Perspectives terapèutiques a la malaltia d Alzheimer · Perspectives terapèutiques a la malaltia...
53
1 Perspectives terapèutiques a la malaltia d’Alzheimer Barcelona 18 de Febrer 2017 Dr. Alberto Lleó Unitat de Memòria Servei de Neurologia Hospital de Sant Pau Barcelona
Transcript of Perspectives terapèutiques a la malaltia d Alzheimer · Perspectives terapèutiques a la malaltia...

1
Perspectives terapèutiques a la
malaltia d’Alzheimer
Barcelona 18 de Febrer 2017
Dr. Alberto Lleó
Unitat de Memòria
Servei de Neurologia
Hospital de Sant Pau
Barcelona
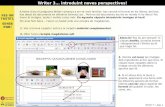
1906 Dr. Alois Alzheimer primera descripción de enfermedad
Auguste D. Alois Alzheimer
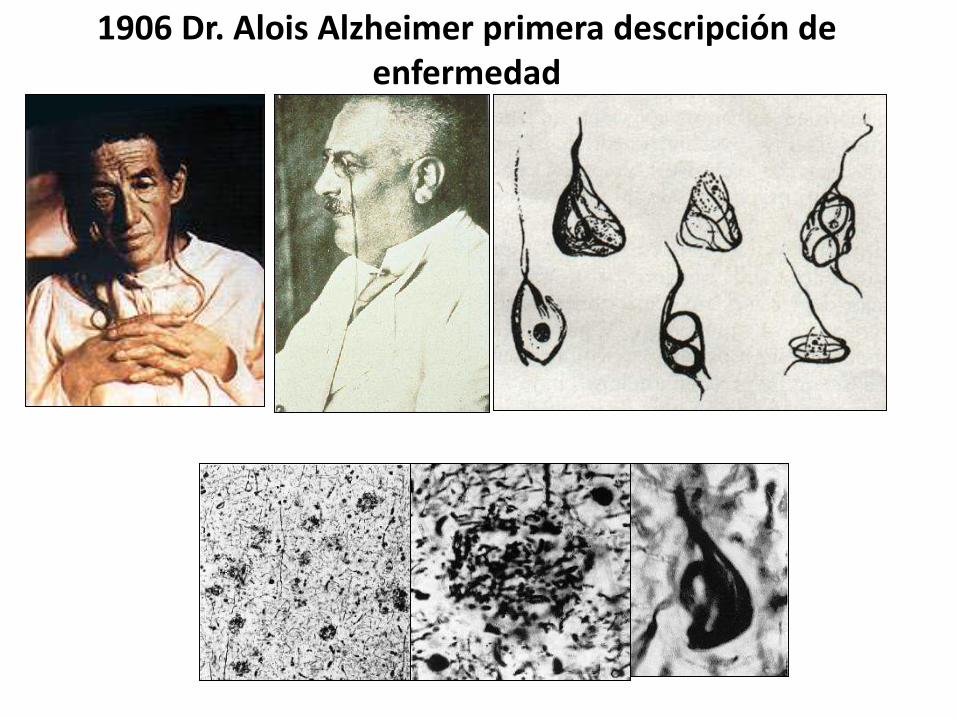
La demencia es el problema de salud más importante
en el siglo XXI
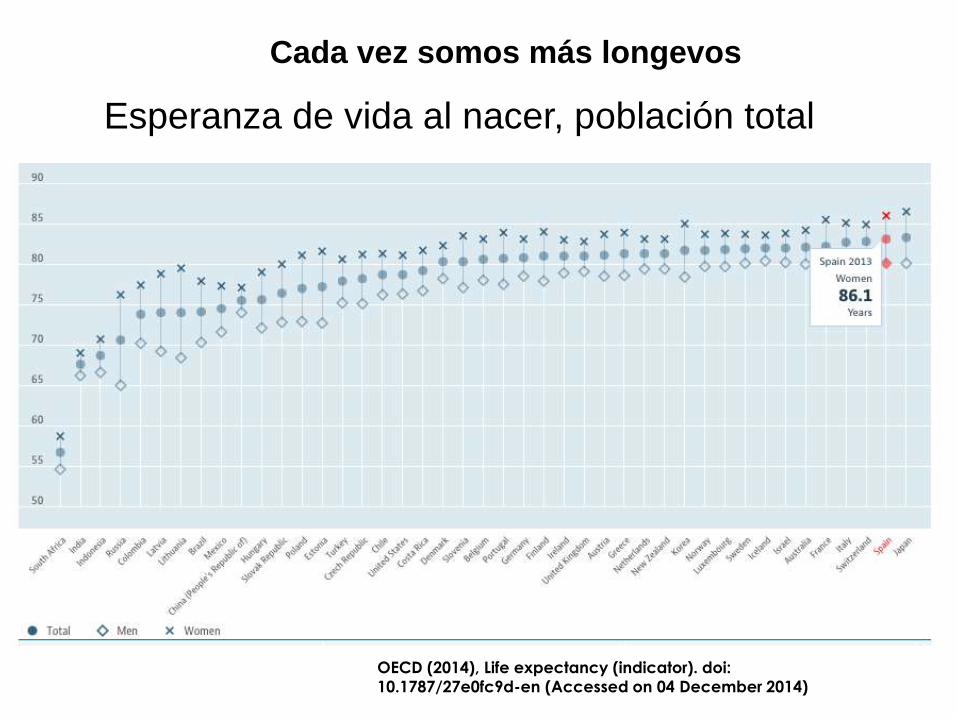
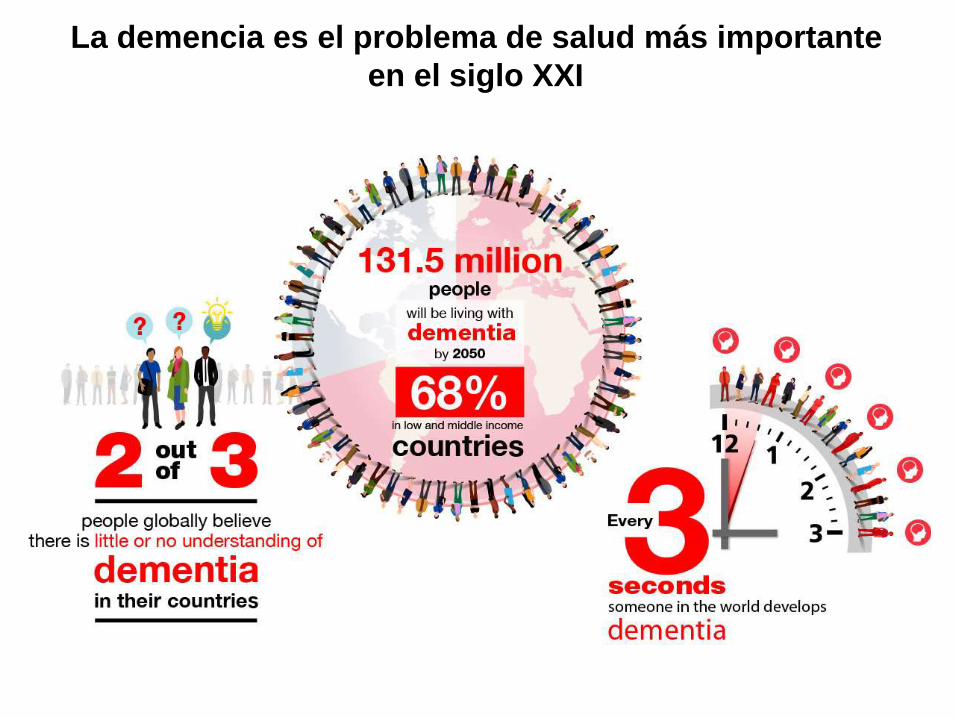
OECD (2014), Life expectancy (indicator). doi:
10.1787/27e0fc9d-en (Accessed on 04 December 2014)
Esperanza de vida al nacer, población total
Cada vez somos más longevos
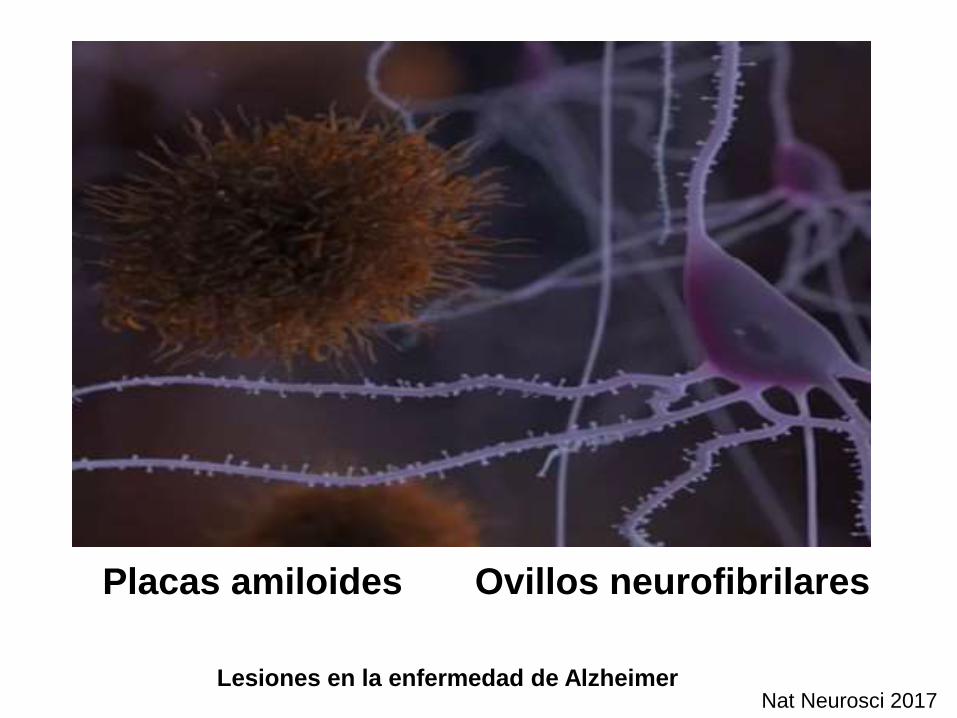
Lesiones en la enfermedad de Alzheimer
Placas amiloides Ovillos neurofibrilares
Nat Neurosci 2017

“El diagnóstico no se puede realizar por test de laboratorio”
“Tests son importantes para identificar otras causas de
demencia”
EA se conceptualiza como demencia
1984 Criterios Alzheimer NINCDS-ADRDA
McKhann, Neurology 1984;34:939-44

2011 – Criterios NIA-AAS

Sd. amnésico característico
(tipo hipocampal)
Memoria episódica
(recuerdo diferido)
Reconocimiento
No mejora con claves
CUADRO CLÍNICO
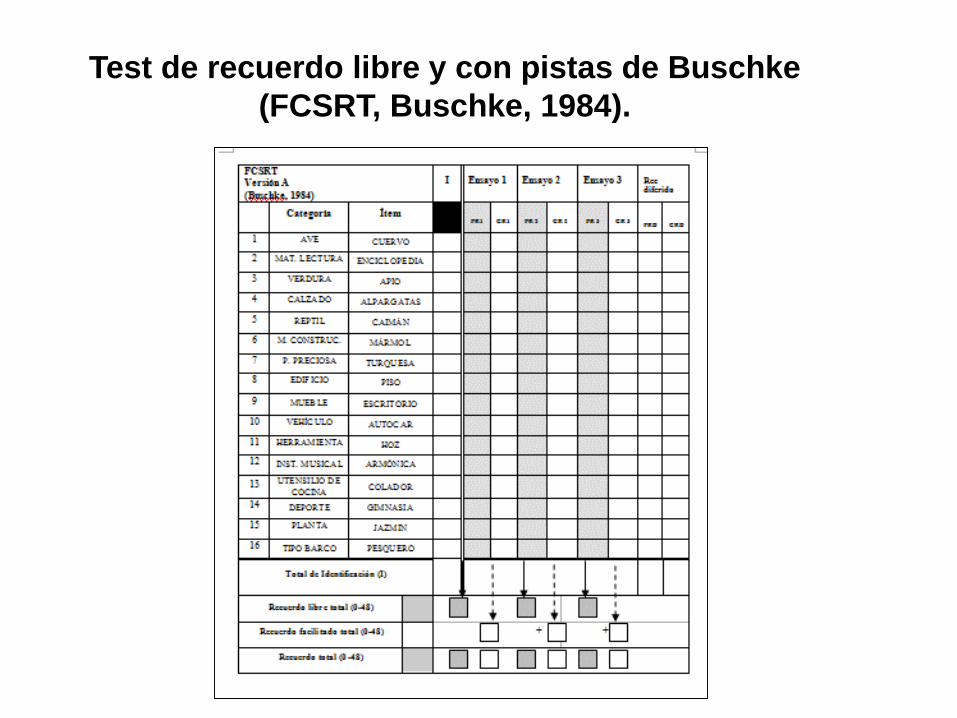
Test de recuerdo libre y con pistas de Buschke
(FCSRT, Buschke, 1984).
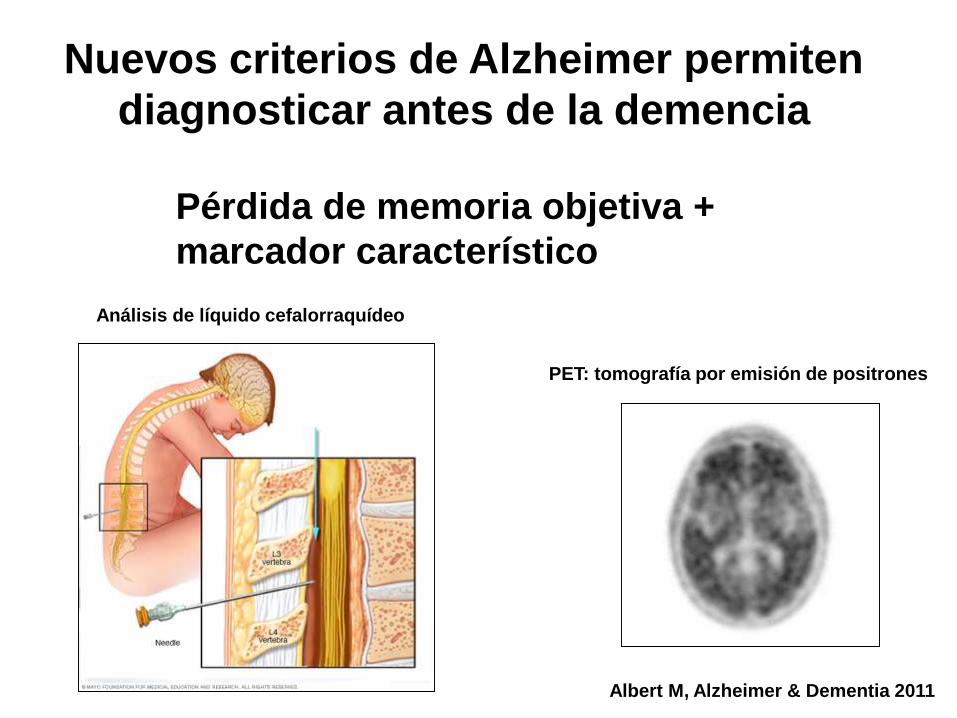
Nuevos criterios de Alzheimer permiten
diagnosticar antes de la demencia
Albert M, Alzheimer & Dementia 2011
Pérdida de memoria objetiva +
marcador característico
Análisis de líquido cefalorraquídeo
PET: tomografía por emisión de positrones
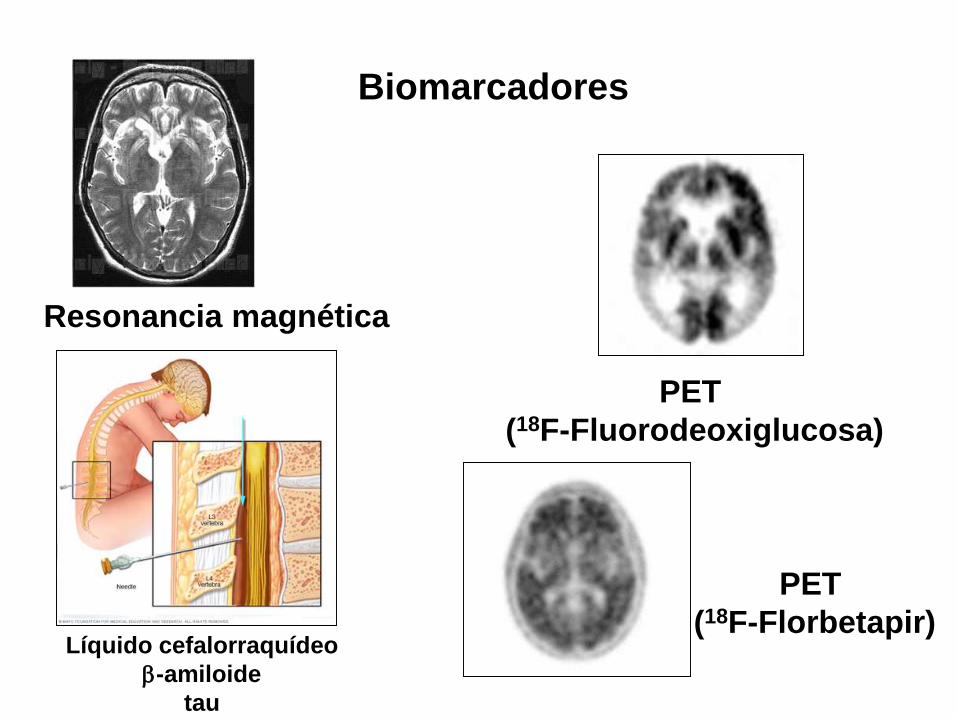
Biomarcadores
Líquido cefalorraquídeo
-amiloide
tau
PET
(18F-Florbetapir)
PET
(18F-Fluorodeoxiglucosa)
Resonancia magnética

