
Craneofaringioma 2012
28
Craneofaringioma Departamento Neurocirugía Pregrado Dr. Gustavo Villarreal Reyna Dr. Carlos Eduardo Salazar Mejía
-
Upload
neurocirugia2012 -
Category
Education
-
view
11.313 -
download
4
description
www.neurocirugiaendovascular.com
Transcript of Craneofaringioma 2012

Craneofaringioma
Departamento Neurocirugía Pregrado
Dr. Gustavo Villarreal Reyna
Dr. Carlos Eduardo Salazar Mejía

Craneofaringioma• Es un tumor disembrioplásico originado a partir de restos de la
bolsa de Rathke, de localización generalmente supraselar.
• Tienden a emerger de los márgenes anterosuperiores de la hipófisis anterior, algunos pueden emerger del III Ventrículo.
• No sufre degeneración maligna, pero la dificultad en su tratamiento puede darle un comportamiento maligno.
Craneofaringioma
CLASIFICACIÓN HISTOLÓGICA:
Craneofaringioma Adamantinomatoso(Los más frecuentes, mayormente pacientes pediátricos)
Craneofaringioma Papilar(Más frecuentes en adultos)
Mixtos

Relaciones anatómicas
Mas frecuentemente surgen cerca del tallo hipofisario, y se proyectan hacia el hipotálamo. Se extienden:
Anteriormente- Hacia la cisterna prequiasmática y espacios subfrontales.Posteriormente- Dentro de las cisternas prepontina e interpeduncular,
ángulo cerebelopontino, tercer ventrículo, fosa posterior y foramen Magno.
Lateralmente- Hacia los espacios subtemporales.
Pueden incluso llegar a la Cisura de Sylvio.
• En raras ocasiones, pueden tener origen extradural o extracraneal(craneofaringiomas nasofaríngeos , de fosa posterior o que se extienden hacia la columna cervical)
• El craneofaringioma puramente intraventricular es usualmente de tipo escamoso papilar, muy raro.

Epidemiología
Incidencia• 0.5- 2 casos por 100,000 habitantes / año.
• 4% de todos los tumores intracraneales en Niños.
(1-3% población general)
• 56% de los tumores selares en Niños. (13% población general).
• Picos de incidencia: Niños de 5-14 años, Adultos 65-74 años.
• No existen diferencias en cuanto a género.

