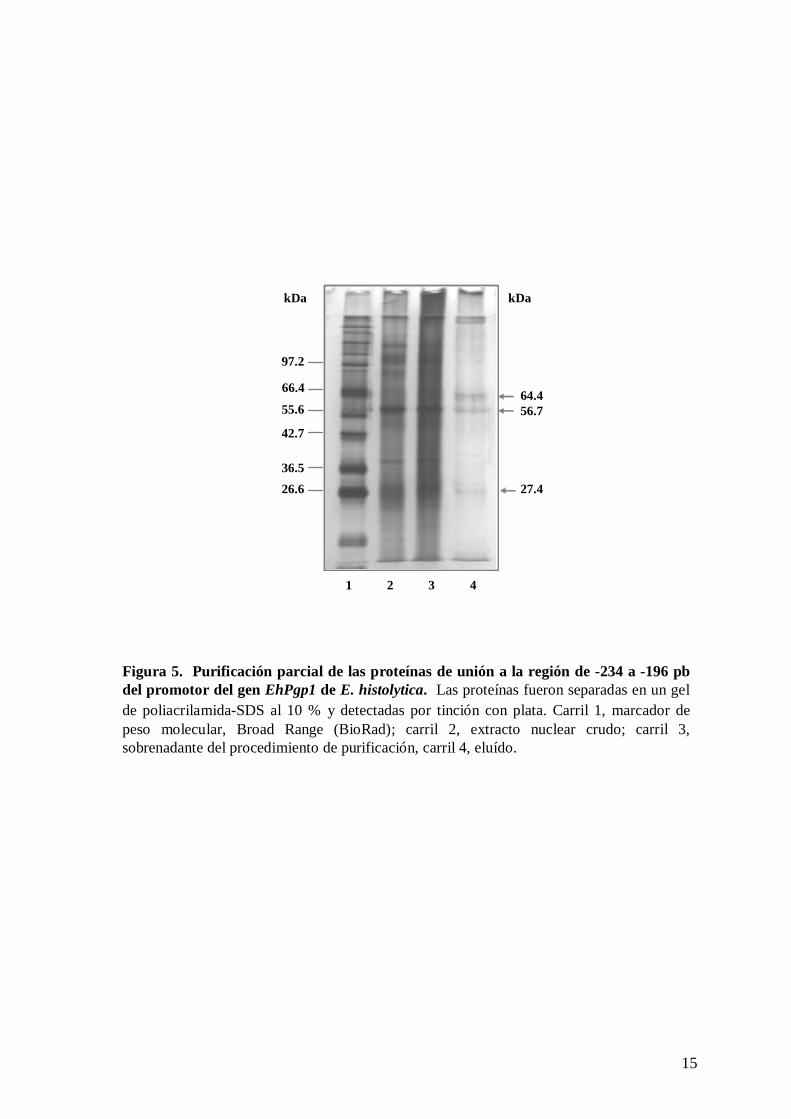
enriquecimiento de tres bandas correspondientes a 64.4. 56.7 y 27.4 kDa (figura 5, carril
4), lo cual apoya la idea de que los complejos DNA-proteína que se forman en está región
están formados por más de una proteína. Por otro lado, cuando se utilizó como DNA sonda
el concatámero conteniendo tres veces la región R9, se detectaron tres bandas de mayor
intensidad de entre 29 y 31 kDa y una banda menos intensa de 63.6 kDa (figura 6, carril 7),
las cuales podrían corresponder con las bandas de 64.4 y 27.4 kDa obtenidas con el
oligonucleótido de 38 pb (figura 5). Esto sugiere que estas proteínas están interactuando en
los elementos R9 y que la proteína de 56.7 kDa probablemente esté reconociendo las
secuencias intermedias (R7).
