







Idiomas
Páginas
Jurídico


FIBROSIS QUÍSTICA* ETIOLOGÍA * EPIDEMIOLOGÍA * FISIOPATOLOGÍA * CUADRO CLÍNICO * MORFOLOGÍA * DIAGNÓSTICO * TRATAMIENTO * PRONÓSTICO *
Equipo #2
• Edgar Alan de Hoyos Flores• César Humberto Mejía Godoy• Carlos Alejandro Palomo
Barrera• Héctor Javier Sandoval
González• Marco Antonio Valdez García
Catedrático
Dra. Brenda Odíl González Sánchez
Patóloga

ETIOLOGÍA

DEFINICIÓN
La fibrosis quística es un trastorno hereditario del transporte de iones, que afecta a la secreción de líquido en las glándulas exocrinas y el revestimiento epitelial de los aparatos respiratorio, digestivo y reproductor.
Neumopatía crónica
secundaria a infecciones de
repetición
Insuficiencia pancreática Esteatorrea Malnutrición
Cirrosis hepática
Obstrucción intestinal
Infertilidad masculina

ETIOLOGÍA
La fibrosis quística es el resultado de un transporte anormal de electrolitos que procede de observaciones de que el sudor de la fibrosis quística contiene concentraciones elevadas de sodio y cloruros.
Se relaciona con una función anormal de una proteína de los canales del cloro epiteliales codificada por el gen del regulador de la conductancia transmembrana de la fibrosis quística (CFTR) en el cromosoma 7q31.2.

FUNCIÓN DEL GEN CFTR
1. Regula múltiples canales iónicos y procesos celulares
Canales de extracción de cloruro rectificado
Canales de introducción de potasio rectificados
Canal epitelial de sodio(ENaC)
Canales de las uniones en hendidura
Los procesos celulares implicados en el transporte de ATP y la secreción del moco.

Interacción entre ENaC y CFTR
El ENaC se encuentra situado en la superficie apical de las células epiteliales exocrinas y es responsable de la captación de sodio desde el líquido luminal.
La actividad del ENaC disminuye como consecuencia de las mutaciones de CFTR.
Se forma un líquido luminal hipertónico que contiene mucho cloruro sódico en sudor.
FUNCIÓN DEL GEN CFTR

2. Las funciones de CFTR son específicas en cada tejido
Conductos de las glándulas sudoríparas es reabsorber los iones cloruro luminales y aumentar la reabsorción de sodio a través del ENaC
En los epitelios intestinal y respiratorio, el CFTR es uno de los sistemas más importantes para la secreción luminal activa de cloruro.
En estos lugares, las mutaciones de CFTR determinan una pérdida o reducción de la secreción de cloruro hacia la luz
La absorción luminal activa de sodio también aumenta
FUNCIÓN DEL GEN CFTR

3. CFTR regula el transporte de iones bicarbonato
La función de CFTR en el transporte de bicarbonato viene mediada por interacciones recíprocas con una familia de intercambiadores aniónicos llamados SLC26
En algunas variantes mutantes de CFTR, existen alteraciones notables en el transporte de bicarbonato.
FUNCIÓN DEL GEN CFTR

La acidez de las secreciones reduce el pH luminal.
Aumento de la precipitación de la mucina
Formación de tapones en los conductos
Incremento de la unión de las bacterias a los tapones de mucina
FUNCIÓN DEL GEN CFTR
Insuficiencia pancreatica

FISIOPATOLOGÍA

ESPECTRO DE MUTACIONES Y CORRELACIÓNGENOTIPO-FENOTIPO
Las mutaciones se pueden agrupar en seis «clases» en función de sus efectos sobre la proteína CFTR:
I. síntesis defectuosa de la proteína. Estas mutaciones se asocian a una ausencia completa de proteína CFTR en las células epiteliales.
II. Plegamiento, procesamiento y circulación anormales de la proteína. Alteración del procesamiento de la proteína desde el retículo endoplásmico al aparato de Golgi; la proteína no se pliega del todo ni se glucosila y se degrada.

