
Patologia del desarrollo craneal
81
PATOLOGIA DEL DESARROLLO CRANEOESPINAL
-
Upload
xlucyx-xxlucyxx -
Category
Health & Medicine
-
view
54 -
download
7
Transcript of Patologia del desarrollo craneal

PATOLOGIA DEL DESARROLLO
CRANEOESPINAL

EMBRIOLOGÍA• El SNC aparece al comienzo de la tercera
semana(18º día) del desarrollo.
25º día
28º día
Cierre

ETAPAS DEL DESARRROLLO DEL S.N.C1. Neurulación primaria.(días 18 -28)3ª semana2. Formación de vesículas cerebrales(días:25- 35)4ªsemana3. Neurulación secundaria.(días: 28 -48)5ª semana
NEURULACIÓN PRIMARIA
DIA 18: Aparición de placa neural ,delimitación de placa neural Formación de surco neuralDIA 21: Cierre del surco neural desde nivel cervical =formación del tubo neural( hasta segmentos lumbares)DIA 25: Cierre del neuroporo anteriorDIA 28: Cierre del neuroporo posterior



Anomalías Espinales
1. SÍNDROMES COMUNES DE DISRRAFISMO ESPINAL,
Manifestaciones neurológicas Manejo

Anomalías congénitas.Disrafias
• Es toda malformación ocasionada por un cierre defectuoso del tubo neural, hueso, piel y/o
musculo supra yacente.

CLASIFICACIÓN DE KOCH
• ESPINA BÍFIDA : Defecto de los arcos óseos vertebrales posteriores (1/1000).
• Tipos :1. Oculta : Simple, Seno dermoide, lipoma,
mielocistocele
2. Manifiesta o quistica: Meningocele (mejor pronostico) Mielomeningocele(mas frecuente) Mielocele(peor pronostico).

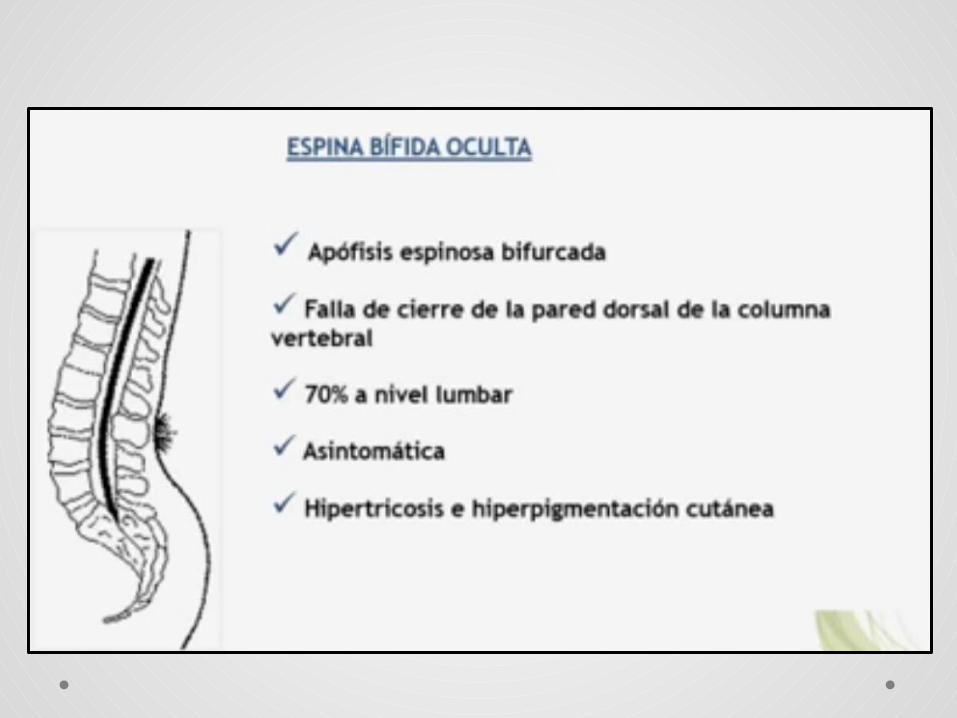

Disrafismo oculto.• Cubierto por piel.• Estigmas cutáneos (hipertricosis , angiomas
capilares)• Complicaciones: Síndrome de medula anclada


DX DIFERENCIAL

TRATAMIENTO• Se recomienda la cirugía a pesar de ser
asintomáticos por el riesgo de:1. Infección 2. Síndrome de medula anclada

