
Qué reportar en la Tecnovigilancia de, Dispositivos …. QUE REPORTAR a: Mal...
43
"Qué reportar en la Tecnovigilancia de, Dispositivos Medicos: Implantables, Descartables y Serializados" Veronica Ferro M. Q.F U.N II Foro Internacional Tecnovigilancia , Junio 17-18 / 2010
Transcript of Qué reportar en la Tecnovigilancia de, Dispositivos …. QUE REPORTAR a: Mal...

"Qué reportar
en la Tecnovigilancia de,
Dispositivos Medicos:
Implantables, Descartables y
Serializados"
Veronica Ferro M. Q.F U.N
II Foro Internacional Tecnovigilancia ,
Junio 17-18 / 2010

ContenidoPrimera Parte
1. Principio Fundamental de la Tecnovigilancia
2. Conceptos básicos en el Reporte de Tecnovigilancia
3. Que Reportar
4. Excepciones – Qué no reportar en la Tecnovigilancia
GHTF/SG2/N54RO:2006
Segunda Parte
1. Experiencia en Colombia: Tendencias de lo Reportado
2. Qué mejorar para realizar un correcto reporte de Eventos Adversos en Tecnovigilancia.
3. Conclusiones

1. Principio Fundamental
El reporte de Eventos Adversos y subsecuente
evaluación tiene como objetivo fundamental
PROTEGER LA SALUD DEL PACIENTE y/o
USUARIO y de OTROS mediante la diseminación
de información que pueda reducir la probabilidad
de, o prevenir repeticiones de eventos adversos, o
aliviar las consecuencias de dicha repetición.

1. Principio Fundamental
El reporte de Eventos Adversos y subsecuente
evaluación tiene como objetivo fundamental
PROTEGER LA SALUD DEL PACIENTE y/o
USUARIO y de OTROS mediante la diseminación
de información que pueda reducir la probabilidad
de, o prevenir repeticiones de eventos adversos, o
aliviar las consecuencias de dicha repetición.
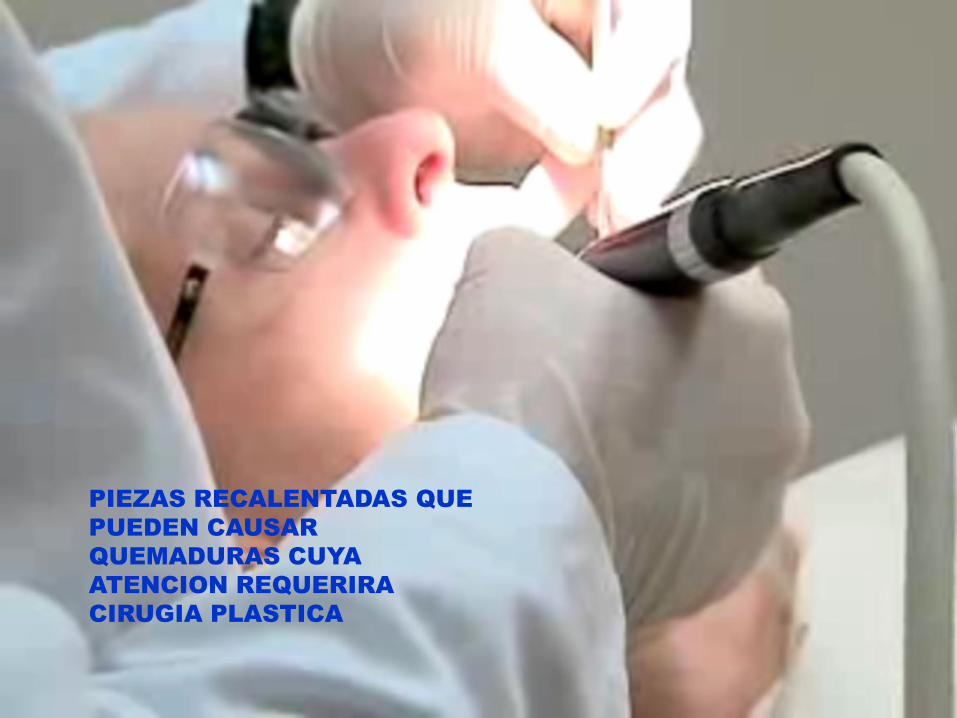
PIEZAS RECALENTADAS QUE
PUEDEN CAUSAR
QUEMADURAS CUYA
ATENCION REQUERIRA
CIRUGIA PLASTICA

QUEMADURAS EN PIEL
PACIENTE - RESONANCIA
MAGNETICA

DATOS ERRONEOS DE
DIAGNOSTICO –
DOSIFICACION INADECUADA,
A LETAL

SOBREEXPOSICION A
RADIACION en CT–
EFECTOS VISIBLES
ERITEMA, CALVICIE.
NO VISIBLES: a largo
plazo

1. Principio Fundamental
El reporte de Eventos Adversos y subsecuente
evaluación tiene como objetivo fundamental
PROTEGER LA SALUD DEL PACIENTE y/o
USUARIO y de OTROS mediante la diseminación
de información que pueda reducir el riesgo,
prevenir la repetición o aliviar las consecuencias de
una repetición.