III. Regulación defectuosa. Las mutaciones de esta clase impiden la activación de CFTR.
IV. Reducción de la conductancia. Se encuentra una cantidad de CFTR normal en la membrana apical, pero con una función reducida.
V. Reducción de la abundancia. Estas mutaciones afectan a los sitios de separación de intrones en el promotor de CFTR, de forma que se reduce la cantidad de proteína normal.
ESPECTRO DE MUTACIONES Y CORRELACIÓNGENOTIPO-FENOTIPO

VI. función alterada en la regulación de los canales iónicos. Las mutaciones de esta clase afectan al papel regulador de CFTR. En algunos casos, una mutación determinada altera la conductancia de CFTR y también la regulación de otros canales iónicos.
ESPECTRO DE MUTACIONES Y CORRELACIÓNGENOTIPO-FENOTIPO

FISIOPATOLOGÍA
En el epitelio intestinal y respiratorio, el CFTR es uno de los sistemas más importantes para la secreción luminal activa de cloruro. En estos lugares, las mutaciones de CFTR determinan una pérdida o reducción de la secreción de cloruro hacia la luz.
La absorción luminal activa de sodio también aumenta y estos dos cambios iónicos aumentan la reabsorción pasiva de sodio epitelial desde la luz.

FISIOPATOLOGÍALa patogenia de las complicaciones respiratorias e intestinales en esta enfermedad parecen deberse a una capa de líquido superficial isotónica, pero de escaso volumen. En los pulmones esta deshidratación determina una acción mucociliar defectuosa y la acumulación de secreciones viscosas hiperconcentradas, que obstruyen las vías aéreas y predisponen al paciente a sufrir infecciones pulmonares de repetición.

EPIDEMIOLOGÍA

EPIDEMIOLOGÍA
Común en caucásicos de Europa, Norte América y Australasia

EUROPA
1 de cada 2000 nacidos vivos
Varia de la Región de cada país
Mutación del gen CFTR (95%)
100% en Islas Faroe de Dinamarca
20% En Turquía
15% Mediterráneo
70% en Europa central, norte y noreste

ÁFRICA
No hay una prevalencia exacta
Mutaciones identificadas en países cerca del Mediterráneo
Nutrición pobre
1 de cada 7056 en Sudáfrica

MEDIO ORIENTE
1 de cada 15,876
Líbano e Israel
Mutación de CFTR F508del
Emiratos Árabes Unidos y Omán
CFTR 548
Arabia Saudita
CFTR 1548del G
ASIA
Muy infrecuente
Mutación del CFTR 508del
India y Pakistan
1 de cada 40,000 nacidos vivos
Japón
1 de cada 100,000 hasta 530,000 nacidos vivos

OCEANÍA
La misma que Europa
Debido a emigración
Mutación de CFTR

NORTE AMÉRICA
Estados Unidos
1 de cada 3500
79.7% por mutación de CFTR en Caucásicos
19.3% por mutación de CFTR 3120+1G A en Afroamericanos
Canadá
Más común en el centro y oeste de Canadá
Mutaciones raras en Etnias

AMÉRICA LATINA
Mutación de CFTR F508
Argentina y Uruguay
90% población Caucásica – Incidencia de 59%
México, Colombia y Chile
57% a 85% Mestizos (Caucásico + Amerindios) – Incidencia de 25%, 14% y 29%
Colombia, Venezuela, Uruguay, Ecuador y Brasil
10% Descendientes Africanos – Incidencia de 4%
AMÉRICA LATINA
Cuba
1 de cada 3900 nacidos vivos
México
1 de cada 8500 nacidos vivos


CUADRO CLÍNICO

SIGNOS Y SÍNTOMAS
Compromete al organismo en su totalidad, su crecimiento, la función respiratoria y digestiva.
SÍNTOMAS INTESTINALES
Dolor Abdominal
Estreñimiento Severo
Aumento de Gases
Abdomen Distendido
Náuseas e Inapetencia
Pérdida de Peso

SIGNOS Y SÍNTOMAS
SINTOMAS PULMONARES
Tos
Aumento de Mucosidad en Senos Paranasales y Pulmones
Congestión Nasal por Pólipos Nasales
Episodios de Neumonía
Fiebre
Dolor o Presión Sinusal por Infección o Pólipos

SIGNOS Y SÍNTOMAS
ENFERMEDAD PULMONAR Y SINUSAL
Inflamación e Infección
Producen cambios estructurales
Dificultad respiratoria crónica
Esputo Sanguinolento
Hemoptisis

ENFERMEDADES GASTROINTESTINAL, HEPÁTICA Y PANCREÁTICA
Impiden el movimiento de las enzimas pancreáticas hacia el intestino
Producen daño irreversible en el páncreas
Proceso de digestión y absorción alterado por la cantidad de H2O, HCO3 y Enzimas en la Luz intestinal
Mal absorción de Grasas y Proteínas
Ausencia de Deposiciones las primeras 24 a 48 hrs. De vida
Heces pálidas o color arcilla, olor fétido, con moco.