Ministerio de Salud. Guía Clínica Disrafias Espinales, diagnóstico y tratamiento. Minsal, 2011.
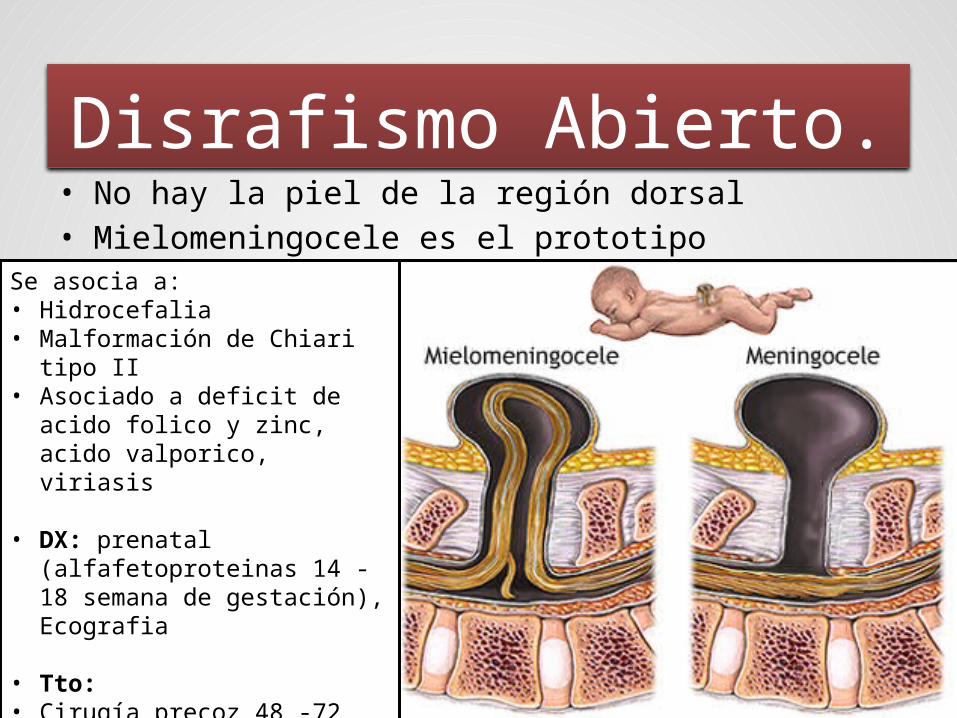
• No hay la piel de la región dorsal• Mielomeningocele es el prototipo
Disrafismo Abierto.
Se asocia a:• Hidrocefalia• Malformación de Chiari tipo
II• Asociado a deficit de acido
folico y zinc, acido valporico, viriasis
• DX: prenatal (alfafetoproteinas 14 -18 semana de gestación), Ecografia
• Tto: • Cirugía precoz 48 -72 horas
(Cesarea ),• -Derivacion(LCR) en
hidrocefalia(80%)



Incidencia: 1/2000

ENCEFALOCELE
El encefalocele es herniación del tejido cerebral, las meninges y cefalorraquídeo fluido (CSF) fuera de los confines del cráneo

Epidemiologia• Los encefaloceles se localizan en la región
occipital en el 75% de los casos y en menor proporción, alrededor del 15%, pueden localizarse en región parietal, frontal y sincipital estos últimos se subclasifican por su localización en: nasofrontal, nasoetmoidal y nasoorbital.

ETIOLOGIA• Causas origen multifactorial:
• Genético (Polimorfismos de la metilentetrahidrofolato reductasa) sustitución de la citosina por la timina en la base 677, la mutación C677T, que origina una sustitución de valina por alanina en la enzima
• Ambiental • El 98% de los casos se debe a un déficit de folatos.• Fármacos: ácido
valproico , etetrinato ,carbamazepina (tratamiento epiléptico)
• Edad materna: madres adolescentes o de más de 35 años.

CLASIFICACIÓN POSTERIOR
Occipital Supratorcular InfratorcularOccipitocervicalParietal Interfrontal Interparietal
ANTERIOR Sincipital Frontoetmoidal Nasofrontales Nasoetmoidal Naso-orbital Interfrontal Hendido CraneofacialBasal Esfenofaringeo Esfeno orbital Esfeno maxilar Esfeno etmoidal Transetmoidal

E.interhemisferico.

