
METODOLOGÍA SINTÉTICA APLICADA A LA SÍNTESIS DE … · 2018-06-13 · Análisis retrosintético...
102
METODOLOGÍA SINTÉTICA APLICADA A LA SÍNTESIS DE FÁRMACOS MIGUEL CARDA
Transcript of METODOLOGÍA SINTÉTICA APLICADA A LA SÍNTESIS DE … · 2018-06-13 · Análisis retrosintético...

METODOLOGÍA SINTÉTICA
APLICADA A LA SÍNTESIS DE
FÁRMACOS
MIGUEL CARDA


Tema 4 Inflamación: síntesis de
antiinflamatorios
Miguel Carda


Tema 4. Inflamación: síntesis de antiinflamatorios
4.1. El proceso inflamatorio 1
4.2. Mediadores de la inflamación 2
4.2.1. Metabolitos del ácido araquidónico 2
4.2.2 Aminas vasoactivas: histamina y serotonina 3
4.2.3. Citoquinas 4
4.2.4. Factor Activador de Plaquetas 4
4.2.5. Óxido nítrico 4
4.2.6. Especies de oxígeno reactivas 5
4.2.7. Constituyentes de los lisosomas de los leuco citos 6
4.2.8. Neuropéptidos 6
4.2.9. Mediadores derivados de proteínas plasmática s 6
4.3. Efectos generales de la inflamación 7
4.3.1. Detención de la respuesta inflamatoria aguda 8
4.3.2. Inflamación crónica 8
4.4. Fármacos antiinflamatorios 9
4.4.1. Ciclooxigenasas 15
4.4.2. Modo de acción de los Antiinflamatorios No E steroideos 23
4.4.2.1. Aspirina 23
4.4.2.2. Ibuprofeno y naproxeno 24
4.4.2.3. Indometacina y flurbiprofeno 25
4.4.2.4. Coxibes: inhibidores selectivos de COX-2 27
4.5. Síntesis de antiinflamatorios 29
4.5.1. Síntesis de ibuprofeno 29
4.5.1.1a. Análisis retrosintético 30
4.5.1.1b. Síntesis 30
4.5.1.1c. Cuestiones 31
4.5.1.2a. Análisis retrosintético de ibuprofeno med iante carbonilación 31
4.5.1.2b. Síntesis de ibuprofeno mediante carbonila ción 31
4.5.1.2c. Cuestiones 32
4.5.1.3a. Análisis retrosintético de ibuprofeno med iante cianohidrina 33
4.5.1.3b. Síntesis de ibuprofeno mediante cianohidr ina 33
4.5.2. Síntesis de flurbiprofeno 34
4.5.2.a. Análisis retrosintético 35
4.5.2.b. Síntesis 35
4.5.2.c. Cuestiones 36
4.5.3. Síntesis de naproxeno 37
4.5.3.1a. Análisis retrosintético 37
4.5.3.1b. Síntesis naproxeno 37
4.5.3.2a. Análisis retrosintético de naproxeno medi ante
acoplamiento organometálico 39
4.5.3.2b. Síntesis de naproxeno mediante acoplamien to organometálico 40

4.5.3.2c. Cuestiones 42
4.5.4. Síntesis de indoprofeno 42
4.5.4.a. Análisis retrosintético 42
4.5.4.b. Síntesis 43
4.5.4.c. Cuestiones 43
4.5.5. Síntesis de indometacina 44
4.5.5.a. Análisis retrosintético 44
4.5.5.b. Síntesis 44
4.5.5.c. Cuestiones 45
4.5.6. Síntesis de sulindac 46
4.5.6.a. Análisis retrosintético 46
4.5.6.b. Síntesis 47
4.5.6.c. Cuestiones 47
4.5.7. Síntesis de etodolaco 48
4.5.7.a. Análisis retrosintético 48
4.5.7.b. Síntesis 48
4.5.7.c. Cuestiones 48
4.5.8. Síntesis de diclofenaco 49
4.5.8.a. Análisis retrosintético 49
4.5.8.b. Síntesis 50
4.5.8.c. Cuestiones 50
4.5.9. Síntesis de ketorolaco 51
4.5.9.1a. Análisis retrosintético 51
4.5.9.1b. Síntesis 52
4.5.9.1c. Cuestiones 53
4.5.9.2. Síntesis enantioselectiva de ketorolaco 53
4.5.10. Síntesis de zomepiraco 54
4.5.10.a. Análisis retrosintético 54
4.5.10.b. Síntesis 55
4.5.10.c. Cuestiones 55
4.5.11. Síntesis de piroxicam 55
4.5.11.a. Análisis retrosintético 55
4.5.6.b. Síntesis 56
4.5.11.c. Cuestiones 56
4.5.12. Síntesis de fenilbutazona 57
4.5.12.a. Análisis retrosintético 57
4.5.12.b. Síntesis 57
4.5.13. Síntesis de ácido flufenámico 58
4.5.13.a. Análisis retrosintético 58
4.5.13.b. Síntesis 58
4.5.13.c. Cuestiones 59
4.5.14. Síntesis de tolmetina 59

4.5.14.a. Análisis retrosintético 59
4.5.14.b. Síntesis 59
4.5.14.c. Cuestiones 60
4.5.15. Síntesis de oxaprocina 61
4.5.15.a. Análisis retrosintético 61
4.5.15.b. Síntesis 61
4.5.15.c. Cuestiones 61
4.5.16. Síntesis de nimesulida 63
4.5.16.a. Análisis retrosintético 63
4.5.16.b. Síntesis 63
4.5.16.c. Cuestiones 64
4.5.17. Síntesis de tenidap 64
4.5.17.a. Análisis retrosintético 64
4.5.17.b. Síntesis 65
4.5.17.c. Cuestiones 66
4.5.18. Síntesis de benzidamina 66
4.5.18.a. Análisis retrosintético 66
4.5.18.b. Síntesis 67
4.5.19. Síntesis de celecoxib (celebrex) 67
4.5.19.a. Análisis retrosintético 69
4.5.19.b. Síntesis 69
4.5.19.c. Cuestiones 70
4.5.20. Síntesis de etoricoxib 70
4.5.20.a. Análisis retrosintético 70
4.5.20.b. Síntesis 71
4.5.20.c. Cuestiones 72
4.5.21. Síntesis de refocoxib (vioxx) 73
4.5.21.1a. Análisis retrosintético 73
4.5.21.1b. Síntesis 73
4.5.21.1c. Cuestiones 74
4.5.21.a.2. Análisis retrosintético de rofexocib me diante
acoplamiento sp2-sp2 74
4.5.21.2b. Síntesis de rofexocib mediante acoplamie nto sp2-sp2 75
4.5.21.2c. Cuestiones 76
4.5.22. Síntesis de lumiracoxib 77
4.5.22.1a. Análisis retrosintético 77
4.5.22.1b. Síntesis 78
4.5.22.1c. Cuestiones 78
4.5.22.2a. Análisis retrosintético de lumiracoxib m ediante homologación 78
4.5.22.2b. Síntesis de lumiracoxib mediante homolog ación 79
4.5.22.2c. Cuestiones 79
4.6. Migraña 80

4.6.1. Factores desencadenantes de la migraña 80
4.6.2. Factores de riesgo 80
4.6.3. Etapas del proceso migrañoso 81
4.7. Fármacos contra la migraña 81
4.8. Síntesis de triptanos 82
4.8.1. Síntesis de sumatriptan 82
4.8.1.a. Análisis retrosintético 82
4.8.1.b. Síntesis 83
4.8.1.c. Cuestiones 85
4.8.2. Síntesis de eletriptan 85
4.8.2.1a. Análisis retrosintético 85
4.8.2.1b. Síntesis 85
4.8.2.2a. Análisis retrosintético del eletriptan me diante síntesis del
indol de Fischer 87
4.8.2.2b. Síntesis 88
4.8.2.2c. Cuestiones 89
4.8.3. Síntesis de zolmitriptan 90
4.8.3.a. Análisis retrosintético 90
4.8.3.b. Síntesis 90
4.8.3.c. Cuestiones 91
4.8.4. Síntesis de naratriptan 91
4.8.4.a. Análisis retrosintético 91
4.8.4.b. Síntesis 92
4.8.4.c. Cuestiones 92
4.8.5. Síntesis de frovatriptan 92
4.8.5.a. Análisis retrosintético 92
4.8.5.b. Síntesis 93