ENFERMEDAD ENDÓCRINA Y CRECIMIENTO:
Provocado por daño en el Páncreas
Perdida de Islotes de Langerhans
Puede producir Diabetes o Pancreatitis
Malformaciones en Manos y Pies (Dedos en Palillo de Tambor)
Piel sabor salado (protina RTFQ)
Esterilidad en hombres



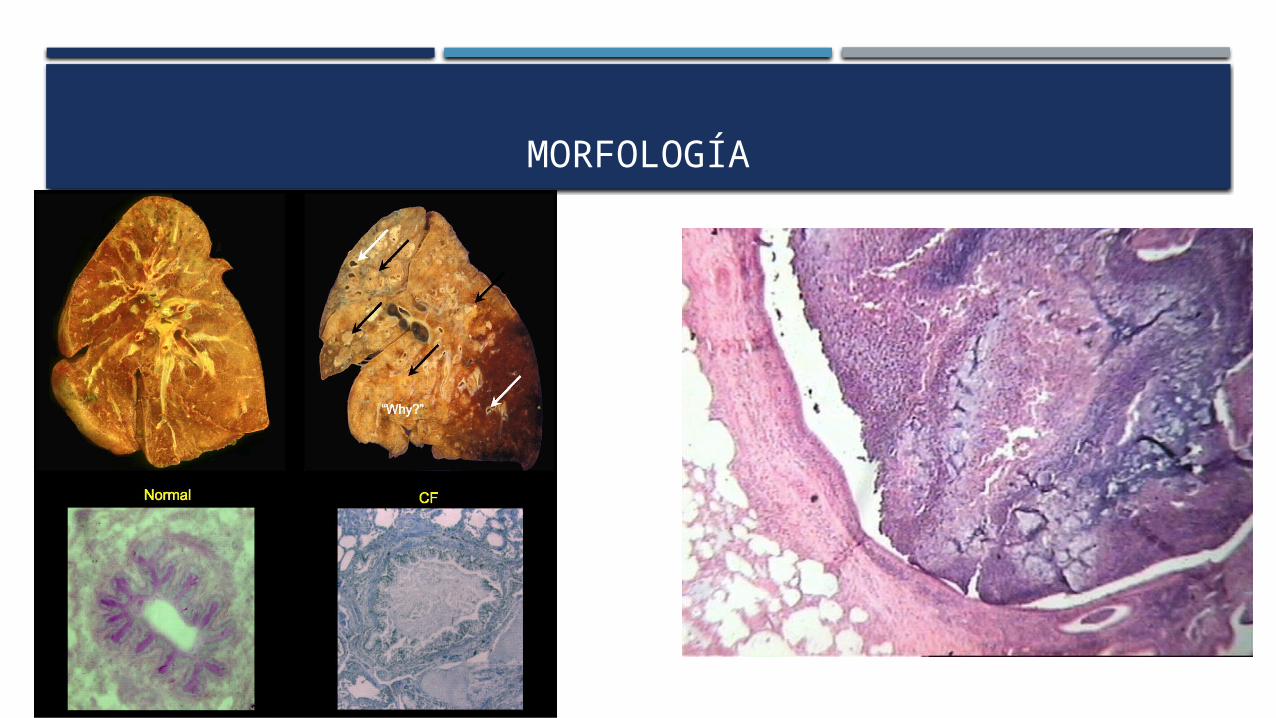
MORFOLOGÍA

Alteraciones pancráticas
85-90% de los pacientes con fibrosis quística.
Acumulación de moco en los conductos pequeños con cierta dilatación de las glándulas exocrinas.
alteraciones graves: atrofia del componente exocrino glandular
Solo persisten los islotes en un estroma fibroadiposo
MORFOLOGÍA

MORFOLOGÍA

En el estómago, intestino delgado, colon y apéndice
Glándulas dilatadas con secreción intraluminal eosinofilica
MORFOLOGÍA

Intestino delgado:
Tapones de moco denso viscoso
Puede provocar obstrucción intestinal
Ileo meconial.
MORFOLOGÍA

Hígado y vías biliares se sigue mismo patrón.
Canalículos biliares:
Taponados por material mucoso
Desarrolla cirrosis biliar focal en un tercio de los pacientes
MORFOLOGÍA

Las glándulas salivales
Cambios histológicos similares a los del páncreas
Dilatación de los conductos, metaplasia escamosa del epitelio de revestimiento y atrofia glandular.
En las vías respiratorias
Bronquiolos distendidos por moco denso
Hiperplasia e hipertrofia de las células secretoras de moco.
Provoca una obstrucción secundaria con infección de las vías aéreas
Pueden ocasionar una bronquitis crónica grave con bronquiectasia.
MORFOLOGÍA

