
Compendio de Medicina II
206
REANIMACION CARDIOPULMONAR Y CEREBRAL. MUERTE SUBITA, PARO CARDIACO Y COLAPSO CARDIOVASCULAR Conjunto de medidas a seguir de modo secuencial para inicialmente sustituir, y posteriormente restablecer, las funciones básicas respiratoria, circulatoria y de prevención del daño cerebral hipoxico. Incluye aspectos referidos al tratamiento y prevención primaria y secundaria del paro cardiorespiratorio: Apoyo vital básico (AVB) ABCD primario, Apoyo vital cardiaco avanzado (AVCA) ABCD secundario, Apoyo prolongado El conjunto de maniobras también se denominan como: 1 . RCP BASICA: No requiere medios especiales y puede ser realizada por cualquier persona capacitada (Permeabilidad de la vía aérea, Respiración boca-boca, Masaje cardiaco, Desfibrilación). 2. RCP AVANZADA: requiere medios especiales y es exclusiva del personal sanitario. Uso de drogas, líquidos IV, monitorización del EKG, desfibrilizacion. 3. RCP en CUIDADOS INTENSIVOS: orientada a la recuperación cerebral. Evaluación del paciente y de su función cerebral. Y cuidados intensivos. PARO CARDIORESPIRATORIO Interrupción brusca, potencialmente reversible, de la ventilación y circulación espontaneas. Se manifiesta por: Perdida de la conciencia, Ausencia de pulso palpable, Ausencia de la respiración. Fisiopatología del PARO cardiopulmonar Paro cardiaco: puede ser ocasionada por: Un fallo primario del corazón, Un fallo secundario. Paro respiratorio: pueden ser de 3 tipos : Neurológicas, Musculares, mecánicas. 1 Medicina Interna Sexto año
-
Upload
fabiola-martinez -
Category
Documents
-
view
39 -
download
1
description
compendio de medicina interna
Transcript of Compendio de Medicina II

REANIMACION CARDIOPULMONAR Y CEREBRAL. MUERTE SUBITA, PARO CARDIACO Y COLAPSO CARDIOVASCULAR
Conjunto de medidas a seguir de modo secuencial para inicialmente sustituir, y posteriormente restablecer, las funciones básicas respiratoria, circulatoria y de prevención del daño cerebral hipoxico.
Incluye aspectos referidos al tratamiento y prevención primaria y secundaria del paro cardiorespiratorio: Apoyo vital básico (AVB) ABCD primario, Apoyo vital cardiaco avanzado (AVCA) ABCD secundario, Apoyo prolongadoEl conjunto de maniobras también se denominan como:
1. RCP BASICA: No requiere medios especiales y puede ser realizada por cualquier persona capacitada (Permeabilidad de la vía aérea, Respiración boca-boca, Masaje cardiaco, Desfibrilación).
2. RCP AVANZADA: requiere medios especiales y es exclusiva del personal sanitario. Uso de drogas, líquidos IV, monitorización del EKG, desfibrilizacion.
3. RCP en CUIDADOS INTENSIVOS: orientada a la recuperación cerebral. Evaluación del paciente y de su función cerebral. Y cuidados intensivos.
PARO CARDIORESPIRATORIO Interrupción brusca, potencialmente reversible, de la ventilación y circulación espontaneas.
Se manifiesta por: Perdida de la conciencia, Ausencia de pulso palpable, Ausencia de la respiración.
Fisiopatología del PARO cardiopulmonar
Paro cardiaco: puede ser ocasionada por: Un fallo primario del corazón, Un fallo secundario.
Paro respiratorio: pueden ser de 3 tipos: Neurológicas, Musculares, mecánicas.
DIAGNOSTICO
Diagnostico de una paro cardiaco: Perdida brusca de la conciencia, Ausencia de pulsos en grandes arterias (carótida, femoral, humeral, etc.) Signos adicionales: apnea, patrón respiratorio ineficaz, cianosis.
Diagnostico de un paro respiratorio: Completa, Parcial
Objetivos de la RCPMantenimiento de las funciones de los órganos vitales hasta que la función cardiocirculatoria y respiratoria han sido restablecidas en una persona con paro cardiaco y/o respiratorio.
Los objetivos del apoyo vital básico: consiste en proporcionar a la persona con PCR una: vía aérea permeable, ventilación adecuada, circulación adecuada.
1 Medicina Interna Sexto año

Los objetivos del apoyo vital cardiaco avanzado: Consiste en restaurar las funciones cardiacas y respiratorias normales mediante técnicas intervencionistas e invasivas avanzadas de ventilación, desfibrilación y terapia medicamentosa posterior a RCP básica e intermedia.
El ABC de la asistencia de emergencia y objetivo prioritario en la evaluación y tratamiento reside siempre en:
Airway… vía aérea: la permeabilidad de la vía aérea
Breathing…. Respiración: la facilitación de la respiración
Circulation… circulación: la conservación de la circulación
Desfibrilación… ABCD
COLAPSO CARDIOVASCULAR, PARO CARDIACO, MUERTE SUBITA
Generalidades y Definiciones
Colapso cardiovascular: perdida repentina del flujo sanguíneo eficaz, que depende del corazón, de la presencia de factores vasculares periféricos o ambas entidades, que puede mostrar reversión espontanea (como en el caso del sincope neurocardiogeno o el sincope vasovagal) o solo con intervenciones (como en el caso del paro cardiaco).
Paro Cardiaco: Interrupción repentina de la función de bomba del corazón que puede revertirse con alguna intervención inmediata, pero que culminara e muerte en caso que no se emprenda.
Muerte: Interrupción irreversible de todas las funciones biológicas.
Etiología, factores iniciadores y Epidemiologia Clínica:
Los trastornos cardiacos constituyen la causa más frecuente de muerte súbita natural.
Tras el máximo inicial de incidencia de muerte súbita que se produce entre el nacimiento y los seis meses de vida (muerte súbita del lactante), la incidencia de muerte súbita desciende de forma brusca y se mantiene de forma baja en la niñez y adolescencia.
Luego comienza a elevarse a partir de los 30 años de edad, hasta alcanzar un segundo máximo entre los 45 y 75 años en los que la incidencia se aproxima a 1 o 2 por cada 1000 por año en una población de adultos de todo tipo.
El incremento de la edad durante este intervalo es un importante factor de riesgo para padecer muerte cardiaca súbita, la proporción de causa cardiaca entre todas las muertes súbitas naturales aumenta de forma espectacular con la edad.
2 Medicina Interna Sexto año

Varones y mujeres jóvenes o de mediana edad presentan una predisposición distinta a las MSC, y las diferencias en función del sexo disminuyen con la edad.
En el grupo de 45 a 64 años de edad el exceso de riesgo de los varones de sufrir una MSC es de casi 7:1. esta proporción desciende alrededor de 2:1 entre 65 y 74 años.
Los factores hereditarios contribuyen al riesgo de MSC. Datos recientes sugieren una predisposición familiar a MSC como forma específica de expresión de coronariopatía.
La cardiopatía ateroesclerótica coronaria constituye la anomalía estructural más común vinculada con MSC en personas de edad media y adultos mayores.
80% dependen de consecuencias ateroescleróticas coronarias, las miocardiopatias comprenden otro 10 a 15%. Los síndromes de arritmias hereditarias son las causas más frecuentes en adolescentes y adultos jóvenes.
ANATOMIA PATOLOGICA
Los datos de las necropsias son concordantes con las observaciones clínicas sobre la prevalencia de la cardiopatía isquémica como principal factor etiológico estructural.
Suele caracterizarse por una combinación de ateroesclerosis extensa y prolongada de las arterias coronarias epicardicas y lesiones coronarias agudas activas del tipo de placas fisuradas o rotas, agregados plaquetarios, hemorragia y trombosis.
Predicción y prevención del paro cardiaco y muerte súbita de origen cardiaco
La muerte cardiaca súbita explica, la mitad de todos los índices de mortalidad cardiovascular. Las estrategias para predecir y prevenir MSC se clasifican en primarias y secundarias, además de las respuestas orientadas a eliminar paros cardiacos.
La prevención primaria : denota los intentos de identificar a pacientes individuales expuestos a riesgos específicos de MSC, y emprender estrategias preventivas.
La prevención secundaria : las medidas emprendidas para evitar el paro cardiaco recurrente o la muerte en sujetos que han sobrevivido a un paro previo.
Manifestaciones Clínicas del Paro Cardiaco
Pródromos, comienzo, paro, muerte.
La MSC se puede presagiar con días, semanas o meses de antelación por un aumento de la angina, disnea, palpitaciones, fatigabilidad fácil y otras molestias inespecíficas; estas molestias prodrómicas, no son específicas de la muerte súbita cardiaca.
3 Medicina Interna Sexto año

El comienzo del suceso terminal, se define como un cambio agudo en el estado cardiovascular que precede al paro hasta en 1 hora; cuando la aparición es instantánea y brusca, la probabilidad de que el paro sea de origen cardiaco es mayor del 95%.
Registros electro cardiográficos continuos, antes de un paro cardiaco, demuestran: cambios en la actividad eléctrica del corazón en los minutos u horas que preceden al suceso.
Existe una tendencia a la taquicardia y a que aparezcan extrasístoles ventriculares de grado avanzado.
La perdida inexplicable y súbita de la circulación eficaz se puede dividir en “fenómenos arrítmicos” e “insuficiencia circulatoria”. Los fenómenos arrítmicos se caracterizan por grandes probabilidades de que los pacientes estén despiertos y activos antes del suceso; su mecanismo eléctrico es predominantemente una fibrilación ventricular y el episodio terminal dura poco tiempo (menos de 1 hora). Por el contrario, las muertes por insuficiencia circulatoria se producen en pacientes inactivos o comatosos, y se caracterizan por mayor incidencia de asistolia que de fibrilación ventricular, en este caso, la enfermedad terminal tiende a ser más duradera y está dominada por fenómenos extra cardiacos anteriores al proceso terminal.
El comienzo del paro cardiaco se puede caracterizar por los síntomas típicos de un suceso cardiaco agudo, como una angina de pecho prolongada o el dolor de un infarto de miocardio, disnea u ortopnea aguda, o la aparición súbita de palpitaciones, taquicardia persistente o sensación de mareo. Sin embargo, el comienzo es súbito y sin síntomas premonitorios.
El paro cardiaco, es brusco por definición.
En el paro cardiaco la pérdida total de la conciencia es un fenómeno constante y obligatorio.
Se acepta de forma general que un paro cardiaco progresa hacia la muerte en el plazo de pocos minutos a menos que se emprendan rápidamente intervenciones activas.
La probabilidad de reanimar a la víctima de un paro cardiaco está relacionada con el tiempo que transcurre entre instauración y comienzo de los esfuerzos de reanimación, ambiente en que se produce el suceso, mecanismo y estado clínico del paciente antes del paro cardiaco.
El retorno de la circulación y los índices de supervivencia como resultado de desfibrilación disminuyen en forma lineal desde el primer minuto hasta los 10 minutos. Hasta los 5 minutos los índices mencionados no son mayores de 25 a 30% en entornos extra hospitalarios.
En las unidades de cuidados intensivos y en los ambientes intrahospitalarios, el desenlace depende considerablemente de la situación clínica previa del paciente.
Cuando el mecanismo es una taquicardia ventricular se obtienen los mejores resultados, la fibrilación ventricular tiene el siguiente mejor pronostico, y la asistolia y la actividad eléctrica sin pulso se asocian con resultados estadísticamente desalentadores.
La edad avanzada influye también en las posibilidades de que la reanimación tenga éxito.
4 Medicina Interna Sexto año

La progresión hacia la muerte biológica depende del mecanismo del paro cardiaco y del retraso previo de las intervenciones.
La fibrilación ventricular o la asistolia tiene un pronóstico sombrío si no se inicia la reanimación cardiopulmonar en los primeros 4 a 6 minutos, y existen muy pocos supervivientes entre los pacientes en quienes no se intento la recuperación durante los primeros 8 min. Después de la instauración.
La encefalopatía anoxia y las infecciones secundarias a una prolongada dependencia del respirador causan 60% de las muertes. Otro 30% se produce como consecuencia de un estado de bajo gasto cardiaco que no responde a las a las intervenciones terapéuticas.
La repetición de las arritmias es una de las causas menos frecuentes de muerte, y causan solo 10% de los fallecimientos intrahospitalarios.
Entre los pacientes con un IAM que sufren un paro cardiaco, hay que separar los primarios de los secundarios.
1. El paro cardiaco primario es el que se produce en inestabilidad hemodinámica.
2. El secundario, el que sobreviene en los pacientes en los que el cuadro clínico previo al paro cardiaco está dominado por las alteraciones hemodinámicas.
El porcentaje de reanimación inmediata satisfactoria en un paro cardiaco primario durante un infarto de miocardio se suele aproximar al 100%.
Los que sufren un paro cardiaco secundario fallecen hasta un 70% inmediatamente o durante la hospitalización.
TRATAMIENTO El individuo que sufre un colapso súbito debe ser tratado en 4 fases:
1. Respuesta inicial y soporte vital básico.2. Soporte vital avanzado.3. Cuidados pos reanimación.4. Tratamiento a largo plazo.
La respuesta inicial confirmara si el colapso súbito es verdaderamente un paro cardiaco.
La observación del estado de conciencia, movimientos respiratorios, color de la piel y presencia o ausencia de pulsos en las arterias femorales y carótidas permitirá determinar de una manera inmediata si se ha producido un paro que ponga en peligro la vida.
El soporte vital básico: RCP, está concebido para mantener la perfusión de los órganos hasta que se pueda llevar a cabo uno intervención definitiva.
Los elementos de la RCP son el establecimiento y mantenimiento de la ventilación pulmonar y la compresión torácica.
5 Medicina Interna Sexto año

Las técnicas de ventilación corriente durante RCP obligan a inflar 2 veces los pulmones en sucesión por cada 15 compresiones torácicas.
La compresión del tórax se basa en la presunción de que el masaje cardiaco permite que el corazón mantenga una función de bomba mediante el llenado y vaciamiento secuencial de sus cavidades, conservando las válvulas componentes la dirección interrogada del flujo. se coloca la palma de una mano sobre la parte inferior del esternón, con la parte inferior de la otra mano sobre el dorso de la mano inferior. Se deprime el esternón manteniendo los brazos estirados, con una frecuencia aprox 100/min. Se aplica una fuerza suficiente de fuerza para deprimir el esternón 4 a 5cm con 1 relajación brusca.
Soporte Vital Avanzado: Pretende conseguir una ventilación adecuada controlar las arritmias cardiacas, estabilizar la presión arterial y el gasto cardiaco, y restablecer la perfusión de los órganos.
Las actividades realizadas para alcanzar las metas en cuestión incluyen:
1. Desfibrilación/cardioversión.2. Estimulación con marcapaso.3. Las 3 técnicas, Intubación con sonda endotraqueal, Colocación de catéter intravenoso.
La desfibrilación inmediata debe hacerse antes de la intubación y colocación de catéter intravenoso. Se realizara RCP mientas se carga el desfibrilador. Se corrobora el diagnostico de fibrilación y taquicardia ventricular se aplica un choque que tenga como mínimo 200 J.
Se prueban mas choques hasta un máximo de 360 J, si con el choque inicial no se corrigieron en forma satisfactoria la taquicardia o fibrilación ventricular. Se aplica 1mg de adrenalina por vía intravenosa después de la desfibrilación fallida, y se repiten los intentos en este sentido. Se puede aplicar una nueva dosis de adrenalina después de intervalos de 3 a 5 min.
Si la persona no está consciente en el momento de la reversión eléctrica o si fracasan 2 o 3 intentos, se pasa a la intubación, la ventilación y se medirán los gases en sangre arterial. La ventilación con O2 a muy corto plazo corregirá la hipoxemia y la acidosis. Si hay persistencia de acidosis después de intentos logrados de desfibrilación e intubación se da 1 meq de solución de NaHCO3/kg de peso inicialmente, y repetir la mitad de la dosis cada 10 a 15 min. Si no se obtuvieron buenos resultados con los intentos iníciales de desfibrilación o persiste o reaparece la inestabilidad eléctrica habrá que emprender la administración de anti arrítmicos.
La amiodarona intravenosa ha surgido como el tratamiento inicial más indicado: 150 mg en un lapso de 10 min, Seguidos de 1 mg/min incluso durante 6 hrs, Después de ese lapso 0.5mg/min. Se puede aplicar en forma rápida 1 mg de lidocaína/kg de peso y se repetirá la dosis en 2 min en pacientes en quienes fue ineficaz la amiodarona y posiblemente en individuos que claramente tuvieron un IAM como mecanismo que desencadeno el paro cardiaco.
6 Medicina Interna Sexto año

El paro cardiaco secundario a una bradiarritmia o una asistolia se trata de forma distinta: Intubar rápidamente al paciente, Continuar la RCP, Intentar controlar la hipoxemia y la acidosis, Se administra adrenalina, atropina IV
Cuidados tras la reanimación: Esta determinada por el ámbito clínico del paro cardiaco.
La fibrilación ventricular primaria en el IAM suele responder muy bien a las técnicas de soporte vital, se controla con facilidad tras el acontecimiento inicial, no suele requerir un apoyo respiratorio, o solo por poco tiempo, la hemodinámica se estabiliza rápidamente tras la desfibrilación o cardioversión.
En fibrilación ventricular secundaria al IAM es menos frecuente que los esfuerzos de reanimación tengan éxito y la tasa de recaída es elevada.
Tratamiento a largo plazo: Los pacientes que no han sufrido una lesión irreversible del SNC y que consiguen quedar hemodinamicamente estables deben someterse a minuciosas pruebas diagnosticas y terapéuticas para proporcionar pautas de tratamiento a largo plazo.
7 Medicina Interna Sexto año
2:
Has

CARDIOPATÍA ISQUÉMICA
Isquemia: Falta de oxigeno por perfusión insuficiente, secundaria a desequilibrio entre el aporte y demanda de oxigeno.
Cardiopatía isquémica: Es la designación genénca aplicada a un grupo de síndromes íntimamente relacionado originados por isquemia miocárdica , un desequilíbrio entre el suministro de sangre oxigenada y La demanda del corazón.
Fisiopatología
Las grandes arterias coronarias epicárdicas pueden contraerse y relajarse; en las personas sanas sirven como conductos y se les denomina vasos de conducción.
Arteriolas Intramiocardicas: normalmente exhiben cambios en el tono por lo tanto se les llama asÍ. La contracción anormal de los vasos de conducción origina isquemia grave en la angina prinzmetal. La contracción anormal o ausencia de dilatación normal en los vasos de resistencia también pueden
originar isquemia; cuando provoca angina se denomina angina microvascular.La circulación coronaria normal es denominada y controlada por las necesidades de origen del miocardio.
Cómo se satisfacen de oxigeno las arterias coronarias?
R/ Estas se satisfacen por la capacidad del lecho vascular coronario para varias considerablemente sus resistencia vascular coronaria y por consiguiente el flujo sanguíneo.
Mientras que el miocardio extrae un porcentaje alto y relativamente fijo del oxigeno.
En condiciones normales las “arteriolas de resistencia intramiocardica“ poseen una gran capacidad de dilatación.
El estrés emocional y el ejercicio: (Px normal)
Aumento la demanda de oxigeno
Aumento de la resistencia vascular coronaria (vasodilatación compensadora)
8 Medicina Interna Sexto año

De esta forma regula el aporte de oxigeno sustratos al miocardio
(regulación metabólica)
¿Finalidad de esto?
R/ Mantener el flujo coronario a niveles apropiados a las necesidades del miocardio (autorregulación)
Estrés emocional y el ejercicio (Px enfermo)
Aumento de la demanda de oxigen y sustratos
Arteroesclerosis limita el incremento correspondiente de la perfusión.
(Reducción de la luz de las coronarias)
Disminución de la perfusión del miocardio en estado basal
Si la destrucción es Si la obstrucción no es
Transitoria: transitoria:
Angina de pecho Infarto de miocardio
El flujo coronario también puede verse limitado por:
1.- trombos
2.- espasmos
9 Medicina Interna Sexto año

3.- emndos coronarios (raro)
4.- Aumento de la demanda de oxigeno por hipertrofia ventricrular izquierda por HTA o estenosis aortica.
Arterosclerosis Coronaria
Las coronarias epicárdicas constituye el sitio principal de la aterosclerosis.
FACTORES DE RIESGO DE LA ARTEROSCLEROSIS
1.- Lipoproteina de baja densidad plasmática elevada.
2.- Lipoproteina de alta densidad plasmática reducida.
3.- Tabaquismo.
4.- Hipertensión.
5.- Diabetes mellitus.
Alteran funciones normales del endotelio vascular
¿Qué funciones?
1.- Regulación local del tono vascular
2.- Conservación de una superficie anticoagulante
3.- Defensa contra células inflamatorias.
Si se pierden estas defensas ¿Qué sucede?
- Contracción, formación de coágulos luminal e interacción con la neonatitos y plaquetas de la sangre serán anormales.
origina
1) Acúmulo en la capa subíntima de grasa, células de músculo liso, fibroblustos y matriz intercelular (placa aterosclerostica)
10 Medicina Interna Sexto año

Reducción segmentaria del área transversal
Más del 90% de los pacientes con Ci presentan aterosclerosis de una o más arterias coronarias. Las manifestaciones clínicas de la aterosclerosis coronaria se deben en general a disminución progresiva de la luz que conduce a estenosis o a rotura aguda de la placa con trombosis que compromete el flujo sanguíneo.
MANIFESTACIONES CLÍNICAS DE CI:
1) Infarto de miocardio2) Angina depecho
ANGINA DE PECHO
Es un complejo de síntomas de CI caracterizado por:
- Crisis paroxisticas y en general recurrente de molestia subesternal o precordial (descrita en forma variable como constricción, apretón, sofocación o puñalada) cansado por isquemia miocardica transitoria (15 segundos a 15 minutos). Sin llegar a producir la necrosis celular que define el infarto.
Patrones superpuestos de angina:
1) Angina estable o típica2) Angina de prinzmetal es variante3) Angina inestable o progresiva4) Angina estable
La forma más común, por eso el nombre de típica parece estar causada por reducción de la profusión coronaria hasta un nivel crítico a causa de la aterosclerosis coronaria estenosante crónica.
La angina típica suele ser aliviada con el reposo (la demanda) o la nitroglicerina que es un vaso dilatador potente y también disminuye el trabajo del corazón.
Generalmente se da en un hombre mayor de 50 años y mujer mayor de 60 años que se queja de una molestia en el torax.
11 Medicina Interna Sexto año

Cuando se le pide al paciente que explique la sensación de opresión, asfixia, compresión, se tocará el esternón, algunas veces con el puño para indicar que le molesta es opresivo central y subesternal (signo de levine). Algunas veces se indica hacia el hombro izquierdo o ambos brazos especialmente las superficies cubitales del antebrazo y mano.
Los episodios de angina habitualmente se desencadenan en condiciones de esfuerzo físico:
- Ejercicios- Prisas- Actividad sexual
La angina de esfuerzo generalmente desaparece con el reposo en un lapso de 1 a 5 minutos.
Exploración física:
Suele ser normal, pero en ocasiones revela evidencias de aterosclerosis, en otros sitios como un aunerisma de la aorta abdominal. Soplo caotídeo y pulso arterial reducido en extremidades inferiores.
Fondo de ojo, revela reflejos luminosos acentuados y muestras arteriovenosas como evidencias de hipertensión.
Palpación: Revela agrandamiento cardíaco y contracción anormal del impulso cardíaco (acinesia o discinesia ventricular izquierda).
Auscultación: Soplos arteriales R3 R4, insuficiencia nitral.
Hay edema
Diagnóstico
A) Laboratorio = El dx de CI puede realizarse por historia clínica.1) Estudiar orina para descartar DM y enfermedades renales (incluye miocroalbuminaria ya que
ambas pueden acelerar la aterosclerosis .2) Medición de lípidos, glucosa, creatinina, HI 3) Rx de tórax (tamaño del corazón), aneurisma ventricular4) Electrocardiograma de 12 derivaciones en reposo es normal, pero algunas veces aparecen signos
de un infarto antiguo de miocardio.
Ciertas anormalidades de la repolarización como:
Cambios del segmento ST y la onda T hipertrofia ventricular izquierda y alteraciones de la conducción intraventricular son sugestiva de CI.
Los cambios típicos del segmento SI y onda T que acompañan a los episodios anginosos y desaparecerán y son más específicos.
5) Prueba de esfuerzo * Electrocardiográfico:
Consiste en el registro electrocardiográfico de 12 derivaciones antes, durante y después del ejercicio por lo general en una banda sin fin. Se realiza en un aumento progresivo de la carga de trabajo externo, mientras se vigilan de forma contínua el ECG, síntomas, PA.
12 Medicina Interna Sexto año

La respuesta isquémica del segmento ST se define como depresión plana del ST superior a 0.1 mv por debajo de la línea basal.
Pronóstico
Si la PA no asciende a 5 por el contrario disminuye con signos de isquemia durante la prueba se considera un signo de mal pronóstico ya que puede reflejar disfunción ventricular izquierda global inducida por isquemia.
Indicadores:
- Edad- Estado funcional del VI- Ubicación de la estenosis coronaria - Tamaño y gravedad o actividad de la isquemia miocárdica- La angina de pecho reciente, angina inestable, angina que parece inmediatamente después del infarto de
miocardio, angina que no responde o responde poco al Tx o bien se acompaña de síntomas de Icc indican un mayor riesgo de padecer eventos coronarios adversos.
- Las placas atedescleroticas segmentadas en las arterias epicardicas con fisuras o defecto de llenado indican un riesgo elevado.
Tratamiento
El plan terapéutico debe consistir:
1) Explicación de la enfermedad y actitud tranquilizadora 2) Identificación y tratamiento de los factores agravantes3) Adaptación de la enfermedad 4) Tx de los factores de riesgo para reducir complicaciones coronarias5) Farmacoterapia de la angina6) Consideración de la revascolarización mecánica
Desarrollo:
1) Los Px con C1 deben entender su enfermedad lo mejor posible y comprender que pueden tener una vida larga y útil.
2) Ciertas enfermedades que no son de naturaleza propiamente cardíaca pueden incrementar la demanda de oxigeno o el aporte de O2al miocardio, precipitando la angina en un Px con CI.
Ejemplo:
- Valvulopatía aórtica
- Miocardiopatía hipertrofica
- Obesidad
- Hipertiroidismo
3) Estos Pxs deben conocer las variaciones diurnas en su tolerancia a ciertas actividades y reducir sus requerimientos energéticos durante la mañana, inmediatamente después de las comidas.
13 Medicina Interna Sexto año

4) Hiperlipidermis, hipertensión y diabetes, el consumo de cigarrillos acelera la ateroesclerosis coronaria.
Tx farmacológico:
A) Nitratos vasodilatación venosa sistémica1) Nitroglicerina sublingual 0.3-0-6 mg
EA= cefalea, mareos
Contraindicaciones = intolerancia a los efectos adversos
2) Dinitrato de isosorbide de liberación retardada oral=10.60 mgC/8 horas.
Sublingual =2.5-10mg c/4 horas
B) Betabloquedores 1) Propanolol = 20-80 mg 4 veces al día2) Metropolol = 25-200 mg 2 veces al día
C) Antagonistas de calcio = nifedipina XL 30-90 mg al día D) Antiplaquetarios = aspirina 75-325 mg vo c/día
ANGINA INESTABLE
Se define como angina de pecho a malestar isquémica equivalente que posee por lo menos una de las tres características siguientes:
1) Surge durante el reposo lo ejercicio mínimo y suele durar más de 10 minutos.2) Es intensa y su comienzo es reciente ( es decir durante las 4 a 6 semanas anteriores)3) Su perfil es de intensificaciones constante.
Fenómenos Fisiopatológicos:
1) Rotar o evasión de una placa ateroesclerótico 2) Obstrucción dinámica (espasmo coronario)3) Obstrucción mecánica progresiva (aterosclerosis coronaria)4) Angina inestable secundaria que depende de la mayor demanda de O2 por el miocardio.
Cuadro clínico:
- Dolor de pecho de localización retroestrizmal típica o a veces en el epigastrio y a menudo irradia a cuello, hombro izquierdo o brazo izquierdo.
- Es precipitado por esfuerzos físico cada vez menores, tiende a ser de duración prolongada.
ECG
- Depresión
14 Medicina Interna Sexto año

- Elevación transitoria del mismo- Inversión de la onda T
Diagnóstico:
1) Anamesis2) ECG3) Indicadores cardíacos (CK-MB y troponina)
Están expuestos a mayor riesgo de muerte o de recurrencia del IM (niveles altos)
4) Prueba de esfuerzo
Tratamiento:
A) NitratosB) BetabloqueadoresNitratos = a sublingual o en aerosol =.3-0.6 mg.
Si la molestia no cede después de 3 dosis aplicadas con una diferencia de 5 min, se recomienda aplicar nitroglicerina IV 5-10 mg /via
Betabloqueadores = se recomienda IV , seguida de su administración oral con el objeto que la FC llegue a 50-60 latidos X’.
Aspirina= inhible aclosigenosa plaquetaria.
Clopidogrel = bloqueo el receptor adnosinico de las plaquetas + leporina de bajo peso molecular.
ANGINA PRINZMETAL
Es un patrón poco común de episodios anginosos que ocurren en reposo y se deben a espasmo arterial coronario.
ECG
Elevador del segmento ST indicador de isquemia transmural.
Cuadro clínico
1. Son Px más jóvenes y tienen menores factores de riesgo coronario (excepto tabaquismo).2. La molestia suele ser intensísima.
Diagníostico
- Detección de elevación transitoria del segmento ST y dolor en reposo.
15 Medicina Interna Sexto año

- Pruebas de esfuerzo = escasa utilidad porque los px pueden mostrar elevación o depresdión del segmento ST o ningún cambio .
Tratamiento
A) NitratosB) Bloqueadores de los canales de Ca.
Pronóstico
- La supervivencia a largo plazo (5 años ) es excelente (90-95%)- Los pxs sin obstrucción coronaria fija o con obstrucción croronaria fija leve siguen una evolución más
benigna que aquelelos con obstrucciones graves.- Los px con este tipo de anginas que terminan por presentar arritmias tienen mayor riesgo de muerte
súbita.
Clasificación de la angina de pecho de la CCS:
Etapa.
I.- Actividad física ordinaria no causa angina:
- caminar o subir escaleras
La angina aparece con el ejercicio laboral rápido o prolongado.
II. Limita ligeramente las actividades ordinarias. La angina aparece al caminar o subir escaleras rápidamente, caminar
Pendiente, cuando hace frío.
III. Limita considerablemente la actividad física ordinaria. La angina aparece al caminar 1 o 2 cuadras y subir un tramo de escaleras a paso normal.
IV. Impide cualquier actividad física, a veces con reposo.
INFARTO DE MIOCARDIO
Conocido también como < <ataque cardíaco>>, consiste en la muerte de musculo cardíaco causada por isquemia.
16 Medicina Interna Sexto año

Tipos:
1. Infarto transmural2. Infarto subendocardico
Infarto transmural La mayoría son de este tipo. La necrosis isquémica afecta el grosor total o casi total de la pared ventricular en la distribución de una
sola arteria.
Infarto SubendocárdicoSe considera por un área de necrosis isquémica limitada al tercio medio o con mucho a la mitad de la pared ventricular.
El IM ocurre a cualquier edad, pero la frecuencia aumenta de forma progresiva con ella y la presencia de factores de riesgo predisponentes a la aterosclerosis.
A lo largo de la vida, los hombres expedrimentan un riesgo significativamente mayor de IM que las mujeres.
Aspectos Fisioopatológicos:
Surge cuando disminuye repentinamente el flujo de sangre por las coronarias después que de un trombo ocluye Una de estas arterias afectadas de aterosclerosis.
La lesión es facilitada o producida por factores como tabaquismo, hipertensión y acumulación de lípidos.
¿Cómo se desencadena el IM?
Cuándo:
Cambio agudo de la placa aterosclerotica (rotura). Se rompe o se ulcera . Trombo mural en el sitio de la rotura y ello culmina con la oclusión de la arteria. Trombosis o vasoespasmo.
Evolución
Primero:
1) Cambio inicial súbito, formación placa atoromatosa (rotura fisura).2) Adhesión plaquetaria, agregación y activación liberación de favorecedores potentes de agregación como
trombosis Az, serotonina y factores plaquetarios 3 y 4.3) Vasoespasmo estimulado por agregación plaquetaria y liberación de mediadores.4) Aumento de volumen del trombo.5) Trombo evoluciona para ocluir por completo la luz del vaso crónico.
17 Medicina Interna Sexto año

Cuadro inicial:
En un 50% de los casos parece haber un factor desencadenante antes de que se manifieste el IM como: ejercicio vigoroso, estrés emocional.
Dolor profundo y visceral (pesado, constrictivo, opresivo) igual al de angina solo que más intenso y duradero.
puede aparecer en región central del tórax o epigástrico y a veces se irradia a brazos. Sitios menos comunes de radiación:1. Espalda 2. abdomen3. Maxilar inferior4. Cuello
Signos
Casi todos los enfermos muestran: ansiedad e inquietud, intentan aplacar el dolor moviéndose, cambiando de postura y estirándose.
Palidez con sudoración abundante y frialdad de las extremidades. Combinaciones de dolor retroesternal que persiste más de 30 min y dioforesis, sugiere netamente la
posibilidad de IM.
El infarto de miocardio evoluciona a tres fases:
1) Agudo (primeras horas a 7 días)2) Recuperación o curación (7 a 28 días)3) Cicatrización (29 o más)
Diagnóstico
1) ECG2) Marcadores cardíacos en sueros (fosfocreatin kinasa)3) Estudio, imagendigicos del corazón4) Indices específicos de necrosis en inflamación nistica.
ECG
Elevación del segmento ST, evolución hasta mostrar ondas Q.
Marcadores cardíacos en suero:
El tejido miocardico y necrotico libera a la sangre grandes cantidades de proteínas llamadas marcadores cardiacos (fosfocreatinkinasa de creatinina).
Mioglobina liberada en la sangre unas horas después de haber comenzado el IM.
18 Medicina Interna Sexto año

Dichos pigmentos es uno de los primeros marcadores cardiacos que aumentan por encima de los límites racionales después del IM.
creatinphosfokinasa o CPK(asciende de 4 a 8 hrs, su max: 24hrs, normaliza a las 48 a 72 hrs: CK > o igual a 2.5 sugiere a IAM, su fracción más específica CKMB que alcanza su max: a alas 8 hrs, y la troponina T e I se mantienen elevadas de 8 a 10 dias.
Estudios imageniológicos
Por medios ecocardiograficos resulta imposible diferenciar el IM agudo de una vieja cicatriz en el miocardio o de izquemia aguda intensa.
- En el pronostico es útil estimar por ecocardiografia la función del ventriculo izquierdo- Infarto en ventrículo derecho, aneurisma ventricular.
Tratamiento
1) Antitrombóticos (heparina de bajo peso pero molecular).2) Betabloqueadores
Tratamiento inicial: Identificartsi el paciente es candidato para reperfusion. Aliviar el dolor Prevenir y tratar arritmias y complicaciones mecanicas
Tratamiento adicional estandar:
Hospitalizarlo y ECG
Canalizar y dar tratamientro para prevenir arritmias
Controlar dolor (sulfato de morfina 2-4 mg IV c/5 a 10 min,(nitroglicerina 0.3 mg sublingual )
Oxigeno 2 a 4 litros/minutos en canula nasal.
Betabloqueantes(disminuyen el consumo de oxigeno por el miocardio , limitan el tamano del infarto y mortalidad)
Complicaciones
- Disfunción ventricular- Arritmias- Taquicardias y fibilación ventriculares- Arritmias supraventriculares- Brodicardio sincel- Tromboembolia
19 Medicina Interna Sexto año

CARDIOPATIA VALVULAR
Estenosis Mitral
Causas y patología
Cerca de 66% de los pacientes con estenosis mitral, es de sexo femenino. La estenosis mitral, estenosis mitral mixta e insuficiencia mitral, son por lo general de origen reumático; rara vez son congénitas. Casi 40% de los pacientes con cardiopatía reumática
padece de estenosis mitral pura o predominante.
En la estenosis reumática, las valvas están engrosadas de forma difusa por tejido fibroso o depósitos calcificados. Las comisuras mitrales se fusionan, las cuerdas tendinosas se fusionan y acortan, y las valvas se tornan rígidas; todos estos cambios, provocan a su vez un estrechamiento del vértice valvular en forma de embudo (boca de pez).
FisiopatologíaEn los adultos normales, el orificio de la válvula mitral mide de 4 a 6 cm2. Cuando existe una obstrucción importante, es decir, cuando el orificio mide menos de 2 cm2, el flujo sólo puede pasarde la aurícula al ventrículo izquierdo si es impulsado por un gradiente de presión auriculoventricular anormalmente elevado, lo que constituye el dato hemodinámico esencial de la estenosis mitral. Cuando el orificio mitral está reducido a 1 cm2, se necesita una presión en la aurícula izquierda de aproximadamente 25 mmHg para mantener el gasto cardíaco.
Sintomas
Cuando la obstrucción valvular es leve, los signos físicos de estenosis mitral no siempre se acompañan de síntomas. Sin embargo, en los pacientes en los que el orificio es lo bastante grande como para albergar un flujo normal sólo con ligeras elevaciones de la presión en la aurícula izquierda, el ejercicio extremo, excitación, fiebre, anemia grave, fibrilación auricular paroxística y otras taquicardias, coito, embarazo y tirotoxicosis pueden desencadenar un aumento notable de la presión en la aurícula izquierda que produzca disnea y tos. A medida que progresa la estenosis, el estrés necesario para desencadenar disnea es menos intenso. La redistribución de la sangre desde las porciones declives al pulmón, que se produce en posición de decúbito, causa ortopnea y disnea paroxística nocturna.Aparece edema pulmonar cuando se produce incremento brusco del flujo a través de un orificio mitral muy estrechado. Cuando ha existido durante años una estenosis mitral moderadamente grave, aparecen arritmias auriculares.
La aparición de fibrilación auricular permanente a menudo señala un cambio en la evolución del paciente y generalmente se asocia a una aceleración de la progresión de los síntomas.
Algunos pacientes con estenosis mitral grave presentan rubor malar y cara congestionada y cianótica. El pulso venoso yugular muestra ondas a prominentes debidas a una enérgica sístole auricular derecha en los pacientes con ritmo sinusal e hipertensión pulmonar importante o estenosis tricuspidea.
Auscultación
20 Medicina Interna Sexto año

El primer ruido cardíaco (S1) por lo general se acentúa y es un chasquido ligeramente retrasado. El componente pulmonar del segundo ruido (P2) es acentuado y los dos componentes del segundo ruido (S2) se desdoblan o se encuentran fijos.En pacientes con estenosis mitral pura habitualmente se oyen soplos sistólicos suaves (grado I o II/VI) en la punta o a lo largo del reborde esternal izquierdo, que no significan necesariamente la presencia de insuficiencia mitral.
Diagnostico
Cateterismo cardíaco y angiocardiografíaEl cateterismo del hemicardio izquierdo resulta útil para aclarar el cuadro cuando existen discrepancias entre los datos clínicos y ecocardiográficos. Ayuda a valorar las lesiones asociadas,como la estenosis o la insuficiencia aórticas. El cateterismo y la coronariografía no suelen ser necesarios para la decisión quirúrgica en pacientes jóvenes con signos típicos de obstrucción grave en la exploración clínica y ecocardiográfica.
Tratamiento
En estos pacientes es muy importante administrar profilaxis con penicilina para las infecciones por estreptococo hemolítico beta (cap. 302) para prevenir la fiebre reumática y la profilaxis de la endocarditis infecciosa.Para reducir la frecuencia ventricular en pacientes con fibrilación auricular. En estos casos también son de utilidad los betabloqueadores o antagonistas de calcio distintos de la dihidropiridina (p. ej., verapamilo o diltiazem). Los pacientes con estenosis mitral que han padecido embolias sistémicas o pulmonares y en aquéllos con fibrilación auricular se administra warfarina hasta obtener una INR de 2 a 3:1 cuando menos durante un año.
Valvulotomía mitral
Si no existe ninguna contraindicación específica, la valvulotomía mitral está indicada para pacientes sintomáticos con estenosis mitral aislada cuyo orificio efectivo sea menor de 1.0 cm2/m2 de superficie corporal o inferior a 1.7 cm2 para adultos de estatura normal.
En pacientes en quienes la valvulotomía percutánea es imposible, carece de éxito o fracasa, es necesario realizar una valvulotomía "abierta" con circulación extracorpórea. Además de abrir las comisuras valvulares, es importante aflojar la fusión subvalvular de los músculos papilares y las cuerdas tendinosas para eliminar los grandes depósitos de calcio, mejorando de esta manera la función valvular, además de extraer los trombos auriculares. La mortalidad es de cerca de 2 por ciento.
Insuficiencia mitral
Causa
La causa de la insuficiencia mitral acentuada es la cardiopatía reumática crónica únicamente en 33% de los casos y esta lesión es más frecuente en varones. El proceso reumático origina rigidez, deformidad y retracción de las valvas, así como fusión de las comisuras y acortamiento, contracción y fusión de las cuerdas tendinosas. También puede haber insuficiencia mitral como anomalía ,congénita, por lo general como defecto de los cojines endocárdicos (defectos de loscojines auriculoventriculares). La insuficiencia mitral también es con frecuencia secundaria a isquemia.
21 Medicina Interna Sexto año

