
Malformaciones congenitas mas frecuentes en la infancia.
142
Anomalías congénitas en cirugía plástica.(labio y paladar hendido, malformaciones en oído externo y malformación de la mano). Dr. Miguel Edgardo Marinero Escobar. Asesor: Dra. Patricia de Calderón. Hospital de niños Benjamín Bloom Agosto 2013.
-
Upload
miguel-marinero -
Category
Education
-
view
1.770 -
download
1
description
Informacion basica sobre malformaciones congenitas de labio y paladar hendido, oido externo y miembros superior. Presentacion en programa de residentado en cirugia pediatrica en Hospital de niños Benjamin Bloom, El Salvador.
Transcript of Malformaciones congenitas mas frecuentes en la infancia.

Anomalías congénitas en cirugía plástica.(labio y paladar hendido, malformaciones en oído externo y malformación de la mano).
Dr. Miguel Edgardo Marinero Escobar.Asesor: Dra. Patricia de Calderón.Hospital de niños Benjamín BloomAgosto 2013.

Labio Hendido.

Embriología.
• En el inicio de la cuarta semana el primordio facial inicia su aparición.
• En el revestimiento ectodérmico, por debajo de extremo anterior del tubo nervioso y por encima del 1º arco branquial, se forma una depresión transversal, poco profunda, que es el estomodeo, o boca primitiva.

Embriología Cinco primordios faciales aparecen como prominencias alrededor
del estomodeo:1.La prominencia frontonasal única.2. La prominencia maxilares pares.3. La prominencia mandibulares pares


Desarrollo normal del paladarDía 35 (semana 5)

Embriología• La fusion de la prominencia nasal medial y las maxilares
originan la continuidad del maxilar con el labio superior y las separacion de la foveas nasales del estomodeo.
• A medida que se fusionan las prominencias nasales mediales se forma el segmento intermaxilar.

Embriología
El segmento intermaxilar origina:• Parte media o filtrum del labio superior.• La parte premaxilar y su encia correspondiente.• El paladar primario.

Estadios del desarrollo del paladar.

Desarrollo normal del paladar.

Desarrollo del Paladar(6ta Sem)El paladar se desarrolla a partir de 2 primordios:• Paladar primario(proceso palatino medial).• Paladar secundario, a partir de proyecciones
mesenquimatosas que se extienden desde la cara interna de las prominencias maxilares(procesos palatinos laterales)

• El tabique nasal se desarrolla como un crecimiento hacia debajo de las partes internas de las prominencias nasales medias unidas; la fusión del tabique nasal y los procesos palatinos comienza en la parte anterior durante la 9 sem y termina en la parte posterior hacia la 12 sem.

8 semanas

10 semanas

Conformación labio superior

Defectos o anormalidades Embriológicas.
• Labio Hendido: falta de fusión de las prominencias maxilares con la prominencias nasales mediales.
• Paladar Hendido: falta de acercamiento y fusión entre si de las masas mesinquematosas de los procesos palatinos laterales.

Labio y paladar hendido. Definición.
• Se conoce así a la entidad clínica congénita caracterizada por la falta de unión de las partes que embriológicamente darán lugar al labio y/o paladar. Incluye a los procesos palatinos, nasales medios y laterales.
• La malformación congénita de labio hendido, es también mal llamada labio leporino como sinónimo de labio de liebre, por el parecido de esta deformación al labio de los lepóridos: familia de los mamíferos roedores que comprende las liebres y los conejos.

Continuación.• Anomalía congénita que afecta labio superior y paladar blando
y duro de la boca.• Frecuencia: 15% de todas las malformaciones.• Incidencia: 0,8 a 1,6 casos por cada 1000 nacimientos.• Sexo masculino más afectado, relación de 7:3• Predominio unilateral sobre el bilateral• Mayor frecuencia el lado izquierdo que el derecho.

Se denomina fisura labial:
Es una hendidura o separación en el labio superior.EFECTO CONGENITO
SE ORIGINAPor el crecimiento descompensado de los
dos lados del labio.
EN EL PRIMER TRIMESTRE
DEL EMBARAZO
Constituye el 15 % de las malformaciones congénitas.
SE ACOMPAÑAGeneralmente del paladar hendido.