Manifestaciones clínicas
Las dependen de la zona del cerebro herniada, siendo las más frecuentes alteraciones visuales,
Microcefalia.Retraso mental .Crisis convulsivas.
Encefaloceles sincipitales tienen además de las alteraciones visuales, manifestaciones nasales y auditivas.

DIAGNOSTICO• La determinación de niveles de alfafetoproteína
materna y la realización de ecografía prenatal, permiten el diagnóstico intraútero .
• La imagen ecográfica del encefalocele consiste en una masa de tejido asociada siempre a un defecto óseo a través del cual se produce la herniación.

TRATAMIENTO• CIRUGIA: Reconstrucción del defecto.• Encefalocele occipital• Escisión quirúrgica del saco y su contenido con un cierre
hermético dural. Se debe tener en mente que las estructuras vasculares están presentes con frecuencia, dentro del saco.
• La hidrocefalia está presente con frecuencia y puede necesitar un tratamiento por separado.
• Encefalocele basal• El abordaje es transnasal.
• PRONOSTICO: Menos del 5% de infantes con encefalocele se desarrollan normalmente.

PREVENCIÓN
• Evitar la ingesta de tóxicos y fármacos teratógenos en el período periconcepcional.
• Dieta equilibrada.• Aporte de folatos desde el período
preconcepcional, al menos tres meses antes de la concepción y hasta doce semanas de la gestación.
• Todas las mujeres que deseen quedar embarazadas deberían tomar un suplemento de 0,4 mg de ácido fólico al día y hasta 4 mg/día aquellas con especial riesgo de tener un hijo con defecto del tubo neural.






Anencefalia• 1/1 500 R.N. más
mujeres• Asociado a
Polihidramnios.• Dx: Amniocentesis:
Niveles de alfa fetoproteína o US «signo ojos de rana»
• Prevención : Acido fólico
• Pronostico: mortalidad 65% intrautero ,casi 100 % en la primera semana.
Exencefalia :Falta del cierre de la porción cefálica del tubo neural

Anomalías Craneales
1. TRASTORNOS DE LA MIGRACIÓN NEURONAL2. HIDROCEFALIA CONGÉNITA Etiopatogenia. Cuadro clínico. Diagnóstico etiológico. Criterios de tratamiento neuroquirúrgico.
2. QUISTES ARACNOIDEOS Y PORENCEFÁLICOS.
3. MALFORMACIONES DE CHIARI Y DANDY-WALKER.


• Citogénesis y la histogénesis • Primer trimestre migran a lo largo del armazón de
glía radial para formar la corteza de múltiples capas.
• Crecimiento y la diferenciación • Segundo y tercer trimestres.
TRASTORNOS DE LA
MIGRACIÓN NEURONAL


Holoprosencefalia( 1/15 000)
Micropoligiria
Lisencefálica
Microgíricos y paquigíricos

HOLOPROSENCEFALIA
• En estos subtipos, los dos hemisferios cerebrales, ya sea por completo o sólo en parte, forman un conglomerado telencefálico único.

LISENCEFALIA(agiria),
• Ausencia de los giros corticales
• Tipo de Bielschowsky
• Tipo de Walker-Warburg HARD
• Pronostico: Malo por problemas respiratorios

HIDROCEFALIA CONGÉNITA
• Por definición Hidrocefalia Congénita "Neonatal o Perinatal" se desarrolla lntrauterinamente durante el embarazo, pero puede detectarse hasta el parto o período de recién nacido, aunque muchas veces las manifestaciones clínicas evolutivas se presentan en períodos posteriores haciendo difícil diferenciar entre una causa congénita y otra adquirida.


QUISTES ARACNOIDEOS Y
PORENCEFÁLICOS

Epidemiologia• 60% a 80% de los quistes aracnoideos son
descubiertos en niños.• Razón hombre-mujer de 2: 1 a 3: 1.• Los quistes son asintomáticos e incidental en la
mayoría de los casos• La > se encuentran en el espacio supratentorial
(90%)

Patogenia• Varios trastornos genéticos están asociados con
una mayor incidencia de quistes aracnoideos, incluyendo:
1. Marfan síndrome, 2. Neurofibromatosis tipo I, 3. Aciduria glutárica,4. Riñón poliquístico
Lo más probable, pueden existir una válvula de ranura o comunicación de válvula de bola entre el espacio subaracnoideo y el quiste, lo que permite la entrada pero no la salida de LCR.

