
CTR-REPSOL, Agustín de Betancourt s/n, 28935 Móstoles...
13
Molecular weight dependence and stereoselective chain cleavage during the early stages of the isotactic polypropylene pyrolysis M.C. Martínez a , R. Benavente b , J.M. Gómez-Elvira b * a CTR-REPSOL, Agustín de Betancourt s/n, 28935 Móstoles, Madrid, Spain b Instituto de Ciencia y Tecnología de Polímeros, ICTP-CSIC, Juan de la Cierva, 3, 28006 Madrid, Spain http://dx.doi.org/10.1016/j.polymdegradstab.2017.06.011 Abstract The comparative study on the pyrolysis of two Polypropylene samples, with a similar high isotactic character but quite different molecular weights, reveals a significant difference in their thermal performances. The detailed qualitative analysis of the parameters, that are expected to govern the thermal degradation, leads to reasonably assume a main role of the molecular weight in the relative early stabilities. Furthermore, the correlation found between the sharp initial build-up of the Eact and the selective split-off of isotactic stereosequences in both samples, suggests the relevance of the stereoregular length into the energy required for chain scission. 1. Introduction Despite the huge effort devoted during decades to the study of thermal stability of polypropylene, there remains a need of going deeper into the topic, because some aspects are not still well understood. This is the case of the low initial activation energy (Eact) value of the pyrolysis and of the strong dependence of the Eact on the conversion into volatiles, during the first stages of the process [ 1,2,3,4], i.e. at low and medium temperatures. The controversy arisen around these issues is usually bypassed in the related bibliography by resorting to the general concept of labile structures, like chemical defect, which must be behind such behaviour. However, the matter is not entirely solved and is not of little importance, since the understanding of the key parameters driving such phenomena has significant technological implications. For instance in chemical recycling, where pyrolysis offers a way of recovering valuable chemicals from polyolefin residues. In fact, the low and medium temperature pyrolysis of polyolefins in general, and of polypropylene (PP) in particular, has a great potential as a process for reducing the molecular weight of polymer waste, up to values that can be thus properly introduced in fluidized bed pyrolysis reactors, where the material undergoes the eventual catalysed conversion into chemicals [5,6]. Of course, another optional utility of the low temperature control of pyrolysis is just the production of high added value oligomers, that can be further used as telechelics or lubricants [7,8,9,10]. The analysis carried out in a previous work on the pyrolysis of a high molecular weight isotactic PP [4], evidenced the two foregoing facts. First, the very early Eact value of the process is abnormally low. In fact, the initial thermal cleavage of PP chains occurs at an Eact as low as the typical value of the oxidation process (about 100 KJ mol -1 ), and second, the Eact depends strongly on the weight loss during the first stages of the process. From that study, the high molecular weight has been proposed as the main factor responsible for the former fact, since a large chain size can produce specific intra-chain interactions that would behave as weak points, under the severe restrictions imposed to chain dynamics in the molten state. Concerning the initial Eact build-up with the pyrolysis conversion, the progressive molecular weight diminution was also proposed as the main reason. The hypothesis for these facts being supported by some theoretical as well as experimental works on the matter [11,12].
Transcript of CTR-REPSOL, Agustín de Betancourt s/n, 28935 Móstoles...

Molecular weight dependence and stereoselective chain cleavage during the early stages of the isotactic
polypropylene pyrolysis
M.C. Martíneza, R. Benaventeb, J.M. Gómez-Elvirab*
a CTR-REPSOL, Agustín de Betancourt s/n, 28935 Móstoles, Madrid, Spain b Instituto de Ciencia y Tecnología de Polímeros, ICTP-CSIC, Juan de la Cierva, 3, 28006 Madrid, Spain
http://dx.doi.org/10.1016/j.polymdegradstab.2017.06.011
Abstract
The comparative study on the pyrolysis of two Polypropylene samples, with a similar high isotactic
character but quite different molecular weights, reveals a significant difference in their thermal performances.
The detailed qualitative analysis of the parameters, that are expected to govern the thermal degradation, leads
to reasonably assume a main role of the molecular weight in the relative early stabilities. Furthermore, the
correlation found between the sharp initial build-up of the Eact and the selective split-off of isotactic
stereosequences in both samples, suggests the relevance of the stereoregular length into the energy required
for chain scission.
1. Introduction
Despite the huge effort devoted during decades to the study of thermal stability of polypropylene, there
remains a need of going deeper into the topic, because some aspects are not still well understood. This is the
case of the low initial activation energy (Eact) value of the pyrolysis and of the strong dependence of the Eact
on the conversion into volatiles, during the first stages of the process [1,2,3,4], i.e. at low and medium
temperatures.
