Grafo Métrico Y No Dirigido Para El Ensamblaje De Novo De ...
18
Grafo Métrico Y No Dirigido Para El Ensamblaje De Novo De Genomas Completos Juan Camilo Bojaca Aguilar 201613162 Universidad de los Andes, Bogotá, Colombia Contenido Grafo Con Pesos Métrico Y No Dirigido Para El Ensamblado De Novo De Genomas Completos ........................................................................................................................................ 1 Marco Teórico ................................................................................................................................ 2 Motivación....................................................................................................................................... 5 Objetivos ......................................................................................................................................... 5 Objetivo general .......................................................................................................................... 5 Objetivos específicos ................................................................................................................... 5 Métodos ........................................................................................................................................... 5 Definición Del Grafo................................................................................................................... 6 Aproximación Al Metric-TSP.................................................................................................. 12 Resultados ..................................................................................................................................... 14 Conclusiones ................................................................................................................................. 16 Referencias .................................................................................................................................... 17
Transcript of Grafo Métrico Y No Dirigido Para El Ensamblaje De Novo De ...

Grafo Métrico Y No Dirigido Para El Ensamblaje De Novo De Genomas Completos
Juan Camilo Bojaca Aguilar
201613162
Universidad de los Andes, Bogotá, Colombia
Contenido
Grafo Con Pesos Métrico Y No Dirigido Para El Ensamblado De Novo De Genomas
Completos ........................................................................................................................................ 1
Marco Teórico ................................................................................................................................ 2
Motivación ....................................................................................................................................... 5
Objetivos ......................................................................................................................................... 5
Objetivo general .......................................................................................................................... 5
Objetivos específicos ................................................................................................................... 5
Métodos ........................................................................................................................................... 5
Definición Del Grafo ................................................................................................................... 6
Aproximación Al Metric-TSP .................................................................................................. 12
Resultados ..................................................................................................................................... 14
Conclusiones ................................................................................................................................. 16
Referencias .................................................................................................................................... 17

Marco Teórico
El ensamblaje de genomas es uno de los principales problemas en el área de biología
computacional. En los últimos años se ha notado mejoras sustanciales en las máquinas de
secuenciación lo que, junto con el desarrollo de algoritmos de ensamblaje, ha permitido el
ensamblaje completo de genomas microbianos [1-2]. Herramientas como Ilumina han logrado
reducir las tasas de errores para lecturas cortas de manera significativa [3]. Por otra parte, se ha
logrado ampliar el tamaño de lecturas gracias a herramientas como Pacbio [4] y MinION [5]. Sin
embargo, el avance más significativo se ve en la reducción de costos de secuenciación, haciendo
de la secuenciación una herramienta accesible. Esto abre la posibilidad de mejorar el ensamblaje
con el uso de grandes volúmenes de información, como lo es el caso de la corrección de errores
en lecturas largas usando lecturas cortas con bajas tasas de error [7].
La secuenciación de genomas arroja como resultado un conjunto de lecturas
pertenecientes a alguna región del genoma. Sin embargo, aunque dos secuencias provengan de la
misma región del genoma no se puede esperar que sean exactamente iguales. Esto debido a la
existencia de tasas de error por parte del propio secuenciador. Estos errores incrementan la
complejidad de reconstruir la secuencia inicial y hacen necesario el manejo apropiado de estos.
Aunque las tasas de error están bajando progresivamente, no se puede aspirar la eliminación por
completo de los mismos.
Dependiendo de los datos disponibles existen dos aproximaciones al problema de
ensamblaje de genomas. Por un lado, si se sabe a qué organismo pertenece el genoma se podría
buscar algún genoma ya armado y usar esa referencia como base para el ensamblaje. Sin
embargo, no siempre se puede contar con esta referencia por lo que resulta necesarios algoritmos
que permitan hacer el ensamblaje desde cero, en un proceso conocido como ensamblaje de novo.
Este tipo de ensamblaje, sin tener una referencia es en el que se centró este trabajo.
Como se definió anteriormente el problema del ensamblaje de novo consiste en la
reconstrucción del genoma de referencia del que se obtuvieron las lecturas. El genoma del que se
obtuvieron las lecturas formalmente se puede ver como una supercadena de las secuencias de
entrada, entiéndase por supercadena a una cadena S tal que toda secuencia de entrada es una
subsecuencia de S. Sin embargo, el hecho de decir que el genoma de referencia es una
supercadena no basta, ya que podría definir una supercadena valida simplemente pegando una