Evaluación=Investigación
Determinar la causa raíz que dió origen al Evento
Adverso:
Muerte
Lesión
Amenaza a la Salud Publica

FALSAS LECTURAS QUE
PUEDEN LLEVAR A
ADMINISTRACIONES
ELEVADAS DE INSULINA QUE
PUEDEN LLEVAR AL SHOCK
O A LA MUERTE.


ELECTRODOS MAL CONECTADOS
O NO ADECUADOS PARA
RESONANCIA

ELECTRODOS Y CABLES
REQUERIDOS PARA MONITOREO
ECG DURANTE EXAMEN, deben ser
EMR compatibles


DETERMINADOS
EXAMENES
REQUIEREN
MAS
EXPOSICION A
LA RADIACION
QUE OTROS

Principio Fundamental
El reporte de Eventos Adversos y subsecuente
evaluación tiene como objetivo fundamental
PROTEGER LA SALUD DEL PACIENTE y/o
USUARIO y de OTROS
mediante la diseminación de información que
pueda reducir el riesgo, prevenir la repetición o
aliviar las consecuencias de una repetición
Electrodos RMVideos

|

1. Principio Fundamental
El reporte de Eventos Adversos y subsecuente
evaluación tiene como objetivo fundamental
PROTEGER LA SALUD DEL PACIENTE y/o
USUARIO y de OTROS mediante la diseminación
de información que pueda reducir el riesgo,
prevenir la repetición o aliviar las consecuencias de
una repetición

El reporte ¿A Quien?
Al Fabricante: realiza la evaluación / nvestigación de evento
Al Responsable de la Distribución: Retroalimenta al Fabricante
A la Autoridad:
como ente verificador de 1. la investigación y 2. la implementación de las Acciones
como parte de la red para la divulgación de la información

2. Conceptos básicos
1. El acto de divulgar un acontecimiento a la Autoridad
no debe ser interpretado como una admisión del
fabricante, del usuario o del paciente por el
acontecimiento y sus consecuencias.

2. Conceptos básicos
El reporte tampoco representa una conclusión de que
este reporte esta completo y ha sido confirmado o que
los dispositivos fallaron.
Tampoco representan la conclusión de que el
Dispositivo Medico causó o contribuyó al evento
adverso. Se debe partir de la negación de lo anterior.

3. QUE REPORTAR
a: Mal funcionamiento/deterioro de las características de
funcionamiento
Un monitor cayo del techo cuando los pernos del techo
que sostienen la pieza giratoria se parte . No hay
lesionados. El equipo fue instalado usado y mantenido
siguiendo las instrucciones
b: Incorrecto o resultado fuera de especificaciones
Revision prematura de un implante ortopédico debido al
aflojamiento. No se conoce la causa.
• c: Descubrimiento de un defecto en el diseño
Falsa lectura tirillas de glucosa.

3. QUE REPORTAR
d: Inexactitud en el etiquetado (incluyen omisiones o deficiencias),
El empaque del DM estéril tiene la leyenda no use si el empaque esta abierto o dañado. La etiqueta por error esta colocada en el interior del dispositivo. El empaque es removido, el dispositivo no es usado en la cirugia y el DM es guardado únicamente con el empaque interno.
e: descubrimiento de una amenza a la salud publica: incluyen eventos significativos no esperados y que generan una alarma:
(VIH, Creutzfeldt-Jacob)

Descubrimiento de una amenaza a la salud publica:
origen y contaminacion cruzada (reuso)
COLOMBIA Decreto 2350 de 2004 prohibición importación de Dispositivos Médicos de
Origen Bovino, Ovino y Caprino hasta no demostrar ausencia de priones
Con el fin de evitar el contagio de virus vacas locas, EEB, variante de la enfermedad de C.Jacob
ESPAÑA FUENTE: Seguridad Industrial y Prevención de Riesgos Laborales -Seguridad Medio
Ambiental y Protección del Entorno
Material médico, se usa pero no se tira El 80% de los hospitales madrileños reutiliza dispositivos desechables. Está prohibido, pero es una práctica generalizada en todo el país por motivos económicos. Centros públicos y privados reciclan sin control
Los expertos piden un consenso para regular el reciclaje pero con estrictos controles de seguridad
El juicio al anestesista Juan Maeso, por el contagio masivo del virus C de la hepatitis a 276 personas, ha revelado el potencial riesgo de contraer una infección que se corre cuando se acude a un hospital.