CLASIFICACION DE MILLARD
Labio leporino cicatricial Labio leporino unilateral (completo e incompleto) Labio leporino bilateral (completo e incompleto)Labio leporino central (forma inusual, agenesia total del prolabio).

tipos

Prolabio.
• Tejido blando al final del componente frontonasal• Acortamiento, sin arco de
cupido,• No columnas filtrales• Comienza cerca de la
columela.• Ausencia de fibras
musculares.

Anatomía patológica• La nariz• Aplanada, chata y con
las fosas nasales abiertas por la ausencia del piso.• Cartílagos alares
ampliamente abiertos.• Columela muy corta( <
1 cm.)

Anatomopatología.• Arcada dentaria:• Dividida en tres
segmentos.• La premaxila esta
protruida hasta 15mm. Y sostenida por el vómer.• Los segmentos
laterales tienden a aplanarse.

Cuando operar.

Técnicas Quirúrgicas• Técnica de Veau.• Técnica de Tenisson Randall.• Técnica de Millard.• Técnica de Bracho.

Complicaciones• Infección de la herida.• Dehiscencia o cicatriz ancha (por aumento de tensión, pero la
infección la complica o inicia).• Retrusión de la premaxila (se previene evitando tracción
excesiva).• Fistulas • Deformidad en silbido (se evita usando colgajos
mucomusculares laterales para aumentar el grosor del prolabio).
• Labio largo (se evita no usando piel lateral).• Colapso de los segmentos laterales (usar férulas acrílicas).

Paladar Hendido

PALADAR DURO, ÓSEO
Paladar duro, óseo.2/3 anteriores del paladar, no hay submucosa ni glándulas palatinas. T. DEL PALADARNO HAY SUBMUCOSAGLÁNDULAS PALATINAS

ANATOMIA DEL PALADAR

Músculos de velo• Elevador del velo del paladar: Eleva el paladar blando o velo
del paladar durante la deglución y el bostezo
• Tensor del velo del paladar: Tensa el paladar blando y abre la trompa durante la deglución y el bostezo para igualar la presión en el oído medio.


Techo de la boca1.- Músculo de la úvula2.- Músculo palatofaríngeo3.- Músculo palatogloso4.- Músculo constrictor
superior de la faringe5.- Rafe pterigomandibular6.- Músculo buccinadorLas fibras del músculo
elevador del velo del paladar que se interdigitan incluyen la mayor parte del paladar blando, junto con el pequeño músculo de la úvula.

FUNCIONES DEL PALADAR
• Masticación• Fonación• Deglución• Respiración

Epidemiología• incidencia de la fisura palatina aislada (FP) es de 1/2000 recién
nacidos vivos.
• predomina el sexo femenino.
• Según raza: orientales, después en caucásicos y finalmente en los de raza negra

Probable origen.• Alcoholismo, fármacos, radiaciones, virus, etc.• La influencia de la herencia es muy grande. (edad
progenitores).

Objetivos quirúrgicos de la palatorrafia.
• Cerrar la comunicación entre la boca y la nariz.• Lograr un cierre velo faríngeo normal.• Evitar un cierre con tensión.• Reposición de la musculatura del paladar.• Alargar el paladar para mayor facilidad del cierre velo faríngeo.• Operar con una mínima perdida de sangre.

Cuando realizar la cirugía.
Dependiendo del caso.• 8 a 12 meses en la mayoría de pacientes no sindromicos y en
aquellos que presenten algún cuadro sindromico es valido operar de los 18 a 24 meses.

Palatoplastia• Palatoplastia de dos colgajos Veau-Wardill-Kilner.• Palatoplastia de Von Langenbeck.• Colgajo de Vómer.• Palatoplastia Furlow.

SECUENCIA QUIRURGICA

COMPLICACIONES• Comunicaciones oronasales• Hipoplasia y colapso maxilar• Maloclusión• Disfunción velofaríngea• Infección• Necrosis• Dehiscencia • Hematoma

Malformaciones congénitas del oído externo.

Oído externo.• Primera porción del oído constituido por:• Pabellón auricular.• Conducto auditivo externo.