secuencia tras otra. Claramente, la solución no contempla lo que se esperaría: que lecturas que se
extrajeron de regiones cercanas en el genoma, se encuentren cercanas en la reconstrucción. Para
reconstruir el genoma es necesario buscar una supercadena que integre las lecturas de la manera
más compacta todas las secuencias. Por lo anterior el problema de ensamblaje de novo
originalmente se definió como: “encontrar una supercadena común más corta (SCS) de las
lecturas de fragmentos dentro de la tasa de error” [9]. Esta definición original no fue utilizada en
la práctica porque es necesario el tomar en cuenta las estructuras repetitivas que se pueden
encontrar en el genoma para generar un ensamblaje que realmente represente el genoma
secuenciado.
A lo largo de los años se han manejado dos estrategias diferentes para dar solución al
problema anteriormente planteado. Por un lado, se tienen algoritmos basados en grafos de bujin y
sus modificaciones. Esta aproximación logra muy buenos resultados consumo de recursos/calidad
usando lecturas cortas con bajas tasas de error, sin embargo, deja de ser efectiva en genomas con
múltiples regiones repetitivas [10]. Para este tipo de genomas y usando de lecturas largas ofrecen
mejores resultados los algoritmos basados en grafos de sobrelapes.
Para definir un grafo de sobrelapes, se debe definir primero el concepto de sobrelape.
Dadas dos secuencias A y B se dice que existe un sobrelape si hay un sufijo de A que sea igual a
un prefijo de B. Resulta especialmente útil definir 𝑠𝑜𝑏𝑟𝑒𝑙𝑎𝑝𝑒(𝐴, 𝐵) como el máximo sobrelape
entre las secuencias. para el conjunto de secuencias se define el grafo sobrelapes como: un grafo
dirigido con cada secuencia representada como un vértice y el peso del enlace entre 𝐴 → 𝐵 es
|𝐵| − 𝑠𝑜𝑏𝑟𝑒𝑙𝑎𝑝𝑒(𝐴, 𝐵) [11]. Bajo esta representación de las secuencias, cada camino en el grafo
define una supercadena que contiene las cadenas en el camino. Por lo cual el problema de la
supercadena común más corta se puede expresar como encontrar el camino de menor costo que
pase por todos los vértices [11]. Este problema es un problema bien conocido en el área de las
ciencias de la computación como el problema del agente viajero, y pertenece a la categoría de
problemas NP-difícil
Usando el grafo de sobrelapes se desarrolló un marco de trabajo para el ensamblaje de
genomas. El método es conocido como Overlap-Layout-Consensus [12] (OLC) y debe su nombre
a las tres fases que lo componen. La operación posee tres fases que le dan su nombre. Overlap es
la fase el descubrimiento de superposiciones entre las lecturas. Debido a la naturaleza de esta fase

se suele realizar en paralelo. Layout consiste en la construcción y manipulación del grafo de
sobrelapes para encontrar el orden en que las secuencias se alinearan. Finalmente, Consensus
consiste en el alineamiento preciso de múltiples secuencias siguiendo el orden dado por él
Layout. La mayoría de los algoritmos basados en este método se diferencian o bien en la forma
en cómo se arma el layout tendiendo a algoritmos avaros para buscar caminos o construir una
solución iterativamente [13].
Con el fin de identificar sobrelapes entre las lecturas de manera eficiente, en este trabajo
se usó un algoritmo basado en Kmers. Un Kmer es una subsecuencia compuesta por k elementos.
Intuitivamente se espera que las lecturas con alta similitud deben compartir una buena cantidad
de Kmers en sus regiones superpuestas. Para la búsqueda de Kmers se optó por el uso de
estructuras de datos especializadas en la búsqueda de cadenas [15] lo cual reduce el costo
computacional de armar el grafo. Esta estrategia de identificación de sobrelapes es sensible
tamaño de los Kmers. Un valor apropiado de K debe ser lo suficientemente grande como para que
la mayoría de las superposiciones falsas no compartan Kmers por casualidad, y lo
suficientemente pequeño como para que la mayoría de las superposiciones verdaderas compartan
Kmers aun con los errores de secuenciación.
Retomando el problema del ensamblaje de genomas se puede plantear como encontrar una
solución al problema del viajero sobre el grafo de sobrelapes. En este trabajo se buscó proponer
una nueva arquitectura del grafo de ensamblajes. Con esta arquitectura se busca simplificar la
representación de los diferentes sobrelapes entre las lecturas y sus reversos complementos.
Además, se busca brindar una mayor variedad de algoritmos a aplicar en la fase de layout [16].
Específicamente creo un grafo no dirigido y métrico para representar el grafo de sobrelapes. Un
grafo este es métrico si sus ejes cumplen la desigualdad del triángulo. Lo que quiere decir que
dados tres vértices cualquiera 𝐴, 𝐵, 𝐶 ∈ 𝑉 la distancia entre A y B nunca será mayor que el
camino pasando por un intermedio C
𝑑𝐴𝐵 ≤ 𝑑𝐴𝐶 + 𝑑𝐶𝐵

Motivación
Debido a la enorme cantidad de lecturas que se manejan y al tamaño de los genomas a
ensamblar, el ensamblado de Novo es computacionalmente costoso. Agregando a lo anterior, el
manejo de tasas de error de las lecturas, las regiones repetitivas entre otros factores dificultan aún
más la labor. Por lo anterior el desarrollo de nuevas técnicas y aproximaciones que permitan
mejores resultados y/o reducción en el consumo de recursos resulta útil por sus aplicaciones en el
campo de la biología. Pero a su vez es un problema interesante que da pie para desarrollo de
software de alta calidad.
Objetivos
Objetivo general
Diseñar e implementar la construcción de un grafo con pesos métrico y no dirigido para el
ensamblado de Novo con el método Overlap-Layout-Consensus (OLC).
Objetivos específicos
• Definición el grafo y sus propiedades.
• Implementación de la construcción del grafo.
• Implementación de un algoritmo aproximado para The Metric travelling salesman
problem (Metric TSP)
Métodos
Este proyecto se desarrolló en el marco de NGSEP como parte del ensamblador de novo.
Gracias al uso del patrón algoritmo se permite probar varias implementaciones de la solución. En
el siguiente diagrama se resaltan en verde las implementaciones llevadas a cabo en este proyecto.

