
TEMA%4% RUTAS%DE%SEÑALIZACIÓN%IMPLICADAS%EN...
35
1 TEMA 4 RUTAS DE SEÑALIZACIÓN IMPLICADAS EN LA PROLIFERACIÓN, DIFERENCIACIÓN Y SUPERVIVENCIA CELULARES El crecimiento, la proliferación, la diferenciación, la movilidad, la supervivencia y la muerte celular están controlados por diversas rutas de señalización. La transducción de una señal desde la membrana plasmática y su propagación en el interior celular se realiza por la activación secuencial de distintas proteínas, que incluyen receptores de la membrana plasmática para hormonas y factores de crecimiento, así como proteínas GTPasas unidas a ella mediante modificaciones lipídicas, las cuales activan distintas proteínas organizadas en lo que se conocen como cascadas de señalización. En concreto, el cáncer se debe a la acumulación de mutaciones que producen, bien la activación constitutiva de proteínas, en el caso de los oncogenes, o la inactivación, en el caso de los genes supresores de tumores, de proteínas implicadas en la señalización celular. El resultado son rutas de señalización constitutivamente activas, que dan órdenes continuas de proliferación celular sin posibilidad de control externo. El número de vías de señalización implicadas en cáncer es creciente. En este capítulo se exponen las mejor caracterizadas, entre las que se encuentran las vías de señalización de las proteínas MAPKs, PI3K/AKT, JAK/STAT, NFκB Y Wnt. ÍNDICE SEÑALIZACIÓN POR PROTEÍNAS G Señalización por proteínas G heterotriméricas Señalización por proteínas G monoméricas VÍA DE SEÑALIZACIÓN RasRAFMEKERK Familia de las proteínas Ras Estructura y funciones de Ras Agentes terapéuticos para Ras oncogénico Inhibidores de la enzima farnesil transferasa Oligonucleótidos antisentido contra Ras Cascada RAFMEKERK Inhibición de las proteínas MEK Proteínas RAF Estructura y funciones de RAF. Implicación de BRAF en cáncer Inhibición de V600E BRAF SEÑALIZACIÓN MEDIADA POR LA VÍA DE PI3KAKTmTOR PI3K y AKT PI3K AKT Efectos biológicos de la activación de PI3KAKT Proliferación celular y crecimiento celular Supervivencia celular mTOR Papel de la vía PI3KAKTmTOR en cáncer Inhibidores de la vía PI3KAKTmTOR Inhibidores de PI3K Inhibidores de AKT Inhibidores mTORC1
Transcript of TEMA%4% RUTAS%DE%SEÑALIZACIÓN%IMPLICADAS%EN...

1
TEMA 4
RUTAS DE SEÑALIZACIÓN IMPLICADAS EN LA PROLIFERACIÓN, DIFERENCIACIÓN Y SUPERVIVENCIA CELULARES
El crecimiento, la proliferación, la diferenciación, la movilidad, la supervivencia y la muerte
celular están controlados por diversas rutas de señalización. La transducción de una señal desde la membrana plasmática y su propagación en el interior celular se realiza por la activación secuencial de distintas proteínas, que incluyen receptores de la membrana plasmática para hormonas y factores de crecimiento, así como proteínas GTPasas unidas a ella mediante modificaciones lipídicas, las cuales activan distintas proteínas organizadas en lo que se conocen como cascadas de señalización. En concreto, el cáncer se debe a la acumulación de mutaciones que producen, bien la activación constitutiva de proteínas, en el caso de los oncogenes, o la inactivación, en el caso de los genes supresores de tumores, de proteínas implicadas en la señalización celular. El resultado son rutas de señalización constitutivamente activas, que dan órdenes continuas de proliferación celular sin posibilidad de control externo. El número de vías de señalización implicadas en cáncer es creciente. En este capítulo se exponen las mejor caracterizadas, entre las que se encuentran las vías de señalización de las proteínas MAPKs, PI3K/AKT, JAK/STAT, NFκB Y Wnt. ÍNDICE SEÑALIZACIÓN POR PROTEÍNAS G
Señalización por proteínas G heterotriméricas Señalización por proteínas G monoméricas
VÍA DE SEÑALIZACIÓN Ras-‐RAF-‐MEK-‐ERK Familia de las proteínas Ras
Estructura y funciones de Ras Agentes terapéuticos para Ras oncogénico
Inhibidores de la enzima farnesil transferasa Oligonucleótidos antisentido contra Ras
Cascada RAF-‐MEK-‐ERK Inhibición de las proteínas MEK
Proteínas RAF Estructura y funciones de RAF. Implicación de B-‐RAF en cáncer Inhibición de V600EB-‐RAF
SEÑALIZACIÓN MEDIADA POR LA VÍA DE PI3K-‐AKT-‐mTOR
PI3K y AKT PI3K AKT Efectos biológicos de la activación de PI3K-‐AKT Proliferación celular y crecimiento celular Supervivencia celular mTOR Papel de la vía PI3K-‐AKT-‐mTOR en cáncer Inhibidores de la vía PI3K-‐AKT-‐mTOR
Inhibidores de PI3K Inhibidores de AKT Inhibidores mTORC1

2
Inhibidores de la actividad quinasa de mTOR Inhibidores duales PI3K/mTOR
Cooperación de las vías Ras-‐RAF-‐MEK-‐ERK y PI3K-‐AKT-‐mTOR en cáncer VÍA DE SEÑALIZACIÓN JAK-‐STAT
Papel de JAK/STAT en cáncer SEÑALIZACIÓN CELULAR POR LA VÍA DE NFκB
Participación de la vía de NFκB en cáncer VÍA DE SEÑALIZACIÓN DE Wnt

3
SEÑALIZACIÓN POR PROTEÍNAS G
La activación de los receptores de membrana se interpreta por la célula mediante la activación de forma simultánea y complementaria de vías de señalización intracelulares. Por una parte, se activan enzimas que generan segundos mensajeros (AMPc, GMPc, Ca2+, inositoles fosfato, etc.), que transmiten la señal al núcleo mediante cascadas de quinasas intracelulares. Paralelamente, se produce la activación directa de quinasas intracelulares que también llevan la señal al núcleo. En la mayoría de los casos, en la transducción de señales actúan las proteínas G, capaces de unir GTP y GDP con alta afinidad y especificidad. Existen dos tipos de proteínas G, que se diferencian en su función y estructura: las heterotriméricas y las monoméricas de tipo Ras. Señalización por proteínas G heterotriméricas
Las proteínas G heteterotriméricas están asociadas a receptores acoplados a proteínas G (GPCRs, G Protein-‐Coupled Receptor), que habitualmente se han relacionado con la modulación de vías de segundos mensajeros «clásicos», como el Ca2+ o el AMPc, y solo recientemente se está prestando atención a su capacidad para regular la proliferación, diferenciación y supervivencia celulares. Teniendo en cuenta la capacidad del AMPc de modular diversas vías mitogénicas en ciertas células y las conexiones encontradas recientemente que conectan los GPCRs con las vías activadas por los receptores con actividad tirosina quinasa (RTKs, tyrosine kinase receptors), no es de extrañar que se hayan encontrado mutaciones y sobreexpresión de diversas proteínas G heterotriméricas en algunos tumores. En concreto, se han observado mutaciones de las proteínas Gαs, Gαi2 y Gα12 en tumores endocrinos adrenales, hipofisarios, ováricos y de tiroides (figura 1).
Sin embargo, es mucho mayor la importancia en carcinogénesis de la superfamilia de proteínas G monoméricas, cuyo prototipo es Ras.
Figura 1. Activación y señalización de las proteínas G heterotriméricas. La unión de una hormona a su receptor recluta las proteínas G heterotriméricas formadas por la subunidad α, β y γ. A continuación se activa la subunidad al producirse el intercambio de GDP por GTP. Una vez activa, la subunidad α se separa del dímero βγ y cada uno de ellos puede activar distintos efectores. Cada una de las proteínas efectoras produce segundos mensajeros que activan a proteínas quinasas, las cuales transmiten señales en el citosol o en el núcleo y producen respuestas celulares.

4
Señalización proteínas G monoméricas
Las proteínas G monoméricas, también llamadas GTPasas pequeñas, comprenden hasta la fecha más de cien miembros identificados en eucariotas. La mayoría de las proteínas G monoméricas se encuentran ampliamente distribuidas en células de mamíferos, aunque los niveles de expresión de los miembros de cada familia varían según el tipo celular. Desde un punto de vista estructural, los miembros de esta superfamilia se clasifican en cinco grandes familias denominadas Ras, Rho/Rac/Cdc42, Rab, Sar1/Arf y Ran, que participan en muchos procesos de señalización intracelulares. La localización subcelular de estas proteínas es variada y se pueden encontrar en la membrana plasmática, en el citosol o en el núcleo, pero siempre unidas a un tipo específico de membranas, ya que sufren modificaciones postraduccionales en su extremo C-‐ terminal mediante la adición de restos lipídicos. Estas modificaciones son necesarias para que se unan a las membranas celulares, donde interaccionan con sus moléculas reguladoras y efectoras. Sobre esta familia de proteínas pueden destacarse, al menos, tres características generales: la diversidad de los receptores de membrana y moléculas reguladoras que pueden activarlas; la gran variedad de sustratos celulares que pueden interactuar con cada una de estas GTPasas y, por último, el extenso entrecruzamiento y cooperación que existe entre las vías de transducción de señales que regulan estas GTPasas.
Las proteínas G monoméricas son GTPasas capaces de unir e hidrolizar nucléotidos trifosfato de guanina o GTP. Estas proteínas existen en su conformación inactiva (unida a GDP) o activa (unida a GTP). El cambio del estado inactivo al activo se produce tras la recepción de una señal estimuladora, lo que provoca cambios estructurales que hacen accesible la región efectora de estas GTPasas a sus proteínas sustrato. En su estado activo, la actividad GTPasa intrínseca de la proteína hidroliza el GTP, volviendo de nuevo al estado inactivo y cerrándose de este modo el ciclo (figura 2).
Figura 2. Ciclo de activación de las proteínas G monoméricas. Las GEFs, después de recibir la señal apropiada, catalizan el intercambio de GTP por GDP de la GTPasa. Las proteínas GAPs estimulan la actividad GTPasa promoviendo la conversión de la forma unida a GTP a la forma unida a GDP. En el caso de las proteínas Rho, participan otras proteínas, denominadas GDIs, que afectan a la disociación del GDP y mantienen a estas GTPasas en el citosol en su estado inactivo.

5
El paso limitante en el proceso de activación es el intercambio de GDP por GTP. Esta reacción es extremadamente lenta y se acelera por un grupo de proteínas denominadas factores intercambiadores de nucléotidos de guanina (GEFs). Estos GEFs estimulan el intercambio de nucleótidos, facilitando la salida del GDP, el cual es rápidamente sustituido por el GTP (Figura 2). Por otra parte, la actividad GTPasa de estas proteínas es bastante lenta, por lo que es necesaria la participación de proteínas activadoras de la función GTPasa (GAPs), que incrementan la tasa de hidrólisis del GTP. En general, hay dos consecuencias del ciclo GDP/GTP sobre las proteínas GTPasas, por una parte, la forma unida a GTP adopta una conformación diferente que la forma unida a GDP, permitiendo la interacción con las moléculas efectoras y, por otra, esta forma unida a GTP se encuentra normalmente asociada de manera más fuerte a la membrana plasmática, o en diferente localización que la forma unida a GDP.
Estas proteínas intervienen en la regulación de funciones celulares muy diversas, entre las que destacan la proliferación, la supervivencia celular, la organización del citoesqueleto, la migración celular, el tráfico intracelular de vesículas y el transporte nuclear.

6
VÍA DE SEÑALIZACIÓN Ras-‐RAF-‐MEK-‐ERK Familia de las proteínas Ras
Estructura y funciones de Ras
Las proteínas Ras son los miembros mejor caracterizados de la superfamilia de proteínas G
monoméricas y merecen un apartado específico debido a su importancia como oncogenes en tumores humanos. En humanos, la familia de proteínas Ras incluye tres proto-‐oncogenes denominados H-‐Ras, N-‐Ras y K-‐Ras, cuyos productos tienen un 85% de homología en su secuencia aminoacídica. En el caso de K-‐Ras, debido a un procesamiento alternativo del cuarto exón de este gen, se generan las formas A y B, siendo la presencia de K-‐Ras4B mucho más abundante que la de K-‐Ras4A en la mayoría de los tipos celulares. Otras proteínas de esta familia son R-‐Ras, las proteínas Ral, las proteínas Rap, la proteína Rheb y las más recientemente descubiertas Rin y Rit. Todas estas proteínas poseen propiedades biológicas similares y están implicadas en procesos de proliferación, diferenciación y supervivencia.
La GTPasa Ras es una proteína de 21 kDa y su molécula contiene 189 aminoácidos, a excepción de K-‐Ras4B, que posee 188. Todos los dominios críticos para la función GTPasa, incluyendo las secuencias con los motivos importantes para la unión a los nucleótidos y la hidrólisis del GTP, están presentes en los 165 aminoácidos del extremo amino terminal. En el carboxilo terminal se encuentra la región hipervariable, que contiene la Caja CAAX: (C, cisteína; A, aminoácido alifático; X, metionina o serina) con los cuatro aminoácidos esenciales para la actividad de éstas proteínas, ya que en esta región es donde se realizan parte de las modificaciones lipídicas postraduccionales (Figura 3).
La actividad biológica de Ras depende de su localización en la membrana plasmática a través
de la unión de farnesilos isoprenoides de 15 carbonos a la caja CAAX de su extremo C-‐terminal. La reacción de farnesilación es catalizada por la enzima farnesil transferasa que incorpora grupos farnesilos al sulfuro de la cadena lateral de la cisteína. Tras la prenilación, los tres aminoácidos AAX del extremo C-‐terminal son hidrolizados por proteólisis y la cisteína prenilada es metilada. Las modificaciones del CAAX convierten al extremo carboxilo de Ras en hidrofóbico. Sin embargo, esta hidrofobicidad no es suficiente para que la unión de Ras a la membrana sea estable y se necesita una segunda señal que consiste en la unión de una secuencia polibásica de seis residuos de lisina adyacentes a la caja CAAX (175-‐180) en el caso de K-‐Ras, o en la unión de un ácido graso, generalmente ácido palmítico de 16 átomos de carbonos, a cisteínas situadas cerca del extremo C-‐terminal en N-‐Ras y H-‐Ras (figura 3).