También hay insuficiencia mitral en el agrandamiento ventricular izquierdo acentuado de cualquier causa donde la dilatación del anillo mitral y el desplazamiento de los músculos papilares en sentido lateral interfieren con la coaptación de las valvas, casi siempre por isquemia.
Fisiopatología
La resistencia al vaciamiento del ventrículo izquierdo está disminuida en la insuficiencia mitral. En consecuencia, hay reflujo de este ventrículo a la aurícula izquierda durante la expulsión y al reducirse el tamaño del ventrículo izquierdo durante la sístole, disminuye rápidamente su tensión.La compensación inicial de la insuficiencia mitral aguda consiste en vaciamiento más completo del ventrículo izquierdo; el volumen de éste, sin embargo, aumenta progresivamente a medida que se eleva la gravedad de la insuficiencia y se deteriora la función del ventrículo. Dicho aumento del volumen se acompaña con frecuencia de una disminución del gasto cardíaco anterógrado. El volumen de reflujo varía directamente con la presión sistólica del ventrículo izquierdo y con el tamaño del orificio insuficiente; este último, a su vez, está profundamente influido por el grado de dilatación ventricular izquierda.
Síntomas
Las molestias más llamativas de la insuficiencia mitral crónica grave son fatiga, disnea de esfuerzoy Ortopnea.
Signos físicos
Generalmente, la presión arterial es normal y la insuficiencia mitral grave se caracteriza por elevación aguda del pulso arterial. El pulso venoso yugular de los pacientes en ritmo sinusal con hipertensión pulmonar notable presenta ondas a con prominencia anormal, mientras que el de aquéllos con insuficiencia triscuspídea grave acompañante muestra ondas v prominentes. Con frecuencia se palpa un frémito sistólico en la punta cardíaca, el ventrículo izquierdo es hiperdinámico y presenta un impulso sistólico brusco y una onda palpable de llenado rápido, y el latido apical suele encontrarse desplazado hacia un lado. En presencia de hipertensión pulmonar notable, se palpa un chasquido del ventrículo derecho y el choque del cierre de la válvula pulmonarEl dato auscultatorio más característico de insuficiencia mitral grave es un soplo sistólico al menosde grado III/VI. Habitualmente es holosistólico
Tratamiento
MÉDICOEl tratamiento no quirúrgico de la insuficiencia mitral grave comenzará con larestricción de las actividades físicas que suelen producir fatiga excesiva y disnea, reducción de la ingestión de sodio y aumento de su eliminación con los diuréticos apropiados.
El nitroprusiato o la nitroglicerina por vía intravenosa, que reducen la poscarga y por consiguiente el volumen de reflujo, son útiles para estabilizar a los pacientes con insuficiencia mitral aguda o grave. Los inhibidores de la enzima convertidora de angiotensina.
QuirurgicoLa cirugía constituye una opción en la insuficiencia mitral acentuada incluso en pacientes asintomáticos o con síntomas leves cuando la disfunción ventricular izquierda es progresiva, la fracción de expulsión del ventrículo
22 Medicina Interna Sexto año

izquierdo desciende por debajo de 60%, la dimensión de la cavidad telesistólica en la ecocardiografía se eleva por arriba de 45 mm o ambas.
Prolapso de la valvula mitral
Exceso y redundancia del tejido de la valva, a menudo afectado por un proceso degenerativo mixomatoso con una gran concentración de mucopolisacáridos ácidos. Se trata de un hallazgo frecuente en los pacientes con trastornos hereditarios del tejido conjuntivo, como el síndrome de Marfan, la osteogénesis imperfecta y síndrome de Ehler-Danlos.
Se limita a la válvula mitral (o con menor frecuencia a la tricuspídea o a la aórtica) y no existe ninguna otra manifestación clínica ni anatomopatológica; por lo general, la valva posterior resulta más afectada que la anterior y el anillo a menudo está muy dilatado. En muchos pacientes, las cuerdas tendinosas alargadas y redundantes causan la insuficiencia o contribuyen a su aparición.
Manifestaciones clínicas
El prolapso de la válvula mitral es más frecuente en las mujeres. Afecta a personas de una amplia gama de edades, principalmente entre los 14 y 30 años. La evolución clínica suele ser benigna. El prolapso de la válvula mitral se observa también entre pacientes mayores (>50 años), de ordinario varones, cuya insuficiencia mitral a menudo tiene carácter grave y precisa tratamiento quirúrgico
Datos de laboratorio
El electrocardiograma suele ser normal, pero a veces muestra ondas T bifásicas o invertidas en lasderivaciones II, III y aVF, así como extrasístoles supraventriculares o ventriculares.
Tratamiento
Sin insuficiencia mitral grave ni arritmias y en prevenir la endocarditis infecciosa con profilaxis antibiótica en los pacientes con soplo sistólico, engrosamiento de las valvas mitrales en la ecocardiografía, o ambos. Se ha comprobado que los betabloqueadores alivian el dolor torácico. Si existen taquiarritmias sintomáticas se administrarán antiarrítmicos de acuerdo con el estudio electrofisiológico.
Estenosis aórtica
23 Medicina Interna Sexto año

La estenosis aórtica afecta aproximadamente a la cuarta parte de todos los pacientes con cardiopatía valvular crónica; cerca del 80% de los adultos con estenosis aórtica sintomática son varones.
CausaLa estenosis aórtica en el adulto es causada por calcificación degenerativa de las cúspides aórticas.Algunas veces es de origen congénito y otras es secundaria a inflamación reumática.
Otras formas de obstrucción del infundíbulo del ventrículo izquierdo
Además de la estenosis aórtica valvular, existen otras tres lesiones que obstruyen el infundíbulo del ventrículo izquierdo:
1. Miocardiopatía hipertrófica 2. Estenosis aórtica subvalvular congénita aislada. 3. Estenosis aórtica supravalvular
Fisiopatología
La obstrucción del infundíbulo del ventrículo izquierdo produce un gradiente de presión sistólica entre el ventrículo izquierdo y la aorta. El ventrículo izquierdo responde con dilatación y reducción del volumen sistólico. Sin embargo, en los pacientes, la obstrucción puede estar presente desde el nacimiento o aumentar gradualmente a lo largo de muchos años, con lo que el gasto ventricular se mantiene por la presencia de hipertrofia concéntrica del ventrículo izquierdo.
Sintomas
La mayoría de los pacientes con estenosis aórtica pura o predominante sufre una obstruccióngradualmente progresiva durante años, aunque no presentan síntomas hasta el sexto u octavodecenio de la vida. Los tres síntomas cardinales son disnea de esfuerzo, angina de pecho y síncope.
Un pulso arterial sistólico palpable doble, el denominado pulso bisferiens, descarta una estenosis aórtica pura o predominante e indica insuficiencia aórtica dominante.
Auscultacion
Ruido protosistólico de expulsión, en realidad el chasquido de apertura de la válvula aórtica. Este ruido habitualmente desaparece cuando la válvula se calcifica y se torna rígida.El soplo de la estenosis aórtica es típicamente un soplo (meso)sistólico. En casi todos los pacientes con obstrucción grave el soplo es al menos de grado III/VI.
Insuficiencia aórtica
Etiología
La insuficiencia aórtica puede obedecer a una valvulopatía primaria o a una enfermedad primaria de la raíz aórtica.Valvulopatía primaria
24 Medicina Interna Sexto año

Aproximadamente tres cuartas partes de los pacientes con insuficiencia aórtica valvular pura o predominante son varones; sin embargo, las mujeres predominan cuando existe una valvulopatía mitral asociada. Casi las dos terceras partes de los casos tienen un origen reumático que determina engrosamiento, deformidad y acortamiento de las valvas de la válvula aórtica, cambios que impiden la abertura apropiada durante la sístole y el cierre durante la diástole.
El origen reumático es menos frecuente en pacientes con insuficiencia aórtica aislada.
Enfermedad primaria de la raíz aórtica
La insuficiencia aórtica, aguda y crónica, puede deberse también en su totalidad a una gran dilatación de la aorta, es decir, a enfermedad de la raíz aórtica, sin afección primaria de las valvas; el ensanchamiento del anillo aórtico y la separación de las valvas causan insuficiencia aórtica.
Fisiopatología
Los pacientes con insuficiencia aórtica presentan un aumento del volumen sistólico total del ventrículo izquierdoA diferencia de la insuficiencia mitral, en la que parte del volumen sistólico del ventrículo izquierdo pasa a la aurícula (sometida a una presión más baja), en la insuficiencia aórtica el volumen sistólico del ventrículo izquierdo es expulsado a una cavidad de alta presión, la aorta.
Signos físicos
En la insuficiencia aórtica grave es posible apreciar la oscilación de todo el cuerpo y las sacudidasde la cabeza con cada sístole, y se ven con claridad la distensión y colapso bruscos de las grandes arterias.
Pulso arterial
Son característicos de la insuficiencia aórtica libre la presencia de una rápida elevación del pulso en"martillo hidráulico", que se colapsa bruscamente conforme disminuye con rapidez la presión arterial al final de la sístole y en la diástole (pulso de Corrigan), así como las pulsaciones capilares, y un rubor y palidez alternativos de la piel de los lechos ungueales cuando se presiona la punta delas uñas (pulso de Quincke). En las arterias femorales se ausculta un ruido explosivo en "pistoletazo" (signo de Traube) y un soplo de vaivén (signo de Duroziez) si se comprime ligeramente la arteria femoral con el estetoscopio.La presión del pulso arterial está ampliada y se observa una elevación de la presión sistólica, a veces hasta 300 mmHg, y una depresión de la diastólica. La medición de la presión arterial diastólica con un esfigmomanómetro puede resultar difícil debido a que los ruidos sistólicos se suelen oír con el manguito completamente desinflado. Sin embargo, el nivel de la presión del manguito cuando se desvanecen los ruidos de Korotkoff (fase IV) se corresponde en gran medida con la presión diastólica intraarterial verdadera.
El soplo de la insuficiencia aórtica generalmente es un soplo diastólico de alta frecuencia, silbante y decreciente, que se oye mejor en el tercer espacio intercostal a lo largo del borde esternal izquierdo.
Estenosis tricuspidea
25 Medicina Interna Sexto año

Generalmente es de origen reumático y más frecuente en las mujeres que en los varones. No suele producirse como una lesión aislada, sino que a menudo se asocia a estenosis mitral. De 5 a 10% de los pacientes con estenosis mitral grave presentan además estenosis tricuspídea de importancia hemodinámica; la estenosis tricuspídea reumática se suele asociar a cierto grado de insuficiencia tricuspídea.
Fisiopatología
Existe un gradiente de presión diastólica entre la aurícula y el ventrículo derechos que puede registrarse mediante un catéter cardíaco de doble luz. Aumenta conforme lo hace el flujo transvalvular durante la inspiración y disminuye durante la espiración.
Un gradiente diastólico medio de 4 mmHg suele bastar para elevar la presión media en la aurícula derecha a cifras que produzcan congestión venosa sistémica y, a menos que se haya restringido la ingestión de sodio o se estén administrando diuréticos, se asocia a ascitis y edema a veces de carácter grave. En pacientes en ritmo sinusal, la onda auricular a puede ser extremadamente alta e incluso alcanzar el nivel de la presión sistólica en el ventrículo derecho. El gasto cardíaco en reposo suele ser muy bajo y no aumenta durante el ejercicio. El gasto cardíaco reducido ocasiona presión sistólica normal o ligeramente elevada en la aurícula izquierda, arteria pulmonar y ventrículo derecho, a pesar de la presencia de estenosis mitral. Por lo tanto, la presencia de estenosis tricuspídea disimula la características hemodinámicas y clínicas de la estenosis mitral que suele acompañarla.
Síntomas
En general, el desarrollo de la estenosis mitral precede al de la estenosis tricuspídea y por tanto muchos pacientes presentan inicialmente síntomas de congestión pulmonar.De forma característica, los pacientes se quejan de disnea relativamente leve en comparación con la hepatomegalia, ascitis y edema. Cuando existe estenosis o insuficiencia tricuspídea son frecuentes la fatiga secundaria al bajo gasto cardíaco y las molestias por edema resistente al tratamiento, ascitis y hepatomegalia marcada.El soplo diastólico de la estenosis tricuspídea tiene muchas de las características del soplo diastólico de la estenosis mitral, y, como la estenosis tricuspídea casi siempre se produce en presencia de estenosis mitral, la lesión valvular menos frecuente puede pasar desapercibida. Sin embargo, el soplo tricuspídeo suele oírse mejor en la parte inferior del borde esternal izquierdo y sobre el apéndice xifoides, y en pacientes en ritmo sinusal es más intenso durante la presístole.
Estudio de laboratorio
Los signos electrocardiográficos de agrandamiento de la aurícula derecha consisten en ondas P altas y picudas en la derivación II y ondas P prominentes y positivas en la derivación V1.
Insuficiencia tricuspídea
26 Medicina Interna Sexto año

Lo más frecuente es que la insuficiencia tricuspídea sea funcional y secundaria a una dilatación notable del ventrículo derecho y del anillo tricúspide.
La insuficiencia tricuspídea funcional puede complicar el agrandamiento del ventrículo derecho producido por cualquier causa, como el infarto de la pared inferior que afecta al ventrículo derecho, y se observa a menudo en los estadios avanzados de la insuficiencia cardíaca por cardiopatías reumáticas o congénitas con hipertensión pulmonar grave, y también en la cardiopatía isquémica, miocardiopatías y cor pulmonale. Es, en parte, reversible si disminuye la hipertensión pulmonar.
Tratamiento
Al igual que en la estenosis tricuspídea, los datos clínicos de la insuficiencia tricuspídea derivan principalmente de la congestión venosa general y de la reducción del gasto cardíaco. Las venas del cuello aparecen distendidas, con ondas v prominentes; son frecuentes la hepatomegalia marcada, ascitis, derrame pleural, edemas, pulsaciones sistólicas del hígado y reflujo hepatoyugular positivo.
Son signos característicos un prominente impulso ventricular derecho en la región paraesternal izquierda y un soplo holosistólico silbante en la parte inferior del borde esternal izquierdo, que puede intensificarse durante la inspiración y disminuir durante la espiración o la fase de tensión de la maniobra de Valsalva; habitualmente existe fibrilación auricular.
COR PULMONALE
Aumento de tamaño del ventrículo derecho secundario a alteraciones del pulmón. Tórax, ventilación o la circulación pulmonar.
FUNCIONAMIENTO NORMAL D ELA CIRCULACION PULMONAR
Fisiología
El volumen sistólico del ventrículo derecho esta regulado por:
1. Precarga2. Contractibilidad3. Poscarga
En condiciones normales es necesaria una presión de 5 cmH2O entre la arteria pulmonar y la aurícula izquierda para impulsar todo el gasto cardiaco en reposo (5l/min.)
CAUSAS QUE AUMENTAN LA POSCARGA DEL VENTRICULO DERECHO
1. Neuropatía Obstructiva Crónica, porque los capilares alveolares se encuentran comprimidos y los vasos pulmonares alargados.
2. Enfermedades pulmonares restrictivas, donde los vasos están comprimidos.3. Vasoconstricción pulmonar inducida por hipoxia o acidosis.
27 Medicina Interna Sexto año

4. Las principales causas son las enfermedades vasculares o parenquimatosas del pulmón.
ENFERMEDADES VASCULARES PULMONARES
La poscarga aumenta como consecuencia de restricción del flujo sanguíneo pulmonar. La hipertensión pulmonar es mas grave que en las enfermedades parenquimatosas del pulmón.
Puede ser consecuencia de:
1. Embolias pulmonares repetidas.2. Vasculitis Pulmonar3. Vasoconstricción pulmonar por grandes alturas.4. Cardiopatías congénitas con comunicación de izquierda a derecha.5. Enfermedad venooclusiva pulmonar.
Cuando se desconoce la causa se denomina Hipertensión Pulmonar Primaria.
COR PULMONAL DEBIDO A EMBOLIAS PULMONARES
Se asocia a dos síndromes diferentes:
1. Cor Pulmonale agudo.2. Cor pulmonale crónico secundario a enfermedad vascular pulmonar.
1. COR PULMONALE AGUDO:
La liberación repentina y masiva de émbolos causa una situación de bajo gasto, debido a la incapacidad del Ventrículo derecho para impulsar la sangre a través del lecho pulmonar.
El ventrículo derecho comienza a fallar cuando la presión sistólica se duplica supera los 50mmHg.
Se sospecha por antecedentes de disnea intensa y colapso cardiovascular en pacientes con antecedentes de trombosis venosa.
Manifestaciones Clínicas
Palidez Diaforesis Hipotensión Pulso rápido y débil. Venas cuello distendidas. Hígado pulsátil, aumentado de tamaño y doloroso a la palpación. Galope presistolico.
28 Medicina Interna Sexto año

PaO2 disminuida.
TRATAMIENTO
Se da tratamiento para embolia pulmonar.
Expansión cuidadosa de la volemia ayuda a mantener el gasto cardiaco.
Inhalación O2, si la vasoconstricción hipoxica es la causa.
2. COR PULMONALE CRONICO SECUNDARIO A ENFERMEDAD VASCULAR PULMONAR
Causas:
1. Émbolos recurrentes de tamaño medio que no se disuelven sino que se organizan.2. Partículas inyectadas por el consumo de drogas.3. Parásitos.4. Tumores.5. Hipertensión Pulmonar primaria.
Manifestaciones Clínicas:
Disnea y taquipnea (molestias durante el ejercicio pequeño y no se alivian sentándose). Tos no productiva. Dolor tórax. Hepatomegalia Edema maleolar. Cianosis. Soplo sistólico o diastólico. Ondas alfa en pulso yugular.
Radiografia
Aumento de tamaño del tronco pulmonar y de los vasos hiliares, asi como de la rama descendente de la arteraia pulmonar derecha.
Electrocardiograma
Onda P pulmonar (alta). Desviación del eje a la derecha. Hipertrofia del Ventrículo derecho.
29 Medicina Interna Sexto año

Ecocardiografia
Grosor ventrículo derecho. Tabique interventricular desplazado a la izquierda. Hipertrofia ventrículo izquierdo.
Resonancia Magnética
Se evalua la masa del ventrículo derecho, espesor de la pared, volumen de la cavidad y fraccion de expulsión.
Cateterismo cardiaco
Medicion de presiones pulmonares.
Biospsia Pulmonar
Vasculitis
Tratamiento Oxigeno Broncodilatadores Antibióticos Diureticos para el edema.
EPOC
30 Medicina Interna Sexto año

Las vías respiratorias, también llamadas vías aéreas, son un conjunto de órganos (cavidad nasal, faringe, laringe, tráquea y bronquios) que transportan el aire hacia los pulmones. Estos están recubiertos internamente por una capa de tejido epitelial (se compone de células muy unidas, con poca sustancia intercelular entre ellas, con lo que protege al cuerpo de lesiones e infecciones) y una mucosa respiratoria. Esta última contribuye de manera importante a que el aire que entra en los pulmones tenga la humedad y la temperatura adecuadas. En la superficie de la mucosa se alternan dos tipos de células: las mucosas, que fabrican y secretan moco hacia laentradas de las vías aéreas, y las ciliadas, que disponen de unos filamentos muy pequeños (cilios) que se mueven constantemente, barriendo el moco y las partículas que se depositan en la mucosa.
Cada pulmón, que se divide en lóbulos, pesa cerca de 800 gramos. El derecho consta de tres (superior, medio e inferior) y el izquierdo, solo de dos (superior e inferior).
Esto último ocurre porque el corazón se encuentra ubicado en el centro del pecho hacia la izquierda, restándole una porción de espacio al pulmón izquierdo.
Dentro de los lóbulos hay una gran cantidad de lobulillos conectados a un bronquiolo, que, a su vez, se divide en cavidades conocidas como vesículas pulmonares.
Acá se forman los alvéolos, que son sacos muy pequeños y elásticos, cuya principal función es producir el intercambio de oxígeno y dióxido de carbono.
En la superficie interna de cada alvéolo existen células de sangre, conocidas como macrófagos, encargadas de comer y destruir las sustancias irritantes que ingresan con el aire, como bacterias, partículas de polvo, entre otros.
NOC: Enfermedad caracterizada por la limitación al flujo aéreo, la cual no es completamente reversible. La limitación al flujo aéreo es comúnmente progresiva y se asocia a una respuesta inflamatoria anormal de los pulmones por la exposición a partículas nocivas o gases.
Los signos caracteristicos del EPOC son: ventilación dispar y desigualdad entre ventilaciony el riego sanguíneo.
31 Medicina Interna Sexto año

Además del EPOC…….. existen otros trastornos de la respiración donde se altera el flujo.
Denominamos apnea al cese de la respiración. La más conocida es la apnea obstructiva del sueño, en la que el flujo de aire no ingresa de manera correcta por las vías aéreas superiores; se suceden una serie de pausas involuntarias durante la respiración, producto de la relajación y posterior bloqueo por parte de los músculos de la garganta, el paladar blando o la lengua.También existe la apnea central, que se relaciona con una falla del centro nervioso respiratorio, el cual no regula de manera exacta el ritmo y continuidad de la respiración. Esta afecta muchas veces a recién nacidos, que mueren al no seguir respirando (muerte súbita).
La hipertensión pulmonar es otra afección que perjudica el intercambio de oxígeno. Se caracteriza por la elevada presión de las arterias pulmonares y por la alteración del trabajo de los vasos sanguíneos alojados en el pulmón.Produce fatiga e insuficiencia cardíaca, ya que, además, incrementa el tamaño del corazón.
La insuficiencia respiratoria aguda es una enfermedad que ocurre a nivel alveolar, en la que los alvéolos se llenan de fluidos y secreciones. Los sacos adaptados para recibir el aire ya no pueden hacerlo, porque su capacidad está totalmente ocupada por el líquido. Al comienzo de la enfermedad, la persona sufre de ahogos, los que se suceden con respiraciones rápidas y poco profundas. Luego, esta evoluciona hasta impedir, casi por completo, el funcionamiento de los pulmones.
32 Medicina Interna Sexto año

Es una afección crónica que destruye la pared alveolar, dificultando la respiración.Los principales factores de riesgo son el humo del cigarro y otros contaminantes, que liberan químicos hacia los pulmones, dañando irreversiblemente estas estructuras.
A los individuos con enfisema se les denomina “sopladores rosados”
33 Medicina Interna Sexto año

A los individuos con Bronquitis crónica se le denomina “ congestivos azules”.
34 Medicina Interna Sexto año

ETIOLOGIA
Aunque la principal causa del EPOC es el tabaquismo, se puede decir que entre un 20% y un 25% de los fumadores desarrollan la enfermedad, pero se desconocen las causas de predisposición al desarrollo.
35 Medicina Interna Sexto año

Fisiopatologia
El signo típico de la EPOC es una:
1. Disminucion persistente del flujo espiratorio forzado.2. Incremento del volumen residual y de la razón volumen residual/capacidad pulmonar total.3. Disminucion desigual de la ventilación.4. Desigualadad en el cociente ventilación/riego.
Obstruccion de las vías respiratorias
A diferencia de lo observado en el asma, la disminución del FEV1rara vez presentan respuestas importantes a los broncodilatadores inhalados, aunque es frecuente advertir mejoría incluso e 15%.
El flujo durante la espiración forzada es resultado del equilibrio entre:
El retroceso elástico de los pulmones en pro del flujo y la resistencia de las vías respiratorias que lo limita.
Hiperinsuflacion
En la EPOC hay “atrapamiento de aire” (incremento del volumen residual y de la razón volumen residual/capacidad pulmonar total). Esta hiperinsunflacion es útil para compensar la obstruccion.
Hiperinsuflacion progresiva (mayor capacidad pulmonar total).
Intercambio de Gases
36 Medicina Interna Sexto año

La hipertensión pulmonar con suficiente intensidad como para causar cardiopatía pulmonar, e insuficiencia del ventrículo derecho por EPOC surge en las personas que muestran una disminución marcada de FEV1 (<25%) e hipoxemia crónica (PaO2 <55mmHg).
Patogenia
La limitación de la corriente de aire, constituye el principal cambio funcional de la EPOC, se debe a una obstruccion de las vías respiratorias finas y al enfisema.
La patogenia del enfermo se puede clasificar en 3 fenomenos interrelacionados:
1. Exposicion de larga duración al humo del tabaco, que podría reclutar células inflamatorias al interior de los espacios aéreos terminales del pulmon.
2. Dichas células inflamatorias liberarían proteinasas elastoliticas que danarian la masa extracelular de los pulmones.
3. Reparacion ineficaz de la elastina y de otros componentes de la matriz extracelular.
Pruebas de Laboratorio
El signo definitorio de EPOC es la obstruccion al flujo del aire. Las pruebas de función pulmonar indican la presencia de este trastorno, con una:
37 Medicina Interna Sexto año

Disminucion del FEV1. Disminucion FEV1/FVC
Al empeorar la enfermedad puede:
Los volúmenes pulmonares. La capacidad pulmonar total. La capacidad residual pulmonar El volumen residual.
La medición de gases en sangre arterial y la oximetría no son métodos sensibles.
El cambio de pH con la Pco2 es de:
0.08u/10 mmHg en la forma aguda. 0.03u/ 10 mmHg en el estado crónico.
Por tal razón, conocer el pH arterial permite certificar la insuficiencia ventilatoria, que se define como una Pco2 > 45 mmHg en los cuadros agudos y crónicos.
La medición de gases en sangre arterial es un componente importante de la evaluación de las personas cuyos síntomas iniciales incluyen la exacerbación.
TRATAMIENTO
38 Medicina Interna Sexto año

TRATAMIENTOS QUIRURGICOS: Trasplante pulmonar
39 Medicina Interna Sexto año

TROMBOEMBOLIA PULMONAR
Definición
Predisposición a la embolia pulmonar
factores adquiridos
viajes aéreos prolongados obesidad Tabaquismo empleo de anticonceptivos orales embarazo restitución hormonal posmenopáusica intervenciones quirúrgicas traumatismos problemas médicos como: síndrome de anticuerpos antifosfolípidos, cáncer, hipertensión arterial
general y neuropatía obstructiva crónica. La trombofilia contribuye de manera notable al riesgo de trombosis venosa.
Factores genéticos:
Las dos mutaciones genéticas autosómicas dominantes más frecuentes son:
La del factor V de Leiden la del gen de la protrombina
Trombosis venosa profunda
Es la presencia de un trombo en una vena profunda y la respuesta inflamatoria que la acompaña .
Los factores que predisponen a la trombosis venosa fueron descritos por virchow en 1965 y son:
Estasis Daño vascular hipercoagulabilidad
La consecuencia mas importante de la trombosis venosa profunda son las embolias pulmonares
FISIOPATOLOGIA
Cuando los trombos venosos son desalojados de su lugar de formación. ↓
40 Medicina Interna Sexto año

embolizan la circulación arterial pulmonar o, paradójicamente, la circulación arterial. ↓
Produciendo asi tromboembolia pulmonar.
Alrededor de la mitad de los pacientes con trombosis de vena pélvica o trombosis venosa profunda proximal de la pierna tienen tromboembolia pulmonar, que suele ser asintomática.
Las trombosis aisladas de la vena de la pantorrilla plantean un riesgo menor, pero son la fuente más común de embolia paradójica.
Fisiología
La embolia pulmonar puede tener los siguientes efectos:
1. Aumento de la resistencia vascular pulmonar debida a la obstrucción vascular o a los agentes neurohumorales, incluida la serotonina.
2. Alteración del intercambio gaseoso debido al aumento del espacio muerto alveolar por obstrucción.
3. Hiperventilación alveolar debida a la estimulación refleja de los receptores de irritación.
4. Aumento de la resistencia de las vías respiratorias por constricción de vías respiratorias distalmente a los bronquios.
5. Distensibilidad pulmonar reducida por un edema pulmonar, una hemorragia pulmonar o la pérdida de tensoactivo.
Disfuncion ventricular derecha
La insuficiencia progresiva del corazón derecho suele ser la causa de muerte por tromboembolia pulmonar.
• Al aumentar la resistencia vascular pulmonar ↓
• Aumenta la tencion de la pared ventricular derecha ↓
• Comprime la arteria coronaria derecha ↓
• Isquemia miocardica e infarto pulmonar derecho
SINTOMAS Y SIGNOS
La disnea es el síntoma más frecuente y la taquipnea el signo más frecuente.
La disnea, el síncope, la hipotensión o la cianosis indican una tromboembolia masiva.
41 Medicina Interna Sexto año

El dolor pleurítico, la tos o la hemoptisis suelen indicar una embolia pequeña situada distalmente cerca de la pleura.
Síndromes clínicos
Tromboembolia masiva: Los pacientes presentan: hipotensión arterial general y suelen tener tromboembolia anatómicamente extensa. La terapia primaria con trombólisis o embolectomía proporciona la mayor posibilidad de supervivencia.
Los pacientes con tromboembolia moderada a grande: en la ecocardiografía presentan una hipocinesia ventricular derecha pero una presión arterial sistémica normal. Estos pacientes pueden beneficiarse con la trombólisis o la embolectomía más que con la anticoagulación administrada aisladamente.
Tromboembolia pequeña a moderada: tienen función cardíaca y presión arterial general normales. Tienen buen pronóstico con cualquier clase de anticoagulación adecuada.
La presencia de infarto pulmonar: suele indicar una tromboembolia pequeña, pero que es particularmente dolorosa porque se alberga en la periferia, cerca de las ramificaciones nerviosas pleurales sensitivas.
La embolia pulmonar no trombótica: puede pasar inadvertida. Entre las posibles causas se incluye la embolia grasa después de traumatismo contuso y las fracturas de los huesos largos, el embolismo tumoral o el gaseoso.
La embolia de líquido amniótico: se produce cuando la membrana fetal tiene pérdidas o se rasga en el borde placentario.
DIAGNOSTICO DIFERENCIAL
Síndrome coronario agudo, incluso angina de pecho e infarto agudo del miocardio Neumonía, bronquitis o exsacervacion del asma, EPOC ICC Pericarditis Pleuresía, osteocondritis Fracturas costales, neumotórax Hipertensión pulmonar primaria Ansiedad
Cuadro 244-1. Sistema de puntuación diagnóstica de Wellsa por la sospecha de tromboembolia pulmonar.
PUNTOS
42 Medicina Interna Sexto año

Signos y síntomas clínicos de DVT (tumefacción y dolor mínimos de la
pierna durante la palpación de las venas profundas)3.0
Es menos probable un diagnóstico alternativo que la PE 3.0
Frecuencia cardíaca >100 latidos por minuto 1.5
Inmovilización o intervención quirúrgica durante las cuatro semanas
previas1.5
Trombosis venosa profunda o tromboembolia previas 1.5
Hemoptisis 1.0
Problemas malignos (bajo tratamiento, sometidos a éste durante los seis meses
previos o paliativo)
1.0
El sistema de puntuación de Wells tiene un máximo de 12.5 puntos. Si la puntuación obtenida es 4 puntos, la posibilidad de PE será de 8% nada más.
Tecnicas de dx sin imagenes
Pruebas sanguíneas: concentración deldimero D medido mediante ELISA, está elevada (>500 ng/ml) en más del 90% de los pacientes con tromboembolia pulmonar , lo que refleja la rotura de la fibrina por la plasmina e indica una trombólisis endógena.
Sin embargo, la prueba del dímero D no es específica y, por este motivo, carece de utilidad para los pacientes que se encuentran hospitalizados de antemano.
43 Medicina Interna Sexto año

Electrocardiograma:
Las anomalías clásicas incluyen taquicardia sinusal; fibrilación o aleteo auricular de nueva presentación y una onda S en la derivación I, una onda Q en la derivación III y una onda T invertida en la derivación III.
Frecuentemente el eje QRS es superior a 90°. La inversión de la onda T en las derivaciones V1 a V4, quizá el cambio más común pero menos conocido,
refleja una tensión ventricular derecha.
Diagnostico de imagenes
Radiografia de torax Ecografia venosa TAC de torax Gammagrafia pulmonary RM Ecocardiografia
Radiografia de torax
Una radiografía de tórax normal o casi normal en un paciente disneico sugiere una tromboembolia pulmonar.
a) Las anomalías bien establecidas incluyen oligoemia focal (signo de Westermark)b) una densidad periférica en forma de cuña encima del diafragma (joroba de Hampton) c) o una arteria pulmonar descendente derecha de mayor tamaño (signo de Palla).
Ecografía venosa
La trombosis venosa profunda (DVT) confirmada puede ser un aspecto relacionado de utilidad de la tromboembolia pulmonar.
Tomografía computadorizada del tórax
La tomografía computadorizada de tórax con material de contraste intravenoso está sobrepasando a la gammagrafía pulmonar como el principal método de imágenes para el diagnóstico de tromboembolia pulmonar.
La TAC de tórax permite diagnosticar con eficacia la tromboembolia central de gran tamaño.
Gammagrafía pulmonar
44 Medicina Interna Sexto año

Se inyectan IV albumina marcados con tenecio –99, la distribucion del radioisotopo sigue la del flujo sanguineo, en la tromboembolia suele haber discordancia entre la ventilacion-perfusion.
Se define como gammagrafía de gran probabilidad de tromboembolia pulmonar la que tiene más de dos defectos segmentarios de perfusión en presencia de ventilación normal
Resonancia magnética (MR)
La RM permite valorar también la función del ventrículo derecho, lo que la vuelve promisoria como prueba única tanto para el diagnóstico de la tromboembolia pulmonar como para valorar su efecto hemodinámico.
Tecnicas de Dx Invasivas
Angiografia Pulmonar: Es el examen mas especifico para establecer el dx definitivo de tromboembolia pulmonar y puede detectar embolos de tan solo 1 a 2mm.
Flebografia de Contraste: Es costosa e incomoda y en ocasiones se acompaña de alergia al material de contraste oflebitis a este agente.
Tratamiento
Tratamiento primario
Consiste en disolver el coagulo con trombolisis o extraer el embolo por embolectomía. Debe reservarse para los pacientes con riesgo elevado de que la evolución clínica sea desfavorable.
Tratamiento secundario La anticoagulación con heparina y warfarina o la colocación de un filtro en la vena cava inferior constituyen la prevención secundaria de una tromboembolia pulmonar recurrente más que el tratamiento primario. Cuando la función ventricular derecha sigue siendo normal se obtienen buenos resultados clínicos con la anticoagulacion exclusivamente.
Trombolisis:
Disuelve gran parte del trombo arterial pulmonar obstructivo;
Evita la liberación continua de serotonina y otros factores neurohumorales que, de otro modo, podrían
exacerbar la hipertensión pulmonar, y
Disuelve buena parte del origen de los trombos en las venas pélvicas o profundas de la pierna, reduciendo
así la probabilidad de PE recurrente.
Heparina
45 Medicina Interna Sexto año

La heparina se une a la antitrombina III y acelera su actividad. La antitrombina III es una enzima que inhibe los
factores de coagulación de trombina (factor IIa), Xa, IXa, XIa y XIIa. De ese modo, la heparina impide la formación
adicional de trombos y permite que los mecanismos fibrinolíticos endógenos lisen el coágulo que ya se ha
formado. Después de cinco a siete días de heparina, el trombo residual empieza a estabilizarse en el endotelio de
la arteria o vena pulmonar. Sin embargo, la heparina no disuelve directamente el trombo ya existente.
Warfarina
Este antagonista de la vitamina K evita la activación por carboxilación gamma de los factores de coagulación II, VII,
IX y X. El efecto completo de la warfarina suele necesitar cinco días, aunque el tiempo de protrombina usado
como control se eleva más rápidamente.
La complicación mas común de estos anticoagulantes es la hemorragia.
ALTERACIONES DE LIQUIDOS Y ELECTROLITOS
SODIO Y AGUA
AGUAElemento que más abunda en el organismo, constituye el 50% del peso corporal en las mujeres y 50% en los varones; la diferencia se debe al tejido adiposo que es mayor en la mujer.
Esta distribuida en 2 grandes compartimientos
1) Intracelular 55-75%2) Extracelular 25-45%
A) Espacio intravascularB) Espacio extravascular
OSMOLALIDADEs la concentración de solutos que tiene un líquido, se expresa en miliosmoles por Kg de agua.
Principales partículas de líquido extracelularCatión: SodioAnión: Cloro y bicarbonato
Liquido intracelular1. Potasio
46 Medicina Interna Sexto año

2. Esteres de los fosfatos orgánicos Trifosfato de adenosina Fostato de creatinina Fosfolipidos
Los osmoles exclusivos son los que determinan la osmolalidad (tonicidad) de cada compartimiento.1. Compartimiento extracelular : Sodio 2. Compartimiento intracelular: Potasio
ADAPTACION OSMOTICAMantiene invariable el volumen de las células Esta mediada por intercambios del el potasio y sodioSe observa en
1) Hiponatremia2) Hipernatremia
OSMOLES INEFICACESSon los que no influyen en el desplazamiento del agua atravez de la membrana celular
1) Urea 2) Glucosa
MOVIMIENTO DE LOS LIQUIDOSSe da entre los espacios intravascular e intersticial y se produce atravez de la pared capilarEstá sometido por las fuerzas de Starling.
BALANCE O EQUILIBRIO HIDRICO
La osmolalidad del plasma es de 275-290mosm/Kg. Para mantener el equilibrio es preciso ingerir y eliminar la mima cantidad de agua Perdidas insensibles por evaporación regulan la temperatura central del cuerpo. Perdidas sensibles o forzosas; Se tiene que eliminar 600mosm diarios La osmolaridad máxima de la orina es 1200mosm/Kg Diuresis tiene que ser 500ml al día.
VÌAS VOLUMEN /DIA PROM.
MÌNIMO (ML)
MÀXIMO (ML)
47 Medicina Interna Sexto año
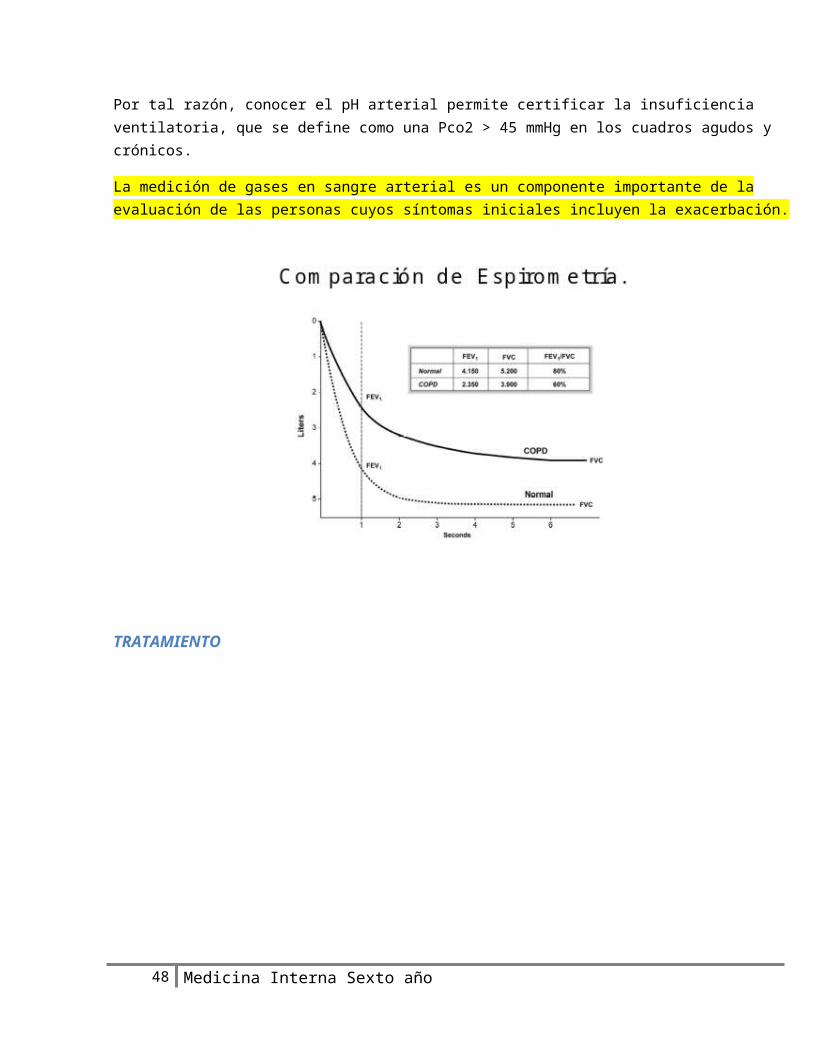
INGRESO SENSIBLE LÌQUIDOS ALIMENTOS INSENSIBLE OXIDACIÒN
800-1500500-700 250
00125
1500/HORA1500800
PÈRDIDAS SENSIBLES ORINA INTESTINOINSENSIBLEPULMON Y PIEL
800-15000-250600
300 ML0600
1400/HORA2500/HORA1500
INGESTION DE AGUA
Principal estimulo para la ingestión de agua es la sed.
Sed surge cuando:1) Aumentar la osmolalidad.2) Disminución del líquido extracelular.3) Disminución de la tensión arterial.
No estimulan la sensación de sed:1) Urea 2) Glucosa
Umbral osmótico de la sed es de 295mosm/kg (varia de un individuo a otro)
ELIMINACION DE AGUA
Regulada estrictamente por factores fisiológicos (la ingestión no). Vasopresina de Arguinina (Hormona anti diurética) es el factor determinante en la eliminación de agua
por el riñón; Es sintetizada por los núcleos supra ópticos y paraventriculares del hipotálamo y es secretado por el lóbulo posterior de la hipófisis.
La hipertonía es el estimulo que aumenta la secrecion de AVP. El umbral osmótico para liberación de AVP es de 280-290mosm/kg; y tiene sensibilidad de suficiente para
que la osmolalidad del plasma no varié mas de 1 a 2%.
Factores no osmóticos que influyen en la secreción AVP1) Volumen circulante (Arterial) eficaz.2) Nauseas3) Estrés 4) Dolor 5) Hipoglicemia6) Muchos fármacos
Para estimular la AVP se necesita una hipovolemia cuya intensidad disminuya la presión arterial media.
48 Medicina Interna Sexto año

