
CENTRO DE INVESTIGACIN Y DE ESTUDIOS … · V.3.Determinaciones del potencial hídrico de hoja y...
168
CENTRO DE INVESTIGACIÓN Y DE ESTUDIOS AVANZADOS DEL INSTITUTO POLITÉCNICO NACIONAL UNIDAD IRAPUATO DEPARTAMENTO DE INGENIERÍA GENÉTICA ANÁLISIS DE LA RESPUESTA A NIVEL FISIOLÓGICO Y DE EXPRESIÓN GÉNICA BAJO SEQUĺA EN TRES MATERIALES CRIOLLOS MEXICANOS DE MAĺZ TESIS QUE PRESENTA M. EN C. ANGELA CORINA HAYANO KANASHIRO PARA OBTENER EL GRADO DE DOCTORA EN CIENCIAS EN LA ESPECIALIDAD DE BIOTECNOLOGÍA DE PLANTAS DIRECTORES DE TESIS: DRA. JUNE KILPATRICK SIMPSON WILLIAMSON DR. LUIS RAFAEL HERRERA ESTRELLA Irapuato, Guanajuato MÉXICO 2009
Transcript of CENTRO DE INVESTIGACIN Y DE ESTUDIOS … · V.3.Determinaciones del potencial hídrico de hoja y...

CENTRO DE INVESTIGACIÓN Y DE ESTUDIOS AVANZADOS DEL INSTITUTO POLITÉCNICO NACIONAL
UNIDAD IRAPUATO
DEPARTAMENTO DE INGENIERÍA GENÉTICA
ANÁLISIS DE LA RESPUESTA A NIVEL FISIOLÓGICO Y DE EXPRESIÓN GÉNICA BAJO SEQUĺA EN TRES MATERIALES CRIOLLOS MEXICANOS
DE MAĺZ
TESIS QUE PRESENTA
M. EN C. ANGELA CORINA HAYANO KANASHIRO
PARA OBTENER EL GRADO DE
DOCTORA EN CIENCIAS
EN LA ESPECIALIDAD DE
BIOTECNOLOGÍA DE PLANTAS
DIRECTORES DE TESIS:
DRA. JUNE KILPATRICK SIMPSON WILLIAMSON
DR. LUIS RAFAEL HERRERA ESTRELLA
Irapuato, Guanajuato
MÉXICO
2009

2
DEDICATORIA
A mis padres, ejemplo de trabajo y perseverancia, a pesar de la distancia gracias por su incondicional apoyo y cariño.
A mis hermanitas Ceci, Pili y Tamiko porque siempre me dieron ánimos y fuerza cuando más lo necesitaba.

3
AGRADECIMIENTOS
- A Dios porque gracias a su ayuda puedo conseguir lo que me
propongo.
- A la Dra. June Simpson Williamson y el Dr. Luis Herrera Estrella por el
apoyo, confianza y enseñanzas durante la realización del presente
trabajo. Pero sobre todo por su paciencia, comprensión y amistad.
- A los Doctores Alfredo Herrera Estrella, Octavio Martinez de la Vega,
Rafael Rivera Busrtamante y Victor Olalde Portugal por sus acertadas
sugerencias durante la realización del presente trabajo, la revisión del
escrito y aceptar ser parte del comité de sinodales. Y al Dr. Jose Luis
Pons Hernández por aceptar ser miembro del Comité de Sinodales y
por sus acertadas sugerencias para el presente trabajo.
- A CONACYT por el apoyo en el otorgamiento de la beca de Doctorado
No. 204749.
- A todos los Doctores que participaron en mi formación académica.
- A mis compañeros de laboratorio (Marcadores moleculares y
Bioinformática): Katy, Moni, Emi, Silvia, Miriam, Rocío, Celso,
Francisco, Yasmín, Alfredo, Juan, Luis, Juan Caballero, Jose, Araceli,
Andrea y Maribel por compartir muy buenos momentos en el lab, la
música, las pláticas divertidas que hemos tenido y la amistad brindada.
- A los compañeros del laboratorio de Ingenieria Metabólica y resistencia
a estrés ambiental: Lenin, Ale, Damar, Rosi, Anahí, Fulgencio, Víctor,
Marco, Araceli, Kike y el Dr. Lenin Sanchez por su amistad y
entretenidas pláticas.
- Al Biólogo Fernando Hernandez G. y el Ing. Agr. Pedro Cervantes por
su ayuda y consejos durante la realización del experimento de sequia
en el invernadero y principalmente por su amistad.

4
- A Emigdia Alfaro por su ayuda en la realización de los experimentos
durante el presente trabajo y por sus buenos consejos durante todo
este tiempo.
- A Alma Rosa y Gerardo por las facilidades brindadas en el trabajo de
invernadero.
- Al Dr. Víctor Olalde por la ayuda prestada para utilizar el IRGA para la
medición de la fotosíntesis y transpiración y al QFB. Iván Gasca por su
ayuda en lo referente al manejo del equipo y sus siempre acertados
consejos.
- A Susana M.L. Fuentes-Guerra y Flor M.X. Zamudio-Hernandez por los
análisis de qRT-PCR.
- A la Dra. Magdalena Segura y QFB. Berenice por su ayuda y
colaboración en la medición de los aminoácidos libres.
- A Letty Chong, Laura Camacho y Eduardo por su siempre oportuna
ayuda y buen trabajo.
- A Dora Elia Anguiano y Paulina Martinez por su ayuda en los trámites
académicos y adminsitrativos y su eficiente trabajo.
- A la Sra. Maricarmen, Carmelita por su ayuda y siempre buena
disposición en conseguir los artículos solicitados.
- A los buenos amigos que he encontrado en todo este tiempo: Pedro,
Christian, Moni, Alex, Miguel, Iván, Vianey, Vero, Alma, Luz, Alba, Nidia,
Jimena, Ernesto, Noé, Vic, Edgar, Frank, César, Gustavo y Xool por los
buenos momentos que he pasado con ellos.
- A las integrantes del equipo de fútbol femenil del CINVESTAV y al
cuerpo técnico por los buenos momentos compartidos.
- A todas las personas que forman parte del CINVESTAV-Irapuato
porque con su ayuda y trabajo podemos salir adelante y finalizar
nuestros proyectos.
- A México y a su gente, por el trato cálido durante todo este tiempo.

5
EL PRESENTE TRABAJO FUE REALIZADO EN EL
LABORATORIO DE MARCADORES MOLECULARES
DEL DEPARTAMENTO DE INGENIERÍA GENÉTICA DE
PLANTAS DEL CENTRO DE INVESTIGACIÓN Y DE
ESTUDIOS AVANZADOS DEL INSTITUTO
POLITÉCNICO NACIONAL, UNIDAD IRAPUATO, BAJO
LA TUTORÍA DE LA DRA. JUNE KILPATRICK
SIMPSON WILLIAMSON Y EL DR. LUIS RAFAEL
HERRERA ESTRELLA. Y LA ASESORIA DEL DR.
ALFREDO HERRERA ESTRELLA, EL DR. OCTAVIO
MARTINEZ DE LA VEGA, EL DR. RAFAEL RIVERA
BUSTAMANTE Y EL DR. VICTOR OLALDE
PORTUGAL.

6
INDICE GENERAL
I. RESUMEN ...................................................................................................................................................... 10
II. RESUMEN EN INGLÉS ............................................................................................................................... 14
III. INTRODUCCIÓN ......................................................................................................................................... 15
IV. ANTECEDENTES ....................................................................................................................................... 19
IV.1.Impacto de la sequía en México ......................................................................................................... 19
IV.2.Efectos fisiológicos a nivel celular que afectan al maíz durante la sequía ................................... 22
IV.3.Efectos de la sequía a nivel de planta completa en maíz ............................................................... 23
IV.4.Mecanismos de tolerancia a la sequía en plantas ........................................................................... 25
IV.5.Etapas críticas del maíz bajo sequía .................................................................................................. 28
IV.6.Rutas de transducción de señales general a estrés ........................................................................ 29
IV.6.1.Percepción y señalización del déficit hídrico ................................................................................. 30
IV.6.2.Inducción del estrés oxidativo debido a la sequía en plantas ..................................................... 34
IV.6.3.El Estrés osmótico activa la señalización por fosfolípidos ........................................................... 36
IV.6.4.Señalización por Ca+2 durante la deshidratación y el estrés salino ........................................... 37
IV.6.5.Otras rutas fosfoproteicas de señalización .................................................................................... 37
IV.7.Respuestas bioquímicas a nivel celular durante la sequía ................................................. 38
IV.8.Estrategias para identificar genes asociados con la tolerancia a sequía en maíz ...................... 40
IV.9.Productos génicos de respuesta a la sequÍa .................................................................................... 43
IV.10. Desarrollo de plantas transgénicas para la tolerancia a estrés abiótico ................................... 47
IV.11.Expresion génica dependiente de ABA durante el déficit hídrico ........................................... 50
IV.12.Expresión génica independiente de ABA durante el déficit hídrico ............................................. 54
IV.12. Estudios de expresión diferencial bajo condiciones de sequía ................................................... 55
IV.12.2.Estudios de expresión diferencial en cereales: trigo y cebada ................................................. 57
IV. OBJETIVOS ................................................................................................................................................. 61
IV.I.GENERAL................................................................................................................................................ 61

7
IV.II.ESPECÍFICOS ....................................................................................................................................... 61
V. MATERIALES Y MÉTODOS ....................................................................................................................... 62
V.1.Material Vegetal ...................................................................................................................................... 62
V.2.Condiciones de crecimiento .................................................................................................................. 63
V.3.Tratamientos de estrés .......................................................................................................................... 63
V.3.Determinaciones del potencial hídrico de hoja y suelo..................................................................... 64
V.4.Extracción de azúcares solubles .......................................................................................................... 65
V.5.Medición de fotosíntesis y conductancia estomatal .......................................................................... 66
v.6.cuantificación de aminoácidos libres .................................................................................................... 67
V.7.Extracción de ARN Total ....................................................................................................................... 67
V.8.Alineamiento del primer T7 y Síntesis de Primera Cadena de cDNA ............................................ 69
V.9.Síntesis de la primera cadena de cADN ............................................................................................. 70
V.10.Síntesis de la segunda cadena .......................................................................................................... 70
V.11.Purificación de cADN ........................................................................................................................... 71
V.12.Transcripción in vitro ............................................................................................................................ 72
V.13.Purificación de ARN modificado ........................................................................................................ 72
V.14.Preparación del colorante Cy3 y Cy5 ................................................................................................ 73
V.15.Unión del fluorocromo al ARN modificado ....................................................................................... 73
V.16.Limpieza de los ARNs modificados ................................................................................................... 74
V.17.HibridaciÓn de los microarreglos ....................................................................................................... 74
V.18.Adquisición de la imagen y extracción de los datos ....................................................................... 78
V.19.Normalización de datos ....................................................................................................................... 78
V.20.Análisis de los genes con expresión diferencial .............................................................................. 79
V.21.Análisis de los perfiles de expresión y rutas metabólicas .............................................................. 82
V.22.Validación de la expresión de transcritos de los microarreglos por medio de RT-PCR tiempo real ...................................................................................................................................................... 82

8
V.23.Asociación de las secuencias de cDNA a QTL's asociados a sequía ......................................... 84
V.24.Número de accesión de los datos de microarreglos ....................................................................... 84
V.25.Anotación funcional y análisis de las rutas metabólicas usando los softwares MapMan y BioMaps ....................................................................................................................................................... 84
V.26.Aplicación de la prueba de Pearson's Chi-cuadrado ...................................................................... 86
VI. RESULTADOS ............................................................................................................................................. 89
VI. 1. Efectos fisiológicos de la sequía ....................................................................................................... 89
VI.1.2.Tasa fotosintética ............................................................................................................................... 91
VI.1.3. Conductancia estomatal .................................................................................................................. 92
VI.2. Variación en la concentración de azúcar .......................................................................................... 94
VI.3. Contenido de prolina ............................................................................................................................ 94
VI.4. Análisis de los microarreglos a partir de las bibliotecas generadas en el Proyecto de Maíz (CINVESTAV) ....................................................................................................................................... 98
VI.5. Análisis de los microarreglos de oligonucleótidos .......................................................................... 99
VI.6. Análisis de la expresión de genes mediante RT-PCR cuantitativo en tiempo real .................. 100
VI.7. Clasificación funcional de los transcritos expresados diferencialmente .................................... 104
VI.8. Fotosíntesis y metabolismo de carbohidratos ................................................................................ 104
VI.9. Metabolismo de aminoácidos ........................................................................................................... 111
VI.10. Genes relacionados a señalización y estrés abiótico ................................................................. 112
VI.11. Respuestas relacionadas a hormonas ........................................................................................ 116
VI.12. Factores de transcripción .............................................................................................................. 118
VI.13. Asociación de genes expresados diferencialmente a QTL’s de maíz ..................................... 121
VII. DISCUSIÓN .............................................................................................................................................. 125
VII.1. Respuestas fisiológicas: fotosíntesis, conductancia estomatal y potencial hídrico de hoja y suelo .................................................................................................................................................. 125
VII.2.Respuestas moleculares comunes a la sequía en los tres genotipos de maíz ........................ 126
VII.3.Respuestas diferenciales entre las 3 variedades bajo estrés y riego de recuperación ........... 131

9
VII.4.Diferencias en el metabolismo de carbohidratos en las 3 variedades bajo sequía y el riego de recuperación ................................................................................................................................. 135
VII.5. El ajuste osmótico por la acumulación de prolina en las tres variedades de maíz bajo estrés y riego de recuperación .................................................................................................................. 136
VII.6. Diferencias en las respuestas moleculares bajo el riego de recuperación en las tres variedades de maíz ..................................................................................................................................... 137
VII.7. Análisis de los datos de sequía usando el programa BioMaps .................................................. 140
VII.8.Asociación de genes expresados diferencialmente a QTL’s de maíz ........................................ 142
VIII. CONCLUSIONES .................................................................................................................................... 144
IX. PERSPECTIVAS ....................................................................................................................................... 146
X. BIBLIOGRAFIA ........................................................................................................................................... 147
Indice de Figuras
Indice de Tablas

10
INDICE DE FIGURAS
Figura 1. Indice de severidad de la sequía………………………………………………………………………………...21
Figura 2. Ruta general para la transducción de señales de frío, sequia y estrés salino en plantas ……………………………………………………….…………..33
Figura 3. Figura 3. Redes regulatorias transcripcionales y FTs involucrados en la expresión génica del estrés osmótico…………………..............................53
Figura 4. Diseño en loop de los microareglos en el presente trabajo……………………………………………………………………………......76
Figura 5. Potencial hídrico de suelo de las diferentes variedades de maíz bajo condiciones de sequía…………………...………………………………………...90
Figura 6. Potencial hídrico de hoja 6 am de las diferentes variedades de maíz bajo condiciones de sequía……………………………………………………….90
Figura 7. Potencial hídrico de hoja 1 pm de las diferentes variedades de maíz bajo condiciones de sequía……………………….……………………………...91
Figura 8. Fotosíntesis a las 11 am de las diferentes variedades de maíz bajo condiciones de sequía……………………………………………………………..93
Figura 9. Conductancia estomatal a las 11 am de las diferentes variedades de maíz bajo condiciones de sequía……………………….………………………..93
Figura 10. Contenido de glucosa en las tres variedades de maíz bajo condiciones de sequía………………………………………………..…………...96
Figura 11. Contenido de Mio-inositol en las tres variedades de maíz bajo condiciones de sequía…………………………………………………………….96
Figura 12. Contenido sacarosa en las tres variedades de maíz bajo condiciones de sequía……………………………………………………………...97
Figura 13. Contenido de prolina en las tres variedades de maíz bajo condiciones de sequía……………………………………………………………...97

11
Figura 14. A. Cambios globales en la expresión génica bajo sequía y riego de recuperación……………………………………………………………………..…102
Figura 15. Validación de los microarreglos por qRT- PCR………..…….…….103
Figura 16. Diagrama general de los transcritos expresados diferencialmente involucrados en los diferentes procesos metabólicos bajo sequía y riego de recuperación en las tres variedades de maíz………………..……………...….105
Figura 17. Diagrama general de los transcritos expresados diferencialmente involucrados en los diferentes procesos metabólicos bajo sequía y riego de recuperación en las tres variedades de maíz……………….………………....106
Figura 18. Diagrama general de los transcritos expresados diferencialmente involucrados en los diferentes procesos metabólicos bajo sequía y riego de recuperación en las tres variedades de maíz……………………………….…107
Figura 19. Expresión diferencial de genes involucrados en la fotosíntesis…………………………………………………………………………109
Figura 20. Clasificación funcional de genes de estrés abiótico bajo 17 días de estrés y riego de recuperación en las tres variedades de maíz……………………………………..............................................................115
Figura 21. Panorama general de las respuestas a la expresión génica en 3 variedades de maíz mexicanos bajo sequía……………………………………127
Figura 22. Panorama general de las respuestas a la expresión génica en 3 variedades de maíz mexicanos bajo riego de recuperación…..……………..139
Figura 23. Genes expresados diferencialmente bajo sequía y riego de recuperación asociados a QTL…………………………………………………..143

12
INDICE DE TABLAS
Tabla 1. Genotipos de maíz utilizados en el presente
estudio……………………………………………………………………………….63
Tabla 2. Secuencias de los oligonucleótidos utilizados para la validación de
algunos genes expresados diferencialmente mediante RT-PCR tiempo
real………………………………………………………………………………...87-88
Tabla 3. Principales familias de factores de transcripción (FT) diferencialmente
reguladas bajo sequía y riego de recuperación en hojas de maíz de tres
variedades diferentes…………………………………………………..………...120
Tabla 4. Lista de los transcritos asociados a QTL’s de maíz que mostraron
mayores cambios de expresión en al menos una de las tres
variedades………………………………………………………………..…...122-124

13
I. RESUMEN
Se estudiaron los cambios en las respuestas fisiológicas y patrones de
expresión génica durante condiciones de sequía y riego de recuperación de 3
variedades mexicanas de maíz, de las cuales dos de ellas son consideradas
tolerantes a sequía (Cajete criollo y Michoacán 21) y la otra susceptible (85-2).
Las condiciones de la sequía fueron obtenidas dejando de regar durante 10 y
17 días y el estudio se complementó con un riego de recuperación. Variables
fisiológicas como fotosíntesis, conductancia estomatal, potencial hídrico de
hoja y de suelo fueron monitoreadas a lo largo del experimento, además de los
análisis de microarreglos obtenidos a los 10 y 17 días de estrés bajo sequía y
después del riego de recuperación. A los 10 días de estrés, 978 genes fueron
identificados como diferencialmente expresados mientras que a los 17 días de
estrés, 2240 transcritos diferenciales fueron identificados (utilizando valores de
cambios de expresión de 2 veces); sin embargo, el riego de recuperación
produjo la mayor cantidad de transcritos diferenciales. Los genes expresados
diferencialmente están involucrados en transducción de señales, metabolismo
de hormonas, respuestas generales a estrés y sequía, incluyendo genes
relacionados a pared celular. Se observó una correlación entre los niveles de
fotosíntesis y los transcritos relacionados bajo estrés. Diferencias en el
número, tipo y niveles de expresión de familias de factores de transcripción
fueron también identificadas bajo sequía y riego de recuperación entre los tres
genotipos de maíz. El análisis de expresión génica sugiere que los genotipos
tolerantes tienen una mayor capacidad para modular rápidamente más genes
durante las condiciones de sequía y el riego de recuperación en comparación
con lo observado con el genotipo susceptible. Diferencias en los patrones de
expresión y respuestas fisiológicas entre las variedades tolerantes, sugiere
que éstas emplean diferentes mecanismos de respuesta. Michoacán 21 fue el
genotipo de mayor respuesta en cuanto al número de genes con expresión
diferencial bajo condiciones de sequía y riego de recuperación.

14
II. RESUMEN EN INGLÉS
Changes in physiological responses and gene expression patterns were
studied under drought stress and recovery in three Mexican maize
landraces which included two drought tolerant materials (Cajete criollo and
Michoacán 21) and one susceptible maize landrace (85-2). Gradual drought
stress was applied by cessation of irrigation for 17 days followed by
recovery irrigation. Photosynthesis, stomatal conductance, soil and leaf
water potentials were monitored throughout the experiment and microarray
analysis was carried out on transcripts obtained at 10 and 17 days following
application of stress and after recovery irrigation. At 10 days stress 978
genes were differentially expressed whereas at 17 days stress, 2,240
differential transcripts were identified and recovery irrigation produced the
highest levels of differential expression. Differentially expressed genes were
found to be involved in signal transduction, hormone metabolism, drought
and general stress responses, including many cell wall related genes. A
correlation between levels of photosynthesis and transcription under stress
was observed and differences in the number, type and expression levels of
transcription factor families were also identified under drought and recovery
between the three maize landraces. Gene expression analysis suggests
that the drought tolerant landraces have a greater capacity to rapidly
modulate more genes under drought and recovery in comparison to the
susceptible landrace. Differences in the patterns of expression and
physiological responses between the tolerant landraces, suggests they
employ different response mechanisms. Landrace Michoacán 21 was the
most responsive in terms of differential gene expression under both drought
and recovery.

15
III. INTRODUCCIÓN
El estrés abiótico es un factor limitante para el crecimiento vegetal y la
producción de alimento en muchas regiones del mundo y sus efectos
empezarán a ser más severos conforme la desertificación se va acrecentando
y se necesite de más tierra apta para la agricultura. Entre los estreses
ambientales, la sequía tiene el efecto más grande en la agricultura a nivel
mundial (Vinocur y Altman, 2005). Un ejemplo de ello es Etiopía que debido a
las escasas lluvias, las pérdidas en las cosechas han alcanzado niveles de
hasta el 75% en algunas de las zonas más castigadas en la temporada
agrícola marzo-abril del 2009
(http://www.fao.org/news/story/es/item/35620/icode/). Asimismo, Nepal y la
República Árabe Siria hacen frente a una situación alimentaria inestable como
consecuencia de las malas cosechas debida a la sequía en el primer semestre
del 2009. Argentina no es la excepción y según los pronósticos iniciales, la
producción de trigo del 2009 en este país podría ser menor a causa de la
sequía (Argentina es considerado uno de los cinco proveedores mundiales
principales de los últimos años y el principal productor de Sudamérica)
(Informe FAO, Julio 2009, ftp://ftp.fao.org/docrep/fao/012/ai484s/ai484s00.pdf).
La sequia ha afectado más del 20% de las áreas tropicales y sub-tropicales
usadas para la producción de maíz (Ribaut y Ragot, 2007). En Kenia por
ejemplo, la cosecha de maíz en los primeros periodos del 2009 se estima que
esté cerca del 28% por debajo de los niveles normales debido a causas como
la escasez de lluvias (http://www.fao.org/news/story/es/item/35620/icode/ ). En
México, cerca del 80% del maíz cultivado es sembrado bajo condiciones de
temporal (Aquino et al, 2001). Se ha mencionado que durante los meses de
julio y agosto del 2009, México registró la sequia más severa de los últimos 60
años; las perdidas en la producción de maíz y frijol fueron de 20 mil millones
de pesos. A inicios de septiembre del 2009 la información que se disponía
sobre el periodo irregular de lluvias en algunas regiones del país era de

16
afectaciones severas en 312 mil 937 hectáreas de maíz, sorgo, trigo y frijol,
principalmente. El 90% de las hectáreas afectadas son de maíz, es decir, que
de los 17.8 millones de toneladas de producción de maíz, 20% se verá
afectada por sequía (el doble de lo que se pierde en un año por merma)
(http://www.cinvestav.mx/Difusi%C3%B3n/Cinvestavenlaprensa/Noticias/Perdi
dasde20milmdpporsequia.aspx). Por lo anterior,, existe una necesidad
urgente de desarrollar variedades tolerantes ya sea por mejoramiento
convencional o por ingeniería genética con la finalidad de afrontar la creciente
demanda del maíz tanto para la alimentación humana como la animal.
Debido a la estructura particular del genoma y la continua selección del
hombre por aproximadamente 7000 años, el maíz es una de las especies
vegetales más plásticas en términos de su adaptación a diferentes condiciones
ambientales, capaz de crecer ya sea en altas y/o bajas altitudes y, en climas
tropicales, sub-tropicales y templados. Esta variabilidad genética ha sido
explotada para producir cultivares de maíz localmente adaptados a regiones
tropicales secas de Indonesia, Kenia, México y Colombia (Pingali y Pandey,
2001). Recientemente, la selección asistida por marcadores (MAS de sus sigla
en inglés) ha sido usada en el desarrollo de germoplasma de maíz con mayor
tolerancia a la sequía (Bruce et al, 2002), basado en QTL’s que afectan la
arquitectura de la raíz, concentración de ABA en hoja y otras características
relacionadas a sequía (Tuberosa et al, 2007). A pesar de estos esfuerzos, los
programas de mejoramiento para la tolerancia a la sequía en maíz han
avanzado lentamente y se necesita la investigación básica para adaptar los
materiales genéticos mejorados para condiciones ambientales particulares
(Pingali y Pandey, 2001), donde estos materiales no solo deben de soportar
niveles mayores de sequía sino también tener altos rendimientos bajo
condiciones óptimas (Ribaut y Ragot, 2007). Además, las accesiones de
variedades locales de maíz pudieran aportar alelos novedosos que
complementarían las estrategias basadas en los mecanismos de adaptación al
estrés (Reynolds et al, 2007).

17
Durante la evolución, las plantas han adquirido una serie de estrategias
metabólicas y de desarrollo para optimizar la toma de agua y balancear
eficientemente ésta con la utilización del agua durante el crecimiento
vegetativo y de reproducción (Parry et al, 2005), haciendo la tolerancia a
sequía una característica multigénica compleja. En la década pasada, los
estudios para esclarecer los procesos moleculares involucrados en la
tolerancia a sequía han recibido atención especial (Chaves et al, 2003, para
revisiones ver Ingram y Bartels, 1996; Bray, 1997; Shinozaki y Yamaguchi-
Shinozaki, 1997). Estudios fisiológicos han mostrado que los azúcares,
alcohol azúcares, aminoácidos y aminas funcionan como osmolitos,
protegiendo funciones celulares de los efectos de la deshidratación y son
conocidos por acumularse bajo condiciones de sequía en diferentes especies
vegetales (Seki et al, 2007). La reducción en el crecimiento vegetativo, el
cierre estomatal y la disminución en la tasa fotosintética (Mahajan y Tuteja,
2005) son las respuestas más tempranas a la sequía, protegiendo a la planta
de la pérdida excesiva de agua (Chaves et al, 2003).
Más recientemente, las tecnologías genómicas han brindado enfoques
integrados de alta calidad para investigar las respuestas globales de expresión
génica no solo a la sequía sino también a otros estreses abióticos (Chaves et
al, 2003). Estudios del perfil de expresión mediante microarreglos bajo
condiciones de sequía ha sido llevado a cabo en diferentes especies vegetales
tales como Arabidopsis (Seki et al, 2002; Oono et al, 2003; Kawaguchi, 2004),
arroz (Rabbani et al, 2003), cebada (Ozturk et al, 2002, Talamé et al, 2007) y
trigo (Mohammadi et al, 2007). Estos estudios identificaron los transcritos
expresados diferencialmente de genes involucrados en fotosíntesis, síntesis
de ABA y señalización, biosíntesis de osmoprotectantes, estabilidad y
protección de proteínas, detoxificación de especies reactivas de oxigeno, toma
de agua y una serie de factores de transcripción que incluyen algunos
miembros de las familias de dedos de zinc, WRKY y bZIP. Hasta el momento
muchos estudios de expresión génica en maíz en respuesta a la sequía se han

18
llevado a cabo para diferentes órganos como raíces (Poroyko et al, 2007) y
granos en desarrollo (Yu y Setter, 2003) o estadíos de desarrollo particular
(Zheng et al, 2004). Sin embargo, no hay reportes dirigidos a la comparación
de las respuestas a sequía entre variedades tolerantes y susceptibles en maíz
o genotipos que han sido reportados como poseedoras de diferentes
mecanismos de tolerancia. Se han reportado genotipos de maíz mexicanos
con mecanismos aparentemente diferentes para alcanzar la tolerancia a
sequía. El objetivo de este estudio fue analizar las diferencias en las
respuestas fisiológicas y la expresión génica de genotipos mexicanos uno
susceptible (85-2) y dos tolerantes (Cajete criollo, CC y Michoacán 21, M21).
Los 3 genotipos fueron sometidos a tratamientos de estrés por sequía
intermedio (10 días sin agua) y severo (17 días sin agua), seguido por un riego
de recuperación y el análisis de expresión global fue evaluado a diferentes
tiempos usando microarreglos de maíz que contienen aproximadamente
56,000 oligonucleótidos. Los resultados confirman las diferentes respuestas
fisiológicas y los diferentes patrones de expresión bajo sequía y en el riego de
recuperación en las 2 variedades tolerantes y ofrece indicios de cómo los
cambios en la expresión génica pudieran conducir a la tolerancia a sequía y al
riego de recuperación en maíz. Los patrones de expresión génica involucrados
en fotosíntesis y metabolismo de carbohidratos y prolina, los que codifican a
factores de transcripción y aquellos conocidos de estar involucrados en otras
respuestas de estrés abiótico fueron estudiados en mayor detalle, aportando
información en la correlación entre las respuestas de expresión génica y
fisiológica de los tres genotipos y, permitiendo la identificación de genes
específicos y los patrones de expresión asociados con rutas metabólicas
particulares en cada una de las 3 variedades.

19
IV. ANTECEDENTES
El agua es esencial para el sostenimiento de la salud humana y la calidad
ambiental. La sequía puede afectar países enteros durante muchos años lo
que resulta en costos serios tanto sociales, como económicos y ambientales
(Parry et al, 2005). La baja disponibilidad de agua es una de las mayores
causas de la reducción en la producción de los cultivos que afecta la mayoría
de las regiones agrícolas alrededor del mundo (Bruce et al, 2002).
Globalmente, aproximadamente 22% de la tierra agrícola es salina y áreas
bajo sequía están expandidas y se prevé que se incrementará aún mas
(Bhatnagar-Mathur et al, 2006). Debido a que el agua como recurso para uso
agrícola empieza a ser más limitante, el desarrollo de líneas tolerantes a
sequía empieza a ser más importante (Bruce et al, 2002). El incremento en la
población mundial y la disminución en las tierras de cultivo disponibles han
conducido a la exploración de métodos que pudieran llevar a la utilización de
tierras no cultivables (Iyer et al, 2006). Más aún los cambios climáticos a nivel
mundial con un particular incremento de la temperatura provocarán un estrés
hídrico en partes no afectadas o no esperadas en diferentes partes del mundo.
La sequía ha mostrado tener un impacto devastador en la productividad de la
mayoría de los cultivos. El este de África sufrió una sequía severa de 2005-
2006 y la ola de sequía y calor que afecto el sur de Europa en el 2003 condujo
a una caída estimada del 20% en la productividad del maíz (Ribaut et al,
2009).
IV.1. Impacto de la sequía en México
La sequía ha jugado un rol significativo en la historia humana de México. Los
arqueólogos han investigado los impactos de la sequía en las poblaciones pre-
colombinas y han debatido la posibilidad de que la sequía pudiera haber
jugado un papel en el colapso de las civilizaciones maya y mesoamericana. La
evidencia es inconclusa y es probable que aunque las fallas en las cosechas
inducidas por la sequía y los recortes de agua pudieran haber afectado las

20
regiones de Yucatán y la parte central de México, las razones del declive
político y demográfico fueron más complejas (Livermann, 2000). En 1930 el
censo agrícola reportó un total de 1.2 millones de hectáreas perdidas por
desastres naturales, un total del 17% del área plantada, con las pérdidas más
serias en los estados del norte y norte-centro del país como Nuevo León (40%
de pérdida) y San Luis (50%). En 1940 un área similar fue perdida en los
estados al norte del país con pérdidas del 20-40% del área sembrada. El
censo de 1950 mostro pérdidas de 1.43 millones de hectáreas y 1.1 millón
sufrió sequía en regiones como El Bajío y Quintana Roo. En la década de los
60, las pérdidas fueron menores, sin embargo, en 1970 se reportaron pérdidas
por sequía en el verano del 22% del área sembrada. Cuatro años de
precipitaciones por debajo de lo normal produjeron condiciones de sequía
severa en el norte de México en 1996. Más de 4.6 millones de hectáreas de
tierra cultivable fueron dañadas y 6 millones de hectáreas quedaron sin cultivar
debido a la sequía (Livermann, 2000).
México tiene una larga y variada experiencia con sequía, descrita por crónicas
históricas tempranas o datos climáticos contemporáneos. Con más del 85%
del área mexicana definida como árida o semiárida, la lluvia interanual es
variable (Liverman, 2000). Cinco áreas del país presentan sequía severa: la de
mayor extensión se ubica sobre el noreste y norte de México y se prolonga
hasta el estado de Guanajuato; otra zona se localiza en el centro norte del
estado de Sonora; una más sobre la costa oriente de Baja California Sur,
mientras que las otras dos áreas, de menor extensión, se ubican sobre la
costa oaxaqueña y en la costa noreste del estado de Yucatán (Hernández y
Valdez,
http://www.atmosfera.unam.mx/editorial/libros/cambio_climatico/sequía.pdf
(Ver Figura 1).
Durante noviembre del 2008, la precipitación a nivel nacional estuvo 68% por
debajo del promedio climatológico. El Servicio Meteorológico Nacional (SMN)

indicó que el mes de Noviembre del 2008 como el más seco para el periodo
1941-2008. La distribución de la lluvia en noviembre se presentó
principalmente sobre el Noroeste, Noreste del país, así como en el norte y
centro de la región costera del Golfo de México. El estado que recibió la mayor
cantidad de precipitación fue Baja California con un total de 168.1% en
contraste, los estados del país que presentaron precipitaciones por debajo de
la media histórica fueron: Distrito Federal, Guanajuato, Michoacán, Morelos,
Querétaro y Tlaxcala
(http://smn.cna.gob.mx/productos/sequía/2008/noviembre/sequía1108.pdf ).
Figura 1. Indice de severidad de la sequía. (http://www.atmosfera.unam.mx/editorial/libros/cambio_climatico/sequía.pdf(htt
p://www.atmosfera.unam.mx/editorial/libros/cambio_climatico/sequía.pdf)
21

22
La heterogeneidad del paisaje mexicano y la alta variabilidad interanual del
clima también ha promovido la diversidad de variedades en los cultivos,
especialmente de maíz y muchos campesinos aún siembran una gran
diversidad de variedades para minimizar el riesgo de la sequía, heladas y
enfermedades. Los campesinos tradicionales en Oaxaca se ajustan a la
sequía seleccionando la variedad de maíz apropiada a las condiciones de
lluvia esperadas, alterando la proporción de frijol y maíz sembrados y
ajustando la densidad de siembra de los cultivos (Liverman, 2000).
IV.2. Efectos fisiológicos a nivel celular que afectan al maíz durante la sequía
La sequía afecta algunas características fisiológicas clave, que se mencionan
a continuación:
a-) Bajo un estrés moderado, la expansión celular es inhibida. Esto se
manifiesta en la reducción en la expansión del área foliar, seguido de una
reducción en el crecimiento de los estilos y en la elongación del tallo.
Finalmente un crecimiento reducido de la raíz, el cual se intensifica cuando el
estrés es mayor.
b-) Bajo un estrés severo, la división celular es inhibida, a pesar de que el
estrés haya cesado, los órganos afectados se ven carentes de células para su
expansión total.
c-) Ajuste osmótico: la mayoría de especies son capaces de producir
sustancias osmóticamente activas en el citoplasma y la vacuola en respuesta
a la sequía. Esto permite compensar el incremento en el potencial osmótico
del suelo y, que la planta tome más agua del suelo y, mantenga la turgencia y
la función celular por un periodo mayor bajo sequía. El ajuste osmótico es
particularmente aparente en sorgo, trigo y arroz (el incremento de la

23
negatividad en el ᴪs es de 1 a 1.7 MPa), pero es mucho menor en maíz (0.3 a
0.5 Megapascales [MPa]).
d-) Acumulación de prolina: la cual ha sido observada a menudo bajo sequía
severa. Podría actuar como un osmolito o proteger las estructuras proteicas
cuando la turgencia se pierde.
e-) Foto-oxidación de la clorofila: la sequía afecta más al fotosistema 2 que al
fotosistema 1 en el mecanismo fotosintético. Estos empiezan a desacoplarse,
resultando en electrones libres de alta energía en la hoja. El transporte de los
electrones no acoplados conduce a la foto-oxidación de la clorofila y la pérdida
de la capacidad fotosintética.
f-) La actividad enzimática: es en general reducida bajo sequía. Por ejemplo, la
conversión de sacarosa a almidón en el grano disminuye debido a que la
actividad de la invertasa acida (una enzima clave que convierte sacarosa a
azucares hexosa) también disminuye (Banziger, M. et al, 2000).
IV.3. Efectos de la sequía a nivel de planta completa en maíz
El cierre estomatal unido a la inhibición del crecimiento de la hoja son las
respuestas más tempranas a la sequía, protegiendo a la planta de la pérdida
excesiva de agua, la cual resultaría en la deshidratación de las células y su
muerte. La apertura y cierre de los estomas resulta de los cambios en la
turgencia de las células guarda relacionadas a las células epidermales, así
como también la energía metabólica y los cambios en la permeabilidad de la
membrana (Chaves et al, 2003). Estudios a finales de 1980s mostraron que
los estomas se cerraban en respuesta al suelo seco incluso cuando el estado
hídrico del tallo era mantenido a una turgencia alta (Chaves et al, 2003).
Evidencia posterior indicó que el cierre estomatal probablemente era mediado
por señales químicas que viajan de las raíces deshidratadas a los tallos
(Chaves et al, 2003). La producción de ABA en las raíces y su transporte a las

24
hojas brinda a la planta un mecanismo para la transmisión de una señal
química que indica el estado hídrico del suelo. El ABA es sintetizado tanto en
raíces como en hojas pero no es muy conocida la localización precisa de esta
síntesis en las raíces, el cual puede influir en cómo la planta percibe y
monitorea el contenido hídrico del suelo (Schachtman y Goodger, 2008). Un
rol importante en la señalización de la raíz al tallo bajo sequía y en la
conductancia estomatal ha sido demostrado en muchos reportes (Schachtman
y Goodger, 2008). La regulación del funcionamiento en el cierre estomatal por
ABA no es simple e involucra como se ha mencionado un transporte a larga
distancia y la modulación de la concentración de ABA en las células guarda a
una dosis dada de la hormona (Chaves et al, 2003). Entre los factores
implicados en esta modulación están la savia del xilema y el pH del tejido
foliar, el cual se podría incrementar en condiciones de alta demanda
evaporativa tales como alto déficit de presión de vapor de agua en el aire, alta
intensidad de luz y alta temperatura de hoja (Chaves et al, 2003).
Cuando el maíz experimenta el déficit hídrico hay una disminución en la
fotosíntesis, ya que la expansión foliar se ve reducida y las hojas senescen;
como consecuencia hay una reducción en la intercepción de la luz, una
reducción en la fijación de carbono por unidad de área foliar, los estomas se
cierran y la foto-oxidación daña el aparato fotosintético (Bruce et al, 2002). El
cierre estomatal en respuesta al déficit hídrico provoca también una
disminución de la tasa fotosintética (Mahajan y Tuteja, 2005). Se sabe que el
estado hídrico de la hoja interactúa con la conductancia estomatal y una buena
correlación entre el potencial hídrico de hoja y la conductancia estomatal
siempre existe, incluso bajo sequía (Reddy et al, 2004). Es probable que los
cambios en el metabolismo celular del carbono son probables de que ocurran
tempranamente en los procesos de deshidratación. La sequía generalmente
reduce la capacidad bioquímica para la asimilación del carbono y su utilización
(Reddy et al, 2004).