7
Figura 3. Modificaciones lipídicas de las proteínas G monoméricas. Las proteínas GTPasas son sintetizadas en ribosomas libres en el citosol. Posteriormente translocan al retículo endoplásmico donde se les adiciona un resto farnesilo de 15C a la cisteína del motivo CAAX del extremo C-‐terminal. A continuación se produce la proteólisis de los tres aminoácidos terminales y la mutilación de la cisteína. Además, también se le une un ácido graso, generalmente palmítico de 16C, que aumenta el anclaje de las GTPasas a la membrana. Posteriormente, las proteínas migran por el aparato de Golgi hasta la membrana plasmática. Las proteínas K-‐Ras4B no se palmitoilan y pasan directamente del retículo endoplásmico a la membrana plasmática por un mecanismo desconocido.
El diagrama inferior muestra el grado de conservación de la secuencia entre las isoformas de la proteína Ras. La región hipervariable contiene el dominio de unión a la membrana con el motivo CAAX del extremo C-‐terminal, común a todas las isoformas, más la secuencia de las señales secundarias: los sitios de cisteína donde se producen las palmitioilaciones (mostrados en rojo) en H-‐,N-‐ y K-‐Ras4A o el dominio polibásico en K-‐Ras4B (mostrado en rojo).
Las proteínas Ras se activan fundamentalmente como respuesta a la unión de un ligando a
un receptor tirosina quinasa de la membrana plasmática (ver tema 3). La fosforilación y activación del receptor crea sitios de unión para proteínas adaptadoras con dominios SH2, como Grb2. Esta proteína adaptadora, a su vez, se une y activa GEFs, entre los que se encuentra Sos. Además, Ras también se puede activar por GEFs regulados por segundos mensajeros producidos después de la activación de GPCRs y por integrinas unidas a la matriz extracelular. Una vez que las proteínas Ras se unen a GTP con la mediación de las proteínas GEFs, actúan a través de la activación de distintas proteínas efectoras, las cuales desempeñan funciones diversas. Los tres efectores más importantes son (figura 4):
• Los miembros de la familia de proteínas serina treonina quinasas RAF, que son los responsables de la activación de la cascada MEK-‐ERK MAPK, implicada en procesos de diferenciación, proliferación y protección frente a apoptosis.
• Algunos miembros de la familia de las Fosfatidil-‐inositol 3 quinasas (PI3K), implicadas en procesos de proliferación y supervivencia.
• Los factores intercambiadores de nucleótidos de guanina (Ral-‐GDS, RGL/Rsb2 y RGL/Rlf), activadores de las GTPasas Ral, implicados en procesos de transformación. Además, Ras también activa otros efectores, como la proteína intercambiadora de
nucleótidos de guanina Tiam-‐1, factores activadores de la actividad GTPasa (p120GAP, NF1),

8
proteínas adaptadoras (AF-‐6), lipasas (PLCε), la serina-‐treonina quinasa PKCξ y la proteína MEKK1, activador de la ruta de JNK (figura 4).
Figura 4. Efectores de Ras. Una vez que Ras es activado activa numerosos sustratos celulares implicados en distintas funciones biológicas. En cuanto a su papel en la proliferación, Ras es necesario para la progresión del ciclo celular
en la fase G1 a través de la síntesis de ciclina D1 por activación de la cascada de ERKs (ver tema 2). Por otra parte, Ras también participa en la protección de la apoptosis, a través de la activación de ERK y de PI3K, por inhibición de las proteínas proapoptóticas Bim, Bad y caspasas (ver tema 5). Además, Ras también participa en la migración y metástasis, a través de las GTPasas de la familia Rho y la expresión de metaloproteasas de la matriz extracelular (ver tema 7).
Además de sus funciones en los procesos fisiológicos celulares, los miembros de la familia de proteínas Ras están involucrados en carcinogénesis. En diversos tumores humanos se ha observado una activación aberrante de Ras, que puede ser directa, por mutaciones oncogénicas, o indirecta, por desregulación de la vía desde el receptor. Así, se han encontrado mutaciones en el gen de ras (H-‐, N-‐, o K-‐Ras) en un 30 % de los tumores humanos, con una alta incidencia de mutaciones en K-‐Ras en carcinomas pancreáticos (50-‐90%), de colon (50%), de ovario (30%), de pulmón (30%), mutaciones de H-‐Ras en carcinomas de mama, riñón y vejiga, de N-‐Ras en hígado, linfomas y melanomas (20%) y mutaciones de las tres isoformas en carcinoma de tiroides (25%) (tabla 1). Entre las mutaciones encontradas, la más abundante es la sustitución de la glicina 12 por valina (V12Ras) y la sustitución de la glutamina 61 por leucina (L61Ras). La importancia de la vía de señalización de Ras en el cáncer humano está totalmente demostrada mediante los análisis mutagénicos de los distintos efectores de Ras, como B-‐RAF, PI3K y RalGEFs, que también intervienen en la transformación celular.
Los mecanismos por los cuales un aumento de la actividad de Ras produce la transformación celular son muy complejos. Por una parte, V12Ras da lugar a un aumento descontrolado de la proliferación celular a través de la desregulación del ciclo; además, participa en los procesos de migración celular a través de la activación de los miembros de la familia de proteínas Rho y protege de la apoptosis celular, promoviendo la capacidad de metastatizar de las células tumorales, a través de la activación de la vía PI3K/AKT.

9
Tabla 1. Mutaciones de Ras encontradas en cáncer Para una información completa ver la base de datos de mutaciones del instituto Sanger:
http://www.sanger.ac.uk/genetics/CGP/cosmic/ Agentes terapéuticos para Ras oncogénico
Inhibidores de la enzima farnesil transferasa La unión de un grupo lipídico farnesilo al extremo C-‐terminal de Ras, mediante la acción de la
enzima farnesil transferasa (FT), es esencial para anclarla a la membrana y, por tanto, necesaria para su actividad. Por ello, se han desarrollado varias estrategias para inhibir la farnesilación de Ras, la más común de ellas ha sido el diseño de compuestos que imitan el motivo CAAX del C-‐terminal Ras para competir por la unión a la farnesil transferasa. Los primeros compuestos fueron péptidos modificados (peptidomiméticos CAAX). Otra estrategia ha sido hacer compuestos que compiten con el grupo farnesil pirofosfato para inhibir a la enzima. Por otra parte, también se han desarrollado otro grupo de fármacos que se conocen como análogos bisustrato, los cuales comparten propiedades tanto del farnesil pirofosfato y de la secuencia CAAX, imitando así un estado de transición en el proceso de farnesilación.
A través del esfuerzo masivo de muchas compañías farmacéuticas, se han identificado un gran número de inhibidores de la farnesiltransferasa (FTI), muy eficaces in vitro y en modelos animales, pero con poco éxito en pacientes humanos. La raíz del problema radica en el hecho de que, aunque H-‐Ras se modifica exclusivamente por farnesilación (15 carbonos), K-‐Ras y, en menor medida N-‐Ras, también se pueden modificar por la unión de geranilgeranilo (20 carbonos), a través de la geranilgeranil transferasa (GGT), lo que les permite mantener su actividad. Por lo tanto, la geranilgeranilación de K-‐Ras y N-‐Ras es importante y mantiene la actividad de estas proteínas cuando se bloquea la farnesilación. Como la inmensa mayoría de mutaciones en Ras en tumores humanos son en K-‐Ras, seguido por N-‐Ras, y muy pocas en H-‐Ras, es probable que la inhibición de la farnesilación de Ras mutante no sea la responsable de los posibles efectos antitumorales de los FTIs. Para evitar este problema también se ha intentado inhibir la función de K-‐Ras y N-‐Ras mediante el uso conjunto de FTIs y GGTIs, pero ha fallado debido a la elevada toxicidad asociada a esta combinación. De hecho, es probable que la falta de toxicidad de los FTIs sea debida al hecho de que no consiguen inhibir eficazmente la función de todas las proteínas Ras endógenas.
Tipo de cáncer % mutación Tipo mutante Páncreas 57-‐90 K y N Pulmón (NSC) 18-‐35 K y N Colorrectal 33-‐45 K y N Tiroides 55 K, N y H Melanoma 15-‐18 K, N y H Vejiga 10 H Hígado 5-‐30 K y N Riñón 10 H Hematológico y linfoide 10-‐30 K y N Síndrome mielodisplásico 40 K y N Ovario 18 K Endometrio 16 K, N y H Próstata 15 K, N y H Estómago 12 K, N y H Mama 6 K y H Tracto biliar 30 K Tracto genital 7 K y H

10
Por otra parte, los FTIs desarrollados para Ras también inhiben la farnesilación de muchas otras proteínas tumorales, por lo que un argumento que se ha planteado en repetidas ocasiones es que la capacidad de los FTIs para inhibir el crecimiento tumoral y la supervivencia celular se debe a la inducción de modificaciones aberrantes de proteínas relacionadas con Ras, u otras proteínas distintas. Por ello, aunque este modelo tiene algunas características convincentes, también existen argumentos sólidos en contra, por lo que esta cuestión sigue sin resolverse en la actualidad. Sin embargo, a pesar de la incertidumbre sobre su mecanismo de acción, los FTIs tienen potencial como drogas antitumorales, ya que poseen marcados efectos sobre el crecimiento y supervivencia de algunas líneas de células tumorales in vitro y en xenoinjertos en ratones desnudos, aunque no necesariamente expresen Ras activado y, además, inhiben el crecimiento de algunas células tumorales humanas que expresan K-‐ras oncogénico, a pesar de que su procesamiento no está bloqueado. Por ello, en la actualidad se están llevando cabo aproximadamente 60 ensayos clínicos utilizando FTIs, aunque su diana real no se conozca (ver http://clinicaltrials.gov/). El éxito o no de estos ensayos determinará si hay un futuro para el uso de los FTIs como terapia contra el cáncer y para qué tipo de tumores se pueden utilizar. Los FTIs utilizados en los ensayos realizados hasta el momento se muestran en la tabla 2. INHIBIDORES DE RAS Inhibidor Diana Tipo cáncer/Observaciones Ensayo clínico R115777 (Zanestra)
Ras Rheb
AML, linfoma, mama, glioma, melanoma
Fase I, II, III
SCH66336 (Sarasar)
Ras No efectos en cáncer páncreas Fase II
L778,123 Ras Sin efectos en NSCLC Fase II BMS-‐214662 Ras Fase I ISIS 2503 HRAS mRNA Sin efecto
Tabla 2. Inhibidores de Ras utilizados en ensayos clínicos
Oligonucleótidos antisentido contra Ras Otro enfoque terapéutico diferente es la inhibición de expresión de H-‐Ras mediante el uso
de oligonucleótidos antisentido cortos sintéticos que son específicos para las secuencias del mRNA de esta proteína. Isis Pharmaceuticals ha desarrollado exitosamente varios derivados de oligonucleótidos que reducen la expresión de H-‐Ras, como el ISIS2503, que actualmente está siendo utilizado en ensayos clínicos. Sin embargo, también existen problemas con su utilización. Por una parte, está el alto nivel de especificidad de estos agentes, que podría significar que no se dirijan a las proteínas tumorales más importantes; por ejemplo, la mutación de H-‐Ras es muy rara en los tumores, por lo que eliminar su expresión puede que sea menos eficaz que eliminar K-‐Ras. Otro problema es la liberación efectiva de estas moléculas relativamente grandes en los tumores y la garantía de que sean captadas por las células malignas. Además, también se han encontrado distintos problemas de toxicidad no específica. A pesar de ello, algunos de estos oligonucleótidos no han mostrado toxicidad en ensayos clínicos, pero todavía no está claro si son una terapia eficaz o no en el tratamiento de tumores. También se ha desarrollado un oligonucleótido antisentido para K-‐Ras, ISIS6957, el cual es eficaz en células en cultivos, pero no se ha analizado su eficacia en ensayos clínicos todavía.
Además de intentar inhibir Ras directamente, las compañías farmacéuticas han hecho grandes esfuerzos para desarrollar inhibidores de las vías activadas por Ras, sobre todo la vía RAF/MEK/ERK y la vía PI3K/AKT. Estos inhibidores se comentarán en los apartados donde se explican estas vías de señalización.

