
PUNTOS DESTACADOS DE LA INFORMACIÓN DE …
16
1 INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: ÍNDICE* ADVERTENCIA: HEPATOTOXICIDAD 1 INDICACIONES Y USO 1.1 Cáncer colorrectal 1.2 Tumores del estroma gastrointestinal 1.3 Carcinoma hepatocelular 2 POSOLOGÍA Y ADMINISTRACIÓN 2.1 Dosis recomendada 2.2 Modificaciones de la dosis 3 FORMAS FARMACÉUTICAS Y CONCENTRACIONES 4 CONTRAINDICACIONES 5 ADVERTENCIAS Y PRECAUCIONES 5.1 Hepatotoxicidad 5.2 Infecciones 5.3 Hemorragia 5.4 Perforación o fístula gastrointestinales 5.5 Toxicidad dermatológica 5.6 Hipertensión 5.7 Isquemia e infarto cardíacos 5.8 Síndrome de leucoencefalopatía posterior reversible 5.9 Riesgo de cicatrización deficiente de las heridas 5.10 Toxicidad embriofetal 6 REACCIONES ADVERSAS 6.1 Experiencia en ensayos clínicos 6.2 Farmacovigilancia 7 INTERACCIONES FARMACOLÓGICAS 7.1 Efecto de los inductores potentes del CYP3A4 en el regorafenib 7.2 Efecto de los inhibidores potentes del CYP3A4 en el regorafenib 7.3 Efecto del regorafenib en los sustratos de la proteína de resistencia al cáncer de mama (PRCM) 8 USO EN POBLACIONES ESPECÍFICAS 8.1 Embarazo 8.2 Lactancia 8.3 Mujeres y hombres con capacidad reproductora 8.4 Uso pediátrico 8.5 Uso geriátrico 8.6 Disfunción hepática 8.7 Disfunción renal 8.8 Raza 10 SOBREDOSIS 11 DESCRIPCIÓN 12 FARMACOLOGÍA CLÍNICA 12.1 Mecanismo de acción 12.2 Farmacodinámica 12.3 Farmacocinética 13 TOXICOLOGÍA PRECLÍNICA 13.1 Carcinogénesis, mutagénesis, alteraciones de la fertilidad 13.2 Toxicología y/o farmacología en animales 14 ESTUDIOS CLÍNICOS 14.1 Cáncer colorrectal 14.2 Tumores del estroma gastrointestinal 14.3 Carcinoma hepatocelular (CHC) 16 PRESENTACIÓN/CONSERVACIÓN Y MANIPULACIÓN 17 INFORMACIÓN DE ASESORAMIENTO A LOS PACIENTES *No se mencionan las secciones o subsecciones omitidas de la información completa de prescripción. PUNTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓN Estos puntos destacados no incluyen toda la información necesaria para usar STIVARGA de manera segura y eficaz. Consulte la información completa de prescripción correspondiente a STIVARGA. STIVARGA ® (regorafenib) en comprimidos, para uso oral Aprobación inicial en los EE. UU.: 2012 ADVERTENCIA: HEPATOTOXICIDAD Consulte la información completa de prescripción para ver la advertencia enmarcada completa. • Se han observado casos de hepatotoxicidad graves y, en ocasiones, mortales en ensayos clínicos. (5.1) • Debe vigilarse la función hepática antes y durante el tratamiento. (5.1) • Interrumpa y luego reduzca o suspenda el uso de STIVARGA en caso de hepatotoxicidad, manifestada por valores elevados en las pruebas de función hepática o necrosis hepatocelular, según la gravedad y la persistencia. (2.2) ----------------------- CAMBIOS IMPORTANTES RECIENTES ---------------------- Advertencias y precauciones (5.9), Riesgo de cicatrización deficiente de las heridas 02/2020 ----------------------------------- INDICACIONES Y USO ---------------------------------- STIVARGA es un inhibidor de la cinasa indicado para el tratamiento de pacientes afectados por: • Cáncer colorrectal (CCR) metastásico, que han recibido previamente quimioterapia basada en fluoropirimidina, oxaliplatino e irinotecán, un tratamiento anti-VEGF y, en los casos con el gen RAS de tipo silvestre, un tratamiento anti- EGFR. (1.1) • Tumor del estroma gastrointestinal (GIST, por sus siglas en inglés) localmente avanzado, irresecable o metastásico, que han recibido previamente tratamiento con mesilato de imatinib y malato de sunitinib. (1.2) • Carcinoma hepatocelular (CHC), que han recibido previamente tratamiento con sorafenib. (1.3) ---------------------------POSOLOGÍA Y ADMINISTRACIÓN -------------------------- • Dosis recomendada: 160 mg por vía oral, una vez al día durante los primeros 21 días de cada ciclo de 28 días. (2.1) • Tome STIVARGA después de una comida de bajo contenido graso. (2.1, 12.3) --------------FORMAS FARMACÉUTICAS Y CONCENTRACIONES ------------- Comprimidos: 40 mg (3) --------------------------------- CONTRAINDICACIONES -------------------------------- Ninguna. ------------------------- ADVERTENCIAS Y PRECAUCIONES ------------------------ • Hepatotoxicidad: Deben vigilarse las pruebas de función hepática. Interrumpa y luego reduzca o suspenda el uso de STIVARGA en función de la gravedad y la duración. (5.1) • Infecciones: Interrumpa el uso de STIVARGA en pacientes con infecciones graves o que empeoran. (5.2) • Hemorragia: Suspenda de manera permanente el uso de STIVARGA en caso de hemorragia intensa o potencialmente mortal. (5.3) • Perforación o fístula gastrointestinales: Suspenda el uso de STIVARGA. (5.4) • Toxicidad dermatológica: Interrumpa y luego reduzca o suspenda el uso de STIVARGA según la gravedad y la persistencia de la toxicidad dermatológica. (5.5) • Hipertensión: Interrumpa de manera temporal o permanente el uso de STIVARGA en caso de hipertensión grave o no controlada. (5.6) • Isquemia e infarto cardíacos: Interrumpa la administración de STIVARGA en caso de isquemia/infarto cardíaco agudo o ante una nueva aparición, y reanude la administración únicamente después de que se resuelvan los eventos de isquemia aguda. (5.7) • Síndrome de leucoencefalopatía posterior reversible (SLPR): Suspenda el uso de STIVARGA. (5.8) • Riesgo de cicatrización deficiente de las heridas: Debe interrumpirse 2 semanas antes como mínimo de cualquier intervención quirúrgica programada. Debe evitarse la administración después de cualquier intervención quirúrgica mayor durante al menos 2 semanas o hasta que las heridas hayan cicatrizado adecuadamente. No se ha establecido la seguridad de reanudar la administración de STIVARGA después de resolver complicaciones de cicatrización de heridas. (5.9) • Toxicidad embriofetal: Puede causar daño fetal. Advierta a las mujeres del posible riesgo para el feto y de que deben usar un método anticonceptivo eficaz durante el tratamiento y durante los 2 meses siguientes a la dosis final. Advierta a los hombres de que deben usar un método anticonceptivo eficaz durante los 2 meses siguientes a la dosis final. (5.10, 8.1, 8.3) ---------------------------------REACCIONES ADVERSAS -------------------------------- Las reacciones adversas más frecuentes (≥20%) son: dolor (incluido dolor gastrointestinal y abdominal), reacción cutánea de manos y pies (hand-foot skin reaction, HFSR), astenia/fatiga, diarrea, reducción del apetito y del consumo de alimentos, hipertensión, infección, disfonía, hiperbilirrubinemia, fiebre, mucositis, pérdida de peso, erupción cutánea y náuseas. (6) Para notificar SOSPECHAS DE REACCIONES ADVERSAS, llame a Bayer HealthCare Pharmaceuticals Inc. al 1-888-842-2937, o comuníquese con la FDA llamando al 1-800-FDA-1088 o visitando www.fda.gov/medwatch. ----------------------- INTERACCIONES FARMACOLÓGICAS ---------------------- • Inductores potentes del CYP3A4: Evite los inductores potentes del CYP3A4. (7.1) • Inhibidores potentes del CYP3A4: Evite los inhibidores potentes del CYP3A4. (7.2) • Sustratos de la PRCM: Vigile atentamente a los pacientes para detectar un aumento de la exposición a los sustratos de la PRCM. (7.3) ------------------------ USO EN POBLACIONES ESPECÍFICAS ----------------------- Madres lactantes: Suspenda la administración del fármaco o la lactancia según la importancia del fármaco para la madre. (8.3) Consulte la sección 17 para ver INFORMACIÓN DE ASESORAMIENTO A LOS PACIENTES y la información sobre el producto para pacientes aprobada por la FDA. Actualizado: 02/2020
Transcript of PUNTOS DESTACADOS DE LA INFORMACIÓN DE …

1
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN: ÍNDICE*ADVERTENCIA: HEPATOTOXICIDAD1 INDICACIONES Y USO 1.1 Cáncer colorrectal 1.2 Tumores del estroma gastrointestinal 1.3 Carcinoma hepatocelular2 POSOLOGÍA Y ADMINISTRACIÓN 2.1 Dosis recomendada 2.2 Modificaciones de la dosis3 FORMAS FARMACÉUTICAS Y CONCENTRACIONES4 CONTRAINDICACIONES5 ADVERTENCIAS Y PRECAUCIONES 5.1 Hepatotoxicidad 5.2 Infecciones 5.3 Hemorragia 5.4 Perforación o fístula gastrointestinales 5.5 Toxicidad dermatológica 5.6 Hipertensión 5.7 Isquemia e infarto cardíacos 5.8 Síndrome de leucoencefalopatía posterior reversible 5.9 Riesgo de cicatrización deficiente de las heridas 5.10 Toxicidad embriofetal6 REACCIONES ADVERSAS 6.1 Experiencia en ensayos clínicos 6.2 Farmacovigilancia7 INTERACCIONES FARMACOLÓGICAS 7.1 Efecto de los inductores potentes del CYP3A4 en el regorafenib 7.2 Efecto de los inhibidores potentes del CYP3A4 en el regorafenib
7.3 Efecto del regorafenib en los sustratos de la proteína de resistencia al cáncer de mama (PRCM)
8 USO EN POBLACIONES ESPECÍFICAS 8.1 Embarazo 8.2 Lactancia 8.3 Mujeres y hombres con capacidad reproductora 8.4 Uso pediátrico 8.5 Uso geriátrico 8.6 Disfunción hepática 8.7 Disfunción renal 8.8 Raza10 SOBREDOSIS11 DESCRIPCIÓN12 FARMACOLOGÍA CLÍNICA 12.1 Mecanismo de acción 12.2 Farmacodinámica 12.3 Farmacocinética13 TOXICOLOGÍA PRECLÍNICA 13.1 Carcinogénesis, mutagénesis, alteraciones de la fertilidad 13.2 Toxicología y/o farmacología en animales14 ESTUDIOS CLÍNICOS 14.1 Cáncer colorrectal 14.2 Tumores del estroma gastrointestinal 14.3 Carcinoma hepatocelular (CHC)16 PRESENTACIÓN/CONSERVACIÓN Y MANIPULACIÓN17 INFORMACIÓN DE ASESORAMIENTO A LOS PACIENTES* No se mencionan las secciones o subsecciones omitidas de la información completa de prescripción.
PUNTOS DESTACADOS DE LA INFORMACIÓN DE PRESCRIPCIÓNEstos puntos destacados no incluyen toda la información necesaria para usar STIVARGA de manera segura y eficaz. Consulte la información completa de prescripción correspondiente a STIVARGA.STIVARGA® (regorafenib) en comprimidos, para uso oralAprobación inicial en los EE. UU.: 2012
ADVERTENCIA: HEPATOTOXICIDADConsulte la información completa de prescripción para ver la advertencia
enmarcada completa.• Se han observado casos de hepatotoxicidad graves y, en ocasiones, mortales
en ensayos clínicos. (5.1)• Debe vigilarse la función hepática antes y durante el tratamiento. (5.1)• Interrumpa y luego reduzca o suspenda el uso de STIVARGA en caso de
hepatotoxicidad, manifestada por valores elevados en las pruebas de función hepática o necrosis hepatocelular, según la gravedad y la persistencia. (2.2)
----------------------- CAMBIOS IMPORTANTES RECIENTES ----------------------Advertencias y precauciones (5.9), Riesgo de cicatrización deficiente de las heridas 02/2020----------------------------------- INDICACIONES Y USO ----------------------------------STIVARGA es un inhibidor de la cinasa indicado para el tratamiento de pacientes afectados por:• Cáncer colorrectal (CCR) metastásico, que han recibido previamente
quimioterapia basada en fluoropirimidina, oxaliplatino e irinotecán, un tratamiento anti-VEGF y, en los casos con el gen RAS de tipo silvestre, un tratamiento anti-EGFR. (1.1)
• Tumor del estroma gastrointestinal (GIST, por sus siglas en inglés) localmente avanzado, irresecable o metastásico, que han recibido previamente tratamiento con mesilato de imatinib y malato de sunitinib. (1.2)
• Carcinoma hepatocelular (CHC), que han recibido previamente tratamiento con sorafenib. (1.3)
---------------------------POSOLOGÍA Y ADMINISTRACIÓN --------------------------• Dosis recomendada: 160 mg por vía oral, una vez al día durante los primeros 21
días de cada ciclo de 28 días. (2.1)• Tome STIVARGA después de una comida de bajo contenido graso. (2.1, 12.3)--------------FORMAS FARMACÉUTICAS Y CONCENTRACIONES -------------Comprimidos: 40 mg (3)--------------------------------- CONTRAINDICACIONES --------------------------------Ninguna.------------------------- ADVERTENCIAS Y PRECAUCIONES ------------------------• Hepatotoxicidad: Deben vigilarse las pruebas de función hepática. Interrumpa y
luego reduzca o suspenda el uso de STIVARGA en función de la gravedad y la duración. (5.1)
• Infecciones: Interrumpa el uso de STIVARGA en pacientes con infecciones graves o que empeoran. (5.2)
• Hemorragia: Suspenda de manera permanente el uso de STIVARGA en caso de hemorragia intensa o potencialmente mortal. (5.3)
• Perforación o fístula gastrointestinales: Suspenda el uso de STIVARGA. (5.4)• Toxicidad dermatológica: Interrumpa y luego reduzca o suspenda el uso de
STIVARGA según la gravedad y la persistencia de la toxicidad dermatológica. (5.5)• Hipertensión: Interrumpa de manera temporal o permanente el uso de STIVARGA
en caso de hipertensión grave o no controlada. (5.6)• Isquemia e infarto cardíacos: Interrumpa la administración de STIVARGA en
caso de isquemia/infarto cardíaco agudo o ante una nueva aparición, y reanude la administración únicamente después de que se resuelvan los eventos de isquemia aguda. (5.7)
• Síndrome de leucoencefalopatía posterior reversible (SLPR): Suspenda el uso de STIVARGA. (5.8)
• Riesgo de cicatrización deficiente de las heridas: Debe interrumpirse 2 semanas antes como mínimo de cualquier intervención quirúrgica programada. Debe evitarse la administración después de cualquier intervención quirúrgica mayor durante al menos 2 semanas o hasta que las heridas hayan cicatrizado adecuadamente. No se ha establecido la seguridad de reanudar la administración de STIVARGA después de resolver complicaciones de cicatrización de heridas. (5.9)
• Toxicidad embriofetal: Puede causar daño fetal. Advierta a las mujeres del posible riesgo para el feto y de que deben usar un método anticonceptivo eficaz durante el tratamiento y durante los 2 meses siguientes a la dosis final. Advierta a los hombres de que deben usar un método anticonceptivo eficaz durante los 2 meses siguientes a la dosis final. (5.10, 8.1, 8.3)
---------------------------------REACCIONES ADVERSAS --------------------------------Las reacciones adversas más frecuentes (≥20%) son: dolor (incluido dolor gastrointestinal y abdominal), reacción cutánea de manos y pies (hand-foot skin reaction, HFSR), astenia/fatiga, diarrea, reducción del apetito y del consumo de alimentos, hipertensión, infección, disfonía, hiperbilirrubinemia, fiebre, mucositis, pérdida de peso, erupción cutánea y náuseas. (6)Para notificar SOSPECHAS DE REACCIONES ADVERSAS, llame a Bayer HealthCare Pharmaceuticals Inc. al 1-888-842-2937, o comuníquese con la FDA llamando al 1-800-FDA-1088 o visitando www.fda.gov/medwatch.----------------------- INTERACCIONES FARMACOLÓGICAS ----------------------• Inductores potentes del CYP3A4: Evite los inductores potentes del CYP3A4. (7.1)• Inhibidores potentes del CYP3A4: Evite los inhibidores potentes del CYP3A4. (7.2)• Sustratos de la PRCM: Vigile atentamente a los pacientes para detectar un
aumento de la exposición a los sustratos de la PRCM. (7.3)------------------------USO EN POBLACIONES ESPECÍFICAS -----------------------Madres lactantes: Suspenda la administración del fármaco o la lactancia según la importancia del fármaco para la madre. (8.3)Consulte la sección 17 para ver INFORMACIÓN DE ASESORAMIENTO A LOS PACIENTES y la información sobre el producto para pacientes aprobada por la FDA.
