
Química de superficies (solido...
40
Química del estado sólido (1615) Sem. 20 - II Alejandro Solano – Peralta, Química del Estado Solido (clave 1615), Química (plan 2004) Facultad de Estudios Superiores - Cuautitlán, UNAM, Mx. Marzo del 2020 Química de superficies (solido – gas) La Química de la superficie se puede definir como el estudio de las reacciones químicas en las interfaces. Está estrechamente relacionado con la ingeniería de superficies, que tiene como objetivo el modificar la composición química de una superficie mediante la incorporación de elementos seleccionados o grupos funcionales que producen diversos efectos deseados o mejoras en las propiedades de la superficie o la interfaz. Esta ciencia es particularmente importante tanto para el estudio de los fenómenos de adsorción importante en muchos procesos industriales de purificación como en reacciones que en condiciones normales tardan mucho en llevarse a cabo. El campo de la química de superficies comenzó con una catálisis heterogénea iniciada por Paul Sabatier en hidrogenación y Fritz Haber en el proceso Haber. Irving Langmuir también fue uno de los fundadores de este campo, y la revista científica sobre ciencias de la superficie, Langmuir, lleva su nombre. La ecuación de adsorción de Langmuir se usa para modelar la adsorción en monocapa en la que todos los sitios de adsorción de superficie tienen la misma afinidad por las especies adsorbentes y no interactúan entre sí. Gerhard Ertl en 1974 describió por primera vez la adsorción de hidrógeno en una superficie de paladio utilizando una técnica novedosa llamada LEED. Estudios similares con platino, níquel, y hierro seguidos. La mayoría de los recientes desarrollos en ciencias superficiales incluyen el 2007 el premio Nobel de Química ganador Gerhard Ertl avances en la química de superficie, específicamente su investigación de la interacción entre moléculas de monóxido de carbono y las superficies de platino. La química de las superficies solidas es de particular importancia para los campos de la catálisis heterogénea, la electroquímica y la geoquímica. Así como en múltiples aplicaciones: fabricación de dispositivos semiconductores, celdas de combustible, monocapas auto ensambladas, la preparación de nuevos materiales, la nanotecnología, los procesos electroquímicos, los análisis cromatográficos, el tratamiento de residuos contaminados, etc. En realidad, en la era de la microelectrónica y nanotecnología, la química de superficies es un área interdisciplinar que contribuye a casi todos los avances tecnológicos. Por todo lo anterior, se espera en este capítulo el adentrar al alumno en los fenómenos que ocurren en la superficie de los sólidos a trabes de si interacción con compuestos en fase gaseosa, siendo todo esto de gran importancia no solo desde el punto de ciencia básica sino también en las múltiples aplicaciones de nuestra vida diaria. Para esto es necesario que el alumno tenga conocimientos básicos tanto de fenómenos de adsorción tanto física como química así como aspectos fundamentales de cinética química y catálisis heterogénea 7.1 Fenómeno de adsorción y desorción en la superficie de un sólido Normalmente se piensa en la superficie de un sólido como una estructura homogénea donde los átomos ocupan posiciones bien definidas y por tanto se repiten regularmente (Figura 7.1). Sin embargo, esto corresponde a una situación ideal muy difícil de conseguir en la práctica. Figura 7.1 Arreglo ideal de las partículas en la superficie de un sólido.
Transcript of Química de superficies (solido...

FES-Cuautitlán
Química del estado sólido (1615) Sem. 20 - II
Alejandro Solano – Peralta, Química del Estado Solido (clave 1615), Química (plan 2004) Facultad de Estudios Superiores - Cuautitlán, UNAM, Mx. Marzo del 2020
Química de superficies (solido – gas)
La Química de la superficie se puede definir como el estudio de las reacciones químicas en las interfaces. Está
estrechamente relacionado con la ingeniería de superficies, que tiene como objetivo el modificar la composición
química de una superficie mediante la incorporación de elementos seleccionados o grupos funcionales que
producen diversos efectos deseados o mejoras en las propiedades de la superficie o la interfaz. Esta ciencia es
particularmente importante tanto para el estudio de los fenómenos de adsorción importante en muchos procesos
industriales de purificación como en reacciones que en condiciones normales tardan mucho en llevarse a cabo.
El campo de la química de superficies comenzó con una catálisis heterogénea iniciada por Paul Sabatier en
hidrogenación y Fritz Haber en el proceso Haber. Irving Langmuir también fue uno de los fundadores de este
campo, y la revista científica sobre ciencias de la superficie, Langmuir, lleva su nombre. La ecuación de
adsorción de Langmuir se usa para modelar la adsorción en monocapa en la que todos los sitios de adsorción de
superficie tienen la misma afinidad por las especies adsorbentes y no interactúan entre sí. Gerhard Ertl en 1974
describió por primera vez la adsorción de hidrógeno en una superficie de paladio utilizando una técnica
novedosa llamada LEED. Estudios similares con platino, níquel, y hierro seguidos. La mayoría de los recientes
desarrollos en ciencias superficiales incluyen el 2007 el premio Nobel de Química ganador Gerhard Ertl avances
en la química de superficie, específicamente su investigación de la interacción entre moléculas de monóxido de
carbono y las superficies de platino.
La química de las superficies solidas es de particular importancia para los campos de la catálisis heterogénea,
la electroquímica y la geoquímica. Así como en múltiples aplicaciones: fabricación de dispositivos
semiconductores, celdas de combustible, monocapas auto ensambladas, la preparación de nuevos materiales, la
nanotecnología, los procesos electroquímicos, los análisis cromatográficos, el tratamiento de residuos
contaminados, etc. En realidad, en la era de la microelectrónica y nanotecnología, la química de superficies es
un área interdisciplinar que contribuye a casi todos los avances tecnológicos.
Por todo lo anterior, se espera en este capítulo el adentrar al alumno en los fenómenos que ocurren en la
superficie de los sólidos a trabes de si interacción con compuestos en fase gaseosa, siendo todo esto de gran
importancia no solo desde el punto de ciencia básica sino también en las múltiples aplicaciones de nuestra vida
diaria. Para esto es necesario que el alumno tenga conocimientos básicos tanto de fenómenos de adsorción tanto
física como química así como aspectos fundamentales de cinética química y catálisis heterogénea
7.1 Fenómeno de adsorción y desorción en la superficie de un sólido
Normalmente se piensa en la superficie de un sólido como una estructura homogénea donde los átomos
ocupan posiciones bien definidas y por tanto se repiten regularmente (Figura 7.1). Sin embargo, esto
corresponde a una situación ideal muy difícil de conseguir en la práctica.
Figura 7.1 Arreglo ideal de las partículas en la superficie de un sólido.

Química del estado sólido (1615) 20 - II
A. S. P. 2 / 40
La superficie de un sólido puede estar llena de defectos estructurales debido al proceso de crecimiento del
cristal o bien a procesos sufridos una vez formado (Figura 7.2). Un tipo común de defecto superficial es un
escalón entre dos capas planas de átomos llamadas terrazas. Un defecto de este tipo puede a su vez tener
defectos, como aristas. Los defectos desempeñan un papel importante en el crecimiento de la superficie y por
ende en los fenómenos que en ella pueda ocurrir como los fenómenos de adsorción – desorción y en su
reactividad (catálisis).
Figura 7.2 Algunos tipos de defectos presenten en la superficie de un solido (Tomado de: Atkins, 2008,
p.910).
Además de sus defectos estructurales, se ha de tener en cuenta cuál será la composición que va a encontrar al
estudiar su superficie. En condiciones normales, las partículas de gas (aire) bombardean continuamente la
superficie del sólido cubriéndola rápidamente. La velocidad de este proceso puede calcularse usando la teoría
cinética de gases. Así, el número de colisiones por unidad de área y unidad de tiempo, zp, es:
𝑍𝑤 =𝑝
(2𝜋𝑚𝑘𝐵𝑇)1/2 (7.1)
Si p= 100 kPa (1.00 bar) y T=300 K, Zw= 3 x1023 cm-2s-1.
Una forma mas practica de esta ecuación es:
𝑍𝑤 =𝑍0(𝑝/𝑃𝑎)
{(𝑇/K)(𝑀/(g mol−1)}1/2 (7.2)
Donde M es la masa molar del gas. El aire (M= 29 g mol-1) a 1 atm y 25 °C con Z0= 2.36 x1024 m-2s-1, la
frecuencia de colisiones es de 3 x 1027 m-2s-1. Puesto que 1m2 de superficie metálica tiene cerca de 1019 atomos,
cada átomo es golpeado unas 108 veces por segundo. Esto se justifica por la densidad en numero de moléculas
N.
Numero de colisiones = 𝑁𝐴Δ𝑡 ∫ 𝑣𝑥𝑓(𝑣𝑥)∞
0dx (7.3)
El flujo de colisiones es el numero de colisiones dividido por A y Δ𝑡 entonces
𝑍𝑤 = 𝑁 ∫ 𝑣𝑥𝑓(𝑣𝑥)∞
0
dx (7.4)
Luego usando la expresión de distribución de velocidades:
∫ 𝑣𝑥𝑓(𝑣𝑥)∞
0
d𝑣𝑥 = (𝑚
2𝜋𝑘𝐵𝑇)
1/2
∫ 𝑣𝑥e−𝑚𝑣𝑥2/2𝑘𝐵𝑇d𝑣𝑥
∞
0
= (𝑘𝐵𝑇
2π𝑚)
1/2
(7.5)
Donde se ha usado la integral estándar

Química del estado sólido (1615) 20 - II
A. S. P. 3 / 40
∫ 𝑥e−𝑎𝑥2dx =
1
2𝑎
∞
0
(7.6)
Por lo tanto,
𝑍𝑤 = 𝑁 (𝑘𝑇
2𝜋𝑚)
1/2
=1
4𝑐𝑁 (7.7)
Donde se ha usado la forma velocidad media o 𝑐 =(8kBT/πm)1/2, lo cual implica que 1
4𝑐= (kBT/2πm)1/2. La
sustitución de N= nNA/ V= p/kBT da la ecuación inicial. La manera obvia para conservar la limpieza es reducir la
presión mediante un sistema convencional de vacio la cual el flujo de colisiones decae (Atkins, 2008), véase
Tabla 7.1.
Tabla 7.1 Efecto del tipo de ambiente en la superficie de un sólido.
Técnicas P (mm Hg) t de Impacto
Vacío típico ≈10-6 1 cada 10 s
Ultra-vacío (UHV, Ultra-High Vacuum) ≈ 10-11 1 cada 106 s (10 días)
La adsorción es el fenómeno de concentración de una especie química en una interfase. De forma general, se
puede ejemplificar de esta manera cuando una molécula de un gas golpea una superficie sólida, esta puede
rebotar o quedar fijada sobre la superficie, es decir, sufrir adsorción. A continuación, la molécula adsorbida
puede difundirse (moverse) sobre la superficie, quedarse fija, sufrir una reacción química o disolverse en el
interior del sólido (proceso conocido como absorción y del cual un ejemplo es el CaCl2 anhidro que se emplea
como desecador donde el agua de la atmósfera es adsorbida y posteriormente forma un hidrato).
Podemos definir el primero de estos procesos de la catálisis heterogénea como se observar en la Figura 7.3:
Figura 7.3 Posibles procesos de adsorción de los gases en sólidos.
a) Adsorción: La adhesión o enriquecimiento de partículas en las proximidades de una superficie. En el
caso de los sistemas de gas/sólido, la adsorción tiene lugar en las proximidades de la superficie sólida y
fuera de la estructura sólida (Figura 7.4). La adsorción puede también definirse como la tendencia de
un componente del sistema a concentrarse en la interfase, donde la composición interfacial es diferente
a las composiciones correspondientes al seno de las fases.

