
SVR-Vol5-2 SVR2-1 copia - svreumatologia.es
25
Actualidad Científica Volumen 5 Número 2 Noviembre 2013 EDITORIAL Vacunas en paciente reumático: colaboración necesa- ria entre la SVR y la SVMPSP Fernández Martínez S ORIGINAL Afectación retroorbitaria de la granulomatosis de Wegener Díaz del Río I, Rueda Cid A, Campos Fernández C, Pastor Cubillo MD, Gonzá- lez-Cruz Cervellera MI, Beltrán Catalán E, Calvo Catalá J REVISION Fármacos anti- TNFα en enferme- dades sistémicas asociables a uveítis Beltrán Catalán E, Martínez-Costa Pérez L CASOS CLÍNICOS Nefropatía IgA y enfermedades reumáticas. Revisión de la literatura a propósito de un caso Montomoli M, de la Morena Barrio I, Ávila Bernabéu A, Oller Rodríguez JE, Vicens Bernabéu E, Ybáñez García D, Valls Pascual E, Martínez Ferrer A, Feced Olmos C, Robustilllo Villarino M, Vizcaíno Castillo B, Serrato Villal- ba A, Alegre Sancho JJ 15 Coxalgia progresiva tras adenomectomía de próstata Senabre-Gallego JM, Salas-Heredia E, Santos-Ramírez C, Santos-Soler G, Ortega-Castro R, Rosas J 19 Neumopatía intersticial linfocítica como manifestación inicial en paciente con síndrome de Sjögren primario: a propósito de un caso Ortega-Castro R, Santos-Ramírez C, Senabre-Gallego JM, Santos-Soler G, Salas-Heredia E, Rosas J, Grupo AIRE-MB 22 Importancia del PET/TAC en el diagnóstico de la aortitis Ferrer Rebolleda J, Calvo Catalá J, Cózar Santiago MA, Campos Fernández C, Sánchez Jurado R, Rueda Cid A GALERÍA DE IMÁGENES Osteopetrosis Santos Ramírez C, Ortega Cas- tro R, Martínez Salinas MP Puente óseo sacroilíaco aislado Andrés M, Fernández-Carballi- do C, Romera C, San Martín A, Sivera F 24 Ecografía musculoes- quelética: Patología trau- matológica en la consulta de reumatología Senabre JM, Salas E, Santos- Soler G, Rosas J 1 3 6 11 ISSN 1133-4800 23
Transcript of SVR-Vol5-2 SVR2-1 copia - svreumatologia.es

Actualidad Científica
Volumen 5 Número 2 Noviembre 2013
EDITORIAL
Vacunas en paciente reumático: colaboración necesa-ria entre la SVR y la SVMPSPFernández Martínez S
ORIGINAL
Afectación retroorbitaria de la granulomatosisde WegenerDíaz del Río I, Rueda CidA, Campos Fernández C,Pastor Cubillo MD, Gonzá-lez-Cruz Cervellera MI,Beltrán Catalán E, CalvoCatalá J
REVISION
Fármacos anti-TNFα en enferme-dades sistémicasasociables a uveítisBeltrán Catalán E,Martínez-Costa Pérez L
CASOS CLÍNICOS
Nefropatía IgA y enfermedades reumáticas.Revisión de la literatura a propósito de un casoMontomoli M, de la Morena Barrio I, Ávila Bernabéu A,Oller Rodríguez JE, Vicens Bernabéu E, Ybáñez García D,Valls Pascual E, Martínez Ferrer A, Feced Olmos C,Robustilllo Villarino M, Vizcaíno Castillo B, Serrato Villal-ba A, Alegre Sancho JJ
15 Coxalgia progresiva tras adenomectomía depróstataSenabre-Gallego JM, Salas-Heredia E, Santos-RamírezC, Santos-Soler G, Ortega-Castro R, Rosas J
19 Neumopatía intersticial linfocítica comomanifestación inicial en paciente con síndromede Sjögren primario: a propósito de un casoOrtega-Castro R, Santos-Ramírez C, Senabre-GallegoJM, Santos-Soler G, Salas-Heredia E, Rosas J, GrupoAIRE-MB
22 Importancia delPET/TAC en eldiagnóstico de laaortitisFerrer Rebolleda J,Calvo Catalá J, CózarSantiago MA, CamposFernández C, SánchezJurado R, Rueda Cid A
GALERÍA DE IMÁGENES
OsteopetrosisSantos Ramírez C, Ortega Cas-tro R, Martínez Salinas MP
Puente óseo sacroilíacoaisladoAndrés M, Fernández-Carballi-do C, Romera C, San Martín A,Sivera F
24 Ecografía musculoes-quelética: Patología trau-matológica en la consultade reumatologíaSenabre JM, Salas E, Santos-Soler G, Rosas J
1
3
6
11
ISSN 1133-4800
23

Revista de la SociedadValenciana de Reumatología
EDITOR
José Rosas Gómez de Salazar
COEDITOR
Juan Antonio Castellano Cuesta
SECRETARIOS DE REDACCIÓN
Miguel Ángel Belmonte SerranoMaría Isabel Tevar Sánchez
COMITÉ EDITORIAL
Juan José Alegre SanchoJavier Calvo Catalá
Cristina Campos FernándezCristina Fernández Carballido
Isabel Ibero DíazJosé Ivorra Cortés
Vega Jovaní CasanoAntonio José Lozano Saez Mauricio Mínguez Vega
José Román IvorraGregorio Santos Soler
Francisca Sivera Mascaró
E-mail: [email protected]
DISEÑO Y COORDINACIÓN EDITORIAL
Ibáñez&Plaza Asociados, [email protected]
www.ibanezyplaza.com
IMPRESIÓN
Alba
DEPÓSITO LEGAL
M-3644-2013
SOPORTE VÁLIDO
SV02/92
ISSN 1133-4800
SOCIEDAD VALENCIANA
DE REUMATOLOGÍA
Presidenta: Pilar Trenor LarrazSecretario: Antonio José Lozano Saez
Tesorera: María Concepción Juliá MollaVicepresidente: Juan A. Castellano Cuesta
Vocal Alicante: Vega Jovani CasanoVocal Castellón: Ana V. Carro Martínez
Vocal Valencia: Luis González PuigPresidente electo: Miguel Ángel Belmonte
Serrano
Avda de la Plata, nº 20 46013 Valencia
http://www.svreumatologia.com
ecesitamos ayuda. Esta frase se hace cada día más patente y es cadavez más pronunciada dada la situación social en general y de lamedicina en particular. En esta sociedad en la que cada día el profe-sional médico se enfrenta a más trabas, en la que la superespeciali-zación limita nuestro campo de actuación y en la que cada vez
resulta más difícil ver al enfermo como un todo, se hace cada vez más necesaria labúsqueda de la colaboración para asegurar la calidad en la atención al paciente.Por tal motivo, surge un proyecto de consenso entre dos sociedades que tieneninterés en una cuestión común y que hasta este momento habían tratado de afron-tar de forma individual. Tanto la Sociedad Valenciana de Reumatología como laSociedad Valenciana de Medicina Preventiva y Salud Pública han aunado esfuer-zos para la elaboración de un documento de consenso sobre la vacunación delpaciente que está recibiendo tratamiento inmunodepresor o va a iniciarlo.Los pacientes afectos de enfermedades autoinmunes tienen más riesgo de padeceruna infección que la población sana. Este aumento del riesgo se debe por un ladoa la propia enfermedad, ya de por si inmunodepresora, a una resistencia disminui-da a las infecciones, conocido como locus minoris resistentiae y al cada vez másfrecuente tratamiento inmunosupresor al que se ven sometidos.Todo este aumento en el riesgo de infección conlleva un aumento tanto de la mor-bilidad como de la mortalidad de los pacientes, de ahí que la toma de conductaspreventivas como la vacunación y la quimioprofilaxis sea esencial.Sin embargo, a pesar de que las vacunas han demostrado ser efectivas en este grupo depacientes inmunodeprimidos, son una medida hoy en día poco utilizada y se calculaque menos de un 40% de estos pacientes están adecuadamente vacunados. Probable-mente sea debido a la falta de concienciación del profesional sanitario y a la infundadadesconfianza que las vacunas han motivado en lo que respecta a dos aspectos primor-diales como son su seguridad y eficacia clínica. Si hablamos de coste-efectividad, elvalor de las vacunas, indicada y administrada adecuadamente, es innegable.Probablemente en la consulta diaria no seamos conscientes de la magnitud delproblema y solo cuando empezamos a medir nos damos cuenta que no son pocoslos pacientes que ingresan cada año en nuestros hospitales por patología infeccio-sa que es, en gran parte, inmunoprevenible.De todo lo anterior surge la necesidad de la colaboración entre nuestras dos Socie-dades, con el objetivo de dilucidar las vacunas que están recomendadas en estosenfermos dependiendo de su situación clínica y los circuitos más apropiados paraasegurar el proceso de vacunación. De un consenso como este deben surgir docu-mentos con alto rigor científico y que resulten útiles en el día a día de la atenciónmédica. Una colaboración, a todas luces, necesaria.
Vacunas en paciente reumático:colaboración necesaria entre laSVR y la SVMPSP
ACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:1
Editorial
FERNÁNDEZ MARTÍNEZ SEspecialista en Medicina Preventiva y Salud Pública. Vicepresidente de la Sociedad Valen-ciana de Medicina Preventiva y Salud Pública (SVMPSP)
Correspondencia: Dr. Sergio Fernández Martínez - Servicio de Preventiva - Hos-pital General de Sagunto - Avda. Ramón y Cajal, s/n - 46520 Sagunto - Valencia [email protected]
N

Normas de publicación de trabajos
La Revista de la Sociedad Valencianade Reumatología (Rev. SociedadVal. Reuma.), es una publicación
semestral, orientada para la formación,información y expresión de los socios de laSVR.
La Rev. Sociedad Val. Reuma., puedeincluir las siguientes secciones:
1. SECCIÓN DE CONTENIDO CIENTÍFICO:• Editorial• Original • Revisión y puesta al día de la SVR• Presentación de casos y dificultades • Cartas al Director• Galería de imágenes• Buzón de la evidencia• Bibliografía comentada• Herramientas y utilidades para la práctica clínica
2. SECCIÓN DEAGENDA/NOTICIAS:• Entrevista• Noticias/Agenda SVR• Grupos de trabajo. Estudios en marcha• Buzón del socio• Biografía. Datos históricos de la SVR • Consulta jurídica• Ocio/Cultura/Viajar y Conocer la Comunidad
NORMAS DE PUBLICACIÓN DE TRABAJOS EN LA REVISTA DE LA SVR:
1.- Los trabajos serán mecanografiadosen español, a doble espacio en hojas DIN-A4 numeradas correlativamente, emplean-do una sola cara. Se remitirán en soporteinformático a la dirección de correo elec-trónico: [email protected] ycopia en papel a la sede de la SVR (Avdade la Plata, nº 20. 46013 Valencia).
2.- En la primera página figurará el títu-lo del trabajo, nombre y apellidos del autoro autores, seguido por el nombre del Cen-tro de Trabajo. En la esquina inferior dere-cha figurará el nombre, la dirección postaly de correo electrónico del autor con quiendebe mantenerse correspondencia.
3.- Originales: Se refiere a trabajossobre cualquier campo de la patología reu-mática. En la segunda hoja figurará unresumen, con un máximo de 200 palabrasen español, describiendo los objetivos,metodología, resultados y conclusiones del
trabajo. En esta misma hoja se incluirántres palabras clave, que faciliten la identifi-cación del trabajo, con las mismas caracte-rísticas de idioma. Los apartados que debeincluir son: introducción; pacientes, mate-rial y método; resultados; discusión; ybibliografía. La extensión máxima será de12 folios de 30 líneas de 70 pulsaciones, adoble espacio y se admitirán hasta unmáximo de 6 figuras y 6 tablas.
4.- Presentación de Casos Clínicos: Enla segunda hoja figurará un resumen delcaso, con un máximo de 100 palabras enespañol. En esta misma hoja se incluirántres palabras clave, que faciliten la identifi-cación del trabajo, con las mismas caracte-rísticas de idioma. A continuación elesquema a seguir incluirá; introducción,descripción del caso y discusión. La biblio-grafía incluirá un máximo de 15 citas. Laextensión máxima será de 5 folios de 30líneas de 70 pulsaciones, a doble espacio yse admitirán hasta un máximo de 3 figurasy 3 tablas. Una vez presentado el caso, seincluirá en formato de tabla o caja, a juiciodel autor, las dificultades del caso y tras ladiscusión del mismo, las llamadas de aten-ción o aprendizaje del mismo.
5.- Cartas al Director: En esta sección sepublicarán objeciones o comentarios rela-tivos a artículos o casos clínicos publica-dos recientemente en la Revista. La exten-sión máxima será de 2 folios como máxi-mo y se admitirá una figura o una tabla. Labibliografía será de 10 citas como máximo.
6.- Revisión y puesta al día: En esteapartado se incluirán en formato de resu-men, las charlas de los ponentes invitados,presentadas en las reuniones, Simposiumy/o Congresos de la SVR. La extensiónmáxima será de 5 folios de 30 líneas de 70pulsaciones, a doble espacio y se admitiránhasta un máximo de 3 figuras y 3 tablas. Elautor incluirá un máximo de 5 aspectosrelevantes de su revisión, que podrán serincluidas en el apartado de Conclusiones.
7.- Galería de Imágenes: Se admitiránimágenes sobre cualquier campo de laReumatología. Se deberá aportar la inter-pretación de la misma, con una extensiónmáxima de 100 palabras.
8.- Buzón de la Evidencia: En esta sec-ción se intentará contestar, según la mejorevidencia posible, a preguntas surgidas enla práctica clínica cotidiana. En la estructu-ra de presentación, quedará al inicio deforma clara la formulación de la pregunta.Posteriormente la contestación, en un
máximo de 3 folios, describirá la ruta debúsqueda realizada, los comentarios y con-clusiones. Se admitirán un máximo de 20citas y hasta 2 tablas. Se podrán remitir pre-guntas con su contestación realizado poralguno de los socios de la SVR, o pregun-tas a contestar en esta sección mediante elapartado de buzón de socio y ser contesta-das por alguno de los socios de la SVR,designado por el Comité Editorial de laRev. Sociedad Val. Reuma.
9.- Herramientas útiles en la asistencia:En este apartado se aceptarán aportacionesde los socios de la SVR, en forma de tablas,formulaciones, árboles de decisión, fraseso axiomas clínicos contrastados, etc, quepuedan ser de utilidad para la práctica clíni-ca cotidiana. La extensión máxima será de1 folio.
10.- Bibliografía comentada: A peti-ción del Comité Editorial de la Rev. Socie-dad Val. Reuma. y por ser considerados deinterés, se publicarán por encargo, comen-tarios o análisis de trabajos publicados anivel nacional o internacional. La exten-sión máxima será de 2 folios.
11.- Grupos de trabajo y Estudios enmarcha: Se incluye la publicación de infor-mación acerca de los grupos de trabajo,dentro de la SVR, en cualquier aspecto dela patología reumática y de los estudios enfase de realización o en fase de diseño, conel ánimo de aumentar la participación enlos mismos.
12.- Buzón del socio: En esta sección sepodrán recibir comentarios, ideas y suge-rencias de los socios de la SVR, en aspec-tos referidos a la propia SVR o a la Revista,en cualquiera de sus apartados.
13.- Resto de secciones, quedará a cri-terio del Comité Editorial de la Rev. Socie-dad Val. Reuma. para su publicación encada número: entrevista, noticias/agenda,biografía y/o datos históricos de la SVR,consulta jurídica, ocio, conocer la Comu-nidad Valenciana.
El Comité Editorial de la Rev. SociedadVal. Reuma. acusará recibo de los trabajosenviados e informará de su aceptación porcorreo electrónico. Este mismo comité sereserva el derecho a rechazar los trabajosenviados, así como proponer modificacio-nes en ellos, cuando lo considere necesa-rio.
Las fechas límite para remitir para valo-rar su publicación en cada número (traba-jos, consultas, imágenes, etc), serán: 15 deJunio y 15 de diciembre, de cada año.
| 2 |