70 años (médico jubilado)
Memoria (olvida nombres, algún
hecho reciente y más reiterativo).
Le cuesta visualizar rutas.
Vida normal.
MMSE: 30/30. FCSRT: Normal. Lista
CERAD: alterada.
Padre con Alzheimer a 79 años.
CASO CLÍNICO
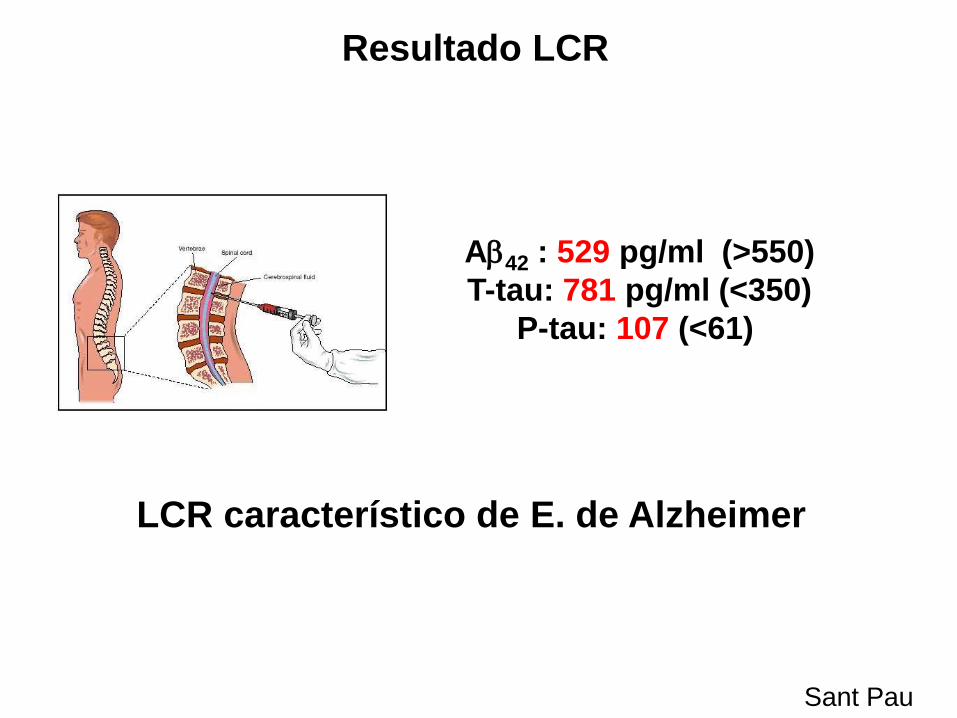
A42 : 529 pg/ml (>550)
T-tau: 781 pg/ml (<350)
P-tau: 107 (<61)
Resultado LCR
Sant Pau
LCR característico de E. de Alzheimer
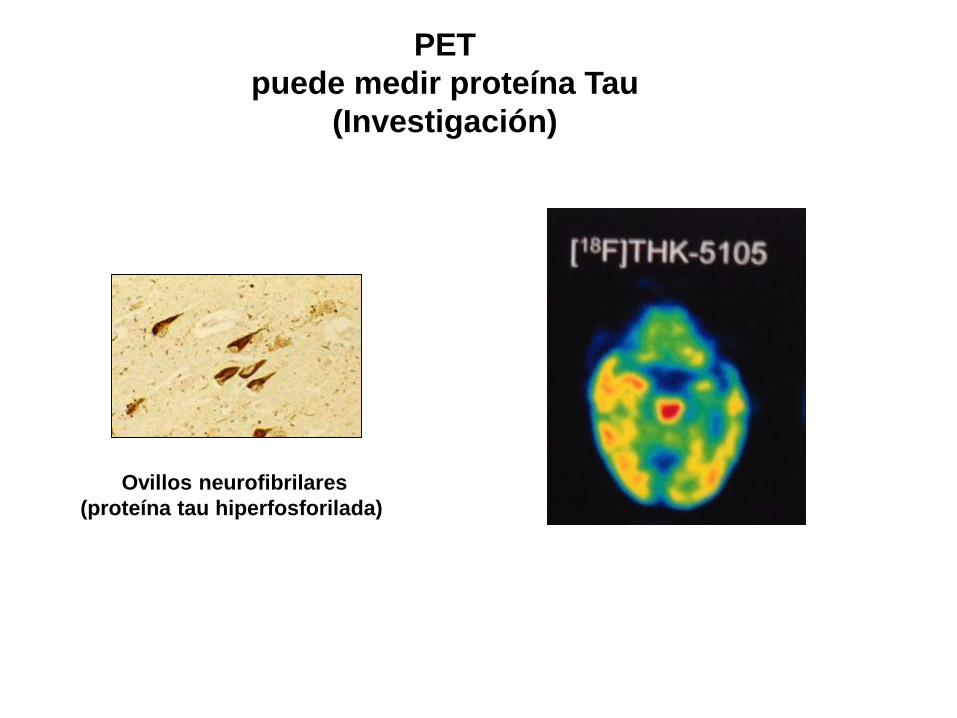
PET
puede medir proteína Tau
(Investigación)
Ovillos neurofibrilares
(proteína tau hiperfosforilada)
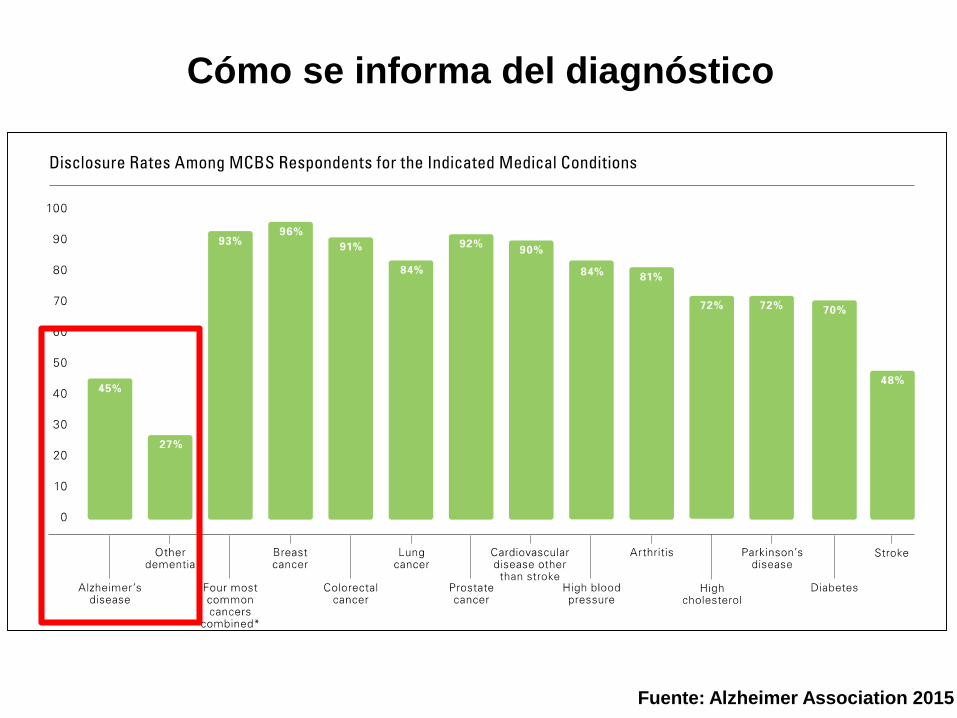
Cómo se informa del diagnóstico
Fuente: Alzheimer Association 2015