3. QUE REPORTAR
f: Error en el uso: Acto u omisión de un acto que da un resultado diferente al propuesto por el fabricante o esperado por el operador.
NOTA: Cuando hay un cambio en la tendencia: aumento en la frecuencia
Una bomba de infusión se entrega el medicamento en bolo cuando la administración debía ser programada dos horas.
Videos luer lock -

3. QUE REPORTAR
h: Uso Anormal: Acto u omisión de un acto por parte del operador o usuario del dispositivo medico que es resultado de una conducta que esta por fuera de cualquier riesgo razonable evaluado por el fabricante.
Empleo de un stent biliar en vascular videos
Uso de una ayuda vascular fuera del cuerpo pero diseñada para ser utilizada al interior del cuerpo videos i

1. Deficiencia encontrada por usuario antes del uso en el paciente El usuario realiza un test de inflación antes de insertar un catéter balón
al paciente, siguiendo las instrucciones que acompañan al DM. Se detecta mal funcionamiento en la inflación. Otro balón es usado. El paciente no es lesionado. IMAGEN
El empaque de un DM esta etiquetado con la advertencia “no use si el paquete esta abierto o dañado, pero el daño del paquete fue obvio se se descubrió y el DM no fue usado.
2. Evento causado por la condición del paciente El paciente murió después de un tratamiento de diálisis. El paciente
tenia una enfermedad renal en estado terminal y murió a causa de la falla renal.
4. QUE NO REPORTAR como
Evento Adverso “Excepciones”

3. Exceder en uso a la vida útil del DM La pérdida de detección de un marcapaso después de que ha
alcanzado su fin de vida útil, indicada en las instrucciones de uso. El indicador de reemplazo lo mostró a su debido tiempo de acuerdo a las especificaciones del DM. Se requirió retirada quirúrgica del marcapaso.
Cemento Oseo después de su fecha de expiración. El usuario no logra fijación de la prótesis.
4. La protección para un mal funcionamiento operó correctamente. Un tomografo se detiene al presionar un brazo del paciente entre su
mesa contra el gantry, éste da señal de alarma apropiada. Cumple con los estándares relevantes y no hubo lesión al paciente.
4. QUE NO REPORTAR como
Evento Adverso “Excepciones”

5. Probabilidad insignificante de la ocurrencia de muerte o lesión seria. Se encontraron partículas en empaques de lentes de contacto.
La posibilidad de ocurrencia de una lesión grave se determino
que es despreciable.
6. Efectos secundarios previstos y previsibles (etiquetados o documentados en práctica clínica) La colocación de un catéter central resulta en una reacción de
ansiedad y dificultad para respirar. Ambas reacciones son conocidas y etiquetadas como efectos colaterales.
4. QUE NO REPORTAR como
Evento Adverso “Excepciones”

7. Acontecimientos adversos descritos en un aviso de
alerta un paciente tiene una reacción tisular indeseable (ej. Alergia al níquel)
previamente conocida y documentada en la información del dispositivo.
8. Excepciones concedidas por la Autoridad a petición
del Fabricante (comunes y bien documentadas) El fabricante emite una noticia de advertencia y avisa de un stent
coronario que ha migrado debido a una inflación inadecuada del
mecanismo del balón adjunto. Ejemplos subsecuentes de migración
fueron resumidos en reportes trimestrales recordando la acción y
eventos adversos individuales no han tenido que ser reportados.
4. QUE NO REPORTAR como
Evento Adverso “Excepciones”

QUEJASEventos Adversos
SISTEMA DE CALIDAD
EXCEPCIONES
1 al 8
-Entradas en Sistema de Administración del Análisis de Riesgo y
-Entradas en Acciones Preventivas y Correctivas

5. EXPERIENCIA EN COLOMBIATendencias de lo reportado ( período 2006-2010 )
( Qué, A quien, Cómo, Cuándo)
Muestra representativa de 37 DM: serializados, consumibles e implantables), de diferentes Fabricantes /Responsables de distribución
Mercado: 150-215 clientes/usuarios
% Reportantes: 15-25%

5. EXPERIENCIA EN COLOMBIATendencias de lo reportado ( período 2006-2010 )
( Qué, A quién, Cómo, Cuándo)
A QUE CORRESPONDE LO REPORTADO?
A. Excepciones 97%
1. Deficiencia encontrada por usuario antes del uso en el paciente
4. La protección para un mal funcionamiento operó correctamente
B. Mal funcionamiento 2%
C. Error en el uso 1%:

Quejas
SISTEMA DE CALIDAD
Eventos Adversos
TECNOVIGILANCIA
5. EXPERIENCIA EN COLOMBIATendencias de lo reportado (período 2006-2010)
( Qué, A quién, cómo, cuándo)
1. Usuario-a la Autoridad Sanitaria
2. Usuario- a la Autoridad Sanitaria y
al Fabricante ó Responsable
3.1 Usuario- al Responsable y/o
Fabricante;
3.2 Responsable y/o Fabricante
a la Autoridad
4. Autoridad Sanitaria-Responsable
y/o Fabricante

5. EXPERIENCIA EN COLOMBIATendencias de lo reportado (periodo 2006-2010)
( Qué, A quién, cómo, cuándo)
UNIVERSALIDAD DE LA INFORMACION
Información Administrativa
Información clínica del evento
Información de la institución
Información del DM
Resultados de la investigación por parte del Fabricante.
Información del paciente
Otra información relevante

5. EXPERIENCIA EN COLOMBIATendencias de lo reportado (periodo 2006-2010)
( Qué, A quién, cómo, cuándo)
1. Información Administrativa Asignación Numero de control del reporte: Usuario, Fabricante, Autoridad
Tipo de reporte: Inicial, Seguimiento, Final, de tendencias.
Fecha del reporte
Fecha de ocurrencia del evento
Fecha de captura del reporte por parte del fabricante.
Fecha esperada del próximo reporte
Persona autorizada para reportar
2. Información clínica del evento Descripción del evento
Número de pacientes involucrados
Numero de DM involucrados
3. Información de la institución Nombre, domicilio, teléfono, fax, persona contacto

5. EXPERIENCIA EN COLOMBIATendencias de lo reportado(periodo 2006-2010)
( Qué, A quién, cómo, cuándo)
4. Información del DM Nombre del manufacturador, persona contacto, teléfono, fax. Correo electrónico
Del Operador en el momento de Evento : paciente, el medico, enfermero, otro, ninguno
De la caracteristica del DM: nuevo, reuso de un unico uso, reuso de un reusable, remanufacutrado/repotenciado, otro (especificar)
Información génerica del DM:Nombre, marca, modelo, #lote/ #serial, código /sistema de nomenclatura
Disposición del DM: Destruido, Permanece en el paciente, Devuelto al manufacturador, Conservado para la investigación
Informacion de aprobacion del DM: #registro sanitario/#documento que aprobó el DM, autoridad que autorizo el DM, otros terceros que aprobaron el DM, # identificación del cuerpo notificador.
5. Resultados de la investigación por parte del Fabricante. Resultados de investigación: especifica, detalles de los métodos de la investigación, resultados,
conclusiones.
Acciones correctivas , Acciones preventivas

5. EXPERIENCIA EN COLOMBIATendencias de lo reportado(periodo 2006-2010)
( Qué, A quién, cómo, cuándo)
6. Información del paciente: edad, sexo, peso (Kg.)
Lista de DM involucrados en el Evento
Condición del paciente en la atención al paciente posterior al Evento:
Acciones correctivas, Evolución
7. Otra información relevante (será incluída únicamente al final del reporte):
eventos similares con igual causa raíz
otra información que puede ser considerada relevante para la industria, para la Autoridad.

EXPERIENCIA EN COLOMBIATendencias de lo reportado (periodo 2006-2010)
( Qué, A quién, cómo, cuándo)
Eventos ocurridos con anterioridad a 6 meses, 3
meses, >1mes
Acumulados: mas de uno
No especificado (no reportada fecha)

Conclusiones
Tecnovigilancia-Eventos Adversos
Sistema de Calidad-Quejas incluye excepciones
Universalidad de la información
Armonización de la información videos marcapasos E.A.

BibliografiaCOLOMBIA
www.invima.gov.co Tecnovigilancia
www.minproteccionsocial.gov.co Legislacion Tecnovigilancia
GHTF
www.ghtf.org/sg2/sg2-final.html Study Group 2 (SG2) - Post-Market Surveillance/Vigilance . Documentos finales sobre vigilancia postmercadeo: SG2-N54R8:2006, SG2-N57R8:2006, SG2-N8R4
FDA
www.fda.gov Informacion sobre Avisos de Alerta, Eventos Adversos y Recalls graves .
www.fda.gov/MedicalDevices/Safety/RecallsCorrectionsRemovals/ListofRecalls/default.htm
www.accessdata.fda.gov/scripts/cdrh/cfdocs/psn/index.cfm
http://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/default.htm
http://www.fda.gov/MedicalDevices/Safety/AlertsandNotices/PatientAlerts/default.htm
http://www.fda.gov/MedicalDevices/Safety/MedSunMedicalProductSafetyNetwork/default.htm
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/psn/viewbroadcasts.cfm
http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfmaude/search.cfm

GRACIAS


