La granulomatosis de Wegener es una enfermedad poco frecuente yque clásicamente presenta una triada clínica clásica: afectación devías respiratorias superior, inferior y renal, pero al tratarse de una vas-culitis de pequeño vaso puede afectar a diversos órganos. La afectación del ojo, órbita o estructuras anexas al ojo ocurre en el 50%
de los casos. La proptosis por la ocupación del espacio retroorbitario porgranulomas, es la manifestación menos frecuente pero la más específica.Presentamos dos casos con afectación retroorbitaria por granuloma-tosis de Wegener, diagnosticándose el primer caso por el estudio ana-tomopatológico y el segundo por criterios clínicos.
Afectación retroorbitaria de la granulomatosisde WegenerDÍAZ DEL RÍO I, RUEDA CID A, CAMPOS FERNÁNDEZ C, PASTOR CUBILLO MD, GONZÁLEZ-CRUZ CERVELLERA MI, BELTRÁN CATALÁN E, CALVOCATALÁ JServicio de Reumatología y Metabolismo Óseo. Consorcio Hospital General Universitario. Valencia
Correspondencia: Dr. Javier Calvo Catalá - Servicio de Reumatología y Metabolismo Óseo - Consorcio Hospital General Universi-tario - Avda. Tres Cruces, 1 - 46014 Valencia [email protected]
/ RESUMEN
INTRODUCCIÓNLa granulomatosis de Wegener (GW)tiene una incidencia que se estima en0,4/100.000 habitantes, generalmenteen personas de mediana edad y siendoalgo más frecuente en hombres (2:1)1.
La triada clínica clásica incluye afecta-ción de vías respiratorias superiores, infe-riores y renal, pero al tratarse de una vascu-litis de pequeño vaso puede afectar a diver-sos órganos. Cuando debuta con afecta-ción de órganos distintos a la tríada clásica,puede retrasarse el diagnóstico2 y con elloel inicio del tratamiento que además sueletener unos resultados excelentes3,4.
La afectación del ojo, órbita o estruc-turas anexas al ojo ocurre en el 50% delos casos y, en ocasiones, es la únicamanifestación de la enfermedad. Semanifiesta desde una simple conjuntivi-tis hasta proptosis debido a la ocupacióndel espacio retroorbitario por granulo-mas, siendo esto último lo menos fre-cuente pero más específico5,6,7,8,9.
Realizamos un estudio descriptivoretrospectivo de los pacientes atendidos en
el Servicio de Reumatología del HospitalGeneral de Valencia en el periodo de 2000-2012 con afectación del espacio retroorbi-
tario debido a la formación de granulomaspor GW. De los trece casos de GW consta-tados en estos años, tres presentaron afec-
ACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:3-5
Original
| 3 |
Palabras clave: granulomatosis de Wegener, lesión retroorbitaria.
FIGURA 1
INFILTRADOS CAVITADOS EN PULMÓN DERECHO
tación ocular y dos de ellos, lesiones retro-oculares. Recogemos las características yanalizamos las pruebas (diagnósticas o decontrol evolutivo) realizadas, los trata-mientos recibidos y su evolución.
CASOS CLÍNICOSCaso clínico 1Varón de 67 años con rinorrea sanguino-lenta diagnosticado en 2004 de GW tras
biopsia nasal. Amputación de falangesdigitales del 4º y 5º dedos mano derechapor isquemia y necrosis. ANCA+. Seinicia tratamiento con corticoides yciclofosfamida (CF) oral, quedando elpaciente asintomático. Se reduce paula-tinamente la CF, dejanto deflazacortcomo tratamiento de mantenimiento. En2007 presentó una crisis focal motora,existiendo infartos lacunares en estudio
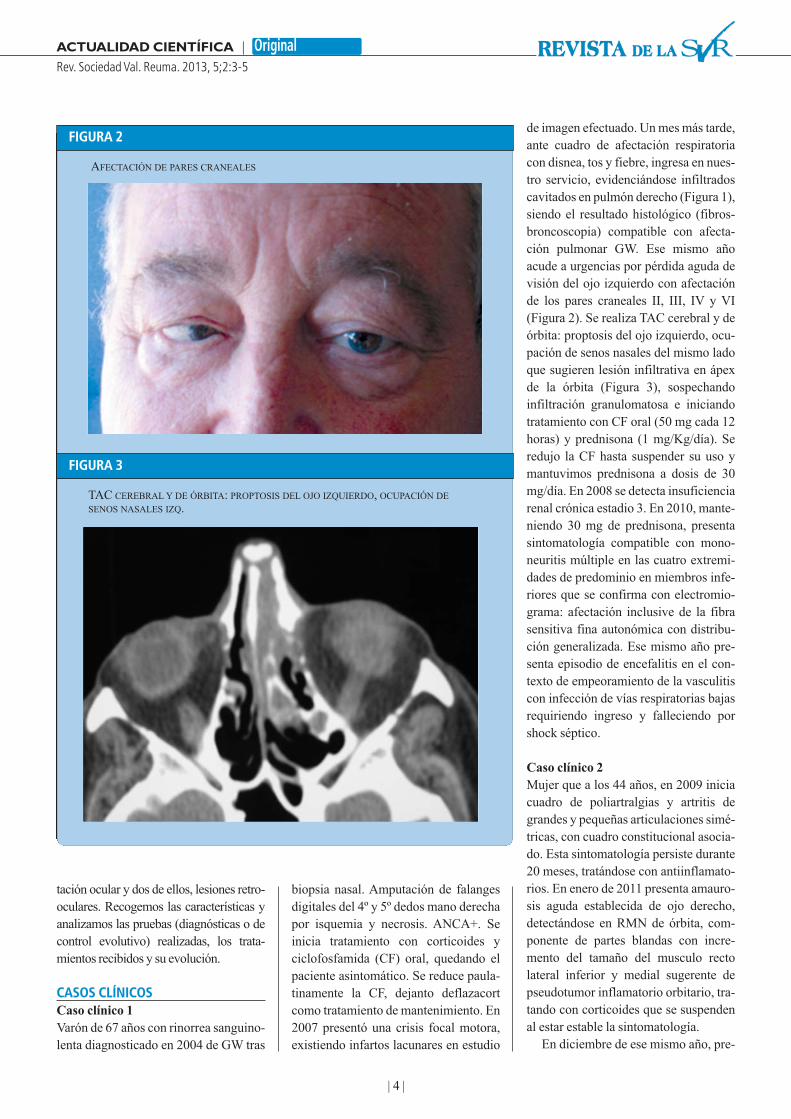
de imagen efectuado. Un mes más tarde,ante cuadro de afectación respiratoriacon disnea, tos y fiebre, ingresa en nues-tro servicio, evidenciándose infiltradoscavitados en pulmón derecho (Figura 1),siendo el resultado histológico (fibros-broncoscopia) compatible con afecta-ción pulmonar GW. Ese mismo añoacude a urgencias por pérdida aguda devisión del ojo izquierdo con afectaciónde los pares craneales II, III, IV y VI(Figura 2). Se realiza TAC cerebral y deórbita: proptosis del ojo izquierdo, ocu-pación de senos nasales del mismo ladoque sugieren lesión infiltrativa en ápexde la órbita (Figura 3), sospechandoinfiltración granulomatosa e iniciandotratamiento con CF oral (50 mg cada 12horas) y prednisona (1 mg/Kg/día). Seredujo la CF hasta suspender su uso ymantuvimos prednisona a dosis de 30mg/día. En 2008 se detecta insuficienciarenal crónica estadio 3. En 2010, mante-niendo 30 mg de prednisona, presentasintomatología compatible con mono-neuritis múltiple en las cuatro extremi-dades de predominio en miembros infe-riores que se confirma con electromio-grama: afectación inclusive de la fibrasensitiva fina autonómica con distribu-ción generalizada. Ese mismo año pre-senta episodio de encefalitis en el con-texto de empeoramiento de la vasculitiscon infección de vías respiratorias bajasrequiriendo ingreso y falleciendo porshock séptico.
Caso clínico 2Mujer que a los 44 años, en 2009 iniciacuadro de poliartralgias y artritis degrandes y pequeñas articulaciones simé-tricas, con cuadro constitucional asocia-do. Esta sintomatología persiste durante20 meses, tratándose con antiinflamato-rios. En enero de 2011 presenta amauro-sis aguda establecida de ojo derecho,detectándose en RMN de órbita, com-ponente de partes blandas con incre-mento del tamaño del musculo rectolateral inferior y medial sugerente depseudotumor inflamatorio orbitario, tra-tando con corticoides que se suspendenal estar estable la sintomatología.
En diciembre de ese mismo año, pre-
ACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:3-5
Original
| 4 |
FIGURA 2
FIGURA 3
AFECTACIÓN DE PARES CRANEALES
TAC CEREBRAL Y DE ÓRBITA: PROPTOSIS DEL OJO IZQUIERDO, OCUPACIÓN DE
SENOS NASALES IZQ.

senta hemoptisis moderada junto conanemia microcítica ferropénica y positi-vidad de p-ANCA. En TAC toracoabdo-minal, se evidencian numerosas adeno-patías diseminadas e inespecíficas detamaño inferior a 1 cm. En marzo de2012, ante un cuadro de insuficienciarespiratoria, se inicia tratamiento conprednisona (1 mg/Kg/día), realizandobiopsia pulmonar que muestra neumo-nía intersticial descamativa. No se pue-den reducir los corticoides por reapari-ción de la clínica respiratoria. Ennoviembre de 2012 es remitida a nuestroservicio, donde se evidencia nariz “ensilla de montar” (Figura 4) y rinorreasanguinolenta, efectuando biopsia quemuestra rinitis ulcerada sin cambiosinflamatorios específicos en los vasos.En ese momento analíticamente desta-ca: p-ANCA+, VSG 120 mmHg y 25hematíes por campo en sedimento uri-nario. Con el diagnóstico de GW, seañadió ciclofosfamida al tratamiento(un primer bolo iv de 1 gr y posterior-mente con 500 mg cada mes hasta untotal de seis infusiones). En RMN maxi-lofacial de control se constata el pseudo-tumor orbitario ya conocido, en faseinflamatoria aguda, junto con engrosa-miento de las celdillas etmoidales y delseno maxilar derecho (Figura 5). Debi-do a que las biopsias previas (pulmón,piel y fosa nasal) mostraron resultadosno concluyentes efectuamos biopsiamuscular y de nervio sural, sin eviden-ciar tampoco lesiones de vasculitis.Actualmente, continuamos el tratamien-to con 10 mg de PDN y completaremoshasta seis infusiones de CF, con resulta-do positivo, permaneciendo la pacienteasintomática en estos momentos.
DISCUSIÓNLa GW es una enfermedad de baja fre-cuencia de aparición que puede afectar acualquier zona del organismo. El diag-nóstico se retrasa cuando la afectaciónno es en las zonas habituales1.
La afectación del ojo, órbita o estruc-turas anexas al ojo ocurre en el 50% de
los casos y, en ocasiones, es la únicamanifestación de la enfermedad. Semanifiesta en forma de conjuntivitis,uveítis, afectación del nervio óptico,oclusión de arterias retinianas o propto-sis debido a la ocupación del espacioretroorbitario por granulomas, siendoesto último lo menos frecuente pero másespecífico5,7.
En el primer caso, la afectación ocu-lar fue posterior al diagnóstico de GW,pero en el segundo caso, al no tenerresultados histológicos compatibles einiciar la clínica de forma más inespecí-fica (artralgias y posterior clínica oftal-mológica), el diagnóstico se retrasó envarios años, siendo importante la defor-midad nasal “en silla de montar” parallegar al mismo. Ambos pacientes cum-plen criterios diagnósticos de la GWsegún la American Collage of Rheuma-tology10.
Debido a que la clínica es similar a laproducida por procesos de otras etiolo-gías como la neoplásica, infecciosa oinflamatoria, es de especial importanciapensar en esta enfermedad para poderllegar al diagnóstico. La alta frecuenciade la afectación ocular de la GW haceque deba estar siempre incluida en eldiagnóstico diferencial de pacientes consíntomas oculares aislados o con pseu-dotumor orbitario.
BIBLIOGRAFÍA1.- Fonollosa V, Vilardell M. Granulomatosisde Wegener. Med Clin (Barc) 1992;99:57-59.2.- Sánchez Ballester E, Lostaunau Costa G,Calvo Catalá J, Campos Fernández J, Gonzá-lez-Cruz MI. Enfermedad de Wegener de pre-sentación atípica. Revista de la SV Reumatolo-gía, 2011; 4(2):32-34.3.- Fauci AS, et al. Wegener’s granulomatosis:Prospective clinical and therapeutic experiencewith 85 patients for 21 years. Clinical review.Ann Intern Med 1983;98:76-85.4.- Michet CJ. Epidemology of vasculitis.Rheum Dis Clin North Am 1990;16:263.5.- Frank P Fechner, et al. Wegener’s Granulo-matosis of the Orbit: A ClinicopathologicalStudy of 15 Patients. The Laryngoscope,112:1945-1950, 2002.6.- Harper SL, el al. Wegener’s granulomatosis:the relationship between ocular and systemicdisease. J. Rheumatol, 2001 May;28 (5):1025-32.7.- Haynes BF, et al. The ocular manifestations
of Wegener’s granulomatosis: fifteen yearsexperience and review of the literature. Am JMed 1977;63:131-141.8.- Woo TL, et al. Australasian orbital andadnexal Wegener’s granulomatosis. Ophthal-mology 2001;108:1535-1543.9.- Bullen CL, et al. Ocular complications ofWegener’s granulomatosis. Ophthalmology1983;90:279-290.10.- Leavitt RY, et al. The American College ofRheumatology 1990 criteria for the classifica-tion of Wegener’s granulomatosis. ArthritisRheum 1990;33:1101-1107.
ACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:3-5
Original
| 5 |
FIGURA 4
FIGURA 5
NARIZ “EN SILLA DE MONTAR”
PSEUDOTUMOR ORBITARIO CON ENGRO-SAMIENTO DE LAS CELDILLAS ETMOIDA-LES Y DEL SENO MAXILAR DERECHO

| 6 |
RevisiónACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:6-10
INTRODUCCIÓNLas enfermedades autoinmunes sistémi-cas constituyen un grupo heterogéneode entidades clínicas con una base fisio-patológica común. Presentan unaamplia diversidad en sus manifestacio-nes, pudiendo afectar a varios órganos ysistemas con una intensidad variable eindividual en cada paciente. Por estemotivo, son consideradas de difícildiagnóstico, clasificación y tratamiento.Se han elaborado unos criterios de clasi-ficación internacionales, que como sunombre indica, nos ayudan a clasificar aestos pacientes definiendo unos perfilesclínico/inmunológicos, pero a veces,resultan insuficientes en la vertiente deldiagnóstico. La manifestación clínicainicial de estas enfermedades es variaday variable, y únicamente un profundoconocimiento de este tipo de patologíaspor parte de los distintos especialistasimplicados, y un alto grado de alerta nospermitirán realizar un diagnóstico pre-coz de estos enfermos, que con seguri-dad conducirá a un mejor pronósticoevolutivo. Estas enfermedades suponenpara nosotros día a día retos diagnósti-co-terapéuticos y conllevan en muchasocasiones decisiones terapéuticasarriesgadas y difíciles. Por este motivo,en muchos centros existen unidadesmultidisciplinares con el objetivo defacilitar la clasificación y el diagnósticode estos pacientes, así como el trata-miento y abordaje integral de sus mani-festaciones clínicas.
La afección ocular se puede conside-rar en ocasiones como una complica-
ción de la enfermedad sistémica, o bienpuede ser una de las primeras manifesta-ciones de una enfermedad sin diagnosti-car. La inflamación del tracto uveal (enforma de uveítis anterior, intermedia oposterior) es una manifestación oftálmi-ca relativamente frecuente de algunasenfermedades autoinmunes sistémicas.De hecho, la inflamación asociada aenfermedad sistémica puede afectar a lapráctica totalidad de las estructuras ocu-lares, órbita y anejos de forma unilateralo bilateral.
En los últimos años, han aparecidonuevas moléculas y terapias biológicascon indicación para el tratamiento dediversas enfermedades reumáticas,cutáneas y digestivas, que también hansido utilizadas fuera de indicación tera-péutica con éxito en el tratamiento dediferentes enfermedades autoinmunessistémicas1. Desafortunadamente, estosbuenos resultados clínicos no han sidocorroborados mediante ensayos de cali-dad. Un reciente meta-análisis realizadopor Pato E et al, otorga un grado de reco-mendación C con nivel de evidencia 4para el tratamiento de uveítis no infec-ciosas con infliximab y adalimumab2.
ESPONDILOARTRITISLos pacientes con espondilitis anquilo-sante (EA), artritis psoriásica, artritisreactiva y enfermedad inflamatoriaintestinal pueden presentar inflamaciónocular, generalmente en forma de uveí-tis anterior aguda (UAA) unilateral,alternante y recidivante. En pacientescon psoriasis está descrita la presencia
de UAA bilateral. La enfermedad sisté-mica que se asocia con mayor frecuen-cia a la presencia de uveítis es la EA. Lamitad de los pacientes que desarrollanuna UAA son HLA-B27 positivos. Lauveítis UAA aparece hasta en un 40% delos pacientes con EA y artritis reactiva alo largo de la evolución de su enferme-dad. Puesto que se presenta en forma debrotes más o menos frecuentes de uveí-tis anterior, se recomienda el tratamien-to de cada episodio mediante corticoste-roides tópicos. La aparición de más detres brotes de UAA al año justifica subirun peldaño terapéutico mediante el usode Salazopirina o Metrotrexato. Sinembargo, los fármacos anti-TNF hansupuesto un gran avance en el trata-miento de la enfermedad sistémicaaxial, en la que los medicamentos inmu-nomoduladores son poco eficaces. Enlos pacientes afectos de EA en trata-miento con anti-TNF se ha objetivadouna menor incidencia de episodios deuveítis3,4. Existen escasas indicacionespara administrar fármacos biológicosúnicamente por la enfermedad ocular enla EA. Entre ellas, se encontrarían lapresencia de múltiples episodios deedema macular inflamatorio, la afeccióndel polo posterior, la cronificación delas uveítis anteriores o la aparición demúltiples brotes poco separados en eltiempo. La reducción del número debrotes con el uso de adalimumab pareceser mayor si el paciente presenta uveítisanterior activa en el inicio del tratamien-to, y menor si existe una historia previade uveítis crónica5.
Fármacos anti-TNFα en enfermedades sistémicasasociables a uveítis BELTRÁN CATALÁN E1, MARTÍNEZ-COSTA PÉREZ L2
1Servicio de Reumatología y Metabolismo Óseo. Consorcio Hospital General Universitario de Valencia2Servicio de Oftalmología Hospital Universitario Doctor Peset. Valencia
Correspondencia: Dra. Emma Beltrán Catalán - Servicio de Reumatología y Metabolismo Óseo - Consorcio Hospital General Universitariode Valencia - Avda. Tres Cruces, 2 - 46014 Valencia [email protected]