CUADRO CLINICO• Depende de la edad, el tamaño y la ubicación • El síntoma más común es el dolor de
cabeza(unilateral en el región supraorbital o temporal que puede ser agravada con el esfuerzo físico).
• Convulsiones focales o generalizadas que puede deberse al efecto de masa locales, alta presión intracraneal (PIC), o hidrocefalia.
• Los bebés puede presentarse con macrocefalia, fontanela tensa ampliada, y suturas extendidos con irritabilidad, falta de crecimiento y desarrollo demora.

INFRATENTORIALES
Angulo pontocerebeloso se puede presentar con tinnitus, vértigo, debilidad facial, pérdida de la sensibilidad facial, pérdida de la audición, o ataxia
Supratentoriales aracnoideos quistesLocalización: Cisura de silvio

Clasificación de Galassi
• FOSA MEDIA O SUPRATENTORIAL.
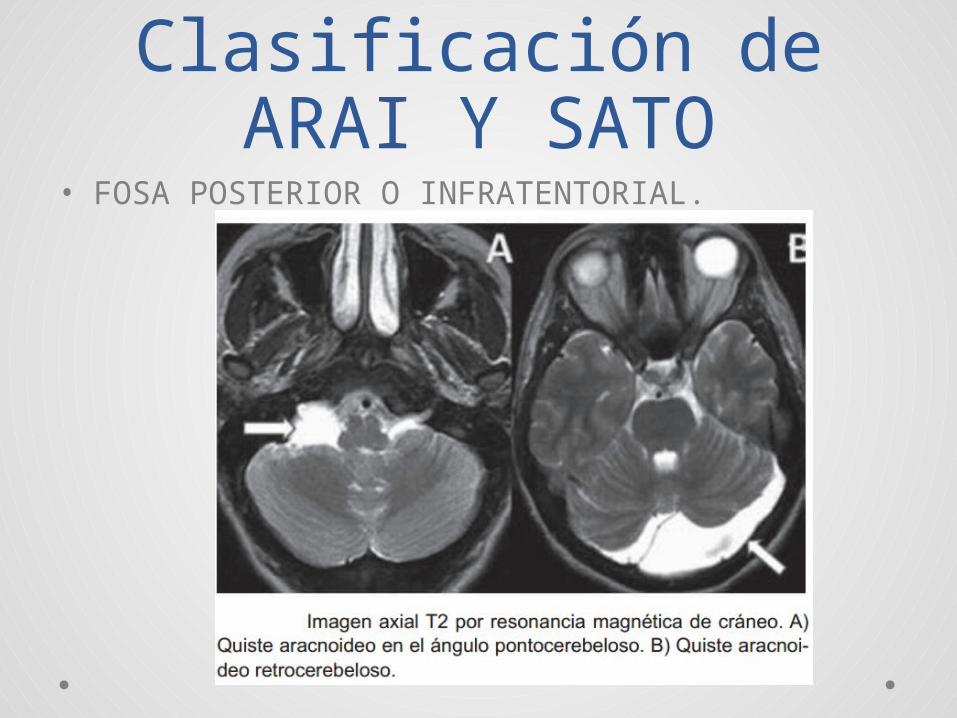
Clasificación de ARAI Y SATO
• FOSA POSTERIOR O INFRATENTORIAL.

Diagnostico• PRENATAL: Ecografía 13ª semana• Ventriculografía por TEM O RM• Espectroscopia de resonancia magnética de protón vivo

TRATAMIENTO• La mayoría de los quistes permanecen
constantes en tamaño, y el tratamiento conservador
Tratamiento conservador• Un enfoque no quirúrgico se recomienda
generalmente en asintomática• Pacientes la mayoría de los pacientes.Cirugía( Marsupializacion o derivación)• Cuando un quiste aracnoideo se documenta con
aumento de la PIC o hidrocefalia,

Definición• Son un conjunto de
anormalidades del cerebro posterior que van desde simples herniación de las amígdalas cerebelosas a través del foramen magnum para completar agenesia del cerebelo.
MALFORMACIÓN DE CHIARI

MALFORMACIÓN DE ARNOLD CHIARI.

Etiología1. Congénita ( la mayoría)2. Secundarias a derivaciones
lumboperitoneales en los niños(sin válvula)

CLASIFICACIÓN
Tipos I: Malformación cerebelobulbar sin meningomielocele Tipo II: Malformación cerebelobulbar con meningomielocele Tipo III: Meningomielocele cervical alto u occipitocervical con hernia cerebelosa Tipo IV: Hipoplasia cerebelosa.