Actualizado: 02/2020

2
INFORMACIÓN COMPLETA DE PRESCRIPCIÓN
ADVERTENCIA: HEPATOTOXICIDAD• Se han observado casos de hepatotoxicidad graves y, en ocasiones, mortales en ensayos clínicos [ver Advertencias y precauciones (5.1)].• Debe vigilarse la función hepática antes y durante el tratamiento [ver Advertencias y precauciones (5.1)].• Interrumpa y luego reduzca o suspenda el uso de STIVARGA en caso de hepatotoxicidad, manifestada por valores elevados en las pruebas de función hepática o
necrosis hepatocelular, según la gravedad y la persistencia [ver Posología y administración (2.2)].
1 INDICACIONES Y USO1.1 Cáncer colorrectalSTIVARGA está indicado para el tratamiento de pacientes con cáncer colorrectal (CCR) metastásico que han recibido previamente quimioterapia basada en fluoropirimidina, oxaliplatino e irinotecán, un tratamiento anti-VEGF y, en los casos con el gen RAS de tipo silvestre, un tratamiento anti-EGFR.
1.2 Tumores del estroma gastrointestinalSTIVARGA está indicado para el tratamiento de pacientes con tumor del estroma gastrointestinal (GIST, por sus siglas en inglés) localmente avanzado, irresecable o metastásico que han recibido previamente tratamiento con mesilato de imatinib y malato de sunitinib.
1.3 Carcinoma hepatocelularSTIVARGA está indicado para el tratamiento de pacientes con carcinoma hepatocelular (CHC) que han recibido previamente tratamiento con sorafenib.
2 POSOLOGÍA Y ADMINISTRACIÓN2.1 Dosis recomendadaLa dosis recomendada es de 160 mg de STIVARGA (cuatro comprimidos de 40 mg) que se toman por vía oral una vez al día durante los primeros 21 días de cada ciclo de 28 días. Continúe el tratamiento hasta que se produzca progresión de la enfermedad o toxicidad inadmisible.Tome STIVARGA a la misma hora todos los días. Trague el comprimido entero con agua después de una comida de bajo contenido graso que contenga menos de 600 calorías y menos de un 30% de grasa [ver Farmacología clínica (12.3)]. No tome dos dosis de STIVARGA el mismo día para compensar una dosis omitida el día anterior.
2.2 Modificaciones de la dosisSi es necesario modificar la dosis, reduzca la dosis en incrementos de 40 mg (un comprimido); la dosis diaria recomendada más baja de STIVARGA es de 80 mg al día.Interrumpa el uso de STIVARGA en las siguientes situaciones:• Reacción cutánea de manos y pies (HFSR) [eritrodisestesia palmo-plantar (EPP)] de grado 2 que sea recurrente o no mejore en el plazo de 7 días a pesar de la reducción
de la dosis; en casos de HFSR de grado 3, interrumpa el tratamiento durante un mínimo de 7 días• Hipertensión sintomática de grado 2• Cualquier reacción adversa de grado 3 o 4• Infección de cualquier grado que empeoreReduzca la dosis de STIVARGA a 120 mg:• Ante la primera aparición de HFSR de grado 2, cualquiera que sea su duración• Después de la recuperación de cualquier reacción adversa de grado 3 o 4, excepto en el caso de infección• En caso de elevación de grado 3 en los valores de aspartato-aminotransferasa (AST)/alanina-aminotransferasa (ALT); reanude la administración únicamente si el posible
beneficio supera el riesgo de hepatotoxicidadReduzca la dosis de STIVARGA a 80 mg:• En caso de reaparición de HFSR de grado 2 con la dosis de 120 mg• Después de la recuperación de cualquier reacción adversa de grado 3 o 4 con la dosis de 120 mg (excepto en caso de hepatotoxicidad o infección)Suspenda el uso de STIVARGA de manera permanente en las siguientes situaciones:• Intolerancia a la dosis de 80 mg• Cualquier valor de AST o ALT más de 20 veces mayor que el límite superior de normalidad (LSN)• Cualquier valor de AST o ALT más de 3 veces mayor que el LSN con un valor concurrente de bilirrubina más de 2 veces mayor que el LSN• Reaparición de un valor de AST o ALT más de 5 veces mayor que el LSN a pesar de la reducción de la dosis a 120 mg• Por cualquier reacción adversa de grado 4; reanude la administración únicamente si el posible beneficio supera los riesgos
3 FORMAS FARMACÉUTICAS Y CONCENTRACIONESSTIVARGA es un comprimido de 40 mg de color rosa claro, forma ovalada, con recubrimiento pelicular, que presenta grabado bajo relieve la inscripción “BAYER” sobre un lado y “40” sobre el otro.
4 CONTRAINDICACIONESNinguna.
5 ADVERTENCIAS Y PRECAUCIONES5.1 HepatotoxicidadSe produjeron casos de muerte por lesión hepática grave inducida por el fármaco en pacientes tratados con STIVARGA en ensayos clínicos. En la mayoría de los casos, la disfunción hepática se produjo dentro de los 2 primeros meses de tratamiento y se caracterizó por un patrón de lesión hepatocelular.En el estudio CORRECT se produjeron casos de insuficiencia hepática mortal en el 1.6% de los pacientes del grupo tratado con regorafenib y en el 0.4% de los pacientes del grupo que recibió placebo. En el estudio GRID se produjeron casos de insuficiencia hepática mortal en el 0.8% de los pacientes del grupo tratado con regorafenib. En el estudio RESORCE no se produjo un aumento de la incidencia de insuficiencia hepática mortal en comparación con el placebo [ver Reacciones adversas (6.1)].Se deben realizar pruebas de función hepática (ALT, AST y bilirrubina) antes de iniciar la administración de STIVARGA y al menos cada dos semanas durante los primeros 2 meses de tratamiento. De allí en adelante se debe realizar el control una vez al mes o con una frecuencia mayor según las indicaciones clínicas. En los pacientes que presenten resultados elevados, las pruebas de función hepática se deben realizar una vez a la semana hasta que se observe una mejora y los valores sean menores que el triple del LSN o el valor basal.Interrumpa la administración temporalmente y luego reduzca o suspenda de manera permanente el uso de STIVARGA según la gravedad y la persistencia de la hepatotoxicidad, manifestada por valores elevados en las pruebas de función hepática o necrosis hepatocelular [ver Posología y administración (2.2) y Uso en poblaciones específicas (8.6)].
5.2 InfeccionesSTIVARGA ocasionó un aumento del riesgo de sufrir infecciones. La incidencia general de infección (grados 1-5) fue más elevada (32% frente al 17%) en 1142 pacientes tratados con STIVARGA en comparación con el grupo de control en ensayos aleatorizados controlados con placebo. La incidencia de las infecciones de grado 3 o mayor en pacientes tratados con STIVARGA fue del 9%. Las infecciones más frecuentes fueron infecciones del aparato urinario (5.7%), rinofaringitis (4.0%), infecciones mucocutáneas y fúngicas sistémicas (3.3%) y neumonía (2.6%). Los casos de muerte por infección se produjeron con más frecuencia en pacientes tratados con STIVARGA (1.0%) en comparación con los pacientes que recibieron placebo (0.3%); las infecciones mortales más frecuentes fueron respiratorias (0.6% en los pacientes tratados con STIVARGA frente al 0.2% en los pacientes que recibieron placebo).

3
Interrumpa el uso de STIVARGA para las infecciones de grado 3 o 4 o para las infecciones de cualquier grado que empeoren. Reanude la administración de STIVARGA con la misma dosis luego de la resolución de la infección [ver Posología y administración (2.2)].
5.3 HemorragiaSTIVARGA ocasionó un aumento en la incidencia de hemorragia. La incidencia general (grados 1-5) fue del 18.2% en 1142 pacientes tratados con STIVARGA y del 9.5% en los pacientes que recibieron placebo en ensayos aleatorizados controlados con placebo. La incidencia de las hemorragias de grado 3 o mayores en pacientes tratados con STIVARGA fue del 3.0%. Los eventos hemorrágicos mortales tuvieron una incidencia del 0.7% y afectaron al sistema nervioso central, las vías respiratorias, el tubo gastrointestinal o el aparato genitourinario.Suspenda de manera permanente el uso de STIVARGA en pacientes con hemorragia intensa o potencialmente mortal. En los pacientes que reciben warfarina, se deben vigilar los niveles del índice internacional normalizado (IIN) con más frecuencia [ver Farmacología clínica (12.3)].
5.4 Perforación o fístula gastrointestinalesSe produjeron casos de perforación gastrointestinal en el 0.6% de 4518 pacientes tratados con STIVARGA en todos los ensayos clínicos de STIVARGA administrado en monoterapia; esto incluyó ocho muertes.Se produjo fístula gastrointestinal en el 0.8% de los pacientes tratados con STIVARGA y el 0.2% de los pacientes del grupo que recibió placebo en todos los ensayos aleatorizados controlados con placebo. Suspenda de manera permanente el uso de STIVARGA en pacientes que presenten perforación o fístula gastrointestinales.
5.5 Toxicidad dermatológicaEn ensayos aleatorizados controlados con placebo se produjeron reacciones adversas en la piel en el 71.9% de los pacientes del grupo tratado con regorafenib y en el 25.5% de los pacientes del grupo que recibió placebo, entre estas: reacción cutánea de manos y pies (HFSR) denominada también eritrodisestesia palmo-plantar (EPP) y erupción cutánea intensa que requirió la modificación de la dosis.En los ensayos aleatorizados controlados con placebo, la incidencia general de HFSR fue más elevada en 1142 pacientes tratados con STIVARGA (53%) que en los que recibieron placebo (8%). La mayoría de los casos de HFSR en los pacientes tratados con STIVARGA aparecieron durante el primer ciclo de tratamiento. La incidencia de HFSR de grado 3 (16% frente a <1%), erupción cutánea de grado 3 (3% frente a <1%), reacciones adversas graves de eritema multiforme (<0.1% frente al 0%) y síndrome de Stevens Johnson (<0.1% frente al 0%) fue también más elevada en los pacientes tratados con STIVARGA [ver Reacciones adversas (6.1)]. En todos los ensayos se observó una incidencia más elevada de HFSR en pacientes asiáticos tratados con STIVARGA (todos los grados: 72%; grado 3: 18%) [ver Uso en poblaciones específicas (8.8)]. Se produjeron casos de necrólisis epidérmica tóxica en el 0.02% de 4518 pacientes tratados con STIVARGA en todos los ensayos clínicos en los que STIVARGA se administró en monoterapia.Interrumpa la administración de STIVARGA, reduzca la dosis o suspenda de manera permanente el uso de STIVARGA según la gravedad y la persistencia de la toxicidad dermatológica [ver Posología y administración (2.2)]. Implemente medidas de apoyo para proporcionar un alivio sintomático.
5.6 HipertensiónEn los ensayos aleatorizados controlados con placebo se produjeron casos de crisis hipertensivas en el 0.2% de los pacientes de los grupos tratados con regorafenib y en ninguno de los pacientes de los grupos que recibieron placebo. STIVARGA causó un aumento de la incidencia de hipertensión (30% frente al 8% en el estudio CORRECT, 59% frente al 27% en el estudio GRID y 31% frente al 6% en el estudio RESORCE) [ver Reacciones adversas (6.1)]. La aparición de hipertensión se produjo durante el primer ciclo de tratamiento en la mayoría de los pacientes que la presentaron (67% en los ensayos aleatorizados controlados con placebo).No inicie la administración de STIVARGA a menos que la presión arterial esté adecuadamente controlada. Debe medirse la presión arterial una vez a la semana durante las primeras 6 semanas de tratamiento y, posteriormente, en cada ciclo o con más frecuencia, según las indicaciones clínicas. Interrumpa de manera temporal o permanente el uso de STIVARGA en caso de hipertensión grave o no controlada [ver Posología y administración (2.2)].
