
Bioquimica Guia Examen Final
40
BIOQUIMICA ll – EXAMEN FINAL Las reacciones pueden ser de tres tipos Exorgonicas, Endorgonicas e Isorgonicas. - Exorgonicas o Son favorables o Liberan energia o La energia se representa con (-) Ejemplo ATP ADP + Pi. - Endogornicas o Son desfavorable o Captan energia o La energia se representa con (+). Ejemplo Glucosa+ P Glucosa-6-P. El ATP es el prototipo historico de un compuesto de alta energia, este posee dos enlaces de alta energia. ATP ADP +Pi AMP + Pi. AMP. Creatinina fosfato tiene enlaces de fosfato los cuales son reserva de energia. Quimica de los carbohidratos Glucosa es el azucar mas prevalente en la naturaleza C6 H12 O6., es un monosacarido, iniciador de diferentes vias metabolicas. Almidon es un polimero de glucosa, el almidon tiene dos componentes, amilosa que es una cadena lineal (1-4) y amilopectina (1-6). Glucogeno es un polimero de glucosas, sintetizado por el humano, tiene tambien cadenas lineales y ramificadas como el almidon. DIGESTION DE CARBOHIDRATOS. La digestion de cabrohidratos se lleva a cabo por hidrólisis, deben ser llevados a monosacaridos (galactosa, fructosa, glucosa) ENZIMA FUENTE SUSTATO PRODUCTO Ptialina Glandula salivales Almidon Glucogeno Oligosacarido
-
Upload
francesco-andres-placencia-squadrito -
Category
Documents
-
view
108 -
download
7
description
ghj
Transcript of Bioquimica Guia Examen Final

BIOQUIMICA ll – EXAMEN FINAL
Las reacciones pueden ser de tres tipos Exorgonicas, Endorgonicas e Isorgonicas.
- Exorgonicas o Son favorables o Liberan energia o La energia se representa con (-)
Ejemplo ATP ADP + Pi.
- Endogornicas o Son desfavorable o Captan energia o La energia se representa con (+).
Ejemplo Glucosa+ P Glucosa-6-P.
El ATP es el prototipo historico de un compuesto de alta energia, este posee dos enlaces de alta energia.
ATP ADP +Pi AMP + Pi. AMP.
Creatinina fosfato tiene enlaces de fosfato los cuales son reserva de energia.
Quimica de los carbohidratos
Glucosa es el azucar mas prevalente en la naturaleza C6 H12 O6., es un monosacarido, iniciador de diferentes vias metabolicas.
Almidon es un polimero de glucosa, el almidon tiene dos componentes, amilosa que es una cadena lineal (1-4) y amilopectina (1-6).
Glucogeno es un polimero de glucosas, sintetizado por el humano, tiene tambien cadenas lineales y ramificadas como el almidon.
DIGESTION DE CARBOHIDRATOS.
La digestion de cabrohidratos se lleva a cabo por hidrólisis, deben ser llevados a monosacaridos (galactosa, fructosa, glucosa)
ENZIMA
FUENTE
SUSTATO
PRODUCTO
Ptialina
Glandula salivales
Almidon Glucogeno
Oligosacarido

Dextrinasa
I. Delgado
Dextrinas
Glucosa.
Lactasa
I. Delgado
Lactosa
Glucosa+Galactosa
Maltasa
I. Delgado
Maltosa
Glucosa+Glucosa
Sacarasa
I. Delgado
Sacarosa
Glucosa+Fructosa
a-Amilasa
Pancreas
Almidon
Oligosacarido.
GLUCOLISIS
Es una via aerobia y anaerobias, que es catalitica para la glucosa, ocurre en el citosol de las celulas.
Como se activa la via alta insulina, bajo ATP , alto ADP + Pi.
Enzimas mas importantes hexoquinasa, fosfofructoquinasa, piruvato quinasa
La enzima mas importante fosfofructoquinasa.
Estas enzimas deben estar fosfatadas para su activacion.
Resultado final:
Glucosa + 2ATP + 2NAD+ + 4ADP + 2Pi 2 piruvatos + 2ATP + H2O + 2 NADH + 2 H
ANAEROBIA
Esta glucolisis se lleva a cabo en ausencia de oxigeno es indispensable la actuacion de la LDH lactato deshidrogenasa.
Como se ativa la enzima niveles bajos de 02 y altos de H+
El lactato se forma apartir de piruvato el proceso puede ser inverso.
El eritrocito maneja glucolisis anaerobia.
Para la sintesis de lactato se necesita NADH y se forma NAD+.
NAD 3 ATP.
FAD 2 ATP.

CICLO DE KREBS
Es el metabolismo del piruvato, proceso es aerobio, se produce CO2 y H20.
Es este proceso de necesita a fuerza OXIGENO.
Este se lleva a cabo en la matriz mitocondrial.
El primer paso de esta via es la conversion de piruvato en ACETIL-CoA.
En este proceso se produce 3NADH , 1FADH, 1 GTP.
Por cada glucosa 40 ATP en el proceso de gastan 2.
C6H1206 + 02 40 ATP- 2 + 6CO2 + 6H20.
INSULINA
Es una proteina pequena, sintetizada por los islotes B del pancreas, se produce como proinsulina y de desdobla como insulina en el aparato de golgi.
En su liberacion se libera el peptido C el cual puede ser checado en sangre.
La insulina se une a los receptores de la tirosina quinasa abren canales de glucosa.
La insulina se libera por la glucemia, se libera por exocitosis, depende de calcio para su liberacion.
Es la hormona mas anabolica.
TRANSPORTADORES DE GLUCOSA
- G1 localizado en casi todo el cuerpo abunda en eritrocitos - G2 higado, intestino, pancreas, rinon - G3 neuronas - G4 depende de insulina (cantidad) tejido adiposo y musculo
esqueletico
Hay transportadores de glucosa que no dependen de insulina como es
- SGLT1 el cual la glucosa y galactosa es transportada como co-transporte en la membrana luminal .
GLUCOGENESIS
Es la formacion de glucogeno a partir de glucosa.
La biosintesis de glucogeno depende de cantidades altas de INSULINA y ATP , son los indicadores de que se lleve la sintesis.

La enzima iniciadora de esta sintesis es : Glucoquinasa, la cual solo se encuentra en citosol del hepatocito.
La enzima reguladora de la biosintesis Glucogeno sintetasa. insulina activa
EL glucogeno se almacena como un polisacarido en higado y musculo.
GLUCOGENOLISIS
Es la ruptura de glucogeno para la formacion de glucosa.
Es una accion catabolica , quien activa la via:
Aumento de : Glucagon, Epinefrina, Cortisol, GH y la dismuncion de ATP.
Como hay una dismnucion de ATP hay aumento de ADP + Pi se fosforila la enzima inciadora de la degradacion
- Glucogeno Fosforilasa.
Enzimas necesarias
- Glucogeno fosforilasa inciadora y reguladora rompe y mete P04. - Transglicosilasa desrramificante - a(1-6) Glucosa ramificada la libera - Fofoglucomutasa - Glucosa 6- Fosfatasa libera como glucosa al torrente.
Si la glucogenolisis sucede en musculo Se va directo a ATP
Si sucede en higado necesita de glucosa-6-fosfatasa se va a torrente.
GLUCONEOGENESIS
Via metabolica en la obtencion de energia apartir de preocursores no glucosidicos como por ejemplo aminoacidos.
Activacion: aumento de Epinefrina, Glucagon, Cortisol.
- Ciclo de cori o Proceso en el cual el lactato es convertido a piruvato o Accion mediada por Deshidrogenasa lactica o Sucede en higado
Los organos glucoNEOgenicos Higado, Musculo, Rinon.