1976
Hipótesis colinérgica de la enfermedad de Alzheimer
disease1403
Letters to the Editor
SELECTIVE LOSS OF CENTRAL CHOLINERGIC
NEURONS IN ALZHEIMER’S DISEASE
SIR,-Alzheimer’s disease is a progressive cerebral degener-ation which continues without remission until death, usuallyin profound dementia. Morphologically, the disease is charac-terised by large numbers of senile plaques and neurofibrillarytangles in the brain, these tangles being especially abundant inthe cerebral cortex. Neurochemical studies are still in their in-
fancy, and we know nothing of the molecular basis of the dis-ease.
We have studied the enzymes associated with the putativeneurotransmitters acetylcholine, y-aminobutyric acid, dopa-mine, noradrenaline and 5-hydroxytryptamine in twenty re-
gions of brains obtained at necropsy from three patients withAlzheimer’s disease and from ten individuals who died without
evidence of neurological or psychiatric disorder. The brains
were removed 24-36 h after death, and each brain was
divided in half down the mid-line. The right half was used forbiochemical analyses, whilst the left half was fixed in formalinfor neuropathological examination. Alzheimer’s disease wasconfirmed histologically. There was no evidence of cerebrovas-cular disease in any of the thirteen cases. The ten controls
ranged in age from 46 to 74; the Alzheimer patients were aged61, 70, and 75. Tissues from the right half of the brain werestored at -190°C. Choline acetyltransferase (C.A.T.) and ace-
tylcholinesterase (A.C.E.) activities were measured by the
methods of Fonnum, and glutamic acid decarboxylase (G.A.D.)activity by the method of Roberts and Simonsen.2
C.A.T. activity in the Alzheimer’s disease brains was muchreduced in the amygdala, hippocampus, and cortex (table I).Only three Alzheimer brains have been studied but the extentof the reduction in these areas strongly suggests this is not achance occurrence. The activity of A.C.E. is dramaticallyreduced in the same areas of the cerebral cortex that show
reductions in C.A.T. activity (table II), and is below the levelsfound in the normal brains in all the other areas. The areas
of the cerebral cortex which show the maximum reductions in
c.A.T. and A.C.E. activity are those which contain the greatestdensity of neurofibrillary tangles.The reductions in the activity of the enzymes involved in the
metabolism of acetylcholine are not a result of non-specificdegenerative process. The activity of G.A.D. in all the areas ofthe Alzheimer’s disease brains studied appears to be well
within the normal range, means ranging from 74% to 121% ofcontrol activities. That this is the case in the cortical areas
which show large losses of c.A.T. and A.C.E. supports the notionthat a selective degenerative process has occurred. The normalvalues obtained for G.A.D. are of special significance because this
enzyme is particularly sensitive to ante-mortem hypoxia.3 It
seems unlikely, therefore, that the decreased activity of
enzymes associated with cholinergic transmission can be
ascribed to this cause; none of the patients with Alzheimer’sdisease had prolonged terminal hypoxic episodes.
Preliminary studies of tyrosine hydroxylase, aromatic amino-acid decarboxylase, dopamine-&bgr;-hydroxylase, and monoamineoxidase indicate no loss of these enzyme activities in Alz-
heimer’s disease and lend weight to the notion that a selectivedestruction of the cortical cholinergic system is an importantfeature of this condition.
We considered the possibility that selective loss of choliner-
gic system components could be due to prolonged drugregimens used in the patients with Alzheimer’s disease. How-
ever, there is no standard drug therapy for Alzheimer’s, and
1. Fonnum, F. Biochem. J. 1969, 115, 465.2. Roberts, E., Simonsen, D. E. Biochem. Pharmac. 1963, 12, 113.3. Bowen, D. M., Davison, A. N. in Biochemistry and Neurological Disease
edited by A. N. Davison); p. 2. Oxford, 1976.
TABLE I--CHOLINE ACETYLASE ([L mol/h/g WET WEIGHT)
TABLE II-ACETYLCHOLINESTERASE (MMOWG WET WEIGHT)
treatment is symptomatic. All three patients were given nitra-
zepam, but this drug was also given to five of the ten control
patients during their terminal illness. Opiates were adminis-tered to two of the Alzheimer patients and five of the con-trols, and phenothiazines were given to one and two patients,respectively. Thus no drug treatment was exclusive to the Alz-heimer’s disease patients, and it seems improbable that thedeficit in C.A.T. and A.C.E. activity in the cortex of these indi-viduals is drug induced.
Expression of results relative to protein, D.N.A., or R.N.A.
content does not alter the pattern of the results significantly.If these data can be confirmed in a larger series of cases the
concept of Alzheimer’s disease as a cholinergic system failure
may have important consequences for research on this condi-tion.
M.R.C. Brain Metabolism Unit,
University Department of Pharmacology,1 George Square,Edinburgh EH8 9JZ
University Department of Pathology,Royal Infirmary of Edinburgh, and
Department of Neuropathology,Western General Hospital,
Edinburgh
P. DAVIES
A. J. F. MALONEY
HEADACHE AFTER LUMBAR PUNCTURE
SiR,—The frequency of headache after lumbar puncture inthe four large series cited by Wolff was 25%.’ The headacheis thought to be due to continued leakage of cerebrospinal fluid
(c.s.F.) through the hole in the theca, the subsequent low pres-sure in the c.s.F. pathways inducing pain by traction on the
pain-sensitive neural endings in the dura and intracranial
venous sinuses and arteries. Aqueous vasopressin injection(’Pitressin’) as a prophylactic measure was popular some years
1. Wolff, H. G. Headache and Head Pain; p. 112. New York, 1963.
N=3
1995 Tacrina aprobada
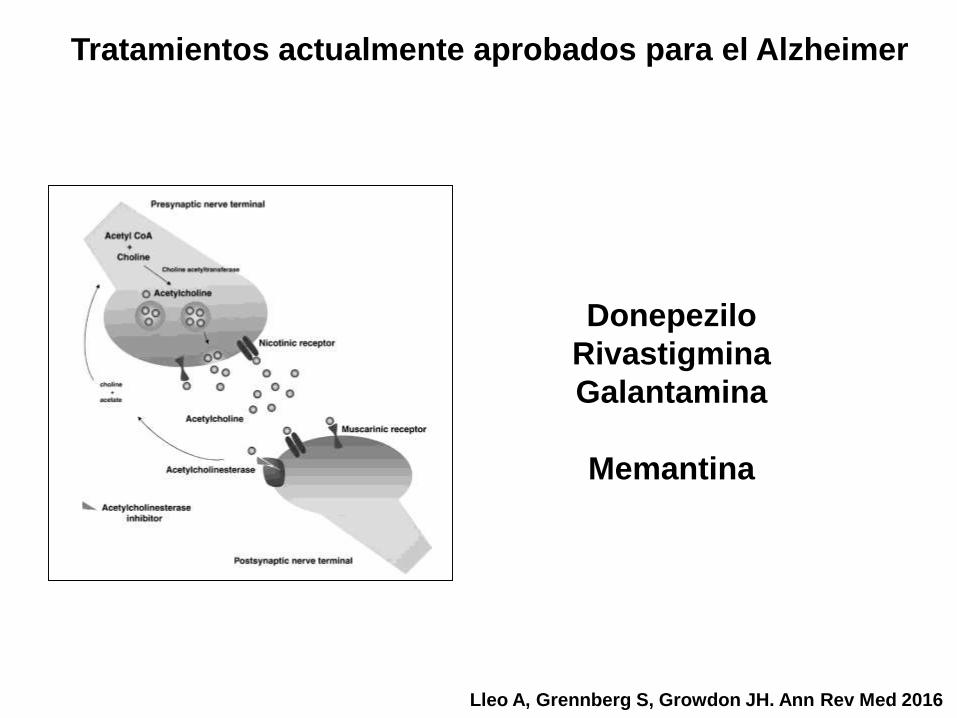
Tratamientos actualmente aprobados para el Alzheimer
Lleo A, Grennberg S, Growdon JH. Ann Rev Med 2016
10 Aug 2005 21:13 AR ANRV262-ME57-07.tex XMLPublishSM(2004/02/24)P1: OKZ /OAH P2:OJO
AR REVIEWS IN ADVANCE10.1146/annurev.med.57.121304.131442
PHARMACOTHERAPY FOR ALZHEIMER’S 7.3
Figure1 Acetylcholine (ACh) biosynthesis, synaptic transmission, and metabolism.
ACh is synthesized from acetyl coenzyme A and choline through the action of the en-
zyme choline acetyltransferase. ACh is concentrated in vesicles and released into the
synaptic cleft where it acts on presynaptic receptors regulating further release of ACh,
and on postsynaptic receptors regulating neurotransmission. Acetylcholinesterase hy-
drolyzes ACh into acetate and choline; choline is then taken up into the presynaptic
neuron for ACh resynthesis. Acetylcholinesterase inhibitors reduce degradation of ACh
and therefore increase its synaptic concentration and postsynaptic neurotransmission
effects.
choline acetyltransferase (see Figure 1), concentrated in vesicles, and released from
the presynaptic cell following depolarization. ACh interacts with muscarinic and
nicotinic cholinergic receptors on pre- and postsynaptic cells. In the case of postsy-
naptic cells, this interaction leads to an activation of biochemical pathways within
the cell. Once released into the synaptic cleft, ACh is rapidly hydrolyzed by local
Ann
u.
Rev
. M
ed. 0
.0:$
{ar
ticl
e.fP
age}
-${ar
ticle
.lP
age}
. D
ow
nlo
aded
fro
m a
rjo
urn
als.
ann
ualr
evie
ws.
org
by
HA
RV
AR
D U
NIV
ER
SIT
Y o
n 0
9/0
1/0
5.
For
per
son
al u
se o
nly
.
Donepezilo
Rivastigmina
Galantamina
Memantina
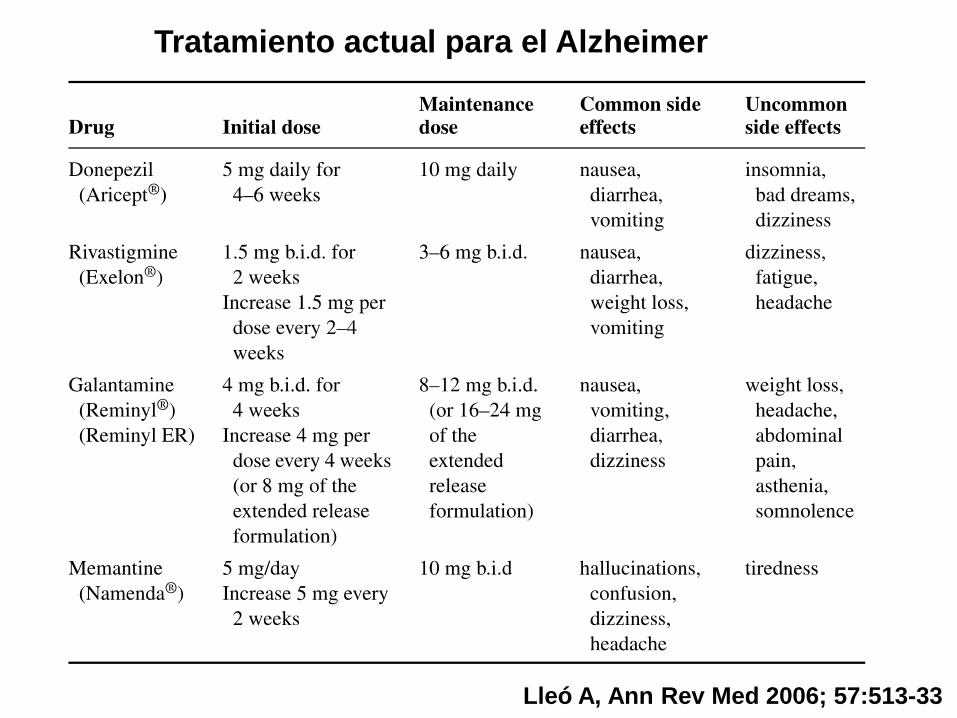
Tratamiento actual para el Alzheimer
Lleó A, Ann Rev Med 2006; 57:513-33

• IACEs muestran beneficios modestos en pacientes
con enfermedad leve, moderada o grave en medidas
de cognición, funcionales, y conductuales.
• El tratamiento estándar es iniciar con IACE y añadir
memantina cuando progresa enfermedad (MMSE <20).
• La combinación se mantiene hasta la fase avanzada a
no ser que aparezcan efectos adversos o se observe
falta de eficacia (GDS 7B).
Resumen tratamiento actual del Alzheimer
Lleó A, Ann Rev Med 2006; 57:513-33