Definición Del Grafo
La entrada para la construcción del grafo consiste en un conjunto 𝐴 de lecturas. Se define
el subconjunto 𝑆 ⊆ 𝐴 como el conjunto de lecturas que no contienen embebidos, esto quiere decir
que ninguna lectura está contenida en otra. Cabe aclarar que las lecturas en 𝐴 ∖ 𝑆 se asocian a
alguna lectura que las contiene para evitar la pérdida de información. El grafo se define como un
grafo no dirigido completo con 2n vértices, donde n corresponde al número de lecturas en S. El
grafo es no dirigido por lo que 𝑊𝑖,𝑗 = 𝑊𝑖,𝑗 donde 𝑊𝑖,𝑗 representa el peso del vértice 𝑉𝑖 al vértice
𝑉𝑗 donde 0 ≤ 𝑖, 𝑗 < 2𝑛.
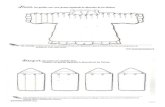
En lugar de la típica representación de una secuencia como un vértice, esta se representa
con dos vértices y un eje. Así pues, para 0 ≤ 𝑖 < |𝑆| se tiene que los vértices 𝑉2𝑖, 𝑉2𝑖+1
representan el inicio y fin de la secuencia 𝑆𝑖 respectivamente. Adicionalmente 𝑊2𝑖, 2𝑖+1 = |𝑆𝑖| el
peso del eje que une el inicio y fin de la secuencia es igual al tamaño de esta. La anterior
representación permite asociar un sentido a la lectura, el sentido original de la secuencia se
representa en el eje 𝑉2𝑖 → 𝑉2𝑖+1 , contrario al eje 𝑉2𝑖+1 → 𝑉2𝑖 que hace referencia al reverso
complemento de la lectura.

Definición: El complemento de un vértice 𝑉𝑖 es el otro eje que fue creado para representar
la lectura 𝑉𝑐𝑜𝑚𝑝(𝑖).
𝑐𝑜𝑚𝑝(𝑖) = 𝑖 − 2(𝑖 𝑚𝑜𝑑 2) + 1
Definición: La función 𝐿(𝑥) El largo de la secuencuencia asociada al vértice 𝑉𝑥.
𝐿(𝑖) = 𝐿(𝑐𝑜𝑚𝑝(𝑖)) = 𝑊𝑖, 𝑐𝑜𝑚𝑝(𝑖) = |𝑆⌊
𝑖2
⌋ |
fig1: definición del grafo de sobrelapes
Finalmente, el peso de los demás ejes del grafo se construyen en base al sobrelape entre
las lecturas. Dado que las lecturas se representan con dos vértices, las posibles formas de
sobrelape entre dos secuencias y sus complementos se representan uniendo los vértices
correspondientes (ver fig1). De esta manera si una lectura 𝑆𝑥 en su sentido original tiene un
sobrelape con el reverso complemento de otra lectura 𝑆𝑦 entonces el sobrelape entre el vértice fin
de la secuencia 𝑆𝑥 (𝑉2𝑥+1) y el inicio de la secuencia 𝑆𝑦 (𝑉2𝑦) corresponde a la longitud de la
subsecuencia del sobrelape. Por último, el peso entre dos vértices 𝑉𝑖, 𝑉𝑗 que no son
complementarios:
𝑊𝑖,𝑗 = 𝐿(𝑖) + 𝐿(𝑗) − 𝑆𝑜𝑏𝑟𝑒𝑙𝑎𝑝𝑒(𝑖, 𝑗) ∀𝑖, 𝑗 | 𝑗 ≠ 𝑐𝑜𝑚𝑝(𝑖)
Entendiéndose 𝑆𝑜𝑏𝑟𝑒𝑙𝑎𝑝𝑒(𝑖, 𝑗) como el sobrelape entre el vértice 𝑉𝑖 y el vertices 𝑉𝑗 o cero en
caso de no existir.