VIA DE LAS PENTOSAS
Via metabolica a partir de la glucosa-6-fosfato, en la cual de obtienen azucares de 5 carbonos requiridos para diversas sintesis ADN, ARN , NAD, FAD.
En esta via ademas de producir ribosa-5-P se produce mucho NADPH, este es necesario para la biosinstesis de colesterol, acidos grasos
Tambien es necesario para la reduccion del glutation y de la toxicidad del oxigeno
Enzimas mas importante Glucosa – 6 – P – deshidrogenasa.
El deficit de esta enzima Anemia Hemolitica.
Esta sintesis de lleva a cabo por : exceso de ATP, carencia de NADPH, mitosis y meiosis para sintesis de PRPP.
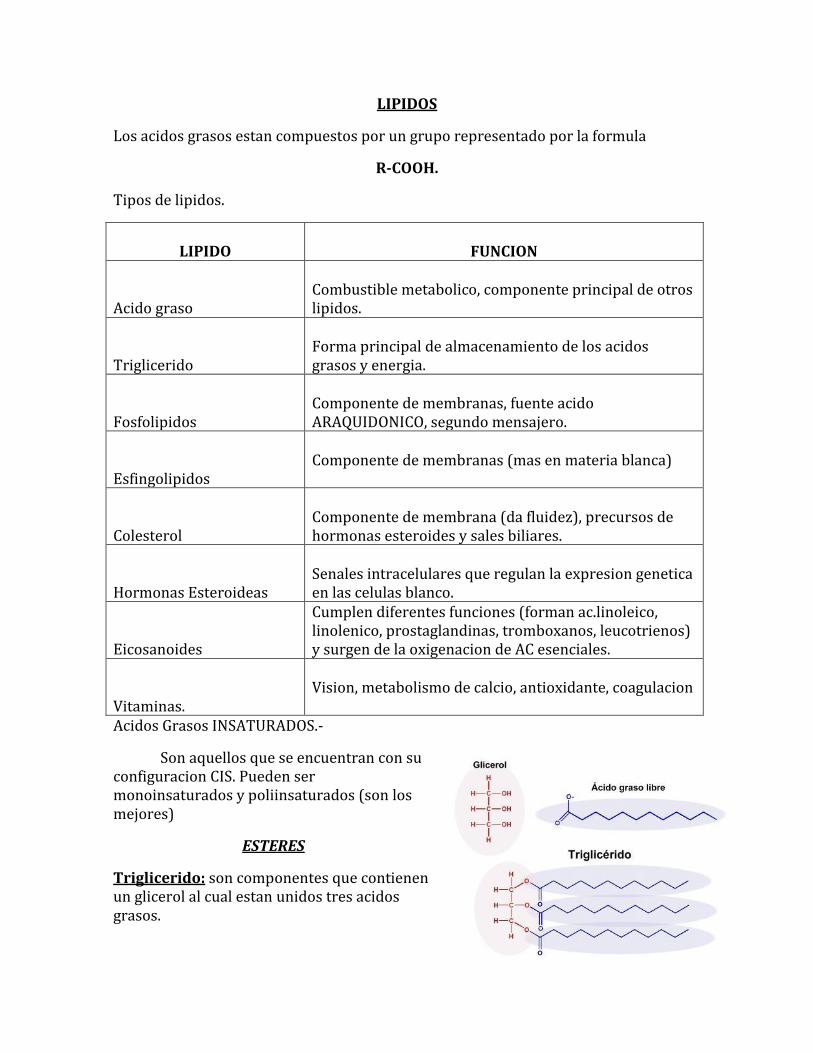
LIPIDOS
Los acidos grasos estan compuestos por un grupo representado por la formula
R-COOH.
Tipos de lipidos.
LIPIDO
FUNCION
Acido graso
Combustible metabolico, componente principal de otros lipidos.
Triglicerido
Forma principal de almacenamiento de los acidos grasos y energia.
Fosfolipidos
Componente de membranas, fuente acido ARAQUIDONICO, segundo mensajero.
Esfingolipidos
Componente de membranas (mas en materia blanca)
Colesterol
Componente de membrana (da fluidez), precursos de hormonas esteroides y sales biliares.
Hormonas Esteroideas
Senales intracelulares que regulan la expresion genetica en las celulas blanco.
Eicosanoides
Cumplen diferentes funciones (forman ac.linoleico, linolenico, prostaglandinas, tromboxanos, leucotrienos) y surgen de la oxigenacion de AC esenciales.
Vitaminas.
Vision, metabolismo de calcio, antioxidante, coagulacion
Acidos Grasos INSATURADOS.-
Son aquellos que se encuentran con su configuracion CIS. Pueden ser monoinsaturados y poliinsaturados (son los mejores)
ESTERES
Triglicerido: son componentes que contienen un glicerol al cual estan unidos tres acidos grasos.

Colesterol: es un alcohol que contiene 27 carbonos, es un esteroide, forma hormonas, le da fluidez ala membrana y forma sales biliares.
FOSFOLIPIDOS
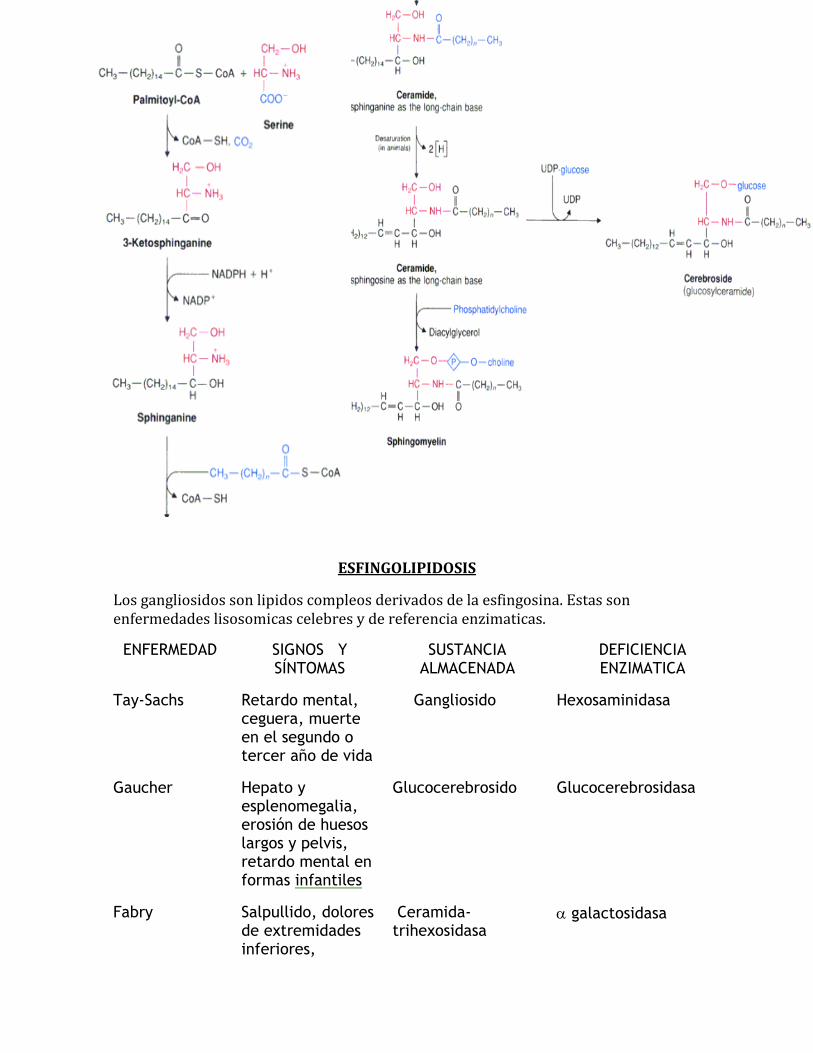
Esfingolipidos: son derivados de la ESFINGOSINA un aminoalcohol, su intermediario es la CERAMIDA (resulta de la acilacion del grupo amino).
Ceramida + fosfatidil colina (C1) ESFINGOMIELINA
Esfingomilina + monosacarido CEREBROSIDO
Esfigomielina + oligosacarido (NANA) GANGLIOSIDO.
DIGESTION Y ABSORCION DE LIPIDOS
Para ayudar a la digestion y absorcion de lipidos, el higado secreta bilis que contiene las sales biliares y fosfatidilcolina, las cuales funcionan como detergentes para solubilizar las grasas y esto facilite la absorcion y digestion.
Enzimas:
Comienza en BOCA, por medio de las glandulas salivales LIPASA SALIVAL, la cual actua en la digestion de lipidos complejos, el estomago produce LIPASA GASTRICA, despues pasa a duodeno la cual el pancreas secreta LIPASA PRANCREATICA , tambien secreta pancreas ESTERASA y FOSFOLIPASA
Todo estos procesos consisten para producir una mezcla de 2-monoglicerido y acidos grasos y tambien para hidrolizar fofolipidos.
Las sales biliares son secretadas para formas MICELAS y asi ayudan a proveer el vehiculo para el transporte de lipidos de la luz intestinal a los enterocitos. Donde ocurre la absorcion de monogliceridos, acidos grasos , colesterol, vitaminas etc.
Despues de que los acidos grasos y monogliceridos son tomados por el epitelio intestinal, son convertidos a trigliceridos y luego estos son liberados ala LINFA como QUILOMICRONES. Despues estos por medio del sistema porta llegan a higado.
LIPOGENESIS
Como su nombre lo dice es la formacion de Lipidos a partir de procursores carbonatados. La lipogenesis se lleva acabo cuando “hay sobrantes de energia” (glucosa en exceso) se lleva acabo cuando el ATP esta alto, insulina es la hormona que propicia ala lipogenesis.
Lugares mas lipogenicos : Higado, Mamas, Tejido Adiposo.
PRECURSOS de lipogenesis: ACETIL CO-A
GLUCOESFINGOLIPIDOS
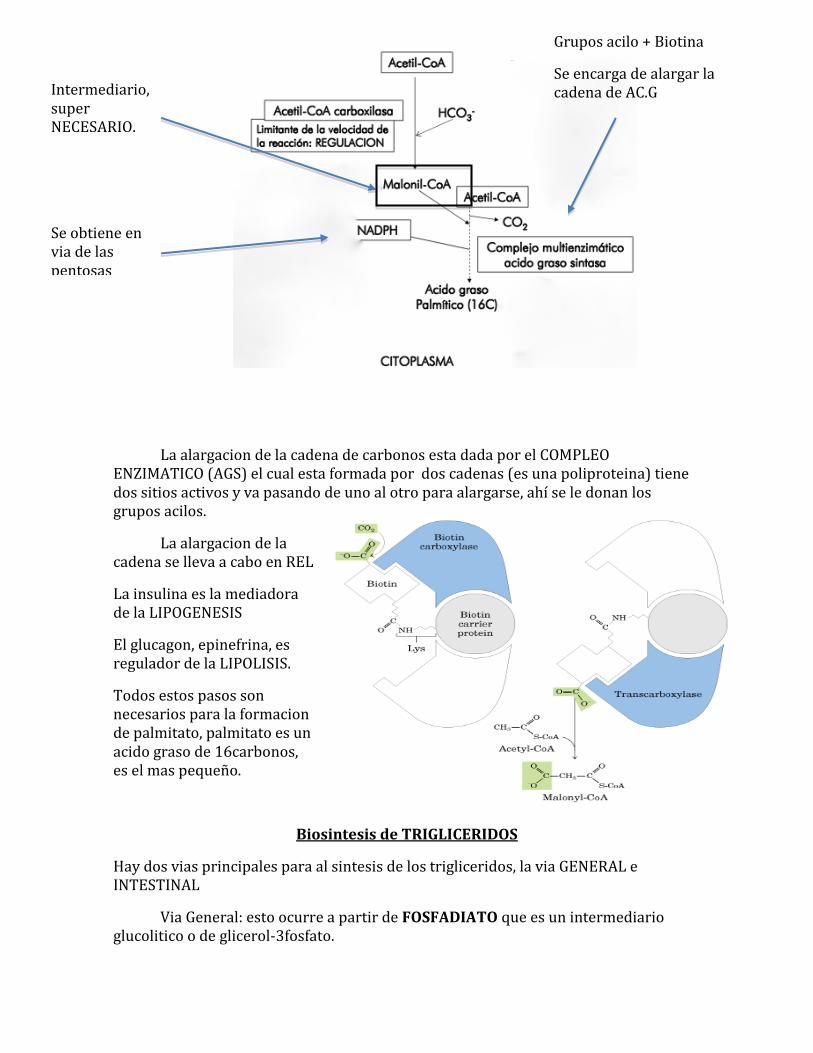
La alargacion de la cadena de carbonos esta dada por el COMPLEO ENZIMATICO (AGS) el cual esta formada por dos cadenas (es una poliproteina) tiene dos sitios activos y va pasando de uno al otro para alargarse, ahí se le donan los grupos acilos.
La alargacion de la cadena se lleva a cabo en REL
La insulina es la mediadora de la LIPOGENESIS
El glucagon, epinefrina, es regulador de la LIPOLISIS.
Todos estos pasos son necesarios para la formacion de palmitato, palmitato es un acido graso de 16carbonos, es el mas pequeño.
Biosintesis de TRIGLICERIDOS
Hay dos vias principales para al sintesis de los trigliceridos, la via GENERAL e INTESTINAL
Via General: esto ocurre a partir de FOSFADIATO que es un intermediario glucolitico o de glicerol-3fosfato.
Se obtiene en via de las pentosas
Intermediario, super NECESARIO.
Grupos acilo + Biotina
Se encarga de alargar la cadena de AC.G