Interacciones DNA-proteína con las proteínas parcialmente purificadas
Con la finalidad de determinar si las proteínas parcialmente purificadas eran capaces de
interactuar con el repetido R9, se llevaron a cabo ensayos de retardamiento utilizando la
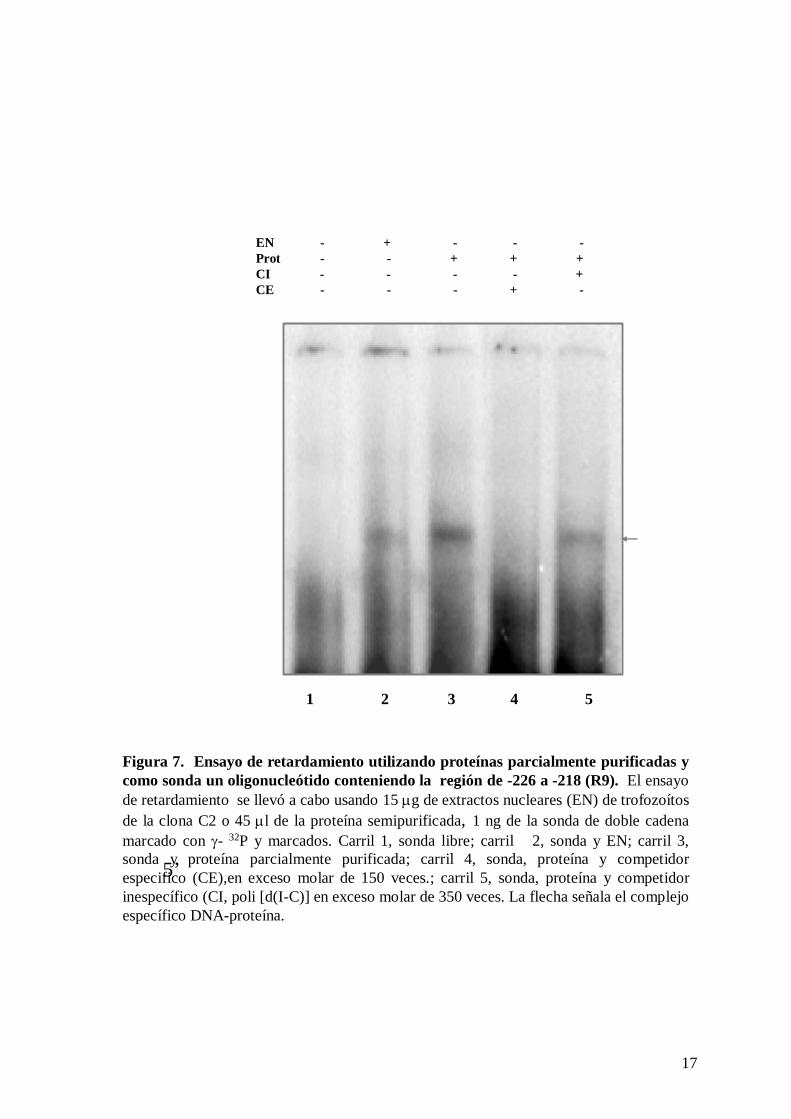
fracción purificada de los EN y el repetido R9 sin concatamerizar. Los resultados
mostraron la formación de un complejo DNA-proteína (figura 7, carril 3), el cual tiene la
misma movilidad electroforetica que el complejo formado con EN de la clona C2 (figura 7,
carril 2), aunque la intensidad del primero es mayor. Este complejo además es competido
por la misma sonda fría (figura 7, carril 4), pero no por el competidor inespecífico poli d(I-

9
C) (figura 7, carril 5). Este experimento demuestra la especificidad de las proteínas
parcialmente purificadas y la afinidad de estas por los repetidos R9.