Comprende cinco subtipos:• - Malformación de Chiari tipo 0: Alteración de la hidrodinámica del LCR a nivel del
foramen magnum. Los pacientes tienen siringomielia con mínimos datos de herniación amigdalar o sin ellos.
• - Malformación de Chiari tipo I: Herniación caudal de las amígdalas cerebelosas mayor de 5 mm por debajo del foramen magnum. Característicamente está asociado a hidrosiringomielia. No suele acompañarse de descenso del tronco del encéfalo o del cuarto ventrículo ni de hidrocefalia.
• - Malformación de Chiari tipo II: herniación caudal a través del foramen magnum del vermis cerebeloso, tronco del encéfalo y cuarto ventrículo. Se asocia con mielomeningocele e hidrocefalia, y de forma menos frecuente, con hidrosiringomielia. Se pueden observar otros tipos de alteraciones intracraneales (hipoplasia del tentorio, craniolacunia, anomalías del conducto de Silvio).
• - Malformación de Chiari tipo III: consiste en un encefalocele occipital con parte de las anomalías intracraneales asociadas al Chiari II.
• - Malformación de Chiari tipo IV: aplasia o hipoplasia del cerebelo asociada con aplasia de la tienda del cerebelo.
CLASIFICACIÓN

CLASIFICACIÓN

Clasificación

MANIFESTACIONES CLINICAS
La clínica muy variable, en función de la :1. Posición. 2. Grado de compresión.3. Nivel de degeneración celular de las amígdalas
cerebelosas. 4. Presencia o no de siringomielos.

MANIFESTACIONES CLINICAS
Malformación Chiari ISÍNTOMAS• Dolores de cabeza occipitocervical, disestesia• Dolor Nonradicular en la espalda, hombros y extremidades• Motrices y sensoriales síntomas• torpeza• disfagia• La disartria• síndrome cerebeloso• Troncal y ataxia apendicularSÍNDROME DE TRONCO CEREBRAL• irregularidades respiratorias• El nistagmo• Disfunción del nervio craneal inferior, incluyendo alteraciones
otológicas• aspiración recurrente• Hipertensión (raro)• Glossal atrofia• Pérdida de la sensibilidad facial• Trigémino o neuralgia del glosofaríngeoSÍNDROME DE MÉDULA ESPINAL• Motrices y sensoriales pérdidas, sobre todo en las manos• Hiporreflexia• hiperreflexia• respuesta BabinskiOTROS SIGNOS• oscilopsia• esotropía• La bradicardia sinusal• ronquera• hipo• La incontinencia urinaria• los ataques de gota• escoliosis

Malformación Chiari IIRECIÉN NACIDOS• (Por lo general asintomática)INFANTES(Los signos de compresión del tronco cerebral)• El estridor secundaria a la parálisis de las cuerdas vocales• La apnea central y obstructiva• La aspiración secundaria a disfagia con neumonía potencial• Retraso en el desarrollo secundario a la disfagia• Espasmos de sollozo con posible pérdida de la conciencia• La hipotonía y tetraparesia• irritabilidadNIÑOS MAYORES Y ADULTOS JÓVENES(Espinal, signos del cerebelo, y oftalmológicos)• dolor occipitocervical• Debilidad en la mano y la pérdida de masa muscular• mielopatía• ataxia• Estrabismo• El nistagmo• Los defectos de los movimientos de seguimiento y convergencia• Los defectos de los movimientos optocinético• escoliosis• La disartria• EMERGENCIA NEUROLÓGICA(Por lo general, <2 años de edad, comúnmente a los 3 meses de edad)• Dolor de cuello Progresista• apnea• disfagia• El estridor• opistótonos• El nistagmo• Disfunción del tronco cerebral progresivo
MANIFESTACIONES CLINICAS

MANIFESTACIONES CLINICAS
• Asintomáticas
• Ataxia • Escoliosis• Siringomielia• SINDROME DE AGUJERO OCCIPITAL• Hidrocefalia con hipertension intracraneal• Disfx aislada de pares craneales
bulbares( estridor o disfagia)• Alteraciones oseas cuello corto , línea de
implantación del cabello muy baja
SINDROME DE AGUJERO OCCIPITAL
• Cefaleas occipitales(por valsalva)• Sincopes• Inestabilidad• Vértigo• Nistagmo vertical( fase rápida hacia abajo )• Signo de Lhermite.• Tetraparesia espástica• Alt. Sensitivas propioceotivas en las piernas y
a veces en manos »pseudoatetosis» y ataxia.