Via Intestinal : aquí ocurre a partir de 2-monoglicerido
Cuerpos cetonicos: ACETOACETATO, ACETONA, B-HIDROXIBUTARATO
LIPOLISIS
Como su nombre lo indica es la “lisis” ruptura de los lipidos, para la obteccion de energia o diversos procesos.
La beta oxidacion es un proceso que sucede en mitocondria, es un proceso basado para la obtencion de energia a partir de acidos grasos.
El primer paso es agregar co-A al palmitato para formar Lipoacil-CoA. Los pasos siguientes consiten en cortar la cadena de dos a dos carbonos y agregandole CoA para llevarlo a la obtencion de energia.
Lipoprotein lipasa: Ayuda a romper TG en ac libres y glicerol de la VLDL
Lipoacil-CoA

BIOSINTESIS DE COLESTEROL
El colesterol es el esterol mas abundante en humanos, es componente de membrana, forma bilis y hormonas.
La sintesis de colesterol ocurre en el citoplasma de las celulas nucleadas. Se forma apartir de ACETIL-COA
Acetil Co-A Hidroximetilglutaril Co-A Mevalonato Pirofosfato de isopentilo
geranilo farnesillo escualeno lanosterol colesterol hormona o bilis.
El lanosterol es el primer nucleo esteroide.
Requiere para su sintesis: 3 ATP (6) = 18 ATP y 18 acetil-CoA.
El colesterol no se puede degradar por que sus anillos esteroides no se pueden romper, por lo cual el colesterol no produce ENERGIA solo suelta precursores.
La enzima reguladora de la sintesis de colesterol es : HMG-CoA reductasa
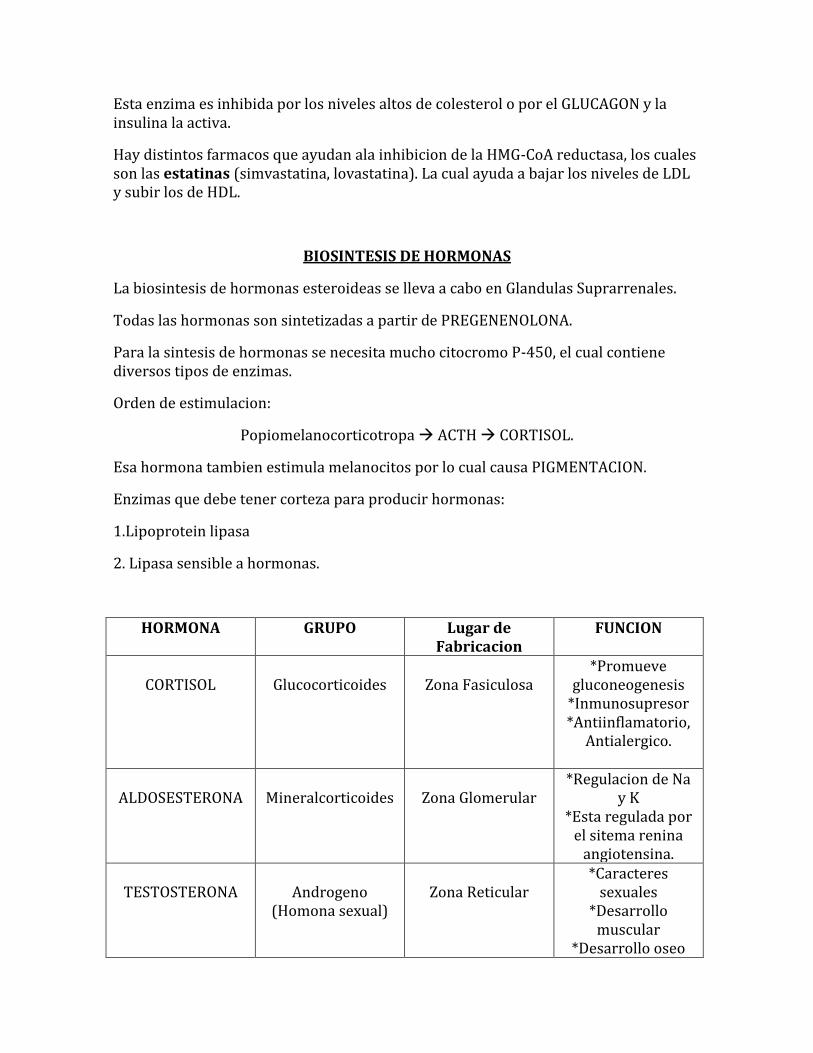
Esta enzima es inhibida por los niveles altos de colesterol o por el GLUCAGON y la insulina la activa.
Hay distintos farmacos que ayudan ala inhibicion de la HMG-CoA reductasa, los cuales son las estatinas (simvastatina, lovastatina). La cual ayuda a bajar los niveles de LDL y subir los de HDL.
BIOSINTESIS DE HORMONAS
La biosintesis de hormonas esteroideas se lleva a cabo en Glandulas Suprarrenales.
Todas las hormonas son sintetizadas a partir de PREGENENOLONA.
Para la sintesis de hormonas se necesita mucho citocromo P-450, el cual contiene diversos tipos de enzimas.
Orden de estimulacion:
Popiomelanocorticotropa ACTH CORTISOL.
Esa hormona tambien estimula melanocitos por lo cual causa PIGMENTACION.
Enzimas que debe tener corteza para producir hormonas:
1.Lipoprotein lipasa
2. Lipasa sensible a hormonas.
HORMONA GRUPO Lugar de Fabricacion
FUNCION
CORTISOL
Glucocorticoides
Zona Fasiculosa
*Promueve gluconeogenesis
*Inmunosupresor *Antiinflamatorio,
Antialergico.
ALDOSESTERONA
Mineralcorticoides
Zona Glomerular
*Regulacion de Na y K
*Esta regulada por el sitema renina
angiotensina.
TESTOSTERONA
Androgeno
(Homona sexual)
Zona Reticular
*Caracteres sexuales
*Desarrollo muscular
*Desarrollo oseo