Anatomía Patológica
• Epitelio tumoral escamoso estratificado.
• Tumoración Sólido-Quística. Se presentan como un quiste simple o múltiples quistes (70-75%) llenos de una sustancia turbia, proteinácea, de color café amarillento debido a su contenido de cristales de colesterol.
Calcificaciones: Microscópicas 50%, RX 90% en niños, 50% adultos
• El subtipo papilar raramente calcifica.
• Otros hallazgos son estructuras hialinas, colágeno, fibroblastos, células gigantes tipo cuerpo extraño.
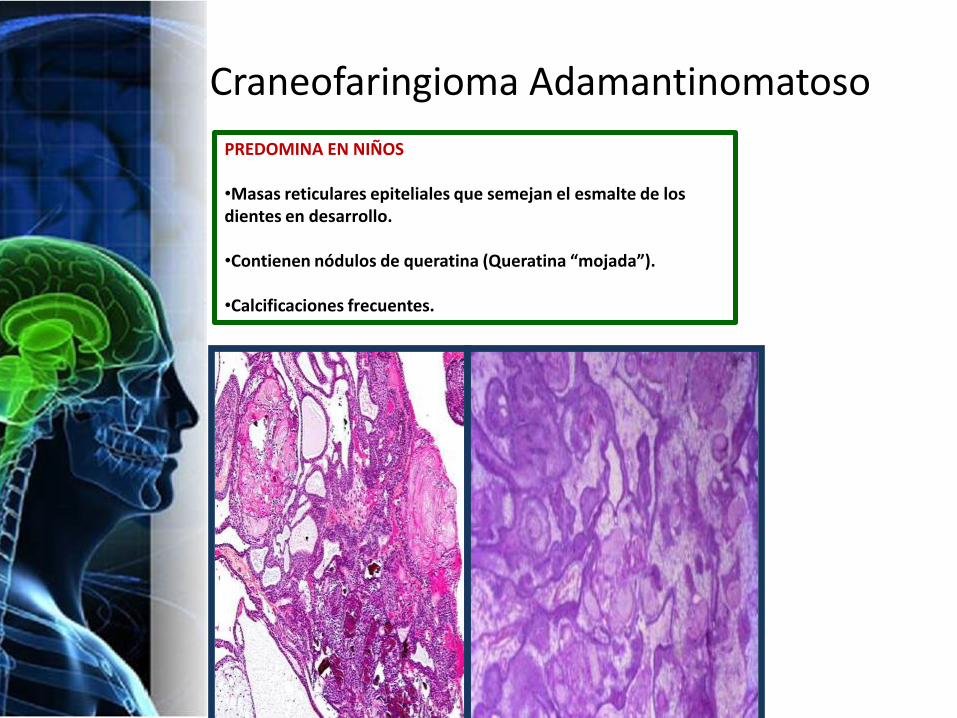
Craneofaringioma Adamantinomatoso
PREDOMINA EN NIÑOS
•Masas reticulares epiteliales que semejan el esmalte de los dientes en desarrollo.
•Contienen nódulos de queratina (Queratina “mojada”).
•Calcificaciones frecuentes.
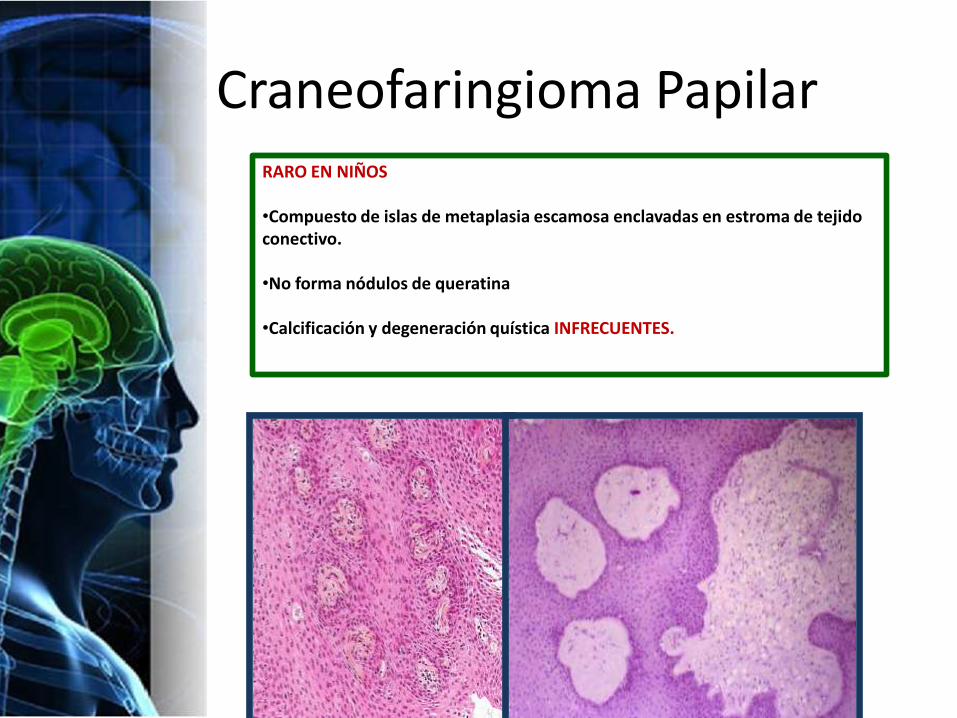
Craneofaringioma PapilarRARO EN NIÑOS
