MIOFIBROBLASTOS CARDIACOS DE RATA ADULTA SON … · 2 AGRADECIMIENTOS Agradezco al profesor Dr....
77
1 UNIVERSIDAD DE CHILE Facultad de Ciencias Químicas y Farmacéuticas Departamento de Química Farmacológica y Toxicológica MIOFIBROBLASTOS CARDIACOS DE RATA ADULTA SON RESISTENTES A AUTOFAGIA INDUCIDA POR ESTIMULACIÓN β 2 - ADRENÉRGICA Memoria para optar al título de Químico Farmacéutico JIMENA ANDREA CANALES URRIOLA Patrocinante : Dr. Guillermo Díaz-Araya. Directores de tesis : Dr. Guillermo Díaz-Araya. Pablo Aránguiz Urroz Santiago, Chile 2010
-
Upload
hoangtuyen -
Category
Documents
-
view
216 -
download
0
Transcript of MIOFIBROBLASTOS CARDIACOS DE RATA ADULTA SON … · 2 AGRADECIMIENTOS Agradezco al profesor Dr....

1
UNIVERSIDAD DE CHILE
Facultad de Ciencias Químicas y Farmacéuticas
Departamento de Química Farmacológica y Toxicológica
MIOFIBROBLASTOS CARDIACOS DE RATA
ADULTA SON RESISTENTES A AUTOFAGIA
INDUCIDA POR ESTIMULACIÓN β2-
ADRENÉRGICA
Memoria para optar al título de Químico Farmacéutico
JIMENA ANDREA CANALES URRIOLA
Patrocinante : Dr. Guillermo Díaz-Araya.
Directores de tesis : Dr. Guillermo Díaz-Araya.
Pablo Aránguiz Urroz
Santiago, Chile
2010

2
AGRADECIMIENTOS
Agradezco al profesor Dr. Guillermo Díaz Araya por darme la oportunidad de
trabajar en su laboratorio y darme a conocer el mundo de la ciencia. También agradezco
su constante apoyo y preocupación, tanto en el ámbito profesional y como en el ámbito
personal.
Al profesor Dr. Hernán Lara por facilitarnos en reiteradas ocasiones
dependencias de su laboratorio y materiales necesarios para realizar algunos
experimentos.
A mis padres, Jorge y Yihecika, por estar siempre presentes y ser responsables
de mucho de lo que he logrado hoy. Por entregar grandes valores y enseñarlos siempre
de la mejor manera: con el ejemplo. Del mismo modo, agradezco a mis hermanos Jorge,
Jessica y Claudia por hacer muchas veces oídos a mis quejas, por ser consejeros y por
ser confidentes. En definitiva, gracias a mi hermosa familia, pues ha sido un pilar
fundamental en mi formación y en el camino que he recorrido para llegar a estas
instancias.
A Mario por su compañía durante este último período. Agradezco la paciencia y
el cariño, como también la demostración que la vida siempre da otras oportunidades.
A mis amigas Sara, Javiera y Marcela por los años de amistad. Este concepto
implica muchos sentimientos y experiencias compartidas que no son simples de resumir
en estas líneas.
A Carlitos (Cartagena y Constenla), Cristina, Nancy y Carmen por su amistad en
el periodo universitario y que ha perdurado hasta hoy, por su apoyo en los momentos
difíciles y por estar siempre dispuestos a compartir.
A Pablo por su entrega de conocimientos durante el período de tesis. A él, a
Raúl, a Miguel, a Deisy y a Coni por hacer mucho más grato el trabajo en el laboratorio.
De la misma manera, agradezco el buen ánimo de Yenni, Gabriela, Diego, Marcelo y
Pedro, con quienes compartí en el último tiempo.
A todos ustedes, muchas gracias.

3
ÍNDICE GENERAL
Página
ÍNDICE GENERAL 3
ÍNDICE DE FIGURAS 8
ÍNDICE DE TABLAS 9
ÍNDICE DE ABREVIATURAS 10
RESUMEN 15
SUMMARY 16
INTRODUCCIÓN 1
Generalidades 1
Estructura del músculo cardiaco. 1
Fibroblastos cardiacos 1

4
Miofibroblastos cardiacos. 2
Comportamiento de fibroblastos y miofibroblastos cardiacos. 3
Sistema adrenérgico en el corazón 4
Sistema adrenérgico en fibroblastos cardiacos 5
Receptores adrenérgicos y sus vías transduccionales en
fibroblastos cardiacos 6
Autofagia 7
Mecanismos moleculares implicados en la autofagia 8
Rol de la autofagia 10
Autofagia en patologías cardiovasculares. 10
HIPÓTESIS 12
OBJETIVOS 13
Objetivo General 13
Objetivos Específicos 13
MATERIALES Y MÉTODOS 14
Reactivos 14
Modelo Animal 14
Aislamiento y cultivo de fibroblastos cardiacos de rata adulta 15

5
Diferenciación de fibroblastos a miofibroblastos 15
Cuantificación de proteínas 16
Preparación de extractos celulares totales 16
Electroforesis de geles de poliacrilamida 17
Electrotransferencia de proteínas 17
Inmunowestern blot 17
Inmunocitoquímica 18
Obtención de membranas celulares 19
Ensayo de desplazamiento y unión de competencia 19
Determinación de AMP cíclico 20
Análisis estadístico 20
RESULTADOS. 21
Caracterización de receptores adrenérgicos en fibroblastos y
miofibroblastos cardiacos de rata adulta 21
Determinación de Bmax y Kd de receptores β2-adrenérgicos en
fibroblastos y miofibroblastos cardiacos de rata adulta 22
Determinación de funcionalidad de receptores β2-adrenérgicos
en fibroblastos y miofibroblastos cardiacos de rata adulta 23
Activación de ERK 1/2 por estímulo con ISO 23
Producción de cAMP por estímulo con ISO 26

6
Determinación de autofagia en fibroblastos y miofibroblastos
cardiacos de rata adulta frente a concentraciones decrecientes
de suero 27
Determinación de autofagia en fibroblastos y miofibroblastos
cardiacos en condiciones basales 29
Determinación de autofagia en fibroblastos y miofibroblastos
cardiacos de rata adulta frente a estimulación β2-adrenérgica 30
Procesamiento de LC3 I a LC3 II inducido por ISO 30
Localización de LC3 endógeno en vesículas autofágicas inducida
por ISO 32
DISCUSIÓN 35
Caracterización de receptores adrenérgicos en fibroblastos y
miofibroblastos cardiacos de rata adulta 35
Determinación de Bmax y Kd de receptores
β2-adrenérgicos en fibroblastos y miofibroblastos cardiacos de rata adulta 35
Determinación de funcionalidad de receptor β2-adrenérgico 37
Autofagia en fibroblastos y miofibroblastos cardiacos de rata adulta 40
Alcances fisiopatológicos 44
CONCLUSIONES 45

7
REFERENCIAS 46

8
ÍNDICE DE FIGURAS
Página
Figura 1: Esquema de autofagia 9
Figura 2: Desplazamiento de [3H]-DHA 21
Figura 3: Ensayo de unión de [3H]-DHA 22
Figura 4: Activación de ERK 1/2 por estimulación β2-adrenérgica 25
Figura 5: Producción de cAMP por estimulación β2-adrenérgica 26
Figura 6: Procesamiento de LC3 I a LC3 II frente a concentraciones
decrecientes de suero 28
Figura 7: Procesamiento de LC3 I a LC3 II en condiciones basales 29
Figura 8: Procesamiento de LC3 I a LC3 II inducido por
estimulación β2-adrenérgica 31
Figura 9: Organización de LC3 endógeno bajo estimulación
β2-adrenérgica 33-34

9
ÍNDICE DE TABLAS
Página
Tabla 1: Kd y Bmax en fibroblastos y miofibroblastos
cardiacos de rata adulta 23

10
ABREVIATURAS
α-SMA : Alfa actina de músculo liso
[3H]-DHA : Dihidroalprenolol tritiado
µg : Microgramo
µl : Microlitro
µm : Micrómetro
µM : Micromolar
AC : Adenilato ciclasa
AMPK : Kinasa activada por Adenosina monofostato
Ang II : Angiotensina II
Atg : Proteína de genes asociados a la autofagia
Bmáx : Número total de receptores expresado en fmoles/mg de
proteína
BSA : Albúmina de suero de bovino
CaCl2 : Cloruro de calcio
Camp: : Adenosina monofosfato ciclico
cel : Célula
cm : Centímetro
cpm : Cuentas por minuto
csp : Cantidad suficiente para

11
DMEM F-12 : Dulbecco's Modified Eagle's Medium formula 12
DMSO : Di metil sulfoxido
EDTA : Acido etilendiaminotetraacético
EGF : Factor de crecimiento epidermal
EGTA : Ácido etilén glicol-bis( γ-aminoetil eter)-N,N,N’,N’-tetracético
EIA : Enzimoinmuno análisis
EMT : Transición epitelial-mesenquimal
ERK : Proteína kinasa activada por señal extracelular
ET-1 : Endotelina 1
FBS : Suero fetal de bovino
FITC : Fluoresceína isotiocianato
fmoles : Femtomoles
FSK : Forskolina
g : Gramos
h : Hora
HEPES : Acido N-2-hidroxietilpiperazina N-2-etanosulfónico
IGF-II : Factor de crecimiento análogo a insulina tipo II
IL 1 : Interleuquina 1
IL-4 : Interleuquina 4
IL-6 : Interleuquina 6
ISO : Isoproterenol

12
JNK : Kinasa N-terminal de c-Jun
KCl : Cloruro de potasio
Kd : Constante de disociación
kDa : Kilo Dalton
LC3 : Cadena liviana 3 de la proteína 1 asociada a microtúbulo
MAPK : Proteína quinasa activada por mitógenos
MEC : Matriz extracelular
mg : Miligramo
min : Minuto
mL : Mililitro
mm : Milímetro
mM : Milimolar
MMP : Metaloproteasa
mRNA : Ácido ribonucleico mensajero
mTOR : mammalian Target of Rapamicin
mTORC1 : Complejo 1 de mTOR
N : Normal
NA : Noradrenalina
Na3VO4 : Ortovanadato de sodio
NaCl : Cloruro de Sodio
NaOH : Hidróxido de sodio

13
NE : Norepinefrina.
ng : Nanogramos
nm : Nanómetros
nM : Nanomolar
nmoles : Nanomoles
PBS : Tampón fosfato salino
PDGF : Factor de crecimiento derivado de plaquetas
PDE : Fosfodiesterasa
PE : Fosfatidiletanolamina
p-ERK : Proteína kinasa activada por señal extracelular fosforilada.
PI3-K : Fosfatidilinositol 3 kinasa.
PKA : Proteína kinasa A
PKB : Proteína kinasa B
pM : Picomolar
PMSF : Fenilmetilsulfonifluoruro
PVDF : Poli fluoruro de vinilideno
rpm : Revoluciones por minuto
s : Segundos
SD : Desviación estándar
SDS : Dodecil sulfato de sodio
SDS-PAGE : Gel de poliacrilamida desnaturante

14
TBS : Tampón tris salino
TCA : Acido tricloro acético
TGF-β : Factor de crecimiento transformante beta
TNF-α : Factor de necrosis tumoral α
Tris : Tris-(hidroximetil)-aminoetano
v/v : Proporción volumen/volumen

15
RESUMEN
Los fibroblastos cardiacos son células que cumplen un rol fundamental en el
mantenimiento de la homeostasis de la matriz extracelular del corazón. Luego de un
daño al miocardio, además, participan activamente del remodelado cardiaco como tal o
diferenciándose a miofibroblasto, un fenotipo celular que presenta características que lo
hacen apto para funciones de cicatrización. Se ha observado que luego de un daño al
miocardio, el corazón está expuesto a un mayor tono adrenérgico con la finalidad de
compensar la disfunción adquirida por la injuria del tejido. Estudios de nuestro
laboratorio han demostrado que la estimulación β2-adrenérgica por Isoproterenol induce
autofagia en fibroblastos cardiacos de rata adulta, un proceso degradativo que se ha
reportado capaz de perder su equilibrio en diversas patologías cardiovasculares. Por
esto, se vuelve interesante determinar si estos mismos estímulos son capaces de inducir
autofagia en miofibroblastos cardiacos, células que aparecen sólo cuando hay daño al
miocardio. Los resultados muestran que tanto fibroblastos como miofibroblastos
cardiacos presentan receptores adrenérgicos sólo del subtipo β2. Miofibroblastos
cardiacos presentaron mayor número de receptores β2-adrenérgicos y con mayor
afinidad por sus ligandos que fibroblastos cardiacos. En ambos fenotipos celulares los
receptores mencionados se encuentran funcionales. En cuanto a la autofagia, los
estímulos clásicos inductores de autofagia (rapamicina y/o privación de nutrientes) y la
estimulación β2-adrenérgica por Isoproterenol inducen autofagia en fibroblastos. De
modo distinto, estos inductores no fueron capaces de inducir autofagia en
miofibroblastos cardiacos de rata adulta, aunque se encontró que en condiciones basales
presentaban mayor nivel de autofagia que los fibroblastos. Los resultados demuestran
que los miofibroblastos cardiacos son resistentes a la inducción de autofagia por
estimulación β2-adrenérgica, lo que puede abrir una puerta para el entendimiento del rol
de este proceso en estados patológicos del corazón.

16
SUMMARY
Cardiac fibroblasts are cells that play a fundamental role in the maintaining of
extracellular matrix homeostasis of the heart. After a myocardial damage, furthermore,
participate actively of cardiac remodelling as itself or differentiated to myofibroblast, a
phenotype that present characteristics which make it suitable for healing functions. It
has been observed that after damage to myocardium the heart is exposed to a higher
adrenergic tone, in order to compensate the acquired dysfunction of tissue injury.
Studies from our laboratory have shown that cardiac fibroblasts of adult rat exposed to
β2-adrenergic stimulation by Isoproterenol, undergo autophagy, a degradative process
have been reported capable of losing their balance in diverse cardiovascular pathologies.
Therefore, it becomes interesting determinate if cardiac myofibroblasts, cells that appear
only when there is myocardial damage, are capable of undergo autophagy by β2-
adrenergic stimulation. The results showed that cardiac fibroblast and myofibroblast
present adrenergic receptor only subtype β2. Cardiac myofibroblasts presented higher
number of β2-adrenergic receptor and with higher affinity that cardiac fibroblasts. In
both cellular phenotypes the receptors are functional. With regard to autophagy, we
showed that cardiac fibroblasts undergo this process by classics stimuli of autophagy
and by β2-adrenergic stimulation. Differently, cardiac myofibroblast of adult rat did not
undergo autophagy under any of de autophagic stimuli, but were found to have a higher
autophagy basal level that cardiac fibroblast. The results show that cardiac
myofibroblasts of adult rat are resistant of undergo autophagy by β2-adrenergic
stimulation, which can open a door for the understanding the role of autophagy in the
pathologic states of heart.