The controversy arisen around these issues is usually bypassed in the related bibliography by resorting to
the general concept of labile structures, like chemical defect, which must be behind such behaviour. However,
the matter is not entirely solved and is not of little importance, since the understanding of the key parameters
driving such phenomena has significant technological implications. For instance in chemical recycling, where
pyrolysis offers a way of recovering valuable chemicals from polyolefin residues. In fact, the low and medium
temperature pyrolysis of polyolefins in general, and of polypropylene (PP) in particular, has a great potential
as a process for reducing the molecular weight of polymer waste, up to values that can be thus properly
introduced in fluidized bed pyrolysis reactors, where the material undergoes the eventual catalysed conversion
into chemicals [5,6]. Of course, another optional utility of the low temperature control of pyrolysis is just the
production of high added value oligomers, that can be further used as telechelics or lubricants [7,8,9,10].
The analysis carried out in a previous work on the pyrolysis of a high molecular weight isotactic PP [4],
evidenced the two foregoing facts. First, the very early Eact value of the process is abnormally low. In fact, the
initial thermal cleavage of PP chains occurs at an Eact as low as the typical value of the oxidation process
(about 100 KJ mol-1), and second, the Eact depends strongly on the weight loss during the first stages of the
process. From that study, the high molecular weight has been proposed as the main factor responsible for the
former fact, since a large chain size can produce specific intra-chain interactions that would behave as weak
points, under the severe restrictions imposed to chain dynamics in the molten state. Concerning the initial Eact
build-up with the pyrolysis conversion, the progressive molecular weight diminution was also proposed as the
main reason. The hypothesis for these facts being supported by some theoretical as well as experimental works
on the matter [11,12].

Notwithstanding the correlation found between the molecular weight and the Eact evolution with the
pyrolysis progression, the comparison of the aforementioned thermal response of a high-molecular weight PP
with that one of a substantially lower molecular weight isotactic PP, would afford enormous support on the
influence of the macromolecular character itself, into the labile nature of some regular C-C bonds, on the one
hand. On the other, this comparative study would allow checking the selective involvement of stereo-regular
chain segments into the first stages of the pyrolysis, whatever the molecular weight of the PP may be. This
specific aspect of the molecular weight reduction was proposed to explain the Eact increase, taking place from
the very beginning of the pyrolysis and up to well advanced stages. The idea is that, in the particular case of
the isotactic configuration, not only the more rigid isotactic sequences are selectively broken at the lowest
pyrolysis temperatures, but also that the length of these stereo-regular configuration drives indeed the energy
demanded for the cleavage to occur.
This study compiles therefore the data already presented before, not only to be compared with the new
ones of another isotactic PP, but to be reanalysed in more detail with the purpose of evaluating the general
validity of the explanations given and identifying other parameter with a potentially detrimental impact on the
stability of the PP.
2. Experimental part
2.1 Material and processing
The samples chosen for this study were two Ziegler-Natta highly isotactic PP (PP2472 and PP2477),
kindly supplied by Repsol. They were used as obtained from the polymerization reactor, without any additive.
The high molecular weight specimen (HPP) was synthesized in the absence of hydrogen, while the low
molecular weight one (LPP) was synthesized under 12 bar of hydrogen. The molecular weights, as they have
been obtained by viscometry (section 2.3), are collected in table I. Their respective melt index values are
shown in table I. The powdery samples were processed by hot pressing at 190 ºC and 20 bar, for 2 min, and
quenched at room temperature to yield films, which were finally used for both analytical and preparative
thermo-gravimetric experiments.
Table I. Characterization of the samples, Mw, melt index, crystallinty and melting temperature of the
quenched samples from the melt
Sample Mw
fc
(wt%)
Tm
(ºC)
Melt index
(g·10 min-1)
HPP 940,000 75 162.4 0.12
LPP 195,000 62 163.0 23.35
2.2 Dynamic pyrolysis
The pyrolysis of the polymer films was carried out by running analytical dynamic TGA as well as in
preparative mode, by using a previously described ad-hoc pyrolysis set-up [3,4]. Thus, large enough quantities
of dynamically pyrolysed samples could be obtained to perform a complete characterization of the pyrolysed
polymer, all along the process. The two series of pyrolysed samples are detailed in table II.
The analytical TGA measurements were carried out from 40 ºC up to 750 ºC in a Thermal
AnalysisTGA-Q-500 device. The samples used were 4mm discs of approximately 5 mg cut from the film with
a calibrated die. Constant heating rates of 2, 5, 10 and 20 ºC/min under a 50 mL/min flow of Helium were
used.

Table II. Series of pyrolysed samples
Final
temperature
of pyrolysis
(ºC)
HPP
(Weight loss %)
LPP
(Weight loss %)
200 0.1 0.1
250 0.2 -
300 - 0.36
330 0.7 -
350 4.7 2.1
375 12.8 5.75
385 15.6 -
390 29.8 10.9
400 40.4 23.2
410 59.0 43.3
420 90.1 68.6
In the case of preparative dynamic pyrolysis, the experiments were run from initial quantities between
1.0 and 1.3g of the film. The heating ramp inside the pyrolysis cell was checked to correspond to the
programmed 2 ºC/min rate in the furnace, by means of a K type thermocouple/data logger (Lascar Electronics),
which tracked the actual temperature value every 10 s. After a 15 min purging stage with Nitrogen (250
mL/min), the pyrolysis cell was let equilibrate at 40 ºC and, then, the heating ramp was started in the Nitrogen
stream. Finally, the sample was cooled down under N2 at room temperature. The weight loss was estimated
by difference weighing. All samples were purified by dissolving them in xylene at 110 ºC under N2 bubbling
and final precipitation in ethanol. After washing with ethanol and drying at room temperature under vacuum,
a white polymer residue was obtained in all cases. The purification step assures the removing of impurities,
i.e. volatile oligomers, which can stay into the polymer bulk, especially at the highest pyrolysis temperatures.