Rivastigmina: parche 13,3 mg
Estudio OPTIMA
Novedades “recientes”
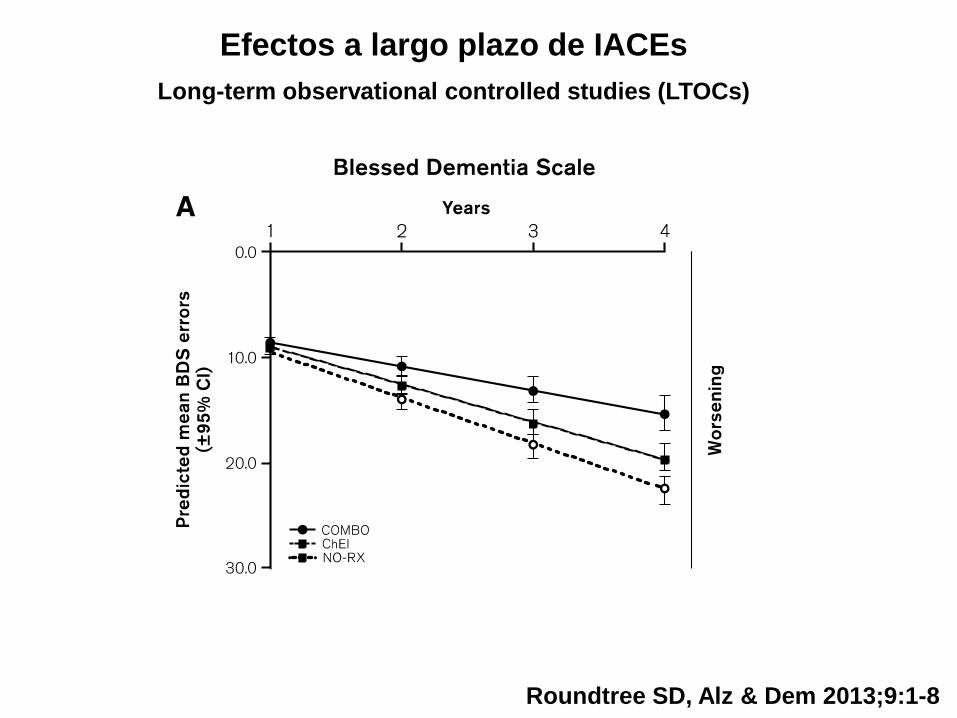
Efectos a largo plazo de IACEs
Long-term observational controlled studies (LTOCs)
Roundtree SD, Alz & Dem 2013;9:1-8
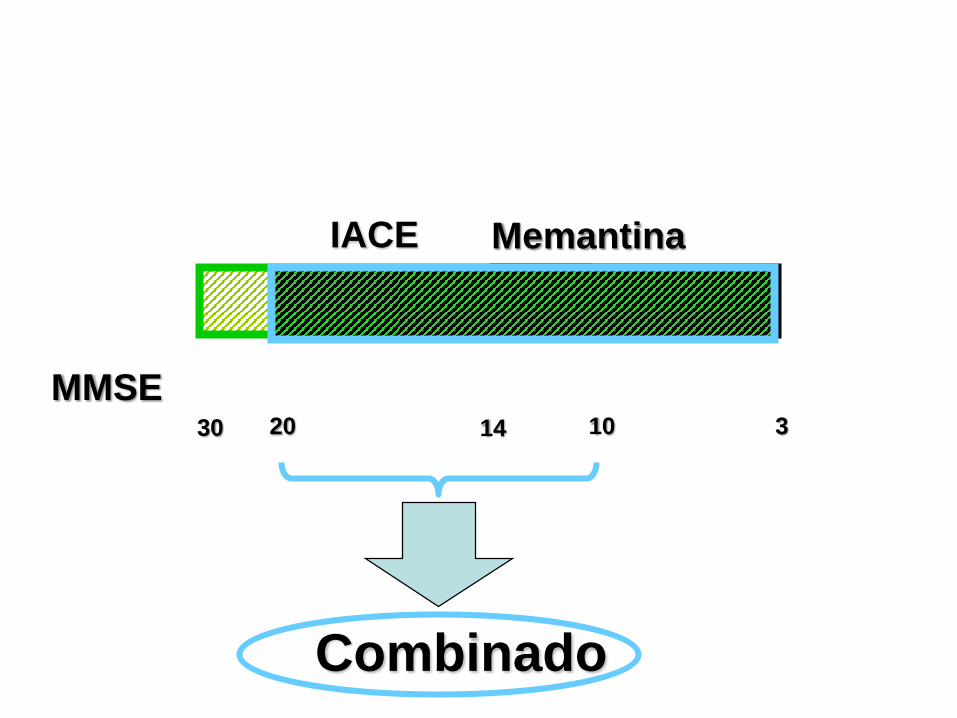
30 10 320
NICE 2006
Memantina
Combinado
MMSE
IACE
14
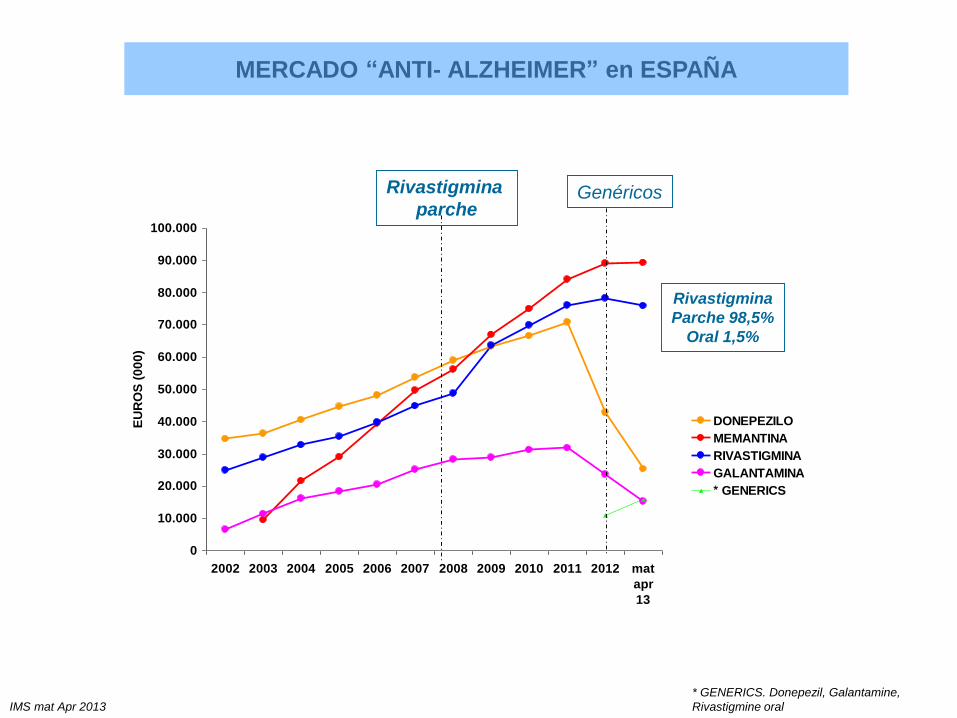
0
10.000
20.000
30.000
40.000
50.000
60.000
70.000
80.000
90.000
100.000
2002 2003 2004 2005 2006 2007 2008 2009 2010 2011 2012 mat
apr
13
EU
RO
S (
00
0)
DONEPEZILO
MEMANTINA
RIVASTIGMINA
GALANTAMINA
* GENERICS
MERCADO “ANTI- ALZHEIMER” en ESPAÑA
IMS mat Apr 2013
Rivastigmina
parcheGenéricos
* GENERICS. Donepezil, Galantamine,
Rivastigmine oral
Rivastigmina
Parche 98,5%
Oral 1,5%
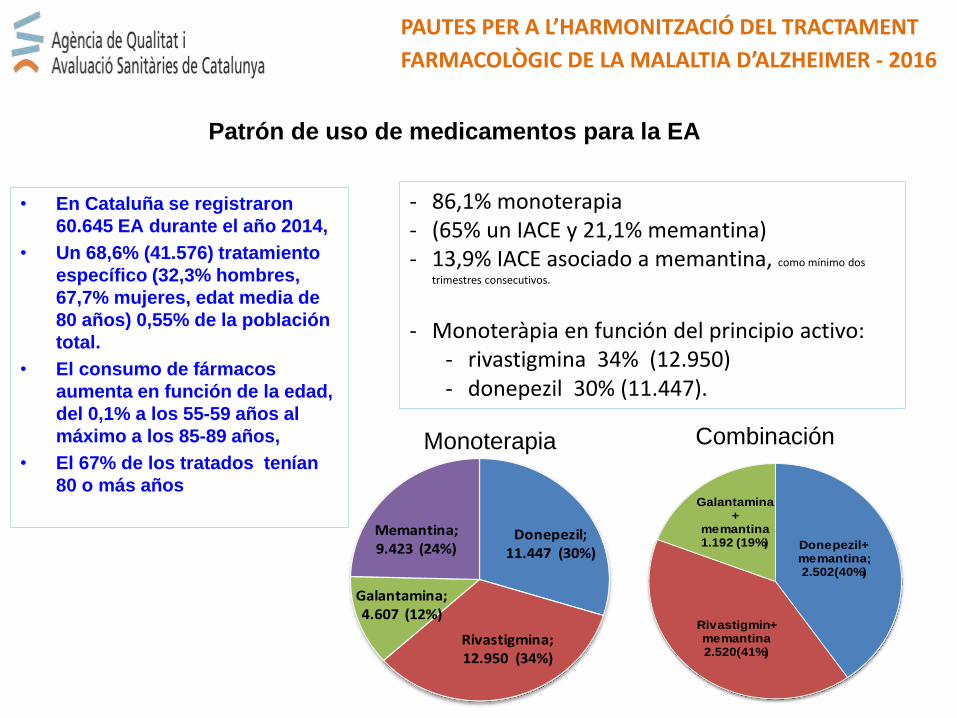
• En Cataluña se registraron
60.645 EA durante el año 2014,
• Un 68,6% (41.576) tratamiento
específico (32,3% hombres,
67,7% mujeres, edat media de
80 años) 0,55% de la población
total.
• El consumo de fármacos
aumenta en función de la edad,
del 0,1% a los 55-59 años al
máximo a los 85-89 años,
• El 67% de los tratados tenían
80 o más años
PAUTES PER A L’HARMONITZACIÓ DEL TRACTAMENT
FARMACOLÒGIC DE LA MALALTIA D’ALZHEIMER - 2016
Patrón de uso de medicamentos para la EA
- 86,1% monoterapia - (65% un IACE y 21,1% memantina) - 13,9% IACE asociado a memantina, como mínimo dos
trimestres consecutivos.
- Monoteràpia en función del principio activo:- rivastigmina 34% (12.950)- donepezil 30% (11.447).
Donepezil; 11.447 (30%)
Rivastigmina; 12.950 (34%)
Galantamina; 4.607 (12%)
Memantina; 9.423 (24%) Donepezil+
memantina; 2.502(40%)
Rivastigmin+memantina2.520(41%)
Galantamina+
memantina 1.192 (19%)
Monoterapia Combinación

Fortasyn Connect
Souvenaid 125ml, contiene Fortasyn® Connect
combinación de ácidos grasos omega3, colina, uridina
monofosfato, fosfolípidos, antioxidantes y vitaminas B.
Food for Special Medical Purpose (FSMP).


1976
Hipótesis colinérgica de la enfermedad de Alzheimer
disease1403
Letters to the Editor
SELECTIVE LOSS OF CENTRAL CHOLINERGIC
NEURONS IN ALZHEIMER’S DISEASE
SIR,-Alzheimer’s disease is a progressive cerebral degener-ation which continues without remission until death, usuallyin profound dementia. Morphologically, the disease is charac-terised by large numbers of senile plaques and neurofibrillarytangles in the brain, these tangles being especially abundant inthe cerebral cortex. Neurochemical studies are still in their in-
fancy, and we know nothing of the molecular basis of the dis-ease.
We have studied the enzymes associated with the putativeneurotransmitters acetylcholine, y-aminobutyric acid, dopa-mine, noradrenaline and 5-hydroxytryptamine in twenty re-
gions of brains obtained at necropsy from three patients withAlzheimer’s disease and from ten individuals who died without
evidence of neurological or psychiatric disorder. The brains
were removed 24-36 h after death, and each brain was
divided in half down the mid-line. The right half was used forbiochemical analyses, whilst the left half was fixed in formalinfor neuropathological examination. Alzheimer’s disease wasconfirmed histologically. There was no evidence of cerebrovas-cular disease in any of the thirteen cases. The ten controls
ranged in age from 46 to 74; the Alzheimer patients were aged61, 70, and 75. Tissues from the right half of the brain werestored at -190°C. Choline acetyltransferase (C.A.T.) and ace-
tylcholinesterase (A.C.E.) activities were measured by the
methods of Fonnum, and glutamic acid decarboxylase (G.A.D.)activity by the method of Roberts and Simonsen.2
C.A.T. activity in the Alzheimer’s disease brains was muchreduced in the amygdala, hippocampus, and cortex (table I).Only three Alzheimer brains have been studied but the extentof the reduction in these areas strongly suggests this is not achance occurrence. The activity of A.C.E. is dramaticallyreduced in the same areas of the cerebral cortex that show
reductions in C.A.T. activity (table II), and is below the levelsfound in the normal brains in all the other areas. The areas
of the cerebral cortex which show the maximum reductions in
c.A.T. and A.C.E. activity are those which contain the greatestdensity of neurofibrillary tangles.The reductions in the activity of the enzymes involved in the
metabolism of acetylcholine are not a result of non-specificdegenerative process. The activity of G.A.D. in all the areas ofthe Alzheimer’s disease brains studied appears to be well
within the normal range, means ranging from 74% to 121% ofcontrol activities. That this is the case in the cortical areas
which show large losses of c.A.T. and A.C.E. supports the notionthat a selective degenerative process has occurred. The normalvalues obtained for G.A.D. are of special significance because this
enzyme is particularly sensitive to ante-mortem hypoxia.3 It
seems unlikely, therefore, that the decreased activity of
enzymes associated with cholinergic transmission can be
ascribed to this cause; none of the patients with Alzheimer’sdisease had prolonged terminal hypoxic episodes.
Preliminary studies of tyrosine hydroxylase, aromatic amino-acid decarboxylase, dopamine-&bgr;-hydroxylase, and monoamineoxidase indicate no loss of these enzyme activities in Alz-
heimer’s disease and lend weight to the notion that a selectivedestruction of the cortical cholinergic system is an importantfeature of this condition.
We considered the possibility that selective loss of choliner-
gic system components could be due to prolonged drugregimens used in the patients with Alzheimer’s disease. How-
ever, there is no standard drug therapy for Alzheimer’s, and
1. Fonnum, F. Biochem. J. 1969, 115, 465.2. Roberts, E., Simonsen, D. E. Biochem. Pharmac. 1963, 12, 113.3. Bowen, D. M., Davison, A. N. in Biochemistry and Neurological Disease
edited by A. N. Davison); p. 2. Oxford, 1976.
TABLE I--CHOLINE ACETYLASE ([L mol/h/g WET WEIGHT)
TABLE II-ACETYLCHOLINESTERASE (MMOWG WET WEIGHT)
treatment is symptomatic. All three patients were given nitra-
zepam, but this drug was also given to five of the ten control
patients during their terminal illness. Opiates were adminis-tered to two of the Alzheimer patients and five of the con-trols, and phenothiazines were given to one and two patients,respectively. Thus no drug treatment was exclusive to the Alz-heimer’s disease patients, and it seems improbable that thedeficit in C.A.T. and A.C.E. activity in the cortex of these indi-viduals is drug induced.
Expression of results relative to protein, D.N.A., or R.N.A.
content does not alter the pattern of the results significantly.If these data can be confirmed in a larger series of cases the
concept of Alzheimer’s disease as a cholinergic system failure
may have important consequences for research on this condi-tion.
M.R.C. Brain Metabolism Unit,
University Department of Pharmacology,1 George Square,Edinburgh EH8 9JZ
University Department of Pathology,Royal Infirmary of Edinburgh, and
Department of Neuropathology,Western General Hospital,
Edinburgh
P. DAVIES
A. J. F. MALONEY
HEADACHE AFTER LUMBAR PUNCTURE
SiR,—The frequency of headache after lumbar puncture inthe four large series cited by Wolff was 25%.’ The headacheis thought to be due to continued leakage of cerebrospinal fluid
(c.s.F.) through the hole in the theca, the subsequent low pres-sure in the c.s.F. pathways inducing pain by traction on the
pain-sensitive neural endings in the dura and intracranial
venous sinuses and arteries. Aqueous vasopressin injection(’Pitressin’) as a prophylactic measure was popular some years
1. Wolff, H. G. Headache and Head Pain; p. 112. New York, 1963.
N=3
1995 Tacrina aprobada
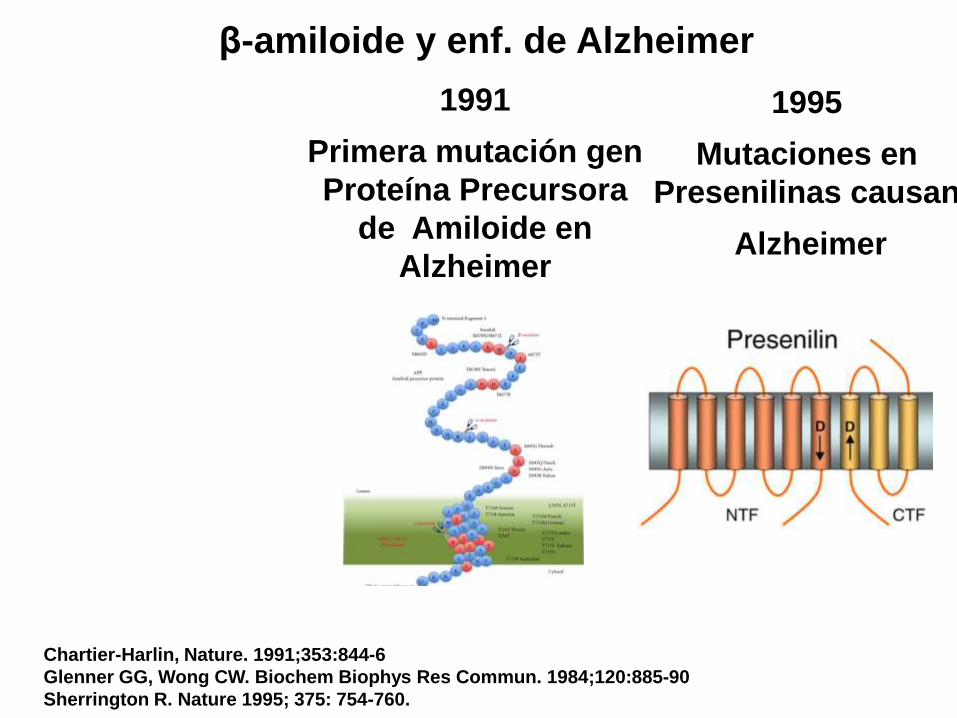
Chartier-Harlin, Nature. 1991;353:844-6
Glenner GG, Wong CW. Biochem Biophys Res Commun. 1984;120:885-90
Sherrington R. Nature 1995; 375: 754-760.
β-amiloide y enf. de Alzheimer
1991
Primera mutación gen
Proteína Precursora
de Amiloide en
Alzheimer
1995
Mutaciones en
Presenilinas causan
Alzheimer
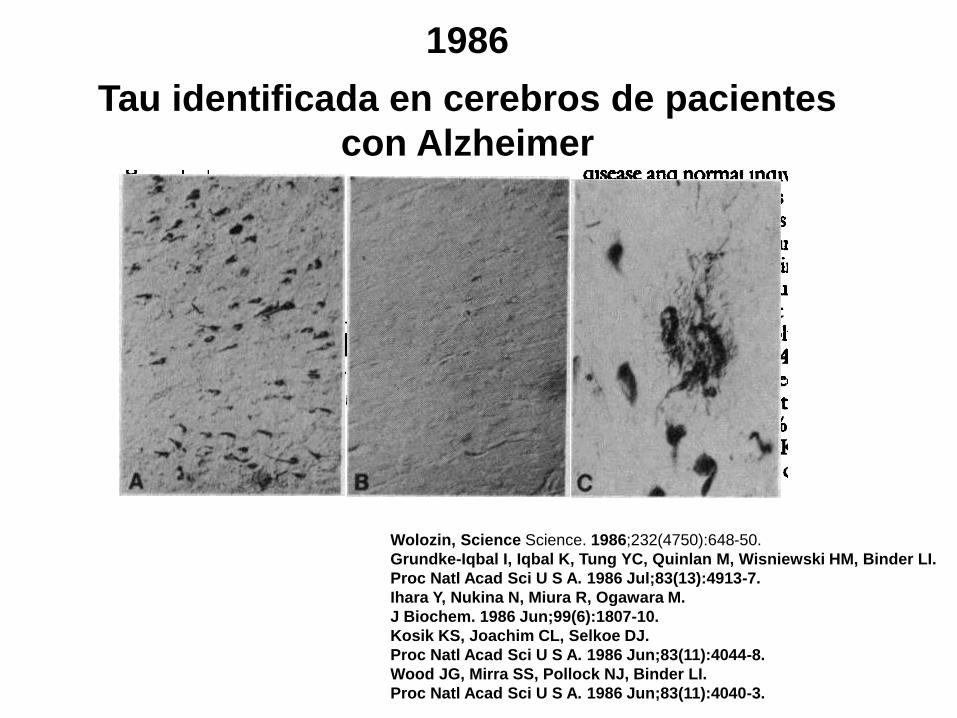
1986
Tau identificada en cerebros de pacientes
con Alzheimer
Wolozin, Science Science. 1986;232(4750):648-50.
Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI.
Proc Natl Acad Sci U S A. 1986 Jul;83(13):4913-7.
Ihara Y, Nukina N, Miura R, Ogawara M.
J Biochem. 1986 Jun;99(6):1807-10.
Kosik KS, Joachim CL, Selkoe DJ.
Proc Natl Acad Sci U S A. 1986 Jun;83(11):4044-8.
Wood JG, Mirra SS, Pollock NJ, Binder LI.
Proc Natl Acad Sci U S A. 1986 Jun;83(11):4040-3.