ESTROGENO
Androgeno
(Hormona sexual)
Zona Reticular
*Incrementa la HDL *Contraresta PTH (Inhibe resorcion)
*Caracteres femeninos
*Estimula Libido *Estimula la formacion de
colageno.
PROGESTERONA
Hormona
Progestacional
-----------------
La accion de esta
hormona esta relacionada con el
ciclo ovarico.
BIOSINTESIS DE VITAMINA D
La vitamina D es precursor de una hormona llamada CALCITRIOL., la cual influye en el metabolismo del Ca y PO4.
Un deficit: Raquitismo y Osteomalacia (en adultos)
Funciones:
*Mantiene concentraciones de calcio y fosfato
*Absorcion de calcio intestinal
*Actua con la PTH en la resocion osea.
Su sintesis comienza en la piel por medio de la luz (UV), despues es llevada al higado donde ocurre una deshidrogenacion y despues es llevada a rinion donde ocurre la ultima deshidrogenacion.
7-Dehidrocolesterol Colecalciferol 25-hidroxicolecalciferol 1,25 Hidroxicolecalciferol
HIPERPLASIA SUPRERRENAL CONGENITA.
Es un transtorno es causado por algun defecto enzimatico de la corteza suprerrenal.
La hiperplasia suprarrenal congenita es una enfermedad recesiva muy frecuente.
La cuasa mas COMUN es un deficit de la : 21-Hidroxilasa., por lo cual la 17-Hidroxilasa estara elevada. (se puede verificar a nivel serico por pruebas)
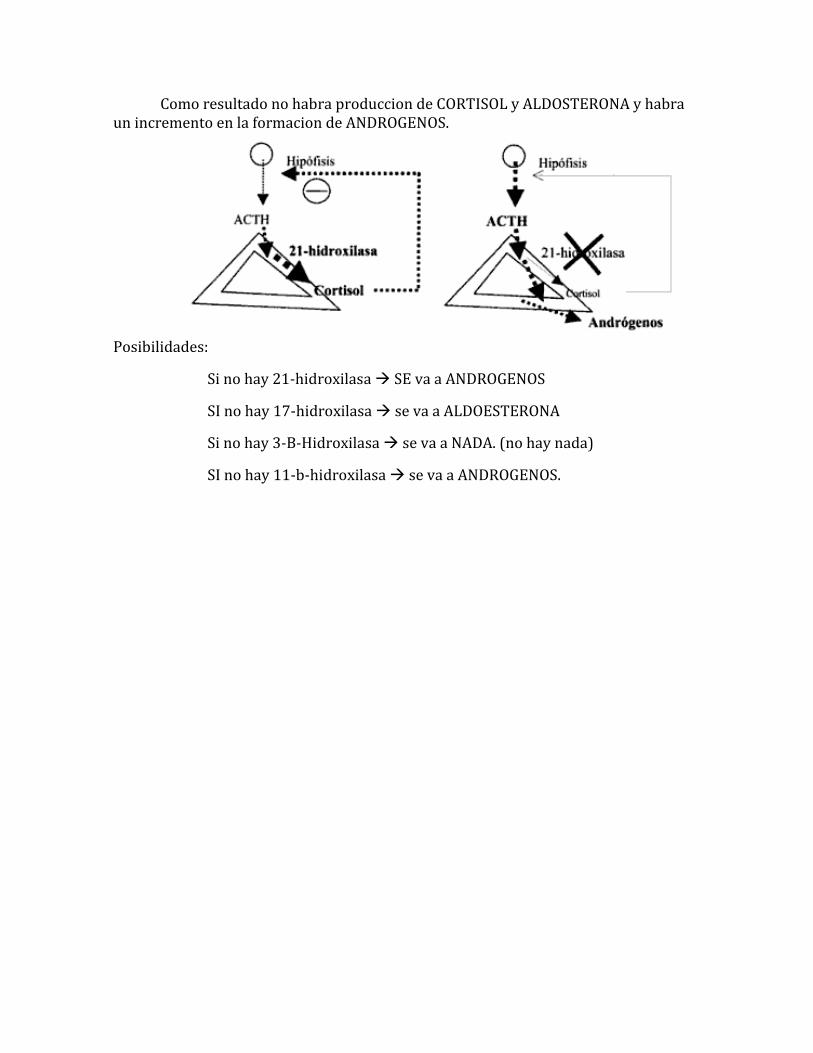
Como resultado no habra produccion de CORTISOL y ALDOSTERONA y habra un incremento en la formacion de ANDROGENOS.
Posibilidades:
Si no hay 21-hidroxilasa SE va a ANDROGENOS
SI no hay 17-hidroxilasa se va a ALDOESTERONA
Si no hay 3-B-Hidroxilasa se va a NADA. (no hay nada)
SI no hay 11-b-hidroxilasa se va a ANDROGENOS.

La 5-a-Reductasa: es una enzima que convierte la testorterona a Dihidrotestosterona, esto lo hace para que no se convierta en estradiol. (es IRREVERSIBLE)
La testosterona es convertida en estradiol por medio de la AROMATASA, es un proceso inverso.
BIOSINTESIS DE ESFIGOLIPIDOS
Los esfingolipidos son derivados del aminoalcohol esfingosina. Hay tres clases de esfingolipidos: esfingomielina, cerebrosidos y gangliosidos.
Pasos:
Palmitoil + CoA Palmitoil-CoA+ Serina CERAMIDA + Fosfatidilcolina ESFINGOMIELINA ceramida + monosacarido cerebrosido + NANA
GANGLIOSIDOS.
ESFINGOLIPIDOSIS
Los gangliosidos son lipidos compleos derivados de la esfingosina. Estas son enfermedades lisosomicas celebres y de referencia enzimaticas.
ENFERMEDAD SIGNOS Y SÍNTOMAS
SUSTANCIA ALMACENADA
DEFICIENCIA ENZIMATICA
Tay-Sachs Retardo mental, ceguera, muerte en el segundo o tercer año de vida
Gangliosido Hexosaminidasa
Gaucher Hepato y esplenomegalia, erosión de huesos largos y pelvis, retardo mental en formas infantiles
Glucocerebrosido Glucocerebrosidasa
Fabry Salpullido, dolores de extremidades inferiores,
Ceramida- trihexosidasa
galactosidasa

problemas renales.
Niemann-Pick Hepato y esplenomegalia, retardo mental.
Esfingomielina Esfingomielinasa
Krabbe Retardo mental, ausencia de mielina
Galactocerebrosido Galactocerebrosidasa
VITAMINAS
VITAMINA
CO-FACTOR DEFICIENCIA FUNCION BIOQUIMICA
Comentario
HIDROSOLUBLES
Tiamina (B1)
Pirofosfato de tiamina
Beri-Beri
-Descaxilizacion Oxidativa.
Riboflavina (B2)
FAD, FMN
Reacciones de oxidoreduccion
Niacina
NAD, NADP
Pelagra
Transfiere hidruros.
Pantoteno
Coenzima A
Transfiere grupos acilo
Prididoxina B6
Piridoxal Fosfato
Reacciones de carboxilizacion
La deficiencia causa convulsiones
Cobalamina B12
Metil-cobalamina
Anemia Perniciosa
Metilmanolil Factor extrinseco
Biotina
Carboxilasa de co-A
Folato
Tetrahidrofolato
Anemia
Biosintesis de purinas
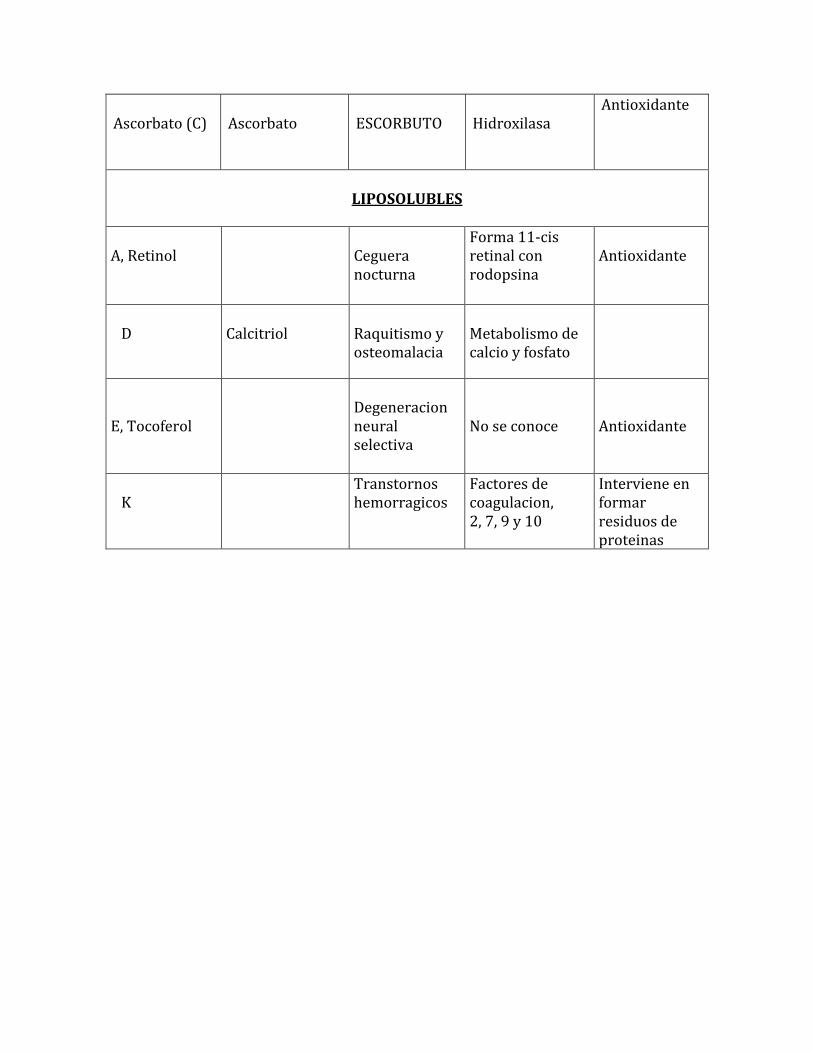
Ascorbato (C)
Ascorbato
ESCORBUTO
Hidroxilasa
Antioxidante
LIPOSOLUBLES
A, Retinol
Ceguera nocturna
Forma 11-cis retinal con rodopsina
Antioxidante
D
Calcitriol
Raquitismo y osteomalacia
Metabolismo de calcio y fosfato
E, Tocoferol
Degeneracion neural selectiva
No se conoce
Antioxidante
K
Transtornos hemorragicos
Factores de coagulacion, 2, 7, 9 y 10
Interviene en formar residuos de proteinas