Embriología.• El oído externo evoluciona armoniosamente con el oído medio
y la trompa acústica lo que explica las patologías de imperforación del conducto sumadas a patologías de caja timpánica y anotias del pabellón.

Embriología.• El pabellón se constituye con mamelones del primer arco
branquial o mandibular y del segundo arco branquial o arco hiodeo, en el primer surco branquial.
• En la cuarta semana de gestación el ectodermo superficial origina la vesícula otica, que posteriormente formara el laberinto membranoso del oído interno.



Embriología(6ta sem.)• La formación del pabellón de la oreja se da a partir de 6
montículos auriculares que resultan de tumefacciones mesenquimatosas. Estos montículos se fusionan para formar la oreja definitiva.

Embriología

ANATOMIA

Esqueleto cartilaginoso

Irrigación

Inervación

Epidemiologia • 1:6000• 1:4000 Japón – 1:900 India• Masculino 2:1• Der – izq 5:3• Puede tener patrón hereditario• Microtia no relacionada con audición• 10% microtia tienen anormalidades de oído interno• En 1500 microtias, 3 ptes sordos

Clasificación TANZER
I. Anotia
II. Completa hipoplasia (microtia)
A. Con atresia del conducto auditivo externo
B. Sin atresia del conducto auditivo externo
III. Hipoplasia del tercio medio de la auricula
IV. Hipoplasia del tercio superior de la auricula
A. Orejas constrictas
B. Criptotia
C. Hipoplasia del tercio superior completo
V. Orejas prominentes

Técnica quirúrgica.• BRENT.• NAGATA.

TECNICA DE BRENT.• Primer tiempo: Implante de cartílago.• Segundo tiempo: Transposición del lóbulo.• Tercer tiempo: Reconstrucción del trago y
excavación de la concha.• Cuarto tiempo: Surco auriculocefalico.

TECNICA DE BRENT• PLANEACION PREOPERATORIA.• Toma de molde de oreja sana• Replica plástica o acrílica• Esterilización de molde• Fotografías• Comparación con canto externo, a la nasal, comisura

1 Tiempo



2° TIEMPO: ROTACIÓN DEL LOBULO

3° tiempo: construcción de trago y definición de la concha


4° tiempo: separación de la región auricular posterior

CRIPTOTIA• Polo superior del cartílago
auricular se encuentra enterrado debajo del cuero cabelludo
• Manejo conservador con férula antes de 6 meses de edad
• Manejo quirúrgico con injertos, Z plastias, colgajos de rotación

Orejas constrictas • Anomalía en que se
observa atrapamiento del helix• Casos leves y moderados • Casos severos manejo de
microtia

Complicaciones • Infección • Seroma• Hematoma• Necrosis• Resorción del cartílago• Exposición del cartílago• Dehiscencia• Neumotorax• Hemotórax

Malformación congénita de la mano.

Embriología• Las yemas de los miembros aparecen al principio como
pequeñas elevaciones de la pared ventrolateral del cuerpo durante la 4 sem. Y el desarrollo se inicia con la activación de un grupo de células mesenquimatosas en el mesodermo.

4 ta semana
• En la punta de cada yema, el ectodermo se engruesa para formar un reborde ectodérmico apical(REA)• El Reborde Ectodérmico Apical ejerce una influencia
inductora en el mesénquima de los miembros que inicia el crecimiento y desarrollo.

• Zona de progreso Población de células no
diferenciadas en proceso de proliferación rápida
• 6 semanas El extremo distal se aplana
y forma las placas de la mano y del pie

Embriología de la mano.• Apoptosis en cresta
celular ectodérmica(moléculas de señalamiento conocidas como proteínas morfo genéticas óseas).
• Séptima semana Miembro superior rota 90° lateral y el M. inferior 90° medial

Vías de señalización

Embriología de la mano.• 6° semana Condensación de
mesenquima: moldes de cartílago hialino.
Osificación endocondral• 12 semana Centros de
osificación primaria en diáfisis

CLASIFICACIÓN SWANSONINTERNATIONAL FEDERATION OF SOCIETIES FOR SURGERY OF THE HAND1968



Tipo 1. defectos de formación
• I.A Déficit transversales• I.B Déficit longitudinales

Defectos de formación.• Déficit transversales. Incluye el primer nivel en el que ocurre la anomalía
• Amelia. • Hemimelia• Adactilia.• Afalangia.