Para determinar el peso molecular de las proteínas unidas a la sonda conteniendo el
elemento R9 repetido tres veces, se realizó una prueba de entrecruzamiento usando EN de
trofozoítos de la clona C2. Las proteínas entrecruzadas se separaron por electroforesis en
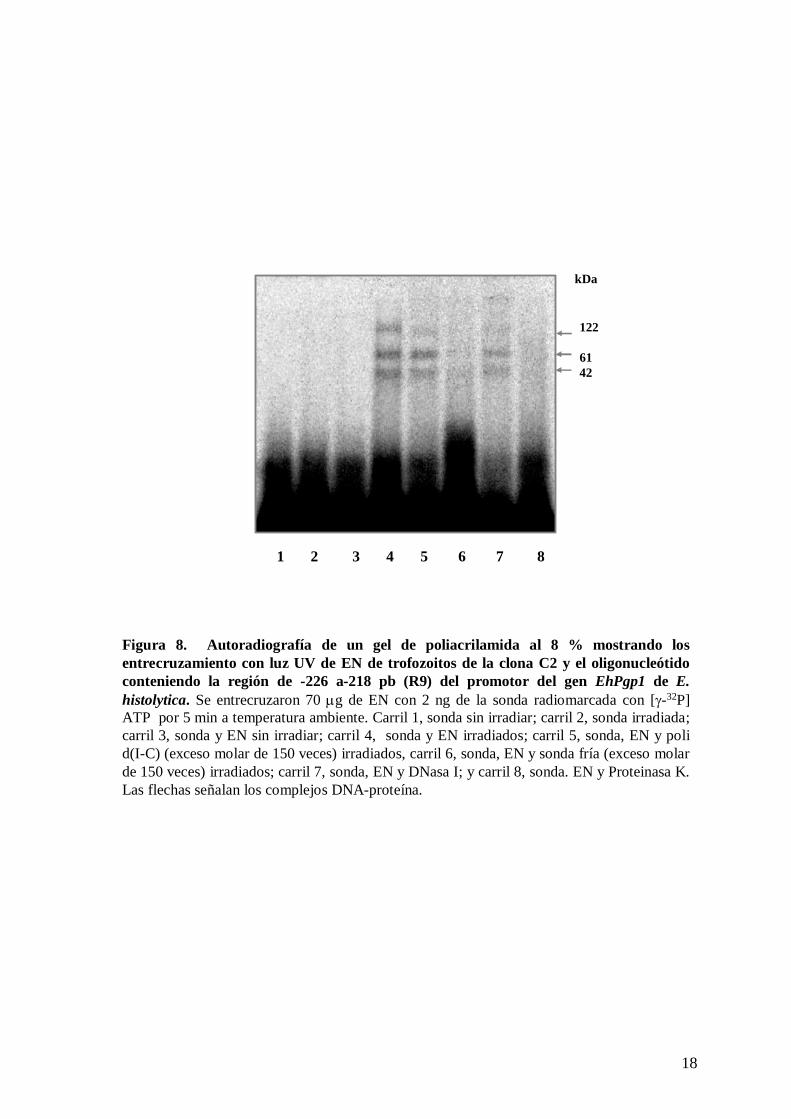
un gel de poliacrilamida al 8 % y fueron analizadas por autoradiografía. En la imagen se
observan tres bandas de alrededor de 146, 85 y 66 kDa (figura 8, carril 4). Debido a que el
peso molecular de la sonda de 38 pb es de 24.66 kDa, el peso molecular de los factores
entrecruzados con el oligonucleótido fue recalculado en alrededor de 122, 61 y 42 kDa.
Estas bandas no se detectaron cuando se utilizó, en la mezcla de unión, un exceso molar de
150 veces de sonda fría (figura 8, carril 6), pero no se afectaron por el competidor
inespecífico (figura 8, carril 5), indicando la especificidad de los complejos. Cuando la
sonda y EN no se irradiaron con luz UV, no se encontraron especies marcadas (figura 8,
carril 2). Cuando a los entrecruzados se les agregó DNasa I, no se observó alteración en la
migración de las bandas, sin embargo, al adicionar proteinasa K, los complejos
desaparecieron (figura 8, carriles 7 y 8), lo cual comprueba que las bandas observadas son
complejos DNA-proteína.
Los pesos moleculares encontrados para estas proteínas no nos proporcionan información
concluyente, aun cuando se ha reportado, para los factores de transcripción cuya secuencia
consenso está presente en esta región, que el gen C/EBPβ codifica para varias isoformas
cuyos pesos moleculares son de 40, 35, 20 y 8.5 kDa (Grigorov y col., 1998). Marchat y
col, 2002 demostraron la presencia de proteínas C/EBP en amiba las cuales son
reconocidas por anticuerpos heterólogos anti-C/EBP de humano cuyos pesos moleculares
son 25 y 65 kDa.