BIOSINTESIS DE SALES BILIARES.
Las sales biliares se dividen en dos clases: primarias y secundarias.
La Primarias son sintetizadas por el hombre.
Las secundarias son sintetizadas por las bacterias, apartir de las primarias.
La biosintesis de sales biliares esta regulada por la : 7a-hidroxilasa de colesterol, en la cual se necesita NADPH, citocromo P-450, etc.
El desoxicolato y litocolato, son sales biliares SECUNDARIAS.
El colesterol no puede ser oxidado a dioxido de cabrono y agua. La unica manera de eliminacion es por heces, en forma de colesterol o sales biliares. El higado sintetiza alrededor de 500mg de sales biliares al dia.
La bilis se excreta en dudodeno y es abosorbido alrededor de un 94% , esta absorcion ocurre en ilieon en su mayoria. Luego estas sales deben de regresar al higado por medio de la circulacion entero-hepatica.
METABOLISMO DE ALCOHOL
El alcohol es una fuente considerable de energia, (calorias vacias) lo que favorece ala desnutricion y la carencia de vitaminas sobre todo las del B
El exceso de NADH contritubuye ala acidosis, reducion de acido urico, inhibe la oxidacion de acidos grasos, con efectos secundarios en el higado.
El metabolismo del alcohol se lleva acabo en HIGADO, por medio de sus peroxisomas y termina en mitocondria.
Enzimas que actuan: Alcohol deshidrogenasa y acetaldehido deshidrogenasa.

SINDROME DE CUSHING
El sindrome de cushing es la expresion clinico de una exposicion excesiva y prologngada a la accion de los glucocorticoides. Es mas comun en mujeres.
Causas;
Hipersecrecion cronica de ACTH (Enfermedad de cushing hipofisiaria) (debida a un adenooma)
Hiperplasia suprarrenal congenita
Iantrogenico
Manifestacion clinica:
Obesidad central (90%), deposito de grasa preferente en peritoneo, abdomen, mediastinoy tejido subcutaneo de cara y cuello. Secundaria a mayor densidad de receptores glucocorticoides a dichos niveles.
*Deposito abdomina: obesidad centripeta.
*Deposito en mejillas: cara de luna
*Depositos en fosa supraclavicular y area dorso cervical: “giba”
Piel : Disminuye division epidermica y sintesis de colageno, hay atrofia de epidermias y tejido conectivo
*Adelgazamiento de piel
*Equimosis: por trauma minimo, con hematomas espontaneos (secundaria ala atrofia de tejido conectivo)
*Estrias rojo-vinosas:, en abdomen principalmente, caderas, muslos, gluteos.
*Hiperpigmentacion: solo si la ACTH esta alta.
Afectacion musculo esqueletica:
*Debilidad muscular, sobre todo en musculatura proximal
* Supresion marcada en la velocidad de crecimiento
* Osteoporosis
Afectacion cardio-vascular:
*Hipertension

*Insuficiencia cardiaca.
Alteraciones endocrino-metabolicas
*Intolerancia ala glucosa o diabetes.
*Habra alteraciones hidroelectroliticas.
Tratamiento:
Ketoconazol: inhibe diferentes enzimas del citocromo P-450
Metapirona: inhibe la 11-beta hidroxilasa
Aminoglutetimida: actua inhibiendo la esterodoigenesis.
Generalidades:
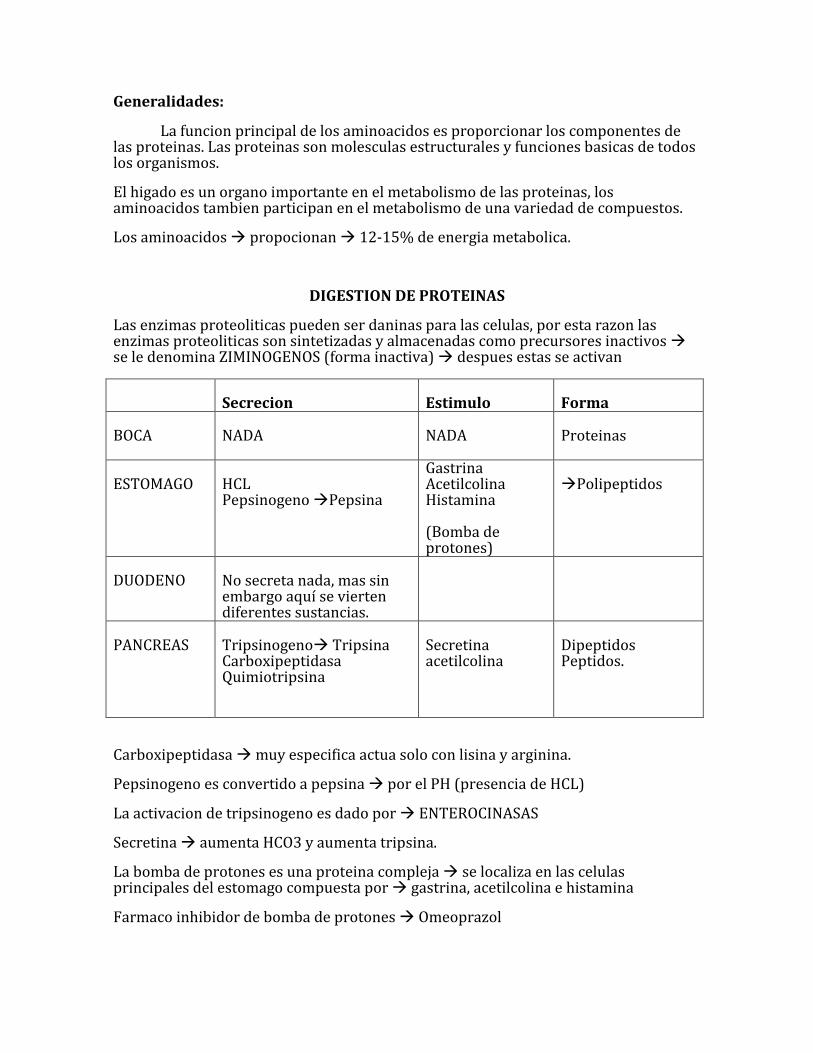
La funcion principal de los aminoacidos es proporcionar los componentes de las proteinas. Las proteinas son molesculas estructurales y funciones basicas de todos los organismos.
El higado es un organo importante en el metabolismo de las proteinas, los aminoacidos tambien participan en el metabolismo de una variedad de compuestos.
Los aminoacidos propocionan 12-15% de energia metabolica.
DIGESTION DE PROTEINAS
Las enzimas proteoliticas pueden ser daninas para las celulas, por esta razon las enzimas proteoliticas son sintetizadas y almacenadas como precursores inactivos se le denomina ZIMINOGENOS (forma inactiva) despues estas se activan
Secrecion
Estimulo
Forma
BOCA
NADA
NADA
Proteinas
ESTOMAGO
HCL Pepsinogeno Pepsina
Gastrina Acetilcolina Histamina (Bomba de protones)
Polipeptidos
DUODENO
No secreta nada, mas sin embargo aquí se vierten diferentes sustancias.
PANCREAS
Tripsinogeno Tripsina Carboxipeptidasa Quimiotripsina
Secretina acetilcolina
Dipeptidos Peptidos.
Carboxipeptidasa muy especifica actua solo con lisina y arginina.
Pepsinogeno es convertido a pepsina por el PH (presencia de HCL)
La activacion de tripsinogeno es dado por ENTEROCINASAS
Secretina aumenta HCO3 y aumenta tripsina.
La bomba de protones es una proteina compleja se localiza en las celulas principales del estomago compuesta por gastrina, acetilcolina e histamina
Farmaco inhibidor de bomba de protones Omeoprazol