5.7 Isquemia e infarto cardíacosSTIVARGA aumentó la incidencia de isquemia e infarto de miocardio (0.9% frente al 0.2%) en ensayos aleatorizados controlados con placebo [ver Reacciones adversas (6.1)]. Interrumpa la administración de STIVARGA en pacientes que presenten casos agudos o una nueva aparición de isquemia o infarto cardíacos. Reanude la administración de STIVARGA únicamente después de que se resuelvan los eventos de isquemia cardíaca aguda y si los posibles beneficios superan los riesgos de otra isquemia cardíaca.
5.8 Síndrome de leucoencefalopatía posterior reversibleEn uno de 4800 pacientes tratados con STIVARGA en todos los ensayos clínicos se produjo el síndrome de leucoencefalopatía posterior reversible (SLPR), el cual es un trastorno asociado a edema vasógeno subcortical diagnosticado mediante hallazgos característicos por resonancia magnética. Realice una evaluación de SLPR en todo paciente que presente convulsiones, cefalea intensa, alteraciones visuales, confusión o alteraciones de la función mental. Suspenda el uso de STIVARGA en pacientes que presenten SLPR.
5.9 CicatrizacióndeficientedelasheridasPueden producirse complicaciones debido a una cicatrización deficiente de las heridas en pacientes tratados con fármacos que inhiban las vías de señalización del VEGF. Por lo tanto, STIVARGA podría afectar de manera adversa la cicatrización de las heridas.Debe interrumpirse 2 semanas antes como mínimo de cualquier intervención quirúrgica programada. Debe evitarse la administración después de cualquier intervención quirúrgica mayor durante al menos 2 semanas o hasta que las heridas hayan cicatrizado adecuadamente. No se ha establecido la seguridad de reanudar la administración de STIVARGA después de resolver complicaciones de cicatrización de heridas.
5.10 Toxicidad embriofetalSobre la base de estudios en animales y su mecanismo de acción, STIVARGA puede causar daño fetal si se administra a una mujer embarazada. No se dispone de información sobre el uso de STIVARGA en mujeres embarazadas. El regorafenib fue embrioletal y teratógeno en ratas y conejos expuestos a concentraciones más bajas que las exposiciones a la dosis recomendada en seres humanos, y produjo un aumento en las incidencias de malformaciones cardiovasculares, genitourinarias y esqueléticas. Advierta a las mujeres embarazadas del posible riesgo para el feto.A las mujeres que tengan capacidad reproductora, adviértales de que deben usar un método anticonceptivo eficaz durante el tratamiento con STIVARGA y durante los 2 meses siguientes a la dosis final. A los hombres que tengan parejas femeninas con capacidad reproductora, adviértales de que deben usar un método anticonceptivo eficaz durante el tratamiento con STIVARGA y durante los 2 meses siguientes a la dosis final [ver Uso en poblaciones específicas (8.1), (8.3)].
6 REACCIONES ADVERSASLas siguientes reacciones adversas graves se analizan en otras partes de la información sobre el producto:
• Hepatotoxicidad [ver Advertencias y precauciones (5.1)]• Infecciones [ver Advertencias y precauciones (5.2)]• Hemorragia [ver Advertencias y precauciones (5.3)]• Perforación o fístula gastrointestinales [ver Advertencias y precauciones (5.4)]• Toxicidad dermatológica [ver Advertencias y precauciones (5.5)]• Hipertensión [ver Advertencias y precauciones (5.6)]• Isquemia e infarto cardíacos [ver Advertencias y precauciones (5.7)]• Síndrome de leucoencefalopatía posterior reversible (SLPR) [ver Advertencias y precauciones (5.8)]
4
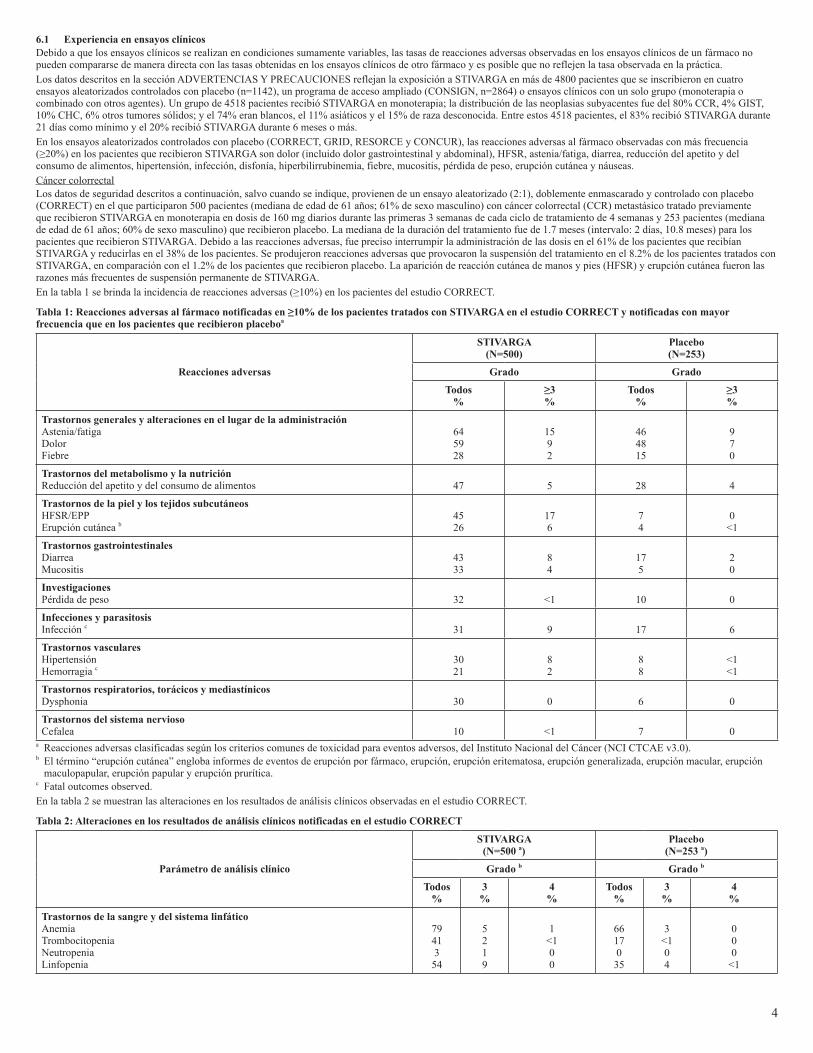
6.1 Experiencia en ensayos clínicosDebido a que los ensayos clínicos se realizan en condiciones sumamente variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden compararse de manera directa con las tasas obtenidas en los ensayos clínicos de otro fármaco y es posible que no reflejen la tasa observada en la práctica. Los datos descritos en la sección ADVERTENCIAS Y PRECAUCIONES reflejan la exposición a STIVARGA en más de 4800 pacientes que se inscribieron en cuatro ensayos aleatorizados controlados con placebo (n=1142), un programa de acceso ampliado (CONSIGN, n=2864) o ensayos clínicos con un solo grupo (monoterapia o combinado con otros agentes). Un grupo de 4518 pacientes recibió STIVARGA en monoterapia; la distribución de las neoplasias subyacentes fue del 80% CCR, 4% GIST, 10% CHC, 6% otros tumores sólidos; y el 74% eran blancos, el 11% asiáticos y el 15% de raza desconocida. Entre estos 4518 pacientes, el 83% recibió STIVARGA durante 21 días como mínimo y el 20% recibió STIVARGA durante 6 meses o más.En los ensayos aleatorizados controlados con placebo (CORRECT, GRID, RESORCE y CONCUR), las reacciones adversas al fármaco observadas con más frecuencia (≥20%) en los pacientes que recibieron STIVARGA son dolor (incluido dolor gastrointestinal y abdominal), HFSR, astenia/fatiga, diarrea, reducción del apetito y del consumo de alimentos, hipertensión, infección, disfonía, hiperbilirrubinemia, fiebre, mucositis, pérdida de peso, erupción cutánea y náuseas.Cáncer colorrectalLos datos de seguridad descritos a continuación, salvo cuando se indique, provienen de un ensayo aleatorizado (2:1), doblemente enmascarado y controlado con placebo (CORRECT) en el que participaron 500 pacientes (mediana de edad de 61 años; 61% de sexo masculino) con cáncer colorrectal (CCR) metastásico tratado previamente que recibieron STIVARGA en monoterapia en dosis de 160 mg diarios durante las primeras 3 semanas de cada ciclo de tratamiento de 4 semanas y 253 pacientes (mediana de edad de 61 años; 60% de sexo masculino) que recibieron placebo. La mediana de la duración del tratamiento fue de 1.7 meses (intervalo: 2 días, 10.8 meses) para los pacientes que recibieron STIVARGA. Debido a las reacciones adversas, fue preciso interrumpir la administración de las dosis en el 61% de los pacientes que recibían STIVARGA y reducirlas en el 38% de los pacientes. Se produjeron reacciones adversas que provocaron la suspensión del tratamiento en el 8.2% de los pacientes tratados con STIVARGA, en comparación con el 1.2% de los pacientes que recibieron placebo. La aparición de reacción cutánea de manos y pies (HFSR) y erupción cutánea fueron las razones más frecuentes de suspensión permanente de STIVARGA.En la tabla 1 se brinda la incidencia de reacciones adversas (≥10%) en los pacientes del estudio CORRECT.
Tabla1:Reaccionesadversasalfármaconotificadasen≥10%delospacientestratadosconSTIVARGAenelestudioCORRECTynotificadasconmayorfrecuencia que en los pacientes que recibieron placeboa
Reacciones adversas
STIVARGA(N=500)
Placebo(N=253)
Grado Grado
Todos%
≥3%
Todos%
≥3%
Trastornos generales y alteraciones en el lugar de la administraciónAstenia/fatigaDolorFiebre
645928
1592
464815
970
Trastornos del metabolismo y la nutriciónReducción del apetito y del consumo de alimentos 47 5 28 4
Trastornos de la piel y los tejidos subcutáneosHFSR/EPPErupción cutánea b
4526
176
74
0<1
Trastornos gastrointestinalesDiarreaMucositis
4333
84
175
20
InvestigacionesPérdida de peso 32 <1 10 0
Infecciones y parasitosisInfección c 31 9 17 6
Trastornos vascularesHipertensiónHemorragia c
3021
82
88
<1<1
Trastornos respiratorios, torácicos y mediastínicosDysphonia 30 0 6 0
Trastornos del sistema nerviosoCefalea 10 <1 7 0
a Reacciones adversas clasificadas según los criterios comunes de toxicidad para eventos adversos, del Instituto Nacional del Cáncer (NCI CTCAE v3.0).b El término “erupción cutánea” engloba informes de eventos de erupción por fármaco, erupción, erupción eritematosa, erupción generalizada, erupción macular, erupción
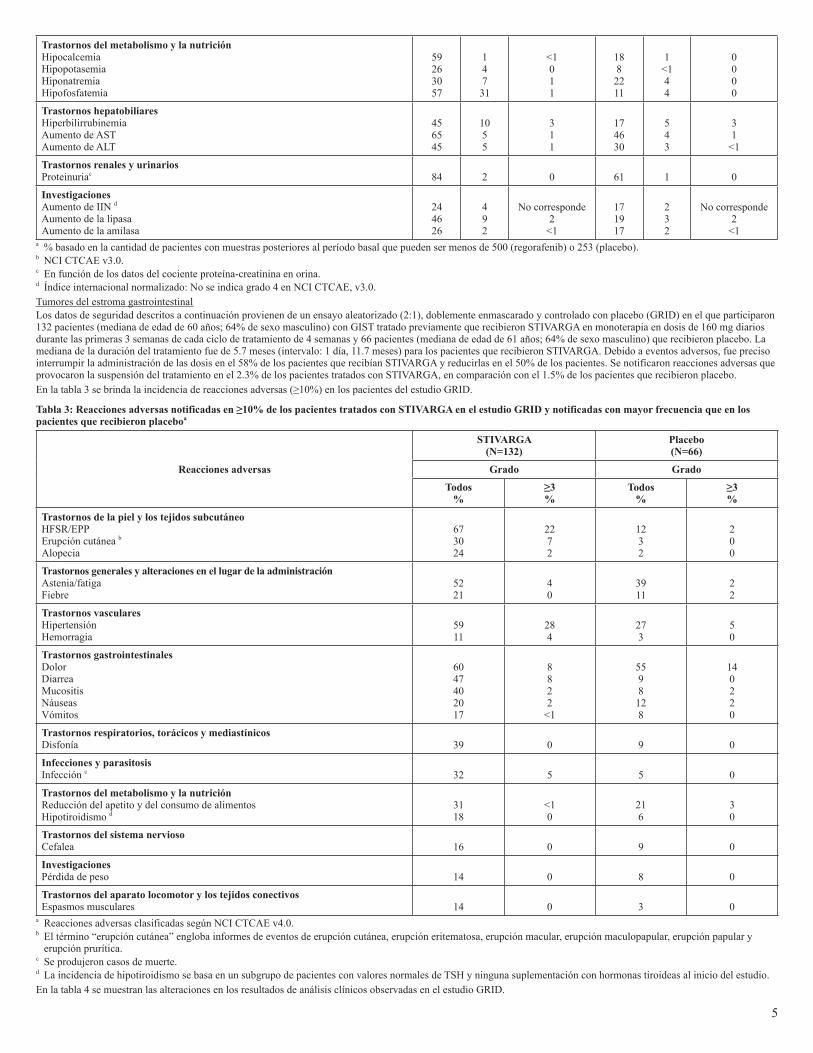
maculopapular, erupción papular y erupción prurítica.c Fatal outcomes observed.En la tabla 2 se muestran las alteraciones en los resultados de análisis clínicos observadas en el estudio CORRECT.