1
1. INTRODUCCIÓN
1.1 Generalidades
Las enfermedades cardiovasculares son, en la actualidad, uno de los principales
problemas de salud que afectan a la población. En los países desarrollados dan cuenta
de un 27% de mortalidad, cifra que se elevaría a 37% en el año 2020 (1). En nuestro
país, concordante con estos datos, las enfermedades cardiovasculares son hoy en día
responsables de un 27% de las muertes (2) . Es por esto que el estudio del sistema
cardiovascular, a nivel molecular, es esencial para entender la causalidad de sus
patologías y, así, vislumbrar nuevas estrategias terapéuticas.
1.2 Estructura del músculo cardiaco
El corazón, a nivel celular, está compuesto principalmente por cardiomiocitos y
fibroblastos. Los cardiomiocitos corresponden al un 30% de las células constituyentes
del corazón, ocupando un 70% del volumen cardiaco, mientras que los fibroblastos,
principales células no musculares del corazón, corresponden a un 70% del total de
células y ocupan un 30% del volumen total del miocardio (3). Menos del 5%
corresponde a otras células no musculares, tales como células endoteliales, células del
músculo liso vascular, mastocitos y células residentes del sistema inmune (4).
1.2.1 Fibroblastos cardiacos
Los fibroblastos son células que, en un principio, se pensaba tenían sólo una función de
soporte estructural en el corazón (4), sin embargo, se ha descubierto que tienen la
capacidad de proliferar, migrar, diferenciarse a miofibroblastos (5) y, además, realizan

2
funciones que anteriormente no se les asociaban. Una de estas es el mantenimiento de la
homeostasis de la matriz extracelular (MEC) (5), al ser responsables de la síntesis y
secreción de proteínas que la conforman (principalmente colágeno tipo I y III y
fibronectina) (6, 7), como también de la secreción de metaloproteasas (MMPs) que
degradan estas proteínas y permiten el recambio de la MEC (8). Por otro lado, los
fibroblastos pueden responder a estímulos mecánicos, eléctricos o químicos para
mantener el normal funcionamiento cardiaco y, además, son capaces de sintetizar y
liberar mediadores que actúan de manera autocrina y/o paracrina, tales como citoquinas
[Factor de Necrosis Tumoral tipo alfa (TNFα), Interleuquina 1 (IL-1) e Interleuquina 6
(IL-6)], factores de crecimiento [factor de crecimiento transformante (TGF-β)] y
también péptidos vasoactivos [Angiotensina II (Ang II) y endotelina 1 (ET-1)] (7, 8).
En condiciones patológicas, tales como hipertensión arterial, insuficiencia cardiaca o
infarto al miocardio, el fibroblasto cardiaco participa activamente en los procesos de
remodelado y, al haber daño tisular, también en los de cicatrización (4, 8). En estos
casos, su función la cumple al diferenciarse a miofibroblasto, un fenotipo especializado
de fibroblasto activado (7). Esta diferenciación ocurre por estímulos mecánicos y
químicos de manera simultánea (9). Los estímulos mecánicos en sí pueden llevar a la
liberación de estímulos químicos, como factores de crecimiento, especialmente TGF-β1,
o bien pueden participar otros mediadores químicos no derivados de la tensión, como
factor de crecimiento derivado de plaquetas (PDGF), factor de crecimiento análogo a la
insulina tipo II (IGF-II) e interleuquina-4 (IL-4) que, junto con la tensión, lograrán la
diferenciación (6, 9).
1.2.2 Miofibroblastos cardiacos
Los miofibroblastos tienen características especiales que los distinguen de los
fibroblastos y que dan cuenta de su rol en los procesos de reparación en que se ven
involucrados. Primero, no son células que residen normalmente en el tejido cardiaco,

3
sino que aparecen en el momento del daño y, funcionalmente, son capaces de secretar
mayores cantidades de proteínas de la MEC (8, 9), siendo los principales responsables
del depósito de éstas en el proceso de reparación del tejido (10). Estos antecedentes,
sumados a que aumentan el recambio de colágeno y la degradación de MMP, los
sindican como células críticas para el proceso de remodelado cardiaco (8). También se
ha demostrado in vivo que tienen mayor propiedad de migración y proliferación que los
fibroblastos, lo que los vuelve aptos para reclutarse en la zona del daño y reparar el
tejido (11). Ahora, en términos estructurales, se caracterizan por expresar proteínas
contráctiles, tales como α-actina del músculo liso (α-SMA), vimentina y desmina, las
cuales formarán un aparato microfilamentoso contráctil que se conectará al espacio
extracelular por adhesiones focales supermaduras, otorgándole a la célula mayor
capacidad de producir fuerza y así contraer el tejido de granulación y, por lo tanto, darle
integridad estructural a la cicatriz (7, 9). En resumen, dadas todas sus características, los
miofibroblastos son capaces de remodelar la MEC y, por anclaje y contracción, limitar
la cicatriz (8).
1.2.3 Comportamiento de fibroblastos y miofibroblastos cardiacos
Fuera de las diferencias estructurales claramente definidas entre fibroblastos y
miofibroblastos, se han observado otras relacionadas con el comportamiento de estos
fenotipos celulares frente a diversos estímulos. Estudios de nuestro laboratorio han
demostrado que estatinas son capaces de inducir muerte celular por apoptosis en
fibroblastos y miofibroblastos cardiacos de ratas neonatas, sin embargo, se observó que
esto ocurre en menor medida en miofibroblastos (12). Otro estudio de nuestro
laboratorio demostró que al sobreexpresar el receptor tipo 1 de Ang II (AT1) en
fibroblastos y miofibroblastos cardiacos de ratas neonatas, al estimular con Ang II éstos
mueren por apoptosis, sin embargo, volvemos a observar que miofibroblasto es más
resistente a este proceso que el fibroblasto (13). Finalmente, se ha observado también
que miofibroblastos resisten mayormente a la privación de suero, pudiendo sobrevivir

4
hasta 7 días bajo esta condición (14). De esta manera, se ha demostrado que en diversas
condiciones desfavorables para la viabilidad del fibroblasto, el miofibroblasto es capaz
de sobrevivir en mayor medida.
Ahora, comparando con otros tipos de miofibroblastos, se ha visto que en la piel, una
vez que la epitelización de una herida ha sido completada, es decir, cuando el tejido
granulomatoso ha evolucionado a una cicatriz madura, los miofibroblastos desaparecen
de la zona mediante una masiva apoptosis (7, 9). Sin embargo, se ha observado que en
la cicatriz madura del infarto, donde ya no deberían residir miofibroblastos, éstos
persisten (15). Esta deficiencia de apoptosis en los miofibroblastos cardiacos podría
asociarlos a procesos ya no de reparación, sino patológicos como la fibrosis cardiaca
(16).
1.3 Sistema adrenérgico en el corazón
Un infarto al miocardio es capaz de progresar y llegar a una disfunción cardiaca, debido
a que la exacerbación del depósito de MEC contribuye al engrosamiento del tejido,
disminuyendo su capacidad de relajación y contracción necesaria para bombear sangre
(17). En este caso, cuando ya se observa una pérdida de función del miocardio, se
encuentra aumentado el tono simpático con la finalidad de compensar esta deficiencia
(18). Así, el sistema adrenérgico se mantiene activado buscando adaptar al corazón a las
necesidades del organismo, pese a su disfunción. Sin embargo, la estimulación
simpática crónica del miocardio contribuye con los cambios morfológicos en el órgano
que finalmente serán deletéreos para su normal funcionamiento (18).
En el corazón, a nivel molecular, la acción del sistema adrenérgico se ejerce a
través de receptores α y β-adenérgicos, siendo estos últimos los más abundantes. En
cuanto a la distribución de estos receptores en las poblaciones celulares del corazón, se
ha descrito que los cardiomiocitos presentan receptores α1, β1 y β2-adrenérgicos, siendo

5
predominantes los receptores β1-adrenérgicos (19). Por otro lado, se ha demostrado que
los fibroblastos sólo presentan un subtipo de receptor adrenérgico: el receptor β2 (20,
21).
Para entender el mecanismo por el cual la estimulación simpática exacerbada
produce efectos nocivos sobre el corazón, es importante saber qué procesos gatilla sobre
sus poblaciones celulares. Estudios han demostrado que catecolaminas provocan
hipertrofia e, incluso, muerte de cardiomiocitos (22). Esto, en parte, podría explicar la
disfunción que adquiere el miocardio bajo estas condiciones, sin embargo, también es
importante saber qué ocurre con la población celular más numerosa del corazón:
fibroblastos.
1.3.1 Sistema adrenérgico en fibroblastos cardiacos
Actualmente existe abundante literatura acerca de los efectos de la estimulación
adrenérgica sobre funciones del fibroblasto cardiaco. En cuanto a la proliferación, es
bien entendido que la estimulación adrenérgica la promueve, sin embargo, la
controversia radica en el mecanismo mediante el cual logra este efecto (8). Fibroblastos
cardiacos humanos han demostrado aumentar su proliferación bajo estímulos con el
agonista β-adrenérgico inespecífico Isoproterenol (ISO), mediante un mecanismo
autocrino, es decir, probablemente este estímulo provoque secreción de factores de
crecimiento u otros mediadores que le otorguen la capacidad de proliferar (20). En
cambio, otro estudio en fibroblastos cardiacos de rata adulta ha demostrado que
Norepinefrina (NE), agonista α- y β- adrenérgico, resulta ser un agente co-mitogénico
en este tipo celular, es decir, este estímulo promueve directamente la proliferación (23).
Ahora, relacionado a la capacidad secretora del fibroblasto, la estimulación con
NE promueve la secreción de TGF-β (un factor de crecimiento sindicado como agente
profibrótico por varios autores (8)), en fibroblastos cardiacos neonatos (24).
Finalmente, en cuanto a la síntesis y secreción de proteínas de la MEC, no hay

6
consenso: mientras hay estudios que indican que Noradrenalina (NA) estimula la
expresión de colágeno tipo I y fibronectina en fibroblastos cardiacos de rata neonata
(25), otros indican que la estimulación con ISO en fibroblastos cardiaco de rata adulta
reduce la síntesis del colágeno (26). A pesar de este último antecedente, la información
global nos da a entender que la estimulación adrenérgica sostenida contribuye al
desarrollo de fibrosis en el corazón bajo condiciones patológicas.
1.3.2 Receptores adrenérgicos y sus vías transduccionales en
fibroblastos cardiacos
Como se mencionó anteriormente, se ha descrito que fibroblastos cardiacos presentan
sólo un subtipo de receptor adrenérgico: el receptor β2 (20, 21). Éste es un receptor de
siete dominios transmembrana acoplado dualmente a proteína Gs y proteína Gi (27).
Dado su acoplamiento a la proteína Gs, los efectos de la estimulación adrenérgica en
estos fibroblastos estarán mediados por la activación de la subunidad Gαs que activará a
adenilato ciclasa (AC), produciendo un aumento en los niveles de AMP cíclico (cAMP)
y, consecuentemente, la activación de la proteína kinasa A (PKA) que es capaz de
activar, mediante fosforilación, diversos intermediarios (28). Sin embargo, ésta no es la
única vía que se activa por estimulación adrenérgica en el fibroblasto, ya que se ha visto
que la subunidad Gβγ también tendría implicancias en los efectos de catecolaminas al
activar la Fosfatidilinositol-3 Kinasa (PI3-K) clase I que llevará a la activación de la vía
de la proteína kinasa B (PKB o Akt) y, consecuentemente, a la activación de mTOR y
p70S6K (29-31). Se han descrito a cAMP y la vía PKA como inhibidores de la
proliferación celular y de la síntesis de proteínas, en cambio la vía PKB/Akt como
activadora de estos fenómenos, por lo que los procesos proliferativos en fibroblastos
cardiacos frente a estimulación adrenérgica se podrían explicar por esta última (29).
Finalmente, la estimulación β-adrenérgica también sería capaz de activar AMPK
mediante el aumento de cAMP y consecuente activación de la vía cAMP-Adenosina
(30). Estas tres vías asociadas a la estimulación β-adrenérgica, PKA, PKB y AMPK,

7
además, convergen en diversos procesos celulares. Uno de ellos es la regulación de la
autofagia: mientras AMPK modula positivamente este proceso, PKB lo inhibe (31) y se
cree que PKA también lo inhibiría (32).
Estudios han demostrado que el estímulo de fibroblastos con TGF-β1, un factor de
crecimiento utilizado para diferenciar in vitro fibroblastos a miofibroblastos, disminuye
el número de receptores β-adrenérgicos, aún manteniendo su afinidad por ISO, en este
tipo celular (33, 34). Sin embargo, las concentraciones y el tiempo de estímulo que se
utilizaron en estos casos fue menor a la necesaria para lograr la diferenciación de
fibroblastos a miofibroblastos. Más allá de estos antecedentes, poco es lo que se conoce
acerca de las vías transduccionales activadas por las catecolaminas en miofibroblastos
cardiacos.
1.4 Autofagia
Autofagia se define como cualquier evento donde material citoplasmático se entregue al
lisosoma para su degradación (35). Se han identificado tres tipos de autofagia:
microautofagia, macroautofagia y autofagia mediada por chaperonas, que difieren entre
sí en su función fisiológica y la manera en que entregan material citoplasmático al
lisosoma (35). Macroautofagia es la forma más prevalente, por lo cual será la que
convoque este trabajo y nos referiremos a ella simplemente como autofagia.
Autofagia es un proceso dinámico, altamente conservado en eucariontes y la principal
vía catabólica mediante la cual las células son capaces de degradar y reciclar tanto
macromoléculas de vida media larga como organelos (35-37). Se caracteriza por el
secuestro de material citoplasmático en una vesícula de doble membrana llamada
autofagosoma. Esta estructura se fusiona con el lisosoma, entregándole su contenido
para que sea degradado y para que los componentes constitutivos puedan ser reciclados
(31).

8
1.4.1 Mecanismos moleculares implicados en la autofagia
El proceso de autofagia consta de varias etapas en las que se ha descrito la participación
de una maquinaria molecular compuesta por diversas proteínas que han sido
mayormente dilucidas en levadura y cuya presencia y función se conserva en
eucariontes. Luego de diversas denominaciones por distintos autores, se ha llegado a su
actual denominación: proteínas Atg (38).
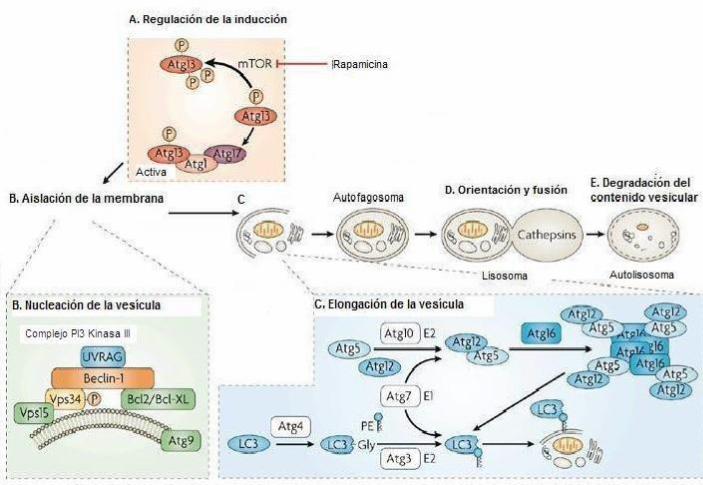
La primera etapa de la autofagia es su inducción, cuya regulación está dada por la
proteína kinasa Target of Rapamicin (TOR, mTOR en mamíferos), sindicada como el
principal inhibidor de este proceso (39). Se han descrito dos formas como ésta puede
inhibir la autofagia: por un lado, mantiene fosforilada a Atg13, lo que disminuye la
afinidad de ésta por Atg1 y, por lo tanto, impide su unión, que es un paso trascendental
para comenzar la señalización del proceso autofágico (Figura 1A); por otro lado, es
capaz de controlar, por señalización río abajo, transcripción y traducción de efectores de
la autofagia (39).
El segundo paso de la autofagia es la nucleación de la vesícula, promovida por el
complejo de la PI3 Kinasa (PI3 Kinasa clase III en mamíferos) y Atg6 (Beclin-1 en
mamíferos) (Figura 1B). Se pensaba que el retículo endoplasmático rugoso y el aparato
de Golgi suplían de membrana durante esta etapa, sin embargo actualmente se indica
que una estructura poco conocida, llamada fagóforo, cumpliría este rol (31).
Un tercer paso es la elongación de la vesícula, donde participan dos sistemas de
conjugación “similares a ubiquitina” (31). El primero de ellos es el sistema de
conjugación de Atg12 a Atg5 mediante ayuda de Atg7 y Atg10. Este nuevo dímero es
capaz de unirse a Atg16 (Atg16 L1 en mamíferos) oligomerizado, formando un
complejo multimérico necesario para la elongación de membranas aisladas (31) (Figura
1C). El segundo sistema de conjugación es el de Atg8 (LC3 en mamíferos), donde esta
proteína es escindida y luego conjugada con Fosfatidiletanolamina (PE) por acción
secuencial de Atg4, Atg7 y Atg3, logrando el paso de LC3-I, que es una proteína

9
soluble, a LC3-II, que es insoluble y se asocia a la membrana del autofagosoma (31, 40)
(Figura 1C).
El cuarto paso es la fusión del autofagosoma al lisosoma, que ocurre por la fusión de la
membrana externa del primero con la membrana del segundo. El autofagosoma,
entonces, entrega su contenido delimitado por su membrana interna (cuerpo autofágico)
al lisosoma, formando así el autolisosoma (Figura 1D), una estructura de membrana
simple que requerirá un pH ácido para poder concretar la última etapa de degradación
del contenido autolisosomal y reciclaje de componentes de interés celular (Figura 1E)
(31, 39).
Figura 1. Esquema de autofagia
A. Regulación de la inducción de autofagia por TOR kinasa (37). B. Aislación de la membrana y participación del
Complejo PI3 Kinasa III y Beclin-1 en la nucleación de la vesícula. C. Elongación de la membrana y participación
de los sistemas de conjugación similares a ubiquitina en este proceso. D. Orientación de autogafosoma al lisosoma y
fusión. E. Degradación del contenido autolisosomal. (Adaptado de Maiuri, 2007 (41)).