Farmaco inhibidor de histamina (de la bomba de protones) Ranitidina la cual inhibe los receptores H2 de histamina.
Organos que ocupan mas proteinas higado, cerebro y musculo.
TRANSAMINACION
Es el proceso en el cual es transferido un grupo amino a otro compuesto, esta accion es catalizada por las transaminasas.
Ejmeplo:
Si ha glutamato le donamos un grupo amino se convierte en GLUTAMINA
Si a a-cetoglutarato le donamos un grupo amino se convierte en GLUTAMATO.
DESAMINACION
Es el proceso en el cual se le quita un grupo amino a un compuesto, esto es regulado por las desaminasas
Ejemplo
Si ha glutamina le quitamos un amino GLUTAMATO.
TRANSDESAMINACION
Son los dos procesos juntos.
Cetoacido corresponde a un aminoacido sin su grupo amino, solo con COOH
Ejemplo:
Glutamato a-cetoglutarato reaccion catalizada por glutamato deshidrogenasa ese grupo AMINO que se libera es llevado a ciclo de urea y la cadena de cabrono al ciclo de krebs. glucogenico.
Acido piruvico Alanina
AMINOACIDOS, CARACTERISTICAS Y CLASIFICACION
Los aminoacidos como sabemos son 20, de los cuales 9 son esenciales y 11 son no esenciales.
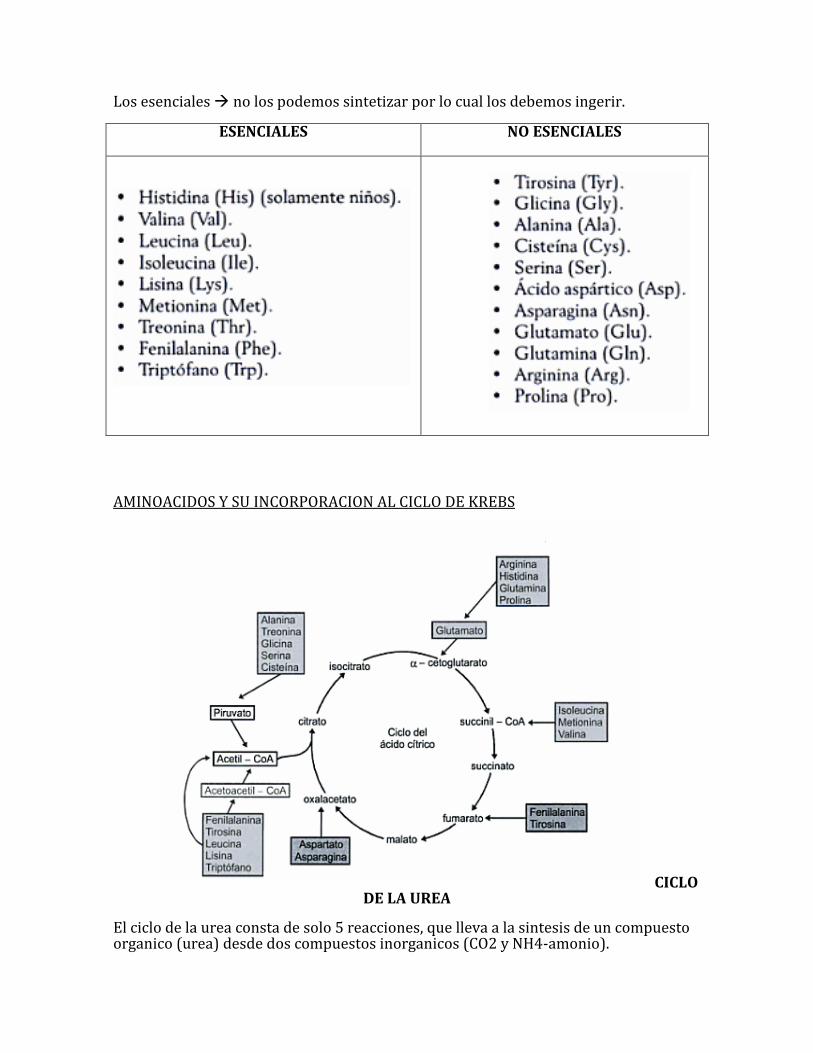
Los esenciales no los podemos sintetizar por lo cual los debemos ingerir.
ESENCIALES NO ESENCIALES
AMINOACIDOS Y SU INCORPORACION AL CICLO DE KREBS
CICLO DE LA UREA
El ciclo de la urea consta de solo 5 reacciones, que lleva a la sintesis de un compuesto organico (urea) desde dos compuestos inorganicos (CO2 y NH4-amonio).

Este se lleva acabo en los hepatocitos celulas periportales.
Tiene lugar en mitocondria y en citosol .
1.- Formacion de carbamoil fosfato
- El paso es irreversible y esta dado por la carbamoil fosfato sintetasa l, es una enzima mitocondrial es la enzima limitante.
2.- Formacion de citrulina
- Grupo carbomil se transfiere a ornitina ornitina transcarbamilasa
3.- Sintesis de arginosuccinato
- Aquí se realiza la conversion de citrulina con acido aspartico
4.- Argininosuccinato
- Aquí se convierte el agininosuccinato a Arginina y Fumarato
5.- Arginina a ornitina y ura.
- La arginina es especifica del higado, lo cual se produce urea.
GLUCOGENICOS GLUCOGENICOS Y CETOGENICOS

CETOGENICOS Glicina
Isoleucina
Leucina
Lisina
Alanina Cisteina Aspartato
Fenilalanina Asparagina Glutamato Glutamina
Tirosina Prolina Histidina
Triptofano Arginina Metionina
Treonina Valina
GLICINA
Aminoacido mas simple
se sintetiza a partir de serina
No esencial
Es glucogenico la serina produce piruvato
Necesita acido folico y lo puede sintetizar tambien
Produce el grupo HEMO de la hemoglobina porfirinas
es un neurotrasmisor
ayuda en la sintesis de purinas

ALANINA
es glucogenico se va a piruvato
No esencial
es un cetoacido de piruvato
participa en ciclo de cori glucoNEOgenesis
es transportador de amonio al higado
LEUCINA
Esencial
cetogenico Enf. De miel de maple
acetoacetil-coA
Cadena ramificada
VALINA
es de cadena ramificada
esencial
glucogenico - entra en succinil-CoA
enfermedad de miel de maple ISOLEUCINA
Glucocetogenico
Forma acetoacetil coA
Esencial
su parte glucogenica se va a succinil-CoA
es de cadena ramificada
FENILALANINA
Esencial
Su unica funcion es formar tirosina
Glucocetogenico
Forma fumarato y acetoacetil-CoA

Aromatico
TIROSINA Forma hormona tiroidea encargada del “metabolismo”
Forma dopamina, adrenalina, noradenalina
La noradrenalina se forma gracias al SAM , dona grupos metilos
Acido vanidilmandelico y metanefrinas asi se eliminan por orina las
catecolaminas
Forma melanina ACTH ayudan a la formacion y MSH
No esencial
glucocetogenica, produce fumarato ya acetoacetato.
Sintetiza melatonina en conjunto del triptofano.
Aromatico
TRIPTOFANO Forma alanina
Formador de serotonina receptores 5TH1, 5TH2, 5HT3, 5TH4, 5TTH5
forma tambien melatonina
Aromatico
Cetoadipato
CISTEINA No esencial Es estructural forma puentes Disulfuro en proteinas terciarias forma taurina
Metionina
Sintetiza homocisteina
Forma el SAM
Se utiliza en sintesis de fosfatidilcolina
Dona grupos metilos en adrenalina – noradrenalina
Forma creatina

es glucogenico se va a succinil-CoA
forma cisteina
Es esencial
Homocisteina
Se ve relacionado con problemas cardiovasculares
Actua junto con la B12
Si no hay B12 aumenta homocisteina
SI no hay SAM no hay noradrenalina, mielina, creatina
metionina + lisina carnitina
LISINA
cetogenico
forma acetoacetil-CoA
junto con metionina carnitina
Produce GH hormona del crecimiento
Cetoadipato
GLUTAMATO
Glutamina principal transportador de amonio Ciclo de Urea
Forma a-cetoglutarato en el CK
Forma GABA
Aminoacidos formadores de glutamato
Histidina, prolina, asparatato, glutamina, arginina
ASPARTATO
se forma desde oxalacetato es su cetoacido
En ins hepatica esta elavado
BUN estructuras no medibles por si solas amonio

PROLINA
Forma colageno, vitamina C
Hidroxiprolina marcador oseo relacionado con osteoporosis
Histidina
Forma histamina
Histidinemia patologia mas comun .
ARGININA
Forma Oxido Nitrico, NO Sintetasa, necesita citrulina
Forma urea
Forma creatina
ASPARAGINA
Forma aspartato
SERINA
Forma fosfatidilserina
Forma esfingolipidos
Se va a piruvato
No esencial
TREONINA
Es glucocetogenico
Forma succinil-CoA
Sirve para la formacion de elastina
BIOSINTESIS DE PURINAS
Las purinas corresponden alas bases nitrogenadas, Guanina y Adenina, se consideran purinas ya que contienen doble anillo en su estructura.
Estas bases nitrogenadas estan presentes en ARN y ADN.