También se consideró la posibilidad de que alguna de estas proteínas pertenezca a la
familia de factores de transcripción GATA, debido a la presencia de secuencias consenso
para algunos de estos factores (GATA1-2, NIT2, GAL4). NIT2 es una proteína
regulatoria de N. crassa cuyos sitios de unión en el promotor de algunos genes, tienen al
menos dos secuencia mínimas GATA para formar un complejo fuerte DNA-proteína, la
distancia efectiva entre estos elementos puede variar entre 5-30 pb sin embargo el espació
óptimo es de 20 pb (Chiang y Marzluf, 1994). En la región de 38 pb que estamos
estudiando (-234 a -196 pb) se localizan tres secuencias GATA las cuales están separadas
por 12 pb, lo cual por lo anterior se consideraría como una distancia efectiva. Además en
nuestros ensayos de funcionalidad encontramos que cuando se elimina uno de estos

10
repetidos (RCIII) la actividad del promotor se abate en un 58%, y en un 87% cuando se
eliminan dos de ellos (RCIII y RCII), estas observaciones nos sugieren que las proteínas
que estamos caracterizando pudieran pertenecer a la familia GATA, o bien que requieran
interactuar con regiones repetidas en el DNA para formar un complejo estable.
Actualmente se están obteniendo las proteínas purificadas y con ellas realizaremos
ensayos de retardamiento y entrecruzamiento. Así mismo, con la finalidad de determinar la
identidad de estas realizaremos ensayos de Western Blot utilizando anticuerpos
heterólogos anti C/EBP y anti GATA1 de humano.
Chiang y Marzluf en 1994, trabajando con el factor de transcripción NIT2 de N. crassa,
encontraron que los sitios de unión al DNA de alta afinidad para este factor son secuencias
repetidas las cuales contienen al menos dos secuencias centrales GATA, y que cuando
cambian un solo nucleótido de alguno de estos sitios se elimina la unión de NIT2,
demostrando así la importancia de estos repetidos. Interesantemente, nosotros
identificamos la secuencia GATA en los repetidos cortos RCIII, RCII y RCI, no obstante
necesitamos dilucidar la relevancia de estas secuencias en el control de la expresión del
gen EhPgp1.
11
Figura 1. Integridad de los extractos nucleares (EN) de trofozoítos de la clona C2. Corrimiento electroforético en un gel de poliacrilamida-SDS al 12 %, de 80 μg de EN de dos diferentes extracciones. Las bandas de proteína se tiñeron con azul de Coomasie.
12
1 2 3 4 5 6 7 8 9
EN - + + + + + + + +CE - - - + CS CA - - -CI - - + - - - - - -GATA1 - - - - - - + - -HOX - - - - - - - + -C/EBP - - - - - - - - +
Figura 2. Unión de proteínas nucleares a la región de -234 a -196 pb del promotor mínimo del gen EhPgp1. El ensayo de retardamiento se llevó a cabo usando 15 μg de extractos nucleares (EN) de trofozoítos de la clona C2, 1 ng del fragmento de -234 a -196 pb de doble cadena marcado con γ- 32P y diferentes competidores no marcados (en exceso molar de 150 veces). Carril 1, sonda libre; carril 2, sin competidor; carril 3, competidor inespecífico (CI, poli [d(I-C)] en exceso molar de 350 veces); carril 4, competidor especifico (CE); carril 5 y 6, competidor específico de cadena sencilla, sentido (CS) y antisentido (CA), respectivamente; y en los carriles 7 a 9, se utilizaron oligonucleótidos de doble cadena conteniendo las secuencias para GATA1, HOX y C/EBP respectivamente. Las flechas indican los complejos específicos DNA-proteína.
13
EN - + + + + +CI - - + - - -CE (3R) - - - + - -CE (2R) - - - - + -CE (1R) - - - - - +
Figura 3. Unión de proteínas nucleares a la región R9(2) (-226 a –218 pb) del promotor mínimo del gen EhPgp1. El ensayo de retardamiento se llevó a cabo usando 15 μg EN de trofozoítos de la clona C2, 1 ng del fragmento de -226 a -218 pb de doble cadena marcado con γ- 32P y diferentes competidores no marcados (en exceso molar de 150 veces). Carril 1, sonda libre; carril 2, sin competidor; carril 3, competidor inespecífico (CI, poli [d(I-C)] en exceso molar de 350 veces); carril 4, competidor especifico (CE) 3R, conteniendo tres veces el elemento R9; carril 5, competidor específico 2R, conteniendo 2 veces el elemento R9; carril 6, competidor especifico 1R, conteniendo una vez el elemento R9. Las flechas indican los complejos específicos DNA-proteína.