Tabla 2: Alteraciones en los resultados de análisis clínicos notificadas en el estudio CORRECT
Parámetro de análisis clínico
STIVARGA(N=500 a)
Placebo(N=253 a)
Grado b Grado b
Todos%
3%
4%
Todos%
3%
4%
Trastornos de la sangre y del sistema linfáticoAnemiaTrombocitopeniaNeutropeniaLinfopenia
7941354
5219
1<100
6617035
3<104
000
<1
5
Trastornos del metabolismo y la nutriciónHipocalcemiaHipopotasemiaHiponatremiaHipofosfatemia
59263057
14731
<1011
1882211
1<144
0000
Trastornos hepatobiliaresHiperbilirrubinemiaAumento de ASTAumento de ALT
456545
1055
311
174630
543
31
<1
Trastornos renales y urinariosProteinuriac 84 2 0 61 1 0
InvestigacionesAumento de IIN d
Aumento de la lipasaAumento de la amilasa
244626
492
No corresponde2
<1
171917
232
No corresponde2
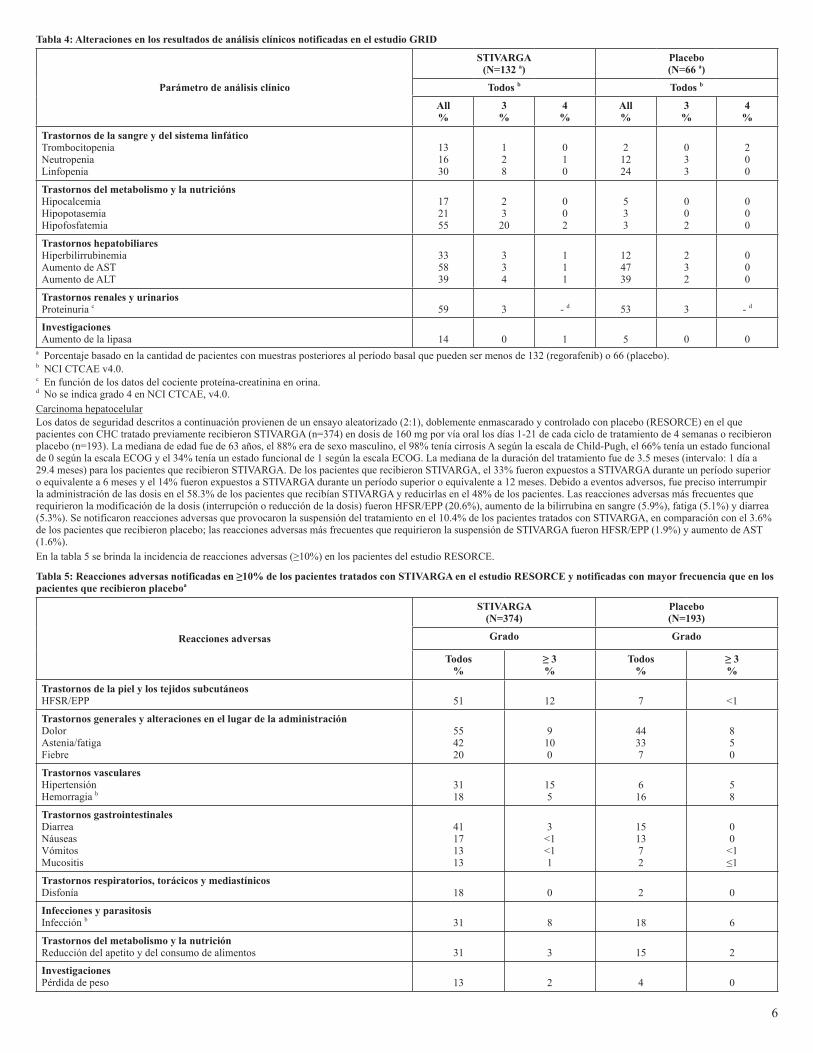
<1a % basado en la cantidad de pacientes con muestras posteriores al período basal que pueden ser menos de 500 (regorafenib) o 253 (placebo).b NCI CTCAE v3.0.c En función de los datos del cociente proteína-creatinina en orina.d Índice internacional normalizado: No se indica grado 4 en NCI CTCAE, v3.0.Tumores del estroma gastrointestinalLos datos de seguridad descritos a continuación provienen de un ensayo aleatorizado (2:1), doblemente enmascarado y controlado con placebo (GRID) en el que participaron 132 pacientes (mediana de edad de 60 años; 64% de sexo masculino) con GIST tratado previamente que recibieron STIVARGA en monoterapia en dosis de 160 mg diarios durante las primeras 3 semanas de cada ciclo de tratamiento de 4 semanas y 66 pacientes (mediana de edad de 61 años; 64% de sexo masculino) que recibieron placebo. La mediana de la duración del tratamiento fue de 5.7 meses (intervalo: 1 día, 11.7 meses) para los pacientes que recibieron STIVARGA. Debido a eventos adversos, fue preciso interrumpir la administración de las dosis en el 58% de los pacientes que recibían STIVARGA y reducirlas en el 50% de los pacientes. Se notificaron reacciones adversas que provocaron la suspensión del tratamiento en el 2.3% de los pacientes tratados con STIVARGA, en comparación con el 1.5% de los pacientes que recibieron placebo.En la tabla 3 se brinda la incidencia de reacciones adversas (≥10%) en los pacientes del estudio GRID.
Tabla3:Reaccionesadversasnotificadasen≥10%delospacientestratadosconSTIVARGAenelestudioGRIDynotificadasconmayorfrecuenciaqueenlospacientes que recibieron placeboa
Reacciones adversas
STIVARGA(N=132)
Placebo(N=66)
Grado Grado
Todos%
≥3%
Todos%
≥3%
Trastornos de la piel y los tejidos subcutáneoHFSR/EPPErupción cutánea b
Alopecia
673024
2272
1232
200
Trastornos generales y alteraciones en el lugar de la administraciónAstenia/fatigaFiebre
5221
40
3911
22
Trastornos vascularesHipertensiónHemorragia
5911
284
273
50
Trastornos gastrointestinalesDolorDiarreaMucositisNáuseasVómitos
6047402017
8822
<1
5598128
140220
Trastornos respiratorios, torácicos y mediastínicosDisfonía 39 0 9 0
Infecciones y parasitosisInfección c 32 5 5 0
Trastornos del metabolismo y la nutrición Reducción del apetito y del consumo de alimentosHipotiroidismo d
3118
<10
216
30
Trastornos del sistema nerviosoCefalea 16 0 9 0
InvestigacionesPérdida de peso 14 0 8 0
Trastornos del aparato locomotor y los tejidos conectivosEspasmos musculares 14 0 3 0
a Reacciones adversas clasificadas según NCI CTCAE v4.0.b El término “erupción cutánea” engloba informes de eventos de erupción cutánea, erupción eritematosa, erupción macular, erupción maculopapular, erupción papular y
erupción prurítica.c Se produjeron casos de muerte.d La incidencia de hipotiroidismo se basa en un subgrupo de pacientes con valores normales de TSH y ninguna suplementación con hormonas tiroideas al inicio del estudio.En la tabla 4 se muestran las alteraciones en los resultados de análisis clínicos observadas en el estudio GRID.
6
Tabla 4: Alteraciones en los resultados de análisis clínicos notificadas en el estudio GRID
Parámetro de análisis clínico
STIVARGA (N=132 a)
Placebo (N=66 a)
Todos b Todos b
All %
3 %
4 %
All %
3 %
4 %
Trastornos de la sangre y del sistema linfáticoTrombocitopeniaNeutropeniaLinfopenia
131630
128
010
21224
033
200
Trastornos del metabolismo y la nutriciónsHipocalcemiaHipopotasemiaHipofosfatemia
172155
2320
002
533
002
000
Trastornos hepatobiliaresHiperbilirrubinemiaAumento de ASTAumento de ALT
335839
334
111
124739
232
000
Trastornos renales y urinariosProteinuria c 59 3 - d 53 3 - d
InvestigacionesAumento de la lipasa 14 0 1 5 0 0
a Porcentaje basado en la cantidad de pacientes con muestras posteriores al período basal que pueden ser menos de 132 (regorafenib) o 66 (placebo).b NCI CTCAE v4.0.c En función de los datos del cociente proteína-creatinina en orina. d No se indica grado 4 en NCI CTCAE, v4.0.Carcinoma hepatocelularLos datos de seguridad descritos a continuación provienen de un ensayo aleatorizado (2:1), doblemente enmascarado y controlado con placebo (RESORCE) en el que pacientes con CHC tratado previamente recibieron STIVARGA (n=374) en dosis de 160 mg por vía oral los días 1-21 de cada ciclo de tratamiento de 4 semanas o recibieron placebo (n=193). La mediana de edad fue de 63 años, el 88% era de sexo masculino, el 98% tenía cirrosis A según la escala de Child-Pugh, el 66% tenía un estado funcional de 0 según la escala ECOG y el 34% tenía un estado funcional de 1 según la escala ECOG. La mediana de la duración del tratamiento fue de 3.5 meses (intervalo: 1 día a 29.4 meses) para los pacientes que recibieron STIVARGA. De los pacientes que recibieron STIVARGA, el 33% fueron expuestos a STIVARGA durante un período superior o equivalente a 6 meses y el 14% fueron expuestos a STIVARGA durante un período superior o equivalente a 12 meses. Debido a eventos adversos, fue preciso interrumpir la administración de las dosis en el 58.3% de los pacientes que recibían STIVARGA y reducirlas en el 48% de los pacientes. Las reacciones adversas más frecuentes que requirieron la modificación de la dosis (interrupción o reducción de la dosis) fueron HFSR/EPP (20.6%), aumento de la bilirrubina en sangre (5.9%), fatiga (5.1%) y diarrea (5.3%). Se notificaron reacciones adversas que provocaron la suspensión del tratamiento en el 10.4% de los pacientes tratados con STIVARGA, en comparación con el 3.6% de los pacientes que recibieron placebo; las reacciones adversas más frecuentes que requirieron la suspensión de STIVARGA fueron HFSR/EPP (1.9%) y aumento de AST (1.6%).En la tabla 5 se brinda la incidencia de reacciones adversas (≥10%) en los pacientes del estudio RESORCE.
Tabla5:Reaccionesadversasnotificadasen≥10%delospacientestratadosconSTIVARGAenelestudioRESORCEynotificadasconmayorfrecuenciaqueenlospacientes que recibieron placeboa
Reacciones adversas
STIVARGA(N=374)
Placebo(N=193)
Grado Grado
Todos%
≥3%
Todos%
≥3%
Trastornos de la piel y los tejidos subcutáneosHFSR/EPP 51 12 7 <1
Trastornos generales y alteraciones en el lugar de la administraciónDolorAstenia/fatigaFiebre
554220
9100
44337
850
Trastornos vascularesHipertensiónHemorragia b
3118
155
616
58
Trastornos gastrointestinalesDiarreaNáuseasVómitosMucositis
41171313
3<1<11
151372
00
<1≤1
Trastornos respiratorios, torácicos y mediastínicosDisfonía 18 0 2 0
Infecciones y parasitosisInfección b 31 8 18 6
Trastornos del metabolismo y la nutriciónReducción del apetito y del consumo de alimentos 31 3 15 2
InvestigacionesPérdida de peso 13 2 4 0

7
Trastornos del aparato locomotor y los tejidos conectivosEspasmos musculares 10 0 2 0
a Reacciones adversas clasificadas según NCI CTCAE v4.0.b Se produjeron casos de muerte.Otras reacciones adversas significativas desde el punto de vista clínico observadas en menos del 10% de los pacientes tratados con STIVARGA fueron: alopecia (7%), hipotiroidismo (6.4%), pancreatitis (1.6%), erupción exfoliativa (1.3%), temblores (1.3%), eritema multiforme (0.8%), isquemia de miocardio (0.8%), fístula gastrointestinal (0.3%) e infarto de miocardio (0.3%).En la tabla 6 se muestran las alteraciones en los resultados de análisis clínicos observadas en el estudio RESORCE.
Tabla 6: Alteraciones en los resultados de análisis clínicos notificadas en el estudio RESORCE
Laboratory Parameter
STIVARGA (N=374 a)
Placebo (N=193 a)
Grado b Grado b
Todos %
3 %
4 %
Todos %
3 %
4 %
Trastornos de la sangre y del sistema linfáticoTrombocitopeniaNeutropeniaLinfopenia
631468
5316
<102
501559
0<111
0<1<1
Trastornos del metabolismo y la nutriciónHipocalcemiaHipopotasemiaHipofosfatemia
2331 70
<1432
0<12
10931
027
000
Trastornos hepatobiliaresHiperbilirrubinemiaAumento de ASTAumento de ALT
789370
13166
32
<1
558459
11175
530
Trastornos renales y urinariosProteinuria c 51 17 - d 37 3 - d
InvestigacionesAumento de IINAumento de la lipasaAumento de la amilasa
444123
<1113
- d 3
<1
352719
282
- d 1
<1a Porcentaje basado en la cantidad de pacientes con muestras posteriores al período basal que pueden ser menos de
374 (regorafenib) o 193 (placebo).b NCI CTCAE v4.0.c Basado en datos de pruebas con tiras reactivas.d No se indica grado 4 en NCI CTCAE, v4.0.
6.2 FarmacovigilanciaDurante el período posterior a la aprobación de STIVARGA se identificó la siguiente reacción adversa. Debido a que estas reacciones son comunicadas de forma voluntaria por una población de tamaño incierto, no siempre es posible calcular de manera fiable su frecuencia ni establecer una relación de causalidad con la exposición al fármaco:
• reacción de hipersensibilidad• síndrome nefrótico• insuficiencia cardíaca
7 INTERACCIONES FARMACOLÓGICAS7.1 Efecto de los inductores potentes del CYP3A4 en el regorafenibLa coadministración de un inductor potente del CYP3A4 con STIVARGA disminuyó las concentraciones plasmáticas del regorafenib, aumentó las concentraciones plasmáticas del metabolito activo M-5 y no provocó ningún cambio en las concentraciones plasmáticas del metabolito activo M-2 [ver Farmacología clínica (12.3)] y puede causar una disminución de la eficacia. Evite el uso concomitante de STIVARGA con inductores potentes del CYP3A4 (p. ej. rifampicina, fenitoína, carbamazepina, fenobarbital e hipérico [hierba de san Juan]).
7.2 Efecto de los inhibidores potentes del CYP3A4 en el regorafenibLa coadministración de un inhibidor potente del CYP3A4 con STIVARGA aumentó las concentraciones plasmáticas del regorafenib y disminuyó las concentraciones plasmáticas de los metabolitos activos M-2 y M-5 [ver Farmacología clínica (12.3)] y puede causar un aumento de la toxicidad. Evite el uso concomitante de STIVARGA con inhibidores potentes del CYP3A4 (p. ej., claritromicina, jugo de pomelo [toronja], itraconazol, ketoconazol, nefazodona, posaconazol, telitromicina y voriconazol).