1992 Hipótesis amiloide marca avance
conceptual clave
Selkoe & Hardy, EMBO Mol Med 2016;8:595-608
< 1% 99%?
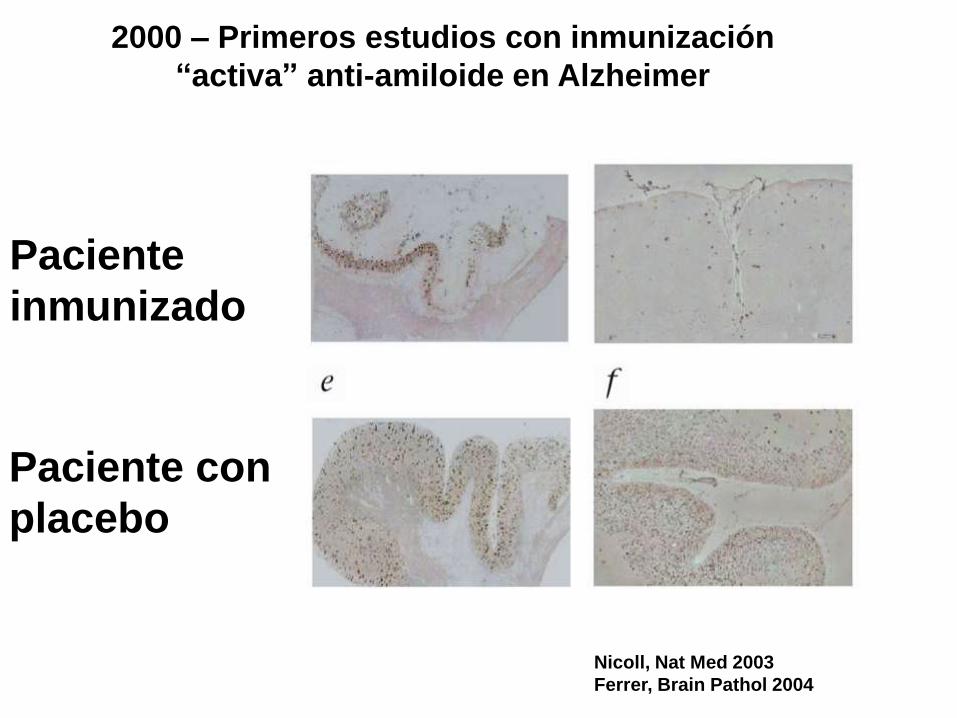
Nicoll, Nat Med 2003
Ferrer, Brain Pathol 2004
2000 – Primeros estudios con inmunización
“activa” anti-amiloide en Alzheimer
Paciente
inmunizado
Paciente con
placebo
NATURE REVIEWS | NEUROLOGY VOLUME 11 | JANUARY 2015 | 47
Nonetheless, a small group of patients underwent CSF
analyses, and their data indicated that AN1792 reduced
levels of tau, but not Aβ1–42
, in antibody responders.97 The
lack of change in CSF Aβ1–42
contrasts with the reductions
in brain Aβ plaque load observed on postmortem assess-
ment.98 This result suggests that that removal of aggre-
gated Aβ does not necessarily affect the pool of soluble Aβ
in CSF, or that clearance of cortical Aβ can occur through
pathways other than CSF.
In agreement with these data, active immunization
with CAD-106, a small Aβ peptide fragment (Aβ1–6
),
had no effect on CSF Aβ1–42
in a phase I trial.99 The
reduction in CSF tau levels by AN1792 might indicate
downstream effects of the treatment on pathology, such
that the intensity of neuronal degeneration is reduced
by Aβ immunization. This notion is supported by post-
mortem neuropathology studies that show amelioration of
neurite abnormalities in patients treated with AN1792.100
In addition, it is possible that the reduction in CSF tau
levels is partly attributable to a mild effect of AN1792
on tau pathology, as has been observed in transgenic
mouse models of AD.101,102 However, autopsy studies of
AN1792-treated patients with AD have reported mild or
no changes in the density of neurofibrillary tangles and
neuropil threads compared with non-treated patients,
suggesting that the effects of Aβ immunization on tau
pathology, at least in patients with dementia, are small.100
Taken together, the data from the AN1792 trial suggest
that removal of cortical Aβ probably ameliorated abnor-
malities in neuronal morphology, which in turn, though
not sufficient to change the clinical course of the disease,
reduced the levels of CSF tau. Other immunization trials
with CAD-106 or ACC-001 in patients with MCI due to
AD, or with mild to moderate AD, are ongoing; these trials
include CSF biomarkers as secondary outcomes (Table 1).
In light of the adverse events observed with active
immunization, most immunotherapy studies in AD now
use passive immunization, which is based on the admin-
istration of monoclonal antibodies directed towards
different regions of Aβ. Bapineuzumab is a human-
ized version of the monoclonal antibody 3D6, which is
directed against the N-terminal region of Aβ and binds to
monomeric, oligomeric and fibrillar forms of Aβ.103 Trials
of bapineuzumab have included CSF biomarkers as sec-
ondary outcome measures of neurodegeneration. In two
phase II trials, bapineuzumab decreased CSF t-tau and
p-tau, but not Aβ1–42
or AβX–42
levels compared with base-
line.104 These data are in general agreement with recent
data from two phase III trials,105 in which bapineuzumab
at two different doses reduced CSF p-tau levels at 71 weeks
relative to placebo in Apo-E ε4 allele (APOE*ε4) carriers.
In noncarriers, however, the antibody only reduced CSF
p-tau levels at the highest dose. Bapineuzumab did not
change CSF Aβ1–42
or Aβ1–40
levels in either the carriers
or the noncarriers, mirroring the results of the active Aβ
immunization trials.
In the phase III trials, bapineuzumab was shown to
modestly decrease the rate of Aβ accumulation in the
brain in APOE*ε4 carriers, suggesting that the concen-
tration of Aβ1–42
in CSF does not always reflect changes
in fibrillar Aβ deposits. Together with the data from
active Aβ immunization trials, these results support a
model in which deposition of fibrillar Aβ decreases CSF
Aβ1–42
, presumably by sequestering Aβ1–42
into plaques,
but removal of fibrillar Aβ does not necessarily have an
effect on the CSF pool of soluble Aβ. The reduction in
CSF p-tau levels in both the phase II and phase III trials
suggests that bapineuzumab reduces tau phosphoryla-
tion in the brain, although this change was not associated
with any clinical benefit. This observation is supported
by data from experiments in rat hippocampal neurons,
which showed that treatment with the 3D6 antibody
prevented tau hyperphosphorylation.103
Clinical development of bapineuzumab was discon-
tinued owing to the lack of clinical benefit. The dose
of bapineuzumab used in these studies, however, was
limited by the emergence of amyloid-related imaging
abnormalities at higher doses. Therefore, it is possible
that despite some target engagement, too little Aβ was
removed to test whether changes in neurodegeneration
markers occur after bapineuzumab treatment.
Solanezumab is a humanized version of the anti-Aβ
monoclonal antibody m266, which has selective affinity
for soluble Aβ. Two large phase III trials (EXPEDITION 1
and 2) have evaluated the efficacy of solanezumab.
Primary end points were not reached, but a pooled analy-
sis showed that solanezumab slowed cognitive decline in
patients with mild AD.106 In a subset of 121 patients who
underwent CSF analyses, total Aβ1–40
and Aβ1–42
levels
were increased after solanezumab treatment, whereas
free (unbound) Aβ1–40
was reduced. These results rep-
licated those observed in phase I and II trials,107,108 and
suggest positive target engagement with mobilization of
the central soluble Aβ pool. Solanezumab is not expected
to bind to fibrillar Aβ109 and, therefore, the observed
increase in total CSF Aβ is unlikely to have come from
interactions with the fibrillar Aβ component. Whether
the effects reflect mobilization within the central soluble
Aβ pool and/or promotion of Aβ efflux from the CNS to
the peripheral circulation remains unclear.110
In addition to Aβ, t-tau and p-tau levels were meas-
ured in CSF after solanezumab treatment, and no
changes were detected.106 This result indicates that mobi-
lization of the Aβ pool did not affect any of the markers
of neurodegeneration. These data seem to support the
Figure 2 | Monoclonal antibodies that target Aβ pathology. One of the main
pathological hallmarks of AD is Aβ plaques, which are formed by progressive
accumulation of Aβ peptides. Solanezumab was designed to bind to monomeric Aβ,
thereby preventing oligomerization and deposition. Bapineuzumab is a humanized
N-terminus-specific monoclonal antibody that binds to Aβ. In animal models of AD,
3D6—the murine form of the antibody—binds to monomeric, oligomeric and fibrillar
forms of Aβ.117 Only the two most investigated antibodies with available cerebrospinal
fluid data have been included. Abbreviations: Aβ, amyloid-β; AD, Alzheimer disease.
Solanezumab
Monomeric Aβ Oligomeric Aβ Protof bril Aβ
Bapineuzumab
Amyloid plaque
Nature Reviews | Neurology
REVIEWS
© 2015 Macmillan Publishers Limited. All rights reserved
Terapias modificadoras de la enfermedad
Terapias anti-amiloide
Lleo A, Nat Rev Neurol 2015; 11:45-55
NATURE REVIEWS | NEUROLOGY VOLUME 11 | JANUARY 2015 | 43
aid clinical trials of β-secretase inhibitors in patients
with AD, as this peptide can reliably reveal drug target
engagement. Apolipoprotein E (Apo-E) levels in CSF have
also been investigated. Although levels are comparable
between patients with AD and controls, they correlate
positively with CSF tau, and lower levels of Apo-E are
associated with cognitive decline and brain atrophy.59,60
A number of tau-independent markers of neuro-
degeneration have also been investigated in patients
with AD, including heart fatty acid-binding protein and
visinin-like protein 1,61,62 which might be useful markers
of neuronal injury in clinical trials. CSF biomarkers of
microglial activation include chitotriosidase, YKL-40
(also known as chitinase-3-like protein 1) and C–C motif
chemokine 2.63,64 Levels of these markers are elevated in
the CSF in patients with AD and other neurodegenera-
tive dementias, but longitudinal studies are required to
determine whether they correlate with disease sever-
ity or clinical progression. CSF biomarkers that reflect
early synaptic damage would be a useful addition to the
current arsenal, as they are likely to correlate with clinical
symptoms or disease progression. A series of presynap-
tic and postsynaptic proteins have been detected in CSF,
and increased levels of the dendritic protein neurogra-
nin have been found in the CSF of patients with AD.65,66
Finally, markers that can detect common copathologies
usually observed in the brain in AD may be of interest.
For example, α-synuclein in CSF is a potential marker for
Lewy body pathology in AD,67 and TAR DNA-binding
protein 43 (TDP-43) in CSF has been explored to detect
TDP-43 pathology.68
Clinicobiological correlations
Clinicopathological studies have shown that low CSF Aβ1–42
levels are associated with fibrillar Aβ deposits and cerebral
amyloid angiopathy postmortem,3,5,69 and with fibrillar Aβ
deposits in cortical biopsies.70 In agreement with these data,
low CSF Aβ1–42
levels are associated with increased cortical
amyloid burden, as assessed by amyloid PET imaging in
patients with AD dementia or MCI and cognitively normal
individuals.71–76 Importantly, the concordance between
CSF Aβ1–42
and amyloid PET is usually ≥90%, regardless
of whether CSF Aβ1–42
is analysed under ideal research
conditions or as part of the clinical routine.77 Exceptions
to this rule, however, are seen in carriers of some rare APP
mutations associated with autosomal dominant AD.78
Together, these data suggest that in the vast majority of
patients, a high correlation exists between soluble CSF
Aβ1–42
and amyloid pathology, and soluble CSF Aβ1–42
levels
decrease as amyloid pathology builds up, presumably due
to sequestration of Aβ1–42
in the brain parenchyma.
Increases in CSF t-tau5 and p-tau79 levels have been
linked to neurofibrillary tangle burden at autopsy. The
majority of studies in patients with AD evaluated the
levels of p-tau181
, although the levels of p-tau231
have been
reported to correlate better with postmortem tangle load
than do p-tau181 levels.
79,80 These data suggest that in AD,
CSF p-tau levels probably reflect neurofibrillary tangle
pathology, whereas CSF t-tau levels are a reflection of
the overall cytoskeletal derangement, and of neuronal
cell damage or death.
In summary, most of the current literature supports the
notion that CSF Aβ1–42
and p-tau levels reflect the main
pathological hallmarks of AD, while t-tau levels are a
measure of the degree of neuronal cell damage. An impor-
tant caveat is that although clinicopathological studies
indicate that tau pathology precedes amyloid pathology
in the disease course of AD, t-tau and p-tau levels become
abnormal in CSF later than does Aβ1–42
, indicating that
a substantial degree of neurofibrillary tangle pathology
and neuronal damage must occur before evidence is seen
in CSF.81 These correlates need further investigation to
facilitate interpretation of the changes in CSF biomarkers
in clinical trials.
Clinical trials in Alzheimer disease
Many of the current clinical trials in patients with AD
include CSF biomarkers as a measure of target engage-
ment. Less frequently, CSF biomarkers are used as second-
ary outcome measures to monitor disease modification,
or for sample enrichment or stratification of patients in
different stages of the pathophysiological process of AD
(Tables 1 and 2). Despite the growing use of these bio-
markers in clinical trials, however, interpretation of the
data is not always straightforward. Whereas the data from
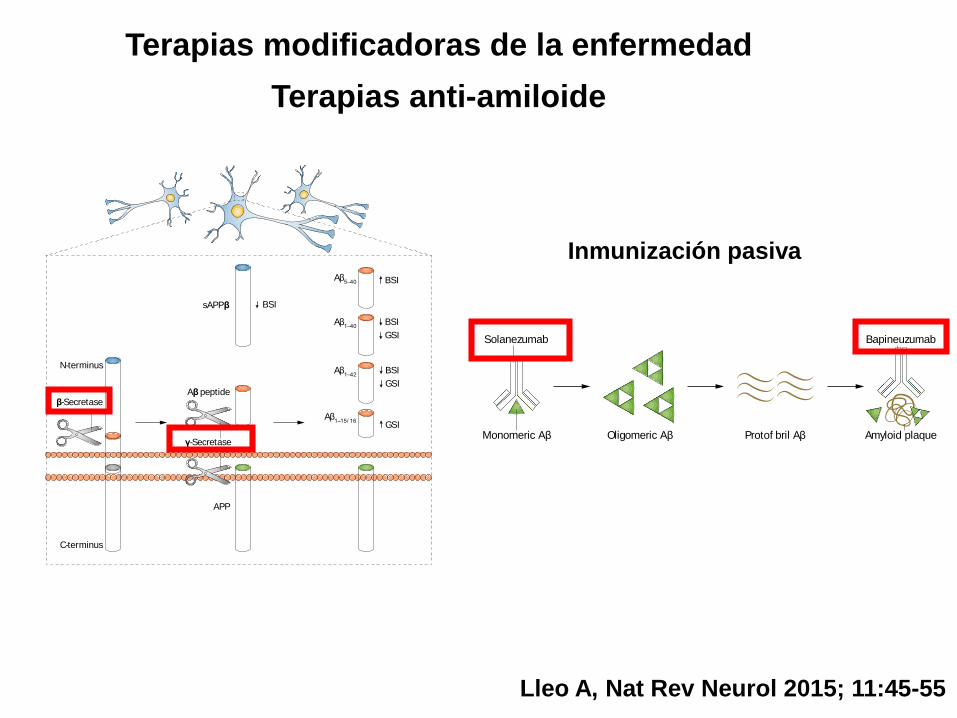
Figure 1 | Main CSF biomarkers used in trials of anti-amyloid drugs in AD. Amyloid
precursor protein is processed sequentially by β-secretase and γ-secretase, and
subsequently aggregates into amyloid plaques in patients with AD. The main CSF
biomarkers used in trials with BACE and γ-secretase inhibitors are shown.
Abbreviations: Aβ, amyloid-β; AD, Alzheimer disease; APP, amyloid precursor protein;
BSI, β-secretase inhibitors; CSF, cerebrospinal fluid; GSI, γ-secretase inhibitors;
sAPPβ, soluble APPβ.
sAPPβ
Aβ peptideβ-Secretase
N-terminus
APP
C-terminus
γ-Secretase
BSI
BSI
BSI
GSI
BSI
GSI
GSI
Aβ5–40
Aβ1–40
Aβ1–42
Aβ1–15/ 16
Nature Reviews | Neurology
REVIEWS
© 2015 Macmillan Publishers Limited. All rights reserved
Inmunización pasiva
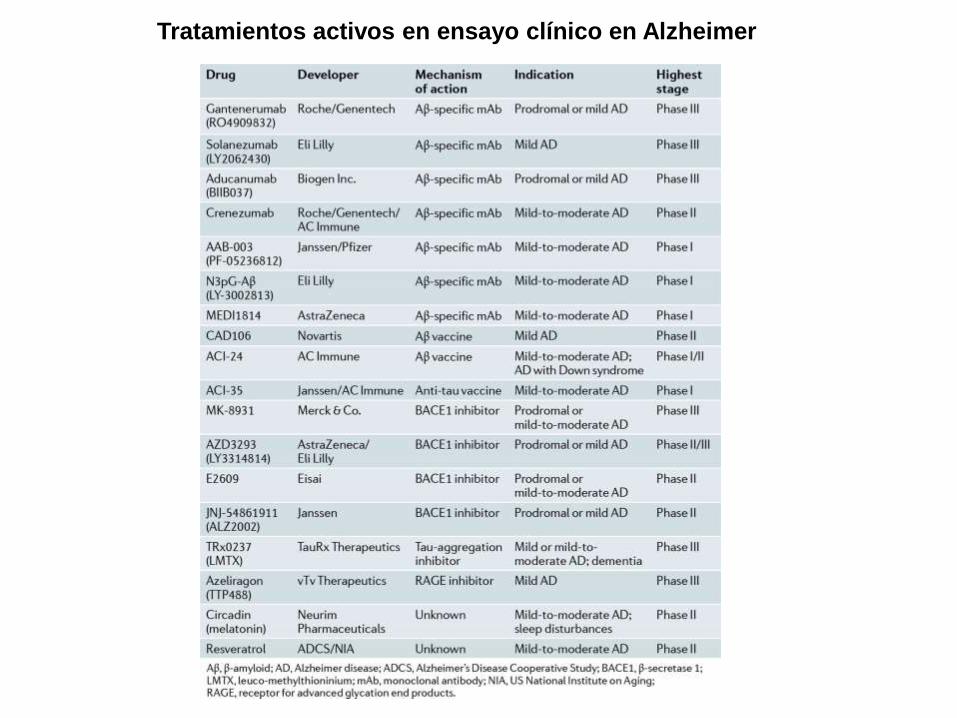
Tratamientos activos en ensayo clínico en Alzheimer
Otros tratamientos en investigación:
inhibidores de producción de amiloide
Nat Neurosci 2017
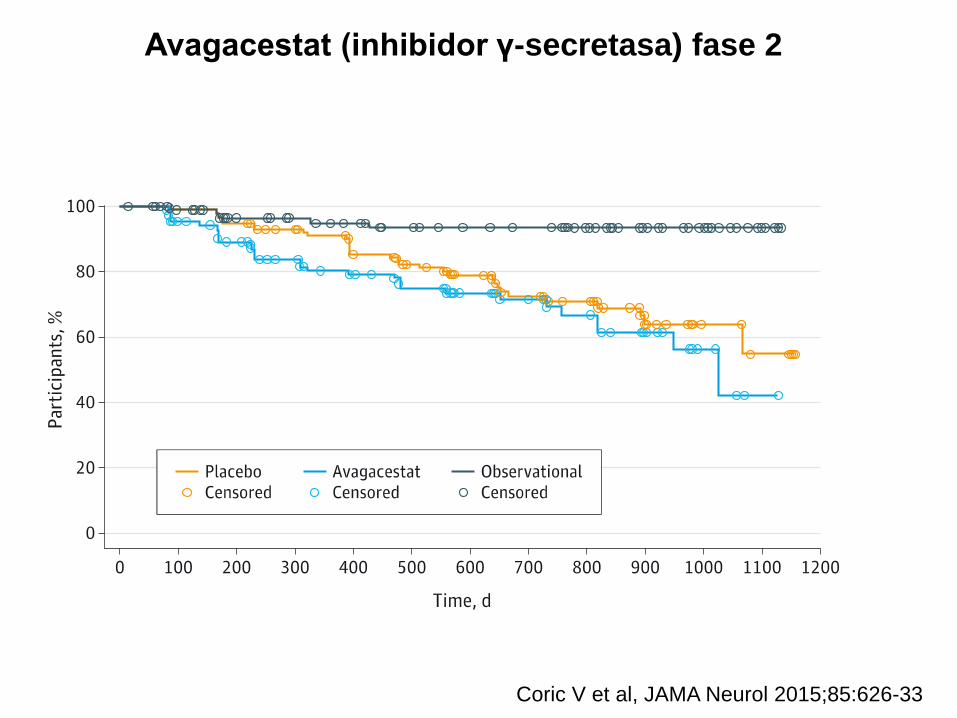
Avagacestat (inhibidor γ-secretasa) fase 2
Coric V et al, JAMA Neurol 2015;85:626-33
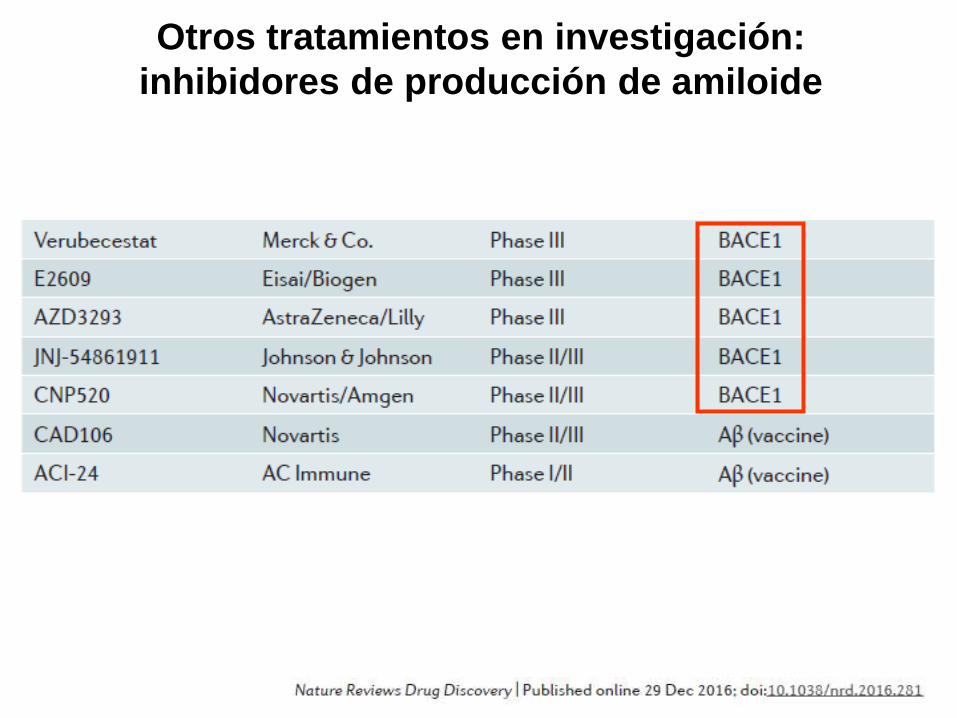
Otros tratamientos en investigación:
inhibidores de producción de amiloide