INCIDENCIA
Deficit transversales
• Hemimelia: 1:20,000• Amelia: 1: 270,000
ETIOLOGIA
• Cigarrillo.• Alcohol.• Disrupción vascular fetal.• Malformación de
vellosidades coriónicas

Déficit longitudinal intersegmental
Focomelia
• La presencia de estructuras digitales esqueléticas lo diferencia del déficit transversal.
INCIDENCIA
• 0.8% • Uso anticoagulantes,
acido valpróico, alcohol ,cigarrillo.

FOCOMELIA

CLASIFICACIÓN FOCOMELIA
Tipo Iausencia completa
de los huesos del miembro próximos a la mano
que se une
directamente al
tronco.
Tipo IIausenci
a de brazo o segmento corto
de brazo-
antebrazo
Tipo IIIausenci
a de antebrazo con
la mano unida
directamente
al humero

Déficit radial longitudinal.• Displasia radial o mano zamba radial.
• El diagnostico incluye:• Hipoplasia o ausencia del pulgar.• Hipoplasia o ausencia radial.

Clasificación Bayne y Klug
Tipo I: radio distal corto.Epifisis radial hipoplasica leve acortamiento del radio, hipoplasia de pulgar
Tipo II: radio hipoplasicoRadio acortado, carpo mal apoyado, cubito arqueado

Clasificación Bayne y Klug
TIPO IIIAusencia parcial del radio. Mas frecuente tercio proximal, carpo sin apoyo
TIPO IVAusencia total del radio. Mano sin apoyo, severo desplazamiento radial

Incidencia mano zamba• 1: 55,000 RNV• Asociada a deficiencia del pulgar• Mas frecuente en sexo masculino que femenino.• Mas común en la raza blanca.• Bilateral en 38-50% de los casos.• Predomina en miembro superior derecho.




Déficit cubital longitudinal • Manifestaciones clínicas variadas
• Incidencia 1: 100,000 RNV
• Sexo femenino 3:2
• Mas frecuente en lado izquierdo

CLASIFICACION DE BAYNE• TIPO I: • hipoplasia del cubito. Desviación mínima de la muñeca, puede
haber ausencia o hipoplasia digital• TIPO II: • aplasia parcial del cubito. Cubito distal ausente, reemplazado por
anillo fibroso.• TIPO III: • ausencia total del cubito. Codo inestable.
• TIPO IV: • sinostosis radio humeral cubito ausente, deformidad evidente, AB
pronación y rotación interna

Anomalías Asociadas:
• Cardiopulmonares• Hematopoyéticas• Gastrointestinales• Contralateral 45%• Deficiencia femoral y peroné

Hipoplasia y ausencia digital.
• Puede ocurrir de manera longitudinal o transversal.• Va desde ausencia transversal simple hasta simbraquidactilia.• Aun no se ha establecido relación directa entre causa y efecto.• Factores ambientales.• Herencia.• Infecciones durante gestación.

Hipoplasia o ausencia de dígitos



Tipo II. Falla en la diferenciación (separación) Compromiso de tejidos blandos:• Diseminada:
• Artrogriposis.contracturas articulares persistentes (neurogena y miopatica).
• A nivel del hombro: • Síndrome de Poland.
• Codo y antebrazo: • Presencia de músculos flexores y extensores aberrantes.
• Muñeca y mano: • Sindactilias.

Artrogriposis.• Se clasifica en neurogena y miopatica.• Las extremidades inferiores se afectan con mayor frecuencia.• Hombros delgados y en rotación interna, codos extendidos,
antebrazos en pronación y semiflexion.


Sindactilias.• Se clasifican de acuerdo con su ubicación en:• Sindactilias cutáneas
• Radial• Central• Cubital• Combinadas• Simples -Complejas -Complicadas
• Camptodactilias (campto= doblado y dactylus= dedo) Dedos desviados sin deformidad esquelética.• Dedo en gatillo congénito.

Sindactilias. Clasificación

Camptodactilia.