Química del estado sólido (1615) 20 - II
A. S. P. 4 / 40
Figura 7.4 Interacciones de una molécula (propanol) en fase gaseosa con la superficie de un sólido (Tomado de:
http://www.posgradoeinvestigacion.uadec.mx/CienciaCierta/CC34/1.html).
b) Desorción: Proceso inverso a la adsorción, en el que la cantidad de moléculas en la superficie
disminuye progresivamente.
Hay una clara diferencia entre el fenómeno de adsorción y absorción, en el segundo existe una penetración
física de una fase en la otra; sin embargo, es factible que ambos sucedan simultáneamente, y en este caso puede
ser muy difícil separar los efectos de ambos fenómenos, inclusive un fenómeno puede afectar al otro.
Algunos absorbentes sólidos comerciales empleados son: carbón activado, zeolitas, tamices moleculares, gel
de sílice, alúmina activada (Figura 7.5), carbonato de calcio, entre otros. Los catalizadores porosos pueden tener
áreas intersticiales muy grandes. Un catalizador de silice-alumina para cracking catalítico puede tener un área
superficial de 300 m2/s.
Figura 7.5 Alúmina activada, empleado principalmente como agente desecante.
Así mismo, se han utilizado absorbentes sólidos para la purificación de gases, purificación de aire,
recuperación de solventes, catalizadores, producción de gases, tratamiento de gases de combustión, celdas de
combustible (Figura 7.6) a estos se les conoce como catalizadores estructurados que son rígidas con grandes
poros o canales que aseguran una baja pérdida de carga y que exponen una elevada área superficial lateral sobre
la que se puede pegar una delgada película de catalizador.
Figura 7.6 Algunos tipos de catalizadores estructurados.
La superficie de un solido
Actualmente, las superficies sólidas comúnmente empleadas son sólidos de orígenes naturales o sintéticos, que
tiene la capacidad de retener sobre su superficie un componente presente en corrientes líquidas o gaseosas. Para

Química del estado sólido (1615) 20 - II
A. S. P. 5 / 40
el caso de la adsorción sobre superficies sólidas, la especie química que resulta adsorbida sobre la superficie de
un material recibe el nombre de adsorbato o soluto en tanto que el sólido recibe el nombre de adsorbente o
substrato.
Figura 7.7 Fenómeno de adsorción en la superficie solido - gas.
Los sólidos adsorbentes se caracterizan por una alta superficie específica y por su inercia química frente al
medio en el que se van a utilizar. Otras características importantes que debe reunir un buen adsorbente son las
siguientes:
• Alta capacidad de adsorción. La relación dé equilibrio entre las fases influye en la eficacia con que se
alcanza la capacidad final y, en muchos casos, controla la capacidad real del soluto. Como quiera que
los mecanismos de unión son muy complejos y no se han determinado con precisión aún, no se
dispone de una norma satisfactoria mediante la cual puedan preverse, a priori las afinidades relativas
entre un material poroso y una sustancia.
• Propiedades físicas y tamaño de partícula adecuados para garantizar la necesaria resistencia mecánica
y facilidad de manejo, produciendo la menor pérdida de carga posible tanto en lechos fijos como en
los móviles o fluidizados.
• Costo bajo, tanto de la materia prima como del proceso de fabricación.
• Fácil regeneración; por desorción, especialmente en el caso de los procesos continuos.
• Alta resistencia a la abrasión,
• Alta estabilidad térmica.
Otra característica que pueden tener los sólidos es ser un medio poroso, es decir, que el sólida en su interior un
sistema de huecos (poros) que pueden o no estar interconectados entre sí. Esta estructura de poros puede
permitir el transporte rápido de las partículas en su interior dado que pueden tener geometrías, tamaños y
topologías variadas, dependiendo del origen de su formación.

Química del estado sólido (1615) 20 - II
A. S. P. 6 / 40
Figura 7.8 Estructuras cristalinas en la que pueden verse los huecos (1), las galerías cilíndricas (2), los canales
(3) y Red de canales tridimensional (4) de una zeolita ZSM-11 por los que pueden pasar las moléculas de
gas.
Cualquier sólido que contenga cavidades, canales o intersticios puede ser considerado como poroso. Por ende,
es necesario definir algunos términos que se emplearan más adelante:
– Sólido poroso: es un sólido con poros, esto es, cavidades, canales e intersticios, que son más profundos
que anchos.
– Volumen de poros (Vp): volumen de los poros medido por un determinado método.
– Tamaño de poro: distancia entre dos paredes opuestas del poro
– Distribución del tamaño de poros: representado por las derivadas (dAp/drp) o (dVp/drp), como una
función de rp, donde Ap, Vp y rp son el área de las paredes de los poros, el volumen y el radio de los
poros, respectivamente.
Así, los poros se pueden clasificar de acuerdo con su disponibilidad a un fluido externo de la siguiente manera:
Poros cerrados: totalmente aislados de sus vecinos. Influencian propiedades macroscópicas como la densidad,
fortaleza mecánica y conductividad térmica, pero son inactivos en procesos como flujo de fluidos y adsorción de
gases.
Poros abiertos: poseen canales continuos de comunicación con la superficie externa del material.
La geometría de un sistema poroso describe las formas y tamaños de sus poros. Las geometrías más frecuentes
de los poros son:
• Poros cilíndricos (por ejemplo, en alúmina y óxido de magnesio).
• Poros en forma de rendija o hendidura (en carbones activados y arcillas).
• Espacios o huecos entre esferas de sólido conectadas (en sílice y en otros sólidos obtenidos a partir de
geles).
• Poros en forma de bote de tinta (ink-bottle shaped): el cuerpo del poro es mayor que su boca.
• Poros en forma de embudo (funnel shaped): el tamaño de la boca del poro es mayor al cuerpo del
poro.

Química del estado sólido (1615) 20 - II
A. S. P. 7 / 40
a. Poros cerrados
b. Poros abiertos
c. Poros cilíndricos
d. Poros en forma de rendija o hendidura
e. Espacios o huecos entre esferas de sólido
conectadas
f. Poros en forma de bote de tinta (ink-bottle shaped)
g. Poros en forma de embudo
Figura 7.9 Estructura de la sección de corte de un sólido poroso.
Figura 7.10 Modelos y tipos de poros.
De las diferentes dimensiones que caracterizan a un dado poro, es de especial interés su dimensión transversal
w, es decir, el diámetro de un poro cilíndrico o la distancia entre las placas en el caso de poros formados por
planos paralelos. Una conveniente clasificación de los poros de acuerdo a su dimensión transversal w, fue
propuesta originalmente por Dubinin y posteriormente adoptada oficialmente por la International Union of Pure
and Applied Chemistry (IUPAC):
Tabla 7.2 Tipos de poros y su intervalo de tamaño.*
Ancho de poro (nm) Nombre
w ≤ 2 Microporo
2 ≤ w ≤ 50 Mesoporo
w > 50 nm Macroporo
* El termino nanoporo abarca las tres categorías de poros anteriores, pero con un límite superior de 100 nm
Esta clasificación se basa en las propiedades que presentan los diferentes poros de acuerdo a su dimensión w
en los procesos de adsorción y que se manifiestan en las isotermas de adsorción. Si bien la mayoría de los
sólidos poseen las tres clases de poros en su red interna, es común referirse como sólidos micros, meso o
macroporosos dependientes de su geometría y de las características de la molécula que se adsorbe. Así, Con esta

Química del estado sólido (1615) 20 - II
A. S. P. 8 / 40
nomenclatura se da cuenta de que la porosidad de dicho solido contiene un determinado rango del tamaño del
poro.
Figura 7.11 Esquema de una estructura de silicatos con inclusión de diversos iones (Tomado de:
https://www.engormix.com/avicultura/articulos/adsorbentes-micotoxinas-eficacia-hscas-t25981.htm).
7.1 Adsorción de un gas por un sólido
Cuando una molécula (adsorbato) está cerca de la superficie de un sólido (adsorbente) puede correr el riesgo
de interactuar y unirse (ser adsorbida) a la superficie sólida. Entre esta interacción entre adsorbato y el
absorbente se pueden distinguir dos comportamientos límites de adsorción: a) La interacción que se produce
puede ser a través de fuerzas de Van der Waals o b) se puede establecer un verdadero enlace químico.
En el primer caso donde hay una ligera interacción entre el adsorbato y el absorbente sin alterar notablemente
la estructura del adsorbato y la del adsorbente es conocida como fisisorción en tanto en el último caso donde se
establece un enlace químico que puede a posteriori conducir a la formación de nuevas especies químicos es
denominado quimisorción y cada una tiene características propias que se mencionaran a continuación.
En la adsorción física o fisisorción, las moléculas del gas se mantienen unidas a la superficie del sólido por
medio de fuerzas de Van der Waals (interacciones dipolares, dispersión y/o inducción). Este hecho define todas
las características de la fisisorción:
• Es una interacción débil.
• Es un proceso exotérmico (las fuerzas de van der Waals son atractivas) en el que los calores liberados,
ΔHads (aprox. 20-40 kJ/mol) son semejantes a las entalpías de condensación de la sustancia adsorbida.
La energía liberada es adsorbida en forma de vibración por la red del sólido y ΔH°ads se puede medir
por el aumento de temperatura de la muestra.
• La molécula fisisorbida mantiene su identidad ya que la energía es insuficiente para romper el enlace,
aunque su geometría puede estar distorsionada.
• La fisisorción es un proceso no especifico ya que las fuerzas que intervienen no lo son y no existe una
selectividad marcada entre adsorbato y adsorbente. En general, los gases muy polarizables son
adsorbidos más fácilmente.
• La fisisorción se produce en multicapas. Sobre una capa de gas fisisorbida puede adsorberse otra. La
ΔHads para la primera capa viene determinada por las fuerzas entre adsorbente (M) y adsorbato (A),
mientras que la ΔHads para las capas siguientes depende de las interacciones A-A (Figura 7.12) y por
tanto es similar a la entalpía de condensación.

Química del estado sólido (1615) 20 - II
A. S. P. 9 / 40
Figura 7.12 Proceso de formación de multicapas.
En la adsorción química o quimisorción, propuesta por Langmuir en 1916, las moléculas de gas se mantienen
unidas a la superficie formando un enlace químico fuerte. Este hecho define las características propias de la
quimisorción:
• Se trata de una interacción más fuerte que la fisisorción.
• Las entalpías de quimisorción son mucho mayores que las de fisisorción y del orden de las que se
liberan en la formación de enlaces químicos, ΔH°ads = -(100-500) kJ/mol. Si en la quimisorción se
produce formación y ruptura de enlaces podrían esperarse valores de ΔH°ads tanto positivos como
negativos (al igual que en las reacciones químicas ordinarias). Sin embargo, la quimisorción es
exotérmica normalmente. La razón es que un proceso espontáneo requiere ΔG<0 y dado que la
libertad de traslación del adsorbato se reduce, ΔS es menor que cero y necesariamente ΔH debe ser
menor que cero. Puede haber excepciones si el adsorbato se disocia y/o tiene una movilidad elevada
sobre la superficie. Ejemplo: el H2 se adsorbe endotérmicamente sobre vidrio ya que aumenta la
entropía H2 (g) ↔2H (vid) ΔS>0.
• La quimisorción es específica. Por ejemplo, el N2 es quimiadsorbido a temperatura ambiente sobre Fe,
W, Ca y Ti, pero no sobre Ni, Zn, Ag, Cu o Pb. El Au(s) quimisorbe O2, C2H2 y CO, pero no H2, CO2
o N2.
• Dado que implica la formación de un enlace entre adsorbato y el adsorbente, el proceso se detiene tras
la formación de una monocapa sobre la superficie. Aunque sólo una capa puede estar quimisorbida
puede producirse adsorción física de nuevas capas de adsorbato sobre la primera.
• En general, la quimisorción implica la rotura y formación de enlaces, por lo que la molécula
quimisorbida no mantiene la misma estructura electrónica (enlaces) que en fase gaseosa.
De forma general, se puede expresar criterio para distinguir un proceso de fisisorción y de quimisorción.
Tabla 7.3. Criterios de distinción de un proceso de fisisorción o de quimisorción (Tomado de: Fuentes, 1997, p.
31).
Fisisorción Quimisorción
Calor de adsorción(-Hads) 4 - 40 KJ / mol 40 - 800 KJ / mol
Energía de activación No hay Si hay
Temperatura Depende del punto de ebullición Depende de la Eact
Número de capas formadas Más de una Una
A continuación, se muestra los valores de las máximas entalpias observadas en fisisorción.