25
La tasa de fotosíntesis en plantas superiores depende de la actividad de la
ribulosa-1,5-bisfosfato carboxilasa/oxigenasa (rubisco) así como la síntesis de
ribulosa bifosfato carboxilasa (RuBP) (Reddy et al, 2004). Se ha sugerido
además que la actividad de la rubisco activasa también disminuye con el
incremento de la sequía. Esta enzima es una proteína abundante que regula la
conformación del sitio activo de rubisco. Como se sabe, las plantas C4 como el
maíz usan el agua más eficientemente que las C3 pero además también
necesitan rubisco para alcanzar una tasa fotosintética dada (Reddy et al,
2004). Se ha propuesto una disminución en las tasas del transporte de
electrones inducida por deshidratación y tratamiento por ABA, debido a la
desactivación de Rubisco. La disminución del CO2 intercelular después del
cierre estomatal bajo condiciones prolongadas de déficit hídrico podría inducir
un ajuste (o represión) de la maquinaria fotosintética y que ésto se asocie con
el sustrato disponible de carbono y con la disminución del crecimiento (Chavez
et al, 2003). Se ha observado también una disminución en la actividad de otras
enzimas del ciclo de Calvin (Chaves et al, 2003) como la fosforibulo-cinasa y la
fructosa-1, 6 bifosfatasa (FBPasa) con la disminución en el contenido relativo
de agua. La primera enzima en las plantas C4 durante la fotosíntesis es la
fosfoenol piruvato carboxilasa (PEP-Casa), la cual es también inhibida bajo
sequía. La sequía también produce cambios en la tasa de los productos finales
de la fotosíntesis, almidón y sacarosa (Reddy et al, 2004). Cuando el estrés
empieza a ser más severo, el crecimiento de la raíz también disminuye y la
toma de nutrientes se ve grandemente reducida (Banziger, M. et al, 2000).
IV.4. Mecanismos de tolerancia a la sequía en plantas
Los mecanismos de tolerancia a sequía en plantas pueden ser categorizados
de la siguiente manera:
a-) Escape: aquel mecanismo en el cual la planta es capaz de completar su
ciclo de vida antes de que el déficit hídrico ocurra (Chaves et al, 2003). Las

26
plantas que escapan a la sequia exhiben un alto grado de plasticidad en el
desarrollo. Este tipo de estrategias se basan en una reproducción exitosa
antes de que se produzca el estrés severo (Chaves et al, 2003). La variación
genética con respecto a la fenología y el tiempo de floración pueden ser
optimizados de tal manera que el ciclo de vida de la planta concuerde con la
disponibilidad de agua en una región ecológica dada. Este mecanismo es
especialmente importante en plantas que producen granos, ya que son más
susceptibles al déficit hídrico durante la fase reproductiva. Genotipos de
floración temprana con periodos cortos reducen la probabilidad de pérdida en
el rendimiento debido a la sequía terminal; sin embargo, estos genotipos
sacrifican rendimientos en años donde el agua está disponible y pudiera
soportar una etapa de crecimiento mayor (Mullet, 2009). Esta estrategia es
importante en regiones áridas, donde las plantas anuales pueden combinar los
ciclos de vida corta con altas tasas de crecimiento, usando al máximo los
recursos disponibles mientras la humedad en el suelo lo permita (Chaves et al,
2003).
b-) Evasión: se basa en mantener un potencial hídrico alto en los tejidos o
tolerar bajos potenciales hídricos bajo sequía. Este mecanismo está asociado
con una variedad de características adaptativas e involucra: a-) minimizar la
perdida de agua y b-) maximizar la toma de agua. La pérdida de agua es
minimizada cerrando los estomas, reduciendo la absorción de luz a través del
enrollamiento de hojas, con una superficie densa de tricomas o reduciendo el
área foliar a través de un reducido crecimiento. La toma de agua está
relacionada con las características del sistema radicular como la variación en
el desarrollo de la raíz, morfología y función (Mullet, 2009). Las características
de evasión tienen un impacto significativo en el rendimiento de grano, debido a
que ellas ayudan a las plantas a mantener un buen estado hídrico, permitiendo
una fotosíntesis, crecimiento y desarrollo continuo (Mullet, 2009).

27
c-) Tolerancia: este mecanismo permite a los tejidos vegetales soportar la
deshidratación, a través de la acumulación de proteínas tales como las
dehidrinas (hidrofilinas) y proteínas de choque térmico y un amplio rango de
solutos compatibles (por ejemplo, polioles, glicina betaína, prolina, inositol).
Las plantas también incrementan el nivel y la actividad de enzimas y rutas que
protegen los tejidos a partir de las especies reactivas de oxigeno (ROS) que
son generadas durante los periodos de limitación de agua y cierre estomatal.
Las características de tolerancia a deshidratación son especialmente
importantes en los forrajes y cultivos para la producción de biocombustibles,
donde la retención de la función foliar, el re-crecimiento seguido de periodos
relativamente severos de déficit hídrico y la acumulación de la biomasa son los
determinantes principales del rendimiento (Mullet, 2009). La tolerancia a
deshidratación es importante durante el establecimiento de la plántula si la
tolerancia ayuda a mejorar su establecimiento y minimiza la necesidad de re-
plantar. La tolerancia incrementada de los tejidos foliares a las especies
reactivas de oxigeno ayudan a minimizar el daño del aparato fotosintético
durante el déficit hídrico y éste debería mejorar la acumulación de biomasa,
manteniendo la parte foliar fotosintéticamente activa. Sin embargo, durante la
fase vegetativa, las hojas más viejas normalmente senescen y sus
constituyentes son movilizados y re-utilizados para la producción de hojas
nuevas. Por lo que las plantas pueden recuperarse de un déficit hídrico
transitorio, que parcialmente dañe un conjunto de hojas, a través de la
producción de hojas adicionales durante el curso normal del desarrollo. Debido
a que los mecanismos de tolerancia son inducidos usualmente bajo
condiciones de déficit hídrico de la planta con cierre estomatal e inhibición de
la fotosíntesis, estos mecanismos podrían proteger las estructuras pre-
formadas y acelerar la recuperación pero sus impactos en la acumulación de
biomasa y el rendimiento pueden ser relativamente pequeños si la limitación
de agua es transitoria. La expresión constitutiva de algunos mecanismos de
tolerancia podrían disminuir la productividad e incrementar el riesgo de la

28
muerte de la planta reduciendo la habilidad de la misma para evitar una
pérdida de agua más severa (por ejemplo previniendo la senescencia foliar y
la pérdida foliar, la cual es un mecanismo clave para disminuir la utilización de
agua en algunas plantas) (Mullet, 2009).
IV.5. Etapas críticas del maíz bajo sequía
El maíz es una de las especies más sensibles al déficit hídrico a pesar de su
metabolismo C4, que le confiere una tasa fotosintética alta combinada con una
tasa de transpiración relativamente baja (Welcker et al, 2007). En el caso del
maíz, los factores que afectan el desarrollo del grano empieza a ser el enfoque
principal en mejorar el rendimiento y la estabilidad del rendimiento en
germoplasmas nuevos (Bruce et al, 2002). Para el maíz, el periodo de
desarrollo crítico para determinar el rendimiento de grano se centra en la
floración (Bruce et al, 2002), especialmente sobre el periodo de tiempo de una
semana antes a una semana después de floración (Welcker et al, 2007) y en
el desarrollo temprano de la semilla.
Numerosos estudios han examinado componentes de respuesta a estrés
durante la etapa de floración. Un estudio reciente mostró que un flujo continuo
de fotoasimilados juega un papel importante tanto en el desarrollo del óvulo
como de la semilla. La interrupción de este flujo debido a la deshidratación, por
ejemplo, causa reducciones en las actividades metabólicas de enzimas clave
involucradas en el metabolismo de carbohidratos (Bruce et al, 2002). Debido a
que el maíz es una especie de polinización cruzada, el polen debe de moverse
de las anteras en la parte superior de la planta a los estigmas expuestos de la
misma planta y de plantas circundantes. Este proceso es riesgoso debido a
que el polen y el delicado tejido estigmático son expuestos a ambientes secos.
Un fenómeno universal observado cuando las flores de maíz están bajo sequía
es el retraso de la aparición de los estigmas en relación a la dispersión del

29
polen, dando lugar al intervalo de la antesis-aparición de estigmas medido en
días (ASI) cuya duración es altamente correlacionada con los factores que
afectan el grano. Bajo estas condiciones el polen puede llegar después de que
se ha secado o cuando los estigmas se han marchitado o después de que los
ovarios hayan extenuado sus reservas de almidón (Bruce et al, 2002). Se ha
propuesto que un ASI más largo está asociado a una mayor inversión en las
estructuras reproductivas masculinas y femeninas, debido a que la variación
en el rendimiento mostró una asociación negativa en el ASI mientras que no
se observó una asociación con los mecanismos de evasión a deshidratación,
lo que sugiere una estrategia evolutiva de sobrevivencia priorizando la
transmisión de genes vía polen (Reynolds y Tuberosa, 2008). La importancia
del tiempo de floración en términos de reducción en el rendimiento bajo
condiciones de sequía está asociado por la correlación mayor del rendimiento
de grano con el número de granos por planta más que el peso de grano
(Tuberosa et al, 2007).
IV.6. Rutas de transducción de señales general a estrés
Una ruta de transducción de señales general inicia con la percepción de la
señal (Xiong et al, 2002), seguida por la generación de los segundos
mensajeros (por ejemplo, especies reactivas de oxígeno (ROS de sus siglas
en inglés). Los segundos mensajeros como los inositol fosfatos (Mahajan y
Tuteja, 2005) pueden modular los niveles de Ca2+, que a menudo inician con
una cascada de fosforilación y terminan con proteínas blanco involucradas
directamente en la protección celular o en factores de transcripción que
controlan un grupo específico de genes regulados por estrés (Xiong et al,
2002). Los productos de estos genes conducirían a la adaptación de la planta
y ayudarla a sobrevivir y afrontar las condiciones desfavorables. Asimismo, los
cambios en la expresión génica podrían participar en la generación de

30
moléculas regulatorias como las hormonas ácido abscísico (ABA), etileno y
ácido salicílico (Xiong et al, 2002; Mahaja y Tuteja, 2005).
La transducción de la señal requiere de una coordinación espacial y temporal
de todas las moléculas de señalización. Por lo que existen ciertas moléculas
que participan en la modificación, ensamble y envío de los componentes de
señalización, pero no recaen directamente en la señal (Xiong et al, 2002).
IV.6.1. Percepción y señalización del déficit hídrico
Numerosos estudios han mostrado que las plantas pueden percibir cambios en
factores abióticos tales como contenido hídrico y oxigeno del suelo, así como
cambios en la composición de los nutrientes del suelo (Schachtman y
Goodger, 2008). Algunos estudios han mostrado que las señales químicas
como ABA son generadas por las reducciones en el contenido de agua en el
suelo y actúan en las hojas para reducir la transpiración y el crecimiento
(Schachtman y Goodger, 2008). Se sabe también que existe un considerable
sobrelapamiento entre las rutas de señalización de estrés abiótico (en parte a
través de la producción de especies reactivas de oxigeno), con la especificidad
al estrés hídrico presente, por ejemplo en la percepción inicial del estrés
(Chaves et al, 2003). La señalización puede ocurrir localmente o a larga
distancia. La señalización de la raíz al tallo requiere que los compuestos
químicos o señales físicas viajen a través de la planta en respuesta al estrés
percibido por las raíces. La importancia relativa de las señales químicas e
hidráulicas para el estrés hídrico influyen en el caso del control de la apertura
estomatal y la regulación de la transpiración donde es probable que ambas
señales actúen (Chaves et al, 2003).
El primer paso en iniciar la respuesta molecular a una señal ambiental (como
el déficit hídrico) es la percepción de la señal por receptores específicos. Dada
la activación, estos receptores inician (o suprimen) una cascada para transmitir

31
la información a través de la ruta de transducción de señales. Siguiendo los
estudios en levadura, fue mostrado que la percepción inicial del déficit hídrico
era mediado a través de una histidina cinasa transmembranal que funciona
como un osmosensor (SLN1) (Chaves et al, 2003; Bartels y Sunkar, 2005).
Estudios posteriores mostraron el déficit hídrico seguido de un estrés
osmótico, condujo a la expresión de un osmosensor putativo AtHTK1 en
Arabidopsis (Chaves et al, 2003). AtHTK1 tiene un dominio de histidina cinasa,
un dominio receptor y dos dominios transmembranales y podría ser el primer
componente en percibir los cambios en el potencial osmótico dentro de la
célula y conducir la cascada de señalización río abajo que resulta de la
expresión génica inducida por deshidratación (Chaves et al, 2003).
Siguiendo la percepción de los cambios osmóticos durante el estrés hídrico
por estimulación del osmosensor, la cascada de transducción de señales
involucra la fosforilación y defosforilación de proteínas mediadas por muchas
proteínas cinasas y fosfatasas cuyos genes han mostrado ser inducidos por
estrés hídrico. Algunas de las proteína cinasas regulatorias más abundantes
involucradas en la señalización por estrés abiótico son proteína cinasas
dependientes de Ca+2 (CDPKs) y proteína cinasas activadas por mitógeno
(MAPK). Muchas MAPKs y CDPKs han sido identificadas en las plantas
sometidas a estrés hídrico y se ha mostrado que están involucradas en
transducir las señales de deshidratación sensadas de la membrana plásmica
al núcleo (Chaves et al, 2003).
Aunque es poco probable que exista un único sensor que perciba el estrés y
controle toda la señalización subsecuente, un sensor podría regular sólo parte
de la cascada de señalización que es iniciada por un aspecto de la condición
del estrés. Por ello debe de existir múltiples sensores primarios que perciban
la señal de estrés abiótico inicial (Xiong et al, 2002). Se sabe que el estrés por
frio, sequia y salinidad inducen un flujo transitorio de Ca+2 en el citoplasma de

32
la celula. Por lo que, los canales responsables para este flujo de Ca+2 podría
representar un tipo de sensor para estas señales de estrés (Xiong et al, 2002).
En la Fig. 2 se presenta un resumen de la ruta de transducción de señales
genérica para la sequía y el estrés en general que empieza con la percepción
de la señal, seguido por la generación de segundos mensajeros. Los
segundos mensajeros pueden modular el Ca+2 intracelular que a menudo
inician una cascada de fosforilación y finalmente involucra directamente las
proteínas que están encargadas de la protección celular o de los factores de
transcripción que controlan los genes específicos regulados por estrés (Xiong
et al, 2002).

Percepción de la señal de
estrésEjemplos de los componentes
Señal
Canales iónicos, histidinacinasa
Receptores de la señal
Ca 2+ Moléculas de deseñalización secundaria
ROS, ABA
Cascadas de fosfoproteinas
CDPK, MAPK, protein fosfatasa
Factores de trascripción
EREBP/AP2, bZIP
Genes de respuesta a estrés
LEA, antioxidantes, enzimasinvolucradas en la osmoprotección y transportadores
Respuestas
Tolerancia a estrés, arresto en el crecimiento o
muerte celular
Figura 2. Ruta general para la transducción de señales de frío,
sequia y estrés salino en plantas (Tomado de Xiong et al., 2002)
33

34
IV.6.2. Inducción del estrés oxidativo debido a la sequía en plantas
Se sabe que la sequía inhibe la actividad fotosintética en tejidos debido al
desbalance entre la captura de la luz y su utilización (Reddy et al, 2004). La
represión de la actividad del fotosistema II (PSII) resulta en un desbalance
entre la generación y la utilización de electrones (Reddy et al, 2004).
Bajo déficit hídrico, la luz absorbida ya sea en la fotosíntesis o en la
fotorespiración y la disipación térmica no es suficiente para afrontar el exceso
de energía. Esto origina una producción de moléculas altamente reactivas
(Chaves et al, 2003). Estas moléculas generadas en el cloroplasto pueden
causar daño oxidativo al aparato fotosintético.
El estrés oxidativo es un término general usado para describir un estado de
daño causado por la especies reactivas de oxigeno (ROS) (Chaves et al,
2003). Estas especies reaccionan con las macromoléculas biológicas más
sensibles en las células deteriorando sus funciones. Sin embargo, bajo
condiciones de estrés intenso y debido a las moléculas dañadas severamente,
una cascada de eventos resultaría en la muerte celular. La sobrevivencia de
las células bajo condiciones estresantes es determinado por la duración del
estrés así como la capacidad protectora de la planta (Reddy et al, 2004).
Las ROS juegan un rol crucial en causar el daño celular bajo sequía. La
secuencia de eventos en el tejido vegetal es: 1) producción incrementada de
ROS, 2) incremento en la expresión de genes para funciones antioxidantes, 3)
incremento en los niveles de sistemas antioxidativos y antioxidantes, 4)
incremento en la capacidad de acarreamiento de ROS, resultando en la
tolerancia a la sequía. Aunque una serie de mecanismos regulatorios han
evolucionado en las células vegetales para limitar la producción de estas
moléculas toxicas, el daño oxidativo sigue siendo un problema, ya que causa
perturbaciones en el metabolismo como la pérdida de la coordinación entre los

35
procesos de producción de energía y utilización de la energía durante la
fotosíntesis en hojas bajo estrés (Reddy et al, 2004).
Durante la sequía se producen especies reactivas de oxigeno como el
superóxido, el peróxido de hidrógeno (H2O2) y los radicales hidroxilo. Estas
especies reactivas de oxígeno necesitan ser destruídas o secuestradas por la
planta (Mahajan y Tuteja, 2005). Las moléculas antioxidantes y enzimas que
están localizadas en diferentes compartimentos celulares pueden secuestrar
los ROS. Estas incluyen las súper óxido dismutasas (SOD), las cuales
catalizan la dismutación de O2- a H2O2; catalasas (CATs) que son
responsables para la remoción de H2O2 (Chaves et al, 2003) y aquellas que
incluyen un grupo de ascorbato y glutatione que controlan la concentración
intracelular de especies reactivas de oxígeno (Mahajan y Tuteja, 2005). Las
peroxiredoxinas son un nuevo tipo de proteínas antioxidantes, no relacionadas
con alguna de las familias de las peroxidasas. Estas proteínas funcionan en la
defensa antioxidante en la fotosíntesis, respiración, respuesta al estrés y
señalización redox. Se ha mostrado su participación en la detoxificación de
ROS y especies reactivas de nitrógeno (Iyer et al, 2006).
El grado con el que se incrementan las actividades de las enzimas
antioxidantes y la cantidad de antioxidantes bajo sequía podría ser
extremadamente variable entre las diversas especies vegetales e incluso entre
dos cultivares de la misma especie. El nivel de respuesta depende de la
especie, el desarrollo y el estado metabólico de la planta, así como la duración
e intensidad del estrés (Reddy et al, 2004). La aclimatación a la sequía esta
generalmente asociada con la actividad de las enzimas antioxidantes,
manteniendo la concentración de ROS relativamente baja (Chaves et al,
2003). Las especies reactivas de oxígeno también actúan como segundos
mensajeros en la transducción de señales REDOX y están implicados en
eventos mediados por hormonas. El peróxido de hidrogeno (H2O2) actúa como
una señal para el cierre de los estomas, la aclimatación de la hoja a alta

36
irradiación y la inducción de proteínas de choque térmico (heat shock
proteins). Las membranas de los cloroplastos son sensibles al estrés oxidativo
causado por la generación de una cantidad excesiva de especies reactivas de
oxigeno en sus membranas. Las especies reactivas de oxigeno pueden causar
una peroxidación y de-esterificación de los lípidos de la membrana así como
conducir a la desnaturalización de las proteínas (Mahajan y Tuteja, 2005).
IV.6.3. El Estrés osmótico activa la señalización por fosfolípidos
La membrana plasmática, como barrera selectiva entre las células y su medio
ambiente, juega un papel clave en la percepción y transmisión de la
información externa. En el estrés osmótico, se han detectado cambios en la
composición de fosfolípidos en las plantas así como en otros organismos.
Durante la exposición al estrés, el principal rol de los fosfolípidos es formar el
esqueleto de las membranas celulares, sin embargo además podrían servir
como precursores para la generación de moléculas que actúan como segundo
mensajeros (Xiong et al, 2002). Las enzimas más relevantes en la degradación
de fosfolípidos son las fosfolipasas A2, C y D; siendo la más estudiada la
fosfolipasa C. La fosfolipasa C hidroliza el fosfotidilinositol 4,5-bifosfato (PIP2)
(Chaves et al, 2003; Xiong et al, 2002) a 1,4,5 trifosfato (IP3) y diacilglicerol
(DAG), los cuales actúan como segundos mensajeros (Chaves et al, 2003). En
plantas, se ha reportado el papel del IP3 exógeno en liberar Ca+2 (Mahajan y
Tuteja, 2005; Xiong et al, 2002). Los niveles de IP3 se incrementan en plantas
de Arabidopsis bajo estrés salino e incrementos transitorios en niveles de IP3
fueron también observados en tejidos vegetales o células cultivadas drante
estrés salino (Xiong et al, 2002). En las células guarda, el IP3 induce el
incremnto de Ca+2 en el citoplasma, conduciendo al cierre estomatal y por lo
tanto a la retención de agua en las células (Mahaja y Tuteja, 2005). El estrés
osmótico activa la fosfolipasa D (PLD), la cual cortalos fosfolipidos de la
membrana para producir ácido fosfatídico (PA). El PA podría sevir como

37
mensajero en plantas. En protoplastos de células guarda la actividad de PLD
media elcierre estomatal inducido por el ácido abscisico (ABA). La sequía y la
hiperosmolaridad activa a la PLD y conduce a un incremnto transitorio en los
niveles de PA en plantas (Xiong et al, 2002). Se ha observado que PLD es
rápidamente activado en respuesta de sequía en 2 especies de plantas:
Craterostigma platagineum y Arabidopsis (Mahajan y Tuteja, 2005).
IV.6.4. Señalización por Ca+2 durante la deshidratación y el estrés salino
El calcio funciona como un segundo mensajero, el cual se acopla a un amplio
rango de estímulos extracelulares y respuestas intracelulares (Bartels y
Sunkar, 2005). Hasta ahora, tres grandes clases de sensores de Ca+2 han
sido caracterizados en plantas. Estas clases son calmodulina, las proteína
cinasas dependientes de Ca+2 (CDPKs) y las proteínas tipo calcineurin B
(CBLs). Muchas líneas de investigación sugieren que estas tres clases de
sensores de Ca+2 están involucradas en la transducción de señales por estrés
y principalmente bajo estrés salino (Bartels y Sunkar, 2005). Se ha sugerido
que las proteínas cinasas dependientes de Ca+2 (CDPKs) son las principales
en acoplar la señal a cascadas específicas de fosforilación. La sobre-
expresión de OsCDPK7 resultó en una tolerancia a estrés osmótico y frío en
arroz. Por lo tanto, las CDPKs de alguna manera juegan un rol en el desarrollo
de la tolerancia a estrés. Una demostración clara del involucramiento de CDPK
en la transducción de señales por estrés ha derivado de experimentos en los
cuales una AtCDPK1 activa indujo la expresión de un promotor de respuesta a
estrés HVA1 gen reportero en protoplastos de hojas de maíz (Xiong et al,
2002).
IV.6.5. Otras rutas fosfoproteicas de señalización
Además de las rutas de proteína cinasas reguladas por Ca+2, las plantas
también usan otros módulos de fosfoproteína para la señalización de estrés

38
abiótico. (Xiong et al, 2002). Las cascadas de proteína cinasa activadas por
mitógeno son módulos de señalización comunes en células eucarióticas
incluyendo plantas. Una característica principal de las cascadas de MAPK es
su composición de tres proteínas cinasas ligadas funcionalmente. Al final de la
cascada, la activación del módulo MAPK citoplásmico a menudo induce la
translocación de la MAPK dentro del núcleo donde la cinasa es capaz de
activar los genes a través de la fosforilación de los factores de transcripción.
IV.7. Respuestas bioquímicas a nivel celular durante la sequía
Las respuestas celulares al déficit hídrico incluye la pérdida de turgencia,
cambios en la fluidez y composición de la membrana plasmática y cambios en
la actividad del agua y/o concentración de solutos (Chaves et al, 2003). Los
solutos compatibles se acumulan en los organismos en respuesta a estrés
osmótico. La función primaria de los solutos compatibles es mantener la
turgencia celular y por consiguiente el manejo del gradiente para la toma de
agua. Existen estudios que indican que los solutos compatibles además
pueden actuar como secuestradores de radicales libres o chaperonas
moleculares, estabilizando las membranas y/o proteínas (Wang et al, 2003),
facilitando el ajuste osmótico. El agua se mueve de un potencial hídrico alto a
un potencial hídrico bajo y la acumulación de estos osmolitos produce un
potencial hídrico bajo dentro de la célula, previniendo la pérdida de agua
intracelular (Mahajan y Tuteja, 2005).
IV.7.1. Osmoprotectantes
Los solutos compatibles se pueden agrupar en 3 clases importantes:
aminoácidos (como por ejemplo prolina), aminas cuaternarias (como por
ejemplo la glicina betaína, y la dimetilsulfonatopropionato) y los poliol/azúcares
(por ejemplo, el manitol, la trealosa) (Wang et al, 2003). Estos compuestos son
llamados solutos compatibles debido a que incluso en altas concentraciones
no inhiben la actividad de las enzimas. Estos compuestos también protegen a

39
las enzimas y las membranas contra efectos deletéreos de los iones tales
como el Na+ y el Cl-. Aunque muchos compuestos osmoprotectantes confieren
protección al estrés en las bacterias, las algas marinas, las células animales y
las plantas, sus rutas sintéticas a menudo difieren en términos de enzimas y
pasos. El osmoprotector es sintetizado en respuesta al estrés y se acumula en
el citoplasma; iones inorgánicos tales como Na+ y Cl- son preferencialmente
secuestrados en la vacuola. Por lo que, esto conduce a mantener la turgencia
de la célula bajo estrés osmótico (Rathinasabapathi, 2002).
Muchos cultivos no tienen la capacidad de sintetizar los osmoprotectores
especiales que son naturalmente acumulados por organismos tolerantes al
estrés (Bhatnagar, 2008). Se cree que la osmorregulación pudiera ser la mejor
estrategia para la tolerancia al estrés abiótico, especialmente si los genes
osmoregulatorios pudiesen ser desencadenados en respuesta a sequía,
salinidad y altas temperaturas. Varias estrategias están siendo llevadas a cabo
para diseñar genéticamente la osmoprotección en plantas. El primer paso
involucrado en obtener plantas transgénicas tolerantes ha sido diseñar los
genes que codifican enzimas para la síntesis de osmolitos seleccionados. Esto
ha resultado en una serie de reportes que involucran osmoprotectantes como
glicina-betaína y prolina. Además, un número de alcohol azúcares han sido
seleccionados para el mejoramiento de la sobre-producción de solutos
compatibles, protegiendo la membrana y los complejos proteicos durante el
estrés (Bhatnagar, 2008).
Los resultados de los estudios en ingeniería genética en las rutas biosintéticas
y metabólicas de osmoprotectores indican que la acumulación de los solutos
compatibles podrían proteger a las plantas contra el daño causado por el
estrés, secuestrando a las especies reactivas de oxigeno (ROS) y por sus
actividades parecidas a chaperonas en mantener las estructuras y funciones
proteicas (Bhatnagar, 2008).

40
IV.8. Estrategias para identificar genes asociados con la tolerancia a sequía en maíz
La identificación de genes o características que mejoran la tolerancia a sequía
en plantas puede darse por diferentes vías: a) identificando los genes que se
encuentran inducidos/reprimidos en respuesta al déficit hídrico; b) analizando
las mutantes que muestren una respuesta modificada a las limitaciones de
agua; c) por expresión ectópica de factores de transcripción y otros genes,
seguido de escrutinios para observar el comportamiento de las plantas bajo
condiciones limitantes de agua, d) analizando las características fisiológicas
seguidas del mapeo de QTL y la clonación de los genes/alelos encontrados en
las colecciones de los germoplasmas y e) mediante un análisis comparativo
entre variedades tolerantes y resistentes a sequía (Mullet, 2009).
IV.8.1. Loci de características cuantitativas (QTL) en los estudios de sequía en maíz
El análisis de QTL es una estrategia complementaria poderosa ayudada por la
genómica para descubrir y aislar los genes de mayor interés para el
agrónomo. La habilidad de los análisis de QTLs es identificar aquellos genes
que regulan o controlan la variación en la expresión fenotípica que requieren la
integración del QTL y los enfoques genómicos para el mejoramiento del cultivo
(Mc Mullen, 2003)
El primer paso crítico es identificar QTLs que juegan un rol mayor en gobernar
la variabilidad genética para la tolerancia a la sequía. En este contexto, la
prioridad debe ser dada a los QTLs caracterizados por una interacción limitada
con el régimen de agua y otras variables ambientales y por consiguiente
asociadas al efecto en el rendimiento. El siguiente paso es verificar la
extensión del efecto del alelo del QTL benéfico que sea consistente con el
fondo genético a ser mejorado (Tuberosa et al, 2007).