Lema 1 El sobrelape entre dos vértices 𝑉𝑖, 𝑉𝑗 es menor o igual al largo de ambas lecturas
asociadas a los vértices. Siendo igual si solo si 𝑗 = 𝑐𝑜𝑚𝑝(𝑖).
0 ≤ 𝑆𝑜𝑏𝑟𝑒𝑙𝑎𝑝𝑒(𝑖, 𝑗) < min(𝐿(𝑖), 𝐿(𝑗)) ∀𝑖, 𝑗| 𝑗 ≠ 𝑐𝑜𝑚𝑝(𝑖)
Teorema 1: El peso de cualquier eje es mayor o igual que la lectura más larga y menor o igual
que su suma.
max(𝐿(𝑖), 𝐿(𝑗)) ≤ 𝑊𝑖,𝑗 ≤ 𝐿(𝑖) + 𝐿(𝑗) ∀ 𝑖, 𝑗
Demostración: Partiendo del lema 1 y de la suposición de que los vértices no son
complementarios 𝑗 ≠ 𝑐𝑜𝑚𝑝(𝑖).
0 ≤ 𝑆𝑜𝑏𝑟𝑒𝑙𝑎𝑝𝑒(𝑖, 𝑗) < min(𝐿(𝑖), 𝐿(𝑗))
0 ≥ −𝑆𝑜𝑏𝑟𝑒𝑙𝑎𝑝𝑒(𝑖, 𝑖) > − min(𝐿(𝑖), 𝐿(𝑗))
𝐿(𝑖) + 𝐿(𝑗) ≥ 𝐿(𝑖) + 𝐿(𝑗) − 𝑆𝑜𝑏𝑟𝑒𝑙𝑎𝑝𝑒(𝑖, 𝑗) > 𝐿(𝑖) + 𝐿(𝑗) − min(𝐿(𝑖), 𝐿(𝑗))
𝐿(𝑖) + 𝐿(𝑗) ≥ 𝑊𝑖,𝑗 > max(𝐿(𝑖), 𝐿(𝑗))
Por otra parte, si 𝑗 = 𝑐𝑜𝑚𝑝(𝑖) entonces 𝑊𝑖,𝑗 = 𝐿(𝑖) = 𝐿(𝑗) por lo que se cumple que:
max(𝐿(𝑖), 𝐿(𝑗)) = max(𝐿(𝑖), 𝐿(𝑖)) = 𝐿(𝑖) ≤ 𝑊𝑖,𝑗 < 2𝐿(𝑖) = 𝐿(𝑗) + 𝐿(𝑖)
Teorema 2: Para todo eje entre los vértices 𝑉𝑖, 𝑉𝑗 no existe un vértice 𝑉𝑘 tal que 𝑊𝑖,𝑘 + 𝑊𝑘,𝑗 <
𝑊𝑖,𝑗. Equivalentemente:
𝑊𝑖,𝑗 ≤ 𝑊𝑖,𝑘 + 𝑊𝑘,𝑗 ∀𝑖, 𝑗, 𝑘
Demostración: se parte de la premisa de que a + b ≤ max(𝑎, 𝑐) + max (𝑏, 𝑐) el máximo de dos
valores es menor que la sumatoria de máximos con un tercero.
𝐿(𝑖) + 𝐿(𝑗) ≤ max(𝐿(𝑖), 𝐿(𝑘)) + max(𝐿(𝑗), 𝐿(𝑘))
Aplicando el teorema 1

𝑊𝑖,𝑗 ≤ 𝐿(𝑖) + 𝐿(𝑗) ≤ max(𝐿(𝑖), 𝐿(𝑘)) + max(𝐿(𝑗), 𝐿(𝑘))
𝑊𝑖,𝑗 ≤ max(𝐿(𝑖), 𝐿(𝑘)) + max(𝐿(𝑗), 𝐿(𝑘))
Aplicando nuevamente el teorema 1 y que 𝑎 ≤ 𝑏 ∧ 𝑐 ≤ 𝑑 ⇒ 𝑎 + 𝑏 ≤ 𝑐 + 𝑑.
𝑊𝑖,𝑗 ≤ max(𝐿(𝑖), 𝐿(𝑘)) + max(𝐿(𝑗), 𝐿(𝑘)) ≤ 𝑊𝑖,𝑘 + 𝑊𝑗,𝑘
𝑊𝑖,𝑗 ≤ 𝑊𝑖,𝑘 + 𝑊𝑗,𝑘
Construcción del Grafo
En la construcción del grafo se utilizó una aproximación basada en índices FM. En primer
lugar, se toman las secuencias de entrada y se concatenan. Lo anterior permite buscar una
secuencia en múltiples lecturas usando un único índice FM. Idealmente se crearía un único índice
con todas las lecturas concatenadas, resultando en búsquedas más rápidas. Sin embargo, el costo
en memoria del algoritmo de construcción del índice podría desbordar la memoria disponible en
la máquina. Se generan múltiples índices FM con varias secuencias concatenadas. Pese a lo
anterior se optó por mantener el algoritmo ya que gracias a DC3 el proceso más lento dentro la
construcción de un índice FM, que es la generación del arreglo se sufijos, se realiza con
complejidad temporal lineal.
Una vez todos los índices fueron creados se procede a pasarlos a una estructura para poder
mapear los índices a las lecturas originales. Con lo anterior en el algoritmo de construcción del
grafo se cuenta con un único manejador de índices FM, que permite la consulta de una cadena
específica sobre todas las lecturas de entrada. Esta estructura retorna los alineamientos, con las
secuencias y las posiciones donde esa cadena se encuentra.