MORFOLOGÍA
MORFOLOGÍA

DIAGNÓSTICO

DIAGNÓSTICO
Resultado Positivo de la Prueba del Sudor
Presencia de 2 Mutaciones de CFTR
Diferencia de Potencial Nasal Transepitelial Anormal

DIAGNÓSTICO
RESPIRATORIOS
Vía Aérea Superior
Pólipos Nasales
Enfermedad de Senos Paranasales
Vía Aérea Inferior
Taquipnea y Tiraje Persistente
Bronquiolitis Recurrente
Obstrucción bronquial recurrente atípica o persistente
Sibilancias con Insuflación Persistente
Aislamiento en Secreciones Bronquiales de:
• Haemophilus Influenzae• Staphylococcus Aureus• Pseudomona Aeruginosa

DIAGNÓSTICO
OTROS
Fallo de Crecimiento
Historia Familiar
Sabor Salado en Sudor
Cristales de Sal en Cuero Cabelludo y Frente
Edema e Hipoproteinemia
Hipocratismo Digital
Azoospermia y Ausencia de Conductos Deferentes
Alcalosis Metábolica
Diabetes
Pesquisa Neonatal

DIAGNÓSTICO
PRUEBA DEL SUDOR
Determinación cuantitativa de cloruros en sudor
Consiste en la estimulación de las glándulas sudoríparas mediante iontoforesis con pilocarpina, la recolección del sudor y la cuantificación de la concentración de cloruros; el sudor se colecta en gasa o papel de filtro.
La tasa de sudoración deber ser superior a 1 g/m2 / min.

TRATAMIENTO

OBJETIVOS DEL TRATAMIENTO
• Prevención y tratamiento de la enfermedadrespiratoria.• Prevención y tratamiento del déficit nutricional.• Prevención y tratamiento de otras manifestaciones o complicaciones.• Cuidado de la salud mental
El objetivo básico del tratamiento consiste en prevenir o controlar la progresión de la enfermedad
respiratoria para evitar o disminuir el dañopulmonar irreversible, marcador principal del pronóstico.

TRATAMIENTO DEL APARATO RESPIRATORIO
Pilares del tratamiento respiratorio
• Terapia inhalatoria.• Kinesiología respiratoria diaria.• Antiinflamatorios.• Antibióticos en las exacerbaciones pulmonares.

TERAPIA INHALATORIA
Para vehiculizar diferentes medicaciones al tracto respiratorio inferior, para aliviar la obstrucción bronquial, ayudar a la depuración mucociliar y tratar o prevenir infecciones.
La administración de los medicamentos es a través de una ampolla nebulizadora de tipo “jet” y se complementa con un propelente de aire.

Antibioticos inhalados
• Erradicación de la infección inicial por Pseudomonas aeruginosa.• Tratamiento de mantenimiento en la infección crónica por Pseudomonas aeruginosa.
Se administrará el antibiótico inhalado junto al antibiótico oral si ya se estaba administrando.
Colistin (infección inicial por Pseudonoma A.)Trobamicina (infección crónica por Pseudomona A.)
Lizinato de Astreonam (infección por Gram Negativas, incluyendo Pseudonoma A.)

ANTIINFLAMATORIOS Macrolidos
• Propiedades ampliamente conocidas, los macrólidos es ampliamente conocida, ya que inhiben la quimiotaxis de neutrófilos y la producción de mediadores inflamatorios y alginato de Pseudo. A.
• Px con infección crónica de esta bacteria que no respondieron a tx inicial
• Disminuyen las complicaciones, asi como también es utilizado como tx profiláctico
Corticoides
• Prednisona, en complicaciones y periodos cortos
• Excepto en lactantes con bronquiolitis grave, ya que su pronostico mejora con tx a largo plazo (meses)

ANTIBIÓTICOS
La infección pulmonar es la principal causa de morbimortalidad en los pacientes con FQ.
Son indicados en:
• Exacerbaciones (tx precoz, oral si es leve o moderada o IV si es grave o hay resistencia)
• Terapia supresiva para disminuir el daño a px con infección crónica
Bacterias mas frecuentes: Staphylococcus aureus, Pseudomonas aeruginosa, Haemophilus
influenzae, Burkholderia cepacia, Achromobacter xylosoxidans y Stenotrophomonas maltophilia
Las exacerbaciones suelen ser desencadenadas
por virus (sincicial respiratorio, adenovirus,
influenza), micoplasma y clamidia

NUEVOS TRATAMIENTOS
Se han desarrollado agentes denominados “correctores” que mejoran la primera anormalidad y “potenciadores” que aumentan la función del canal.
Una sustancia potenciadora, el IVACAFTOR ha demostrado mejoría de la función pulmonar, el estado nutricional y reducción de los valores de cloro en el sudor, en pacientes con al menos una copia de la mutación G551D, que ocurre en el 4% de los pacientes con FQ a nivel mundial y en el 0,2% en nuestro país.