Dedo en gatillo.
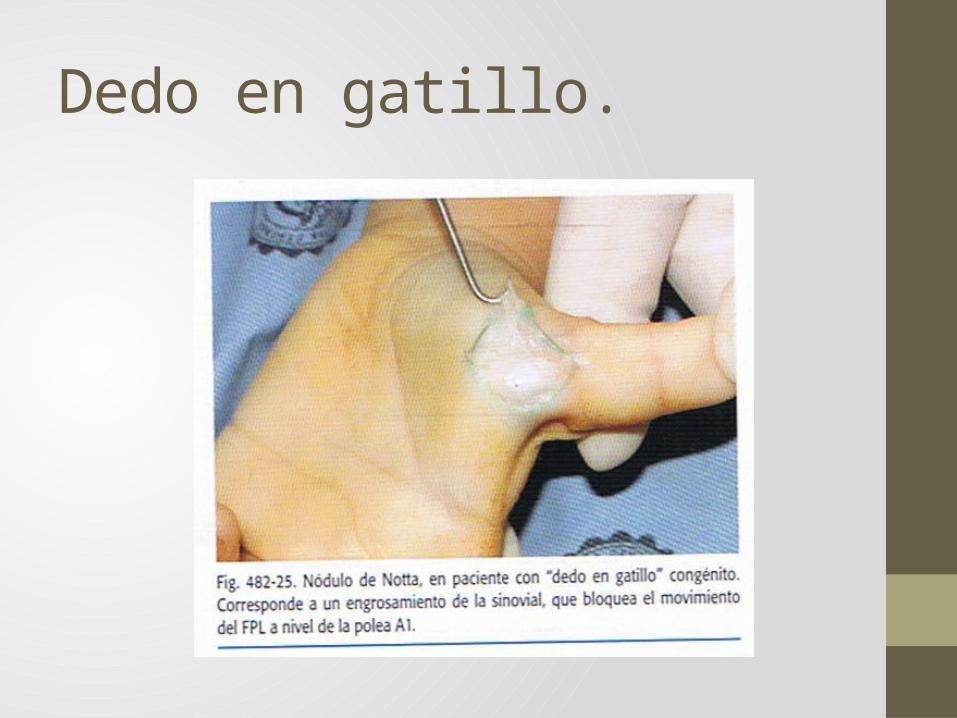
Dedo en gatillo.

Tipo II. Falla en la diferenciación (separación)Compromiso óseo.• A nivel del hombro: humero varo congénito.• Codo: sinostosis humero-radial, humero-cubital, anquilosis del
codo.• Antebrazo: sinostosis radio-cubital• Muñeca y mano:• Sinostosis de los huesos del carpo.• De metacarpianos.• De falanges( sindactilia ósea)• Sinfangilismo. ( syn=con y phalanx= hueso)• Clinodactilias. (clino= desviado y dactylos=dedo)• Hipersegmentacion: pulgar trifalangico.

Clinodactilia.

Tipo III. Duplicación.• Constituyen las anomalías mas comunes.• De acuerdo a su ubicación se clasifican en tres tipos:• Preaxiales o radiales:
• La mas común es la duplicación de pulgar.• Mas común en raza blanca( 1:3,000 RNV)
• Centrales:• Menos del 10% de todas las duplicaciones.• Se acompañan de membranas interdigitales (sinpolidactilias)
• Postaxiales o cubitales:• Son las mas frecuentes en la raza negra (1: 300 RNV)

Duplicaciones del pulgar. Clasificación Iowa

Duplicación Pulgar Tipo I



Tipo IV




Pulgar trifalangico

Mano en espejo.

Duplicaciones cubitales.• 3 categorias• Mas frecuente en
raza negra





Sobrecrecimiento.• Macrodactilia o gigantismo.• Macros: grande dactylos: dedo• Todas las estructuras del digito están aumentadas de tamaño.• Causas:• Aporte nervioso anormal.• Anomalías vasculares.• Mecanismos humorales no establecidos


Macrodactilia
Anomalias asociadas:• Sindactilia 10%• Criptorquidia• Lipofibromatosis


Tipo V. Síndrome de anillos constrictivos.
• Síndrome de banda amniótica, constricción anular o displasia de Streeter.
• Ocasionado por secuencia de disrupción• Factores de riesgo• Oligohidramnios.• Prematuridad.