11
Cascada RAF-‐MEK-‐ERK
Las cascadas de proteínas quinasas activadas por mitógenos (MAPK, Mitogen Activated Protein Kinases) transducen las señales desde la membrana celular al núcleo, en respuesta a un amplio rango de estímulos. Hasta el momento se han descrito 6 familias de MAPKs en mamíferos, de las cuales las más estudiadas son las que incluyen las proteínas quinasas reguladas por señales extracelulares (ERK1/2, Extracellular Signal-‐Regulated protein Kinases), las proteínas quinasas del extremo N-‐terminal de c-‐Jun (JNKs, c-‐Jun N-‐terminal Kinases) y las p38 MAPKs. Además, existen otras familias de MAPKs, las ERK3/4, la ERK5 y la ERK7, cuyas funciones no se conocen en detalle. Las MAPKs intervienen en gran número de funciones celulares en respuesta a diferentes estímulos, y median respuestas celulares específicas, que incluyen la expresión génica, proliferación, diferenciación, movilidad, metabolismo y supervivencia o muerte celulares (figura 5).
Figura 5. Cascadas de MAPKs. La activación de las MAPKs está regulada por una cascada jerárquica conocida como el módulo de MAPK. Mitógenos como factores de crecimiento, citoquinas, y estrés ambiental activan una GTPasa, que conduce a la activación de las MAPKKK, que a su vez fosforilan y activan a las MAPKKs. Las MAPKKs activan las MAPKs Erk1/2, JNK o p38 y JNK / SAPK a través de la fosforilación de residuos específicos de treonina y tirosina. La activación de estas MAPKs activa varias de dianas citosólicas o nucleares que regulan diversos eventos celulares, incluyendo la supervivencia celular, la proliferación, diferenciación y apoptosis.
Las MAPKs son una familia de proteínas serina/treonina quinasas organizadas en una cascada
de tres niveles. Las MAPKs se activan por hormonas, factores de crecimiento, citoquinas, e incluso a través del estrés ambiental producido por radiaciones, choques osmóticos y/o daño isquémico, estímulos que pueden actuar sobre receptores tirosina quinasa (RTKs), receptores acoplados a proteínas G (GPCRs), receptores de citoquinas o receptores serina/treonina quinasa (ver tema 3). La activación de la cascada se inicia cuando se activa una proteína G monomérica, la cual interacciona con una serina/treonina quinasa, MAPK quinasa quinasa (MAPKKK, MAPK Kinase Kinase), que es

12
activada por fosforilación. La MAPKKK fosforila y activa una MAPK quinasa (MAPKK, MAPK Kinase) de especificidad dual, que a su vez activa a la MAPK por fosforilación doble en residuos de treonina y tirosina. A continuación, las MAPKs fosforilan a sus sustratos en residuos de serina o treonina (figura 5). Las MAPKs tienen sustratos tanto a nivel citosólico como en el núcleo, donde fosforilan factores de transcripción y regulan la expresión de un gran número de genes.
Las proteínas ERKs MAPK responden primordialmente a factores de crecimiento y mitógenos y en menor medida a ligandos de los GPCRs, citoquinas, y estrés. Éstos transmiten la señal al interior de la célula, activando la proteína Ras que, unida a GTP, recluta a la membrana plasmática y activa a su proteína quinasa efectora RAF (MAPKKK). Las proteínas RAF activan a una segunda quinasa llamada MEK (MAPK-‐ERK Kinase, MAPKK), que a su vez activa a una tercera proteína quinasa llamada ERK (Extracelular signal-‐Regulated Kinase, MAPK) (figura 6).
Figura 6. Vía de señalización Ras-‐RAF-‐MEK-‐ERK.
Tras la activación de los receptores de membrana por señales externas se produce la activación de Ras, la cual recluta a RAF a la membrana, ésta se fosforila e inicia la cascada de fosforilación MEK y ERK. ERK puede activar proteínas citosólicas como las quinasas RSK o MSK1, o las fosfatasas MKP1/2, o bien translocarse al núcleo y regular múltiples factores de transcripción entre los que se encuentran Elk-‐1, Myc, Fos y Jun. La gran diversidad de sustratos de ERK hace que esta vía este implicada en multitud de procesos celulares como supervivencia, apoptosis, diferenciación y migración.
Las proteínas RAF son una familia de proteínas citosólicas compuesta por tres miembros: A-‐RAF, B-‐RAF y C-‐RAF; las proteínas MEK son dos, MEK1 y MEK2 y las proteínas ERK también son dos, conocidas como p44 y p42 por sus respectivos pesos moleculares (ERK1, 44 kDa y ERK2, 42 kDa). Las ERKs tienen actividad serina/treonina quinasa y, una vez activas, pueden fosforilar más de 150 posibles sustratos entre proteínas nucleares y citosólicas con diversas funciones. Algunos ejemplos de estos sustratos son las proteínas quinasas MSK1/2 (Mitogen and Stress-‐activated Kinase), RSK (también conocida como p90 RSK S6 quinasa), fosfatasas como MKP1/2, proteínas implicadas en

13
apoptosis como BAD y BIM y factores de transcripción como Elk-‐1, c-‐Fos, c-‐Jun, c-‐Myc y p53 (figura 6). De esta forma ERK controla los procesos de proliferación, transformación, supervivencia y diferenciación celular.
Esta vía puede encontrarse desregulada en varios de sus niveles, lo que la ha vinculado ampliamente al proceso de carcinogénesis. Concretamente, muchos receptores que activan la vía están mutados en cáncer, las proteínas Ras y RAF son protooncogenes, con una tasa de mutación de un 30 % en el caso de Ras y de un 10 % en el caso de B-‐RAF, y la actividad ERK está aumentada en un aproximadamente 30 % de tumores. Inhibición de las proteínas MEK
Aunque las proteínas MEK no se han encontrado mutadas en ningún tipo de célula tumoral, fueron las primeras proteínas de la cascada utilizadas como dianas terapéuticas en diferentes tipos de cáncer. Por ello, se han desarrollado distintos inhibidores para esta quinasa, entre los que se encuentran PD98059, CI-‐1040 (SelleckChem), PD0325901 (Pfizer), U0126 (DuPont), Selumetinib (ARRY-‐142886, AZD6244) (Astra-‐Zeneca), MEK162/ARRY-‐162 (Novartis), GDC-‐0973 (Genentech), Refametinib/DEA119 (Ardea Biosciences/Bayer), GSK112012 (GlaxoSmithKlein), TAK-‐733 (Takeda San Diego, Millennium Pharmaceuticals, Inc), RO4987655 (Roche) y AS703026 (EMD Serono). Los inhibidores de MEK se diferencian de la mayoría de los inhibidores de otras quinasas en que no compiten por la unión del ATP, lo que les confiere una alta especificidad. Por ello, la mayoría de los inhibidores de MEK son específicos y no inhiben otras proteínas quinasas.
En la actualidad hay aproximadamente 84 ensayos clínicos con inhibidores de MEK en la lista de la página web (ver http://clinicaltrials.gov/) para cáncer de pulmón, de páncreas, de colon, leucemias, cánceres cerebrales y cáncer de mama (tabla 3). Curiosamente, no existe ningún ensayo para cáncer de próstata. Sin embargo, los resultados iniciales de los ensayos clínicos no han dado los resultados deseados para el uso de inhibidores de MEK como un único agente terapéutico en pacientes con cáncer que no han sido previamente seleccionados para la activación pre-‐existente de la vía Ras/RAF/MEK/ERK. INHIBIDORES DE MEK Inhibidor Diana Tipo cáncer Ensayo clínico Compañía CI-‐1040 (PD-‐184352)
MEK1, MKK5 colorrectal, NSCLC, páncreas, riñón, melanoma, mama
Fase I, II (discontinuo)
Pfizer
PD0325901 MEK1/2 mama, colon, NSCLC, melanoma
Fase I, II (discontinuo)
Pfizer
XL518 MEK Fase I Exelixis Selumetinib(AZD6244, ARRY-‐142886)
MEK melanoma, HCC, páncreas, colon, pulmón, mama
Fase I, II Astra Zeneca/ Array BioPharma
RDEA119 (BAY 869766)
MAP2K1 (MAPK/ ERK kinase 1)
tumores avanzados Fase I, II Ardea/Bayer
PD098059 MEK1/2 cánceres hematológicos avanzados y cánceres sólidos avanzados
Preclínico ParkeDavis/Pfizer
U0126 MEK1/2 cánceres hematológicos avanzados y cánceres sólidos avanzados
Preclínico DuPont Pharmaceuticals
SL-‐327 MEK1/2 No evaluado para su uso en el tratamiento de cáncer
Preclínico DuPont Pharmaceuticals
Tabla 3. Inhibidores de MEK utilizados en ensayos clínicos

14
Proteínas RAF Estructura y funciones de RAF. Implicación de B-‐RAF en cáncer
La familia de proteínas RAF consta de tres miembros A-‐RAF, B-‐RAF y C-‐RAF, cuya activación comienza a partir de su interacción directa con la proteína GTPasa Ras. Sin embargo, la unión a Ras no es suficiente para la activación completa de RAF, y según las diferentes isoformas, se requieren algunas modificaciones adicionales, como interacciones proteína-‐proteína, unión a lípidos y sobre todo fosforilación.
Además de tener funciones propias, los miembros de la familia RAF pueden regular la activación de la vía MEK/ERK a través de la formación de homodímeros y heterodímeros. En este sentido, se ha observado que, en respuesta a la estimulación por factores de crecimiento y a la activación de Ras, C-‐RAF y B-‐RAF forman heterodímeros cuya actividad quinasa sobre MEK es más alta que la de los respectivos monómeros u homodímeros. El modelo propuesto describe que, en condiciones basales, B-‐RAF posee una conformación “cerrada” que impide la interacción con C-‐RAF. Cuando B-‐RAF es activado, bien por un estímulo a través de Ras o por mutaciones en el segmento de activación, adopta una conformación “abierta” permitiendo la interacción con C-‐RAF. De esta forma B-‐RAF activa a C-‐RAF y éste transmite la señal a MEK y ERK. La consecuencia más importante de este modelo de activación es que B-‐RAF puede mediar respuestas celulares a través de C-‐RAF, lo que supone una mayor flexibilidad en la regulación de la vía MEK-‐ERK y un nuevo mecanismo de B-‐RAF para transmitir las señales tumorogénicas.
Las proteínas RAF solo se consideraban importantes en carcinogénesis por su posición debajo de Ras, sin embargo, esta idea cambió al descubrirse que existen mutaciones oncógenicas de B-‐RAF en aproximadamente el 10 % de cánceres humanos. Por el contrario, las mutaciones de C-‐RAF en cáncer son raras y sólo aparecen con una frecuencia de 1 %, y no se han encontrado mutaciones en A-‐RAF. Concretamente se ha encontrado B-‐RAF mutado en el 30-‐60 % de melanomas, 30-‐50 % de tumores tiroideos, 5-‐20 % de cánceres colorrectales, 30-‐40 % de cánceres de ovario y en menor porcentaje (1-‐3 %) en otros tipos de cáncer (ver web (http://www.sanger.ac.uk/genetics/CGP/cosmic).
En total se han descrito unas 100 mutaciones para B-‐RAF, sin embargo existe una predominante: el cambio del nucleótido 1799 de timidina a adenosina, convirtiendo la valina 600 del segmento de activación en un glutamato (V600E). Esta mutación representa aproximadamente el 90 % de las mutaciones encontradas para B-‐RAF en melanoma y cáncer de tiroides, lo que convierte a V600EB-‐RAF en el mutante más importante de todos los encontrados (figura 7). El mutante V600EB-‐RAF tiene una actividad quinasa in vitro 500 veces mayor que la de B-‐RAF normal y es capaz de estimular constitutivamente la actividad de ERK. Este mutante induce la proliferación y transformación de células en cultivo y permite a estas células crecer como tumores en ratones inmunodeprimidos. Además, la inhibición de V600EB-‐RAF en células tumorales humanas disminuye su proliferación, aumenta su apoptosis basal e impide la formación de tumores xenotransplantados, por lo tanto, se considera un oncogén. Sin embargo, V600EB-‐RAF no es el único mutante de B-‐RAF con capacidad transformante. Muchos de los otros mutantes encontrados también son capaces de transformar células. El caso más sorprendente es el de los mutantes que tienen menor actividad quinasa que B-‐RAF normal, como D594V, y aún así son capaces de activar MEK-‐ERK a través de la formación de dímeros con C-‐RAF.
Las mutaciones en B-‐RAF se dan sobre todo en etapas tempranas de tumores papilares tiroideos (PTCs) y melanomas, ya que se han encontrado tanto en micro-‐PTCs, como en nevus melanocíticos, ambos considerados como precursores de PTCs y melanomas, respectivamente. Por ello, se considera una de las primeras mutaciones adquiridas por las células tumorales para dotarlas de las distintas características oncogénicas y que éstas puedan progresar a estados tumorales más avanzados. Los efectos de la mutación de B-‐RAF en cáncer son muy variados e incluyen alteraciones en la proliferación, supervivencia, invasión celular y metástasis.