Buenas noticias en 2016
La Vanguardia 2016
3 de noviembre de 2016
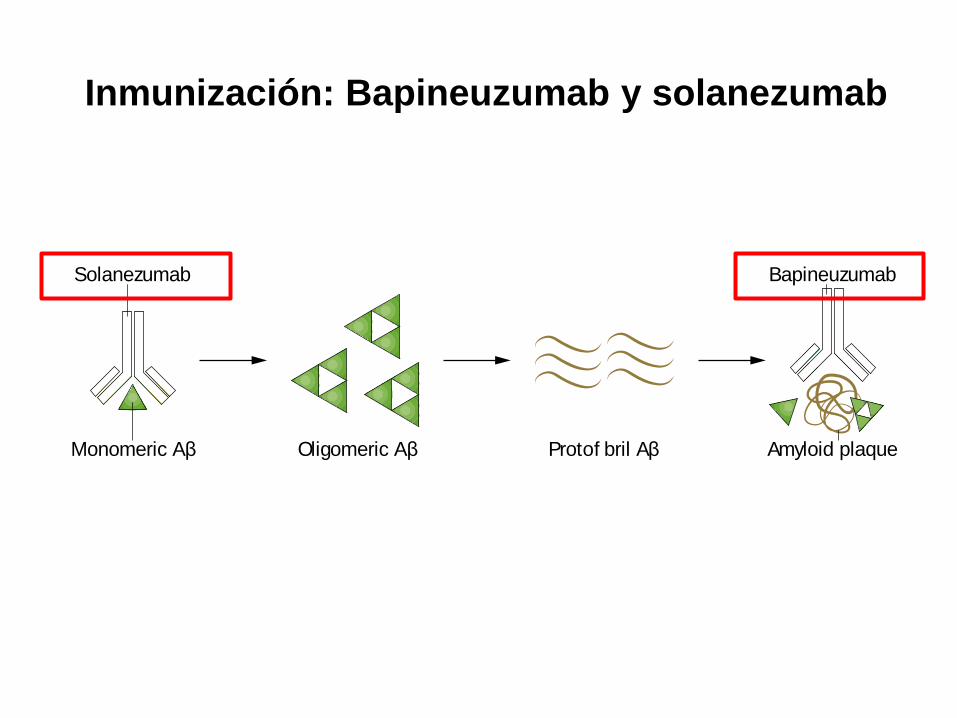
Inmunización: Bapineuzumab y solanezumab
NATURE REVIEWS | NEUROLOGY VOLUME 11 | JANUARY 2015 | 47
Nonetheless, a small group of patients underwent CSF
analyses, and their data indicated that AN1792 reduced
levels of tau, but not Aβ1–42
, in antibody responders.97 The
lack of change in CSF Aβ1–42
contrasts with the reductions
in brain Aβ plaque load observed on postmortem assess-
ment.98 This result suggests that that removal of aggre-
gated Aβ does not necessarily affect the pool of soluble Aβ
in CSF, or that clearance of cortical Aβ can occur through
pathways other than CSF.
In agreement with these data, active immunization
with CAD-106, a small Aβ peptide fragment (Aβ1–6
),
had no effect on CSF Aβ1–42
in a phase I trial.99 The
reduction in CSF tau levels by AN1792 might indicate
downstream effects of the treatment on pathology, such
that the intensity of neuronal degeneration is reduced
by Aβ immunization. This notion is supported by post-
mortem neuropathology studies that show amelioration of
neurite abnormalities in patients treated with AN1792.100
In addition, it is possible that the reduction in CSF tau
levels is partly attributable to a mild effect of AN1792
on tau pathology, as has been observed in transgenic
mouse models of AD.101,102 However, autopsy studies of
AN1792-treated patients with AD have reported mild or
no changes in the density of neurofibrillary tangles and
neuropil threads compared with non-treated patients,
suggesting that the effects of Aβ immunization on tau
pathology, at least in patients with dementia, are small.100
Taken together, the data from the AN1792 trial suggest
that removal of cortical Aβ probably ameliorated abnor-
malities in neuronal morphology, which in turn, though
not sufficient to change the clinical course of the disease,
reduced the levels of CSF tau. Other immunization trials
with CAD-106 or ACC-001 in patients with MCI due to
AD, or with mild to moderate AD, are ongoing; these trials
include CSF biomarkers as secondary outcomes (Table 1).
In light of the adverse events observed with active
immunization, most immunotherapy studies in AD now
use passive immunization, which is based on the admin-
istration of monoclonal antibodies directed towards
different regions of Aβ. Bapineuzumab is a human-
ized version of the monoclonal antibody 3D6, which is
directed against the N-terminal region of Aβ and binds to
monomeric, oligomeric and fibrillar forms of Aβ.103 Trials
of bapineuzumab have included CSF biomarkers as sec-
ondary outcome measures of neurodegeneration. In two
phase II trials, bapineuzumab decreased CSF t-tau and
p-tau, but not Aβ1–42
or AβX–42
levels compared with base-
line.104 These data are in general agreement with recent
data from two phase III trials,105 in which bapineuzumab
at two different doses reduced CSF p-tau levels at 71 weeks
relative to placebo in Apo-E ε4 allele (APOE*ε4) carriers.
In noncarriers, however, the antibody only reduced CSF
p-tau levels at the highest dose. Bapineuzumab did not
change CSF Aβ1–42
or Aβ1–40
levels in either the carriers
or the noncarriers, mirroring the results of the active Aβ
immunization trials.
In the phase III trials, bapineuzumab was shown to
modestly decrease the rate of Aβ accumulation in the
brain in APOE*ε4 carriers, suggesting that the concen-
tration of Aβ1–42
in CSF does not always reflect changes
in fibrillar Aβ deposits. Together with the data from
active Aβ immunization trials, these results support a
model in which deposition of fibrillar Aβ decreases CSF
Aβ1–42
, presumably by sequestering Aβ1–42
into plaques,
but removal of fibrillar Aβ does not necessarily have an
effect on the CSF pool of soluble Aβ. The reduction in
CSF p-tau levels in both the phase II and phase III trials
suggests that bapineuzumab reduces tau phosphoryla-
tion in the brain, although this change was not associated
with any clinical benefit. This observation is supported
by data from experiments in rat hippocampal neurons,
which showed that treatment with the 3D6 antibody
prevented tau hyperphosphorylation.103
Clinical development of bapineuzumab was discon-
tinued owing to the lack of clinical benefit. The dose
of bapineuzumab used in these studies, however, was
limited by the emergence of amyloid-related imaging
abnormalities at higher doses. Therefore, it is possible
that despite some target engagement, too little Aβ was
removed to test whether changes in neurodegeneration
markers occur after bapineuzumab treatment.
Solanezumab is a humanized version of the anti-Aβ
monoclonal antibody m266, which has selective affinity
for soluble Aβ. Two large phase III trials (EXPEDITION 1
and 2) have evaluated the efficacy of solanezumab.
Primary end points were not reached, but a pooled analy-
sis showed that solanezumab slowed cognitive decline in
patients with mild AD.106 In a subset of 121 patients who
underwent CSF analyses, total Aβ1–40
and Aβ1–42
levels
were increased after solanezumab treatment, whereas
free (unbound) Aβ1–40
was reduced. These results rep-
licated those observed in phase I and II trials,107,108 and
suggest positive target engagement with mobilization of
the central soluble Aβ pool. Solanezumab is not expected
to bind to fibrillar Aβ109 and, therefore, the observed
increase in total CSF Aβ is unlikely to have come from
interactions with the fibrillar Aβ component. Whether
the effects reflect mobilization within the central soluble
Aβ pool and/or promotion of Aβ efflux from the CNS to
the peripheral circulation remains unclear.110
In addition to Aβ, t-tau and p-tau levels were meas-
ured in CSF after solanezumab treatment, and no
changes were detected.106 This result indicates that mobi-
lization of the Aβ pool did not affect any of the markers
of neurodegeneration. These data seem to support the
Figure 2 | Monoclonal antibodies that target Aβ pathology. One of the main
pathological hallmarks of AD is Aβ plaques, which are formed by progressive
accumulation of Aβ peptides. Solanezumab was designed to bind to monomeric Aβ,
thereby preventing oligomerization and deposition. Bapineuzumab is a humanized
N-terminus-specific monoclonal antibody that binds to Aβ. In animal models of AD,
3D6—the murine form of the antibody—binds to monomeric, oligomeric and fibrillar
forms of Aβ.117 Only the two most investigated antibodies with available cerebrospinal
fluid data have been included. Abbreviations: Aβ, amyloid-β; AD, Alzheimer disease.
Solanezumab
Monomeric Aβ Oligomeric Aβ Protof bril Aβ
Bapineuzumab
Amyloid plaque
Nature Reviews | Neurology
REVIEWS
© 2015 Macmillan Publishers Limited. All rights reserved