| 7 |
RevisiónACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:6-10
ARTRITIS IDIOPÁTICA JUVENIL (AIJ)Los fármacos anti-TNF se han mostradoeficaces tanto en la artritis idiopáticajuvenil como en la uveítis asociada aartritis psoriásica en niños6,7,8. La tasa deeficacia a corto y medio plazo parece sersimilar entre infliximab y adalimumab(70-80%)9-11. En los niños, el adalimu-mab está siendo ampliamente utilizadodebido a que se trata de un fármacohumanizado y a que se administra porvía subcutánea.
SARCOIDOSISLa sarcoidosis ocular es una entidad pro-bablemente infradiagnosticada. Se creeque muchas uveítis clasificadas como“idiopáticas” son en realidad sarcoidosiscon pocas o ninguna manifestaciónextraocular. La sarcoidosis es la segundacausa más frecuente de enfermedad sis-témica asociada a uveítis en Norteaméri-ca, tras la EA12. Con frecuencia, la mani-festación inicial de una sarcoidosis esocular. La incidencia de uveítis posterioren los enfermos con sarcoidosis llegahasta el 30% de los casos, y probable-mente es aún mayor para las uveítis ante-riores, en las cuales hay un aumento deTNF intracelular a partir de linfocitosT13. La vasculitis retiniana es una carac-terística importante de la sarcoidosis,aunque la vasculitis sistémica no es típi-ca de esta enfermedad.
La producción de TNFα, la activa-ción de macrófagos y la presencia de uninfiltrado de células CD4+T activadases crucial en la patogénesis de la forma-ción de granulomas y la afección mul-tiórgánica en la sarcoidosis14. El inflixi-mab, se ha recomendado como primeraopción terapéutica en los casos de sar-coidosis pulmonar severa o refracta-ria15,1, y probablemente es también efec-tivo en la sarcoidosis ocular, aunque, eneste caso la evidencia se limita a casosaislados en series de pacientes16-18. Exis-te un ensayo clínico doble ciego contro-lado con placebo sobre la eficacia deletanercept en la sarcoidosis ocular cró-nica19 en el cual se concluye que el eta-nercept no fue superior al placebo. Elinfliximab y el adalimumab20-22 sí que sehan mostrado eficaces el tratamiento de
la sarcoidosis multisistémica y refracta-ria, al menos en estudios de series decasos prospectivas y en estudios abier-tos reducidos.
Paradójicamente, se han descrito 38casos de sarcoidosis inducida por laadministración de fármacos anti-TNF,de los cuales, 3 tenían afección ocular1.Se ha estimado en un 0,04% la prevalen-cia de sarcoidosis inducida por anti-TNFα23.
ENFERMEDAD DE BEHÇETLa enfermedad de Behçet se caracterizapor la aparición de episodios recurrentesy remitentes de uveítis y manifestacio-nes mucocutáneas fundamentalmente.Aunque en su evolución, también puedepresentar manifestaciones musculoes-queléticas, neurológicas, gastrointesti-nales, vasculares y respiratorias. En estaenfermedad, se ha objetivado un incre-mento de los niveles de TNFα, y cito-quinas. El infliximab, ha demostradogran eficacia en la enfermedad de Beh-çet mucocutánea. La afección ocular esfrecuente, en ocasiones agresiva, y se haconsiderado clásicamente un criteriomayor de diagnóstico. En muchoscasos, la aparición de una uveítis poste-rior con vasculitis oclusiva (Figura 1)condiciona un cambio terapéutico enestos pacientes. La EULAR (EuropeanLeague Against Rheumatism) ha elabo-rado unas directrices para el tratamientode la enfermedad de Behçet basadas enel consenso de un comité de expertos yuna revisión sistemática de la literatu-ra24. Se considera que la enfermedad deBehçet ocular debe tratarse con azatio-prina y corticosteroides sistémicoscomo primera opción terapéutica. En loscasos refractarios, se recomienda aso-ciar azatioprina con infliximab o biencon ciclosporina. Una consideración deinterés es la mayor rapidez de acción dela terapia endovenosa con infliximabfrente a ciclosporina (Figura 2). En casode utilizarse este último fármaco, acon-sejan asociar corticoides a dosis altaspuesto que tarda semanas en producir unefecto terapéutico. Se propone el inter-ferón α endovenoso como alternativaterapéutica a estos regímenes. Éste no
debe asociarse nunca a azatioprina porexistir un riesgo elevado de mielosupre-sión.
En un reciente análisis sobre el usode anti-TNFα en la enfermedad de Beh-çet25 se encontraron 88 artículos sobre eluso de infliximab, 12 sobre etanercept y13 sobre adalimumab. En un 89% de loscasos el infliximab produjo una respues-ta mantenida en el tiempo, con reduc-ción o desaparición de recurrencias deuveítis. Los 3 estudios prospectivos,abiertos y controlados sobre los efectosa largo plazo de las infusiones intrave-nosas repetidas de infliximab concluyenque se consigue la prevención de lasrecurrencias, el mantenimiento de laagudeza visual y la reducción de la tera-pia inmunosupresora. La asociación deinfliximab con metotrexato, azatioprinao ciclosporina parece ser más eficaz quela monoterapia.
Todos los estudios que se localizaronsobre adalimumab y etanercept eranseries de casos retrospectivos. La remi-sión de la enfermedad se consiguió entodos los casos publicados para adali-mumab y en más de un 50% de los deetanercept.
Como conclusión, aunque se requeri-rían más estudios prospectivos y contro-lados, los anti-TNFα parecen ser efica-ces en el tratamiento de las manifesta-ciones oculares asociadas a la enferme-dad de Behçet.
ENFERMEDADES SISTÉMICAS ASO-CIABLES A PATOLOGÍA DE CORNEAY ESCLERALa mitad de los pacientes que padecenescleritis son diagnosticados de unaenfermedad sistémica. Las enfermeda-des autoinmunes asociadas con mayorfrecuencia a escleritis son, la artritis reu-matoide, las vasculitis sistémicas, ellupus, la enfermedad inflamatoria intes-tinal, el síndrome de Cogan y la policon-dritis recidivante.
El bloqueo del TNFα se muestracomo una terapia prometedora en lasescleritis no infecciosas refractarias altratamiento convencional, tanto en loscasos puramente oculares como sobretodo en las escleritis asociadas a enfer-

| 8 |
RevisiónACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:6-10
FIGURA 1
VASCULITIS OCLUSIVAEN UN CASO DE ENFERMEDAD DEBEHÇET
FIGURA 2
2.1. SE OBSERVAN 2 FOCOS DE RETINITIS SUPERIORES EN
UN PACIENTE CON ENFERMEDAD DE BEHÇET
2.2. REMISIÓN TRAS LA PRIMERA INFUSIÓN DE INFLIXIMAB
medad sistémica16,26. La artritis reumatoide evolucionada es laenfermedad sistémica que con mayor frecuencia se asocia aescleritis, seguida probablemente por la granulomatosis conpoliangeítis (Wegener) en la que puede presentarse comomanifestación inicial (Figura 3). Los anticuerpos monoclona-les contra el TNFα son los fármacos más utilizados en lospacientes afectos de escleritis asociada a artritis reumatoide.En las vasculitis asociadas a ANCA, el rituximab, un anticuer-po monoclonal dirigido al CD20 de los linfocitos B, parecetener una mayor eficacia al inducir respuesta y mantener laremisión clínica1,27.
Policondritis recidivanteLa policondritis recidivante es una enfermedad autoinmunesistémica que se caracteriza por episodios inflamatorios deltejido cartilaginoso. Entre sus manifestaciones clínicas puedeasociar artritis, manifestaciones oculares y audiovestibulares,cardiovasculares, renales, dermatológicas y neurológicas.Los brotes de inflamación pueden conllevar una destrucciónpermanente de las estructuras afectas. Con cierta frecuenciapuede asociarse a otras enfermedades autoinmunes sistémicascomo el lupus, la artritis reumatoide y el Sindrome de Sjögreny a vasculitis como la granulomatosis con poliangeítis (Wege-ner), la panarteritis nodosa y la enfermedad de Behçet. La aso-ciación de policondritis recidivante y enfermedad de Behçetse denomina síndrome MAGIC (Mouth And Genital ulcerswith Inflamed Cartilage). En la policondritis recidivante lasmanifestaciones oculares pueden aparecer hasta en un 65% delos casos, e incluyen proptosis, edema de párpados, epiescleri-tis, escleritis, parálisis de músculos extraoculares, conjuntivi-tis, infiltrados corneales, queratitis ulcerativa periférica, uveí-tis anterior, desprendimiento exudativo de la retina y neuritisóptica28. En una revisión sistemática recientemente publicadasobre el uso de anti-TNF para policondritis recidivante selocalizaron 30 artículos, todos ellos sobre series de casos ocasos aislados. En 43 pacientes se había utilizado anti-TNFα(31 infliximab, 8 etanercept y 4 adalimumab), en 11 rituxi-mab, en 5 anakinra, en 2 tocilizumab y en 1 abatacept. Los fár-macos biológicos fueron efectivos en 27 pacientes, parcial-mente efectivos en 5 y no efectivos en 29 pacientes29. La expe-riencia es, por tanto, limitada, precisándose estudios aleatori-zados y controlados para definir sus indicaciones y eficacia.
Síndrome de CoganEl síndrome de Cogan es una enfermedad inflamatoria de ori-gen desconocido caracterizado por queratititis intersticial noinfecciosa, escleritis y déficit vestibuloautitivo. Puede asociaraortitis y vasculitis sistémica. El infliximab se ha utilizado enalgunos casos aislados reportados en la literatura30,31.
Vasculitis sistémicasLas vasculitis sistémicas en raras ocasiones asocian vasculitisretiniana, siendo por el contrario frecuente que a nivel ocularproduzcan escleritis y patología corneal periférica. Se clasifi-

| 9 |
RevisiónACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:6-10
can según el tamaño del vaso afecto envasculitis de vaso pequeño, mediano ygrande. Las vasculitis de vaso pequeñoasociadas a ANCA incluyen la granulo-matosis con poliangeítis (Wegener), elsíndrome de Churg-Strauss y la polian-geítis microscópica. Todas ellas puedenasociar en mayor o menor grado escleri-tis, queratitis ulcerativa periférica, irido-ciclitis, vasculitis retiniana y afecciónde órbita y vías lagrimales.
Las vasculitis de vaso mediano ygrande incluyen la arteritis de célulasgigantes (ACG), la arteritis de Takaya-su, la poliarteritis nodosa y la enferme-dad de Kawasaki. La arteritis de Taka-yasu produce sobre todo un síndrome deisquemia ocular. La poliarteritis nodosano suele asociarse con manifestacionesoculares, aunque raramente se han des-crito síndrome de isquemia ocular,infartos coroideos y escleritis. La arteri-tis de células gigantes o arteritis de latemporal es una entidad diagnosticadacon frecuencia por sus manifestacionesoculares en forma sobre todo de neuro-patía óptico-isquémica anterior. Laenfermedad de Kawasaki es una vascu-litis aguda multisistémica que afectageneralmente a niños menores de 5años. Se caracteriza por una inflamaciónpredominante de las arterias de medianotamaño complicada en ocasiones con lapresencia de aneurismas coronarios. Laconjuntivitis no purulenta bilateral, pre-dominantemente en conjuntiva bulbar,sin fotofobia ni dolor ocular, se presentaprecozmente y es uno de los criteriosiniciales de diagnóstico. En un porcen-taje elevado de pacientes se detecta unauveítis anterior como síntoma asociado.
Los anti-TNFα pudieran tener unpapel en el tratamiento de la arteritis deTakayasu refractaria o de difícil manejocontribuyendo a la remisión y a lareducción de la dosis de corticoidescomo se describe en estas series decasos de pacientes en tratamiento coninfliximab y etanercept32,33. Sin embar-go, se considera que los anti-TNFα noson efectivos como ahorradores de cor-ticoides o para prevenir recidivas en laarteritis de células gigantes. Hoffman etal.34 describen en un ensayo randomiza-
do y controlado con 44 pacientes con(ACG) que el tratamiento adyuvantecon infliximab no ayuda a la reducciónde la dosis de corticoides sistémicos nidisminuye las recidivas comparado conel corticoide en monoterapia y pautadescendente durante 6 meses. Martínez-Taboada et al.35 utilizan tratamientoadyuvante con etanercept en 17 pacien-tes con ACG en un estudio doble ciegocontrolado con placebo y concluyen queno existen diferencias significativasentre los dos grupos de tratamiento encuanto a la posibilidad de retirar los cor-ticoides después de un año. Por tantopor el momento podemos afirmar queno existe evidencia para el uso de anti-TNFα en el tratamiento de la ACG. Enel caso de la enfermedad de Kawasaki,existe información de que el infliximabpuede ser usado como una alternativa ala gammaglobulina por vía intravenosaen pacientes sin respuesta a una primeradosis de ésta. En el momento actual ycon los estudios de los que se dispone nose puede recomendar el uso de antiTNF-α en las vasculitis sistémicas aso-ciadas a ANCA36.
BIBLIOGRAFÍA1.- Ramos-Casals M, Brito-Zeron P, Munoz S, etal. A systematic review of the off-label use ofbiological therapies in systemic autoimmunediseases. Medicine (Baltimore) 2008;87(6):345-364.2.- Pato E, Muñoz-Fernández S, Francisco F,Abad MA, Maese J, Ortiz A, Carmona L; Uvei-tis Working Group from Spanish Society ofRheumatology. Systematic review on the effec-tiveness of immunosuppressants and biologicaltherapies in the treatment of autoimmune pos-terior uveitis. Semin Arthritis Rheum 2011Feb;40(4):314-23.3.- Braun J, Baraliakos X, Listing J, et al.Decreased incidence of anterior uveitis inpatients with ankylosing spondylitis treatedwith the anti-tumor necrosis factor agents infli-ximab and etanercept. Arthritis Rheum2005;52(8):2447-2451.4.- Guignard S, Gossec L, Salliot C, et al. Effi-cacy of tumour necrosis factor blockers inreducing uveitis flares in patients with spondy-larthropathy: a retrospective study. Ann RheumDis 2006;65(12):1631-1634.5.- Rudwaleit M, Rodevand E, Holck P, et al.Adalimumab effectively reduces the rate ofanterior uveitis flares in patients with activeankylosing spondylitis: results of a prospectiveopen-label study. Ann Rheum Dis 2009May;68(5):696-701.6.- Ardoin SP, Kredich D, Rabinovich E, et al.Infliximab to treat chronic noninfectious uvei-tis in children: retrospective case series withlong-term follow-up. Am J Ophthalmol2007;144(6):844-849.e1.
FIGURA 3
ESCLERITIS NECROTIZANTE (FLECHAS NEGRAS) Y ADELGAZAMIENTO CORNEAL PERI-FÉRICO (FLECHA BLANCA) EN UN PACIENTE AFECTO DE GRANULOMATOSIS CON
POLIANGEÍTIS (WEGENER)