15
Figura 7. Mutaciones del dominio catalítico de B-‐RAF asociadas a cáncer y su incidencia.
La mayoría de las mutaciones de B-‐RAF se producen en el segmento rico en glicina, responsable de la unión al ATP, o en el segmento de activación, dando lugar a una conformación “abierta” permanentemente activa. Las posiciones de las mutaciones que tienen lugar en cáncer humano están indicadas sobre la estructura. La longitud de las barras rojas indica la frecuencia relativa con que se producen dichas mutaciones. Los aminoácidos por los que se sustituyen se indican en rojo sobre la secuencia.
En cuanto a la proliferación, se ha observado que V600EB-‐RAF regula algunos componentes del
ciclo celular produciendo una proliferación incontrolada. Por una parte, V600EB-‐RAF induce la hiperfosforilación de retinoblastoma (Rb), lo que da lugar a la liberación del factor de transcripción E2F y a un aumento de la expresión de ciclina D1, promoviendo el paso de la fase G1 a la fase S del ciclo celular (ver tema 2) (figura 8). Además, V600EB-‐RAF disminuye los niveles de p27, un inhibidor de quinasas dependientes de ciclinas (CDKs), que produce la parada del ciclo celular en la fase G1 (figura 8).
La presencia de la mutación V600EB-‐RAF también confiere a las células que la poseen resistencia a morir por apoptosis. Así, células de melanoma con V600EB-‐RAF sufren arresto del ciclo en G1 y apoptosis al inhibir B-‐RAF (figura 8). Por otra parte, V600EB-‐RAF ejerce protección contra la apoptosis inducida por cisplatino, actinomicina-‐D o daunorubicina en células de melanoma. Además, V600EB-‐RAF también desempeña un papel importante en la invasión tumoral y en las interacciones con el estroma, en la promoción de la angiogénesis y en la creación de un microambiente inmunológico privilegiado. V600EB-‐RAF aumenta la expresión de varias proteínas que regulan la migración celular, como Rdn3, PlexinB1 o Minerva/FAM129B, favoreciendo el anclaje y la metástasis de células de melanoma (figura 8). Asimismo, V600EB-‐RAF modifica el ambiente tumoral induciendo la expresión de los factores angiogénicos HIF-‐1, VEGF y VEGFR, las metaloproteasas MMP1, MMP2, MMP3, MMP9 y MMP13, implicadas en el aumento de la invasión celular, la expresión de genes involucrados en la composición y regulación de la MEC, así como la sobreexpresión de IL-‐8 e IL-‐10 en numerosas líneas celulares tumorales (figura 8). Por todo esto, en PTCs humanos que poseen esta mutación existe una estrecha relación entre B-‐RAF mutado y la

16
progresión a estados avanzados del tumor, invasión extratiroidea y metástasis en ganglios linfáticos. Por otra parte, V600EB-‐RAF aumenta la invasión de melanomas.
Otro mecanismo de regulación de la tumorogéneis por V600EB-‐RAF es a través de la inhibición de la expresión de genes por metilación. En este sentido, el inhibidor tisular de MMP3 (TIMP3), la proteína quinasa asociada a muerte (DAPK) y el receptor β2 del ácido retinoico (RARβ2) son ejemplos de genes, cuya metilación se ha asociado a la expresión de V600EB-‐RAF (ver tema 10).
V600EB-‐RAF también parece regular la senescencia. En consonancia con estos datos, V600EB-‐RAF se encuentra con frecuencia en hiperplasia benigna melanocítica (nevus), lesiones cuyas células tienen detenido el crecimiento y se encuentran en un estado similar al de senescencia. Esta senescencia inducida por V600EB-‐RAF puede ser contrarrestada por cooperación con otros mecanismos, entre los que encontramos la pérdida de supresores de tumores, en particular, p16INK4a, p53 y PTEN, y la sobreexpresión de protooncogenes como Myc. Además, se ha observado que las células epiteliales de pulmón con V600EB-‐RAF no progresan a adenocarcinoma a menos que se inhiba la expresión de p53 o de p16INK4a.
Figura 8. Efecto de V600EB-‐RAF sobre la tumorogénesis. V600EB-‐RAF tiene un efecto sobre el ciclo células al promover la transición de la fase G1 a la fase S a través de la disminución de los niveles de p27 y el aumento de los niveles de Ciclina D1. V600EB-‐RAF protege de la apoptosis, ya que promueve la degradación de IκB aumentando así la actividad de NFκB nuclear. NFκB induce la expresión de proteínas antiapoptóticas como XIAP, c-‐IAP1 y c-‐IAP2. V600EB-‐RAF aumenta la angiogénesis a través de la regulación de la expresión de factores angiogénicos e inhibición de antioangiogénicos. V600EB-‐RAF favorece la invasión y la metástasis modificando la expresión de genes relacionados con la transición epitelio-‐mesénquima (EMT) y con la matriz extracelular. V600EB-‐RAF favorece la tumorogénesis mediante la regulación del silenciamiento de genes supresores tumorales producidos por alteraciones epigenéticas de la metilación de sus promotores.

17
V600EB-‐RAF como diana terapéutica
Debido a la implicación de V600EB-‐RAF en la progresión del cáncer se han realizado muchos estudios para investigar la utilidad del mutante V600EB-‐RAF como diana terapéutica. Por ello, se han desarrollado un gran número de moléculas inhibidoras de V600EB-‐RAF durante la última década. Algunos inhibidores, por ejemplo Sorafenib (NexavarTM, Bayer), ideado inicialmente para inhibir específicamente RAF, se ha demostrado posteriormente que inhiben múltiples proteínas, por ejemplo, VEGF-‐R, Flt-‐3, c-‐Kit, PDGF-‐R). Sin embargo, ello no excluye su utilidad en la terapia del cáncer y, actualmente, está aprobado para el tratamiento de ciertos tipos de cáncer, como carcinoma de células renales y pacientes con hepatocarcinoma no resecable. Sin embargo, el Sorafenib no se considera efectivo para el tratamiento de la mayoría de los melanomas con la mutación V600E B-‐RAF.
Más recientemente se han desarrollado nuevos inhibidores de la actividad quinasa de B-‐RAF, como Vemurafenib (PLX-‐4032, Plexxikon/Roche), PLX-‐4720 (Plexxikon/Roche), Dabrafenib (GSK2118436, GlaxoSmithKlein, GSK), GDC-‐0879 (Genentech) y Raf-‐265 (Novartis), que están dando excelentes resultados en ensayos clínicos, algunos de los cuales se muestran en la tabla 4 (para una información completa ver (http://clinicaltrials.gov/). Algunos de estos compuestos, como el Vemurafenib, ya ha tenido resultados positivos en ensayos clínicos en pacientes de melanoma y ha sido aprobado por la Food and Drug Administration (FDA) de EEUU para el tratamiento de pacientes con melanoma metastásico irresecable con la mutación V600EB-‐RAF. Sin embargo, se ha observado que algunos de los pacientes tratados con estos inhibidores desarrollan lesiones de la piel asociadas a hiperproliferación, llegando en algunos casos a producirse carcinomas de células escamosas. Estos efectos paradójicos se han interpretado recientemente al observar que estos compuestos, en ciertas condiciones celulares, son capaces de producir la dimerización entre las distintas isoformas de las proteínas RAF. En este sentido, se ha demostrado que en células con K-‐Ras mutado, el inhibidor selectivo de B-‐RAF 885-‐A se une a B-‐RAF normal y promueve la formación de heterodímeros B-‐RAF/C-‐RAF, activándose este último y aumentando la actividad de la vía MEK-‐ERK (figura 9). Asimismo, se ha observado que los mutantes de B-‐RAF catalíticamente inactivos se comportan de la misma manera, uniéndose a C-‐RAF, activando MEK/ERK e induciendo melanomas en ratones transgénicos.
Por otra parte, otros estudios han demostrado que los inhibidores Vemurafenib, GDC-‐0879 y PLX4720 inhiben la actividad de V600EB-‐RAF, pero también activan la vía RAF-‐MEK-‐ERK en células con Ras mutado y B-‐RAF normal, a través de la formación de dímeros B-‐RAF/C-‐RAF, B-‐RAF/A-‐RAF y C-‐RAF/C-‐RAF (figura 9). Por último, también se ha demostrado que estos inhibidores no solo activan MEK-‐ERK en células tumorales con mutaciones en Ras, sino que también lo hacen en células en la que la vía Ras-‐RAF-‐MEK-‐ERK está hiperactivada por otros oncogenes como HER2.
Aunque los mecanismos precisos por los que los distintos inhibidores activan la vía RAF-‐MEK-‐ERK pueden variar, estos estudios demuestran que la inducción de ERK en respuesta a los inhibidores es dependiente de la actividad de Ras y de la transactivación de C-‐RAF producida por la subunidad B-‐RAF unida a la droga. Estos descubrimientos han ayudado a explicar los efectos secundarios producidos por estos inhibidores en pacientes con melanoma y ponen de manifiesto la importancia de seleccionar a los pacientes para una terapia determinada en función de las mutaciones que contengan sus células tumorales.

18
Tabla 4. Inhibidores de RAF utilizados en ensayos clínicos
Figura 9. Efectos de los inhibidores de V600EB-‐RAF sobre distintos tipos celulares. A) En células con el mutante V600EB-‐RAF la hiperactivación de la vía RAF-‐MEK-‐ERK promueve el crecimiento tumoral. B) En células tumorales con V600EB-‐RAF el tratamiento con inhibidores selectivos del mutante (esferas verdes) inhibe la vía y revierte el crecimiento tumoral. C) Sin embargo, en células con WTB-‐RAF y mutaciones en la proteína Ras, los inhibidores producen la formación de dímeros B-‐RAF/B-‐RAF o C-‐RAF/C-‐RAF que, junto con Ras mutado, causan una señalización excesiva que provoca efectos secundarios como el carcinoma de células escamosas.
INHIBIDORES DE RAF Inhibidor Diana Tipo cáncer Ensayo
clínico Compañía
Sorafenib (BAY-‐43-‐9006, Nexavar®)
RAF, VEGFR2, VEGFR3, PDGFR, c-‐Kit, c-‐Fms, Flt-‐3
carcinoma de células renales, HCC, melanoma, leucemias
Fase I, II, III Bayer
AAL-‐881 RAF tiroides, glioma Preclínico Novartis LBT-‐613 RAF glioma, tiroides Preclínico Novartis RAF265 B-‐RAF, C-‐RAF, A-‐RAF,
V600EB-‐RAF, VEGFR-‐2 melanoma Fase I Novartis
XL281 B-‐RAF, C-‐RAF, V600EB-‐RAF
colorrectal, tiroides papilar, ovario, próstata, melanoma
Fase I Exelixis/Bristol Myers Squibb
SB-‐590885 RAF, V600EB-‐RAF melanoma Preclínico GlaxoSmithKline PLX-‐4720 RAF, V600EB-‐RAF melanoma Preclínico Plexxikon/Roche PLX-‐4032 (Vemurafenib)
RAF, V600EB-‐RAF melanoma, tiroides, ovario, tumores sólidos
Fase I Plexxikon/Roche
L-‐779,450 RAF leucemia Preclínico Merck GW5074 C-‐RAF melanoma,
glioblastoma Preclínico GlaxoSmithKline
SB-‐699393 RAF Preclínico GlaxoSmithKline

19
SEÑALIZACIÓN MEDIADA POR LA VÍA DE PI3K-‐AKT-‐mTOR PI3K y AKT PI3K
Las proteínas PI3K (PhosphatidylInositol-‐3-‐Kinase) forman una familia de enzimas capaces de fosforilar el grupo 3-‐OH del anillo de inositol del fosfoinositol (PI) y generar fosfatidilinositoles PIP3, a los que debe su nombre. La familia de PI3K está constituida por tres clases diferentes: la clase I (subdividida a su vez en IA y IB), la clase II y la clase III. La clasificación está basada en su estructura primaria, su regulación y su diferente especificidad in vitro por los sustratos lipídicos. De todas, sólo la clase IA ha sido vinculada a la carcinogénesis.
Las proteínas PI3K son heterodímeros formados por una subunidad reguladora y otra catalítica. La clase IA tiene cinco isoformas de la subunidad reguladora, de las cuales p85 es la más estudiada. Ésta contiene dos dominios SH2, a través de los cuales se une a residuos de fosfotirosina de RTKs y un dominio de unión a la subunidad catalítica p110. La subunidad catalítica p110 tiene tres isoformas diferentes con la misma estructura básica, que incluye varios dominios responsables de la interacción con p85 y Ras, un dominio C2, importante en el anclaje a la membrana, y el dominio catalítico quinasa.
El complejo p85/p110, en ausencia de señales, se encuentra inactivo en el citoplasma. Su activación comienza por la unión del ligando apropiado a un RTK, que da lugar a la transfosforilación de su dominio citoplásmico. Seguidamente, el dímero p85/p110 se une al receptor a través del dominio SH2 de p85 y esta subunidad sufre cambios conformacionales que activan la subunidad p110. Así, p110 activa se sitúa junto a sus sustratos lipídicos en la membrana celular (figura 10). Los receptores RTKs también pueden activar a PI3K de forma indirecta a través de Ras, la cual se une directamente a la subunidad catalítica p110, activándola, sin necesidad de la participación de la subunidad reguladora p85 (figura 4).
Figura 10. Mecanismo de activación de PI3K y AKT. Tras la activación del RTK por unión de su sustrato, se produce la transfosforilación en residuos de tirosina de los
dominios citosólicos del mismo, la subunidad p85 de PI3K se une a dichos residuos de forma que se produce un cambio conformacional que activa la subunidad p110. La subunidad catalítica p110 transfiere un grupo fosfato a los PIP2 situados en la membrana y forma PIP3. La fosfatasa PTEN revierte este proceso. Una vez que PI3K genera PIP3, AKT se une a ellos a través de su dominio PH. A continuación es fosforilada en los residuos S473 y T308 por mTORC2 y PDK1 respectivamente, de forma que queda completamente activa.