Química del estado sólido (1615) 20 - II
A. S. P. 10 / 40
Tabla 7.4 Valores de entalpia de adsorción observadas para algunos gases (Datos tomados de: Atkins, P.,
2008, p. 917).
Adsorbato ΔHѳ/ KJ mol-1
CH4 -21
H2 -84
H2O -59
N2 -21
Tabla 7.5 Entalpias de quimisorción, Δad H°/(kJ mol-1) de algunos gases en distintos metales (Datos tomados
de: Atkins, P., 2008, p. 917).
Adsorbato Cr Fe Ni
CH4 -427 -285 -243
CO
-192
H2 -188 -134
NH3
-188 -155
En las curvas de potencial, al acercarse una molécula gaseosa y ser adsorbida por el sólido cambia en ambos
procesos, fisisorción y quimisorción, y la distancia para la cual la energía del sistema respecto a la molécula a
ser adsorbida es mínima conoce como distancia de adsorción. En el caso de la fisisorción, la distancia de
adsorción es menor que la de la quimisorción (dfisisorción < dquimisorción) debido a que en la fisisorción solo hay
interacciones débiles tipo Van der Waals y por ende no se necesita la molécula gaseosa acercarse mucho a la
superficie del sólido, cosa contraria con la quimisorción donde se necesita un mayor acercamiento para poder
hacer una mayor atracción (enlace) con la molécula adsorbida así el potencial de atracción para la especie
quimisorbida es mayor que la fisisorbida dado que la molécula estará más cerca de la superficie. En general, la
fisisorción es una etapa previa y necesaria para que ocurra la quimisorción, que tiene una mayor interacción con
la superficie y, por lo tanto, menor distancia de adsorción.
Figura 7.13 Evolución de la energía potencial de una molécula de gas acercándose a una superficie plana, a)
fisisorción, b) fisisorción de una molécula disociada, c) fisisorción seguida de quimisorción.
Como ejemplo se pone, en la Figura 7.14 donde el eje horizontal representa la distancia desde la superficie, a
la molécula de O3 la cual puede ser fisisorbido hasta una distancia con energía de unión, Ep. Ahí, puede
desorberse térmicamente para regresar a la fase gaseosa con una vida de desorción de nanosegundos, o puede

Química del estado sólido (1615) 20 - II
A. S. P. 11 / 40
acercarse más a la superficie, superar una barrera de activación (Ea, pc), pasar por un estado de transición (ET de
energía potencial) y entrar en un estado disociado con una energía de enlace Ec dando como producto de
descomposición una molécula de dioxígeno y un átomo de oxígeno, o bien, el proceso puede revertirse
superando la barrera de activación desde la quimisorción hasta la fisisorción (Ea, cp).
Figura 7.14 Curvas de potencial para la adsorción de ozono (O3) en los procesos de fisisorción y quimisorción
(Tomado de: Shiraiwa, 2011, pp. 291–295).
En este punto es importante mencionar que, tanto en el proceso de fisisorción como en el de quimisorción hay
implícito un equilibrio termodinámico, por lo que el proceso de adsorción y desorción puede expresarse en
términos de una constante de equilibrio de la siguiente manera:
Adsorción Kads
Adsorbato (g) + Adsorbente (sup) Adsorción Keq = Kdes
Desorción
Isotermas de adsorción
Un parámetro importante en las curvas de adsorción de un gas sobre un sólido es el denominado grado de
adsorción el cual es el grado de recubrimiento superficial y se expresa normalmente como fracción de
recubrimiento, θ, definido como:
θ =número de sitios de adsorción ocupados
números de sitios de adsorción disponibles (7.8)
Esta cobertura fraccional de un recubrimiento, también, por lo general se expresa en términos del volumen, V,
adsorbido a una cierta presión (P) de la fase fluiva como:
(7.9)
=V
V

Química del estado sólido (1615) 20 - II
A. S. P. 12 / 40
Donde V es el volumen de adsorbato correspondiente a una cobertura de una sola capa (monocapa) en la
superficie del sólido (Atkins, 2008).
La variación de θ con la presión del adsorbato a una temperatura dada se denomina isoterma de adsorción En
las isotermas de adsorción de sólido se representa la presión de gas en equilibrio (P) en el eje X, mientras que en
el eje Y se representa la cantidad adsorbida. Esta magnitud puede darse de diferentes formas: moles
adsorbidos/gramos de adsorbente (x/m) y volumen de gas adsorbido/gramos de adsorbente (v), que es
proporcional a la cantidad anterior. Esto gráficos se usan con frecuencia como modelos experimentales, que no
hacen afirmaciones sobre los mecanismos subyacentes y las variables medidas.
Figura 7.15 Gráfico típico de una isoterma de adsorción.
Actualmente la IUPAC (International Union of Pure and Applied Chemistry) reconoce seis tipos de isotermas
de adsorción los cuales se muestran en la siguiente representación esquemática. Figura 7.16.
Figura 7.16 Tipos de isotermas experimentales.
La isoterma tipo I, se caracteriza porque la adsorción se produce a presiones relativas baja. Característica de
los sólidos microporosos (adsorción en monocapa). I(a): Adsorbentes microporosos con tamaños de poro ≤1 nm
y I(b): Materiales con distribuciones de poros que incluyen microporos más anchos y mesoporos estrechos ≤2.5
nm. Denominada como isoterma de Langmuir caracteristico de un proceso de quimisorción.
La isoterma tipo II Isoterma reversible con un punto de inflexión que resulta de la adsorción mono-
multicapa. Es característica de sólidos macroporosos o no porosos, tales como el carbón activado.

Química del estado sólido (1615) 20 - II
A. S. P. 13 / 40
La isoterma tipo III ocurre cuando la interacción adsorbato-adsorbente es baja y las moléculas adsorbidas se
agrupan alrededor de los sitios más favorables en la superficie de un sólido macroporoso (física de multicapa).
Ejemplo: adsorción de agua en negros de carbón grafitizados.
La isoterma tipo IV es característica de multicapas sobre sólidos mesoporosos. Similar a la isoterma tipo II,
pero a presiones grandes ocurre el fenómeno de condensación capilar. Se caracterizan por tener una meseta de
saturación final. Presenta un incremento de la cantidad adsorbida importante a presiones relativas intermedias, y
ocurre mediante un mecanismo de llenado en multicapas. IV(a): A partir de determinado diámetro crítico de
poro la condensación capilar viene acompañada de histéresis. IV(b): Por debajo del diámetro critico o en poros
cerrados en uno de sus extremos en forma cónica.
La isoterma tipo V, al igual que la isoterma tipo III, es característica de interacciones adsorbato-adsorbente
débiles, pero se diferencia de la anterior en que el tramo final no es asintótico.
La isoterma tipo VI es poco frecuente. Este tipo de adsorción en escalones ocurre sólo para sólidos con una
superficie no porosa muy uniforme. Ejemplo: adsorción de gases nobles en carbón grafitizados.
Una forma de obtener información de las isotermas de adsorción es mediante el empleo de modelos o
planteamientos que describan el fenómeno que se está observando. En este sentido, hay una gran variedad de
modelos matemáticos de las isotermas, aquí solo se presentarán los más frecuentemente usados y son las
denominadas:
1) Isoterma lineal (Henry),
2) Isoterma de Langmuir,
3) Isoterma de BET,
4) Isoterma de Freundlich,
5) Isoterma de Temkin,
Cada uno de ellos tienen planteamientos o suposiciones diferentes entre sí y haciendo el mejor ajuste
estadístico se puede llegar a concluir cual es el mejor modelo, y sus consideraciones implícitas, que describan
mejor al sistema que se esté estudiando. A continuación, se describirán de forma general estos modelos de
isoterma.
La isoterma lineal o de Henry muestra un comportamiento formalmente idéntico a la ley de Henry
(adsorción de gases en líquidos). La isoterma de Henry, establece que la cantidad de adsorbato de una superficie
es directamente proporcional a la presión parcial, p, del gas de adsorción (figura 7.17). Se considera válido para
coberturas superficiales bajas y la energía de adsorción es independiente de la cobertura (falta de homogeneidad
en la superficie).
Figura 7.17 Comportamiento (lineal) de la isoterma de Henry.

Química del estado sólido (1615) 20 - II
A. S. P. 14 / 40
Este modelo, linealiza la primera parte de una isoterma experimental del tipo I o del tipo II y, por lo tanto, sólo
se aplica a muy bajas concentraciones (C). Este comportamiento puede ser observado a relativamente altas
temperaturas y muy bajas presiones.
θH = KH . p (7.10)
KH = limp→0
(θ
1 − θ)
1
p= N. K (7.11)
K =Kads
Kdes (7.12)
Donde N es el número de sitios totales y KH es la constante de adsorción de Henry.
Isoterma de Langmuir, dedujo la isoterma tipo I. explica la adsorción al suponer que un adsorbato se
comporta como un gas ideal en condiciones isotérmicas. En estas condiciones, los adsorbatos de presión parcial,
PA, está relacionado con el volumen de éste, V, adsorbido en un adsorbente sólido. El adsorbente, como se
indica en la figura, se supone que es una superficie sólida ideal compuesta por una serie de sitios distintos
capaces de unirse al adsorbato. La unión del adsorbato se trata como una reacción química (Quimisorción) entre
la molécula de adsorbato. A(g) y un sitio vacío, S. Esta reacción produce un complejo adsorbido Aads con una
constante de equilibrio asociada keq.
A(g) + M(sup) 𝑘𝑎𝑑𝑠
𝑘𝑑𝑒𝑠 AM(sup)
Donde la variación de grado de adsorción, θ, con el tiempo es descrito de la siguiente manera:
dθ
dt= kadspN(1 − θ) (7.13)
dθ
dt= −kdesNθ (7.14)
dθ
dt= 0 (Equilibrio) (7.15)
θ =Kp
(1 + Kp) (7.16)
K = kads
kdes (7.17)
Donde p es la presión parcial de A, N (1-) es el número de sitios vacantes y N es el número de sitios totales.
Para adsorción de una molécula y su disociación se tiene;
A2
(g) 2A(sup)
dθ
dt= kadsp[N(1 − θ)]2 (7.18)
dθ
dt= −kdes(Nθ)2 (7.19)

Química del estado sólido (1615) 20 - II
A. S. P. 15 / 40
θ =(Kp)1/2
1 + (Kp)1/2 (7.20)
Donde K es conocida como la constante de Langmuir.
Figura 7.18 Isotermas de Langmuir para adsorción (izq.) no-disociativa y (dcha.) disociativa con varios
valores de K (Tomado de: Atkins, 2008, p. 919).
Para isotermas del tipo I la ecuación de Langmuir se linealizada de la siguiente manera:
P
a=
1
amK+
P
am (7.21)
Donde a es la cantidad de soluto adsorbida, P presión del adsorbato y K la constante de Langmuir. am o θm
como la adsorción máxima.
Figura 7.19 Tendencia de la isoterma de Langmuir linealizada.
Isoterma de BET. La ecuación de Brunauer-Emmett-Teller se emplea rutinariamente para la determinación
del área específica superficial total de un adsorbente. Este modelo de adsorción linealiza la isoterma del tipo II
considerando:
• Moléculas de gas se adsorben físicamente en un sólido en capas infinitamente;
• No hay interacción entre cada capa de adsorción y
• La teoría de Langmuir se puede aplicar a cada capa.