ASPECTOS NUTRICIONALES Y GASTRONENTEROLOGICOS
El estado nutricional adecuado en pacientes con FQ ha sido relacionado con una menor progresión de la enfermedad respiratoria, mejor calidad de vida y mayor sobrevida
Es necesario considerar que no existen razones para aceptar el déficit nutricional como parte de la enfermedad, ya que los genotipos de FQ no codifican para talla baja, retraso de la pubertad o falla de crecimiento

VALORACIÓN DEL ESTADO NUTRICIONAL
1. Datos antropométricos
2. Análisis de la composicióncorporal
3. Estado de desarrollo puberal
4. Evaluación bioquímica
5. Aspectos Psicosociales de la Conducta Alimentaria
6. La adherencia del paciente a las recomendacionesnutricionales y a la ingesta de enzimas pancreáticas
Dicha evaluación se realizará al
menos cada 3 meses hasta los dos años de
edady luego, 1 vez al año en forma independiente a
los controles pediátricos habituales

RECOMENDACIONES NUTRICIONALES
El aporte calórico total se establecerá entre el 120 y 150% de la ingesta dietética recomendada, con un aporte de grasas que represente el 40% del total de calorías y una ingesta proteica un 15-20% mayor que la Recomendada.

USO DE ENZIMAS PANCREÁTICAS
La indicación de enzimas y el manejo de las dosis deben ser evaluados por el gastroenterólogo o nutricionista de un centro de referencia
Debe comenzarse con:• 1000 U lipasa/kg peso/comida en
menores de4 años.
• 500 U lipasa/kg peso/comida en mayores de
4 años.
Los lactantes deberán recibir entre 2000 y 4000
unidades de lipasa por cada 120 ml de fórmula o
en cada toma de leche materna, lo que equivale
a 450-900 unidades de lipasa por gramo de grasa ingerido.

RESPUESTA INADECUADA AL TX CON ENZIMASFactores dietéticos:
• Pobre ajuste de la dosis de enzimas
sobrecomidas ricas en
grasas.• Excesivo numero de colaciones con
inadecuada toma de enzimas.
• Toma excesiva de jugos.
• La creencia familiar de que no
son necessárias con algunos alimentos
Pobre adherencia al tratamiento
• Dificultades en la administración a lactantes.
• Factores psicológicos-sociales en la etapa
escolar.• Bajo apoyo de instituciones
educacionales.• Negación al tratamiento durante la adolescencia.
• La necesidad de no mostrarse diferente a sus
pares.• Deseo de perder peso
Presencia de patologías
gastrointestinalesasociadas a la FQ
• Hiperacidez gástrica.• Reflujo
gastroesofágico.• Malabsorción de
lactosa.• Parasitosis,
especialmente giardiasis.• Síndrome del intestino
corto.• Enfermedad celíaca.
• Enfermedades inflamatorias intestinales.
• Sobredesarrollo bacteriano.
• Enfermedad biliar.

PRONÓSTICO

PRONÓSTICO
En conjunto, las mejoras del tratamiento de la fibrosis quística han permitido aumentar la mediana de esperanza de vida hasta casi 40 años, y la que era una enfermedad mortal de la infancia está pasando a convertirse en una enfermedad crónica de adultos.

BIBLIOGRAFÍA
Robbins & Cotran “Las Bases Patológicas de la Enfermedad 9na. Edición. “ Enfermedades de la Infancia” Cap.10 pag: 466 – 471
World Health Organization (OMS), The Molecular Genetic Epidemiology Cystic Fibrosis (La Epidemiología Molecular y Genética de la Fibrosis Quística). Reino Unido 2012. Disponible en: http://www.who.int/genomics/publications/en/HGN_WB_04.02_report.pdf
Guía de diagnóstico y tratamiento de pacientes con firosis quística. Comités Nacionales de Neumología, Nutrición, Gastroenterología y Grupo de Trabajo de Kinesiología. Disponible en: http://www.sap.org.ar/docs/profesionales/consensos/consenso_fq_2014.pdf
Comité Nacional de Neumologia, Gastroenterología y Kinesologiía. “Guía del Diagnóstico y Tratamiento de Pacientes con Fibrosis Quistica”. Disponible en: http://www.sap.org.ar/docs/profesionales/consensos/consenso_fq_2014.pdf
Top Related