2.3 Viscometry
The weight average molecular mass of both virgin PP films, as well as those ones of samples pyrolysed
up to 330 ºC and 350 ºC, for HPP and LPP respectively, were estimated by means of the intrinsic viscosity in
tetralin stabilized with Irganox 1010 (1 g/L), at 135 ºC, by using the relationship [] = 1.66·10-4 · Mw0.733 [13].
Viscometry is not suitable for low molecular weights of samples pyrolysed beyond the abovementioned
temperatures; hence, the 1HNMR analysis was employed in these cases to obtain the number average
molecular weight (section 2.6). In the case of sample HPP pyrolysed at 330ºC and sample LPP pyrolysed at
350ºC, both viscometry and 1HNMR analysis could be used to get the weight and the number average of the
molecular weight.
2.4 FTIR characterization

The FTIR spectra of the virgin HPP and LPP films were recorded by performing 10 scans in a Perkin-
Elmer Spectrum One, in the 4000 to 450 cm-1 range, at a resolution of 2 cm-1. The spectra have been base line
corrected and normalized to the 2722 cm-1 peak.
2.5 Thermal analysis
The thermal properties were analyzed in a TA Q100 Instrument calorimeter connected to a cooling
system and calibrated with Indium and Zinc. The films of the samples were heated from -45 º C to 220 ºC at
a heating rate of 20 ºC/min. The values of Tm and crystallinity (fc) were taken from the maximum and the
enthalpy of fusion, respectively, of the endotherm peak obtained. For the estimation of the crystallinity, a
value of 162 J/g [14] was taken as the melting enthalpy of a 100% crystalline iPP. The results are shown in
table I.
2.6 NMR characterization
Chain microstructure of samples, i.e. molecular weight, tacticity and anomalous groups produced
during the pyrolysis process, was analyzed by means of 13C and 1H NMR spectroscopy. In the former case,
the spectra were obtained from 1,1,2,2- tetrachloroethane-d2 (10 wt%) in a Bruker Avance III HD (125 MHz)
at 100ºC, using the solvent signal at 74.00 ppm as an internal reference. A minimum of 8000 scans were
recorded with broad band proton decoupling, an acquisition time of 0.59 s, a relaxation delay of 5 s and a
pulse angle of 30º. The 1H NMR spectra were previously recorded in the same spectrometer, at the same
temperature, in order to accurately assess the relative content of chemical groups generated during the
pyrolysis. This way, any transformation due to long residence times at high temperature in the 13C NMR
analysis is thus prevented.
While 13CNMR has been employed to measure the evolution of the tacticity distribution with the
pyrolysis temperature [3,4], 1HNMR provides accurate results on the chain-end group content and,
consequently, on the molecular weight from about 50,000 dalton downwards. Terminal double bonds analyzed
include vinylidene, vinylene and isobutenyl groups. The structures of these species, their characteristic signals
and the details about the calculus of the molecular weight are reported in previous works [3,4].
3. Results and discussion
Non-isothermal methods (iso-conversional or not) have been used for long in the estimation of the Eact of
polymer degradation, by using the TGA technique, either under oxidative or inert atmosphere
[1,2,15,16,17,18]. More recently, a comparative study on the thermal and thermooxidative stability of several
polymers, performed by TGA, DSC and CL [19], has shown that the multiple heating rate is the recommended
approach for the Eact assessment, especially when the TGA is used, even though the sensitivity of the loss
volatiles measurement is much lower than that one of the CL signal. Despite this, some care is needed when
interpreting TGA results in terms of the energy required for chain bond scission to take place. In fact, values
of the Eact obtained at very high weight losses can be influenced by some other processes than the mere chain
scission. This is the case, for instance, of the volatilization of relatively high molecular weight molecules,
either in the form of radical or neutral species. Consequently, although the Eact has been calculated all along
the complete weight loss, only conversions up to 50% have been considered in order to withdraw consistent
conclusions.
The TGA curves for the two PP samples studied, obtained at the different heating rates, are shown in figure
1. All of them correspond to one-single step processes that can been described through an apparent first order
mechanisms, where the only variable is the mass conversion into volatiles [1,15,19]. This feature can be also
remarked in the DTGA curves shown in figure 2, where one single peak is detected. The most important
difference between the two PP samples is the quite different stability found at low heating rates. It is absolutely

clear that the lowest molecular weight PP exhibits higher values of both Tonset (figure 1) and Tpeak (figure
2), especially at 2 ºC·min-1 (see table I) and at low and medium conversion degrees (roughly below 50 weight
loss %). On the contrary, an inversion of the thermal stability is found at 10 and 20ºC·min-1.