10
1.4.2 Rol de la autofagia
Autofagia es un proceso que se encuentra activo en condiciones normales, pues es
necesario para mantener la homeostasis celular y para recambiar proteínas y organelos
(35, 40). Sin embargo, es capaz de aumentar en condiciones específicas: al aumentar el
requerimiento energético, en el remodelado estructural celular o al aumentar
componentes dañados en la célula (35). Por su función, se entiende que la autofagia
sirva como un mecanismo de adaptación de la célula a condiciones de estrés en pos de
su supervivencia, sin embargo, es capaz también de provocar un tipo de muerte celular
programada distinta a la apoptosis, llamada Muerte Celular Programada Tipo II (31). De
este modo, no hay consenso en cuanto al beneficio o perjuicio que la autofagia pueda
significar a la célula.
1.4.3 Autofagia en patologías cardiovasculares
Se ha observado que la autofagia se encuentra alterada en diversos estados patológicos
cardiovasculares: aumentada en cardiomiopatía e insuficiencia cardiaca (36) y
disminuida en hipertrofia cardiaca (40). Además, se ha encontrado aumentada en
condiciones de isquemia y reperfusión (36). Sin embargo, nuevamente nos encontramos
frente a la misma interrogante: ¿aumenta o disminuye para promover la sobrevida bajo
una condición patológica o para llevar a muerte celular?. Estudios de autofagia en
células cardiacas están orientados mayoritariamente al cardiomiocito, donde se ha
indicado que el proceso autofágico sirve como respuesta al estrés y esta respuesta
participa en la patogénesis de la enfermedad (42). Sin embargo, poco se sabe en cuanto
a autofagia en fibroblastos cardiacos.
Datos de nuestro laboratorio han determinado que la estimulación β-adrenérgica con
ISO en fibroblastos cardiacos de rata adulta induce autofagia (43). Este dato es
importante para comenzar a entender la función de autofagia en patologías
cardiovasculares, ya que asocia la estimulación adrenérgica, aumentada en diversas

11
patologías cardiacas, con el proceso autofágico en la población celular más numerosa
del corazón.
La interrogante que nace a partir de todos los antecedentes presentados es si existe
alguna relación entre la autofagia y la estimulación adrenérgica en miofibroblastos
cardiacos, fenotipo celular inducido por daño tisular al corazón y que presenta mayor
resistencia a condiciones desfavorables para la célula.

12
2. HIPÓTESIS
Los miofibroblastos cardiacos de rata adulta son más resistentes a la autofagia inducida
por estimulación β2-adrenérgica que los fibroblastos cardiacos.

13
3. OBJETIVOS
3.1 Objetivo general
Determinar in vitro si el proceso de autofagia promovido por estimulación
adrenérgica en los miofibroblastos cardiacos de rata adulta está disminuido respecto a
los fibroblastos cardiacos de rata adulta.
3.2 Objetivos específicos
3.2.1 Determinar presencia y caracterizar receptores β-adrenérgicos en fibroblastos y
miofibroblastos cardiacos de rata adulta.
3.2.2 Demostrar funcionalidad de receptores β-adrenérgicos en fibroblastos y
miofibroblastos cardiacos de rata adulta.
3.2.3 Determinar si estimulación con ISO induce autofagia en fibroblastos y
miofibroblastos cardiacos de rata adulta.

14
4. MATERIALES Y MÉTODOS
4.1 Reactivos
De Sigma Chemical Co. (St. Louis, MO, EEUU): Tritón X-100, azul de tripán.
De Gibco BRL (Carlsbad, California EEUU): tripsina-EDTA, estándares para
masas moleculares de proteínas pre-teñidas, suero fetal de bovino (FBS).
De MERCK (Darmstadt, Alemania): compuestos inorgánicos y orgánicos, sales, ácidos
y solventes.
De PerKin Elmer Life Sciences, Inc. (Boston, MA, EEUU): reactivo
quimioluminiscente para Western Blot (Western Lightning), [3H]-DHA.
De Cell Signaling Technology (Beverly, MA): anticuerpo fosfo-p44/42 MAPK (p-
ERK1/2) y anticuerpo LC3B.
De Santa Cruz Biotechnology Inc. (Heidelberg, Germany): anticuerpo ERK 1.
De Falcon (USA): material de plástico estéril para la obtención y cultivo de fibroblastos
cardiacos.
De Chemicon (Tamecula, USA): TGF-β1. De Calbiochem (La jolla, CA, EEUU):
anticuerpos secundarios anti-IgG de conejo y ratón conjugado a peroxidasa.
4.2 Modelo animal
Ratas Sprague-Dawley macho adultas de 200 - 300 g de peso, provenientes del bioterio
de la Facultad de Ciencias Químicas y Farmacéuticas, Universidad de Chile, en
cumplimiento de todas las normas éticas referidas a la utilización de animales.

15
4.3 Aislamiento y cultivo de fibroblastos cardiacos de rata
adulta
Las ratas fueron anestesiadas con 300 μl de una solución de Ketamina:Xilacina
(2:1) para removerles el corazón desde la aorta. El corazón fue canulado por la aorta y
se perfundió una solución de CaCl2 2 mM para promover el latido y eliminar la sangre
desde el tejido. Luego se perfundió una solución de EGTA 2 mM para quelar el calcio
remanente. Finalmente se perfundió una solución de Colagenasa A 0,13% por 30 min.
Después de esto, se retiraron las aurículas y se homogenizó el ventrículo para llevar a
tres sucesivas digestiones con colagenasa A por 10 min cada una en baño
termorregulado a 37ºC con agitación. El producto de las digestiones fue centrifugado a
500 rpm por 2 min, se recuperó el sobrenadante y se centrifugó a 1000 rpm por 10 min
para obtener los fibroblastos en el pellet. Los fibroblastos fueron plaqueados con
DMEM-F12 + 10%FBS, luego de 2 h se lavaron 3 veces con PBS, se repuso DMEM-
F12 + 10% FBS y se dejaron proliferar hasta llegar a confluencia. Los cambios de
pasaje se realizaron mediante tripsinización (hasta pasaje 1 como máximo).
4.4 Diferenciación de fibroblastos a miofibroblastos
Se utilizaron fibroblastos cardiacos adultos en pasaje 1, los cuales se cultivaron por 84 h
en medio DMEM-F12 suplementado con TGF-β1 5 ng/mL. En estas condiciones cerca
del 100% de los fibroblastos se diferenciaron a miofibroblastos. Una vez cumplido el
tiempo se retiró el medio suplementado y los miofibroblastos se mantuvieron por 6 h
con DMEM-F12 suplementado con FBS 10%. Luego, los miofibroblastos obtenidos se
utilizaron para los ensayos posteriores.

16
4.5 Cuantificación de proteínas
Se cuantificó proteínas, tanto de extractos celulares como de membranas
celulares, por el método de Lowry: se preparó el reactivo A, que consiste en CTC
(sulfato de cobre 0,1%, ácido tartárico 0,2% y carbonato de sodio 10%), dodecilsulfato
de sodio 10%, hidróxido de sodio 0,8 N y agua nanopura en una proporción de 1:1:1:1.
400 μl del reactivo A se mezclaron con 395 μl de agua nanopura y con 5 μl de muestra,
se dejó reaccionar por 10 min y luego se le agregó 200 μl del reactivo B (Stock de Folin
& Ciocalteu en una dilución de 1:5 con agua nanopura) y se incubó a 40º C por 30 min.
Finalmente la lectura de absorbancia se midió en espectrofotómetro a una longitud de
onda de 750 nm.
Para la curva de calibración se siguió el mismo procedimiento, utilizando como
estándar de proteínas Albúmina de Suero Bovino y tampón de lisis RIPA como
disolvente.
4.6 Preparación de extractos celulares totales
Se prepararon extractos de proteínas totales para evaluar el procesamiento de LC3
(LC3-I/LC3-II) y la activación de ERK (fosfo-p44/42 MAPK o pERK 1/2) en
fibroblastos y miofibroblastos adultos. Se sembraron en placas de 60 mm a una
densidad de 1x104 cel/cm2 para fibroblastos y de 5x103 cel/cm2 para miofibroblastos. Al
finalizar los estímulos correspondientes, las células se lavaron tres veces con PBS frío y
luego se lisaron con 100 µL de tampón de lisis RIPA (Tris-HCl 10 mM pH 7,2; EDTA
5 mM; NaCl 150 mM; Tritón X-100 1% v/v; SDS 0.1% v/v; deoxicolato 1% v/v;
leupeptina 2 µg/mL; benzamidina 10mM; PMSF 1 mM y Na3VO4 100 µM). El
homogeneizado se centrifugó a 15.000 rpm durante 10 min a 4°C. El sobrenadante se
recuperó en un tubo nuevo, se determinó la concentración de proteínas por el método de
Lowry y se desnaturó en tampón SDS-PAGE 4X (glicerol 20 mL, 2-mercaptoetanol 10

17
mL, SDS 5 g, Tris base 1,51 g, Azul de bromofenol 0,01 g, Agua csp. 100 mL, pH 6.8),
para ser almacenado a –20°C.
4.7 Electroforesis en geles de poliacrilamida
La separación de las proteínas de acuerdo a su masa molecular se realizó mediante
electroforesis en geles de poliacrilamida según Laemmli, 1970. Para la detección se
cargaron 50 µg de extracto proteico para LC3 I/LC3 II y fosfo-p44/42 MAPK. Los geles
concentrador y separador fueron al 5 y 15%, respectivamente, para LC3I/LC3 II; y al 5
y 12%, respectivamente, para pERK 1/2. La electroforesis se realizó a un voltaje
constante de 90 Volt en tampón de electroforesis (Tris base 30,25 g, glicina 144 g, SDS
10 g, agua 1000 mL para tampón de electroforesis 10X).
4.8 Electrotransferencia de proteínas
Una vez realizada la electroforesis, las proteínas se electrotransfirieron a una membrana
de nitrocelulosa (BioRad) para pERK 1/2 y a una membrana de PVDF para LC3 I/LC3
II, a 350 mili-Amperes en tampón de transferencia durante 90 min para la primera y 45
min para la segunda.
4.9 Inmunowestern blot
Una vez transferidas, la membranas se bloquearon con tampón de bloqueo (TBS;
Tween-20 0,1% (TBS-T); leche sin grasa 5% p/v) durante 1 h a temperatura ambiente y
posteriormente se incubaron con los anticuerpos primarios correspondientes según
ensayo. LC3 B y pERK 1/2 se utilizaron en tampón de incubación (TBS-T 0,1%) a una
dilución 1:1000 toda la noche a 4°C con agitación suave. Posterior a la incubación, las

18
membranas se lavaron 3 veces por 10 min en TBS–T al 0,1%, e incubadas durante 2 h a
temperatura ambiente con el segundo anticuerpo anti-IgG de conejo o ratón conjugado
con peroxidasa, a un título de 1:5000 en tampón de bloqueo TBS-T al 0,1%.
Para detectar las proteínas, las membranas, previamente lavadas, se incubaron durante 1
min con el sustrato quimioluminiscente “Western Lightning” y se expusieron a la
película de fotografía Kodak-Biomax. Las películas se digitalizaron y las imágenes
fueron sometidas a densitometría con ayuda de los programas computacionales Corel ®
Paint Shop Pro Photo X2 y USI. Después de realizar los ensayos de inmunowestern
blot, las membranas se incubaron por 45 min en una solución de rojo Ponceau (rojo
Ponceau 2%, TCA 30%, ácido sulfosalicílico 30%) para desprender los anticuerpos,
posteriormente se lavaron en TBS–T al 0,1% por tres veces. Luego de este tratamiento,
las membranas pudieron ser reutilizadas para ensayos de Western blot con tubulina para
LC3I/LC3 II y con ERK-1 para pERK 1/2 como controles de carga.
4.10 Inmunocitoquímica
Se sembraron las células en placas de 35 mm con cubreobjeto a una densidad de
1x104 cel/cm2 para fibroblastos y de 5x103 cel /cm2 para miofibroblastos. Al finalizar
los estímulos correspondientes, las células fueron lavadas dos veces con PBS frío, luego
fueron fijadas al cubreobjeto con Formaldehído 4% por 15 min en frío. Se lavaron 2
veces con PBS frío y fueron permeabilizadas con Tritón X-100 en PBS al 0,2% por 5
min en frío. Se volvieron a lavar con PBS frío y fueron bloqueadas con una solución de
Albúmina de Suero Bovino (BSA) en PBS al 3% por 1 h a temperatura ambiente. Luego
fueron lavadas y se les expuso al anticuerpo LC3 B en una dilución 1:100 en BSA al 3%
durante toda la noche a 4º C. Luego se lavaron tres veces con PBS frío y se les expuso
al anticuerpo secundario Anti-IgG de conejo conjugado con el fluoróforo FITC, al
marcador de núcleos Hoescht y al marcador de esqueleto de actina
Rhodamina/Phalloidin en una dilución de 1:800, 1:1000 y 1:600 en BSA 3%,

19
respectivamente, durante 2 h a temperatura ambiente y oscuridad. Finalmente fueron
lavadas tres veces con PBS frío y los cubreobjetos que contenían las células fueron
montados a un portaobjeto con el reactivo Dako, que permitió fijar el vidrio y mantener
las células en perfecto estado. Se almacenaron en oscuridad y se observaron y
fotografiaron por microscopía confocal.
4.11 Obtención de membranas celulares
Las células se sembraron en placas de 100 mm a una densidad de 1x104 cel/cm2
para fibroblastos y de 5x103 cel /cm2 para miofibroblastos. Al finalizar los estímulos
correspondientes, fueron lavadas con PBS frío 3 veces y se lisaron con tampón de lisis
para binding. (Tris-HCl 1mM pH 7,5 y EGTA 2 mM). Obtenido el homogenizado, se
centrifugó a 15.000 rpm por 30 min a 4º C, se descartó el sobrenadante y el pellet se
resuspendió en tampón de resuspensión (glucosa 0,25 M, EDTA 1 mM, Tris-HCl 5mM)
y se almacenó a -80º C. Para la cuantificación de proteínas mediante el método de
Lowry y posterior ensayo de binding, el pellet fue descongelado, resuspendido en
tampón de binding (Tris-HCl 50 mM pH 7,4, NaCl 120 mM, KCl 4 mM, CaCl2 1mM,
bacitracina 0,1 mM y BSA 0,25%) y sonicado.
4.12 Ensayo de desplazamiento y Unión de competencia
Para el ensayo de desplazamiento se creó una batería experimental donde el
control llevó 20 μl de Dihidroalprenolol tritiado ([3H]-DHA) 150 nM y 200 μg de
proteína total del homogenizado de membranas. Además, a los otros tubos se les agregó
20 μl del desplazante correspondiente (propanolol 10 mM, atenolol 10 mM o ICI-
118551 10 mM). Todos los tubos se llevaron a un volumen final de 200 μl con tampón
Tris-Mg y se incubaron en baño termorregulado a 37º C con agitación por 30 min.
Luego de esto, la reacción se detuvo agregando 2 mL de tampón Tris-Mg a cada tubo y