Química del estado sólido (1615) 20 - II
A. S. P. 16 / 40
En la teoría BET se amplía la teoría del modelo de monocapa de Langmuir mediante la introducción de ciertas
suposiciones, que incluyen adsorción en multicapa y la condensación capilar.
V
Vmono=
cz
(1 − z)[1 − (1 − c)z]; z =
p
p0 (7.22)
c = exp [(∆𝐻𝑑𝑒𝑠
0 − ∆𝐻𝑣𝑎𝑝0 )
𝑅𝑇] (7.23)
si c >> 1 (c≈ 102)
(7.24)
La adsorción en la primera capa tiene lugar sobre sitios en la superficie de energía homogénea. Las moléculas
adsorbidas en la primera capa actúan como sitios de adsorción de la segunda capa y así sucesivamente, lo que en
el caso más simple se aproxima a un espesor infinito conforme la presión de vapor Pv, se aproxima a la presión
de vapor de saturación del líquido, P0 (presión de vapor de líquido puro). Las características de condensación y
evaporación son idénticas en todas las capas excepto en la primera. El calor de adsorción en la segunda y demás
capas son iguales al calor de condensación del gas (Hv).
( )
−+=
− 0
monomono
0
Total p
p
cV
1c
cV
1
ppV
p
(7.25)
Donde Vmono es el volumen que corresponde a la cobertura en monocapa, c es una constante que relaciona el
calor de adsorción de la primera capa y el calor de adsorción de las multicapas capas (Hv).
Figura 7.20 Isoterma de BET con varios valores de K (Tomado de: Atkins, 2008, p. 920).
Isoterma de Freundlich, expresa una variación logarítmica. Este modelo considera interacciónes sustrato –
sustrato. Representa la variación isotérmica de la adsorción de una cantidad de gas adsorbido por unidad de
( )z1
1
V
V
)10(c 1 c si
mono
2
−=

Química del estado sólido (1615) 20 - II
A. S. P. 17 / 40
masa de adsorbente sólido con presión, es una relación empírica entre la concentración de un soluto en la
superficie de un adsorbentey la concentración del soluto en el líquido con el que está en contacto.
𝜃 = 𝑐1𝑝1
𝑐2⁄ o tambien 𝑥
𝑚= 𝐾𝑝
1𝑛⁄ (7.26)
También está escrito como;
log𝑥
𝑚= 𝑙𝑜𝑔𝐾 +
1
𝑛log 𝑃 (7.27)
O:
𝑥
𝑚= 𝐾𝑐
1𝑛⁄ (7.28)
log𝑥
𝑚= 𝑙𝑜𝑔𝐾 +
1
𝑛log 𝑐 (7.29)
Dónde x es masa de adsorbato m es la masa de adsorbente (o en volúmenes), p como Presión de equilibrio del
adsorbato c la Concentración de equilibrio de adsorbato en solución. K y n son constantes para un adsorbato y
adsorbentes dados a una temperatura particular. A alta presión 1 / n = 0, por lo tanto, el grado de adsorción se
vuelve independiente de la presión.
Figura 7.21 Tendencia de la isoterma de Freundlich.
Se utiliza en los casos en que no se conoce la identidad real del soluto, como la adsorción de material
coloreado.
Aunque la isoterma de Freundlich linealiza la isoterma del tipo I, no predice la adsorción máxima y presenta
desviaciones a valores elevados de adsorción. Experimentalmente, se determinó que la extensión de la adsorción
de gas varía directamente con la presión y luego varía directamente con la presión elevada a la potencia 1 / n
hasta que se alcanza la presión de saturación Ps. Más allá de ese punto, la tasa de adsorción se satura incluso
después de aplicar una presión más alta. Por lo tanto, la isoterma de adsorción de Freundlich falla a mayor
presión
Isoterma de Temkin. Fueron observadas experimentalmente por Temkin en 1940 que se dio cuenta de que
los calores de adsorción disminuían más frecuentemente que aumentaban con el aumento de la cobertura. Las
isotermas de Temkin supone que la entalpia de adsorción cambia linealmente con la presión. son las isotermas
de adsorción que presentan la siguiente forma:
θT = A ∙ ln(Bp) (7.30)
Donde A y B son dos constantes empíricas.

Química del estado sólido (1615) 20 - II
A. S. P. 18 / 40
Se utiliza para sistemas en los cuales la entalpía de adsorción es inversamente proporcional a θ, es decir,
decrece linealmente con θ; este factor no se tiene en consideración en la isoterma de Langmuir. La
representación lineal de θ frente ln P en base a la siguiente ecuación:
θ = A lnB + A lnP (7.31)
Permite calcular A de la pendiente de la recta y B de la ordenada en el origen.
Se puede agregar otra Isoterma denominada de Prausnitz - Radke que está dado como:
−+
=
bP1
PaRP
(7.32)
En este punto es importante reiterar el realizar los correspondientes ajustes de las isotermas a los modelos a
considerar y evaluar por el método estadístico adecuado considerando para todos los casos que las isotermas que
mejor se ajustaron a los datos experimentales son aquellas que presentan un coeficiente de correlación cercano a
1.
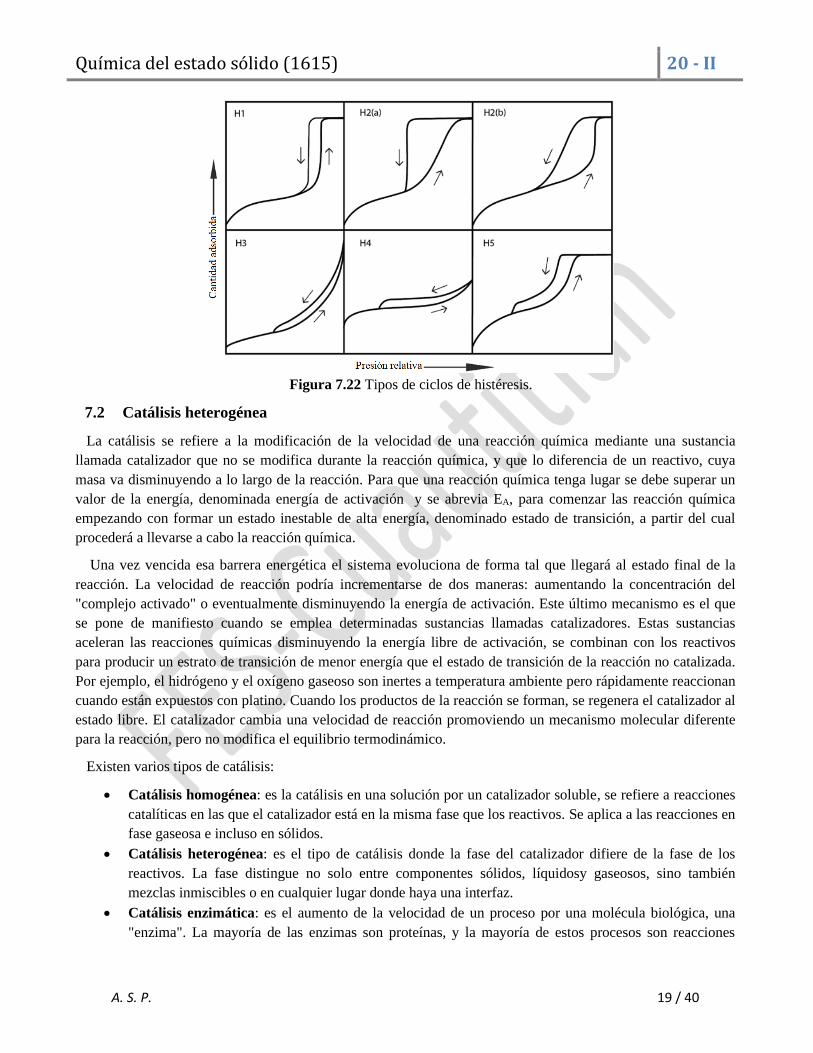
Ciclos de histéresis
La histéresis es el fenómeno de inercia por el cual un material ofreciendo resistencia a un cambio, tiene una
tendencia a conservar sus propiedades. Es decir, el fenómeno de histéresis se manifiesta cuando la isoterma de
adsorción no coincide con la isoterma de desorción. Esto se debe a la presencia de microporos en la superficie,
que provoca condensación capilar del adsorbato y entonces, la desorción se lleva a cabo bajo la presión del
líquido puro, Pº. Haciendo que el proceso de variación sea distinto en un sentido que en el contrario. La
histéresis que aparece en el rango de multicapa de las isotermas de fisisorción se asocia normalmente con la
condensación capilar en la estructura de mesoporos.
Se han observado distintos tipos de histéresis de adsorción, y sus correspondientes asociaciones con el tipo de
poro presente, los cuales se presentan a continuación:
H1: Materiales que exhiben un rango estrecho de mesoporos uniformes. Histéresis por condensación retardada
en la rama de adsorción. Las características del enrejado no influyen.
H2: Estructuras donde las características del enrejado son importantes. H2(a): Percolación en un intervalo
estrecho de cuellos de poro o evaporación inducida por cavitación. H2(b): También asociado con el bloqueo de
poros, pero la distribución del tamaño de los anchos de cuello es ahora mucho más grande.
H3: Característico de las isotermas tipo II. El límite inferior de la rama de desorción se encuentra
normalmente en la presión relativa inducida por cavitación. Característico de agregados flexibles de partículas
laminares como las arcillas.
H4: Similar a H3, pero para isotermas tipo I y II características de materiales microporosos.
H5: Característico de materiales porosos que contienen mesoporos abiertos y parcialmente cerrados.
Las características graficas de estos tipos de isoterma se muestran a continuación.
Química del estado sólido (1615) 20 - II
A. S. P. 19 / 40
Figura 7.22 Tipos de ciclos de histéresis.
7.2 Catálisis heterogénea
La catálisis se refiere a la modificación de la velocidad de una reacción química mediante una sustancia
llamada catalizador que no se modifica durante la reacción química, y que lo diferencia de un reactivo, cuya
masa va disminuyendo a lo largo de la reacción. Para que una reacción química tenga lugar se debe superar un
valor de la energía, denominada energía de activación y se abrevia EA, para comenzar las reacción química
empezando con formar un estado inestable de alta energía, denominado estado de transición, a partir del cual
procederá a llevarse a cabo la reacción química.
Una vez vencida esa barrera energética el sistema evoluciona de forma tal que llegará al estado final de la
reacción. La velocidad de reacción podría incrementarse de dos maneras: aumentando la concentración del
"complejo activado" o eventualmente disminuyendo la energía de activación. Este último mecanismo es el que
se pone de manifiesto cuando se emplea determinadas sustancias llamadas catalizadores. Estas sustancias
aceleran las reacciones químicas disminuyendo la energía libre de activación, se combinan con los reactivos
para producir un estrato de transición de menor energía que el estado de transición de la reacción no catalizada.
Por ejemplo, el hidrógeno y el oxígeno gaseoso son inertes a temperatura ambiente pero rápidamente reaccionan
cuando están expuestos con platino. Cuando los productos de la reacción se forman, se regenera el catalizador al
estado libre. El catalizador cambia una velocidad de reacción promoviendo un mecanismo molecular diferente
para la reacción, pero no modifica el equilibrio termodinámico.
Existen varios tipos de catálisis:
• Catálisis homogénea: es la catálisis en una solución por un catalizador soluble, se refiere a reacciones
catalíticas en las que el catalizador está en la misma fase que los reactivos. Se aplica a las reacciones en
fase gaseosa e incluso en sólidos.
• Catálisis heterogénea: es el tipo de catálisis donde la fase del catalizador difiere de la fase de los
reactivos. La fase distingue no solo entre componentes sólidos, líquidosy gaseosos, sino también
mezclas inmiscibles o en cualquier lugar donde haya una interfaz.
• Catálisis enzimática: es el aumento de la velocidad de un proceso por una molécula biológica, una
"enzima". La mayoría de las enzimas son proteínas, y la mayoría de estos procesos son reacciones