Figure 1. Series of TGA curves for the HPP (black) and LPP (blue) films at 2 (solid thick lines), 5 (solid thin
lines), 10 (dashed lines) and 20 ºC/min (dotted lines).
Figure 2. Series of the dW/dT curves for the pyrolysis of the HPP (black) and LPP (blue) samples at 2 (solid
thick lines), 5 (solid thin lines), 10 (dashed lines) and 20 ºC/min (dotted lines).
The fact confirming that the thermal stability of the two PP samples is really different, is what it is found
when the weight loss is tracked during the preparative pyrolysis. Figure 3 displays the weight loss evolution
at 2ºC·min-1 in both samples, under nitrogen, and the corresponding analytical TGA curves at the same heating
rate. It is quite apparent that preparative and analytical curves are not that different and then, that the two
specimens reflect an identical qualitative behaviour under these different conditions, being the low-molecular

weight PP more stable. The inspection of the pyrolysis progression in both polymers, shown in table II, reveals
that the weight loss reached by LPP is about a half of that one of HPP, just from low conversions at 350ºC,
and this difference keeps up almost constant for conversions under 50 wt loss %.
Figure 3. Comparison of the weight loss at 2 ºC/min for analytical (lines) and preparative (circles) TGA
runs. Data for HPP are in black and in blue for LPP.
On account of the fact that little differences between the two samples, arising from distinctive
characteristics of the PP samples, can make the development of the early stages of pyrolysis to be kinetically
different, it is imperative to consider, at least from a qualitative point of view, which influence would be
expected for each one of them.
Some data in related literature [19] have evidenced the influence of air traces adsorbed in the amorphous
phase in the initial degradation states of pyrolysis. For this reason, this study has been carried out with films
obtained under same processing conditions (see experimental part). Furthermore, the crystallinity of the
samples has been compared in order to assess the fraction of amorphous phase, where air might be trapped at
a molecular level. The results obtained by means of the DSC are shown in table 1 and allow reasonably
concluding that LPP can retain a higher oxygen content and, consequently, its thermal stability should be
lower than that one of HPP. However, the actual fact is that LPP is more stable.
The difference in melt viscosity is a second parameter that must be taken into account. The large difference
in the melt index of both samples (table I) is expected to make the loss of volatiles more difficult in HPP and,
then, to show this sample as apparently more stable. However, the reverse fact occurs.
Other factor that could be involved is the quantity and/or quality of anomalous groups, mainly oxygen-
containing functions and double bonds. On this issue, the role of terminal double bonds in the different thermal
stability is ruled out, because of the smaller content of such groups in the highest molecular weight HPP. In
addition, the fact that the Eact increases in the initial states of pyrolysis, despite the continuous double bond
build-up, allows attributing a low relevance to these species as a pro-degradant under pyrolytic conditions.
A second type of anomalous groups are oxygen-containing chain units. In this case, a meticulous
inspection of the very small 1HNMR signals, i.e. those ones whose intensity is equal or below the olefinic
protons in the virgin polymers, reveals that the only peak with a vanishing evolution with pyrolysis conversion

seems to be that one at 2.15 ppm, that can be tentatively assigned to methyl ketone groups [20]. This is what
can be inferred from the evolution of such signal for HPP and LPP in figures 4 and 5 respectively. In both
figures, the increasingly intensity of the vinylidene signals at 4.73 ppm, 4.81 ppm and from 2.10 to 2.15 ppm,
allows tracking the advance of the pyrolysis. Certainly, the removal of methylketone groups seems to occur
at low conversions but, once again, the results contradict what it would be expected if these carbonyl species
had a great impact in the very early thermal stability of the samples. Actually, the less stable HPP sample
shows a lower content of these carbonyl species than the LPP sample, as it can be also confirmed by comparing
the normalized FTIR windows corresponding to the CO stretching region. This can be checked in figure 6,
where the signals of CO groups of either saturated ketones and acids (1708 cm-1) or α,β-unsaturated ketones
(1688 cm-1) are more intense in the normalized spectrum of LPP.
Figure 4. Evolution of the 1H NMR spectral window of protons associated to vinylidene and methylketone
end-capping groups, for HPP, with the weight loss %.