Tema 4. Inflamación
1
4.1. El proceso inflamatorio
La inflamación es la respuesta del organismo frente a las agresiones del medio y está
generada por los agentes inflamatorios. La respuesta inflamatoria ocurre sólo en tejidos
conectivos vascularizados y surge con el fin defensivo de aislar y destruir al agente dañino,
así como reparar el tejido u órgano dañado. Se considera a la inflamación un mecanismo de
inmunidad innata, en contraste con la reacción inmune adaptativa, que es específica para
cada tipo de agente infeccioso.
La inflamación se denomina en medicina con el sufijo -itis: faringitis, laringitis, colitis,
conjuntivitis, etc.
Los agentes o condicionantes que pueden provocar la respuesta inflamatoria son los
siguientes:
a) Las bacterias, virus, parásitos y hongos. Estos agentes infecciosos expresan compuestos
patógenos que se unen a los RTT (receptores de tipo Toll, en inglés TLRs de Toll-like
receptors), proteínas transmembrana de tipo I que forman parte del sistema inmunitario
innato del organismo. Los TLRs detectan la presencia de agentes patógenos y
desencadenan vías de señalización que estimulan la producción de diferentes mediadores,
provocando en última instancia la respuesta inflamatoria (véase la figura 4.1).
Figura 4.1. Representación del modo de acción de lo s TLR
b) Los agentes que producen necrosis de los tejidos. Cuando estos agentes provocan la
necrosis se produce la liberación de metabolitos, como ácido úrico, ADP o incluso ADN, que
activan la respuesta inflamatoria. Los agentes capaces de necrosar tejidos son:
.- Agentes físicos, como radiaciones, frío, calor, rayos UV.
.- Agentes químicos, como venenos y toxinas.
.- Traumatismos y cuerpos extraños, que producen inflamación porque dañan los tejidos
(necrosis) o aportan microbios.
.- Alteraciones vasculares, como por ejemplo las que producen isquemia.
.- Alteraciones inmunitarias, como las respuestas de hipersensibilidad o las autoinmunes. En
estos casos es la propia respuesta inmunitaria la que induce la inflamación, que es la causa
principal del daño tisular.

Síntesis de antiinflamatorios
2
4.2. Mediadores de la inflamación
Los mediadores de la inflamación son pequeñas moléculas como prostaglandinas,
leucotrienos y tromboxanos, aminoácidos modificados (histamina, serotonina) y pequeñas
proteínas (citoquinas, factores de crecimiento, interleuquinas, etc) que provocan una
respuesta en aquellas células que contienen receptores específicos en su membrana
plasmática.
4.2.1. Metabolitos del ácido araquidónico
Los derivados del ácido araquidónico, también denominados eicosanoides, sirven como
señales intra o extracelulares en la inflamación y en otros procesos biológicos, como en el
caso de la hemostasis (conjunto de mecanismos que utiliza el organismo para detener los
procesos hemorrágicos).
El ácido araquidónico (AA) es un derivado del ácido linoleico, que se encuentra
normalmente esterificado en forma de fosfolípido en las membranas celulares. El AA se
libera por acción de las fosfolipasas celulares, a partir de cualquier célula activada
(plaquetas), estresada o a punto de morir por necrosis. Una vez liberado, el AA puede
metabolizarse en leucotrienos por acción de las lipooxigenasas, y en tromboxanos,
prostaciclinas o prostaglandinas por acción de las ciclooxigenasas (figura 4.2).
Fosfolípidos Fosfolípidos
Ácido araquidónico
LeucotrienosHPETE
Lipoxinas
TromboxanosProstaciclinas
Prostaglandinas
Factor activador de plaquetas
Citoquinasproinflamatorias
Fosfolipasa A2
Lipoxigenasa Ciclooxigenasa
Transcripción de fosfolipasa A2
Figura 4.2. Representación de la ruta metabólica de oxidación del ácido araquidónico
La vía de la lipoxigenasa (LOX) convierte al ácido araquidónico en leucotrienos, HPETE
(ácidos hidroxiperoxieicosatetraenoicos) y lipoxinas, mientras que la vía de la ciclooxigenasa
(COX) convierte al ácido araquidónico en prostaglandinas, prostaciclinas y tromboxanos.
Estas dos enzimas no actúan sobre el ácido araquidónico esterificado, por lo que primero
debe ser liberado de los fosfolípidos de la membrana mediante hidrólisis mediada por
fosfolipasas.

Tema 4. Inflamación
3
En la figura 4.3 se indican las estructuras de los metabolitos resultantes de las vías
enzimáticas de oxidación del ácido araquidónico (en esta figura se ha dibujado
arbitrariamente la estructura de un representante de cada familia de metabolitos).
COOH
Ácido araquidónico HO
HO OH
Prostaglandinas (prostaglandina F2 )
COOH
LOX
OH OH
COOH
Leucotrienos (leucotrieno LTB4)
HPETE (ácido 5-Hidroperoxieicosatetraenoico)
OOH
COOH
OH
OH
OHCOOH
Lipoxinas (lipoxina B4)
O
OHCOOH
HP
OH
Tromboxanos (tromboxano B2)
COXO
HO OH
HOOC
Prostaciclina PGI2
Figura 4.3. Metabolitos resultantes de la oxidación del ácido araquidónico
4.2.2 Aminas vasoactivas: histamina y serotonina
La histamina y la serotonina son las dos principales aminas vasoactivas, llamadas así
por su importante acción sobre los vasos sanguíneos. Se almacenan preformadas en
gránulos, dentro de las células que las producen, por lo que son mediadores precoces de la
inflamación.

Síntesis de antiinflamatorios
4
Figura 4.4. Estructuras de la histamina y de la ser otonina
4.2.3. Citoquinas
Las citoquinas son pequeñas proteínas (entre 5 y 20 kD) que permiten el intercambio de
información entre las células durante el proceso de inflamación, la hematopoyesis1 y las
respuestas inmunes. Los factores de crecimiento que utilizan las células epiteliales para
estimular su renovación son asimismo citoquinas.
4.2.4. Factor Activador de las Plaquetas
El Factor Activador de Plaquetas (en inglés Platelet Activating Factor, PAF) es un
derivado de fosfolípidos mediador de la inflamación. Las principales acciones del PAF se
enfocan a la agregación de las plaquetas, la vasoconstricción y broncoconstricción, la
adhesión leucocitaria al endotelio, la quimiotaxis, la desgranulación, el estallido oxidativo y
la activación de la síntesis de eicosanoides.
Figura 4.5. Estructura del Factor de Agregación de Plaquetas
4.2.5. Óxido nítrico
El óxido nítrico (NO) es un gas soluble producido en algunas neuronas del cerebro,
macrófagos y células endoteliales. Actúa de forma paracrina (acción corta y local) sobre las
células diana a través de la inducción de GMPc (guanosín monofosfato cíclico), el cual inicia
una serie de sucesos intracelulares que acaban provocando la relajación del músculo liso
(vasodilatación). La vida media in vivo del NO es muy corta, por lo que sólo actúa sobre las
células muy próximas a su lugar de producción.
El NO se sintetiza a partir de L-arginina por la enzima NO-sintasa (NOS). Hay tres tipos
de NOS: endotelial (eNOS), neuronal (nNOS) e inducible (iNOS). Las dos primeras son
constitutivas, se expresan a niveles bajos y pueden activarse rápidamente aumentando los
niveles de calcio intracelular. Sin embargo, la iNOS se activa solamente cuando los
macrófagos y otras células son activados por citoquinas (como IFN-γ) o productos
microbianos.
1 Proceso de formación, desarrollo y maduración de los elementos formes de la sangre (eritrocitos, leucocitos y plaquetas) a partir de un precursor celular común e indiferenciado conocido como célula madre hematopoyética pluripotencial.