7.3 Efecto del regorafenib en los sustratos de la proteína de resistencia al cáncer de mama (PRCM)La coadministración de STIVARGA con un sustrato de la PRCM aumentó las concentraciones plasmáticas del sustrato de la PRCM [ver Farmacología clínica (12.3)]. Vigile atentamente a los pacientes para detectar signos y síntomas de toxicidad relacionada con la exposición al sustrato de la PRCM (p. ej., metotrexato, fluvastatina, atorvastatina). Consulte la información del producto del sustrato de la PRCM concomitante a la hora de considerar la administración de dichos productos junto con STIVARGA.
8 USO EN POBLACIONES ESPECÍFICAS8.1 EmbarazoResumen de los riesgosSobre la base de estudios en animales y su mecanismo de acción, STIVARGA puede causar daño fetal si se administra a una mujer embarazada. No se dispone de información sobre el uso de STIVARGA en mujeres embarazadas. La administración de regorafenib fue embrioletal y teratógena en ratas y conejos expuestos a concentraciones más bajas que las exposiciones a la dosis recomendada en seres humanos, y produjo un aumento en las incidencias de malformaciones cardiovasculares, genitourinarias y esqueléticas [ver Datos]. Advierta a las mujeres embarazadas del posible peligro para el feto. Se desconoce el riesgo espontáneo estimado de defectos congénitos importantes y aborto espontáneo para la población indicada. En la población estadounidense en general, el riesgo de fondo estimado de defectos congénitos importantes y aborto espontáneo en embarazos clínicamente validados es del 2 al 4% y del 15 al 20%, respectivamente.

8
DatosDatos de animalesEn estudios sobre el desarrollo embriofetal, se observó una pérdida total de la gestación (reabsorción del 100% de las crías) en ratas que recibieron dosis de tan solo 1 mg/kg (aproximadamente el 6% de la dosis recomendada en seres humanos basada en el área de superficie corporal) y en conejos que recibieron dosis de tan solo 1.6 mg/kg (aproximadamente el 25% de la exposición a la dosis clínica recomendada en seres humanos medida según el área bajo la curva [ABC]).En un estudio sobre distribución de dosis únicas realizado en ratas preñadas, hubo un aumento de la penetración del regorafenib a través de la barrera hematoencefálica en los fetos, en comparación con lo observado en las madres. En un estudio con administración diaria de regorafenib durante la organogénesis en ratas preñadas, los hallazgos incluyeron retraso de la osificación en los fetos en presencia de dosis ≥0.8 mg/kg (aproximadamente el 5% de la dosis recomendada en seres humanos sobre la base del área de superficie corporal) y aumentos dependientes de la dosis en las malformaciones esqueléticas, que incluyeron fisura palatina y agrandamiento de fontanelas, en presencia de dosis ≥1 mg/kg (aproximadamente el 10% de la exposición clínica sobre la base del ABC). Con dosis ≥1.6 mg/kg (aproximadamente el 11% de la dosis recomendada en seres humanos sobre la base del área de superficie corporal), se observaron aumentos dependientes de la dosis en la incidencia de malformaciones cardiovasculares, anomalías externas, hernia diafragmática y dilatación de la pelvis renal.En conejas preñadas que recibieron regorafenib diariamente durante la organogénesis, se observaron defectos del tabique interventricular, que se hicieron evidentes con la dosis de prueba más baja de 0.4 mg/kg (aproximadamente el 7% del ABC en pacientes que reciben la dosis recomendada). Con dosis ≥0.8 mg/kg (aproximadamente el 15% de la exposición a la dosis recomendada en seres humanos sobre la base del ABC), la administración de regorafenib provocó aumentos dependientes de la dosis en la incidencia de malformaciones cardiovasculares y anomalías esqueléticas adicionales, así como efectos adversos significativos en el aparato urinario, que incluyeron ausencia de riñón/uréter; riñón de tamaño reducido, deformado y mal posicionado e hidronefrosis. En dos estudios de toxicidad embriofetal en conejos, la proporción de fetos viables machos disminuyó al incrementar la dosis.
8.2 LactanciaResumen de riesgosNo hay datos sobre la presencia del regorafenib o sus metabolitos en la leche humana ni de los efectos del regorafenib en los bebés lactantes o en la producción de leche. En ratas, el regorafenib y sus metabolitos se excretan en la leche. Dada la posibilidad de que se produzcan reacciones adversas graves en los bebés lactantes a causa de STIVARGA, no se debe amamantar durante el tratamiento con STIVARGA y durante 2 semanas después de la dosis final.
8.3 Mujeres y hombres con capacidad reproductoraAnticoncepciónMujeresUse un método anticonceptivo eficaz durante el tratamiento y hasta 2 meses después de terminar el tratamiento.HombresA los pacientes masculinos que tengan parejas femeninas con capacidad reproductora, adviértales de que deben usar un método anticonceptivo eficaz durante el tratamiento y durante los 2 meses siguientes a la dosis final de STIVARGA [ver Toxicología preclínica (13.1)].InfertilidadNo hay datos sobre el efecto de STIVARGA en la fertilidad de los seres humanos. Los resultados obtenidos de los estudios en animales indican que el regorafenib puede deteriorar la fertilidad masculina y femenina [ver Toxicología preclínica (13.1)].
8.4 Uso pediátricoNo se han establecido la seguridad ni la eficacia de STIVARGA en pacientes pediátricos menores de 18 años.Datos de animalesEn estudios de 28 días con dosis repetidas en ratas, se observaron hallazgos dependientes de la dosis que consistieron en alteración de la dentina y angiectasia. Estos hallazgos se observaron con dosis de regorafenib de tan solo 4 mg/kg (aproximadamente el 25% del ABC en seres humanos con la dosis recomendada). En estudios de 13 semanas con dosis repetidas administradas a perros, se observaron hallazgos similares que consistieron en alteración de la dentina con dosis de tan solo 20 mg/kg (aproximadamente el 43% del ABC en seres humanos con la dosis recomendada). La administración de regorafenib en estos animales también ocasionó el crecimiento y engrosamiento persistentes del cartílago de crecimiento epifisario femoral.
8.5 Uso geriátricoDe los 1142 pacientes tratados con STIVARGA inscritos en los ensayos aleatorizados controlados con placebo, el 40% tenía 65 años y más, mientras que el 10% tenía 75 años y más. No se observaron diferencias generales en la eficacia entre estos pacientes y los de menor edad. Hubo un aumento de la incidencia de hipertensión de grado 3 (18% frente al 9%) en los ensayos controlados con placebo entre los pacientes tratados con STIVARGA de 65 años de edad y mayores en comparación con los pacientes de menor edad. Además, se notificó un evento de hipertensión de grado 4 en el grupo de 65 años y más y ninguno en el grupo de menor edad.
8.6 Disfunción hepáticaNo hay recomendaciones de ajuste de la dosis en pacientes con disfunción hepática leve (bilirrubina total ≤LSN y AST >LSN o bilirrubina total >LSN a ≤1.5 veces LSN) o moderada (bilirrubina total >1.5 a ≤3 veces LSN y cualquier valor de AST), [ver Farmacología clínica (12.3)]. Debe vigilarse atentamente a los pacientes con disfunción hepática para detectar reacciones adversas [ver Advertencias y precauciones (5.1)].No se recomienda el uso de STIVARGA en pacientes con disfunción hepática grave (bilirrubina total >3x LSN), dado que STIVARGA no se ha estudiado en esta población.
8.7 Disfunción renalNo hay recomendaciones de ajuste de la dosis en pacientes con disfunción renal. No se ha estudiado la farmacocinética del regorafenib en pacientes sometidos a diálisis, y no hay recomendaciones de dosis para esta población de pacientes [ver Farmacología clínica (12.3)].
8.8 RazaSobre la base de los datos combinados de tres ensayos controlados con placebo (CORRECT, GRID y CONCUR), se produjo una incidencia más elevada de HFSR y anomalías en los valores de las pruebas de función hepática en los pacientes asiáticos tratados con STIVARGA en comparación con los de raza blanca [ver Advertencias y precauciones (5.1, 5.5)]. No es necesario ajustar la dosis inicial en función de la raza.
10 SOBREDOSISLa dosis más alta de STIVARGA estudiada clínicamente es de 220 mg por día. Las reacciones adversas al fármaco observadas con más frecuencia con esta dosis fueron eventos dermatológicos, disfonía, diarrea, inflamación de la mucosa, boca seca, reducción del apetito, hipertensión y fatiga. No existe un antídoto conocido para la sobredosis de STIVARGA. En caso de sospecha de sobredosis, interrumpa el uso de STIVARGA, administre tratamiento de apoyo y mantenga bajo observación hasta la estabilización clínica.

9
11 DESCRIPCIÓNSTIVARGA (regorafenib) es un inhibidor de multicinasas con el nombre químico de monohidrato de 4-[4-({[4-cloro-3-(trifluorometil)fenil]carbamoíl}amino)-3-fluorofenoxi]-N-metilpiridina-2-carboxamida. El regorafenib tiene la siguiente fórmula estructural:
El regorafenib es un monohidrato que tiene la fórmula molecular C21H15ClF4N4O3 • H2O y un peso molecular de 500.83. El regorafenib es prácticamente insoluble en agua, levemente soluble en acetonitrilo, metanol, etanol y acetato de etilo, y moderadamente soluble en acetona.Los comprimidos de STIVARGA para administración oral están formulados como comprimidos de color rosa claro con forma ovalada y presentan grabado bajo relieve la inscripción “BAYER” sobre un lado y “40” sobre el otro. Cada comprimido contiene 40 mg de regorafenib en estado anhidro, que corresponden a 41.49 mg de monohidrato de regorafenib, y los siguientes ingredientes inactivos: celulosa microcristalina, croscarmelosa sódica, estearato de magnesio, povidona y dióxido de silicio coloidal. El recubrimiento pelicular contiene los siguientes ingredientes inactivos: óxido férrico rojo, óxido férrico amarillo, lecitina (soya), polietilenglicol 3350, alcohol polivinílico, talco y dióxido de titanio.
12 FARMACOLOGÍA CLÍNICA12.1 Mecanismo de acciónEl regorafenib es una molécula pequeña que actúa como inhibidor de múltiples cinasas intracelulares y unidas a la membrana que intervienen en las funciones celulares normales y en los procesos patológicos como la oncogénesis, la angiogénesis tumoral, la metástasis y la inmunidad antitumoral. En ensayos celulares o bioquímicos in vitro, el regorafenib o sus principales metabolitos activos en seres humanos (M-2 y M-5) inhibieron la actividad de RET, VEGFR1, VEGFR2, VEGFR3, KIT, PDGFR-alfa, PDGFR-beta, FGFR1, FGFR2, TIE2, DDR2, TrkA, Eph2A, RAF-1, BRAF, BRAF V600E, SAPK2, PTK5, Abl y CSF1R en concentraciones de regorafenib que se han alcanzado clínicamente. En modelos in vivo, el regorafenib demostró actividad antiangiogénica en un modelo tumoral de rata e inhibición del crecimiento tumoral en varios modelos de xenoinjertos de ratón, incluidos algunos correspondientes al carcinoma colorrectal humano, carcinoma del estroma gastrointestinal y hepatocelular. El regorafenib también exhibió actividad antimetastásica en un modelo de xenoinjerto de ratón y dos modelos ortotópicos de ratón de carcinoma colorrectal humano.
12.2 FarmacodinámicaElectrofisiología cardíacaSe evaluó el efecto de múltiples dosis de STIVARGA (160 mg una vez al día durante 21 días) en el intervalo QTc en un estudio abierto de un solo grupo con 25 pacientes que tenían tumores sólidos avanzados. No se detectó ninguna variación considerable en la media del intervalo QTc (es decir, >20 ms) en el estudio.
12.3 FarmacocinéticaAbsorciónLuego de una dosis única de 160 mg de STIVARGA en pacientes con tumores sólidos avanzados, el regorafenib alcanza una media geométrica del nivel plasmático máximo (Cmáx) de 2.5 µg/ml en una mediana de tiempo de 4 horas y una media geométrica del área bajo la curva (ABC) de concentración plasmática en función del tiempo de 70.4 µg*h/ml. A dosis mayores de 60 mg, el ABC del regorafenib en estado de equilibrio aumenta de manera menos que proporcional a las dosis. En estado de equilibrio, el regorafenib alcanza una media geométrica de la Cmáx de 3.9 µg/ml y una media geométrica del ABC de 58.3 µg*h/ml. El coeficiente de variación del ABC y la Cmáx está entre el 35% y el 44%.La biodisponibilidad relativa media de los comprimidos en comparación con una solución oral está entre el 69% y el 83%.En un estudio sobre el efecto de los alimentos, 24 hombres sanos recibieron una dosis única de 160 mg de STIVARGA en tres momentos distintos: en condiciones de ayuno, con una comida de alto contenido graso y con una comida de bajo contenido graso. La comida de alto contenido graso (945 calorías y 54.6 g de grasa) aumentó la media del ABC del regorafenib en un 48% y disminuyó la media del ABC de los metabolitos M-2 y M-5 en un 20% y un 51%, respectivamente, en comparación con la administración en condiciones de ayuno. La comida de bajo contenido graso (319 calorías y 8.2 g de grasa) aumentó la media del ABC del regorafenib, el M-2 y el M-5 en un 36%, 40% y 23%, respectivamente, en comparación con la administración en condiciones de ayuno. En los estudios CORRECT y GRID, STIVARGA se administró con una comida de bajo contenido graso [ver Posología y administración (2.1), Estudios clínicos (14)].DistribuciónEl regorafenib entra en la circulación enterohepática y presenta múltiples picos de concentración plasmática en el transcurso de un intervalo de administración de 24 horas. El regorafenib tiene una alta afinidad de unión (99.5%) a proteínas plasmáticas humanas.EliminaciónLuego de una dosis oral única de 160 mg de STIVARGA, la media geométrica (mínimo a máximo) de las semividas de eliminación para el regorafenib y el metabolito M-2 en el plasma es de 28 horas (14 a 58 horas) y de 25 horas (14 a 32 horas), respectivamente. El M-5 tiene una semivida de eliminación media (mínimo a máximo) más prolongada de 51 horas (32 a 70 horas).MetabolismoEl regorafenib es metabolizado por el CYP3A4 y la UGT1A9. Los principales metabolitos del regorafenib en circulación medidos en estado de equilibrio en el plasma humano son el M-2 (N-óxido) y el M-5 (N-óxido y N-desmetilo). Ambos metabolitos presentan actividad farmacológica y concentraciones en estado de equilibrio in vitro similares a las del regorafenib. El M-2 y el M-5 tienen una alta afinidad de unión a proteínas (99.8% y 99.95%, respectivamente).ExcreciónAproximadamente el 71% de una dosis radiomarcada se excretó en las heces (el 47% como compuesto original y el 24% como metabolitos) y el 19% de la dosis se excretó en la orina (el 17% como glucurónidos) en los 12 días siguientes a la administración de una solución oral radiomarcada con una dosis de 120 mg.Poblaciones específicasLa edad, el sexo, la raza y el peso no tuvieron un efecto clínicamente significativo en la farmacocinética del regorafenib.Disfunción hepáticaSobre la base de un análisis de población farmacocinética, no se observaron diferencias clínicamente significativas en la exposición media total del regorafenib, incluidos los metabolitos M-2 y M-5, entre los pacientes con función hepática normal (bilirrubina total y AST ≤ LSN, n=744), disfunción hepática leve (bilirrubina total ≤LSN y AST >LSN o bilirrubina total >LSN a ≤1.5x LSN, n=437) y disfunción hepática moderada (bilirrubina total >1.5x a ≤3x LSN y cualquier valor de AST, n=36). El análisis combinado incluyó 391 pacientes con CHC, de los cuales 116, 249 y 26 fueron categorizados por tener función hepática normal y disfunción hepática leve y moderada, respectivamente. No se evaluó la farmacocinética del regorafenib en pacientes con disfunción hepática grave (bilirrubina total >3x LSN).Disfunción renalSe evaluó la farmacocinética del regorafenib, M-2 y M-5 en 6 pacientes afectados por disfunción renal grave (CLcr 15-29 ml/min) y 18 pacientes con función renal normal/leve (CLcr ≥60 ml/min) luego de la administración de 160 mg diarios de STIVARGA durante 21 días. No se observaron diferencias en la exposición media en estado de equilibrio del regorafenib, el M-2 o el M-5 en los pacientes con disfunción renal grave, en comparación con los pacientes con función renal normal. No se ha estudiado la farmacocinética del regorafenib en pacientes con enfermedad renal terminal que se someten a diálisis.