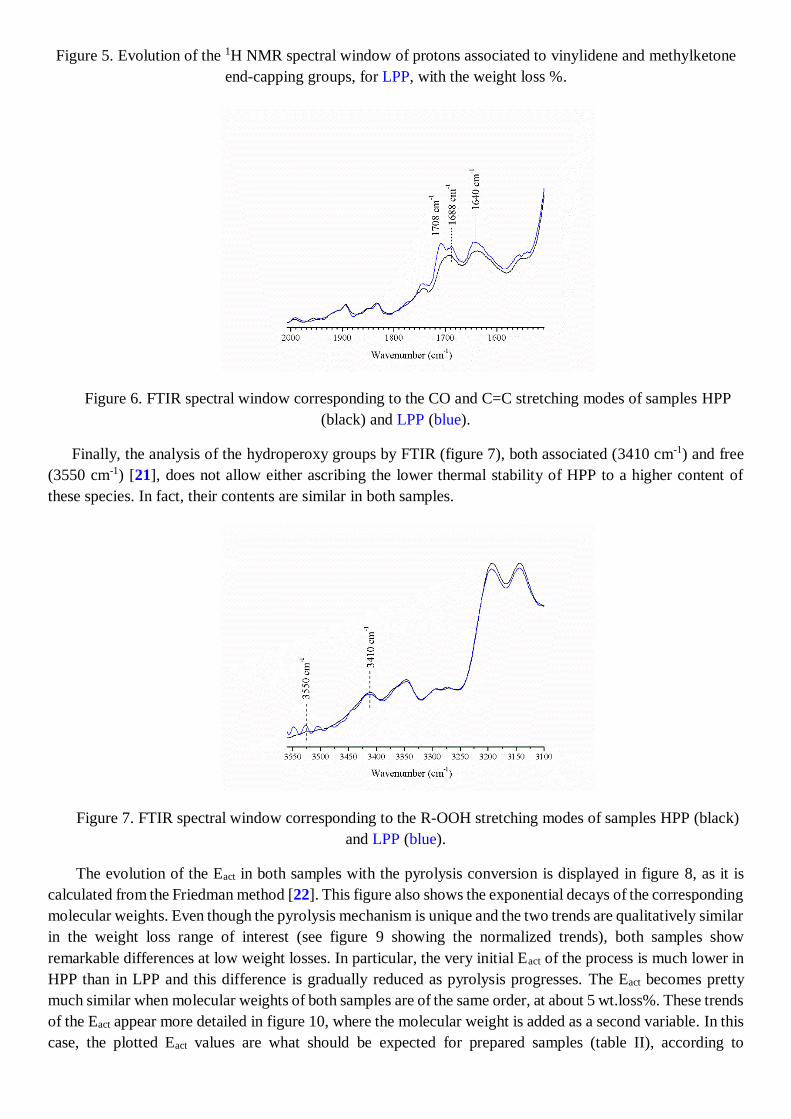
Figure 5. Evolution of the 1H NMR spectral window of protons associated to vinylidene and methylketone
end-capping groups, for LPP, with the weight loss %.
Figure 6. FTIR spectral window corresponding to the CO and C=C stretching modes of samples HPP
(black) and LPP (blue).
Finally, the analysis of the hydroperoxy groups by FTIR (figure 7), both associated (3410 cm-1) and free
(3550 cm-1) [21], does not allow either ascribing the lower thermal stability of HPP to a higher content of
these species. In fact, their contents are similar in both samples.
Figure 7. FTIR spectral window corresponding to the R-OOH stretching modes of samples HPP (black)
and LPP (blue).
The evolution of the Eact in both samples with the pyrolysis conversion is displayed in figure 8, as it is
calculated from the Friedman method [22]. This figure also shows the exponential decays of the corresponding
molecular weights. Even though the pyrolysis mechanism is unique and the two trends are qualitatively similar
in the weight loss range of interest (see figure 9 showing the normalized trends), both samples show
remarkable differences at low weight losses. In particular, the very initial Eact of the process is much lower in
HPP than in LPP and this difference is gradually reduced as pyrolysis progresses. The Eact becomes pretty
much similar when molecular weights of both samples are of the same order, at about 5 wt.loss%. These trends
of the Eact appear more detailed in figure 10, where the molecular weight is added as a second variable. In this
case, the plotted Eact values are what should be expected for prepared samples (table II), according to

extrapolations based on curves shown in figure 8. In such figure, the projection of the 3D curves on the
Eact/Mw plane illustrates how the above mentioned convergence of the Eact takes place in a roughly unique
linear way, once the pyrolysis has left the initial stages.
Figure 8. Evolutions of Eact (solid circles) and molecular weight (open circles) with the weight loss %. HPP
(black), LPP (blue)
Figure 9. Evolutions of Eact with the weight loss % in a normalized scale. HPP (black), LPP (blue)
To sum up, two main results are well worth standing out. Firstly, despite the fact that the mechanism of
the PP is unique, there is a significant difference in the Eact of processes occurring at the very beginning of the
pyrolysis, i.e. when the molecular weight difference in both PP samples is very important. Secondly, the Eact
reach similar and increasingly values for further conversions, when molecular weight become similar.
As far as the initial Eact gap is concerned, some causes have been argued to account for it without reaching
a definitive conclusion, because of the practical difficulties in evidencing their influence. The most invoked
one has been the presence of very low contents of anomalous groups in the virgin [1,2,23] polymer, as it is
obtained from the polymerization reactor. On this matter, the recent work of Tobita et al [24] is a smart way
of proving that some chemical defects are inevitably produced during the propene polymerization. In
particular, the content of foreign groups seems to be closely related to the catalyst activity and, therefore, to
very high local temperatures in the polymerization medium, which would be ultimately the reason for the
production of anomalous chemical structures.