Tema 4. Inflamación
5
4.2.6. Especies de oxígeno reactivas
Las especies de oxígeno reactivas (en inglés ROS, de Reactive Oxigen Species)
pueden liberarse al medio extracelular por los leucocitos después de que hayan sido
activados por la presencia de microbios, quimioquinas, complejos inmunes, o después de la
fagocitosis. Su producción depende de la activación del sistema NADPH oxidasa. Las
principales especies producidas intracelularmente son el anión superóxido (O2-), el peróxido
de hidrógeno H2O2 y el radical hidroxilo (·OH).
El anión superóxido puede combinarse con el óxido nítrico para formar especies
reactivas del nitrógeno. Estas sustancias atacan todos los materiales biológicos (ADN,
proteínas, lípidos, etc), ya sea arrancando electrones, arrancando átomos de hidrógeno o
adicionándose sobre los enlaces dobles y reaccionando como potentes oxidantes. La
consecuencia de estos procesos oxidantes es la alteración y la posterior pérdida de función
de las moléculas afectadas.
La liberación extracelular de radicales libres de oxígeno (RLO) activa quimioquinas,
citoquinas y moléculas de adhesión leucocitaria endotelial, amplificando la respuesta
inflamatoria. En estas respuestas inflamatorias se provoca:
a) Daño de las células endoteliales, lo que consecuentemente produce un aumento de la
permeabilidad vascular.
b) Daño a otras células, como glóbulos rojos o células del parénquima.
c) Inactivación de antiproteasas, como la α1-antitripsina, lo cual provoca un incremento de la
destrucción tisular, como ocurre en el enfisema pulmonar.
El efecto negativo de los ERO se deja sentir cuando se produce un desequilibrio debido
a una producción exagerada, o a una disminución de los sistemas de defensa, enzimáticos
y no enzimáticos. El plasma, los fluidos tisulares y las células poseen enzimas y
mecanismos antioxidantes que les permiten protegerse de los radicales libres de oxígeno.
Entre estos se encuentran:
a) La enzima superóxido dismutasa, que convierte el anión superóxido en peróxido de
hidrógeno.
b) La enzima catalasa, que destoxifica el peróxido de hidrógeno.
c) El glutatión peroxidasa, otro potente destoxificador del H2O2.
d) El ácido úrico, un potente antioxidante presente en el plasma en una concentración
mucho mayor que el ascorbato (vitamina C).
e) La proteína ceruloplasmina, la principal transportadora de cobre en el suero.
f) La fracción plasmática libre de hierro de la proteína transferrina.
También existen compuestos de origen alimentario con capacidad antioxidante que
intervienen en la neutralización de ERO como:
a) El α-tocoferol (vitamina E), compuesto liposoluble con capacidad de protección de las
membranas celulares.
b) Los carotenoides (como el β-caroteno) y los polifenoles (como el ácido caféico y la
quercetina).

Síntesis de antiinflamatorios
6
c) El ascorbato (vitamina C), compuesto hidrosoluble capaz de regenerar los demás
antioxidantes, como el glutatión o el α-tocoferol.
4.2.7. Constituyentes de los lisosomas de los leuco citos
Los neutrófilos y los monocitos contienen gránulos lisosomiales necesarios para la
digestión de los materiales fagocitados. Si estos compuestos se vierten al exterior, pueden
amplificar la respuesta inflamatoria, ya que tienen un efecto destructor sobre los tejidos
(elastasas, colagenasas, proteasas, etc). Para contrarrestar su efecto, existen antiproteasas
en el suero, fundamentalmente la α1-antitripsina, que es el principal inhibidor de la elastasa.
Otra antiproteasa importante es la α2-macroglobulina.
4.2.8. Neuropéptidos
Los neuropéptidos son sustancias segregadas por los nervios sensoriales y por varios
tipos de leucocitos, y juegan un papel en la propagación de la respuesta inflamatoria. Entre
ellos se encuentran la sustancia P y la neurocinina A, pertenecientes a la familia de los
taquininos producidos en el SNC y periférico. Los pulmones y el tracto gastrointestinal son
ricos en fibras que contienen sustancia P. Este compuesto tiene, entre otras funciones, la de
la transmisión de las señales dolorosas, la regulación de la presión sanguínea, la
estimulación de la secreción de las células endocrinas y el aumento de la permeabilidad
vascular.
4.2.9. Mediadores derivados de proteínas plasmática s
Una gran variedad de fenómenos de la respuesta inflamatoria están mediados por
proteínas plasmáticas que pertenecen a tres sistemas interrelacionados:
a) El sistema del complemento:2 las proteínas de este sistema están presentes en el plasma
en forma inactiva, y cuando se activan se convierten en enzimas proteolíticas que degradan
otras proteínas del complemento, formando una cascada. Los elementos que participan en
el proceso inflamatorio se les conoce con el nombre de anafilotoxinas y son el C3a, C5a y
en menor medida C4a. Estas enzimas estimulan la liberación de histamina por los
mastocitos y, por tanto, producen vasodilatación. El C5a además tiene capacidad
quimiotáctica y activa la lipooxigenasa, generando leucotrienos.
b) La coagulación: la inflamación aumenta la producción de algunos factores de la
coagulación y convierte al endotelio en trombogénico. En contrapartida, la trombina
promueve la inflamación mediante la activación de receptores denominados PAR (protease-
activated receptors), que activan diferentes respuestas como la movilización de selectina-P,
la producción de quimioquinas y citoquinas, la expresión de receptores para integrinas en el
endotelio, la inducción de la COX-2 y la producción de prostaglandinas, la producción de NO
y PAF, y cambios en la forma endotelial. Como la coagulación y la inflamación pueden
iniciar un círculo vicioso de amplificación, la interferencia con la coagulación puede ser una
estrategia terapéutica para reducir la inflamación en algunas patologías.
2 El sistema del complemento es uno de los componentes fundamentales de la respuesta inmunitaria defensiva ante un agente hostil. Consta de una serie de moléculas plasmáticas, las cuales constituyen un 15% de la fracción de inmunoglobulina del suero, y cuyas funciones son potenciar la respuesta inflamatoria, facilitar la fagocitosis y dirigir la lisis de células incluyendo la apoptosis.
Tema 4. Inflamación
7
c) Las quininas son péptidos vasoactivos derivados de proteínas plasmáticas, denominadas
quininógenos, por la acción de enzimas específicas denominadas calicreínas. El sistema de
quininas está íntimamente ligado a la coagulación. Así, la forma activa del factor XII, FXIIa,
convierte la precalicreína del plasma en calicreína, que corta una proteína del plasma de
alto peso molecular para generar bradiquinina. La bradiquinina aumenta la permeabilidad
vascular y causa contracción del músculo liso, dilatación de los vasos y dolor, efectos
similares a los de la histamina. Por otro lado, la calicreína tiene efecto quimiotáctico, ya que
convierte C5 del sistema del complemento en C5a (también quimiotáctico) y convierte el
plasminógeno en plasmina para degradar el coágulo secundario. Los mediadores de la
inflamación más importantes del conjunto de los tres sistemas son la bradiquinina el C3a, el
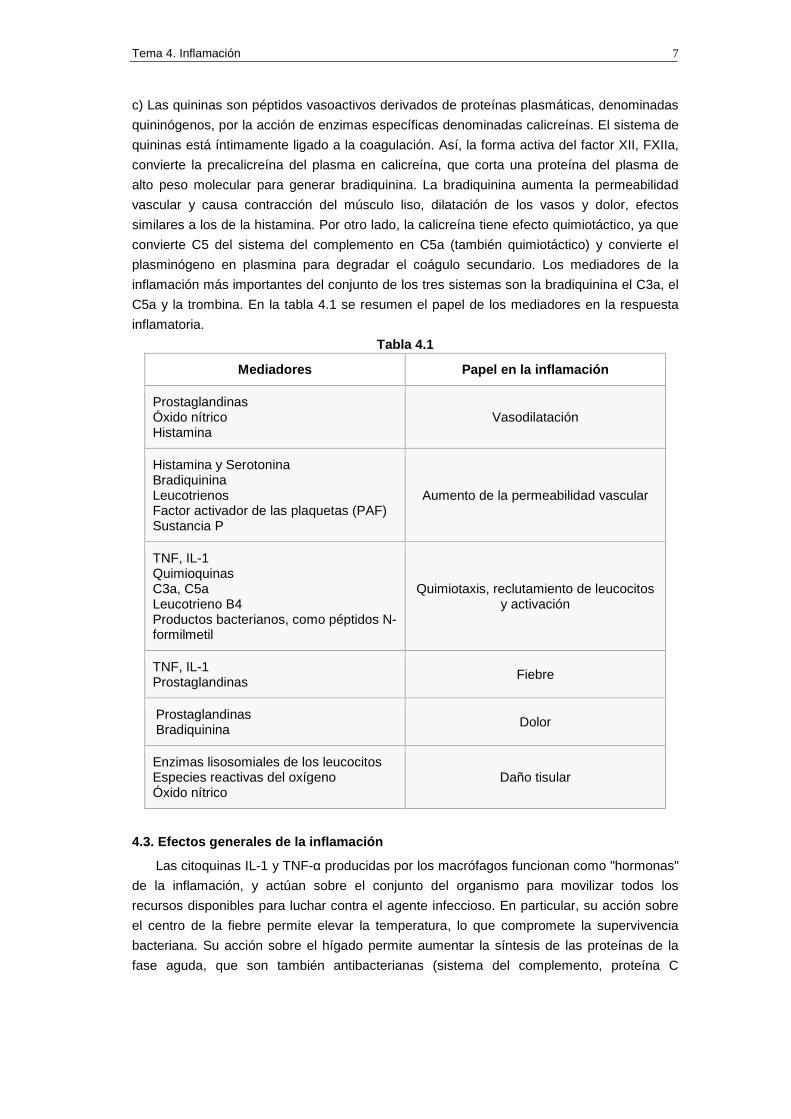
C5a y la trombina. En la tabla 4.1 se resumen el papel de los mediadores en la respuesta
inflamatoria.
Tabla 4.1
Mediadores Papel en la inflamación
Prostaglandinas Óxido nítrico Histamina
Vasodilatación
Histamina y Serotonina Bradiquinina Leucotrienos Factor activador de las plaquetas (PAF) Sustancia P
Aumento de la permeabilidad vascular
TNF, IL-1 Quimioquinas C3a, C5a Leucotrieno B4 Productos bacterianos, como péptidos N- formilmetil
Quimiotaxis, reclutamiento de leucocitos y activación
TNF, IL-1 Prostaglandinas
Fiebre
Prostaglandinas Bradiquinina
Dolor
Enzimas lisosomiales de los leucocitos Especies reactivas del oxígeno Óxido nítrico
Daño tisular
4.3. Efectos generales de la inflamación
Las citoquinas IL-1 y TNF-α producidas por los macrófagos funcionan como "hormonas"
de la inflamación, y actúan sobre el conjunto del organismo para movilizar todos los
recursos disponibles para luchar contra el agente infeccioso. En particular, su acción sobre
el centro de la fiebre permite elevar la temperatura, lo que compromete la supervivencia
bacteriana. Su acción sobre el hígado permite aumentar la síntesis de las proteínas de la
fase aguda, que son también antibacterianas (sistema del complemento, proteína C