•Compuesto de islas de metaplasia escamosa enclavadas en estroma de tejido conectivo.
•No forma nódulos de queratina
•Calcificación y degeneración quística INFRECUENTES.
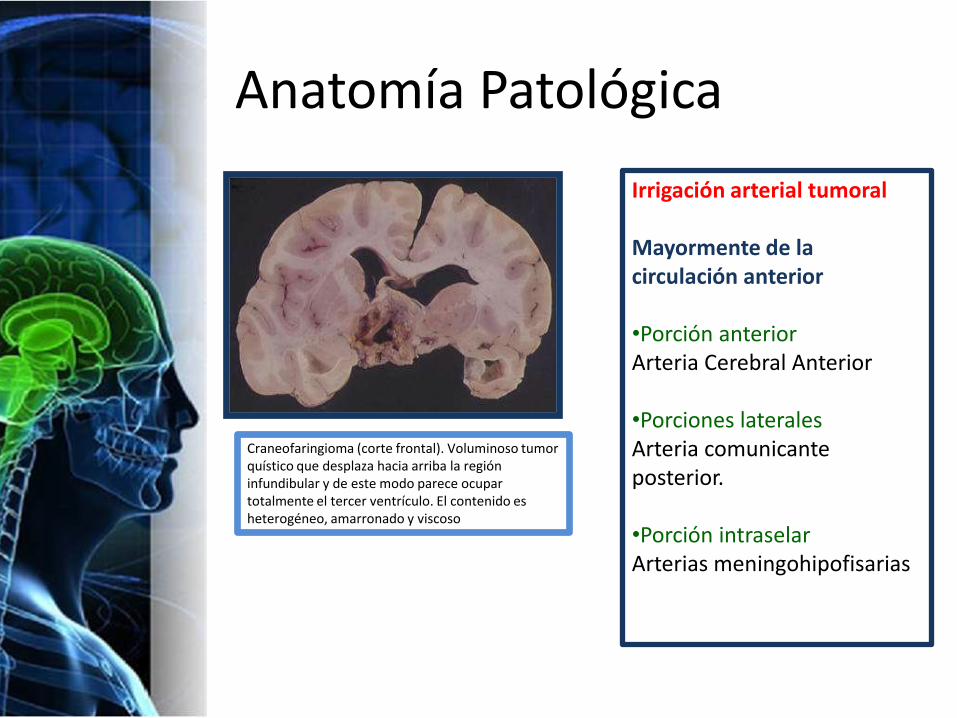
Anatomía Patológica
Craneofaringioma (corte frontal). Voluminoso tumor quístico que desplaza hacia arriba la región infundibular y de este modo parece ocupar totalmente el tercer ventrículo. El contenido es heterogéneo, amarronado y viscoso
Irrigación arterial tumoral
Mayormente de la circulación anterior
•Porción anteriorArteria Cerebral Anterior
•Porciones lateralesArteria comunicante posterior.
•Porción intraselarArterias meningohipofisarias
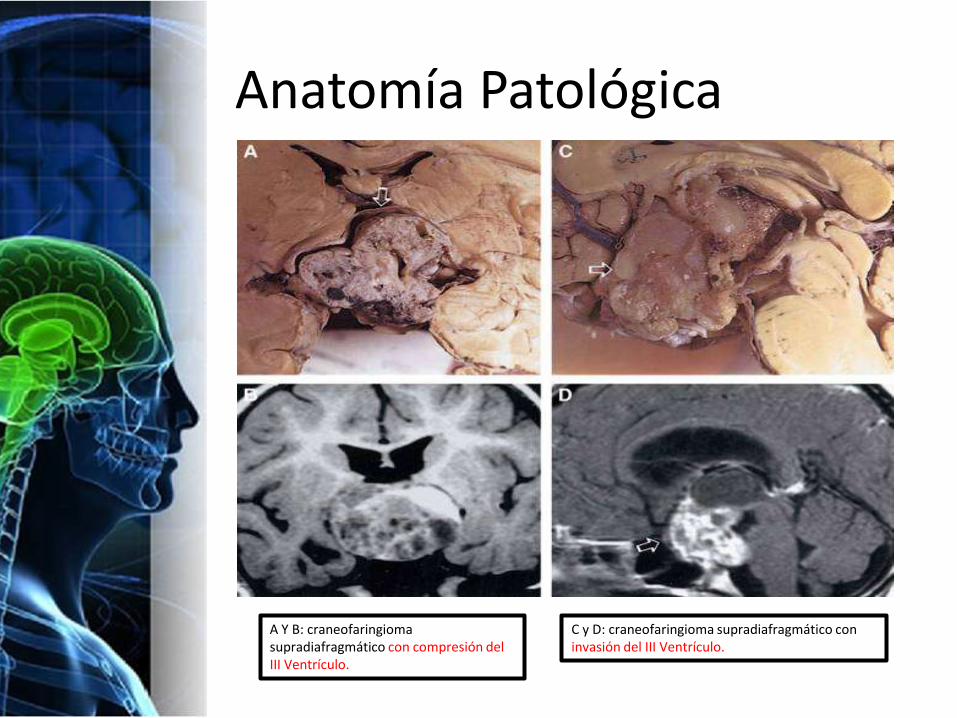
Anatomía Patológica
A Y B: craneofaringiomasupradiafragmático con compresión del III Ventrículo.
C y D: craneofaringioma supradiafragmático con invasión del III Ventrículo.