Se necesitan 3 pasos para que el riñón pueda eliminar una sobrecarga de agua1) Filtración y paso de agua y electrolitos a los sitios de la nefrona donde se diluya la orina2) Resorción activa de sodio y cloro, sin agua en la porción gruesa de la rama ascendente del asa de Henle y
en menor grado en la nefrona distal.3) Mantenimiento de la dilución de la orina gracias a la impermeabilidad del conducto colector al agua
cuando falta la acción de la AVP.
BALANCE (EQUILIBRIO) DEL SODIO
El 85-90% de todo el sodio es extracelular.Los cambios en la concentración de sodio reflejan trastornos de la hemostasia del agua.Alteración en la cantidad de sodio se manifiestan por contracción o expansión del volumen extracelular.
Ingestión de SodioLa ingestión diaria de sodio produce un mayor volumen del liquido extracelular y esto a su vez estimula la eliminación renal de sodio.
Eliminación de sodio El déficit o el exceso de sodio se manifiesta por descenso o aumento de la volemia Los cambios en la volemia originan cambios en la TFG. La resorción tubular de sodio: Es el principal mecanismo regulador de la eliminación de sodio. Casi 2/3 del sodio que se filtra en los glomérulos se resorben en el túbulo contorneado proximal,
fenómeno que es isosmotico y electroneutro. En la porción gruesa de la rama ascendente del asa de Henle se resorbe el 25 a 30% del sodio. En el túbulo contorneado distal se resorbe el 5%. La resorción final de sodio tiene lugar en los conductos colectores de la corteza y medula renal, y se
elimina una cantidad aproximadamente a la que se ingiere.
HIPOVOLEMIA
Consiste en una pérdida de sodio y agua que supera la cantidad ingerida.Las perdidas pueden ser renales y extra renales.
Perdidas Renales1) Administración de diuréticos.
(Manitol es un diurético que produce diuresis osmótica)2) Necrosis tubular aguda.3) Después de una resolución de la obstrucción bilateral de las vías urinarias.4) Déficit de mineralocorticoides (hiperaldosteronismo).
(Origina perdida de sodio aunque la función renal intrínseca sea normal)
La reducción del LEC es menos intensa, ya que los 2/3 del líquido que se pierde proceden del agua intracelular.Entidades que causan pérdida urinaria de agua
1) Diabetes insípida central2) Diabetes insípida nefrogena
Se deben a una menor secreción de AVP y falta de respuesta del riñón a la AVP.
49 Medicina Interna Sexto año

Perdidas Extrarenales
Comprenden pérdidas de líquidos por:1. Tubo digestivo2. Piel3. Aparato respiratorio4. Acumulación de liquido en el tercer espacio
Quemaduras Pancreatitis Peritonitis
1. En tubo digestivo se acumulan 9 litros diarios de liquido: 2 son líquidos digeridos y 7 son secreciones digestivas.
2. El 98% de esta cantidad se resorbe por lo que las pérdidas son de 100-200ml al día; Si aumenta o disminuye se produce deshidratación.
3. Perdidas insensibles 500ml al día.4. La concentración de sodio en el sudor es de 20 a 50mol/L; esta disminuye cuando hay sudación intensa
por acción de la aldosterona.5. El sudor es hipotónico (se pierde más H2O que sodio)6. En la hiperventilación aumenta la perdida de agua que se evapora por el aparato respiratorio.
Tercer Espacio
Este compartimiento es extracelular pero no se intercambia con el LEC ni con el LIC.Es líquido que procede del LEC y por lo tanto puede ocasionar hipovolemia. Ejemplos de este espacio.
1. La luz intestinal cuando hay obstrucción del tubo digestivo.2. El tejido subcutáneo en las quemaduras graves3. El espacio retro peritoneal en pancreatitis aguda4. Cavidad peritoneal en la peritonitis.
Fisiopatología de la Hipovolemia.Las pérdidas del LEC se manifiestan por reducción del volumen del plasma e hipotensión arterial.La hipotensión se debe a:
1. Disminución del retorno venoso (precarga).2. Disminución del gasto cardiaco.3.
Lo anterior estimula los barorreceptores del seno carotideo y del cayado aórticoLuego se activa el SNS y el Sistema Renina Angiotensina.El objetivo es mantener la presión arterial media y la circulación coronaria cerebral.
Cuadro Clínico
La mayor parte son inespecíficos y dependen del desequilibrio hidroelectrolitico.1. Cansancio2. Debilidad3. Calambres
50 Medicina Interna Sexto año

4. Sed5. Mareos ortostastico
Si las pérdidas son más graves hay isquemia y se manifiesta por:1. Oliguria2. Cianosis3. Dolores abdominales y torácicos4. Confusión mental5. Obnubilación
No son parámetros fieles de la disminución del liquido intersticial1. L a menor turgencia cutánea.2. Sequedad de mucosas.
Signos de Hipovolemia1. Descenso de la presión venosa yugular.2. Hipotensión postural.3. Taquicardia postural.
Choque Hipovolemico1. Taquicardia2. Vasoconstricción periférica3. Deficiencia del riego periférico
Cianosis Miembros fríos y sudorosos Oliguria Alteración de la conciencia.
Diagnostico Aumento del BUN. Aumento de la creatinina. En sujetos normales el cociente BUN/Creatinina es de 10:1. En la hiperazoemia prerenal el cociente es de 20:1. concentración de sodio en la orina <20mmol/L. Osmolalidad > 450mosm/L. Densidad urinaria 1.015.
Tratamiento1) Natremia normal o poco disminuida: cloruro de sodio al 0.9%2) Hiponatremia intensa: cloruro de sodio al 3%3) Hipernatriemia :cloruro de sodio al 0.45% (es una solución hipotonica)
HIPONATREMIA
Es la concentración plasmática de sodio menor de 135mmol/L.
51 Medicina Interna Sexto año

En casos de Hiponatremia la osmolalidad del plasma puede resultar normal o alta.
Hiponatremia isotónica o ligeramente hipotonica puede observarse como complicación de la extirpación de la vejiga, próstata atravez de la uretra, cuando se absorbe gran cantidad de soluciones isosmotico como el manitol o hipoosmoticas (sorbitol, glicina).
Hiponatremia hipertónica puede deberse a hiperglicemia o a la administración de manitol.
En la diabetes mellitus mal compensada la glucosa es un osmol o un soluto eficaz que retira agua de las células musculares y ocasiona Hiponatremia.La concentración de sodio en plasma desciende 1.4mmol/L por cada 100mg/100ml que se eleva la concentración de glucosa en plasma.La Hiponatremia hipotonica se debe a aumento primario del agua (y a perdida secundaria de sodio) o a perdida primaria de sodio (con aumento secundario de agua)La Hiponatremia secundaria a diuréticos casi siempre se debe a tiazidas.
Síndrome de secrecion inadecuada de Hormona Antidiueretica
Es la causa más importante de Hiponatremia normovolemicaSe debe a la liberación anormal de AVP por el lóbulo posterior de la hipófisis o síntesis ectópica de esta hormona.La eliminación de H2O libre por el riñón disminuye mientras que la regulación de sodio no se altera.Causas mas frecuentes:
1) Cuadros neuropsiquiatricos2) Cuadros pulmonares3) Tumores malignos4) Intervenciones de cirugía mayor5) Algunos fármacos
El dolor intenso y las nauseas son estímulos fisiológicos para la liberación de AVP, pero son insuficientes si no hay hipovolemia o hiperosmolaridad.
El Síndrome de secrecion inadecuada de hormona Antidiueretica se clasifica en 4:1) Secrecion de AVP autónoma o aberrante (ectópica)2) Regulación normal de la liberación de AVP ajustada a un nivel más bajo de la osmolalidad (caquexia y
malnutrición)3) Respuesta normal de la AVP a la hipertonía que no se inhibe del todo ante una osmolalidad baja (sección
incompleta del tallo hipofisiario)4) Secrecion normal de AVP con aumento de la sensibilidad a sus efectos o secrecion de algún otro factor
antidiuretico (raro)
Las hormonas pueden causar Hiponatremia por exceso o por déficit.
El hipertiroidismo se manifiesta con Hiponatremia no confundir con Síndrome. De secrecion inadecuada AVP.El hipotiroidismo produce Hiponatremia mediante varios mecanismos:
1) Reducción gasto cardiaco2) Reducción de la taza de filtración glomerular3) Aumento de la secrecion de AVP por reacción a los estímulos hemodinámicos.
52 Medicina Interna Sexto año

Puede aparecer Hiponatremia sin que exista AVP ni insuficiencia renal si el riñón es incapaz de eliminar un gran volumen de agua ingerida.Capacidad de eliminación del riñón 12/L al día; la polidipsia psicógena o primaria puede sobrepasar esta cantidad.La eliminación urinaria máxima depende de la osmolalidad urinaria mínima; el metabolismo de una dieta normal produce 600mosm/día. Y la osmolalidad mínima del ser humano es de 50mom/Kg. La eliminación urinaria máxima será de 12L. (600÷50).El déficit de cortisol induce indirectamente la hipersecreción de AVP.
Potomania de la cervezaLos bebedores ingieren pocas proteínas y electrolitos y grandes cantidades de esta cerveza, que excede la capacidad de eliminación del riñón y se produce Hiponatremia.
Cuadro Clínico
Turgencia de las células cerebrales o edema cerebral.Los síntomas son principalmente neurológicos
1) Nauseas y malestar2) Cefalalgia3) Letargo4) Confusión mental5) Obnubilación
No suele haber estupor, convulsiones o coma salvo que las concentraciones de sodio en plasma sean menores de 120mmol/L.
Diagnostico
La Hiponatremia no es una enfermedad sino una manifestación de distintos trastornos.El diagnostico diferencial debe incluir insuficiencia cardiaca congestiva, cirrosis hepática y síndrome. Nefrotico.Variables de laboratorio que proporcionan información.
1) Osmolalidad del plasma2) Osmolalidad urinaria3) Concentración de sodio en orina4) Concentración de potasio en orina.
En la Hiponatremia la osmolalidad plasmática esta disminuida por la disminución de sodio.La orina tiene una osmolalidad y densidad menores de 100mosm/Kg y de 1.003.El síndrome. De secrecion inadecuada de AVP se caracteriza por Hiponatremia hipoosmoticas y orina inapropiadamente concentrada (osmolalidad urinaria mayor de 100mosm/L); El paciente suele tener normovolemia y balance normal de sodio.
Tratamiento1) Elevar la concentración plasmática de sodio, al restringir la ingestión de agua y facilitar la perdida de agua.2) Corregir el trastorno primario.
Al corregir la Hiponatremia con demasiada rapidez se corre el riesgo de que aparezca el síndrome de desmielinizacion osmótica; es un cuadro neurológico que se caracteriza por parálisis flácida, disartria y disfagia.
53 Medicina Interna Sexto año

HIPERNATREMIA
Se define como la concentración de sodio en plasma mayor de 145mmol/L; constituye un estado de hiperosmolaridad.Se debe a un aumento primario de sodio o déficit de agua; la mayor parte se debe a déficit de agua.El agua está repartida en una proporción de 2:1 en el LIC y LEC.Una pérdida de determinada cantidad de líquidos; en el LIC será del doble que en el LEC.Ejemplo: Si se pierde 1 L de H2O el volumen del líquido intracelular disminuirá en 667ml, mientras que en el extracelular será de 333ml.El grado de hiperosmolaridad suele ser leve salvo que el mecanismo de la sed no funcione o no exista el acceso al agua para beber como en lactantes, minusválidos, paciente con trastorno mental y los enfermos entubados en UCI.
Hipernatremia Hipodipsica primaria.Causada por la lesión de los osmorreceptores hipotalámicos que regulan la sed(Es raro que la falta de sed se deba a hipodipsia primaria).La hipodipsia puede depender de varias lesiones como: enfermedad granulomatosa, oclusión vascular y tumores.
Hipernatremia esencialNo mejora con la ingestión forzada de agua.Parece deberse a defectos específicos de los osmorreceptores, lo cual produce secrecion de AVP no regulada por mecanismos osmóticos.
Las pérdidas insensibles aumentan con: 1) Fiebre2) Ejercicio3) Exposición al calor4) Quemaduras graves5) Paciente sometido a ventilación mecánica
La concentración de Na en el sudor disminuye cuando hay sudación profusa y de este modo aumenta la perdida de agua sin solutos.
La diarrea es la causa de Hipernatremia de origen digestivo. Diarreas osmóticas y las gastroenteritis víricas producen perdidas de agua que superan a las de sodio y
potasio. Las diarreas secretoras (cólera, carcinoide y VIPomas) tienen una osmolalidad fecal parecida a la del
plasma; y se manifiestan por disminución del volumen de los LEC con sodio normal en plasma o por Hiponatremia.
Las pérdidas de agua por el riñón son la causa más frecuente de Hipernatremia y se debe a:1) Diuresis osmótica o inducida por fármacos.2) Diabetes insípida.
Hipernatremia secundaria a pérdidas no osmóticas.1) Diabetes insípida central (se caracteriza por menor secrecion de AVP).2) Diabetes insípida nefrogena (se debe a resistencia del riñón a la acción de la vasopresina).
Un aumento primario de sodio puede causar también Hipernatremia pero es raro.
54 Medicina Interna Sexto año

Cuadro Clínico
La hipertonía causa que el agua salga de las células y con ello que disminuya el LIC, también aminora el volumen de las células cerebrales y aumenta el riesgo de hemorragia sub aracnoides.Los principales síntomas son neurológicos y consisten en:
1) Alteración del estado de la conciencia2) Debilidad 3) Irritabilidad neuromuscular4) Déficit neurológico focal5) Coma 6) Convulsiones7) Poliuria 8) Sed
La Hipernatremia crónica produce menos síntomas gracias a los mecanismos compensadores que tratan de mantener sin cambios el volumen celular.
Diagnostico 1) Hiperosmolaridad.2) Volumen mínimo de orina (<500ml/día).3) Osmolalidad urinaria > 800 mosm/kg.4) Sodio en orina > 100 mmol/L (asegura que existe un exceso primario de sodio).
La eliminación diaria de solutos mayores de 750 mosm equivale a diuresis osmótica.
Tratamiento
1) Detener la pérdida progresiva de agua.2) Corregir al trastorno causal subyacente.3) Reponer el déficit de agua.
En la Hipernatremia por perdida de agua, el agua corporal total abarca alrededor del 50% de la masa corporal magra en varones y 40% en mujeres.El déficit de agua debe corregirse lentamente, durante un periodo de 48-72 horas como mínimo.La mejor vía para administrar el agua es la oral o sonda nasogástrica.Se puede usar solución salina glucosada al 5% o solución salina semiisotonica, por vía intravenosa.El tratamiento mas idóneo para la diabetes insípida central es la vasopresina intranasal.
Fármacos que estimulan la secrecion de AVP y potencia sus efectos en el riñón:1) Clorpropamida2) Clofibrato3) Carbamazepina4) AINES
POTASIO
55 Medicina Interna Sexto año

Principal catión del medio intracelular con una concentración de 150mmol/L. En el plasma la concentración de potasio es de 3.5-5 mmol/L. Cantidad de potasio en el LEC (30-70mmol/L ) constituye menos del 2% del potasio corporal total. Potasio corporal total 2500-4000mmol. El cociente de la concentración de potasio en el LIC y el LEC es de 38:1. Según el régimen de alimentación la persona ingiere entre 40-120mmol/día de potasio. El 90% de esta cantidad se absorbe en el tubo digestivo. El exceso de potasio se elimina por la orina.
Eliminación del potasio
Riñón principal vía.La cantidad de potasio que se filtra es de 10 a 20 veces mayor que la cantidad de potasio que está en el LEC; parte de esa proporción de 90% del potasio filtrado se resorbe en el túbulo contorneado proximal y el asa de Henle.La secrecion de potasio está regulada por 2 mecanismos fisiológicos:
1) Aldosterona (secretada por las células de la zona glomerulosa de la corteza suprarrenal; se secreta cuando aumenta el nivel de renina y Angiotensina II o cuando hay hipercalemia).
2) Hipocalemia.
HIPOCALEMIA
Se define como la concentración del potasio en plasma <3.5mmol/L.
Se debe a:1) Ingestión disminuida (rara vez es la única causa).2) Penetración en las células.3) Aumento final de las perdidas.
La restricción de potasio en la dieta puede agravar la Hipocalemia debida a las pérdidas de este catión.Causa poco frecuente de ingestión de escasa de potasio es el habito ce comer arcilla (geofagia) porque ella retiene el hierro y el potasio de la dieta.
Perdidas Extrarenales de potasio 1) Sudación excesiva2) Hiperaldosteronismo secundario
En condiciones normales el potasio que se pierde en las heces es de 5-10mmol/día en un volumen de 100 a 200ml.
Pueden ocasionar Hipocalemia1) Diarreas profusas2) Adenomas vellosos3) VIPomas4) Abuso de laxantes
La concentración del potasio en el liquido del estomago es de 5-1ommol/L.La Hipocalemia generalmente es por perdidas renales.
56 Medicina Interna Sexto año

Perdidas renales de potasio1) Exceso de mineralocorticoides.2) Hiperaldosteronismo primario.3) Tumores del aparato yuxtaglomerular que secretan renina.4) Tumores que secretan renina.
a) Carcinoma de células renales.b) Cáncer de ovario.c) Tumor de Wills.
5) Hiperplasia suprarrenal congénita.
Síndrome de LiddleEnfermedad familiar rara autosomica dominante.Se caracteriza por hipertensión, alcalosis metabólica, Hipocaliemia, pérdidas renales de potasio, e inhibición de la secrecion de renina y aldosterona.
Síndrome de BartterSe caracteriza por hipocaliemia, alcalosis metabólica, hiperaldosteronismo hiporreninemico secundario a disminución del LEC.
Cuadro clínico
Pocas veces hay síntomas solo que descienda la concentración de potasio en plasma <3mmol/L.1) Cansancio2) Mialgias3) Debilidad muscular de los miembros inferiores.4) Debilidad progresiva.5) Hipoventilacion6) Parálisis completa.
Alteraciones electrocardiografías en la hipocaliemia
Se deben al retraso de la repolarizacion ventricular y no guarda estrecha relación con la cantidad de potasio en plasma.
1) Aplanamiento e inversión de la onda T2) Onda U prominente3) Depresión del segmento ST4) Intervalo QU prolongado
Disminución intensa provoca:1) Intervalo PR largo2) Voltaje disminuido3) Ensanchamiento del complejo QRS
Tratamiento1) Corregir el déficit de potasio2) Reducir al mínimo las perdidas3) Suplementos intravenosos de potasio
a) Vena periférica 40mmol/L
57 Medicina Interna Sexto año

b) Vena central 60mmol/L4) Vigilar con frecuencia la concentración de potasio
HIPERCALIEMIA
Se define como la concentración de potasio en plasma >5mmol/L.El fenómeno de adaptación al potasio provoca una eliminación rápida del mismo cuando aumenta su ingestión.
Hipercaliemia YatrogenaCuando se repone potasio con demasiado entusiasmo por vía parenteral.
SeudohipercaliemiaConsiste en la concentración artificialmente de potasio en plasma consecutiva a la salida del potasio de la célula poco antes o después de una punción venosa.Factores que la favorecen
1) Torniquete prolongado2) Hemolisis 3) Leucocitosis4) Trombositosis intensa.
La administración de betas bloqueadores causa rara de Hipercaliemia, pero pueden favorecer el aumento de potasio en plasma.
Parálisis periódica hipercaliemica.Raro cuadro autosomico dominante.Se caracteriza por episodios de parálisis, debilidad muscular.
La Hipercaliemia crónica se debe a decremento de la eliminación renal.
Hipoaldosteronismo Hiporreninemico
Síndrome caracterizado por normovolemia o aumento del LEC junto con niveles inhibidos de renina y aldosteronaSe observa en:
1) Nefropatía diabética2) Insuficiencia renal leve3) Nefropatía diabética
El paciente reacciona con menor potasuria a la administración de glucorticoides exógenos.
Pacientes con mayor riesgo de sufrir Hipercaliemia inducida por IECA son los que tienen1) Diabetes mellitus2) Insuficiencia renal3) Estenosis bilateral de las arterias renales4) Que usen ahorradores de potasio.
El síndrome de Gordon ocasiona Hipercaliemia.
58 Medicina Interna Sexto año

Cuadro clínico1) Debilidad 2) Parálisis flácida3) Hipoventilacion4) Acidosis metabólica
Electrocardiografía1) Aumento de amplitud de ondas T2) Ondas T altas y acuiformes
Hipercaliemia intensificada1) Prolongación del intervalo PR y QRS2) Fusión con la onda T que produce un trazo ondulante por ondas sinusoides (monofásicas)
Diagnostico1) Anamnesis2) Secreción renal de potasio
La respuesta adecuada al riñón en la Hipercaliemia es la eliminación de 200mosm diarios de potasio como mínimo
TratamientoHay Hipocalemia capaz de causar la muerte salvo que la concentración de potasio en plasma supere los 7.5mmol/L. y se acompañe de:
1) Profunda debilidad2) Ausencia de ondas P3) Ensanchamiento del QRS4) Arritmias ventriculares en el electrocardiograma.
El tratamiento se debe enfocar en 1) Reducir al mínimo la despolarización de la membrana y la entrada de potasio en las células
a) Gluconato cálcico en las dosis habituales de 10ml de una solución al 10% que se inyecta en goteo de 2-3 minutos. Disminuye la excitabilidad de la membrana.
b) La insulina estimula la entrada de potasio a las células.
2) Interrumpir el aporte exógeno de potasio.3) Facilitar la perdida de potasio.
a) Diuréticos, resinas de intercambio de cationes o la diálisis.b) Diurético con acción en asa de Henle y las tiazidas suelen administrarse combinados y pueden
aumentar la eliminación de potasio.
SOLUCIONES DE TERAPIA INTRAVENOSA (Composición y propiedades)
Principales soluciones de terapia intravenosa
59 Medicina Interna Sexto año

Soluciones Cristaloides o Solución Salina Isotónica (Suero Fisiológico)
o Soluciones Salinas Hipertónicas
o Solución Glucosada Isotónica (Suero Glucosado al 5%)
o Soluciones Glucosadas Hipertónicas
o Soluciones Glucosalinas
o Solución Polielectrolítica de Ringer Lactato
o Soluciones Alcalinizantes
o Soluciones Acidificantes
Soluciones Coloidales
o Albúmina
o Fracciones Proteicas de Plasma Humano
o Soluciones Coloidales Artificiales
1. SOLUCIONES CRISTALOIDES
Las soluciones cristaloides son aquellas que contienen agua, electrolitos y/o azúcares en diferentes proporciones y que pueden ser hipotónicas, hipertónicas o isotónicas respecto al plasma.Su capacidad de expander volumen va a estar relacionada con la concentración de sodio de cada solución.Las soluciones cristaloides isotónicas respecto al plasma se van a distribuir por el fluído extracelular, presentan un alto índice de eliminación y se puede estimar que a los 60 minutos de la administración permanece sólo el 20% del volumen infundido en el espacio intravascular.La perfusión de grandes volúmenes de estas soluciones puede derivar en la aparición de edemas periféricos y edema pulmonar.Las soluciones hipotónicas se distribuyen a través del agua corporal total. Consisten fundamentalmente en agua isotonizada con glucosa para evitar fenómenos de lisis hemática. Sólo el 8% del volumen perfundido permanece en la circulación, ya que la glucosa entra a formar parte del metabolismo general generándose CO2 y H2O y su actividad osmótica en el espacio extracelular dura escaso tiempo. Debido a la mínima o incluso nula presencia de sodio en estas soluciones, su administración queda prácticamente limitada a tratamientos de alteraciones electrolíticas (hipernatremia), otros estados de deshidratación hipertónica y cuando sospechemos la presencia de hipoglucemia.
1.1. Solución Salina Isotónica (Suero Fisiológico)
Composición Suero Fisiológico al 0,9%
pH: 5,5Osmolaridad: 308 mOsm/LSodio: 154 mEq/LCloro: 154 mEq/L
La solución salina al 0.9% también denominada Suero Fisiológico, es la sustancia cristaloide estándar, es levemente hipertónica respecto al líquido extracelular y tiene un pH ácido.La normalización del déficit de la volemia es posible con la solución salina normal, aceptando la necesidad de grandes cantidades, debido a la libre difusión entre el espacio vascular e intersticial de esta solución.
60 Medicina Interna Sexto año

Después de la infusión de 1 litro de suero salino sólo un 20-30% del líquido infundido permanecerá en el espacio vascular después de 2 horas. Como norma general es aceptado que se necesitan administrar entre 3 y 4 veces el volumen perdido para lograr la reposición de los parámetros hemodinámicos deseados.
1.2. Soluciones Salinas Hipertónicas
Composición Suero Fisiológico al 3%
pH: 5,5Osmolaridad: 684 mOsm/LSodio: 342 mEq/LCloro: 342 mEq/L
Su mecanismo de actuación se debe principal y fundamentalmente, al incremento de la concentración de sodio y aumento de la osmolaridad que se produce al infundir el suero hipertónico en el espacio extracelular (compartimento vascular). Así pues, el primer efecto de las soluciones hipertónicas sería el relleno vascular. Habría un movimiento de agua del espacio intersticial y/o intracelular hacia el compartimento intravascular. Una vez infundida la solución hipertónica, el equilibrio hidrosalino entre los distintos compartimentos se produce de una forma progresiva y el efecto osmótico también va desapareciendo de manera gradual.
1.3. Solución Glucosada Isotónica (Suero Glucosado al 5%)
Composición del Suero Glucosado al 5%
pH: 4Osmolaridad: 278 mOsm/LGlucosa: 5 gr/100mLCalorías: 200 Kcal/L
Es una solución isotónica (entre 275-300 mOsmol/L) de glucosa, cuya dos indicaciones principales son la rehidratación en las deshidrataciones hipertónicas (por sudación o por falta de ingestión de líquidos) y como agente aportador de energía.La glucosa se metaboliza en el organismo, permitiendo que el agua se distribuya a través de todos los compartimentos del organismo, diluyendo los electrolitos y disminuyendo la presión osmótica del compartimento extracelular. El desequilibrio entre las presiones osmóticas de los compartimentos extracelular e intracelular, se compensa por el paso de agua a la célula. En condiciones normales, los osmoreceptores sensibles al descenso de la presión osmótica, inhiben la secreción de hormona antidiurética y la sobrecarga de líquido se compensa por un aumento de la diuresis.El suero glucosado al 5% proporciona, además, aporte calórico. Cada litro de solución glucosada al 5% aporta 50 gramos de glucosa, que equivale a 200 kcal. Este aporte calórico reduce el catabolismo protéico, y actúa por otra parte como protector hepático y como material de combustible de los tejidos del organismo más necesitados (sistema nervioso central y miocardio).
1.4. Soluciones Glucosadas Hipertónicas
Las soluciones de glucosa hipertónicas, al igual que la solución de glucosa isotónica, una vez metabolizadas desprenden energía y se transforman en agua. A su vez, y debido a que moviliza sodio desde la célula al espacio extracelular y potasio en sentido opuesto, se puede considerar a la glucosa como un proveedor indirecto de potasio a la célula.
Composición Suero Glucosado al 10%
pH: 4Osmolaridad: 555 mOsm/L
61 Medicina Interna Sexto año

Glucosa: 10 gr/100mLCalorías: 400 Kcal/L
Composición Suero Glucosado al 20%
pH: 4Osmolaridad: 1100 mOsm/LGlucosa: 20 gr/100mLCalorías: 800 Kcal/L
Composición Suero Glucosado al 30%
pH: 4Osmolaridad: 1655 mOsm/LGlucosa: 30 gr/100mLCalorías: 1200 Kcal/L
Composición Suero Glucosado al 50%
pH: 4Osmolaridad: 2775 mOsm/LGlucosa: 50 gr/100mLCalorías: 2000 Kcal/L
Composición Suero Glucosado al 70%
pH: 4Osmolaridad: 3890 mOsm/LGlucosa: 70 gr/100mLCalorías: 2800 Kcal/L
1.5. Soluciones Glucosalinas
Las soluciones glucosalinas son eficaces como hidratantes y para cubrir la demanda de agua y electrolitos con aporte de glucosa.
Composición Suero Glucosalino (Glucosa 3,3% + NaCl 0,3%)
Osmolaridad: 270 mOsm/LSodio: 51 mEq/L
62 Medicina Interna Sexto año

Cloro: 51 mEq/LGlucosa: 3,3 gr/100mLCalorías: 132 Kcal/L
Composición Suero Glucosalino (Glucosa 5% + NaCl 0,9%)
Osmolaridad: 560 mOsm/LSodio: 154 mEq/LCloro: 154 mEq/LGlucosa: 5 gr/100mLCalorías: 200 Kcal/L
1.6. Solución Polielectrolítica de Ringer Lactato
Composición Ringer Lactato
pH: 6Osmolaridad: 272 mOsm/LSodio: 130 mEq/LPotasio: 4 mEq/LCloro: 109 mEq/LCalcio: 0.75 mEq/LLactato: 28 mmol/l
La mayoría de las soluciones cristaloides son acidóticas y por tanto pueden empeorar la acidosis tisular que se presenta durante la hipoperfusión de los tejidos ante cualquier agresión. Sin embargo, la solución de Ringer Lactato contiene menos cloro que el suero fisiológico, causando menos posibilidad de causar acidosis. Es de preferencia cuando debemos administrar cantidades masivas de soluciones cristaloides.El efecto de volumen que se consigue es muy similar al de la solución fisiológica normal.La vida media del lactato plasmático es de más o menos 20 minutos, pudiéndose ver incrementado este tiempo a 4 ó 6 horas en pacientes con shock.
1.7. Soluciones alcalinizantes
Se utilizan en aquellas situaciones que exista o se produzca una acidosis metabólica.El bicarbonato sódico fue el primer medicamento que se utilizó como tampón.Las de utilización más habitual son la solución de bicarbonato 1 Molar ( 1 M = 8.4% ), que sería la forma preferida para la corrección de la acidosis metabólica aguda, y la solución de bicarbonato 1/6 Molar ( 1.4% ) con osmolaridad semejante a la del plasma.
Composición Bicarbonato 1 Molar
Bicarbonato: 1000 mEq/LSodio: 1000 mEq/L
Composición Bicarbonato 1/6 Molar
Bicarbonato: 166 mEq/LSodio: 166 mEq/L
63 Medicina Interna Sexto año

Otra solución isotónica correctora de la acidosis es el Lactato sódico. El lactato de sodio es transformado en bicarbonato sódico y así actuaría como tamponador, pero como esta transformación previa implica un metabolismo hepático, se contraindica su infusión en pacientes con insuficiencia hepática así como en la situación de hiperlactasemia.
1.8. Soluciones acidificantes
El cloruro amónico 1/6 Molar es una solución isotónica (osmolaridad = 334), acidificante, de utilidad en el tratamiento de la alcalosis hipoclorémica. El ión amonio es un dador de protones que se disocia en H+ y NH3+, y su constante de disociación es tal que en la gama de pH de la sangre el NH4+ constituye el 99% del amoníaco total. La acción acidificante depende de la conversión de los iones amonio en urea por el hígado, con generación de protones. Por ello, las soluciones de sales de amonio están contraindicadas en la insuficiencia hepática. Además, el cloruro de amonio posee toxicidad cuando es administrado de forma rápida, y puede desencadenar bradicardia, alteraciones respiratorias y contracciones musculares.
2. SOLUCIONES COLOIDALES
Las soluciones coloidales contienen partículas en suspensión de alto peso molecular que no atraviesan las membranas capilares, de forma que son capaces de aumentar la presión osmótica plasmática y retener agua en el espacio intravascular. Así pues, las soluciones coloidales incrementan la presión oncótica y la efectividad del movimiento de fluídos desde el compartimento intersticial al compartimento plasmático deficiente. Es lo que se conoce como agente expansor plasmático. Producen efectos hemodinámicos más rápidos y sostenidos que las soluciones cristaloides, precisándose menos volumen que las soluciones cristaloides, aunque su coste es mayor.
2.1. Albúmina
La albúmina se prepara mediante la precipitación con alcohol de plasma humano, y posteriormente es sometida a pasteurización durante 10 horas a 60 grados centígrados para la inactivación viral. Es el derivado plasmático más seguro desde el punto de vista de la transmisión de enfermedades infecciosas conocidas.La albúmina se produce en el hígado y es responsable de aproximadamente un 70-80% de la presión oncótica del plasma, constituyendo un coloide efectivo.La albúmina se distribuye entre los compartimentos intravascular (40%) e intersticial (60%).Su síntesis es estimulada por el cortisol y hormonas tiroideas, mientras que su producción disminuye cuando aumenta la presión oncótica del plasma. La concentración sérica normal en suero es de 3.5 a 5.0 g/dL y está correlacionado con el estado nutricional del sujeto. Si disminuyese la concentración de albúmina en el espacio intravascular, la albúmina del intersticio pasaría al espacio vascular a través de los canales linfáticos o bien por reflujo transcapilar.La capacidad de retener agua que tiene la albúmina viene determinada tanto por su cantidad como por la pérdida de volumen plasmático que se haya producido. Un gramo de albúmina incrementa el volumen plasmático aproximadamente en 18 mL, y 100 mL de albúmina al 25% incrementan el volumen plasmático una media de más o menos 465 ± 47 mL.La albúmina administrada se distribuye completamente dentro del espacio intravascular en dos minutos y tiene aproximadamente una vida media entre 4 y 16 horas. El 90% de la albúmina administrada permanece en el plasma unas dos horas tras la administración, para posteriormente equilibrarse entre los espacios intra y extravascular durante un período de tiempo entre 7 a 10 días. Un 75% de la albúmina comienza a desaparecer del plasma en 2 días. Su catabolismo tiene lugar en el tracto digestivo, riñones y sistema fagocítico mononuclear.Los preparados tienen generalmente una concentración del 5% (isooncóticos) o del 20% (hiperoncóticos), con un pH de 6.9.Principales indicaciones: Hipovolemia (cuando existe contraindicación de coloides y cristaloides), Hipoalbuminemia (si albúmina inferior a 20g/l,en periodos cortos).Ritmo de infusión: Velocidad individualizada.Recomendación:- Albúmina 20%: 1-2 mL por minuto.- Albúmina 5%: 5 mL por minuto.
2.2. Fracciones Proteicas de Plasma Humano
Las fracciones proteicas del plasma, al igual que la albúmina, se obtienen por fraccionamientos seriados del plasma humano.
64 Medicina Interna Sexto año

La fracción proteica debe contener al menos 83% de albúmina y no más de un 1% de g-globulina, el resto estará formado por a y b-globulinas. Esta solución de fracciones proteicas está disponible como solución al 5% en suero fisiológico y estabilizado con caprilato y acetiltrifosfanato sódico.Presentan propiedades similares a la albúmina y la principal ventaja de esta solución consiste en su fácil manufacturación y la gran cantidad de proteínas aportadas.
2.3. Soluciones Coloidales Artificiales
- Dextranos: Polisacáridos de origen bacteriano producidos por el Leuconostoc mesenteroides.- Hidroxietil-almidón (HEA): Almidón sintético preparado a partir de la amilopectina mediante la introducción de grupos hidroxietil éter en sus residuos de glucosa.- Pentaalmidón: Preparado con formulación semejante al hetaalmidón, pero con un peso molecular de 280.000 daltons y un número molecular medio de 120.000 daltons, por lo que también puede ser llamado hetaalmidón de bajo peso molecular.- Derivados de la gelatina: Polipéptidos obtenidos por desintegración del colágeno.
CÁLCULO DE LA VELOCIDAD DE PERFUSIÓN EN SUEROTERAPIA
1 mL = 1 cc = 20 gotas = 60 microgotas
Número de gotas por minuto = volumen para administrar en cc x 20 gotas / tiempo de perfusión en minutos
Número de microgotas por minuto = volumen para administrar en cc x 60 microgotas / tiempo de perfusión en minutos
ALTERACIONES ACIDO-BASICAS
Homeostasis acido básica normal
El pH arterial sistémico se mantiene entre 7.35 y 7.45 debido al amortiguamiento químico extracelular e intracelular y a los mecanismos reguladores que aportan los pulmones y los riñones. El control de la tensión arterial de CO2 (Paco2) por el sistema nervioso central y el aparato respiratorio, y el control del bicarbonato plasmático por los riñones, estabilizan el pH arterial mediante la eliminación o la retención de ácidos o álcalis. Los componentes metabólico y respiratorio que regulan el pH sistémico están descritos por la ecuación de Henderson-Hasselbalch:
En casi todas las circunstancias, la producción y la eliminación de CO2 están equilibradas, y la Paco2 habitual en situación estable se mantiene en 40 mmHg. El déficit de eliminación de CO2 produce hipercapnia, mientras que su eliminación excesiva ocasiona hipocapnia.la Paco2 está regulada principalmente por factores respiratorios o nerviosos y no por la velocidad de producción de CO2. La hipercapnia suele ser consecuencia de la hipoventilación y no de una mayor producción de CO2.
65 Medicina Interna Sexto año