Síntesis de antiinflamatorios
8
reactiva). Durante la fase reparadora juegan un papel clave en la activación y movilización
de los leucocitos polimorfonucleares (leucocitos PMN) a partir de la médula ósea, así como
en la activación de los fibroblastos
4.3.1. Detención de la respuesta inflamatoria aguda
Puesto que este potente proceso de defensa puede producir daños importantes en los
tejidos del huésped, es importante mantenerlo bajo un estricto control. En parte, la
inflamación desaparece simplemente porque los mediadores se producen en estallidos
rápidos (sólo mientras persiste el estímulo), tienen vidas medias cortas, y son degradados
tras su liberación. Los neutrófilos también tienen una vida media corta y mueren por
apoptosis unas pocas horas después de dejar la sangre. Además, durante el desarrollo del
proceso inflamatorio se disparan una serie de señales de STOP que sirven para terminar la
reacción de forma activa. El proceso de parada se debe al cambio en el tipo de metabolitos
producidos a partir del ácido araquidónico, deteniéndose la producción de leucotrienos
proinflamatorios por lipoxinas antiinflamatorias.
Por otro lado, los macrófagos y otras células liberan citoquinas antiinflamatorias, como
TGF-β e IL-10, produciendo mediadores lípidicos antiinflamatorios (como resolvinas y
protectinas) derivados de ácidos grasos poliinsaturados, generando impulsos nerviosos
(descargas colinérgicas) que inhiben la producción de TNF (Tumor Necrosis Factor) por los
macrófagos.
4.3.2. Inflamación crónica
Cuando la inflamación se mantiene durante un tiempo prolongado (semanas o meses),
se habla de inflamación crónica, en la que coexisten el daño tisular y los intentos de
reparación, en diversas combinaciones. La inflamación crónica puede producirse por
mantenimiento de la inflamación aguda (si no se resuelve la causa), o bien empezar de
manera progresiva y poco evidente, sin las manifestaciones de la inflamación aguda. Este
segundo caso es el responsable del daño tisular de algunas de las enfermedades humanas
más invalidantes, como la artritis reumatoide, la aterosclerosis, la tuberculosis o la fibrosis
pulmonar. Además, es importante en el desarrollo del cáncer y en enfermedades que
anteriormente se consideraban exclusivamente degenerativas, como el Alzheimer. Entre las
causas de la inflamación crónica se pueden distinguir:
a) Infecciones persistentes producidas por microbios difíciles de erradicar, como
micobacterias, ciertos hongos, virus y parásitos.
b) Enfermedades mediadas por el sistema inmune debido a una sobredimensión de la
respuesta inmunitaria.
c) Exposición prolongada a agentes tóxicos

Tema 4. Inflamación
9
4.4. Fármacos antiinflamatorios
Muchos medicamentos antiinflamatorios deben su modo de acción a la inhibición de la
síntesis de prostaglandinas, sustancias de carácter lipídico derivadas del ácido araquidónico
(véase la figura 4.6).
Figura 4.6. Estructuras de prostaglandinas de la se ries E y F (subserie 2)
Las series de las protaglandinas vienen determinadas por el tipo de sustitución que
éstas exhiben en el anillo ciclopentánico. La subserie la determina el grado de insaturación
de las cadenas laterales. En la figura 4.7 se representan algunas series y subseries de
prostaglandinas.
Figura 4.7. Estructuras de series y subseries de pr otaglandinas

Síntesis de antiinflamatorios
10
Los antiinflamatorios naturales, segregados por el propio organismo, son los derivados
de los corticoides, sustancias de origen esteroideo de potente acción antiinflamatoria, pero
que causan importantes efectos secundarios.
Los fármacos antiinflamatorios no esteroideos (AINEs) se denominan de esta forma en
oposición a los corticoides. Los AINEs disminuyen la inflamación, el dolor y la fiebre
inhibiendo la acción de las ciclooxigenasas, enzimas que participan en la biosíntesis de las
prostaglandinas. Las funciones de las prostaglandinas se pueden resumir en cinco puntos:
a) Intervienen en la respuesta inflamatoria provocando la vasodilatación, el aumento de la
permeabilidad de los tejidos permitiendo el paso de los leucocitos y actuando como
antiagregante plaquetario estimulando las terminaciones nerviosas del dolor.
b) Aumentan la secreción de mucus gástrico y disminuyen la secreción de ácido gástrico.
c) Provocan la contracción de la musculatura lisa, lo que es especialmente importante en la
zona uterina. De hecho, en el semen humano hay cantidades pequeñas de prostaglandinas
que favorecen la contracción del útero y, como consecuencia, la ascensión de los
espermatozoides a las trompas uterinas (trompas de falopio). Del mismo modo, durante la
menstruación se produce la liberación de protaglandinas para favorecer el desprendimiento
del endometrio. Los dolores menstruales son tratados muchas veces con inhibidores de la
liberación de prostaglandinas.
d) Intervienen en la regulación de la temperatura corporal.
e) Controlan el descenso de la presión arterial al favorecer la eliminación de sustancias en
el riñón.
Los AINEs se pueden clasificar en:
a) Salicilatos y derivados, como la aspirina (ácido acetilsalicílico), el benorilato, la
salicilamida, el diflunisal, el clonixinato de lisina o el etersalato.
Figura 4.8. Estructuras de salicilatos AINEs
b) Derivados indol-acéticos, como el sulindac, la indometacina, la acemetacina, la
oxametacina, o la glucametacina.