fig2: construcción del índice
Posteriormente se procede a la identificación de alineamientos, para cada secuencia y su
reverso complemento se recorren los Kmers de tamaño 𝐾 con distancia entre ellos 𝑁 y se buscan
en el índice FM para múltiples secuencias. Los alineamientos se agrupan por lectura para su
posterior revisión. Es necesario almacenar todos los alineamientos con la lectura para poder
determinar si la cadena de la que se extrajeron los Kmers y la lectura presentan sobrelape o no.
Seudocódigo Armar grafo
𝑭𝒐𝒓 𝒍𝒆𝒄𝒕𝒖𝒓𝒂 𝒊𝒏 𝑨:
𝒊𝒇 𝒆𝒔𝑬𝒎𝒃𝒆𝒃𝒊𝒅𝒂(𝒍𝒆𝒄𝒕𝒖𝒓𝒂): 𝒄𝒐𝒏𝒕𝒊𝒏𝒖𝒆
𝑯 ≔ { }
𝑭𝒐𝒓 𝒑𝒐𝒔 𝒊𝒏 [𝟏 𝒕𝒐 |𝒍𝒆𝒄𝒕𝒖𝒓𝒂 | 𝒔𝒌𝒊𝒑 𝑵]:
𝑯 ∶= 𝑯 ∪ 𝑭𝑴𝒊𝒏𝒅𝒆𝒙. 𝒃𝒖𝒔𝒄𝒂𝒓(𝒍𝒆𝒄𝒕𝒖𝒓𝒂 [𝒑𝒐𝒔: 𝒑𝒐𝒔 + 𝒌])
𝒇𝒐𝒓 𝒑𝑳𝒆𝒄𝒕𝒖𝒓𝒂 𝒊𝒏 𝑨 ∖ {𝒍𝒆𝒄𝒕𝒖𝒓𝒂}:
𝒓𝒆𝒗𝒊𝒔𝒂𝒓({(𝒍𝒆𝒄, 𝒑𝒐𝒔, 𝒂𝒍𝒏) ∈ 𝑯 |𝒍𝒆𝒄 = 𝒑𝑳𝒆𝒄𝒕𝒖𝒓𝒂})
𝒍𝒆𝒄𝒕𝒖𝒓𝒂 ∶= 𝒍𝒆𝒄𝒕𝒖𝒓𝒂 . 𝐫𝐞𝐯𝐞𝐫𝐬𝐨𝐂𝐨𝐦𝐩𝐥𝐞𝐦𝐞𝐧𝐭𝐨()
𝑯 ≔ { }
𝑭𝒐𝒓 𝒑𝒐𝒔 𝒊𝒏 [𝟏 𝒕𝒐 |𝒍𝒆𝒄𝒕𝒖𝒓𝒂 | 𝒔𝒌𝒊𝒑 𝑵]:
𝑯 ∶= 𝑯 ∪ 𝑭𝑴𝒊𝒏𝒅𝒆𝒙. 𝒃𝒖𝒔𝒄𝒂𝒓(𝒍𝒆𝒄𝒕𝒖𝒓𝒂 [𝒑𝒐𝒔: 𝒑𝒐𝒔 + 𝒌])
𝒇𝒐𝒓 𝒑𝑳𝒆𝒄𝒕𝒖𝒓𝒂 𝒊𝒏 𝑨 ∖ {𝒍𝒆𝒄𝒕𝒖𝒓𝒂}:
𝒓𝒆𝒗𝒊𝒔𝒂𝒓({(𝒍𝒆𝒄, 𝒑𝒐𝒔, 𝒂𝒍𝒏) ∈ 𝑯 |𝒍𝒆𝒄 = 𝒑𝑳𝒆𝒄𝒕𝒖𝒓𝒂})

Una vez se identificaron los alineamientos con una lectura se procede a revisar si efectivamente
las secuencias sobrelapan o si una esta embebida dentro de otra. Lo primero que se hace es
calcular la posición relativa del inicio de la lectura referencia, de donde se extrajeron los Kmers,
sobre 𝑝𝐿𝑒𝑐𝑡𝑢𝑟𝑎. Lo anterior con la formula 𝑝𝑜𝑠𝑅𝑒𝑙 = 𝑎𝑙𝑛 − 𝑝𝑜𝑠 , si el valor de 𝑝𝑜𝑠𝑅𝑒𝑙 es
positivo quiere decir que la lectura referencia alinea en alguna posición de 𝑝𝐿𝑒𝑐𝑡𝑢𝑟𝑎. Una vez se
computan todos los valores de posición relativa para los alineamientos encontrados se pasa a la
fase de conteo. Se agrupan los alineamientos por posición en la lectura de referencia. Luego se
recorren los grupos de alineamientos y en cada iteración se buscan las posiciones relativas más
cercanas de la iteración anterior. Posteriormente del grupo de las posiciones más cercas se
selecciona la que tiene más Kmers se le suma uno y se asigna al conteo de Kmers que soportan
ese alineamiento. Finalmente, los alineamientos de la iteración anterior que no son cercanos a
ninguno de la iteración actual son agregados. En (Fig 3) se representa el proceso de conteo de
Kmers siendo cada tupla la posición relativa y el conteo de Kmers respectivamente. Dado que un
alineamiento en una iteración puede tener mas de un hit cercano en la iteración anterior, se pasa a
usar el que mayor conteo de Kmers tenga. Cada columna de la tabla se encuentra almacenada
como un árbol binario, ya que facilita la consulta de rangos en la siguiente iteración.
Fig3: conteo de alineamientos.
Como se puede observar en la Fig3 en la cada iteración se mantiene llave el alineamiento más
reciente, para mantener la información del primer alineamiento este se almacena en una variable
dentro del nodo. Al finalizar la fase de conteo de Kmres, solo se consideran aquellos que superen
una tasa de kmers, esto con el fin de eliminar los alineamientos que se dan probabilísticamente.