DIAGNOSTICO• Imagen Tomografía computarizada Resonancia magnética(Gold starndar)

MANEJOMALFORMACIÓN
CHIARI I ASINTOMÁTICA
Excluir hidrocefalia , compresión ventral e inestabilidad cervical
Siringe No Siringe
Descompresión de chiari
Descenso >7 mm
Descenso <7 mm
Ejercicio juicio clínico
Observación

MALFORMACIÓN CHIARI I
SINTOMÁTICA
Excluir hidrocefalia , compresión ventral e inestabilidad cervical
Siringe No Siringe
Descenso >7 mm
Descenso 3- 7 mm
Ejercicio juicio clínico
Observación con reevaluaciones frecuentes de gravedad de los síntomas
Descompresión de chiari
Descenso < 3mm

MALFORMACIÓN CHIARI II
ASINTOMÁTICA
Verificar funcionamiento de la derivación y la estabilidad cervical
Siringe No Siringe
Verificar funcionamiento de la derivación
Larga siringe
Considere la laminectomía cervical y shunt syringopleural
Observación
Pequeño siringe
Observación

MALFORMACIÓN CHIARI II
SINTOMÁTICA
Verificar funcionamiento de la derivación y la estabilidad
cervical
No siringe o pequeña a moderada siringe (no expansión del cable)
Large siringe (ampliando el cable
Laminectomía cervical ± limitado fosa posterior descompresión
Descompresión Chiari más shunt syringopleural

SINDROME DE DANDY WALKER
Un conjunto de síntomas y signos secundarios a una disembriogenesis del cerebro medio y por tanto las alteraciones anatómicas que aparecen dependen de la severidad del trastorno malformativo.

• DEFINICION:• El síndrome de Dandy-Walker (DWS) representa una
malformación congénita que se caracteriza por :
1. Agenesia o hipoplasia del vermis cerebeloso, 2. Dilatación quística del cuarto ventrículo, y 3. Ampliación de la fosa posterior con o sin hidrocefalia
4. Elevados senos transversales5. Obstrucción de las salidas de la forámenes de
Magendie y Luschka (no se ve en todos los casos
MALFORMACIÓN DE DANDY-
WALKER.


Epidemiologia• En 1 de 25.000 a 30.000 recién nacidos• 7% de los casos de hidrocefalia congénita. • Más frecuente en el sexo femenino • Un 70% se diagnostica durante la lactancia • Se han reportan casos incidentales en la adultez.



PATOGENIA• Es un trastorno en el desarrollo embriológico del
cerebro medio que ocurre durante el primer trimestre del embarazo.

ETIOLOGIA• Es desconocida. • Sin embargo se mencionan algunos factores
predisponentes como la rubéola, las infecciones por citomegalovirus y toxoplamosis así como el uso de la warfarina, el alcohol y de isotretinoina .

Cuadro clínico• La presentación postnatal más común es
macrocrania.• Gran fosa posterior, convulsiones, espasticidad,
letargo, hitos con retraso, insuficiencia respiratoria, apnea, aumento de la presión intracraneal, e hidrocefalia(90%).
• La anormalidad del SNC más comúnmente asociado es la agenesia del cuerpo calloso, mientras que por vía sistémica, hemangiomas capilares y defectos cardíacos representan las asociaciones más frecuentes.

• El examen revela macrocefalia con una abombamiento de la fontanela, diástasis de sutura, y, a menudo, llena de sangre venas del cuero cabelludo. La fosa posterior es normalmente grande y occipital transiluminación es positiva

Diagnostico• Niños menores de 1 año.• Adolescencia o incluso la edad adulta.
• Ecografía fetal o postnatal o MRI O TC. • Evaluación citogenética

TRATAMIENTO• Actualmente, el desvío de LCR a través de la
derivación es el tratamiento estándar universalmente aceptado de DWS
• Tipos de derivación.• (1)Derivación del compartimiento supratentorial,
o lateral• (2)Derivación del quiste cerebelosa, o 4to
ventriculo • (3)Derivación de ambos compartimentos (shunt)

PRONOSTICO• 75-100% de posibilidades de supervivencia. • Sólo el 50% tiene IQ nornal. • Ataxia, espasticidad, y el poco control motor son
menos comunes. Los ataques ocurren en el 15%
GRACIAS.


















