
RABDOMIOLISIS Dr. Mario Arévalo 4to año de la residencia de Emergentologia HCIPS Año 2013
Upload
nguyenliemCategory
view
215download
0
Enfermedades neuromusculares
Dr. Piero Canepa L.


PCR POR SUCCI • Succi se introdujo en 1951. Primer PCR 1953.
• Mecanismo: Hiperkalemia (1967)
• Factores predisponentes: Quemados, trauma muscular, denervación, UTI prolongada (atrofia muscular + denerv. por RM)
• 2 mecanismos: Upregulation
Rabdomiolisis
• Sin relación a Hipertemia Maligna

UPREGULATION

RABDOMIOLISIS • Pérdida función sarcolema
• Fuga de contenido intracelular: • Mioglobina • Potasio • Creatinkinasa
• Causas farmacológicas y no farmacológicas
• Generalmente tienen miopatía de base, conocida o no


RECOMENDACIÓN FDA
“Tenerla siempre, usarla nunca”

PCR INESPERADO SIN SUCCI
• 1987: Adenoidectomía bajo halotano s/relajantes.
Rabdomiolisis en recuperación ⇒ DMD no dg.
• 1990: PCR inesperado en recup. en DMD.
Rabdomiolisis severa dp isoflurano s/succi
• 1999: DMD anestesiado con sevo s/RM
Rabdomiolisis en recup. sin PCR
• 2005: Sevo + Iso s/succi. Rabdomiolisis severa en recup. PCR ⇒ DMD no diagnosticada

PCR INESPERADO SIN SUCCI
• 8a. Distrofia Becker leve. Anestesia 35’. A los 10’ en recup. PCR. K=12 mEq/l. RCP 2 hrs. Paraplejia T7
• PCR 10’ postinducción. RCP exitosa tras 90’.
• 6a. 80’ postinducción. K=7,9 mEq/l. CK=200.000 U/l. RCP=Muerte cerebral. Asintomático preop., pero historia pigmenturia y CK reposo 13.000
• 6a. Probable DMD ⇒ Bp muscular. CK basal=15.000. Anestesia con N20 + halotano. PCR en PACU 15’. RCP exitosa. K=7,9 mEq/l. CK>25.000 U/l

MIOPATIAS
I. Hereditarias
II. Inflamatorias
III. Endocrinológicas/metabólicas
IV. Tóxicas
V. Tumorales

MIOPATIAS HEREDITARIAS
A. Distrofias musculares
B. Miopatías congénitas
C. Miopatías metabólicas 1. Glucógeno 2. Metabolismo lípidos 3. Purinas 4. Mitocondriales
D. Trastornos de la excitabilidad de membrana 1. Miotonía 2. Parálisis periódica
• Hipokalémica • Hiperkalémica

DISTROFIAS MUSCULARES
• Miopatías más frecuentes
• Causa genética. Herencia mendeliana diversa
• Período de función normal temprano en la vida, luego deterioro progresivo
• Afectan grupos musculares característicos
• Sin tratamiento

DISTROFIAS MUSCULARES • Distrofia muscular de Duchenne y Becker
• Distrofia de Emery-Dreifuss
• Distrofia en cinturón de los miembros (Limb-girdle)
• Distrofia facioescapular
• Distrofia muscular distal
• Distrofia oculofaríngea

DISTROFIAS MUSCULARES

DISTROFIA DE DUCHENNE-BECKER
• Distrofia más frecuente (1 : 3.500 niños)
• Herencia recesiva ligada al X (sólo sexo masculino)
• Afecta formación de proteína distrofina
• Inicio en infancia temprana, debilidad progresiva proximal en piernas y brazos (signo de Gower)
• Pantorrillas pseudohipertróficas
• 70% tienen compromiso miocárdico
• Requieren silla de ruedas ± 12 años
• Muerte ± 20 años
• Distrofia Becker: curso más benigno (Debuta 12a, muerte a los 40 – 50 años)

DISTROFIA DE DUCHENNE-BECKER

DISTROFIA DE DUCHENNE-BECKER

SIGNO DE GOWER

ANESTESIA EN DMD • Reporte de casos creciente de rabdomiolisis en DMD sometidos
a halogenados con y sin succinicolina
• Varios PCR en este grupo. Algunos fatales
• No está clara la fisiopatología (numerosos reportes de anestesia inhalatoria sin incidentes)
• Succinilcolina está contraindicada en DMD
• Todavía no hay consenso en contraindicar halogenados
• Problema de casos no diagnosticados
• Búsqueda de antecedentes en entrevista (<10% distróficos no tienen historia familiar o personal)
• Screening con CPK ante la duda

MIOPATIAS HEREDITARIAS
A. Distrofias musculares
B. Miopatías congénitas
C. Miopatías metabólicas 1. Glucógeno 2. Metabolismo lípidos 3. Purinas 4. Mitocondriales
D. Trastornos de la excitabilidad de membrana 1. Miotonía 2. Parálisis periódica
• Hipokalémica • Hiperkalémica

MIOPATIAS CONGENITAS • Herencia mendeliana
• Rasgos miopatológicos específicos
• Hipotonía desde el nacimiento. Curso no progresivo, a veces mejoran. Elevación mínima o nula de CPK.
• Relación con Hipertermia Maligna: • Miopatía de Evans (MH myopathy) • Síndrome King-Denborough • Enfermedad del núcleo central (Central-core disease) • Parálisis periódica hipokalémica