Salloway S, 2014
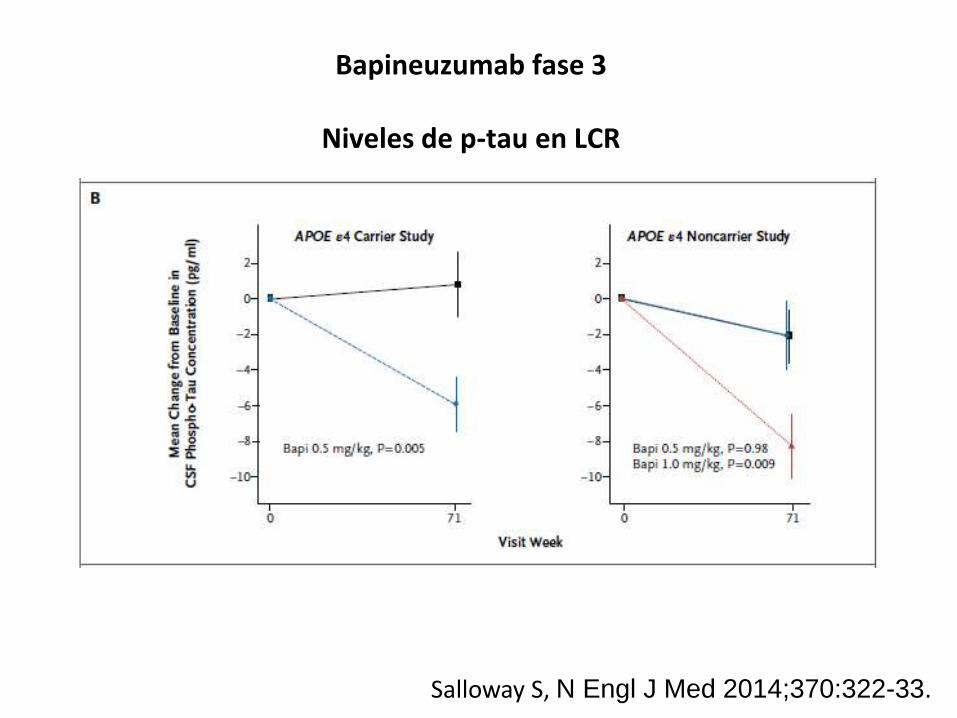
Bapineuzumab fase 3
Niveles de p-tau en LCR
Salloway S, N Engl J Med 2014;370:322-33.

Doody R, N Engl J Med 2014;370:311-21.