| 10 |
RevisiónACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:6-10
7.- Foeldvari I, Nielsen S, Kummerle-DeschnerJ, et al. Tumor necrosis factor-alpha blocker intreatment of juvenile idiopathic arthritis-asso-ciated uveitis refractory to secondline agents:results of a multinational survey. J Rheumatol2007;34(5):1146-1150.8.- Gallagher M, Quinones K, Cervantes-Cas-taneda RA, et al. Biological response modifiertherapy for refractory childhood uveitis. Br JOphthalmol 2007 Oct; 91(10):1341-1344.9.- Saurenmann RK, Levin AV, Rose JB, ParkerS, Rabinovitch T, Tyrrell PN, Feldman BM,Laxer RM, Schneider R, Silverman ED.Tumour Necrosis Factor {Alpha} Inhibitors inthe Treatment of Childhood Uveitis. Rheuma-tology (Oxford). 2006 Aug; 45(8):982-989.10.- Vazquez-Cobian LB, Flynn T, Lehman TJ.Adalimumab therapy for childhood uveitis. JPediatr 2006;149(4):572-575.11.- Biester, S, Deuter C, Michels H, et al. Ada-limumab in the therapy of uveitis in childhood.Br J Ophthalmol 2007;91(3):319-324.12.- Gary S. Firestein, MD, Ralph C. Budd,MD, Edward D. Harris, Jr., MD, Iain B. McIn-nes, Shaun Ruddy, MD, John S. Sergent, MD.Kelley's Textbook of Rheumatology, 8th Edi-tion. Elservier Mosby Saunders ISBN: 978-1-4160-3285-4.13.- Murphy CC, Duncan L, Forrester JV, et al.Systemic CD4+ T cell phenotype and activa-tion status in intermediate uveitis.Br J Ophthal-mol 2004 Mar;88(3):412-416.14.- Ziegenhagen MW, Muller-Quernheim J.The cytokine network in sarcoidosis and its clini-cal relevance. J Intern Med 2003;253(1):18-30.15.- Baughman RP, Drent M, Kavuru M, et al.Infliximab therapy in patients with chronic sar-coidosis and pulmonary involvement. Am JRespir Crit Care Med 2006;174(7):795-802.16.- Murphy CC, Ayliffe WH, Booth A, et al.Tumor necrosis factor [alpha] blockade withinfliximab for refractory uveitis and scleritis.Ophthalmology 2004;111(2):352-356.17.- Suhler EB, Smith JR, Wertheim MS, et al.A prospective trial of infliximab therapy for
refractory uveitis: preliminary safety and effi-cacy outcomes. Arch Ophthmol 2005Jul;123(7):903-912.18.- Cruz BA, Reis DD, Araujo CA; MinasGerais Vasculitis Study Group. Refractory reti-nal vasculitis due to sarcoidosis successfullytreated with infliximab. Rheumatol Int. 2007Oct;27(12):1181-3. Epub 2007 May 23.19.- Baughman RP, Lower EE, Bradley DA,Raymond LA, Kaufman A. Etanercept forrefractory ocular sarcoidosis: results of a dou-ble-blind randomized trial. Chest 2005;128:1062-47.20.- Croft AP, Situnayake D, Khair O, GiovanniG, Carruthers D, Sivaguru A, Gordon C. Refrac-tory multisystem sarcoidosis responding to infli-ximab therapy. Clin Rheumatol 2012 Jan 17.21.- Erckens RJ, Mostard RL, Wijnen PA,Schouten JS, Drent M. Adalimumab successfulin sarcoidosis patients with refractory chronicnon-infectious uveitis. Graefes Arch Clin ExpOphthalmol 2011 Nov 27.22.- Doty JD, Mazur JE, Judson MA. Treat-ment of sarcoidosis with infliximab. Chest2005 Mar;127(3):1064-71.23.- Daïen CI, Monnier A, Claudepierre P,Constantin A, Eschard JP, Houvenagel E, et al,Club Rhumatismes et Inflammation (CRI).Sarcoid-like granulomatosis in patients treatedwith tumor necrosis factor blockers: 10 cases.Rheumatology (Oxford) 2009;48: 883-6. 24.- Hatemi G, Silman A, Bang D, Bodaghi B,Chamberlain AM, Gul A, Houman MH, Kötter I,Olivieri I, Salvarani C, Sfikakis PP, Siva A, Stan-ford MR, Stübiger N, Yurdakul S, Yazici H;EULAR Expert Committee. EULAR recom-mendations for the management of Behçet disea-se. Ann Rheum Dis 2008 Dec;67(12):1656-62.25.- Arida A, Fragiadaki K, Giavri E, SfikakisPP. Anti-TNF agents for Behçet's disease:analysis of published data on 369 patients.Semin Arthritis Rheum 2011 Aug;41(1):61-70.26.- Doctor P, Sultan A, Syed S, Christen W,Bhat P, Foster CS. Infliximab for the treatmentof refractory scleritis. Br J Ophthalmol 2010
May;94(5):579-83. Epub 2009 Dec 2.27.- de Menthon M, Cohen P, Pagnoux C,Buchler M, Sibilia J, Détrée F, GayraudM,Khellaf M, Penalba C, Legallicier B, Mou-thon L, Guillevin L. Infliximab orrituximab forrefractory Wegener's granulomatosis: long-term follow up. A prospective randomised mul-ticentre study on 17 patients. Clin Exp Rheu-matol 2011Jan-Feb;29(1 Suppl 64):S63-71.28.- Yoo JH, Chodosh J, Dana R. Relapsingpolychondritis: systemic and ocular manifesta-tions, differential diagnosis, management, andprognosis. Semin Ophthalmol 2011 Jul-Sep;26(4-5):261-9.29.- Kemta Lekpa F, Kraus VB, Chevalier X.Biologics in Relapsing Polychondritis: A Litera-ture Review. Semin Arthritis Rheum 2011 Nov 7.30.- Fricker M, Baumann A, Wermelinger F,Villiger PM, Helbling A. A novel therapeuticoption in Cogan diseases? TNF-alpha blockers.Rheumatol Int 2007 Mar;27(5):493-5.31.- Beccastrini E, Emmi G, Squatrito D, Vannuc-chi P, Emmi L. Infliximab and Cogan's syndrome.Clin Otolaryngol 2010 Oct;35(5):441-2.32.- Hoffman GS, Merkel PA, Brasington RD,et al. Antitumor necrosis factor therapy inpatients with difficult to treat Takayasu arteri-tis. Arthritis Rheum 2004;50:2296-2304.33.- Molloy ES, Langford CA, Clark TM, et al.Antitumour necrosis factor therapy in patientswith refractory Takayasu arteritis: long-termfollow-up. Ann Rheum Dis 2008;67:1567-1569.34.- Hoffman GS, Cid MC, Rendt-Zagar KE, etal. Infliximab for maintenance of glucocorti-costeroid-induced remission of giant cell arteri-tis: a randomized trial. Ann Intern Med 2007;146:621-630.35.- Martínez-Taboada VM, Rodríguez-Val-verde V, Carreño L, et al. A double-blind place-bo controlled trial of etanercept in patients withgiant cell arteritis and corticosteroid sideeffects. Ann Rheum Dis 2008;67:625-630.36.- Díaz-Orta MA, Rojas-Serrano J. [Biologictherapies in the systemic vasculitides]. Reuma-tol Clin 2011 Dec;7 Suppl 3:S33-6.

INTRODUCCIÓNLa práctica rutinaria de un sedimento deorina forma parte del protocolo de moni-torización de ciertas patologías, como elLupus Eritematoso Sistémico. La necesi-dad y periodicidad de esta determinaciónestá menos clara en otras patologías arti-culares inflamatorias como la ArtritisReumatoide (AR) o las Espondiloartropa-tías. Recientemente se ha publicado unarevisión de la Nefropatía IgA en la que serepasa, entre otros aspectos, su asociacióna patología reumática1. A continuación,presentamos un caso clínico ilustrativo yprocedemos a realizar una revisión deesta patología y de su impacto en el con-junto de las enfermedades reumáticas.
CASOPaciente varón de 18 años controlado ennuestras consultas de Reumatología poruna Espondiloartropatía indiferenciadaHLA B27 positiva, con afección exclusi-vamente periférica en forma de una oli-goartritis-entesitis con afección demiembros inferiores. El cuadro se iniciócon una monoartritis de tobillo, con afec-ción aditiva posterior del tobillo contrala-teral, tenosinovitis de tibial posterior yperoneos, y entesitis aquílea. La clínicase inició tras un cuadro autolimitado dediarreas acuosas sin productos patológi-cos. En la anamnesis dirigida no destacóningún otro antecedente personal nifamiliar de significación, ni se evidencia-ron lesiones sugestivas de psoriasis en laexploración física. Analíticamente desta-caba una elevación de reactantes de fase
aguda y el HLA B27 fue positivo. Elresto de analítica con FR, anti-CCP,ANA, bioquímica completa, hemogramay coagulación no mostraron hallazgos designificación. En el sedimento de orina seidentificó una microhematuria, siendo elresto de sedimento normal. El estudioanalítico se completó con un cultivo delíquido sinovial y serologías de artritisreactivas, que fueron todos negativos.Las radiografías de ambos pies, sacroilía-cas y tórax fueron, igualmente, normales.Las pruebas de Mantoux y Booster tam-bién fueron negativas. Ante el fracaso deltratamiento inicial con AINE, se iniciótratamiento con glucocorticoides orales yMetotrexato en escalada rápida hasta 20mg semanales, consiguiendo la remisióndel cuadro. A pesar de la normalizaciónanalítica, ha persistido la microhematuriaaislada con normalidad de la funciónrenal y sin evidencia clínica de HTA. Enuna ocasión se solicitó un estudio enorina de 24 h, en la que detectó una pro-teinuria de 160 mg/24 h. La ecografíarenal fue normal. Valorado por Nefrolo-gía, se solicitaron niveles plasmáticos deIgA que fueron de 527 mg/dl (RN: 70-400). Se completó el estudio con la deter-minación de ANCA y crioglobulinas, quefueron negativas. De momento, siguecontroles simultáneos por Nefrología,con la sospecha de Nefropatía IgA enrelación con su Espondiloartropatía.Hasta la fecha, la presentación clínica yla evolución de la misma no han hechonecesaria la práctica de una biopsia renalde confirmación.
NEFROPATÍA IgAEpidemiologíaLa nefropatía IgA es la glomerulonefritisprimaria más común. Su verdadera pre-valencia, sin embargo, es desconocida, ylos datos disponibles en la literatura sonmuy variables. Ello es debido a la varia-bilidad en la indicación de biopsia renal,necesaria para la confirmación diagnósti-ca, y a la práctica rutinaria a toda lapoblación de sedimentos urinarios seria-dos en algunos países, lo que incrementasu detección precoz en los mismos2. Cabedestacar que el hallazgo de depósitos deIgA en la biopsia renal puede darse hastaen un 3-16% de la población sin padecerla enfermedad3,4. Su prevalencia es máselevada en población asiática y caucási-ca, siendo menos frecuente en la razanegra. Afecta con mayor frecuencia avarones (relación 2:1) y, a pesar de quepuede debutar a cualquier edad, es másfrecuente en la segunda y tercera décadasde la vida5,6. La incidencia anual en Espa-ña, según el Registro Nacional de Glo-merulonefritis, se estima en 6-7 casos pormillón de habitantes.
EtiopatogeniaLa nefropatía IgA es considerada unaenfermedad sistémica en base, principal-mente, a dos hechos: 1) recidiva de lamisma tras el trasplante renal, y 2) reso-lución del daño renal tras trasplante de unriñón afecto a pacientes sanos7. En apoyoa la teoría de la enfermedad sistémica seencuentra su similitud con la patogeniade la púrpura de Henoch-Schonlein (H-
| 11 |
Casos ClínicosACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:11-14
Nefropatía IgA y enfermedades reumáticas.Revisión de la literatura a propósito de un casoMONTOMOLI M1, DE LA MORENA BARRIO I2, ÁVILA BERNABÉU A1, OLLER RODRÍGUEZ JE2, VICENS BERNABÉU E2, YBÁÑEZ GARCÍA D2, VALLSPASCUAL E2, MARTÍNEZ FERRER A2, FECED OLMOS C2, ROBUSTILLLO VILLARINO M2, VIZCAÍNO CASTILLO B1, SERRATO VILLALBA A1, ALEGRESANCHO JJ2
1Servicio de Nefrología2Sección de Reumatología. Hospital Universitario Dr Peset. Valencia
Correspondencia: Dr. Juan José Alegre Sancho - Sección Reumatología - Hospital Dr. Pesset - Avda. Gaspar Aguilar, 90 - 46017 Valencia [email protected]