Química del estado sólido (1615) 20 - II
A. S. P. 20 / 40
químicas. Dentro de la enzima, generalmente la catálisis ocurre en un sitio localizado, llamado sitio
activo.
Para realizar catálisis, se necesita un catalizador. Existen varias definiciones de catalizador. Entre ellas se
encuentran las debidas a:
1) Ostwald (1901) define catalizador como cualquier sustancia que modifica la velocidad de una reacción
química sin experimentar cambio alguno en sí misma.
2) Bell (1941), el catalizador de una reacción es cualquier sustancia cuya concentración figura en la
ecuación de velocidad elevada a una potencia superior a la que le correspondería en la ecuación
estequiométrica de dicha reacción.
Por su parte, un inhibidor, lo que hace es aumentar la energía de activación, haciendo, de esa manera, que la
velocidad de la reacción disminuya. En otras palabras, sería una catálisis negativa.
Otras posibles clasificaciones de los catalizadores presentes en la literatura se basan en su estado físico, es así
como el catalizador puede ser gaseoso, líquido o sólido. Dependiendo de la sustancia sobre la cual se ha
manufacturado, el catalizador puede ser orgánico (enzimas o ácidos orgánicos) o inorgánicos (metales, óxidos
metálicos, etc.). Finalmente, basado en la acción la catálisis puede clasificarse en enzimática, acido-base,
fotocatalítica, etc.
En el caso de la química del estado sólido, el catalizador es en estado sólido por lo que se estudiara en este
capítulo la catálisis heterogénea, en ella, el catalizador está presente en la reacción en una fase diferente a la de
los reactivos. Generalmente el catalizador es un sólido y los reactivos son líquidos o gases. La separación más
simple y completa del catalizador del producto provoca que la catálisis heterogénea sea más atractiva
económicamente. Uno de los inconvenientes que presenta los catalizadores heterogéneos es la desactivación,
esta puede originarse por sinterizado de la superficie, envenenamiento irreversible provocado por alguna
sustancia o ensuciamiento provocado por la deposición de carbón u otras sustancias.
Para que la reacción se produzca, uno o más de los reactivos deben difundir a la superficie del catalizador y
adsorberse en él. Después de la reacción, los productos deben desorberse de la superficie y difundir lejos de la
superficie del sólido. Con frecuencia, este transporte de reactivos y productos de una fase a otra desempeña un
papel dominante en la limitación de la velocidad de reacción. La comprensión de estos fenómenos de transporte
y la química de superficies, como p. ej. La dispersión, es un área importante de investigación de los
catalizadores heterogéneos.
7.2.1 Fenómenos de catálisis
El poder atribuir a una especie o especies la propiedad de catalizador, dependen de una serie de características
que se especifican a continuación:
1) El catalizador se recupera sin haberse consumido al completarse la reacción.
2) Pequeñas cantidades de catalizador pueden producir un efecto considerable sobre una gran cantidad de
reactivos.
3) No son capaces de provocar o de iniciar esa reacción
4) Tampoco pueden hacer variar el valor de la constante de equilibrio.
5) La acción de los catalizadores puede ser general o específica.
Para cada proceso químico debe diseñarse un tipo de catalizador especial, con funcionalidades que promuevan
preferencialmente las reacciones deseadas. Un catalizador además debe tener suficiente resistencia mecánica, al

Química del estado sólido (1615) 20 - II
A. S. P. 21 / 40
igual que un área específica grande, sin que esto impida la difusión de reactivos al interior del catalizador. Los
catalizadores que se utilizan en la mayoría de los procesos deben ser sólidos porosos de alta área específica,
térmicamente estables y que cumplan con los requerimientos de alta reactividad y selectividad, por lo tanto, las
características y propiedades básicas que un catalizador debe presentar para tener una aplicación industrial son:
1) Actividad
2) Selectividad
3) Estabilidad
Características del catalizador;
• Actividad; Medida de que tan rápido un reactivo clave se consume en una reacción química. La
actividad puede explicarse de un punto de vista estrictamente cinético como:
(RA) =Cantidad convertida de una sustancia o reactivo dado
(masa del catalizador)(unidad de tiempo) (7.33)
Con la ecuación anterior se tienen tres posibilidades de expresar la actividad de la siguiente manera:
a) Expresión de velocidad de reacción
b) Coeficiente cinético
c) Energía de activación
Otra medida de la actividad es el TON; Turn Over Number (numero de recambio). Está basado en
el número de sitios activos presentes; por lo tanto,
TON =numero de moles obtenidos de producto
mole de catalizador (7.34)
En catálisis heterogénea la actividad depende de la superficie expuesta. De manera alternativa se
tiene el TOF; Turn over Frequency (frecuencia de repetición);
TOF =numero de moles obtenidos de producto
(tiempo x moles de catalizador) (7.35)
• Selectividad y rendimiento; Sea el proceso la donde puede haber 2 o más reacciones en
competencia.
B (producto deseado)
A
C
La selectividad representa cuanto del producto deseado se forma respecto al no deseado.
SB/C =moles formadas de B
moles formadas de C (7.36)
En cambio, el rendimiento de esta reacción está basado en la cantidad de producto formado u la
cantidad alimentada de reactivo, es decir:
θB/C =moles formadas de B
moles consumidas de A (7.37)
• Sitios activos y área activa del catalizador heterogéneo; Fase activa: El compuesto y/o fase
cristalográfica que presenta las propiedades catalíticas. En 1926 Taylor sugiere que una reacción
Química del estado sólido (1615) 20 - II
A. S. P. 22 / 40
catalítica no se presenta en toda la superficie de la fase activa, sino que únicamente en ciertos sitios
activos o centros activos.
Sitio activo del catalizador: Lugar donde ocurre la reacción catalítica, puede visualizarse como átomos
no saturados que existen en los sólidos; como son las orillas, dislocaciones o irregularidades existentes
en los nanocristales de fase activa.
• Dispersión; Con la preparación de catalizadores se busca aprovechar al máximo la cantidad de fase
activa que se presenta propiedades catalíticas. Se introduce entonces el concepto de dispersión (de la
fase activa), como una medida de la “efectividad “de la preparación; Como definición general se
puede tomar la siguiente:
Dispersión =cantidad de fase activa que actúa como sitio activo
cantidad de fase activa utilizada en la preparacion (7.38)
Para obtener una buena dispersión se requiere en principio de una gran área del soporte
Área > = + dispersión = + actividad;
Se utilizan soportes con grandes áreas, es decir, que presenten una estructura porosa. Por ejemplo, los silicatos
mesoporosos han encontrado utilidad como catalizadores debido a que sus áreas superficiales pueden ser de más
de 1000 m2/g, lo que aumenta la probabilidad de que una molécula de reactivo en solución entre en contacto con
la superficie del catalizador, se adsorba y se lleve a cabo la reacción de interés.
Algunos soportes comunes aparecen en la Tabla 7.6. El soporte es necesario para dispersar la fase activa y
presentar grandes áreas activas.
Tabla. 7.6 Algunos catalizadores sólidos de uso común (Datos tomados de: Wheeler, 1950).
Catalizador Área de
superficie (m2/g)
Volumen de
Poro (cm3/g)
Radio medio de
poro (Å)
Carbón activado 500-1500 0.6-0.8 10-20
Silica gel 200-600 0.4 15-100
SiO3-Al2O3 catálisis de ruptura 200-500 0.2-0.7 33-150
Arcilla activada 150-225 0.4-0.52 100
Alúmina activada 175 0.39 45
Celita 4.2 1.1 11000
Catálisis sintética de amonio, Fe 4-11 0.12 200-1000
Una reacción química puede llevarse a cabo en una, dos o más etapas denominadas elementales, durante las
cuales participan las moléculas de los reactivos. En general, existirá una etapa más lenta que las otras y será ésta
la que determine la velocidad global de la transformación. El catalizar una reacción implica reemplazar este
paso por varias etapas más rápidas que se llevan a cabo sólo en presencia del catalizador. Esto significa que la
intervención del catalizador abre un camino nuevo a la reacción, compuesto de reacciones elementales con
energía de activación menor (Figura 7.23).

Química del estado sólido (1615) 20 - II
A. S. P. 23 / 40
Figura 7.23 Curva de la energía potencial a lo largo de la coordenada de la reacción para un proceso catalítico
heterogéneo (Tomado de: Fuentes, 1997, p. 12).
Se puede analizar el efecto del catalizador utilizando la teoría de colisiones (aunque no es la única forma). En
la Figura 7.23, la reacción sin catalizador entre una molécula gaseosa de A y una de B se lleva a cabo por el
camino marcado con una línea continua. Al introducir el catalizador (K), A y B interaccionan con él, si el
catalizador es heterogéneo (sólido), se dice que A y B se adsorben en la superficie, formando un complejo
superficial ABK inestable (línea punteada). Este complejo superficial reaccionará al suministrarle energía de
manera que formará los productos que aún quedan fijos (línea de guiones) sobre la superficie. Para sacar los
productos adsorbidos, es necesario otra pequeña energía que nos conduce al estado final productos +K (doble
línea).
El proceso homogéneo en una sola etapa ha sido substituido por tres etapas que son:
Figura 7.24 Procesos de una reacción química realizados en la superficie de un sólido (Tomado de: Fuentes,
1997, p. 12).

Química del estado sólido (1615) 20 - II
A. S. P. 24 / 40
En estos esquemas los signos x representan los "sitios activos" del catalizador y el aumento en la velocidad de
la reacción (número de moléculas de A o B transformadas por unidad de tiempo) es proporcional a la diferencia
entre E-Ecat.
Figura 7.25 Ejemplo de un sistema con posibles choques efectivos, aumenta la velocidad de reacción
(Tomado de: Fuentes, 1997, p. 12).
Etapas de la catálisis heterogénea
El proceso global de una reacción heterogénea procede a través de una serie de etapas representadas en la
Figura 7.26:
Figura 7.26 Proceso de la catálisis heterogénea.
1. Transporte de reactivos hasta desde el fluido a la superficie externa del catalizador,
2. Difusión (Quimisorción) de los reactivos desde la boca del poro a la vecindad de la superficie
catalítica,
3. Adsorción de los reactivos sobre el catalizador,
4. Reacción sobre la superficie catalítica,
5. Desorción de los productos de reacción,
6. Difusión de los productos desde el interior de los poros a la superficie externa y
7. Transporte de los productos hacia el seno del gas.
Usualmente los pasos 1 y 5 son rápidos por lo tanto cualquiera de los pasos 2, 3 o 4 puede ser el paso limitante
(el más lento) en cualquier reacción heterogénea. Langmuir asumió que el paso 3, la reacción en superficie es el

Química del estado sólido (1615) 20 - II
A. S. P. 25 / 40
paso lento del proceso, por lo que no es de extrañar que se utilice la isoterma de Langmuir para estimar la
concentración de especies adsorbidas.
En este capítulo las etapas de adsorción, reacción superficial y la desorción serán consideradas para
determinar la ecuación de velocidad de la reacción catalítica. Para ello será necesario disponer de información
experimental obtenida en condiciones tales que las etapas de difusión interna y externa sean lo suficientemente
altas como para que puedan ser despreciadas dado que, si no se tienen en cuenta las velocidades de difusión, las
velocidades de reacción para varias reacciones en las superficies dependen únicamente de las constantes de
velocidad y las concentraciones de los reactivos.
Mecanismos generales
Como se ha dicho anteriormente, las especies adsorbidas son estables en las condiciones experimentales de
trabajo por lo que su desorción suele implicar una cierta energía de activación. Aunque no suele ser la etapa más
lenta, puede producir cierto retraso o inhibición si el catalizador queda bloqueado temporalmente por los
productos no desorbidos. Puesto que estamos suponiendo que las velocidades de adsorción y desorción son
mucho mayores que la de la reacción química superficial, se considera que el equilibrio de adsorcion‐desorción
se mantiene durante el tiempo que transcurre la reacción y, por tanto, suponer que la isoterma de Langmuir es
válida para describir el equilibrio de adsorción previo a la etapa determinante. La etapa 3 es propiamente la
etapa de reacción, porque en ella se produce la transformación de reactivos en productos y suele conllevar una
energía de activación mayor al tener una mayor energía de activación, suele ser la etapa determinante de la
velocidad. Si esta etapa tiene lugar entre especies adsorbidas sobre la superficie del catalizador, se dice que la
reacción tiene lugar a través de un mecanismo tipo Langmuir-Hishelwood. En cambio, si la etapa 3 involucra
una especie adsorbida que reacciona con otra especie no adsorbida, el mecanismo se denomina de Eley-Rideal.
Los mecanismos de Langmuir-Hishelwood son más comunes que los de Eley-Rideal.
Así, vamos a analizar el caso de procesos controlados por la etapa de reacción considerando, además, esta
etapa como elemental e irreversible. Distinguiremos dos tipos de procesos (Macías, N., 2017):
Procesos unimoleculares (Modelo de Langmuir-Hinshelwood); Supongamos una reacción elemental e
irreversible de una especia A es adsorbida y que ninguno de los productos queda adsorbido:
Figura 7.27 Ejemplo del mecanismo de adsorción unimolecular.