2 manifestaciones:• Necrosis focal
• Anillos de constricción parciales o circunferenciales complejos, superficiales o profundos, hasta periostio.
• Amputaciones intrauterinas• A cualquier nivel o en combinación de lesiones digitales



TipoVII. Sindrome de Apert.• Craneosinostosis coronal y lambdoidea.• 1:100,000 NV• Sindactilias complejas y complicadas en pies y manos.• Generalmente NO se acompaña de retardo mental


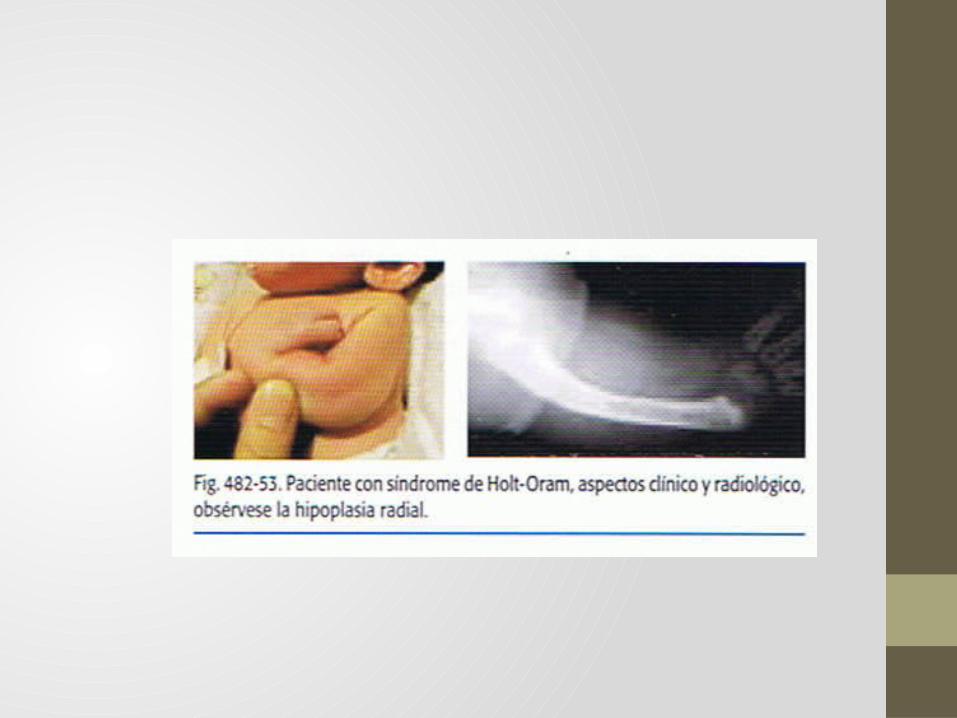
Síndrome de Holt- Oram.• Descrito a finales de años 50, por Holt (Filadelfia) y Oram
(Londres).• Consiste en displasia radial y malformaciones cardiacas
congénitas.• En ocasiones se acompaña de sinostosis radiocubital y pulgar
hipoplásico.

Síndrome de Poland.• Descrito por Alfred Poland, 1849.• Ausencia de cabeza esternal de musculo pectoral mayor, mano
hipoplásica y sinbraquidactilia.• Los trastornos torácicos se corrigen en la adolescencia y la
sindactilia antes del primer año.



BIBLIOGRAFIA• EMBRIOLOGIA MEDICA• Langman, WT Sadler, 8° edición, 2009, cap 15
• PLASTIC SURGERY• McCarthy, tomo IV, WB Saunders, 1990. caps 52
• LABIO Y PALADAR HENDIDO• Isaac Rozen Fuller, 2° edición, 2005. caps 2 y 3
• CIRUGÍA PLÁSTICA, RECONSTRUCTIVA Y ESTÉTICA• Felipe Coiffman, Tomo III, Amolca.2008, Pags 2647-2658
• GREEN´S OPERATIVE HAND SURGERY• Green, Elsevier, 5° edición, tomo II, caps 39 - 42

•Gracias…..


