10
Estudios de interacción farmacológicaEfecto del regorafenib en los sustratos del citocromo P450: Los estudios in vitro sugirieron que el regorafenib inhibe la acción del CYP2C8, CYP2C9, CYP2B6, CYP3A4 y CYP2C19; el M-2 inhibe la acción del CYP2C9, CYP2C8, CYP3A4 y CYP2D6; y el M-5 inhibe la acción del CYP2C8. Los estudios in vitro sugirieron que el regorafenib no induce la actividad enzimática del CYP1A2, CYP2B6, CYP2C19 y CYP3A4.Se administró a pacientes con tumores sólidos avanzados una dosis oral única de sustratos del CYP: 2 mg de midazolam (CYP3A4), 40 mg de omeprazol (CYP2C19) y 10 mg de warfarina (CYP2C9) o 4 mg de rosiglitazona (CYP2C8) una semana antes y dos semanas después de STIVARGA en una dosis de 160 mg una vez al día. No se observó ningún efecto clínicamente significativo en la media del ABC de la rosiglitazona (N=12) ni en la media de las concentraciones plasmáticas de omeprazol (N=11) medidas 6 horas después de la administración o en el ABC medio del midazolam (N=15). El ABC medio de la warfarina (N=8) aumentó en un 25% [ver Advertencias y precauciones (5.2)].Efecto de los inductores potentes del CYP3A4 en el regorafenib: Veintidós hombres sanos recibieron una dosis única de 160 mg de STIVARGA en monoterapia y, posteriormente, 7 días después de iniciar la administración de rifampicina. La rifampicina, un inductor potente del CYP3A4, se administró en dosis de 600 mg diarios durante 9 días. La media del ABC del regorafenib disminuyó en un 50% y la media del ABC del M-5 aumentó en un 264%. No se observó ningún cambio en la media del ABC del M-2 [ver Interacciones farmacológicas (7.1)].Efecto de los inhibidores potentes del CYP3A4 en el regorafenib: Dieciocho hombres sanos recibieron una dosis única de 160 mg de STIVARGA en monoterapia y, posteriormente, 5 días después de iniciar la administración de ketoconazol. El ketoconazol, un potente inhibidor del CYP3A4, se administró en dosis de 400 mg diarios durante 18 días. La media del ABC del regorafenib aumentó en un 33%, y la media del ABC tanto del M-2 como del M-5 disminuyó en un 93% [ver Interacciones farmacológicas (7.2)].Efecto de la neomicina en el regorafenib: Veintisiete hombres sanos recibieron una dosis única de 160 mg de STIVARGA y, posteriormente, 5 días después de iniciar la administración de neomicina. La neomicina, un antibiótico no absorbible, se administró en dosis de 1 gramo tres veces al día durante 5 días. No se observó ningún efecto clínicamente significativo en la media del ABC del regorafenib; sin embargo, la media del ABC del M-2 disminuyó un 76% y la media del ABC del M-5 disminuyó un 86%. La disminución de la exposición del M-2 y el M-5 puede traducirse en una disminución de la eficacia de STIVARGA. No se han estudiado los efectos de otros antibióticos en la exposición del regorafenib y sus metabolitos activos.Efecto del regorafenib en los sustratos de la UGT1A1: En estudios in vitro se demostró que el regorafenib, el M-2 y el M-5 inhiben de manera competitiva la acción de la UGT1A9 y la UGT1A1 en concentraciones terapéuticamente adecuadas. Once pacientes recibieron quimioterapia combinada que contenía irinotecán junto con STIVARGA en dosis de 160 mg. Cuando se administró el irinotecán 5 días después de la última de las 7 dosis diarias de STIVARGA, la media del ABC del irinotecán aumentó en un 28% y la media del ABC del SN-38 aumentó en un 44%.Efecto del regorafenib en los sustratos de la PRCM: La administración de regorafenib (160 mg durante 14 días) antes de la administración de una dosis única de rosuvastatina (5 mg), un sustrato de la PRCM, tuvo como resultado un aumento de 3.8 veces la exposición media (ABC) de rosuvastatina y un aumento de 4.6 veces la Cmáx [ver Interacciones farmacológicas (7.3)].
13 TOXICOLOGÍA PRECLÍNICA13.1 Carcinogénesis, mutagénesis, alteraciones de la fertilidadNo se han llevado a cabo estudios que examinen el potencial carcinógeno del regorafenib. El regorafenib en sí no demostró genotoxicidad en los ensayos in vitro o in vivo; sin embargo, un importante metabolito activo del regorafenib en seres humanos (el M-2) produjo resultados positivos de clastogenicidad, y provocó anomalías cromosómicas en células V79 de hámster chino.No se han llevado a cabo estudios destinados específicamente a examinar los efectos del regorafenib en la fertilidad; sin embargo, se observaron hallazgos histológicos de atrofia tubular y degeneración en los testículos, atrofia en la vesícula seminal y residuos celulares y oligospermia en el epidídimo de ratas machos que recibieron dosis similares a la dosis clínica recomendada en humanos en función del ABC. En ratas hembras, hubo un aumento de los hallazgos de cuerpo lúteo necrótico en los ovarios ante las mismas exposiciones. Se observaron hallazgos similares en perros de ambos sexos en los estudios con dosis repetidas en exposiciones de aproximadamente el 83% de la exposición a la dosis recomendada en seres humanos en función del ABC. Estos hallazgos sugieren que es posible que el regorafenib provoque efectos adversos en la fertilidad de los seres humanos.
13.2 Toxicología y/o farmacología en animalesEn un estudio de 26 semanas con dosis repetidas crónicas en ratas, se observó un aumento dependiente de la dosis en el hallazgo de engrosamiento de la válvula auriculoventricular. Con una dosis que generó una exposición de aproximadamente el 12% de la exposición a la dosis recomendada en seres humanos, este hallazgo estuvo presente en la mitad de los animales examinados.
14 ESTUDIOS CLÍNICOS14.1 Cáncer colorrectalLa eficacia y la seguridad clínicas de STIVARGA se evaluaron en un ensayo internacional, multicéntrico, aleatorizado (2:1), doblemente enmascarado y controlado con placebo [estudio “Patients with metastatic COloRectal cancer treated with REgorafenib or plaCebo after failure of standard Therapy” (CORRECT); NCT 01103323)] en el que participaron 760 pacientes con cáncer colorrectal metastásico que habían recibido tratamiento previo. El principal criterio de evaluación de la eficacia fue la supervivencia general (SG); otros criterios de evaluación de la eficacia fueron la supervivencia sin progresión (SSP) y la tasa de respuesta tumoral general.Se asignó aleatoriamente a los pacientes para recibir 160 mg de regorafenib por vía oral una vez al día (N=505) más la mejor atención médica de apoyo (best supportive care, BSC), o recibir placebo (N=255) más BSC durante los primeros 21 días de cada ciclo de 28 días. STIVARGA se administró con un desayuno de bajo contenido graso que contenía menos de un 30% de grasa [ver Posología y administración (2.1), Farmacología clínica (12.3)]. El tratamiento se continuó hasta la aparición de progresión de la enfermedad o toxicidad inadmisible.Las características demográficas basales eran: mediana de la edad de 61 años, 61% de sexo masculino, 78% de raza blanca y todos los pacientes tenían un estado funcional de 0 o 1 según la escala ECOG. Los principales lugares afectados por enfermedad eran el colon (65%), el recto (29%) o ambos (6%). Se informaron los antecedentes de la evaluación de KRAS correspondientes a 729 (96%) pacientes; se informó que 430 (59%) de estos pacientes tenían una mutación KRAS. La mediana de la cantidad de líneas terapéuticas anteriores para enfermedad metastásica era de 3. Todos los pacientes recibieron tratamiento previo con quimioterapia basada en fluoropirimidina, oxaliplatino e irinotecán, y con bevacizumab. Salvo uno, todos los pacientes con tumores negativos para la mutación KRAS recibieron panitumumab o cetuximab.La adición de STIVARGA a la BSC provocó una mejora estadísticamente significativa en la supervivencia, en comparación con el placebo más BSC (ver tabla 7 y figura 1).
Tabla 7: Resultados de eficacia del estudio CORRECT
STIVARGA(N=505)
Placebo(N=255)
Supervivencia general
Cantidad de muertes (%) 275 (55%) 157 (62%)
Mediana de la supervivencia general (meses) 6.4 5.0
IC del 95% a (5.8, 7.3) (4.4, 5.8)
Cociente de riesgos instantáneos (HR) (IC del 95%) 0.77 (0.64, 0.94)
Valor p de la prueba de rangos logarítmicos estratificada b, c 0.0102
Supervivencia sin progresión
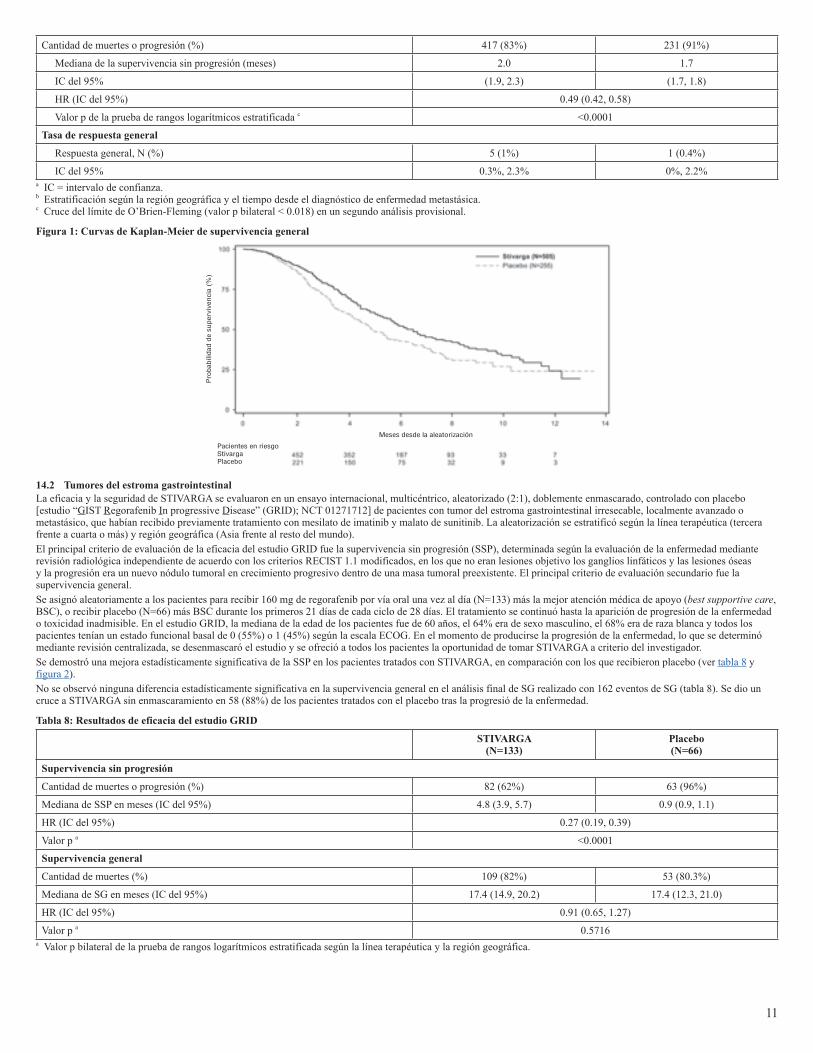
11
Cantidad de muertes o progresión (%) 417 (83%) 231 (91%)
Mediana de la supervivencia sin progresión (meses) 2.0 1.7
IC del 95% (1.9, 2.3) (1.7, 1.8)
HR (IC del 95%) 0.49 (0.42, 0.58)
Valor p de la prueba de rangos logarítmicos estratificada c <0.0001
Tasa de respuesta general
Respuesta general, N (%) 5 (1%) 1 (0.4%)
IC del 95% 0.3%, 2.3% 0%, 2.2%a IC = intervalo de confianza.b Estratificación según la región geográfica y el tiempo desde el diagnóstico de enfermedad metastásica.c Cruce del límite de O’Brien-Fleming (valor p bilateral < 0.018) en un segundo análisis provisional.