1 2 3 4 5 6
CE(3R) AAAAATGTTCCGATCAAAAATGTTC CGATC AAAAATGTTCE(2R) AAAAATGTTCCGATCAAAAATGTTCCGATCCE(1R) AAAAATGTTCCGATC
14
Figura 4 . Formación de los concatámeros de la región de -234 a -196 pb y de -226 a -218 pb por la técnica self-primed PCR. Geles de agarosa al 1%, A) carril 1, marcador PhiX digerido con HaeIII; carril 2, 10 μl del producto de PCR de la región de 38 pb (-234 a -196 pb); carril 3, marcador Lambda digerido con HindIII. B) carril 1, marcador PhiX digerido con HaeIII; carriles 2, 10 μl de producto de PCR del fragmento correspondiente a la región -226 a -218 pb; carril 3, marcador Lambda digerido con HindIII.
1 2 3 1 2 3
A B
13531078
310
231309416
pb pb pb pb
1353872
31023130
6557
4361
1 2 3 1 2 3
A B
13531078
310
231309416
pb pb pb pb
1353872
31023130
6557
4361
15
Figura 5. Purificación parcial de las proteínas de unión a la región de -234 a -196 pb del promotor del gen EhPgp1 de E. histolytica. Las proteínas fueron separadas en un gelde poliacrilamida-SDS al 10 % y detectadas por tinción con plata. Carril 1, marcador de peso molecular, Broad Range (BioRad); carril 2, extracto nuclear crudo; carril 3, sobrenadante del procedimiento de purificación, carril 4, eluído.
97.2
64.456.7
27.4
66.4
55.6
42.7
36.526.6
1 2 3 4
kDa kDa

16
63.6
29-31
Figura 6. Purificación parcial de las proteínas nucleares que se unen a la región de-226 a –218 pb (R9), del promotor del gen EhPgp1 de E. histolytica. Las proteínas
fueron separadas en geles de poliacrilamida-SDS al 10% y detectadas por tinción con plata. Carril 1, marcador de peso molecular, Broad Range BioRad; carril 2, extracto nuclear crudo; carril 3, sobrenadante del procedimiento de purificación, carril 4-6, lavados del procedimiento de purificación; carril 7, eluído.
1 2 3 4 5 6 7
kDa
1169766
45
31
21
kDa
17
EN - + - - -Prot - - + + +CI - - - - + CE - - - + -
5’
1 2 3 4 5
Figura 7. Ensayo de retardamiento utilizando proteínas parcialmente purificadas y como sonda un oligonucleótido conteniendo la región de -226 a -218 (R9). El ensayo de retardamiento se llevó a cabo usando 15 μg de extractos nucleares (EN) de trofozoítosde la clona C2 o 45 μl de la proteína semipurificada, 1 ng de la sonda de doble cadena marcado con γ- 32P y marcados. Carril 1, sonda libre; carril 2, sonda y EN; carril 3, sonda y proteína parcialmente purificada; carril 4, sonda, proteína y competidor especifico (CE),en exceso molar de 150 veces.; carril 5, sonda, proteína y competidor inespecífico (CI, poli [d(I-C)] en exceso molar de 350 veces. La flecha señala el complejo específico DNA-proteína.
18
Figura 8. Autoradiografía de un gel de poliacrilamida al 8 % mostrando los entrecruzamiento con luz UV de EN de trofozoitos de la clona C2 y el oligonucleótidoconteniendo la región de -226 a-218 pb (R9) del promotor del gen EhPgp1 de E. histolytica. Se entrecruzaron 70 μg de EN con 2 ng de la sonda radiomarcada con [γ-32P] ATP por 5 min a temperatura ambiente. Carril 1, sonda sin irradiar; carril 2, sonda irradiada; carril 3, sonda y EN sin irradiar; carril 4, sonda y EN irradiados; carril 5, sonda, EN y poli d(I-C) (exceso molar de 150 veces) irradiados, carril 6, sonda, EN y sonda fría (exceso molar de 150 veces) irradiados; carril 7, sonda, EN y DNasa I; y carril 8, sonda. EN y Proteinasa K. Las flechas señalan los complejos DNA-proteína.