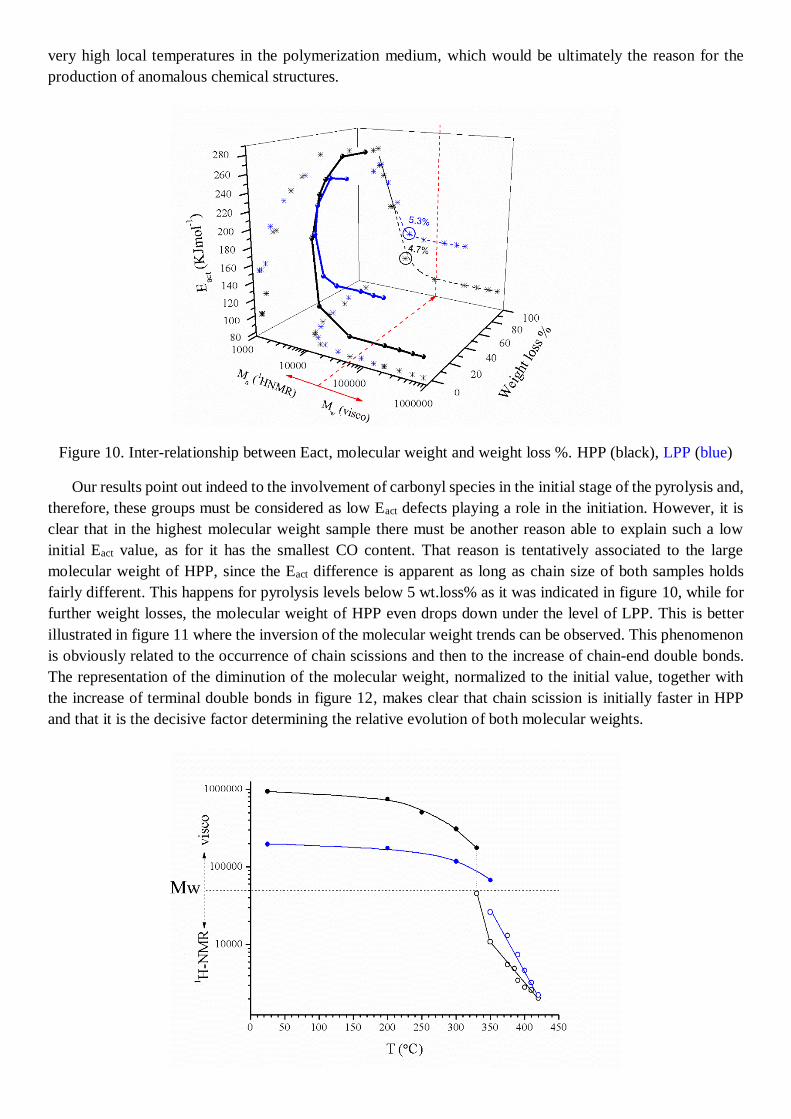
Figure 10. Inter-relationship between Eact, molecular weight and weight loss %. HPP (black), LPP (blue)
Our results point out indeed to the involvement of carbonyl species in the initial stage of the pyrolysis and,
therefore, these groups must be considered as low Eact defects playing a role in the initiation. However, it is
clear that in the highest molecular weight sample there must be another reason able to explain such a low
initial Eact value, as for it has the smallest CO content. That reason is tentatively associated to the large
molecular weight of HPP, since the Eact difference is apparent as long as chain size of both samples holds
fairly different. This happens for pyrolysis levels below 5 wt.loss% as it was indicated in figure 10, while for
further weight losses, the molecular weight of HPP even drops down under the level of LPP. This is better
illustrated in figure 11 where the inversion of the molecular weight trends can be observed. This phenomenon
is obviously related to the occurrence of chain scissions and then to the increase of chain-end double bonds.
The representation of the diminution of the molecular weight, normalized to the initial value, together with
the increase of terminal double bonds in figure 12, makes clear that chain scission is initially faster in HPP
and that it is the decisive factor determining the relative evolution of both molecular weights.

Figure 11. Evolution of the molecular weight with the final pyrolysis temperature for HPP (black) and LPP
(blue): solid circles correspond to weight averages from viscometry and open circles to number averages
from 1H NMR.
Figure 12. Evolutions of the molecular weight (normalized for values from viscosity) (solid circles) and
terminal double bond content (open circles) with the final pyrolysis temperature: HPP (black) and LPP
(blue)
On the basis of the above commented considerations, the high molecular weight is proposed to be in the
origin of the lower initial thermal stability of HPP with respect to LPP. The higher chain size of the former is
expected to generate more severe restrictions to chain dynamics in the molten state by producing intra-chain
interactions which, under high thermal stress, would behave as weak points. The work of Nyden et al [11] on
the simulation of the iPP pyrolysis predicts indeed an initial value for the Eact as low as 100 KJ·mol-1 and
points out to an effective role of the macromolecular character itself in the initiation of pyrolysis. In our case,
considering that the isotacticity degree is similar in both samples, the especially high molecular weight of HPP
is a factor that would justify a lower Eact value.