Química del estado sólido (1615) 20 - II
A. S. P. 26 / 40
La velocidad de adsorción se describe como:
va = ka[A][S] (1- θA) (7.39)
Donde ka es la constante de velocidad para la adsorción y [S](1-θA) es la fracción de la superficie libre. La
velocidad de desorción es entonces:
vd = kd[S]θA (7.40)
Donde kd es la constante de desorción y [S]θA corresponde a la fracción de la superficie cubierta por las
moléculas de A.
En el equilibrio:
ka[A][S] (1- θA) = kd [S]θA (7.41)
𝜃𝐴
1 − 𝜃𝐴=
𝑘𝑎
𝑘𝑑
[𝐴] = 𝐾𝐴[𝐴] (7.42)
Por tanto
𝜃𝐴 =𝐾𝐴[𝐴]
1 + 𝐾𝐴[𝐴] (7.43)
Si la [A] se expresa en función de la presión se obtiene:
𝜃𝐴 =𝐾𝐴𝑃𝐴
1 + 𝐾𝐴𝑃𝐴 (7.44)
Conocida como la ecuación de la isoterma de Langmuir. Así, la velocidad de reacción por unidad de
superficie será proporcional al número de moléculas adsorbidas o, lo que es equivalente, a la fracción de centros
ocupados por moléculas de A:
𝑣 =−𝑑𝐴(𝑎𝑑𝑠)
𝑑𝑡= 𝑘2[𝐴(𝑎𝑑𝑠)] = 𝑘2[𝑆]𝜃𝐴 (7.45)
Es decir,
𝑣 =k2[S]KAPA
1 + KAPA=
k2[S]KA[A]
1 + KA[A] (7.46)
De la figura 7.28, la dependencia de la velocidad con la concentración de A en bajas presiones se cumple que
v ≈ k2KA [S] PA, por lo que la ecuación es de primer orden, en donde la velocidad depende linealmente de la
presión, KAPA << 1. La fracción de superficie cubierta es muy baja, mientras que para altas presiones se cumple
que: v ≈ k2 [S], la ecuación es de orden cero, con la velocidad de reacción independe de la presión, KAPA>>1
involucrando un porcentaje de la superficie cubierta alto.

Química del estado sólido (1615) 20 - II
A. S. P. 27 / 40
Figura 7.28 Isoterma de adsorción Langmuir.
La cinética se complica si alguno de los productos queda adsorbido sobre la superficie, reduciendo el área del
catalizador disponible para A. Al igual que con la ecuación de Michaelis - Menten se puede aplicar el
tratamiento propuesto por Lineweaver-Burk y determinar la k2 o khet mediante una gráfica de 1/v en función de
1/PA, así se tiene la siguiente ecuación:
1
𝑣=
1
𝑘2[𝑆]+
1
𝑘2[𝑆]𝐾𝐴(
1
𝑃𝐴) (7.47)
La cual da una representación lineal donde la pendiente es 1
𝑘2[𝑆]𝐾𝐴, el punto de corte del eje de las ordenadas es
el inverso de k2[S], 1
𝑘2[𝑆].
En muchos casos la adsorción está acompañada por la disociación de la molécula adsorbida sobre la
superficie. Por ejemplo: H2 se adsorbe sobre muchos metales en forma de átomos, cada átomo ocupando un sitio
sobre la superficie.
El proceso de adsorción se describe como:
vads = ka [A][S] (1- θA)2 (7.48)
La velocidad de desorción involucra dos átomos adsorbidos, por lo tanto, la velocidad es proporcional al
cuadrado de la superficie cubierta.
𝑣𝑑 = 𝑘𝑑[𝑆]θA2
En el equilibrio:
𝑣𝑎 = 𝑣𝑑 (7.49)
Es decir,
𝑘𝑑[𝑆]𝜃𝐴2 = 𝑘𝑎[𝐴][𝑆](1 − 𝜃𝐴)2 (7.50)
𝜃𝐴
1 − 𝜃𝐴= (
𝑘𝑎
𝑘𝑑[𝐴])
1/2
= 𝐾𝐴1/2
[𝐴]1/2 (7.51)

Química del estado sólido (1615) 20 - II
A. S. P. 28 / 40
Por tanto
𝜃𝐴 =𝐾𝐴
1/2[𝐴]1/2
1 + 𝐾𝐴1/2
[𝐴]1/2; 𝑣 = 𝑘2[𝑆]𝜃𝐴 (7.52)
Procesos bimoleculares
Los procesos bimoleculares son reacciones del tipo: A + B → productos y se pueden explicar por dos
mecanismos que a continuación son indicados:
Modelo de Langmuir-Hinshelwood. A y B reaccionan cuando están adsorbidos sobre la superficie:
A(g) + B(g) ⇌ A(ads) + B(ads) → Productos(g)
Las moléculas adsorbidas sobre la superficie pueden difundirse de un centro activo a otro hasta que se
encuentren en posiciones vecinas y reaccionen.
Figura 7.29 Ejemplo del mecanismo de adsorción Bimolecular.
Asumiendo como etapa lenta la reacción y que la adsorción se encuentra en equilibrio, la velocidad de
reacción será proporcional al producto de las concentraciones superficiales o de forma equivalente, al producto
de las fracciones de centros ocupados, de la forma: θA y θB fracción de la superficie cubierta por las moléculas de
A y B respectivamente
Velocidad = khet [S]2 θA θB (7.53)
Donde θA y θB fracción de la superficie cubierta por las moléculas de A y B respectivamente.
Las velocidades de adsorción de A y B son:
𝑣𝑎𝑑𝑠𝐴 = 𝑘𝑎
𝐴[𝐴][𝑆](1 − 𝜃𝐴 − 𝜃𝐵) (7.54)
Y
𝑣𝑎𝐵 = 𝑘𝑎
𝐵[𝐵][𝑆](1 − 𝜃𝐴 − 𝜃𝐵) (7.55)
Las velocidades de desorción de A y B son:
𝑣𝑑𝐴 = 𝑘𝑑
𝐴[𝑆]𝜃𝐴 y 𝑣𝑑𝐵 = 𝑘𝑑
𝐵[𝑆]𝜃𝐵 (7.56)

Química del estado sólido (1615) 20 - II
A. S. P. 29 / 40
En el equilibrio, para la especie A se tiene:
𝑣𝑎𝐴 = 𝑣𝑑
𝐴
𝑘𝑎𝐴[𝐴][𝑆](1 − 𝜃𝐴 − 𝜃𝐵) = 𝑘𝑑
𝐴[𝑆]𝜃𝐴 (7.57)
𝜃𝐴
1 − 𝜃𝐴 − 𝜃𝐵=
𝑘𝑎𝐴
𝑘𝑑𝐴
[𝐴] = 𝐾𝐴[𝐴] (7.58)
𝜃𝐴 =𝐾𝐴[𝐴]
1 + 𝐾𝐴[𝐴] + 𝐾𝐵[𝐵] (7.59)
Haciendo un tratamiento para la especie B se tiene:
𝜃𝐵 =𝐾𝐵[𝐵]
1 + 𝐾𝐴[𝐴] + 𝐾𝐵[𝐵] (7.60)
Así, la ecuación de velocidad para la reacción A—B será:
𝑣 =𝑘ℎ𝑒𝑡[𝑆]2𝐾𝐴[𝐴]𝐾𝐵[𝐵]
(1 + 𝐾𝐴[𝐴] + 𝐾𝐵[𝐵])2 (7.61)
Expresada en presiones:
𝑣 =𝑘ℎ𝑒𝑡[𝑆]2𝐾𝐴𝑃𝐴𝐾𝐵𝑃𝐵
(1 + 𝐾𝐴𝑃𝐴 + 𝐾𝐵𝑃𝐵)2 (7.62)
Figura 7.30 Descripción del efecto de la velocidad para una reacción bimolecular donde los dos reactivos
compiten por los sitios activos en un catalizador.
Aquí se tienen 2 casos.
Caso I: La adsorción tanto de A como de B es débil
KAPA << 1
KBPB << 1
Por lo tanto, se tiene la condición
(1 + 𝑥) ≈ 1

Química del estado sólido (1615) 20 - II
A. S. P. 30 / 40
Quedando la ecuación:
𝑣 = 𝑘ℎ𝑒𝑡[𝑆]2𝐾𝐴𝑃𝐴𝐾𝐵𝑃𝐵 (7.63)
Se tiene, entonces, que la reacción es de segundo orden total y de primer orden con respecto a A y a B.
Caso II: Un reactivo, A, es adsorbido con más fuerza que el otro,
De tal manera que:
KAPA >> 1 + KBPB
Se tiene entonces
𝑣 =𝑘ℎ𝑒𝑡𝐾𝐴𝐾𝐵[𝑆]2𝑃𝐴𝑃𝐵
(𝐾𝐴𝑃𝐴)2=
𝑘ℎ𝑒𝑡𝐾𝐵[𝑆]2𝑃𝐵
𝐾𝐴𝑃𝐴 (7.64)
Aquí, la velocidad de la reacción es inversamente proporcional a la concentración de la especie adsorbida
con más fuerza, A en este caso, y la reacción ocurre entre una molécula en fase gas, molécula B, y una molécula
adsorbida sobre la superficie del catalizador, molécula A.
Figura 7.31 Esquema donde el reactivo, A, es adsorbido con más fuerza que el otro.
A bajas presiones de A, KAPA << 1, se tiene
𝑣 ≈ 𝑘ℎ𝑒𝑡[𝑆]𝐾𝐴𝑃𝐵𝑃𝐴 (7.65)
La reacción es de primer orden en A.
𝑣 ≈ 𝑘𝑜𝑏𝑠𝑃𝐴 (7.66)
En altas presiones de A, KAPA>>1, se tiene:
𝑣 ≈ 𝑘ℎ𝑒𝑡[𝑆]𝑃𝐵 (7.67)

Química del estado sólido (1615) 20 - II
A. S. P. 31 / 40
Y la reacción es orden cero en A. Esto se puede ver en la siguiente figura donde a bajas presiones de A la
velocidad de reacción es directamente proporcional (primer orden) con respecto a A y a altas presiones de A, la
velocidad se mantiene constante, es decir, no depende (orden cero) de la presión de A.
Figura 7.32 Efecto de la presión de A en el orden de una reacción bimolecular.
Mecanismo de Eley-Rideal: Surge de la interacción entre una molécula adsorbida y una molécula en fase
gas. Supongamos las reacciones:
A(g) ⇌ A(ads)
A(ads)+B(g)→ Productos(g)
Esquemáticamente (figura 7.33), la reacción tendría lugar a través de un estado de transición de la forma:
Figura 7.33 Esquema donde el reactivo, A, es adsorbido y ahí interacciona con el reactivo B.
De acuerdo con el mecanismo, y asumiendo la etapa de reacción como etapa lenta, podemos escribir la
siguiente ecuación de velocidad tal y como se aprecia en la figura 7.34:
𝑣𝑠 =𝑘𝐾𝐴𝑃𝐴𝑃𝐵
1 + 𝐾𝐴𝑃𝐴 (7.68)
Figura 7.34 Evolución de la velocidad de conversión por unidad de superficie del catalizador en función de la
presión parcial de A para un mecanismo tipo Eley-Rideal.