| 12 |
Casos ClínicosACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:11-14
S), con la que comparte lesiones histoló-gicas y ciertos rasgos, como son la pre-sencia de IgA1 deficitaria en la glicosila-ción, la activación de la vía alternativadel complemento y la aparición de anti-cuerpos específicos. Se han descrito,incluso, casos de gemelos monocigotosdonde uno estaba afecto de NefropatiaIgA y otro de púrpura de H-S8.
La hipótesis más aceptada comomecanismo patogénico se basa en la exis-tencia de una deficiencia en la glicosila-ción de IgA, subtipo 1, tanto a nivel de lasmucosas como en la médula ósea9. EstasIgA1 ven expuesto su extremo N-acetil-galactosamina terminal, que contiene epí-topos con secuencias similares a los con-tenidos en ciertos virus y bacterias, comoson el citomegalovirus, el Haemophylusparainfluenzae, el Staphylococcus aureusy el Toxoplasma10,11. Por mecanismos demimetismo molecular se formarían anti-cuerpos de la clase IgG e IgA contra estosfragmentos, con la subsiguiente forma-ción de inmunocomplejos12. Estos inmu-nocomplejos se depositarían en el mesan-gio renal y estimularían su proliferaciónmediante la activación de dos mediadoresdistintos: el CD71, como inductor de laactivación de la célula mesangial, y el RRFc alfa linfomonocitario, como mediadorde la infiltración intersticial13-16. El acúmu-lo de estos inmunocomplejos se veríapotenciado por un aclaramiento insufi-ciente de los mismos.
Se han investigado posibles factoresgenéticos asociados al desarrollo de laenfermedad y se ha descrito su asocia-ción con cinco locus en los cromosomas1, 6 y 2217. La existencia de estos locus desusceptibilidad ha sido confirmada en unmetaanálisis en el que se incluyó pobla-ción asiática, caucásica y africana18. Losprimeros 3 locus se localizan en el brazocorto del cromosoma 6 (6p21), responsa-ble de la síntesis del complejo mayor dehistocompatibilidad, lo que podría apo-yar una patogenia autoinmune para laenfermedad. El cuarto se sitúa en el locusde interés de la síntesis del factor H(1q32), responsable del control inhibito-rio de la regulación de la vía alternativade la activación del complemento. Resul-ta de interés comprobar en la histología
renal de la nefropatía IgA la presencia deproperdina (vía alternativa) y ausencia deC1q (vía clásica)19. En estos mismos estu-dios se ha podido evidenciar que hasta un5% de los pacientes con Nefropatía IgAposeen un familiar afecto, por lo que seha hipotetizado una posible transmisióndominante con penetrancia incompleta.
ClínicaLa presentación clínica más común, en el95% de los casos, es la hematuria. En un78% de los casos se trata de una microhe-maturia. Sin embargo, hasta un 54% delos pacientes pueden presentar macrohe-maturia, en general durante pocos días ycon el hallazgo persistente de microhe-maturia entre estos episodios. La hematu-ria macroscópica suele presentarse aso-ciada a infecciones respiratorias, en con-creto a faringitis (hematuria sinfaringiti-ca), o a infecciones gastrointestinales.
Otro signo presente en el 30-40% delos casos es la proteinuria, que suele serinferior a 1 g/24 h. Sin embargo, hastaun 5-10% de los pacientes pueden pre-sentar una proteinuria grave o, incluso,desarrollar un síndrome nefrótico.
Un 15% de los pacientes puede desarro-llar hipertensión arterial, asociada o no a undeterioro de la función renal.
DiagnósticoPara la confirmación diagnóstica en unpaciente con los hallazgos anteriormentereseñados, es necesaria la biopsia renal.Las indicaciones para biopsia, sin embar-go, pueden diferir entre países. En general,se indicaría ésta cuando a los hallazgosprevios se añadan una proteinuria sosteni-da >1 gr/24 hr, un aumento significativo dela creatinina o un aumento de la las cifrastensionales respecto a las propias basales20.La biopsia renal en la Nefropatía IgA secaracteriza por los siguientes hallazgos:hipercelularidad mesangial, glomeruloes-clerosis segmental, hipercelularidad endo-capilar, fibrosis intersticial y atrofia tubu-lar. En base a estos hallazgos puede esta-blecerse el diagnóstico de acuerdo con laClasificación de Oxford21. Junto a estoshallazgos, debemos encontrar en las mues-tras depósitos de IgA del subtipo IgA1,junto a los cuales podemos hallar diversos
mediadores de la vía alternativa del com-plemento, como properdina y/o lecitina.En cualquier caso, debemos recordar queel diagnóstico de certeza de la NefropatíaIgA se basa en la observación de depósitosde IgA en la inmunofluorescencia, ya quela apariencia a microscopía óptica puedeser muy variable.
Se han propuesto diversos marcado-res biológicos que pudieran sustituir lanecesidad de biopsia para el diagnósti-co. Entre ellos destacan la cuantifica-ción de la IgA1 deficitaria en la glicosi-lación, que posee una baja sensibilidad yespecificidad, o la determinación deanticuerpos específicos contra estasIgA1 deficientes en su glicosilación,considerados por algunos autores comomarcadores pronósticos, más que diag-nósticos, de la enfermedad12,22. El futuroparece venir marcado por la búsquedade marcadores diagnósticos a través delanálisis proteómico de la orina23,24.
Evolución y pronósticoEsta patología suele tener una evoluciónlenta y progresiva que conduce en el 50%de los casos a diálisis en un plazo de 20-25 años25. En muchos casos puede haberremisiones espontáneas, pero éstas sólopasan a ser sostenidas en un 10% decasos. La probabilidad de entrar en diáli-sis depende del grado de proteinuria, delas características de las lesiones histoló-gícas, y de la gravedad de la hipertensiónarterial, siendo la proteinuria el factorpronóstico más importante26. Teniendoen cuenta estos 3 parámetros se puedecalcular el ARR (ABSOLUTE RENALRISK), que predice la probabilidad detratamiento renal sustitutivo en los 10-20años siguientes al diagnóstico27.
TratamientoAnte la escasez de ensayos clínicos ade-cuadamente diseñados, las recomenda-ciones actuales de tratamiento se basanen consenso de expertos, fundamental-mente el KDIGO 201228. El objetivo delmismo sería enlentecer la progresión dela enfermedad. Sólo en casos seleccio-nados llega a plantearse la regresión dela misma mediante el uso de fármacosinmunosupresores.

No existe una recomendación clarasobre quién podría beneficiarse del trata-miento. En general, no se recomienda tra-tar a pacientes con microhematuria aislada,proteinuria inferior a 0,5-1gr/24 horas, ocon filtrado glomerular normal29. En elresto de casos, se recomienda iniciar trata-miento con IECAS o ARA II con el objeti-vo de mantener una TAS <130 mmHg, ouna proteinuria <1gr/24h30. No ha podidodemostrarse una mayor eficacia del trata-miento combinado. Si no fuera posibleobtener el objetivo de proteinuria <1gr/24h en un plazo de 6 meses, se reco-mienda asociar al tratamiento 3 gr/d de áci-dos grasos omega 3. Suele recomendarsela asociación de estatinas a pesar de que noexisten evidencias de que éstas puedanmodificar la evolución de la enfermedad.
Aunque no existe todavía un consensoal respecto, se recomienda iniciar trata-miento con glucocorticoides en lospacientes con un deterioro rápido o pro-gresivo del filtrado glomerular a pesar deltratamiento con IECAs, con una proteinu-ria en rango nefrótico, o en los que la biop-sia renal muestra lesiones difusas prolife-rativas mesangiales. A pesar de los hallaz-gos clínicos, no se recomienda iniciar tra-tamiento corticoideo cuando la biopsiarenal muestre signos de fibrosis o atrofiatubulointersticial o glomerular31. La pautamás recomendada es la de 0,5 mg/kg/48horas de prednisona oral durante 6 meses,a lo que se añaden pulsos intermitentes de1g de metilprednisolona ev durante 3 díasal principio de los meses 1, 3 y 5. Unapauta alternativa consiste en iniciar el tra-tamiento con prednisona 1 mg/Kg/ddurante dos meses, y proceder a su reduc-ción paulatina durante otros 4 meses.
En los pacientes con síndrome nefróticoy lesiones mínimas en la biopsia no existeduda acerca de la necesidad de tratamiento,para lo cual se utilizan pautas de esteroidessimilares a las utilizadas en casos de enfer-medad de cambios mínimos. Cuando seevidencia en la biopsia una extensa forma-ción de semilunas, debe considerarse la adi-ción de Ciclofosfamida o Azatioprina enpauta similar a las glomerulonefritis extra-capilares32. Se ha ensayado también, enestos casos, el uso de Micofenolato Mofeti-lo, con resultados contradictorios33,34. No se
ha demostrado, en cambio, la utilidad de losantiagregantes, las inmunoglobulinas o laamigdalectomía en estos casos35,36.
Nefropatía IgA en las enfermedadesreumáticasLa nefropatía IgA puede presentarse ais-ladamente o asociada a otras enfermeda-des. Dentro de éstas, las más comunes sonlas enfermedades hepáticas graves, comola cirrosis hepática, debido a una reduc-ción en la eliminación de IgA por parte delas células de Kupffer37,38. Otra enferme-dad clásicamente asociada es la celiaquía,donde parece jugar un papel común lasIgA anti-Gliadina39,40. De todas formas, elaumento de IgA, ya sea por reducción delcatabolismo, como en la patología hepáti-ca, o por su aumentada producción, comoen la celiaquía, por sí solo no explicaría lapatogenia de la Nefropatía IgA. Ademásde su relación con estas patologías, en laliteratura pueden encontrarse casos deNefropatía IgA en pacientes con infec-ción por VIH y asociada a diversas enfer-medades reumáticas.
Nefropatía IgA en las espondiloartro-patíasLa espondiloartropatías son las enfer-medades reumáticas que se asocian conmayor frecuencia a la nefropatía IgA, enespecial la Espondilitis Anquilosante(EA). Debemos recordar que la afecciónrenal en la EA es infrecuente, si exclui-mos la causada por el uso crónico deAINE. Tras ella, la afección más comúnen pacientes con EA activa y de largaevolución es la amiloidosis renal secun-daria, descrita en un 6% de pacientescon EA41. En cuanto a la Nefropatía IgA,no existen datos acerca de su incidenciay/o prevalencia real42. Su aparición noparece guardar relación, a diferencia dela amiloidosis, con la gravedad de laenfermedad articular. Así, se ha descritosu desarrollo tanto de forma previa,como simultáneamente o con posteriori-dad al diagnóstico de EA. A pesar de queen los pacientes con EA es frecuente lapresencia de concentraciones séricaselevadas de IgA y de depósitos cutáneosde la misma, su hallazgo tampoco serelaciona con la afección renal.
Desde un punto de vista práctico,tanto la presentación clínica como loshallazgos en la biopsia renal son indis-tinguibles de la forma primaria. Para sudiagnóstico y/o monitorización se con-sidera suficiente un análisis semestral oanual del sedimento urinario.
El tratamiento de la Nefropatía IgA enel contexto de la EA no difiere tampocodel recomendado en las formas prima-rias. Se ha ensayado el uso de terapiasbiológicas con resultados decepcionan-tes43. En este sentido, destaca una publi-cación reciente según la cual los pacien-tes tratados con Etanercept obtuvieronuna importante mejora de la clínica arti-cular, pero no de la afección renal44.
Nefropatía IgA en otras enfermedadesreumáticasSe han descrito casos en pacientes con AR,granulomatosis con poliangeítis (Wege-ner) o policondritis recidivante. Su asocia-ción, en muchos casos, puede ser mera-mente casual. En otros casos, se ha asocia-do su desarrollo a la inmunosupresióninducida por el tratamiento de las mismas.Se ha postulado que la situación de inmu-nodeficiencia predispondría a infeccionesde la vías aéreas, las cuales serían final-mente las iniciadoras de los fenómenos deactivación a nivel renal. Así, se han descri-to casos de granulomatosis con poliangeí-tis en los que la suspensión del tratamientoinmunosupresor se asoció a una resolucióndel daño renal, lo que descarta una posibleasociación con la propia vasculitis45. Estosmismo argumentos han sido empleados enun caso de un paciente con AR que des-arrolló una Nefropatia IgA mientras reci-bía tratamiento con Abatacept46.
CONCLUSIONESLa Nefropatía IgA es una enfermedad sis-témica con afección renal de etiopatoge-nia desconocida que puede asociarse,aunque de forma rara y ocasional, a pato-logía reumática, en especial a las espondi-loartropatías. Su aparición no guarda rela-ción con la evolución de la enfermedadinflamatoria y su hallazgo más frecuentees la hematuria, en general de caráctermicroscópico. Por ello, se recomienda elcontrol rutinario del sedimento urinario
| 13 |
Casos ClínicosACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:11-14

| 14 |
Casos ClínicosACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:11-14
cada 6 meses. Con frecuencia evolucionalentamente al fallo renal. El objetivo de sutratamiento es enlentecer el deterioro dela función renal mediante el control de lascifras tensionales, de la dislipemia, y de laproteinuria, mediante el uso de IECAS,ARAII, estatinas y ácidos omega 3. Encasos seleccionados puede ser necesarioel uso de glucocorticoides y ciclofosfami-da. Es, por ello, necesario un diagnósticoprecoz, ya que el tratamiento es ineficazuna vez establecido el daño renal.
BIBLIOGRAFÍA1.- Wyatt RJ, Julian BA. IgA nephropathy. NEngl J Med 2013;368:2402.2.- Suzuki K, Honda K, Tanabe K, et al. Inci-dence of latent mesangial IgA deposition inrenal allograft donors in Japan. Kidney Int2003;63:2286.3.- Waldherr R, Rambausek M, Duncker WD,Ritz E. Frequency of mesangial IgA deposits ina non-selected autopsy series. Nephrol DialTransplant 1989;4:943.4.- Barratt J, Feehally J. IgA nephropathy. J AmSoc Nephrol 2005;16:2088.5.- Wyatt RJ, Kritchevsky SB, WoodfordSY, etal. IgA nephropathy: long-termprognosis forpediatric patients. J Pediatr 1995;127:913-9.6.- Wyatt RJ, Julian BA, Baehler RW, et al. Epi-demiology of IgA nephropathy in central andeastern Kentucky for the period 1975 through1994. J Am Soc Nephrol 1998;9:853-8. 7.- Silva FG, Chander P, Pirani CL, Hardy MA.Disappearance of glomerular mesangial IgAdeposits after renal allograft transplantation.Transplantation 1982;33:241-6.8.- Davin JC. Henoch-Schönlein purpura neph-ritis: pathophysiology, treatment, and futurestrategy. Clin J Am Soc Nephrol 2011;6:679-89.9.- Allen AC, Harper SJ, Feehally J. Galactos-ylation of N- and O-linked carbohydrate moie-ties of IgA1 and IgG in IgA nephropathy. ClinExp Immunol 1995;100:470-4.10.- Suzuki S, Nakatomi Y, Sato H, et al. Hae-mophilus parainfluenzae antigen and antibodyin renal biopsy samples and serum of patientswith IgA nephropathy. Lancet 1994;343:12.11.- Novak J, Julian BA, Mestecky J, RenfrowMB. Glycosylation of IgA1 and pathogenesisof IgA nephropathy. Semin Immunopathol2012;34:365-82.12.- Suzuki H, Fan R, Zhang Z, et al. Aberrantlyglycosylated IgA1 in IgA nephropathy patients isrecognized by IgG antibodies with restricted hete-rogeneity. J Clin Invest 2009;119:1668-77.13.- Amore A, Conti G, Cirina P, et al. Abe-rrantly glycosylated IgA molecules downregu-late the synthesis and secretion of vascularendothelial growth factor in human mesangialcells. Am J Kidney Dis 2000;36:1242-52.14.- Lai KN. Pathogenesis of IgA nephropathy.Nat Rev Nephrol 2012;8:275-83.15.- Moura IC, Benhamou M, Launay P,Vrtovsnik F, Blank U, Monteiro RC. The glo-merular response to IgA deposition in IgA
nephropathy. Semin Nephrol 2008;28:88-95. 16.- Launay, P, Grôssetete B, Arcos Fajardo M,et al. Fc alpha Receptor (CD89) Mediates theDevelopment of Immunoglobulin A (IgA)Nephropathy (Berger's Disease). Evidence forpathogenic soluble receptor-IgA complexes inpatients and CD89 transgenic mice. J Exp Med2000;191:1999-2010. 17.- Gharavi AG, Kiryluk K, Choi M, et al.Genome-wide association study identifies sus-ceptibility loci for IgA nephropathy. Nat Genet2011;43:321-7. 18.- Kiryluk K, Li Y, Sanna-Cherchi S, et al.Geographic differences in genetic susceptibi-lity to IgA nephropathy: GWAS replicationstudy and geospatial risk analysis. PLoS Genet2012;8:e1002765.19.- Miyamoto H, Yoshioka K, TakemuraT,Akano N, Maki S. Immunohistochemical studyof the membrane attack complex of comple-ment in IgA nephropathy. Virchows Arch APathol Anat Histopathol 1988;413:77-86.20.- Hall CL, Bradley R, Kerr A, et al. Clinicalvalue of renal biopsy in patients with asymptoma-tic microscopic hematuria with and without low-grade proteinuria. Clin Nephrol 2004;62:267.21.- Cattran DC, Coppo R, Cook HT, et al. TheOxford classification of IgA nephropathy:rationale, clinicopathological correlations,andclassification. Kidney Int 2009;76:534-45.22.- Moldoveanu Z, Wyatt RJ, Lee JY, et al.Patients with IgA nephropathy have increasedserum galactose-deficient IgA1 levels. KidneyInt 2007;71:1148-54. 23.- Haubitz M, Wittke S, Weissinger EM, et al.Urine protein patterns can serve as diagnostictools in patients with IgA nephropathy. KidneyInt 2005;67:2313-20.24.- Julian BA, Wittke S, Novak J, et al. Elec-trophoretic methods for analysis of urinarypolypeptides in IgA-associated renal diseases.Electrophoresis 2007;28:4469-83.25.- Radford MG Jr,Donadio JV Jr, Bergstral EJ,Grande JP. Predicting renal outcome in IgA neph-ropathy. J Am Soc Nephrol 1997;8:199-207.26.- Gutiérrez E, González E, Hernández E,Morales E, Martínez MA, Usera G, Praga M.Factors that determine an incomplete recoveryof renal function in macrohematuria-inducedacute renal failure of IgA nephropathy. Clin JAm Soc Nephrol 2007;2:51.27.- Berthoux F, Mohey H, Laurent B, MariatC, Afiani A, Thibaudin L. Predicting the riskfor dialysis or death in IgA nephropathy.J AmSoc Nephrol 2011;22:752-61.28.- Immunoglobulin A nephropathy. Generalprinciples in the management of glomerulardisease. KDIGO Clinical Practice Guidelinefor Glomerulonephritis. Kidney Int Suppl2012;2:209-217.29.- Appel GB, Waldman M.The IgA nephro-pathy treatment dilemma. Kidney Int. 2006Jun;69:1939-44.30.- Praga M, Gutiérrez E, González E, MoralesE, Hernández E. Treatment of IgA nephropathywith ACE inhibitors: a randomized and controlledtrial. J Am Soc Nephrol 2003;14:1578.31.- Floege J, Eitner F. Present and future the-rapy options in IgA-nephropathy. J Nephrol.2005 Jul-Aug;18:354-61.
32.- Daniel C Cattran, MD, Gerald B Appel,MD. Treatment and prognosis of IgA nephro-pathy UptoDate. Literature review currentthrough: Sep 2013. This topic last updated: feb11,2013.33.- Maes BD, Oyen R, Claes K, Evenepoel P,Kuypers D, et al. Mycophenolate mofetil inIgA nephropathy: results of a 3-year prospecti-ve placebo-controlled randomized study. Kid-ney Int 2004;65:1842.34.- Tang, et al. Long-term study of mycophe-nolate mofetil treatment in IgA nephropathy.Kidney Int 2010;77: 543-9.35.- Komatsu H, Fujimoto S, Hara S, Sato Y,Yamada K, Kitamura K. Effect of tonsillec-tomy plus steroid pulse therapy on clinicalremission of IgA nephropathy: a controlledstudy. Clin J Am Soc Nephrol 2008;3:1301.36.- Rostoker G, Desvaux-Belghiti D. High-dose immunoglobulin therapy for severe IgAnephropathy and Henoch-Schönlein purpura.Ann Intern Med 1994;120:476.37.- Amore A, Coppo R, Roccatello D, et al.Experimental IgA nephropathy secondary tohepatocellular injury induced by dietary defi-ciencies and heavy alcohol intake. Lab Invest1994;70: 68.38.- Van de Wiel A, Valentijn RM, SchuurmanHJ, et al. Circulating IgA immune complexesand skin IgA deposits in liver disease. Relationto liver histopathology. Dig Dis Sci1988;33:679.39.- Sategna-Guidetti C, Ferfoglia G, Bruno M,Pulitano R, Roccatello D, Amore A, Coppo R.Do IgA antigliadin andIgA antiendomysiumantibodies show there is latent coeliac disease inprimary IgA nephropathy? Gut 1992;33: 476-8.40.- Almroth G, Axelsson T, Mussener E,Grodzinsky E, Midhagen G, Olcen P. Increasedprevalence of anti-gliadin IgA-antibodies withaberrant duodenal histopathological findings inpatients with IgA-nephropathy and relateddisorders. Ups J Med Sci 2006;111:339-52.41.- Lance NJ, Curran JJ. Amyloidosis in a caseof ankylosing spondylitis with a review of theliterature.J Rheumatol 1991;18:100.42.- Gratacós J, Collado A, SanmartíR, Poch E,Torras A, Muñoz-Gomez J. Coincidental amy-loid nephropathy and IgA glomerulonephritisin a patient with ankylosing spondylitis. JRheumatol 1993;20:1613.43.- Jacquet A, Francois H, Frangie C, YahiaouiY, Ferlicot S, et al. IgA nephropathy associatedwith ankylosing spondylitis is not controlled byinfliximab therapy. Nephrol Dial Transplant2009;24:3540-2.44.- Sang-Hoon Lee, Eun Jung Lee. Renal invol-vement in ankylosing spondylitis: prevalence,pathology, response to TNF-a blocker Rheuma-tology International 2013;33:1689-1692.45.- Andrassy K, Waldherr R, Erb A, Ritz E. Denovo glomerulonephritis in patients duringremission from Wegener's granulomatosis.Clin Nephrol 1992;38:295.46.- Michel M, Henri P, Vincent FB, Leon N, Mar-celli C. Mesangial immunoglobulin (Ig)A glome-rulonephritis in a patient with rheumatoid arthritistreated with abatacept. Joint Bone Spine 2013 Jun28. pii: S1297-319X(13)00143-7. doi: 10.1016/j.jbspin.2013.05.003. [Epub ahead of print].