Figura 1: Curvas de Kaplan-Meier de supervivencia general
Pro
bab
ilidad
de
super
vive
nci
a (%
)
Meses desde la aleatorización
Pacientes en riesgoStivargaPlacebo
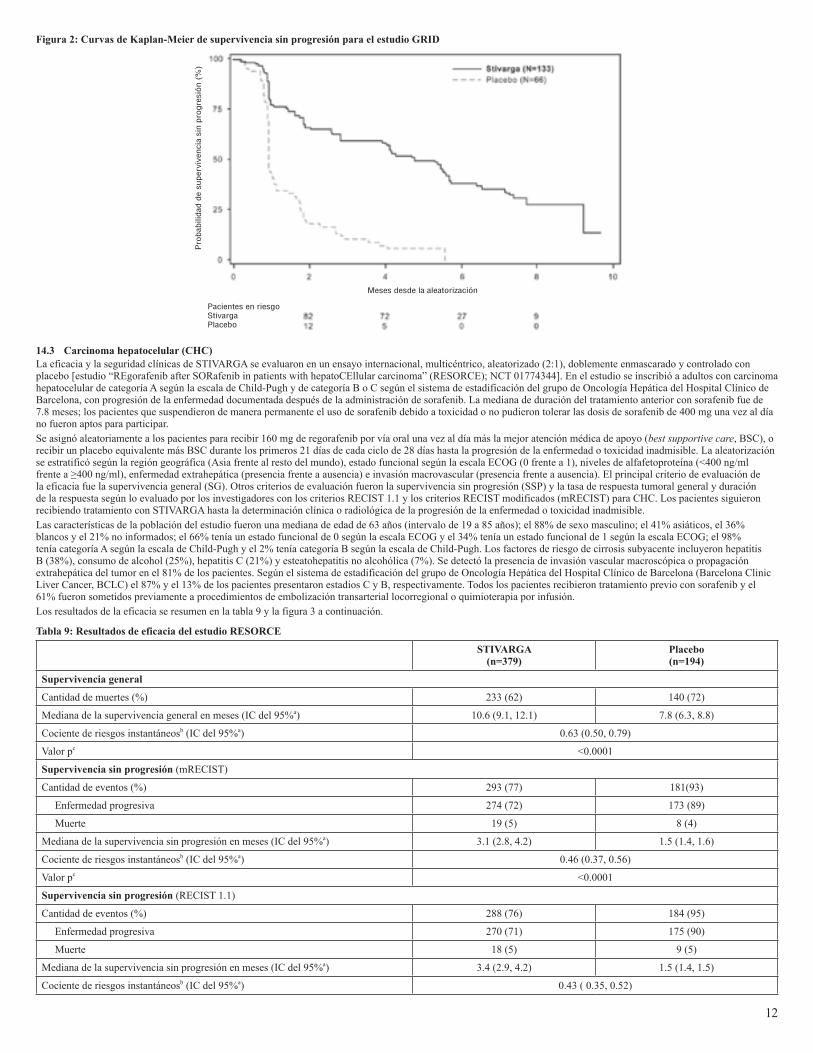
14.2 Tumores del estroma gastrointestinalLa eficacia y la seguridad de STIVARGA se evaluaron en un ensayo internacional, multicéntrico, aleatorizado (2:1), doblemente enmascarado, controlado con placebo [estudio “GIST Regorafenib In progressive Disease” (GRID); NCT 01271712] de pacientes con tumor del estroma gastrointestinal irresecable, localmente avanzado o metastásico, que habían recibido previamente tratamiento con mesilato de imatinib y malato de sunitinib. La aleatorización se estratificó según la línea terapéutica (tercera frente a cuarta o más) y región geográfica (Asia frente al resto del mundo).El principal criterio de evaluación de la eficacia del estudio GRID fue la supervivencia sin progresión (SSP), determinada según la evaluación de la enfermedad mediante revisión radiológica independiente de acuerdo con los criterios RECIST 1.1 modificados, en los que no eran lesiones objetivo los ganglios linfáticos y las lesiones óseas y la progresión era un nuevo nódulo tumoral en crecimiento progresivo dentro de una masa tumoral preexistente. El principal criterio de evaluación secundario fue la supervivencia general.Se asignó aleatoriamente a los pacientes para recibir 160 mg de regorafenib por vía oral una vez al día (N=133) más la mejor atención médica de apoyo (best supportive care, BSC), o recibir placebo (N=66) más BSC durante los primeros 21 días de cada ciclo de 28 días. El tratamiento se continuó hasta la aparición de progresión de la enfermedad o toxicidad inadmisible. En el estudio GRID, la mediana de la edad de los pacientes fue de 60 años, el 64% era de sexo masculino, el 68% era de raza blanca y todos los pacientes tenían un estado funcional basal de 0 (55%) o 1 (45%) según la escala ECOG. En el momento de producirse la progresión de la enfermedad, lo que se determinó mediante revisión centralizada, se desenmascaró el estudio y se ofreció a todos los pacientes la oportunidad de tomar STIVARGA a criterio del investigador. Se demostró una mejora estadísticamente significativa de la SSP en los pacientes tratados con STIVARGA, en comparación con los que recibieron placebo (ver tabla 8 y figura 2).No se observó ninguna diferencia estadísticamente significativa en la supervivencia general en el análisis final de SG realizado con 162 eventos de SG (tabla 8). Se dio un cruce a STIVARGA sin enmascaramiento en 58 (88%) de los pacientes tratados con el placebo tras la progresió de la enfermedad.
Tabla 8: Resultados de eficacia del estudio GRID
STIVARGA(N=133)
Placebo(N=66)
Supervivencia sin progresión
Cantidad de muertes o progresión (%) 82 (62%) 63 (96%)
Mediana de SSP en meses (IC del 95%) 4.8 (3.9, 5.7) 0.9 (0.9, 1.1)
HR (IC del 95%) 0.27 (0.19, 0.39)
Valor p a <0.0001
Supervivencia general
Cantidad de muertes (%) 109 (82%) 53 (80.3%)
Mediana de SG en meses (IC del 95%) 17.4 (14.9, 20.2) 17.4 (12.3, 21.0)
HR (IC del 95%) 0.91 (0.65, 1.27)
Valor p a 0.5716a Valor p bilateral de la prueba de rangos logarítmicos estratificada según la línea terapéutica y la región geográfica.
12
Figura 2: Curvas de Kaplan-Meier de supervivencia sin progresión para el estudio GRID
Pro
bab
ilid
ad d
e su
per
vive
nci
a si
n p
rog
resi
ón
(%
)
Meses desde la aleatorización
Pacientes en riesgoStivargaPlacebo
14.3 Carcinoma hepatocelular (CHC)La eficacia y la seguridad clínicas de STIVARGA se evaluaron en un ensayo internacional, multicéntrico, aleatorizado (2:1), doblemente enmascarado y controlado con placebo [estudio “REgorafenib after SORafenib in patients with hepatoCEllular carcinoma” (RESORCE); NCT 01774344]. En el estudio se inscribió a adultos con carcinoma hepatocelular de categoría A según la escala de Child-Pugh y de categoría B o C según el sistema de estadificación del grupo de Oncología Hepática del Hospital Clínico de Barcelona, con progresión de la enfermedad documentada después de la administración de sorafenib. La mediana de duración del tratamiento anterior con sorafenib fue de 7.8 meses; los pacientes que suspendieron de manera permanente el uso de sorafenib debido a toxicidad o no pudieron tolerar las dosis de sorafenib de 400 mg una vez al día no fueron aptos para participar.Se asignó aleatoriamente a los pacientes para recibir 160 mg de regorafenib por vía oral una vez al día más la mejor atención médica de apoyo (best supportive care, BSC), o recibir un placebo equivalente más BSC durante los primeros 21 días de cada ciclo de 28 días hasta la progresión de la enfermedad o toxicidad inadmisible. La aleatorización se estratificó según la región geográfica (Asia frente al resto del mundo), estado funcional según la escala ECOG (0 frente a 1), niveles de alfafetoproteína (<400 ng/ml frente a ≥400 ng/ml), enfermedad extrahepática (presencia frente a ausencia) e invasión macrovascular (presencia frente a ausencia). El principal criterio de evaluación de la eficacia fue la supervivencia general (SG). Otros criterios de evaluación fueron la supervivencia sin progresión (SSP) y la tasa de respuesta tumoral general y duración de la respuesta según lo evaluado por los investigadores con los criterios RECIST 1.1 y los criterios RECIST modificados (mRECIST) para CHC. Los pacientes siguieron recibiendo tratamiento con STIVARGA hasta la determinación clínica o radiológica de la progresión de la enfermedad o toxicidad inadmisible.Las características de la población del estudio fueron una mediana de edad de 63 años (intervalo de 19 a 85 años); el 88% de sexo masculino; el 41% asiáticos, el 36% blancos y el 21% no informados; el 66% tenía un estado funcional de 0 según la escala ECOG y el 34% tenía un estado funcional de 1 según la escala ECOG; el 98% tenía categoría A según la escala de Child-Pugh y el 2% tenía categoría B según la escala de Child-Pugh. Los factores de riesgo de cirrosis subyacente incluyeron hepatitis B (38%), consumo de alcohol (25%), hepatitis C (21%) y esteatohepatitis no alcohólica (7%). Se detectó la presencia de invasión vascular macroscópica o propagación extrahepática del tumor en el 81% de los pacientes. Según el sistema de estadificación del grupo de Oncología Hepática del Hospital Clínico de Barcelona (Barcelona Clinic Liver Cancer, BCLC) el 87% y el 13% de los pacientes presentaron estadios C y B, respectivamente. Todos los pacientes recibieron tratamiento previo con sorafenib y el 61% fueron sometidos previamente a procedimientos de embolización transarterial locorregional o quimioterapia por infusión.Los resultados de la eficacia se resumen en la tabla 9 y la figura 3 a continuación.
Tabla 9: Resultados de eficacia del estudio RESORCE
STIVARGA(n=379)
Placebo(n=194)
Supervivencia general
Cantidad de muertes (%) 233 (62) 140 (72)
Mediana de la supervivencia general en meses (IC del 95%a) 10.6 (9.1, 12.1) 7.8 (6.3, 8.8)
Cociente de riesgos instantáneosb (IC del 95%a) 0.63 (0.50, 0.79)
Valor pc <0.0001
Supervivencia sin progresión (mRECIST)
Cantidad de eventos (%) 293 (77) 181(93)
Enfermedad progresiva 274 (72) 173 (89)
Muerte 19 (5) 8 (4)
Mediana de la supervivencia sin progresión en meses (IC del 95%a) 3.1 (2.8, 4.2) 1.5 (1.4, 1.6)
Cociente de riesgos instantáneosb (IC del 95%a) 0.46 (0.37, 0.56)
Valor pc <0.0001
Supervivencia sin progresión (RECIST 1.1)
Cantidad de eventos (%) 288 (76) 184 (95)
Enfermedad progresiva 270 (71) 175 (90)
Muerte 18 (5) 9 (5)
Mediana de la supervivencia sin progresión en meses (IC del 95%a) 3.4 (2.9, 4.2) 1.5 (1.4, 1.5)
Cociente de riesgos instantáneosb (IC del 95%a) 0.43 ( 0.35, 0.52)

13
Respuesta general (mRECIST)
Tasa de respuesta general 11% 4%
IC del 95%a (8%, 14%) (2%, 8%)
Respuesta completa 0.5% 0
Respuesta parcial 10% 4%
Respuesta general (RECIST 1.1)
Tasa de respuesta general 7% 3%
IC del 95%a (4%, 10%) (1%, 6%)
Respuesta completa 0 0
Respuesta parcial 7% 3%a IC = intervalo de confianza.b Calculado con el modelo de riesgo proporcional de Cox estratificado por región geográfica, estado funcional según la escala ECOG, nivel de alfafetoproteína, presencia
frente a ausencia de enfermedad extrahepática y presencia frente a ausencia de invasión macrovascular.c Prueba de rangos logarítmicos estratificada por región geográfica, estado funcional según la escala ECOG, nivel de alfafetoproteína, presencia frente a ausencia de
enfermedad extrahepática y presencia frente a ausencia de invasión macrovascular.
Figura 3: Curva de Kaplan-Meier de la supervivencia general del estudio RESORCE
Pro
babil
idad d
e s
uperv
ivenci
a (
%)
Meses desde la aleatorizaciónPacientes en riesgo
STIVARGA
Placebo
16 PRESENTACIÓN/CONSERVACIÓN Y MANIPULACIÓNPresentaciónLos comprimidos se presentan en envases que contienen tres frascos de 28 comprimidos cada uno, lo que suma un total de 84 comprimidos por envase (NDC 50419-171-03).Conservación y manipulaciónConserve STIVARGA a 25 °C (77 °F); se permiten oscilaciones de 15 a 30 °C (59 a 86 °F) [ver Temperatura ambiente controlada, USP]. Conserve los comprimidos en el frasco original y no retire el desecante. Mantenga el frasco bien cerrado después de haberlo abierto por primera vez. Deseche los comprimidos no utilizados 7 semanas después de abrir el frasco. Elimine los comprimidos no utilizados de acuerdo con los requisitos locales.
17 INFORMACIÓN DE ASESORAMIENTO A LOS PACIENTESAdvierta al paciente de que debe leer el prospecto para pacientes aprobado por la FDA (Información para el paciente).HepatotoxicidadAdvierta a los pacientes de que deberán someterse a controles para detectar daño hepático, y que deberán notificar de inmediato cualquier signo o síntoma de daño hepático grave a su profesional de la salud [ver Advertencias y precauciones (5.1), Uso en poblaciones específicas (8.6)].InfeccionesAdvierta a los pacientes de que deben comunicarse con su proveedor de atención médica si presentan signos y síntomas de infección [ver Advertencias y precauciones (5.2)].HemorragiaAdvierta a los pacientes de que deben comunicarse con su proveedor de atención médica si presentan inusual sangrado, formación de moretones o síntomas de sangrado, como aturdimiento [ver Advertencias y precauciones (5.3)].Perforación o fístula gastrointestinalesAdvierta a los pacientes de que deben comunicarse inmediatamente con un proveedor de atención médica si presentan dolores intensos en el abdomen, hinchazón persistente del abdomen, fiebre alta, escalofríos, náuseas, vómitos o deshidratación [ver Advertencias y precauciones (5.4)].Toxicidad dermatológicaAdvierta a los pacientes de que deben comunicarse con su proveedor de atención médica si presentan cambios en la piel, como HFSR, erupción cutánea, dolor, ampollas, sangrado o hinchazón [ver Advertencias y precauciones (5.5)].HipertensiónAdvierta a los pacientes de que deberán someterse a controles de la presión arterial y que deberán comunicarse con su proveedor de atención médica si presentan presión arterial elevada o síntomas de hipertensión, incluidos dolor de cabeza intenso, aturdimiento o síntomas neurológicos [ver Advertencias y precauciones (5.6)].Isquemia e infarto cardíacosAdvierta a los pacientes de que deben buscar de inmediato asistencia de emergencia si presentan dolor de pecho, falta de aliento o mareos, o si sienten que se van a desmayar [ver Advertencias y precauciones (5.7)].