20
El principal sustrato in vivo de PI3K IA es el PIP2, que es fosforilado y convertido en PIP3. Estos PIP3 actúan como segundos mensajeros y activan vías por debajo de ésta, como AKT y otras proteínas. En células de mamífero sin estimular los niveles de PIP3 son casi indetectables, debido a la fina regulación de PI3K y, sobre todo, a la acción de la fosfatasa PTEN. Esta fosfatasa se encarga de eliminar los grupos fosfato de los PIP3 y así regular negativamente los efectos de PI3K impidiendo la activación de AKT (figura 10).
AKT
De todas las proteínas activadas por PI3K, la mejor caracterizada y más directamente implicada en cáncer es AKT. AKT es una familia de proteínas formada por 3 miembros: AKT1, AKT2 y AKT3, también conocidos como PKBα, PKBβ y PKBγ (Protein Kinase B α, β y γ). Existen algunas características específicas de cada isoforma, por ejemplo AKT1 está principalmente implicada en supervivencia, AKT2 en el transporte de glucosa y el papel de AKT3 es menos claro, pero parece que regula el crecimiento celular en cerebro.
La regulación de la actividad de AKT comienza por su translocación desde el citoplasma a la membrana plasmática. Los PIP3 generados por PI3K se sitúan en la cara interna de la membrana plasmática y AKT se une de forma directa a éstos, a través de su dominio PH, situándose en la membrana. Este evento es crítico para la activación de AKT, ya que su relocalización en la membrana le confiere proximidad a las proteínas quinasas encargadas de fosforilarla y activarla, además de producir un cambio conformacional en AKT que facilita su fosforilación (figura 11).
Figura 11. Efectores de AKT.
Una vez que AKT es activada interviene en diversos procesos celulares por fosforilación de numerosas proteínas, activándolas o inhibiéndolas, involucradas en supervivencia, síntesis de proteínas, metabolismo y ciclo celular.
La fosforilación de los residuos T308 y S473 es fundamental para su actividad. La T308 es
fosforilada por la proteína serina/treonina quinasa PDK1 (3-‐Phosphoinositide-‐Dependent Kinase-‐1). Su fosforilación parece ser suficiente para la activación de AKT, sin embargo la activación máxima se alcanza tras la fosforilación de la Ser473 por PDK2. La identidad de PDK2 no está clara, aunque recientemente se ha propuesto que el complejo mTORC2 media la fosforilación de la S473 in vivo (figura 11).

21
Efectos biológicos de la activación de PI3K-‐AKT
Una vez activo, AKT fosforila numerosos sustratos implicados en diversos procesos biológicos. De todos ellos, los más relevantes en carcinogénesis son la supervivencia, la proliferación y el crecimiento celular (figura 11). Proliferación celular y crecimiento celular
La mayoría de estudios sobre las funciones de AKT se han centrado en su papel en supervivencia celular, mientras que la regulación de la proliferación celular está mayoritariamente asociada a la vía Ras/ERK-‐MAPK. Sin embargo, AKT también interviene en el proceso de proliferación regulando la maquinaria del ciclo celular. En ese sentido se ha descrito que el bloqueo de la actividad de PI3K o AKT, farmacológicamente o mediante estrategias genéticas, da lugar a la parada del ciclo celular en varias líneas celulares tumorales. Concretamente, AKT inhibe la proteína GSK-‐3β (Glycogen Synthase Kinase 3β) mediante fosforilación de la misma. GSK-‐3β controla gran número de eventos críticos del ciclo celular a través de la fosforilación inhibitoria de reguladores de éste, como Myc, ciclina D1 y ciclina E. Además, AKT, directamente o a través de los factores de transcripción de la familia FOXO, también fosforila inhibidores del ciclo celular como p21WAF1/CIP1 y p27KIP1, modificando su transcripción, estabilidad y su localización subcelular (figura 11).
Por otra parte, AKT regula el crecimiento celular por inhibición de la actividad de TSC2 (Tuberous Sclerosis 2, también conocida como Tuberin). La proteína TSC2 inhibe la vía Rheb/mTOR, responsable de la activación del crecimiento celular en respuesta a la disponibilidad de nutrientes, por tanto, la inhibición de TSC2 por AKT activa la vía Rheb/mTOR (figura 11). Supervivencia celular
El mecanismo por el que AKT protege a las células de la apoptosis es multifactorial, ya que AKT fosforila directamente a diversos componentes de la maquinaria de la muerte celular, pero también es capaz de regular de forma indirecta la actividad de distintos factores de transcripción de la maquinaria apoptótica (ver tema 5). Así, AKT produce la fosforilación e inactivación de proteínas que median la apoptosis, como Bim, Bad y la caspasa 9, o la activación de proteínas antiapoptóticas de la familia Bcl-‐2. En cuanto a los factores de transcripción, AKT fosforila e inactiva directamente miembros de la familia de los factores de transcripción proapoptóticos FOXO, como FoxO1, secuestrándolos en el citoplasma e impidiendo su translocación al núcleo y, por tanto, evitando la activación de sus genes diana, entre los que se encuentran las proteínas proapoptóticas BIM y ligando FAS. Además, AKT regula a NFκB, que induce supervivencia celular en respuesta a numerosos estímulos apoptóticos. Por último, AKT disminuye la actividad proapoptótica de p53 por fosforilación de su inhibidor MDM2, de forma que éste se transloca al núcleo y se une a p53, dando lugar a su degradación (figura 11)(ver tema 6). mTOR
La proteína mTOR (Mammalian Target Of Rapamycin) se descubrió hace casi 20 años al estudiar el mecanismo de acción de rapamicina. Se trata de una proteína con actividad serina/treonina quinasa que integra las señales de factores de crecimiento, mitógenos e insulina, y actúa como un sensor de los niveles de nutrientes, energía y oxígeno celulares. mTOR es la subunidad catalítica de dos complejos moleculares diferentes de las células, mTORC1 y mTORC2 (mTOR Complex 1 y mTOR Complex 2), cada uno con sus propios sustratos y, por tanto, con diferentes funciones.
El complejo mTORC1 está formado por mTOR, la proteína reguladora LST8 y la proteína RAPTOR (Regulatory Associated Protein of mTOR). mTORC1 regula el crecimiento celular en respuesta a factores de crecimiento y nutrientes, modulando la transcripción, la síntesis de proteínas, el inicio de la traducción, la captación de nutrientes y la autofagia. El complejo mTORC1

22
está regulado negativamente por el complejo TSC1/2 (también conocidas como Hamartin y Tuberin, respectivamente), que inhibe el activador de mTORC1 Rheb. TSC1/2 puede ser inhibido mediante fosforilación directa de AKT, ERK y RSK in vivo. Los dos sustratos de mTORC1 mejor conocidos son las proteínas reguladoras de la traducción p70S6K (también conocida como S6K1, Ribosomal S6 Kinase 1) y el factor de iniciación de la traducción 4EBP1 (Eukaryotic Initiatior Factor 4E (eIF4E) Binding Protein 1) (figura 12).
Figura 12. Vía de señalización PI3K-‐AKT-‐mTOR.
Entre todas las proteínas reguladas por AKT, se encuentra mTOR. AKT fosforila e inhibe a TSC2, un inhibidor de Rheb, que activa el complejo mTORC1. Este complejo proteico activa, entre otros, a la proteína p70S6K, factores de iniciación de la traducción y factores de transcripción, que activan la síntesis de proteínas.
El complejo mTORC2 está formado por mTOR, LST8, RICTOR (Rapamycin-‐Insensitive
Companion of mTOR) y mSin1 (también conocida como MK1 (MAPK-‐Associated Protein 1). La regulación y funciones de este complejo son menos conocidas que las de mTORC1 aunque, como se comentó anteriormente, se ha descrito recientemente que este complejo activa directamente a AKT por fosforilación de la S473 (figura 12).
La sensibilidad a rapamicina de ambos complejos también es diferente, en concreto, la rapamicina inhibe fuertemente mTORC1 por inhibición de su actividad quinasa, mientras que mTORC2 se ve afectado por rapamicina sólo en determinados contextos celulares y tras tratamientos prolongados, ya que en este caso sólo impide la formación de nuevos complejos mTORC2. Por otra parte, los inhibidores de PI3K LY294002 y Wortmanina también son capaces de bloquear la activación de mTOR por AKT. Papel de la vía PI3K-‐AKT-‐mTOR en cáncer
El interés sobre PI3K en cáncer comenzó a mitad de los años 80, al asociarse la actividad enzimática de PI3K con la actividad transformante de oncogenes virales de la famila Src. En los últimos años la vía de señalización de PI3K se ha implicado directamente en carcinogénesis, pues esta

23
vía se encuentra desregulada en el 30 % de los tumores humanos esporádicos, lo que hace pensar que la vía PI3K es una de las vías centrales para el desarrollo y mantenimiento del cáncer. Su activación aberrante confiere un aumento de la proliferación y de la supervivencia celular, así como el mantenimiento del metabolismo celular en condiciones de escasez de factores de crecimiento.
La activación de esta vía de señalización en cáncer se puede producir por varios mecanismos diferentes: amplificaciones de los genes codificantes de los distintos componentes de la cascada, mutaciones puntuales en los mismos o activación por otros oncogenes. En concreto una de las primeras evidencias de la desregulación de PI3K en cáncer humano fue el descubrimiento de la amplificación de genes codificantes para la subunidad catalítica p110α de PI3K (PI3KCA) y para AKT2 en cáncer de ovario, mama y páncreas. Por otra parte, la secuenciación de los exones de los genes de PI3K en tumores humanos ha revelado la existencia de mutaciones somáticas en el gen PIK3CA en el 20-‐30 % de tumores gástricos, de mama, colon, endometrio, tracto urinario y cerebro examinados. Las investigaciones sobre las mutaciones más frecuentes de p110α han mostrado que éstas incrementan la actividad de PI3K y conducen a la transformación celular in vitro e in vivo. También se han encontrado amplificaciones y mutaciones en los genes que codifican para la subunidad reguladora p85α en tumores primarios de colon y ovario, AKT1, AKT2, AKT3 y PDK1 en cáncer de mama, colon, ovario, pulmón y gástricos. Respecto al papel de mTOR, se ha demostrado que la tumorogénesis inducida por la hiperactivación de la vía PI3K/AKT requiere de mTORC1, ya que su inhibición con rapamicina bloquea la transformación de células tumorales. Sin embargo, su uso no ha dado los resultados esperados en pacientes, probablemente porque este fármaco no inhibe el complejo mTORC2, de forma que AKT permanece activa pudiendo activar otros efectores distintos de mTOR.
A pesar de la sorprendente tasa de mutaciones activadoras de p110α, la pérdida de la fosfatasa de lípidos PTEN sigue pareciendo el mecanismo de activación de la vía de PI3K más frecuente en cánceres humanos incluidos próstata, mama y cerebro. De hecho, se piensa que PTEN es el segundo supresor de tumores más comúnmente mutado en humanos, tras p53.
Inhibidores de la vía PI3K-‐AKT-‐mTOR
En la actualidad existen numerosos inhibidores de cada uno de los componentes de esta vía, algunos de los cuales están dando resultados prometedores en ensayos clínicos para el tratamiento de distintos cánceres. A continuación se presenta un breve resumen de los más significativos (tabla 5).
Inhibidores de PI3K Los primeros inhibidores de PI3K que se desarrollaron son el inhibidor de origen fúngico
LY294002 (Lilly) y la Wortmanina. Estos inhibidores bloquean la actividad enzimática de PI3K por diferentes mecanismos; la Wortmanina es un inhibidor irreversible, mientras que el LY294002 es un inhibidor reversible competitivo del ATP clásico. Sin embargo, a pesar de que estos inhibidores no son muy específicos e inhiben otras quinasas, por ejemplo, LY294002 también inhibe mTOR, la caseína quinasa 2 (CK2), la proteína quinasa dependiente de ADN (DNA-‐PK) y otras, así como sus propiedades farmacéuticas desfavorables, ambos inhibidores se han utilizado herramientas importantes en la investigación desde hace más de una década para determinar el papel de PI3K en el cáncer humano.
Actualmente se han desarrollado otros inhibidores más específicos que están siendo utilizados en ensayos clínicos. Entre estos se encuentran la Wortmanina modificada, PX-‐866 (Oncothyreon), GDC-‐0941 (Genentech), IC87114 y CAL-‐101 (Calistoga farmaceuticals Gilead Sciences), XL-‐147 XL 765 (Exelixis/Sanofi-‐Aventis), BKM120 y BYL719 (Novartis), ver tabla 5 de sus uso en ensayos clínicos, (ver http://clinicaltrials.gov/).