Química del estado sólido (1615) 20 - II
A. S. P. 32 / 40
En el límite de adsorción débil,
KAPA<<1
se obtiene:
vs≈ kKAPAPB (7.69)
la cual es una ecuación de orden dos global y en el límite de adsorción fuerte,
KAPA>>1
vs≈kPB (7.70)
es una ecuación de orden uno con respecto a la presión de B. Por ejemplo, la combinación de dos átomos de H
para dar hidrógeno molecular, 2H→H2. Experimentalmente se observa que es de orden 1 respecto a la presión
de hidrógeno atómico a temperaturas bajas y de orden 2 a temperaturas altas. Esta observación se puede explicar
con un mecanismo:
H (g) ↔ H (ads)
H (ads) + H (g) → H2 (g)
La ley de velocidad que corresponde a este mecanismo es:
𝑣𝑠 =𝑘𝐾𝐻𝑃𝐻
2
1 + 𝐾𝐻𝑃𝐻 (7.71)
Como ΔHads<0, temperaturas bajas, KH es grande y vs≈kPH, (orden 1); mientras que a temperaturas altas la
constante de adsorción disminuye, cumpliéndose que KHPH << 1 y, por tanto, vs≈k’PH2 (orden 2).
Ejemplos de catálisis heterogénea
La naturaleza química de los catalizadores es tan diversa como la catálisis misma, aunque pueden hacerse
algunas generalizaciones. Los ácidos próticos son probablemente los catalizadores más ampliamente usados,
especialmente para muchas reacciones que involucran agua, incluyendo la hidrólisis y su inversa. Los sólidos
multifuncionales a menudo suelen ser catalíticamente activos, por ejemplo, las zeolitas, la alúmina y ciertas
formas de carbono grafítico. Los metales de transición son utilizados a menudo para catalizar reacciones redox
(oxigenación, hidrogenación). Muchos procesos catalíticos, especialmente los que involucran hidrógeno,
requieren metales del grupo del platino.
Algunos de los llamados catalizadores son, en realidad, precatalizadores. Los precatalizadores se convierten
en el catalizador en el transcurso de la reacción. Por ejemplo, el catalizador de Wilkinson RhCl(PPh3)3 pierde un
ligando trifenilfosfina antes de entrar en el verdadero ciclo catalítico. Los precatalizadores son más fáciles de
almacenar, pero son fácilmente activados in situ. Debido a esta etapa de preactivación, muchas reacciones
catalíticas involucran un período de inducción.
Las especies químicas que mejoran la actividad catalítica son denominadas co-catalizadores o promotores,
en la catálisis cooperativa. La catálisis heterogénea es de suma importancia en muchas áreas de las industrias
químicas y energéticas. La catálisis heterogénea ha atraído premios Nobel para Fritz Haber en 1918, Carl Bosch
en 1931, Irving Langmuir en 1932 y Gerhard Ertl en 2007.

Química del estado sólido (1615) 20 - II
A. S. P. 33 / 40
Con todo lo anterior, se muestran a continuación la gran importancia que tiene la catálisis en la industria, ya
que hasta el 90% de los productos químicos que hoy en día se sintetizan en las industrias se producen a través de
procesos catalíticos. Algunos de los ejemplos más comunes son:
El petróleo
Los petróleos crudos que se extraen de los diferentes campos petrolíferos de la Tierra son de naturaleza muy
variada incluso en su apariencia externa. El petróleo bruto es una mezcla de diferentes hidrocarburos (la mayor
parte saturados). El gas natural, por ejemplo, consiste en una mezcla de moléculas ligeras como metano, etano,
propano y butano. Además, en su composición podemos encontrar ácido sulfhídrico que es corrosivo, por lo que
es necesaria una purificación para eliminarlo. Tradicionalmente el gas natural es utilizado como combustible
para uso doméstico e industrial. Sin embargo, en la última década su consumo para la producción de hidrógeno
se ha elevado. El hidrógeno puede ser utilizado en procesos como la síntesis del amoniaco o como combustible
no contaminante en motores de combustión interna.
El petróleo, una vez extraído, es enviado por oleoductos hacia las refinerías. Los crudos son separados
inicialmente por destilación. En este proceso aproximadamente el 75% de los compuestos son volátiles,
quedando un residuo llamado asfáltico en el fondo. La fracción volátil se separa como sigue en orden
decreciente de punto de ebullición: 1) hidrocarburos gaseosos (metano o butano), 2) gasolina ligera, 3) gasolina
pesada o nafta; 4) queroseno, 5) gasóleo ligero, y 6) gasóleo pesado. Generalmente, los productos obtenidos en
este proceso no son suficientes en calidad ni cantidad para los requerimientos actuales de consumo. Por lo tanto,
se requiere transformar estos productos en otros de uso más conveniente mediante procesos químicos que
habitualmente conllevan el empleo de catalizadores.
El objetivo de los procesos catalíticos del petróleo es el de modificar las fracciones del mismo para la
obtención de productos en cantidad y calidad acordes con los requisitos del mercado. Podemos destacar los
siguientes:
a) Fragmentación. Muchas de las moléculas orgánicas de pequeño tamaño utilizadas en la industria química
se obtienen cortando hidrocarburos de cadena larga procedentes del petróleo. La fragmentación de estos
hidrocarburos inducida catalíticamente se denomina cracking y se realiza sobre alúmino-silicatos
(SiO2/Al2O3, zeolitas) para formar moléculas más ramificadas. Estos isómeros, más cortos, se queman de
forma más suave y eficaz en los motores de combustión interna.
b) Reforming. Es la transformación de la estructura molecular de las naftas. Las naftas extraídas directamente
de la destilación primaria suelen tener moléculas lineales por lo que tienden a detonar por presión. Así, el
reforming se encarga de transformar dichas moléculas lineales en ramificadas y cíclicas, que, al ser más
compactas, no detonan por efecto de la presión. El reforming utiliza un catalizador de doble función,
mezcla de platino y alúmina. El platino proporciona la función metal, catalizando la hidrogenación y
deshidrogenación y la alúmina aporta la función ácida. Primero el hidrocarburo se quimisorbe sobre el
platino. El hidrocarburo pierde dos átomos de hidrógeno formando un alqueno. El alqueno migra a una
posición ácida donde acepta un protón y se une a la superficie como ión carbonio. Este ión se puede
romper, isomerizarse a formas más ramificadas o formar anillos. El efecto general es que el producto
contiene hidrocarburos con formas moleculares más compactas que dan valores más altos de octanaje que
los hidrocarburos presentes en la nafta. Estos productos son parte de la gasolina de alto octanaje o
gasolina comercial.
Biocombustibles

Química del estado sólido (1615) 20 - II
A. S. P. 34 / 40
Los biocombustibles son combustibles de origen biológico alternativos al petróleo, obtenidos a partir de
restos orgánicos. Estos restos orgánicos proceden habitualmente del azúcar, trigo, maíz o semillas oleaginosas.
El uso de biocombustibles tiene impactos ambientales negativos y positivos. Los impactos negativos hacen que,
a pesar de ser una energía renovable, no sea considerada por muchos expertos como una energía no
contaminante y, en consecuencia, tampoco una energía verde. Una de las causas negativas es que, a pesar de que
inicialmente en su producción sólo se usan restos de actividades agrícolas, con su generalización en los países
desarrollados, muchos países subdesarrollados, especialmente el sureste asiático, están destruyendo sus espacios
naturales, incluyendo selvas y bosques, para crear plantaciones para biocombustibles. La consecuencia de esto
es justo la contraria de lo que se desea conseguir con los biocombustibles: los bosques y selvas limpian más el
aire de lo que lo hacen los cultivos que se ponen en su lugar.
Los biocombustibles líquidos proporcionan actualmente la energía equivalente a 20 millones de toneladas de
petróleo. Los biocombustibles que más se utilizan son el biodiesel (ésteres metílicos de ácidos grasos) y el
bioetanol (etanol de origen biológico). El biodiesel es un biocombustible sintético líquido que se obtiene a partir
de lípidos naturales como aceites vegetales o grasas animales, nuevas o usadas, mediante procesos industriales
de esterificación y transesterificación y que se aplica en la preparación de sustitutos totales o parciales del
petrodiésel o gasóleo obtenido del petróleo. Los vegetales más utilizados para la obtención del bioetanol suelen
poseer elevadas cantidades de sacarosa (caña de azúcar, remolacha, …), almidón (maíz, patata, etc) o celulosa
(madera, residuos agrícolas, y demás.). El proceso de fabricación del bioetanol a partir del almidón es más
complejo que a partir de la sacarosa, pues el almidón debe ser hidrolizado previamente para convertirlo en
azúcares. Para ello se mezcla el vegetal triturado con agua y con una enzima (o catalizador), y se calienta la
papilla obtenida a 120-150 °C. Posteriormente, se cuela la masa en un proceso llamado escarificación y se envía
a los reactores de fermentación. La fermentación de los azúcares es llevada a cabo por microorganismos
(levaduras o bacterias) y produce etanol, así como grandes cantidades de CO2. Además, produce otros
compuestos oxigenados indeseables como el metanol, alcoholes superiores, ácidos y aldehídos.
El proceso de fabricación a partir de celulosa es aún más complejo, ya que primero hay que pre-tratar la
materia vegetal para que la celulosa pueda ser luego atacada por las enzimas hidrolizantes que la convierten en
los azúcares precursores del bioetanol. El pretratamiento puede consistir en una combinación de trituración,
pirólisis y ataque con ácidos y otras sustancias. Esto es uno de los factores que explican por qué los
rendimientos en etanol son altos para la caña de azúcar, mediocres para el maíz y bajos para la madera.
Productos químicos de síntesis
Algunos de los productos químicos obtenidos a gran escala se producen a través de la oxidación catalítica, a
menudo usando oxígeno. Algunos ejemplos son el ácido nítrico (a partir de amoníaco), el ácido sulfúrico (a
partir de dióxido de azufre a trióxido de azufre por el proceso de las cámaras de plomo) y el acrilonitrilo a partir
de propano y amoníaco. No obstante, otros muchos productos químicos son generados por reducción a gran
escala, a menudo a través de la hidrogenación. Un ejemplo de estos procesos de reducción es la fabricación
industrial del metanol a partir de monóxido de carbono. La reacción global, catalizada mediante una mezcla de
NiO/Cu, es:
CO( g) + 2H2( g) → CH3OH( g)
Este proceso sigue formalmente un mecanismo de tipo Langmuir-Hinshelwood, donde tanto el CO como el H2
sufren quimisorción (ésta última disociativa). Los reactivos quimisorbidos reaccionan en un proceso que
contiene varias etapas:

Química del estado sólido (1615) 20 - II
A. S. P. 35 / 40
CO(g) → CO(ads)
H2(g) → 2H(ads)
CO(ads) + 2H(ads) → CHOH(ads)
CHOH(ads) + 2H(ads) → CH3OH(ads)
Este mismo proceso puede conducir a la formación de hidrocarburos, dependiendo de las condiciones
experimentales. Si el CO adsorbido reacciona con otras moléculas de CO adsorbido genera carbono adsorbido.
Éste puede reaccionar fácilmente con el hidrógeno monoatómico para dar una cadena de reacciones que
conducen a la formación de hidrocarburos:
CO(ads) +CO(ads) →C(ads) +CO2(ads)
C(ads) + H(ads) → CH(ads )
C(ads) + H(ads) → CH(ads)
Química fina
Muchos productos de química fina también se preparan a través de la catálisis. Son procesos especializados
que serían prohibitivamente caros a gran escala, tales como la síntesis de algunos fármacos y reactivos
biomédicos.
Debido a que la mayoría de los compuestos bioactivos son quirales, muchos productos farmacéuticos son
producidos mediante procesos catalíticos asimétricos. El primer proceso catalítico asimétrico comercializado fue
la síntesis de la L-DOPA (L-3,4-dihidroxifenilalanina) por hidrogenación catalítica asimétrica. La L-DOPA es
el aminoácido precursor inmediato de la dopamina y el fármaco disponible más eficaz en el tratamiento de la
enfermedad de Parkinson. En el tratamiento del Parkinson se administra L-DOPA en lugar de dopamina porque
la dopamina no puede atravesar la barrera hematoencefálica (la que forman las meninges entre los vasos
sanguíneos y el líquido cefalorraquídeo), en cambio la L-DOPA sí que puede atravesarla. William S. Knowles y
Ryoji Noyori recibieron el Premio Nobel de Química en 2001 por sus estudios sobre las reacciones de
hidrogenación mediante catalizadores quirales, conocida como hidrogenación catalítica asimétrica. El
mecanismo de la reacción de hidrogenación catalítica asimétrica empleada en la síntesis de la L-DOPA viene
descrito en la Figura 7.35.
Figura 7.35 Mecanismo de catálisis asimétrica empleado en la síntesis de la L-DOPA.

Química del estado sólido (1615) 20 - II
A. S. P. 36 / 40
Procesamiento de alimentos
Un ejemplo de las aplicaciones de la catálisis en el sector alimentario es la hidrogenación de las grasas.
Frecuentemente se usa níquel metálico como catalizador para producir la margarina. En este caso, la
hidrogenación de los dobles enlaces se utiliza para obtener grasas comestibles a partir de aceites vegetales.
El mecanismo de este proceso está detallado en la Figura 7.41, donde el grupo químico alqueno (1) se
adsorbe formando dos enlaces con la superficie (2), sobre la cual se ha producido también quimisorción
disociativa de H2. Cuando se forma un enlace alqueno- H se rompe uno de los enlaces alqueno- superficie (3, 4).
La formación de un nuevo enlace alqueno- H libera la grasa con los dobles enlaces saturados (5).
Figura 7.36 Mecanismo general de hidrogenación.
Producción de amoniaco
El amoniaco (NH3) un gas incoloro con un olor característico, es un elemento químico fundamental y un
componente clave en la fabricación de muchos productos de uso diario. Por ejemplo, Alrededor del 90 por
ciento del amoníaco producido se utiliza en fertilizantes para mantener la producción agrícola de alimentos. Se
usa como ingrediente activo en muchos productos de limpieza para el hogar como bañeras, lavabos y retretes,
para encimeras y azulejos de baño y cocina. A nivel industrial, usa como un gas refrigerante y en equipos de aire
acondicionado, para purificar los suministros de agua y como un elemento fundamental en la fabricación de
muchos productos como plásticos, explosivos, telas, pesticidas y tinturas entre muchos otros. Erisman y
colaboradores (Erisman, 2008) publica un artículo titulado Cómo un siglo de la síntesis del amoniaco cambió el
mundo. Sus autores ofrecen algunas cifras que destacan la importancia del NH3.
No obstante, la importancia del amoniaco, los principios del siglo XX, la producción de amoniaco era un
proceso extremadamente complicado, debido a altas presiones y temperaturas requeridas para realizar el
proceso, y a los bajos rendimientos de este, aproximadamente del.
3H2 + N2 → 2NH3 H= -92 KJ/mol
En 1910, Friz Haber patento un proceso el cual fue mejorado posteriormente por Carl Bosch realizándose
actualmente a temperaturas (400 – 500 C) y presiones (150 - 300 atm) más accesibles con rendimientos del 10 -
20 % rend. En este proceso, denominado Haber - Bosch, se emplean catalizadores que son elementos metálicos
de hierro (Ertl, 1983) y óxidos de aluminio (Al2O3) y potasio (K2O) permitiendo que el equilibrio se alcance con
mayor rapidez.

Química del estado sólido (1615) 20 - II
A. S. P. 37 / 40
Figura 7.37 Datos técnicos del proceso Haber-Bosch para la producción de amoniaco.
Figura 7.38 Proceso de adsorción para amoniaco, explicado (Tomado de:
https://elpais.com/diario/2007/10/17/futuro/1192572005_740215.html).
Esto puede verse esquemáticamente en el siguiente diagrama.

Química del estado sólido (1615) 20 - II
A. S. P. 38 / 40
Figura 7.39 Diagrama esquemático de los diversos pasos que ocurren en la conversión de dinitrogeno en
amoniaco durante el proceso Haber-Bosch.
Recientemente, Ertl propuso el mecanismo de reacción para de la síntesis catalítica de amonio (Proceso Haber
– Bosch) el cual se muestra a continuación.
Figura 7.40 Mecanismo de la síntesis catalítica de amonio o Proceso Haber – Bosch (Tomado de: Ertl,
G., et al., 1980, p.201).
En este mecanismo se puede observar primeramente la adsorción de ambas moléculas diatómicas (nitrógeno
e hidrogeno) para posteriormente ser separadas en átomos individuales los cuales reaccionan entre si para dar
lugar ya a la formación del amoniaco (producto) el cual es finalmente desorbido y purificado.
Convertidor catalítico
El uso del automóvil y la contaminación del aire son uno de los grandes problemas medioambientales a los
que nos enfrentamos hoy en día, y uno de los más preocupantes de nuestra sociedad, sobre todo para las grandes
ciudades. A diario se vierten a la atmósfera cantidades elevadas de contaminantes que proceden de nuestra vida
cotidiana, así como de las industrias, siendo buena parte de dicho vertido, procedente de los coches que circulan
por nuestras calles.
Para la reducción de la contaminación en los coches, todos los coches en la actualidad incluyen entre sus
piezas un convertidor catalítico que sirve para transformar hidrocarburos, monóxido de carbono y óxidos de
nitrógeno en gases menos dañinos para el medio ambiente.

Química del estado sólido (1615) 20 - II
A. S. P. 39 / 40
En muchos procesos de combustión se generan óxidos de nitrógeno, que son muy contaminante (son uno de
los principales causantes de la lluvia ácida). Para solucionarlo, se suelen utilizar catalizadores a 500°C para
oxidarlos a nitrógeno, que es el gas mayoritario del aire.
El catalizador está compuesto de platino, rodio y paladio y cuando los gases nocivos se ponen en contacto
con él, se generan y aceleran las reacciones químicas que descomponen y oxidan estos gases transformándolos
en gases inocuos para el medio ambiente.
Figura 7.41 Corte de un convertidor catalítico de contacto, que se utiliza en los tubos de escape de los
automóviles.
Así, las principales reacciones químicas que tienen lugar, y en las que participa el catalizador, son:
2 CO (g) + O2 (g) → 2CO2 (g)
2NO (g) + 2 CO (g) → N2 (g) + 2 CO2 (g)
2 C2H6 (g) + 7 O2 (g) → 4 CO2 (g) + 6 H2O (g)
Una variable fundamental para el buen funcionamiento y la vida útil del sistema catalítico, es la temperatura.
Hasta una temperatura de unos 300ºC, el catalizador no funciona óptimamente, siendo la zona óptima para
trabajar está entre los 400 y 800ºC; cuando la temperatura es superior se deteriora lentamente, lo que puede
conllevar a la inutilización del sistema debido al envejecimiento térmico.
7.3 Bibliografía
Adamson, A., & Gast, A. (1997). Physical chemistry of surfaces. New York: Wiley.
Aguilar Rios, G., & Salmones Blasquez, J. (2002). Fundamentos de catalisis. Mexico: Alfaomega, Instituto
Politecnico Nacional.
Arias, J., Paternina, E., & Barragán, D. (2009). Adsorción física sobre sólidos: aspectos termodinámicos.
Química Nova, 32(5), 1350-1355.
Atkins, P., & De Paula, J. (1998). Physical chemistry (6a ed.). Oxford: Oxford University Press.
Castellan, G., Amador Bedolla, C., Costas Basin, M., & Rodriguez Flores, M. (1998). Fisicoquimica. Mexico:
Addison-Wesley Iberoamericana.
Dubinin, M. (1960). The Potential Theory of Adsorption of Gases and Vapors for Adsorbents with Energetically
Nonuniform Surfaces. Chemical Reviews, 60(2), 235-241.
Dumeignil, F., Paul, J., & Paul, S. (2017). Heterogeneous Catalysis with Renewed Attention: Principles,
Theories, and Concepts. Journal of Chemical Education, 94(6), 675-689.
Erisman, J., Sutton, M., Galloway, J., Klimont, Z., & Winiwarter, W. (2008). How a century of ammonia
synthesis changed the world. Nature Geoscience, 1(10), 636-639.

Química del estado sólido (1615) 20 - II
A. S. P. 40 / 40
Ertl, G. (1983). Surface characterization of ammonia synthesis catalysts. Journal of Catalysis, 79(2), 359-377.
Fuentes, S., & Díaz, G. (2016). Catalizadores, ¿la piedra filosofal del siglo XX? (La Ciencia para Todos 59),
México D.F., Fondo de Cultura Económica.
Hanson, R. (2012). A Unified Graphical Representation of Chemical Thermodynamics and Equilibrium.
Journal of Chemical Education, 89(12), 1526-1529.
Heveling, J. (2012), Heterogeneous Catalytic Chemistry by Example of Industrial Applications, Journal of
Chemical Education, 89 (12), 1530-1536.
Levine, I. (1996). Fisicoquimica (4th ed.). McGraw-Hill.
Liu, L., & Corma, A. (2018). Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters
and Nanoparticles. Chemical Reviews, 118(10), 4981-5079.
Liu, Z., Li, J., & Woo, S. (2012). Recent advances in the selective catalytic reduction of NOx by hydrogen in
the presence of oxygen. Energy & Environmental Science, 5(10), 8799.
Lyklema, J. (2005). Fundamentals of interface and colloid science. Amsterdam: Elsevier.
Manual IUPAC de Símbolos y Terminología, Apéndice 2, Pt. 1, (1972), coloide y química de superficies, Pure
Appl. Chem. 31, 578
Mattson, B. M., Foster, W., Greimann, J., Hoette, T., Le, N., Mirich, A., ... Schwanke, E. (2013). Heterogeneous
catalysis: The horiuti-polanyi mechanism and alkene hydrogenation. Journal of Chemical Education, 90(5),
613-619.
Mizuno, N., & Misono, M. (1998). Heterogeneous Catalysis. Chemical Reviews, 98(1), 199-218.
Mu, Q., Shiraiwa, M., Octaviani, M., Ma, N., Ding, A., & Su, H. et al. (2018). Temperature effect on phase state
and reactivity controls atmospheric multiphase chemistry and transport of PAHs. Science Advances, 4(3),
eaap7314.
Schloegl, R., Schoonmaker, R., Muhler, M., & Ertl, G. (1988). Bridging the material gap? Between single
crystal studies and real catalysis. Catalysis Letters, 1(6-7), 237-241


