On the other hand, the aforementioned authors have also shown how the Eact of the poly-isobutylene
pyrolysis increases when the molecular weight diminishes [12], because of the removal of non-bonded
interactions in polymer chains. The loss of the macromolecular character of chains makes the Eact to gradually
approach the theoretical value corresponding to the C-C dissociation energy. This is what it is found with the
pyrolysis progression in both PP samples. Anyway, the measured Eact values must be considered to reflect the
steric characteristic of links between propylene units that, in our case, is isotactic in both samples. In fact, the
Eact evolution of the more flexible syndiotactic PP chains has been reported to be appreciably over the values
presented here [3,25]. A difference that is ascribed to the energy consumption needed for the distinctive GGTT
helix in sPP chains to be uncoiled, before the C-C scission occurs [25]. On the contrary, the ability of the
characteristic GTGT helix to dissipate energy by deformation is quite limited. Therefore, isotactic chain
segments are expected to be especially prone to be broken over the temperature build-up.
This hypothesis can be assumed from the detailed evaluation of the tacticity distribution at the pentad level
in the iPP samples, all over the conversion range. Figure 13 reveals indeed, that the production of terminal
double bonds takes place at the expense of C-C bonds in (mmmm) pentads exclusively, while the content of
any other pentad does not change in the initial conversion range (only the total of the (non-mmmm) pentads

has been plotted for the purpose of clarity). This happens up to a content of roughly 2 mol% of terminal double
bonds, which corresponds to 23 and 30 wt.loss % for LPP and HPP respectively. Within these initial weight
loss periods, the molecular weight of both PP samples has already collapsed up to values in the order of a few
thousand Dalton (4600 and 3700 for LPP and HPP respectively), and the Eact is about to leave the initial stage
of fast growth. This finding suggests that the increase of the Eact could be related with the variations in length
of the isotactic segments involved, in such a manner that the shorter the isotactic length is the more difficult
the chain scission would occur. A hypothesis that needs confirmation by additional work.
Figure 13. Evolution of the mmmm (solid circles) and non-mmmm (open circles) pentad contents with the
percentage of terminal double bonds. HPP (black), LPP (blue)
4. Conclusions
Despite the fact that the two PP studied exhibit similar values of isotacticity, their thermal stabilities are
proven to be quite different. The negative impact of some factors like oxygen adsorbed in the amorphous
phase and the physical restrictions imposed by the viscosity of the molten state for the volatiles to evolve, are
inferred not to be important, as for they would contribute the most in the more stable LPP specimen.
In addition, although methylcarbonyl groups are likely involved in the very early stages of the pyrolysis,
they do not explain the particularly low values of both the Tonset and the initial Eact of the HPP sample. On
account of the fact that the largest difference between both PP lies in their molecular weights, a very large
chain size is proposed to produce an enhanced content of specific chain interactions in the molten state, which
would behave as weak points under high thermal stress. Bringing to mind again the secondary role of other
factors, the higher thermal stability of the low molecular weight sample suggests that a shorter average chain
length is the main cause of a higher performance, since the likelihood of producing weak points by intrachain
distortions is lower.
The further Eact increase with the progression of the pyrolysis, can be related not only to the diminution of
the molecular weight, as some theoretical works predict, but to the decrease of the length of isotactic segments
where C-C scission selectively occurs. The linear relationship between the vanishing of (mmmm) pentads and
the initial build-up of terminal double bonds, i.e. during the diminution and collapse of the PP molecular
weight, without changing that one of the (non-mmmm) pentads, supports this hypothesis.

Acknowledgements
The authors acknowledge the financial support of MINECO Spain (project MAT2016-79869-C2-1-P), as
well as the kindly assistance of Repsol for the supply of samples.
References
1 Chan JH, Balke ST. The thermal degradation kinetics of polypropylene: part III. Thermogravimetric analyses. Polym Deg Stab
1997;57:135-49. 2 Peterson JD, Vyazovkin S, Wight CA. Kinetics of the thermal and thermooxidative degradation of polystyrene, polyethylene and
poly(propylene). Macromol Chem Commun 2001;202:775-84. 3 Gómez-Elvira JM, Benavente R, Martínez MC. Unravelling the contribution of chain microstructure in the mechanism of the
syndiotactic polypropylene pyrolysis. Polym Deg Stab 2013;98:1150-63. 4 Gomez-Elvira JM., Benavente R, Martínez MC, Correlation between chain microstructure and activation energy in the pyrolysis
of a high molecular weight isotactic polypropylene, Polym Deg Stab 117 (2015) 46-57. 5 Marcilla A, Gómez-Siurana A, Berenguer D, Study of the influence of the characteristics of acid solids in the catalytic pyrolysis
of different polymers. App Cat: A General 2006;301:222-231. 6 Marcilla A, Gómez-Siurana A, Berenguer D, Study of the early deactivation in pyrolysis of polymers in the presence of catalysts.