Tema 4. Inflamación
11
Figura 4.9. Estructuras de derivados indol-acéticos AINEs
El sulindac inhibe la producción de prostaglandinas, por lo que se indica para el alivio
del dolor, fiebre y la inflamación. Aparte de la inhibición de la ciclooxigenasa, el sulindac
inhibe el crecimiento de pólipos y lesiones precancerosas del colon, especialmente en
pacientes con poliposis adenomatosa familiar.
c) Derivados aril-acéticos, como el etodolaco, que se utiliza para reducir la inflamación y
para tratar dolores leves a moderados relacionados con la osteoartritis o la artritis
reumatoide. En la figura 4.10 se indican las estructuras de otros derivados aril-acéticos con
actividad antiinflamatoria.
Figura 4.10. Estructuras de derivados aril-acéticos AINEs

Síntesis de antiinflamatorios
12
d) Ácidos enólicos.
d.1) Oxicames, como el piroxicam, que se emplea en tratamiento de los síntomas de la
artritis reumatoide, osteoartritis, dolor menstrual primario y dolor posoperatorio.
Figura 4.11. Estructuras de oxicames AINEs
d.2) Pirazolonas, como la fenilbutazona, que se prescribe para el tratamiento del dolor
crónico, incluyendo los síntomas de la artritis. Sin embargo, su uso es limitado en humanos
por sus efectos adversos severos tales como la supresión de los glóbulos blancos y la
anemia aplásica. En la figura 4.12 se indican las estructuras de otras pirazolonas con
actividad antiinflamatoria.
Figura 4.12. Estructuras de pirazolonas AINEs
e) Derivados arilpropiónicos como el ibuprofeno, el flurbiprofeno, el naproxeno, el
fenoprofeno, el benoxaprofeno o el suprofeno. Todos estos compuestos compiten con el
ácido araquidónico por el sitio activo de la ciclooxigenasa.

Tema 4. Inflamación
13
Figura 4.13. Estructuras de derivados arilpropiónic os AINEs
f) Fenematos, como el ácido meclofenámico, analgésico indicado para el tratamiento del
dolor leve o moderado, y también indicado como antiinflamatorio y antipirético. En la figura
4.14 se indican las estructuras de otros fenematos con actividad antiinflamatoria.
Figura 4.14. Estructuras de fenematos AINEs
g) Coxibes, como el valdecoxib, que es un inhibidor selectivo de COX-2 y se prescribe para
el tratamiento de los dolores mentruales, artritis y osteroartritis.
Figura 4.15. Estructuras de coxibes AINEs

Síntesis de antiinflamatorios
14
h) Otros, como la nimesulida, el paracetamol, la namubetona, la diacereina, la tolmetina o la
oxaprocina. La nimesulida es un antiinflamatorio relativamente COX-2 selectivo, con efectos
analgésicos y antipiréticos. Está aprobado como indicación para el tratamiento del dolor
agudo, la sintomatología de la osteoartritis y dismenorrea en adolescentes y adultos, por
encima de los 12 años de edad. Sin embargo, este fármaco ha sido retirado del mercado
debido a su potencial hepatotoxicidad.
N CH3
COOH
O
CH3Tolmetina
Nimesulida
HO
HN
O
CH3
Paracetamol
NO2
O
NHS
H3C
O O
MeO
O
CH3
Nabumetona
OH3C
O
O CH3
O
O
O
COOH
Diacereina
N
OCOOH
Oxaprocina
N
N
O
Proquazona
NN O N
Benzidamina
N ON
NH2 O O
Morniflumato
N
O
S
ONH2
OH
Cl
Tenidap
O
OH
HOHO
H2NOH
Glucosamina
Figura 4.16. Estructuras de otros fármacos AINEs
El paracetamol también se incluye entre los AINEs, a pesar de su poca acción
antiinflamatoria.
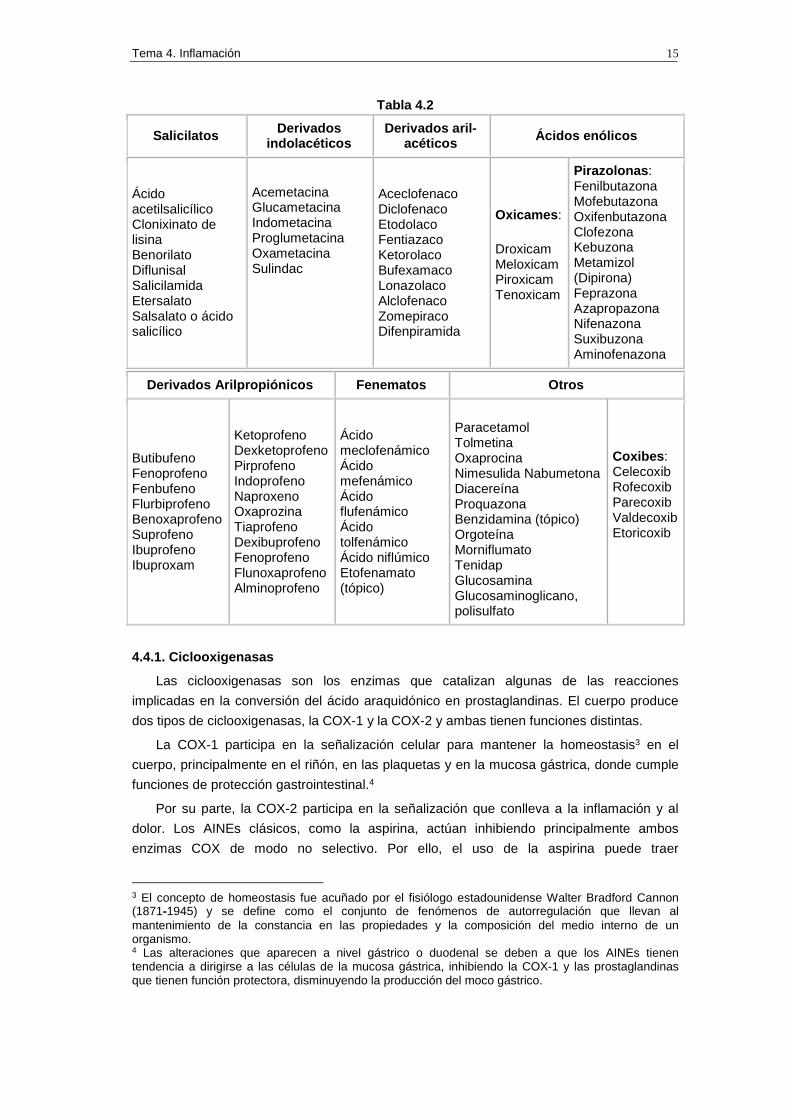
En la tabla 4.2 se resume la clasificación de los AINEs.
Tema 4. Inflamación
15
Tabla 4.2
Salicilatos Derivados indolacéticos
Derivados aril -acéticos Ácidos enólicos
Ácido acetilsalicílico Clonixinato de lisina Benorilato Diflunisal Salicilamida Etersalato Salsalato o ácido salicílico
Acemetacina Glucametacina Indometacina Proglumetacina Oxametacina Sulindac
Aceclofenaco Diclofenaco Etodolaco Fentiazaco Ketorolaco Bufexamaco Lonazolaco Alclofenaco Zomepiraco Difenpiramida
Oxicames :
Droxicam Meloxicam Piroxicam Tenoxicam
Pirazolonas : Fenilbutazona Mofebutazona Oxifenbutazona Clofezona Kebuzona Metamizol (Dipirona) Feprazona Azapropazona Nifenazona Suxibuzona Aminofenazona
Derivados Arilpropiónicos Fenematos Otros
Butibufeno Fenoprofeno Fenbufeno Flurbiprofeno Benoxaprofeno Suprofeno Ibuprofeno Ibuproxam
Ketoprofeno Dexketoprofeno Pirprofeno Indoprofeno Naproxeno Oxaprozina Tiaprofeno Dexibuprofeno Fenoprofeno Flunoxaprofeno Alminoprofeno
Ácido meclofenámico Ácido mefenámico Ácido flufenámico Ácido tolfenámico Ácido niflúmico Etofenamato (tópico)
Paracetamol Tolmetina Oxaprocina Nimesulida Nabumetona Diacereína Proquazona Benzidamina (tópico) Orgoteína Morniflumato Tenidap Glucosamina Glucosaminoglicano, polisulfato
Coxibes : Celecoxib Rofecoxib Parecoxib Valdecoxib Etoricoxib
4.4.1. Ciclooxigenasas
Las ciclooxigenasas son los enzimas que catalizan algunas de las reacciones
implicadas en la conversión del ácido araquidónico en prostaglandinas. El cuerpo produce
dos tipos de ciclooxigenasas, la COX-1 y la COX-2 y ambas tienen funciones distintas.
La COX-1 participa en la señalización celular para mantener la homeostasis3 en el
cuerpo, principalmente en el riñón, en las plaquetas y en la mucosa gástrica, donde cumple
funciones de protección gastrointestinal.4
Por su parte, la COX-2 participa en la señalización que conlleva a la inflamación y al
dolor. Los AINEs clásicos, como la aspirina, actúan inhibiendo principalmente ambos
enzimas COX de modo no selectivo. Por ello, el uso de la aspirina puede traer
3 El concepto de homeostasis fue acuñado por el fisiólogo estadounidense Walter Bradford Cannon (1871-1945) y se define como el conjunto de fenómenos de autorregulación que llevan al mantenimiento de la constancia en las propiedades y la composición del medio interno de un organismo. 4 Las alteraciones que aparecen a nivel gástrico o duodenal se deben a que los AINEs tienen tendencia a dirigirse a las células de la mucosa gástrica, inhibiendo la COX-1 y las prostaglandinas que tienen función protectora, disminuyendo la producción del moco gástrico.