Etiología
Teoría Dual (Teoría embriogénica + Teoría metaplásica)
• El tipo adamantinomatoso surge de los remamentesepiteliales del conducto craneofaríngeo de Rathke.
• El tipo escamoso papilar surge como resultado de la metaplasia de restos de células escamosas epiteliales, remanentes del estomodeo.

Manifestaciones Clínicas
• Tumores de lento crecimiento. Usualmente se vuelven sintomáticos al llegar a los 3cms de tamaño.
• Intervalo de 1-2 años desde inicio de síntomas hasta diagnóstico.
Elevación de la presión intracraneal Cefalea progresiva (55-68%), náuseas y vómitos. Papiledema.
Disfunción endócrina (66-90%)
Hipofunción: Hipotiroidismo (40%), hipotensión ortostática (25%), talla baja (23-45%), diabetes insípida (20%), amenorrea, impotencia .
Hiperfunción: Pubertad precoz en niñosObesidad en adultos (11-18%).

Manifestaciones ClínicasAlteraciones visuales (40-70% en adultos, 20-30% en niños):
Hemianopsia heterónima bitemporal, homónima, escotoma o atrofia óptica.
POR LOCALIZACIÓN TUMORAL:Prequiasmática: Atrofia del nervio óptico (agudeza, campos visuales)Retroquiasmática: Hidrocefalia con signos de hipertensión
intracraneal.Intraselar: Cefalea y endocrinopatía.
OTROS SÍNTOMASMeningitis química (por ruptura quística)Crisis comicialesBajo desarrollo intelectualLabilidad emocional

Diagnóstico
Hallazgos clínicos
Hallazgos radiológicos
Confirmación: Histológica

ImagenologíaLesión selar/paraselar parte sólida, parte quística, calcificada.
75% Supraselares20% Supra e infraselar5% Intra/Infraselares.
• La calcificación se aprecia mejor con TAC.• Extensión tumoral se aprecia mejor con RM.
Elección.
• Angio RM delimita vasos cererbrales y su relación con el tumor, ayuda a diferenciar la lesión de una posible malformación vascular.
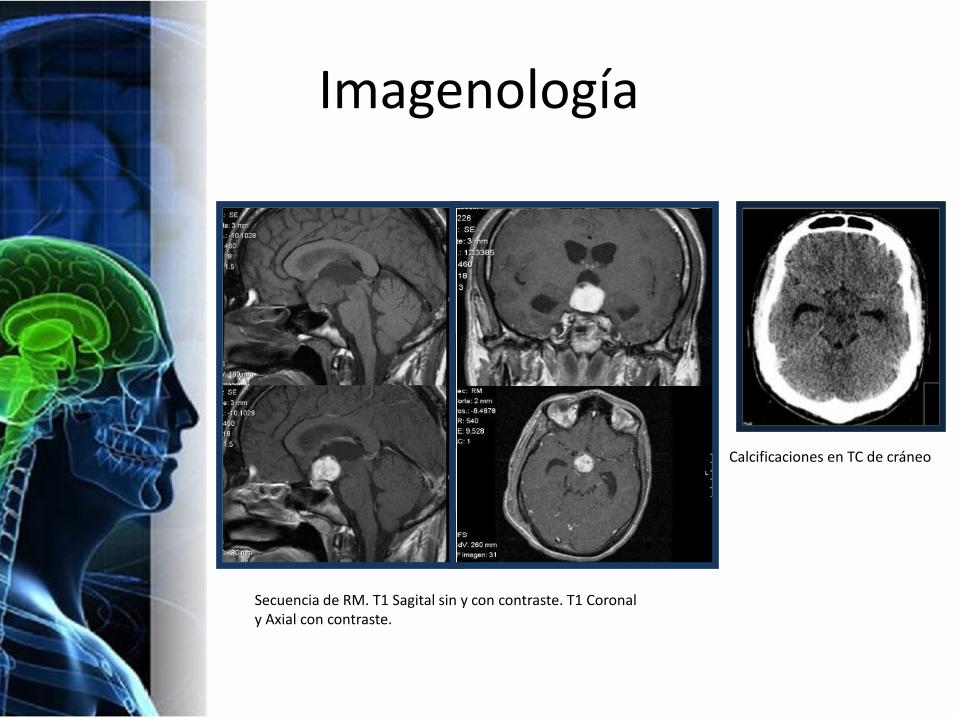
Imagenología
Calcificaciones en TC de cráneo
Secuencia de RM. T1 Sagital sin y con contraste. T1 Coronal y Axial con contraste.
Estudios Hormonales
• Niveles de GH, TSH, LH, FSH, Cortisol y osmolaridadplasmática y urinaria.
• Toda anomalía hormonal debe ser tratada