41
En el maíz, durante la última década se ha aplicado el uso de QTLs en lo que
respecta a tolerancia a sequía (Ribaut et al, 2009). Existe un gran número de
QTLs que gobiernan la variación en la concentración de ABA debido a las
diferentes poblaciones estudiadas, diferentes condiciones de crecimiento,
dinámica del déficit hídrico y en el estadio de crecimiento a la cual las plantas
fueron sometidas a la sequía (Tuberosa et al, 2002). Dentro de ellos, se
encontró un QTL mayor que originalmente mostró ser afectado por la
concentración de ABA en hoja y después que estaba involucrado en la
arquitectura radicular y otras características relacionadas a sequía. Los
resultados sugirieron un efecto primario del QTL en la arquitectura de la raíz
seguido por un efecto secundario en el ABA acumulado en la hoja (Tuberosa
et al, 2007). Asimismo, se ha encontrado QTLs que influyen en el potencial
hídrico de hoja localizados en las regiones cerca de los QTLs para
concentración de ABA en hoja y en xilema (Tuberosa et al, 2002).
Por otro lado, se ha demostrado que las características de las raíces son un
factor importante que influye en la evasión a la sequía. En el maíz, la fuerza de
la raíz es una de las características que determina la capacidad exploratoria a
gran escala de las raíces debajo de la superficie del suelo. Se han identificado
QTLs para el número de raíces seminales, numero de raíces del nódulo en la
base del tallo y QTLs que regulan otras características de la raíz como longitud
de raíz primaria, diámetro de la raíz primaria, peso de la raíz primaria y el peso
de las raíces seminales adventicias (Tuberosa et al, 2002).
La implementación de mejoramiento asistido por marcadores (MAS) basados
en QTLs ha sido aplicado satisfactoriamente en el mejoramiento del maíz
(Ribaut y Ragot, 2007). El enfoque del mejoramiento asistido por marcadores
(MAS) se ha desarrollado para tener estimaciones exactas de los efectos de
los QTLs en una población de referencia relativamente reducida, después de

42
monitorear que los efectos de los QTLs deseables no variarán después de un
número de ciclos de selección (Tuberosa et al, 2007). La primera aplicación de
MAS a gran escala para la tolerancia a sequía en maíz fue realizada en el
CIMMYT para mejorar líneas elite y poblaciones de polinización abierta. Con la
finalidad de reducir el intervalo entre la emergencia de la flor femenina y
masculina (ASI). El ASI bajo regímenes de déficit hídrico está negativamente
asociado con el rendimiento de grano (Tuberosa et al, 2007). Estos estudios
de QTL identifican frecuentemente loci relacionados a tiempo de floración y la
mayoría más fuertemente asociados con la adaptación a sequía. Sin embargo,
sólo unos pocos QTLs de efecto mayor han sido bien documentados, como
aquellos QTLs asociados con un alto rendimiento con probabilidades de ser
optimizados a través de mejoramiento convencional en materiales elite
(Reynolds y Tuberosa, 2008). La disponibilidad de estos marcadores
moleculares ligados a QTLs para el ASI permiten una selección más efectiva
bajo sequía así como cuando la sequía ocurre en la floración. Sin embargo,
esta ventaja se ve disminuída cuando la intensidad del estrés es más baja o el
estrés desaparece reduciendo el rendimiento a menos del 40%, aunque en
algunas líneas seleccionadas bajo condiciones de riego no mostraron alguna
penalidad en el rendimiento comparado con las líneas control (Tuberosa et al,
2007). El uso de MAS se ve limitado además por los costos comparado con
las prácticas de mejoramiento convencional (Tuberosa et al, 2007).
La eficiente identificación de genotipos con tolerancia a estrés abiótico
requiere un manejo cuidadoso de la sequía para exponer las variaciones
genéticas en fenotipos tales como ASI y fecundidad (Bruce et al, 2002). Las
pruebas en campo necesitan ser establecidas donde la probabilidad de sequía
es alta, y el tiempo, la duración e intensidad del estrés puede ser modulado
por irrigación y donde hay similitudes obvias al área de adaptación del
germoplasma bajo estudio. Los tres elementos importantes para la
caracterización de la sequía para un mejoramiento exitoso en la tolerancia a
sequía son: tiempo, duración e intensidad del estrés. La mayoría de los

43
periodos de estrés que se encuentran en las áreas de crecimiento de grano
alrededor del mundo son transitorios, impredecibles e imprecisamente
medibles, conduciendo a una dificultad en esfuerzos de mejoramiento para la
tolerancia a sequía (Bruce et al, 2002).
La experiencia en el CIMMYT y en Pioneer Hi-Bred indica que las
características secundarias clave bajo sequía son fertilidad reducida, intervalo
antesis-aparición de estilos, “staygreen” y en menos medida el enrollamiento
de hoja bajo sequía. Los sistemas radiculares obviamente juegan un papel
importante en la adquisición para las plantas y son un componente significativo
de tolerancia al déficit hídrico (Bruce et al, 2002).
IV.9. Productos génicos de respuesta a la sequÍa
Las plantas responden y se adaptan al déficit hídrico tanto a nivel celular como
molecular. Una serie de genes con diversas funciones son inducidos o
reprimidos por el estrés. La mayoría de estos productos génicos podrían
funcionar en la respuesta al estrés y la tolerancia a nivel celular. El análisis de
las funciones de estos genes es crítico para un mayor entendimiento de los
mecanismos moleculares que gobiernan la respuesta y tolerancia del estrés en
plantas (Shinozaki y Yamaguchi-Shinozaki, 2007).
IV.9.1. Proteínas LEA y genes de choque térmico (HSP)
Las proteínas LEA representan una categoría de proteínas de alto peso
molecular que son abundantes durante la embriogénesis tardía y se acumulan
durante la desecación de la semilla y en respuesta a estrés hídrico (Bhatnagar,
2008). Entre los grupos de proteínas LEA, aquellos que pertenecen al grupo 3
se sabe que juegan un papel en el secuestramiento de iones que están
concentrados durante la deshidratación celular. El grupo 1 de las proteínas
LEA se predice que tienen de tener una capacidad de unión al agua, mientras
el grupo 5 de las proteínas LEA se piensa que secuestran los iones durante la
pérdida de agua. La sobre-expresión constitutiva de HVA1, un miembro del

44
grupo 3 de las proteínas LEA de cebada confirió tolerancia al déficit hídrico y
estrés salino en plantas transgénicas de arroz (Bhatnagar, 2008). La expresión
inducida por estrés o constitutiva del gen HVA1 resultó en el mejoramiento de
las características de crecimiento y tolerancia a estrés en términos de
integridad celular en trigo y arroz bajo condiciones de estrés salino e hídrico
(Bhatnagar, 2008). Además se ha propuesto una función protectora tipo
chaperona de las proteínas LEA que actúan contra el daño celular, indicando
el rol de las proteínas LEA en la anti-agregación de enzimas en plantas
sometidas a estrés por desecación y frio (Bhatnagar, 2008).
La respuesta a choque térmico, definida como la transcripción incrementada
de un grupo de genes en respuesta a la exposición a un agente tóxico o en
respuesta a calor es una respuesta biológica altamente conservada que ocurre
en todos los organismos (Bhatnagar, 2008). La respuesta es mediada por un
factor de transcripción (HSF), el cual se presenta en una forma monomérica en
células no estresadas y es activado por estrés formando un complejo capaz de
unirse a los promotores de genes de choque térmico. La inducción de los
genes que codifican proteínas de choque térmico (Hsps) es uno de las
respuestas más prominentes observadas a nivel molecular de organismos
expuestos a alta temperatura (Bhatnagar, 2008). Existen reportes que han
relacionado positivamente los niveles de proteínas de choque térmico y la
tolerancia a altas temperaturas (Bhatnagar, 2008).
IV.9.2. Genes de transporte
Una estrategia importante para alcanzar la tolerancia al estrés abiótico es
ayudar a las plantas a re-establecer la homeostasis bajo condiciones
ambientales estresantes, restaurando tanto la homeostasis iónica y osmótica
(Bhatnagar, 2008). La tasa de flujo de agua dentro o fuera de las células está
determinada por el gradiente de potencial hídrico que actúa como una fuerza
directriz para el transporte y por la permeabilidad de agua de la membrana
(Bartels y Sunkar, 2005). Bajo condiciones no estresantes, las plantas

45
mantienen su balance hídrico, ajustando la conductancia del agua en sus
tejidos. Los tejidos vasculares y las células guarda juegan un papel importante
en este proceso. Un componente significante en el transporte de agua celular
son las proteínas denominadas aquaporinas (Ramanjulu y Bartels, 2002).
IV.9.3. Aquaporinas
Las aquaporinas son una familia compleja de canales proteicos que facilitan el
transporte de agua a través de los gradientes de potencial hídrico
transmembranales. Estas aquaporinas pueden regular la conductividad
hidráulica de las membranas y potenciar de 10 a 20 veces el incremento en la
permeabilidad de la membrana al paso del agua (Ramanjulu y Bartels, 2002).
Las aquaporinas son miembros de una gran familia de proteínas conformadas
por las Proteínas Intrínsecas Mayores (MIPs) (Bartels y Sunkar, 2005). Hasta
el momento, 14 aquaporinas han sido identificados en mamíferos, mientras
que en plantas existe una gran abundancia de homólogos de aquaporinas. La
mayor diversidad de aquaporinas en plantas se ha atribuído al grado mayor de
compartamentalización de las células vegetales y una mayor necesidad para
el control fino de transporte de agua y adaptación a los cambios en el medio
ambiente (Kaldenhoff et al, 2008). En plantas, las aquaporinas se pueden sub-
dividir en 4 grandes grupos indicando la localización del tejido vegetal o su
posible localización sub-celular: proteínas intrínsecas de membrana
plasmática (PIPs), proteínas intrínsecas de tonoplasto (TIPs), proteínas
intrínsecas tipo NOD-26 (Kaldenhoff et al, 2008), las cuales se establecen a
partir de la simbiosis entre las plantas leguminosas y las bacterias como
Rhizobium y Bradyrhizobium, formándose los nódulos de la raíz. Estas
bacterias se encuentran encerradas en un organelo específico conocido como
simbiosoma, el cual está delimitado por una membrana de origen vegetal,
conocido como la membrana del simbiosoma, la cual regula el flujo de
nitrógeno fijado del endosimbionte al citosol de la planta. Durante el desarrollo

46
de los nódulos, la expresión de un número de proteínas especificas de nodulo
(nodulinas) se inducen (Rivers et al., 1997).
Las respuestas fisiológicas a la sequía mencionadas anteriormente tales como
cierre estomatal, disminución de la fotosíntesis y disminución en el transporte
de asimilados son procesos que probablemente sean influenciados por la
función de las aquaporinas (Kaldenhoff et al, 2008). Sin embargo los estudios
que tratan de relacionar las respuestas fisiológicas al estrés hídrico con
patrones de expresión de diferentes aquaporinas han conducido a resultados
contradictorios, principalmente debido a que los patrones de expresión son
complejos y diferentes formas de aquaporinas podrían inducir distintas
respuestas. Actualmente las respuestas de las aquaporinas al déficit hídrico
podría involucrar la expresión tanto inducida como reprimida de los genes o no
mostrar cambios dependiendo del tiempo y la intensidad del estrés. Aunque se
piensa que las aquaporinas juegan un papel en ayudar a las plantas a
mantener la homeostasis y el balance de agua bajo condiciones de déficit
hídrico (Kaldenhoff et al, 2008).
IV.9.4. Factores de transcripción
Una categoría atractiva para la manipulación y regulación génica involucrada
en las respuestas a estrés abiótico, es el grupo pequeño de factores de
transcripción (FT) que han sido identificados que unen a los elementos
regulatorios de genes que son regulados por estreses abióticos (Bhatnagar-
Mathur et al, 2008). Los FT activan cascadas de genes que actúan en conjunto
para conferir la tolerancia hacia múltiples estreses. Docenas de estos FT están
involucradas en la respuesta de la planta a sequía. La mayoría de estos caen
dentro de distintas familias de FT, tales como APETALA2/FACTOR DE
RESPUESTA A ETILENO (AP2/ERF), ZIPPER DE LEUCINA (bZIP), NAC,
MYB, MYC, la familia dedos de zinc Cysteina2/Histidina2 (C2H2) y WRKY.
Miembros individuales de la misma familia a menudo responden de manera
diferente a varios estímulos de estrés. Por otro lado, algunos genes de

47
respuesta a estrés podrían compartir los mismos FT, como lo indican los
perfiles de expresión génicos que se traslapan y que son inducidos en
respuesta a diferentes estreses (Bhatnagar-Mathur, 2008).
El análisis de la expresión de genes inducibles bajo déficit hídrico ha sugerido
la existencia de una ruta independiente y dependiente de ABA (Yamaguchi-
Shinozaki y Shinozaki, 2006). Se ha hipotetizado la existencia de al menos
cuatro rutas de señalización independiente en la activación de genes
inducibles de estrés bajo condiciones de deshidratación: dos son dependientes
de ABA y dos independientes de ABA (Shinozaki & Yamaguchi-Shinozaki,
1997). Los mecanismos moleculares que regulan la expresión génica en la
respuesta a la deshidratación y el estrés por frío han sido estudiados
analizando los elementos que actúan en cis y trans que funcionan en la
expresión génica dependiente e independiente de ABA durante el estrés en
Arabidopsis (Yamaguchi-Shinozaki y Shinozaki, 2006) (Ver Fig. 3).
IV.10. Desarrollo de plantas transgénicas para la tolerancia a estrés abiótico
Gracias al rápido progreso en la secuenciación genómica y la biotecnología,
ahora existe una diversidad de herramientas para la identificación de genes
potencialmente involucrados en procesos específicos, incluyendo la respuesta
a sequia (Ribaut et al, 2009). El desarrollo de plantas diseñadas por ingeniería
genética mediante la introducción y/o sobreexpresión de genes seleccionados
parece ser una opción viable para la producción de plantas mejoradas, así
como una vía más rápida para insertar genes benéficos, comparado con el
mejoramiento convencional. Asimismo, la ingeniería genética permite controlar
el tiempo, la especificidad del tejido y el nivel de expresión de los genes
introducidos para su función óptima. Esto es una consideración importante si
la acción de un gen dado o un factor de transcripción es deseado solo en un
tiempo especifico, en un órgano específico o bajo condiciones específicas de
estrés. Los hallazgos básicos en la estructura de los promotores de genes

48
asociados con estrés han conducido a un cambio mayor en el paradigma para
diseñar genéticamente los cultivos en años recientes. Los promotores más
ampliamente usados en generar las plantas transgénicas son expresados
constitutivamente (Bhatnagar et al, 2008).
Entre las estrategias para mejorar la tolerancia al estrés abiótico de los cultivos
ha sido la obtención de plantas transgénicas cuyos genes codifican a enzimas
para la síntesis de osmolitos seleccionados como glicina betaína, prolina,
alcohol azúcares y poliaminas en plantas modelo como Arabidopsis, tabaco y
arroz. Los resultados de las modificaciones transgénicas de las rutas
biosintéticas y metabólicas en la mayoría de los casos indican que se puede
obtener una mayor tolerancia al estrés y que la acumulación de los solutos
compatibles podrían proteger a las plantas contra el daño por las especies
reactivas de oxígeno. Sin embargo, algunos reportes han mostrado un efecto
negativo del ajuste osmótico en el rendimiento. Las manipulaciones genéticas
de las enzimas que sintetizan los solutos compatibles no siempre conducen a
una acumulación significativa de los compuestos. Aunque algunos estudios
indican que el ajuste osmótico no provee un efecto benéfico del rendimiento
bajo sequía, los cambios generados en un proceso metabólico dado, podrían
brindar un ligero efecto benéfico para el rendimiento (Bhatnagar et al, 2008).
Por otro lado, el mejoramiento para la tolerancia al estrés también ha incluído
la sobre expresión de enzimas involucradas en la protección oxidativa tales
como glutatión peroxidasa, superóxido dismutasa, ascorbato peroxidasa y
glutatión reductasa en tabaco y alfalfa. Se ha generado tabaco transgénico
sobre expresando el gen de la superóxido dismutasa (SOD) en el cloroplasto,
mitocondria y citosol, resultando en una tolerancia oxidativa. La sobre
expresión del gen de Cobre/Zinc superóxido dismutasa (SODCu/Zn) de
cloroplasto mostró un mejoramiento dramático en la actividad fotosintética bajo
estrés por frío en tabaco transgénico. Sin embargo, plantas de tabaco
transgénico (Manganeso superóxido dismutasa (MnSOD) mostraron una

49
tolerancia al estrés oxidativo sólo en presencia de otras enzimas antioxidantes
y sustratos, mostrando que el genotipo y la composición de la isoenzima
también tienen un efecto profundo en la tolerancia relativa de las plantas
transgénicas al estrés abiótico (Bhatnagar et al, 2008).
Las proteínas LEA también han sido objeto de estudio en este campo. La
expresión constitutiva o inducida del gen HVA1 resultó en el mejoramiento de
las características de crecimiento y la tolerancia al estrés en términos de
integridad celular en trigo y arroz bajo condiciones de sequía (Bhatnagar et al,
2008).
Un número de plantas transgénicas tolerantes a estrés abiótico han sido
producidas incrementando los niveles celulares de proteínas (tales como
proteínas antiporter vacuolares) que controlan las funciones del transporte. Por
ejemplo, plantas de melón y tomate transgénico expresando el gen HAL1
mostraron un cierto nivel de tolerancia a salinidad como resultado de retener
más K que las plantas control bajo salinidad. Además, plantas transgénicas de
Arabidopsis y tomate que sobre-expresan AtNHX1 (un gen vacuolar Na+/H+
antiporter) acumularon cantidades abundantes de transportador en el
tonoplasto y exhibieron una tolerancia estimulada a salinidad (Bhatnagar et al,
2008).
Un aspecto importante de la tecnología transgénica es la expresión regulada
de los transgenes. La capacidad del promotor y la posibilidad de usar
promotores inducibles por estrés, de estadio de desarrollo determinado y de
tejido específico es un punto importante para la respuesta de la planta a los
estreses. Algunos productos génicos por ejemplo, se necesitan en grandes
cantidades por lo que se necesita de un promotor fuerte. Los promotores que
han sido más comúnmente usados en la producción de plantas tolerantes a
estrés incluyen 35S CaMV, ubiquitina 1 y actina. Estos promotores siendo
constitutivos expresan los transgenes rio abajo en todos los órganos y en
todos los estadios. Sin embargo, la sobreproducción constitutiva de moléculas

50
tales como trealosa o poliaminas causan anormalidades en las plantas
crecidas bajo condiciones normales. Además, la producción de las moléculas
arriba descritas puede ser metabólicamente cara. En estos casos, el uso de
promotores inducibles por estrés son los más convenientes, con un nivel basal
de expresión génica bajo en la ausencia de agentes inductores pero la
expresión debería de ser reversible y dosis dependiente. Los hallazgos en los
promotores de genes inducidos por estrés han conducido a un cambio mayor
para diseñar cultivos tolerantes (Bhatnagar et al, 2008).
IV.11. Expresion génica dependiente de ABA durante el déficit hídrico
El ABA es producido bajo condiciones de déficit hídrico y juega un papel
importante en la respuesta de las plantas a la tolerancia a la sequía y la
salinidad. El ABA induce la expresión de muchos genes a través de los
factores que actúan en cis y trans de respuesta a ABA y proteína-cinasas y
fosfatasas que interactúan con Ca+2 en una cascada de señalización. Se sabe
que el ABA puede inducir cascadas de señalización de adentro hacia fuera de
la célula y múltiples receptores de ABA han sido propuestos. Muchos genes
que son inducidos bajo condiciones de sequía contienen un elemento
conservado de respuesta a ABA (ABRE) en sus regiones promotoras (Chaves
et al, 2003), el cual es el elemento más importante que actúa en cis en la
expresión génica de respuesta a ABA (Yamaguchi-Shinozaki y Shinozaki,
2006). Sin embargo, un número de genes inducidos ambientalmente,
diferentes de los regulados por ABA contienen un elemento conservado que
actúa en cis llamado caja G. Se ha sugerido que al menos un elemento que
actué en cis además de la caja G se requiere para una apropiada activación
transcripcional (Yamaguchi-Shinozaki y Shinozaki, 2006).
Muchos factores de transcripción tipo zipper de leucina (bZIP) que actúan en
cis han sido clonados en Arabidopsis y en muchos casos su transcripción es
regulada por sequía y salinidad en un mecanismo probablemente basado en el

51
incremento del potencial osmótico generado por estos estreses (Chaves et al,
2003). Se ha sugerido que las proteínas bZIP inducibles por ABA están
también involucradas en una de las rutas dependientes de ABA. Muchos
genes inducibles por el ABA y diferentes estreses codifican factores de
transcripción y se piensa que funcionan en la regulación de genes inducibles
por ABA, los cuales responden al estrés hídrico poco después de la
producción de factores de transcripción inducibles por ABA (Shinozaki &
Yamaguchi-Shinozaki, 1997).
cDNAs de Arabidopsis que codifican para los factores de transcripción bZIP,
referidos como proteínas de unión a ABRE (AREB) o factores de unión a
ABRE, fueron aislados usando el método de escrutinio por un híbrido. Entre
estas proteínas AREB/ABF, la expresión de AREB1/ABF2, AREB2/ABF4 y
ABF3 fue inducida por ABA, estrés por deshidratación y alta salinidad. Se ha
sugerido que los AREBs/ABFs regulan la expresión génica, dependiente de
ABRE y mediada por el ABA que estimula la tolerancia a sequía en tejidos
vegetativos y los eventos de fosforilación/defosforilación juegan un rol
importante en la señalización de ABA y en la activación de las proteínas
AREB/ABF (Yamaguchi-Shinozaki y Shinozaki, 2006).
Los motivos “ABRE like” no están involucrados en la regulación por el ABA de
algunos genes inducibles a estrés como RD22. La inducción de RD22 por
deshidratación es mediada por ABA. Los sitios de reconocimiento MYC y MYB
en el promotor RD22 funcionan como elementos que actúan en cis en la
expresión inducible por deshidratación de RD22. Los factores de transcripción
MYB y MYC se unen a estos elementos en cis en el promotor RD22 y activan
cooperativamente la expresión de RD22. Estos dos factores de transcripción
son sintetizados después de la acumulación de ABA endógeno, indicando que
juegan un papel en los estadios tardíos de la respuesta de la planta a
diferentes estreses (Yamaguchi-Shinozaki y Shinozaki, 2006). Estudios
realizados han revelado que plantas transgénicas que sobreexpresan MYC y

52
MYB tienen una hipersensibilidad mayor a ABA y la tolerancia a estrés
osmótico (Yamaguchi-Shinozaki y Shinozaki, 2006) (Ver Figura 4).
El gen RD26 de Arabidopsis codifica la proteína NAC que es inducida no sólo
por deshidratación sino también por ABA. Las plantas transgénicas que sobre-
expresan RD26 fueron altamente sensibles a ABA, mientras que las que
reprimían este gen fueron insensibles (Yamaguchi-Shinozaki y Shinozaki,
2006). La introducción de FT en la ruta de señalización de ABA puede ser
también un mecanismo de mejoramiento genético vegetal a la tolerancia a
estrés (Bhatnagar, 2008).

Figura 3. Redes regulatorias transcripcionales y FTs involucrados en la expresión génica del estrés osmótico. Los FTs controlan la expresión de
genes inducibles a estrés, los cuales son mostrados en los elipses de colores
y los elementos que actúan en cis involucrados en la respuesta al estrés de la
transcripción son mostrados en cuadrados. Las flechas gruesas indican las
rutas mayores de señalización y estas rutas regulan muchos genes río abajo.
53

54
IV.12. Expresión génica independiente de ABA durante el déficit hídrico
Hay muchos genes inducibles por deshidratación que no responden a frío o al
tratamiento de ABA, sugiriendo la existencia de una ruta independiente de
ABA en la respuesta a la deshidratación (Yamaguchi-Shinozaki y Shinozaki,
2006).
Estudios realizados han demostrado la existencia de un elemento que actúa
en cis esencial para regular la inducción del gene RD29A en la respuesta
independiente de ABA a la deshidratación y al frío. RD29A es inducido por
sequía, frío y ABA (Yamaguchi-Shinozaki y Shinozaki, 2006). Aunque estudios
posteriores indicaron que su regulación es tanto dependiente como
independiente de ABA bajo estrés por sequía y frío (Yamaguchi-Shinozaki y
Shinozaki, 2006). Este elemento en cis llamado DRE, consta de una secuencia
conservada de 9 pb, TACCGACATA. El DRE también se encuentra en las
regiones promotoras de muchos genes inducibles por sequía y frío
(Yamaguchi-Shinozaki y Shinozaki, 2006). Estos resultados sugieren que los
motivos relacionados a DRE, están involucrados en la respuesta por frío y
sequía pero su expresión génica es independiente de ABA (Shinozaki y
Yamaguchi-Shinozaki, 1997). A este respecto, dos clonas de cDNA que
codifican a las proteínas de unión a DRE, DREB1A y DREB2A, fueron
descubiertas por medio del análisis de un híbrido. Las secuencias deducidas
de aminoácidos de DREB1A y DREB2A mostro una similitud de la secuencia
con dominios de unión a DNA conservados encontrados en las proteínas
Factor de respuesta a etileno (ERF de sus siglas en inglés) y APETALA2 que
funciona en la morfogénesis (Yamaguchi-Shinozaki et al, 2002).
Posteriormente se encontró que DREB2A y DREB2B son considerados los
factores transcripcionales más importantes que funcionan bajo condiciones de
deshidratación y alta salinidad. Sin embargo, la sobre-expresión de DREB2A
en plantas transgénicas no causó una tolerancia al estrés mejorada y tuvo un

55
crecimiento normal, sugiriendo que la proteína DREB2A requiere una
modificación post-traduccional como una fosforilación para su activación
(Yamaguchi-Shinozaki y Shinozaki, 2006). El análisis del dominio de DREB2A
usando protoplastos de Arabidopsis, reveló que existe un dominio regulatorio
negativo en la región central de DREB2A y que la deleción de esta región
transforma a DREB2A a una forma activa constitutiva. La sobreexpresión de
esta forma activa constitutiva de DREB2A resultó en un retardo en el
crecimiento de las plantas transgénicas de Arabidopsis, con una tolerancia
significativa a la sequía pero sólo una leve tolerancia al frío (Yamaguchi-
Shinozaki y Shinozaki, 2006).
Otra ruta independiente de ABA, de expresión génica activada en respuesta al
estrés hídrico y la senescencia involucra al gen ERD. El análisis del promotor
de este gen identificó dos elementos nuevos que actúan en cis involucrados
en la inducción por deshidratación y la senescencia inducida por oscuridad
(Bhatnagar, 2008).
Por otro lado, los represores transcripcionales que reprimen la expresión
génica bajo condiciones de estrés han sido usados para mejorar la tolerancia a
sequía. Ejemplo de ello es AtMYB60 (un represor transcripcional R2R3-MYB)
en Arabidopsis, que funciona en la regulación de movimientos estomatales
siendo específicamente expresado en las células guarda y regulado
negativamente durante la sequía. Interesantemente, la mutación atmyb60-1
resulta en defectos específicos de las células guarda, sin un efecto deletéreo
aparente sobre procesos fisiológicos y de desarrollo (Umezawa et al, 2008).
IV.12. Estudios de expresión diferencial bajo condiciones de sequía
El avance de la tecnología de análisis genómico, el incremento en las bases
de datos disponibles y los perfiles globales de expresión han sido usados para

56
identificar genes involucrados en las respuestas adaptativas a sequia y otros
estreses abióticos (Talame et al, 2007).
Sin embargo, los procedimientos experimentales que han sido aplicados en los
estudios de de perfiles de expresión y que pretenden imitar las condiciones de
sequia difieren grandemente en términos de la dinámica y/o intensidad de los
tratamientos de estrés aplicados. Por lo que es importante verificar la
correspondencia de los cambios en los perfiles de expresión que ocurren bajo
diferentes condiciones experimentales en el laboratorio, invernadero ó el
campo. En la mayoría de los casos, el perfil transcripcional para la respuesta
al déficit hídrico ha sido realizado en muestras colectadas de platas sometidas
a una intensidad de estrés frecuentemente desarrollada en un corto tiempo, lo
que impide la identificación de aquellos genes de respuesta tardía que podrían
jugar un papel importante en la adaptación a la sequia bajo condiciones de
campo. Y menos numerosos han sido aquellos estudios donde el tratamiento
de estrés se ha aplicado progresivamente para investigar los efectos de la
sequia en los perfiles de transcripción (Talamé et al, 2007).
IV.12.1. Estudios de expresión diferencial en Arabidopsis thaliana y arroz
La tecnología de microarreglos utilizando cADNs u oligonucleótidos es una
herramienta poderosa para analizar los perfiles de expresión de genes en
plantas expuestas a estreses abióticos como sequía, salinidad, frio o
tratamiento de ABA (Shinozaki y Yamaguchi-Shinozaki, 2007). Los primeros
estudios realizados en perfiles de expresión fueron en Arabidopsis. Por medio
de los microarreglos, se identificaron 299 genes inducibles por la sequía en
Arabidopsis. Mas de la mitad de ellos fueron también inducidos bajo
tratamientos de salinidad y/o ABA, implicando una relación significativa entre
las respuestas a sequia, salinidad y ABA (Shinozaki y Yamaguchi-Shinozaki,
2007). Unos años después, se llevaron a cabo análisis de microarreglos en
arroz bajo diferentes condiciones de estrés (frio, sequia, y salinidad). La

57
expresión de cerca del 40% de los genes inducibles bajo sequía o salinidad
fue también afectada por frio. Por otro lado, la expresión de más del 98% de
los genes inducibles por salinidad y 100% de los genes inducibles por ABA
fueron inducidos por sequía. Los resultados sugieren la existencia de un
sistema regulatorio común o una relación muy significativa entre la
señalización asociada con el estrés por la sequia y el estrés salino, y entre la
sequia y las rutas inducidas por ABA. Los autores también indican que existe
una débil relación entre las rutas de señalización activadas en respuesta a frio
versus sequia o entre frio versus el tratamiento de ABA (Shinozaki y
Yamaguchi-Shinozaki, 2007). Asimismo, los autores mencionan que existe una
relación consistente en la respuesta de la expresión génica de sequía y
salinidad entre arroz y Arabidopsis (Shinozaki y Yamaguchi-Shinozaki, 2007).
IV.12.2. Estudios de expresión diferencial en cereales: trigo y cebada
En lo que respecta a los cereales, el estudio de perfiles de expresión bajo
condiciones de sequia se ha realizado principalmente en trigo y cebada. Un
estudio de expresión diferencial fue llevado a cabo en hojas de cebada
sometidas a condiciones graduales de secado del suelo durante 7 y 11 días,
así como la imposición a 6 horas de deshidratación colocándolos en papel. En
total, encontraron 173 transcritos que cambiaron su nivel de expresión en
respuesta a al menos uno de los tratamientos. Muchas de las categorías
funcionales fueron representadas en los grupos inducidos o reprimidos,
aunque hubo una ocurrencia mayor de los cambios en los transcritos inducidos
comparado con los reprimidos que codifican a proteínas involucradas en
respuesta a estrés abiótico, tales como proteínas LEA, proteínas relacionadas
a la biosíntesis de osmoprotectantes o de respuesta a jasmonato. Asismismo,
compararon las respuestas transcripcionales entre el tratamiento rápido de
deshidratación y la imposición gradual de sequía, aproximadamente 57% de
los transcritos regulados diferencialmente cambiaron en su expresión
exclusivamente bajo el tratamiento rápido de deshidratación y solo 10% fueron

58
regulados de manera común para ambos tratamientos. El 33% restante
comprendió transcritos diferencialmente expresados exclusivos para 7 días de
estrés (6%), 11 dias de estrés (14%), re-hidratación (6%) y comunes para
sequia y deshidratación (6%) y aquellos comunes para el tratamiento rápido de
deshidratación y re-hidratacion (1%). En este estudio se reportó 23 transcritos
expresados diferencialmente a los 7 días y 42 transcritos a los 11 días de
estrés gradual (Talame et al, 2007).
En el caso del trigo, estudios de la respuesta a 36 horas sin riego de las raíces
de la variedad Opata, una variedad sensible a sequía, identificaron 394 genes
distintos, de los cuales 190 transcritos fueron más abundantes en tejidos
deshidratados y 204 transcritos disminuyeron en abundancia (Mohammadi et
al., ,2008). Además, se realizó un análisis transcripcional comparativo entre
esta variedad (Opata) y un genotipo sintético más tolerante a sequía derivado
de esta variedad como la abundancia del transcrito o la relación estrés/control
y se identificó que 525 transcritos diferían significativamente en sus niveles de
expresión después de 36 horas de estrés (Mohammadi et al., 2008).
IV.12.3. Estudios de expresión diferencial en maíz
El impacto de la expresión bajo déficit hídrico en maíz ha sido estudiado por
los análisis de microarreglos para: a) dar una visión amplia de las categorías
de genes regulados por el estrés en base a los cambios fisiológicos,
bioquímicos y regulatorios involucrados en la respuesta y las diferencias entre
diferentes tejidos y genotipos, b) identificar genes altamente responsivos, los
cuales podrían ser candidatos para clonación y un análisis funcional profundo
y c) identificar promotores putativos involucrados en la expresión génica bajo
estrés hídrico (Ribaut et al, 2009). En ese sentido, Yu y Setter (2003)
analizaron la expresión diferencial en el endospermo en desarrollo y tejidos de
placenta en maíz bajo déficit hídrico y recuperación. La mayoría de genes de
expresión diferencial en la placenta fueron inducidos bajo estrés, mientras que
la mayoría de genes afectados en el endospermo fueron reprimidos (Yu y

59
Setter, 2003). Genes relacionados a estrés como chaperoninas y proteínas de
choque térmico fueron las clases mayoritarias en la placenta, mientras que
este grupo fue el minoritario para los genes afectados en el endospermo (Yu y
Setter, 2003).
Por otro lado, Spollen et al. (2008) realizó un estudio para identificar los
transcritos expresados diferencialmente en diferentes regiones de la zona de
elongación de la raíz del maíz bajo condiciones de potenciales hídricos bajo.
La zona de expansión longitudinal mostró un mayor número de genes
expresados diferencialmente involucrados en el estrés, donde se ve
incrementado el ensanchamiento de pared, el ajuste osmótico, regulación por
ABA y cambios en el transporte de membrana, comparado con la región donde
la expansión longitudinal disminuye de una tasa máxima a una tasa
progresivamente más lenta bajo estrés.
Como se ha observado, diversos estudios de microarreglos han sido llevados
a cabo en maíz bajo condiciones de sequía conduciendo a la identificación de
múltiples genes como parte de la red de respuesta a sequía (Ribaut et al,
2009). Sin embargo, hasta el momento no se han llevado a cabo estudios en
maíz comparando genotipos que posean diferentes mecanismos de tolerancia
a estrés y/o que se comparen genotipos susceptibles y tolerantes a sequía.
Los perfiles de expresión génica como los microarreglos son enfoques útiles
para identificar un gran número de transcritos y rutas relacionadas a los
mecanismos de tolerancia al estrés (Sreenivasulu et al, 2007). Las propuestas
enfocadas en combinar y entender los aspectos moleculares, fisiológicos y
metabólicos de la tolerancia a la sequía aportarían un conocimiento importante
de los efectos de los genes entre un periodo corto y largo bajo estrés
(Bhatnagar, 2008). Aunque se han realizados varios estudios de expresión
génica en maíz, la respuesta de los genes varía entre genotipos (Sreenivasulu
et al, 2007) e incluso entre diferentes tejidos. La identificación de un grupo de
genes clave de respuesta a estrés y durante el riego de recuperación es un

60
paso importante en la caracterización funcional de genes posiblemente
involucrados en los mecanismos de tolerancia a sequía en maíz.