| 15 |
Casos ClínicosACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:15-18
INTRODUCCIÓNLa osteomielitis pubis es una infección
localizada en el tejido óseo de las ramas
púbicas que cursa con dolor en la región
suprapúbica, dificultad para caminar y
aumento de la base de sustentación a la
deambulación1. Las osteomielitis pélvi-
cas se han relacionado con la utilización
de catéteres o dispositivos implantados,
los traumatismos, las úlceras por pre-
sión, la cirugía pélvica o urológica, las
lesiones obstétricas o el aborto, el cate-
terismo cardíaco y el uso de drogas
intravenosas2.
El cuadro clínico es indistinguible de
la osteítis del pubis, una inflamación
“estéril” de la sínfisis púbica, que es fre-
cuente en deportistas, pero también se
relaciona con traumatismos, embarazo y
cirugía pélvica3.
Esta dificultad en el diagnóstico dife-
rencial hace que en casos sospechosos
sea necesario utilizar pruebas de ima-
gen, como la resonancia magnética,
para descartar infección.
CASO CLÍNICOHombre de 71 años que acude a consul-
ta de reumatología por coxalgia dcha. El
dolor se había iniciado 6 meses antes,
empeoraba con la bipedestación y el
esfuerzo físico, y aumentó progresiva-
mente hasta impedir la deambulación y
aparecer también en reposo. En el
momento de la consulta el dolor se irra-
diaba hasta ingle izquierda, sin pareste-
sias. La anamnesis fue negativa para fie-
bre o clínica infecciosa.
Como antecedentes de interés estaba
en tratamiento con hipolipemiantes
(ezetimiba) y se había sometido a ciru-
gía en varias ocasiones: hernia crural
derecha (11 meses antes) y adenomecto-
mía de próstata (7 meses antes).
La exploración física reveló dolor en
cadera derecha con limitación de la
movilidad en todos los planos y dolor a
la palpación en hipogastrio y pubis, sin
signos de irritación peritoneal ni eviden-
cia de visceromegalias o masas. La
exploración de la cadera izquierda fue
normal, así como la palpación de cade-
nas ganglionares y la auscultación car-
dio-pulmonar. Las constantes vitales:
temperatura, tensión arterial, frecuencia
cardíaca y frecuencia respiratoria se
encontraban dentro de los límites nor-
males.
Ante la sospecha de artritis de cadera
se realizó ecografía articular objetivando
distensión de cápsula articular de conte-
nido hipoecoico y colección hipoecoica
compresible en partes blandas, compati-
ble con artritis y absceso ilio-psoas.
(Figura 1-A) Se realizó punción del abs-
ceso con guía ecográfica obteniendo
líquido inflamatorio (89.000 leucoci-
tos/mm3) que se cultivo.
Con el diagnóstico de monoartritis de
cadera y absceso ilio-psoas se extraje-
ron analítica y cultivos de sangre y
orina, y se inició tratamiento antibiótico
empírico con cloxacilina 2 g IV cada 8
horas y ceftriaxona 2 g IV cada 24
horas. Para completar el estudio se soli-
citó radiografía pélvica, resonancia
magnética y gammagrafía ósea.
La analítica de sangre detectó anemia
(Hb 9,1 g/dl) microcítica hipocrómica,
trombocitosis (plaquetas 743 mil/ul) y
aumento de reactantes de fase aguda
(PCR 11,8 mg/dl; VSG 120 mm/h). El
estudió microbiológico del líquido sino-
vial mostró bacilos gram-negativos en
tinción de Gram por los que se sustituyó
el antibiótico por meropenem 1 g IV cada
8 horas. A las 48 el cultivo fue positivo
Coxalgia progresiva tras adenomectomía de próstataSENABRE-GALLEGO JM1, SALAS-HEREDIA E1, SANTOS-RAMÍREZ C2, SANTOS-SOLER G1, ORTEGA-CASTRO R2, ROSAS J1
1Sección de Reumatología del Hospital Marina Baixa. Villajoyosa. Alicante2Sección de Reumatología del Hospital de Denia. Alicante
Correspondencia: Dr. José Miguel Senabre - Sección Reumatología - Hospital Marina Baixa - Avda. Jaime Botella Mayor, 7 -
03570 Villajoyosa (Alicante)
La similitud clínica entre osteítis púbica y osteomielitis infecciosa dalugar a dificultades diagnósticas que requieren pruebas de imagenavanzadas como la resonancia magnética. Presentamos el caso clínico
de un paciente con coxalgia tras cirugía de próstata en el que las prue-bas de imagen fueron fundamentales para confirmar la osteomielitis,conseguir el diagnóstico etiológico y pautar el tratamiento adecuado.
/ RESUMEN
Palabras clave: osteítis pubis, osteomielitis pubis, artritis séptica, resonancia magnética, gammagrafía ósea.

| 16 |
Casos ClínicosACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:15-18
para Pseudomonas aeruginosa y el anti-biograma multisensible por lo que se sus-tituyó el antibiótico por ceftazidima, conmejor posología para una futura adminis-tración domiciliaria. Tanto hemocultivoscomo urocultivos fueron negativos.
La radiografía simple de pelvis mos-tró aumento del espacio articular en lasínfisis púbica, de bordes irregulares ymal definidos. Ambas articulacionescoxo-femorales fueron normales (Figu-ra 2).
La RM confirmó los hallazgos dederrame en articulación coxo-femoralderecha y bursitis ilio-psoas. Ademásdetectó edema óseo en hueso esponjosoen ambas láminas cuadriláteras delpubis, con signos de osteolisis, y edemaóseo en la mitad anterior de ramasisquiopubianas (Figura 3).
La gammagrafía ósea con tecnecio-99mmostró hipercaptación en sínfisis púbi-ca, predominante en fase tardía, comoproceso osteoblástico activo. La capta-ción de galio-67, aunque de menorintensidad, corroboró la sospecha deinflamación activa en pubis (Figura 4).
El paciente fue mejorando progresiva-mente, tanto clínica como analíticamente(Tabla 1). La siguiente exploración eco-gráfica al décimo día de ingreso mostróresolución del absceso ilio-psoas (Figura1-B). El paciente fue dado de alta hospi-talaria e ingresó en la Unidad de Asisten-cia en Domicilio para continuar con trata-miento intravenoso hasta completar unmes. La mejoría se mantuvo en lossiguientes controles ambulatorios y secontinuó la antibioterapia con ciproflo-xacino 750 mg VO cada 12 horas. Enmay-11, tras 6 meses de tratamiento anti-biótico, al observar mejoría clínica y enlos controles analíticos y de imagen,(Figura 2-B) se decide interrumpir el tra-tamiento antibiótico. El paciente seencuentra asintomático hasta la fecha.
DISCUSIÓNEl diagnóstico definitivo fue artritis sép-tica de cadera por Pseudomonas aerugi-nosa con absceso ilio-psoas y osteomie-litis pubis.
En nuestro caso, la ausencia de sínto-mas sistémicos y el inicio insidioso de la
clínica podría haber orientado el diag-nóstico inicial hacia la osteítis no infec-ciosa. Pero la concomitancia de mono-artritis y absceso en proximidad hicie-ron necesario descartar osteomielitis.
Los hallazgos de la radiografía simple(ensanchamiento de la sínfisis púbica,reabsorción ósea y osteopenia) suelen sertardíos y pueden estar presentes en las dosentidades por lo que no es una técnica útilen el diagnóstico diferencial. Sin embar-go la RMN puede mostrar cambios de
osteomielitis, como el edema óseo o elderrame articular, que no se observan enla osteítis “aséptica”. Asimismo estándescritos la formación de abscesos en lostejidos blandos adyacentes a la pelvisósea o disecando áreas a distancia4.
Cuando no es posible realizar unaRM, por ejemplo en pacientes portado-res de implantes metálicos, la pruebaindicada es la TC. La TC es capaz demostrar el hueso cortical y trabecular, lareacción perióstica, el gas o los secues-
FIGURA 1
FIGURA 2
ECOGRAFÍA DE CADERA DERECHA. A: DÍA DEL INGRESO. DISTENSIÓN DE CÁPSULAARTICULAR
DE CONTENIDO HIPOECOICO (FLECHA) Y COLECCIÓN HIPOECOICA COMPRESIBLE EN PARTES
BLANDAS, COMPATIBLE CON ARTRITIS YABSCESO ILIO-PSOAS. B: RESOLUCIÓN DELABSCESO Y
PERSISTENCIA DISTENSIÓN DE CAPSULAARTICULAR TRAS 10 DÍAS DE EVOLUCIÓN
RADIOGRAFÍA DE CADERAS. A. RADIOGRAFÍA SIMPLE DE PELVIS (PROYECCIÓN ANTERO-POSTE-RIOR) A FECHA DEL INGRESO EN REUMATOLOGÍA: AUMENTO DE ESPACIO ARTICULAR EN SÍNFISIS
PÚBICA DE BORDES IRREGULARES Y MAL DEFINIDOS. AMBAS ARTICULACIONES COXO-FEMORA-LES NORMALES. B. RADIOGRAFÍA SIMPLE DE PELVIS TRAS 6 MESES DE SEGUIMIENTO: AUMENTO
DEL ESPACIO ARTICULAR EN SINFISIS PÚBICA (MENOR RESPECTO A EXPLORACIÓN PREVIA) YESCLEROSIS DE BORDES ÓSEOS. C. RADIOGRAFÍA SIMPLE DE PELVIS TRAS 1 AÑO DE SEGUIMIEN-TO: IRREGULARIDAD ARTICULAR Y ESCLEROSIS DE BORDES ÓSEOS EN SÍNFISIS PÚBICA

| 17 |
Casos ClínicosACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:15-18
tros óseos. Sin embargo los metalestambién artefactan las imágenes obteni-das, dificultando su interpretación.
La gammagrafía ósea es de eleccióncuando la TC o RM no se pueden utili-zar. Los isótopos más empleados son el
tecnecio-99m en tres fases y el galio-67,más sensible y específico para detectarinflamación. Los leucocitos marcadosse utilizan con menor frecuencia por seruna técnica más laboriosa. Los hallaz-gos deben ser interpretados con cauteladado que el aumento del remodeladoóseo en patologías degenerativas pue-den dar lugar a falsos positivos5. Sinembargo una gammagrafía con Ga-67negativa descarta el diagnóstico de oste-omielitis6.
La ecografía músculo-esquelética esla técnica de elección para detectar artri-tis de cadera y puede servir de guía parala aspiración del líquido sinovial7. Suaccesibilidad en la consulta de reumato-logía evita demoras en el diagnóstico y eltratamiento. Por ejemplo, en nuestrocaso, la punción se realizó inmediata-mente tras el diagnóstico del absceso y seobtuvo muestra antes de iniciar trata-miento antibiótico, mejorando así el ren-dimiento del cultivo. Obtener una mues-tra para cultivo es necesario para el diag-nóstico etiológico, y permite realizar unantibiograma que indique la sensibilidada diferentes antibióticos. La biopsia óseaestá indicada para descartar infeccióncuando los cultivos son negativos. Ennuestro caso no se realizó por disponer deun cultivo positivo del absceso y por labuena evolución. Si la sospecha clínicaes alta, el inicio de tratamiento antibióti-co de forma empírica está recomendado8.
La importancia del diagnóstico deosteomielitis, en el presente caso, es queprolonga la duración recomendada de laantibioterapia. En casos de artritis sépti-ca (sin osteomielitis) se recomiendanregímenes de 14 días de tratamientoparenteral seguidos de otros 14 días detratamiento oral. Sin embargo, la dura-ción de la vía parenteral se puede redu-cir en gérmenes sensibles a antibióticoscon buena biodisponibilidad oral, comolas fluoroquinolonas, y se debe prolon-gar en otros casos de microorganismosresistentes como Enterobacter o Pseu-domonas9,10.
Cuando existe osteomielitis la mayo-ría de los expertos están a favor de con-tinuar con tratamiento intravenoso hastaque el hueso desbridado se recubre de
RESONANCIA MAGNÉTICA DE PELVIS, CORTES A NIVEL DE SÍNFISIS PÚBICA. AMPLIACIÓN DE
LA INTERLÍNEAARTICULAR DE SÍNFISIS PÚBICA CON CARILLAS ARTICULARES IRREGULARES DE
MARGEN HIPOINTENSO EN SECUENCIAT1 POR ESCLEROSIS ÓSEA (A-B); ASOCIA CAMBIOS DE
LA SEÑAL ÓSEA VECINA EN PUBIS, CON HIPERINTENSIDAD T2-SUPRESIÓN GRASA (C-D), EHIPOINTENSIDAD T1 CON REALCE DIFUSO, POR EDEMA ÓSEO, SIN INVOLUCRO INTRAMEDU-LAR ÓSEO. COLECCIÓN EN VIENTRE MUSCULAR DE PSOAS ILÍACO CON PROLONGACIÓN HACIA
LA REGIÓN INGUINAL QUE SE COMUNICA CON DERRAME ARTICULAR COXO-FEMORAL
FIGURA 3
GAMMAGRAFÍA ÓSEA. HIPERCAPTACIÓN DE TC-99 EN SÍNFISIS PÚBICA, PREDOMINANTE EN
FASE TARDÍA, COMO PROCESO OSTEOBLÁSTICO ACTIVO. LA CAPTACIÓN DE GA-67, AUNQUE
DE MENOR INTENSIDAD, CORROBORÓ LA SOSPECHA DE INFLAMACIÓN ACTIVA EN PUBIS
FIGURA 4