Por último, se determina el sentido del sobrelape con el 𝑝𝑜𝑠𝑅𝑒𝑙 y junto con el tamaño de las
cadenas si es embebido o no y se agrega al grafo los ejes correspondientes.
Seudocódigo Contar Kmers
𝐇 ∶= {(𝒑𝒐𝒔, 𝒂𝒍𝒏, 𝒑𝒐𝒔𝑹𝒆𝒍)|(𝒍𝒆𝒄, 𝒑𝒐𝒔, 𝒂𝒍𝒏) ∈ 𝐇}
𝑮𝒓𝒖𝒑𝒐𝒔 = { {(𝒂, 𝒃, 𝒄) ∈ 𝑯 | 𝒂 = 𝒑𝒐𝒔} |∃ (𝒊, 𝒋, 𝒌) 𝒊𝒏 𝑯 ∶ 𝒊 = 𝒑𝒐𝒔}
𝑨𝒍𝒏𝒔 ∶= {}
𝑭𝒐𝒓 𝑮𝒓𝒖𝒑𝒐 𝒊𝒏 𝑮𝒓𝒖𝒑𝒐𝒔. 𝒑𝒐𝒔𝒊𝒔𝒊𝒐𝒏𝒆𝒔𝑶𝒓𝒅𝒆𝒏𝒂𝒅𝒂𝒔( ):
𝒂𝒈𝒓𝒆𝒈𝒂𝒓, 𝒓𝒆𝒎𝒐𝒗𝒆𝒓 ∶= { }, { }
𝑭𝒐𝒓 (𝐩𝐨𝐬, 𝐚𝐥𝐧, 𝒑𝒐𝒔𝑹𝒆𝒍) 𝒊𝒏 𝑮𝒓𝒖𝒑𝒐:
𝒏𝒐𝒅𝒐𝒔 ∶= {(𝒍𝒍𝒂𝒗𝒆, 𝒄𝒐𝒏𝒕𝒆𝒐) ∈ 𝑨𝒍𝒏𝒔| 𝒍𝒍𝒂𝒗𝒆 − 𝒓 ≤ 𝒑𝒐𝒔𝑹𝒆𝒍 ≤ 𝒍𝒍𝒂𝒗𝒆 + 𝒓}
𝒊𝒇 |𝒏𝒐𝒅𝒐𝒔| = 𝟎
𝒂𝒈𝒓𝒆𝒈𝒂𝒓 ∶= 𝒂𝒈𝒓𝒆𝒈𝒂𝒓 ∪ {(𝒑𝒐𝒔𝑹𝒆𝒍, 𝟏)}; 𝒄𝒐𝒏𝒕𝒊𝒏𝒆
𝒓𝒆𝒎𝒐𝒗𝒆𝒓 ∶= 𝒓𝒆𝒎𝒐𝒗𝒆𝒓 ∪ 𝒏𝒐𝒅𝒐𝒔
𝒎𝒆𝒋𝒐𝒓 ≔ 𝒎𝒂𝒙((𝒌𝒆𝒚, 𝒄𝒐𝒏𝒕𝒆𝒐) ∈ 𝒏𝒐𝒅𝒐𝒔|: 𝒄𝒐𝒏𝒕𝒆𝒐)
𝒂𝒈𝒓𝒆𝒈𝒂𝒓 ∶= 𝒂𝒈𝒓𝒆𝒈𝒂𝒓 ∪ {(𝒑𝒐𝒔𝑹𝒆𝒍, 𝒎𝒆𝒋𝒐𝒓 + 𝟏)};
𝑨𝒍𝒏𝒔 ∶= (𝑨𝒍𝒏𝒔 ∖ 𝒓𝒆𝒎𝒐𝒗𝒆𝒓) ∪ 𝒂𝒈𝒓𝒆𝒈𝒂𝒓
𝒂𝒄𝒕𝒖𝒂𝒍𝒊𝒛𝒂𝒓𝑮𝒓𝒂𝒇𝒐()
Aproximación Al Metric-TSP
Para el algoritmo de layout se implementará El algoritmo de Christofides [16] El cual es un
algoritmo aproximado para encontrar el árbol de expansión mínima. El algoritmo garantiza que
en el peor de los casos el camino no será mayor a 1.5 el camino óptimo. El seudo código del
algoritmo se presenta a continuación.

Seudocódigo Layout
𝐓 ∶= 𝐀𝐫𝐛𝐨𝐥𝐄𝐱𝐩𝐚𝐧𝐬𝐢𝐨𝐧𝐌𝐢𝐧𝐢𝐦𝐚(𝐆)
𝑶 ∶= {𝒗 𝒊𝒏 𝑽| 𝒈𝒓𝒂𝒅𝒐(𝒗)𝒎𝒐𝒅𝟐 = 𝟏}
𝑴 ∶
= 𝑬𝒎𝒑𝒂𝒓𝒆𝒋𝒂𝒎𝒊𝒆𝒏𝒕𝒐𝑷𝒆𝒓𝒇𝒆𝒄𝒕𝒐𝑫𝒆𝑴𝒊𝒏𝒊𝒎𝒐𝑷𝒆𝒔𝒐(𝑮. 𝒔𝒖𝒃𝒈𝒓𝒂𝒇𝒐𝑰𝒏𝒅𝒖𝒄𝒊𝒅𝒐(𝑶))
𝑯 ∶= 𝑮(𝑽, {𝑴. 𝑬 ∪ 𝑻. 𝑬})
𝒑𝒂𝒕𝒉 ∶= 𝒄𝒊𝒄𝒍𝒐𝑬𝒖𝒍𝒆𝒓𝒊𝒂𝒏𝒐(𝑯)
𝒑𝒂𝒕𝒉 ∶= 𝒒𝒖𝒊𝒕𝒂𝒓𝑵𝒐𝒅𝒐𝒔𝒓𝒆𝒑𝒆𝒕𝒊𝒅𝒐𝒔(𝒑𝒂𝒕𝒉)
𝒑𝒂𝒓𝒕𝒊𝒓𝑬𝒋𝒆𝒔𝑺𝒊𝒏𝑺𝒐𝒃𝒓𝒆𝒍𝒂𝒑𝒆(𝒑𝒂𝒕𝒉)
Como entrada al algoritmo se maneja una matriz de adyacencia ya que todos los ejes están
conectados, el peso es el descrito en la sección de arriba, y se asigna inclusive si no hay
sobrelape, en cuyo caso el sobrelape es igual a cero.
Para la implementación de este algoritmo se tuvieron que tomar decisiones en cada fase,
para calcular el árbol de expansión mínima se opto por usar el algoritmo de Kruskal con una
estructura de conjuntos disjuntos basada en árboles. Gracias a que el eje con menor peso para un
vértice es su complemento, el árbol de expansión mínima necesariamente incluye todos los ejes
complementos y por lo tanto se garantiza para cada conjunto con solo un nodo el primer conjunto
con el que se va a unir contiene su complemento.
El emparejamiento perfecto de peso mínimo se implementó con el algoritmo de Emmons
Blossoms[17]. Se optimizo la función dual, con una aproximación de pesos en los nodos, se usaron
arboles alternantes para manejar los bolsones. Sin embargo, se omitió por simplicidad el uso de
múltiples colas de prioridad. Como esquema de actualización de la variable dual se optó por la
aproximación de múltiples árboles.
Finalmente, para el camino euleriano se implementó el algoritmo de Hierhozer. Aunque en
el algoritmo original no se explica cual ejes cortar se opto por dejarlos ejes con menor grado, los
cuales normalmente corresponden al vértice complemento. En la fase final del algoritmo se parte
el camino encontrado si el sobrelape entre los nodos es cero. De esta manera se parte un único
camino de layout forzado en múltiples contigs.