J Anal App Pyr (2007);79:443-449. 7 Sawaguchi T, Ikemura T, Seno M. Preparation of α,-Diisopropenyloligopropylene by thermal degradation of isotactic
polypropylene. Macromolecules 1995;28:7973-8. 8 Sawaguchi T, Suzuki Y, Sakaki A, Saito H, Yano S, Seno M. Chemical recycling of commodity vinyl polymers: selective
preparation of end-reactive oligomers by controlled thermal degradation. Polym Int 2000;49:921-5. 9 C. Janiack. Metallocene and related catalysts for olefin, alkyne and silane dimerization and oligomerization. Coord Chem Rev
2006;250:66–94. 10 C. Janiak, F. Blank. Metallocene catalysts for olefin oligomerization. Macromol Symp 2006;236:14–22. 11 Nyden MR, Stoliarov SI, Westmoreland PR, Guo ZX, Jee C. Applications of reactive molecular dynamics to the study of the
thermal decomposition of polymers and nanoscale structures. Mat Sci Eng 2004;A365:114-21. 12 Stoliarov SI, Lyon RE, Nyden MR. A reactive molecular dynamics model of thermal decomposition in polymers. II.
polyisobutylene. Polymer 2004;45: 8613-21. 13 Shiomura T, Kohno M, Inoue N, Asanuma T, Sugimoto R, Iwatani T, Uchida O, Kimura S, Harima S, Zenkoh H, Tanaka E.
Recent advances in olefin polymerization Macromol Symp 1996;101:289-99. 14 Krache R, Benavente R, López-Majada JM, Pereña JM, Cerrada ML, Pérez E. Competition between α, β, and polymorphs in a
β-nucleated metallocenic isotactic polypropylene. Macromolecules 2007;40:6871-8. 15 Hatakeyama T, Quinn FX. Thermal analysis: Fundamentals and applications to polymer science. 2nd Ed. Chichester: Wiley; 1999. 16 Ito M, Inayoshi S, Moriyama K. A simple method for the evaluation of heat resistant property of elastomers. Polym Deg Stab
2008;93:1935-38. 17 Simoes PN, Coelho J, Gonçalves P, Gil MH. Comparative non-isothermal kinetic analysis of termal degradation of poly(vinyl
chloride) prepared by living and conventional free radical polymerization methods. Eur Polym J 2009;45:1949-1959. 18 Arranz-Andrés J, López FA, Benavente R. The addition of aluminum nanoparticles to polypropylene increases its thermal
stability. Int J Eng Res Appl 2017;7:5-12. 19 Rychlý J, Matisová-Rychlá L, Csmorová K, Janigová I. Non-isothermal thermogravimetry, differential scanning calorimetry and
chemiluminescence in degradation of polyethylene, polypropylene, polystyrene and poly(methyl methacrylate). Polym Deg Stab
2011;96:1573-1581. 20 Spectral Database for Organic Compounds, SDBS. http://riodb01.ibase.aist.go.jp/sdbs/cgi-bin/cre_search.cgi. 21 Chien JCW, Vanderberg EJ, Jabloner H. Polymer reactions. III. Structure of polypropylene hydroperoxide. J Polym.Sci A1
1968;6:381-392. 22 Friedman H. Kinetics of thermal degradation of char-forming plastic from thermogravimetry. Application to a phenolic plastic. J
Polym Sci C 1964;C6: 183-95. 23 Kruse T, Wong HW, Broadbelt LJ. Mechanistic modeling of polymer pyrolysis: polypropylene. Macromolecules 2003;36:9594-
607. 24 Tobita T, Chammingkwan P, Terano M, Taniike T, Initiation of oxidative degradation in polypropylene reactor powder produced
by Ziegler-Natta catalyst. Polym Deg Stab (2017);137:131-137. 25 He P, Xiao Y, Zhang P, Xing Ch, Zhu N, Zhu X, Yan D. Thermal degradation of syndiotactic polypropylene and the influence of
stereoregularity on the thermal degradation behaviour by in situ FTIR spectroscopy. Polym Deg Stab 2005;88:473-9.

![Protocolo CSI CO2: Hacia un lenguaje común...16 Puzzolana [t/yr, dry weight] 17 Others (e.g., CKD added to cement mill) [t/yr, dry weight] 18 Total MIC consumed for Portland and blended](https://static.fdocuments.ec/doc/165x107/610cec8f0b585204937e2fe5/protocolo-csi-co2-hacia-un-lenguaje-comn-16-puzzolana-tyr-dry-weight.jpg)













![Bobina Rogowski como sensor de corriente para monitoreo … · 6. Referencias. [1] Luka Ferković,DamirIlićand Roman Malarić, Mutual inductance of a precise Rogowski coil in dependence](https://static.fdocuments.ec/doc/165x107/5ae7f1cc7f8b9acc268f5d3d/bobina-rogowski-como-sensor-de-corriente-para-monitoreo-referencias-1-luka.jpg)