| 18 |
Casos ClínicosACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:15-18
tejido vascularizado, como mínimohasta 6 semanas después de la cirugía.La monitorización de los marcadoresinflamatorios séricos, como la VSG o laPCR pueden ser útiles, si no se han nor-malizado al finalizar el plan de antibio-terapia, se deben investigar clínica yradiológicamente los motivos. En nues-tro caso el desbridamiento quirúrgico nofue necesario por la buena evolución clí-nica con el antibiótico pautado, que semantuvo hasta que se confirmó remi-sión clínica y analítica completa.
En conclusión, el presente caso deberecordarnos mantener la sospecha diag-nóstica de procesos infecciosos enpacientes sometidos a cirugía pélvica,incluso cuando la clínica de dolor se ini-cia varios meses después de la interven-ción. La realización de una anamnesisexhaustiva y una exploración clínicaminuciosa va a orientar el diagnósticoinicial, que se debe completar con prue-bas de imagen y cultivos, para obtener eldiagnóstico etiológico y pautar el trata-miento adecuado.
Agradecimientos: Este trabajo ha sidoapoyado con una beca de la Asociaciónpara la Investigacióin en Reumatologíade la Marina Baixa (AIRE-MB).
Componentes del Grupo AIRE-MB:José Rosas, Esteban Salas, José MiguelSenabre-Gallego, Gregorio Santos-Soler,Catalina Cano, Mª Luisa Betoret, AnaPons (S. Reumatología, Hospital Marina
Baixa), Francisca LLinares-Tello, JuanMolina (S. Laboratorio, Hospital MarinaBaixa); Carlos Santos-Ramírez, RafaelaOrtega Castro (S. Reumatología HospitalMarina Alta. Denia), Xavier Barber (Cen-tro de Operaciones e Investigación de laUniversidad Miguel Hernández, Elche),Mabel Sánchez-Barrioluengo (INGENIO(CSIC-UPV), Universitat Politècnica deValència). .
BIBLIOGRAFÍA1.- Mader R, Yeromenco E. Pseudomonas Oste-omyelitis of the symphysis Pubis After InguinalHernia Repair. Clin Rheumatol (1999)18:167-169.2.- Sexton DJ. Pelvic osteomyelitis. In: UpToDa-te, Basow, DS (Ed), UpToDate, Waltham, MA,2013.3.- Rodríguez Montero S. Sinfisitis púbica. Revi-sión bibliográfica. Seminarios de la FundaciónEspañola de Reumatología 2007;8:145-53.4.- Erdman WA, Tamburro F, Jayson HT, Weathe-rall PT, Ferry KB, Peshock RM. Osteomyelitis:
characteristics and pitfalls of diagnosis with MRimaging. Radiology 1991;180(2):5335.- Tumeh SS, Aliabadi P, Weissman BN, McNeilBJ. Chronic osteomyelitis: bone and gallium scanpatterns associated with active disease. Radiology1986;158(3):685.6.- Pineda C, Vargas A, Rodríguez AV. Imaging ofosteomyelitis: current concepts. Infect Dis ClinNorth Am 2006;20(4):789.7.- Ho G, Jue SJ, Cook PP. Artritis producida porbacterias o sus componentes. En Harris E, BuddRC, Genovese MC, Firestein GS, Sergent JS ySledge CB. Kelley Tratado de Reumatología. 7ªEdición. Madrid: Elsevier España; 2006. p. 1638.8.- Lalani T. Overview of osteomyelitis in adults.In: UpToDate, Basow, DS (Ed), UpToDate, Wal-tham, MA, 2013.9.- Goldenberg DL, Sexton DJ. Septic arthritis inadults. In: UpToDate, Basow, DS (Ed), UpToDa-te, Waltham, MA, 2013.10.- González Ferrández JA, Noguera Pons JR,Tovar Beltrán JV, Navarro Blasco FJ. Artritisinfecciosa. En: Belmonte Serrano MA, Castella-no Cuesta JA, Román Ivorra JA, Rosas Gómez deSalazar J. Enfermedades reumáticas: Actualiza-ción SVR. 2ª Edición. Valencia: Ibáñez y Plaza;2013. p. 659-679.
TABLA 1
EVOLUCIÓN DE LOS PARÁMETROS ANALÍTICOS
Ingreso 10 días 1 mes 6 meses
Hb (g/dl) 9,1 10,1 12,1 13,0
Plaquetas (miles/ul) 743 604 300 160
Leucocitos (miles/ul) 5,7 6,1 6,5 4,0
PCR (mg/dl) 11,8 4,0 0,6 0,1
VSG (mm/h) 120 ND ND 7

| 19 |
Casos ClínicosACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:19-21
INTRODUCCIÓNEl síndrome de Sjögren (SS) es una exo-crinopatía crónica autoinmune caracteri-zado por la infiltración de las glándulasexocrinas por linfocitos y células plasmá-ticas. Entre las manifestaciones extra-glandulares puede afectar diferentes órga-nos cómo la piel, pulmón, corazón, riñón,sistema nervioso y hematopoyético. Lascomplicaciones pulmonares incluyensequedad de la vía aérea, diversos tipos deafectación intersticial, linfoma no Hodg-kin, engrosamiento pleural y raramenteenfermedad tromboembólica e hiperten-sión pulmonar. El tratamiento de la afec-tación pulmonar se basa en la identifica-ción anatomopatológica, la severidad delos síntomas y la extensión radiográfica.A continuación se presenta el caso de unapaciente con SS primario que debuta conafectación pulmonar tipo neumonitisintersticial linfocítica.
DESCRIPCIÓN DEL CASOMujer de 70 años con antecedentes dehipertensión arterial, hipercolesterole-
mia, síndrome seco (xeroftalmia y xeros-tomía) y adenocarcinoma de mama inter-venido en 2002, actualmente libre deenfermedad. La paciente ingresa condiagnóstico de bronconeumonía infec-ciosa en contexto de cuadro de disnea deesfuerzo de un mes de evolución que haempeorado en la última semana asociadoa malestar general y un pico febril de38ºC, sin otra clínica asociada, salvoastenia progresiva desde hace 4 meses.
A su ingreso la paciente presentaregular estado general, saturación basalde oxígeno de 88% con crepitantes biba-sales. En la analítica destacan 11.200leucocitos (72% de neutrófilos), proteí-na C reactiva (PCR) de 56 mg/L, veloci-dad de sedimentación globular (VSG)de 28 mm/h, resto de hemograma y bio-química normales. En la radiografía detórax se aprecian infiltrados alveolaresen ambos lóbulos inferiores, língula yperiferia del lóbulo superior izquierdosugestivo de bronconeumonía bilateral(Figura 1). Se inicia tratamiento conoxígeno y antibioterapia empírica con
ertapenem y claritromicina. Ante la tór-pida evolución clínica y radiográfica seamplía estudio mediante tomografíaaxial computarizada de alta resolución(TACAR) de tórax que muestra múlti-ples infiltrados confluentes, distribuidosde forma difusa por ambos hemitórax,predominando en lóbulos inferiores,compatible con focos de bronconeumo-nía (Figura 2). Se realiza broncoscopíaque resulta normal, siendo el BAS nega-tivo para malignidad, sin flora patológi-ca y con presencia de moderados leuco-citos. Todos los cultivos y serologíasinfecciosas realizadas fueron negativas,salvo un esputo aislado positivo parapseudomona, sensible a piperazilinatazobactam, por lo que se modifica anti-bioterapia. Ante la escasa respuesta altratamiento antibiótico se amplía analí-tica y se solicita estudio de autoinmuni-dad (previamente negativa) con positi-vidad del factor reumatoide, los anti-cuerpos antinucleares a título 1/320 yAnti-Ro. En el proteinograma se obser-va una hipergammaglobulinemia poli-
Neumopatía intersticial linfocítica como manifestacióninicial en paciente con síndrome de Sjögren primario:a propósito de un casoORTEGA-CASTRO R1, SANTOS-RAMÍREZ C1, SENABRE-GALLEGO JM2, SANTOS-SOLER G2, SALAS-HEREDIA E2, ROSAS J2, GRUPO AIRE-MB1Sección de Reumatología del Hospital de Denia. Alicante2Sección de Reumatología del Hospital Marina Baixa. Villajoyosa. Alicante
Correspondencia: Dra. Rafaela Ortega Castro - Reumatología - Hospital de Denia - Partida Beniadlá s/n - 03700 Denia (Alicante) [email protected]
El síndrome de Sjögren (SS) es una enfermedad autoinmune, crónica,inflamatoria, que se caracteriza por la infiltración de las glándulas exo-crinas por linfocitos y células plasmáticas. La destrucción glandular dalugar a sus principales manifestaciones clínicas: queratoconjuntivitisseca y xerostomía. Sin embargo, puede tener afectación sistémica a
través de la infiltración linfoide de diferentes órganos, cómo el pulmón.La afectación pulmonar asociada con el SS es más frecuente en lasformas primarias. Suele ocurrir entre 5-10 años después del comienzode la enfermedad, aunque puede ocasionalmente preceder el diagnós-tico de la misma, como en el caso que presentamos a continuación.
/ RESUMEN
Palabras clave: síndrome de Sjögren primario, afectación extraglandular, neumopatía intersticial.

| 20 |
Casos ClínicosACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:19-21
clonal. En el TACAR de control no se aprecian cambios res-pecto al primero, objetivándose condensaciones pulmonaresparcheadas bilaterales, de predominio en campos postero-infe-riores, engrosamiento intersticial inter e intralobulillar y bron-quiectasias cilíndricas por tracción, todo ello sugestivo defibrosis, ya presente en el TAC previo y dados los antecedentesde la paciente sugestivo de enfermedad pulmonar intersticialen relación con enfermedad de Sjögren. Ante la sospecha clíni-ca inicial de bronconeumonía infecciosa, previo al inicio detratamiento se decide completar estudio con biopsia pulmonarabierta siendo el diagnóstico histopatológico de neumonitisintersticial linfocitaria moderada asociada a bronquiolitis foli-cular con mínima fibrosis. Finalmente, con diagnóstico de sín-
drome de SS primario con neumopatía intersticial, se inicia tra-tamiento con prednisona 1 mg/Kg/día más suplementos de cal-cio-vitamina D y bifosfonatos con importante mejoría clínica.
DISCUSIÓNEl SS es una exocrinopatía crónica autoinmune, de progresiónlenta y etiología desconocida, caracterizado por infiltraciónlinfocítica glandular y extraglandular asociado a producción deautoanticuerpos. Puede existir cómo un desorden primario ais-lado (SS primario) o asociado con otros procesos autoinmunescomo la artritis reumatoide, lupus eritematoso sistémico oesclerodermia (SS secundario)1-3. La destrucción de las glándu-las da lugar a las principales manifestaciones clínicas: querato-conjuntivitis seca por disminución de la secreción lacrimal yxerostomía por disminución de la secreción de saliva, aunqueson estos los síntomas predominantes también puede tenerafectación sistémica a través de la infiltración linfoide de dife-rentes órganos cómo los pulmones, riñones, vasos sanguíneos,músculos o transformarse en una enfermedad proliferativa delas células B4.
La afectación pulmonar asociada con el SS es más frecuenteen las formas primarias, predominantemente en mujeres conuna edad media de 60 años5. Suele ocurrir entre 5-10 años des-pués del comienzo de la enfermedad, aunque puede ocasional-mente preceder el diagnóstico de la misma cómo es el caso denuestra paciente. La proporción de pacientes con SS que des-arrollan afectación pulmonar está estimada en un 11%6, dife-renciando: la vía aérea, el parénquima pulmonar, pleura y tras-tornos linfoproliferativos (Tabla 1). Los síntomas típicos son latos y la disnea, pero la pérdida de peso, fiebre y dolor pleuríticotambién pueden estar presentes. Encontramos crepitantes biba-sales en la exploración física aproximadamente en el 60% depacientes7,8. En el caso que presentamos, la paciente acude aurgencias por clínica de malestar general, tos seca y disneajunto con crepitantes bibasales a la auscultación cardiorrespira-toria. Existe una buena correlación entre el resultado delTACAR pulmonar y los hallazgos histológicos, que evita enmuchos casos la realización de una biopsia pulmonar abierta, lacual estaría indicada cuando existe sospecha de malignidad,incremento de los síntomas respiratorios, deterioro de las prue-bas de función respiratoria, o empeoramiento en el TACAR oradiografía simple9,10. En nuestro caso ante la sospecha clínicainicial de etiología infecciosa con tórpida evolución clínico-radiológica finalmente llevamos a cabo la biopsia pulmonarcon resultado histológico compatible con neumopatía intersti-cial linfocítica asociada a bronquiolitis folicular. Finalmente,dada la presencia de síndrome seco ANA, FR y anti-Ro positi-vo y neumopatía intersticial, la paciente fue diagnosticada deSS primario.
El tratamiento de la afectación pulmonar se basa en la iden-tificación anatomopatológica, la severidad de los síntomas y laextensión radiográfica. Las formas histológicas principales dela enfermedad pulmonar intersticial (EPI) en los pacientes conSS primario son4:
FIGURA 1
RADIOGRAFÍA DE TÓRAX: INFILTRADOS ALVEOLARES EN AMBOS
LÓBULOS INFERIORES, LÍNGULA Y PERIFERIA DEL LÓBULO SUPE-RIOR IZQUIERDO
FIGURA 2
TOMOGRAFÍA AXIAL COMPUTARIZADA DE ALTA RESOLUCIÓN
(TACAR) DE TÓRAX: MÚLTIPLES INFILTRADOS CONFLUENTES,DE DISTRIBUCIÓN DIFUSA POR AMBOS HEMITÓRAX, PREDOMI-NANDO EN LÓBULOS INFERIORES