20
el contenido de cada uno se depositó en su respectivo pocillo del sistema de filtración al
vacío donde previamente fueron colocados filtros Whitman GF/C. Cada uno de los
tubos se lavó 2 veces con 2 mL de tampón Tris HCl, contenido que también se vertió en
el correspondiente papel filtro de sistema. Los papeles filtros se recuperaron en viales de
centelleo, se agregó a cada uno 0,5 ml de agua destilada y 4 ml de mezcla de centelleo y
se agitaron vigorosamente en vórtex. Para obtener las cuentas totales (Ct) se agregó a un
vial de centelleo 20 μl de [3H]-DHA 150 nM, 0,5 ml de agua destilada y 4 ml de mezcla
de centelleo. Para el blanco se agregó a un vial de centelleo 0,5 ml de agua y 4 ml de
mezcla de centelleo. La radioactividad de cada tubo se midió en cuentas por minuto
(cpm) en el contador de centelleo.
Para el ensayo de unión de competencia se procedió de igual manera, con la
diferencia que se utilizó un único desplazante (propanolol) a distintas concentraciones
(concentraciones finales de 1nM, 10 nM, 100 nM, 1 μM, 10 μM, 100 μM y 1mM). Para
determinar la cantidad de receptores (Bmax) y la afinidad del receptor (Kd) por
propanolol se utilizó el método de Scatchard.
4.13 Determinación de AMP cíclico
Se determinó AMP cíclico (cAMP) mediante un enzimoinmuno análisis (EIA)
de competencia con Cyclic AMP EIA Kit de Cayman Chemical Company.
4.14 Análisis estadístico
Los resultados mostrados corresponden al promedio ± SD de, al menos, tres
ensayos independientes. Los datos se analizaron por ANOVA y la prueba Tuckey para
determinar la significancia estadística de los resultados. Para comparar dos grupos se
utilizó la prueba t Student.
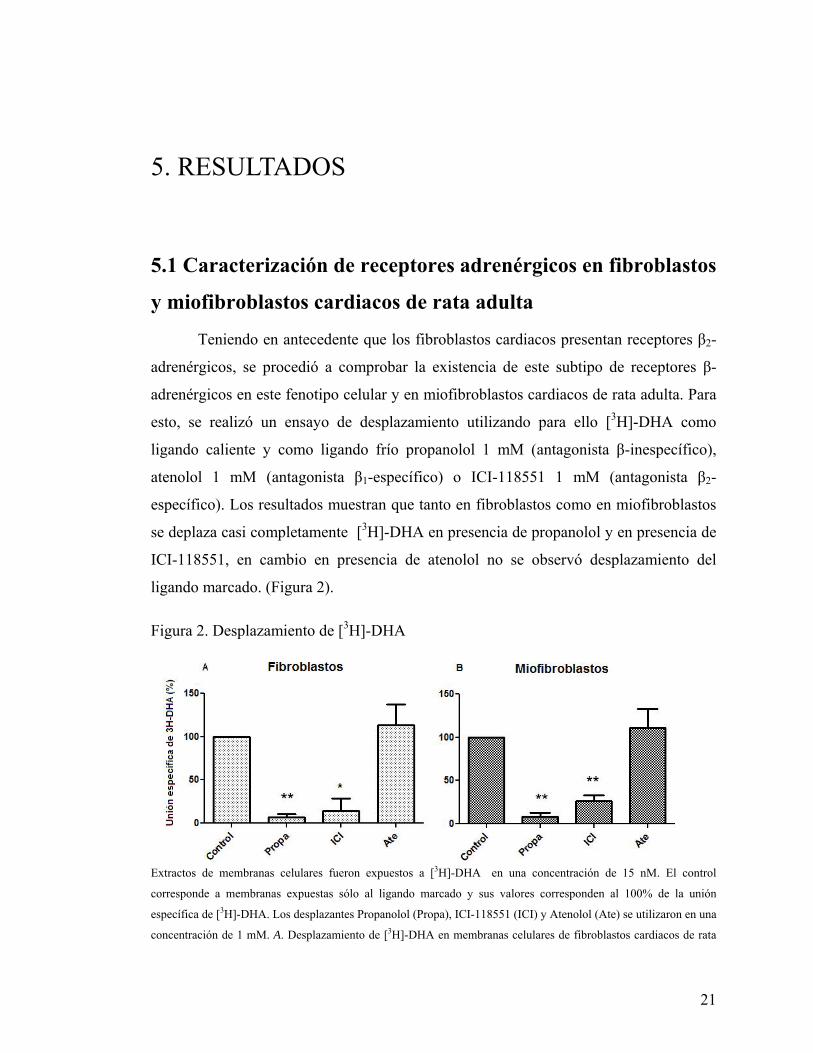
21
5. RESULTADOS
5.1 Caracterización de receptores adrenérgicos en fibroblastos
y miofibroblastos cardiacos de rata adulta
Teniendo en antecedente que los fibroblastos cardiacos presentan receptores β2-
adrenérgicos, se procedió a comprobar la existencia de este subtipo de receptores β-
adrenérgicos en este fenotipo celular y en miofibroblastos cardiacos de rata adulta. Para
esto, se realizó un ensayo de desplazamiento utilizando para ello [3H]-DHA como
ligando caliente y como ligando frío propanolol 1 mM (antagonista β-inespecífico),
atenolol 1 mM (antagonista β1-específico) o ICI-118551 1 mM (antagonista β2-
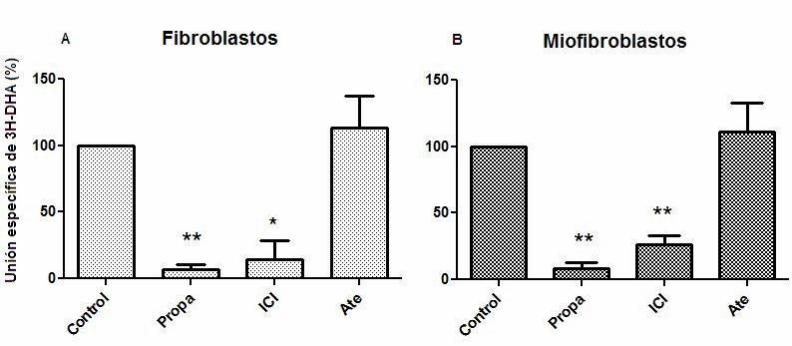
específico). Los resultados muestran que tanto en fibroblastos como en miofibroblastos
se deplaza casi completamente [3H]-DHA en presencia de propanolol y en presencia de
ICI-118551, en cambio en presencia de atenolol no se observó desplazamiento del
ligando marcado. (Figura 2).
Figura 2. Desplazamiento de [3H]-DHA
Extractos de membranas celulares fueron expuestos a [3H]-DHA en una concentración de 15 nM. El control
corresponde a membranas expuestas sólo al ligando marcado y sus valores corresponden al 100% de la unión
específica de [3H]-DHA. Los desplazantes Propanolol (Propa), ICI-118551 (ICI) y Atenolol (Ate) se utilizaron en una
concentración de 1 mM. A. Desplazamiento de [3H]-DHA en membranas celulares de fibroblastos cardiacos de rata
22
adulta. B. Desplazamiento de [3H]-DHA en membranas celulares de miofibroblastos cardiacos de rata adulta. Los
resultados corresponden al promedio de tres experimentos independientes ± SD. (* p < 0,05 , ** p< 0,01)
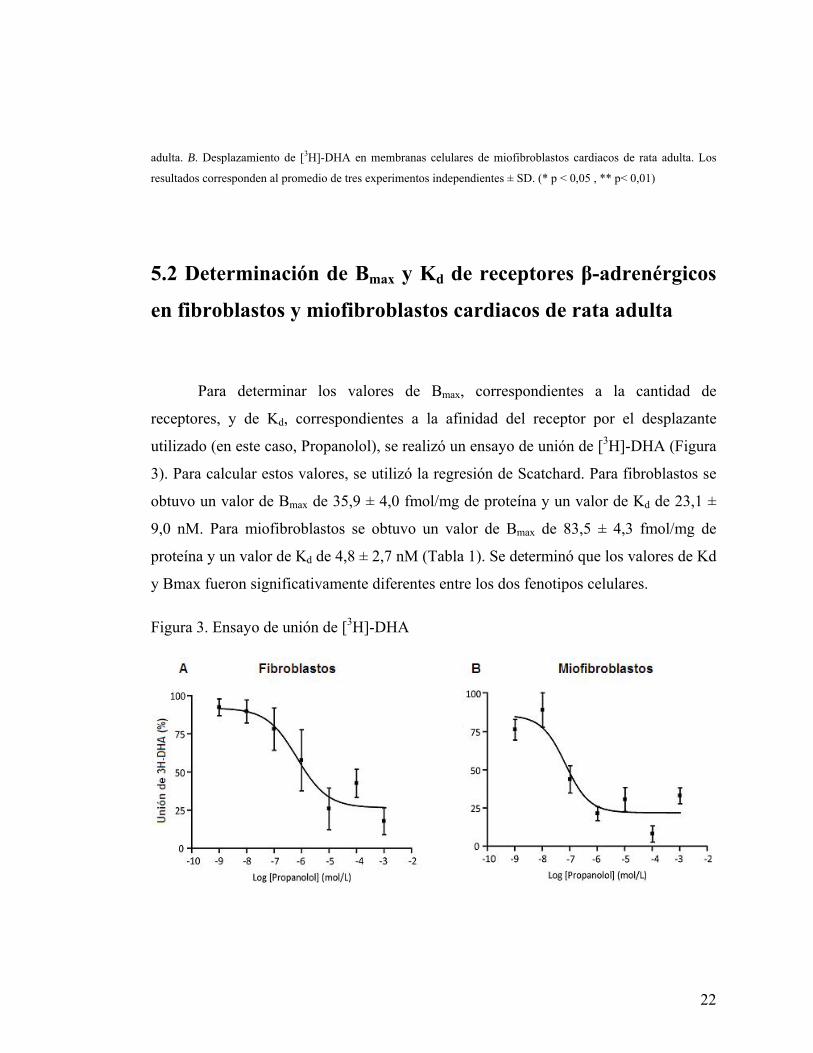
5.2 Determinación de Bmax y Kd de receptores β-adrenérgicos
en fibroblastos y miofibroblastos cardiacos de rata adulta
Para determinar los valores de Bmax, correspondientes a la cantidad de
receptores, y de Kd, correspondientes a la afinidad del receptor por el desplazante
utilizado (en este caso, Propanolol), se realizó un ensayo de unión de [3H]-DHA (Figura
3). Para calcular estos valores, se utilizó la regresión de Scatchard. Para fibroblastos se
obtuvo un valor de Bmax de 35,9 ± 4,0 fmol/mg de proteína y un valor de Kd de 23,1 ±
9,0 nM. Para miofibroblastos se obtuvo un valor de Bmax de 83,5 ± 4,3 fmol/mg de
proteína y un valor de Kd de 4,8 ± 2,7 nM (Tabla 1). Se determinó que los valores de Kd
y Bmax fueron significativamente diferentes entre los dos fenotipos celulares.
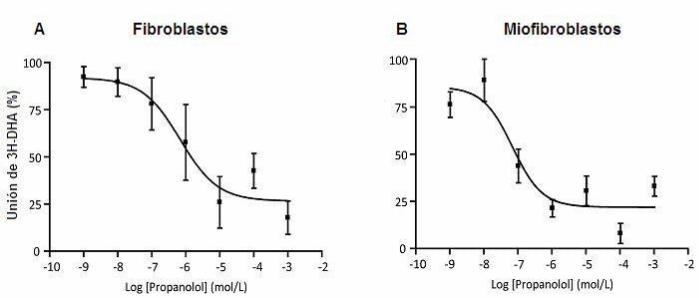
Figura 3. Ensayo de unión de [3H]-DHA

23
Extractos de membranas celulares fueron expuestas a [3H]-DHA en una concentración de 15 nM y como desplazante
fue utilizado Propanolol en concentraciones de 1nM, 10 nM, 100 nM, 1 μM, 10 μM, 100 μM y 1mM. A. Unión de
[3H]-DHA frente a concentraciones crecientes de propanolol en membranas celulares de fibroblastos cardiacos de rata
adulta. B. Unión de [3H]-DHA frente a concentraciones crecientes de propanolol en membranas celulares de
miofibroblastos cardiacos de rata adulta. Datos del promedio ± SD de tres experimentos independientes.
Tabla 1. Kd y Bmax en fibroblastos y miofibroblastos cardiacos de rata adulta
Fibroblastos Miofibroblastos.
Kd (nM) 23,1 ± 9,0 4,8 ± 2,7 *
Bmax (fmol / mg proteína) 35,9 ± 4,0 83,5 ± 4,3 **
Los datos obtenidos del ensayo de unión de [3H]-DHA se analizaron por Scatchard y se obtuvo los valores de la
constante de disociación (Kd) y la cantidad de receptores (Bmax) para los receptores β2-adrenérgicos en fibroblastos y
miofibroblastos cardiacos de rata adulta. Los resultados corresponden al promedio de tres experimentos
independientes ± SD. (* p < 0,05 , ** p< 0,01)
5.3 Determinación de funcionalidad de receptor β2-
adrenérgico en fibroblastos y miofibroblastos de rata adulta
5.3.1 Activación de ERK 1/2 por ISO
Luego de determinar la presencia del receptores β2-adrenérgicos en fibroblastos
y miofibroblastos cardiacos de rata adulta, evaluamos la funcionalidad de éstos. Para