61
IV. OBJETIVOS
IV.I. GENERAL
Determinar las respuestas fisiológicas y moleculares de dos variedades de
maíz tolerantes y una susceptible, bajo sequía y riego de recuperación.
Determinar si las respuestas fisiológicas y transcripcionales durante la sequía
y el proceso de recuperación son diferentes entre las tres variedades de maíz
y que diferencias podrían conferir la tolerancia o la susceptibilidad a la sequía.
IV.II. ESPECÍFICOS
Identificar y comparar los cambios en el comportamiento fisiológico de las tres
variedades de maíz durante la sequía y el riego de recuperación.
Determinar los patrones de expresión génica de las tres variedades de maíz.
Respecto a los diferentes tratamientos de sequía.
Analizar aquellos genes asociados a la respuesta por sequía y correlacionarlos
con su respuesta fisiológica.
Determinar los cambios en contenido de algunos azúcares y aminoácidos en
las tres variedades de maíz bajo condiciones de sequía y riego de
recuperación.
Identificar los genes particularmente involucrados en la tolerancia a sequía y al
riego de recuperación en las tres variedades de maíz bajo estudio.
Desarrollar un modelo general de las respuestas fisiológicas, moleculares y
bioquímicas de las tres variedades de maíz bajo condiciones de sequía y de
riego de recuperación.

62
V. MATERIALES Y MÉTODOS
V.1. Material Vegetal
Se utilizaron los siguientes materiales de maíz: 85-2, Cajete criollo y
Michoacán 21. Las características de cada una de estos maíces criollos se
encuentran además en Hayano-Kanashiro (2004). La variedad susceptible a
sequía es 85-2 (desarrollada por el Dr. Jose Luis Pons). Las variedades
tolerantes son: Cajete criollo (CC) y Michoacán 21 (M21). CC es cultivado
principalmente en el estado de Oaxaca, México, tiene una alta tolerancia al
bajo contenido de humedad en el suelo y un ciclo vegetativo largo con un
crecimiento lento hasta la llegada de lluvias y con una rápida respuesta en
términos de crecimiento y con la subsecuente recuperación (Pérez, 1979).
M21 proveniente de la sierra Purépecha en el estado de Michoacán (México)
fue descrita como una colección que tiene una clara respuesta a la sequía y al
frio (Fischer et al, 1983). El mecanismo de tolerancia de M21 fue denominado
“latencia” y consiste en prolongar el estadio vegetativo bajo condiciones de
sequía y un rápido retorno al crecimiento normal y finalización del ciclo
reproductivo cuando las lluvias llegan. M21 es más resistente a una marchitez
permanente en plántula en comparación a otros genotipos de maíz y tiene una
mayor tasa de transpiración bajo condiciones normales de riego comparado
con condiciones limitantes de agua (Fischer et al, 1983).

V.2. Condiciones de crecimiento
Las semillas de maíz se desinfectaron con hipoclorito de sodio comercial al
10% por 30 min y se enjuagaron varias veces con agua destilada. Las semillas
se colocaron a una profundidad de 7cm directamente en las macetas
conteniendo una mezcla de arena (92.56%) y arcilla (7.44%). Esta mezcla fue
previamente lavada con agua varias veces para eliminar tierra y desechos.
Después de ello, fue esterilizada con bromuro de metilo. La siembra de las
variedades de maíz fue realizada bajo condiciones de invernadero en el
CINVESTAV – Unidad Irapuato. Las temperaturas promedio durante el
transcurso del experimento estuvieron entre 19°C y de 32°C como máximo
durante el mediodía, la humedad relativa fue de 60±5%. Las plantas de maíz
fueron regadas diariamente hasta antes de aplicar el estrés. La fertilización fue
aplicada usando un fertilizante de liberación (Triple 17, Profer Mix, 4g para
cada maceta-17:17:17 NPK). Adicionalmente, se aplicó una vez a la semana la
solución Long Ashton (Phillips and Jennings, 1976) hasta antes de la
aplicación del estrés.
V.3. Tratamientos de estrés
Los tratamientos de estrés se dieron de la siguiente manera:
1) Riego contínuo: las plantas estuvieron con riego continuo a lo largo del
63
Tabla 1. Genotipos de maíz utilizados en el presente estudio
Característica Pais Genotipo Estado de origen
Raza
Susceptible a sequía México 85-2 Guanajuato Celaya y Tuxpeño
Tolerante a sequía México Cajete criollo Oaxaca Bolita
Tolerante a sequía México Michoacán 21 Michoacán Cónico y Celaya

64
experimento.
2) Estrés moderado por suspensión de riego durante 10 días.
3) Estrés severo por suspensión de riego durante 17 días.
4) Riego de recuperación, el cual se aplico después de los 17 días de estrés.
El estrés se dió a los 30 dias después de la germinación de la semilla. Y se
realizaron 5 repeticiones por cada variedad y cada tratamiento, tanto los que
estuvieron bajo riego como los de estrés hídrico. Los experimentos se
realizaron durante dos años consecutivos.
V.3. Determinaciones del potencial hídrico de hoja y suelo
V.3.1. Potencial hídrico de hoja
Las mediciones del potencial hídrico en las hojas de las plantas de maíz se
realizaron utilizando una cámara C-30 (WESCOR, INC.) con 2 discos de 1.4
cm. de diámetro por planta, tomadas de éstas con la ayuda de un
sacabocados. Se consideró un tiempo de equilibrio de 3 horas y una
temperatura de 37°C, para lo cual los psicrómetros fueron mantenidos en
baño maría. Se realizaron dos mediciones, la primera a las 6 am y la segunda
medición a la 1 pm.
V.3.2. Potencial hídrico de suelo
Las mediciones del potencial hídrico del suelo se realizaron colocando los
psicrómetros a una profundidad de 15.5 cm en el centro de las macetas. La
medición se realizó a las 2 pm. Las lecturas de los potenciales hídricos de
suelo y hoja se realizaron mediante un microvoltímetro (modelo HR-33T,
WESCOR.Inc., Logan Utah).

65
V.4. Extracción de azúcares solubles
El método para la extracción de carbohidratos solubles se realizó de acuerdo a
lo utilizado por Altamirano-Hernández (2007). Se realizó la extracción de
azúcares solubles en hojas de maíz bajo riego y bajo condiciones de sequía
partiendo de 500 mg de tejido molido con 3 repeticiones. Se colocaron en
tubos de vidrio de 50 mL y se adicionó 15 mL de etanol al 80%. Los viales
fueron incubados a 95°C por 30 min y centrifugados a 15000 rpm por 15 min.
El sobrenadante se colocó en otro vial limpio y se adicionó a la pastilla 15 mL
de etanol al 80% y se dejó incubando a 95°C por 30 min. Este proceso se
repitió otras dos veces. Las tres fracciones se unieron y se evaporaron a
sequedad mediante rotavapor (Caframo, WB2000. Wiarton, Ontario, Canadá).
Los sólidos resultantes fueron recuperados en 1 mL de etanol 80 % y
almacenados a 4°C hasta ser procesados.
V.4.1. Cuantificación de carbohidratos solubles
Los extractos obtenidos como se mencionó anteriormente fueron derivatizados
para obtener la forma aldonitrilo-peracetilada de los carbohidratos de acuerdo
a Altamirano-Hernández (2007). La derivatización se realizó en campana de
extracción de la siguiente manera: a partir del extracto etanólico mencionado
anteriormente, se transfirió 100 μL de cada una de las muestras a reactiviales
de 2 mL. Se adicionaron 25 μg de perseitol (utilizado como estándar interno),
se evaporó a sequedad a 65°C con flujo de N2 y los sólidos fueron
resuspendidos en 1 mL de diclorometano y evaporados a sequedad
nuevamente bajo las mismas condiciones, este proceso se realizó tres veces.
Seguidamente, a la muestra se le adicionó una solución de piridina/cloruro de
hidroxilamina (3 mL x 53 mg v/w) preparada al momento del análisis. Las
muestras fueron sonicadas por media hora, después se agitó en vortex y se
incubó 1 h a 85°C (se agitó a los 30 min nuevamente). Después de este
tiempo se dejó enfriar y se adicionó 1 mL de anhidro acético y 500 μL de
piridina y se incubó 30 min a 85°C. Se dejaron enfriar las muestras y fueron

66
transferidas a tubos de ensayo de 50 mL, se adicionaron 2 mL de cloroformo y
2 mL de agua desionizada. Se agitó en vortex y se dejó reposar hasta la
separación de las fases acuosa y orgánica. Posteriormente se eliminó la fase
acuosa y a la muestra se le adicionó 1 mL de agua desionizada y se agitó
nuevamente (este proceso de lavado se realizó en total 10 veces). La fase
obtenida fue filtrada a través de columnas que contenían sulfato de sodio
anhidro y se eluyeron con 2 mL de cloroformo (para asegurarse que las
muestras no contengan agua). Este filtrado se evaporó a sequedad y fueron
almacenadas para su posterior análisis. Este análisis se realizó mediante
cromatografía de gases acoplada a espectrometría de masas (GC, HP6850;
MS, HP5873). Las muestras derivatizadas se resuspendieron en el momento
del análisis en 100 μL de cloroformo grado espectrofotométrico. Los
parámetros utilizados para la corrida de las muestras en el GC-MS fueron
iguales a los descritos por Altamirano-Hernández (2007), siendo las
siguientes:
Inyector: 300°C
Inyeccion 1 μL con split 2:1
Columna: HP-5MS (30 m, D.I. 250 um, Film 0.25 um)
Gas acarreador: Helio
Flujo: constante 1 mL min-1, post-corrida 1.5 mL min-1.
La cuantificación de cada uno de los carbohidratos se realizó en base a la
comparación de los espectros de masas de los estándares cuantificados
anteriormente por Altamirano-Hernández (2007). Esta cuantificación se realizó
mediante la medición del área bajo la curva a partir de los cromatogramas
obtenidos.
V.5. Medición de fotosíntesis y conductancia estomatal
La tasa de asimilación neta de CO2 fue registrada a los 0, 10 y 17 días de
estrés y finalmente en el riego de recuperación, realizándose lecturas cada 2

67
horas durante cada tratamiento de estrés, desde las 7 am hasta las 7 pm;
usando un analizador de gases infrarrojo en un sistema cerrado (IRGA) (LI-
6200 Portable Photosynthesis System. LI-COR, INC.)
V.6. Cuantificación de aminoácidos libres
Se utilizaron muestras de hoja de maíz molidas (aproximadamente 250 mg de
muestra) bajo riego de las tres variedades y muestras de 10 y 17 días de
estrés, así como de riego de recuperación para todas las variedades de maíz.
Para el análisis de aminoácidos libres suspendidos en 1 mL de la solución de
ácido tricloroacético (TCA) al 5% y homogenizado por 30 min. Las muestras
fueron dejadas toda la noche en un refrigerador a 4°C. Se siguió la técnica
modificada por Harrigan y colaboradores (2007).
V.7. Extracción de ARN Total
El ARN total fue extraído a partir de hojas sometidas a estrés hídrico y riego de
las diferentes variedades utilizando el reactivo TRIZOL (Invitrogen), mediante
el siguiente protocolo: por cada gramo de tejido molido se adicionaron 10 mL
del reactivo TRIZOL (frío) en un tubo Falcon de 50 mL, mezclándose
lentamente. Se incubó por 5 min a temperatura ambiente y se colocó en
posición horizontal en un agitador a velocidad lenta. Se centrifugó a 12,000
rpm por 10 min a 4°C. Luego de ello se tomó el sobrenadante y se llevó a un
tubo nuevo, y se adicionó 3 mL de cloroformo. Se dió una ligera agitación con
las manos y se incubó por 2 min. Después de ello se centrifugó a 12,000 rpm
por 15 min a 4°C. Se tomó el sobrenadante y se colocó en un tubo nuevo, se
adicionó 1 volumen de isopropanol y se dejó en agitación a velocidad lenta por
10 min a temperatura ambiente. Se centrifugó a 12,000 rpm por 10 min y se
decantó el sobrenadante. A la pastilla se le adicionó 1 mL de etanol al 75%. Se
centrifugó a 7500 rpm por 5 min, se dejó secar la pastilla y se adicionó 100 μL
de agua desionizada estéril libre de ARNasas (tratada con dietil pirocarbonato
al 0.01%). Con el fin de que las muestras se encuentren lo más limpias

68
posibles, se procedió a realizar una segunda extracción con el reactivo Plant
RNA Concert (Invitrogen). Se adicionó 533 μL de este reactivo a la muestra
extraída en el paso anterior y se adicionó 106 μL de NaCl 5M, se mezcló bien
y se añadió 318 μL de cloroformo. Se centrifugó a 12,000 rpm por 15 min a
4°C. Se tomó el sobrenadante y se colocó en un tubo limpio. Se adicionó un
volumen de isopropanol y se incubó por 10 min a temperatura ambiente en
agitación lenta. Se centrifugó por 10 min a 12,000 rpm a 4°C y se decantó el
sobrenadante. Se adicionó 500 μL de etanol al 75% y se centrifugó a 7500
rpm por 5 min. Se decantó el sobrenadante y se dejó secar la pastilla.
Finalmente, la pastilla se disolvió en 50 μL de agua desionizada estéril
(adicionada con dietil pirocarbonato al 0.01%).
V.7.1. Cuantificación e integridad del ARN total
Para la cuantificación del ARN total se tomó 2 μL de cada muestra y se
cuantificó en el espectrofotómetro NanoDrop (ND-1000). Se consideró que la
relación de absorbancia 260/280 nm fuera mayor a 2. La integridad del ARN
total se analizó por medio de electroforesis, por medio de un gel de agarosa al
1% con TAE 1X estéril. En algunos casos, se utilizó el método de
electroforesis capilar utilizando el equipo Agilent 2100 Bionanalyzer, el cual
asigna una calificación de integridad que se obtiene a partir de la comparación
del electroferograma obtenido con una base de datos de ARNs íntegros
(Calderón-Vásquez, 2008).
V.7.2. Purificación del ARN total
El ARN total después de las extracciones descritas anteriormente, se procedió
a purificar las muestras de la siguiente manera, utilizando el kit de RNeasy
Minielute Cleanup (QIAGEN): 20 μg de RNA total se ajustaron a un volúmen
de 100 μL con agua libre de RNasas. Se adicionó 350 μL del amortiguador
RLT y se mezcló completamente. Enseguida se adicionó 250 μL de etanol al
100% y se mezcló cuidadosamente por pipeteo. Todo este volúmen de
aproximadamente 700 μL de muestra se llevó a la columna que se encuentra

69
en un tubo colector de 2 mL. Se centrifugó por 15 seg a 10,500 rpm. El líquido
fue descartado, se cambió a un tubo colector nuevo y se agregaron 500 μL de
amortiguador RPE a la columna. Se centrifugó por 15 seg a 10,500 rpm. Y
seguidamente se adicionó 500 μL de etanol al 80%. Se centrifugó nuevamente
2 min a 10,500 rpm. Se transfirió la columna a un tubo colector nuevo y se
centrifugó por 5 min a 12,000 rpm; manteniendo la tapa del tubo abierta. Se
colocó la columna en un tubo colector nuevo y se adicionó 15 μL de agua a
37°C en el centro de la membrana de la columna y se centrifugó a 12,000 rpm
por 1 min. Se repitió el paso anterior con 15 μL más de agua con un volúmen
final de aproximadamente 30 μL.
V.7.3. Amplificación del ARN
La amplificación de muestras de ARN es un método standard para análisis de
arreglos. Este método fue desarrollado en el laboratorio de James Eberwine
(Van Gelder, et al, 1990). Este procedimiento consistió en obtener el cDNA
utilizando un oligo dT con un promotor viral T7, por medio de una transcripción
in vitro, de tal manera que se producirán rendimientos mayores de cDNA de la
primera cadena comparado con otras enzimas de transcripción reversa
(Manual del kit MessageAmpTM II aRNA Amplification-AMBION). Para realizar
este paso se utilizó el kit MessageAmpTM II aRNA Amplification (AMBION).
Este sistema incorpora UTP modificado (aminoallyl-dUTP, Ambion), que
permitirá acoplar los fluorocromos Cy3 o Cy5 posteriormente.
V.8. Alineamiento del primer T7 y Síntesis de Primera Cadena de cDNA
Se colocó aprox. 1.5 μg de ARN total en un tubo libre de ARNasa de 0.2 mL.
Seguidamente se adicionó 1 μL de Oligo dT primer T7. Se adicionó agua libre
de nucleasas a un volúmen final de 6 μL. Se incubó 10 min a 70°C en un
termociclador. Luego de ello, se removió las muestras de ARN del

70
termociclador a 70°C y se centrifugó brevemente y se transfirió
inmediatamente a hielo.
V.9. Síntesis de la primera cadena de cADN
Se realizó la mezcla de reacción de la primera cadena:
Mezcla de reacción Cantidad (μL)
10X Amortiguador de primera cadena
1
Inhibidor Ribo. 0.5
dNTP 2
Enzima Array Script 0.5
Volúmen total 4
Total/ Reacción 4
Se añadió 4 μL de la mezcla de reacción a las muestras arriba mencionadas y
se mezcló suavemente por pipeteo y colocar los tubos en el termociclador a
42°C (de preferencia se recomienda usar una temperatura de tapa a 48°C). Se
incubó durante 2 h. Se centrifugaron los tubos brevemente y se colocaron los
tubos en hielo y a continuación se procede a la síntesis de la segunda cadena
de cDNA.
V.10. Síntesis de la segunda cadena
Se realizó la mezcla de reacción de la segunda cadena:
Mezcla de reacción Cantidad (μL)
Agua DEPC 31.5

71
10X Amortiguador Segunda cadena 5
dNTP 2
ADN polimerasa 1
ARNasa 0.5
Total/ Reacción 40
Se realizó una mezcla de reacción para esta parte del procedimiento y se tomó
40 μL de esta mezcla y se adicionó al tubo de reacción de la primera cadena.
Se mezcló suavemente por pipeteo y se centrifugó los tubos. Se incubó a 16°C
por 2 h en el termociclador y ensequida se continuó con la purificación del
cDNA y se almacenó a -20°C.
V.11. Purificación de cADN
Se utilizó el kit DNA clear (AMBION) for cDNA purifications (Ambion Cat. Nro.
1756). Antes de empezar, el agua hay que calentarlo a 50°C por un mínimo de
10 min. Siguiendo los pasos que se dan a continuación: se colocó los tubos
con filtro para cDNA en tubos de 2 mL y se pipeteó 50 μL de amortiguador de
unión a cDNA en el filtro en el tubo de cDNA. Se incubó a temperatura
ambiente por 5 min. Se adicionó 250 μL del amortiguador de unión de cDNA a
cada muestra de cDNA a partir de la síntesis de la segunda cadena y se
mezcló repetidamente por pipeteo. Se pipeteó la muestra de
cDNA/amortiguador de unión cDNA al centro del tubo con filtro equilibrado. Se
centrifugó por 1 min a 12,000 rpm. Se descartó el liquido y aplicar 500 μL de
amortiguador de lavado de cDNA y se centrifugó por 1 min a 12000 rpm. Se
descartó el líquido y se centrifugó por 1 min adicional para remover las trazas
de etanol. Se transfirió el tubo con filtro a un tubo de elución nuevo. Al centro
del filtro se aplicó 6 μL de agua libre de nucleasa, previamente calentada a

72
50°C. Se dejó a temperatura ambiente por 2 min y centrifugar 2 min a 12000
rpm. Se repitió el paso anterior con otros 6 μL de agua libre de RNasa
previamente calentada. El cDNA de doble cadena estará ya eluido. Se
procedió a cuantificar la concentración de cDNA en el Nanodrop.
V.12. Transcripción in vitro
Se preparó la mezcla de reacción y centrifugar a 3000 rpm por 30 seg:
Mezcla de reacción Cantidad (μL)
aaUTP (50mM) 1.5
Mezcla de ATP, CTP, GTP 6
Solución UTP (75mM) 0.5
T7 10X Amortiguador de reacción 2
Enzima T7 2
Volúmen total 12
Se incubó el tubo a 37°C en el termociclador (la tapa del termociclador debe
de estar a 40°C) por 14 horas. Se paró la reacción adicionando 80 μL de agua
libre de ARNasa a cada muestra de cARN (ARN complementario) y con un
volúmen de 100 μL. Mezclar bien el contenido del tubo con vortex. Proceder
directamente a la purificación del cARN o guardar a -20°C.
V.13. Purificación de ARN modificado
Este paso es importante para remover todos los nucleótidos no incorporados
del ARN modificado. Se realizó lo siguiente: Se adicionó 350 μL de
amortiguador de unión a ARN a cada muestra de ARN y proseguir
inmediatamente al siguiente paso. Se adicionó 250 μL de etanol 100% a cada

73
muestra de ARN y mezclar por pipeteo hasta tres veces. Se prosiguió
inmediatamente después que se haya agregado el etanol a cada muestra.
Cualquier retraso en el procedimiento resultaría en la pérdida de ARN ya que
el ARN podría formar un precipitado. Se pipeteó cada muestra del paso 2 al
centro del filtro del tubo. Y se centrifugó por 1 min a 12000 rpm o se continuó
hasta que la mezcla ha pasado por el filtro. Se descartó la elución. Y aplicó
650 μL del buffer de lavado a cada tubo con el filtro y se centrifugó por 1 min a
12000 rpm. Se descartó la elución. Se centrifugó 3 min adicionales a 12000
rpm para remover cualquier traza del buffer de lavado. Se transfirió los tubos
con filtro a un tubo nuevo colector y se adicionó al centro del filtro 30 μL de
agua libre de ARNasa (previamente calentada a 50°C). Se dejó a temperatura
ambiente por 2 min y se centrifugó a 12000 rpm por 2 min. Se repitió el paso
anterior con otros 30 μL de agua libre de ARNasa. El ARN modificado estará
en 60 μL de agua libre de nucleasas. Posteriormente se procedió a determinar
la concentración de ARN usando el Nanodrop.
V.14. Preparación del colorante Cy3 y Cy5
Se disolvió el contenido de cada tubo de colorante en 45 μL DMSO, agitando
varias veces y dejando 30 min a temperatura ambiente protegido de la luz. Se
centrifugó a 2000 rpm por 30 seg y se guardó a -20°C, siempre protegido de la
luz envolviéndolo en papel aluminio.
V.15. Unión del fluorocromo al ARN modificado
Se utilizó 8 μg de ARN modificado para la hibridación de los microarreglos (o
su equivalente a 350 picomoles por cada fluorocromo). Estas muestras se
secaron en un evaporador tipo Speedvac a temperatura ambiente. Luego de
ello, se adicionó 5 μL del amortiguador de carbonatos (Na2CO3 200 mM con
NaHCO3 200 mM, pH 9.0). Una vez que la muestra estaba hidratada, se dió
una centrifugación de 30 seg y se adicionó 5 μL del colorante Cy3 o Cy5
correspondiente a la muestra en DMSO, se mezcló cuidadosamente y se dejó

74
incubando toda la noche protegiéndolos de la luz a temperatura ambiente.
Pasado el tiempo, se adicionó 2.25 μL de hidroxilamina 4M (Sigma) se mezcló
suavemente y se dejó incubando 15 min a temperatura ambiente y se adicionó
6.125 μL de LiCl 7.5M, se mezcló suavemente y se dió finalmente una
centrifugación de 30 seg. Se incubó a -20°C con un mínimo de 8 h o máximo
14 horas.
V.16. Limpieza de los ARNs modificados
Las muestras se centrifugaron a 12,000 rpm a 4°C por 30 min. Se retiró el
sobrenadante cuidadosamente con pipeta. Se adicionó a cada muestra 500 μL
de etanol al 75%. Se mezcló suavemente para lavar el sedimento y se
centrifugó a 12,000 rpm por 3 min. Se repitió el lavado con etanol al 75% y la
centrifugación 2 veces más. Las muestras se dejaron secar a temperatura
ambiente. Una vez secas las muestras se adicionaron 50 μL de agua libre de
ARNasas y se procedió a la cuantificación en el espectrofotómetro NanoDrop.
V.17. Hibridación de los microarreglos
V.17.1. Diseño de los microarreglos
En el presente estudio se utilizó el arreglo de oligonucleotidos de maíz (MOA
de sus siglas en inglés) de la página WEB
http://www.maizearray.org.http://www.maizearray.org. Estos oligos contienen
cerca de 57000 puntos individuales divididos en 2 láminas (A y B) y contienen
putativamente todos los genes de maíz identificados cuando las láminas
fueron obtenidos. La anotación del arreglo y su composición está disponible en
www.maizearray.org.www.maizearray.org. Con la finalidad de contrastar las
diferencias de expresión génica entre los genotipos bajo las diferentes
condiciones de estrés, se utilizó un diseño en loop (Fig. 4). El diseño en loop
se caracteriza en contrastar los pares de muestras en una manera que se
puede controlar la variación de las manchas y los patrones de distribución de

75
las muestras usando modelos estadísticos (Simon y Dobbin, 2003). Asimismo,
cada variedad es marcada con cada colorante. Este balance hace que los
efectos de los colorantes no interfieran con los efectos de la variedad. Por lo
que cualquier comportamiento anómalo de los genes con respecto a los
colorantes no sesgará las estimaciones para el resultado final. Balanceando
las variedades con respecto a los colorantes se producen datos, en el cual los
efectos “colorante y gen” pueden ser detectados (Kerr y Churchill, 2001).
Las muestras analizadas consistieron en dos réplicas biológicas y dos réplicas
técnicas. Las réplicas biológicas fueron obtenidas juntando 4 ó 5 plantas
representativas de cada cultivar para cada tratamiento en particular (0, 10 y 17
días de estrés, así como el de riego de recuperación). Un total de 24 juegos
(48 láminas) de hibridaciones de microarreglos fueron llevados a cabo
incluyendo comparaciones directas e intercambio de colorantes. La elección
del diseño en loop para este estudio se basó en la facilidad que ofrece el
mismo para hacer las comparaciones directas entre las tres variedades de
maíz en cada tratamiento de estrés, además de alternar los dos colorantes
para cada variedad y poder observar las diferencias de expresión entre cada
genotipo por cada tratamiento.

76
L1
L2 L3
L1
L2 L3
L1
L2 L3
L1
L2 L3
T0 T1 T2 T3
L1= 85-2L2= Cajete criolloL3= Michoacán 21
T0= Tiempo0 (0 diasde estrés)T1= Tiempo1 (10 diasde estrés)T2= Tiempo2 (17 diasde estrés)T3= Tiempo3 (Riegode recuperación)
Figura 4. Diseño en loop de los microareglos en el presente trabajo. Las flechas en diferente orientación signifcan el marcaje con los dos colorantes (dye
swap)
V.17.2. Preparación de los vidrios
Las sondas fueron rehidratadas y unidas a los vidrios con los microarreglos
impresos. Los vidrios fueron re-hidratados en baño maría a 55° C por 5 seg.
Los vidrios se mantuvieron con las impresiones hacia abajo sobre el vapor de
agua. Seguidamente se secaron en una placa caliente a 45°C por 5 seg y se
dejaron temperar por 1 min. Estos pasos se repitieron 4 veces más. Para fijar
los puntos a los vidrios se aplicó luz ultravioleta a 180 mJ en un entrecruzador
(Stratalinker). A continuación se lavaron los vidrios en SDS al 1% por 5 min a
temperatura ambiente en una cámara con agitación continua a 120 rpm. Los
vidrios se enjuagaron 10 veces en agua desionizada. Inmediatamente se
transfirieron los vidrios en etanol al 100% y se sumergieron 5 veces y se
incubaron 3 min en agitación. Luego se dieron una centrifugación de no más
de 200 x g por 1 min para eliminar cualquier resto de líquido que pudieran
tener los vidrios. Si en el vidrio se observaban manchas, se repitió el lavado
con etanol y sus respectivas centrifugaciones.

77
V.17.3. Pre-hibridación
Cada vidrio se colocó en un tubo con 50 mL de solución de pre- hibridación:
SSC 5X, SDS 0.1% y BSA 1%, el cual se esterilizó por filtración. Esta solución
se calentó a 42°C por al menos 30 min antes de ser usada. Se agitó por
inversión durante 1 h como mínimo ó 3 h como máximo, manteniendo la
temperatura entre 42-50°C. Luego de ello, se lavó con SSC 0.1X durante 3-4
min, dos veces. Luego de ello, se lavó con SSC 0.01X durante 1-2 min. Se
centrifugó por 1 min a 2000 g para quitar los restos de líquido que pudieran
quedar y además se trató con aire comprimido para quitar restos de partículas.
V.17.3. Hibridación
Cuatro μg de cada muestra marcada con Cy3 y la respectiva muestra marcada
con Cy5 fueron mezcladas en un mismo tubo y llevadas a secar en un
evaporador tipo Speedvac. Luego de ello se preparo el amortiguador de
hibridación que consistió en 15 μL de SSC 20X, 0.6 μL de SDS 10%, 30 μL de
formamida, 10.8 μL de agua, 2.4 μL de tRNA (10ug/ul, Sigma) y 1.2 μL de
DNA de esperma de salmón (10 μg/μL, Sigma). Todo ello fue de un volumen
final de 60 μL de amortiguador de hibridación 1X por muestras. Las muestras
fueron desnaturalizadas a 95°C por 3 min y luego llevadas a hielo por 30 seg.
Las muestras se centrifugaron brevemente por 2 min y se mantuvieron a
temperatura ambiente siempre en oscuridad hasta su aplicación al vidrio. Al
vidrio una vez listo para hibridar se le colocó un cubreobjetos. La muestra se
aplicó lentamente en un extremo del cubreobjeto y se dejó que se dispersara
evitando que se formen burbujas. A los pozos de la cámara de hibridación se
le adicionaron 25 μL de agua y fue sellada. La cámara de hibridación se
incubó a 42°C por 14 h.
V.17.4. Lavado de los vidrios
El vidrio fue removido de la cámara y colocado en la solución de lavado: SSC
2X, SDS 0.1% precalentado a 45°C en agitación constante por 5 min. Luego

78
de ello se lavaron los vidrios en otra solución: SSC 0.1X, SDS 1% por 5 min,
dos veces en agitación constante. Después de estos lavados siguieron dos
lavados más con SSC 0.1X por 5 min y el lavado final de SSC 0.01X por 3
min. Se centrifugó por 1 min a 200 g para eliminar los residuos líquidos. En el
caso que los vidrios presentaban manchas, se dió un lavado adicional y se
volvieron a centrifugar como se mencionó anteriormente. Los vidrios se
guardaron a temperatura ambiente y en oscuridad para adquirir la imagen.
V.18. Adquisición de la imagen y extracción de los datos
El escáner Axon GenePix 4100 fue usado para adquirir la imagen de cada
vidrio a una resolución de 10 μm utilizando el software GenePix Pro 6.0
(Axon). Los parámetros de intensidad de laser y ganancia para obtener niveles
globales de intensidad de fluorescencia en ambos canales fueron ajustados
(Calderón-Vásquez, 2008).
V.19. Normalización de datos
Los datos crudos provenientes de archivos con terminación GPR fueron
importados en el paquete LIMMA (Smyth et al, 2003) del software R 2.2.1
(www.R-project.org). En el caso de los arreglos de oligonucleótidos, la
corrección de la intensidad de los puntos fue realizada con el algoritmo RMA
(Irizarry et al, 2003). La normalización de las señales de cada punto fue
realizada utilizando el método “printtiploess” (Yang et al, 2002). Las
intensidades de cada vidrio fueron analizadas en gráficas para asegurar que la
distribución de las intensidades de cada canal Cy3 y Cy5 fuera proporcional
dentro de cada vidrio así como entre los vidrios utilizados en el experimento. Si
la intensidad global de cada vidrio no fue proporcional se realizó una
normalización entre vidrios. Luego de ello, el valor numérico de cada punto fue
exportado a archivos de texto delimitados por tabuladores para su posterior
análisis estadístico (Calderón Vázquez, 2008).