24
INHIBIDORES DE PI3K/AKT/mTOR Inhibidor Diana Tipo cáncer Ensayo
clínico Compañía
UCN-‐01 PDK-‐1, Chk1, isoformas PKC
leucemia, linfoma, ovario, cavidad peritoneal, trompa falopio
Fase I, II Kyowa Hakko Kogyo Co., Ltd./Keryx Biopharmaceuticals
(NVP)-‐BAG956 PDK, p110 PI3Ks (exc. isoforma β)
leucemia, melanoma Preclínico Novartis
Celecoxib (Celebrex®)
PDK-‐1, COX-‐2 pulmón, próstata, H&N Fase I, II Pfizer
OSU-‐03012
PDK-‐1 próstata, glioma, leucemia, HCC, mama
Preclínico Arno Therapeutics/ Ohio State University
BX-‐795
PDK-‐1, ERK8, TBK1, IKK-‐ε
mama, próstata, colon, melanoma, páncreas, cervical
Preclínico Berlex/Bayer
BX-‐912 PDK-‐1 mama, próstata, colon, melanoma, páncreas, cervical
Preclínico Berlex/Bayer
BX-‐320
PDK-‐1 mama, próstata, colon, melanoma, páncreas, cervical
Preclínico Berlex/Bayer
AR-‐12 PDK-‐1, PI3K, Akt
mama, colon, pulmón, próstata, linfoma
Fase I Arno Therapeutics
KP372-‐1 PDK-‐1, Akt, Flt3 AML, tiroides, glioblastoma Preclínico Kinetek Pharmaceutic LY294002 PI3K, otras
quinasas cánceres hematológicos y cánceres sólidos avanzados
Preclínico Lilly
PWT-‐458 PI3K NSCLC, glioblastoma, renal Preclínico Wyeth/Pfizer PX-‐866
PI3K glioma, mama, colon, próstata, NSCLC, tumores sólidos pancreáticos avanzados
Fase I Oncothyreon Inc.
CAL-‐101 PI3K (p110δ) leucemia, linfoma, mieloma Fase I Calistoga Pharmaceuticals
XL-‐147 PI3Ks NSCLC, tumores sólidos Fase I Exelixis/Sanofi Aventis
ZSTK474 PI3Ks NSCLC, melanoma, ovario, próstata
Preclínico Zenyaku Kogyo Co. Ltd
GDC-‐0941 PI3K (p110α), Flt3 linfoma, NSCLC, mama, tumores sólidos
Fase I PIramed Pharma/ Roche/Genetech
(NVP)-‐BEZ235 PI3K, mTOR mama, glioma, melanoma, páncreas
Fase I, II Novartis
AS-‐252424 PI3Ks (p110γ) Preclínico Merck Serono TGX-‐221
PI3K (p110β) No evaluado para su uso en tratamiento de cáncer
Preclínico Alexis/Enzo Life Sciences, Inc.
XL-‐765 PI3K, mTOR glioma, NSCLC Fase I Exelixis/Sanofi-‐Aventis
Wortmannin PI3K, mTOR, DNA-‐PK, MAPK
cánceres hematológicos y cánceres sólidos avanzados
Preclínico
PI-‐103 p110 PI3K, mTORC1/2, DNA -‐PK
glioma, próstata, colon, NSCLC
Preclínico Piramed Pharma/ Roche
Perifosine (KRX-‐0401)
Akt, MEK 1/2, ERK 1/2, JNK
mieloma múltiple, leucemia, NSCLC, tumores sólidos avanzados
Fase I, II Æterna Zentaris Inc./Keryx Biopharmaceuticals
Triciribine (API-‐2)
Akt 1, 2, 3
AML, cánceres hematológicos avanzados
Fase I VioQuest Pharmaceuticals

25
Tabla 5. Inhibidores de la vía PI3K/AKT/mTOR utilizados en ensayos clínicos
Inhibidores de AKT Durante muchos años se han hecho muchos intentos para desarrollar inhibidores de AKT,
pero en la mayoría de los casos éstos carecen de especificidad o tienen efectos secundarios perjudiciales, probablemente debido a las numerosas funciones críticas que tiene AKT en la fisiología normal. Entre éstos se encuentran la Triciribina (API-‐2 o VQD-‐002, VioQuest Pharmaceuticals), MK-‐2206 (Merck), GSK2141795 y GSK690693 (GlaxoSmithKlein), A-‐443654 (Abbott Laboratories), KP372-‐1 (QLT Inc),y Perifosina (KRX-‐0401, Aeterna Zentaris/Keryx Pharmaceuticals Inc), entre otros (tabla 5). Un inhibidor dual de la proteína quinasa C-‐β (PKC-‐β) y AKT es el Enzasturin (LY317615), desarrollado por Lilly. Este inhibidor está siendo usado en aproximadamente 48 ensayos clínicos bien solo o en combinación con otros agentes en pacientes con diversos tipos de cáncer, entre los que se incluyen cerebro, NSCLC, CRC, así como otros tipos de cáncer (ver http://clinicaltrials.gov/).
Inhibidores mTORC1 La Rapamicina (Pfizer) es un inhibidor de mTORC1 que fue aprobado por la FDA en el año
1999 para prevenir el rechazo en el trasplante de órganos. La Rapamicina y otras rapamicinas modificadas (rapálogos) actúan como inhibidores alostéricos de mTORC1 y no afectan directamente a la actividad catalítica de mTOR. La Rapamicina se está ensayando en pacientes con distintos tipos de cáncer: cerebro, mama, hepático, leucemia, linfoma, NSCLC, pancreático, próstata y renal. Los rapálogos Torisel® (CCI-‐779, Pfizer) y Afinitor® (Novartis) fueron aprobados en 2007 y 2009, respectivamente, para tratar pacientes con carcinoma renal, linfoma de las células del manto, astrocitomas y tumores pancreáticos neuroendocrinos (tabla 5) (ver http://clinicaltrials.gov/).
Inhibidores de la actividad quinasa de mTOR También se han desarrollado inhibidores de la actividad quinasa de mTOR, los cuales tienen
ventajas sobre la Rapamicina y rapálogos porque inhiben mTORC1 y mTORC2, mientras que los anteriores sólo inhiben mTORC1. Además, estos inhibidores de las quinasas mTOR también inhiben la
INHIBIDORES DE PI3K/AKT/mTOR (Continuación) Inhibidor Diana Tipo cáncer Ensayo
clínico Compañía
SR13668 Akt mama, próstata, ovario Preclínico SRI International AR-‐67 (DB-‐67) Akt tumores sólidos avanzados Fase I, II Arno Therapeutics AR-‐42 Akt Preclínico Arno Therapeutics GSK690693 Akt1, 2, 3 leucemia, linfoma Fase I GlaxoSmithKline KP372-‐1 Akt, PDK-‐1, Flt3
leucemia, tiroides, H&N, glioma
Preclínico QLT Inc
VQD-‐002 (API-‐2) Akt NSCLC, leucemia, linfoma, próstata
Fase I, II VioQuest Pharmaceuticals
A-‐443654 Akt cánceres hematológicos y sólidos
Preclínico Abbott Laboratories
MK-‐2206 Akt tumores sólidos Fase I Merck Rapamycin (Sirolimus)
mTORC1
cánceres hematológicos y cánceres sólidos avanzados , HIV
Fase I, II Wyeth/Pfizer
CCI-‐779 (Torisel®, Temsirolimus)
mTORC1 leucemia, linfoma, NSCLC, próstata, colorrectal, renal
Fase I, II Wyeth/Pfizer
RAD001 (Afinitor®, Everolimus)
mTORC1, mTORC2
cervical, renal, HCC, leucemia, linfoma
Fase I, II Novartis
AP-‐23573 (Ridaforolimus, Deforolimus)
mTORC1 cánceres hematológicos avanzados, próstata, endometrio
Fase I, II Ariad/Merck

26
vía de activación de AKT por mTORC2. Entre éstos se encuentran: OSI-‐027 (Astellas Pharma Inc), PP-‐242 e INK-‐128 (Intellikine), OXA-‐01 (OSI Pharmaceuticals) y TORKi CC223 (Celgene) (tabla 6) (ver http://clinicaltrials.gov/). INHIBIDORES DEL SITIO ACTIVO DE mTOR Inhibidor Diana Tipo cáncer Ensayo
clínico Compañía
AZD-‐8055 mTORC1/mTORC2 tumores sólidos avanzados, linfomas, HCC Fase I, II AstraZeneca OSI-‐027 mTORC1/mTORC2 tumores sólidos avanzados, linfomas Fase I OSI
Pharmaceuticals INK-‐128 mTORC1/mTORC2
cánceres avanzados, mieloma múltiple, macroglobulinemia de Waldenstrom
Fase I Intellikine
PP-‐242 mTORC1/mTORC2 Fase I UCSF
Tabla 6. Inhibidores del sitio catalítico de mTOR utilizados en ensayos clínicos
Inhibidores duales PI3K/mTOR Los sitios catalíticos de PI3K y mTOR comparten un alto grado de homología de secuencia.
Esta característica ha permitido que se sinteticen inhibidores competitivos del ATP que se unen al sitio catalítico de estas quinasas. Se han estudiado varios inhibidores duales de PI3K y mTOR en preclínica que han mostrado una fuerte reducción de la tasa de proliferación y mayor citotoxicidad en células leucémicas y en modelos in vivo de cáncer humano xenoinjertado en ratones, que cualquiera de los inhibidores de PI3K o los inhibidores alostéricos de mTOR, rapamicina o rapálogos. Su uso para el tratamiento con pacientes puede ser beneficioso, ya que se puede reducir la toxicidad y los efectos secundarios que pueden producir el uso de varios inhibidores conjuntos para estas proteinas.
El PI-‐103, desarrollado en la UCSF en el año 2006, fue el primer inhibidor dual de PI3K/mTOR. Además otros están en ensayos clínicos, como monoterapia o en combinación con otros inhibidores. Entre estos están NVP-‐BEZ235 (Novartis), PKI-‐587 (Pfizer), XL-‐147 y XL-‐765 (Exelixis/Sanofi-‐Aventis), GNE-‐477 (Genentech) y GDC-‐0980 (GlaxoSmithKlein) (ver http://clinicaltrials.gov/). Cooperación de las vías Ras-‐RAF-‐MEK-‐ERK y PI3K-‐AKT-‐mTOR en cáncer
La vía de PI3K puede ser activada por Ras y por algunos RTKs, como EGFR, HER2 y PDGFR, que
están activados en muchos cánceres. Es frecuente encontrar mutaciones activadoras de PIK3CA junto con otras alteraciones genéticas que activan la vía de PI3K, como son la pérdida de PTEN (en mama, endometrio y colon), mutaciones activadoras de Ras (colon) y amplificaciones de HER2 (mama). Aunque las mutaciones en la familia de Ras y la pérdida de PTEN son mutuamente excluyentes en algunos tumores, en melanoma sí se han encontrado mutaciones simultáneas en RAF y PTEN.
Muchos estudios han demostrado que las vías Ras/RAF/MEK/ERK y PI3K/AKT/mTOR cooperan estrechamente en la transducción de señales de supervivencia. Así, se ha demostrado que AKT3, la principal isoforma de AKT activada en el melanoma, coopera con B-‐RAF en la protección de la apoptosis. En este sentido, se ha observado que la activación de la vía PI3K/AKT media la resistencia a la apoptosis inducida por la inhibición de B-‐RAF con los inhibidores Sorafenib, PLX4720 o SB 590885 y, de hecho, se ha observado que la inhibición de B-‐RAF en células tumorales neuroendocrinas y de melanoma aumenta la activación de AKT. Estos hechos indican la existencia de mecanismos compensatorios por los que se activa una vía cuando la otra es inhibida. Por lo tanto, la inhibición dual de las vías Ras/RAF/MEK/ERK y PI3K/AKT/mTOR tiene razón de ser en diferentes tipos de cáncer, ya que existen evidencias que muestran que la inhibición simultánea de ambas rutas de señalización es más eficaz in vitro y en modelos animales, que la inhibición de cada una de estas rutas individualmente. Por ello, en la actualidad hay muchos ensayos clínicos en marcha con inhibición doble.