24
ello, primero evaluamos la activación de las proteínas Kinasas Reguladas por Señal
Extracelular 1 y 2 (ERK 1/2 o p44/42 MAPK), ya que es un reflejo de la activación de
receptores acoplado a proteína G por actividad de la subunidad Gβγ. Se determinó la
activación temprana (considerada menor a 20 min) de esta vía por el estímulo β2-
adrenérgico. De este modo, se evaluó la activación de ERK 1/2 a 5 y 15 min de estímulo
con ISO.
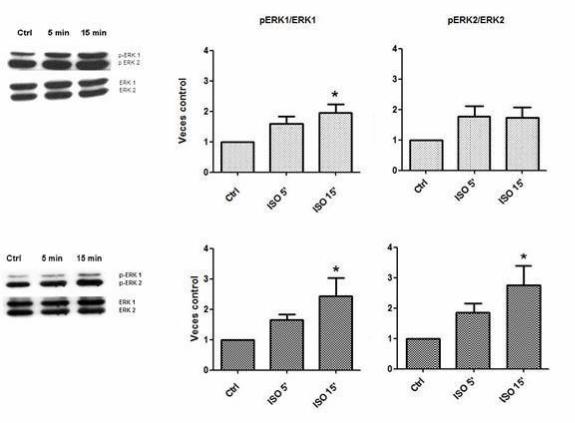
Se obtuvo que en fibroblastos hubo fosforilación significativa sólo de ERK 1 a
los 15 minutos de estímulo con ISO 10 μM, en cambio en miofibroblastos se fosforiló
significativamente ERK 1 y ERK 2 a igual tiempo de estímulo (Figura 4).
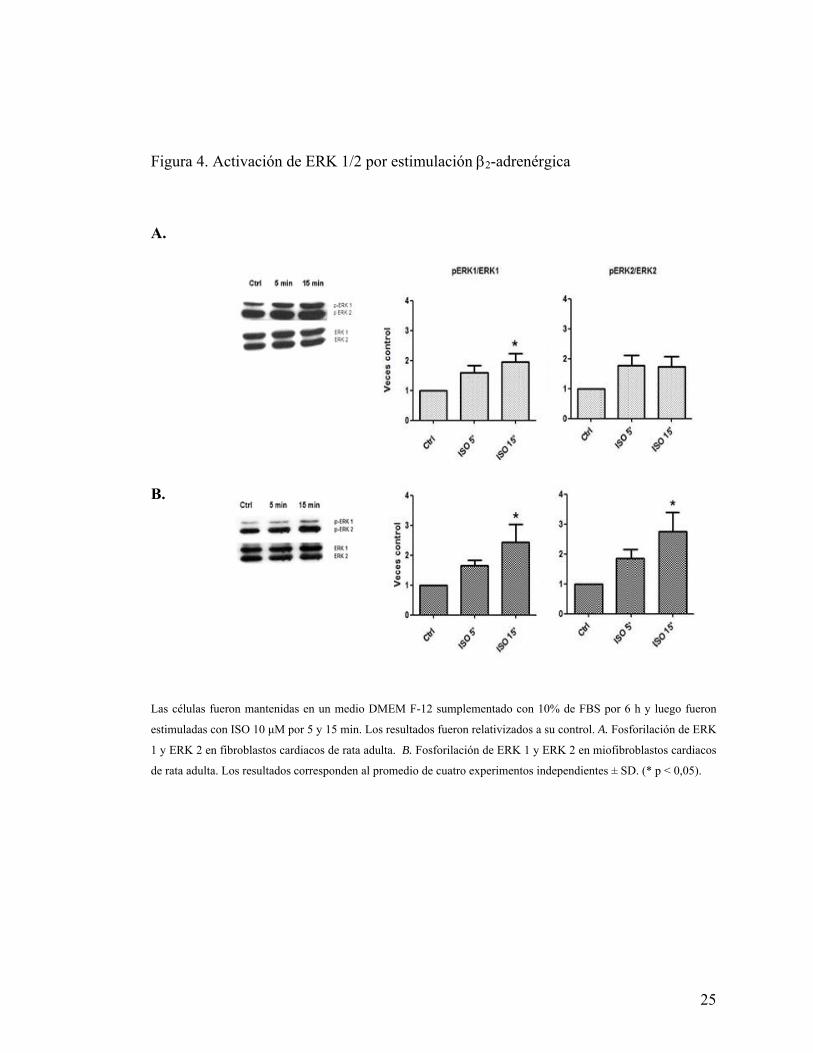
25
Figura 4. Activación de ERK 1/2 por estimulación β2-adrenérgica
A.
B.
Las células fueron mantenidas en un medio DMEM F-12 sumplementado con 10% de FBS por 6 h y luego fueron
estimuladas con ISO 10 μM por 5 y 15 min. Los resultados fueron relativizados a su control. A. Fosforilación de ERK
1 y ERK 2 en fibroblastos cardiacos de rata adulta. B. Fosforilación de ERK 1 y ERK 2 en miofibroblastos cardiacos
de rata adulta. Los resultados corresponden al promedio de cuatro experimentos independientes ± SD. (* p < 0,05).
26
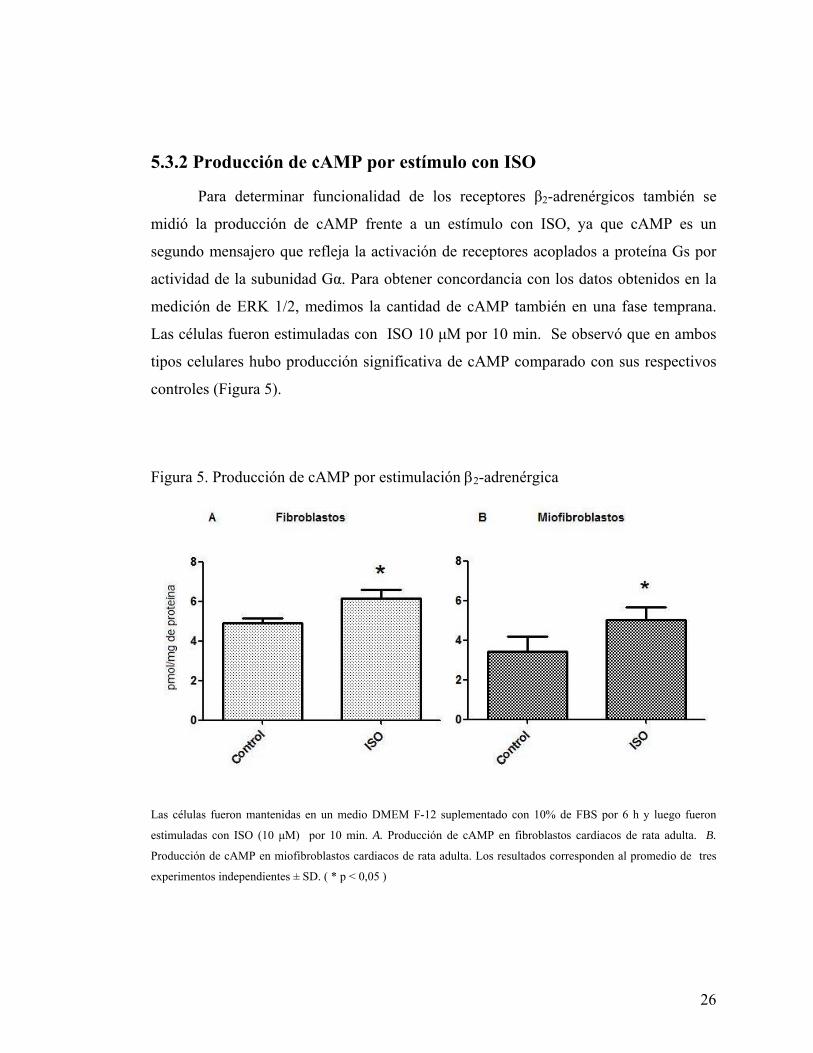
5.3.2 Producción de cAMP por estímulo con ISO
Para determinar funcionalidad de los receptores β2-adrenérgicos también se
midió la producción de cAMP frente a un estímulo con ISO, ya que cAMP es un
segundo mensajero que refleja la activación de receptores acoplados a proteína Gs por
actividad de la subunidad Gα. Para obtener concordancia con los datos obtenidos en la
medición de ERK 1/2, medimos la cantidad de cAMP también en una fase temprana.
Las células fueron estimuladas con ISO 10 μM por 10 min. Se observó que en ambos
tipos celulares hubo producción significativa de cAMP comparado con sus respectivos
controles (Figura 5).
Figura 5. Producción de cAMP por estimulación β2-adrenérgica
Las células fueron mantenidas en un medio DMEM F-12 suplementado con 10% de FBS por 6 h y luego fueron
estimuladas con ISO (10 μM) por 10 min. A. Producción de cAMP en fibroblastos cardiacos de rata adulta. B.
Producción de cAMP en miofibroblastos cardiacos de rata adulta. Los resultados corresponden al promedio de tres
experimentos independientes ± SD. ( * p < 0,05 )

27
5.4 Determinación de autofagia en fibroblastos y
miofibroblastos cardiacos de rata adulta frente a
concentraciones decrecientes de suero
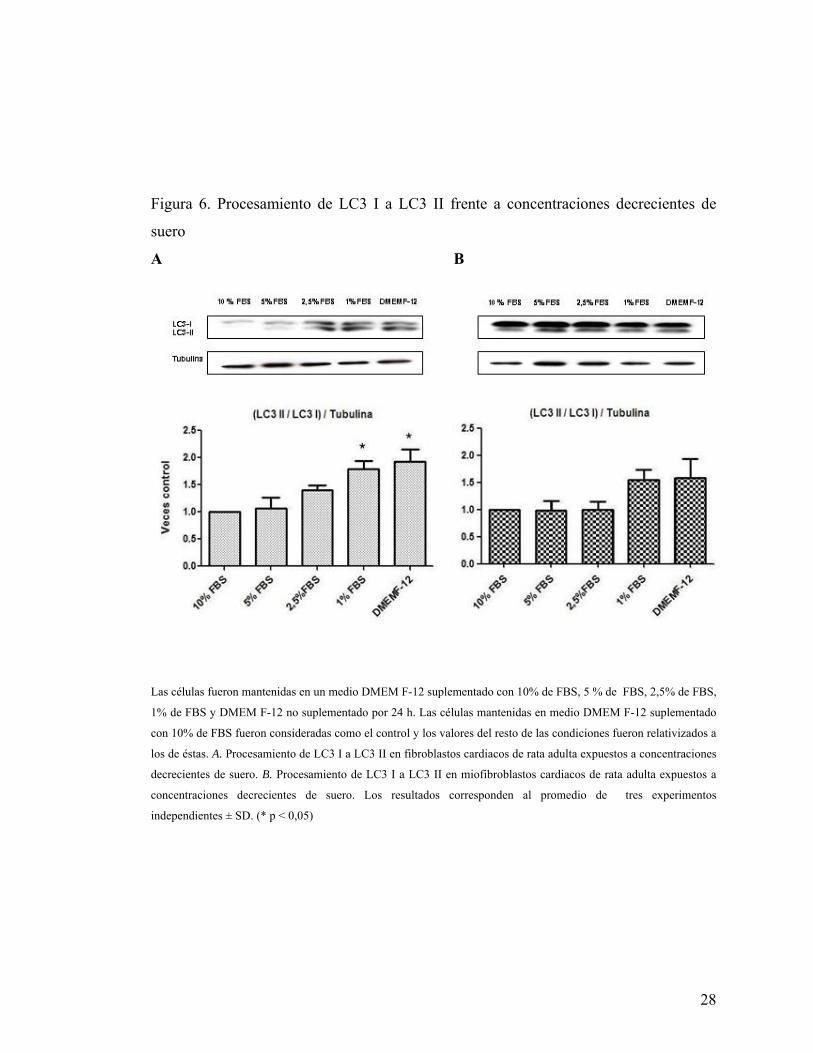
Como se sabe que el proceso autofágico es promovido, entre otras condiciones,
por estrés nutricional, se observó, por Inmunowestern blot, el procesamiento de la
proteína LC3 I a LC3 II en fibroblastos y miofibroblastos cardiacos expuestos a un
medio suplementado con concentraciones decrecientes de suero por 24 horas. Se
observó que fibroblastos comenzaron a procesar la proteína LC3 I a LC3 II de manera
significativa, comparada con su control, ya en un medio DMEM F-12 suplementado con
1% de FBS (Figura 6A). De manera diferente, miofibroblastos no presentan un
procesamiento significativo de la proteína LC3 I a LC3 II, respecto a su control, ni
siquiera en la privación total de suero (Figura 6B). Es destacable el hecho que,
aparentemente, en miofibroblastos la condición basal presenta mayor procesamiento de
la proteína LC3 I a LC3 II que fibroblastos (Figura 6, Western Blot).
28
Figura 6. Procesamiento de LC3 I a LC3 II frente a concentraciones decrecientes de
suero
A B
Las células fueron mantenidas en un medio DMEM F-12 suplementado con 10% de FBS, 5 % de FBS, 2,5% de FBS,
1% de FBS y DMEM F-12 no suplementado por 24 h. Las células mantenidas en medio DMEM F-12 suplementado
con 10% de FBS fueron consideradas como el control y los valores del resto de las condiciones fueron relativizados a
los de éstas. A. Procesamiento de LC3 I a LC3 II en fibroblastos cardiacos de rata adulta expuestos a concentraciones
decrecientes de suero. B. Procesamiento de LC3 I a LC3 II en miofibroblastos cardiacos de rata adulta expuestos a
concentraciones decrecientes de suero. Los resultados corresponden al promedio de tres experimentos
independientes ± SD. (* p < 0,05)
29
5.5 Comparación de autofagia entre fibroblastos y
miofibroblastos cardiacos de rata adulta en condiciones
basales
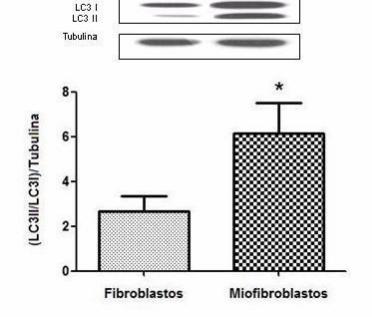
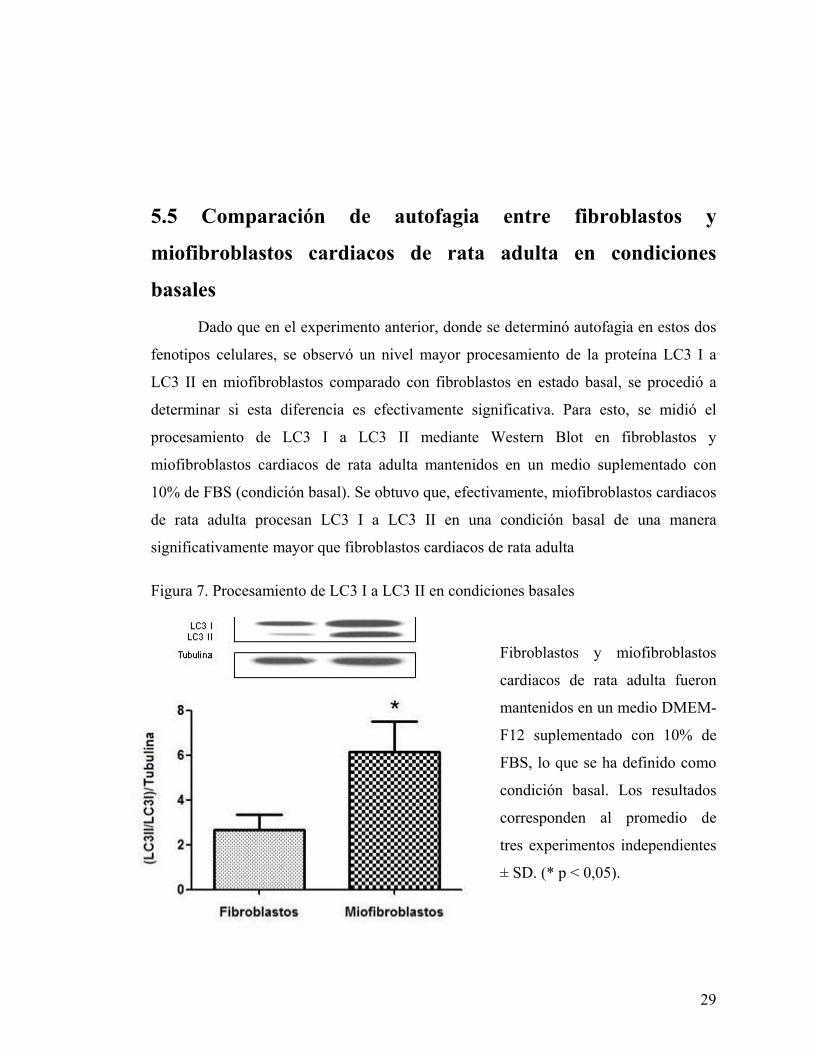
Dado que en el experimento anterior, donde se determinó autofagia en estos dos
fenotipos celulares, se observó un nivel mayor procesamiento de la proteína LC3 I a
LC3 II en miofibroblastos comparado con fibroblastos en estado basal, se procedió a
determinar si esta diferencia es efectivamente significativa. Para esto, se midió el
procesamiento de LC3 I a LC3 II mediante Western Blot en fibroblastos y
miofibroblastos cardiacos de rata adulta mantenidos en un medio suplementado con
10% de FBS (condición basal). Se obtuvo que, efectivamente, miofibroblastos cardiacos
de rata adulta procesan LC3 I a LC3 II en una condición basal de una manera
significativamente mayor que fibroblastos cardiacos de rata adulta
Figura 7. Procesamiento de LC3 I a LC3 II en condiciones basales
Fibroblastos y miofibroblastos
cardiacos de rata adulta fueron
mantenidos en un medio DMEM-
F12 suplementado con 10% de
FBS, lo que se ha definido como
condición basal. Los resultados
corresponden al promedio de
tres experimentos independientes
± SD. (* p < 0,05).

30
5.6 Determinación de autofagia en fibroblastos y
miofibroblastos cardiacos de rata adulta frente a estimulación
β2-adrenérgica
5.6.1 Procesamiento de LC3 I a LC3 II inducido por ISO
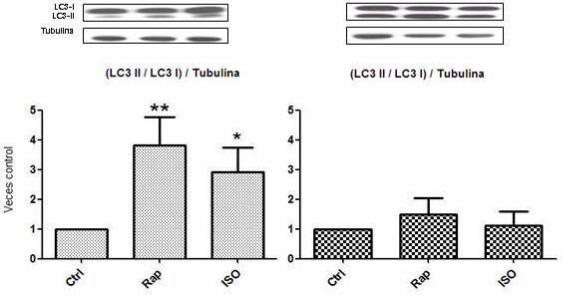
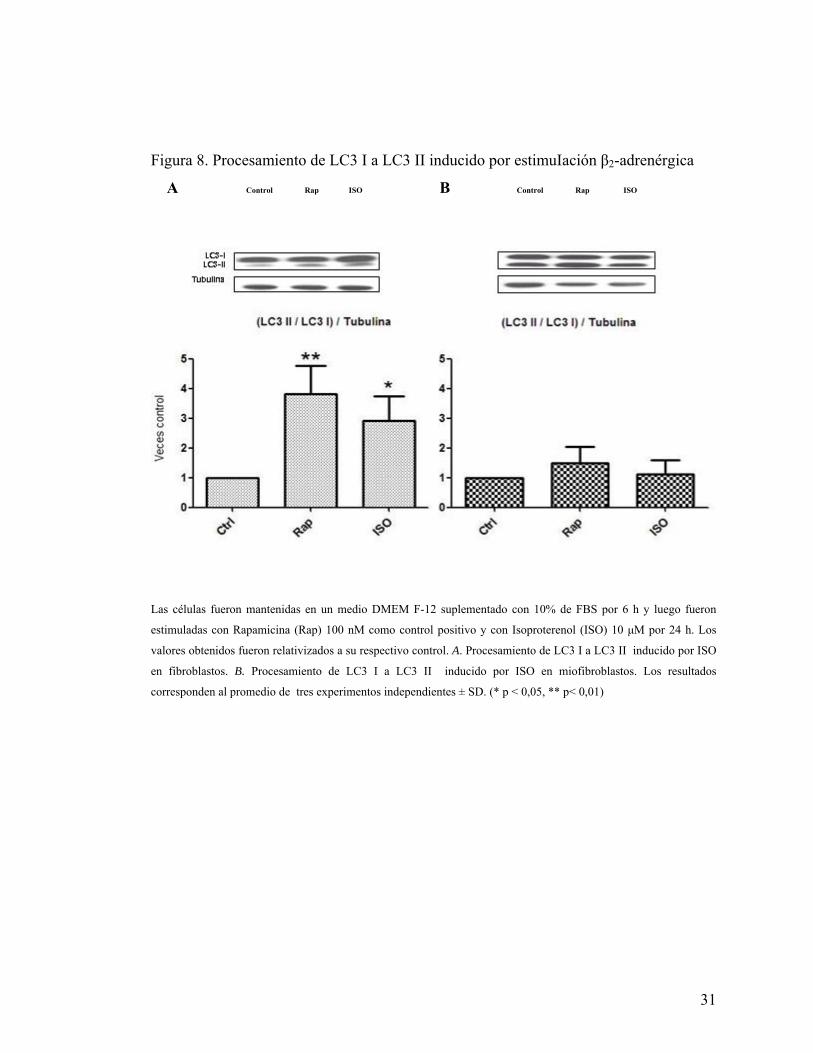
Se determinó el procesamiento de LC3 I a LC3 II por Inmunowestern blot en
fibroblastos y miofibroblastos cardiacos de rata adulta expuestos a ISO 10 μM para
observar si la activación β-adrenérgica es capaz de promover autofagia, y se utilizó
rapamicina 100 nM, inhibidor de mTOR y estímulo clásico de autofagia, como control
positivo. Los resultados muestran que en fibroblastos hay un significativo
procesamiento de la proteína LC3 I a LC3 II bajo estímulo con ISO y rapamicina
respecto a su control (Figura 8A). En miofibroblastos no hay una significancia de dicho
proceso bajo ninguno de los estímulos respecto a su control (Figura 8B). Es destacable
que en miofibroblastos, como ya se ha comprobado, la condición basal tiene un
procesamiento mayor de la proteína LC3 I a LC3 II respecto a la condición basal de
fibroblastos (Figura 8A y 8B, Western blot).
31
Figura 8. Procesamiento de LC3 I a LC3 II inducido por estimuIación β2-adrenérgica
A Control Rap ISO B Control Rap ISO
Las células fueron mantenidas en un medio DMEM F-12 suplementado con 10% de FBS por 6 h y luego fueron
estimuladas con Rapamicina (Rap) 100 nM como control positivo y con Isoproterenol (ISO) 10 μM por 24 h. Los
valores obtenidos fueron relativizados a su respectivo control. A. Procesamiento de LC3 I a LC3 II inducido por ISO
en fibroblastos. B. Procesamiento de LC3 I a LC3 II inducido por ISO en miofibroblastos. Los resultados
corresponden al promedio de tres experimentos independientes ± SD. (* p < 0,05, ** p< 0,01)