Síntesis de antiinflamatorios
16
complicaciones adversas, como sangramiento en el estómago debido a la destrucción de la
mucosa gástrica.
Las ciclooxigenasas COX-1 y COX-2 se encuentran ancladas a la superficie de la
membrana celular y contienen alrededor de 600 aminoácidos. Su centro activo se encuentra
en el fondo de un estrecho túnel o canal hidrofóbico. Tres de las hélices alfa del dominio de
unión a la membrana están en la entrada de este túnel. Las paredes del túnel están
definidas por cuatro hélices alfa formadas por los residuos 106-123, 325-353, 379-384 y
520-535. Al fondo de este canal hidrofóbico se encuentra un importante residuo catalítico: la
Tyr-385.
Figura 4.17. Representación de la enzima COX-2 y de los grupos hemo
Se piensa que la inhibición de la COX-2 es la responsable de la acción antiinflamatoria,
analgésica y antipirética de los AINEs. Sin embargo, los inhibidores selectivos de COX-2
(Coxibes) no están exentos de riesgos secundarios porque la inhibición de COX-2 rompe el
balance entre el efecto antitrombótico y el protrombótico (TxA2), incrementándose la
posibilidad de una trombosis cardiovascular. Experimentalmente se ha demostrado que la
inhibición selectiva de COX-2 en ratones produce trombogénesis acelerada y presión
arterial elevada.
Las ciclooxigenasas tienen acción enzimática dual, puesto que tienen actividad
peroxidasa y actividad ciclooxigenasa. El centro activo peroxidasa incluye una parte hemo,
con el átomo de Fe(III) coordinado con la His-388 y la His-207. En la figura 4.17 se
representa el dímero COX-2 y los grupos hemo que éste contiene.
Las ciclooxigenasas COX-1 y COX-2 son muy similares. Las principales diferencias, por
lo que hace a sus centros activos, son el reemplazo de la isoleucina-434 y 523 de COX-1
por la menos voluminosa valina en COX-2, y la sustitución de la arginina-513 de COX-1 por
histidina en COX-2. Este cambio genera un bolsillo lateral en el canal de acceso al centro
activo de COX-2, en el cual interaccionan los AINEs que inhiben selectivamente a esta
enzima.
En la figura 4.18 se representa una superposición de las enzimas COX-1/COX-2. La
enzima COX-1 se representa en amarillo y la COX-2 en magenta. El sitio peroxidasa está en

Tema 4. Inflamación
17
el lado opuesto al del canal de entrada al centro activo ciclooxigenasa, que es la zona
marcada con un asterisco en la figura 4.18. Se puede apreciar que la superposición de las
dos enzimas es prácticamente total.
Figura 4.18. Superposición de las enzimas COX-1 y C OX-2
En la figura 4.19 se representa la estructura del dímero de COX con indicación del
dominio EGF (del inglés Epidermal Growth Factor) y el dominio MBD (del inglés Membrane
Binding Domains), que es el que se ancla en la superficie de la membrana celular. Existe
una sustancial diferencia en la zona MBD entre las dos isoformas de COX.
Figura 4.19. Dímero de COX mostrando los dominios d e enlace a membrana (MBD)
Las enzimas periféricas de membrana se anclan a la superficie de las membranas
celulares pero no atraviesan completamente la membrana biológica uniéndose a ésta de un
solo lado, con un extremo de la proteína sobresaliendo fuera de la célula (véase la figura
4.20).

Síntesis de antiinflamatorios
18
Figura 4.20. Anclaje de COX a la membrana celular
La unión de la proteína a la membrana se lleva a cabo mediante enlaces no covalentes,
de tipo enlaces de hidrógeno y fuerzas electrostáticas. No intervienen fuerzas hidrófobas, ya
que las proteínas periféricas sólo interactúan con la zona exterior de la membrana, nunca
con su interior hidrófobo. Se unen a la cabeza polar de los lípidos de la bicapa o a
determinadas regiones de las proteínas transmembrana. Las enzimas COX son globulares e
hidrofílicas, ya que al estar en la periferia de la membrana están expuestas al medio acuoso
extracitosólico.
La actividad peroxidasa hemo-dependiente de la COX está implicada en la formación de
un radical Tyr-385 (radical tirosinilo), que es necesario para desempeñar la actividad
ciclooxigenasa.
En el esquema 4.1 se describe la conversión del ácido araquidónico en la
prostaglandina PF2α. El proceso se inicia con la abstracción de un átomo de hidrógeno del
carbono C-13 del ácido araquidónico por parte del radical tirosinilo, situado en el centro
activo del enzima ciclooxigenasa. Esta reacción genera un radical que es capturado por el
oxígeno molecular. El resultado del proceso es la formación de un radical hidroperóxido
(intermedio I) que captura un átomo de hidrógeno del hidroxilo fenólico de la tirosina y se
convierte en un hidroperóxido intermedio denominado ácido 11R-
hidroperoxieicosatetraenoico (11R-HPETE). A continuación, la abstracción de hidrógeno por
parte del radical tirosinilo y la subsiguiente ciclación intramolecular forman un radical
endoperóxido (intermedio III). Una reacción de ciclación oxidante sobre este radical forma
un intermedio radicalario (intermedio IV) que ya contiene el anillo ciclopentánico
característico de las prostaglandinas. Este intermedio se convierte en el hidroperóxido PGG2
por reacción con el residuo de tirosina. La reducción de la función hidroperóxido forma la
PGH2, la cual, por apertura reductora del puente peróxido, se transforma en la
prostaglandina PF2α.

Tema 4. Inflamación
19
Esquema 4.1
En el esquema 4.2 se describen con más detalle los pasos de formación del radical 15-
peroxi-PGG2 (compuesto IV del esquema 4.1). El proceso contiene las siguientes etapas:
1) Abstracción por parte del radical tirosinilo del átomo de hidrógeno en C-13 del ácido
araquidónico y formación del radical 11-araquidonilo.
2) Oxidación del radical 11-araquidonilo y formación del radical 11-peroxi (intermedio II
del esquema 4.1).
3) Etapa de ciclación oxidante y formación del radical 15-peroxi-PGG2 (intermedio IV del
esquema 4.1).

Síntesis de antiinflamatorios
20
Esquema 4.2
En la figura 4.21 se indica la estructura del enzima COX y la colocación del ácido
araquidónico en el centro activo. El centro activo peroxidasa (centro POD) está colocado en
la parte superior de la figura 4.21 y se encuentra expuesto al disolvente, de este forma el
peróxido y los substratos exógenos tienen un fácil acceso a este centro enzimático.
Figura 4.21. Colocación del ácido araquidónico en e l centro activo de la enzima COX

Tema 4. Inflamación
21
El centro activo COX se sitúa en el interior de la enzima y sólo es accesible a lo largo de
un surco hidrofóbico con una longitud de 12Å y una anchura de 6Å. El ácido araquidónico
accede al centro activo del enzima y adquiere allí una conformación doblada que expone el
hidrógeno pro-S del átomo de carbono C-13 a la abstracción por la tirosina-385 (véase el
esquema 4.2).
En la figura 4.22 se muestra la colocación del araquidonato en el centro activo de COX-
2. El grupo caboxilato forma un par iónico con la Arg-120 y está enlazado mediante puente
de hidrógeno con la Tyr-355. El átomo de hidrógeno pro-S del C-13 del araquidonato se
coloca en la proximidad del agente oxidante Tyr-385, colocándose el grupo metilo C-20
cerca de la Gly-533. Otros residuos claves son la Ser-530, que es el centro de acetilación de
la aspirina, y los aminoácidos Val-523 y la Arg-513, cuyos restos permiten la unión de los
grupos sulfonamida y sulfona de los fármacos de tipo diarilheterocíclico.
Figura 4.22. Colocación del araquidonato en el cent ro activo de COX-2
Las estructuras de los aminoácidos clave en los centros activos de las enzimas COX se
indican en la figura 4.23:
Figura 4.23. Estructuras de los aminoácidos clave d el centro activo de COX