La alteración primaria de la Paco2 origina amortiguamiento celular y adaptación renal, fenómeno lento que aumenta su eficacia con el tiempo. El cambio primario en la [HCO3 –] plasmática a causa de factores metabólicos o renales induce cambios compensadores en la ventilación, que amortiguan las variaciones del pH sanguíneo que de otra forma se producirían. Tales modificaciones respiratorias se denominan secundarias o compensadoras, ya que son una reacción a cambios metabólicos primarios.
Los riñones regulan la [HCO3 –] plasmática mediante tres acciones principales: "resorción" del HCO3 – filtrado. formación de ácido titulable y Eliminación de NH4 + por la orina. Los riñones filtran alrededor de 4 000 mmol de HCO3 – al día.
Para resorber la carga filtrada de HCO3 –, los túbulos renales deben secretar, por tanto, 4 000 mmol de hidrogeniones. En el túbulo proximal se resorbe 80 a 90% del HCO3 –. La nefrona distal resorbe el resto y secreta protones, como los producidos por el metabolismo, para mantener el pH sistémico. Aunque esta cantidad de protones, 40 a 60 mmol/día, es pequeña, debe ser secretada para evitar que se produzca un balance positivocrónico de H+ y acidosis metabólica. Esta cantidad de protones secretados está representada en la orina como ácido titulable y NH4 +. La acidosis metabólica que surge en el marco de la función renal normal aumenta la producción y la eliminación de NH4 +, producción y eliminación que están alteradas en caso de insuficiencia renal crónica, hipercaliemia y acidosis tubular renal.En resumen, estas respuestas reguladoras, como el amortiguamiento químico, la regulación de la Paco2 por el aparato respiratorio y la de [HCO3 –] por los riñones, actúan de forma concertada para mantener el pH arterial sistémico entre 7.35 y 7.45.
Diagnóstico de los tipos generales de alteraciones
Las alteraciones clínicas más frecuentes son los trastornos acido-básicos simples, es decir, acidosis o alcalosis metabólicas, y acidosis o alcalosis respiratorias. Dado que la compensación no es completa en los trastornos simples, el pH es anormal. Las situaciones clínicas más complicadas causanalteraciones acido- básicas.
Trastornos acido-básicos simples
Las alteraciones respiratorias primarias (cambios primarios de la Paco2) ocasionan respuestas metabólicas compensadoras (cambios secundarios de la [HCO3 –]), y los trastornos metabólicos primarios desencadenan respuestas respiratorias compensadoras previsibles.
Los cambios primarios de la Paco2 o de la [HCO3 –] modifican el pH sistémico y ocasionan acidosis o alcalosis. Como ejemplo, la acidosis metabólica debida a un aumento de los ácidos endógenos (p. ej., cetoacidosis) disminuye la [HCO3 –] del líquido extracelular y el pH extracelular.
Trastornos acidobásicos mixtos
Los trastornos acidobásicos mixtos, definidos como trastornos independientes coexistentes y no como respuestas meramente compensadoras, se observan a menudo en los pacientes de las unidades de cuidados intensivos y pueden dar origen a cifras extremas peligrosas de pH. Un enfermo con cetoacidosis diabética (acidosis metabólica) puede sufrir un problema respiratorio independiente causante de acidosis o alcalosis respiratoria. Los individuos con enfermedad pulmonar primaria quizá no reaccionen ante la acidosis metabólica con una respuesta
66 Medicina Interna Sexto año

ventilatoria adecuada, debido a su insuficiente reserva respiratoria. Esta superposición de una acidosis respiratoria a una acidosis metabólica puede ocasionar acidemia intensa de mal pronóstico.
Cuadro 42-1. Datos que pronostican respuestas compensatorias a trastornos acido-básicos simple
Trastorno Dato que pronostica la compensación
Acidosis metabólica Paco2 = (1.5 x HCO3 –) + 8
o
Paco2 1.25 mmHg por mmol/L en [HCO3–]
o
Paco2 = [HCO3 –] + 15
o
Paco2 = [HCO3 –] + 15 Paco2 0.75 mmHg por mmol/L de en [HCO3 –]
Alcalosis metabólica
O
Paco2 6 mmHg por 10 mmol/L de en [HCO3 –]
o
Paco2 = [HCO3 –] + 15
Alcalosis respiratoria
Aguda
Crónica
[HCO3 –] 2 mmol/L por 10 mmHg de en Paco2
[HCO3–] 4 mmol/L por 10 mmHg de en Paco2
Acidosis respiratoria
67 Medicina Interna Sexto año

Aguda
Crónica
[HCO3 –] 1 mmol/L por 10 mmHg de en Paco2
[HCO3 –] 4 mmol/L por 10 mmHg de en Paco2
Método escalonado para el diagnóstico de trastornosAcido básicos:
1. Medir simultáneamente gases y electrólitos en sangre arterial [ABG (arterial blood gases)].2. Comparar [HCO3 -] en ABG y electrólitos para verificar su exactitud.3. Calcular la brecha aniónica (AG).4. Identificar cuatro causas de acidosis por incremento de AG (cetoacidosis, acidosis por ácido láctico, insuficiencia renal y toxinas).5. Identificar dos causas de acidosis hipoclorémica alta o que no depende de brecha aniónica (pérdida de bicarbonato por vías gastrointestinales, acidosis tubular renal).6. Estimar la respuesta compensatoria. 7. Comparar la AG y la HCO3 –.8. Comparar los cambios en cloruros al cambiar el sodio.
Brecha aniónica
En todas las evaluaciones de los trastornos acidobásicos debe realizarse un cálculo simple de la brecha aniónica (anion gap, AG), que representa los aniones no medidos en el plasma (normalmente 10 a 12 mmol/L) y se calcula: AG = Na+ – (Cl– + HCO3 –). Los aniones no medidos comprenden proteínas aniónicas, fosfato, sulfato y aniones or-gánicos.
Cuando en el líquido extracelular se acumulan aniones ácidos, como el acetoacetato y el lactato, la AG aumenta y origina acidosis con gran AG. la AG puede aumentar secundariamente a la elevación de la albúmina iónica, ya sea por una mayor concentración de albúmina o por alcalosis, que altera la carga de la albúmina. La disminución de la AG puede deberse a: 1) aumento de los cationes no medidos; 2) adición a la sangre, de cationes anormales, como el litio (intoxicación por litio) o las inmunoglobulinas catiónicas (discrasias de células plasmáticas); 3) disminución de la concentración de albúmina, el principal ion plasmático (síndrome nefrótico); 4) disminución de la carga aniónica eficaz de la albúmina causada por la acidosis, o 5) hiperviscosidad e hiperlipidemia intensa, que puede dar lugar a una infravaloración de las concentraciones de sodio y cloruro. Un descenso de 1 g/100 ml en la albúmina sérica respecto del valor normal (4.5 g/100 ml) reduce la brecha aniónica en 2.5 meq/litro.
Acidosis metabólica
La acidosis metabólica puede producirse por efecto del aumento de la producción endógena de ácido (como el lactato o los cetoácidos), por la pérdida de bicarbonato (como ocurre en la diarrea) o por la acumulación de ácidos endógenos (como ocurre en la insuficiencia renal). La acidosis metabólica ejerce efectos notables en los
68 Medicina Interna Sexto año

aparatos respiratorio y cardíaco y el sistema nervioso. El descenso del pH sanguíneo conlleva un aumento característico de la ventilación, en especial del volumen de ventilación pulmonar (respiración de Kussmaul).
Existen dos categorías principales de acidosis metabólica clínica: con brecha aniónica alta, o con tal variable normal, o la acidosis hiperclorémica
Causas de acidosis metabólica por brecha aniónica alta Acidosis láctica Cetoacidosis
Diabética
Alcohólica
Por inanición
Toxinas Etilenglicol
Metanol
Salicilatos
Insuficiencia renal (aguda y crónica)
Causas de acidosis no dependiente de la brecha aniónicaI. Pérdida de bicarbonato por vías gastrointestinales
A. Diarrea
B. Drenaje externo de jugo pancreático o intestinal
C. Ureterosigmoidostomía, asa de yeyuno o de íleon
D. Fármacos
1. Cloruro de calcio (agente acidificante)
2. Sulfato de magnesio (diarrea)
3. Colestiramina (diarrea por ácidos biliares)
II. Acidosis renal
III. Hipercaliemia farmacoinducida (con insuficiencia renal)
A. Diuréticos que ahorran potasio (amilorida, triamtereno, espironolactona)
B. Trimetoprim
C. Pentamidina
D. Inhibidores de la enzima convertidora de angiotensina y bloqueadores de receptores AT-II
E. Antiinflamatorios no esteroideos
F. Ciclosporina
IV. Otras
A. Pérdida de ácido (cloruro de amonio,
69 Medicina Interna Sexto año

A. Hipocaliemia
1. RTA proximal (tipo 2)
2. RTA distal (clásica) (tipo 1)
B. Hipercaliemia
1. Disfunción generalizada de la nefrona distal (RTA de tipo 4)
a. Deficiencia de mineralocorticoides
b. Resistencia de mineralocorticoides
c. Menor ingreso de sodio a la porción distal de la nefrona
d. Enfermedad tubulointersticial
e. Defecto en la excreción de amonio
hiperalimentación)
B. Pérdida posible de bicarbonato: cetosis con excreción de cetonas
C. Acidosis por expansión (administración rápida de solución salina)
D. Ácido hipúrico
E. Resinas de intercambio catiónico
Acidosis con brecha aniónica alta
Existen cuatro causas principales de acidosis con AG alta: 1) acidosis láctica, 2) cetoacidosis, 3) ingestión de toxinas y 4) insuficiencia renal aguda y crónica .
La detección inicial para diferenciar las acidosis con AG alta debe comprender:1) Revisión de los datos de la anamnesis en busca de pruebas de ingestión de fármacos y toxinas y medición de gases en sangre arterial para detectar alcalosis respiratoria coexistente (salicilatos) 2) Identificación de la posible presencia de diabetes mellitus (cetoacidosis diabética).3) Búsqueda de pruebas de alcoholismo o de valores altos de hidroxibutirato beta (cetoacidosis alcohólica). 4) Observación en busca de signos clínicos de uremia, y medición del nitrógeno de la urea sanguínea ( blood urea nitrogen, BUN) y creatinina (acidosis urémica). 5) Inspección de la orina en busca de cristales de oxalato (etilenglicol). 6)Diagnóstico de las múltiples situaciones clínicas en las que pueden estar aumentados los valores de lactato (hipotensión, choque, insuficiencia cardíaca, leucemia, cáncer, e ingestión de fármacos o toxinas).
1. Acidosis láctica
La acumulación de L-lactato en el plasma puede ser secundaria a riego hístico deficiente (tipo A): insuficiencia circulatoria (choque, insuficiencia cardíaca), anemia intensa, defectos de las enzimas mitocondriales, e inhibidores
70 Medicina Interna Sexto año

(monóxido de carbono, cianuro), o a trastornos aerobios (tipo B): cánceres, diabetes mellitus, insuficiencia renal o hepática, infecciones graves (cólera, paludismo), convulsiones, SIDA, o ingestión de fármacos o toxinas (biguanidas, etanol, metanol, isoniazida, análogos de la zidovudina y fructosa). La isquemia o el infarto intestinal no diagnosticados en un paciente con aterosclerosis o descompensación cardíaca graves tratado con vasopresores es causa frecuente de acidosis láctica. La acidosis por D-lactato, que suele acompañar a la derivación yeyunoileal o a la obstrucción intestinal, y que se debe a la formación de D-lactato por las bacterias intestinales, puede producir aumento de la AG e hipercloremia.
TratamientoEn primer lugar es preciso corregir el trastorno primario que interrumpe el metabolismo del lactato; se restablecerá el riego hístico cuando sea insuficiente. Si es posible, se evitará el uso de vasoconstrictores, ya que pueden disminuir todavía más el riego hístico. Se suele aconsejar el tratamiento alcalinizante en caso de acidemia aguda intensa (pH <7.15), a fin de mejorar la función cardíaca y la utilización del lactato. Sin embargo, la administración de NaHCO3 puede deprimir, paradójicamente, el rendimiento cardíaco y exacerbar la acidosis al estimular la producción de lactato (el HCO3– estimula a la fosfofructocinasa).
2. Cetoacidosis
A. Cetoacidosis diabéticaLa entidad mencionada es causada por aumento del metabolismo de los ácidos grasos y la acumulación de cetoácidos (acetoacetato e hidroxibutirato beta). La cetoacidosis diabética suele aparecer en pacientes con diabetes mellitus insulinodependiente, en que cesa la aplicación de insulina o surge una enfermedad intercurrente, como infección, gastroenteritis, pancreatitis o infarto del miocardio, que aumentan de forma temporal y aguda las necesidades de insulina.
B. Cetoacidosis alcohólicaLos alcohólicos crónicos pueden presentar cetoacidosis cuando se restringe súbitamente su consumo de alcohol; suele acompañar a situaciones como intoxicación etílica, vómito, dolor abdominal, inanición y disminución de volumen. La concentración de glucosa es baja o normal, y la acidosis puede ser intensa debido al mayor nivel de cetonas, en particular hidroxibutirato beta. Aveces coexiste una acidosis láctica leve por alteración del estado oxirreductor.Es característico que los valores de insulina sean bajos, y están aumentadas lasconcentraciones de triglicéridos, cortisol, glucagon y hormona del crecimiento.
TratamientoConsiste en la reposición intravenosa del líquido extracelular mediante la administración de soluciones salina y glucosada (glucosa al 5% en NaCl a 0.9%). A veces coexisten hipofosfatemia, hipocaliemia e hipomagnesemia, que deben ser corregidas.
3. Acidosis inducida por fármacos y toxinasA. Salicilatos
La intoxicación por salicilatos en el adulto suele producir alcalosis respiratoria, alteración mixta de acidosis metabólica y alcalosis respiratoria, o acidosis metabólica pura con AG alta. En este último caso, menos frecuente,
71 Medicina Interna Sexto año

tan sólo una parte de la brecha aniónica se debe a los salicilatos. A menudo también está aumentada la producción de ácido láctico.
Tratamiento
Lavado gástrico enérgico con solución salina isotónica (no con NaHCO3), seguido de la administración de carbón vegetal activado. Para facilitar la eliminación del salicilato, se administra NaHCO3 en cantidades adecuadas para alcalinizar la orina y mantener la diuresis (pH urinario >7.5).Se administra glucosa debido al riesgo de hipoglicemia.Diálisis: si la IR evita la eliminación del salicilato.
Etilenglicol
La ingestión de etilenglicol (que se emplea comúnmente como anticongelante) causa acidosis metabólica y lesiones graves del sistema nervioso central, el corazón, los pulmones y los riñones.
El diagnóstico se deriva por la observación de cristales de oxalato en la orina, la presencia de la diferencia osmolar en el suero y la acidosis con AG alta.
Tratamiento
Instauración rápida de diuresis salina u osmótica y la administración de suplementos de tiamina y piridoxina, fomepizol o etanol, y práctica de diálisis.
Metanol
La ingestión de metanol (alcohol de madera) ocasiona acidosis metabólica, y sus metabolitos, formaldehído y el ácido fórmico, producen graves lesiones del nervio óptico y del sistema nervioso central.
Tratamiento
Es similar al de la intoxicación por etilenglicol, y comprende medidas generales de sostén, administración de fomepizol o etanol, y hemodiálisis.
4. ACIDOSIS POR INSUFICIENCIA RENAL
La acidosis hiperclorémica de la insuficiencia renal moderada se convierte al final en la acidosis con la AG alta, propia de la insuficiencia renal avanzada.
Por tanto, la acidosis urémica se caracteriza por disminución de la producción y eliminación de NH4+, debida predominantemente a la disminución de la masa renal.
72 Medicina Interna Sexto año

Acidosis metabólicas hiperclorémicas
El organismo puede perder álcalis por el aparato digestivo, en caso de diarrea, o por los riñones (acidosis tubular renal [RTA]).Los cambios recíprocos de la [Cl–] y de la [HCO–] dan por resultado AG normal. En la acidosis hiperclorémica pura, por tanto, el aumento de [Cl–] por encima de los valores normales es igual a la disminución de [HCO–].
La pérdida de parénquima renal funcional por enfermedad renal progresiva ocasiona acidosis hiperclorémica cuando la tasa de filtración glomerular se sitúa entre 20 y 50 ml/min, y acidosis urémica con AG alta cuando la GFR desciende hasta <20 ml/ min.
ALCALOSIS METABOLICA
pH arterial alto.
Aumento de la [HCO3–] sérica.
Aumento de la Paco2 a causa de la hipoventilación alveolar compensadora.
A menudo conlleva hipocloremia e hipocaliemia.
Todo paciente con [HCO3–] alta y [Cl–] baja presenta alcalosis metabólica o acidosis respiratoria crónica.
Patogenia
La alcalosis metabólica se produce a causa de ganancia neta de [HCO3 –] o de pérdida de ácidos no volátiles (en general HCl, por el vómito) procedentes del líquido extracelular. Dado que es poco habitual que se añadan alcalinizantes al organismo, el trastorno entraña una fase generadora, en la que la pérdida de ácido suele causar alcalosis, y una fase de "mantenimiento", en la que el riñón es incapaz de compensar mediante la eliminación de HCO3 –, debido a la contracción de volumen, a una GFR baja o a disminución de Cl– o de potasio.
Manifestaciones clínicas
Alteraciones de la función de SNC: confusión mental, embotamiento y predisposición a las convulsiones, parestesias, calambres, tetania, agravamiento de las arritmias e Hipoxemia en la enfermedad pulmonar obstructiva crónica.
Las alteraciones electrolíticas en estos casos consisten en hipocaliemia e hipofosfatemia.
Causas de alcalosis metabólica
I. "Cargas" exógenas de bicarbonatoA. Administración muy rápida de alcalinos.B. Síndrome de leche y alcalinos.
73 Medicina Interna Sexto año

II. Contracción efectiva del volumen extracelular; normotensión, deficiencia de potasio e hiperaldosteronismo hiperreninémico secundarioA. Originada en vías gastrointestinales1. Vómito2. Broncoaspiración de material gástrico.3. Cloridorrea congénita4. Adenoma velloso5. Administración mixta de sulfonato de poliestireno sódico (Kayexalate) e hidróxido de aluminio.
B. De origen renal1. Diuréticos2. Estados edematosos.3. Estado poshipercápnico.4. Hipercalcemia o hipoparatiroidismo.5. Recuperación tras acidosis láctica o cetoacidosis.6. Aniones no resorbibles, como penicilina y carbenicilina.7. Deficiencia de magnesio.8. Depleción de potasio.9. Síndrome de Bartter (pérdida de mutación de función en TALH).10. Síndrome de Gitelman (pérdida de la mutación funcional en el transportador de sodio y cloruro en el túbulo contorneado distal)
ACIDOSIS RESPERATATORIA
Aumento de la Paco2.
La disminución del PH.
Puede deberse a enfermedad pulmonar grave, fatiga de los músculos de la respiración o alteraciones en el control de la ventilación.
CAUSAS DE ACIDOSI RESPIRATORIA
AcidosisA. Central
1.Fármacos(anestésicos,
morfina, sedantes)
2. Accidente cerebro vascular
3. Infección
B. Vías respiratorias
D. Neuromusculares
1. Poliomielitis
2. Cifoescoliosis
3. Miastenia
4. Distrofias musculares
E. Diversas
74 Medicina Interna Sexto año

1. Obstrucción
2. Asma
C. Parénquima
1. Enfisema
2. Neumoconiosis
3. Bronquitis
4. Síndrome disneico del adulto
5. Barotraumatismo
1. Obesidad
2. Hipoventilación
Un aumento rápido de la Paco2 puede ocasionar ansiedad, disnea, confusión, psicosis y alucinaciones, e incluso evolucionar y llegar al coma.
Grados menores de disfunción en caso de hipercapnia crónica comprenden alteraciones del sueño, pérdida de memoria, somnolencia diurna, alteraciones de la personalidad, deterioro de la coordinación y alteraciones motoras como temblor, contracciones mioclónicas y asterixis.
Las cefalalgias y otros signos que remedan hipertensión intracraneal, como el edema de papila, las alteraciones de los reflejos y la debilidad muscular focal, se deben a la vasoconstricción secundaria a la pérdida de los efectos vasodilatadores del dióxido de carbono.
Tratamiento
El tratamiento de la acidosis respiratoria depende de su intensidad y de su rapidez de aparición. La forma aguda puede ser peligrosa para la vida, y las medidas para corregir la causa subyacente se deben tomar al mismo tiempo que se inicia la restauración de la ventilación alveolar adecuada.
ALCALOSIS RESPIRATORIA
Disminución de la Paco2
Aumenta la relación HCO3–/ Paco2.
Incremento del pH
Aparece hipocapnia
75 Medicina Interna Sexto año

Manifestaciones clínicas
Los efectos de la alcalosis respiratoria varían con base en su duración e intensidad, pero corresponden principalmente a las de la enfermedad subyacente.
La disminución del flujo sanguíneo cerebral a causa del descenso rápido de la Paco2 puede ocasionar inestabilidad, confusión mental y convulsiones, incluso en ausencia de hipoxemia.
En los pacientes con cardiopatía, pueden aparecer arritmias cardíacas, a causa de las alteraciones en la "descarga" de oxígeno en la sangre, debidas al desplazamiento hacia la izquierda de la curva de disociación del oxígeno y la hemoglobina (efecto Bohr).
El síndrome de hiperventilación puede ser incapacitante. La existencia de parestesias, insensibilidad peribucal, opresión o dolor en la pared torácica, mareos, incapacidad para respirar adecuadamente y, en raras ocasiones, tetania, pueden tener por sí mismos la suficiente gravedad, para perpetuar el trastorno.
CAUSAS DE ALCALOSIS RESPIRATORIA
Alcalosis
A. Estimulación del SNC:
1. Dolor
2. Ansiedad, psicosis
3. Fiebre
4. Accidente cerebrovascular
5. Meningitis, encefalitis
6. Tumor
7. Traumatismo
B. Hipoxemia o hipoxia hística:
1. Grandes alturas; disminución PaCO2
2. Neumonía, edema pulmonar
3. Broncoaspiración
4. Anemia intensa
C. Fármacos u hormonas:
D. Estimulación de receptores torácicos:
1. Hemotórax
2. Tórax flácido
3. Insuficiencia cardíaca
4. Embolia pulmonar
E. Diversas
1. Septicemia
2. Insuficiencia hepática
3. Hiperventilación mecánica
4. Exposición al calor
5. Recuperación después de acidosis metabólica
76 Medicina Interna Sexto año

1. Embarazo, progesterona
2. Salicilatos
Tratamiento
El tratamiento de la alcalosis respiratoria va dirigido a corregir el trastorno primario.
Los pacientes con síndrome de hiperventilación mejoran si se les tranquiliza, se les hace respirar en una bolsa de papel durante las crisis sintomáticas y se presta atención a la tensión psicológica subyacente.
No se recomiendan los antidepresivos ni los sedantes.
Los bloqueadores de los receptores beta-adrenérgicos (betas bloqueadores o betabloqueantes) a veces mejoran las manifestaciones periféricas del estado hiperadrenérgico.
ALCOHOL Y ALCOHOLISMO
En dosis bajas el alcohol pudiera tener algunos efectos beneficiosos como disminución en la frecuencia de infarto del miocardio, de accidente cerebrovascular, de cálculos vesiculares y posiblemente de demencias vasculares o de Alzheimer, pero el consumo de más de dos bebidas al día agrava el peligro de problemas de salud en muchos aparatos y sistemas. Beber de manera repetitiva grandes cantidades de alcohol, como se observa en el abuso y la dependencia de esta droga, acorta la esperanza de vida unos 10 años en los dos géneros, en todos los grupos culturales y en todos los estratos socioeconómicos.
Incluso cantidades pequeñas de alcohol tienen un efecto significativo en muchos aparatos y sistemas, agravan una gran parte de los cuadros patológicos preexistentes, y alteran la eficacia o los valores sanguíneos de muchos de los fármacos de venta libre y de prescripción.
EFECTOS CONDUCTUALES, TOLERANCIA Y DEPENDENCIA
Aunque la definición de "intoxicación legal" requiere una concentración de alcohol en sangre de al menos 80 a 100 mg/100 ml, los cambios de comportamiento, psicomotores y cognitivos aparecen ya con concentraciones tan bajas como 20 a 30 mg/100 ml (es decir, después de una o dos bebidas) (cuadro 372-1). Se puede observar un sueño profundo, aunque alterado, con una concentración del doble de la que marca la intoxicación legal, y la muerte puede sobrevenir con concentraciones entre 300 y 400 mg/100 ml. Es probable que el etanol sea responsable de más muertes tóxicas por sobredosis que ninguna otra droga.
77 Medicina Interna Sexto año
OSMOLARIDAD

Los efectos intoxicantes del alcohol parecen estar relacionados con las acciones sobre neurotransmisores, receptores y transportadores específicos. El alcohol potencia los receptores del ácido aminobutírico gamma A (gamma-aminobutyric acid A, GABAA), e inhibe los receptores de N-metil-D-aspartato (NMDA). Las neuronas se adaptan rápidamente a estas acciones, y por ello puede haber diferentes efectos durante la administración crónica y durante la abstinencia. Después de una exposición repetida a la sustancia, el organismo realiza al menos tres tipos de compensaciones para poder tolerar concentraciones más altas de etanol.
En primer lugar, después de una o dos semanas de consumo diario de alcohol, ocurre tolerancia metabólica o farmacocinética, con aumento de 30% en la velocidad del metabolismo hepático del etanol. Esta alteración desaparece casi con la misma rapidez con que se presenta.
En segundo lugar, ocurre tolerancia farmacodinámica o celular a través de cambios neuroquímicos que también pueden contribuir a la dependencia física.
En tercer lugar, las personas pueden aprender a adaptar su comportamiento, de manera que funcionan mejor de lo previsible bajo la influencia de la droga (tolerancia de comportamiento). Una vez que las células se han adaptado a la exposición crónica al etanol, los cambios estructurales o bioquímicos a veces no vuelven a la normalidad durante al menos varias semanas. Mientras tanto, las neuronas necesitan etanol para funcionar de forma óptima y se dice que la persona tiene dependencia física. Ésta es diferente de la dependencia psicológica, concepto que señala que la persona se siente psicológicamente incómoda sin la droga.
EFECTOS DEL ETANOL EN APARATOS Y SISTEMAS
SISTEMA NERVIOSO CENTRAL
78 Medicina Interna Sexto año

Alrededor de 35% de los bebedores presentan a veces un lapso de "ausencia", un episodio de amnesia anterógrada temporal, en la cual la persona olvida parte o todo lo que ocurrió durante una sesión etílica. Otro problema frecuente que surge incluso luego de beber unas cuantas copas es que el alcohol altera las etapas del sueño y causa alguna deficiencia en el sueño de movimientos oculares rápidos y el profundo, de lo que resultan ensoñaciones intensas y a veces perturbadoras más tarde en la noche. Por último, el alcohol relaja los músculos de la faringe, lo cual ocasiona ronquidos y exacerba la apnea durante el sueño, y en 75% de los varones alcohólicos mayores de 60 años surgen tales síntomas.
El efecto del alcohol en el sistema nervioso es todavía más intenso entre los individuos que dependen del alcohol. Las dosis altas por largo tiempo originan neuropatía periférica en 5 a 15% de los alcohólicos: los pacientes muestran insensibilidad de ambas extremidades, hormigueos y parestesias, todo ello más intenso en sentido distal.
En menos de 10% de los alcohólicos se observa síndrome de Wernicke (oftalmoparesia, ataxia y encefalopatía) y síndrome de Korsakoff como resultado de deficiencia de tiamina, en particular en sujetos con deficiencia de transcetolasa. Alrededor de 1% de los alcohólicos terminan por sufrir degeneración cerebelosa, un síndrome en que hay inestabilidad progresiva del bipedismo y la marcha y que a menudo se acompaña de nistagmo leve; los estudios neuroimagenológicos indican atrofia del vermis del cerebelo.
Los alcohólicos pueden presentar graves problemas cognitivos que incluyen deficiencias de la memoria reciente y remota durante semanas o meses después de una borrachera. Por último, durante lapsos de consumo intenso o la abstinencia ulterior pueden presentarse temporalmente casi todos los síndromes psiquiátricos; incluyen tristeza intensa que dura días o semanas en medio de una borrachera en 40% de los alcohólicos, y se le clasifica como un trastorno del ánimo inducido por alcohol; ansiedad profunda y temporal en 10 a 30% de los alcohólicos, que suele comenzar durante la etapa de abstinencia y persiste a veces meses después de interrumpir el consumo de bebidas alcohólicas (trastorno de ansiedad inducido por alcohol); y alucinaciones auditivas, delirios paranoides o ambos trastornos en la persona lúcida (trastorno psicótico inducido por alcohol) que aparecen temporalmente en 1 a 10% de los alcohólicos. El tratamiento de todas las formas de psicopatología inducida por el alcohol incluye abstinencia y medidas de apoyo; es de esperar la recuperación completa en cuestión de días a cuatro semanas.
VÍAS GASTROINTESTINALES
Esófago y estómagoLa ingestión "aguda" de alcohol (esto es, en grandes cantidades y a breve plazo) puede causar inflamación del esófago y el estómago y ocasionar molestias epigástricas y hemorragia gastrointestinal. La ingestión "crónica" (por largo tiempo) en abundancia, si se acompaña de vómitos violentos, puede originar la lesión de Mallory-Weiss, que es un desgarro longitudinal en la mucosa de la unión gastroesofágica.
Páncreas e hígadoLa incidencia de pancreatitis aguda (alrededor de 25 por 1 000 por año) es casi del triple que en la población general, lo cual explica la estimación de 10% o más de casos de este trastorno. El alcohol disminuye la gluconeogénesis en el hígado y, como consecuencia, aminora la cantidad de glucosa obtenida del glucógeno; aumenta la producción de ácido láctico y disminuye la oxidación de ácidos grasos, con lo cual aumenta la acumulación de grasa en los hepatocitos. En el sujeto sano los cambios mencionados son reversibles, pero con la exposición repetitiva al etanol ocurren a veces cambios más intensos que incluyen acumulación de grasas, hepatitis inducida por alcohol, esclerosis perivenular y cirrosis; esta última se observa en un estimado de 15 a 20% de los alcohólicos.
79 Medicina Interna Sexto año

CáncerEl consumo de apenas 1.5 bebidas al día incrementa 1.4 veces el peligro de cáncer mamario en la mujer. Para ambos géneros, cuatro bebidas al día incrementan el peligro de cáncer de boca y esófago unas tres veces, y de cánceres del recto, en un factor de 1.5; siete, ocho o más bebidas al día elevan en un factor de cinco los riesgos de muchos cánceres.
SISTEMA HEMATOPOYÉTICO
El etanol hace que aumente el tamaño de los eritrocitos [volumen corpuscular medio], lo cual refleja los efectos en los hemoblastos. Si el consumo intenso se acompaña de deficiencia de ácido fólico, puede haber también neutrófilos hipersegmentados, reticulocitopenia e hiperplasia de médula ósea; si la persona está mal nutrida se observan cambios sideroblásticos. El consumo intenso por largo tiempo puede disminuir la producción de muchos leucocitos, la movilidad y la adherencia de los granulocitos y la respuesta de hipersensibilidad tardía a nuevos antígenos (con la posibilidad de resultados negativos falsos de la cutirreacción con tuberculina). Por último, muchos alcohólicos tienen trombocitopenia leve que por lo regular muestra resolución en término de siete días de abstinencia, salvo que haya cirrosis hepática o esplenomegalia congestiva.
SISTEMA CARDIOVASCULAR
De forma aguda, el etanol disminuye la contractilidad miocárdica y causa vasodilatación periférica, dando como resultado un descenso suave de la tensión arterial y un aumento compensador del gasto cardíaco. El alcohol en grandes cantidades es un factor que contribuye a la hipertensión leve o moderada. El consumo crónico y excesivo de alcohol puede causar miocardiopatía con síntomas que van desde arritmias inexplicables en presencia de alteración ventricular izquierda a insuficiencia cardíaca con dilatación de las cuatro cavidades e hipocontractilidad del músculo cardíaco. Tal vez un tercio de los casos de miocardiopatía son inducidos por el alcohol.
Se pueden formar trombos murales en la aurícula o el ventrículo izquierdos, mientras que un aumento de tamaño del corazón superior al 25% puede causar insuficiencia mitral. Son posibles arritmias auriculares o ventriculares, sobre todo taquicardia paroxística, después de un exceso alcohólico en personas que no tienen otros signos de cardiopatía, síndrome denominado "corazón del día de fiesta". La ingestión de cantidades pequeñas de alcohol por largo tiempo puede tener algunos efectos beneficiosos. Beber como máximo una o dos copas al día puede disminuir el peligro de muerte de origen cardiovascular, tal vez por un incremento del nivel de colesterol-lipoproteínas de alta densidad (HDL) o cambios en los mecanismos de coagulación. En un gran estudio a nivel nacional, en personas que señalaron haber consumido una o dos copas al día disminuyó 30 a 40% el índice de mortalidad cardiovascular, en comparación con quienes no bebieron, y la mortalidad global fue la más baja entre quienes consumieron aproximadamente una bebida al día. Datos recientes han corroborado también la disminución del riesgo de accidente isquémico (pero no el hemorrágico) si la persona consume alcohol regularmente en cantidades moderadas.
ALTERACIONES EN APARATO GENITOURINARIO, FUNCIONAMIENTO SEXUAL Y DESARROLLO FETAL
80 Medicina Interna Sexto año

Las dosis moderadas de etanol (p. ej., concentración de alcohol en sangre de 100 mg/100 ml o menos) pueden aumentar el impulso sexual en los varones así como disminuir la capacidad eréctil. Incluso en ausencia de trastorno hepático, una minoría significativa de varones alcohólicos crónicos presentan atrofia testicular irreversible con reducción concomitante de los túbulos seminíferos, disminución del volumen eyaculado y descenso del recuento de espermatozoides.
La ingestión repetida de dosis elevadas de etanol en mujeres puede producir amenorrea, disminución del tamaño de los ovarios, ausencia de cuerpos amarillos con esterilidad asociada y abortos espontáneos.
El consumo excesivo de alcohol durante el embarazo produce una transferencia placentaria rápida de etanol y acetaldehído, que pueden acarrear graves consecuencias para el desarrollo fetal. El síndrome de alcoholismo fetal se manifiesta como una combinación de cualesquiera de los fenómenos siguientes: cambios faciales con pliegues oculares epicánticos, conchas auriculares poco formadas y dientes pequeños con defectos del esmalte; comunicaciones interauriculares o interventriculares cardíacas, surco palmar aberrante y limitación del movimiento articular, y microcefalia con retraso mental. La cantidad de etanol o el momento específico de vulnerabilidad durante el embarazo (o ambas cosas) no se han definido, por lo que se aconseja a las mujeres embarazadas que se abstengan totalmente de beber.
OTROS EFECTOS DEL ETANOL
Entre la mitad y dos tercios de los alcohólicos muestran disminución de la fuerza muscular esquelética causada por miopatía alcohólica aguda; este trastorno mejora pero puede no desaparecer con la abstinencia.
Los efectos sobre el sistema esquelético del exceso de alcohol consisten en trastorno del metabolismo del calcio, menor densidad ósea, y menor crecimiento de las epífisis, con aumento del riesgo de fracturas óseas y osteonecrosis de la cabeza femoral.
Los cambios hormonales comprenden aumento de la concentración de cortisol, que puede permanecer elevada durante todo el tiempo que dure el consumo abundante; inhibición de la secreción de vasopresina al ascender la concentración de alcohol en sangre y el fenómeno opuesto cuando aquélla desciende (con el resultado final de que la mayoría de los alcohólicos suelen tender a una ligera sobrehidratación); decremento moderado y reversible de la tiroxina sérica (T4) y un descenso más acusado de la triyodotironina sérica (T3).
81 Medicina Interna Sexto año

CIRROSIS
Es una entidad definida histopatologicamente que se acompaña de un amplio espectro de manifestaciones clínicas.
Los datos patológicos cardinales consisten en:
Fibrosis extensa Formación de nódulos de regeneración
Tipos de cirrosis
1. Alcohólica2. Criptogena y poshepática3. Biliar4. Cardiaca5. Metabólica, hereditaria y producida por medicamentos
82 Medicina Interna Sexto año

Cirrosis alcohólica (Laennec)
Se caracteriza por cicatrización difusa y sutil, perdida homogénea de células hepáticas y aparición de nódulos de regeneración de pequeño tamaño (cirrosis micronodular).
Patogenia
La cantidad y la duración de la ingesta de alcohol son los factores de riesgo mas importantes en el desarrollo de hepatopatía alcohólica.
Las mujeres son más predispuestas que los hombres en desarrollar enfermedad hepática con un consumo de alcohol menor.
El umbral para desarrollar una hepatopatía alcohólica grave en varones es de 60-80 g/día durante 10 años y en mujeres 20-40 g/día.
Una cerveza; 113.6 ml de vino o 28.4 ml de licor con 80% de alcohol tienen aproximadamente 12 g de alcohol.
La infección crónica con virus de hepatitis “C” es un cuadro coexistente importante en la evolución de la hepatopatía alcohólica hasta originar cirrosis.
Ingerir más de 50 g de alcohol/día duplica el riesgo de cirrosis en personas infectadas con hepatitis “C”.
La ingestión de alcohol en sujetos cirróticos infectados por HCV agrava el peligro de Ca. Hepatocelular.
Anatomía patológica
Si se mantiene la ingesta de alcohol y destrucción de los hepatocitos, aparecen fibroblastos (incluidas las cls. Estrelladas) en el lugar de la lesión y depositan colágeno en la periportales y pericentrales y aparecen tabiques reticulares de tejido conjuntivo que terminan por conectar triadas portales y las venas centrolobulillares. Esta fina red rodea hepatocitos supervivientes, que se regeneran y forman nódulos.
La destrucción de hepatocitos y acumulación de colágeno determinan que el hígado se reduzca de tamaño, adquiere un aspecto nodular y se endurezca conforme se va desarrollando la etapa final de cirrosis.
Manifestaciones clínicas
Clínicamente silenciosa en 10-40%, diez o más años de consumo excesivo de alcohol suelen aparecer los síntomas.
La anorexia y la malnutrición originan pérdida de peso y decremento de masa muscular.
83 Medicina Interna Sexto año

1. Equimosis2. Debilidad creciente y astenia3. Pueden haber manifestaciones de hipertensión portal: ictericia progresiva, hemorragia por varices
gastroesofagicas, ascitis y encefalopatía.
Signos
84 Medicina Interna Sexto año

1. Palpación de hígado duro y nodular2. Ictericia3. Eritema palmar4. Angiomas en araña5. Aumento de tamaño de glándulas
parótidas y lagrimales6. Acropaquias7. Esplenomegalia8. Pérdida de masa muscular
9. Ascitis con o sin edema periférico10. Atrofia testicular11. Aumento de la producción periférica
de estrógenos12. Contractura de dupuytren (fibrosis
de fascia palmar y que originan contractura en flexión de los dedos, es consecuencia del alcoholismo pero no es especifica de cirrosis).
Datos de laboratorio
Anemia Déficit nutricionales
Hiperesplenismo
85 Medicina Interna Sexto año

Efecto mielosupresor directo del alcohol sobre la medula osea
Anemia hemolítica ( efecto de la hipercolesterinemia sobre las membranas eritrocitarias que forman acantocitosis)
Hiperbilirrubinemia ligera o intensa
Aumento de fosfatasa alcalina AST aumentada Tiempo de protrombina aumentado Albumina disminuida Globulina aumentada Aumento de la concentración de
amoniaco.
Diagnostico
1. Antecedentes de consumo de alcohol excesivo y prolongado2. Manifestaciones clínicas3. Datos de laboratorio4. Biopsia hepática
Pronostico
La abstinencia y atención médica precoz y adecuada pueden reducir la morbimortalidad a largo plazo y retrasar o impedir la aparición de complicaciones.
Si ha presentado complicaciones graves de la cirrosis y sigue bebiendo tienen supervivencia a los 5 años de 50%. Si se mantienen abstemios tienen un pronóstico mejor. La mayoría mueren por hemorragia masiva por varices o encefalopatía hepática grave.
Tratamiento
Prohibición radical del alcohol. Es fundamentalmente de sostén. Glucocorticoides en dosis moderadas en pacientes con hepatitis alcohólica grave y
encefalopatía no aplican en cirrosis ya establecidas. S_ adenosilmetionina ( disminuye la concentración de citocinas proinflamatorias)
Cirrosis poshepatica (macronodular o multilobulillar) y criptogena
Es la vía final común de muchos tipos de lesión hepática crónica.
Etiología
Pruebas epidemiológicas sugieren que el 25-75% hay antecedentes de hepatitis vírica (B o C), en zonas endémicas de hepatitis B hasta 15% de la población pueden adquirir la infección al principio de la infancia, la cirrosis se desarrolla en la cuarta parte de los portadores crónicos.
En pacientes con hepatitis “C” más del 20% desarrollan cirrosis.
Manifestaciones clínicas
Ascitis Esplenomegalia
86 Medicina Interna Sexto año

Hiperesplenismo Encefalopatías Hemorragia por varices esofágica
Alteraciones de la conciencia
Tratamiento
Suele limitarse a las complicaciones de la hipertensión portal. La ascitis con supresión de medicamentos o aporte proteico excesivo que puedan
desencadenar coma hepático. Tratamiento precoz de la infección.
Cirrosis biliar
Se debe a lesión u obstrucción prologada del sistema biliar intrahepatico o extrahepatico. Se acompaña de trastornos de la excreción biliar, destrucción del parénquima hepático y fibrosis progresiva.
Cirrosis biliar primaria
Etiología y patogenia
La causa es desconocida. Puede estar implicada un trastorno de la respuesta inmunitaria ocurre con frecuencia en asociación a diversos trastornos como el síndrome de CREST (calcinosis, fenómeno de Raynaud, alteraciones de la motilidad esofágica, esclerodactilia, y telangectasias, el síndrome seco), tiroiditis autoinmunitarias, DM tipo I y déficit de IgA.
Se ha detectado en más del 90% de los sujetos con cirrosis biliar primaria (PBC) anticuerpos IgG antimitocondrial (AMA)
Anatomía patológica
Se divide en cuatro estadios:
Estadio I: Colangitis destructiva no supurativa crónica: es un proceso inflamatorio y necrosante de espacios portales.
Estadio II: El infiltrado inflamatorio resulta menos evidente, el número de conductos biliares se reduce y proliferan conductillos biliares de menor calibre.
Estadio III: Si la evolución es por meses o años origina disminución de los conductos interlobulillares, con pérdida de hepatocitos y expansión de la fibrosis periportal para formar una red cicatrizal de tejido conjuntivo.
Estadio IV: cirrosis micronodular o macronodular.
Manifestaciones clínicas
87 Medicina Interna Sexto año

Muchos son asintomáticos y generalmente se detectan por concentraciones elevadas de fosfatasa alcalina. 90% son mujeres entre 35 y 60 años.
Prurito (síntoma inicial) generalizado o en palmas y plantas.
Astenia (muy destacado) Ictericia Melanosis Esteatorrea
Malabsorción de vitaminas liposolubles
Xantelasmas y xantomas Signos de insuficiencia de
insuficiencia hepatocelular, hipertensión portal y desarrollar ascitis.
Examen físico
Ictérica intensa Hiperpigmentacion de zonas
expuestas Xantelasma y xantomas Hepatomegalia moderada o
evidente Esplenomegalia
Acropaquias Dolor oseo con la presión Signos de compresión vertebral Equimosis Glositis Dermatitis
Datos de laboratorio
Aumento al doble de la fosfatasa alcalina Gammaglutamil_transpeptidasa en suero aumentada Bilirrubina es normal al final de aumenta hasta 30 mg/100 ml Aminotransferasa mínimamente elevada AMA positiva Hiperlipidemia Concentración elevada de cobre en el hígado
Diagnostico
Mujeres de mediana edad con prurito inexplicable positivo, concentraciones elevadas de fosfatasa alcalina.
AMA negstivos Biopsia hepática
Tratamiento
Ursodiol 13-15 mg/kg/dia
88 Medicina Interna Sexto año

Trasplante de hígado Glucocorticoides, colquicina, metotrexato, azatioprina, ciclospora, tracolimo. Colestiramina 12-16 gr/dia (reduce prurito) y hipercolesterolemia. Vitaminas ADEK Calcio 1,500 gr/dia Vitamina “D” 1000 UI/día
Cirrosis biliar secundaria
Etiología
Consecuencia de obstrucción prolongada parcial o completa del colédoco o sus ramas en adultos generalmente por estenosis posoperatorias o cálculos biliares, generalmente colangitis con infección sobreañadida.
Puede ser complicación de la colangitis primaria esclerosante, también de tumores malignos de colédoco o de páncreas
Manifestaciones clínicas
Ictericia Prurito Fiebre
Dolor en hipocondrio derecho Antecedentes de cirugía biliar u
obstrucción prolongada.
Tratamiento
En caso de colangitis dar tratamiento con antibióticos
Cirrosis cardiaca
La Insuficiencia cardiaca derecha congestiva prolongada puede originar una lesión crónica del hígado y cirrosis cardiaca.
Etiología y patogenia
La ICC, la transmisión retrograda de la presión venosa, aumenta a través de la cava inferior y de las venas hepáticas originan congestión hepática , esta congestión y la isquemia debida a mala perfusión produce necrosis de los hepatocitos centrolobulillares y fibrosis microscópica del hígado en nuez moscada.
89 Medicina Interna Sexto año

ManifestacionesBilirrubina sérica elevada tanto la conjugada como la no conjugada
Aumento de la fosfatasa alcalina y tiempo de protrombina Aumento AST Hígado pulsátil Hígado grande, duro y no duele Ascitis y edema periférico.
Diagnostico
Hígado duro, aumentado de tamaño más signos de hepatopatía crónica en un paciente con valvulopatia, pericarditis constrictiva o Cor pulmonale de larga evolución (> 10 años).
Biopsia ( contraindicada en ascitis y coagulopatia) Se debe hacer diagnostico diferencial con síndrome de Budd-Chiari: oclusión de venas
hepáticas o vena cava inferior puede confundirse con hepatomegalia congestiva aguda, en este síndrome el hígado es grande y doloroso y existe ascitis y resistencia al tratamiento.
Tratamiento
Depende de la enfermedad cardiovascular subyacente.
HIPERTENSION PORTAL Y ENCEFALOPATIA HEPATICA
Hipertension Portal
La presion normal en la vena porta es baja ( 5-10 mmHg) porque la resistencia vascular en los sinusoides es minima.
Síndrome muy frecuente caracterizado por un aumento patológico de la presión hidrostática en el territorio venoso portal (mayor de 10 mmHg). Dado que el sistema venoso porta carece de válvulas, la resistencia ejercida en cualquier nivel entre las cavidades cardiacas derechas y los vasos esplacnicos, induce la trasnsmision retrograda de una presión elevada.
La resistencia puede aumentar a 3 niveles con respecto a los sinusoides hepáticos:
Presinusoidal Sinusoidal Postsinusoidal
Etiologia
Se divide en: Prehepaticas, hepáticas y post hepáticas.
1. Prehepáticas: La trombosis portal y/o esplénica (etiología: idiopática, neoplasias de páncreas, postesplenectomía, S. de hipercoagulabilidad etc.) y los síndromes de hiperaflujo, secundarios a esplenomegalias gigantes.