79
Para la normalización de los arreglos de oligonucleótidos se utilizó el programa
R usando LIMMA siendo los scripts los siguientes:
library (limma) targets <- readTargets( "TargetsB.txt") f <- function(x) as.numeric(x$Flags > -99) RG <- read.maimages(targets$FileName, wt.fun=f, source="genepix") RG$genes <- readGAL("MOB-1-9.anno.gal") RG$printer <- getLayout(RG$genes) spottypes <- readSpotTypes("SpotTypes.txt") RG$genes$Status <- controlStatus(spottypes, RG) plotMA(RG) RG <- backgroundCorrect(RG) plotMA(RG) MA <- normalizeWithinArrays(RG) plotMA(MA) boxplot(MA$M~col(MA$M),names=colnames(MA$M)) MA <- normalizeBetweenArrays(MA, method = "Aquantile") boxplot(MA$M~col(MA$M),names=colnames(MA$M)) ###### convertir M normalizados en RG ###### RG <- RG.MA(MA) write.table(cbind(MA$genes, RG$R,RG$G), row.names=FALSE, "DIFB.txt", sep="\t", quote=TRUE) > write.table(cbind(MA$genes, MA$M,MA$A), row.names=FALSE, "allMA.txt", sep="\t", quote=TRUE)
V.20. Análisis de los genes con expresión diferencial
Los datos normalizados fueron transformados a logaritmo base 2 usando el
procedimiento Mixed (SAS 9.0 software, SAS Institute Inc., Cary, NC, USA)
considerando como efectos fijos el tratamiento (Tiempo 0 y días de estrés sin
agua) y el fluorocromo, así como la interacción variedad*tratamiento. Los
efectos del vidrio y la interacción arreglo*fluorocromo fueron considerados
como efectos aleatorios. Las Pruebas F tipo 3, así como los valores de
probabilidad de los efectos del tratamiento y de la interacción
variedad*tratamiento fueron analizadas y los valores de significancia fueron
ajustados por el método llamado: False Discovery Rate (FDR) (Benjamini y
Hochberg, 1995). En base a este parámetro, los genes con un valor de FDR ≤

80
0.05 fueron considerados como diferenciales. Además, se tomó en cuenta los
cambios en la intensidad de la señal de los genes diferenciales entre el
Tiempo 0 y el tratamiento respectivo de al menos 2 veces, los cuales fueron
considerados como expresados diferencialmente.
Para determinar los genes estadísticamente diferenciales se utilizó el
programa SAS, cuyos scripts fueron los siguientes:
data a01; infile "C:\Foldername\Subfolder\Aq\slideA\277.txt" delimiter='09'x ; array="B"; slide=1 ;L="1"; T="1"; cy=5 ; input id Y x; Todo esto para el vidrio 277, el arreglo A, el vidrio 1, la variedad 1= 85-2, el tratamiento 1=10 días de estrés y marcado con Cy5 data a13; infile "C:\ Foldername\Subfolder\Aq\slideA\277.txt" delimiter='09'x ; array="B"; slide=1 ;L="3"; T="1"; cy=3 ; input id x Y; Todo esto para el vidrio 277, el arreglo A, el vidrio 1, la variedad 3= M21, el tratamiento 1=10 días de estrés y marcado con Cy3. data b01; set a01 a02 a03 a04 a05 a06 a07 a08 a09 a10;drop x; data b02; set a11 a12 a13 a14 a15 a16 a17 a18 a19 a20;drop x; data b03; set a21 a22 a23 a24;drop x; data slideA; set b01 b02 b03; run; Enseguida de ello, los valores se convierten a logaritmo de base 2 y se ajusta al modelo y se extraen los datos, data slideal; set slidea; y=log2(y); run; proc sort data=slideal; by ID ; run;

81
proc mixed data=Slideal; class slide cy; model y = cy / outp=rfi; random slide slide*cy; run; ods exclude all; ods noresults; proc mixed data=rfi; by ID; class slide cy L T; model resid = T L T*L cy / outp=rfir; random slide; lsmeans cy T L L*T / diff; ods output covparms=covparms tests3=tests3 lsmeans=lsms diffs=diffs; run; ods exclude none; ods results; data t3ap; set tests3; rename ProbF = raw_p; run; proc sort data=t3ap; by effect id; run; ods exclude all; ods noresults; proc multtest pdata=t3ap fdr out=t31; by effect; run; ods exclude none; ods results; data LT; set lsms; where effect = "L*T"; run; data signif; set t31; where fdr_p<0.05 and fdr_p ne . and effect="L*T"; run; PROC EXPORT DATA= WORK.signif

82
OUTFILE= "C:\ Foldername\Subfolder\resultados por Tratamiento\T1\LTsignifA.txt" DBMS=TAB REPLACE; RUN; data mediasLTsignifA; set LT; where id =(aqui se agrega el ID de cada gen de la tabla LTsignifA.txt) DMBS=TAB REPLACE; run; De los datos obtenidos de las tablas se realizó un filtro de aquellos genes que
tuvieran un FDR≤0.05; los cuales fueron considerados para los posteriores
análisis.
V.21. Análisis de los perfiles de expresión y rutas metabólicas
Los perfiles de expresión de los transcritos expresados diferencialmente bajo
condiciones de sequía fueron agrupados utilizando el software Genespring 7.0
(Silicon Genetics, Redwood City, CA), tomando en cuenta el FDR y los
cambios de expresión. Para una mejor visualización de los cambios de
expresión de los transcritos y sus respectivos nombres se utilizó el macro de
Excel (Microsoft) FiRe 2.2 (Garcion, et al, 2006). Debido a que la anotación de
Arabidopsis está mejor categorizada que la de maíz, se utilizó el software
MapMan para tener un mejor panorama de las diferencias entre los tres
maíces criollos bajo condiciones de sequía en las diferentes rutas metabólicas
existentes.
V.22. Validación de la expresión de transcritos de los microarreglos por medio de RT-PCR tiempo real
Con la finalidad de validar los resultados de los análisis de microarreglos se
utilizó la técnica de retro transcripción por medio de PCR en tiempo real. Para
ello, se diseñaron los oligonucleótidos de los genes seleccionados siguiendo
las recomendaciones del programa Primer Express 3.0 (Applied Biosystems).
Para el diseño de los oligonucleótidos se utilizó como templado las secuencias
originales de los cuales, fueron impresos en el microarreglo

83
(www.maizearray.org). Para ello se necesitan tomar en cuenta las siguientes
recomendaciones: a-) el amplicón debe de tener una longitud promedio de 100
pares de bases, b-) los oligonucleótidos deben de tener una temperatura de
alineamiento (Tm) de 60-65°C, c-) el tamaño de los oligonucleótidos debe de
ser en promedio de 20 pares de bases, d-) el porcentaje de GC debe de estar
entre 30-80%, e-) que la secuencia del oligonucleótido no tenga más de 4G
seguidas y finalmente f-) que en los últimos 5 residuos del extremo 3’ no haya
más de 2 G ó C (Calderón-Vázquez, 2008). Una vez obtenidos los posibles
oligonucleótidos, se verificó que las secuencias presentaran una mínima
formación de estructuras secundarias. Las secuencias de los oligonucleótidos
se encuentran en la Tabla 2.
Se utilizó 10 µg de RNA total para realizar el PCR tiempo real y la transcripción
reversa se realizó con la enzima Superscript III (Invitrogen). El equipo utilizado
para realizar el PCR cuantitativo fue 7500 Real time PCR System (Applied
Biosystems) usando el reactivo SYBR Green PCR Master mix (Applied
Biosystems). Con la finalidad de definir la temperatura adecuada de cada
primer se realizaron gradientes de temperatura y a la vez verificar que los
productos amplificados sean específicos, para ello se necesita que un sólo
producto de amplificación se genere en la reacción para cada gen a validar y
que la fluorescencia generada por la incorporación del reactivo SYBR green a
las moléculas de DNA corresponda al producto y no a productos inespecíficos
en la reacción. Para ello se realiza un ensayo de disociación, en el cual la
temperatura se incrementa gradualmente; de esta manera se monitorea la
curva de disociación de los productos amplificados. Una reacción de PCR en
tiempo real que utiliza SYBR green debe validarse mediante este análisis que
debe presentar un sólo pico en la grafica de disociación para garantizar su
especificidad. Para validar los ensayos de PCR en tiempo real, se realizaron
curvas estándar seriales diluidas en órdenes de 10, tanto de las muestras
como del control interno, la cuales se amplifican y determinan por
cuadruplicado. Todo ello para determinar la eficiencia de amplificación y

84
sensibilidad de la técnica (Manual de entrenamiento de PCR en tiempo real
(Applied Biosystems). La expresión génica fue determinada por el método del
Ct (treshold cycle, que es inversamente proporcional a la cantidad de cDNA
original presente en la muestra) (Calderón-Vázquez, 2008). Este método
compara los valores de Ct de las muestras de interés (muestras bajo sequía)
contra el Ct de un control (control en condiciones de riego). Los valores Ct
tanto del calibrador como de las muestras de interés fueron normalizados con
un gen control interno constitutivo apropiado (UBIQUITIN 2, UBQ2,
TC305418). Los valores correspondientes del valor Ct de UBQ2 fueron
sustraídos del valor de Ct de cada gen analizado (Calderón-Vázquez, 2008).
Hay que tomar en cuenta de que las eficiencias de amplificación de cada gen
deben tener un valor mínimo de 92%.
V.23. Asociación de las secuencias de cDNA a QTL's asociados a sequía
Se realizó un BLAST a los transcritos expresados diferencialmente derivados
de los análisis de microarreglos con los ESTs mapeados en la base de datos
de Maize GDB (www.maizegdb.org). Existen 3039 ESTs mapeados en la base
de datos y los parámetros para realizar el análisis fueron principalmente que
tengan un valor esperado menor o igual a 1E-10. Seguido de ello, se analizó
manualmente el resultado del BLAST y se ligó estos resultados con los datos
respectivos de los QTL’s.
V.24. Número de accesión de los datos de microarreglos
Los datos de microarreglos han sido depositados en el NCBI en el Gene
Expression Omnibus (Edgar et al, 2002) y son accesibles a través del número
de accesión de las series GEO: GSE14728
(http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE14728).
V.25. Anotación funcional y análisis de las rutas metabólicas usando los softwares MapMan y BioMaps

85
Los análisis de rutas metabólicas y de anotación funcional fueron realizadas
como se describe en Calderón-Vázquez et al., 2008. Los genes expresados
diferencialmente de acuerdo a los parámetros seleccionados (FDR < 0.05 y
Fold ±2) fueron visualizados y agrupados con el método de correlación
estándar usando el software GeneSpring 7.0 (Silicon Genetics, Redwood City,
CA). El FiRe 2.2 macro Excel® (Microsoft) (Garcion et al, 2006) fue usado para
facilitar el manejo de la información de los microarreglos. Debido a la limitada
anotación funcional en maíz, la clasificación funcional que se encuentra
organizada para los genes de Arabidopsis a partir del arreglo de Affymetrix
ATH1 en diferentes procesos metabólicos y celulares fue aprovechado para
los datos de maíz usando el programa MapMan (Thimm et al., 2004). Los
genes de maíz expresados diferencialmente fueron anotados funcionalmente
realizando un alineamiento mediante BLAST a la base de datos liberada de
TAIR Arabidopsis 6.0 (www.arabidopsis.org) y la base de datos de PLANTA
(TIGR).
Los mejores apareamientos con las anotaciones provenientes de Arabidopsis
a la base de datos de proteínas TAIR (con al menos un valor esperado de
1.0E-10) fueron asignados a su correspondiente ortólogo en maíz. El software
MapMan (Thimm et al., 2004) fue utilizado para mostrar las diferencias en la
expresión génica en diferentes procesos metabólicos y celulares. Las
proporciones fueron expresadas en una escala de log2 para importarlo en el
software y los cambios en la expresión fueron mostrados por medio de un
código de color (Thimm et al., 2004).
Además del software MapMan, los datos de microarreglos fueron analizados
usando una herramienta llamada BioMaps (Gutiérrez et al., 2007) en la página
web de Virtual-Plant (www.virtual plant.org). Esta herramienta ayuda a
relacionar los datos de expresión diferencial con las categorías funcionales
basadas en la clasificación funcional por la anotación del Centro de
Información de Múnich para secuencias proteicas MIPS), tomando en cuenta

86
el mejor apareo a la base de datos de proteínas TAIR y fue utilizado para
identificar las categorías funcionales comunes relacionadas a la sequía entre
las tres variedades y/o las variedades tolerantes.
V.26. Aplicación de la prueba de Pearson's Chi-cuadrado Con la finalidad de verificar las diferencias estadísticamente significativas entre
las tres variedades de maíz de los genes expresados diferencialmente con los
análisis de microarreglos fue utilizada la prueba de Pearson chi cuadrado
usando el software R versión 2.7.1 (2008-06-23) (http://www.R-project.org)
para algunas categorías funcionales y para cada tratamiento de estrés y riego
de recuperación.

87
Tabla 2. Secuencias de los oligonucleótidos utilizados para la validación de algunos genes expresados diferencialmente mediante RT-PCR tiempo real
ID del arreglo Accesión No. Secuencia del oligonucleótido
Inicial Reverso
MZ00019427 TC279430 CGCCCAACTTGGAGAAGATGA CTGGCAAGTCGGCTGGTTTA
MZ00038553 CD952060 GCCCATGCCGCTCTCACTTCTT ACTTCAGCGCAACGTTCTTCCTCA
MZ00043812 AZM5_14615 CTACATCAGCCAGGGCCAAA AACGATTTTCCCTGGCCTTT
MZ00040421 CF628075_root CACCGCATCCAGTTCAAGCT AATACAAAGCACGCCAACAAAG
MZ00024481 TC260113 CCCAAGGCGAGCACGGAGTA GGGCTGGATGATGACGGACACA
MZ00041103 TC191404 TGTTCTCCATGTTCGGCTTCT ATCATGCCCTCGCTCACTTG
MZ00041136 TC312355 AGACCCCAACGCAACAATCA CGGCTCACCGCGAAGTC
MZ00016752 AZM5_46378 TGGTGATGCGTCGGTTGTGCTAA GGCGGCTTTGGGAGTTTGACAG
MZ00032083 TC335117 GCAGGAAGGCGTAGGCGTAGTTG ATCGGCGGCGGAGGAATG
MZ00040626 NP161441|AF099387.1|AAF04662.1
TACCTGCGCCCCGACAT CGATTGCCGACCATCTGTT
MZ00043312 TC333246 CTCGAGGCGCCCATCAAAAT CAAGACCCCCAAAAGGCAAAGA

88
MZ00047950 TC318956 CCGGCCTCCACTTCGTCAA CTGGCTGGGGTGTGGGTAAACT
MZ00024833 DR802497 CAGTTCGCCCTGATGCCATA GCGCATAGAGATCCCAGCAA
MZ00029485 TC301702 CTGATCTGATGCGCCATGTG TGTCAAAGGGCCGAAAGCA
MZ00016926 TC310514 CCGCATCAGCTACCAGGAGTT TTTCTCCCTTTTGCCGACACT
MZ00022903 TC296224 ACGGAAACGGTATCGCAAAC GCTGGGAATAGGCACCTTATGT
MZ00041182 TC321656 CCCGGACTTCGCCACCATT GCAGATCCACGAGAGCAAAACATT
MZ00041251 TC191823 GCCACCATCCTTAGCTACCTCACC CACGCCACTTCGCTCACTTGT
MZ00029816 TC283096 CCGCGGCGCAGGAGAAC TCGGCGACATCGGATCAACTAAC
MZ00008750 CD961341 GGCGCTGCTCTACCCCTACT CATGTACGCGGCGACTATTG

89
VI. RESULTADOS
VI. 1. Efectos fisiológicos de la sequía VI.1.1. Cambios en el potencial hídrico de hoja y suelo durante los tratamientos de sequía
Para asegurar que las plantas crecieran en condiciones de sequía, los
potenciales hídricos de las hojas y el suelo fueron monitoreados durante el
experimento. Como se muestra en la Figura 5, el potencial hídrico de suelo fue
similar en las tres variedades de maíz a lo largo del experimento. Para
determinar si el estado hídrico de la planta difería entre las tres variedades, el
potencial hídrico de la hoja fue monitoreado a los 10 y 17 días de estrés y
después del riego de recuperación. Las plantas irrigadas de los tres genotipos
mantuvieron potenciales hídricos de hoja relativamente constantes entre -0.52
a -0.53 MPa (Figura 6) a lo largo del experimento a las 6 am y de -0.53 a -0.69
MPa a la 1pm (Figura 7). Los potenciales hídricos en plantas estresadas
empiezan a ser más negativas mientras el nivel de estrés se incrementa, -0.98
a -1.17 MPa a los 10 días de estrés y de -1.06 a -1.23 MPa a los 17 días de
estrés a las 6 am y -1.3 a -1.49 MPa a los 17 días de estrés a la 1 pm. Los dos
genotipos tolerantes mostraron niveles ligeramente más altos de potencial
hídrico comparado con el genotipo susceptible. A los 17 días de estrés, 85-2 y
M21 mostraron una disminución similar en el potencial hídrico de hoja,
mientras que CC aun mantenía un potencial hídrico de hoja mayor. Después
del riego de recuperación, los genotipos mostraron un incremento similar en el
potencial hídrico a niveles ligeramente menores que los resultados antes de
suspender la irrigación.

-4.50-4.00-3.50-3.00-2.50-2.00-1.50-1.00-0.500.00
0 días 10 días estrès 17 días estrèsRiego de
recuperación
Pote
ncia
l híd
rico
de s
uelo
(M
Pa)
85-2 Control 85-2 Estrès CC Control
CC Estrès M21 Control M21 Estrès
Dìas de estrès
Figura 5. Potencial hídrico de suelo de las diferentes variedades de maíz bajo condiciones de sequía. CC: Cajete
criollo, M21: Michoacán 21.
-1.60
-1.40
-1.20
-1.00
-0.80
-0.60
-0.40
-0.20
0.000 días 10 dias estrès 17 días estrès Riego de
recuperación
Po
ten
cial
híd
rico
de
ho
ja (ψ
h
oja)
85-2 Control 85-2 Estrès CC Control
CC Estrès M21 Control M21 Estrès
Días de estrès
Figura 6. Potencial hídrico de hoja 6 am de las diferentes variedades de maíz bajo condiciones de sequía. CC:
Cajete criollo, M21: Michoacán 21.
90

-1.80-1.60-1.40-1.20-1.00-0.80-0.60-0.40-0.200.00
0 días 10 días estrées
17 días estrés Riego de recuperación
Pot
enci
alhí
dric
o de
hoj
a (ψ
hoj
a)
85-2 Control 85-2 EstrésCC Control CC EstrésM21 Control M21 Estrés
Días de estrés
Figura 7. Potencial hídrico de hoja 1 pm de las diferentes variedades de maíz bajo condiciones de
sequía. CC: Cajete criollo, M21: Michoacán 21.
VI.1.2. Tasa fotosintética
Para determinar el efecto de la sequía sobre la actividad fotosintética en los
tres genotipos, las tasas fotosintéticas fueron determinadas a los 10 y 17 días
de estrés y después del riego de recuperación con sus controles respectivos a
partir de las 7 am cada dos horas hasta las 7 pm. Bajo condiciones de riego,
Michoacán 21 mostró la tasa fotosintética más alta seguida por Cajete criollo y
85-2. Los tratamientos de estrés causaron una reducción en las tasas
fotosintéticas en los tres genotipos. Michoacán 21 mostró la reducción más
rápida de la tasa fotosintética a los 10 días de estrés en comparación con los
niveles en las plantas bajo riego (71.4%). Cajete criollo mostró una
disminución más lenta en la tasa fotosintética (34.9%) comparado con 85-2
(41.3%) y Michoacán 21. Después del riego de recuperación M21 mostró una
91

92
gran recuperación de fotosíntesis seguido de Cajete criollo y 85-2. Los
resultados de la tasa fotosintética se muestran en la Figura 8.
VI.1.3 . Conductancia estomatal
El estrés por sequía también causó una disminución gradual en la
conductancia estomatal en los 3 genotipos de maíz mientras el estrés
comenzó a ser más severo. Los valores de las plantas bajo riego antes del
estrés a las 11 am fueron muy similares para las 3 variedades, aunque
Michoacán 21 mostró un valor ligeramente más alto. Los valores de
conductancia estomatal en los controles de cada variedad en los días de
estrés siempre estuvieron superiores comparados con las plantas estresadas.
A los 10 días de estrés, Michoacán 21 mostró una caída rápida (75%) en la
conductancia estomatal comparado con las plantas irrigadas, mientras para
Cajete criollo y 85-2 la conductancia estomatal disminuyó 33.3% y 62.5%
respectivamente. A los 17 días de estrés las 3 variedades de maíz mostraron
una disminución significativa en la conductancia estomatal (90% para 85-2,
93.33% para Cajete criollo y 77.77% para Michoacán 21) comparado con el
valor correspondiente al control de riego en el tratamiento de estrés. Durante
el riego de recuperación los 3 genotipos mostraron un rápido incremento en la
conductancia estomata. Los resultados de la conductancia estomatal se
muestran en la Figura 9.

Figura 8. Fotosíntesis a las 11 am de las diferentes variedades de maíz bajo condiciones de sequía. CC: Cajete criollo, M21: Michoacán 21
Figura 9. Conductancia estomatal a las 11 am de las diferentes variedades de maíz bajo condiciones de sequía. CC: Cajete criollo,
M21: Michoacán 21
93

94
VI.2. Variación en la concentración de azúcar
En respuesta al estrés, los carbohidratos en hoja se alteran y podría servir
como una señal metabólica en respuesta al estrés. La síntesis de almidón está
normalmente bajo una fuerte inhibición incluso bajo déficit hídrico moderado, la
concentración de azúcares solubles en general se incrementa o al menos
permanece constante bajo condiciones de estrés. Se ha observado que la
acumulación de azúcares simples como glucosa y fructosa siguen en la
actividad de la invertasa en hojas bajo sequia (Mahajan y Tuteja, 2005). Es por
ello que se decidió realizar las mediciones de algunos azúcares para observar
si existen diferencias bajo sequia entre las diferentes variedades. El análisis
del contenido de glucosa y Mio-inositol (Figuras 10 y 11) mostró un incremento
en ambos azúcares a los 17 días de estrés en Michoacán 21, mientras que
Cajete criollo mostró un aumento en el nivel de glucosa a los 10 días de estrés
pero una disminución a los 17 días de estrés. 85-2 mostró una disminución de
los niveles de glucosa comparado con los controles respectivos en cada
tratamiento de estrés. Cajete criollo mostró los niveles más altos de Mio-
inositol a los 10 días de estrés. Michoacán 21 mostró un aumento en los
niveles de Mio-inositol conforme el estrés fue más severo. Durante el riego de
recuperación, los niveles de glucosa disminuyeron en 85-2 y Cajete criollo
comparado con sus respectivos controles, mientras que los niveles de Mio-
inositol disminuyeron durante el riego de recuperación en las tres variedades
de maíz. Con respecto a la sacarosa, se encontró una disminución de sus
niveles con respecto a su respectivo control para cada tratamiento de estrés y
cada variedad de maíz. Sin embargo, se observo un ligero aumento de los
niveles del mismo a los 17 días de estrés en todas las variedades (Ver Figura
12).
VI.3. Contenido de prolina
La sequía causa cambios en el metabolismo de aminoácidos en general y en
particular en la acumulación de prolina que ha sido correlacionada con la

95
osmoprotección en muchas especies vegetales (Chaves et al, 2005; Bartels y
Sunkar, 2005). La determinación de los niveles de prolina en los 3 genotipos
de maíz bajo sequía y en recuperación mostró un incremento de 3 veces a los
10 días de estrés en 85-2 y de 3.4 a 3.8 veces de incremento en Cajete criollo
y Michoacán 21 respecto a sus respectivos controles. A los 17 días de estrés,
el contenido de prolina en 85-2 se incremento en 5.7 veces y
aproximadamente 4.5 y 3.5 veces en Cajete criollo y Michoacán 21. Sin
embargo, Michoacán 21 mostró el nivel más alto de prolina a los 17 días de
estrés. En el riego de recuperación, los niveles de prolina disminuyeron en
todas las variedades a 3.4 veces más, comparado con los valores controles en
ese mismo tratamiento en 85-2 y cerca de 2.3 y 2.7 veces más en Cajete
criollo y Michoacán 21 (Figura 13). Siendo Michoacán 21, a pesar del riego de
recuperación la variedad que tuvo un mayor nivel de prolina. Estos resultados
sugieren que la protección osmótica por acumulación de prolina pudiera ser un
factor importante para la tolerancia a la sequía ya que es compartido por
ambos genotipos tolerantes.

Figura 11. Contenido de Mio-inositol en las tres variedades de maíz bajo condiciones de sequía. CC: Cajete criollo y M21: Michoacán 21.
Figura 10. Contenido de glucosa en las tres variedades de maíz bajo condiciones de sequía. CC: Cajete criollo y M21: Michoacán 21.
96

Figura 12. Contenido sacarosa en las tres variedades de maíz bajo condiciones de sequía. CC: Cajete criollo y M21: Michoacán
21.
Figura 13. Contenido de prolina en las tres variedades de maíz bajo condiciones de sequía. CC: Cajete criollo y M21: Michoacán
21.
97

98
VI.4. Análisis de los microarreglos a partir de las bibliotecas generadas en el Proyecto de Maíz (CINVESTAV)
Con la finalidad de evaluar los transcritos diferencialmente expresados
obtenidos de bancos sustractivos de cADN realizados anteriormente (Hayano
Kanashiro, 2004) e identificar secuencias candidatas potenciales involucradas
en la tolerancia a sequía, se llevó a cabo hibridaciones entre ARN de hoja y
raíz de la variedad Cajete criollo y microarreglos de los ensamblados de
cADNs realizados bajo condiciones de carencia de fósforo (Calderón Vázquez,
2008) y los generados anteriormente bajo condiciones de sequía (Hayano
Kanashiro, 2004). La impresión de los vidrios se realizó en Cinvestav Irapuato,
en el Laboratorio de Microarreglos y constó de 814 genes con 9 réplicas cada
uno. Se utilizó ARN extraído de la variedad Cajete criollo de hoja y raíz bajo
condiciones de sequía (10 días de estrés) y el riego de recuperación cada uno
con sus respectivos controles bajo riego. Se obtuvieron 83 secuencias
diferenciales con un valor de ±1.5 de cambio en la expresión y con un valor de
probabilidad (ANOVA) ≤ 0.05 para los 10 días de estrés; de los cuales 26
fueron reprimidos y 57 inducidos. Para el riego de recuperación, 60 secuencias
diferenciales fueron encontradas, de las cuales 7 fueron reprimidas y 53
inducidas. Los datos de las secuencias diferenciales como anotación
funcional, valores de expresión y valor de probabilidad se encuentran en la
Tabla Suplementaria 1. De estas 60 secuencias diferenciales 6.8% se
encontraban inducidas a los 10 días de estrés y reprimidas en el riego de
recuperación. Mientras que 4.9% de las secuencias diferenciales se
encontraban inducidas tanto a los 10 días de estrés como en el riego de
recuperación, sin embargo, se observó una ligera disminución en el nivel de
expresión durante el riego de recuperación. Mientras que 2.9% no tuvieron
cambio a los 10 días de estrés ni en el riego de recuperación.

99
VI.5. Análisis de los microarreglos de oligonucleótidos
El arreglo de oligonucleótidos de maíz (MOA de sus siglas en ingles) se utilizó
para analizar las diferencias en la expresión génica global bajo sequía entre
los dos genotipos tolerantes y el susceptible. Las diferencias en el número,
nivel de expresión y el tipo de genes de respuesta a estrés se observaron
entre las diferentes variedades y bajo los diferentes tratamientos de sequía. En
general, los cambios en la expresión génica (inducidos y reprimidos) fueron
mayores en Michoacán 21. Cajete criollo mostró una respuesta intermedia y
85-2 el número más bajo de genes expresados diferencialmente.
Una comparación numérica de los transcritos expresados diferencialmente
entre los 3 genotipos bajo condiciones de estrés se muestra en las Figuras
14B y 14C. A los 10 días de estrés, 103 genes inducidos y 92 reprimidos son
comunes a las 3 variedades. Michoacán 21 muestra un número mayor de
transcritos específicos expresados diferencialmente (106 inducidos y 86
reprimidos) comparado con 85-2 (90 inducidos y 79 reprimidos) y Cajete criollo
(70 inducidos y 86 reprimidos) (Figura 14B). Sin embargo, conforme el nivel de
estrés se incrementa, el número de genes expresados diferencialmente
también se incrementa. A los 17 días de estrés, un total de 246 genes
inducidos y 106 reprimidos fueron comunes a las 3 variedades. Al mismo
tiempo, la variedad susceptible (85-2) mostró el número más bajo de genes
expresados diferencialmente (51 inducidos y 57 reprimidos), Cajete criollo
mostró un número intermedio (123 inducidos y 143 reprimidos) y Michoacán
21 el número más alto (611 inducidos y 411 reprimidos) (Figura 14B). A los 17
días de estrés, el número de transcritos diferenciales compartidos por las
variedades tolerantes (141 inducidos y 198 reprimidos) es también mayor que
aquellos compartidos entre cada variedad tolerante con la susceptible.
En total un mayor número de transcritos expresados diferencialmente fueron
observados en el riego de recuperación en comparación con aquellos
encontrados a los 10 y 17 días de estrés (Figuras 14B y 14C); bajo estas

100
condiciones; 444 transcritos están inducidos y 653 reprimidos en las tres
variedades. Un patrón de expresión similar observado a los 17 días de estrés
fue también observado en el riego de recuperación, con 85-2 mostrando un
nivel más bajo de genes diferenciales específicos de este genotipo (218
inducidos y 139 reprimidos), mientras que Cajete criollo mostró un
comportamiento intermedio (460 inducidos y 237 reprimidos) y Michoacán 21
el nivel más alto (662 inducidos y 1064 reprimidos). El número de transcritos
expresados diferencialmente compartidos por las variedades tolerantes (746
inducidos y 548 reprimidos) es también más alto que aquellos compartidos
entre cada una de las tolerantes y la 85-2. Además, se encontró que 65.49%
de los genes inducidos a los 17 días de estrés fueron reprimidos en el riego de
recuperación; mientras que el 55.68% de los genes reprimidos a los 17 dias de
estrés fueron inducidos en el riego de recuperación. Tambien se encontró que
el 8.13% de los genes fueron inducidos tanto a los 10 y 17 días de estrés.
VI.6. Análisis de la expresión de genes mediante RT-PCR cuantitativo en tiempo real
El patrón de expresión diferencial observado en el experimento de
microarreglos fue corroborado para 16 genes usando qRT-PCR (Figura 15). La
mayoría de los genes seleccionados mostraron el mismo patrón de expresión
en el microarreglo y en el análisis de qRT-PCR. Las diferencias fueron
principalmente observadas a nivel cuantitativo, con el análisis de qRT-PCR
mostrando en general un cambio mayor de inducción o represión que el
análisis de microarreglos. Los genes se seleccionaron en base a: 1) la
intensidad en sus niveles de expresión (los más inducidos y/o más reprimidos)
bajo las condiciones de estrés y 2) reportados anteriormente de respuesta a
déficit hídrico o sequía. Aunque en algunos de los casos los datos de los
microarreglos no eran significativos en determinado tratamiento de estrés
(FDR ≥0.05) para algunos de los genes analizados; debido a que se
seleccionaron principalmente los de respuesta durante los 17 días de estrés.

101
La mayoría de los cambios en la expresión de estos genes fueron muy
similares con respecto al qRT-PCR. Diferencias cuantitativas similares entre el
qRT-PCR y los datos de microarreglos han sido reportados previamente
(Marcuende et al, 2007 y Calderón-Vazquez et al, 2008).

51 123
611
246
23
14177
85‐2 Cajete criollo
Michoacán 21
57 143
411
106
25
19828
85‐2 Cajete criollo
Michoacán 21
C Genes inducidos (17 días de estrés)
Genes reprimidos (17 días de estrés
218 460
662
444
88
74671
85‐2 Cajete criollo
Michoacán 21
139 237
1064
653
49
548197
85‐2 Cajete criollo
Michoacán 21
D Genes reprimidos(Riego de recuperación)
Genes reprimidos (Riego de recuperación)
Michoacán 21 Michoacán 21
90 70
106
103
32
5457
85‐2 Cajete criollo
79 86
86
92
33
4446
85‐2 Cajete criollo
B Genes inducidos (10 días de estrés)
Genes reprimidos (10 días de estrés
Riego de recuperación
17 días de estrés
A 10 días de estrés
1 2 3 1 2 3 1 2 3
Figura 14. A. Cambios globales en la expresión génica bajo sequía y riego de recuperación. 1: 85-2, 2: Cajete criollo, 3: Michoacán 21. Barras rojas: genes inducidos, barras verdes: genes reprimidos y barras amarillas: genes sin cambio de expresión. Diagramas de Venn de genes inducidos y reprimidos bajo: B. 10 días de estrés, C. 17 días de estrés y D. riego de recuperación entre las diferentes variedades de maíz (Zea mays L.).
102

0.0100
0.0200
0.0400
0.0800
0.1600
0.3200
0.6400
1.2800
2.5600
5.1200
10.2400
20.4800
40.9600
81.9200
TC279430
CD952060
AZM5_14615
CF628075_root
TC260113
TC191404
TC312355
AZM5_46378
NP161441
TC318956
DR802497
TC310514
TC296224
TC321656
TC191823
TC283096
85‐2
CC
M21
qRT‐PCR (10 DÍAS DE ESTRÉS)
0.0100
0.1000
1.0000
10.0000
100.0000
TC279430
CD952060
AZM5_14615
CF628075_root
TC260113
TC191404
TC312355
AZM5_46378
NP161441
TC318956
DR802497
TC310514
TC296224
TC321656
TC191823
TC283096
85‐2
CC
M21
qRT‐PCR (17 DÍAS DE ESTRÉS)
0.010
0.100
1.000
10.000
100.000
1000.000
TC279430
CD952060
AZM5_14615
CF628075_root
TC260113
TC191404
TC312355
AZM5_46378
NP161441
TC318956
DR802497
TC310514
TC296224
TC321656
TC191823
TC283096
85‐2
CC
M21
qRT‐PCR (RIEGO DE RECUPERACIÓN)
0.0001
0.001
0.01
0.1
1
10
100
1000
10000
TC279430
CD952060
AZM5_14615
CF628075_root
TC260113
TC191404
TC312355
AZM5_46378
NP161441
TC318956
DR802497
TC310514
TC296224
TC321656
TC191823
TC283096
85‐2
CC
M21
MICROARREGLOS (RIEGO DE RECUPERACIÓN)
* * *
0.010
0.100
1.000
10.000
100.000
TC279430
CD952060
AZM5_14615
CF628075_root
TC260113
TC191404
TC312355
AZM5_46378
NP161441
TC318956
DR802497
TC310514
TC296224
TC321656
TC191823
TC283096
85‐2
CC
M21
MICROARREGLOS (17 DÍAS DE ESTRÉS)
* *
0.010
0.100
1.000
10.000
100.000
TC279430
CD952060
AZM5_14615
CF628075_root
TC260113
TC191404
TC312355
AZM5_46378
NP161441
TC318956
DR802497
TC310514
TC296224
TC321656
TC191823
TC283096
85‐2
CC
M21
MICROARREGLOS (10 DÍAS DE ESTRÉS)
* * * * * * * * * * *
103
Figura 15. Validación de los microarreglos por qRT-PCR. (*) Genes que no tienen valores significativos en los microarreglos (FDR ≥ 0.05). TC279430 :Nitrato reductasa, CD952060: proteína putativa ERD4, AZM5_14615: Ribulosa bisosfato carboxylasa/oxygenasa activasa, CF628075_root: proteína regulada por frio, TC260113: probable photosystema II oxygen-evolving complex protein 2 precursor, TC191404: Proteína chlorophyll a-b binding, precursor de cloroplasto, TC312355: ferredoxina, AZM5_46378: receptor ser/thr protein precursor, NP161441|AF099387.1|AAF04662.1: R2R3MYB-dominio proteico, TC318956: proteína like de transporte de prolina, DR802497: fructosa-1, 6-bifosfatasa, TC310514: protein cinasa putativa dependendiente de Ca+2, TC296224: Protein cinasa putative de repetido de transmembrana rico en leucina, TC321656: Triosafosfato isomerasa putativa, TC191823: mio-inositol 1-fosfate sintasa, TC283096: proteína de choque térmico 26.