1 2 3 4 5 6 7 8
122
6142
kDa

19
Impacto.- Los estudios de la expresión de los genes en organismos eucariontes se ha
centrado principalmente en animales, plantas y hongos, mientras que poca información hay
disponible para protozoarios parásitos.
Entamoeba histolytica es un protozoario anaerobio responsable de la amibiasis humana. El
conocer la estructura de su DNA y los mecanismos que regulan la transcripción de sus
genes es un paso hacia el entendimiento de su virulencia. Además, nos permitirá ampliar
nuestro conocimiento respecto al fenómeno MDR en E. histolytica, el cual es un
mecanismo que el parásito ha desarrollado para sobrevivir en presencia de los fármacos que
se usan para combatir el problema de la amibiasis, y de esta forma poder contribuir a la
erradicación y prevención de esta enfermedad.
Bibliografía.
• Espinosa-Cantellano, M. and Matínez-Palomo, A. 2000. Pathogenesis of intestinal
amoebiasis: from molecules to disease. Clin. Microbiol. Rev. 13 (2): 318-331.
• Petri, W. A. Jr., Haque, R., Lyerly, D., and Vines, R. R. 2000. Estimating the
impact of amebiasis on health. Parasitol. Today 16 (8): 320-321.
• http://www.dgepi.salud.gob.mx/boletin/2006/sem9/pdf/cua4.3.
• Samarawickrema, N; Brown, D; Upcroft, J; Thammapalerd, N; Upcroft, P. 1997.
Involvement of superoxide dismutase and pyruvate: Ferredoxin oxidoreductase in
mechanisms of metronidazole resistance in E. histolytica. J. Antimicrobiol.
Chemother. 40: 833-840.
• Gómez, C., Pérez, G., López-Bayghen E., and Orozco, E. 1998. Transcriptional
analysis of the EhPgp1 promoter of Entamoeba histolytica multidrug resistant
mutant. J. Biol. Chem. 273: 7277-7284.
• Marchat, L. A., Gomez, C., Perez, D. G., Paz, F., Mendoza, L., and Orozco, E.
2002. Two CCAAT/enhancer binding protein sites are cis-activator elements of the
Entamoeba histolytica EhPgp1 (mdr-like) gene expression. Cell Microbiol. 4 (11):
725-737.
• Nickel, R., and Tannich, E. 1994. Transfection and transient expression of
chloramfenicol acetyltransferase gene in the protozoan parasite Entamoeba
histolytica. Proc. Natl. Acad. Sci. USA 91: 7095-7098.
• Sambrook, J., Fritsch, E. F., and Maniatis, T. 1989. Molecular cloning. A
laboratotory manual. 2º Ed.

20
• Neumann, J. R., Morency, C. A., and Russian, K. O. 1987. A novel rapid assay for
chloramphenicol acetyltransferase gene expression. Biotechniques 5: 444-446.

21
México, D. F. a 11 de enero de 2008.
Secretaría de Investigación y Posgrado
Instituto Politécnico Nacional
A la comisión evaluadora de los proyectos de investigación SIP
Por medio de este documento quiero hacer de su conocimiento que en mi informe
técnico final del proyecto de investigación 2007 y en mi ficha de productividad no
reporto ningún tipo de producto en el rubro III, que corresponde a Formación de
Recursos Humanos, sin embargo en este momento cuento con un estudiante PIFI a nivel
licenciatura y una estudiante que esta haciendo su tesis de maestría y que se graduará en
el transcurso del presente año. Además quiero informarle que con los resultados hasta
ahora obtenidos hemos escrito un artículo y lo enviamos a una revista internacional, de
la cual estamos esperando una respuesta para su publicación.
Sin más por el momento, le agradezco la atención prestada al presente documento.
Atentamente,
_______________________________________
Dra. María Esther Ramírez Moreno Profesor Asociado ENMyH-IPN


