2. Hepáticas: que distinguiremos entre las pre y postsinusoidales.a. Presinusoidal: Fibrosis portal no cirrótica, fibrosis hepática congénita, esquistosomiasis, etc.b. Sinusoidal o post sinusoidal: cirrosis hepática de diferentes etiologías (post-hepatitis, alcohólica, etc.).
3. Post-hepáticas: S. de Bud-Chiari (trombosis de suprahepáticas, membranas congénitas relativamente frecuente en Japón, trombosis de vena cava inferior,etc.). Pericarditis constrictiva, miocardiopatía,etc.).
Manifestaciones Clinicas
Hemorragia por varices esofágicas Esplenomegalia con hiperesplenismo Ascitis Encefalopatia hepática aguda y crónica.
Cortocircuitos portosistemicos: venas rectales y peri rrectales ( hemorroides), union cardioesofagica (varices esofagicas), el retroperitoneo y ligamento falciforme del higado ( venas colaterales periumbilicales y de la pared abdominal, que estas emergen radialmente del ombligo y se dirigen hacia el apéndice xifoides y el reborde costal por lo que se le llama caput medusae).
Diagnostico
Varices esofágicas ( esofagoscopia fibrooptica).
Circulación colateral ( Resonancia magnetica y tomografía computarizada con medio de contraste intravenoso.
Presión dentro de la vena porta ( cateterismo percutáneo y transhepatico con aguja fina, o indirectamente por cateterismo transyugular de las venas supra hepática).
Tratamiento
Esta orientado a las complicaciones especificas de la hipertencion portal, a veces se intenta reducir la presion en el sistema venoso portal.
Bloqueo adrenergico beta ( propanolol, atenolol.. etc.)
Colocacion percutanea de una derivacion porto sistemica ( cortocircuito portosistemico intrahepatico transyugular).
Trasplante Hepatico.

Encefalopatia Hepatica
Sindrome neuropsiquiatrico caracterizado por alteraciones de la conciencia y de la conducta, cambios de la personalidad, signos neurologicos fluctuantes, asterixis y alteraciones electroencefalograficas caracteristicas.
Patogenia
No se conoce la causa especifica de la encefalopatía hepática.
Los factores mas importantes en su patogenia son: La disfunción hepatocelular avanzada y el cortocircuito intrahepatico o extrahepatico de la sangre venosa portal.
Como consecuencia de estos trastornos, una seria de productos toxicos absorbidos en el intestino no son desintoxicados por el hígado y ocasionan alteraciones metabolicas en el SNC.
El amoniaco es el producto que mas se ha implicado en la patogenia. Muchos pxs con encefalopatía tienen concentraciones elevadas de amoniaco en la sangre.Otras sustancias o metabolitos que pueden colaborar a la aparición de la encefalopatía son los metacaptanos ( que derivan del metabolismo intestinal de la metionina), y acidos grasos de cadena corta y fenol.
Tambien pueden intervenir falsos transmisores neuroquímicos (la octopamina) que resultan en parte de las alteraciones de las concentraciones plasmáticas de los aminoacidos aromaticos.
Un factor adicional implicado en la patogenia de la encefalopatía hepática podría ser: el aumento de la permeabilidad de la barrera hematoencefalica para algunas de estas sustancias.
Hay datos que sugieren que las concentraciones excesivas del neurotransmisor inhbidor GABA en el SNC influyen en la reducción del nivel de conciencia que se observa en la encefalopatía hepática.Apoyando esta teoría hay datos que sugieren que las benzodiacepinas endógenas, que actúan a través del receptor GABA, pueden colaborar a la aparición de la encefalopatía hepática.
Factores Desencadenantes

Manifestaciones Clinicas
Son de dos clases:
1. Alteraciones neuropsiquiátricas : Síndrome confusional, y pueden desarrollarse en forma rápida o lenta.
2. Alteraciones neuromusculares: Consisten en asterixis, rigidez y signos piramidales (paresia, hiperreflexia, clonus y signo de Babinski), los cuales pueden ser unilaterales y complican el diagnóstico diferencial.
Etapas tempranas à alteraciones del sueño.
Inversión del ciclo sueño vigilia. Trastornos de personalidad y del estado de animo. Confusión y deterioro del cuidado personal y en la escritura. Cambios en el estado mental Hedor hepático Olor a moho o a lugar cerrado y húmedo
(aliento y orina) Asterixis “aleteo hepático”
Breves claudicaciones asimétricas y arrítmicas del control voluntario del mantenimiento de la postura de las extremidades, cabeza y el tronco.

Diagnostico
Suele hacerce por exclusion, no hay alteraciones especificas de las pruebas de funcion hepatica, aunque las concentraciones sericas elevadas de amoniaco en un contexto clinico adecuado son muy sugerentes del diagnostico.
Tratamiento
Objetivos
• En el intestino: disminuir la formación y el paso a la circulación de sustancias nitrogenadas intestinales.
• En el hígado: Potenciar el metabolismo de las toxinas.
• la barrera hematoencefálica : Disminuir el paso de amoníaco.
• Cerebro: contrarrestar las alteraciones en los diferentes sistemas neurotransmisores.
Dieta:
Proteínas 0,5 g/kg/día Aporte calórico mediante carbohidratos y lípidos.
Farmacologico:
En el periodo agudo, se puede administrar jarabe de lactulosa en dosis de 30-60ml/hr hasta que ocurra diarrea; a continuacion la dosis se ajusta ( 15-30 ml tres veces al dia) para que el paciente haga de 2 – 4 deposiciones blandas.
Se puede reducir la produccion intestinal de amoniaco administrando neominicina v.o. (0.5-1g. cada 6 hrs).
Antagonistas de accion corta de las benzodiazepinas ( Flumazenilo).

PANCREATITIS AGUDA
CONSIDERACIONES GENERALES
El páncreas secreta de 1 500 a 3 000 ml de líquido isoosmótico alcalino (pH >8) al día, que contiene cerca de 20 enzimas y cimógenos. Las secreciones pancreáticas proporcionan las enzimas necesarias para la mayor parte de la actividad digestiva del aparato digestivo y aportan un pH óptimo para la función de estas enzimas.
REGULACIÓN DE LA SECRECIÓN PANCREÁTICA
La función exocrina del páncreas recibe influencias de los sistemas hormonal y nervioso en íntima interacción. La secreción exocrina del páncreas recibe la influencia de neuropéptidos inhibidores como somatostatina, polipéptido pancreático, péptido YY, neuropéptido Y, encefalina, pancreastatina, péptidos vinculados con el gen de calcitonina, glucagon y galanina.
SECRECIÓN DE AGUA Y ELECTRÓLITOS
El bicarbonato es el ion de importancia fisiológica capital en la secreción pancreática.El bicarbonato ayuda a neutralizar el ácido estomacal y crear así el pH apropiado para que actúen las enzimas pancreáticas.
SECRECIÓN ENZIMÁTICA
La célula acinar es una estructura dividida en compartimientos precisos y se ocupa de secretar las enzimas pancreáticas. El páncreas secreta enzimas amilolíticas, lipolíticas y proteolíticas. Las primeras (amilolíticas), como la amilasa, hidrolizan los almidones en oligosacáridos y el disacárido maltosa. Las segundas (lipolíticas) comprenden lipasa, fosfolipasa A y esterasa de colesterol. Las sales biliares inhiben la lipasa aislada; sin embargo, la colipasa, que es otro constituyente de la secreción pancreática, se fija a la lipasa e impide su inhibición. Las sales biliares activan la fosfolipasa A y la esterasa de colesterol. Las terceras (proteolíticas) incluyen endopeptidasas (tripsina, quimotripsina), las exopeptidasas (carboxipeptidasas, aminopeptidasas) y la elastasa. Las enzimas proteolíticas son secretadas en la forma de precursores inactivos (cimógenos).
AUTOPROTECCIÓN DEL PÁNCREAS
La autodigestión del páncreas se evita por la envoltura de precursores de proteasas y por la síntesis de inhibidores de proteasa, es decir, inhibidor de tripsina secretoria pancreática (pancreatic secretory trypsin inhibitor, PSTI) e inhibidor de proteasa de serina, tipo 1 de Kasal (serine protease inhibitor, Kasal type 1, SPINK1). La pérdida de cualesquiera de estos mecanismos protectores conduce a la activación de cimógeno, autodigestión y pancreatitis aguda.
EJE ENTEROPANCREÁTICO E INHIBICIÓN POR REALIMENTACIÓN
La secreción enzimática del páncreas es controlada, al menos en parte, por un mecanismo de realimentación (retroalimentación) negativa inducido por la presencia de proteasas de serina activas en el duodeno.

CONSIDERACIONES GENERALES DE LA PANCREATITIS AGUDA
La enfermedad inflamatoria del páncreas puede clasificarse como 1) pancreatitis aguda y 2) pancreatitis crónica. El espectro anatomopatológico de la pancreatitis aguda varía desde la pancreatitis intersticial, que suele ser un trastorno leve y de evolución limitada, hasta la pancreatitis necrosante, en la cual el grado de necrosis del páncreas guarda relación con la gravedad del ataque y con sus manifestaciones generales.
La incidencia de la pancreatitis varía según los países y depende de la causa, por ejemplo consumo de alcohol, cálculos biliares, factores metabólicos y fármacos.
CUADRO 294-1 CAUSAS DE PANCREATITIS AGUDA
Causas comunes
Litiasis vesicular (incluida la microlitiasis)
Alcohol (alcoholismo agudo y crónico)
Hipertrigliceridemia
Colangiopancreatografía retrógrada endoscópica (ERCP), en particular después de la práctica de manometría de vías biliares
Traumatismo (en particular el no penetrante del abdomen)
Estado posoperatorio (estado ulterior a operaciones abdominales y no abdominales)
Fármacos (azatioprina, 6-mercaptopurina, sulfonamidas, estrógenos, tetraciclina, ácido valproico, fármacos contra VIH)
Disfunción del esfínter de Oddi
Causas poco comunes
Causas vasculares y vasculitis (estados de isquemia-hipoperfusión después de operaciones del corazón)
Enfermedades de tejido conjuntivo y púrpura trombocitopénica trombótica
Cáncer de páncreas
Hipercalciemia
Divertículo periampollar
Páncreas dividido
Pancreatitis hereditaria
Fibrosis quística
Insuficiencia renal
Causas raras
Infecciones (parotiditis, por virus coxsackie o citomegalovirus, echovirus y parásitos)
Autoinmunitarias (como síndrome de Sjögren)
Causas por considerar en personas con crisis recurrentes de pancreatitis aguda sin un origen evidente
Enfermedad oculta de vías biliares o conductos pancreáticos, en particular microlitiasis, sedimento
Fármacos
Hipertrigliceridemia
Páncreas dividido
Cáncer pancreático
Disfunción del esfínter de Oddi
Fibrosis quística
Causas idiopáticas
ETIOLOGÍA Y PATOGENIALa pancreatitis aguda tiene innumerables causas, pero no se han identificado los mecanismos por los cuales estas situaciones anormales desencadenan la inflamación del páncreas. Los cálculos vesiculares siguen siendo la causa principal de pancreatitis aguda en muchas series (30 a 60%). El alcohol constituye la segunda causa y origina de 15 a 30% de los casos de pancreatitis. La incidencia de pancreatitis en alcohólicos es sorprendentemente baja (5/100000), lo cual denota que además del volumen del alcohol ingerido, otros factores desconocidos afectan la susceptibilidad de la persona a sufrir lesión del páncreas. No se conoce a fondo el mecanismo de la lesión. La hipertrigliceridemia es la causa de pancreatitis aguda en 1.3 a 3.8% de los casos; los valores de triglicéridos séricos por lo común son mayores de 11.3 mmol/L (>1 000 mg/100 ml). La pancreatitis aguda se observa en 5 a 20% de las personas que han sido sometidas a colangiopancreatografía retrógrada endoscópica (endoscopic retrograde cholangiopancreatography, ERCP). Se sabe que 2 a 5% de los casos de pancreatitis aguda son causados por fármacos; el mecanismo causal puede ser una reacción de hipersensibilidad o la generación de un metabolito tóxico.
La autodigestión es una teoría sobre la patogenia, según la cual ocurre pancreatitis cuando las enzimas proteolíticas (como tripsinógeno, quimotripsinógeno, proelastasa y fosfolipasa A) son activadas en el páncreas y no en el interior del intestino. Se piensa que las proenzimas mencionadas son activadas por diversos factores (como endotoxinas, exotoxinas, infecciones víricas, isquemia, anoxia y traumatismo directo). Las enzimas proteolíticas activadas y en particular la tripsina, además de digerir los tejidos pancreáticos y peripancreáticos, también activan otras enzimas como elastasa y fosfolipasa.
ACTIVACIÓN DE ENZIMAS PANCREÁTICAS EN LA PATOGENIA DE LA PANCREATITIS AGUDAEstudios recientes han sugerido que la pancreatitis es una enfermedad que surge y evoluciona en tres fases.
La primera o inicial se caracteriza por la activación intrapancreática de enzimas digestivas y por la lesión de células acinares.
La segunda fase comprende la activación, quimioatracción y secuestro de neutrófilos en el páncreas, que origina una reacción inflamatoria intrapancreática de intensidad variable.

La tercera fase de la pancreatitis se debe a los efectos de las enzimas proteolíticas y de mediadores activados, liberados por el páncreas inflamado, en órganos distantes.
El daño y la muerte de las células hacen que se liberen péptidos de bradicinina, sustancias vasoactivas e histamina, que originarán vasodilatación, mayor permeabilidad vascular y edema, con profundos efectos en muchos órganos, en particular el pulmón. Pueden ocurrir como consecuencia de la cascada de efectos locales y a distancia el síndrome de respuesta inflamatoria generalizada (systemic inflammatory response syndrome, SIRS), el síndrome de insuficiencia respiratoria aguda (acute respiratory distress syndrome, ARDS) y la falla de múltiples órganos.
ESTUDIO DEL PACIENTE: Cuadro clínico
El dolor abdominal es el síntoma principal de la pancreatitis aguda. El dolor puede variar desde una molestia leve y tolerable hasta un dolor intenso, constante e incapacitante. De manera característica el dolor, que es constante y terebrante, se localiza en el epigastrio y la región periumbilical y a menudo se irradia hacia espalda, tórax, flancos y región inferior del abdomen. El dolor suele ser más intenso cuando el paciente se encuentra en decúbito supino y aliviarse cuando se sienta con el tronco flexionado y las rodillas recogidas.
También son frecuentes náusea, vómito y distensión abdominal, debidos a la hipomotilidad gástrica e intestinal y a la peritonitis química.
La exploración física suele mostrar un paciente angustiado e inquieto. Son bastante frecuentes febrícula, taquicardia e hipotensión. No es raro el choque.
La ictericia es rara y cuando se presenta suele deberse a edema de la cabeza del páncreas, que comprime la porción intrapancreática del conducto colédoco.
Pueden aparecer nódulos eritematosos en la piel por necrosis de la grasa subcutánea. En 10 a 20% de los pacientes existen signos pulmonares, como estertores basales, atelectasias y derrame pleural; este último es más frecuente en el lado izquierdo. Hay diversos grados de hipersensibilidad y rigidez muscular en el abdomen, pero pueden resultar insignificantes en comparación con el intenso dolor. Los ruidos intestinales suelen estar disminuidos o ausentes. En la región superior del abdomen se puede palpar un seudoquiste pancreático. A veces se observa una coloración azul pálido alrededor del ombligo (signo de Cullen) debida al hemoperitoneo y una coloración azul, roja o morada o verde-parda en los flancos (signo de Turner) secundaria al catabolismo hístico de la hemoglobina. Estos dos signos son poco frecuentes y revelan pancreatitis necrosante grave.

EXAMENES DE LABORATORIO La concentración sérica de amilasa se usa ampliamente como prueba de detección sistemática para la pancreatitis aguda en el paciente que presenta dolor abdominal agudo o dolor de espalda. Los valores tres veces mayores que el límite superior de lo normal prácticamente confirman el diagnóstico si se descarta perforación o infarto intestinal. En caso de no encontrarse datos objetivos de pancreatitis por medio de ecografía abdominal, tomografía computadorizada, colangiopancreatografía retrógrada endoscópica o ecografía endoscópica, las elevaciones leves a moderadas de la amilasa( el valor normal es de 60-180 U/L) o de la lipasa (el valor normal es de 0-160 U/L) o de ambas enzimas a la vez, son problemáticas para establecer un diagnóstico de pancreatitis. La amilasa se eleva a las 24h y permanece elevada durante uno a tres días. Las cifras retornan a la normalidad en tres a cinco días, salvo en el caso de que exista necrosis pancreática extensa, obstrucción incompleta de los conductos o formación de un seudoquiste. Alrededor de 85% de los pacientes con pancreatitis aguda presentan un aumento de la amilasa sérica. No obstante, puede haber valores normales si: 1) se retrasa (de dos a cinco días) la obtención de muestras de sangre; 2) el trastorno subyacente es una pancreatitis crónica en vez de una pancreatitis aguda, y 3) hay hipertrigliceridemia. Se ha observado que los pacientes con hipertrigliceridemia y pancreatitis comprobada presentan valores falsamente bajos de amilasa y de actividad de lipasa.
Resulta útil tambien recurrir a un análisis del tripsinógeno sérico (realizado por varios laboratorios comerciales). Dado que esta enzima es secretada específicamente por el páncreas, un valor de tripsinógeno sérico normal en un paciente con elevación mínima de la amilasa séricaprácticamente excluye la pancreatitis aguda
La lipasa puede ser ahora la enzima más indicada para establecer un diagnóstico de pancreatitis aguda, la lipasa es mas especifica pero menos sensible. La otra eventualidad donde la lipasa puede ser superior a la amilasa para apoyar el diagnóstico clínico de pancreatitis aguda es en casos de

presentación tardía a la consulta; ya que la vida media de la amilasa es tan sólo de 48 horas y de la lipasa es de 5 a 8 días .
Ninguna prueba sanguínea aislada es fiable para el diagnóstico de pancreatitis aguda en los pacientes con insuficiencia renal.
ADEMAS PUEDE HABER:
Con frecuencia existe leucocitosis (15 000 a 20 000 leucocitos/ l).
En los casos más graves puede haber hemoconcentración con valores de hematócrito >44%. La hemoconcentración puede ser el precursor de una enfermedad más grave, como necrosis pancreática.
Es frecuente la hiperglucemia secundaria a múltiples factores, entre los que están la menor producción de insulina, el aumento de liberación de glucagon y la mayor producción de glucocorticoides y de catecolaminas suprarrenales.
Alrededor de 25% de los casos presentan hipocalciemia y no se conoce bien su patogenia.
Hay hiperbilirrubinemia [bilirrubina sérica >68 mol/L (>4 mg/100 ml)] en casi 10% de los pacientes.
Las concentraciones de fosfatasa alcalina y de aminotransferasa de aspartato (aspartate aminotransferase, AST) en el suero también se encuentran elevadas de manera transitoria.
Los valores muy altos de deshidrogenasa láctica (lactic dehydrogenase, LDH) en suero [>8.5 mol/L (>500 U/100 ml)] indican un mal pronóstico.
Alrededor de 10% de los casos presentan disminución de los valores séricos de albúmina a 30 g/L (3 g/100 ml), que se asocia a pancreatitis más grave.
En 15 a 20% de los casos hay hipertrigliceridemia y los valores séricos de amilasa en estos pacientes a menudo son falsamente normales.
PRUEBAS RADIOGRÁFICAS
Las radiografías simples de abdomen proporcionan información útil en los pacientes con pancreatitis aguda. Radiografia de torax y abdomen: En todos los pacientes con el diagnóstico clínico de pancreatitis aguda se les debe practicar rutinariamente una radiografía de tórax y abdomen simple para tenerlas como patrón de base y para excluir otras patologías como la presencia de una víscera perforada o de un cuadro de obstrucción intestinal.
Las alteraciones más frecuentes son: 1) íleo localizado que suele afectar el yeyuno ("asa centinela"); 2) íleo generalizado con niveles hidroaéreos; 3) "signo del colon interrumpido", que se debe a la distensión aislada del colon transverso; 4) distensión duodenal con niveles hidroaéreos, y 5) una tumoración que con frecuencia es un seudoquiste. En la pancreatitis crónica, un signo radiográfico importante es la calcificación pancreática, que de manera característica se localiza adyacente o superpuesta a la segunda vértebra lumbar

La ecografía puede aportar información importante en casos de pancreatitis aguda, pancreatitis crónica, calcificación pancreática, seudoquiste y carcinoma de páncreas. Las imágenes ecográficas pueden indicar la presencia de edema, inflamación y calcificación.
La TAC es el mejor método de diagnóstico por imagen para la evaluación inicial de un posible trastorno pancreático crónico y de las complicaciones de la pancreatitis aguda y crónica.La tomografía computadorizada (computed tomography, CT) puede confirmar la sospecha clínica de pancreatitis aguda incluso en caso de valores normales de amilasa sérica. Es de destacar el hecho de que la CT es bastante útil para indicar la gravedad de la pancreatitis aguda y el riesgo de morbilidad y de mortalidad.
La tomografia con medio de contraste (contrast-enhanced CT, CECT), proporciona información valiosa sobre la gravedad y el pronóstico de la pancreatitis aguda. En particular, una CECT permite estimar la presentación y la magnitud de la necrosis pancreática.
Es razonable reservar la CECT para los pacientes con pancreatitis grave o en quienes se sospechan complicaciones sépticas locales.
Índice de gravedad en la pancreatitis aguda.Segun TAc de Abdomen (Balthazar)
DIAGNÓSTICO diferencial
El diagnóstico diferencial debe hacerse con los siguientes trastornos: 1) víscera perforada, sobre todo úlcera péptica, 2) colecistitis aguda y cólico biliar, 3) obstrucción intestinal aguda, 4) oclusión vascular mesentérica, 5) cólico renal, 6) infarto de miocardio, 7) aneurisma disecante de aorta, 8)

enfermedades del tejido conjuntivo con vasculitis, 9) neumonía y 10) cetoacidosis diabética.
EVOLUCIÓN DE LA ENFERMEDAD Y COMPLICACIONES
Es importante identificar a las personas con pancreatitis aguda que tienen un mayor riesgo de fallecer. Son difíciles de utilizar los sistemas de puntuación de múltiples factores (Ranson, Imrie, Apache II), tienen escasa capacidad predictiva y los clínicos no los han aceptado unánimemente. En el cuadro 294-2 se presentan los indicadores básicos de un ataque grave de pancreatitis;
CUADRO 294-2 FACTORES DE RIESGO QUE AFECTAN DE MANERA ADVERSA LA SOBREVIDA EN LA PANCREATITIS AGUDA
Pancreatitis aguda grave
1. Asociada a falla orgánica y/o complicaciones locales como necrosis
2. Manifestaciones clínicas
a. Obesidad con BMI >30
b. Hemoconcentración (hematócrito >44%)
c. Edad >70
3. Falla orgánicaa
a. Choque
b. Insuficiencia pulmonar (PO2 <60)
c. Insuficiencia renal (CR >2.0 mg%)
d. Hemorragia gastrointestinal
4. 3 criterios de Ransom (no completamente utilizables hasta las 48 h)
5. Calificación APACHE II >8 (complicada)
a Por lo general se manifiesta poco después del inicio.
CUADRO 294-3 COMPLICACIONES DE LA PANCREATITIS AGUDA
Locales

Necrosis
Estéril
Infectada
Organizada
Acumulación de líquido pancreático
Absceso pancreático
Seudoquiste pancreático
Dolor
Rotura
Hemorragia
Infección
Obstrucción del tubo digestivo (estómago, duodeno, colon)
Ascitis pancreática
Rotura del conducto pancreático principal
Seudoquiste con escape
Afección de órganos vecinos por pancreatitis necrosante
Hemorragia intraperitoneal masiva
Trombosis de vasos sanguíneos (vena esplénica, vena porta)
Infarto intestinal
Ictericia obstructiva
Generales
Pulmonares
Derrame pleural
Atelectasia
Absceso mediastínico
Neumonitis
Síndrome apneico del adulto
Cardiovasculares
Hipotensión
Hipovolemia
Muerte súbita

Cambios inespecíficos de ST-T en el electrocardiograma que simulan infarto de miocardio
Derrame pericárdico
Hematológicas
Coagulación intravascular diseminada
Hemorragia digestiva
Úlcera péptica
Gastritis erosiva
Necrosis pancreática hemorrágica con erosión hacia grandes vasos sanguíneos
Trombosis de la porta, várices hemorrágicas
Renales
Oliguria
Hiperazoemia
Trombosis de arteria o vena renal
Necrosis tubular aguda
Metabólicas
Hiperglucemia
Hipertrigliceridemia
Hipocalciemia
Encefalopatía
Ceguera súbita (retinopatía de Purtscher)
Sistema nervioso central
Psicosis
Embolia grasa
Necrosis grasa
Tejido subcutáneo (nódulos eritematosos)
Hueso
Varios (mediastino, pleura, sistema nervioso)
Muchos de los enfermos que tienen hipertrigliceridemia persistente tras recuperarse de la pancreatitis y son propensos a padecer episodios recurrentes de pancreatitis.
PANCREATITIS RECURRENTE

Cerca de 25% de los enfermos que han presentado un ataque de pancreatitis aguda sufren recidivas. Los dos factores etiológicos más comunes son alcoholismo y colelitiasis. En individuos con pancreatitis recurrente sin una causa manifiesta se debe descartar una enfermedad oculta de vías biliares, incluyendo microlitiasis, hipertrigliceridemia, farmacoterapia, cáncer de páncreas, disfunción del esfínter de Oddi, páncreas dividido, fibrosis quística y cáncer pancreático .
PANCREATITIS EN PACIENTES CON SIDA
La incidencia de pancreatitis aguda es mayor en los pacientes con SIDA por dos razones: 1) la gran incidencia de infecciones que afectan al páncreas, como las causadas por citomegalovirus, Cryptosporidium y el complejo Mycobacterium avium y 2) el uso frecuente de medicamentos como didanosina, pentamidina, trimetoprim-sulfametoxazol e inhibidores de proteasa en los pacientes con SIDA (cap. 182).
TRATAMIENTO
En la mayoría de los pacientes (85 a 90%) con pancreatitis aguda, la enfermedad es de evolución limitada y cede espontáneamente, en general tres a siete días después de instaurado el tratamiento. Las medidas habituales son: 1) analgésicos para el dolor, 2) líquidos y coloides intravenosos para mantener un volumen intravascular normal y 3) no dar alimentos por vía oral.
El paciente con pancreatitis leve a moderada por lo general requiere tratamiento con líquidos intravenosos y ayuno. Entre el tercer y sexto días a menudo se comienza a implementar una dieta a base de líquidos claros y entre el quinto y el séptimo días una dieta normal. La decisión de reiniciar la ingesta por la boca suele basarse en los criterios siguientes: 1) decremento o resolución del dolor abdominal; 2) el paciente siente hambre y, 3) alivio de la disfunción de órganos en caso de estar presente. La elevación de los valores de amilasa-lipasa séricos o la persistencia de cambios inflamatorios detectados en la CT no deben ser elementos en contra de alimentar a un sujeto asintomático y hambriento. En este sentido, quizá durante semanas o meses no se normalicen los cambios inflamatorios en la CT y persistan las elevaciones en la amilasa-lipasa sérica.
El individuo con pancreatitis fulminante sin remisión por lo común necesita volúmenes extraordinarios de líquidos y atención muy precisa de complicaciones como colapso cardiovascular, insuficiencia respiratoria e infección pancreática; esta última se tratará con una combinación de técnicas radiográficas y quirúrgicas.
Hay que realizar el desbridamiento quirúrgico intensivo del páncreas (extirpación de tejido necrótico) poco después de que se confirme la presencia de necrosis infectada y a veces se necesitan múltiples cirugías. El índice de mortalidad en la pancreatitis necrosante aguda estéril es de alrededor de 10%, por lo cual habrá que pensar en la realización de laparotomía con drenaje adecuado y extracción del tejido necrótico, si con las medidas ordinarias no se frena ni evita el deterioro del paciente.
El empleo de nutrición parenteral total (total parenteral nutrition, TPN) permite el apoyo y sostén nutricionales en casos de pancreatitis grave, aguda o tardía, en sujetos que no pueden ingerir normalmente alimentos. Se ha sugerido que la alimentación enteral con un tubo nasoyeyunal es preferible a la nutrición parenteral total en virtud de que disminuye la tasa de infección. Los alimentos enterales menos costosos satisfacen casi sólo 50% de los requerimientos nutricionales en tanto que la nutrición parenteral total satisface 90% de tales necesidades.
Los pacientes con pancreatitis grave debida a cálculos biliares pueden mejorar de manera

impresionante si se realiza una papilotomía en las primeras 36 a 72 h del inicio. Estudios recientes indican que sólo los pacientes con pancreatitis por cálculos biliares que pertenecen al grupo de los muy graves deben ser considerados candidatos para una ERCP de urgencia.
Por último, el tratamiento de los pacientes con pancreatitis asociada a hipertrigliceridemia consiste en las siguientes medidas: 1) pérdida de peso hasta el ideal, 2) dieta baja en grasas, 3) ejercicio, 4) evitar el alcohol y los fármacos que puedan elevar los triglicéridos séricos (es decir, estrógenos, vitamina A, tiazidas y propranolol) y 5) control de la diabetes.
UTILIDAD DE LOS ANTIBIÓTICOS
El beneficio de la profilaxis con antibióticos en el tratamiento de la pancreatitis aguda necrosante sigue siendo controversial.
Si bien no se han definido por completo los medicamentos óptimos y la duración del tratamiento, en la actualidad en los pacientes con pancreatitis aguda necrosante se recomienda el empleo de un antibiótico general como imipenem cilastatina, 500 mg tres veces por día durante siete días.
NECROSIS PANCREÁTICA INFECTADA, ABSCESO Y SEUDOQUISTE DE PÁNCREAS
Es preciso diferenciar del absceso pancreático.
La necrosis pancreática infectada: Es una infección difusa de un páncreas necrótico con inflamación aguda, que ocurre en las dos primeras a cuatro semanas siguientes al inicio de una pancreatitis. La necrosis pancreática infectada debe tratarse mediante desbridamiento quirúrgico. En 20 a 35% de los pacientes el páncreas necrótico contrae una infección secundaria, generalmente por bacterias gramnegativas de origen alimentario.
El absceso pancreático es una acumulación líquida de pus de escasa definición que evoluciona a lo largo de un periodo más prolongado, a menudo en cuatro a seis semanas. Tiende a ser menos grave para el paciente y se asocia a una tasa inferior de mortalidad quirúrgica. El absceso pancreático puede tratarse mediante cirugía o, en casos concretos, mediante drenaje percutáneo.. Los signos característicos del absceso son fiebre, leucocitosis, íleo y rápido deterioro de un paciente que se recuperaba de la pancreatitis. Sin embargo, en ocasiones las únicas manifestaciones son fiebre persistente y signos de inflamación pancreática ininterrumpida.
Se sospecha de una infección pancreática, basándose en la existencia de fiebre, leucocitosis y CT anormal (seudoquiste o acumulación de líquido extrapancreático). La mayoria de estas infecciones se produjeron en las tres primeras semanas.
Los seudoquistes del páncreas son acumulaciones de tejido, líquido, detritos, enzimas pancreáticas y sangre que se forman en cuatro a seis semanas después del inicio de la pancreatitis aguda y que aparecen en casi 15% de los pacientes. A diferencia de los verdaderos quistes, los seudoquistes carecen de revestimiento epitelial. Los seudoquistes están precedidos de pancreatitis en 90% de los casos y de un traumatismo en 10%. Alrededor de 85% de los seudoquistes se localizan en el cuerpo o la cola del páncreas y 15% en la cabeza. Algunos pacientes tienen dos o más de ellos. El síntoma de presentación habitual es dolor abdominal con irradiación a la espalda o sin ella. En la porción media o izquierda de la región superior del abdomen suele encontrarse una tumoración palpable e hipersensible. La amilasa sérica aumenta

en 75% de los pacientes en algún momento de su enfermedad y puede presentar notables fluctuaciones.
En los casos en que no se produce la resolución espontánea pueden aparecer complicaciones graves, como: 1) dolor causado por la expansión de la lesión y la presión sobre otras vísceras, 2) rotura, 3) hemorragia y 4) absceso. La rotura de un seudoquiste pancreático es una complicación particularmente grave. Casi siempre sobreviene choque y la mortalidad varía desde 14% si la rotura no se asocia a hemorragia, hasta más de 60% si se produce esta asociación. La rotura y la hemorragia son las principales causas de mortalidad en el seudoquiste pancreático.
ASCITIS PANCREÁTICA Y DERRAMES PLEURALES DEL PÁNCREAS
La ascitis pancreática suele deberse a la rotura del conducto pancreático principal, que a menudo se asocia a una fístula interna entre el conducto y la cavidad peritoneal o a un seudoquiste que deja escapar su contenido. Debe sospecharse ascitis pancreática en un paciente que presenta aumento de la amilasa sérica, que también tiene cifras elevadas de albúmina [>30 g/L (>3 g/100 ml)] y un aumento notable de amilasa en el líquido ascítico. El líquido de la ascitis pancreática verdadera suele tener concentración de amilasa >20 000 U/L . En el diagnóstico diferencial es preciso descartar carcinomatosis intraperitoneal, peritonitis tuberculosa, pericarditis constrictiva y síndrome de Budd-Chiari.
SANGRADO DIGESTIVO ALTO
La hemorragia de vías digestivas altas puede manifestarse de cinco formas:
Hematemesis o vómito con sangre roja. Los vómitos en "posos de café" se producen por sangrado más lento o que ha cesado, con conversión de la Hb roja en hematina parda por el ácido gástrico.
Melena. Las heces negras que son negativas en la prueba para sangre oculta pueden deberse a la ingestión de hierro, bismuto o diversos alimentos.
La hematoquezia es la evacuación de sangre de color rojo vivo o granate por el recto. Como pérdidas ocultas de sangre. Otros síntomas (palidez, astenia, adinamia, mareo, síncope, angina de pecho o disnea).
Existen tres factores clínicos independientes que señalan peligro de muerte en pacientes hospitalizados por hemorragia de vías digestivas altas son:
Senectud. Coexistencia de otras enfermedades. Deterioro hemodinámica (taquicardia o hipotensión).
Estudio del paciente
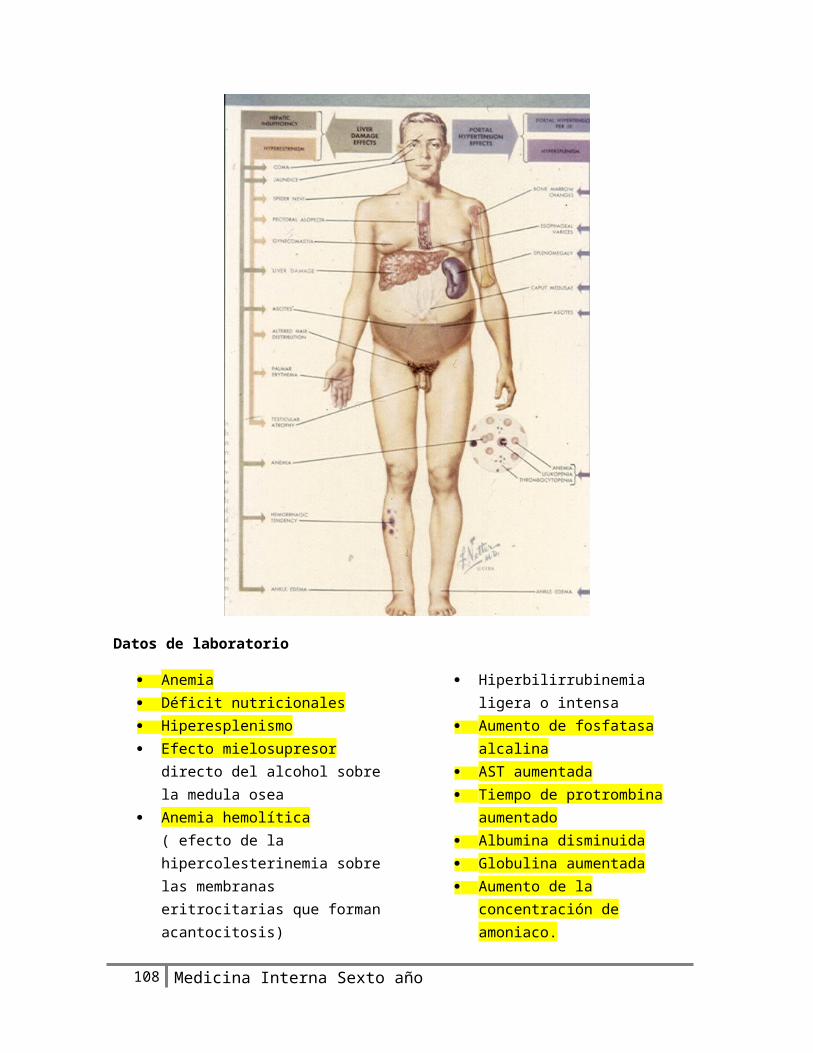
La medición de la frecuencia cardíaca y de la tensión arterial es la mejor manera de iniciar el estudio de un paciente con sangrado digestivo.
Las hemorragias de alcance clínico modifican esas constantes al cambiar de postura al paciente; después aparece taquicardia y, al final, hipotensión, incluso con el sujeto en decúbito.
En cambio, la hemoglobina no desciende inmediatamente después de un sangrado digestivo agudo, porque el volumen del plasma y el de los eritrocitos disminuyen proporcionalmente (es decir, "las personas pierden sangre entera").
A medida que los líquidos extravasculares se desplazan hacia el espacio vascular para restablecer la volemia, la hemoglobina desciende, pero este proceso puede tardar 72 h en producirse. Los pacientes con sangrado digestivo lento y prolongado pueden tener cifras muy bajas de hemoglobina aunque tengan tensión arterial y frecuencia cardíaca normales. Con la aparición de anemia ferropénica, el volumen corpuscular medio disminuye y la anisocitosis aumenta, con mayor número de formas inmaduras.
Diferenciación entre sangrado digestivo alto y bajo
La hematemesis indica que el origen del sangrado es alto (por encima del ligamento de Treitz). La melena indica que hubo sangre derramada en el tubo digestivo por lo menos durante 14 h. Por tanto, cuanto más proximal esté situado el punto de hemorragia, más probable es que haya melena. La hematoquezia suele depender de sangrado digestivo procedente de una localización más baja, aunque también una lesión alta puede sangrar tan rápidamente que la sangre no permanezca el tiempo suficiente en el intestino como para asumir la forma de melena.
Cuando la hematoquezia es la primera manifestación de un sangrado digestivo alto conlleva deterioro hemodinámico y descenso del nivel de hemoglobina. Las lesiones hemorrágicas del intestino delgado pueden originar melena o hematoquezia.
Otras pistas que orientan hacia la sospecha de sangrado digestivo alto son los ruidos intestinales intensos y la elevación del nitrógeno ureico en sangre (BUN), que se produce al disminuir la volemia y absorberse las proteínas sanguíneas derramadas en el intestino.
La aspiración nasogástrica extrae material sin sangre incluso en 16% de los casos de los sangrados digestivos altos (casi siempre de origen duodenal). Cuando ese material está teñido de bilis no es posible excluir una lesión pospilórica hemorrágica, ya que se sabe que en 50% de los casos, aproximadamente, detectar bilis en el material aspirado no es fidedigno. Las pruebas de hemorragia oculta que se realizan con el material de aspiración en el que no existe sangre macroscópica carecen de valor clínico.
Evaluación diagnóstica de los pacientes con sangrado digestivo alto
Es raro que los datos de la anamnesis y la exploración física permitan identificar el origen de un sangrado digestivo.