Síntesis de antiinflamatorios
22
Es interesante señalar que a pesar de las grandes similitudes entre los enzimas COX-1
y COX-2, el ácido araquidónico se une de forma diferente en el centro activo de cada uno de
estos dos enzimas.
En la parte izquierda de la figura 4.24 se representa la enzima COX-1 con el ácido
araquidónico en su centro activo (parte inferior central de la estructura en modelo space-
filling en color gris). En la parte superior central de la enzima COX-1 se puede observar la
estructura, en modelo space-filling, del grupo hemo. En el recuadro izquierdo de la figura
4.24 se observa que el ácido araquidónico adopta una conformación alargada en el centro
activo de COX-1.
En el recuadro derecho de la figura 4.24 se observa (parte inferior central en gris y en
modelo space-filling) que el ácido araquidónico adopta una conformación más doblada en el
centro activo de COX-2 (en la parte superior central se observa el grupo hemo).
Figura 4.24. Colocación de araquidonato en COX-1 y COX-2
Las diferentes propiedades y funciones de las enzimas COX-1 y COX-2 se pueden
explicar por las pequeñas diferencias entre los centros activos de ambas enzimas. Así, la
isoleucina-434 e isoleucina-523 de COX-1 son reemplazadas en COX-2 por el aminoácido
valina, menos voluminoso, mientras que la arginina-513 de COX-1 es sustituida por histidina
en COX-2. Estas diferencias estructurales entre los centros activos de ambas enzimas se
han empleado en el desarrollo de inhibidores selectivos de éstos, en particular de COX-2.
En la figura 4.25 se indican de forma esquemática las diferencias en los centros activos
de las enzimas COX. Esta diferencia se debe, fundamentalmente, a la presencia de la
isoleucina en la posición 523 de COX-I y de valina en la posición 523 de COX-2.

Tema 4. Inflamación
23
Figura 4.25. Representación esquemática de los cent ros activos de COX-1 y COX-2
4.4.2. Modo de acción de los Antiinflamatorios No E steroideos
4.4.2.1. Aspirina
La inhibición de COX-1 por aspirina es 170 veces mayor que la inhibición de COX-2. La
aspirina bloquea la ciclooxigenasa mediante un mecanismo completamente distinto al del
ibuprofeno. Así, después de la unión de la aspirina a la ciclooxigenasa se produce la
transferencia del resto acilo de aquélla a un grupo hidroxilo de un residuo de serina (serina
530 de COX-1 o serina 516 de COX-2). Esta acetilación genera una forma catalíticamente
inactiva de la COX-1, mientras que la COX-2 es incapaz de convertir el ácido araquidónico
en PGH2. En su lugar se forma el ácido 15R-hidroxieicosatetraenoico (15R-HETE).
Figura 4.26. Interacción COX-1/aspirina mostrando l a Ser-530 acetilada
En la figura 4.26 se indica la estructura de una molécula de COX-1 inactivada por la
aspirina. La aspirina, la molécula gris más pequeña, acetila la serina de la posición 530 en
cada uno de los monómeros de la COX-1. En la figura 4.26 se aprecia también el cofactor
hemo con un átomo de hierro (la molécula gris con el hierro de color marrón).

Síntesis de antiinflamatorios
24
En la figura 4.27 se representa esquemáticamente la acción antiinflamatoria de la
aspirina mediante acetilación del residuo de serina-530 del centro activo de COX-1.
Figura 4.27. Inactivación del centro activo de COX por acetilación con aspirina
4.4.2.2. Ibuprofeno y naproxeno
El ibuprofeno y el naproxeno ejercen su acción antiinflamatoria ocupando el centro
activo de la ciclooxigenasa e impidiendo el acceso al mismo del ácido araquidónico.
Figura 4.20. Estructuras del ibuprofeno y naproxeno
En la figura 4.28 se indica la interacción de ibuprofeno con COX-1.
Figura 4.28. Interacción COX-1/ibuprofeno

Tema 4. Inflamación
25
Estudios sobre COX-2 mutada han demostrado que los grupos naftilo del naproxeno
(ácido (S)-6-metoxi-2-metil-2-naftalenacético) son esenciales para la inhibición de la enzima.
La mutación de Trp-387 por Phe reduce significativamente la inhibición por el naproxeno. El
cambio del átomo de oxígeno del naproxeno por un átomo de azufre incrementa la
selectividad del fármaco hacia COX-2.
Figura 4.29. Estructuras del naproxeno y tionaproxe no
En la figura 4.30 se indica la interacción del naproxeno con COX-2:
Figura 4.30. Interacción COX-2/naproxeno
4.4.2.3. Indometacina y flurbiprofeno
La indometacina y el flurbiprofeno provocan la inhibición lenta de COX-1 y COX-2
mediante formación de un puente salino entre el grupo carboxilato del fármaco y el residuo
Arg-120, que se encuentra en el túnel del acceso al centro activo.
Figura 4.31

Síntesis de antiinflamatorios
26
En la figura 4.32 se detalla la interacción de la indometacina (estructura en color verde y
amarillo) con el centro activo de la COX.
Figura 4.32. Indometacina en el centro activo de CO X
En la figura 4.33 se indica la interacción del flurbiprofeno (en amarillo) con el centro
activo de COX-1, con indicación en modelo space-filling de los residuos Ile-434 (color
cobre), His-513 (color verde), Phe-518 (color cobre) e Ile-523 (color cobre).
Figura 4.33. Interacción del flurbiprofeno con COX- 1

Tema 4. Inflamación
27
4.4.2.4. Coxibes: inhibidores selectivos de COX-2
En la figura 4.34 se muestran las estructuras de algunos inhibidores selectivos de COX-
2. El rofecoxib, comercializado como Vioxx por la compañía Merck & Co, ha sido uno de los
fármacos más recetados para combatir la osteoartritis, el dolor agudo y la dismenorrea.
Figura 4.34. Estructuras de inhibidores selectivos de COX-2
En el año 2004 la compañía Merck retiró del mercado el rofecoxib, ante la posible
relación entre el aumento de infartos y derrames cerebrales y el consumo de este fármaco
en pacientes que tomaban rofecoxib durante periodos prolongados de tiempo y en dosis
relativamente elevadas.
A priori la inhibición selectiva de COX-2 es beneficiosa porque COX-2 participa en la
biosíntesis de las prostaglandinas malas, responsables del dolor y la inflamación, mientras
que COX-1 interviene en la biosíntesis de prostaglandinas buenas, responsables de la
protección de la mucosa estomacal. De hecho los fármacos que inhiben la COX-1, como la
aspirina, pueden provocar úlceras estomacales.
Las principales diferencias entre las dos isoformas de COX son el intercambio de
isoleucina-434, isoleucina-523 y arginina-513 en COX-1 por valina-434, valina-523 e
histidina-513 en COX-2.
En la figura 4.25 se han indicado de forma esquemática las diferencias en los centros
activos de las enzimas COX debido, fundamentalmente, a la presencia de la isoleucina en la
posición 523 de COX-1 y de valina en la posición 523 de COX-2. El aminoácido valina es
más pequeño que la isoleucina y el vioxx puede entrar en el bolsillo de COX-2 ocupado por
la valina, pero no puede entrar en el bolsillo enzimático de COX-1 porque esta enzima
contiene isoleucina, cuyo mayor volumen impide la entrada del fármaco.
En la figura 4.35 se muestra en modelo space-filling la superposición del vioxx con los
centros activos de los enzimas COX-1 (en color magenta) y COX-2 (en color gris). En la
figura se puede apreciar la colisión entre el anillo fenílico del vioxx con la cadena lateral del
aminoácido isoleucina del centro activo de COX-1.