14
Síndrome de leucoencefalopatía posterior reversibleAdvierta a los pacientes de que deben comunicarse con su proveedor de atención médica si presentan signos y síntomas de SLPR [ver Advertencias y precauciones (5.8)].Riesgo de cicatrización deficiente de las heridasAdvierta a los pacientes que STIVARGA puede afectar negativamente la cicatrización de las heridas y que se recomienda interrumpir su administración temporalmente antes de cualquier intervención quirúrgica programada [ver Advertencias y precauciones (5.9)].Toxicidad embriofetalAdvierta a los pacientes de que el regorafenib puede causar daño fetal. Advierta a las mujeres embarazadas del posible riesgo para el feto [ver Advertencias y precauciones (5.10), Uso en poblaciones específicas (8.1, 8.3)].Mujeres y hombres con capacidad reproductora• Advierta a las mujeres con capacidad reproductora de que deben usar un método anticonceptivo eficaz durante el tratamiento con STIVARGA y durante los 2 meses
siguientes a haberlo finalizado. Advierta a las mujeres con capacidad reproductora de que deben comunicarse inmediatamente con su proveedor de atención médica en caso de sospecha o confirmación de un embarazo durante el tratamiento con STIVARGA o durante los 2 meses siguientes de haberlo finalizado [ver Advertencias y precauciones (5.10) y Uso en poblaciones específicas (8.1, 8.3)].
• Advierta a los hombres con capacidad reproductora de que deben usar un método anticonceptivo eficaz durante el tratamiento con STIVARGA y durante los 2 meses siguientes a haberlo finalizado [ver Uso en poblaciones específicas (8.3)].
LactanciaAdvierta a las madres lactantes de que no se sabe si el regorafenib está presente en la leche materna, y analice si es conveniente suspender la lactancia o suspender el uso de regorafenib [ver Uso en poblaciones específicas (8.2)].Administración• Advierta a los pacientes de que deben tragar el comprimido de STIVARGA entero con agua a la misma hora todos los días luego de consumir una comida de bajo
contenido graso. Informe a los pacientes de que la comida de bajo contenido graso debe contener menos de 600 calorías y menos del 30% de grasa [ver Posología y administración (2.1)].
• Advierta a los pacientes de que deben conservar el medicamento en el envase original. El medicamento no debe colocarse en pastilleros diarios o semanales. Deseche los comprimidos sobrantes 7 semanas después de abrir el frasco. El frasco debe quedar bien cerrado después de cada apertura y el desecante debe mantenerse dentro del frasco [ver Presentación (16)].
Instrucciones para la administraciónAdvierta a los pacientes de que deben tomar STIVARGA después de consumir una comida de bajo contenido graso. Advierta a los pacientes de que deben tomar cualquier dosis omitida ese mismo día, en cuanto lo recuerden, y que no deben tomar dos dosis el mismo día para compensar una dosis omitida el día anterior [ver Posología y administración (2.1)].
Fabricado para:Bayer HealthCare Pharmaceuticals Inc.Whippany, NJ 07981 EE. UU.
Información para el paciente:STIVARGA
(regorafenib)comprimidos
¿Cuál es la información más importante que debo conocer acerca de STIVARGA?STIVARGA puede causar efectos secundarios graves, tales como:Problemas del hígado. STIVARGA puede causar problemas al hígado que pueden ser graves y, en ocasiones, provocar la muerte. Su proveedor de atención médica le hará análisis de sangre para evaluar la función hepática antes de que usted comience a tomar STIVARGA y durante su tratamiento con STIVARGA a fin de detectar problemas del hígado. Informe de inmediato a su proveedor de atención médica si durante el tratamiento presenta alguno de estos síntomas de problemas del hígado:• amarilleo de la piel o del blanco de los ojos (ictericia)• náuseas o vómitos• orina oscura “del color del t锕 cambios en el patrón de sueño
¿Qué es STIVARGA?STIVARGA es un medicamento de venta con receta utilizado para tratar a personas que tienen:• cáncer de colon o de recto que se ha diseminado a otras partes del cuerpo y para el cual han recibido tratamiento previo con determinados medicamentos de
quimioterapia• un cáncer poco frecuente del estómago, los intestinos o el esófago llamado GIST (siglas en inglés de “tumores del estroma gastrointestinal”) no tratable con una
operación o que se ha diseminado a otras partes del cuerpo y para el cual han recibido tratamiento previo con determinados medicamentos• un tipo de cáncer de hígado llamado carcinoma hepatocelular (CHC) en personas que han sido tratadas previamente con sorafenibNo se sabe si STIVARGA es seguro y eficaz en niños menores de 18 años de edad.
Antes de tomar STIVARGA, informe a su proveedor de atención médica sobre todas sus afecciones médicas, lo que incluye si:• tiene problemas hepáticos aparte del cáncer de hígado• tiene problemas de sangrado• tiene presión arterial alta• tiene problemas del corazón o dolor de pecho• tiene previsto someterse ha alguna cirugía o ha tenido alguna cirugía reciente. Debe dejar de usar STIVARGA al menos 2 semanas antes de cualquier cirugía
programada. Consulte “¿Cuáles con los posibles efectos secundarios de STIVARGA?”.• está embarazada o buscando el embarazo. STIVARGA puede ser perjudicial para su bebé en gestación. Las mujeres deben usar un método anticonceptivo eficaz durante el tratamiento con STIVARGA y durante los 2 meses siguientes a la última dosis de STIVARGA.
Informe de inmediato a su proveedor de atención médica si usted queda embarazada durante el tratamiento con STIVARGA o durante los 2 meses siguientes a la última dosis de STIVARGA.
Los hombres con parejas femeninas que puedan quedar embarazadas deben usar un método anticonceptivo eficaz durante el tratamiento con STIVARGA y durante los 2 meses siguientes a la última dosis de STIVARGA.
• alimenta a su bebé con leche materna o piensa hacerlo. No se sabe si STIVARGA pasa a la leche materna. No amamante durante el tratamiento con STIVARGA y durante las 2 semanas siguientes a la última dosis de STIVARGA.
Informe a su proveedor de atención médica acerca de todos los medicamentos que tome, incluidos los medicamentos de venta con receta y de venta libre, las vitaminas y los suplementos a base de hierbas. STIVARGA puede afectar la manera en que actúan otros medicamentos, y otros medicamentos pueden afectar la acción de STIVARGA.

15
¿Cómo debo tomar STIVARGA?• Tome STIVARGA exactamente como se lo indique su proveedor de atención médica.• Por lo general, tomará STIVARGA 1 vez al día durante 21 días (3 semanas) y luego dejará de hacerlo durante 7 días (1 semana). Esto constituye 1 ciclo de tratamiento.
Repita este ciclo cuantas veces se lo indique su proveedor de atención médica.• Trague los comprimidos de STIVARGA enteros con agua luego de consumir una comida de bajo contenido graso.• Tome STIVARGA a la misma hora todos los días con una comida de bajo contenido graso que contenga menos de 600 calorías y menos del 30% de grasa.• Si omite una dosis, tómela en cuanto lo recuerde ese mismo día. No tome dos dosis el mismo día para compensar una dosis omitida.• Si toma demasiado STIVARGA, llame a su proveedor de atención médica o diríjase a la sala de emergencia más cercana inmediatamente.
¿Qué debo evitar mientras tome STIVARGA?• Durante el tratamiento con STIVARGA evite consumir jugo de pomelo (toronja) e hipérico (hierba de San Juan). Estos pueden afectar la acción de STIVARGA.
¿Cuáles son los posibles efectos secundarios de STIVARGA?STIVARGA puede causar efectos secundarios graves, tales como:• Consulte “¿Cuál es la información más importante que debo conocer acerca de STIVARGA?”• Infección. STIVARGA puede aumentar el riesgo de infecciones especialmente del aparato urinario, la nariz, la garganta y los pulmones. STIVARGA puede también
aumentar el riesgo de infecciones fúngicas de la membrana mucosa, la piel o el cuerpo. Informe de inmediato a su proveedor de atención médica si presenta: • fiebre • sensación de quemazón o dolor al orinar • tos intensa con o sin aumento • secreción vaginal o irritación inusuales
de producción de mucosidad • enrojecimiento, hinchazón o dolor en (esputo) cualquier parte del cuerpo
• dolor de garganta intenso • falta de aliento• sangrado profuso. STIVARGA puede causar sangrado que puede ser grave y, en ocasiones, provocar la muerte. Informe a su proveedor de atención médica si presenta
cualquier signo de sangrado durante el tratamiento con STIVARGA, tales como: • vómito con sangre o de aspecto • sangrado vaginal inusual similar a café molido • sangrado nasal frecuente • orina de coloración rosada o marrón • formación de moretones • heces de color rojo o negro • aturdimiento (con aspecto de alquitrán) • sangrado menstrual más abundante • tos con sangre o coágulos de sangre de lo normal• desgarro de la pared estomacal o intestinal (perforación del intestino). STIVARGA puede causar un desgarro en la pared del estómago o de los intestinos
(perforación intestinal), que puede ser grave y, en ocasiones, provocar la muerte. Informe de inmediato a su proveedor de atención médica si presenta: • dolor intenso en la zona del estómago • escalofríos (abdomen) • náuseas • hinchazón del abdomen • vómitos • fiebre • deshidratación• un problema de la piel denominado reacción cutánea de manos y pies, y erupción cutánea intensa. Las reacciones cutáneas de manos y pies son frecuentes y a
veces pueden ser graves. Informe de inmediato a su proveedor de atención médica si presenta enrojecimiento, dolor, ampollas, sangrado o hinchazón en las palmas de las manos o las plantas de los pies, o erupción cutánea intensa.
• presión arterial alta. Se le debe medir la presión arterial cada semana durante las primeras 6 semanas de haber comenzado a tomar STIVARGA. Se le debe medir la presión arterial con regularidad, y usted debe recibir tratamiento para cualquier episodio de presión arterial alta durante el tratamiento con STIVARGA. Informe a su proveedor de atención médica si tiene dolores de cabeza intensos, aturdimiento o alteraciones de la visión.
• disminución de la circulación de la sangre hacia el corazón y ataque cardíaco. Obtenga de inmediato asistencia de emergencia si presenta síntomas como dolor de pecho, falta de aliento o mareos, o si siente que se va a desmayar.
• una afección denominada síndrome de leucoencefalopatía posterior reversible (SLPR). Llame de inmediato a su proveedor de atención médica si presenta dolores de cabeza intensos, convulsiones, confusión, alteraciones de la visión o dificultad para pensar.
• riesgo de tener problemas de cicatrización de heridas. Es posible que las heridas no cicatricen bien durante el tratamiento con STIVARGA. Informe a su proveedor de atención médica si tiene previsto someterse a cirugía durante el tratamiento con STIVARGA o antes del inicio del mismo.
Debe dejar de tomar STIVARGA al menos 2 semanas antes de una cirugía planificada. Su profesional de atención médica deberá indicarle cuándo puede empezar a usar STIVARGA después de la cirugía.Los efectos secundarios más frecuentes de STIVARGA son:• dolor, incluida la zona del estómago (abdomen) • aumento de ciertos valores en las• cansancio, debilidad, fatiga pruebas de función hepática• deposiciones frecuentes o sueltas (diarrea) • fiebre• reducción del apetito • hinchazón, dolor y enrojecimiento• infección del revestimiento en la boca, la• pérdida de peso garganta, el estómago y los • cambios en la voz o ronquera intestinos (mucositis)Si usted presenta ciertos efectos secundarios, su proveedor de atención médica podría cambiarle la dosis o suspenderle temporal o permanentemente el tratamiento con STIVARGA.Estos no son todos los efectos secundarios posibles de STIVARGA.Llame a su médico para obtener recomendaciones médicas acerca de los efectos secundarios. Para comunicar los efectos secundarios a la FDA, llame al 1-800-FDA-1088.
¿Cómo debo conservar STIVARGA?• Conserve los comprimidos de STIVARGA a temperatura ambiente entre 68 y 77 °F (20 y 25 °C).• Mantenga STIVARGA dentro del frasco en el que se presenta. No coloque los comprimidos de STIVARGA en un pastillero diario o semanal.• El frasco de STIVARGA contiene un desecante que ayuda a mantener seco el medicamento. Mantenga el desecante en el frasco.• Mantenga el frasco de STIVARGA bien cerrado.• Elimine (deseche) de manera segura cualquier comprimido sin utilizar de STIVARGA 7 semanas después de abrir el frasco.Mantenga STIVARGA y todos los medicamentos fuera del alcance de los niños.

16
Información general acerca del uso seguro y eficaz de STIVARGA.En ocasiones, los medicamentos se recetan con fines distintos de los que se mencionan en un folleto de Información para el paciente. No use STIVARGA para tratar una afección para la cual no haya sido recetado. No proporcione STIVARGA a otras personas, ni siquiera si tienen los mismos síntomas que usted. Podría perjudicarlas. Puede solicitar a su proveedor de atención médica o farmacéutico que le proporcionen información sobre STIVARGA que ha sido escrita para profesionales de atención médica.
¿Cuáles son los ingredientes de STIVARGA?Principio activo: regorafenibIngredientes inactivos: celulosa microcristalina, croscarmelosa sódica, estearato de magnesio, povidona y dióxido de silicio coloidal.Recubrimiento pelicular: óxido férrico rojo, óxido férrico amarillo, lecitina (soya), polietilenglicol 3350, alcohol polivinílico, talco y dióxido de titanio.Fabricado para Bayer HealthCare Pharmaceuticals Inc., Whippany, NJ 07981 USA. © 2017 Bayer HealthCare Pharmaceuticals Inc.Para obtener más información, visite www.STIVARGA-US.com o llame al 1-888-842-2937.
Esta información para el paciente ha sido aprobada por la Administración de Alimentos y Medicamentos de los Estados Unidos. Actualizado: 02/2020
6708309MS1





![PUNTOS DESTACADOS DE LA FICHA TÉCNICA DEL ......tratamientos antidiabéticos para los pacientes con antecedentes de pancreatitis (consulte Advertencias y precauciones [5.2]) . •](https://static.fdocuments.ec/doc/165x107/5e8b821299818f7830407534/puntos-destacados-de-la-ficha-tcnica-del-tratamientos-antidiabticos.jpg)