Buenas noticias en 2016
La Vanguardia 2016
1 de septiembre de 2016
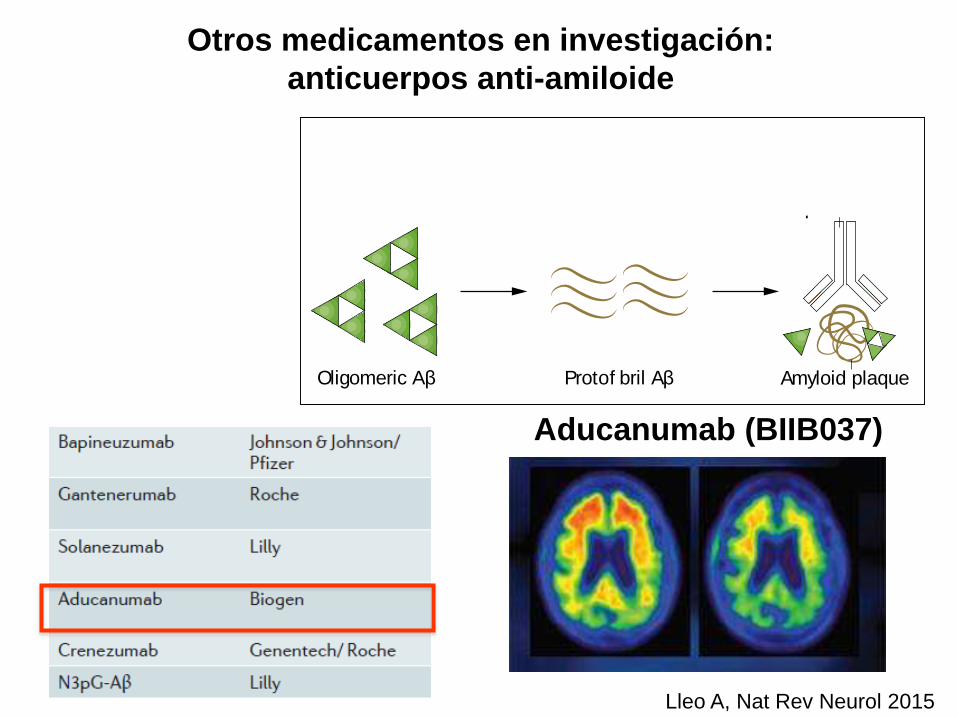
Otros medicamentos en investigación:
anticuerpos anti-amiloide
Lleo A, Nat Rev Neurol 2015
NATURE REVIEWS | NEUROLOGY VOLUME 11 | JANUARY 2015 | 47
Nonetheless, a small group of patients underwent CSF
analyses, and their data indicated that AN1792 reduced
levels of tau, but not Aβ1–42
, in antibody responders.97 The
lack of change in CSF Aβ1–42
contrasts with the reductions
in brain Aβ plaque load observed on postmortem assess-
ment.98 This result suggests that that removal of aggre-
gated Aβ does not necessarily affect the pool of soluble Aβ
in CSF, or that clearance of cortical Aβ can occur through
pathways other than CSF.
In agreement with these data, active immunization
with CAD-106, a small Aβ peptide fragment (Aβ1–6
),
had no effect on CSF Aβ1–42
in a phase I trial.99 The
reduction in CSF tau levels by AN1792 might indicate
downstream effects of the treatment on pathology, such
that the intensity of neuronal degeneration is reduced
by Aβ immunization. This notion is supported by post-
mortem neuropathology studies that show amelioration of
neurite abnormalities in patients treated with AN1792.100
In addition, it is possible that the reduction in CSF tau
levels is partly attributable to a mild effect of AN1792
on tau pathology, as has been observed in transgenic
mouse models of AD.101,102 However, autopsy studies of
AN1792-treated patients with AD have reported mild or
no changes in the density of neurofibrillary tangles and
neuropil threads compared with non-treated patients,
suggesting that the effects of Aβ immunization on tau
pathology, at least in patients with dementia, are small.100
Taken together, the data from the AN1792 trial suggest
that removal of cortical Aβ probably ameliorated abnor-
malities in neuronal morphology, which in turn, though
not sufficient to change the clinical course of the disease,
reduced the levels of CSF tau. Other immunization trials
with CAD-106 or ACC-001 in patients with MCI due to
AD, or with mild to moderate AD, are ongoing; these trials
include CSF biomarkers as secondary outcomes (Table 1).
In light of the adverse events observed with active
immunization, most immunotherapy studies in AD now
use passive immunization, which is based on the admin-
istration of monoclonal antibodies directed towards
different regions of Aβ. Bapineuzumab is a human-
ized version of the monoclonal antibody 3D6, which is
directed against the N-terminal region of Aβ and binds to
monomeric, oligomeric and fibrillar forms of Aβ.103 Trials
of bapineuzumab have included CSF biomarkers as sec-
ondary outcome measures of neurodegeneration. In two
phase II trials, bapineuzumab decreased CSF t-tau and
p-tau, but not Aβ1–42
or AβX–42
levels compared with base-
line.104 These data are in general agreement with recent
data from two phase III trials,105 in which bapineuzumab
at two different doses reduced CSF p-tau levels at 71 weeks
relative to placebo in Apo-E ε4 allele (APOE*ε4) carriers.
In noncarriers, however, the antibody only reduced CSF
p-tau levels at the highest dose. Bapineuzumab did not
change CSF Aβ1–42
or Aβ1–40
levels in either the carriers
or the noncarriers, mirroring the results of the active Aβ
immunization trials.
In the phase III trials, bapineuzumab was shown to
modestly decrease the rate of Aβ accumulation in the
brain in APOE*ε4 carriers, suggesting that the concen-
tration of Aβ1–42
in CSF does not always reflect changes
in fibrillar Aβ deposits. Together with the data from
active Aβ immunization trials, these results support a
model in which deposition of fibrillar Aβ decreases CSF
Aβ1–42
, presumably by sequestering Aβ1–42
into plaques,
but removal of fibrillar Aβ does not necessarily have an
effect on the CSF pool of soluble Aβ. The reduction in
CSF p-tau levels in both the phase II and phase III trials
suggests that bapineuzumab reduces tau phosphoryla-
tion in the brain, although this change was not associated
with any clinical benefit. This observation is supported
by data from experiments in rat hippocampal neurons,
which showed that treatment with the 3D6 antibody
prevented tau hyperphosphorylation.103
Clinical development of bapineuzumab was discon-
tinued owing to the lack of clinical benefit. The dose
of bapineuzumab used in these studies, however, was
limited by the emergence of amyloid-related imaging
abnormalities at higher doses. Therefore, it is possible
that despite some target engagement, too little Aβ was
removed to test whether changes in neurodegeneration
markers occur after bapineuzumab treatment.
Solanezumab is a humanized version of the anti-Aβ
monoclonal antibody m266, which has selective affinity
for soluble Aβ. Two large phase III trials (EXPEDITION 1
and 2) have evaluated the efficacy of solanezumab.
Primary end points were not reached, but a pooled analy-
sis showed that solanezumab slowed cognitive decline in
patients with mild AD.106 In a subset of 121 patients who
underwent CSF analyses, total Aβ1–40
and Aβ1–42
levels
were increased after solanezumab treatment, whereas
free (unbound) Aβ1–40
was reduced. These results rep-
licated those observed in phase I and II trials,107,108 and
suggest positive target engagement with mobilization of
the central soluble Aβ pool. Solanezumab is not expected
to bind to fibrillar Aβ109 and, therefore, the observed
increase in total CSF Aβ is unlikely to have come from
interactions with the fibrillar Aβ component. Whether
the effects reflect mobilization within the central soluble
Aβ pool and/or promotion of Aβ efflux from the CNS to
the peripheral circulation remains unclear.110
In addition to Aβ, t-tau and p-tau levels were meas-
ured in CSF after solanezumab treatment, and no
changes were detected.106 This result indicates that mobi-
lization of the Aβ pool did not affect any of the markers
of neurodegeneration. These data seem to support the
Figure 2 | Monoclonal antibodies that target Aβ pathology. One of the main
pathological hallmarks of AD is Aβ plaques, which are formed by progressive
accumulation of Aβ peptides. Solanezumab was designed to bind to monomeric Aβ,
thereby preventing oligomerization and deposition. Bapineuzumab is a humanized
N-terminus-specific monoclonal antibody that binds to Aβ. In animal models of AD,
3D6—the murine form of the antibody—binds to monomeric, oligomeric and fibrillar
forms of Aβ.117 Only the two most investigated antibodies with available cerebrospinal
fluid data have been included. Abbreviations: Aβ, amyloid-β; AD, Alzheimer disease.
Solanezumab
Monomeric Aβ Oligomeric Aβ Protof bril Aβ
Bapineuzumab
Amyloid plaque
Nature Reviews | Neurology
REVIEWS
© 2015 Macmillan Publishers Limited. All rights reserved
Aducanumab (BIIB037)
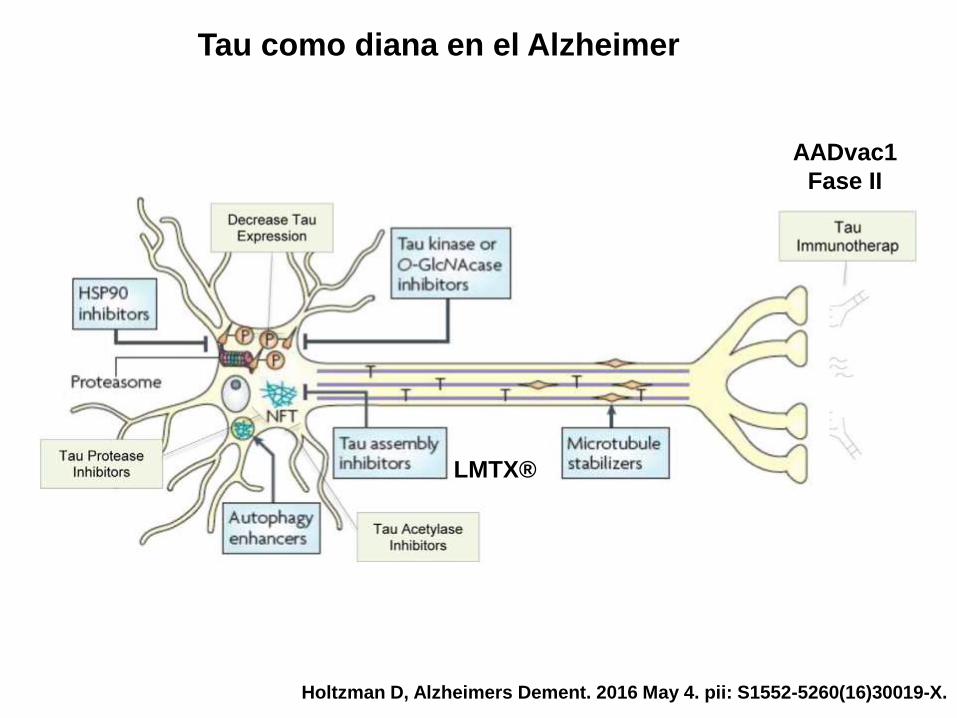
Tau como diana en el Alzheimer
Holtzman D, Alzheimers Dement. 2016 May 4. pii: S1552-5260(16)30019-X.
AADvac1
Fase II
LMTX®
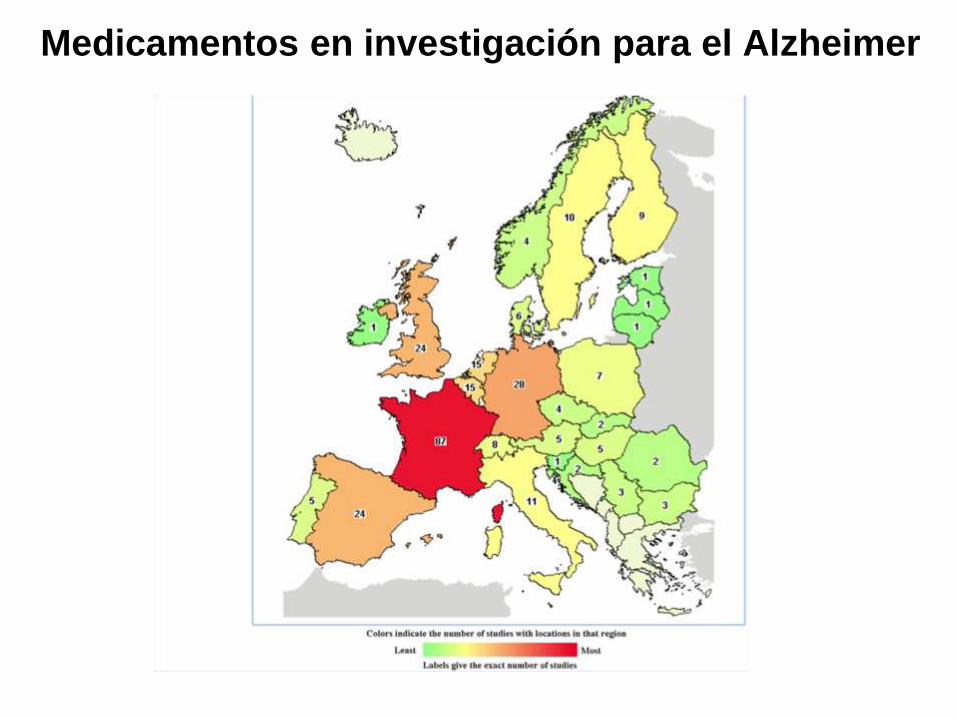
Medicamentos en investigación para el Alzheimer
Cortesía: Roser Ribosa

Estudios activos según enfermedad
(2017)
Cortesía: Roser Ribosa
12.409 Cáncer
5.498 Enf. cardiovasculares
369 Alzheimer

Otras demencias: demencia con cuerpos de Lewy

Se puede prevenir el Alzheimer?

Avance en prevención en 2016
22-44% reducción de casos
nuevos de demencia en 3
décadas

The Lancet 2015 385, 2255-2263DOI: (10.1016/S0140-6736(15)60461-5) Copyright © 2015 Elsevier Ltd Terms and Conditions
Estudio FINGER - Finlandia
1260 personas 60-77 años
2 años
Intervención intensiva
- Dieta
- Ejercicio físico (2-5 veces por semana)
- Estimulación cognitiva
- Control factores de riesgo: diabetes, colesterol, etc
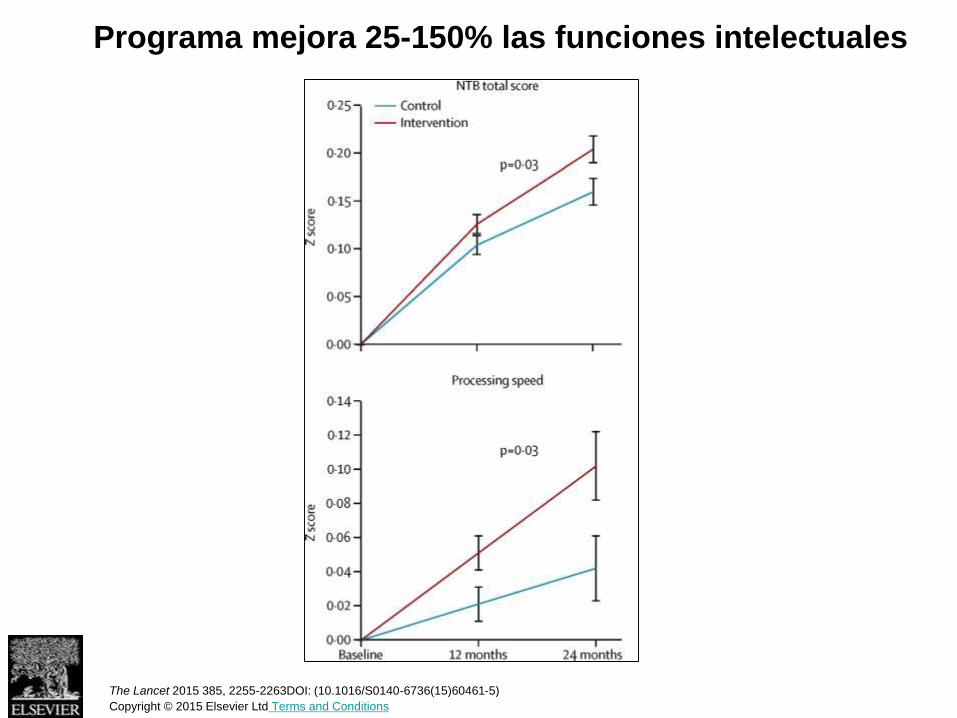
Programa mejora 25-150% las funciones intelectuales
The Lancet 2015 385, 2255-2263DOI: (10.1016/S0140-6736(15)60461-5)
Copyright © 2015 Elsevier Ltd Terms and Conditions
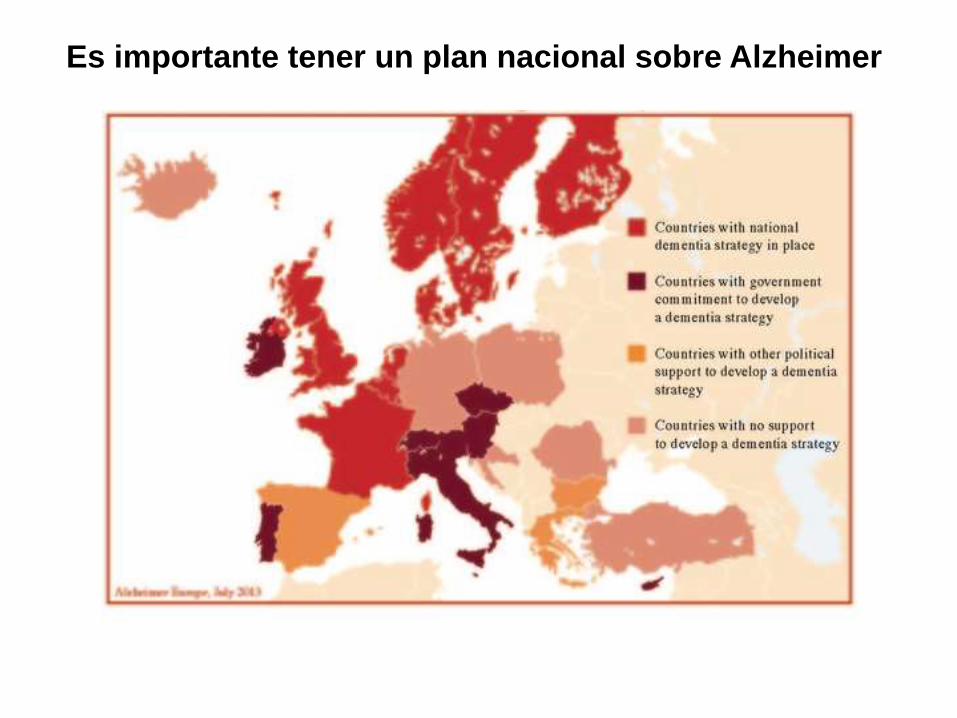
Es importante tener un plan nacional sobre Alzheimer

Mensajes finales
√ Biomarcadores han abierto una nueva era
√ Diagnóstico precoz e información son clave
√ Terapias anti-amiloide en el horizonte
√ Otras estrategias todavía en su infancia
√ El Alzheimer merece un diagnóstico y
tratamiento agresivo como hacemos en otras
enfermedades (ictus o cáncer)