32
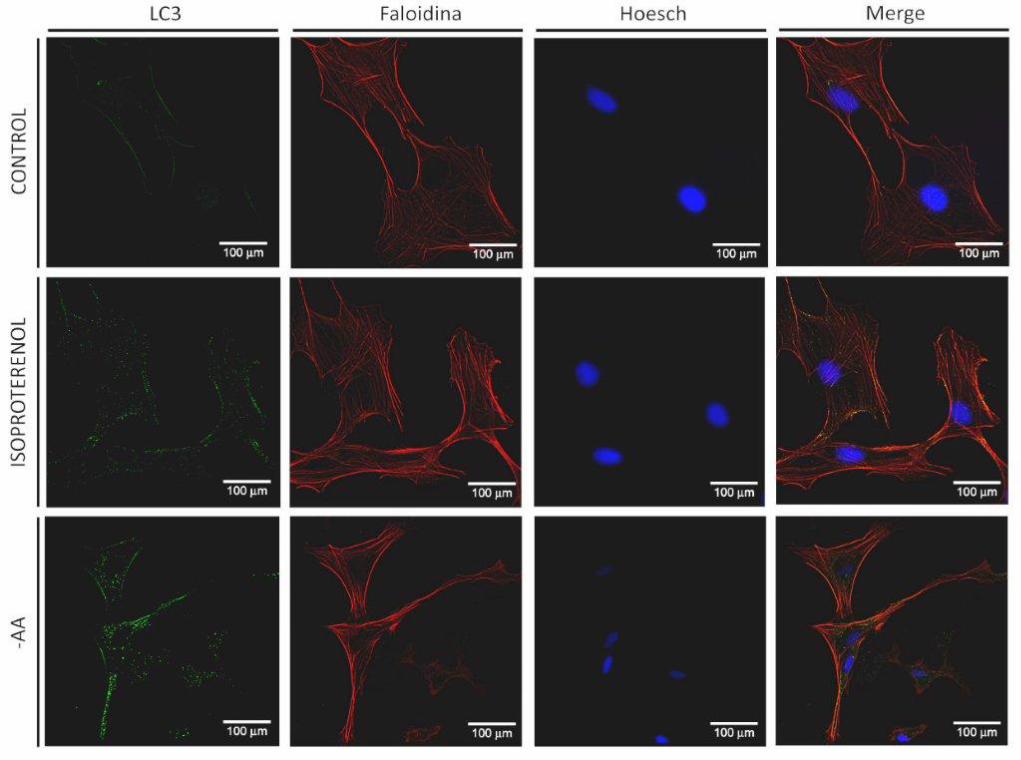
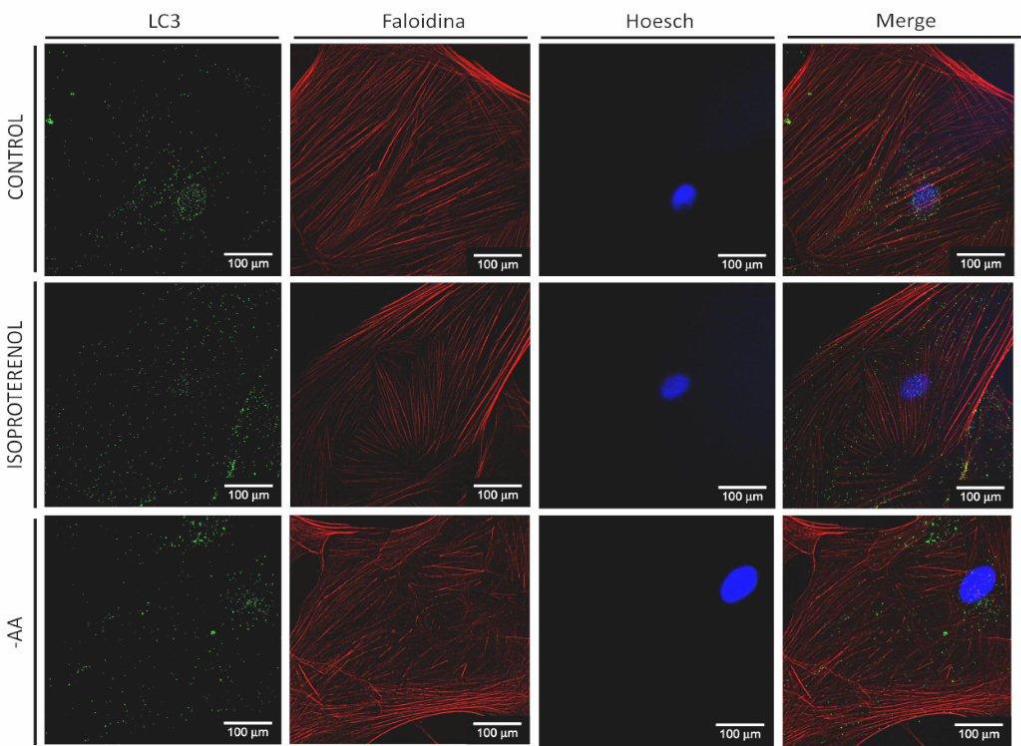
5.6.2 Localización de LC3 endógeno en vesículas autofágicas inducida
por ISO
Por otro lado, se determinó autofagia por Inmunocitoquímica. Mediante esta
técnica observamos la organización de LC3 endógeno bajo distintos estímulos. Cuando
el proceso autofágico es inducido, LC3 se localiza en las vesículas autofágicas y, por lo
tanto, se observa un patrón punteado con mayor intensidad de marca y claramente
definido que puede distinguirse del patrón observado en una célula en la que no se ha
inducido autofagia.
Fibroblastos y miofibroblastos cardiacos de rata adulta fueron expuestos a
privación de aminoácidos, estímulo clásico de la autofagia utilizado como control
positivo, y a ISO 10 μM como estímulo de interés para este trabajo.
En fibroblastos se observa que el control no presenta marca verde
correpondiente a LC3 endógeno en la forma e intensidad que ocurre frente al estímulo
con ISO o con privación de aminoácidos (Figura 9A). Estas dos últimas condiciones
muestran una marca verde claramente definida en un patrón punteado que indica la
localización de LC3 en las vesículas autofágicas, siendo mayor la marca en privación de
aminoácidos que en estímulo con ISO. Por otro lado, vemos que en miofibroblastos
control aparece un patrón punteado de la marca verde con mayor intensidad que en el
control de los fibroblastos, sin embargo, entre miofibroblastos control, estimulados con
ISO o en privación de aminoácidos, no hubo diferencias que denotaran mayor o menor
autofagia entre ellos (Figura 9B).
Finalmente, es importante destacar que todas las imágenes presentadas se
encuentran en la misma escala de aumento. En un campo visual se pueden observar más
de un fibroblasto, en cambio, en miofibroblastos, células diferenciadas de mayor
tamaño, no alcanza un campo para cubrir la visión total de al menos una célula. Esto
demuestra las diferencias morfológicas ampliamente descritas entre fibroblastos y
miofibroblastos.

33
Figura 9. Organización de LC3 endógeno bajo estimulación β2-adrenérgica
A
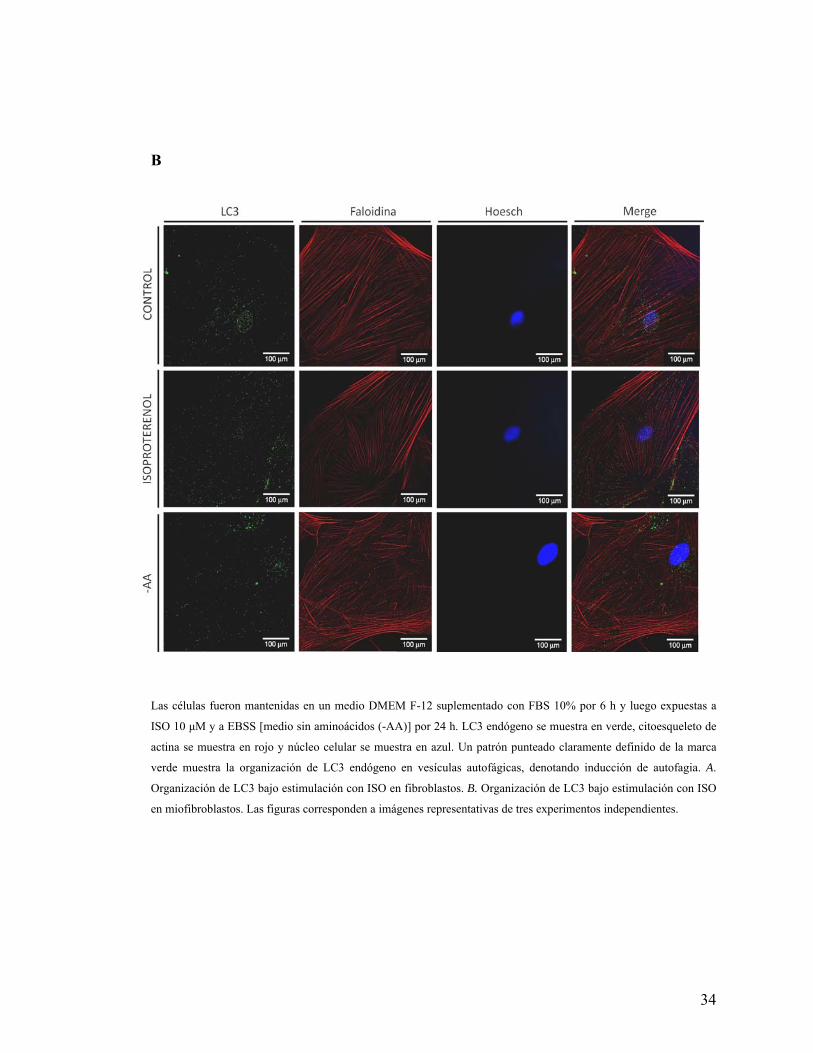
34
B
Las células fueron mantenidas en un medio DMEM F-12 suplementado con FBS 10% por 6 h y luego expuestas a
ISO 10 μM y a EBSS [medio sin aminoácidos (-AA)] por 24 h. LC3 endógeno se muestra en verde, citoesqueleto de
actina se muestra en rojo y núcleo celular se muestra en azul. Un patrón punteado claramente definido de la marca
verde muestra la organización de LC3 endógeno en vesículas autofágicas, denotando inducción de autofagia. A.
Organización de LC3 bajo estimulación con ISO en fibroblastos. B. Organización de LC3 bajo estimulación con ISO
en miofibroblastos. Las figuras corresponden a imágenes representativas de tres experimentos independientes.

35
6. DISCUSIÓN
6.1 Caracterización de receptores adrenérgicos en fibroblastos
y miofibroblastos cardiacos de rata adulta
El primer objetivo de la presente memoria fue caracterizar los receptores
adrenérgicos en miofibroblastos cardiacos de rata adulta teniendo en cuenta que en
fibroblastos cardiacos, células de las cuales se originan los miofibroblastos cardiacos,
sólo existen receptores del tipo β2-adrenérgicos. De este modo, realizamos un ensayo de
desplazamiento utilizando sólo ligandos del receptor β-adrenérgico: β-adrenérgico
inespecífico (propanolol), β1-específico (atenolol) y β2-específico (ICI-118551). Los
resultados mostraron, en ambos fenotipos, un desplazamiento de la marca radiactiva
sólo frente a propanolol e ICI-118551, lo que confirma que los fibroblastos y
miofibroblastos cardiacos de rata adulta presentan receptores adrenérgicos sólo del
subtipo β2. Esto ya había sido descrito por otros autores en fibroblastos cardiacos
humanos y de rata adulta (20, 21), sin embargo, en los miofibroblastos no ha sido
descrito previamente en la literatura la presencia única de este subtipo de receptores
adrenérgicos.
6.2 Determinación de Bmax y Kd de receptores β2-adrenérgicos
en fibroblastos y miofibroblastos cardiacos de rata adulta
En cuanto al número de receptores β2-adrenérgicos (representado por el valor
Bmax) y la afinidad de éstos por el desplazante utilizado (representado por el valor Kd),
se observó significativas diferencias entre fibroblastos y miofibroblastos cardiacos de
rata adulta. Los miofibroblastos presentaron mayor número de receptores β2-
adrenérgicos y mayor afinidad de estos receptores por propanolol que los fibroblastos.

36
En la literatura se ha encontrado que el TGF-β1, factor de crecimiento utilizado
para diferenciar fibroblastos a miofibroblastos in vitro, modula el número de receptores
β-adrenérgicos en distintos tipos celulares: fibroblastos cardiacos de rata adulta, en
fibroblastos pulmonares de embrión humano (células HEL 299) y en células del
músculo liso (33, 34, 44). En todos estos estudios se observó que TGF- β1 tiene la
capacidad de disminuir el número de receptores β-adrenérgicos aún manteniendo la
afinidad de éstos por el ligando utilizado en cada caso, lo que se contrapone a lo
determinado en el actual estudio. Sin embargo, existen diferencias experimentales que
darían cuenta de los disímiles resultados: las concentraciones y el tiempo de estímulo
con TGF-β1 en esos casos son considerablemente menores a las condiciones utilizadas
para lograr la diferenciación del fibroblasto a miofibroblasto (1ng/ml, 2 ng/ml ó 1,5 pM
de TGF-β1 por 24 h como tiempo máximo contra 5 ng/ml de TGF-β1 por 84 h utilizados
para la diferenciación in vitro). Hasta la fecha no hay antecedentes del efecto en función
del tiempo de TGF-β1 en fibroblastos cardiacos. De acuerdo a esos resultados, se haría
necesario determinar el efecto de TGF-β1 dependiente de la dosis y dependiente del
tiempo sobre el número de receptores β-adrenérgicos. De todas maneras, hay que
atender al hecho que nuestro objetivo es el miofibroblasto cardiaco y para ello el efecto
de TGF-β1 se debe evaluar al tiempo y concentración en los cuales la diferenciación
total de ellos ha ocurrido, esto es 84 h y 5 ng/ml.
Por otro lado, se ha visto que en el corazón de ratas transgénicas que
sobreexpresan TGF-β1 el número de receptores β-adrenérgicos aumenta, aunque se
mantiene la afinidad de éstos por el ligando utilizado (45). A pesar que aquel estudio de
unión de ligandos se realizó en membranas de un homogenizado de corazón y no sobre
membranas de fibroblastos purificados, por lo que se observa también el efecto sobre
cardiomiocitos, es destacable el hecho que, al sobreexpresar TGF-β1, el corazón esté
siendo expuesto a este estímulo de manera crónica y, por lo tanto, podría dar luz de lo
que ocurre con el número de receptores β-adrenérgicos frente a un estímulo sostenido en
el tiempo con este factor de crecimiento.