La endoscopia alta es la técnica más indicada en estos pacientes y debe realizarse urgentemente si hay inestabilidad hemodinámica (hipotensión, taquicardia o modificaciones de la frecuencia cardíaca o de la tensión arterial con los cambios de postura). Cuando la hemorragia es más leve también debe efectuarse tempranamente endoscopia sistemática.
Causas de la hemorragia de vías digestivas altas
Cuadro 1.Punto de origen de hemorragia en pacientes
Hospitalizados por hemorragia aguda de vías digestivas altas
Punto de origen
Úlceras
Varices
Desgarros de Mallory-Weiss
Erosiones gastroduodenales
Esofagitis erosiva
pX(%)
35–62
4–31
4–13
3–11
2–8

Cánceres
No se identificó el punto de origen
1–4
7–25
1. Úlcera Péptica
Una úlcera se define como la pérdida de la integridad de la mucosa del estómago o del duodeno que produce un defecto local o excavación a causa de inflamación activa o bien como una rotura de la superficie de la mucosa >5 mm de tamaño, que en profundidad alcanza la submucosa.
Las úlceras se producen en el estómago o el duodeno, y con frecuencia son de naturaleza crónica.
La prevalencia de la úlcera péptica es de aproximadamente12% en varones y 10% en mujeres.
Tipo de ulcera Localización Epidemiología Síntomas y signos
El dolor epigástrico, descrito como quemante o lacerante, se puede presentar
tanto con la úlcera duodenal como con la gástrica.
La hiperestesia epigástrica
Relación con CA
Ulcera duodenal Asientan sobre todo en la primera porción del duodeno (>95%) Por
lo común miden 1 cm. de diámetro
6 a 15% de la población occidental.
El dolor aparece de 90 min. a 3 h antes de una comida y a menudo se alivia con antiácidos o alimentos. despierta al paciente por la noche (2/3de los pacientes presentan esta molestia)
Las úlceras duodenales malignas son extraordinariamente raras.

Ulcera gástrica Área prepilórica o cuerpo
Tienden a aparecer más tarde en la vida, con un pico
de incidencia durante el sexto decenio
Las molestias pueden desencadenarse con la ingestión de alimentos. Las náuseas y la pérdida de peso son más frecuentes en estos pacientes.
Las úlceras gástricas pueden ser malignas.
Fisiopatología
Causada por un desbalance entre los factores protectores de la mucosa y la acción del ácido y la pepsina.
Ahora se sabe que H. pylori y los NSAID son la causa de la inmensa mayoría de las úlceras pepticas.
Otras enfermedades que pueden causar úlceras pépticas: Síndrome de Zollinger-Ellison, Mastocitosis sistémica, Trauma, Quemaduras, Tensión fisiológica mayor.
H. pylori.

Enfermedad inducida por NSAID
Los NSAID se encuentran entre los fármacos más utilizados. El espectro de complicaciones inducidas por dichos fármacos varió desde náuseas y dispepsia (la prevalencia notificada llegó a 50 a 60%) hasta una complicación gastrointestinal grave como úlcera péptica franca (3 a 4%) complicada por hemorragia o perforación, hasta en 1.5% de los usuarios, por año.
Incluso 75 mg de aspirina diariamente pueden ocasionar graves úlceras gastrointestinales, y por ello ninguna cantidad de NSAID es totalmente innocua.
Complicaciones relacionadas con la úlcera péptica
1. Hemorragia digestiva (2 y 3: perforación y obstrucción)La hemorragia digestiva es la complicación más frecuente de la úlcera péptica. Se produce en casi 15% de los pacientes, y con mayor frecuencia en individuos de más de 60 años.
La incidencia más elevada en los ancianos tal vez se deba a que en este grupo de edad el empleo de NSAID es más frecuente. Hasta 20% de los pacientes con hemorragia relacionada con una úlcera sangran sin presentar ningún signo o síntoma de alarma previo.
Entre los factores de riesgo definidos están:
1. ancianidad2. antecedente de úlcera

3. empleo concomitante de glucocorticoides4. dosis altas de NSAID y empleo concomitante de anticoagulantes y 5. enfermedad grave o que afecta múltiples órganos.
Entre los posibles factores de riesgo están infección concomitante por H. pylori, tabaquismo y consumo de bebidas alcohólicas.
En la exploración física la taquicardia y el ortostatismo (en el pulso (un cambio >10 latidos/min) o en la PA (una caída 10 mm Hg). sugieren deshidratación secundaria a los vómitos o una hemorragia digestiva activa.
Tratamiento y pronóstico
Las características de la úlcera en la endoscopia también son datos importantes para el pronóstico. Hasta 33% de los pacientes con hemorragias activas o con un vaso visible que no sangra volverá a sangrar y necesitará una intervención urgente si sigue un tratamiento conservador.
Estos pacientes mejoran claramente si se tratan por vía endoscópica con electrocoagulación bipolar, sondas de calor, o con inyecciones esclerosantes (p. ej., de alcohol absoluto, adrenalina al 1:10 000.
En cambio, en los pacientes con una úlcera de base limpia, la posibilidad de que repitan las hemorragias es casi nula. Si no hay otra razón para la hospitalización, estos pacientes deben ser dados de alta el mismo día de su ingreso en el hospital, una vez estabilizada su situación.
Los pacientes en los que el fondo de la úlcera contiene sangre deben permanecer hospitalizados durante tres días, ya que la mayor parte de las hemorragias recidivantes se produce durante ese período.
Los estudios comparativos con asignación aleatoria han corroborado que el goteo intravenoso constante de dosis altas de omeprazol (administración intravenosa rápida de 80 mg y goteo a razón de 8 mg/h), utilizado para incrementar el Ph intragástrico entre 6 y 7 y mejorar la estabilidad del coágulo, disminuye la persistencia de la hemorragia (aunque no el riesgo de muerte), incluso después de practicar endoscopia terapéutica apropiada en individuos con úlceras de alto riesgo.
Cerca de 33% de los pacientes con hemorragia ocasionada por úlcera vuelven a sangrar en un período de uno a dos años. La profilaxis de las nuevas hemorragias se dirige a los tres factores patogénicos principales de la úlcera:
1. Helicobacter pylori (Los pacientes con úlceras hemorrágicas disminuyen extraordinariamente las recidivas de la hemorragia a <5%).

Cuadro 2. Regímenes recomendados para erradicar la infección por H. pylori.
Fármaco Dosis
Tratamiento TRIPLE
1. Subsalicilato de bismuto mas Metronidazol más
Tetraciclina
2. Ranitidina o citrato de bis más
Tetraciclina más
Claritromicina o metronidazol
3. Omeprazol (lansoprazol) mas Claritromicina más
Metronidazol o
Amoxicilinac
Tratamiento CUÁDRUPLE
Omeprazol (lansoprazol) Subsalicilato de bismuto Metronidazol
Tetraciclina
2 tabletas/6 h
250 mg/6 h
500 mg/6 h
400 mg/12 h
500 mg/12 h
500 mg/12 h
20 mg/12 h (30 mg/12 h)
200 o 500 mg/12 h
500 mg/12 h
1 g/12 h
20 mg (30 mg)/día
2 tabletas/6 h
250 mg/6 h
500 mg/6 h

2. Los NSAID y el ácido estomacal. Se debe interrumpir su consumo, siempre que sea posible. Si es necesario continuar el uso de antiinflamatorios no esteroideos, el tratamiento inicial debe incluir un inhibidor de la bomba de protones. Más tarde se pasará de un NSAID no selectivo a otro que sea inhibidor específico de la ciclooxigenasa (COX-2) o se agregarán otros tratamientos para las vías digestivas.
Los inhibidores de la bomba de protones y el misoprostol son agentes terapéuticos concurrentes eficaces.
Los sujetos con riesgo muy alto (p. ej., ancianos con una úlcera que ha sangrado antes) quizá deban recibir un inhibidor específico de COX-2 y un inhibidor de la bomba de protones. Los que presentan úlceras sangrantes no causadas por H. pylori ni por NSAID deben seguir recibiendo por tiempo indefinido antisecretorios en dosis completas.
2.Desgarro mucoso o síndrome de Mallory-Weiss
Éste se debe en general a vómitos, eructos y tos enérgica; suele afectar la mucosa gástrica vecina a la unión escamocilíndrica (unión gastroesofágica). Los pacientes presentan hemorragia digestiva alta que puede ser intensa.
Los datos clásicos en el interrogatorio consisten en vómito, arcadas o tos, seguidos de hematemesis, ante todo en los sujetos alcohólicos.
En 80 a 90% de los casos se cohíbe espontáneamente y recidiva sólo en 0 a 5%.
Tratamiento
El tratamiento endoscópico resulta eficaz durante la fase de hemorragia activa. En ciertos casos se requiere angiografía terapéutica con goteo endoarterial de vasopresina o embolización, y tratamiento quirúrgico con sobrehilado del desgarro.
La hemorragia continua puede responder al tratamiento con vasopresina o a la embolización angiográfica. Rara vez es necesaria la cirugía.
3. Varices esofágicas
Patogenia
El asiento más común de esta complicación son las varices situadas en la unión gastroesofágica. Los factores que colaboran a la hemorragia por varices gastroesofágicas no se conocen por completo, pero incluyen:
el grado de hipertensión portal (>12 mmHg) y el tamaño de las varices.

Síntomas y signos:
hematemesis indolora pero masiva. con o sin melena. taquicardia ortostática leve.
Puede haber choque profundo, dependiendo de la cantidad de sangre perdida y del grado de hipovolemia. La endoscopia es el mejor modo de evaluar la hemorragia digestiva alta en pacientes con hipertensión portal conocida o presunta.
Tratamiento y Pronóstico
Los pacientes con hemorragia de varices tienen peor pronóstico que los que sangran por otras causas.
El tratamiento endoscópico realizado en ese momento disminuye la repetición de las hemorragias, y las sesiones endoscópicas repetidas para suprimir las varices disminuyen en grado considerable la reaparición de la hemorragia y la mortalidad.
La técnica más idónea es la ligadura endoscópica de las varices esofágicas, porque la hemorragia reaparece menos veces, la mortalidad disminuye, hay menos complicaciones locales, y se necesitan menos sesiones terapéuticas para erradicar las varices, que con la escleroterapia.
El tratamiento inmediato con la administración intravenosa rápida de 50 g de octreótido, seguida de goteo intravenoso a razón de 50 g/h por 2 a 5 días, puede ayudar a cohibir la hemorragia aguda, y ha sustituido a la vasopresina como el agente terapéutico médico más indicado contra la hemorragia aguda por varices, han mostrado eficacia también agentes, como la somatostatina y la terlipresina. A largo plazo, la administración de betabloqueadores no selectivos disminuye la hemorragia recidivante por varices esofágicas. Por lo regular se administran betabloqueadores, junto con el tratamiento endoscópico de largo plazo.
Está indicado un criterio más enérgico cuando las hemorragias persisten o recidivan a pesar del tratamiento médico y endoscópico. La derivación portosistémica intrahepática a través de la vena yugular TIPS) disminuye las recidivas de la hemorragia con más eficacia que el tratamiento endoscópico.
4. Gastropatía ("gastritis") hemorrágica y erosiva
La gastropatía o gastritis hemorrágica y erosiva es la denominación que se utiliza para designar a las hemorragias y erosiones subepiteliales que se identifican en la endoscopia. Se trata de lesiones de la mucosa y que por tanto no producen hemorragias importantes.

Aparecen en varias situaciones clínicas:
1. la ingestión de NSAID Cerca de 50% de los pacientes que ingieren prolongadamente NSAID tiene erosiones.
2. Alcohol, 20% de los alcohólicos bebedores activos que tienen síntomas de sangrado digestivo alto presentan signos de hemorragia o de erosiones subepiteliales.
3. el estrés. Las lesiones de la mucosa gástrica relacionadas con el estrés sólo aparecen en pacientes muy graves, como los que han sufrido traumatismos intensos, intervenciones de cirugía mayor, quemaduras de extensión superior a un tercio de la superficie corporal, enfermedades intracraneales graves o alguna otra enfermedad importante (dependencia de un ventilador mecánico, coagulopatías).
La profilaxis farmacológica de las hemorragias puede estar indicada en esos pacientes de alto riesgo. Los mejores datos clínicos indican que el tratamiento más indicado es la administración de antagonistas de los receptores H2 por vía intravenosa, aunque el sucralfato también es eficaz.
El tratamiento profiláctico disminuye las hemorragias, pero no reduce la mortalidad.
Otras causas:
Otras causas menos frecuentes de sangrado digestivo alto son:
1. la duodenitis erosiva2. las neoplasias 3. las fístulas aortointestinales 4. las lesiones vasculares [como la telangiectasia hemorrágica hereditaria (de Osler-Weber-
Rendu)] 5. las ectasias vasculares del antro gástrico ("estómago en sandía cortada") 6. la lesión de Dieulafoy (en la que un vaso anómalo de la mucosa sangra por un punto
defectuoso de ésta) 7. la gastropatía de prolapso (prolapso del estómago proximal en el esófago provocado por
los esfuerzos del vómito, ante todo en los alcohólicos) 8. hemobilia y9. la hemorragia del colédoco o del conducto pancreático.
Diagnostico Diferencial
Una historia de episodios hemorrágicos (p. ej., púrpura, equimosis, hematuria) puede indicar la presencia de una diátesis hemorrágica (p. ej., hemofilia). La presencia de diarrea sanguinolenta, fiebre y dolor abdominal es consistente con una enfermedad intestinal inflamatoria (colitis ulcerosa, enfermedad de Crohn) o una colitis infecciosa (p. ej., por Shigella, Salmonella, Campylobacter, amebiasis). La hematoquecia o la sangre oculta en las heces pueden ser el primer signo de un cáncer o un pólipo de colon, particularmente en pacientes >45 años de edad.

Infecciones estafilocócicas: introducciónStaphylococcus aureus, la más virulenta de las muy diversas especies de estafilococos, es un patógeno pluripotente que origina enfermedad por mecanismos tanto mediados como no mediados por toxinas. Este microorganismo origina infecciones nosocomiales y de origen comunitario que varían desde procesos relativamente menores de la piel y partes blandas hasta trastornos generalizados que pueden ser mortales.Los "otros" estafilococos, llamados globalmente coagulasanegativos (CoNS), son mucho menos virulentos que S.aureus.
Aspectos microbiológicos y taxonómicosLas 33 especies de estafilococos son patógenos pertenecientes a la familia Micrococcaceae. En la figura 1-1 se incluye una estrategia sencilla para identificar las especies de mayor importancia clínica. Un método que ha demostrado ser fiable para diferenciar una especie de otra es el análisis del RNA ribosómico de 16S. Con pocas excepciones, S. aureus se distingue de las demás especies de estafilococos por su producción de coagulasa, una enzima superficial que transforma el fibrinógeno en fibrina. La mayor parte de los demás estafilococos de importancia clínica son coagulasanegativos. S. aureus también fermenta el manitol, posee proteína A y produce DNasa. En las placas de agar-sangre, S. aureus tiende a formar colonias betahemolíticas doradas; por lo contrario, los CoNS producen colonias no hemolíticas de color blanco.FIGURA 1-1.

INFECCIONES S. AUREUS
Aspectos epidemiológicos
S. aureus forma parte de la flora normal del ser humano. El sitio más frecuente de colonización es la zona anterior de las vías nasales, aunque también puede colonizar la piel (en particular si está lesionada), la vagina, las axilas, el perineo y la bucofaringe. Se sabe que 25 a 50% de los sujetos sanos pueden estar colonizados por S. aureus de manera persistente o transitoria. La frecuencia de colonización es mayor entre los diabéticos insulinodependientes, los sujetos infectados por el VIH, los usuarios de drogas inyectables, los pacientes sometidos a hemodiálisis y los individuos con lesiones cutáneas.
En general, S. aureus es una causa importante de infecciones nosocomiales. Es la causa más frecuente de infección en las incisiones quirúrgicas y ocupa el segundo lugar, después de los CoNS, como causa de bacteriemia primaria. A nivel comunitario, S. aureus sigue siendo una causa importante de infecciones cutáneas y de partes blandas, de infecciones respiratorias y (en las personas que consumen drogas inyectables) de endocarditis infecciosa.
Varios informes han descrito infecciones comunitarias (en medios tanto rurales como urbanos) causadas por S. aureus resistente a meticilina ( MRSA) en sujetos sin exposición previa de tipo médico. A diferencia de las cepas de MRSA de origen nosocomial, estos microorganismos aislados en la comunidad han seguido siendo sensibles a muchos antibióticos no betalactámicos.
Un aspecto preocupante ha sido la aparente capacidad que poseen las cepas comunitarias de MRSA para causar cuadros graves en personas inmunocompetentes; tal facultad quizá dependa de la presencia de diferentes genes toxígenos en estas especies, y también del empleo de agentes betalactámicos como tratamiento empírico de los pacientes infectados por ellas.
Patogenia
S. aureus es un patógeno piógeno conocido por su capacidad de formar abscesos en los focos de infección tanto locales como metastásicos. Las bacterias de este tipo desencadenan una reacción inflamatoria que se caracteriza al principio por una respuesta intensa de leucocitos polimorfonucleares (PMN) y una infiltración ulterior de macrófagos y fibroblastos.
Si la respuesta celular del hospedador (incluido el depósito de fibrina y colágena) no frena la infección, ésta se propaga a los tejidos vecinos o al torrente circulatorio. En el síndrome de choque tóxico (TSS) por estafilococos bastan condiciones que permitan la síntesis de la toxina en los sitios de colonización (p. ej., la presencia de un tampón superabsorbente) para que surja la enfermedad clínica.
El genoma de S. aureus

Se conoce la secuencia de todo el genoma de varias cepas de S. aureus. Entre los datos más interesantes están los siguientes: 1) se advierte un alto grado de semejanza en cuanto a secuencias de nucleótidos entre las distintas cepas. 2) Se adquiere una cantidad bastante grande de información genética por transferencia horizontal a partir de otras especies bacterianas. 3) S. aureus contiene diversos islotes de "patogenicidad" o "genómicos" característicos; se trata de elementos genéticos móviles que contienen cúmulos de genes de enterotoxinas y exotoxinas o determinantes de resistencia antimicrobiana. 4) Entre los genes que están dentro de los islotes en cuestión figuran los que portan mecA, el gen encargado de la resistencia a la meticilina; los islotes que contienen esta resistencia reciben el nombre de cromosomas en casete estafilocócicos (staphylococcal cassette chromosomes, SCCmec) y su tamaño varía de 20 a 60 kb. El SCCmec de tipo 4 se ha vinculado con las cepas de MRSA de origen comunitario que han ocasionado muchos brotes.
Patogenia de la infección invasora por S. aureus
Los estafilococos son microorganismos oportunistas; para que invadan al hospedador y causen infección, se necesitan algunos de los factores siguientes o todos ellos: inoculación y colonización local de las superficies hísticas, invasión, evasión de la respuesta del hospedador y propagación metastásica. El comienzo de la infección por estafilococos obliga a que haya algún punto de transgresión o penetración de las barreras cutáneas o mucosas. Las cepas o subespeciescolonizantes transferidas de una persona a otra se inoculan en la piel lesionada, las heridas o la corriente sanguínea.
Colonización de las vías nasales
La porción anterior de las vías nasales constituye el sitio principal de colonización por estafilococos en el ser humano. Al parecer comprende la adherencia de S. aureus a la mucina nasal y las células del epitelio queratinizado que están en la porción anterior de las vías nasales. Otros factores que pueden contribuir a la colonización son la influencia de otros miembros de la flora residente en las fosas nasales y el número de bacterias, los daños de la mucosa nasal (p. ej, después de la inhalación de drogas), las propiedades antimicrobianas de las secreciones nasales y los factores genéticos del hospedador [p. ej., el tipo de antígeno leucocitario humano (HLA)].
Inoculación y colonización de las superficies hísticas
Los estafilococos pueden penetrar en los tejidos a raíz de excoriaciones leves, de la aplicación de medicamentos como la insulina o de la colocación de una vía intravenosa. Una vez dentro del foco hístico, las bacterias se replican y colonizan las superficies de los tejidos del hospedador.Intervienen de manera importante como mediadores de la adherencia a dichos focos los miembros de una familia de proteínas de superficie de S. aureus con semejanza estructural, denominados MSCRAMM (componentes microbianos superficiales reconocedores de moléculas adherentes de la matriz). Los MSCRAMM, como la proteína de unión de la fibronectina, el factor de agrupamiento y la proteína de unión de la colágena, permiten a las bacterias colonizar diferentes superficies hísticas;

las proteínas en cuestión contribuyen a la patogenia de infecciones invasoras como la endocarditis y la artritis, al facilitar la adherencia de S. aureus a las superficies en que están al descubierto la fibronectina, el fibrinógeno o la colágena.
Invasión
Después de la colonización, los estafilococos se replican en el foco inicial de la infección y elaboran enzimas, proteasas, hialuronidasas, termonucleasas y lipasas, que facilitan la supervivencia bacteriana y se propagan localmente por las superficies hísticas, aunque no se ha establecido con exactitud su participación precisa en las infecciones. Las lipasas también pueden facilitar la supervivencia en las zonas con abundantes lípidos, como los folículos pilosos, en que suelencomenzar las infecciones por S. aureus. La leucocidina Panton-Valentine, una toxina de S. aureus, tiene acción citolítica sobre los polimorfonucleares, los macrófagos y los monocitos. Desde el punto de vista epidemiológico, las cepas que elaboran dicha toxina se han vinculado con infecciones cutáneas como los furúnculos y el ántrax, y también con infecciones pulmonares graves en adolescentes. La pared del microorganismo, que consta de unidades alternas de ácido N-acetil murámico y N-acetil glucosamina en combinación con un componente adicional de la pared, que es el ácido lipoteicoico, puede desencadenar una reacción inflamatoria, incluido un síndrome séptico.
Evasión de los mecanismos de defensa del hospedador
Un factor de máxima importancia para la invasión es la evasión de los mecanismos de defensa del hospedador. Los estafilococos poseen una microcápsula de polisacáridos antifagocíticos. La cápsula de S. aureus parece desempeñar un papel importante en la inducción de abscesos. Los polisacáridos capsulares se caracterizan por un perfil de carga zwitteriónico (presencia de moléculas de carga negativa y positiva al mismo tiempo) que es de máxima importancia para la formación de abscesos. La proteína A, un MSCRAMM característico de S. aureus, actúa como receptor de Fc. Dicha proteína puede unirse a la porción Fc de las subclases 1, 2 y 4 de la inmunoglobulina G, impidiendo que los polimorfonucleares ejerciten su capacidad opsónica y de fagocitosis.Otro mecanismo más para evadir la respuesta del hospedador por parte de S. aureus es su capacidad de supervivencia intracelular. La internalización de estafilococos por las células endoteliales crea un santuario que protege a las bacterias contra las defensas del hospedador; también origina cambios celulares, como la expresión de receptores de integrinas y Fc y la liberación de citocinas. Los cambios celulares en cuestión pueden contribuir a las manifestaciones sistémicas de la enfermedad, incluidas la septicemia y las vasculitis. Por último, S. aureus sobrevive dentro de los polimorfonucleares y puede utilizarlos para propagarse y sembrar otros tejidos.
Respuesta del hospedador a la infección por S. aureus
La respuesta primaria del hospedador a la infección por S. aureus es la proliferación de polimorfonucleares. Los PMN son atraídos a los focos de infección por componentes bacterianos como los péptidos formilados o los peptidoglucanos. También son atraídos por el factor de necrosis tumoral (TNF) y las interleucinas (IL) 1 y 6, citocinas que son liberadas por los macrófagos activados y las células endoteliales.

Patogenia de la enfermedad mediada por toxinas
S. aureus produce tres tipos de toxinas: citotoxinas, superantígenos, que son toxinas pirógenas, y toxinas exfoliativas. Los datos epidemiológicos y los obtenidos de animales sugieren que la presencia de anticuerpos antitoxinas protege de la enfermedad en el TSS, la intoxicación alimentaria por estafilococos y el síndrome de dermatitis exfoliativa causado por dicho microorganismo (staphylococcal scalded-skin syndrome, SSSS). El cuadro patológico surge después de la síntesis y la absorción de la toxina, y de que ésta inicie la respuesta del hospedador.
Enterotoxina y toxina 1 del síndrome de choque tóxico( TSST-1 )
Los superantígenos toxínicos de acción pirógena son una familia de proteínas de tamaño molecular pequeño estructuralmente semejantes que originan dos enfermedades: el síndrome de choque tóxico (TSS) y la intoxicación alimentaria. El primero es consecuencia de la facultad que tienen las enterotoxinas y la TSST-1 de actuar como mitógenos en los linfocitos T. En el proceso de presentación de antígenos normal, éstos se procesan al principio en el interior de la célula y,después, los péptidos son presentados en el surco del complejo de histocompatibilidad mayor (MHC) de clase II, lo que desencadena una respuesta medida en los linfocitos T, las enterotoxinas se unen directamente a la región invariable del MHC. Después, las enterotoxinas se unen a los receptores del linfocito T a través de la cadena v, lo que da lugar a una drástica sobreexpansión de los clones de células T (hasta un 20% de la población total de células T).A consecuencia de dicha expansión celular surge el equivalente de una "tormenta de citocinas", en la que se liberan mediadores de la inflamación como el interferón (IFN) gamma, las interleucinas 1 y 6, y los factores de necrosis tumoral alfa y beta. El resultado es un cuadro multisistémico en el que aparece toda una constelación de signos y síntomas, como mialgias, fiebre, erupciones e hipotensión. Estos datos son similares a los que aparecen en el choque endotoxínico; sin embargo, los mecanismos patógenos son diferentes. Se ha planteado la hipótesis de que uno de los factores que participan en el TSS sería la liberación de endotoxina desde las vías gastrointestinales, lo que podría intensificar por sinergismo los efectos de la toxina.Otra región de la molécula de enterotoxina es la que ocasiona los síntomas de intoxicación alimentaria. Las enterotoxinas son termoestables y sobreviven en entornos que destruirían a las bacterias. El cuadro patológico aparece por la ingestión de la toxina preformada y, en consecuencia, el período de incubación es breve (1 a 6 h). La toxina estimula el nervio neumogástrico y el centro del vómito en el encéfalo. También parece estimular la actividad peristáltica del intestino.
Toxinas exfoliativas y síndrome exfoliativo estafilocócico
Las toxinas con acción exfoliativa son las que causan el síndrome exfoliativo estafilocócico. Las toxinas que ocasionan enfermedad en los seres humanos se han dividido en dos serotipos: ETA y ETB. Estas toxinas rompen los desmosomas que unen a las células vecinas. El resultado es la dehiscencia de la epidermis a nivel de la granulosa, fenómeno que desencadena la descamación superficial de la piel que es típica de la enfermedad.
Diagnóstico

Las infecciones por S. aureus pueden diagnosticarse fácilmente por medio de la tinción de Gram y por el examen microscópico del contenido del absceso o del tejido infectado. El cultivo sistemático del material infectado suele generar resultados positivos, y los cultivos de sangre son a veces positivos incluso cuando la infección se localiza en zonas extravasculares. Para el diagnóstico rápido de la infección por el microorganismo mencionado se han aplicado métodos basados en la reacción en cadena de la polimerasa (PCR).
Síndromes clínicos
Infecciones de piel y partes blandasS. aureus origina infecciones cutáneas de diversa índole. Entre los factores predisponentes más frecuentes están las dermatosis, los daños de la piel (como el causado por picaduras de insectos o traumatismos menores), las inyecciones (como en el caso de la diabetes o el consumo de drogas inyectables) y la falta de aseo personal. Las infecciones en cuestión se caracterizan por la formación de vesículas pustulosas que suelen comenzar en los folículos pilosos y se propagan a los tejidos vecinos. La foliculitis es una infección superficial que afecta el folículo piloso en la que existe una zona central de purulencia (pus) rodeada de induración y eritema. Los forúnculos son lesiones más extensas y dolorosas que suelen aparecer en las regiones pilosas y húmedas del cuerpo, y que se extienden desde el folículo piloso hasta transformarse en un absceso verdadero con una zona de purulencia central. El ántrax se sitúa con mayor frecuencia en la mitad inferoposterior del cuello y es todavía más doloroso y grave, porque surge de la coalescencia de otras lesiones que abarcan la capa más profunda del tejido subcutáneo. En términos generales, los forúnculos y el ántrax se identifican fácilmente porque de ellos mana pus al comprimirlos o espontáneamente.
Enfermedades causadas frecuentemente por Staphylococcus Aureus
Infecciones de piel y partes blandasFoliculitis. Furúnculo, ántrax. Celulitis. Impétigo. Mastitis. Infecciones de incisiones quirúrgicas. Hidradenitis supurativa(infecciones foliculares repetitivas en regiones como la axila).
Infecciones musculoesqueléticasArtritis séptica. Osteomielitis. Piomiositis. Abscesos del psoas
Infecciones de vías respiratoriasNeumonía por ventiladores o nosocomial. Émbolos pulmonares sépticosNeumonía posvírica (como influenza). Empiema
Bacteriemia y sus complicacionesSepsis, choque séptico. Focos metastásicos de infección (en riñones, articulaciones, huesos, pulmones). Endocarditis infecciosa
Endocarditis infecciosaPor drogas inyectables. En válvulas originales. En prótesis valvulares. Nosocomial
Infecciones en dispositivos (como catéteres intravasculares o prótesis articulares)

Enfermedades mediadas por toxinasSíndrome de choque tóxico. Intoxicación alimentaria. Síndrome exfoliativo estafilocócico
En 1 a 3% de las mujeres que amamantan a sus hijos surge mastitis. El cuadro infeccioso, que suele aparecer a las dos a tres semanas después del parto, se caracteriza por manifestaciones que van desde la celulitis hasta la formación de abscesos. En los casos más graves suelen surgir signos de tipo general, como fiebre y escalofríos.S. aureus es uno de los microorganismos patógenos más frecuentes en las infecciones de las incisiones quirúrgicas.
Infecciones de tejidos musculoesqueléticosS. aureus es uno de los microorganismos que con mayor frecuencia origina infecciones óseas (por diseminación hematógena y también por propagación de una parte blanda vecina).En los niños, la osteomielitis hematógena afecta muy a menudo a los huesos largos. El cuadro inicial incluye fiebre y dolor óseo o aversión del pequeño a soportar pesos. Suele aumentar el número de leucocitos y la tasa de eritrosedimentación. Los cultivos de sangre son positivos en cerca de 50% de los casos. Las radiografías corrientes pueden resultar normales incluso 14 días después de comenzar los síntomas. La gammagrafía con fosfonato de 99mTc suele identificar los primeros signos de la infección. La resonancia magnética es más sensible que las otras técnicas para confirmar el diagnóstico radiológico.En los adultos, la forma mencionada de osteomielitis que afecta los huesos largos es menos frecuente. Sin embargo, uno de los cuadros clínicos iniciales más comunes es la osteomielitis vertebral. Este trastorno suele observarse en personas con endocarditis, en las sometidas a hemodiálisis, en los diabéticos y en los adictos a drogas inyectables. Las infecciones de las vértebras pueden debutar con dorsalgia y fiebre intensas, pero su presentación también puede ser menos grave, con dorsalgia crónica y febrícula. S. aureus es el patógeno que con mayor frecuencia causa abscesos epidurales, complicación que puede originar una afección del sistema nervioso. La persona se queja de dificultad para orinar o caminar, y de dolor radicular, además de los síntomas propios de la osteomielitis. Por medio de la resonancia magnética se confirma con bastante certeza el diagnóstico.
Las infecciones óseas que son producto de la propagación desde tejidos vecinos tienden a surgir de infecciones de partes blandas, como en el caso de las úlceras diabéticas o vasculares, las operaciones y los traumatismos. La afección ósea se puede corroborar mediante el cultivo de fragmentos de hueso y el estudio histopatológico. La contaminación del material de cultivo por microorganismos de los tejidos vecinos dificulta el diagnóstico de osteomielitis cuando no se ha logrado la confirmación por métodos histopatológicos. Además, con los medios radiográficos resulta a veces difícil distinguir la osteomielitis de la infección de las partes blandas suprayacentes, con osteítis subyacente.
Staphylococcus aureus es la causa más frecuente de artritis séptica en los niños. El cuadro se presenta con fiebre, hinchazón articular y dolor intenso al mover la articulación afectada. El líquido aspirado de la articulación es turbio, tiene más de 50000 polimorfonucleares/ml y presenta cocos grampositivos en cúmulos en el estudio con tinción de Gram. En los adultos, la artritis puede deberse a traumatismos, operaciones o diseminación hematógena. Las articulaciones afectadas con mayor frecuencia son las rodillas, los hombros, las caderas y las interfalángicas. El cuadro suele comenzar en articulaciones que ya estaban dañadas por una artrosis o una artritis reumatoide.