104
VI.7. Clasificación funcional de los transcritos expresados diferencialmente
Debido a la limitada anotación funcional que se encuentra disponible hasta el
momento para los transcritos de maíz, la clasificación funcional de los transcritos
diferencialmente expresados fue llevada a cabo usando el software de ontología
jerárquica MapMan (Thimm et al, 2004) como se describió en Materiales y Métodos.
Una visión general de los procesos celulares y metabólicos en los que los genes
expresados diferencialmente participan de acuerdo al programa MapMan se
muestra en las Figuras 16A, 17A y 18A para 85-2, Cajete criollo y Michoacán 21
respectivamente para 17 días de estrés y la Figura 16B, 17B y 18B para 85-2,
Cajete criollo y Michoacán 21 para el riego de recuperación. Estos mapas globales
de genes expresados diferencialmente ilustran que las variedades tolerantes
muestran una mayor respuesta metabólica y celular que 85-2 a los 17 días de
estrés y en el riego de recuperación.
VI.8. Fotosíntesis y metabolismo de carbohidratos
Para determinar si los cambios observados en las tasas fotosintéticas descritas
anteriormente correlacionaron con los cambios en la expresión génica, el efecto de
la sequía y el riego de recuperación en genes que codifican para componentes de la
maquinaria fotosintética fueron analizados en detalle. A los 10 días de estrés, 4, 1 y
2 genes inducidos (estas diferencias indican tendencias entre los cultivares pero no
son estadísticamente significativas) y 13, 17 y 22 reprimidos relacionados a
fotosíntesis fueron encontrados en 85-2, Cajete criollo y Michoacán 21
respectivamente (Tabla suplementaria 2). A los 17 días de estrés, todos los genes
diferencialmente expresados relacionados a fotosíntesis, fueron reprimidos en los 3
genotipos (85-2: 11 genes, Cajete criollo: 28 genes y Michoacán 21: 45 genes,
Tabla suplementaria 3). La única excepción notable fue un transcrito para una
fructosa-bifosfato aldolasa, la cual fue inducida en Michoacán 21. Una vista general
de estos datos es mostrada en la Fig. 19A, 19B y 19C para cada miembro de cada

105
Figura 16. Diagrama general de los transcritos expresados diferencialmente involucrados en los diferentes procesos metabólicos bajo sequía y riego de recuperación en las tres variedades de maíz. (A) Genes que se expresan a los 17 días en 85-2, (B) Genes que se expresan en el riego de recuperación en 85-2. Los cuadros de color rojo son aquellos transcritos inducidos, los de color verde los reprimidos
B
A

A
B
Figura 17. Diagrama general de los transcritos expresados diferencialmente involucrados en los diferentes procesos metabólicos bajo sequía y riego de recuperación en las tres variedades de maíz. (A) Genes que se expresan a los 17 días en Cajete criollo, (B) Genes que se expresan en el riego de recuperación en Cajete criollo. Los cuadros de color rojo son aquellos transcritos inducidos, los de color verde los reprimidos.
106

A
B
Figura 18. Diagrama general de los transcritos expresados diferencialmente involucrados en los diferentes procesos metabólicos bajo sequía y riego de recuperación en las tres variedades de maíz. (A) Genes que se expresan a los 17 días en
Michoacán 21, (B) Genes que se expresan en el riego de recuperación en Michoacán 21. Los
cuadros de color rojo son aquellos transcritos inducidos, los de color verde los reprimidos.
107

108
familia relacionada a la fotosíntesis. Interesantemente, Cajete criollo y Michoacán
21 mostraron más familias de genes que estaban reprimidos comparado con 85-2.
Por ejemplo, 3 genes relacionados con el ciclo de Calvin fueron reprimidos en 85-2
comparado con 7 en Cajete criollo y 12 en Michoacán 21. Los transcritos reprimidos
de los genes relacionados al ciclo de Calvin como triosafosfato isomerasa (TPI),
fructosa-1, 6 bifosfatasa (FBPasa), RBCS (RuBisCO sub-unidad pequeña) y
Rubisco activasa fueron identificadas.
En el riego de recuperación el patrón de expresión fue revertido, con un incremento
en la expresión diferencial de los genes relacionados a la fotosíntesis en los tres
genotipos (Tabal suplementaria 4). Las respuestas de las variedades fueron baja
(17), intermedia (61) y alta (81) en términos de número y niveles de inducción de
genes específicos para 85-2, Cajete criollo y Michoacán 21 respectivamente (Fig.
19D, 19E y 19F). Con respecto a los genes relacionados al ciclo de Calvin, 6, 19 y
21 genes fueron inducidos en 85-2, Cajete criollo y Michoacán 21 respectivamente.
La limitación de la fotosíntesis bajo sequia es un fenómeno complejo y los cambios
en el metabolismo del carbono ocurren en los procesos de deshidratación
tempranos. La sequia generalmente reduce la capacidad bioquímica para la
asimilación del acarbono y su utilización (Reddy et al, 2004). En base a ello, en el
presente trabajo se observó diferencias en la expresión de genes relacionados al
metabolismo de carbohidratos entre los genotipos. En este contexto, 8, 15 y 28
genes relacionados al metabolismo de carbohidratos son inducidos en el riego de
recuperación en 85-2, Cajete criollo y Michoacán 21 respectivamente. Un transcrito
para la ß-amilasa es inducido en los 3 genotipos a los 10 y 17 días de estrés y en
un mayor grado en M21 a los 17 días de estrés (Tabla suplementaria 3), mientras
que en el riego de recuperación, tres transcritos para la ß-amilasa fueron reprimidos
en M21 (Tabla suplementaria 4). Dos transcritos para hexocinasa (HXK) fueron
inducidas a los 17 días de estrés en Michoacán 21 y uno en 85-2 mientras en
Cajete criollo estos transcritos no alcanzaron el nivel de 2 (1.84 y 1.4). En el riego

Figura 19. Expresión diferencial de genes involucrados en la fotosíntesis. 17 días de estrés (A: 85-2, B: Cajete criollo and C: Michoacán 21). Riego de
recuperación (D: 85-2, E: Cajete criollo and F: Michoacán 21). Los cuadros de
color rojo son aquellos transcritos inducidos, los de color verde los reprimidos.
109

110
de recuperación tres transcritos de hexocinasa fueron reprimidos en Michoacán 21,
pero en 85-2 una expresión constitutiva fue observada mientras que en Cajete
criollo se observó una familia relacionada a la fotosíntesis. Interesantemente, Cajete
criollo y Michoacán 21 mostraron más familias de genes que estaban reprimidos
comparado con 85-2 variación de 0.64 sugiriendo que los cambios ocurren en el
metabolismo de la glucosa bajo estrés que son revertidos o reprimidos durante la
recuperación.
Tal vez sorprendentemente, los genes que codifican a las enzimas involucradas en
la síntesis de mio-inositol (Ins(3) P sintasa y MI monofosfatasa) son reprimidas o
mantenidas constantes bajo estrés en los 3 genotipos a pesar de las fluctuaciones
de los niveles de mio-inositol arriba descritos, sugiriendo que estos cambios son
probablemente regulados a nivel traduccional o post-traduccional. En el riego de
recuperación, sin embargo, Michoacán 21 muestra un ligero incremento en la
expresión de estos genes.
La inducción y represión de los genes asociados con proteínas involucradas en las
rutas de señalización dependientes e independientes de HXK fue observado como
se ha propuesto para A. thaliana. Por ejemplo, genes asociados con la ruta de
señalización de la glucosa dependiente de HXK tales como, CAB y Rbc fueron
reprimidas bajo estrés pero inducidas en el riego de recuperación, mientras que la
fosfolipasa D (PLD) fue inducida bajo sequía y reprimida en el riego de recuperación
para las 3 variedades. Para la ruta independiente HXK, ADP-glucosa pirofosforilasa
(AGPasa) y la fenilalanina amonia-liase (PAL), donde se encuentran reprimidas o
constitutivas en Cajete criollo y 85-2 pero inducidos en Michoacán 21 a los 17 días
de estrés, mientras que todos o están reprimidos o constitutivos en las 3 variedades
en el riego de recuperación.
Los genes que codifican las enzimas involucradas en el metabolismo de sacarosa
fueron principalmente constitutivas o reprimidas a los 17 días de estrés, sin
embargo, se encontró 1 y 4 transcritos inducidos para sacarosa sintasa (Susy) en
Cajete criollo y Michoacán 21 respectivamente a los 17 días de estrés. En el riego

111
de recuperación, la mayoría de genes relacionados al metabolismo de sacarosa
fueron inducidos en Cajete criollo y Michoacán 21, mientras que los niveles de
expresión de estos genes en 85-2 permanecieron constantes.
VI.9. Metabolismo de aminoácidos
VI.9.1. Metabolismo de prolina
Los patrones de expresión de genes que codifican enzimas involucradas en la
síntesis y degradación de prolina se encuentran acorde con los niveles de prolina
determinados. El gen de la pirrolina 5-carboxilasa sintasa fue inducido bajo estrés
en Cajete criollo y constitutiva en Michoacán 21 y 85-2 bajo 17 días de estrés. En
contraste, la ornitina aminotransferasa involucrada en una ruta biosintética diferente
de prolina fue inducido solo en Michoacán 21 bajo estrés (Tabla suplementaria 3).
Esto podría indicar un uso preferencial de uno u otra ruta biosintética de prolina en
las variedades tolerantes bajo sequía. Un transcrito para la prolina oxidasa
involucrados en la degradación de prolina mostró una disminución ligera (0.95 y
0.58 como valor de cambio) bajo sequía en Cajete criollo y Michoacán 21
respectivamente pero inducido en 85-2. En el riego de recuperación, un transcrito
para la pirrolina 5 carboxilato dehidrogenasa (P5CDH) involucrado en la
degradación de prolina fue inducido en los genotipos tolerantes pero constitutivo en
85-2. Dos transcritos para prolina oxidasa fueron inducidos en Cajete criollo y uno
en 85-2 (Tabla suplementaria 3) durante el riego de recuperación, pero ningún
cambio fue detectado para esta familia de genes en Michoacán 21. Por otro lado, se
encontró un transcrito que codifica para la pirrolina 5 carboxilato sintasa (P5CS),
involucrado en la síntesis de prolina que estaba reprimido en el riego de
recuperación en Michoacán 21, aunque 85-2 y Cajete criollo mostraron 0.79 y 0.54
como valor de cambio en la expresión respectivamente.
VI.9.2. Metabolismo de otros aminoácidos
Genes expresados diferencialmente relacionados a la degradación de leucina,
arginina, tirosina, triptófano, histidina, serina y síntesis de GABA (ácido gama-

112
aminobutírico) fueron identificados a los 10 días de estrés (Tabla suplementaria S1).
Un transcrito para glutamato decarboxilasa fue inducido solo en Michoacán 21 a los
10 días de estrés, pero a los 17 días de estrés, el mismo transcrito fue inducido en
85-2 y en un nivel mayor en Michoacán 21. Un gen para una putativa fosfoglicerato
dehidrogenasa fue reprimido en 85-2 y Cajete criollo a los 10 días de estrés pero
inducido en Michoacán 21 a los 17 días de estrés mientras las otras dos variedades
mostraron un valor de cambio de 0.7. Además, una fumarilacetoacetasa putativa
involucrada en la degradación de tirosina fue reprimida en 85-2 y Michoacán 21 a
los 10 días de estrés pero inducida solo en Michoacán 21 a los 17 días de estrés. A
los 17 días de estrés, los genes relacionados con tirosina, metionina, valina,
arginina, triptófano, alanina, serina, síntesis de corismato y GABA fueron
expresados diferencialmente (Tabla suplementaria 3). Interesantemente, 2
transcritos para el gen putativo de shikimato sintasa fueron inducidos sólo en
Michoacán 21. En el riego de recuperación, se observaron más genes
diferencialmente expresados relacionados con el metabolismo de tirosina,
asparagina, leucina, corismato, fenilalanina, triptófano, lisina, metionina, alanina y
aspartato en comparación con las obtenidos bajo estrés (Tabla suplementaria 4).
Dos transcritos reprimidos para fosfoglicerato deshidrogenasa a los 17 días de
estrés, fueron inducidos en Cajete criollo en el riego de recuperación, sin embargo,
en 85-2 y Michoacán 21 sólo uno fue inducido.
VI.10. Genes relacionados a señalización y estrés abiótico
Las respuestas metabólicas a diferentes tipos de estrés abiótico son a menudo
compartidas. Por lo que con la finalidad de comparar los cambios en la expresión de
genes relacionados entre las 3 variedades bajo sequía, los transcritos anotados
funcionalmente fueron agrupados en diferentes clases: metabolismo de hormonas,
señalización, transporte, detoxificación, proteínas de choque térmico (incluyendo
dehidrinas y LEA) y genes relacionados a estrés abiótico. En general, un número
menor de genes fueron diferencialmente expresados a los 10 días de estrés,
comparado con los 17 días de estrés; entre 40 y 100 genes fueron expresados

113
diferencialmente (tanto inducidos como reprimidos) a los 17 días de estrés para
cada clase de genes tomando en cuenta los 3 genotipos (Tabla suplementaria 2,
Tabla suplementaria 3). Aunque patrones similares de inducción y represión fueron
observados para todas las clases de genes en las 3 variedades, Michoacán 21
mostró los cambios más altos en la abundancia del transcrito de genes expresados
diferencialmente en todas las clases y muchos genes expresados diferencialmente
fueron únicos a Michoacán 21 (Tabla suplementaria 3). En el riego de recuperación,
en general el número de clases de genes inducidos fue más bajo comparado con el
número de genes reprimidos. Cajete criollo y Michoacán 21 mostraron patrones muy
similares de inducción, aunque el número de genes observados en cada clase fue
ligeramente mayor para Michoacán 21 con la excepción de los “genes de estrés
abiótico”, donde hay más transcritos inducidos en comparación a las otros
genotipos. La mayor diferencia entre las 3 variedades se observó para los
transcritos reprimidos en el riego de recuperación. Cajete criollo y 85-2 mostraron
patrones muy similares de represión donde los números de transcritos en cada
clase no excedieron de 40. En contraste para Michoacán 21, todas las clases con la
excepción de “genes de estrés abiótico” mostraron represión entre 40 a más de 70
transcritos que representan casi el doble transcritos en comparación con las otras 2
variedades (Fig. 20).
Algunos patrones de expresión bien definidos pudieron ser determinados para
genes específicos, en las clases definidas. Por ejemplo, genes asociados con
transducción de señales, tales como, proteína cinasas dependientes de calcio
(CDPKs), proteínas G y receptores cinasas fueron inducidas y reprimidas bajo
sequía. Michoacán 21 probó ser el genotipo con mayor número de genes
relacionados a señalización expresados diferencialmente a los 17 días de estrés en
comparación a las otras 2 variedades, con 42 genes expresados diferencialmente,
sólo 7 fueron comunes a las 2 variedades tolerantes y 27 genes fueron únicos a
Michoacán 21 incluyendo: proteína de unión a calcio, proteína cinasas dependientes
de calcio (CDPKs), calmodulina, proteína de unión a GTP y fosfoinositidos (Tabla
Suplementaria 3). Para el proceso de recuperación, 170 genes expresados

114
diferencialmente relacionados a señalización fueron identificados. De estos, 32
fueron comunes a las 2 variedades tolerantes incluyendo genes que codifican a
receptor cinasas, proteínas G, señalización de calcio y fosfoinositidos. Cabe
mencionar que Cajete criollo y Michoacán 21 compartieron 17 genes inducidos
(Tabla suplementaria 4).
En cuanto a los genes de choque térmico, un gran número de genes que codifican a
proteínas de choque térmico (HSP) fueron inducidos a los 17 días de estrés,
especialmente en las variedades tolerantes de maíz (24 y 31 para Cajete criollo y
Michoacán 21 respectivamente) en comparación a 85-2 (16). Tal vez
sorprendentemente solo los transcritos para HSP y LEA fueron significativamente
inducidos bajo estrés y en común con HSP18, HSP70, DnaJ, HSF y dehidrinas
estos transcritos fueron fuertemente reprimidos en el riego de recuperación. Otros
genes relacionados a estrés abiótico, tales como estrés a frio, sequía y salinidad
estaban en general inducidos bajo estrés y reprimidos en el riego de recuperación
como se esperaba. Muchos transcritos asociados a transporte, mostraron un
cambio pequeño bajo sequía pero mostraron tanto inducción como represión en el
riego de recuperación incluyendo aquellos asociados con aminoácidos o metales,
transportadores ABC, transportadores de Pi y metabolitos y transporte de cationes
H+ATPasas. Un patrón de inducción bajo estrés y represión en el riego de
recuperación fue observado para los genes que codifican aquaporinas. A los 10
días de estrés, 2 transcritos inducidos para las proteínas intrínsecas de tonoplasto
(TIPs) fueron identificados solo en las variedades tolerantes. Uno de estos TIPs fue
también inducido a los 17 días de estrés solo en las 2 variedades tolerantes. Dos
transcritos para la proteína intrínseca nodulin-like (NIPs) fueron inducidos a los 17
días de estrés y en el riego de recuperación fueron reprimidos. Otros genes
asociados a transporte fueron principalmente responsivos en el riego de
recuperación. Interesantemente los genes asociados al transporte de azucares
fueron inducidos y reprimidos en ambos estadios, reflejando cambios en el
metabolismo de azúcares y transporte que ocurre en relación a los niveles de

Figura 20. Clasificación funcional de genes de estrés abiótico bajo 17 días de estrés y riego de recuperación en las tres variedades de maíz. (A): Genes inducidos bajo 17 dias de estrés, (B): Genes reprimidos bajo 17 dias de estrés, (C): Genes inducidos bajo riego de recuperación, (D): Genes reprimidos bajo riego de recuperación.
115

116
fotosíntesis bajo sequía y recuperación. Los genes relacionados al transporte de
sulfato fueron reprimidos durante el estrés e inducidos en el riego de recuperación.
En relación a los genes asociados con detoxificación, solo aquellos para
peroxidasas y tioredoxinas fueron inducidos. Estos genes mostraron tanto inducción
como represión en el riego de recuperación, así como los genes asociados con el
metabolismo de ascorbato, glutatione, las dismutasas y catalasas. Además de los
genes asociados al transporte de sulfato, los únicos transcritos que mostraron
claramente una represión bajo sequía e inducción en el riego de recuperación
fueron aquellos asociados con las periredoxinas. Michoacán 21 y Cajete criollo
mostraron el número más alto de genes para periredoxinas expresados
diferencialmente en el riego de recuperación relacionado a este grupo comparado
con 85-2.
VI.11. Respuestas relacionadas a hormonas
Muchos genes relacionados al metabolismo y señalización por hormonas fueron
expresados diferencialmente bajo sequía y en el riego de recuperación, tales como
aquellos relacionados a ácido abscísico (ABA), auxinas, citocininas y metabolismo
de etileno. La hormona vegetal ABA juega un papel central en muchos aspectos de
respuesta a varias señales de estrés (Bartels y Sunkar, 2005; Mahajan y Tuteja,
2005) y ha sido mostrado que participa en respuesta a sequía y mecanismos de
tolerancia a salinidad (Wasilewska et al, 2008). En este estudio, la expresión
diferencial de genes involucrados en el metabolismo de ABA (síntesis, degradación
y/o transducción de señales) fue observada bajo sequía y riego de recuperación. Un
transcrito para 9-cis epoxicarotenoide dioxigenasa fue inducido en 85-2 y
Michoacán 21, un gen de zeaxantina epoxidasa fue también inducido en 85-2 y
Cajete criollo bajo 10 días de estrés. A los 17 días de estrés, los transcritos para los
genes ABA insensitive 3 (ABI3) y ABA responsive HVA22 fueron inducidos solo en
Michoacán 21. En el riego de recuperación, sólo tres transcritos relacionados a ABA
fueron inducidos: HVA22 solo en 85-2, ABI3 sólo en Cajete criollo y 9-cis
epoxicarotenoide dioxigenasa en los dos genotips tolerantes. Asimismo, 11

117
transcritos reprimidos relacionados al metabolismo de ABA fueron identificados.
Durante el riego de recuperación, los transcritos para AREB2, ABRE y una proteína
fosfatasa 2C involucrados en la transducción de señales por ABA fueron reprimidos
en las 3 variedades y 5 transcritos para HVA22 fueron reprimidos en al menos una
de las variedades. Sin embargo, un transcrito para HVA22 que fue inducido en
Michoacán 21 a los 17 días de estrés fue reprimido en el riego de recuperación,
pero 85-2 y Cajete criollo no mostraron un cambio marcado.
Las citocininas son hormonas que juegan un papel importante en el crecimiento y
desarrollo vegetal (Brugiére et al, 2003). En este estudio, los genes relacionados a
las citocininas mostraron pocos cambios bajo sequía pero fueron fuertemente
inducidos en el riego de recuperación con pocos genes reprimidos. Bajo 10 días de
estrés, un transcrito para citocinina oxidasa fue inducido solo en 85-2. A los 17 días
de estrés fueron identificados transcritos expresados diferencialmente relacionados
con la transducción de señales de citocininas. Una histidina fosfotranferasa que
contiene el factor 5 (HPt) fue inducida solo en Michoacán 21 y tres genes fueron
reprimidos en al menos una de las 3 variedades. Sin embargo, en el riego de
recuperación se observaron muchos transcritos inducidos incluyendo 14, 17 y 18 en
85-2, Cajete criollo y Michoacán 21 respectivamente (aunque estas diferencias en el
numero de transcritos no son estadísticamente significativas). La mayoría fueron
genes reguladores de respuesta (ARR de sus siglas en ingles) relacionados con la
transducción de señales de citocininas. Sólo tres genes fueron reprimidos en al
menos una de las 3 variedades, 2 de los cuales codifican citocinina oxidasas
putativas que estaban reprimidas en los 3 genotipos y una citocinina oxidasa
reprimida solo en 85-2 y Cajete criollo.
Genes relacionados a auxinas mostraron una inducción de 2, 1 y 5 transcritos
(estas diferencias en el número de transcritos no son estadísticamente
significativas) a los 10 días de estrés en 85-2, Cajete criollo y Michoacán 21
respectivamente. A los 17 días de estrés, 7 genes inducidos y 6 reprimidos para el
metabolismo de auxinas fueron identificados y un transcrito para la enzima GH3-like

118
protein; que conjuga los aminoácidos al ácido indol-3 acético (Park et al, 2007), fue
inducido en mayor grado en Michoacán 21. En el riego de recuperación, el número
de genes expresados diferencialmente fue de 18 y 28 genes inducidos y reprimidos
respectivamente en al menos una de las 3 variedades. Notablemente, el gen GH3
mostró represión solo en Michoacán 21.
El etileno es otra hormona que está relacionada a las respuestas de estrés abiótico.
Sin embargo, se encontró que los genes relacionados a la síntesis y señalización
del etileno no mostraron una respuesta fuerte bajo estrés aunque un transcrito que
codifica una oxidoreductasa fue inducida en Cajete criollo y Michoacán 21 a los 10
días de estrés y a los 17 días de estrés solo en Michoacán 21. El gen
correspondiente a la ACC oxidasa fue reprimido solo en Michoacán 21 mientras que
en Cajete criollo, este gen se encontró constitutivo y en 85-2 el valor de expresión
fue de 0.81. Un gen correspondiente al factor de respuesta a etileno fue también
reprimido en Cajete criollo y Michoacán 21 a los 17 días de estrés. En el riego de
recuperación, muchos genes relacionados al metabolismo de etileno fueron
reprimidos. El gen de la oxidoreductasa dependiente de Fe/ascorbato fue reprimido
bajo este tratamiento en Cajete criollo y Michoacán 21 pero no en 85-2. Transcritos
de ACC oxidasa fueron expresados constitutivamente en Michoacán 21 pero
reprimidos en Cajete criollo. Un transcrito reprimido para el factor de respuesta a
etileno (ARF) fue identificado en 85-2 y otro en 85-2 y Michoacán 21. Sin embargo,
un transcrito de ARF fue también inducido en Cajete criollo y Michoacán 21 en el
riego de recuperación.
VI.12. Factores de transcripción
La Tabla 3 compara el número de genes expresados diferencialmente que codifican
a diferentes familias de factores de transcripción (FT) para cada genotipo. Todas las
familias de FT analizadas mostraron expresión diferencial en las 3 variedades con
diferencias en los patrones de inducción/represión. En general, la respuesta de
cada variedad mostro un patrón parecido al descrito previamente para los números
de transcritos expresados diferencialmente: 85-2, bajo, Cajete criollo, intermedio y

119
Michoacán 21 alto. Un total de 121 genes expresados diferencialmente que
codifican a FT fueron identificados bajo estrés. Entre éstos, las variedades
tolerantes CC y M21 mostraron más genes inducidos y reprimidos (24 y 78
respectivamente) para las familias bHLH, WRKY, C2C2 y C2H2, HB y CCAAT
(HAP2) comparado con 85-2. Además, más miembros de las familias AP2/EREBP,
HB y MADS estaban inducidos específicamente en Michoacán 21 bajo estrés.
Tanto CC y M21 tenían un mayor número de FT inducidos y reprimidos en el grupo
no clasificado comparado con 85-2. En el riego de recuperación, 202 genes
expresados diferencialmente codifican a FT. Las variedades tolerantes mostraron
respuestas significativamente diferentes con más genes reprimidos e inducidos
(Cajete criollo 104 y M21 139 FTs respectivamente), comparado con la variedad
susceptible para AP2, NAC, MADS, CCAAT, HB, bHLH y bZIP, los cuales la
mayoría de ellos fueron inducidos bajo estrés pero reprimidos en el riego de
recuperación. Un miembro de la familia HAP3 para el cual se ha mostrado
previamente que confiere tolerancia a sequía, por la sobreexpresión de NF-YB
(HAP3) en Arabidopsis y maíz (Nelson et al, 2007) es reprimido específicamente
durante la recuperación en Michoacán 21, mientras los transcritos para HAP2 y
HAP5 fueron inducidos en Cajete criollo y Michoacán 21, también bajo riego de
recuperación. Michoacán 21 mostró tener el mayor número de familias de genes de
FTs diferenciales, sugiriendo que ésta es la variedad con una mayor respuesta a
nivel de expresión diferencial bajo sequía, así como bajo riego de recuperación.

Tabla 3. Principales familias de factores de transcripción (FT) diferencialmente reguladas bajo sequía y riego de recuperación en hojas de maíz de tres variedades diferentes
SEQUIA RIEGO DE RECUPERACION 85‐2 85‐2 Cajete
criollo Cajete criollo
Michoacán 21
Michoacán 21
85‐2 85‐2 Cajete criollo
Cajete criollo
Michoacán 21
Michoacán 21
IND R IND R IND R IND R IND R IND R
AP2/EREBP 1 0 2 0 4 1 1 2 3 1 5 6 bHLH 1 0 2 2 6 3 1 2 6 5 10 8 bZIP/Putative bZIP 2 0 0 0 2 1 3 1 5 2 5 6 C2C2 0 1 2 1 3 0 0 2 5 5 6 5 C2H2 0 2 0 3 0 1 1 5 7 5 7 5 CCAAT: 0 0 1 0 1 1 0 0 2 1 2 3-HAP2 0 0 1 0 1 1 0 0 1 0 1 0 -HAP3 0 0 0 0 0 0 0 0 0 0 0 3 -HAP5 0 0 0 0 0 0 0 0 1 1 1 0 DNA/Putative DNA binding protein
1 1 0 2 3 0 3 3 6 5 9 5
Finger/Putative Finger TF
0 2 0 2 0 2 0 2 1 2 1 2
G2 like 1 3 1 4 1 2 1 4 3 5 3 3 HB 3 0 3 2 5 1 4 5 5 7 8 6 HD leucine zipper 1 0 1 0 1 0 1 0 1 1 1 1 MADS 3 0 2 0 6 0 3 4 2 4 6 7 MYB 3 3 2 4 7 5 5 6 7 8 11 12 NAC 0 0 1 1 0 0 1 2 1 3 2 3 Unclassified 2 2 5 3 6 5 7 7 18 11 24 16 WRKY 0 2 0 3 0 1 2 2 1 4 1 3 Zinc finger/Zinc Finger HD
1 0 0 2 1 2 3 2 9 2 9 6
Total 19 16 22 29 46 24 36 49 83 71 111 98
120

121
VI.13. Asociación de genes expresados diferencialmente a QTL’s de maíz
Para explorar la posibilidad que algunos de los genes expresados diferencialmente
pudieran estar asociados con QTL’s reportados previamente para tolerancia a
sequía, se llevó a cabo una comparación entre los genes expresados
diferencialmente bajo sequía y en el riego de recuperación con QTL’s y ESTs de
maíz mapeados disponibles en www.maizegdb.org. Se identificaron 62 genes
expresados diferencialmente relacionados al menos a un locus de QTL (Tabla
suplementaria 4). Sin embargo, 24 de estos genes mostraron un nivel de inducción
y/o represión más alta en al menos una de las 3 variedades (Tabla 4). Los QTL’s
identificados estaban asociados con rendimiento de grano, peso de grano, intervalo
de antesis y floración (ASI), días a antesis, días a floración, altura de planta, número
de filas del grano y número de mazorcas por planta entre otros, sugiriendo que los
genes en estas regiones pudieran ser importantes para la tolerancia a sequía
(Figura suplementaria 2). Los genes que codifican a una invertasa vacuolar, una
proteína de la familia de respuesta a deshidratación, proteína cinasas y algunos
genes sin anotación funcional también fueron identificados. Sin embargo, una
relación directa entre los genes diferencialmente regulados y los QTL’s de tolerancia
a sequía requiere de experimentación adicional para ser demostrada.

122
Tabla 4. Lista de los transcritos asociados a QTL’s de maíz que mostraron mayores cambios de expresión en al menos una de las tres variedades
Identificador Bin QTL’s Tratamiento de estres
Categoria funcional MapMan
MZ00035814
1.09
Peso de grano, rendimiento de grano, contenido de proteína
17 días Inducido/RR-Reprimido
MZ00019574
1.09
Peso de grano, rendimiento de grano, contenido de proteína
RR-Inducido
MZ00044388
1.11
Peso de grano, longitud de mazorca y días a antesis
17 días Inducido/RR-Reprimido
Inhibidor de proteasa
MZ00035691
2.02
Peso de grano, rendimiento de grano numero de granos por fila, altura de planta
17 días Inducido/ RR-Reprimido
Metabolism degradation.sucrose
MZ00021386
2.09
Rendimiento de grano, peso de grano, numero de hojas, concentración de fitoglicógeno
17 días Inducido/ RR-Reprimido
Invertasa/pectin metilesterasa
MZ00016917
2.09
Dias a antesis RR-Reprimido
Proteínas de pared celular
MZ00030352
3.04
Peso de grano, intervalo ASI, peso de mazorca, longitud de mazorca, peso de grano, longitud de grano, peso de grano, altura de planta, concentración de almidón, contenido de proteína, concentración de sacarosa
RR-Inducido Modificación post-traduccional
MZ00028427 3.08 Peso de grano, altura de planta, 17 días Inducido/ RR-
Biodegradación de

123
contenido proteico Reprimido xenobióticos
MZ00025313
3.09
Altura de planta, contenido de aceite
Inducido RR
MZ00035263
4.04
Peso de grano 17 días Inducido/ RR-Reprimido
MZ00035502
4.04
Peso de grano 17 días Inducido/ RR-Reprimido
Glicólisis.
MZ00035802
4.05
Altura de planta, número de hileras de grano, rendimiento de grano
17 días Inducido
MZ00018721
4.06
días a crecimiento de estilo RR-Reprimido
Transporte transportadores ABC
MZ00017224
4.06
Peso de grano, altura de planta Peso de grano, altura de planta,, crecimiento de estilo
RR-Inducido
estrés abiotico sequía/salinidad
MZ00021105
4.09
Mazorcas por planta, número de hileras de grano, peso de grano, humedad de grano
RR-Reprimido
MZ00020352
5.08
Altura de planta, actividad de ADP-glucosa pirofosforilasa, sacarosa fosfato sintasa
17 días Inducido/ RR-Reprimido
Sin ontología
MZ00021059
6.02
Rendimiento de grano, número de hojas
RR-Reprimido
Degradación de proteína
MZ00028407
6.05
Peso de grano, intervalo de ASI, días a antesis diámetro de mazorca, longitud de mazorca, mazorcas por planta, peso de grano, rendimiento de grano, longitud de grano, altura de planta, contenido proteico, concentración de almidón,
RR-Inducido
Degradación de proteína

124
rendimiento de almidón
MZ00003720
7.01
Mazorcas por planta, peso de grano
17 días Inducido/ RR-Reprimido
Desarrollo
MZ00041800
8.03
Peso de grano, intervalo de ASI, días a crecimiento de estilo, rendimiento de grano, peso de grano, altura de –planta, stay green, concentración de almidón y rendimiento de almidón
17 días Inducido/ RR-Reprimido
Metabolismo de hormonas
MZ00035827
8.04
Altura de planta, contenido de proteína, peso de grano, días a crecimiento de estilo, días a antesis
RR-Reprimido
MZ00001326
9.05
Días a antesis, días a crecimiento de estilo, diámetro de mazorca, altura de planta, concentración de almidón, rendimiento de almidón
17 días Inducido
Desconocido
MZ00018811
10.02
Peso de grano, intervalo de ASI, rendimiento de grano
RR-Inducido
MZ00036787
10.04
Intervalo de ASI, diámetro de ASI, diámetro de mazorca, rendimiento de grano
RR-Inducido

125
VII. DISCUSIÓN
El objetivo de este estudio fue comparar los cambios a nivel fisiológico y de
expresión global génica de 2 variedades de maíz tolerantes y una susceptible en
respuesta a la aplicación gradual del estrés por sequía, con la finalidad de identificar
las respuestas generales de maíz bajo estas condiciones y las posibles diferencias
en los mecanismos empleados para alcanzar la tolerancia. Aunque existe alguna
variación genética entre las variedades estudiadas, con la repetición del
experimento de sequía en 2 diferentes años y el uso de réplicas de cada variedad
para obtener los datos fisiológicos y de expresión génica se produjeron datos
consistentes específicos para cada variedad. El monitoreo de los potenciales
hídricos de suelo a través de la aplicación de la sequía y en el riego de recuperación
aseguró que los niveles de estrés fueran adecuados y equivalentes para las 3
variedades.
VII.1. Respuestas fisiológicas: fotosíntesis, conductancia estomatal y potencial hídrico de hoja y suelo
Está bien documentado que durante el déficit hídrico, la mayoría de las plantas
responden rápidamente con el cierre estomatal para evitar la pérdida excesiva de
agua. Además, llevan a cabo cambios moleculares y fisiológicos para prevenir el
daño irreversible a la maquinaria fotosintética (para una revisión, ver Mahajan y
Tuteja, 2005). Estos procesos están ligados ya que el cierre estomatal resulta en
una disminución en la tasa de fotosíntesis (Pelleschi et al, 1997; Foyer et al, 1998).
Por lo tanto, los potenciales hídricos de hoja, la conductancia estomatal y la tasa de
fotosíntesis fueron monitoreados a diferentes tiempos a lo largo del experimento.
Esto reveló diferentes respuestas en las 3 variedades para los parámetros
fisiológicos evaluados. Cajete criollo, en general, mostró una disminución gradual y
menos pronunciada en el potencial hídrico de hoja, tasa fotosíntetica y conductancia
estomatal, mientras que Michoacán 21, incluso a los 10 días de estrés, mostró una
disminución significativa de la tasa fotosintética y conductancia estomatal. La
variedad susceptible, 85-2, respondió más lentamente que Michoacán 21 pero más