37
6.3 Determinación de funcionalidad de receptor β2-
adrenérgico
Para determinar la funcionalidad del receptor β2-adrenérgico, un receptor
asociado a proteína Gs, se evaluaron elementos que reflejan la actividad de la subunidad
Gαs y de la subunidad Gβγ: producción de cAMP y activación de ERK 1/2,
respectivamente. En fibroblastos y miofibroblastos cardiacos de rata adulta se observó
un aumento significativo de la producción de cAMP frente al estímulo β2-adrenérgico
(ISO) respecto a sus controles. De la misma manera, se observó que en ambos tipos
celulares se activó ERK a los 15 min de estímulo con ISO; la diferencia radicó en que
fibroblastos sólo presentó activación de ERK 1 y miofibroblastos presentó activación de
ERK 1/2. Dado los resultados, podemos decir que los receptores β2-adrenérgicos son
funcionales en fibroblastos y miofibroblastos cardiacos de rata adulta.
Es destacable el hecho que, a pesar que miofibroblastos presentaron mayor
número de receptores y mayor afinidad de éstos por su ligando, esto no se vio reflejado
en la producción de cAMP, que es una de las formas más directas de evaluar
funcionalidad de receptores acoplados a proteína Gs. De partida, al comparar los niveles
de cAMP en condiciones basales entre fibroblastos y miofibroblastos, se obtuvo que la
producción de cAMP fue significativamente menor en miofibroblastos respecto a
fibroblastos (dato no mostrado). Esto nos da a entender que TGF-β1, además de modular
el número de receptores y la Kd, podría modular las vías transduccionales implicadas en
la síntesis y degradación de cAMP.
En la literatura existe abundante información acerca de los efectos antagónicos
de cAMP y TGF-β, donde el primero inhibiría efectos del segundo en diversos tipos
celulares (46-48), incluyendo fibroblastos cardiacos de rata adulta (26, 49). Actualmente
existe poca información acerca de la situación inversa, donde TGF-β influya sobre los
niveles de cAMP, que es lo que podría explicar el fenómeno observado en este estudio.
En cardiomiocitos, se ha observado que TGF-β1 disminuye la acumulación de cAMP

38
estimulada por el factor de crecimiento epidermal (EGF) (50), y en células del músculo
liso de arteria pulmonar se observó el mismo efecto de TGF-β1 sobre el aumento de
cAMP inducido por activación del receptor de prostaciclina (51). Lo importante de estos
estudios es que en ambos se determinó que, al menos en parte, la disminución de la
producción de cAMP estaba asociada a una disfunción de la adenilato ciclasa (AC). Más
aún, en células del músculo liso de arteria pulmonar se determinó que una disminución
de la expresión del mRNA de las isoformas 1,2 y 4 de AC y un aumento en la proteína
Gi estaría asociada, entre otros efectos, a la disminución de producción de cAMP por
TGF-β1 (51). Consistente con todos estos datos, se ha documentado en células HEL 299
que TGF-β1 es capaz de disminuir el número de receptores β2-adrenérgicos y por esto se
ve disminuido la producción de cAMP frente a un estímulo con un agonista β2, sin
embargo, en el mismo trabajo se vio que luego del pretratamiento con TGF-β1, el
estímulo con forskolina (FSK), un activador directo de AC, no es capaz de aumentar la
acumulación de cAMP (34).
Por lo demás, también se debe considerar la participación de fosfodiesterasa
(PDE), una enzima que cumple la función de hidrolizar cAMP y/o cGMP, en el control
de los niveles de cAMP. Se ha reportado que TGF- β1, al inducir la transición epitelial-
mesenquimal (EMT) en una línea de células humanas de epitelio alveolar tipo II, es
capaz de regular la expresión y actividad de distintas isoformas de PDE (52). En este
trabajo se observó que TGF- β1 fue capaz de aumentar la expresión de PDE4A y
PDE4D a nivel proteico y de mRNA, así como también aumentó la actividad enzimática
específica sobre cAMP dos veces por sobre el control. Además, ellos demostraron que
la isoforma PDE4 fue el principal contribuyente a la actividad específica de degradación
de cAMP, lo que corroboró la relación entre un aumento específico de isoformas PDE4
y una disminución específica del cAMP frente al pretratamiento con TGF- β1.
De este modo, se observa que el efecto de TGF-β1 sobre la acumulación de
cAMP podría no estar relacionado exclusivamente a su efecto sobre la cantidad de

39
receptores que se presentan en la célula, sino también a un efecto directo de éste sobre
AC y/o PDE.
Finalmente, existen tres puntos a considerar en el modelo experimental utilizado
en nuestro trabajo: primero, la diferenciación de fibroblastos a miofibroblastos implica
la exposición prolongada a TGF-β1 por 84 h; segundo, el estímulo adrenérgico ISO se
hace en medio suplementado con suero FBS 10%, el cual en sí lleva TGF-β1; tercero, se
ha demostrado que los miofibroblastos secretan TGF-β1 y que expresan un mayor
número de receptores para este factor de crecimiento. Considerando estas condiciones,
es posible esperar un efecto mantenido de TGF-β1 sobre la actividad de la AC y la PDE.
Por lo tanto, aunque aquí no se evaluó la actividad de la AC y la PDE, el escaso
aumento de cAMP estimulado por ISO podría, en nuestro caso, ser explicado por esos
hallazgos.
En cuanto a la activación de ERK 1/2, es destacable notar que es similar en
ambos tipos celulares, aunque en fibroblastos no hubo una activación de ERK 2 a los 15
min de estímulo con ISO. Fuera de este resultado, podemos ver que no hay diferencias
significativas entre el nivel de activación de ERK 1 entre fibroblastos y miofibroblastos
(dato no mostrado), lo que nuevamente aparece como una contradicción frente a la
diferencia del número de receptores β2-adrenérgicos y su afinidad, entre ambos
fenotipos celulares. Se ha reportado en la literatura que agentes que aumentan los
niveles de cAMP son capaces de inhibir la activación de ERK promovida por distintos
estímulos en distintos modelos celulares (26, 53-55). Orientando hacia nuestro modelo,
se ha descrito en fibroblastos cardiacos de rata adulta que ISO, un agente que en sí
mismo es capaz de activar la vía de las ERKs, bloquea la activación de ésta cuando es
promovida por TGF-β en etapas tempranas (20 min de estímulo). Con estos
antecedentes, se podría inferir que, en el actual estudio, el estímulo con ISO causa un
efecto dual en que, a tiempos menores, cAMP esté inhibiendo la expresión de ERK y
luego este efecto se pueda revertir. De este modo, tendríamos que los miofibroblastos,
que expresan menor cantidad de cAMP basal y frente a un estímulo β2-adrenérgico, son

40
capaces de activar ERK 1/2 a los 15 min de estímulo con ISO, en cambio los
fibroblastos, que expresan mayor cantidad de cAMP basal y bajo un estímulo β2-
adrenérgico, son capaces de activar sólo ERK 1 a los 15 min de estímulo con ISO. De
todas formas, es importante determinar qué ocurre en cuanto a la activación de ERK 1/2
en fibroblastos a mayor tiempo de estímulo con ISO.
6.4 Autofagia en fibroblastos y miofibroblastos cardiacos de
rata adulta
Para determinar autofagia en estos fenotipos celulares, se evaluó mediante la
técnica de Western blot el procesamiento de la proteína LC3 I a LC3 II. Dado que
autofagia se ha asociado ampliamente a condiciones de privación de nutrientes (31, 39),
en un primer lugar, se observó este proceso en fibroblastos y miofibroblastos frente a
concentraciones decrecientes de suero por 24 h. Se obtuvo que los fibroblastos
comenzaron a sufrir autofagia significativamente por sobre su control en un medio
suplementado con 1% de FBS y, como se esperaba, también en un medio libre de suero.
Por otro lado, miofibroblastos, aunque mostraron una tendencia similar a la de los
fibroblastos, no fueron capaces de sufrir el mismo efecto bajo ninguna de las
condiciones.
En cuanto al procesamiento de LC3 I a LC3 II, al estimular con rapamicina
(inhibidor de mTOR, por lo tanto, inductor de la autofagia) y con ISO como estímulo de
receptores β2-adrenérgicos, se observó que en fibroblastos cardiacos de rata adulta hubo
una clara inducción de la autofagia. En este caso, se obtuvo que la estimulación β2-
adrenérgica logró inducir autofagia en menor grado que rapamicina. Por
inmunocitoquímica se confirmó lo mismo que por Western blot: LC3 endógeno
prácticamente no localiza en vesículas autofágicas en fibroblastos controles, pero sí lo
hace bajo estímulo con ISO y, en mayor medida, en un medio privado de aminoácidos.

41
En cuanto a miofibroblastos, nuevamente se obtuvo, por ambas técnicas, un nulo
incremento de la respuesta autofágica frente a los estímulos indicados.
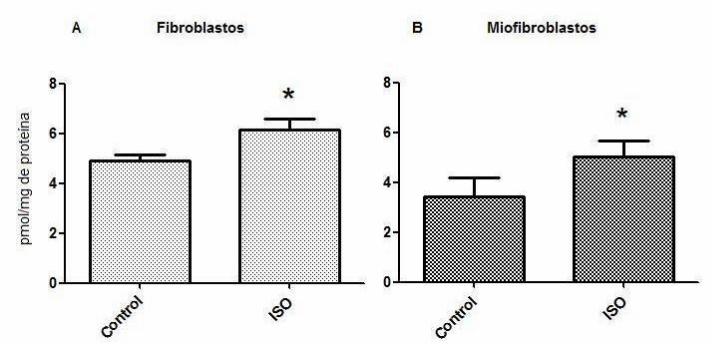
Cabe destacar que los miofibroblastos no fueron capaces de sufrir mayor
autofagia que su propio control bajo ninguna de las condiciones a las cuales fueron
expuestos, sin embargo, los niveles basales de procesamiento de la proteína LC3 I a
LC3 II detectados por Western blot aparentaron ser considerablemente mayores que en
fibroblastos. Del mismo modo, se observó por inmunocitoquímica que en
miofibroblastos el nivel basal de localización de LC3 endógeno en vesículas autofágicas
fue mayor al del nivel basal en fibroblastos, sin embargo, no hubo diferencias en el
patrón autofágico entre miofibroblastos controles y los sujetos a estímulos para inducir
autofagia. Esta virtual mayor autofagia en los miofibroblastos cardiacos respecto a los
fibroblastos cardiacos fue confirmada comparando el procesamiento de LC3 I a LC3 II
en ambos fenotipos en una condición basal.
Todos estos antecedentes muestran la dualidad del comportamiento de
miofibroblastos frente al proceso de autofagia: por un lado, en una condición basal
sufren de dicho proceso, sin embargo, frente a estímulos clásicos y frente a nuestro
estímulo de interés, no existe un aumento por sobre su control.
La explicación de este fenómeno se puede entender a través de los efectos que
produce TGF-β1 y que son adicionales a aquél que promueve la diferenciación de
fibroblastos a miofibroblastos. Se ha determinado que TGF-β1 induce autofagia en
células epiteliales mamarias, en células epiteliales renales y en células de carcinoma
hepatocelular y mamario (56-58). Se dilucidó, en células de carcinoma hepatocelular,
que la vía se señalización por la cual TGF-β1 induce la autofagia involucraría a las vías
de Smad y de JNK (58). Esto antecedentes podrían servir, en parte, para explicar la
visualización de un mayor procesamiento de la proteína LC3 I a LC3 II en
miofibroblastos cardiacos en su estado basal, así como la mayor localización de LC3
endógeno en vesículas autofágicas. A pesar de esto, los miofibroblastos cardiacos de

42
rata adulta no fueron capaces de aumentar esta autofagia frente a estímulos clásicos del
proceso ni bajo estimulación β2-adrenérgica.
En diversos estudios se ha determinado que TGF-β1 realiza muchos de sus
efectos por una vía no canónica, es decir, una vía alternativa a aquella que tiene relación
con las proteínas Smad. Esta vía no canónica involucraría a la proteína PI3-K y, por lo
tanto, indirectamente a mTOR, una proteína clave en la regulación de la autofagia. En
fibroblastos de riñón se ha determinado que TGF-β1 activa la vía de PI3-K y que esto
lleva a una activación de mTOR a través de la fosforilación de Akt y tuberina (TSC2)
(59). En este estudio, además, se asoció la activación del Complejo 1 de mTOR
(mTORC1) con la proliferación de miofibroblastos en el intersticio renal y con la mayor
expresión de proteínas de la MEC, ambos efectos inducidos por TGF-β1. Además, los
autores explican que este efecto producido por TGF-β1 se ha observado en fibroblastos,
pero no en células epiteliales. Por otro lado, se ha visto que TGF-β1 regula una variedad
de procesos en el desarrollo del cáncer, entre éstos la EMT (60). Recientemente se ha
descrito en células epiteliales que la EMT inducida por TGF-β1 induce tanto el aumento
del tamaño celular como la capacidad invasiva de células que han sufrido esta transición
a través de la activación de mTOR, y que esta activación ocurre por la vía PI3-K y Akt
(61). Finalmente, en fibroblastos embriónicos humanos se ha observado TFG-β activa a
mTORC1 en este tipo de células y no en células epiteliales en un forma dependiente de
la vía PI3-K/Akt/TSC2, y que rapamicina, un inhibidor del complejo mTORC1, sólo
disminuyó levemente la diferenciación de fibroblasto a miofibroblasto por TGF-β (62).
Cabe destacar que en todos estos estudios citados, la participación de mTOR se evaluó
al inhibir a esta proteína (sola o en el complejo mTORC1) con rapamicina a distintas
concentraciones por distintos tiempos. Lo importante es recalcar que rapamicina inhibió
los efectos asociados a TGF-β en un pretratamiento con el este agente inhibidor de
mTOR, lo que difiere con nuestras condiciones experimentales (primero utilizamos
TGF-β1 para diferenciar y luego se estimuló con rapamicina). Por esto, seguramente, en
nuestro estudio no vemos que rapamicina pueda revertir el efecto de diferenciación
ejercido por TGF-β1.

43
Lo interesante de los últimos antecedentes expuestos, es que todos apuntan a una
asociación entre TGF-β1, el agente utilizado para diferenciar fibroblastos a
miofibroblastos, y mTOR, la proteína clave en el proceso de autofagia. Como se ha
indicado, la autofagia se induce al inhibir la proteína mTOR, por esto se eligió
rapamicina como control positivo de algunos experimentos. Si TGF-β1 es capaz de
activar mTOR por su vía no canónica, PI3-K/Akt, es razonable esperar que el proceso
de autofagia se vea disminuido en miofibroblastos que han logrado su diferenciación in
vitro. Ahora, como ya se ha mencionado previamente, nuestras condiciones
experimentales conservarían los efectos de TGF-β1 (la exposición a TGF-β1 por tiempo
prolongado, por el suero y el efecto autocrino), indicando que su vía de señalización se
mantiene activa durante este período. Considerando esto último, se podría explicar
porqué ni siquiera el estímulo con rapamicina por 24 h pueda revertir la situación y, por
lo tanto, los miofibroblastos no puedan sufrir autofagia en mayor medida que su propio
control.
En resumen, podemos decir que TGF-β1 podría estar ejerciendo un efecto dual
en cuanto al proceso de autofagia. Por un lado, estaría activando la vía Smad y JNK y,
por lo tanto, manteniendo un nivel aparentemente elevado de autofagia, seguramente
por modulación de la expresión de proteínas implicadas en el proceso de autofagia, ya
que ambas actúan a nivel de transcripción. Sin embargo, por otro lado estaría
estimulando la vía PI3-K/Akt/mTOR y, por lo tanto, volviendo a las células poco
susceptibles a estímulos autofágicos que se dirijan, directa o indirectamente, a la
inhibición de la proteína mTOR. Por todo lo anteriormente expuesto, se vuelve
interesante evaluar la asociación y efecto de las vías de señalización asociadas a TGF-β1
en el proceso de autofagia en fibroblastos y miofibroblastos cardiacos de rata adulta.