Diagnóstico Diferencial
Anomalías congénitas.• Quiste aracnoideo.• Quiste de la bolsa de Rathke.
Otros tumores.• Tumores hipofisarios.• Metástasis.• Meningioma.• Dermoides y epidermoides.• Gliomas óptico-hipotalámicos.
Proceso infeccioso/inflamatorio.• Granuloma eosinofílico.• Hipófisis linfocitaria.• Sarcoidosis.• Tuberculosis.

Diagnóstico Diferencial en PatologíaCraneofaringioma VS Quiste de la Bolsa de Rathke
Característica Craneofaringioma Quiste de la Bolsa de Rathke
Sitio de origen Margen anterosuperiorde la hipófisis.
Pars intermedia de la hipófisis.
Línea celular Epitelio escamoso estratificado
Epitelio cuboidal noestratificado
Contenido quístico Cristales de colesterol Semeja aceite de motor
Tratamiento quirúrgico Remoción total es el objetivo.
Escisión parcial y drenaje.
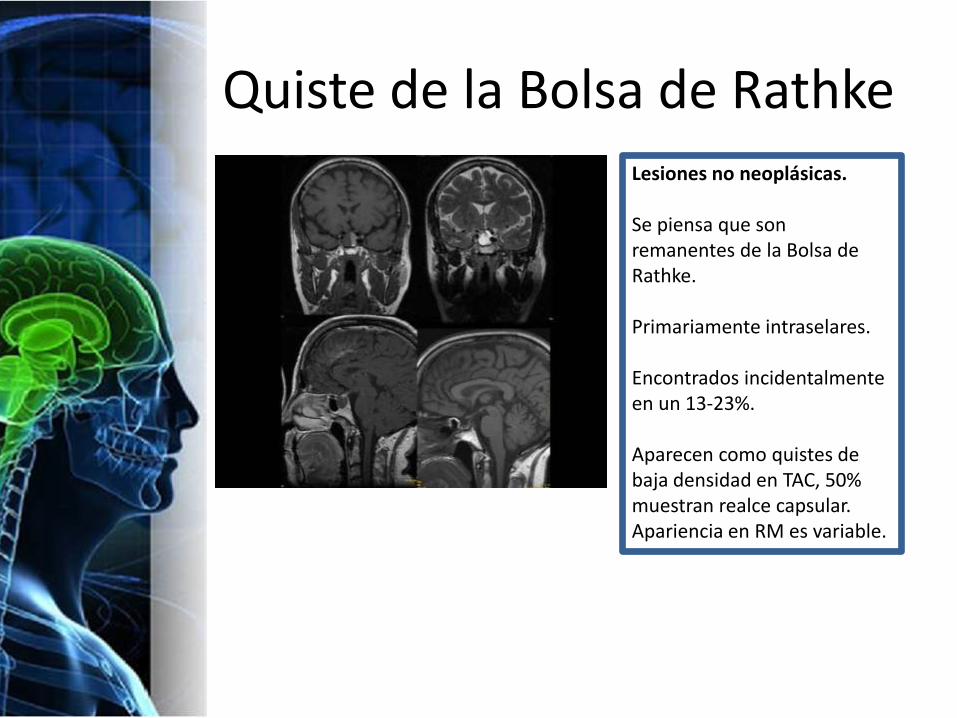
Quiste de la Bolsa de RathkeLesiones no neoplásicas.
Se piensa que son remanentes de la Bolsa de Rathke.
Primariamente intraselares.
Encontrados incidentalmente en un 13-23%.
Aparecen como quistes de baja densidad en TAC, 50% muestran realce capsular.Apariencia en RM es variable.