Resultados
Al armar el grafo se probaron distintas configuraciones de los parámetros tanto para
datos simulados como reales. Los parámetros son: el tamaño del kmer, la distancia desde el inicio
de un kmer hasta el inicio del siguiente. La tasa mínima de kmers que soportan un alineamiento y
la diferencia máxima entre las posiciones relativas. Se realizo pruebas con datos simulados con
datos de 2% de cambios por sustitución y 1% de indels. Para verificar se usó como medida la
precisión y el recall. La precisión es el número de elementos, sobrelapes o alineamientos,
encontrados que también están el grafo esperado sobre el total de elementos encontrados. El
recall se calcula como el numero de elementos encontrados que también están en el grafo
esperado sobre la cantidad total de esos elementos en el grafo esperado. Para calcular el grafo
esperado se usaron los índices de inicio desde donde se copio la lectura de la secuencia que se
uso para simular. Además, no se agregaron los alineamientos que corresponden a menos de 100
bases.
parámetros de entonación resultados
tamaño
del
kmer
distancia
entre
kmers
tasa de
kmers
minima
máxima
distancia
posiciones
relativas
tiempo de
ejecución
(milisegundos)
embebidos sobrelapes
precision recall presision recall
30 30 0,02 10 190143 100 86,094 92,3280 72,4576
30 30 0,02 5 243216 100 76,275 92,3453 59,3487
30 30 0,02 20 144887 100 93,546 92,6170 84,1324
30 30 0,02 40 114412 100 99,285 93,1806 94,0822
30 30 0,02 80 109853 100 99,719 93,1278 94,7596
30 30 0,02 160 109584 100 99,696 92,9331 94,7358
30 30 0,01 80 108640 100 99,719 92,2473 94,7664
30 30 0,04 80 108989 100 99,708 95,6404 93,9574
30 30 0,08 80 137026 100 93,707 98,0719 68,5649
30 30 0,16 80 579135 100 0,3122 94,4575 1,7522
30 15 0,02 80 185761 100 99,6884 92,6957 96,9649
30 10 0,02 80 243963 100 99,6807 92,6157 97,6201
25 30 0,02 80 131512 100 99,6923 93,0395 96,9756
35 30 0,02 80 87318 100 99,6154 94,6292 90,3005
20 30 0,02 80 166374 100 99,6461 93,2617 98,3148
15 30 0,02 80 459951 100 99,5844 92,4830 99,2687

Tabla1: resultados sobre datos simulados sobre bacterias
tamaño
del
Kmer
distancia
entre
kmers
tasa de
kmers
mínima
máxima
distancia
posiciones
relativas
sobrelapes Tiempo
calcular
el índice
FM (ms)
Tiempo
armar el
grafo
(ms) recall precisión
15 5 30 0,36 99,995 98,185 496246 326131
15 10 30 0,36 99,998 97,994 560535 294231
15 15 30 0,36 99,998 98,068 697352 289961
15 15 10 0,36 99,795 66,095 534183 512346
15 15 20 0,36 99,998 98,067 488001 237411
15 15 30 0,28 99,998 95,322 643481 268089
15 15 30 0,32 99,998 96,598 484081 215807
30 10 60 0,14 99,979 89,893 489756 146665
30 20 60 0,14 100 88,822 479564 129549
30 30 40 0,14 99,953 90,127 541459 116748
30 30 50 0,14 99,953 90,102 565588 113649
30 30 60 0,1 99,926 93,287 447874 80632
30 30 60 0,12 99,979 92,357 458145 95533
30 30 60 0,14 99,953 90,114 527896 115174
Tabla2: ajuste de parámetros con corrección
90
92
94
96
98
100
80 85 90 95 100
PR
ECIS
ION
RECALL
Kmer length (15-35) Distance between kmers (30-10)
Distance between relative positions (5- 160) Min % kmers (1%-8%)