44
6.5 Alcances fisiopatológicos
Los alcances fisiopatológicos de los resultados obtenidos en este trabajo tiene
diversas aristas. Por un lado, en los miofibroblastos cardiacos, a pesar de haber un
mayor número de receptores β2-adrenérgicos, esto no se refleja en la producción de
cAMP y en la activación de ERK 1/2. Así, pareciera existir una autorregulación en la
señalización intracelular de modo de no verse mayormente afectados por el ya
aumentado tono adrenérgico en estados de daño al miocardio.
Por otro lado, se obtuvo que los miofibroblastos sufrieron autofagia basalmente.
Esto podría encontrar explicación en el hecho que el miofibroblasto es un fenotipo
celular asociado a procesos de reparación tisular y, por lo tanto, es extremadamente
secretor de proteína de la MEC, por lo que seguramente aumenta su autofagia en un
estado basal con la finalidad de obtener precursores y energía para la síntesis de
proteínas MEC, esencialmente colágeno. En este sentido, nuestro laboratorio ha
determinado que la autofagia en fibroblastos cardiacos de rata adulta estaría siendo
activada por estimulación β2-adrenérgica, en parte, con la finalidad de degradar
colágeno (43), probablemente, a fin de utilizar esos aminoácidos y energía en promover
la proliferación celular, un efecto que sí es mediado por estimulación adrenérgica en
fibroblastos, pero no en los miofibroblastos pues ellos no tienen la capacidad de
proliferar in vitro (63). Si entendemos que el miofibroblasto repara el tejido en gran
medida por depósito de proteínas de la MEC, el estímulo adrenérgico aumentado en
estados de daño al miocardio, sería contraproducente para su función. De este modo,
disminuiría su respuesta al estímulo β2-adrenérgico para evitar aumentar la degradación
de proteínas de la MEC por autofagia, sin embargo, recurriría a un nivel basal que le
permita obtener precursores y energía para procesos de síntesis proteica.

45
7. CONCLUSIONES
1. Fibroblastos y miofibroblastos cardiacos de rata adulta presenta sólo un subtipo de
receptor adrenérgico: el receptor β2-adrenérgico.
2. Miofibroblastos cardiacos de rata adulta presentan mayor número de receptores β2-
adrenérgicos que fibroblastos cardiacos de rata adulta. Del mismo modo, los receptores
β2-adrenérgicos de miofibroblastos cardiacos de rata adulta son más afines por sus
ligandos que los de fibroblastos cardiacos de rata adulta.
3. Los receptores β2-adrenérgicos de fibroblastos y miofibroblastos cardiacos de rata
adulta son funcionales, ya que son capaces de activar a proteínas ERK y aumentar la
producción de cAMP frente a un estímulo β2-adrenérgico.
4. Fibroblastos cardiacos de rata adulta sufren autofagia en un medio con baja o nula
suplementación de suero, mientras que los miofibroblastos no son capaces de sufrir
mayor autofagia en un medio con baja o nula suplementación con suero.
5. Fibroblastos cardiacos de rata adulta sufren autofagia por estímulos clásicos de dicho
proceso, como lo son rapamicina o privación de aminoácidos, y de manera menos
potente por estímulo β2-adrenérgico. Por otro lado, los miofibroblastos cardiacos de rata
adulta no son capaces de sufrir autofagia por estímulos clásicos del proceso, como
tampoco por estímulo β2-adrenérgico, sin embargo, sus niveles basales de autofagia son
mayores a los niveles basales de fibroblastos.

46
8. REFERENCIAS
1. Organización Mundial de la Salud. 55ª Asamblea Mundial de la Salud , A55/16,
punto 13.11. In: 2002.
2. Szot MJ. Demographic-epidemiologic transition in Chile, 1960-2001. Rev Esp
Salud Publica 2003;77:605-13.
3. Camelliti P, Borg TK, Kohl P. Structural and functional characterisation of
cardiac fibroblasts. Cardiovasc Res 2005;65:40-51.
4. Weber KT. Fibrosis in hypertensive heart disease: focus on cardiac fibroblasts. J
Hypertens 2004;22:47-50.
5. Booz GW, Baker KM. Molecular signalling mechanisms controlling growth and
function of cardiac fibroblasts. Cardiovasc Res 1995;30:537-43.
6. MacKenna D, Summerour SR, Villarreal FJ. Role of mechanical factors in
modulating cardiac fibroblast function and extracellular matrix synthesis. Cardiovasc
Res 2000;46:257-63.
7. Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast:
therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol
2005;45:657-87.
8. Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial
remodeling. Pharmacol Ther 2009;123:255-78.
9. Gabbiani G. The myofibroblast in wound healing and fibrocontractive diseases.
J Pathol 2003;200:500-3.
10. Chai Q, Krag S, Chai S, Ledet T, Wogensen L. Localisation and phenotypical
characterisation of collagen-producing cells in TGF-beta 1-induced renal interstitial
fibrosis. Histochem Cell Biol 2003;119:267-80.
11. Sun Y, Weber KT. RAS and connective tissue in the heart. Int J Biochem Cell
Biol 2003;35:919-31.

47
12. Copaja M. Participación de Rho en la muerte inducida por estatinas de
fibroblastos y miofibroblastos cardiacos de rata. Tesis Doctoral. Universidad de Chile.
2010.
13. Vivar R. Apoptosis dependiente de Angiotensina II en fibroblastos cardiacos que
sobreexpresan el receptor AT1: Participación de Fosfolipasa C y Proteinakinasa C.
Memoria de Título. 2007.
14. Soto C. Muerte celular de fibroblastos y miofibroblastos cardiacos neonatos por
sobre-expresión de los receptores tipo 1 y 2 de Angiotensina II. Memoria de Título.
Universidad de Chile. 2006.
15. Sun Y, Weber KT. Infarct scar: a dynamic tissue. Cardiovasc Res 2000;46:250-
6.
16. Zhang HY, Phan SH. Inhibition of myofibroblast apoptosis by transforming
growth factor beta(1). Am J Respir Cell Mol Biol 1999;21:658-65.
17. Swaney JS, Patel HH, Yokoyama U, Lai NC, Spellman M, Insel PA, Roth DM.
Adenylyl cyclase activity and function are decreased in rat cardiac fibroblasts after
myocardial infarction. Am J Physiol Heart Circ Physiol 2007;293:H3216-H3220.
18. Hasking GJ, Esler MD, Jennings GL, Burton D, Johns JA, Korner PI.
Norepinephrine spillover to plasma in patients with congestive heart failure: evidence of
increased overall and cardiorenal sympathetic nervous activity. Circulation
1986;73:615-21.
19. Woodcock EA. Cardiac alpha 1-adrenergic drive in pathological remodelling.
Cardiovasc Res 2008;77:452-62.
20. Turner NA, Porter KE, Smith WH, White HL, Ball SG, Balmforth AJ. Chronic
beta2-adrenergic receptor stimulation increases proliferation of human cardiac
fibroblasts via an autocrine mechanism. Cardiovasc Res 2003;57:784-92.
21. Meszaros JG, Gonzalez AM, Endo-Mochizuki Y, Villegas S, Villarreal F,
Brunton LL. Identification of G protein-coupled signaling pathways in cardiac
fibroblasts: cross talk between G(q) and G(s). Am J Physiol Cell Physiol
2000;278:C154-C162.

48
22. Scheuer J. Catecholamines in cardiac hypertrophy. Am J Cardiol 1999;83:70H-
4H.
23. Leicht M, Greipel N, Zimmer H. Comitogenic effect of catecholamines on rat
cardiac fibroblasts in culture. Cardiovasc Res 2000;48:274-84.
24. Fisher SA, Absher M. Norepinephrine and ANG II stimulate secretion of TGF-
beta by neonatal rat cardiac fibroblasts in vitro. Am J Physiol 1995;268:C910-C917.
25. Akiyama-Uchida Y, Ashizawa N, Ohtsuru A, Seto S, Tsukazaki T, Kikuchi H,
Yamashita S, Yano K. Norepinephrine enhances fibrosis mediated by TGF-beta in
cardiac fibroblasts. Hypertension 2002;40:148-54.
26. Liu X, Sun SQ, Hassid A, Ostrom RS. cAMP inhibits transforming growth
factor-beta-stimulated collagen synthesis via inhibition of extracellular signal-regulated
kinase 1/2 and Smad signaling in cardiac fibroblasts. Mol Pharmacol 2006;70:1992-
2003.
27. Rosenbaum DM, Rasmussen SG, Kobilka BK. The structure and function of G-
protein-coupled receptors. Nature 2009;459:356-63.
28. Xiao RP, Cheng H, Zhou YY, Kuschel M, Lakatta EG. Recent advances in
cardiac beta(2)-adrenergic signal transduction. Circ Res 1999;85:1092-100.
29. Colombo F, Gosselin H, El-Helou V, Calderone A. Beta-adrenergic receptor-
mediated DNA synthesis in neonatal rat cardiac fibroblasts proceeds via a
phosphatidylinositol 3-kinase dependent pathway refractory to the antiproliferative
action of cyclic AMP. J Cell Physiol 2003;195:322-30.
30. Dubey RK, Gillespie DG, Mi Z, Jackson EK. Endogenous cyclic AMP-
adenosine pathway regulates cardiac fibroblast growth. Hypertension 2001;37:1095-
100.
31. Yang YP, Liang ZQ, Gu ZL, Qin ZH. Molecular mechanism and regulation of
autophagy. Acta Pharmacol Sin 2005;26:1421-34.
32. Budovskaya YV, Stephan JS, Deminoff SJ, Herman PK. An evolutionary
proteomics approach identifies substrates of the cAMP-dependent protein kinase. Proc
Natl Acad Sci U S A 2005;102:13933-8.

49
33. Iizuka K, Sano H, Kawaguchi H, Kitabatake A. Transforming growth factor
beta-1 modulates the number of beta-adrenergic receptors in cardiac fibroblasts. J Mol
Cell Cardiol 1994;26:435-40.
34. Mak JC. Transforming growth factor beta-1 inhibits beta-2-adrenoceptor gene
transcription. Naunyn-Schmiedeberg's Arch Pharmacol 2010;362:520-5.
35. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell
2008;132:27-42.
36. Gustafsson AB, Gottlieb RA. Autophagy in ischemic heart disease. Circ Res
2009;104:150-8.
37. Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular
degradation. Science 2000;290:1717-21.
38. Klionsky DJ, Cregg JM, Dunn WA, Jr., Emr SD, Sakai Y, Sandoval IV, Sibirny
A, Subramani S, Thumm M, Veenhuis M, Ohsumi Y. A unified nomenclature for yeast
autophagy-related genes. Dev Cell 2003;5:539-45.
39. Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms
and biological functions of autophagy. Dev Cell 2004;6:463-77.
40. Nishida K, Kyoi S, Yamaguchi O, Sadoshima J, Otsu K. The role of autophagy
in the heart. Cell Death Differ 2009;16:31-8.
41. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing:
crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 2007;8:741-52.
42. Zhu H, Rothermel BA, Hill JA. Autophagy in load-induced heart disease.
Methods Enzymol 2009;453:343-63.
43. Aranguiz-Urroz P, Canales J, Copaja M, Troncoso R, Vicencio JM, Carrillo C,
et al. Beta(2)-adrenergic receptor regulates cardiac fibroblast autophagy and collagen
degradation. Biochim Biophys Acta 2010.
44. Nogami M. TGF- beta 1 modulates beta-adrenergic receptor number and
function in cultured human tracheal smooth muscle cells. Am J Physiol Lung Cell Mol
Physiol 1994;266:L187-L191.

50
45. Rosenkranz S, Flesch M, Amann K, Haeuseler C, Kilter H, Seeland U, Schlüter
KD, Böhm M. Alterations of beta-adrenergic signaling and cardiac hypertrophy in
transgenic mice overexpressing TGF-beta(1). Am J Physiol Heart Circ Physiol
2002;283:H1253-H1262.
46. Greenberg RS, Bernstein AM, Benezra M, Gelman IH, Taliana L, Masur SK.
FAK-dependent regulation of myofibroblast differentiation. FASEB J 2006;20:1006-8.
47. Liu X, Ostrom RS, Insel PA. cAMP-elevating agents and adenylyl cyclase
overexpression promote an antifibrotic phenotype in pulmonary fibroblasts. Am J
Physiol Cell Physiol 2004;286:C1089-C1099.
48. Xing D, Bonanno JA. Effect of cAMP on TGFbeta1-induced corneal keratocyte-
myofibroblast transformation. Invest Ophthalmol Vis Sci 2009;50:626-33.
49. Swaney JS, Roth DM, Olson ER, Naugle JE, Meszaros JG, Insel PA. Inhibition
of cardiac myofibroblast formation and collagen synthesis by activation and
overexpression of adenylyl cyclase. Proc Natl Acad Sci U S A 2005;102:437-42.
50. Nair BG, Yu Y, Rashed HM, Sun H, Patel TB. Transforming growth factor-beta
1 modulates adenylyl cyclase signaling elements and epidermal growth factor signaling
in cardiomyocytes. J Cell Physiol 1995;164:232-9.
51. El-Haroun H, Clarke DL, Deacon K, Bradbury D, Clayton A, Sutcliffe A, Knox
AJ. IL-1beta, BK, and TGF-beta1 attenuate PGI2-mediated cAMP formation in human
pulmonary artery smooth muscle cells by multiple mechanisms involving p38 MAP
kinase and PKA. Am J Physiol Lung Cell Mol Physiol 2008;294:L553-L562.
52. Kolosionek E, Savai R, Ghofrani HA, Weissmann N, Guenther A, Grimminger
F, Seeger W, Banat GA, Schermuly RT, Pullamsetti SS. Expression and activity of
phosphodiesterase isoforms during epithelial mesenchymal transition: the role of
phosphodiesterase 4. Mol Biol Cell 2009;20:4751-65.
53. Cospedal R, Lobo M, Zachary I. Differential regulation of extracellular signal-
regulated protein kinases (ERKs) 1 and 2 by cAMP and dissociation of ERK inhibition
from anti-mitogenic effects in rabbit vascular smooth muscle cells. Biochem J 1999;342
( Pt 2):407-14.

51
54. Graves LM, Bornfeldt KE, Raines EW, Potts BC, Macdonald SG, Ross R, Krebs
EG. Protein kinase A antagonizes platelet-derived growth factor-induced signaling by
mitogen-activated protein kinase in human arterial smooth muscle cells. Proc Natl Acad
Sci U S A 1993;90:10300-4.
55. Wu J, Dent P, Jelinek T, Wolfman A, Weber MJ, Sturgill TW. Inhibition of the
EGF-activated MAP kinase signaling pathway by adenosine 3',5'-monophosphate.
Science 1993;262:1065-9.
56. Gajewska M, Gajkowska B, Motyl T. Apoptosis and autophagy induced by
TGF-B1 in bovine mammary epithelial BME-UV1 cells. J Physiol Pharmacol 2005;56
Suppl 3:143-57.
57. Koesters R, Kaissling B, Lehir M, Picard N, Theilig F, Gebhardt R, et al.
Tubular Overexpression of Transforming Growth Factor-beta1 Induces Autophagy and
Fibrosis but not Mesenchymal Transition of Renal Epithelial Cells. Am J Pathol 2010.
58. Suzuki HI, Kiyono K, Miyazono K. Regulation of autophagy by transforming
growth factor-beta (TGFbeta) signaling. Autophagy 2010;6.
59. Wang S, Wilkes MC, Leof EB, Hirschberg R. Noncanonical TGF-beta
pathways, mTORC1 and Abl, in renal interstitial fibrogenesis. Am J Physiol Renal
Physiol 2010;298:F142-F149.
60. Zavadil J, Bottinger EP. TGF-beta and epithelial-to-mesenchymal transitions.
Oncogene 2005;24:5764-74.
61. Lamouille S, Derynck R. Cell size and invasion in TGF-beta-induced epithelial
to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol
2007;178:437-51.
62. Rahimi RA, Andrianifahanana M, Wilkes MC, Edens M, Kottom TJ, Blenis J,
Leof EB. Distinct roles for mammalian target of rapamycin complexes in the fibroblast
response to transforming growth factor-beta. Cancer Res 2009;69:84-93.
63. Petrov VV, van Pelt JF, Vermeesch JR, Van Duppen VJ, Vekemans K, Fagard
RH, Lijnen PJ. TGF-beta1-induced cardiac myofibroblasts are nonproliferating
functional cells carrying DNA damages. Exp Cell Res 2008;314:1480-94.