La piomiositis es una infección poco común de los músculos de fibra estriada que se observa más bien en los climas tropicales. El cuadro inicial incluye fiebre, hinchazón y dolor sobre el músculo afectado. La aspiración de líquido del tejido lesionado indica la presencia de pus, que contiene innumerables leucocitos y bacterias grampositivas en cúmulos.
Infecciones de vías respiratoriasS. aureus es la causa de infecciones graves en neonatos y lactantes; estas infecciones se manifiestan al principio por disnea, fiebre e insuficiencia respiratoria. En las radiografías de tórax se advierten neumatoceles (cavidades de pared delgada y contornos filamentosos). El neumotórax y el empiema son complicaciones conocidas de este tipo de infección.En los adultos, las infecciones pulmonares de tipo nosocomial por S. aureus suelen surgir en las personas intubadas (tráquea) o en las atendidas en unidades de cuidados intensivos. Los pacientes generan volúmenes crecientes de esputo purulento y terminan por mostrar un cuadro disneico con fiebre y nuevos infiltrados pulmonares.
Las infecciones de vías respiratorias por S. aureus contraídas en la comunidad suelen surgir después de cuadros víricos o por efecto de émbolos pulmonares sépticos (p. ej., en usuarios de drogas inyectables). La influenza es la causa más común del primer tipo de cuadro inicial. Las manifestaciones iniciales son la fiebre, la generación de esputo sanguinolento y la presencia de neumatoceles en 50% de los campos pulmonares o de múltiples infiltrados pulmonares irregulares.El diagnóstico se hace por tinción de Gram y cultivo del esputo.
Bacteriemia, sepsis y endocarditis infecciosaLa bacteriemia por S. aureus puede complicarse con cuadros como sepsis, endocarditis, vasculitis o siembra metastásica (aparición de acumulaciones de pus o supuración en tejidos de otras localizaciones). Entre los tejidos afectados con mayor frecuencia están los huesos, las articulaciones, los riñones y los pulmones.
Entre los trastornos coexistentes que suelen aparecer junto con la bacteriemia por S. aureus y que agravan el riesgo de complicaciones están la diabetes, la infección por VIH y la insuficiencia renal.Desde el punto de vista clínico, el cuadro inicial de la sepsis por S. aureus es semejante al observado en las sepsis por otras bacterias. Normalmente se observa la secuencia de cambios hemodinámicos clásicamente descrita (que comienza con alcalosis respiratoria y manifestaciones clínicas como hipotensión y fiebre). El diagnóstico microbiológico se corrobora por la positividad de los cultivos de sangre.
Según la serie publicada, en la actualidad S. aureus origina un 25 a 35% de todos los casos de endocarditis bacteriana; este incremento se debe, por lo menos en parte, al empleo creciente de dispositivos intravasculares; la incidencia de la endocarditis infecciosa en individuos con bacteriemia por S. aureus y catéteres intravasculares fue de 25% en un estudio mediante ecocardiografía transesofágica. Otros factores vinculados con un mayor riesgo de endocarditis son el consumo de drogas inyectables, la hemodiálisis, la presencia de prótesis intravasculares y la inmunodepresión. Entre las complicaciones de la endocarditis por S. aureus están la insuficiencia valvular cardíaca, los émbolos periféricos, la siembra metastásica y la afección del sistema nervioso central. El

absceso cerebral por S. aureus es una complicación conocida de la endocarditis de la mitad izquierda del corazón.
La endocarditis por S. aureus se observa en cuatro situaciones clínicas: 1) endocarditis de la mitad derecha del corazón, vinculada con el consumo de drogas inyectables; 2) endocarditis de las válvulas originales de la mitad izquierda; 3) endocarditis de las prótesis valvulares, y4) endocarditis nosocomial, el diagnóstico se confirma al identificar los estigmas clínicos que sugieren endocarditis, que comprenden manifestaciones cardíacas, como soplos nuevos o cambiantes en las válvulas del corazón; manifestaciones cutáneas de la endocarditis, como lesiones vasculíticas, nódulos de Osler o lesiones de Janeway; manifestaciones de la enfermedad embólica, y antecedentes que sugieran riesgo de bacteriemia por S. aureus.
La ecocardiografía transtorácica, que es menos sensible que la transesofágica, es menos invasiva y suele señalar la presencia de vegetaciones valvulares.La endocarditis aguda derecha de la válvula tricúspide por S. aureus suele observarse en los adictos a drogas inyectables. El cuadro clásico inicial consiste en fiebre alta, aspecto clínico de intoxicación, dolor pleurítico y generación de esputo purulento (a veces sanguinolento). Las radiografías de tórax muestran datos indicativos de émbolos pulmonares sépticos (lesiones circulares pequeñas y periféricas que con el tiempo pueden mostrar cavidades). En el comienzo de la enfermedad, el individuo puede mostrar sólo fiebre, sin signos cardiovasculares o de cualquier otra localización.
Las personas con antecedentes de daño valvular cardíaco suelen mostrar al principio una endocarditis valvular izquierda previamente dañada. Tienden a ser de más edad que los que presentan endocarditis del lado derecho, su pronóstico es peor y la incidencia de complicaciones es mayor (émbolos periféricos, descompensación cardíaca y siembra metastásica).S. aureus es uno de los patógenos que con mayor frecuencia origina endocarditis en las prótesis valvulares. En casi todos los casos, las medidas médicas no son suficientes por sí solas y es necesario sustituir la válvula urgentemente.
Los enfermos tienden a presentar insuficiencia valvular o abscesos de miocardio que nacen de la región del implante valvular.La mayor frecuencia de endocarditis nosocomial (15 a 30% de los casos, según el estudio) refleja en parte el uso cada vez más frecuente de dispositivos intravasculares. La endocarditis causada por ellos suele depender de S. aureus.
Infecciones por dispositivos protésicosS. aureus origina una gran proporción de las infecciones por dispositivos protésicos; éstas suelen afectar a catéteres intravasculares, prótesis valvulares, aparatos ortopédicos, catéteres peritoneales o intraventriculares e injertos vasculares.
Componentes importantes para establecer el diagnóstico son la aspiración de dichas acumulaciones y la realización de cultivos de sangre. Las infecciones por S. aureus tienden a aparecer con mayor frecuencia en la etapa temprana después del implante, salvo que el dispositivo se utilice como vía de acceso a vasos y otros órganos (catéteres intravasculares o de hemodiálisis). En este último caso, las infecciones persisten durante todo el tiempo que se emplea el dispositivo.

Como ocurre con casi todas las infecciones de prótesis, suele ser necesario extraerlas para obtener buenos resultados. Si se deja en su sitio, el dispositivo es un posible foco de infecciones repetitivas o persistentes.
Enfermedades mediadas por toxinas
Síndrome de choque tóxico (TSS)
Antes de 1985 ya había despertado la atención a nivel nacional en Estados Unidos, donde se detectó un gran brote en mujeres jóvenes, por lo demás sanas, que tenían la menstruación. Las investigaciones epidemiológicas señalaron que tales casos guardaban un vínculo importante con la menstruación y con el uso de un tampón muy absorbente que se había comercializado en fecha reciente. Estudios ulteriores demostraron la participación de la TSST-1 en la enfermedad. Al retirarse el tampón absorbente del comercio disminuyó rápidamente la incidencia de la enfermedad. Sin embargo, no cesó la notificación de casos menstruales y no menstruales.
El cuadro clínico inicial es semejante en las formas menstrual y no menstrual de TSS, aunque hay una diferencia clara en cuanto a la naturaleza del riesgo. No es necesaria la presencia previa de una infección clínica por S. aureus para que surja la enfermedad. El TSS es consecuencia de la elaboración de una enterotoxina o de TSST-1, una toxina parecida a la enterotoxina y estructuralmente similar. Más de 90% de los casos menstruales son causados por la TSST-1, en tanto que un alto porcentaje de los no menstruales son causados por las enterotoxinas.
El TSS comienza con síntomas bastante inespecíficos, similares a los del resfriado. En la variedad menstrual, el cuadro comienza dos a tres días después del inicio de la menstruación con fiebre, hipotensión y eritrodermia de intensidad variable. Es frecuente la afección de las mucosas (p. ej., hiperemia conjuntival). La enfermedad puede evolucionar rápidamente hacia síntomas como vómito, diarrea, confusión, mialgias y dolor abdominal, que reflejan la naturaleza multisistémica del problema y la afección del hígado, los riñones, las vías digestivas, el sistema nervioso central o varios de estos órganos.
En la convalecencia, que tiene lugar una o dos semanas después de comenzar la enfermedad, se descama la piel. Las pruebas de laboratorio muestran hiperazoemia, leucocitosis, hipoalbuminemia, trombocitopenia y anormalidades de la función del hígado.
El diagnóstico del TSS depende aún de la identificación de un conjunto de signos y manifestaciones, más que de un dato específico (cuadro 120-2). El trastorno afecta sólo a las personas que no generan anticuerpos contra la TSST-1. Es posible que el cuadro reaparezca si no hay síntesis de anticuerpos después de la enfermedad.
Cuadro 1-2. Definición del síndrome de choque tóxico por S. aureus1. Fiebre: temperatura de 38.9°C o mayor2. Hipotensión: tensión sistólica de 90 mmHg o menor, o hipotensión ortostática (descenso ortostático de 15 mmHg o mayor en la tensión diastólica, síncope o mareo ortostático)3. Erupción macular difusa con descamación una a dos semanas después del comienzo (incluidas palmas y plantas)4. Afección de diversos órganos y sistemas a. Hígado: niveles de bilirrubina o aminotransferasas dos veces mayores que lo normal (o más)

b. Sangre: recuento plaquetario; 100 000 trombocitos/ l o menos c. Riñones: nitrógeno ureico en sangre o nivel sérico de creatinina dos veces mayor que el límite superior de lo normal o más d. Mucosas: hiperemia en vagina, bucofaringe o conjuntivas e. Vías gastrointestinales: vómito o diarrea desde el comienzo de la enfermedad f. Aparato muscular: mialgias intensas o nivel sérico de fosfocinasa de creatinina dos veces mayor que el límite superior de lo normal, o más g. Sistema nervioso central: desorientación o alteración de la conciencia sin signos neurológicos focales y sin fiebre ni hipotensión5. Negatividad de pruebas serológicas o de otro tipo, como las del sarampión, la leptospirosis o la fiebre moteada de las Montañas Rocosas, y también negatividad de los cultivos de sangre o líquido cefalorraquídeo en cuanto a microorganismos distintos de S. aureus.
Intoxicación alimentaria
S. aureus constituye una de las principales causas de brotes de infección de origen alimentario en Estados Unidos; en estos casos, si el microorganismo patógeno es S. aureus, proviene de la inoculación de una cepa toxígena en los alimentos por manipuladores colonizados. La toxina queda después incluida en productos promotores del crecimiento, como natillas, ensaladas de patata o carnes preparadas. Aunque se destruya la bacteria por calentamiento, la toxina termoestable se conserva. La enfermedad comienza en forma rápida y "explosiva", y sus manifestaciones surgen en el plazo de 1 a 6 h después de la ingestión del alimento contaminado.
El cuadro se caracteriza por náusea y vómito, aunque también pueden surgir diarrea, hipotensión y deshidratación. Entre las entidades a incluir en el diagnóstico diferencial está la diarrea por otras causas, en particular la causada por toxinas similares (como las elaboradas por Bacillus cereus). La rapidez del comienzo, la ausencia de fiebre y la naturaleza epidémica del cuadro inicial deben hacer sospechar la presencia de esta entidad. Los síntomas por lo común se resuelven en un plazo de 8 a 10 h. El diagnóstico se puede corroborar al demostrar la presencia de bacterias o de la enterotoxina en el alimento al que se atribuye el brote. El tratamiento incluye sólo medidas de sostén.
Síndrome exfoliativo estafilocócico (SSSS)
Este síndrome suele afectar a neonatos y niños. El trastorno varía desde la formación de ampollas delimitadas hasta la exfoliación de gran parte de la superficie cutánea. La piel suele ser frágil, duele a la palpación y presenta ampollas de paredes finas llenas de líquido. Se rompen con una presión suave, quedando al descubierto las capas subyacentes (signo de Nikolsky; fig. 1-4) El trastorno no afecta normalmente a las mucosas. En la infección más generalizada hay síntomas de carácter general, como fiebre, letargo e irritabilidad con inapetencia. En los casos más extensos se pierden cantidades notables de líquido. La enfermedad suele surgir después de una infección localizada en alguno de los diversos sitios posibles. El SSSS es menos frecuente en adultos, pero puede aparecer después de infecciones causadas por cepas toxígenas exfoliativas.
Prevención

Prevenir que se propaguen las infecciones por S. aureus en el entorno hospitalario entraña lavarse las manos y prestar atención a los métodos más apropiados deaislamiento. Se ha investigado en diversas situaciones clínicas el empleo de antimicrobianos tópicos (como la mupirocina) para eliminar la colonización de las vías nasales por S. aureus y así evitar infecciones ulteriores. La eliminación del estado de portador de S. aureus en las vías nasales ha disminuido la incidencia de infecciones en los individuos sometidos a hemodiálisis y diálisis peritoneal.
Infecciones por estafilococos coagulasanegativos
Los CoNS, a pesar de ser mucho menos virulentos que S. aureus, constituyen algunos de los microorganismos que con mayor frecuencia originan infecciones en las prótesis. Alrededor de 50% de las 32 especies identificadas de CoNS se han vinculado con infecciones en seres humanos. De las especies en cuestión, S. epidermidis ha sido el patógeno por lo general más común en el ser humano; este integrante de la flora humana aparece en la piel (en la que representa la especiebacteriana más abundante) y también en la bucofaringe y la vagina. S. saprophyticus, que es una especie resistente a novobiocina, es un patógeno presente en infecciones de las vías urinarias.
PatogeniaEntre los CoNS, S. epidermidis es la especie que con mayor frecuencia origina infecciones en los dispositivos protésicos. La infección es un fenómeno bifásico en el que primero se produce la adherencia al dispositivo y después su colonización. S. epidermidis está especialmente adaptado para colonizar dichos dispositivos, gracias a su capacidad de elaborar el polisacárido extracelular (sedimento) que facilita la formación de una biocapa protectora en la superficie del dispositivo.El material protésico implantado suele estar recubierto de constituyentes hísticos o del suero del hospedador, como fibrinógeno o fibronectina. Estas moléculas actúan como ligandos capaces de tender puentes de unión y de facilitar la adherencia de la bacteria a la superficie del dispositivo. La autolisina, una enzima estafilocócica de superficie (AtlE), podría intervenir en la adherencia a otras superficies protésicas, modificadas o no. Además de la AtlE, otras moléculas de superficie, como la proteína de unión del fibrinógeno y el ácido teicoico de la pared celular, parecen intervenir en la adherencia al fibrinógeno y la fibronectina, respectivamente. La adhesina, un polisacárido intercelular, facilita la colonización ulterior por estafilococos y su acumulación en la superficie del dispositivo. También están en S. aureus los genes encargados de la síntesis de dicho polisacárido(genes ica), aunque su papel podría diferir entre las dos especies. En S. epidermidis, los genes mencionados aparecen con mayor frecuencia en las cepas que infectan los dispositivos que en las que colonizan las superficies de las mucosas. La biocapa parece que actúa como una barrera capaz de proteger a las bacterias de los mecanismos de defensa del hospedador y de los antibióticos mientras crean un entorno idóneo para sobrevivir.Otras dos especies más de estafilococos, S. lugdunensis y S. schleiferi, originan infecciones más graves (endocarditis en válvulas originales y osteomielitis) que las producidas por otros estafilococos coagulasanegativos. No se tiene explicación de esta virulencia mayor, aunque las dos especies parecen compartir con S. aureus más determinantes de virulencia (p. ej., el factor de agrupación y la lipasa) que otros estafilococos coagulasanegativos.La capacidad de S. saprophyticus para causar infecciones de vías urinarias en las mujeres jóvenes parece vinculada con una mayor capacidad de adherirse a las células uroepiteliales. Pudiera contribuir a esta afinidad la hemaglutinina-adhesina de 160 kilodaltones.
Diagnóstico

No es difícil detectar estafilococos coagulasanegativos en los focos de infección o en la corriente sanguínea por los métodos de cultivo microbiológicos comunes, pero la interpretación de los resultados suele ser problemática. Se ha calculado que sólo un 10 a 25% de los cultivos de sangre en que se identifican CoNS reflejan una bacteriemia verdadera. Entre los datos clínicos que sugieren una bacteriemia verdadera están la fiebre, los signos de infección local (eritema o drenaje purulento en el sitio del catéter intravenoso), la leucocitosis y los signos sistémicos de sepsis. Entre los resultados de laboratorio que sugieren una bacteriemia verdadera están los múltiples cultivos separados positivos para la misma cepa (es decir, la misma especie con el mismo antibioticograma o con una huella de DNA muy similar), la proliferación de la cepa en el plazo de 48 h y la proliferación de las bacterias en recipientes para aerobios y anaerobios.
Síndromes clínicos
Los CoNS pueden originar infecciones de diversa índole en las prótesis de válvulas cardíacas y articulares, los injertos vasculares, los dispositivos intravasculares y las derivaciones en el sistema nervioso central. Los signos de infección delimitados suelen ser sutiles, la evolución de la enfermedad es lenta y los signos sistémicos suelen ser escasos. A veces se advierten signos manifiestos de infección, como drenaje purulento, dolor en el sitio afectado o aflojamiento de la prótesis implantada. Es frecuente que surja fiebre, pero no siempre aparece y a veces hay leucocitosis leve.
Son poco frecuentes las infecciones no vinculadas con dispositivos protésicos, aunque los estafilococos coagulasanegativos serían responsables de cerca de 5% de las endocarditis de válvulas originales en algunas revisiones. S. lugdunensis parece un patógeno más enérgico en tales circunstancias; origina una mayor mortalidad y una destrucción más rápida de la válvula, con formación de abscesos.
Tratamiento
Principios generalesAdemás de la selección de antimicrobianos apropiados contra las infecciones por estafilococos, se necesita la incisión quirúrgica y el drenaje de todas las acumulaciones de pus. Existen pocas probabilidades de obtener buenos resultados con el tratamiento en las infecciones de prótesis si no se las extrae.
Como el riesgo de complicaciones de la bacteriemia de S. aureus es bien conocido, el tratamiento es normalmente prolongado (cuatro a ocho semanas), salvo si se detecta que el paciente pertenece al pequeño porcentaje de individuos que tienen un riesgo pequeño de complicaciones, como son los enfermos inmunocompetentes y aquéllos cuya infección por S. aureus se vincula con un foco eliminable (como un catéter intravenoso) y cuyo dispositivo se extrae en muy breve plazo.
Duración del tratamiento antimicrobianoSubsisten los debates en cuanto a la duración del tratamiento contra las infecciones bacteriémicas por S. aureus.Entre las manifestaciones que se vinculan con mayor riesgo de bacteriemia complicada están la persistencia de cultivos sanguíneos positivos a las 48 a 96 h de emprender el tratamiento, el contagio de la infección en la comunidad, la falta de retiro de focos eliminables de infección (p. ej.,

catéteres intravasculares) y las manifestaciones cutáneas y embólicas de la infección. En los individuos inmunocompetentes en que se planee un tratamiento de corta duración estaríajustificada la práctica de un ecocardiograma transesofágico para descartar una endocarditis, ya que ni los datos clínicos ni los de laboratorio bastan para corroborar la afección del corazón.
Selección de los antimicrobianosCada vez ha resultado más difícil escoger los antimicrobianos para combatir las infecciones por estafilococos coagulasapositivos y CoNS, ante la prevalencia de las cepas resistentes a múltiples fármacos. Esta tendencia fue mucho más manifiesta en el caso de los CoNS: más de 80% de los aislados en hospitales son resistentes a la meticilina, y estas cepas de MRSA también son normalmente resistentes al resto de los antibióticos en su mayoría. La selección de los agentes antimicrobianos para tratar la infección por S. aureus es semejante a la que se hace en las infecciones por CoNS; por ambas causas, se exponen juntas las opciones terapéuticas contra dichos patógenos.
En caso de sospecha de infección por S. aureus de origen comunitario o nosocomial, se recomienda usar vancomicina, con un aminoglucósido o sin él, ante la mayor frecuencia con que se identifican cepas resistentes a meticilina en la comunidad a Las dosis recomendadas corresponden a adultos con función renal y hepática normal.
La vía de administración es la intravenosa, salvo que se señale otra distinta. La dosis debe ajustarse en los individuos con menor aclaramiento de creatinina.
En el tratamiento de la endocarditis de prótesis valvulares se recomienda agregar gentamicina (1 mg/kg de peso cada 8 h) y rifampicina (300 mg PO cada 8 h), ajustando la dosis del primer antibiótico si disminuye la eliminación de creatinina.
En fecha reciente se ha señalado la aparición de cepas de S. aureus resistentes a vancomicina en las infecciones clínicas.
Nota: TMP-SMX, trimetoprim-sulfametoxazol; VISA, S. aureus con resistencia intermedia a vancomicina; VRSA, S. aureus resistente a vancomicina; mU, millones de unidades; PO, vía oral.
Como los plásmidos que contienen la enzima penicilinasa están ampliamente diseminados, son pocas las cepas de estafilococos (<5%) que siguen siendo sensibles a la penicilina. Sin embargo, contra estas cepas sensibles, la penicilina sigue siendo el fármaco preferente. Los aislados resistentes a penicilina se tratan con penicilinas semisintéticas resistentes a la penicilinasa (semisynthetic penicillinase-resistant penicillins, SPRP), como la oxacilina o la nafcilina. En la actualidad se usa poco la meticilina, que fue la primera de las SPRP. Otros agentes terapéuticos a los que cabe recurrir en estas infecciones son las cefalosporinas; las de segunda y tercera generación no brindan ventaja terapéutica alguna en comparación con las de primera generación en el tratamiento de las infecciones por estafilococos. El carbapenem imipenem posee una actividad excelente contra el S. aureus sensible a meticilina (methicillin-sensitive S. aureus, MSSA), pero no contra el S. aureus resistente a meticilina.En muchos hospitales, 40 a 50% de los aislados de S. aureus son resistentes a meticilina, y este fenómeno denota resistencia a todos las SPRP y también a todas las cefalosporinas. Muchos aislados de MRSA también son resistentes a otras familias de antimicrobianos, como los aminoglucósidos, las quinolonas y los macrólidos.

La resistencia a la meticilina se explica por la aparición de una nueva proteína de unión a la penicilina ([penicillin-binding protein, PBP]) esta proteína es sintetizada por el gen mecA, que forma parte de un gran elemento genético móvil (una isla de patogenicidad o genómica), el llamado cromosoma en casete estafilocócico (SCCmec). La expresión fenotípica de la resistencia a la meticilina puede ser constitutiva (expresada en todos los microorganismos de una población) o heterogénea (expresada por una parte del total de la población de microorganismos). Además de las técnicas de PCR, recientemente se han creado métodos rápidos para detectar la resistencia a la meticilina.
La vancomicina es el fármaco preferente para tratar las infecciones por estafilococos resistentes a meticilina. Como su acción bactericida es menor que la de los betalactámicos sólo debe utilizarse después de un análisis cuidadoso y en sujetos con antecedentes de alergia a estos últimos antibióticos. No se ha identificado el mecanismo de resistencia propio de los VISA, pero parece consistir en una anomalía de la pared celular, lo que se observó por primera vez mediante microscopia electrónica. La vancomicina queda atrapada en los enlaces cruzados anormales entre peptidoglucanos y no puede llegar al sitio donde actúa normalmente. En el año 2002 se publicó la identificación del primer aislado clínico de S. aureus totalmente resistente a la vancomicina. La resistencia de este y de otro aislado clínico posterior se debía a la presencia de vanA, el gen encargado de la expresión de la resistencia a vancomicina en los enterococos. Los enfermospresentaban MRSA y también enterococos resistentes a vancomicina en los cultivos de material obtenido de los focos de infección. Los microorganismos siguieron siendo sensibles a fármacos como el cloranfenicol, el linezolid, la minociclina, la quinupristina-dalfopristina y el trimetoprim sulfametoxazol (TMP-SMX).
Otros fármacos que pueden utilizarse en el lugar de los betalactámicos y la vancomicina tienen menor actividad antiestafilocócica. Las quinolonas muestran una actividad in vitro razonable contra dichos microorganismos, pero ha aumentado poco a poco la frecuencia con que surge la resistencia a las fluoroquinolonas, en particular entre los microorganismos resistentes a la meticilina. Los estafilococos sensibles a meticilina han seguido siendo más sensibles a las fluoroquinolonas que las cepas resistentes a dicho antibiótico. Las nuevas quinolonas tienen una mayor actividad in vitro contra los estafilococos, pero no se sabe si ello se podría traducir en una intensificación de la actividad in vivo.
Se han utilizado con buenos resultados otros antibióticos, como la minociclina y el TMP-SMX, para combatir las infecciones por estafilococos resistentes a meticilina en los casos de toxicidad o intolerancia a la vancomicina.Entre los nuevos agentes antiestafilocócicos la estreptogramina parenteral quinupristinadalfopristina presenta actividad bactericida frente a todos los estafilococos, incluidas las cepas VISA. Dicho fármaco se ha utilizado con buenos resultados para combatir infecciones graves por MRSA. En los casos de resistencia a la eritromicina o la clindamicina, tiene efecto bacteriostático contra los estafilococos.El linezolid (el primer miembro de una nueva familia de fármacos, las oxazolidinonas) es bacteriostático frente a los estafilococos, se tolera bien y tiene la ventaja de que su biodisponibilidad es similar después de su administración oral o parenteral. Los casos de resistencia a dicho fármaco han sido escasos, aunque se ha notificado, como mínimo, una cepa clínica resistente.

No se ha determinado la eficacia del linezolid en el tratamiento de las infecciones profundas, como la osteomielitis. No hay datos suficientes sobre la eficacia de la quinupristina-dalfopristina y el linezolid en el tratamiento de la endocarditis infecciosa. La daptomicina, un nuevo agente bactericida por vía parenteral con actividad antiestafilocócica. Este fármaco rompe la membrana citoplásmica. Está siendo sometida a ensayos clínicos la oritavancina, que es un nuevo glucopéptido.A veces se utilizan combinaciones de antiestafilocócicos para intensificar la actividad bactericida en el tratamiento de infecciones como la endocarditis o la osteomielitis. En ciertos casos (como la endocarditis de la mitad derecha del corazón) se utilizan combinaciones medicamentosas para acortar la duración del tratamiento. Una de ellas es las de rifampicina, aminoglucósidos (como gentamicina) y ácido fusídico (no se encuentra fácilmente en Estados Unidos).Los estudios in vitro han demostrado que las siguientes combinaciones presentan sinergismo contra los estafilococos: 1) betalactámicos y aminoglucósidos; 2) vancomicina y gentamicina; 3) vancomicina, gentamicina y rifampicina (contra los CoNS), y 4) vancomicina y rifampicina. En varios casos, estas observaciones in vitro se han visto respaldadas por estudios que han utilizado modelos experimentales de endocarditis en animales.
Antimicrobianos en situaciones especialesEn las infecciones cutáneas y de partes blandas no complicadas, se obtienen buenos resultados con los agentes antiestafilocócicos orales. En otras infecciones conviene el tratamiento parenteral.La endocarditis por S. aureus suele ser una infección aguda y potencialmente fatal. Por tal razón, es necesario realizar hemocultivos lo antes posible y emprender de inmediato un tratamiento antimicrobiano empírico. En el caso de la endocarditis valvular por S. aureus, suele utilizarse una combinación de antimicrobianos. Como consecuencia, muchos clínicos comienzan el tratamiento de las infeccionespotencialmente mortales con un betalactámico y un aminoglucósido (gentamicina, 1 mg/kg por vía intravenosa cada 8 h) durante tres a cinco días. Si se aísla una cepa de MRSA, habrá que administrar vancomicina, 30 mg/kg cada 24 h en dos dosis iguales, hasta alcanzar un total de 2 g.Los pacientes reciben tratamiento durante cuatro a seis semanas, según su respuesta clínica.En la endocarditis de prótesis valvulares suele ser necesaria una operación, además de la antibioticoterapia. Se recomienda combinar un agente betalactámico (o en caso de resistencia del microorganismo, vancomicina, 30 mg/kg cada 24 h, en dos dosis iguales, hasta alcanzar un total de 2 g) con un aminoglucósido (gentamicina, 1 mg/kg por vía intravenosa cada 8 h) y rifampicina oral,300 mg cada 8 h. Esta combinación se utiliza para que no surja una posible resistencia a la rifampicina durante el tratamiento si se usan sólo dos fármacos.
En caso de osteomielitis hematógena o artritis séptica en los niños, por lo común se obtienen resultados satisfactorios con un ciclo de cuatro semanas de tratamiento. En los adultos, este ciclo suele ser más largo. En las formas crónicas de osteomielitis se necesita un desbridamiento quirúrgico en combinación con los antimicrobianos. En las infecciones articulares, un componente crítico de tratamiento es la aspiración repetida o la artroscopia de la articulación afectada, para evitar el daño causado por los leucocitos. Se ha utilizado con buenos resultados la combinación de rifampicina y ciprofloxacina, en particular para tratar las infecciones de las prótesis articulares y cuando resulta imposible extraer el dispositivo. Cada vez más, la vancomicina (en combinación con un aminoglucósido o rifampicina contra las infecciones graves) es el fármaco preferente para las infecciones de origen tanto comunitario como nosocomial.

Tratamiento del síndrome de choque tóxicoEl elemento básico del tratamiento del TSS son las medidas de sostén, para revertir la hipotensión. Se necesitan líquidos y vasopresores. Los tampones y demás materiales de taponamiento deben extraerse a corto plazo. Algunos investigadores recomiendan combinar la clindamicina con una penicilina semisintética. La primera se recomienda porque, al ser inhibidora de la síntesis de proteínas, aminora la síntesis de toxinas in vitro. Se sugiere el uso de una penicilina semisintética para eliminar cualquier foco posible de infección y para erradicar el estado de portador, que podría aumentar la probabilidad de una recidiva. Informes aislados corroboran el empleo provechoso de la inmunoglobulina intravenosa para combatir el TSS.
Tratamiento de otras enfermedades mediadas por toxinasEl tratamiento de la intoxicación alimentaria por estafilococos es totalmente de apoyo. En el SSSS, el tratamiento antiestafilocócico se orienta hacia el foco primario de infección.

SHOCK SEPTICO
Los signos cardinales de la respuesta sistémica son fiebre o hipotermia, leucocitosis o leucopenia, taquipnea y taquicardia, llamados síndrome de respuesta inflamatoria, SIRS). Puede tener causas infecciosas o no infecciosas. Si se sospecha infección, se dice que la persona con SIRS tiene septicemia. Si la septicemia se acompaña de disfunción de órganos distantes al sitio de la infección, la persona tiene septicemia grave que puede acompañarse de hipotensión o manifestaciones de deficiencia circulatoria.
Cuando es imposible corregir la hipotensión por la administración de soluciones, la entidad recibe el nombre de choque séptico. La septicemia por lo común es reversible en tanto que las personas en choque séptico suelen morir a pesar del tratamiento intensivo.
Definiciones utilizadas para describir el estado de los pacientes sépticos…
- Bacteriemia Presencia de bacterias en la sangre como lo manifiestan los hemocultivos positivos
- Septicemia Presencia de microbios o sus toxinas en la sangre- Síndrome de respuesta inflamatoria sistémica (SIRS)
Dos o más de las manifestaciones siguientes:1) fiebre (temperatura en la boca >38°C) o hipotermia (<36°C); 2) taquipnea (más de 24 rpm); 3) taquicardia (frecuencia cardíaca mayor de 90 lpm) 4) leucocitosis (>12 000 leucocitos/ l), leucopenia (<4 000 leucocitos/ l) o más de 10% de formas
en banda; pudiera tener un origen no infeccioso- Septicemia SIRS puede tener un origen microbiano probado o sospechado- Septicemia grave (similar al "síndrome de septicemia") Septicemia con uno o más signos de
disfunción de órganos; por ejemplo:1. Aparato cardiovascular: tensión sistólica arterial 90 mmHg o tensión arterial media 70 mmHg.
que mejora con la administración de soluciones intravenosas.2. Renal: diuresis <0.5 ml/kg/h durante 1 h a pesar de fluidoterapia adecuada.3. Aparato respiratorio: PaO2/FIO2 250 o si el pulmón es el único órgano con disfunción, 200.4. Sangre: recuento plaquetario <80 000 trombocitos/ l o disminución del 50% en el número de
plaquetas, en relación con la cifra más alta cuantificada en los tres días anteriores.5. Acidosis metabólica no explicada: pH 7.30 o déficit alcalino 5.0 meq/L y concentración de
lactato en plasma >1.5 veces el límite superior de lo normal, respecto al laboratorio que hace el estudio.
6. Fluidoterapia adecuada: presión capilar pulmonar de enclavamiento 12 mmHg o presión venosa central 8 mmHg.
- Choque séptico Septicemia con hipotensión (tensión arterial <90 mmHg sistólica, o 40 mmHg menor que la tensión normal del paciente) durante 1 h como mínimo, a pesar de fluidoterapia adecuada; o Necesidad de vasopresores para conservar la tensión sistólica 90 mmHg o la presión arterial media 70 mmHg
- Choque séptico resistente al tratamiento Choque séptico que dura más de 1 h y que no mejora con administración de soluciones ni presores

- Síndrome de insuficiencia multiorgánica Disfunción de varios órganos que obliga a intervención para conservar la homeostasia
FACTORES ETIOLÓGICOS
La septicemia grave puede ser una reacción a cualquier tipo de microorganismos. - No es esencial para que surja dicha forma de septicemia la invasión de la corriente sanguínea
por microbios, porque la inflamación local también desencadena disfunción de órganos distantes e hipotensión.
- Los cultivos de sangre se identifican bacterias u hongos, sólo en 20 a 40%, aproximadamente, de los individuos con septicemia grave y 40 a 70% de los casos de choque séptico.
- Las bacterias grampositivas o gramnegativas representan 70% de los microorganismos aislados.
- En los pacientes con hemocultivos negativos, el agente etiológico suele identificarse de un foco local.
- En sujetos con positividad de los cultivos de sangre el peligro de que surja septicemia grave es mayor en personas que tienen más de 50 años de edad y en los que el sitio primario de infección se localiza en pulmones, órganos abdominales o sistema nervioso y meninges.
Microorganismos identificados en episodios de septicemia grave - Bacterias gramnegativas: Enterobacterias, pseudomonas, Haemophilus sp., entre otras.- Bacterias grampositivas: Staphylococcus aureus, estafilococos coagulasa negativos,
enterococos, Streptococcus pneumoniae, entre otras.- Hongos - Infección polimicrobiana - Patógenos clásicos: Neisseria meningitidis, S. pneumoniae, H. influenzae y Streptococcus
pyogenes
Cuadros patológicos predisponen a la aparición de infecciones con positividad en los hemocultivos- BACILOS GRAMNEGATIVOS : Diabetes Mellitus, Enfermedades linfoproliferativas, Cirrosis
hepática, Quemaduras, métodos o dispositivos cruentos, neutropenia, sonda apermanencia en vejiga, diverticulitis o perforación de víscera hueca.
- BACTERIAS GRAMPOSITIVAS : Catéteres intravasculares, dispositivos mecánicos a resistencia, quemaduras, neutropenia, uso de fármacos intravenosos, infección Streptococcus pyogenes productor de superantigeno, hongos, neutropenia y empleo de antimicrobianos de amplio espectro.
EPIDEMIOLOGÍA- Septicemia es causa en Estados Unidos más de 200 000 muertes anuales. - La incidencia de septicemia grave y choque séptico ha aumentado a >300 000 casos cada año.- Más o menos dos terceras partes de los casos se producen en enfermos hospitalizados por
otras enfermedades. - La incidencia creciente es atribuible al envejecimiento de la población, a la mayor longevidad
de los pacientes con enfermedades crónicas y en pacientes que padecen SIDA, uso

generalizado de agentes antimicrobianos, glucocorticoides, catéteres permanentes y dispositivos mecánicos, y la ventilación artificial.
ASPECTOS FISIOPATOLÓGICOSCasi todos los casos de septicemia grave son originados por bacterias u hongos que en circunstancias corrientes no causan enfermedad sistémica en hospedadores inmunocompetentes. A diferencia de ello, los microbios patógenos pueden "esquivar" las defensas innatas, al elaborar toxinas u otros factores de virulencia. En ambos casos, el cuerpo no destruye los invasores a pesar de desencadenar una reacción inflamatoria intensa que a veces llega a la septicemia grave. La respuesta séptica también puede ser inducida por exotoxinas microbianas que actúan como superantígenos (toxina 1 del síndrome de choque séptico).
MECANISMOS DEL HOSPEDADOR PARA CAPTAR LA PRESENCIA DE MICROORGANISMOS
Poseemos mecanismos de sensibilidad para identificar y reaccionar a moléculas microbianas conservadas. Ejem: La fracción del lípido A de lipopolisacárido (LPS, también conocida como endotoxina;). El lípido A es el centro bioactivo de los LPS de todas las bacterias gramnegativas en la naturaleza. Una proteína del hospedador (proteína que se fija a LPS se liga al lípido A y transfiere LPS a CD14 en las superficies de monocitos, macrófagos y neutrófilos.) En tales sitios LPS y CD14 interactúan con un receptor similar a toll 4 y MD-2, para formar un complejo molecular que transduce la señal LPS al interior de la célula. La señal en cuestión desencadena rápidamente la producción y la liberación de mediadores como el factor de necrosis tumoral (TNF), que amplifican la señal de LPS y la transmiten a otras células y tejidos. El peptidoglucano bacteriano, los ácidos lipoteicoicos, DNA, algunos polisacáridos y los pelos (fimbrias) desencadenan respuestas, semejantes a las inducidas por LPS; en tanto que algunas de las moléculas mencionadas también se ligan a CD14, e interactúan con TLR diferentes.
En consecuencia, CD14 atrae a innumerables moléculas "no autóctonas" a las superficies de las células, lo cual intensifica enormemente la sensibilidad con la que dichas moléculas son reconocidas por el hospedador. Poseer innumerables complejos de receptor basados en TLR permite a las células reconocer innumerables moléculas microbianas conservadas. Otras proteínas de reconocimiento de perfiles moleculares importantes para "percibir" la infección microbiana incluyen el complemento (en particular la vía alternativa), la lectina que se liga a manosa, la proteína que incrementa la permeabilidad bactericida y la proteína C reactiva.
RESPUESTAS LOCALES Y SISTÉMICAS DEL HOSPEDADOR A LOS MICROORGANISMOS INVASORES
El reconocimiento de las moléculas microbianas por parte de los fagocitos hísticos desencadena la producción, liberación o ambos fenómenos, de innumerables moléculas del hospedador, lo que incrementa la corriente sanguínea al tejido infectado, la permeabilidad de los vasos locales, reclutamiento de neutrófilos al sitio de la infección y aparición de dolor. Todos conocidos elementos de la inflamación local, que constituyen el mecanismo inmunitario innato principal que posee el organismo para eliminar a los microbios invasores. Las respuestas sistémicas pueden ser activadas por la comunicación nerviosa, humoral o de ambos tipos, con el hipotálamo y el tallo encefálico.

CITOCINAS Y OTROS MEDIADORES
Las citocinas ejercen efectos endocrinos, paracrinos y autocrinos. TNF- estimula a los leucocitos y las células del endotelio vascular para liberar otras citocinas (y también más TNF-), para expresar moléculas de superficie celular, que incrementan la adherencia de neutrófilos al endotelio en los sitios de infección y también aumentan la producción de prostaglandinas y leucotrienos. Las concentraciones sanguíneas de TNF- aumentan sujetos con septicemia grave o choque séptico. La administración intravenosa de TNF- desencadena muchas de las anormalidades característicasde la septicemia, que incluyen fiebre, taquicardia, taquipnea, leucocitosis, mialgias y somnolencia.
TNF- es un mediador con actividad central, pero es solamente una de las muchas moléculas proinflamatorias que contribuyen a las defensas innatas del hospedador. Las quimiocinas, predominantemente la interleucina (IL) 8, atrae neutrófilos circulantes al sitio de la infección. IL-1 posee muchas de las mismas actividades que TNF- . Es probable que actúen sinérgicamente TNF- , IL-1,interferón gamma, IL-12 y otras citocinas entre sí y con otros mediadores. Pruebas recientes sugieren que las citocinas proinflamatorias que circulan en la sangre suelen provenir del sitio local inflamado.
FACTORES DE COAGULACIÓN
La trombosis intravascular, signo característico de la respuesta inflamatoria local, permite "contener" a los microorganismos invasores y evitar que la infección e Inflamación se propaguen a otros tejidos. El depósito intravascular de fibrina, trombosis y DIC son características importantes de la respuesta sistémica.
La IL 6 y otros mediadores estimulan la coagulación intravascular induciendo inicialmente la expresión del factor hístico por los monocitos sanguíneos. Cuando estas células expresan el factor hístico, éste se une al factor VIIa para formar un complejo activo que puede convertir los factores IX y X en sus formas enzimáticamente activas. El resultado es la activación de las vías de la coagulación, tanto intrínseca como extrínseca, lo cual culmina en formación de fibrina. La alteración funcional de la vía inhibidora de la proteína C-proteína S y el agotamiento de antitrombina y proteína C también favorecen la coagulación, en tanto que la elevación de las concentraciones plasmáticas de inhibidor 1 del activador del plasminógeno previene la fibrinólisis. Por tanto, existe una llamativa tendencia al depósito intravascular de fibrina, a la trombosis y a la hemorragia.
MECANISMOS DE SUPRESIÓN
Las pruebas disponibles sugieren que la respuesta sistémica del organismo a la lesión y la infección, normalmente impiden que surja inflamación en órganos que están alejados del sitio de la infección.