27
VÍA DE SEÑALIZACIÓN JAK-‐STAT
La vía de señalización JAK-‐STAT desempeña un papel esencial en la transmisión de las acciones de citoquinas y factores de crecimiento, y comparte en su estrategia de señalización muchos elementos con las vías de los RTKs. Esta cascada regula muchos aspectos críticos del crecimiento, diferenciación y supervivencia celular, y su desregulación es frecuente en muchos neoplasmas mieloproliferativos, que pueden progresar y producir los linfomas de células T, como la leucemia mieloide aguda (figura 13).
Figura 13. Vías de señalización activadas por JAKs. La unión de un ligando a los receptores de citoquinas induce su dimerización. Las proteínas JAK se unen al
receptor y se transfosforilan. Una vez activas fosforilan los factores de transcripción STATs. Los STATs activados dimerizan y translocan al núcleo donde activan o reprimen la expresión de genes diana. Además, JAKs también fosforilan al receptor y crean otros sitios de unión a STATs. Por otra parte, el receptor de citoquinas también puede activar la vía de ERK y la vía PI3K-‐AKT
Los receptores de citoquinas, tales como el de la eritropoyetina (Epo), trombopoyetina (TPO),
factor estimulador de la formación de colonias de granulocitos (G-‐CSF), los interferones tipo I y tipo II (IFN) y casi todas las interleuquinas (IL) pertenecen a la superfamilia de receptores de citoquinas. Estos receptores no presentan ninguna actividad catalítica intrínseca en sus dominios citosólicos y se unen, a través ellos, a uno o varios miembros de la familia de proteínas quinasas citosólicas Janus (JAK). La familia de las tirosinas quinasas citosólicas JAK tiene cuatro miembros, JAK1, JAK2, JAK3 y Tyk2. La unión de una citoquina al dominio extracelular de su receptor específico provoca un cambio conformacional que promueve la formación de dímeros u oligómeros de monómeros activados. Esta conformación se transmite al dominio citosólico del receptor, donde se unen las proteínas JAK. El resultado de esta unión es la activación de JAK, ya sea a través de su autofosforilación y/o transfosforilación por otro miembro de la familia. Las JAKs activadas fosforilan entonces al receptor en residuos de tirosina que sirven de sitios de unión para proteínas con dominios SH2. Los sustratos más destacados son los miembros de la familia de los factores de transcripción Transductores y Activadores de la Transcripción (STATs). Hasta el momento se han descubierto 7 genes que codifican

28
para distintas STATs. Estos factores de transcripción, una vez unidos al receptor son fosforilados bien por el propio receptor, o bien por las proteínas JAK o por Src. La fosforilación de las STATs permite su homodimerización o heterodimerización con otro miembro de la familia y, una vez dimerizados, se translocan al núcleo, donde regulan la expresión de genes específicos, entre los que se encuentran Myc y otras citoquinas (figura 13).
Sin embargo, la fosforilación de los receptores de citoquinas permite, además, la interacción de éstos con otras proteínas que contienen dominios SH2, tales como las quinasas Src, proteínas fosfatasas y otras proteínas adaptadoras de señalización como Shc, Grb2 y PI3K, que activan varias vías de señalización (figura 13). Un ejemplo de este tipo de activación múltiple consiste en la fosforilación de STAT3 en células T, que controla la progresión de la fase G1 a S, la expansión clonal y la diferenciación funcional. La interacción de IL-‐2 con su receptor supone la activación de JAK1 y JAK3 y la consecuente fosforilación de STAT3 y, por otra parte, favorece también su interacción con Shc, que da lugar a la activación de Ras y la vía ERK-‐MAPK, que también fosforila a STAT3. Papel de JAK/STAT en cáncer
En cuanto al papel de la vía JAK/STAT en cáncer, el gen más comúnmente alterado es el que codifica para JAK2. Así, se han encontrado dos tipos de alteraciones, translocaciones cromosómicas del locus y mutaciones puntuales del gen.
En cuanto a las translocaciones cromosómicas, se han observado reordenamientos del gen JAK2, que dan lugar a proteínas con una actividad tirosina quinasa constitutivamente activada con propiedades oncogénicas. Estas translocaciones producen unas gran variedad de transcritos quiméricos de JAK2, con la consiguiente expresión de sus proteínas de fusión que, a menudo, conducen al desarrollo de leucemias de orígenes mieloides y linfoides.
En cuanto a las mutaciones puntuales activadoras del gen JAK2, su relevancia clínica se demostró en 2005 cuando se descubrió la presencia de la mutación V617F (sustitución G1849T) en pacientes con neoplasias mieloproliferativas (MPN). Las mutaciones en JAK2 son los principales eventos moleculares en neoplasias hematológicas humanas y, en concreto, la mutación V617F se encuentra en más del 90% de pacientes con policitemia vera y en una gran proporción de pacientes con trombocitemia, mielofibrosis idiopática y leucemia linfoblástica aguda (LLA).
Además, también se han encontrado mutaciones somáticas en JAK1 y JAK3 en pacientes con leucemia aguda megacarioblástica y en aproximadamente un 20% de leucemia linfoblástica aguda de células T.
En noviembre de 2011 la FDA aprobó Ruxolitinib® (Novartis), inhibidor de JAK1 y JAK2, para el tratamiento de pacientes con mielofibrosis con riesgo medio-‐alto. Además, actualmente se están llevando a cabo diferentes ensayos clínicos con este inhibidor para otros tipos de tumores, entre los que se incluyen linfomas y tumores de próstata, páncreas y mama (ver http://clinicaltrials.gov/).

29
SEÑALIZACIÓN CELULAR POR LA VÍA DE NFκB
El factor de transcripción NFκB fue descubierto en 1986 por Baltimore y colaboradores como un factor del núcleo de las células B que se une al potenciador de la cadena ligera kappa de las inmunoglobulinas. Posteriormente, se ha comprobado que NFκB se expresa de manera ubicua en el citoplasma de todos los tipos celulares. Este factor se encuentra en estado basal en el citosol y es capaz de translocar al núcleo sólo cuando está activo, donde regula la expresión de más de 200 genes relacionados con el sistema inmune, el crecimiento, la inflamación y la apoptosis.
NFκB representa a un grupo de proteínas relacionadas estructuralmente y conservadas evolutivamente con 5 miembros en mamíferos: Rel, p65/RelA, RelB, p50/NFκB1 y p52/NFκB2. En células sin estimular, el complejo NFκB está secuestrado en el citoplasma por unión a miembros de la familia de inhibidores IκB. Como consecuencia de un estímulo apropiado (citoquinas, lipopolisacáridos, lípidos oxidados, bacterias, virus o mitógenos), se produce la activación del complejo de quinasas de IκB (IKK-‐ IκB Kinases). Este complejo está formado por 3 subunidades, las catalíticas IKK1 y 2 (también conocidas como IKKα y β), que tienen actividad quinasa y fosforilan a IκB, y la reguladora IKK3, IKKγ o NEMO (NFκB Esencial Modulator). La fosforilación de IκB favorece su ubiquitinación y su degradación mediada por el proteasoma, lo que produce la liberación del heterodímero NFκB seguida de su rápida translocación al núcleo. Una vez en el núcleo, NFκB se une a secuencias específicas del ADN de genes diana (figura 14).
Figura 14. Vía de activación de NFκB. Las proteínas IKKs activadas por señales procedentes de distintos receptores extracelulares fosforilan a IκB, lo que induce su degradación por el proteasoma, dejando libre el complejo p50-‐p65/RelA (NFκB). Una vez libre, NFκB se transloca al núcleo donde se unen a secuencias -‐κB de sus genes diana, regulando su expresión.

30
Se han descrito una gran cantidad de inductores e interacciones con proteínas que afectan a la regulación de la vía de NFκB; sin embargo, como se ha comentado anteriormente, los reguladores de la actividad de NFκB mejor conocidos son aquellos que actúan a través de los receptores de la familia de TNFα, receptores similares a Toll (TLRs) y el receptor de IL-‐1. Además, también están implicadas en su activación otras vías de transducción de señales, por ejemplo, las proteínas MAPK como ERK o p38 MAPK y proteínas de la familia de Rho. También se ha observado que la sobreexpresión de la proteína AKT o de la subunidad catalítica p110 de la PI3K regula la activación de NFκB.
NFκB regula la transcripción de unos 200 genes. Entre ellos se incluyen genes de proteínas que controlan la apoptosis, la adhesión, la proliferación, respuestas del sistema inmune, la inflamación, respuestas al estrés celular y el remodelado tisular. En concreto, NFκB regula la expresión de numerosos componentes del sistema inmune, que incluyen citoquinas proinflamatorias, quimioquinas, moléculas de adhesión y enzimas inducibles como la ciclooxigenasa-‐2 o la óxido nítrico sintasa, que regulan la respuesta inmune innata, así como proteínas que regulan la respuesta inmune específica, como el complejo mayor de histocompatibilidad y citoquinas como las interleukinas IL-‐2, IL-‐12 y el interferón-‐γ, que controlan la proliferación y diferenciación de los linfocitos. Por otra parte, NFκB participa también en la regulación de la expresión de una variedad de proteínas que inhiben la apoptosis y promueven la supervivencia y proliferación celular como c-‐IAP-‐1, c-‐IAP-‐2, XIAP, el ligando de Fas, así como miembros antiapoptóticos de la familia de Bcl-‐2, como Bcl-‐XL y FLIP, por lo que tiene un papel preventivo de la apoptosis (ver tema 5).
En condiciones fisiológicas la activación de NFκB es un fenómeno transitorio y la expresión de estos genes está finamente coordinada por otras vías de señalización, por tanto, el resultado de la activación de NFκB depende de la naturaleza y el contexto celular de su inducción. Así, una activación inapropiada de NFκB puede contribuir en patologías como inflamación y carcinogénesis.
Participación de la vía de NFκB en cáncer
Aunque no se han encontrado mutaciones activadoras en ninguno de los componentes de la vía de señalización de NFκB en cáncer, aparte de alguna mutación esporádica de p65 en algunos linfomas malignos, sí se ha observado un aumento de la actividad de la vía en melanomas, cáncer de mama, pulmón, colon, páncreas, mielomas, linfomas y distintos tipos de leucemias. La presencia de NFκB activo en cáncer da lugar a la alteración de la proliferación, supervivencia, migración y angiogénesis (figura 15). La supresión de NFκB en distintos tipos de células tumorales inhibe la proliferación y da lugar a apoptosis. La inhibición del crecimiento de células tumorales por inhibición de NFκB se ha observado en pulmón, melanoma, etc. Además, mediante estudios genéticos en ratones, se ha demostrado que la eliminación de IKKβ disminuye la progresión de cáncer de colon y mielomas, entre otros, y que el cáncer de mama inducido por HER2 requiere de IKKα, ya que su eliminación retarda el crecimiento de estos tumores.
Este factor de transcripción participa en carcinogénesis porque entre sus genes diana se encuentran los miembros del ciclo celular ciclina D1, ciclina E, CDK2 y el proto-‐oncogen Myc y se ha visto que la supresión de NFκB en determinadas células tumorales causa la parada de este ciclo. Además, se ha observado que en fibroblastos transformados por Ras o RAF oncogénicos hay un aumento de la expresión de genes inducidos por NFκB. Por otra parte, el papel principal de NFκB en el desarrollo del cáncer parece ser su función antiapoptótica. Como se ha comentado anteriormente, NFκB participa en la regulación de la expresión de numerosas proteínas antiapoptóticas y se ha demostrado que la resistencia a la apoptosis de numerosas líneas celulares tumorales se debe a un aumento de la actividad de NFκB, ya que cuando ésta se inhibe, se produce un aumento de la muerte celular. Por último, NFκB no solo está implicado en la iniciación del cáncer, ya que se ha observado que su activación también puede mediar la invasión celular y promover metástasis. NFκB induce la transición epitelio-‐mesenquima (EMT) a través de la regulación de la transcripción de E-‐cadherina, las metaloproteasas MMP2 y MMP9, y las moléculas de adhesión ICAM-‐1, VCAM-‐1 y

31
ELAM-‐1. También puede inducir angiogénesis, a través de la regulación del factor de crecimiento de células endoteliales (VEGF) y otros reguladores angiogénicos como CXCL1 e IL-‐8. Además, recientemente se ha observado una relación directa entre la activación de NFκB y el potencial metastático de las células tumorales prostáticas, a través de la inhibición del gen supresor de metástasis Maspin. Por último, la activación de NFκB aumenta la expresión de citoquinas inflamatorias que favorecen la inflamación del microambiente tumoral, proceso que favorece el desarrollo de los tumores (figura 15).
En cuanto al uso de NFκB como diana terapéutica, no existen ensayos clínicos con inhibidores específicos de componentes de esta vía. En estudios in vitro se han ensayado dos compuestos, BMS-‐345541, que inhibe la subunidad catalítica de IKK, y el péptido NBD (Nemo binding domain), con resultados prometedores. Por el momento sólo se están empleando los compuestos Bortezomib, un inhibidor del proteasoma que estabiliza IκB, y la curcumina, especie extraída de la planta Curcuma longa y componente del curry, que interfiere en la actividad de NFκB, como inhibidores indirectos en ensayos clínicos de melanoma, cáncer de páncreas, colon, mieloma, linfoma de células T, osteosarcoma y cabeza y cuello (ver http://clinicaltrials.gov/).
Figura 15. Genes regulados por NFκB que pueden participar en cáncer.
La activación de NFκB puede ser regulada por la MAPKs ERK y p38, o por la vía PI3K-‐AKT. Una vez activa en el núcleo NFκB regula la expresión de una gran cantidad de genes implicados en distintos procesos celulares que pueden participar en la tumorogénesis.