SINDROME KING-DENBOROUGH

CENTRAL-CORE DISEASE

HIPERTERMIA MALIGNA
• Trastorno genético de la membrana muscular con desregulación del calcio intracelular
• Principal mutación afecta al gen del receptor de rianodina (RyR1). 50% familias.
• Genética heterogénea: 100 mutaciones distintas. Alteración de otras proteínas también generan HM.
Múltiples genes, múltiples alelos
• Gatillantes anestésicos y no anestésicos


HIPERTERMIA MALIGNA

HIPERTERMIA MALIGNA

HIPERTERMIA MALIGNA • Más de 100 mutaciones conocidas en gen RyR1.
22 son reconocidas como causales de MH. Existen además otros locus (Subunidad α1 de DHPR, canal Na)
• 30% MHS tienen genética compatible en RyR1
• Si familiar de MHS comparte el gen ⇒ MHS sin biopsia
• Si familiar de MHS no tiene el gen ⇒ IVCT
• El unico mecanismo para certificar MHN es el IVCT

HM NO ANESTESICA
• Golpe de calor (ejercicio + calor ambiente)
• Rabdomiolisis (stress, tóxica, espontánea)
• Síndrome neuroléptico maligno
• Sd. muerte súbita infantil

MIOPATIAS HEREDITARIAS
A. Distrofias musculares
B. Miopatías congénitas
C. Miopatías metabólicas 1. Glucógeno 2. Metabolismo lípidos 3. Purinas 4. Mitocondriales
D. Trastornos de la excitabilidad de membrana 1. Miotonía 2. Parálisis periódica
• Hipokalémica • Hiperkalémica

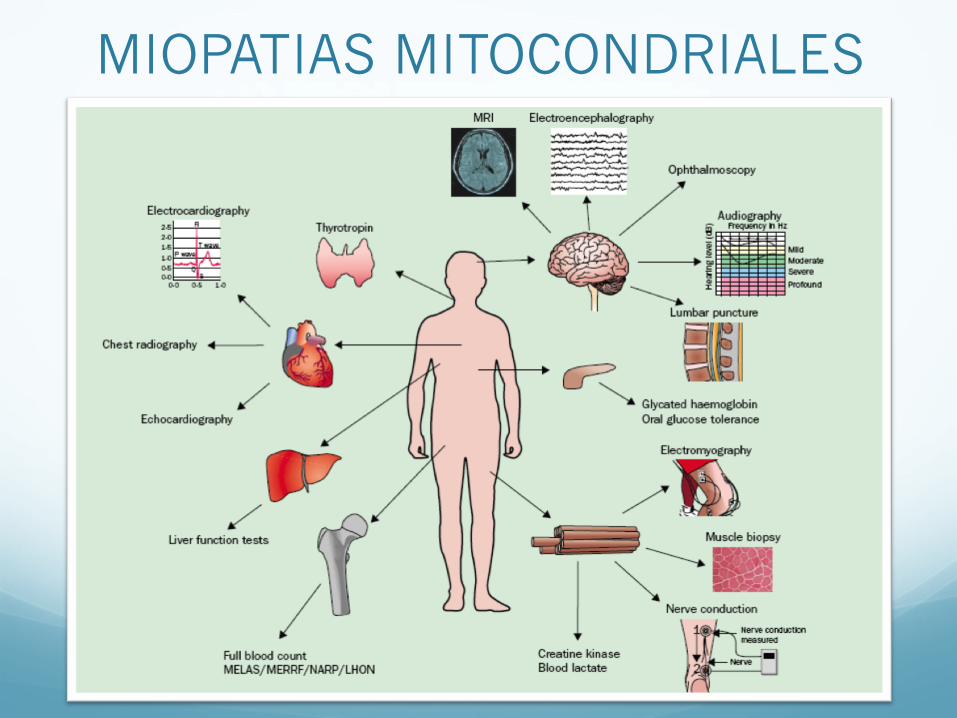
MIOPATIAS MITOCONDRIALES
• Enfermedades de la cadena respiratoria
• 10-15 casos por 100.000
• Pueden ser causadas por mutaciones de mtDNA o nDNA
Genética mitocondrial o mendeliana
• Frecuente compromiso muscular y encefálico ⇒ Encefalomiopatías mitocondriales
• Genética mitocondrial: Características o Herencia materna o Heteroplasmía y efecto umbral o Segregación mitótica

MIOPATIAS MITOCONDRIALES

MIOPATIAS MITOCONDRIALES

ENFERMEDADES mtDNA
• Generalmente multisistémicas
• Expresión variable entre órganos e intraórgano
• Presencia de portadores oligo o asintomáticos
• Preferencia por tejidos de alto consumo energético
• Idéntica mutación puede manifestarse de manera distinta (distribución durante embriogénesis)

ENFERMEDADES nDNA
• Herencia mendeliana
• Expresión en todas las células (y mitocondrias)
• Mutaciones dirigidas a: o Proteínas estructurales de la cadena respiratoria o Proteínas de ensamblaje o Proteínas de replicación y transcripción de mtDNA o Metabolismo de cardiolipinas
MIOPATIAS MITOCONDRIALES

CONSIDERACIONES ANESTESICAS
• Enfermedades multisistémicas: • SNC: Encefalopatía, RM, disf. autonómica, apnea • Musculo esquelético: Miopatía • Corazón: MCP dilatada/hipertrófica, defectos conducción • Otras: Anemia, infecciones recurrentes, hipoglicemia
• Descompensación severa metabólica y neurológica por stress, infección, cirugía y anestesia
• Reporte de MHS en enf. mitocondriales

CONSIDERACIONES ANESTESICAS


