Los mejores resultados obtenidos rondan el 99.99% de recall y el 98% de precisión.
Conclusiones
Con datos simulados usando como base genomas sencillos, se logró una buena precisión y
recall en un tiempo competitivo. Cabe aclarar que en los genomas probados la tasa de repetición
no es muy alta por lo que no se probó si la solución escala bien en estos casos.
Al definir el grafo como métrico y no dirigido, permite el uso de algoritmos con la mejor
relación de aproximación para esta clase de grafos. Sin embargo, hay que tener en cuenta los
vértices complementarios a la hora de implementar las partes para evitar que en el layout los
vértices complementarios queden desconectados entre ellos.
Discusión
Aunque el algoritmo de layout garantiza que la solución se aproxime al TSP real, hay
que tener en cuenta que el grafo no es tal cual el grafo de sobrelapes perfecto, ya que debido a
errores en la secuenciación y en la tasa de aceptación de parámetros, En el grafo existen ejes que
realmente no poseen un sobrelape real. De la misma hay ejes faltantes. Estos problemas pueden
llegar a afectar la calidad del layout de manera significativa.
15|5|30|0,3615|10|30|0,36
15|15|20|0,36
15|15|30|0,28
15|15|30|0,32
15|15|30|0,36
30|10|60|0,14
30|20|60|0,14
30|30|60|0,12
88
90
92
94
96
98
100
99,97 99,975 99,98 99,985 99,99 99,995 100 100,005 100,01

Referencias
[1] S. Goldstein, L. Beka, J. Graf, and J. L. Klassen, “Evaluation of strategies for the assembly of
diverse bacterial genomes using MinION long-read sequencing,” BMC Genomics, vol. 20, no. 1,
Sep. 2019.
[2] S. Koren and A. M. Phillippy, “One chromosome, one contig: complete microbial genomes
from long-read sequencing and assembly,” Current Opinion in Microbiology, vol. 23, pp. 110–
120, 2015.
[3] A. E. Minoche, J. C. Dohm, and H. Himmelbauer, “Evaluation of genomic high-throughput
sequencing data generated on Illumina HiSeq and Genome Analyzer systems,” Genome Biology,
vol. 12, no. 11, 2011.
[4] A. Rhoads and K. F. Au, “PacBio Sequencing and Its Applications,” Genomics, Proteomics &
Bioinformatics, vol. 13, no. 5, pp. 278–289, 2015.
[5] M. Jain, H. E. Olsen, B. Paten, and M. Akeson, “The Oxford Nanopore MinION: delivery of
nanopore sequencing to the genomics community,” Genome Biology, vol. 17, no. 1, 2016.
[6] S. Goodwin, J. D. Mcpherson, and W. R. Mccombie, “Coming of age: ten years of next-
generation sequencing technologies,” Nature Reviews Genetics, vol. 17, no. 6, pp. 333–351,
2016.
[7] E. Bao and L. Lan, “HALC: High throughput algorithm for long read error correction,” BMC
Bioinformatics, vol. 18, no. 1, May 2017.
[8] P. J. A. Cock, C. J. Fields, N. Goto, M. L. Heuer, and P. M. Rice, “The Sanger FASTQ file
format for sequences with quality scores, and the Solexa/Illumina FASTQ variants,” Nucleic
Acids Research, vol. 38, no. 6, pp. 1767–1771, 2009.
[9] E. W. Myers, “Toward Simplifying and Accurately Formulating Fragment Assembly,”
Journal of Computational Biology, vol. 2, no. 2, pp. 275–290, 1995.
[10] Z. Li, Y. Chen, D. Mu, J. Yuan, Y. Shi, H. Zhang, J. Gan, N. Li, X. Hu, B. Liu, B. Yang,
and W. Fan, “Comparison of the two major classes of assembly algorithms: overlap-layout-
consensus and de-bruijn-graph,” Briefings in Functional Genomics, vol. 11, no. 1, pp. 25–37,
2011.

11) N. Nagarajan and M. Pop, “Parametric Complexity of Sequence Assembly: Theory and
Applications to Next Generation Sequencing,” Journal of Computational Biology, vol. 16, no. 7,
pp. 897–908, 2009.
[12] J. R. Miller, S. Koren, and G. Sutton, “Assembly algorithms for next-generation sequencing
data,” Genomics, vol. 95, no. 6, pp. 315–327, 2010.
[13] S. Koren, B. P. Walenz, K. Berlin, J. R. Miller, N. H. Bergman, and A. M. Phillippy, “Canu:
scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation,”
Genome Research, vol. 27, no. 5, pp. 722–736, 2017.
[15] Ferragina P, Manzini G 2000. Opportunistic data structures with applications. In
Proceedings of the 41st Annual Symposium on Foundations of Computer Science, pp. 390–398.
IEEE Computer Society, Washington, DC.
[16] Goodrich, Michael T.; Tamassia, Roberto (2015), "18.1.2 The Christofides Approximation
Algorithm", Algorithm Design and Applications, Wiley, pp. 513–514.
[17] Kolmogorov, Vladimir (2009), "Blossom V: A new implementation of a minimum cost
perfect matching algorithm", Mathematical Programming Computation, 1 (1): 43–67