Funciones de las purinas:
- Formadoras de ADN y ARN - Moleculas de alta energia ATP y ADP - Mediadores celulares cAMP y cGMP - Componentes de coenzimas NAD, FAD - Portadores de intermediarios UDP-glucosa - Efectores alostericos Regula vias metabolicas cantidades altas de ATP
inhiben glucolisis y activa otras vias por ejemplo.
La biosintesis de purinas se debe llevar a cabo previo a ala division celular, en la interfase, en la fase S se produce la biosintesis.
Las celulas que mas producen mitosis habra mas biosintesis
En pacientes que tienen algun cancer, se le s da inhibidores de esta via como puede ser
- Metotrexato inhibidor de tetrahidrofolato., actua a nivel de la dihidro reductasa
SUSTRATOS NECESARIOS
- Glicina - Glutamina sale como glutamato en la via (donador de NH3) - Aspartato - C02 - Acido folico en forma de Tetrahidrofolato - Ribosa 5-fosfaton proviene de via de pentosas en forma de PRPP
PRPP fosforibosilpirofosfato dona la pentosa en esta biosintesis
Formacion de PRRP
Ocurre atraves de la enzima PRPP sintetasa.
Un aumento de PRPP es un activador de la sintesis.
¿Cómo se controla la sintesis de purinas?
-Enzima reguladora PRPP amido transferasa es la enzima reguladora de la sintesis.
- PRPP alto, activa la via.
- Cantidades altas de IMP son inhibidores de la via.

Como producto final de la interaccion de los sustratos se necesita
- I glicina, 5 ATP, 2 glutamina, 1 C02, 2 formiatos, 1 aspartato IMP - En la produccion se libera un fumarato.
El intermediario de la sintesis es IMP, este se convierte en AMP y GMP
DEGRADACION DE PURINAS
Un exceso de purinas generara una degracion, un exceso de purinas es activador de la PURINA NUCLEOTIDO FOSFORILASA degrada las purinas
El AMP se degrada a HIPOXANTINA y luego a XANTINA ACIDO urico
El GMP se degrada directo a XANTINA ACIDO URICO
La enzima encargada de producir el paso a acido urico es XANTINA OXIDASA.
RECAPTACION DE PURINAS
Las purinas se pueden recaptar, la enzima encargada de la recaptacion es la HGPRTasa, en cuando las bases vuelven a ser IMP una patologia con una disminucion de esta ezima Leschn Nyhan presentan hiperuricemia (tipo gota)
ENFERMEDAD DE GOTA
Es una enfermedad relacionado con una sobreproduccion de purinas es una enfermedad ligada al cromosoma X
Esta enfermedad es caracterizada por una hiperuricemia se divide en gota primaria y secundaria secundaria corresponde a ins.renal, dano hepatico, etc.
La deficiencia primaria es de tipo congenita, causas:
- Deficiencia de la retrohibicion de la PRPP amido transferasa - Aumento de la actividad de la PRPP sintetasa - Deficiencia de la glucosa 6- fosfatasa.
Manifestaciones clinicas
- Presentan artritis por depositos de uratos (cristales de acido urico)

o Presentan generalmente en dedo gordo del pie a esto se le conoce como podagra.
- Al deposito masivo de uratos TOFOS - Leucocitos tratan de fagocitar quimiotaxis inflamacion y dolor. - Produce nefropatia por obstruccion nefropatia gotosa
Farmacos utilizados para gota
Coquicina se da en gota inflamatoria, es inhibidor de la fagocitosis, y eh inhibe la division celular en metafase retraccion del huso acromatico
Alopurinol Inhibidor de la xantina oxidasa, este famarco se para que no se produzca acido urico y quede en xantina xantina es mas soluble.
Mercaptopurinas inhibidores de enzimas especificas para produccion de pirimidinas y purinas.
BIOSINTESIS DE PIRIMIDINAS
Las pirimidinas corresponden a las bases nitrogenas que contienen un solo anillo de nitrogeno en su estructura corresponden a CITOSINA, URACILO Y TIMINA (CUT). Las pirimidinas no se sintetizan como nucleotidos se sintetizan como NUCLEOSIDO La unica diferencia es que los nucleosidos no tienen el grupo PO4. La biosintesis empieza a partir de bicarbonato+amonio y asparato Sustratos
- Bicarbonato - Amonio, NH4 es cual es donado por GLUTAMINA - Aspartato - PRPP donador de ribosa
La primera enzima que debe actuar y la cual es reguladora de la biosintesis de pirimidinas es Carbamoil Fosfato Sintetasa ll
El primer ciclo que se forma en la biosintesis es OROTATO pero el intermedario para biosintesis de pirimidinas es UMP.
Cantidadades elevadas de UMP inhibe la sintesis
La division celular y el PRPP alto Activa.

ENFERMEDAD DE LESCH – NYHAN
Es una enfermedad ligada al cromosoma X se trasmite de forma recesiva, mas comun en hombres que en mujeres
Esta enfermedad de trata de un transtorno por una hiperuricemia y problemas neurologicos
Esta enfermedad se ve ligada a deficiencia pacial o comepleta de la hipoxantina-guanina- fosforribosiltransferasa (HGPRTasa).
Al estar incontrolada esta enzima no se lleva acabo la recaptacion de purinas, por lo que se realiza una sobreproduccion de IMP es cual es llevado a acido urico y presentan URICEMIA.
Manifestaciones clinicas: son muy similares a gota
- Formacion de tofos - Formacion de critales de acido urico en articulaciones - Nefropatias obstructiva - Deficiencia neurologica explicada por un dano en ganglios basales que
se explican por problemas dopaminergicos y de otros neurotrasmisores. - Automutilacion - No viven despues de los 20.
Tratamiento:
- Alopurinol - Diazepam.
ENFERMEDAD DE HARTNUP
Es una enfermedad genetica autosomica recesiva Resulta de un transtorno en la absorción gastrointestinal de aminoácidos neutros , (valina, isoleucina, leucina, tirosina, fenilalanina y triptófano) Es causado por un defecto en el transporte de aminoácidos en i.delgado y rinones El dx depende de encontrar hiperaminoaciduria La poca capacidad de absorber triptófano conduce a menudo a las 3D de pelagra dermatitis, demencia y diarrea. Estos sintomas son por lo general respodientes de niacina tx Niacina

FENILCETONURIA La forma mas comun de esta patologia es causada por una deficiencia de la fenilalanina hidroxilasa Esta es una enfermedad autosomica recesiva Ligada a cromsoma X. 12q. El aumento de fenilalanina en sangre produce un transtorno osmotico a nivel de la membrana de diferentes aminoácidos Cuadro clinico
- Convulsiones - Albinismo - Fotosensibilidad - Olor a heces de raton
En esta patologia se ven afectados procesos como
- Producción de tirosina o Si no se produce tirosina no hay
Dopamina Adrenalina Noradrenalina Tiroxina Melanina Melatonina
ALCOPTONURIA
Es una enfermedad rara hereditaria, causada por la deficiencia de homogentisato dioxigenasa. El homogentisato, producido durante el metabolismo de la fenilalanina y de la tirosina, no puede ser metabolizado. Este es secretado en orina. La orina se vuelve gradualmente oscura, cuando el acido es oxidado por oxigeno disuelto par producir compuesto de color obscuro. Los individuos con esta patologia presentan en tejido conectivo OCRONOSIS. Triada de alcoptonuria
- Ocronosis - Orina oscura - Artritis
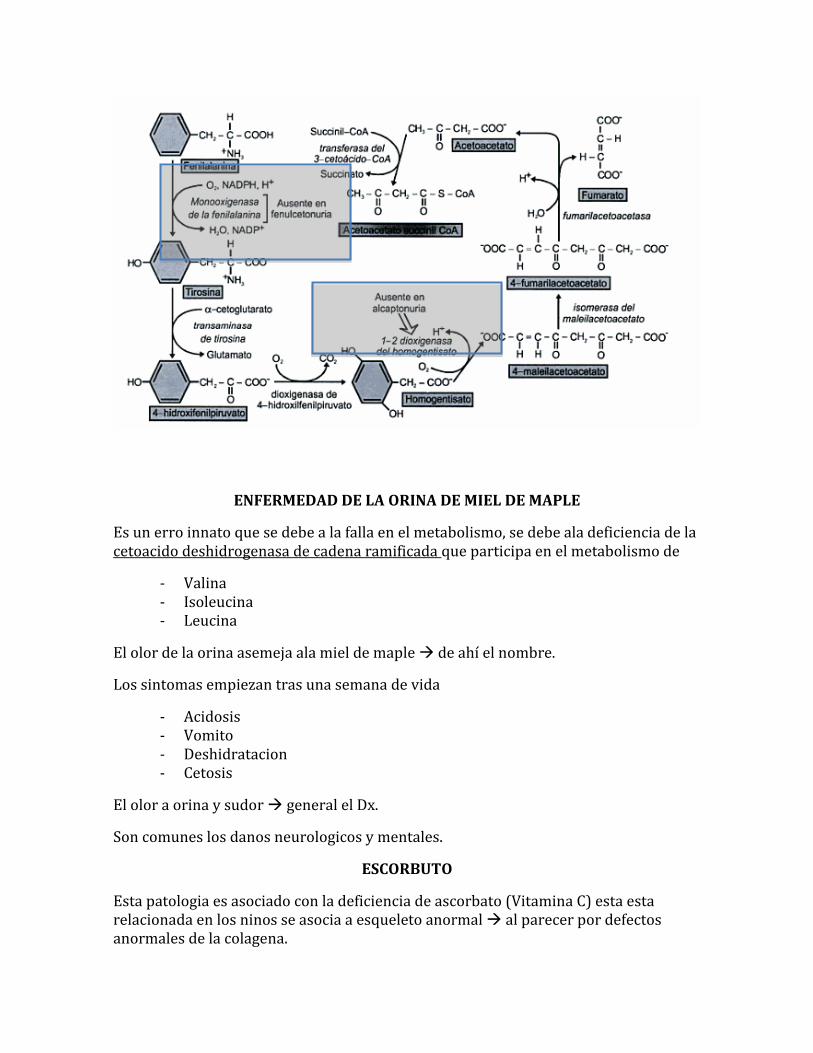
ENFERMEDAD DE LA ORINA DE MIEL DE MAPLE
Es un erro innato que se debe a la falla en el metabolismo, se debe ala deficiencia de la cetoacido deshidrogenasa de cadena ramificada que participa en el metabolismo de
- Valina - Isoleucina - Leucina
El olor de la orina asemeja ala miel de maple de ahí el nombre.
Los sintomas empiezan tras una semana de vida
- Acidosis - Vomito - Deshidratacion - Cetosis
El olor a orina y sudor general el Dx.
Son comunes los danos neurologicos y mentales.
ESCORBUTO
Esta patologia es asociado con la deficiencia de ascorbato (Vitamina C) esta esta relacionada en los ninos se asocia a esqueleto anormal al parecer por defectos anormales de la colagena.