| 21 |
Casos ClínicosACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:19-21
1. Neumopatía intersticial no especí-fica: Es la forma más común de EPI, conuna prevalencia del 60%. Presenta unbuen pronóstico con una supervivenciaa los 5 años del 83%. Para pacientesasintomáticos con pruebas de funciónrespiratoria (PFR) normales o mínima-mente alteradas se aconseja seguimien-to clínico, funcional y radiológico.
2. Neumopatía intersticial usual: Laprevalencia alcanza el 6% de los casos.Cursa de forma insidiosa con tos y dis-nea progresiva y no suele acompañarsede síntomas sistémicos. Más común enSS secundario.
3. Neumopatía intersticial linfocítica:Es la forma histopatológica de nuestrocaso clínico. Se trata de una prolifera-ción bronquial linfoide benigna. Su pre-valencia es del 1%, caracterizada poruna extensa proliferación de linfocitos Ty B y un número variable de célulasplasmáticas. A menudo se asocia conbronquiolitis folicular. La mayoría depacientes están sintomáticos y respon-den bien al tratamiento con esteroides,aunque su pronóstico es peor, con unamortalidad a los 5 años del 50% de lospacientes. Se estima que el 5% evolu-ciona a linfoma9-11.
4. Bronquiolitis folicular: Caracteri-zada por Hiperplasia Linfoide de lamucosa asociada a tejido linfoide(MALT). Puede asociarse con neumo-nía intersticial linfocítica, en cuyo casose trata la misma.
Por tanto el tratamiento inicial de laEPI en los pacientes con SS primariosintomáticos se basa en el uso de predni-sona, a dosis de 1-2 mg/Kg/día, entre 1 a2 meses para posteriormente reevaluarcon controles de TACAR y PFR condifusión (DLCO). Si ha habido respues-ta se puede iniciar una pauta descenden-te de corticoides con controles clínicos.Si no ha habido respuesta clínica (pro-gresión en el TACAR o disminuciónmayor del 10% en DLCO), se aconsejaintroducir tratamiento inmunosupresor,inicialmente ciclofosfamida en boluscomo inductor de la remisión¹², paraposteriormente continuar con azatiopri-na de mantenimiento¹³. Se han descritocasos con buena respuesta a rituximab,
especialmente en pacientes con otrasmanifestaciones extraglandulares delSS primario14,15.
En nuestro caso comenzamos trata-miento con prednisona a dosis de 1mg/Kg/día con importante mejoría clí-nica, actualmente pendiente de reeva-luar respuesta.
Agradecimientos: Este trabajo ha sidoapoyado con una beca de la Asociaciónpara la Investigación en Reumatologíade la Marina Baixa (AIRE-MB).
Componentes del Grupo AIRE-MB:José Rosas, Esteban Salas, José MiguelSenabre-Gallego, Gregorio Santos-Soler, Catalina Cano, Mª Luisa Betoret,Ana Pons (S. Reumatología, HospitalMarina Baixa), Francisca LLinares-Tello, Juan Molina (S. Laboratorio,Hospital Marina Baixa); Carlos Santos-Ramírez, Rafaela Ortega Castro (S.Reumatología Hospital Marina Alta.Denia), Xavier Barber (CIO de la Uni-versidad Miguel Hernández, Elche),Mabel Sánchez-Barrioluengo (INGE-NIO (CSIC-UPV), Universitat Politèc-nica de València).
BIBLIOGRAFÍA1.- Rosas J, Ramos-Casals M, Fernández-Car-ballido C, Santos-Soler G, Senabre-GallegoJM, Cano C, et al. Síndorme de Sjögren. En,Belmonte MA, Castellano JA, Román JA,Rosas J, eds. Enfermedades reumáticas: actua-lización SVR. Madrid, Editorial Ibáñez yPlaza, 2013:207-38.2.- Morillas López L. Síndrome de Sjögren pri-mario. En: Cañete JD, Gómez-Reino JJ, Gon-zález-Gay MA, Herrero-Beaumont G, MorillasL, Pablos JL, et al. editores. Manual SER de lasenfermedades reumáticas. Madrid: EditorialMédica Panamericana; 2008. p. 252-8. 3.- Larché M. A short review of the pathogene-sis of Sjögren’s syndrome. Autoimmunity Rev2006; 5:132-135.4.- Rosas Gómez de Salazar J, Senabre GallegoJM, Santos Ramírez C. Manejo de las manifes-taciones extraglandulares del síndrome de Sjö-gren primario. Reumatología Clin 2010;6:6-11.5.- Watanabe M, Naniwa T, Hara M, et al. Pul-monary manifestations in Sjögren´s syndrome:correlation analysis between chest computedtomographic findings and clinical subsets withpor prognosis in 80 patients. J Rheumatol2010;37:365.6.- Ramos-Casals M, Solans R, Rosas J, CampsMT, Gil A, del Pino-Monteset J, and theGEMESS study group. Primary Sjögrensyndrome in Spain. Clinical and immunologic
expression in 1010 patients. Medicine (Balti-more) 2008;87:210-9.7.- Ramos-Casals M, Tzioufas A, Font J. Pri-mary Sjögren’s syndrome: new clinical andtherapeutic concepts. Ann Rheum Dis 2005;64:347-358.8.- Lahdensuo A, Korpela M. Pulmonary fin-dings in patients with primary Sjögren’ssyndrome. Chest 1995;108:316-319.9.- Parambil JG, Myers JL, Lindell RM, MattesonEL, Ryu JH. Interstitial lung disease in primarySjögren syndrome. Chest 2006;130:1489-95.10.- Ito I, Nagai S, Kitaichi M, Nicholson AG,Johkoh T, Noma S, et al. Pulmonary manifesta-tions of primary Sjögren’s syndrome: a clinical,radiologic, and pathologic study. Am J RespirCrit Care Med 2005;171:632-8.11.- Parke AL. Pulmonary manifestations ofprimary Sjögren’s syndrome. Rheum Dis ClinNorth Am 2008;34:907-20.12.- Shi JH, Liu HR, Xu WB, Feng RE, ZhangZH, Tian XL, et al. Pulmonary Manifestationsof Sjögren’s Syndrome. Respiration2009;78:377-86.13.- Deheinzelin D, Capelozzi VL, KairallaRA, Barbas Filho JV, Saldiva PH, de CarvalhoCR. Interstitial lung disease in primary Sjögre-n’s syndrome. Clinical pathological evaluationand response to treatment. Am J Respir CritCare Med 1996;154:794-9.14.- Seror R, Sordet C, Guillevin L, HachullaE, Masson C, Ittah M, et al. Tolerance and effi-cacy of rituximab and changes in serum B cellbiomarkers in patients with systemic complica-tions of primary Sjögren’s syndrome. AnnRheum Dis 2007;66:351-7.15.- Devauchelle-Pensec V, Pennec Y, MorvanJ, Pers JO, Daridon C, Jousse-Joulin S, et al.Improvement of Sjögren’s syndrome after twoinfusions of rituximab (Anti-CD20). ArthritisRheum 2007;15;57:310-7.
TABLA 1
1. Vía aérea- Sequedad vías aéreas- Bronquitis/bronquiolitis folicular linfocítica
- Obstrucción
2. Enfermedad pulmonar intersticial (EPI)
- Neumopatía intersticial no específica- Neumopatía intersticial usual- Neumopatía intersticial linfocítica
23. Pleura- Pleuritis- Derrame pleural
24. Enfermedad linfoproliferativa- Infiltración linfocítica folicular- Pseudolinfoma/linfoma- Amiloidosis
AFECTACIÓN PULMONAR EN EL SÍNDROME
DE SJÖGREN

| 22 |
Casos ClínicosACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2012, 4;3:22
CASOLa aortitis es una vasculitis que se carac-teriza por la inflamación y alteraciónestructural de la pared aórtica, indepen-dientemente de su causa. Su sintomato-logía es inespecífica y suelen estar ele-vados los reactantes de fase aguda1,2.Podemos clasificarlas en aortitis infec-ciosas, secundarias a enfermedades deltejido conectivo y aortitis primarias.Diversas técnicas de imagen nos permi-ten evaluar tanto la luz vascular como lapared: tomografía computerizada, reso-nancia magnética, angiografía y másrecientemente la PET-TAC.
La introducción de la tomografía poremisión de protones (PET), permitedetectar el aumento de metabolismo en lapared aórtica por acúmulo de fluorodeo-xiglucosa en fases agudas, siendo ade-más útil tanto para el seguimiento de laenfermedad como para evaluar la res-puesta terapéutica3. Se combina con TACpara mejorar la precisión anatómica,alcanzando así una especificidad del 89-100% y una sensibilidad del 77-92%.
Presentamos una paciente de 51 añoscon cuadro poliarticular de 6 meses deevolución sin sinovitis y con alteraciónde reactantes de fase aguda (VSG: 110mmHg y PCR 4,3 mg/dl). No existendatos analíticos (factor reumatoide yanticuerpos anti-péptidos citrulinados,negativos) ni radiológicos que sugieranla existencia de artritis reumatoide.Exploración vascular con auscultaciónnormal y pulsos simétricos tanto enmiembros superiores como inferiores.Normalidad de tensión arterial. Negati-
vidad de autoanticuerpos (ANA yENA). En estudio TAC, se aprecia dila-tación de aorta ascendente de hasta 41mm de diámetro. Cayado (26 mm) ydescendente (25 mm), normales.
Con la sospecha de aortitis, se realizóestudio PET/TAC corporal total quemuestra: 1) Afectación de la pared de laaorta de carácter inflamatorio compati-ble con la sospecha clínica de aortitis. 2)Marcada captación del trazador enpared de la aorta, que se extiende desdela raíz de la misma hacia cayado y aortatorácica y de menor intensidad en aortaabdominal. 3) Mínima captación, nosignificativa en ramificación de las ilía-cas (Figura 1). Al realizar renderizadovolumétrico (Figura 2), evidenciamosuna reconstrucción demostrativa delárea afectada en tres dimensiones.
Las imágenes de PET/TAC son muydemostrativas, siendo una técnica deimagen que ha mejorado el diagnósticode las aortitis. Tras el diagnóstico deaortitis, la paciente inicia tratamientocon prednisona (1 mg/kg/día) y 10 mgde metotrexato con ácido fólico, que-dando clínicamente asintomática y sien-do la VSG de 30 mm Hg tras 45 días detratamiento.
BIBLIOGRAFÍA1.- Gornik H, Creager M. Aortitis. Circulation2008;117:3093-3051. 2.- Kuehl H, Eggebrecht H, Boes T, Antoch G,Rosenbaum S, Ladd S, Bockisch A, Barkhau-sen J, Erbel R. Detection of inflammation inpatientswithacuteaortic síndrome: comparisonof FDD-PET/CR imaging and serologicalmar-kersoginflammation. Heart 2008;94:1472-1477.
3.- James O, Christensen J, Wong T, Borges-Neto S,Koweek L. Utility of FDG PET/CT ininflammatory cardiovascular disease. Radio-graphics 2011;31:1271-1286.
Importancia del PET/TAC en el diagnóstico de laaortitisFERRER REBOLLEDA J1, CALVO CATALÁ J2, CÓZAR SANTIAGO MA1, CAMPOS FERNÁNDEZ C2, SÁNCHEZ JURADO R1, RUEDA CID A2
1Servicio de Medicina Nuclear ERESA, Servicio de Reumatología y Metabolismo Óseo2Consorcio Hospital General Universitario de Valencia
Correspondencia: Dr. Javier Calvo Catalá - Consorcio Hospital General Universitario de Valencia - Servicio de Reumatología y Metabo-lismo Óseo - Avda. Tres Cruces, 1 - 46014 Valencia [email protected]
FIGURA 1
FIGURA 2
DILATACIÓN AORTA ASCENDENTE CON
AFECTACIÓN INFLAMATORIA DE LA PARED
IMAGEN DE LA ZONA AFECTADA EN 3D

| 23 |
GALERÍA DE IMÁGENES
Osteopetrosis
Puente óseo sacroilíaco aislado
Imagen 1. Radiografía lateral decolumna lumbar: se observa lapresencia de bandas escleróticasbien definidas adyacentes a losplatillos vertebrales con una zonacentral radiolucente, mostrando laimagen de “vertebra en sandwich”característica de la osteopetrosis.
Imagen 2. RM lumbar: secuenciaT1 en que se objetiva hipointensi-dad de la médula ósea con morfo-logía en franjas horizontales enrelación con osteopetrosis.
Imagen 3. TC lumbar: osteoescle-rosis difusa con aumento de den-sidad de las vertebras de la colum-na lumbo-sacra de forma hetero-genea, afectando tanto los cuerposvertebrales como los elementosposteriores.
Interpretación:Hallazgo mediante radiografía (imagen superior) y TAC (imagen inferior)de puente óseo prominente a nivel de la articulación sacroilíaca derecha,que no se acompaña de disminución del espacio articular ni otros cambiosde tipo degenerativo. La imagen recuerda a los sindesmofitos de la hiperos-tosis esquelética idiopática difusa (en inglés, DISH), que en ocasionesafectan a articulaciones de la pelvis, pero en este paciente llama la atenciónla práctica ausencia de afectación a otros niveles. Sólo presentaba muyincipientes sindesmofitos en la articulación sacroilíaca izquierda y entrelos cuerpos vertebrales T11 y T12.
Andrés M, Fernández-Carballido C, Romera C, San Martín A, Sivera F. Sección de Reumatología, Hospital General Universitario de Elda.Correspondencia: Mariano Andrés. Sección de Reumatología, HospitalGeneral Universitario de Elda. Carretera Elda-Sax s/n. CP 03600. Elda(Alicante). [email protected]
Santos Ramírez C1, Ortega Castro R1, Martínez Salinas MP2
1 Sección de Reumatología. 2 Servicio de Diagnóstico por la Imagen. Hos-pital de Denia. Alicante.Correspondencia: Carlos Santos RamírezHospital de Denia. Partida de Beniadla s/n. 03700. Denia. Alicante. [email protected]
Se presentan imágenes de radiografíasimple, TC y RM de columna lumbar deuna misma paciente con los hallazgoscaracterísticos de la osteopetrosis.
1 2 3
Galería de imágenesACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:23-24
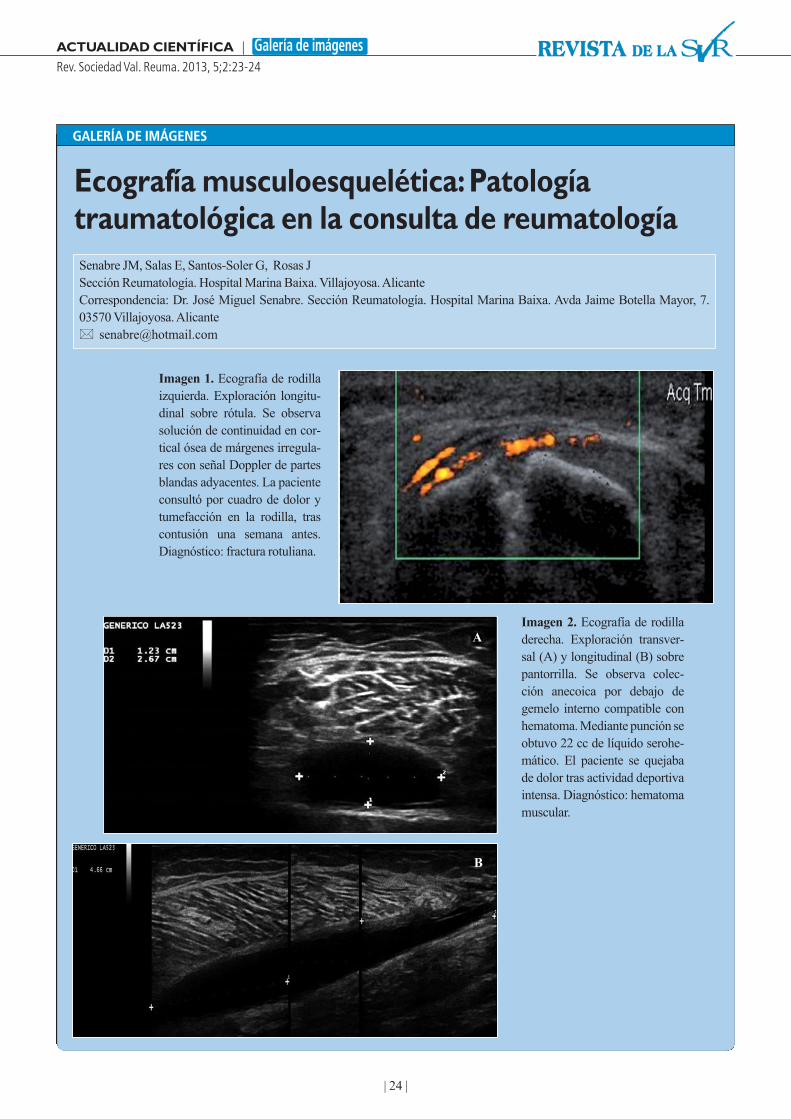
| 24 |
GALERÍA DE IMÁGENES
Ecografía musculoesquelética: Patología traumatológica en la consulta de reumatología
Imagen 1. Ecografía de rodillaizquierda. Exploración longitu-dinal sobre rótula. Se observasolución de continuidad en cor-tical ósea de márgenes irregula-res con señal Doppler de partesblandas adyacentes. La pacienteconsultó por cuadro de dolor ytumefacción en la rodilla, trascontusión una semana antes.Diagnóstico: fractura rotuliana.
Senabre JM, Salas E, Santos-Soler G, Rosas JSección Reumatología. Hospital Marina Baixa. Villajoyosa. AlicanteCorrespondencia: Dr. José Miguel Senabre. Sección Reumatología. Hospital Marina Baixa. Avda Jaime Botella Mayor, 7.03570 Villajoyosa. Alicante [email protected]
Imagen 2. Ecografía de rodilladerecha. Exploración transver-sal (A) y longitudinal (B) sobrepantorrilla. Se observa colec-ción anecoica por debajo degemelo interno compatible conhematoma. Mediante punción seobtuvo 22 cc de líquido serohe-mático. El paciente se quejabade dolor tras actividad deportivaintensa. Diagnóstico: hematomamuscular.
A
B
Galería de imágenesACTUALIDAD CIENTÍFICA |
Rev. Sociedad Val. Reuma. 2013, 5;2:23-24


