Tratamiento Quirúrgico
Síntomas de hipertensión intracraneal o deterioro progresivo de la función visual: Emergencia neuroquirúrgica
Debe realizarse cirugía de descompresión quística o tratamiento de la hidrocefalia previo al tratamiento definitivo.
Evaluación Prequirúrgica, debe descartar:• Hipocortisolismo• HipotiroidismoElevan la mortalidad quirúrgica

Resección Completa
• Tratamiento de elección.
• Índices de morbilidad 20%, mortalidad 12%
El abordaje puede ser:
• Vía pterional (frontotemporal)
• Orbitocraneal
• Subfrontal
• Transesfenoidal
• Transcalloso
Resección CompletaMoribilidades perioperatorias potenciales:• Convulsiones• Déficits visuales-Ceguera• Lesión hipotalámica• Stroke• Fuga de LCR
Las adherencias tumorales a las estructuras vasculares circundantes son la causa mas común de remoción tumoral incompleta

Resección Completa
Evaluación Postoperatoria
80-90% de los pacientes post-operados, requerirán terapia de remplazo hormonal.
Obesidad: 50% de los pacientes postoperados.
Diabetes insípida:68-75% en adultos, 80-93% en niñosDebe ser manejada inicialmente con terapia de reemplazo de líquidos. Use ADH de acción corta si es necesario.
La recurrencia/progresión tras esta cirugía es del 75%Al agregar radioterapia, la recurrencia es de 4-25%
Cirugía Limitada Seguida de Radioterapia
Reducir efecto de masa tumoral sobre aparato óptico y/o restablecer circulación de LCR
+• Radioterapia craneal externa (5400-5500 cGy, 180 cGy/Fracción)• Radiocirugía• Radioterapia con bomba de protones
Desarrollado por la alta frecuencia de secuelas que produce la cirugía radical.
Progresión tras esta intervención: 12-25%

Otras Consideraciones Terapéuticas
Tumor puramente quístico• Colocación de catéter de manera estereotáctica para
aspiración.• Radioterapia intraquística con Ytrio-90 o Fósforo-32• Quimioterapia intraquística con Bleomicina.
La terapia con Interferón Alfa 2a ha mostrado resultados interesantes en tumores progresivos o recurrencias.

Pronóstico
Supervivencia media a los 5 años es del 80%.
Mayor supervivencia en edad pediátrica.
99% a los 5 años en menores de 20 años.
38% a los 5 años en mayores de 65 años.
Recurrencia
La mayoría ocurren en el sitio de origen primario.
Generalmente, dentro del primer año tras el tratamiento.
Incrementa la morbilidad/mortalidad.

Bibliografía
• Handbook of Neurosurgery, 6th Edition. Mark S. Greenberg, Nicolas Arredondo. Thieme New York; ISBN-10 / ASIN: 1588904571; 2005.
• Craneofaringiomas. Julián Castro, Jesús Agulleiro, Juan Villa, Ana Pastor. Neurocirugía contemporánea. ISSM 1988-2661; Volúmen4, Número 6, Junio 2010.
• Influence of tumor location on the presentation and evolution of craniopharyngiomas. Meuric S, Brauner R, Trivin C et. Al.. J Neurosurg. Nov 2005; 103 (5 suppl):421-6.





![[2012] Informe Inversión Publicidad 2012](https://static.fdocuments.ec/doc/165x107/558a0052d8b42a33628b470b/2012-informe-inversion-publicidad-2012.jpg)