El aparato de señales que vincula el reconocimiento microbiano con las respuestas celulares disminuye enormemente en la sangre. A pesar de que LBP interviene en el reconocimiento de la presencia de LPS. Las concentraciones hemáticas de LBP y CD14 soluble aumentan durante la respuesta a la infección, y mejoran la capacidad del organismo para evitar que se activen las células sanguíneas, por acción de lipopolisacáridos. Las respuestas sistémicas a la infección también reducen las respuestas celulares a las moléculas microbianas.
DISFUNCIÓN DE ÓRGANOS Y CHOQUE
A pesar de que se observan moléculas proinflamatorias y antiinflamatorias en grandes concentraciones, el "balance" neto de mediadores en el plasma en estos enfermos extraordinariamente graves, en realidad se podría orientar hacia las moléculas antiinflamatorias. Además, los leucocitos de individuos con septicemia grave suelen mostrar respuesta deficiente a los agonistas como LPS. No se sabe la forma en que las fuerzas proinflamatorias y antiinflamatorias contribuyen a lahipotensión y a la disfunción de órganos que están en puntos distantes respectoal sitio de la infección.
LESIÓN ENDOTELIAL
La lesión amplísima del endotelio vascular constituye el mecanismo principal de disfunción de múltiples órganos. Existe un gran número de células del endotelio vascular en la sangre periférica de enfermos sépticos. Los mediadores de origen leucocítico y los trombos de plaquetas-leucocitos-fibrina pueden contribuir a la lesión vascular, pero al parecer también interviene de modo activo el endotelio vascular. Estímulos como TNF- inducen a las células del endotelio vascular a producir y liberar citocinas, moléculas procoagulantes, factor activador plaquetario (platelet-activating factor, PAF), óxido nítrico y otros mediadores. Además, las moléculas de adherencia celular reguladas estimulan la adherencia de neutrófilos a células del endotelio. Las respuestas mencionadas atraen fagocitos a los sitios infectados y activan sus arsenales antimicrobianos, pero la activación de las células endoteliales también incrementa la permeabilidad vascular, trombosis microvascular, DIC e hipotensión. CHOQUE SÉPTICO
El signo característico del choque séptico es la disminución de la resistencia vascular periférica que surge a pesar de que haya mayores concentraciones de catecolaminas vasopresoras. El gasto cardíaco aumenta y también la corriente sanguínea a tejidos periféricos. La utilización de oxígeno por parte de los tejidos puede disminuir notablemente, por la distribución desigual de la corriente sanguínea y también por la disfunción microcirculatoria.
Entre las moléculas notables que generan hipotensión están el óxido nítrico, betaendorfina, bradicinina, PAF y prostaciclina. Los agentes que inhiben la síntesis o la acción de cada uno de los mediadores mencionados evitan o revierten el choque endotóxico. Los sujetos en choque séptico suelen mostrar disminución de las respuestas vasoconstrictoras a las catecolaminas. Tal pérdida de la sensibilidad se ha atribuido a la regulación descendente de los receptores adrenérgicos y a un incremento en las concentraciones de óxido nítrico. En personas en choque séptico las concentraciones plasmáticas de vasopresina no aumentan para intensificar la vasoconstricción; en algunas series pequeñas de individuos en choque séptico, la administración intravenosa de

vasopresina incrementó la tensión arterial y disminuyó la necesidad de catecolaminas en goteo intravenoso.
Septicemia grave: ¿un solo mecanismo patógeno?
Las bacterias circulantes y sus productos desencadenan la disfunción de múltiples órganos e hipotensión, al estimular de manera directa respuestas inflamatorias dentro del árbol vascular.
En personas con meningococemia fulminante, por ejemplo, la mortalidad guardó relaciónprecisa con las concentraciones de endotoxinas en sangre y con la aparición de CID. A diferencia de ello, en muchos enfermos de infecciones nosocomiales el número de bacterias o moléculas bacterianas circulantes puede reflejar infección incontrolable en el tejido local y pudiera tener escaso o nulo impacto directo en órganos distantes; en tales enfermos, los mediadores inflamatorios que provienen del sitio local al parecer son los elementos básicos que desencadenan la septicemia grave y el choque séptico. Los datos que apoyan este concepto se han obtenido de un estudio en que se indujo la bacteriemia por Pseudomonas aeruginosa en conejos, por goteo intravenoso constante de bacterias o por inoculación endotraqueal hasta producir neumonía. Solamente aquellos con neumonía terminaron por mostrar choque séptico. El tercer mecanismo fisiopatológico pudiera ser representado por la septicemia grave causada por Staphylococcus aureus o Streptococcus pyogenes, que producen superantígenos, dado que la activación de linfocitos T inducida por tales toxinas, genera un perfil de citocinas que difiere sustancialmente del desencadenado en la infección por bacterias gramnegativas.
En resumen, la patogenia de la septicemia grave puede diferir con base en el microorganismo infectante, sitio de la infección primaria, presencia o ausencia de defectos inmunitarios y estado funcional previo del hospedador. También pueden ser importantes los factores genéticos.
MANIFESTACIONES CLÍNICAS
Las manifestaciones de la respuesta séptica suelen superponerse a los síntomas ysignos de la enfermedad subyacente y de la infección primaria del paciente. La hiperventilación a menudo es un signo precoz. La desorientación, confusión y otras manifestaciones de encefalopatía también pueden aparecer de manera temprana en la respuesta séptica, especialmente en ancianos y en personas con alteraciones neurológicas previas. Los signos neurológicos focales son raros, pero los déficit focales preexistentes pueden agravarse.
La hipotensión y DIC predisponen al desarrollo de acrocianosis y necrosis isquémica de los tejidos periféricos, sobre todo de los dedos. Cuando las bacterias o los hongos se diseminan por la piel o por los tejidos blandos subyacentes por vía hematógena, pueden aparecer celulitis, pústulas, ampollas o lesiones hemorrágicas. Las toxinas bacterianas también pueden diseminarse por vía hematógena y desencadenar reacciones cutáneas difusas. En ocasiones, las lesiones cutáneas hacen sospechar la presencia de determinados agentes patógenos. Por ejemplo, cuando la septicemia va acompañada de púrpura o petequias cutáneas, debe sospecharse una infección por Neisseria meningitidis(o, con menos frecuencia, por Haemophilus influenzae); cuando un paciente ha sido picado por una garrapata en una zona endémica, las lesiones petequiales deben hacer pensar también en la fiebre moteada de las Montañas Rocosas.

Una lesión cutánea que aparece casi exclusivamente en pacientes neutropénicos es la ectima gangrenosa, causada por lo regular por Pseudomonas aeruginosa; se trata de una lesión ampollosa con un halo edematoso y una zona central de hemorragia y necrosis. Las lesiones hemorrágicas o ampollosas en un paciente con septicemia que ha consumido recientemente ostras crudas deben hacer sospechar una bacteriemia por Vibrio vulnificus, mientras que en un paciente que ha sido mordido por un perro, esas mismas lesiones pueden indicar una infección sanguínea por Capnocytophaga canimorsus o por C. cynodegmi. Una eritrodermia generalizada en un paciente con septicemia debe hacer sospechar un síndrome de choque tóxico debido a Staphylococcus aureus o a Streptococcus pyogenes.Las manifestaciones gastrointestinales como náuseas, vómitos, diarrea e íleo hacen sospechar una gastroenteritis aguda.
Las úlceras de estrés pueden provocar una hemorragia digestiva alta. A veces, la ictericia colestásica, con concentraciones elevadas de bilirrubina sérica (conjugada en su mayoría) y de fosfatasa alcalina, aparece antes que otros signos de septicemia. Al parecer en la mayor parte de los casos existen alteraciones funcionales hepatocelulares o canaliculares subyacentes, y las pruebas funcionales hepáticas vuelven a normalizarse tras la curación de la infección. La hipotensión intensa o prolongada puede provocar una lesión hepática aguda o necrosis isquémica del intestino.Las concentraciones de lactato en sangre ascienden precozmente, en parte por el aumento de la glucólisis en los tejidos periféricos asociado a disminución del aclaramiento hepático y renal del lactato y piruvato resultantes. A medida que disminuye el riego sanguíneo de los tejidos, la hipoxia hística genera más ácido láctico, contribuyendo al desarrollo de acidosis metabólica. La glucemia a menudo aumenta, especialmente en pacientes diabéticos, aunque el deterioro de la gluconeogénesis y liberación excesiva de insulina pueden, en ocasiones, producir hipoglucemia.
COMPLICACIONES PRINCIPALES
COMPLICACIONES CARDIOPULMONARESEl desequilibrio ventilación-perfusión produce un descenso de la PO2 arterial al comienzo de la evolución del proceso. El aumento de la permeabilidad capilar alveolar da lugar a un aumento del contenido de agua de los pulmones, lo cual disminuye la distensibilidad pulmonar y dificulta el intercambio de oxígeno. La aparición progresiva de infiltrados pulmonares difusos y la hipoxemia en sangre arterial (PaO2 /FIO2 <200) indican el desarrollo de un síndrome apneico agudo (acute respiratory distress syndrome, ARDS).
Este síndrome aparece en cerca del 50% de los pacientes con septicemia grave o choque séptico. La insuficiencia de los músculos respiratorios puede agravar la hipoxemia y la hipercapnia. Una elevación de la presión capilar pulmonar de enclavamiento (>18 mmHg) sugiere una sobrecarga de volumen o insuficiencia cardíaca y no ARDS. Las neumonías por virus o por Pneumocystis carinii pueden ser indistinguibles, desde el punto devista clínico, del síndrome apneico agudo.La hipotensión inducida por septicemia suele ser ocasionada por mala distribución generalizada del riego sanguíneo y del volumen de sangre, y por hipovolemia causada, en parte, por la extravasación capilar difusa del líquido intravascular. Otros factores que pueden reducir el volumen intravascular efectivo son la deshidratación secundaria a una enfermedad previa o a pérdidas insensibles de líquidos, vómitos o diarrea, y poliuria.

Durante las primeras fases del choque séptico la resistencia vascular sistémica suele estar elevada y el gasto cardíaco puede estar disminuido. El deterioro de la función miocárdica, que se manifiesta por aumento de los volúmenes telediastólico y sistólico ventriculares con descenso de la fracción de expulsión, aparece durante las primeras 24 h en la mayoría de los pacientes con septicemia grave.
COMPLICACIONES RENALESSon frecuentes la oliguria, hiperazoemia, proteinuria y cilindros urinarios inespecíficos. Muchos pacientes presentan una poliuria inadecuada; la hiperglucemia puede acentuar esta tendencia. La insuficiencia renal se debe en gran parte a la necrosis tubular aguda provocada por la hipotensión o la lesión capilar, aunque algunos pacientes sufren también glomerulonefritis, necrosis cortical renal o nefritis intersticial.
COAGULACIÓNAunque en 10 a 30% de los enfermos existe trombocitopenia, no se conocen bien los mecanismos subyacentes. En pacientes con DIC suelen encontrarse cifras muy bajas de plaquetas (menos de 50 000/ l), lo que habitualmente refleja una lesión endotelial difusa o trombosis microvascular.
COMPLICACIONES NEUROLÓGICAS Cuando la enfermedad séptica se prolonga a lo largo de semanas o meses, la polineuropatía "de la enfermedad crítica" puede impedir la retirada del respirador y producir una debilidad motora distal. Los estudios electrofisiológicos son diagnósticos. Hay que descartar el síndrome de Guillain-Barré, trastornos metabólicos y actividad de toxinas.DATOS DE LABORATORIOEn las primeras fases de la respuesta séptica suelen encontrarse las siguientes alteraciones:
- leucocitosis con desviación a la izquierda- trombocitopenia- hiperbilirrubinemia- proteinuria.
A veces hay leucopenia. Los neutrófilos pueden contener gránulos tóxicos, cuerpos de Döhle o vacuolas citoplásmicas. Al comienzo de la septicemia, la hiperventilación provoca alcalosis respiratoria. Más adelante, junto al cansancio de la musculatura respiratoria y acumulación de lactato, normalmente sobreviene una acidosis metabólica (con aumento de la brecha aniónica). La gasometría en sangre arterial revela hipoxemia, corregible inicialmente con la administración de oxígeno pero más tarde resistente al tratamiento aunque se administre oxígeno al 100%, lo que indica la formación de un cortocircuito de derecha a izquierda. La radiografía de tórax puede ser normal o mostrar neumonía subyacente, sobrecarga de volumen o infiltrados difusos de ARDS. El electrocardiograma puede mostrar únicamente una taquicardia sinusal ociertas alteraciones inespecíficas del segmento ST o de la onda T. La mayoría de los pacientes diabéticos con septicemia presenta hiperglucemia. La infección grave puede precipitar cetoacidosis diabética, que empeora la hipotensión (cap. 323). Rara vez hay hipoglucemia. La concentración de albúmina sérica, en límites normales al principio, desciende a medida que avanza la septicemia. La concentración de lípidos séricos suele elevarse, mientras que la hipocalcemia es rara.
DIAGNÓSTICO

No existen pruebas específicas para la respuesta a la septicemia. Los hallazgos de significado diagnóstico en un paciente en el que se sospecha o se ha demostrado una infección consisten en fiebre o hipotermia, taquipnea, taquicardia y leucocitosis o leucopenia; otros datos que tambiénsugieren el diagnóstico son la alteración aguda del estado mental, trombocitopenia o hipotensión.
Las etiologías no infecciosas del SIRS son pancreatitis, quemaduras, traumatismos, insuficiencia suprarrenal, embolia pulmonar, aneurisma aórtico disecante o roto, infarto de miocardio, hemorragia oculta, taponamiento cardíaco, síndrome posderivación cardiopulmonar, anafilaxis y sobredosis de fármacos o drogas.
El diagnóstico etiológico definitivo requiere el aislamiento del microorganismo en la sangre o en un foco infeccioso local. Para los cultivos deben extraerse al menos dos muestras de sangre (10 ml cada una), procedentes de punciones venosas realizadas en lugares distintos.
TRATAMIENTO
El tratamiento debe ser enérgico si se sospecha septicemia. Esta tarea es llevada a cabo de manera ideal en una unidad de cuidados intensivos por personal con experiencia en el cuidado de pacientes gravemente enfermos. Para que el tratamiento tenga éxito es preciso tomar medidas urgentes dirigidas a combatir la infección del foco local, proporcionar soporte hemodinámico y respiratorio, y erradicar el microorganismo causal. El pronóstico también depende de la enfermedad subyacente, que debe ser tratada enérgicamente.
AGENTES ANTIMICROBIANOSLa elección inicial de los antibióticos debe basarse en el conocimiento de los microorganismospatógenos más probables según su localización. También deben tenerse en cuenta los patrones de sensibilidad antimicrobiana de las bacterias aisladas en la población, en el hospital y en el paciente. Es importante instaurar un tratamiento antimicrobiano empírico que sea eficaz contra bacterias grampositivas y gramnegativas mientras se esperan los resultados de los cultivos.
Deben administrarse las dosis máximas recomendadas de los fármacos antimicrobianos por vía intravenosa, realizando, cuando sea preciso, los ajustes correspondientes en caso de insuficiencia renal.Casi todos los pacientes necesitan tratamiento antimicrobiano durante al menos una semana; la duración del tratamiento depende de factores como el lugar de la infección, idoneidad del drenaje quirúrgico, enfermedad subyacente del paciente y sensibilidad a los antimicrobianos de las bacterias aisladas.
ELIMINACIÓN DEL FOCO DE INFECCIÓN PRIMARIOEs esencial eliminar o drenar el foco de infección primario. Hay que retirar los catéteres intravenosos permanentes, introducir la punta en una placa de agar-sangre para realizar un cultivo cuantitativo y colocar a continuación un nuevo catéter en un lugar distinto. Deben retirarse las sondas de Foley y catéteres de drenaje. Hay que tener en cuenta la posibilidad de una sinusitis paranasal (causada con frecuencia por bacterias gramnegativas) en cualquier paciente que haya sido sometido a una intubación nasal. En sujetos con normalidades extensas en las radiografías de tórax, la tomografía computadorizada (computed tomography, CT) del tórax puede identificar ataque no sospechado del parénquima, mediastino o pleura.

En pacientes neutropénicos deben buscarse regiones de piel que presenten hiperalgesia y eritema,especialmente en la región perianal. En pacientes con úlceras de decúbito sacras o isquiáticas debe excluirse la posibilidad de acumulaciones de pus en la pelvis o en otros tejidos blandos utilizando si es preciso la tomografía computadorizada o la resonancia magnética.
En pacientes con septicemia grave procedente de las vías urinarias se recurrirá a la ecografía o a tomografía computadorizada para descartar una obstrucción ureteral y abscesos perirrenales o renales
SOPORTE HEMODINÁMICO, RESPIRATORIO Y METABÓLICO
El objetivo fundamental consiste en restablecer un aporte adecuado de oxígeno y de sustratos a los tejidos. Es esencial mantener una perfusión adecuada de los órganos. La reducción del volumen intravascular efectivo es frecuente en pacientes con septicemia; el tratamiento inicial de la hipotensión debe incluir la administración de líquidos intravenosos, normalmente 1 a 2 L de solución salina normal en 1 a 2 h. Para evitar el edema pulmonar, la presión capilar pulmonar de enclavamiento debe mantenerse entre 12 y 16 mmHg, o bien la presión venosa central permanecer entre 8 y 12 cmH2O. La diuresis debe mantenerse superior a 0.5 ml/kg/h, para lo cual se administran líquidos de forma continua; en caso necesario puede ser útil añadir un diurético del tipo de la furosemida.
En alrededor de la tercera parte de los pacientes la hipotensión y la hipoperfusión orgánica responden a la reposición de líquidos; un objetivo razonable consiste en mantener una presión arterial media superior a 65 mmHg (presión sistólica >90 mmHg) y un índice cardíaco igual o superior a 4 L/min/m2.
Si no es posible mantener estos parámetros mediante el aporte de volumen, está indicado el tratamiento inotrópico y vasopresor. El estado circulatorio también se valora mediante parámetros clínicos (estado mental, diuresis, perfusión cutánea) y, siempre que sea posible, mediante determinaciones del aporte y del consumo de oxígeno. En caso de hipoxemia, hipercapnia, insuficiencia de los músculos respiratorios o deterioro neurológico progresivos está indicada la asistencia respiratoria.
Una taquipnea sostenida (frecuencia respiratoria >30 respiraciones por minuto [rpm]) suele ser un anuncio de un colapso respiratorio inminente; a menudo se inicia la ventilación mecánica para garantizar una oxigenación adecuada, desviar la sangre de los músculos de la respiración, evitar la aspiración del contenido orofaríngeo y reducir la poscarga cardíaca. Si existen dificultades para el aporte de oxígeno por una baja concentración de hemoglobina (<7 g/100 ml), está indicada la transfusión de hematíes.En caso de acidosis metabólica grave (pH arterial <7.2) a veces se adm bicarbonato. La DIC, si se complica con hemorragias importantes, debe ser tratada con transfusiones de plasma fresco congelado y plaquetas. El tratamiento satisfactorio de la infección subyacente es esencial para controlar la acidosis y la coagulación intravascular diseminada.
SOPORTE GENERALEn pacientes con septicemia grave prolongada (duración superior de dos a tres días), la administración de complementos nutricionales puede reducir el impacto del hipercatabolismo proteínico; los datos disponibles respaldan la vía entérica. También se facilita la recuperación

mediante la prevención de la maceración cutánea, trombosis venosa profunda, infecciones nosocomiales y úlceras deestrés.
OTRAS MEDIDASA pesar de un tratamiento enérgico, muchos pacientes con septicemia grave o choque séptico acaban muriendo. Existen tres tipos de agentes que están siendo investigados y que pueden ayudar a evitar estas muertes:
1) Fármacos que neutralizan la endotoxina bacteriana y que, por tanto, podrían resultarbeneficiosos para pacientes con septicemia (aproximadamente la mitad) que sufren infecciones por bacterias gramnegativas;
2) Fármacos antiinflamatorios que interfieren con uno o más de los mediadores de la respuesta inflamatoria,
3) Anticoagulantes para evitar o revertir la microtrombosis.
PRONÓSTICO
Alrededor de 20 a 35% de los pacientes con septicemia grave y entre 40 y 60% de los que tienen un choque séptico fallecen en 30 días. Otros lo hacen en los seis meses siguientes. Las muertes tardías suelen ser causadas por infecciones mal controladas, complicaciones de los cuidados intensivos, insuficiencia multiorgánica o a la enfermedad subyacente.Sistemas de estratificación pronóstica como APACHE II indican que la ponderación de factores como la edad, cuadro subyacente y diversas variables fisiológicas del paciente proporcionan estimaciones del riesgo de muerte por septicemia grave.
PREVENCIÓN
Por medio de la prevención se tiene la mayor oportunidad de disminuir la morbilidad y mortalidad. En países desarrollados, muchos de los episodios de septicemia grave y choque séptico han sido complicaciones de infecciones de origen nosocomial. Los casos en cuestión pudieron haberse evitado al disminuir el número de procedimientos cruentos practicados; al limitar el uso (y duración) de catéteres a permanencia en vasos y sondas vesicales, al disminuir la incidencia y la duración de neutropenia profunda (<500 neutrófilos/ l) y por el tratamiento más intensivo de infecciones nosocomiales localizadas.
Es importante evitar el uso indiscriminado de antimicrobianos y glucocorticoides, y emprender medidas óptimas de erradicación de infecciones.
TUBERCULOSIS
Enfermedad causada por bacterias pertenecientes al complejo Mycobacterium tuberculosis. Esta enfermedad suele asentar en los pulmones, pero en 33% de los casos afecta a otros órganos. Sin tratamiento más de la mitad de los enfermos (50-65%) pueden morir en un plazo de 5 años.

Agente Causal
Pertenecen a la familia Mycobacteriaceae y al orden Actinomycetales. El complejo incluye:
M. tuberculosis (más importante). M. bovis M. africanus M. microti (bacilo de los roedores y menos virulento).
M. tuberculosis:
bacteria anaerobia fina no esporogena, cilíndrica mide de 0.5 por 3 µm. bacilos acidorresistentes: se debe a que en su pared celular tienen gran cantidad de ácidos
micólicos, de ácidos grasos de cadena larga y enlaces cruzados. ineficacia a la mayoría de antibióticos: los lípidos están unidos a los arabinogalactanos y a
los peptidoglucanos subyacentes. supervivencia en el macrófago: lipoarabinomanan. proteína característica: PPD
Epidemiología
En el 2001 se informó a la OMS de mas de 3.8 millones de casos nuevos de tuberculosis (pulmonar y extrapulmonar), y 90% venían de países en desarrollo. En el año 2000 se produjeron 1.8 millones de fallecimientos por tuberculosis, 98% de los cuales se produjeron en países en desarrollo.
En Estados Unidos, la tuberculosis es también una enfermedad propia de adultos infectados por el VIH, de inmigrantes, o de una población indigente/marginada.
De la exposición a la infección
Se transmite casi siempre desde un paciente con tuberculosis pulmonar contagiosa a otras personas por medio de las gotitas respiratorias que la tos, el estornudo o la fonación convierten en aerosol. Las menores de < de 10 µm de diámetro pueden permanecer suspendidas en el aire durante horas y alcanzar las vías respiratorias terminales al ser inhaladas. Con cada golpe de tos se pueden expulsar nada menos que 3000 gotitas contagiosas.
Otras vías de contagio: piel y placenta.
Factores importantes de transmisión:
Probabilidades de entrar en contacto con un caso de TB.

Duración e intimidad de ese contacto Grado de contagiosidad Ambiente (muy importante porque apiñamiento en espacios mal ventilaos intensifica el
contacto con el enfermo).
Paciente con bacilos visibles en su esputo son los más contagiosos, y suelen tener una TB cavitaria o de las vías respiratorias (endotraqueal o laríngea) y eliminan esputos que contienen nada menos que 10⁵ bacilos/ml. Paciente con esputo negativo y cultivo positivo es menos contagioso y paciente con esputo y cultivo negativo carece de contagiosidad.
Infección depende de factores exógenos.
De la infección a la enfermedad
Enfermedad depende factores endógenos:
Predisposición natural de la enfermedad Eficacia funcional de la inmunidad celular Edad (incidencia máxima al final de la adolescencia y comienzos de la edad adulta). Sexo (femenino entre 25 y 34 años, pero en edades avanzadas: masculino). Infección simultanea de VIH (factor más importante).
TB Primaria: manifestaciones que aparecen inmediatamente después de la infección, se observa con frecuencia en niños de hasta 4 años, y suele ser no contagiosa.
Pueden quedar bacilos en estado latente durante años antes de que se reactiven y produzcan: TB secundaria, que suele ser contagiosa.
Evolución Natural de la Enfermedad
La TB no tratada suele ser letal. 33% de los pacientes fallecía en el primer año tras el diagnostico, y la mitad en los cincos años posteriores al mismo. Los pacientes con frotis de esputo positivo tuvieron una mortalidad a los 5 años de 65%. Alrededor de 60% de quienes sobrevivían a los cinco años consiguió una remisión espontanea, pero los demás seguían expulsando bacilos tuberculosos.
Patogenia e Inmunidad

Interacción comienza cuando las gotitas infecciosas de los pacientes contagiosos son inhaladas por alguna persona. La mayor parte de los bacilos quedan atrapados en las vía respiratorias superiores y son expulsadas por el barrido ciliar de las células de la mucosa, pero por lo general menos del 10% llegan hasta los alveolos. Allí son englobados inespecíficamente por los macrófagos alveolares. Esta invasión se debe:
Unión de del C2a a la pared celular bacteriana Opsonización de las bacterias por C3b Reconocimiento de macrófagos.
El equilibrio entre la actividad bactericida del macrófago y la virulencia del bacilo (ligada a la riqueza en lípidos de la pared celular y a su capsula glucolipídica) es el que determina los fenómenos que siguen a la fagocitosis.
En la primera fase de interacción:
Puede ocurrir que los macrófagos con bacilos englobados inhiban su multiplicación gracias a la producción de enzimas proteolíticas y citocinas o que los bacilos comiencen a multiplicarse.
Si los bacilos se multiplican, lisan y matan los macrófagos. Monocitos no activados llegan al sitio de la lesión y fagocitan a los bacilos.

Esta etapa suele ser asintomática.
2-4 semanas después de la infección se producen 2 respuestas:
Respuesta de lesión hística: se debe a la reacción de hipersensibilidad retardada, y destruye los macrófagos con bacilos. Única capaz de contrarrestar la proliferación de las micobacterias dentro del macrófago. Causa necrosis solida precoz en el centro del tubérculo. Los bacilos pueden seguir vivos, pero su proliferación queda inhibida en este ambiente necrótico debido a la escasa tensión de oxigeno y al pH bajo. Unas lesiones curan por fibrosis y calcificación y otras evolucionan.
Reacción de activación de macrófagos: les confiere la capacidad de destruir y digerir a los bacilos tuberculosos.
Cuando se adquiere la inmunidad específica y se acumulan muchos macrófagos activados en el sitio de la lesión primaria aparecen lesiones granulomatosas (tubérculos) que contienen:
Linfocitos activados Macrófagos activados Células epiteliales Células gigantes
La inmunidad celular es esencial en esta primera fase y confiere protección parcial frente a M. tuberculosis. En la mayoría de las personas infectadas los macrófagos locales se activan cuando los antígenos bacilares procesados por los macrófagos estimulan los linfocitos T para que liberen diversas linfocinas. Estas células activadas se acumulan rodeando la lesión y neutralizan eficazmente los bacilos tuberculosos sin provocar nuevas destrucciones hísticas. En el centro de la lesión, el material necrótico se asemeja al queso blando (necrosis caseosa). Aunque se produzca la curación hay bacilos viables que permanecen en estado latente dentro de los macrófagos durante años.
En una minoría de casos, la respuesta de activación de macrófagos es débil y produce:
Lesión aumenta de tamaño El material caseoso se licua Invasión y destrucción de las paredes bronquiales y de los vasos sanguíneos Formación de cavidades Material se expulsa a través de los bronquios
Muchos macrófagos transportan a los bacilos los ganglios linfáticos regionales, desde donde se difunden ampliamente a muchos órganos y tejidos.
Hay 2 clases de células que son esenciales:
Macrófagos que fagocitan directamente a los bacilos tuberculosos. Células T, principalmente CD4+, que ejerce un papel protector.

Después de la infección:
Macrófagos alveolares segregan varias citocinas: IL-1 que contribuye a la fiebre; IL-6 contribuye a la hiperglobulinemia y el factor de necrosis tumoral-α que favorece la destrucción de micobacterias, la formación de granulomas, fiebre y adelgazamiento. Los macrófagos son esenciales para procesar y presentar los antígenos a los linfocitos T; el resultado es una proliferación de los linfocitos CD4+.
Los CD4+ reactivos generan citocinas de tipo TH₁ que participan en la destrucción de las células infectadas por M. tuberculosis.
Las células CD4+TH₁ producen interferon gamma e IL-2 que estimulan la inmunidad celular.
Las células TH₂ generan IL-4, IL-5 e IL-10 y favorecen la inmunidad humoral.
Coincidiendo con la aparición de la inmunidad, surge la reacción de hipersensibilidad retardada a M. tuberculosis. Esta reactividad sirve de base a la prueba cutánea del PPD que hoy en día, es la única prueba fidedigna que detecta la infección en personas asintomáticas.
Manifestaciones Clínicas
La TB se divide en pulmonar y extrapulmonar. Antes del VIH, más del 80% de todos los casos de TB se localizaban en los pulmones. ⅔ de los pacientes pueden padecer una enfermedad tuberculosa pulmonar y extrapulmonar, o solo extrapulmonar.
La TB pulmonar puede ser primaria o posprimaria.
Tuberculosis Primaria
Suele afectar a niños. Se localiza en campos medios e inferiores de los pulmones. La lesión cura espontáneamente y mas tarde puede descubrirse por un pequeño nodulo
calcificado (lesión de Ghon).
En pacientes con VIH O desnutridos:
La TB pulmonar primaria puede agravarse rápidamente y producir manifestaciones clínicas:
Derrame pleural Adenopatías hiliares o mediastinicas: los ganglios pueden comprimir los bonquios. Si la
obstrucción es parcial puede aparecer un efisema obstructivo seguido de atelectasias. Diseminación hematogena: manifestación más grave.
Tuberculosis Posprimaria
Llamada también TB secundaria, de reactivación, o de tipo adulto.

Suele localizarse en los segmentos apicales y posteriores de los lóbulos superiores, donde la gran concentración de oxigeno favorece el crecimiento de las micobacterias, y los segmentos superiores de los lóbulos inferiores.
Cuando debido a la confluencia de varias lesiones, se afecta masivamente un segmento o lóbulo pulmonar, el resultado es una neumonía tuberculosa.
Consunción: evolución crónica cada vez mas debilitante. Síntomas inespecíficos:
o Fiebreo Sudores nocturnoso Perdida de pesoo Anorexiao Malestar generalo Debilidad o Tos o Hemoptisiso Aneurisma de Rasmussen: hemoptisis masiva consecutiva a la rotura de un vaso
dilatado en una caverna.o Dolor precordial de tipo pleurítico
En las formas extensas hay:
Disnea Síndrome de Insuficiencia Respiratoria Aguda
Signos:
Estertores inspiratorios Roncus: debido a obstrucción de bronquios Soplo anfórico
Datos hematológicos:
Anemia Leucositosis
Tratamiento
Los dos objetivos principales del tratamiento son:
Interrumpir la transmisión de la tuberculosis Evitar las complicaciones y la muerte
Agentes de primera línea:
Isionazida

Rifampicina Pirazinamida Etambutol
Son los de elección porque:
Actividada bactericida Actividad esterilizante Cifra pequeña de inducción de resistencia a fármacos.
Fármaco Dosis Diaria Dosis 3 veces/semana
Isionazida 5 mg/kg, máximo 300 mg 15 mg/kg máximo 900 mg
Rifampicina 10 mg/kg, máximo 600 mg 10 mg/kg, máximo 600 mg
Pirazinamida 20-25 mg/kg, máximo 2 g 30-40 mg/kg, máximo 3 g
Etambutol 15-20 mg/kg 25-30 mg/kg
2 fases:
I FASE INICIAL O BACTERICIDA: 2 meses, dar los 4 medicamentos
II FASE DE CONTINUACION: 4 meses, dar isionazida y rifampicina (puede ser diario o intermitentes (3 veces/semana)).
Si al cabo de 2 meses el esputo sigue positivo, se ampliara el tratamiento a 3 meses más. Para evitar la neuropatía por isionazida, habrá que agregar al régimen 10-25 mg/dia de
piridoxina, en personas muy propensas a sufrir deficiencia de vitamina B₆. El incumplimiento de los regímenes terapéuticos, constituye el impedimento más
importante para la curación. Con el régimen recomendado de 6 meses, el esputo se negativiza en más del 80% de los
pacientes al final del segundo mes. Los frotis positivos pasados 5 meses indican que el tratamiento ha fracasado. Reacciones secundarias:
o Hepatitis (mas importante)o Reacciones de hipersensibilidado Hiperuricemia: causada por pirazinamida. o Artralgias: causada por pirazinamida.o Trombocitopenia autoinmunitaria secundaria: causada por rifampicina.o Neuritis óptica: causada por etambutol.o Lesión del octavo par craneal: causada por estreptomicina.
Agentes de segunda línea:
Estreptomicina

Kanamicina Amikacina Capreomicina Etionamida Cicloserina Actualmente también fluoroquinolonas.
Los sujetos que requieren tratamiento de TB latente se descubren mediante la prueba cutánea del PPD:
Debe inyectarse por vía intradérmica 5 U de tuberculina del PPD estabilizado con polisorbatos en al superficie de flexión del antebrazo (técnica de Mantoux).
Las reacciones se leen a las 48-72 horas midiendo en milímetros el diámetro de la induración.
Grupo de Riesgo Diámetro de la reacción a la tuberculina
Infección por VIH o tratamiento con inmunosupresores. ≥5
Contacto muy estrecho con tuberculosos ≥5
Lesiones fibróticas en las radiografías de tórax ≥5
Infección reciente (dos años o menos) ≥10
Trastornos médicos de alto riesgo ≥10
Bajo riesgo de TB ≥15
a. En las formas latentes se da isionazida diariamente en dosis de 5 mg/kg durante 9 meses. b. Otro régimen es dar rifampicina diario por cuatro meses.
Prevención
Vacunación con BCG: es innocua y no causa complicaciones graves.
TUBERCULOSIS EXTRAPULMONAR
Localización por orden de frecuencia: ganglios linfáticos, pleura, genitourinario, huesos, articulaciones, meninges y peritoneo.Frecuente en pacientes con VIH.

Tb ganglionar (adenitis tuberculosa)
Forma mas frecuente de tb extrapulmonar. La manifestación inicial es la inchazon indolora de los ganglios linfáticos (frecuentemente los cervicales y supraclaviculares: trastorno llamado escrófula) que pueden llegar a inflamarse y formar fistulas donde se expulsa material caseoso.
Diagnostico: mediante BAAF o biopsia quirúrgica. El cultivo es positivo en 80% de los casos y hay ABF (bacilos acidoresistentes) en 50% de los casosAl examen histológico solo los pacientes con VIH no revelan granulomasDiagnóstico diferencial: linfomas y metástasis de un carcinoma
Tb pleural
Las lesiones de la pleura son frecuentes en la tb primaria por penetración de los bacilos tuberculosos al espacio pleural. El derrame pleural ocurre según el sistema inmunitario del paciente puede pasar desde inadvertido y resolverse espontáneamente hasta sintomático con matidez a la percusión y abolición del murmullo vesicular.
Características: Liquido color pajizo (puede ser hemorrágico), concentración de proteínas mayor del 50% que las del suero, glucosa normal o disminuida, ph <7.2 y con 500 a 2500 leucocitos/ μl.
Diagnóstico: biopsia pleural con aguja donde se encuentran granulomas y el cultivo es positivo en 70% de los casos.
Complicaciones: Empiema tuberculoso por la rotura de una caverna al espacio pleural o por la formación de una fistula pleropulmonar. El derrame es espeso, purulento, rico en linfocitos y hay ABF positivos.Puede llegar a producir fibrosis pleural intensa con insuficiencia respiratoria restrictivaEs necesario dar tratamiento farmacológico y drenaje quirúrgico.
Tb de las vías respiratorias superiores
Es una complicacion de la tuberculosis pulmonar cavitaria avanzada y puede afectar la laringe, faringe y epiglotis.
Síntomas: ronquera, disfagia, tos y expectoraciones crónicas.El esputo contiene ABF, pero a veces es necesario confirmar el diagnóstico con una biopsia.
Tb genitourinaria
Se debe a siembra hematógena. Síntomas frecuentes son polaquiuris, disuria, hematuria y dolor en fosa renal. La enfermedad puede descubrirse cuando ya hay lesiones avanzadas

destructivas en los riñones. En 90% de los casos el examen de orina revela piuria y hematuria.Ante un examen de orina con piuria y cultivo negativo debe hacerse siempre la sospecha de una tuberculosis.
La pielografia intensa ayuda al diagnóstico y son signos sospechosos la presencia de calcificaciones y estrechez uretral. El diagnóstico seguro se obtiene con el cultivo positivo 3 muestras de la primera orina de la mañana.La tb genital es mas común en mujeres, afecta trompas de Falopio y endometrio pudiendo causar esterilidad, dolores pélvicos y trastornos menstruales. En hombres afecta el epidídimo preferentemente pero puede causar protatitis y orquitis.
Tb osteoarticular
La patogenia guarda relación con la reactivación de focos hematógenos o con diseminación procedente de los ganglios linfáticos paravertebrales próximos. Las articulaciones mas afectadas son las que soportan el peso. La enfermedad de Pott (tb vertebral) afecta con frecuencia la región dorsal baja y las vertebras lumbares supeiores. La lesión alcanza el cuerpo vertebral y el disco intervertebral.En fases avanzadas hay cifosis y paraplejia. Sin tratamiento las articulaciones pueden quedar destruidas.
Meningitis tuberculosa
Representa cerca de 5% de los casos de tuberculosis extrapulmonares. Es mas frecuente en los niños pequeños y en adultos infectados por VIH. La meningitis tuberculosa se debe a una diseminación hematogena de la lesión pulmonar primaria o postprimaria, o a la rotura de un tuberculos subependimario en el espacio subaracnoideo
La enfermedad puede manifestarse en forma sutil por cefalalgia y trastornos mentales o de forma aguda con confusión mental, letargo, ateración del sensorio y rigidez de nuca. La enfermdad suele evolucionar en una a dos semanas. Es frecuente la paresia de los pares craneales oculomotore y la afección de las arterias cerebrales puede producir isquemia focal.
El cultivo de lLCR establece el diagnostico en 80% de los casos.Exiten seguelas en el 25% de los casos no tratados, la administración de glucocorticoides reduce el riesgo de secuelas.
El tuberculoma es una forma poco frecuente de tb que consiste en la formación de una o mas lesiones ocupantes de espacio y pueden manifestarse por convulsiones y signos neurológicos focales.
Tuberculosis digestiva

El aparato gastrointestinal se afecta por diversos mecanismos: deglución de los esputos por lo consiguiente siembra directa o por diseminación hematógena .
Los sitios de mayor afección son el ileon terminal y el ciego.Los síntomas son dolor abdominal parecido al de la apendicitis, diarrea, obstrucción, hematoquezia, una tumoración palpable en el abdomen, fiebre, perdida de peso y sudores nocturnos.
La peritonitis tuberculosa aparece después de una siembra directa de bacilos tuberculosos procedentes de los órganos intrabdominales o de unos ganglios linfáticos rotos o bien por siembra hematógena.
La peritonitis tuberculosa debe de sospecharse siempre en presencia de solores abdominales inespecíficos, fiebre y ascitis
Tuberculosis pericardica
Se da por extensión directa de un poco primario situado en el pericardio, por reactivación de un foco latente o por la rotura de un ganglio linfático contiguo.
Puede tener un comienzo subagudo o se puede manisfestar de forma aguda con fiebre, dolor sordo retroesternal y roce pericardico. Puede producirse en derrame que causa síntomas de taponamiento cardiaco. El derrame es un exudado rico en leucocitos con predominio de mononucleares y es común el derrame hemorrágico.La corticoterapia es útil para tratar los síntomas agudos.
Tuberculosis miliar o diseminada
Consiste en una siembre hematogena de bacilos tuberculosos.Las manifestaciones clínicas son inespecíficas y variadas dependediendo de la localización y el órgano afectado. Generalmente los primeros síntomas son fiebre, sudores nocturnos, anorexia, debilidad y perdidad de peso.Al examen físico hay viveromegalia en 30% de los casos.Descubrir tuberculos en la coroides es patoneumonico de la tuberculosis miliar.Pueden haber alteraciones hematológicas como anemia, leucopenia, leucocitosis y policitemia.
Diagnostico
El diagnóstico definitivo de tuberculosis sólo puede establecerse cuando se cultiva M. tuberculosis.
Prueba tuberculínica:

Es una reacción cutánea de hipersensibilidad que indica la existencia de infección tuberculosa previa. La prueba se lleva a cabo con un extracto proteico purificado (PPD) de M. tuberculosis.
Las reacciones se leen midiendo el diámetro transverso de la zona de induración a las 48-72 horas.
La prueba se considere positiva a partir de 5 mm.
La prueba tuberculínica puede ser positiva si el paciente ha tenido contacto con otras micobacterias no tuberculosas.
En los pacientes que han sido vacunados contra la tuberculosis con BCG, la prueba tuberculínica puede ser positiva durante un período aproximado de 10 años.
Examen microscópico:
Recoger 3 muestras de esputos a primera hora por la mañana. Buscar ABF las muestras son teñidas con coloración Ziel – Neelsen. Se hace un cultivo microbiológico.
Cultivo de micobacterias:
Se obtienen de la muestra de esputo de tos productiva. Se detecta el crecimiento a las 4 a 8 semanas. Medio de cultivo: Lowensten Jensen.
Rayos X
Infiltrados y cavernas de los lobulo superiores. Cavitaciones. Signos atipicos en px con VIH. Nodulo solitario. Infiltrados alveolares. Radiografias normales.



