126
rápidamente, y en mayor grado, que Cajete criollo. Por otro lado, mientras que
Cajete criollo y Michoacán 21 mostraron un incremento rápido y fuerte en la
fotosíntesis con el riego de recuperación, la respuesta de 85-2 fue más débil. Esto
sugiere, como se observó previamente a partir de las observaciones en el campo,
los genotipos tolerantes a sequía probados poseen diferentes mecanismos para
poder afrontar el déficit hídrico. La estrategia de Michoacán 21, podría ser ventajosa
cuando se experimentan periodos cortos de sequía severa, pero la rápida reducción
en la tasa de fotosíntesis pudiera tener un efecto negativo bajo sequía prolongada
aunque menos severa. Bajo estas últimas premisas, la estrategia de Cajete criollo
de implementar una reducción gradual podría permitir a la planta sobrevivir periodos
más largos de baja disponibilidad de agua. De manera similar, la represión bajo
sequía de genes involucrados en la fotosíntesis fue previamente reportada en
cebada (Tálame et al, 2007), Arabidopsis (Seki et al, 2003), trigo (Xue et al, 2008) y
arroz (Degenkolbe et al, 2009).
VII.2. Respuestas moleculares comunes a la sequía en los tres genotipos de maíz
Un análisis general de los cambios globales en la expresión génica en respuesta a
sequía para las variedades tolerantes y susceptibles, muestra que existen muchas
alteraciones comunes, además de un grado significativamente diferente en términos
del número de familias de genes, número de miembros en cada familia y el valor de
cambio en la expresión, en el nivel de expresión de genes involucrados en
diferentes procesos metabólicos y celulares (Figura 21). La primera respuesta a la
sequía es el cierre de los estomas para proteger a la planta de la pérdida de agua
(Chaves et al, 2003; Mahajan y Tuteja, 2005) y, consecuentemente, la inhibición de
la fotosíntesis (Chaves et al, 2003). A este respecto, la primera respuesta común
obvia entre las 3 variedades fue la disminución en los niveles del transcrito de
genes asociados a la fotosíntesis durante la sequía y su incremento durante el riego
de recuperación. En particular, los genes que codifican a los componentes del
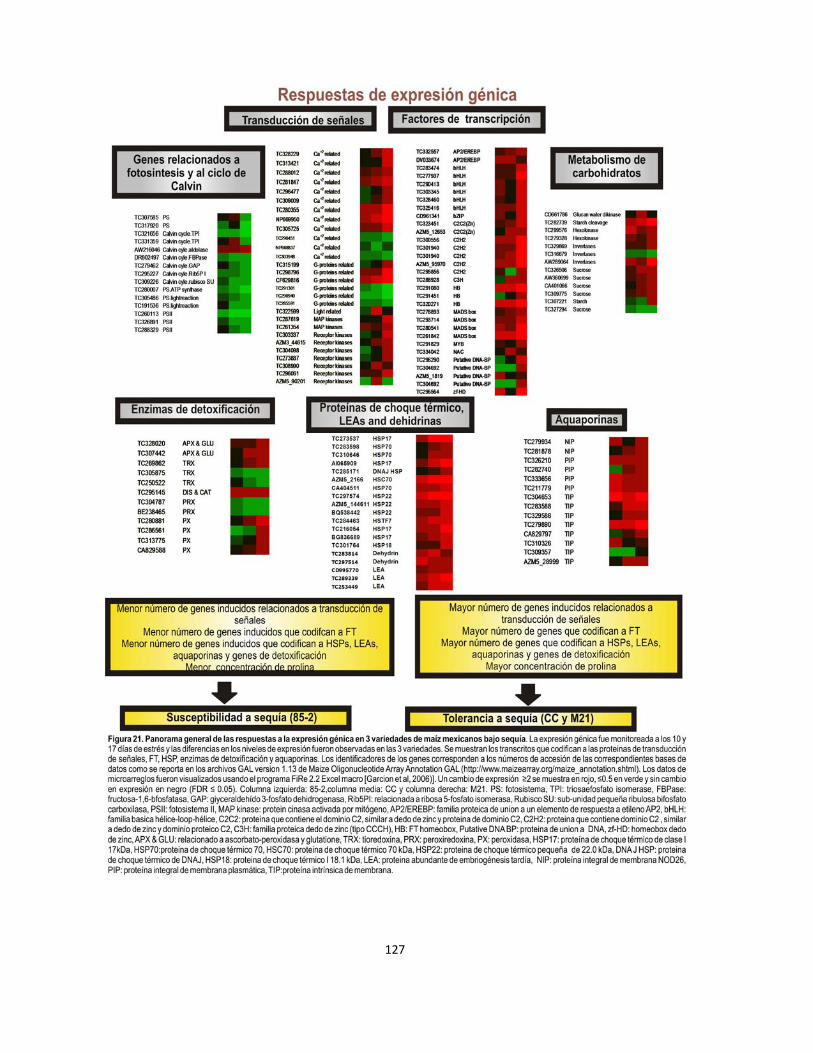
127

128
Fotosistema II y en menos grado del Fotosistema I y la enzima del Ciclo de Calvin
triosa fosfato isomerasa estuvieron reprimidas durante la sequía e inducidas durante
el riego de recuperación. Una reducción en los componentes de los Fotosistemas I y
II prevendrían la foto-oxidación del aparato fotosintético y la formación de radicales
libres que son dañinos para la célula, aunque como se discute más adelante existen
diferencias significativas en la expresión de genes asociados a la fotosíntesis que
son observados solo en los genotipos tolerantes de sequía o específicos a cada uno
de ellos. Otra característica esperada en el patrón general de expresión génica en
maíz bajo sequía fue la inducción de genes que codifican a HSPs y proteínas LEA.
Los genes que codifican a HSP 17, 22, 70 y 90 son inducidos en los 3 genotipos
bajo sequía, así como un factor de transcripción “heat shock”. Estas proteínas
previenen de los efectos detrimentales de estrés previniendo la agregación de
proteínas, protegiendo las enzimas no nativas de la degradación y ayudando en el
re-plegamiento de proteínas (Wang et al, 2003). La inducción de estos genes bajo
sequía fue observado en estudios previos sobre sequía en cebada (Talamé et al,
2007) y arroz (Rabanni et al, 2003), deshidratación en Arabidopsis (Seki et al, 2002)
y estrés por PEG en maíz (Jia et al, 2006). Interesantemente, sólo dos genes para
proteínas LEA que actúan como moléculas de unión a agua y están involucradas en
el secuestro de iones y en la estabilización de macromoléculas y membranas fueron
identificadas como inducidos en los 3 genotipos, sugiriendo que estas proteínas
inducibles por estrés podrían no jugar un rol importante en la respuesta general de
maíz bajo sequía.
La respuesta y el crecimiento de la planta en condiciones de estrés está bajo el
control de hormonas (Mahajan y Tuteja, 2005). En particular, el ABA ha sido
asociado con la apertura estomatal y juega un papel importante en la respuesta a la
tolerancia de las plantas a sequía y salinidad (Yamaguchi-Shinozaki y Shinozaki,
2006). En el presente estudio, los genes que codifican a enzimas relacionadas con
la síntesis de ABA (ZEP y NCED) fueron inducidas a los 10 días de estrés en las
tres variedades. Estos mismos genes fueron inducidos bajo deshidratación en
Arabidopsis (Yamaguchi-Shinozaki y Shinozaki, 2006). Como se mencionó

129
anteriormente, NCED es una enzima clave en la biosíntesis de ABA; At-NCED3 fue
fuertemente inducida por deshidratación y salinidad y su sobre-expresión mejoró la
tolerancia a deshidratación en las plantas transgénicas, indicando el rol importante
en la acumulación de ABA durante la deshidratación. La inducción de HVA22 bajo
diferentes estreses ambientales como frío y sequía y además por ABA ha sido
reportado en cebada (Shen et al, 2001). También fue reportado que la expresión
ectópica de ABI3, confirió una tolerancia a frío en plantas de Arabidopsis
transgénicas (Tamminen et al, 2001). La inducción de los transcritos de ABI3 y
HVA22 exclusivamente en Michoacán 21 bajo estrés, sugiere la existencia de una
respuesta de señalización de ABA específica a estrés en esta variedad que pudiera
ser importante para la tolerancia a sequía.
La reducción en la fijación de carbono y la inhibición de la actividad fotosintética por
sequía también altera el equilibrio metabólico de los carbohidratos. Para las plantas,
la regulación basada en los carbohidratos representa un mecanismo valioso para
ajustarse al cambio ambiental (Koch, 1996). Un incremento en los transcritos de ß-
amilasa en las tres variedades bajo sequía sugiere que cuando los niveles de
fotosíntesis caen, los carbohidratos almacenados como almidón pudieran ser
movilizados de los cloroplastos. Esto podría conducir al incremento en los niveles
de glucosa observados para Michoacán 21 y 85-2.
La glucosa además de tener un rol estructural, funciona como una molécula señal
tanto en la ruta dependiente como independiente de hexocinasa (Xiao et al, 2000).
Aunque los transcritos de hexocinasa fueron inducidos bajo sequía, los genes que
codifican para otras enzimas necesarias para producir mio-inositol a partir de
glucosa fueron reprimidos sugiriendo que el incremento en el contenido de mio-
inositol observado a los 10 días de estrés en Cajete ciollo y 85-2 y a los 10 y 17 días
de estrés en Michoacán 21 pudiera ser el resultado de cambios regulados a nivel
traduccional o post-traduccional. El hecho de que Michoacán 21 tenga un alto
contenido de mio-inositol, pudiera indicar otra estrategia de tolerancia a sequía, ya
que mio-inositol está implicado en muchos aspectos del metabolismo incluyendo:

130
osmoregulación, fisiología de auxinas, estructura de pared celular, metabolismo de
membrana y señalización entre otros (Hegeman et al, 2001). Los incrementos en
los niveles de transcrito en respuesta a sequía tanto a nivel transcripcional y/o post-
transcripcional, requiere la participación de componentes de rutas de señalización
que activan la transcripción y/o la estabilización del ARN mensajero. En este
estudio, los componentes de las rutas de transducción de señales relacionados a la
señalización por Ca+2 y proteínas G (CDPK, Ca+2-binding EF, Rho e inhibidor de
disociación de Rab GDP) fueron los únicos identificados como regulados
diferencialmente en los 3 genotipos bajo estrés y el proceso de riego de
recuperación. La señalización por Ca+2 en respuesta a estrés osmótico y iónico ha
sido bien documentado (Bartels and Sunkar, 2005). Las redes de transducción de
señales usualmente incluyen factores de transcripción y elementos que actúan en
cis (Yamaguchi-Shinozaki y Shinozaki, 2006), los cuales activan la cascada de
genes que codifican a proteínas y enzimas que podrían actuar juntas para estimular
la tolerancia a múltiples estreses (Bhatnagar-Mathur et al, 2007). Estudios previos
han revelado que docenas de factores de transcripción están involucrados en
respuestas a la sequía. La mayoría de estos factores de transcripción se clasifican
en varias familias tales como AP2/ERF. bZIP, NAC, MYB, dedos de zinc CysHis2 y
WRKY (Umezawa et al, 2006) y muchas plantas muestran diferencias en la
expresión de estas familias génicas bajo sequía. Los 3 genotipos de maíz
analizados en este estudio muestran una inducción de genes que codifican a FTs
de las familias C2H2, G2-like, HB, MADS, MYB y la proteína Hox7 homeodominio
zipper de leucina, los cuales pudieran ser considerados factores de transcripción
inducidos en la respuesta general de maíz en sequía. Algunos de los genes que
codifican a FTs comunes a las tres variedades como MADS y FTs homeodominio
zipper de leucina, fueron también inducidos en trigo (Mohammadi et al, 2007), MYB
en Arabidopsis (Seki et al, 2002) y C2H2 bajo estrés con PEG en maíz (Jia et al,
2006). Esto sugiere que algunas de las respuestas en la expresión génica
diferencial a sequía son probablemente moduladas por los mismos tipos de FTs e
involucran rutas de señalización similares.

131
VII.3. Respuestas diferenciales entre las 3 variedades bajo estrés y riego de recuperación
Con la finalidad de elucidar las diferencias entre las variedades tolerantes y la
susceptible, se identificaron familias de genes que están sobre-representadas o
tienen diferencias en su nivel de expresión en los genotipos tolerantes con respecto
a la susceptible. De hecho uno podría esperar más cambios en los cultivares
tolerantes, los cuales deberían poseer alelos de los genes que contribuyen al
incremento de la tolerancia (Degenkolbe et al, 2009). Aunque es posible que
algunos de los genes responsables para la tolerancia a sequía pudieran no ser
inducibles o reprimibles durante el estrés, la identificación de genes expresados
diferencialmente en los genotipos tolerantes a sequía, podría brindar información
acerca de los procesos metabólicos y celulares que finalmente son responsables de
la tolerancia al estrés. A este respecto, los factores de transcripción juegan un papel
importante en la regulación de la expresión diferencial génica y el análisis de los
cambios en la expresión de las familias de genes de los factores de transcripción en
si puede brindar aportaciones importantes en las respuestas a sequía. 18 genes
que codifican a factores de transcripción se encontraron expresados
diferencialmente bajo sequia en los genotipos tolerantes pero no en 85-2 como los
miembros de las familias AP2, bHLH, C2C2, C2H2, C3H, dedos de zinc, el factor de
unión CCAAT (HAP2) y WRKY (Tabla suplementaria 2 y 3). De éstos, un gen que
codifica para un AP2/EREBP, un C2C2 (factor de unión 2b al promotor de la
proteína H), un dominio C2 que contiene la familia dedo de zinc C2H2 y un dedo de
zinc tipo CCCH y la proteína de unión CCAAT NF-YA fueron inducidas bajo estrés
solo en los genotipos tolerantes. Las familias C2C2 y CCAAT han sido propuestas
como importantes en la tolerancia a sequía (Spollen et al, 2008). También fue
reportado que la expresión de NFYA5, un miembro de NF-YA (HAP2) fue inducido
fuertemente por sequía o tratamientos de ABA en Arabidopsis y se sugirió que
juega un papel importante en controlar la apertura estomatal y la resistencia a
sequía (Li et al, 2008). Por otro lado, la familia de los FT AP2/EREPB incluyen a las
proteínas DREB o CBF, las cuales se unen a los elementos de respuesta a

132
deshidratación DRE o los repetidos de C (Bartels y Sunkar, 2005), los cuales
también mostraron estar involucrados en mejorar la tolerancia a sequía, salinidad y
frio en Arabidopsis (Yamaguchi-Shinozaki y Shinozaki, 2006) y arroz (Shinozaki,
Yamaguchi-Shinozaki, 2007). Adicionalmente, 37 y 7 genes para FT fueron
específicamente inducidos bajo estrés para Michoacán 21 y Cajete criollo
respectivamente. En Michoacán 21 algunos de estos genes mostraron un cambio
de 3 a 8 veces mayor comparado con 85-2 y CC (Tabla Suplementaria 2 y 3).
Los FT identificados en este estudio podrían ser importantes para la regulación de
los genes de respuesta a sequia y podrían estar involucrados en los mecanismos de
tolerancia. El mayor número de genes de FT con expresión diferencial observados
para Michoacán 21, podrían explicar las rápidas respuestas y de amplio número de
genes observados en esta variedad y por lo tanto su mecanismo de tolerancia.
En CC una diferencia marcada fue la inducción del gen que codifica a un FT NAC.
Las proteínas NAC son conocidas como activadores transcripcionales en
cooperación con las proteínas ZFHD (homeodominio dedo de zinc) y la sobre-
expresión de estos genes se incrementó significativamente en la tolerancia al estrés
(Yamaguchi-Shinozaki y Shinozaki, 2006). Diferentes alelos de los mismos genes
de FT podrían producir diferentes respuestas entre las variedades tolerantes y entre
las variedades tolerantes y susceptibles, así como una coordinación cuidadosa de la
expresión génica conduciendo a la tolerancia o susceptibilidad en un mayor o menor
grado. Tomado en conjunto, estos resultados sugieren que algunos mecanismos de
tolerancia son similares mientras otros son específicos a cada variedad.
La tolerancia a la sequía requiere cambios en el transporte de agua para permitir a
las células y tejidos que se adapten a la situación de estrés. Las aquaporinas
facilitan la ósmosis, formando poros de agua específicos como una alternativa a la
difusión de agua a través de la bicapa lipídica incrementando la permeabilidad de la
membrana (Bartels y Sunkar, 2005). Aunque muchas aquaporinas fueron
expresadas diferencialmente en los tres genotipos, CC y particularmente M21
tuvieron más genes inducidos que codifican a NIP, TIP y PIP comparado con 85-2.

133
Estos resultados sugieren que en genotipos tolerantes la activación de un mayor
número de genes de aquaporinas podrían facilitar el flujo de agua para mantener la
homeostasis celular.
Aunque algunos reportes indican que los genes de aquaporinas inducidos por
estrés podrían desencadenar una mayor permeabilidad y facilitar el flujo de agua
(Bartels and Sunkar, 2005), otros reportes sugieren que la represión de los genes
de aquaporinas resulta en una permeabilidad de la membrana disminuida y por lo
tanto, permite la conservación del agua celular. A partir de este último punto, se
esperaría que los genotipos tolerantes activarían un número mayor de genes de
aquaporinas (Bartels y Sunkar, 2005). La coordinación en la regulación de la
inducción y represión de los genes de aquaporinas para facilitar el flujo de agua,
mantener la homeostasis celular y conservar el agua podría ser una estrategia
importante de las plantas tolerantes a la sequía.
Un efecto secundario de deshidratación es el incremento en las especies reactivas
de oxígeno (ROS) (Bartels y Sunkar, 2005) y consecuentemente la actividad
estimulada de enzimas antioxidantes (Chaves et al, 2003) con la finalidad de
proteger las células de daño oxidativo bajo sequía (Bhatnagar-Mathur et al, 2007).
En este caso, niveles de transcritos incrementados para tioredoxina fueron
encontrados en ambas variedades tolerantes y transcritos para peroxidasas fueron
inducidos principalmente en Michoacán 21, sin cambio significativo en el genotipo
susceptible. Estos resultados sugieren que los genotipos tolerantes activan la
expresión de genes que pudieran permitirles afrontar mejor loslaslos ROS.
Aunque fue observado que la inducción de genes que codifican a HSPs es una
respuesta común en maíz, un número mayor de genes que codifican HSPs fueron
inducidos bajo sequía en las variedades tolerantes (24 y 31 para Cajete criollo y
Michoacán 21 respectivamente) comparado con la susceptible. Particularmente, un
número mayor de miembros de la familia de HSP17 fue inducido en los genotipos
tolerantes. Una correlación positiva entre los niveles de muchos HSPs y la
tolerancia al estrés ha sido reportado en otros estudios (Sun et al, 2002; Vinocur y

134
Altman, 2005; Bartels y Sunkar, 2005). Dado que los HSP pequeños tienen una vida
media larga, se ha propuesto que podrían jugar un papel importante durante el
proceso de recuperación (Sun et al, 2002), por lo que, las rutas de señalización que
conducen a una expresión incrementada de HSP pequeños podrían ser activadas
preferencialmente en los genotipos de maíz tolerantes en comparación a la
susceptible.
Aunque los transcritos para genes relacionados a fotosíntesis fueron reprimidos en
las 3 variedades, un mayor número de transcritos fueron reprimidos en las 2
variedades tolerantes y especialmente en Michoacán 21. Una situación similar fue
también observada en cultivares de arroz tolerantes a la sequía bajo estrés por
sequía, donde se reportó que en particular un mayor número de miembros de
familias de genes relacionados al fotosistema I y fotosistema II fueron reprimidos en
las variedades tolerantes que en la susceptible (Degenkolbe et al, 2009). En nuestro
estudio, se encontró también que los genes relacionados al fotosistema I y
fotosistema II fueron reprimidos principalmente en las variedades tolerantes. La
reducción de la fotosíntesis bajo sequía, es atribuida a la disminución en los niveles
de expresión de los genes del Ciclo de Calvin, donde las reducciones en la
conductancia estomatal y las actividades de Rubisco, conducen a la reducción en la
fijación de carbono en el primer paso del Ciclo de Calvin como se observó en trigo
(Xu et al, 2008). El presente estudio también encontró que los genes relacionados al
Ciclo de Calvin, tales como transcritos para Rubisco, fosfoglicerato cinasa, GADPH,
TPI, y FBPasa fueron reprimidos bajo estrés y en un mayor grado en Michoacán 21,
sugiriendo que el maíz, trigo y arroz podrían mostrar respuestas similares bajo
sequía y que las diferencias podrían ser debidas al número de genes específicos
expresados diferencialmente en una manera especie-genotipo dependiente. Los
transcritos Cab y Rbcs fueron reprimidos bajo sequía principalmente en las
variedades tolerantes. Diez transcritos reprimidos a los 17 días de estrés fueron
inducidos en el riego de recuperación en al menos una de las tres variedades y la
mayoría de ellos lo compartieron las variedades tolerantes. Estos resultados
sugieren que los genotipos tolerantes de maíz analizados en este estudio reducen

135
más ampliamente el transporte de electrones en los sistemas PSI y PSII,
probablemente para reducir el efecto de la foto-oxidación y la síntesis de las
enzimas involucradas en la fijación del carbono para evitar gastar energía y
recursos bajo condiciones de baja disponibilidad de CO2.
VII.4. Diferencias en el metabolismo de carbohidratos en las 3 variedades bajo sequía y el riego de recuperación
La reducción en la fijación del carbono y la inhibición de la actividad fotosintética por
sequía también altera el equilibrio metabólico de carbohidratos (Xue et al, 2008).
Para las plantas, la regulación basada en los carbohidratos representa un
mecanismo especialmente valioso para ajustar a los cambios ambientales (Koch,
1996). La glucosa además de su rol estructural, funciona como una molécula señal
tanto en la ruta dependiente como la ruta independiente de hexocinasa (Xiao et al,
2000). El mio-inositol está también implicado en muchos aspectos del metabolismo
incluyendo: osmoregulación, fisiología de auxinas, metabolismo de la membrana y
pared celular y señalización entre otros (Hegeman et al, 2001). Un incremento en
los transcritos de ß-amilasa en las 3 variedades bajo sequía sugiere que cuando los
niveles de fotosíntesis caen, los carbohidratos almacenados como almidón podrían
ser movilizados de los cloroplastos. Esto pudiera conducir al incremento en los
niveles de glucosa observados para Michoacán 21 y 85-2. Aunque la transcripción
de ß-amilasa también se incrementa en Cajete criollo, los niveles de glucosa caen
sugiriendo que el almidón podría ser procesado diferentemente en esta variedad,
por ejemplo a través de la maltosa más que la glucosa.
Los patrones de expresión de genes asociados con señalización de glucosa
dependiente de hexocinasa fueron también consistentes. Aunque los transcritos de
hexocinasa están inducidos bajo sequía, los genes que codifican a otras enzimas
que necesitan producir mio-inositol a partir de glucosa son reprimidas, sugiriendo
que el incremento en el contenido de mio-inositol observado a los 10 días en Cajete
criollo y 85-2 y a los 10 días y 17 días en Michoacán 21 pudiera ser el resultado de
los cambios probablemente regulados a nivel traduccional y post-traduccional. La

136
expresión de transcritos para los genes que codifican a enzimas directamente
involucradas en el metabolismo de la sacarosa correlaciona con una disminución en
la actividad fotosintética bajo sequía: la mayoría están reprimidos o
constitutivamente expresados en las 3 variedades bajo estrés. Sin embargo, bajo
condiciones de riego, 85-2 acumula una mayor concentración de sacarosa en
comparación de las variedades tolerantes sugiriendo una diferencia fundamental en
el metabolismo de sacarosa entre los genotipos.
VII.5. El ajuste osmótico por la acumulación de prolina en las tres variedades de maíz bajo estrés y riego de recuperación
La prolina es probablemente el osmolito mas ampliamente distribuido en plantas
(Bartels y Sunkar, 2005) y está implicado en las respuestas a varios estreses
ambientales (Mahaja y Tuteja, 2005). Además del ajuste osmótico, otros roles tales
como la protección de la integridad de la membrana plasmática, como fuente de
energía o poder reductor, fuente de carbono o nitrógeno o un acarreador de radical
hidroxilo (Bartels y Sunkar, 2005), así como la preservación de la estructura y
actividad enzimática han sido propuestos para la prolina en tejidos vegetales bajo
estrés osmótico. En este estudio solo la inducción del transcrito P5CS en Cajete
criollo y un transcrito involucrado en la ruta biosintética alternativa (via ornitin
aminotransferasa) en Michoacán 21 fue detectada. Esto pudiera indicar que las dos
rutas biosintéticas están activas en maíz y requiere una mayor investigación. No se
observó una fuerte correlación entre la acumulación de prolina y la expresión de
genes asociados con el metabolismo de prolina y esto está acorde con un reporte
donde se encontró que la acumulación de prolina no es proporcional con la
inducción de genes relacionados al metabolismo de prolina en hojas de papa bajo
sequía (Schafleitner et al, 2007). Los autores sugirieron la posibilidad de que la
regulación de los niveles de prolina sea a nivel post-transcripcional o post-
traduccional y una situación similar podría ocurrir en maíz. Sin embargo, las dos
variedades tolerantes mostraron un incremento significativo en el contenido de
prolina bajo sequía pero la variedad 85-2 no. La incapacidad para acumular prolina

137
a niveles altos podría por lo tanto, ser un factor importante en distinguir el
germoplasma tolerante y susceptible.
En el riego de recuperación una fuerte correlación entre el contenido de prolina y la
inducción de transcritos de los genes involucrados en la degradación de prolina:
pirrolina-5-carboxilato deshidrogenasa (P5CDH) y la represión de la síntesis de
prolina: pirrolina-5-carboxilato sintetasa (P5CS) fue observada en las 3 variedades
de maíz y sugiere la degradación de prolina durante este proceso. Además, se ha
sugerido que la degradación de prolina después del estrés, podría ofrecer carbón,
nitrógeno y energía para la recuperación (Hare et al, 1998) y un alto contenido de
prolina fue asociado con una capacidad de recuperación incrementada (Schafleitner
et al, 2007). Esta observación apoya la hipótesis de que Michoacán 21 tiene una
mejor capacidad de recuperación.
VII.6. Diferencias en las respuestas moleculares bajo el riego de recuperación en las tres variedades de maíz
Las respuestas de las plantas son importantes no solo bajo sequía sino también en
el riego de recuperación. En el riego de recuperación 2689 y 2887 transcritos
inducidos y reprimidos fueron identificados respectivamente. De ellos, 1466 genes
mostraron ser regulados inversamente entre el estrés y el riego de recuperación
(inducidos durante el estrés y reprimidos durante el riego de recuperación o
viceversa). La Figura 22 muestra un esquema general de las respuestas
observadas en las tres variedades durante el proceso de recuperación. La
observación de que el número más grande de genes expresados diferencialmente
fue encontrado en el riego de recuperación, sugiere una re-activación general del
metabolismo después de un estrés severo. El número de genes reprimidos que
codifican a las HSPs fue mayor en las variedades tolerantes que en la susceptible,
particularmente en Michoacán 21. Asimismo, las dos variedades tolerantes
compartieron genes expresados diferencialmente relacionados a señalización en la
recuperación incluyendo receptor cinasas, proteínas G, señalización por Ca+2 y
fosfoinositidos. El hecho de que Cajete criollo y Michoacán 21 tienen un número

138
similar de genes de señalización inducidos en el riego de recuperación (48 y 47
respectivamente) presenta la posibilidad de que ellos responden más
eficientemente durante el proceso de recuperación en comparación al cultivar
susceptible. Genes relacionados al metabolismo del carbono fueron inducidos en
las variedades tolerantes, sin embargo, en 85-2 la mayoría de estos genes se
encontraron constitutivos y esto podría ser una de las diferencias clave entre las
respuestas tolerantes y susceptibles. Los genes que codifican a las enzimas del
Ciclo de Calvin, fotosistema I y II y enzimas relacionadas a fotosíntesis fueron
también inducidas principalmente en los genotipos tolerantes en el riego de
recuperación. La inducción de los genes involucrados en la fotosíntesis y el ciclo de
Calvin está en relación con el incremento en la tasa de fotosíntesis y conductancia
estomatal observada después del riego de recuperación en CC y M21. En este
contexto, se sabe que la inducción de los genes de peroxiredoxina protegen el DNA,
las membranas y ciertas enzimas contra el daño removiendo el H2O2 y los radicales
hidroxilo (Bartels y Sunkar, 2005), durante el riego de recuperación en las dos
variedades tolerantes, lo que podría representar un mecanismo protector contra la
producción de ROS durante una rápida re-activación de la actividad fotosintética. La
represión durante el riego de recuperación de genes que codifican a aquaporinas
apoya su importancia en la tolerancia al estrés. Los patrones de expresión de genes
que codifican a FT fueron distintos para las tres variedades, sugiriendo las
diferentes respuestas durante el riego de recuperación. Un mayor número de genes
que codifican a factores de transcripción incluyendo bHLH, MADS y MYB fueron
encontrados inducidos ó reprimidos en las variedades tolerantes en comparación
con 85-2. Dos transcritos para la familia de factores de transcripción CCAAT fueron
inducidos en las variedades tolerantes.
Como se observó el proceso de recuperación involucra respuestas muy complejas.
Aunque Michoacán 21 y 85-2 muestren respuestas muy diferentes bajo sequía y
recuperación ambos poseen un fondo genético compartido que proviene de la raza
de maíz “Celaya”. Aunque Michoacán 21 mostró los genes más diferencialmente
expresados en casi todas las categorías funcionales (excepto para señalización),

139

140
Cajete criollo también mostró cambios significativos en expresión aunque en un
menor grado al de Michoacán 21. Esta observación podría indicar diferentes
mecanismos para la recuperación y esta posibilidad debería ser estudiada en mayor
detalle.
VII.7. Análisis de los datos de sequía usando el programa BioMaps
Además de utilizar el software MapMan, los datos de microarreglos fueron
analizados usando una herramienta llamada BioMaps en el sitio Virtual-Plant
(www.virtualplant.org). Esta herramienta ayuda a relacionar los datos de expresión
diferencial con categorías funcionales basado en la clasificación funcional por la
anotación del Centro de Información Munich para secuencias proteicas (MIPS),
tomando en cuenta la mejor correlación con la base de datos de proteína TAIR y fue
utilizada para identificar las categorías funcionales comunes relacionadas a la
sequía entre las 3 variedades y/o las variedades tolerantes. BioMaps permite un
análisis estadístico no paramétrico-riguroso de sobre-representación usando una
distribución hiper geométrica y genera un valor P. Mientras más bajo sea el valor P,
mayor es la significancia de la sobre representación (Gutiérrez et al, 2006). Con
esta herramienta 121 genes inducidos y 67 genes reprimidos comunes a las 3
variedades a los 17 días de estrés, en categorías funcionales relacionados al
censado celular, la respuesta al estimulo externo, quimopercepción, proteínas de
pared celular, respuesta a choque térmico, regulación de hormona vegetal y
desarrollo vegetal fueron identificados para la respuesta de inducción mientras en la
respuesta de represión las principales categorías funcionales fueron energía,
fotosíntesis, transporte celular y metabolismo de tetraterpenos, lo cual relaciona
bien con los datos obtenidos usando el sistema MapMan (Tabla suplementaria 4 y
Tabla suplementaria 5). La respuesta común de inducción a los 17 días de estrés en
las variedades tolerantes mostró categorías adicionales a las arriba mencionadas
que están relacionadas al ciclo del glioxilato y la regulación del crecimiento celular
direccional (Tabla suplementaria 6). Sin embargo, ambas variedades tolerantes
comparten más genes inducidos, indicando una respuesta común para las

141
variedades tolerantes. Una mayor diferencia fue observada en la respuesta a las
variedades tolerantes en términos de genes reprimidos a los 17 días de estrés,
incluyendo categorías funcionales adicionales tales como los relacionadas a
compuestos de carbono, transporte de carbohidratos, metabolismo de azúcares y
nucleótidos, respuesta a estrés y metabolismo de porfiirinas (Tabla suplementaria
7). En el riego de recuperación, se encontraron 287 genes inducidos involucrados
en fotosíntesis, energía y transporte celular, la mayoría de los cuales, estaban
reprimidos bajo estrés en las 3 variedades (Tabla suplementaria 8, Tabla
suplementaria 9). Los genes reprimidos comunes (342) en las tres variedades de
maíz fueron identificadas en categorías denominadas como estrés osmótico y
salino, respuesta a estrés oxidativo, censado celular, unión a calcio, regulación de
hormona vegetal, transducción de señales mediada por hormonas y respuesta a
choque térmico entre otros (Tabla suplementaria 10). Durante la recuperación, una
re-activación del metabolismo vegetal debido a un mayor número de genes
inducidos (694) que conformaron diferentes categorías funcionales como
metabolismo secundario, compuestos transportados, metabolismo de vitaminas,
grupos de co-factores y prostéticos, glicolisis, aminoácidos, metabolismo de
carbohidratos y glutamato, censado celular a estimulo externo así como, genes
relacionados al metabolismo de cloroplastos y biogénesis fue también observado
(Tabla suplementaria 11). Muchos genes reprimidos comunes (632) a las
variedades tolerantes fueron relacionados con ciclo celular, transporte celular,
respuesta a choque por frio y aquellos involucrados en la regulación del
metabolismo y función proteica (Tabla suplementaria 12). Muchos genes reprimidos
en el riego de recuperación fueron inducidos durante la sequía, una situación
también observada previamente en Arabidopsis y cebada (Talamé et al, 2007).

142
VII.8. Asociación de genes expresados diferencialmente a QTL’s de maíz
En maíz, la floración es el estadio más crucial en términos de efectos negativos de
la sequía para el rendimiento. Por lo que, la falla reproductiva es el factor más
importante relacionado a la sequía que contribuye a pérdidas de rendimiento en
maíz. La importancia del tiempo de floración en términos de reducción en el
rendimiento bajo condiciones de sequía está correlacionada con el rendimiento de
grano (Tuberosa et al, 2007). En este estudio, de los 62 genes expresados
diferencialmente asociados a QTL, la mayoría de ellos están asociados con QTLs
involucrados en la productividad de maíz tales como ASI, rendimiento de grano,
peso de grano, días a la aparición de estilos y “stay green” (Tabla Suplementaria 13
y Fig. 23) Además, la mayoría de los genes asociados a QTLs están inducidos en
las variedades tolerantes durante la sequía y algunos en el riego de recuperación.
Los genes asociados con estos QTLs pudieran ser candidatos importantes para
estudios funcionales futuros tales como un gen que codifica a una familia proteica
de respuesta a deshidratación (similar a ERD3) inducido solo en Cajete criollo a los
17 días de estrés, una invertasa vacuolar que fue inducida a un nivel mayor en
Michoacán 21, así como a una enzima inhibitoria/pectinesterasa y dos genes para
G3PDH inducidos solo en Michoacán 21 a los 17 días de estrés. Los genes sin
anotación funcional fueron también inducidos principalmente en las variedades
tolerantes y podrían ser candidatos importantes para el desarrollo de la tolerancia a
sequía. Aunque la asociación putativa de los genes expresados diferencialmente a
partir de genotipos tolerantes con QTL’s es relevante a la tolerancia a la sequía, una
asociación formal deberá ser determinada.