32
VÍA DE SEÑALIZACIÓN DE Wnt
La cascada de señalización Wnt desempeña diversas funciones que incluyen el mantenimiento de células troncales, proliferación, diferenciación y apoptosis, por lo que tiene un papel fundamental en la embriogénesis y el cáncer. El término “Wnt” deriva de la fusión de los nombres del gen de polaridad de Drosophila “wingless” (sin alas) y del proto-‐oncogen Int-‐1 del virus de tumor mamario de ratón (MMTV).
Los ligandos secretados Wnt actúan como factores de crecimiento pleiotrópicos en al menos tres cascadas de señalización intracelular diferentes: la vía canónica Wnt/β-‐catenina, la vía Wnt/Ca2+
y la vía de polaridad celular planar Wnt/PCP. La vía mejor caracterizada es la cascada de señalización de Wnt/β-‐catenina, cuya desregulación está implicada en una variedad de cánceres humanos y otras enfermedades.
Esta vía canónica está regulada principalmente por los niveles de β-‐catenina en el citosol, los cuales son bajos debido a la existencia de un complejo de degradación. Este complejo contiene la proteína supresora de tumores adenomatosis poliposis coli (APC), dos quinasas, la caseína quinasa 1 (CK1) y la glucógeno sintasa quinasa 3β (GSK-‐3β), y Axin2, que mantiene el complejo unido. En ausencia de ligandos Wnt, el complejo receptor de membrana, formado por frizzled (Fzd) y el receptor de lipoproteínas de baja densidad relacionado con la proteína 5/6 (LRP5/6) no está activado, y las CK1 y GSK-‐3β fosforilan a la β-‐catenina, marcándola para su degradación vía proteasoma (figura 16). Tras la unión de ligandos a los receptores de Wnt, el complejo de destrucción se desestabiliza, lo que impide la degradación de β-‐catenina. Esta se acumula en el citosol y, posteriormente, sufre una translocación al núcleo. Allí, desplaza al correpresor Groucho, recluta a la proteína histona acetilasa de unión CREB (CBP/p300) y otros coactivadores, como Pygopus (PYG) y BCL9. Posteriormente, se asocia con los factores de la familia LEF/TCF y activa la expresión de genes implicados en proliferación y diferenciación celular, entre los que se incluyen la ciclina D1 y Myc (figura 16).
Figura 16. Señalización por la vía canónica de Wnt. A) En ausencia de estimulación por Wnt, los receptores Fdz y Lpr5/6 están inactivos, en el citosol los niveles de β-‐catenina son mínimos debido a que se mantiene unida a un complejo de destrucción compuesto de APC, Axin2, GSK3 y CK1. GSK3 y CK1 fosforilan a β-‐catenina, que se une a β-‐TrCP, que favorece su degradación por el proteasoma. En el núcleo, la expresión de muchos genes está inhibida por la presencia de represores como Groucho. B) Tras la estimulación de los receptores por Wnt, el complejo de destrucción se desestabiliza y β-‐catenina se acumula en el núcleo, donde se une a los factores de transcripción Tcf/LEF y coactivadores como CBP, para activar la transcripción de genes diana.
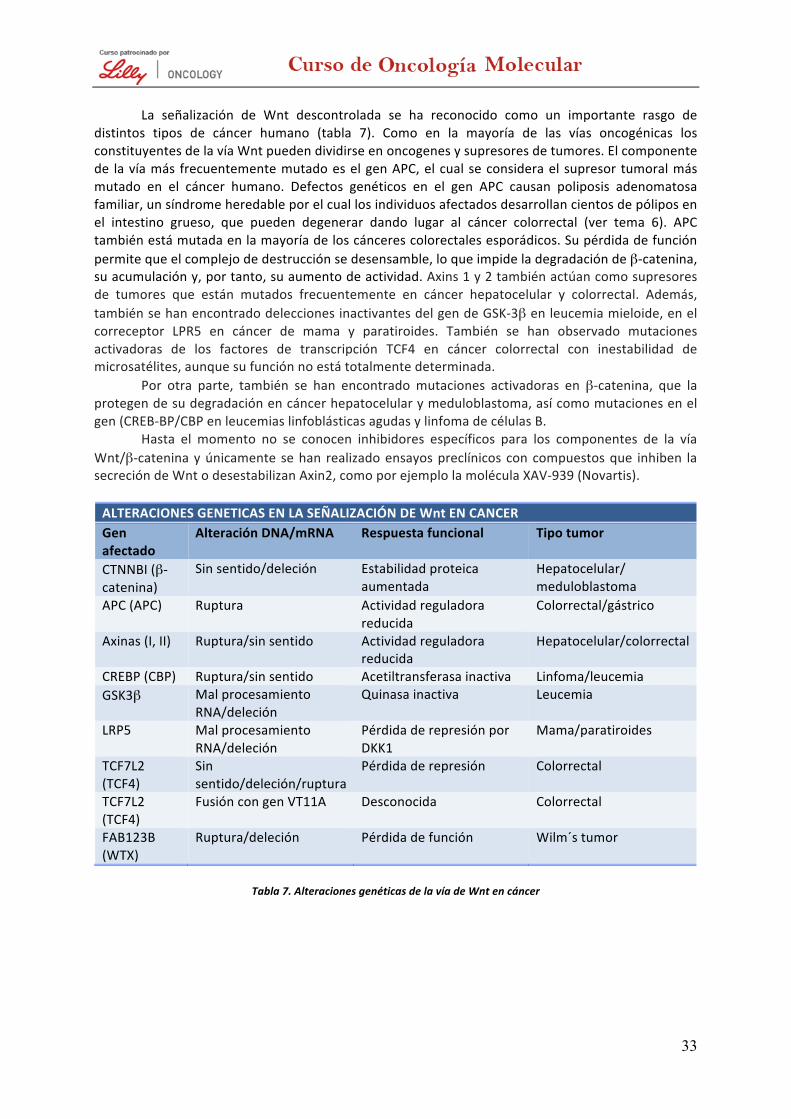
33
La señalización de Wnt descontrolada se ha reconocido como un importante rasgo de distintos tipos de cáncer humano (tabla 7). Como en la mayoría de las vías oncogénicas los constituyentes de la vía Wnt pueden dividirse en oncogenes y supresores de tumores. El componente de la vía más frecuentemente mutado es el gen APC, el cual se considera el supresor tumoral más mutado en el cáncer humano. Defectos genéticos en el gen APC causan poliposis adenomatosa familiar, un síndrome heredable por el cual los individuos afectados desarrollan cientos de pólipos en el intestino grueso, que pueden degenerar dando lugar al cáncer colorrectal (ver tema 6). APC también está mutada en la mayoría de los cánceres colorectales esporádicos. Su pérdida de función permite que el complejo de destrucción se desensamble, lo que impide la degradación de β-‐catenina, su acumulación y, por tanto, su aumento de actividad. Axins 1 y 2 también actúan como supresores de tumores que están mutados frecuentemente en cáncer hepatocelular y colorrectal. Además, también se han encontrado delecciones inactivantes del gen de GSK-‐3β en leucemia mieloide, en el correceptor LPR5 en cáncer de mama y paratiroides. También se han observado mutaciones activadoras de los factores de transcripción TCF4 en cáncer colorrectal con inestabilidad de microsatélites, aunque su función no está totalmente determinada.
Por otra parte, también se han encontrado mutaciones activadoras en β-‐catenina, que la protegen de su degradación en cáncer hepatocelular y meduloblastoma, así como mutaciones en el gen (CREB-‐BP/CBP en leucemias linfoblásticas agudas y linfoma de células B.
Hasta el momento no se conocen inhibidores específicos para los componentes de la vía Wnt/β-‐catenina y únicamente se han realizado ensayos preclínicos con compuestos que inhiben la secreción de Wnt o desestabilizan Axin2, como por ejemplo la molécula XAV-‐939 (Novartis). ALTERACIONES GENETICAS EN LA SEÑALIZACIÓN DE Wnt EN CANCER Gen afectado
Alteración DNA/mRNA Respuesta funcional Tipo tumor
CTNNBI (β-‐catenina)
Sin sentido/deleción Estabilidad proteica aumentada
Hepatocelular/ meduloblastoma
APC (APC) Ruptura Actividad reguladora reducida
Colorrectal/gástrico
Axinas (I, II) Ruptura/sin sentido Actividad reguladora reducida
Hepatocelular/colorrectal
CREBP (CBP) Ruptura/sin sentido Acetiltransferasa inactiva Linfoma/leucemia GSK3β Mal procesamiento
RNA/deleción Quinasa inactiva Leucemia
LRP5 Mal procesamiento RNA/deleción
Pérdida de represión por DKK1
Mama/paratiroides
TCF7L2 (TCF4)
Sin sentido/deleción/ruptura
Pérdida de represión Colorrectal
TCF7L2 (TCF4)
Fusión con gen VT11A Desconocida Colorrectal
FAB123B (WTX)
Ruptura/deleción Pérdida de función Wilm´s tumor
Tabla 7. Alteraciones genéticas de la vía de Wnt en cáncer

34
BIBLIOGRAFÍA
Basseres DS. y Baldwin AS. (2006). Nuclear factor-‐kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene 25(51):6817-‐6830.
Berndt N., Hamilton AD. y Sebti SM.(2011). Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 11(11):775-‐791.
Chappell WH., Steelman LS., Long JM., Kempf RC., Abrams SL., Franklin RA., Bäsecke J., Stivala F., Donia M., Fagone P., Malaponte G., Mazzarino MC., Nicoletti F., Libra M., Maksimovic-‐Ivanic D., Mijatovic S., Montalto G., Cervello M., Laidler P., Milella M., Tafuri A., Bonati A., Evangelisti C., Cocco L., Martelli AM. y McCubrey JA. (2011). Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget . 17(3):135-‐164.
Engelman JA. (2009). Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat. Rev. Cancer 9(8):550-‐562.
Garnett MJ. y Marais R. (2004). Guilty as charged: B-‐RAF is a human oncogene. Cancer Cell 6(4):313-‐319.
Jamie NA. y Moon RT. (2013). Wnt signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 13:11-‐26.
Jancík S., Drábek J., Radzioch D. y Hajdúch M. (2010). Clinical relevance of KRAS in human cancers. J. Biomed. Biotechnol. 2010:150960.
Jatiani SS., Baker SJ., Silverman LR. y Reddy EP. (2011). JAK/STAT Pathways in Cytokine Signaling and Myeloproliferative Disorders: Approaches for Targeted Therapies. Genes & Cancer 1(10):979–993.
McDonald PC., Fielding AB., Dedhar S. (2008). Integrin-‐linked kinase-‐essential roles in physiology and cancer biology. J. Cell. Sci. 121(Pt 19):3121-‐3132.
De Luca A., Maiello MR., D'Alessio A., Pergameno M. y Normanno N. (2012). The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets 16(S2):S17-‐27.
McCubrey JA., Steelman LS., Chappell WH., Abrams SL., Franklin RA., Montalto G., Cervello M., Libra M., Candido S., Malaponte G., Mazzarino MC., Fagone P., Nicoletti F., Bäsecke J., Mijatovic S., Maksimovic-‐Ivanic D., Milella M, Tafuri A., Chiarini F., Evangelisti C., Cocco L. y Martelli AM. (2012). Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR Cascade Inhibitors: How Mutations Can Result in Therapy Resistance and How to Overcome Resistance. Oncotarget 3(10):1068-‐111.
Maurer G., Tarkowski B. y Baccarini M. (2011). Raf kinases in cancer-‐roles and therapeutic opportunities. Oncogene 30(32):3477-‐3488.
Polakis P. (2007). The many ways of Wnt in cancer. Curr. Opin. Genet. Dev. 17(1):45-‐51.
Raman M., Chen W. y Cobb MH. (2007). Differential regulation and properties of MAPKs. Oncogene 26(22):3100-‐3112.

35
Polakis P. (2012). Wnt Signaling in Cancer. Cold Spring Harb. Perspect. Biol. 2012:1-‐13. Sabatini DM. (2006). mTOR and cancer: insights into a complex relationship. Nat. Rev. Cancer 6(9):729-‐734.
Santarpia L., Lippman SM. y El-‐Naggar AK. (2012). Targeting the MAPK-‐RAS-‐RAF signaling pathway in cancer therapy. Expert Opin. Ther. Targets 16(1):103-‐119.
Shaw RJ., Cantley LC. (2006). Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 441(7092):424-‐430. Sudarsanam S. y Johnson DE. (2010). Functional consequences of mTOR inhibition. Curr. Opin. Drug Discov. Devel. 13:31–40. Wong KK., Engelman JA. y Cantley LC. (2010). Targeting the PI3K signaling pathway in cancer. Curr. Opin. Genet. Dev. 20(1):87-‐90. Naugler WE, Karin M. NF-‐kappaB and cancer-‐identifying targets and mechanisms. Curr Opin Genet Dev 2008; 18(1):19-‐26. Yuan TL.y Cantley LC. (2008). PI3K pathway alterations in cancer: variations on a theme. Oncogene 27:5497–5510.


