En adultos desarrollan hemorragias llamadas equimosis y petequias
Presentan debilidad, inflamacion de las encias y aflojamiento de los dientes, El almacenamiento de vitamina c dura 3 a 4 meses.
ENFERMEDAD DE PARKINSON
Este transtorno se caracteriza por marcha o andar tambaleante, temblor, rigidez y se debe ala degeneracion de las neuronas de la via dopaminergica nigroestriada del encefalo. Ocurre un 70% de disminucion de dopamina.
Los individuos afectados son tratados amenudo con inhibidores de la MAO-B, como el deprenil.
la inhibicion de la MOA-B, se encarga de la catalizacion de la dopamina (y otras aminas biogenicas), esto hace un aumento de la concentracion eficaz de la dopamina.
La enfermedad de parkinson tambien es trataa con administracion bucal de L-Dopa.
Esta sustancia es transportada atraves de la barrera hematoencefalica y sirve como sustrato para la biosintesis de dopamina en las celulas estriadas.
L-Dopa se cataliza fuera del cerebro por los aminoacidos aromaticos se da carbidopa esta no cruza barrera hematoencefalica.
Aumento de dopamina esquizofrenia.
LEVOTIROXINA
La levotiroxina es el principal componente de la secrecion tiroidea, del que depende el buen funcionamiento del tiroides.
Un gran numero de pacientes con hipotiroidismo L-tiroxina
Su efecto consiste en incrementar el metabolismo basa y el consumo de oxigeno, se trata de un profarmaco que se metaboliza a T3.
ENCEFALOPATIA HEPATICA
Este es un sindrome neuropsiquiatrico complejo secundario a una insuficiencia hepatica y a cortocircuitos vasculares. Es una de las complicaciones mas frecuentes de la cirrosis hepatica.
Fisiopatologia:

Es causado por una hiperamonemia esto es causado por que no se lleva acabo el ciclo de la urea, por lo que el amonio se estanca y no se produce ura.
El aumento de amonio en sangre es toxico para el SNC.
El aumento de amonio produce un aumento de la osmolaridad, por lo cual los astrocitos son los principales afectador esto produce el edema.
Como hay aumento de amonio el a-cetoglutarato se junta con amonio Glutamato este hace lo mismo y forma Glutamina.
El proceso anterior indica que porque no se lleva el ciclo de kreb completo provocara un desceso del ATP Por lo cual se presentara somnolencia , convulsiones , etc.
Como sabemos el amonio es de carácter acido presentan acidosis
Presentan asterexis, ya que al glutamato convertirse en glutamina no se produce GABA , el cual es un neurotrasmisor inhibitorio.
Tratamiento:
- Lactulosa: Es fermentado por bacterias el cual cambia el pH del la luz intestinal No se absorbe amonio.
- Hemodialisis - Antibioticos No absorbibles, matan bacterias del tracto (bacterias
producen amonio). o Neomicina o Paramomicina
ANEMIA MEGALOBLASTICA
Hay dos tipos de anemias megaloblasticas: una causada por deficiencia de folato y otra por una carencia de la vitamina B12.
Ambas vitaminas son necesarias para la sintesis de ADN por lo tanto sus efectos sobre su carencia sobre la eritropoyesis son muy similares.
La caracteristica morfologica de esta anemia es el aumento del tamano de los precursores eritroides (megaloblastos) que producen eritrocitos exageradamente grande (macrocitos).
El trastorno subyacente al gigantismo celulas es una alretacion de la sintesis de ADN que provoca un retraso de la maduracion nuclear y de la division celular.
Como la sintesi de ARN y sus elementos prosigue a su ritmo normal superan el crecimiento del nucleo, entonces se muestra asincronia nucleo-citoplasma
Anemia por Vitamina B12 Anemia Perniciosa.

RELACION ACIDO FOLICO – VITAMINA B12
La produccion de tetrahidrofolato esta ligada ala vitamina B12, dado que el folato se cataboliza, el ciclo requiere una fuente, que es 5-Metiltetrahidrofolato. Este libera un grupo metilo, que forma el tetrahidrofolato y lo cerde para destoxificar la homocisteina y la convierte METIONINA.
El cofactor que transfiere el grupo metilo es Vitamina B12.
HIDROXIPROLINA
La hidroxiprolina representa el 13% del contenido de aminoacidos en la molecula de colageno. Esta presente en otras estructuras similaes.
La hidroxiprolina liberada durante la degradacion del colageno no puede ser reutilizada en una nueva forma por lo que refleja la RESORCION OSEA.
El 95% de hidroxiprolina esta en tejido oseo la determinacion de hidroxiprolina puede ser atraves de orina el cual significa marcador oseo osteoporosis.

DEPRANOCITOSIS
O conocida también como anemia falciforme.
Es causada por una mutacion puntual unica en el gen que codifica la cadena B-globina de la
hemoglobina A. El resultado de esta mutacion es una cadena de B-globina anormal
El dano esta en que el aminoacido valina ah remplazado al aminoacido glutamato
Los pacientes con depranocitos por lo general cursan con cuadros de hemolisis ya que estos
eritrocitos son mas fragiles, cursan con obstrucciones de vasos y cursan con apoplejia (ACV)
Este es un transtorno genetico homocigoto recesivo.
ENZIMAS HEPATICAS
ALT Alanina- aminotrasaminasa, es una enzima hepatica, se eleva en procesos agudos como
hepatitis, y se disminuye en procesos cronicos como alcoholismo
La enzima cataliza la transferencia de un grupo amino desde la alanina al alfa-cetoglutarato,
los productos de esta transaminación reversible de piruvato y glutamato.
glutamato + piruvato ⇌ α-cetoglutarato + alanina
AST Aspartato-aminotransaminasa se incrementa en IAM, hepatopatia aguda
Esta enzima cataliza la reacción de transferencia de un grupo amino desde el L-aspartato al 2-
oxoglutarato formándose L-glutamato y oxaloacetato
L-aspartato + 2-oxoglutarato oxaloacetato + L-glutamato
NEUROTRASMISORES Y SUS RECEPTORES
Dopamina:
-Es un neurotransmisor catecolaminergico mas importante del SCN, participa
en la regulación de la conducta motora, emotividad, y la afectividad. Se sintetiza a
partir de tirosina.
Su biosíntesis es en células estriadas de la sustancia nigra y en medula suprarrenal.
Receptores : Se le conocen al menos 5 D1, D2, D3, D4, D5. acoplados a Proteína G

Familia de receptores D1 Incluye D1 y D5
Estimulan la formación de cAMP.
Familia de receptores D2 Incluye D2, D3, D4
Inhiben formación de cAMP, activan canales de K+ y educen entrada de ion Ca+
GABA: Es el neurotransmisor inhibitorio central deriva del glutamato
La glicina tiene un efecto similar, solo que solamente en ME.
La células gabanergicas: abundan en ganglios basales.
Hay fármacos depresoras del SNC que aumentan gaba Benzodiacepinas
Actuan uniendose a una localizacon reguladora especifica del receptor GABA y
potenciando el efecto inhibidor gaba
Diazepam Conlvusiones, epilepsia, ansiedad
Serotonina: o conocida como 5-hidroxitriptamina es una amina que se localiza
fundamentalmente en SNC, pero tambien esta en gastrointesinal y plaquetas, es
sintetizada a partir de triptófano MAO la metaboliza.
SNC sensibilidad al dolor, sueno, tono postural
Vasos vasocontriccion en vasos del SNC.
Inhibidores de la recaptura de serotonina Receptores 5HT1-2-3-4-5
Fluoxatina inhibe la recaptación, se utiliza como antidepresivo., en hombres causa perdida
del libido y no producen ereccion.
5TH2 Procineticos mueven musculo liso para mover intestino Cinitrapida
5TH3 regula canales Na y K causa nausea andrositron (antagonista)














![Guia de Lab Oratorio de Bioquimica 2011 Salud]](https://static.fdocuments.ec/doc/165x107/5571fac64979599169931548/guia-de-lab-oratorio-de-bioquimica-2011-salud.jpg)