Figura 23. Genes expresados diferencialmente bajo sequía y riego de recuperación asociados a QTL
143

144
VIII. CONCLUSIONES
1) Diferencias en las tasas de fotosíntesis, conductancia estomatal, la concentración
de azúcares solubles y prolina y los patrones de expresión fueron identificados en
las 3 variedades de maíz. En muchos casos, en comparación con las variedades
tolerantes.
2) La variedad 85-2 falló en responder, o respondió más débilmente, durante el
tratamiento de sequía. Diferencias importantes fueron también notadas entre las
variedades tolerantes que probablemente recaen en diferentes mecanismos para
alcanzar la tolerancia: CC podría tener una ventaja bajo condiciones prolongadas de
sequía debido a la gradual reducción de la fotosíntesis y conductancia estomatal,
mientras que en M21 con la capacidad de latencia, la rápida reducción de
fotosíntesis y la eficiente respuesta de recuperación podría comportarse mejor bajo
periodos cortos de una sequía severa.
3) Se encontraron diferencias en los mecanismos de respuesta apoyadas por los
cambios detallados en los patrones de expresión génica bajo condiciones de
sequía.
4) La modulación de un gran número de genes expresados diferencialmente de
diferentes familias de genes que codifican a FT podría ser una característica
importante en las variedades tolerantes, muchas de las cuales pertenecen a familias
previamente implicadas en las respuestas a estrés tales como los miembros de las
familias AP2/EREBP, bHLH, HB, CCAAT y MYB.
5-) Los genes que codifican a enzimas responsables de la síntesis, degradación y
transducción de señales de las hormonas, aquellos que codifican a proteínas de
transporte como las aquaporinas, las proteínas de choque térmico y aquellos que
codifican a las enzimas de detoxificación fueron inducidos en un mayor grado en las
variedades tolerantes, lo que sugiere que las respuestas moleculares involucradas

145
en estos grupos de genes responden más rápido en las variedades tolerantes y la
eficiencia podría deberse a esta rapidez para afrontar el estrés .
6-) En el caso del riego de recuperación después del tratamiento de sequía, la
característica más importante fue la velocidad y los cambios en la expresión génica,
los cuales difirieron entre los 3 genotipos.
7-) Este trabajo enfatiza las diferencias más importantes entre los genotipos
tolerantes y susceptibles e identifica reguladores potenciales de la sequía y
procesos de recuperación en maíz.

146
IX. PERSPECTIVAS
La tarea ahora es caracterizar los patrones de expresión y las respuestas únicas de
cada variedad y los genes o alelos específicos involucrados con la finalidad de
comparar con otro germoplasma comercial de maíz y sugerir posibles estrategias de
mejoramiento tradicional o por ingeniería genética para mejorar la tolerancia a
sequía. La modulación de la expresión de genes de FT ha sido probado
exitosamente para mejorar la tolerancia a sequía (Wang et al, 2003). En este
reporte muchas familias de FT adicionales fueron identificadas y los efectos
regulatorios de estos genes en particular deberían de ser estudiados en más
detalle. Asimismo, un estudio más amplio del sistema radicular de cada variedad
aumentaría el conocimiento de los mecanismos de tolerancia a sequía.

147
X. BIBLIOGRAFIA
Altamirano-Hernández, J., M. López, et al. (2007). “Influence of Soluble Sugars on Seed Quality in Nodulated Common Bean (Phaseolus vulgaris L.): The Case of Trehalose.” Crop Science 47: 1193-1205.
Aquino, P., F. Carrion, et al. (2001). Selected Maize Statistics. Part 4. CIMMYT 1999-2000 World Maize Facts and Trends. Meeting World Maize Needs Technological Opportunities and Priorities for the Public Sector. P. L. Pingali. Mexico, D.F., CIMMYT: 45-57.
Bartels, D. a. S., R. (2005). “Drought and Salt Tolerance in Plants.” Critical Reviews in Plant Sciences 24: 23-58.
Benjamini, Y. and Y. Hochberg (1995). “Controlling the false discovery rate: a practical and powerful approach to multiple testing.” Journal of the Royal Statistical Society B 57: 289-300.
Bhatnagar-Mathur, P., V. Vadez, et al. (2008). “Transgenic approaches for abiotic stress tolerance in plants:retrospect and prospects.” Plant Cell Reports 27(3): 411-424.
Bray, E. A. (1997). “Plant responses to water deficit.” Trends in Plant Science 2(2): 48-54.
Bruce, W. B., G. O. Edmeades, et al. (2002). “Molecular and physiological approaches to maize improvement for drougth tolerance”. Journal of Experimental Botany 53(366): 13-25.
Brugiére, N., Jiao, S., Hantke, S., Zinselmeier, C., Roessler, J.A., Niu, X., jones, R.J., Habben, J. (2003). “Cytokinin oxidase genes expression in maize is localized to the vasculature, and is induced by cytokinins, abscisic acid, and abiotic stress.” Plant Physiology 132: 1228-1240.
Calderon-Vazquez, C., E. Ibarra-Laclette, et al. (2008). “Transcript profiling of Zea mays roots reveals gene responses to phosphate deficiency at the plant- and species-specific levels.” Journal of Experimental Botany 59: 2479-2497.
Chaves, M. M., J. P. Maroco, et al. (2003). “Understanding plant responses to drought-from genes to the whole plant.” Functional Plant Biology 30: 239-264.
Degenkolbe, T., Do, P.T., Zuther, E., Repsilber, D., Walther, D., Hincha, D.K., Köhl, K.I. (2009). “Expression profiling of rice cultivars differing in their tolerance to long-term drought stress.” Plant Molecular Biology 69(1-2): 133-153.
Fischer, K. S., E. C. Johnson, et al. (1983). Breeding and Selection for Drought Resistance in Tropical Maize. Mexico City (Mexico), CIMMYT: 20.

148
Foyer, C., M. Valadier, et al. (1998). “Drought-Induced Effects on Nitrate Reductase Activity and mRNA and on the Coordination of Nitrogen and Carbon Metabolism in Maize Leaves.” Plant Physiology 117: 283-292.
Garcion, C., F. Applimath, et al. (2006). “FiRe and microarrays: a fast answer to burning questions.” Trends in Plant Science 11: 320-322.
Gutiérrez, R. A., Gifford, M.L., Poultney, C., Wang. R., Shasha, D.E., Coruzzi, G.M., Crawford, N.M. (2007). “Insights into the genomic nitrate response using genetics and the Sungear Software System.” Journal of Experimental Botany 58: 2359-2367.
Hare, P. D., Cress, W.A., Van Staden, J. (1998). “Dissecting the roles of osmolyte accumulation during stress.” Plant, Cell and Environment 21: 535-553.
Hayano-Kanashiro, A. C. (2004). “Obtencio y caracterizacion de bibliotecas susractivas de cDNA de maiz (Zea mays L.) bajo condiciones de sequía. Tesis de Maestria en Ciencias. Ingenieria Genetica. CINVESTAV-IPN. Irapuato. Guanajuato.”
Hegeman, C., L. Good, et al. (2001). “Expression of D-myo-Inositol-3-Phosphate Synthase in Soybean. Implications for Phytic Acid Biosynthesis.” Plant Physiology 125: 1941-1948.
Ingram, J. and D. Bartels (1996). “The molecular basis of dehydration tolerance in plants ” Annual Review of Plant Physiology and Plant Molecular Biology 47: 377-403.
Irizarry, R., Hobbs, B., Collin, F., Beazer-Barclay, Y.D., Antonellis, K.J., Scherf, U. y Spedd, T.P. (2003). “Exploration, normalization, and summaries of high density oligonucleotide array probe level data.” Biostatistics 4: 249-264.
Jia, J., J. Fu, et al. (2006). “Annotation and expression profile analysis of 2073 full-length cDNAs from stress-induced maize (Zea mays L.) seedlings.” The Plant Journal 48: 710-727.
Kaldenhoff, R., Ribas-Carbo, M., Flexas-Sans, J., Lovisolo, C., Heckwolf, M. and Uehlein, N. (2008). “Aquaporins and plant water balance.” Plant, Cell and Environment 31: 658-666.
Kawaguchi, R., T. Girke, et al. (2004). “Differential mRNA translation contributes to gene regulation under non-stress and dehydration stress conditions in Arabidopsis thaliana.” The Plant Journal 38: 823-839.
Kerr, M.K. y Churchill, G.A. (2001). Experimental design for gene expression microarrays. Biostatistics 2,2, pp. 183-201.

149
Koch, K. E. (1996). “Carbohydrate-modulated gene expression in plants.” Annual Review of Plant Physiology and Plant Molecular Biology 47: 509-540.
Lee, K. H., Piao, H.L., Kim, H.Y., Choi, S.M., Jiang, F., Hartung, W., Hwang, I., Kwak, J.M., Lee, I.J. and Hwang, I. (2006). “Activation of Glucosidase via stress-induced polymerization rapidly increases active pools of abscisic acid.” Cell 126: 1109-1120.
Mahajan, S. and N. Tuteja (2005). “Cold, salinity and drought stresses: An overview.” Archives of Biochemistry and Biophysics 444: 139-158.
Mohammadi, M., Kav, N. N. V. y Deyholos, M.K. (2007). “Transcriptional profiling of hexaploid wheat (Triticum aestivum L.) roots identifies novel, dehydration-responsive genes.” Plant, Cell and Environment 30: 630-645.
Mullet, J. (2009). “Traits and Genes for Plant Drought Tolerance.” Biotechnology in Agriculture and Forestry. Molecular Genetic Approaches to Maize Improvement 63: 55-64.
Nelson, D., Repetti, P.P., Adams, T.R., Creelman, R.A., Wu, J., Warner, D.C., Anstrom, D.C., Bensen, R.J., Castiglioni, P.P., Donnarummo, M.G., Hinchey, B.S., Kumimoto, R.W., Maszle, D.R., Canales, R.D., Krolikowski, K.A., Dotson, S.B., Gutterson, N., Ratcliffe, O.J., Heard, J.E. (2007). “Plant nuclear factor Y (NF-Y) B subunits confer drought tolerance and lead to improved corn yields on water-limited acres.” Proceedings of the National Academy of Sciences 104(42): 16450-16455.
Oono, Y., M. Seki, et al. (2003). “Monitoring expression profiles of Arabidopsis gene expression during rehydration process after dehydration using ca. 7000 full-length cDNA microarray.” The Plant Journal 34: 868-887.
Ozturk, Z. N., V. Talame, et al. (2002). “Monitoring large-scale changes in transcript abundance in drought- and salt-stressed barley.” Plant Molecular Biology 48: 551-573.
Park, J. E., Park, J.Y., Kim, Y.S., Staswick, P.E., Jeon, J., Yun, J., Kim, S.Y., KLim, J., Lee, Y.H., Park, C.M. (2007). “GH3-mediated Auxin Homeostasis links growth regulation with stress adaptation response in Arabidopsis.” The Journal of Biological Chemistry 282(13): 10036-10046.
Parry, M. A. J., J. Flexas, et al. (2005). “Prospects for crop production under drought: research priorities and future directions.” Annals of Applied Biology 147: 211-226.
Pelleschi, S., J. Rocher, et al. (1997). “Effect of water restriction on carbohydrate metabolism and photosynthesis in mature maize leaves.” Plant, Cell and Environment 20: 493-503.

150
Perez, J. G. (1979). Comportamiento de los maíces de Cajete bajo diversos niveles de humedad. Texcoco, Estado de Mexico, Colegio de Postgraduados, Chapingo (Mexico). Maestro en Ciencias. 193 pp.
Phillips, R. D. and D. H. Jennings (1976). “Succulence, cations and organic acids in leaves of Kalanchoe daigremontiana grown in long and short days in soil and water culture.” New Phytologist 77: 599-611.
Pierik, R., Sasidharan, R., Voesenek, L.A.C.J. (2007). “Growth control by ethylene: adjusting phenotypes to the environment.” Journal of Plant Growth Regulation 26: 188-200.
Pingali, P. L. a. P., S. (2001). Meeting World Maize Needs:Technological Opportunities and Priorities for the Public Sector. CIMMYT 1999-2000 World Maize Facts and Trends. Meeting World Maize Needs Technological Opportunities and Priorities for the Public Sector.
Poroyko, V., W. G. Spollen, et al. (2007). “Comparing regional transcript profiles from maize primary roots under well-watered and low water potential conditions.” Journal of Experimental Botany 2: 279-289.
Rabbani, M. A., K. Maruyama, et al. (2003). “Monitoring Expression Profiles of Rice Genes under Cold, Drought, and High-Salinity Stresses and Abscisic Acid Application Using cDNA Microarray and RNA Gel-Blot Analyses.” Plant Physiology 133: 1755-1767.
Ramanjulu, S. B., D. (2002). “Drought-and desiccation-induced modulation of gene expression in plants.” Plant, Cell and Environment 25: 141-151.
Rathinasabapathi, B. (2000). “Metabolic engineering for stress tolerance: installing osmoprotectant synthesis.” Annals of Botany 86: 709-716.
Reddy, A., K. Chaitanya, et al. (2004). “Drought-induced responses of photosynthesis and antioxidant metabolism in higher plants.” Journal of Plant Physiology 161(11): 1189-1202.
Reynolds, M., F. Dreccer, et al. (2007). “Drought-adaptive traits derived from wheat wild relatives and landraces.” Journal of Experimental Botany 58(2): 177-186.
Ribaut, J. M. and M. Ragot (2007). “Marker-assisted selection to improve drought adaptation in maize: the backcross approach, perspectives, limitations,and alternatives.” Journal of Experimental Botany 58(2): 351-360.
Rivers, R.L., Dean, R.M., Chandy, G., Hall, J.E., Roberts, D.M. y Zeidel, M.L. (1997). Functional analysis of Nodulin 26, an aquaporin soybean root nodule symbiosomes. The Journal of Biological Chemistry. Vol. 272. Nro. 26. Issue June27: 16256-16261.

151
Schachtman, D. P. y Goodger., J.Q.D. (2008). “Chemical root to shoot signaling under drought.” Trends in Plant Science 13(6): 281-287.
Schafleitner, R., Gutierrez-Rosales, R.O., Gaudin, A., Alvarado-Aliaga, C.A., Nombeo-Martinez, G., Tincopa-Marca, L.R., Avila-Bolivar, L., Mendiburu-Delgado, F., Simon, R. and Bonierbale. (2007). “Capturing candidate drought tolerance traits in two native Andean potato clones by transcription profiling of field grown plants under water stress.” Plant Physiology and Biochemistry 45: 673-690.
Seki, M., Kamei, A., Yamaguchi-Shinozaki, K. & Shinozaki, K. (2003). “Molecular responses to drought, salinity and frost: common and different paths for plant protection.” Current Opinion in Biotechnology 14(2): 194-199.
Seki, M., M. Narusaka, et al. (2002). “Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray.” The Plant Journal 31(3): 279-292.
Seki, M., Umezawa, T., Urano, K., Shinozaki, K. (2007). “Regulatory metabolic networks in drought stress responses.” Current Opinion in Plant Biology 10: 296-302.
Shinozaki, K., Yamaguchi-Shinozaki, K. (1997). “Gene expression and signal transduction in water-stress response ” Plant Physiology 115(2): 327-334.
Simon, R.M. y Dobbin, K. (2003). Experimental design of DNA microarray experiments. Bio Techniques 34:S16-S21 (March 2003).
Spollen, W. G., Tao, W., Valliyodan, B., Chen, K., Hejlek, L.G., Kim, J.L., LeNoble, M.E., Zhu, J., Bohnert, H., Henderson, D., Schachtman, D.P., Davis, G.E., Springer, G.K., Sharp, R.E., Nguyen, H.T. (2008). “Spatial distribution of transcript changes in the maize primary root elongation zone at low water potential.” BMC Plant Biology 3: 8-32.
Sreenivasulu, N., Sopory, S.K. and Kavi Kishor, P.B. (2007). “Deciphering the regulatory mechanisms of abiotic stress tolerance in plants by genomic approaches.” Gene 388(1-2): 1-13.
Sun, W., M. Van Montagu, et al. (2002). “Small heat shock proteins and stress tolerance in plants ” Biochimica et Biophysica Acta (BBA) - Gene Structure and Expression 1577(1): 1-9.
Talamé, V., Z. N. Ozturk, et al. (2007). “Barley transcript profiles under dehydration shock and drought stress treatments: a comparative analysis.” Journal of Experimental Botany 58: 229-240.
Thimm, O., O. Blasing, et al. (2004). “MAPMAN:a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes.” The Plant Journal 37: 914-939.

152
Triphaty, J. N., J. Zhang, et al. (2000). “QTLs for cell-membrane stability mapped in rice (Oryza sativa L.) under drought stress.” Theoretical and Applied Genetics 100: 1197-1202.
Tuberosa, R., S. Salvi, et al. (2007). “Genome-wide Approaches to Investigate and Improve Maize Response to Drought.” Crop Science 47 (S3): S120-141.
Tuberosa, R., Slavi, S., Sanguineti, M.C., Landi, P., Maccaferri, M. and Conti, S. (2002). “Mapping QTLs regulating morpho-physiological traits and yield:case studies, shortcomings and perspectives in drought-stressed maize.” Annals of Botany 89: 941-963.
Umezawa, T., M. Fujita, et al. (2006). “Engineering drought tolerance in plants: discovering and tailoring genes to unlock the future.” Current Opinion in Biotechnology 17(2): 113-122.
Vinocur, B. and A. Altman (2005). “Recent advances in engineering plant tolerance to abiotic stress: achievements and limitations.” Current Opinion in Biotechnology 16: 123-132.
Wasilewska, A., Vlad, F., Sirichandra, C., Redko, Y., Jammes, F., Valon, C., Frei dit Frey, N., Leung, J. (2008). “An update on Abscisic acid signaling in plants and more...” Molecular Plant 1(2): 198-217.
Xiao, W., Sheen, J., Jang, J.C. (2000). “The role of hexokinase in plant sugar signal transduction and growth and development.” Plant Molecular Biology 44: 451-461.
Xiong, L., Schumaker, K.S. and Zhu, J.K. (2002). “Cell signaling during Cold, Drought, and Salt stress.” The Plant Cell 14(Suppl): S165-S183.
Xue, P., C. McIntyre, et al. (2008). “Use of expression analysis to dissect alterations in carbohydrate metabolism in wheat leaves during drought stress.” Plant Molecular Biology 67(3): 197-214.
Yamaguchi-Shinozaki, K., M. Kasuga, et al. (2002). “Biological mechanisms of drought stress response.” JIRCAS Working report: 1-8.
Yamaguchi-Shinozaki, K. and K. Shinozaki (2006). “Transcriptional Regulatory Networks in Cellular Responses and Tolerance to Dehydration and Cold Stresses.” Annual Review of Plant Biology 57: 781-803.
Yang, Y., S. Dudoit, et al. (2002). “Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation.” Nucleic Acids Research 30(4): e15.
Yu, L. and T. L. Setter (2003). “Comparative Transcriptional Profiling of Placenta and Endosperm in Developing Maize Kernels in Response to Water Deficit.” Plant Physiology 131: 568-582.

153
Zheng, J., J. Zhao, et al. (2004). “Isolation and analysis of water stress induced genes in maize seedlings by subtractive PCR and cDNA macroarray.” Plant Molecular Biology 55: 807-823.

154
TABLAS SUPLEMENTARIAS

155
Tabla Suplementaria 5. Análisis de BioMaps análisis de los genes inducidos comunes a las tres variedades de maíz a, los 17 días de estrés
Clasificación Frequencia observada
Frecuencia esperada
Valor P
Sensado cellular y respuesta a estímulo externo
25 genes, 20.7% 4.7% 3.81E-08
INTERACCION CON EL AMBIENTE 25 genes, 20.7% 5.3% 3.92E-07
Quimopercepción y respuesta 18 genes, 14.9% 2.8% 8.16E-07
Pared celular 9 genes,7.4% 0.9% 0.00019
Respuesta a estrés 16 genes, 13.2% 2.9% 4.48E-05
Percepción de temperatura y respuesta
9 genes, 7.4% 0.9% 0.00016
RESCATE CELULAR, DEFENSA Y VIRULENCIA
20 genes, 16.5% 4.9% 0.00019
Respuesta a choque térmico 6 genes, 5% 0.3% 0.00021
Regulación hormonal de la planta 12 genes, 9.9% 2% 0.00049
INTERACCION SISTEMICA CON EL AMBIENTE
13 genes, 10.7% 2.4% 0.0008
Desarrollo de fruto y maduración 7 genes, 5.8% 0.6% 0.00149
Componente de matriz extracelular 5 genes, 4.1% 0.3% 0.00669
Otras moleculas de senalizacion vegetal (acido jasmónico, acido
6 genes, 5% 0.6% 0.01214

156
salicílico, etc.)
BIOGENESIS DE LOS COMPONENTES CELULARES
18 genes, 14.9% 5.5% 0.01215
Desarrollo vegetal 11 genes, 9.1% 2.4% 0.01895
Regulador de las proteínas-G de senalizacion
2 genes, 1.7% 0% 0.04163
Tabla suplementaria 6. Análisis de BioMaps de los genes comunes reprimidos entre las 3 variedades de maíz a los 17 días de estrés
Clasificación Frequencia observada
Frecuencia esperada
Valor P
Energía 10 genes, 14.9% 1.5% 5.14E-06
Fotosíntesis 5 genes, 7.5% 0.2% 1.73E-05
Conversión de energía y regeneración 4 genes, 6% 0.2% 0.0014
TRANSPORTE CELULAR, FACILITACION DEL TRANSPORTE Y RUTAS DE TRANSPORTE
17 genes, 25.4% 8.6% 0.00322
Metabolismo de tetraterpenos 3 genes, 4.5% 0.1% 0.00354

157
Tabla Supplementaria 7. Análisis de BioMaps de los genes inducidos comunes of a las variedades tolerantes a los 17 días de estrés
Clasificación Frequencia observada
Frecuencia esperada
Valor P
Respuesta a estrés 31 genes, 15.3% 2.9% 5.39E-12
Sensado celular y respuesta a estímulo externo
39 genes, 19.2% 4.7% 8.38E-12
INTERACCION CON EL AMBIENTE 39 genes, 19.2% 5.3% 2.96E-10
RESCATE CELULAR, DEFENSA Y VIRULENCIA
37 genes, 18.2% 4.9% 7.45E-10
Respuesta a choque térmico 11 genes, 5.4% 0.3% 5.30E-09
Percepción de temperatura y respuesta 16 genes, 7.9% 0.9% 8.42E-09
Quimopercepción y respuesta 24 genes, 11.8% 2.8% 4.64E-07
Pared celular 16 genes, 7.9% 1.6% 2.15E-05
INTERACCION SISTEMICA CON EL AMBIENTE
19 genes, 9.4% 2.4% 8.41E-05
Desarrollo vegetal 19 genes, 9.4% 2.4% 9.00E-05
Desarrollo de fruto y maduración 9 genes, 4.4% 0.6% 0.00111
DESTINO CELULAR 12 genes, 5.9% 1.4% 0.00406

158
Ciclo del glioxilato
Regulación hormonal vegetal
Regulacion del crecimiento celular direccional
3 genes, 1.5%
14 genes, 6.9%
2 genes, 1%
0%
2%
0%
0.00684
0.0077
0.02422
Tabla Supplementaria 8. Análisis de BioMaps de los genes reprimidos comunes a las variedades tolerantes a los 17 días de estrés.
Clasificación Frequencia observada
Frecuencia esperada
Valor P
Energía 22 genes, 12% 1.5% 1.06E-11
Fotosíntesis 9 genes, 4.9% 0.2% 1.23E-08
Conversión de energía y regeneración 7 genes, 3.8% 0.2% 2.97E-05
Metabolismo 59 genes, 32.2% 17.6% 0.00015
Rescate celular, defensa y virulencia 25 genes, 13.7% 4.9% 0.00056
Plastido 44 genes, 24% 12.3% 0.00117
Transporte celular, facilitación de transporte y rutas de transporte
34 genes, 18.6% 8.6% 0.00226
159
Compuestos‐C y transporte de carbohidratos
7 genes, 3.8% 0.5 % 0.0042
Metabolismo de compuestos C‐3 6 genes, 3.3% 0.3% 8.41E-05
Metabolismo de tetraterpenos 4 genes, 2.2% 0.1% 0.00491
Cloroplasto 42 genes, 23% 12.2% 0.00519
Azúcar, glucósido, catabolismo de polioles y carboxilatos
6 genes, 3.3% 0.3% 0.00539
Compuestos‐C y metabolismo de carbohidratos
24 genes, 13.1% 5.9% 0.03133
Metabolismo de azúcares y nucleótidos
7 genes, 3.8% 0.7% 0.03573
Metabolismo de porfirinas 4 genes, 2.2% 0.2% 0.04211
Respuesta a estrés 15 genes, 8.2% 2.9% 0.04216

160
Tabla suplementaria 9. Análisis de BioMaps de los genes inducidos comunes a las tres variedades de maíz bajo riego de recuperación.
Clasificación Frequencia observada
Frecuencia esperada
Valor P
Plastido 84 genes, 29.3% 12.3% 2.06E-12
Cloroplasto 83 genes, 28.9% 12.2% 4.24E-12
Fotosintesis 8 genes,2.8% 0.2 % 1.62E-05
METABOLISMO 87 genes, 30.3% 17.6% 1.63E-05
ENERGÍA 18 genes, 6.3% 1.5% 8.18E-05
LOCALIZACIÓN SUBCELULAR 144 genes, 50.2% 37.6% 0.00171
Conversión de energía y regeneración 6 genes, 2.1% 0.2% 0.00939
TRANSPORTE CELULAR, FACILITACION DEL TRANSPORTE Y RUTAS DE TRANSPORTE
45 genes, 15.7% 8.6% 0.01243
Metabolismo secundario 14 genes, 4.9% 1.5% 0.02147
Metabolismo de porfirinas 5 genes, 1.7% 0.2% 0.02637

161
Tabla Suplementaria 10. Análisis de BioMaps de los genes reprimidos comunes entre las 3 variedades de maíz bajo riego de recuperación
Clasificación Frequencia observada
Frecuencia esperada
Valor P
Respuesta a estrés 49 genes, 14.3% 2.9% 3.70E-18
RESCATE CELULAR, DEFENSA Y VIRULENCIA
59 genes, 17.3% 4.9% 6.39E-15
Sensado celular y respuesta a estímulo externo
57 genes,16.7% 4.7% 1.48E-14
INTERACCION SISTEMICA CON EL AMBIENTE
58 genes, 17% 5.3% 5.64E-13
Respuesta a choque térmico 16 genes, 4.7% 0.3% 1.52E-12
Percepción de temperatura y respuesta 22 genes, 6.4% 0.9% 1.43E-10
Quimopercepción y respuesta 37 genes, 10.8% 2.8% 4.74E-10
Respuesta a estrés hídrico 13 genes, 3.8% 0.4% 4.33E-07
Compuestos‐C y metabolismo de carbohidratos
50 genes, 14.6% 5.9% 6.35E-07
METABOLISMO 104 genes, 30.4% 17.6% 7.80E-07
Respuesta a ácido abscísico 13 genes, 3.8% 0.6% 1.69E-05

162
Respuesta a estrés osmótico y salino 13 genes, 3.8% 0.7% 0.00028
INTERACCION SISTEMICA CON EL AMBIENTE
24 genes, 7% 2.4% 0.00067
Unión a Calcio 11 genes, 3.2% 0.6% 0.00216
Regulación hormonal vegetal
20 genes, 5.8% 2% 0.00296
Transducción de señales mediada por segundos mensajeros
15 genes, 4.4% 1.2% 0.00323
Respuesta y sensado sistémico especifico de plantas / hongos
21 genes, 6.1% 2.2% 0.00433
Transducción de señales mediada por hormonas
11 genes, 3.2% 0.7% 0.00494
DESTINO CELULAR 15 genes, 4.4% 1.4% 0.01677
Pared celular 16 genes, 4.7% 1.6% 0.02019
Respuesta a estrés oxidativo 10 genes, 2.9% 0.7% 0.03395
Ciclo del glioxilato 3 genes, 0.9% 0% 0.03974

163
Tabla suplementaria 11. Analisis de BioMaps de los genes inducidos communes a las variedades tolerantes de maiz bajo riego de recuperación
Clasificación Frequencia observada
Frecuencia esperada
Valor P
No anotado 2 genes, 0.3% 0% 0
Plastido 201 genes, 29% 12.3% 3.08E-30
Cloroplasto 197 genes, 28.4% 12.2% 1.00E-28
LOCALIZACIÓN SUBCELULAR 380 genes, 54.8% 37.6% 4.06E-18
METABOLISMO 215 genes,31% 17.6% 6.93E-16
ENERGÍA 47 genes, 6.8% 1.5% 2.28E‐15
Fotosintesis 17 genes, 2.4% 0.2% 3.04E‐12
Metabolismo de porfirinas 16 genes, 2.3% 0.2% 6.65E‐12
Metabolismo de productos secundarios derivados de glicina, L‐serina and L‐alanina
16 genes, 2.3% 0.2% 1.86E‐10
Compuestos‐C y metabolismo de carbohidratos
85 genes, 12.2%
5.9%
4.7E‐08
Absorción de luz 9 genes, 1.3% 0.1% 4.7E‐08

164
Metabolismo secundario 32 genes, 4.6% 1.5% 5.65E‐06
Metabolismo de azúcar, glucósido, polioles y carboxilatos
50 genes, 7.2% 3.1% 8.01E‐06
Conversión de energía y regeneración 12 genes, 1.7% 0.2% 1.37E‐05
Fotopercepción y respuesta 21 genes, 3% 0.9% 0.0003
Biosíntesis of vitaminas, cofactores y prostéticos
14 genes, 2% 0.4% 0.00078
Compuestos transportados (substratos) 79 genes, 11.4% 6.7% 0.00085
Catabolismo de azucares, glucosidos, polioles y carboxilatos
12 genes, 1.7% 0.3% 0.00107
Citoplasma 51 genes, 7.3% 3.8% 0.00245
Union a complejos de cofactores/cosubstratos/vitaminas
13 genes, 1.9% 0.4% 0.00254
Sensado celular y respuesta a estímulo externo
59 genes, 8.5% 4.7% 0.00272
TRANSPORTE CELULAR, FACILITACION DEL TRANSPORTE Y RUTAS DE TRANSPORTE
93 genes, 13.4% 8.6% 0.00385
Glicólisis and gluconeogénesis 10 genes, 1.4% 0.3% 0.00402
Plastido 8 genes, 1.2% 0.2% 0.00412
Metabolismo de glutamato 7 genes, 1% 0.1% 0.0052

Chromoplasto 6 genes, 0.9% 0.1% 0.00823
INTERACCION CON EL AMBIENTE 62 genes, 8.9% 5.3% 0.01119
Metabolismo de tetraterpenos (carotenoides)
6 genes, 0.9% 0.1% 0.01643
Percepción de temperatura y respuesta 18 genes, 2.6% 0.9% 0.01811
Transporte de electrones 35 genes, 5% 2.5% 0.0185
Metabolismo de compuestos C‐3 10 genes, 1.4% 0.3% 0.02295
Metabolismo de aminoácidos 21 genes, 3% 1.2% 0.03802
Metabolismo de isoprenoides 14 genes, 2% 0.6% 0.03937
Ruta de pentosa‐fosfato 7 genes, 1% 0.2% 0.03984
Biosíntesis of vitaminas, cofactores y prostéticos
17 genes, 2.4% 0.9% 0.04692
165

166
Tabla Supplementaria 12. Análisis de BioMaps de los genes reprimidos comunes en las variedades tolerantes bajo riego de recuperación
Clasificación Frequencia observada
Frecuencia esperada
Valor P
No anotado 3 genes, 0.5% 0% 0
Respuesta a estrés 66 genes, 10.4% 2.9% 4.35E‐17
Sensado celular y respuesta a estímulo externo
81 genes, 12.8% 4.7 % 6.91E‐14
RESCATE CELULAR, DEFENSA Y VIRULENCIA
82 genes, 13% 4.9% 2.87E‐13
INTERACCION CON EL AMBIENTE 83 genes, 13.1% 5.3% 4.57E‐12
Respuesta a choque térmico 18 genes, 2.8% 0.3% 1.77E‐10
Percepción de temperatura y respuesta 29 genes, 4.6% 0.9% 1.99E‐10
METABOLISMO 183 genes, 29% 17.6% 2.16E‐10
Quimopercepción y respuesta 15 genes, 2.4% 0.7% 1.216E‐09
Compuestos‐C y metabolismo de carbohidratos
83 genes, 13.1% 5.9% 2.02E‐09
Respuesta a estrés osmótico y salino 21 genes, 3.3% 0.7% 2.23E‐06
Respuesta a estrés hídrico 16 genes, 2.5% 0.4% 2.55E‐06

167
Respuesta a ácido abscísico 16 genes, 2.5% 0.6% 0.00018
Ciclo del glioxilato
5 genes, 0.8% 0% 0.00035
INTERACCION SISTEMICA CON EL AMBIENTE
35 genes, 5.5% 2.4% 0.00141
TRANSPORTE CELULAR, FACILITACION DEL TRANSPORTE Y RUTAS DE TRANSPORTE
87 genes, 13.8% 8.6% 0.00232
Respuesta a estrés oxidativo 16 genes, 2.5% 0.7% 0.00328
Union a complejos de cofactores/cosubstratos/vitaminas
12 genes, 1.9% 0.4% 0.00449
REGULACIÓN OF METABOLISMO Y FUNCIÓN DE PROTEÍNA
31 genes, 4.9% 2.2% 0.00877
Respuesta a frío 13 genes, 2.1% 0.6% 0.01477
Envejecimiento celular 7 genes, 1.1% 0.2% 0.01488
Regulación hormonal vegetal
28 genes, 4.4% 2% 0.01508
Respuesta y sensado sistémico especifico de plantas / hongos
30 genes, 4.7% 2.2% 0.01663
DESTINO CELULAR 22 genes, 3.5% 1.4% 0.01889
Transducción de señales mediada por 14 genes, 2.2% 0.7% 0.03202

168
hormonas
Compuestos transportados (substratos) 67 genes, 10.6% 6.7% 0.03706
Unión NAD/NADP 7 genes, 1.1% 0.2% 0.03914
Unión a calcio 13 genes, 2.1% 0.6% 0.04791
Respuesta a estímulo biótico 15 genes, 2.4% 0.8% 0.04906


