Síntesis de antiinflamatorios
28
Figura 4.35. Superposición de vioxx con los centros activos de COX-1 y COX-2
La cardiotoxicidad del rofecoxib (vioxx) radica en la supresión de la biosíntesis de
prostaciclinas, aunque también se ha propuesto que la cardiotoxicidad de este fármaco
puede estar asociada con los metabolitos producidos durante su ionización en condiciones
fisiológicas.

Tema 4. Inflamación
29
4.5. Síntesis de antiinflamatorios
4.5.1. Síntesis de ibuprofeno
El ibuprofeno es un antiinflamatorio no esteroideo (AINE), utilizado frecuentemente para
el alivio sintomático del dolor de cabeza (cefalea), dolor dental (odontalgia), dolor muscular
o mialgia, molestias de la menstruación (dismenorrea), dolor neurológico de carácter leve,
síndrome febril y dolor tras cirugía (postquirúrgicos). También se usa para tratar cuadros
inflamatorios, como los que se presentan en artritis, artritis reumatoide (AR) y artritis gotosa.
El ibuprofeno es un inhibidor no selectivo de COX-1 y COX-2. El efecto antiinflamatorio y
analgésico está relacionado con la inhibición de COX-2 mientras que la inhibición de COX-1
bloquea la formación de tromboxanos. La inhibición prolongada de COX-1 puede causar
toxicidad gástrica ya que la actividad de COX-1 está relacionada con el mantenimiento de la
mucosa gástrica.
El ibuprofeno fue desarrollado por la división de investigación de los laboratorios Boots y
fue patentado en 1961. Este medicamento forma parte del listado de la Organización
Mundial de la Salud de fármacos indispensables.
El ibuprofeno se administra como racemato. El diasteroisómero (-)-R es
enzimáticamente isomerizado al (+)-S, pudiendo considerarse como un profármaco de este
último. El mecanismo de isomerización implica una conversión inicial del enantiómero (-)-R
en el tioéster de la coenzima A (compuesto 4.1 del esquema 4.3). Este intermedio, mediante
tautomería ceto-enólica vía enol 4.2, probablemente mediada por un enzima, se epimeriza
al intermedio 4.3, que por hidrólisis enzimática se transforma en el (+)-S-ibuprofeno.
Esquema 4.3
El hecho de que el enantiómero S no parezca sufrir una epimerización similar puede
explicarse atendiendo a la estereoselectividad del enzima CoA-sintetasa que actúa
preferentemente sobre el enantiómero (-)-R.

Síntesis de antiinflamatorios
30
4.5.1.1a. Análisis retrosintético
En el esquema 4.4 se indica un análisis retrosintético del ibuprofeno que se inicia con la
interconversión de la función carboxilo en nitrilo. Esta operación genera el cianocompuesto
4.4 el cual, mediante una nueva operación IGF, se transforma en la oxima 4.5, que a su vez
deriva del aldehído 4.6. En este punto del análisis retrosintético se lleva a cabo la escisión
del grupo formilo. Esta operación genera el sintón aniónico no natural 4.7 y el sintón
catiónico 4.8. El equivalente sintético del sintón aniónico 4.7 se explicará en el parte de
síntesis, mientras que para el sintón catiónico 4.8 se empleará como equivalente sintético la
aril metil cetona 4.9.
COOH
Ibuprofeno
CN
4.4 4.5
4.74.8 4.6
NOH
O
H
O
H
+
O
4.9
AGFIGF
IGF
Esquema 4.4
4.5.1.1b. Síntesis
En el esquema 4.5 se describe la síntesis del ibuprofeno según el análisis retrosintético
indicado en el esquema anterior. Esta síntesis es la que patentó los laboratorios Boots en
1961.
Esquema 4.5
La secuencia sintética se inicia con obtención de la aril metil cetona 4.9 por reacción de
acilación SEAr del isobutilbenceno 4.10 con anhídrido acético en presencia de AlCl3. La
cetona 4.9 se convierte en el α,β-epoxiéster 4.11 mediante reacción de Darzens con
cloroacetato de etilo en presencia de etóxido sódico. Cuando el epoxiéster 4.11 se calienta

Tema 4. Inflamación
31
en medio ácido se provoca una reacción de hidrólisis, con descarboxilación concomitante,
que conduce a la obtención del aldehído 4.6. Este compuesto se convierte en la oxima 4.5,
la cual se deshidrata al nitrilo 4.6. La hidrólisis del nitrilo proporciona el ibuprofeno.
4.5.1.1c. Cuestiones
1) Proponga un mecanismo la formación del α,β-epoxiéster 4.11 mediante reacción de
Darzens.
2) El cloroacetato de etilo actúa en la síntesis del ibuprofeno como equivalente sintético del
anión de formilo. Después de la formación del α,β-epoxiéster 4.11, por reacción de la cetona
con el cloroaceato de etilo, se obtiene el aldehído 4.6 mediante hidrólisis y descarboxilación
del epoxiéster. Explique mecanísticamente la conversión del α,β-epoxiéster 4.11 en el
aldehído 4.6.
3) Un método que permite la conversión de oximas en nitrilos se indica a continuación:5
Proponga un mecanismo para la reacción anterior.
4.5.1.2a. Análisis retrosintético mediante carbonil ación
En el esquema 4.6 se describe un segundo análisis retrosintético para el ibuprofeno. La
primera operación retrosintética desconecta el grupo carbonilo del fármaco y conduce al
alcohol bencílico 4.12. El aumento del estado de oxidación de este compuesto proporciona
la cetona 4.9, cuya síntesis ya se ha descrito en el esquema 4.4.
Esquema 4.6
4.5.1.2b. Síntesis de ibuprofeno mediante carbonila ción
La síntesis del ibuprofeno, según el análisis retrosintético del esquema anterior, se
describe en el esquema 4.7 y es la que desarrolló la empresa Hoechst para la producción
del fármaco. La secuencia sintética comienza con la preparación de la aril metil cetona 4.9,
que en este caso se lleva a cabo mediante reacción SEAr del isobutilbenceno con anhídrido
acético en presencia de cantidades catalíticas de HF. La reducción del carbonilo cetónico,
por hidrogenación molecular de la cetona 4.9 en presencia del catalizador Ni-Raney,
conduce al alcohol bencílico 4.12 que se convierte en ibuprofeno mediante reacción de
carbonilación catalizada por paladio en presencia de HI como promotor.
5 E-C. Wang, K-S. Huang, H-M. Chen, C-C. Wu, G-J. Lin. J. Chin. Chem. Soc. 2004, 51, 619-627.

Síntesis de antiinflamatorios
32
Esquema 4.7
4.5.1.2c. Cuestiones
1) La síntesis del ibuprofeno de Hoechst es superior a la de Boots porque únicamente
requiere de tres etapas, contra seis que necesita la de Boots, y porque todas las reacciones
de la síntesis de Hoechst son catalíticas.
Explique por qué la reacción de acilación del isobutilbenceno 4.10 con anhídrido acético
necesita cantidades estequiométricas de AlCl3 (síntesis de Boots, esquema 4.8), mientras
que la acilación en presencia de HF (síntesis de Hoechst, esquema 4.8) es catalítica en el
ácido protónico.
Esquema 4.8
2) La última etapa en la síntesis del ibuprofeno de Hoechst es la reacción de carbonilación
catalizada por paladio del alcohol bencílico 4.12 (véase el esquema 4.6). No se disponen de
datos más precisos sobre esta reacción puesto que la síntesis del ibuprofeno de Hoechst ha
sido patentada y nunca ha sido publicada. No obstante, en la literatura científica se pueden
encontrar métodos de carbonilación de alcoholes bencílicos, como el que han descrito Lin y
Yamamoto,6 que consiguen la transformación directa de esta clase de alcoholes en los
ácidos carboxílicos homólogos mediante reacción de carbonilación en medio acuoso en
presencia de HI y de Pd(PPh3)4.
Proponga un mecanismo que explique la reacción anterior.
6 Y-S Lin, A. Yamamoto. Bull. Chem. Soc. Jpn. 1988, 71, 723-734.

Tema 4. Inflamación
33
4.5.1.3a. Análisis retrosintético de ibuprofeno med iante cianohidrina
En el esquema 4.9 se indica un análisis retrosintético para el ibuprofeno que se inicia
con el intercambio del grupo de ácido carboxílico por el grupo nitrilo. Este proceso genera el
nitrilo 4.4 que por adición del grupo funcional hidroxilo se transforma en cianohidrina 4.13.
La cianohidrina derivará de la aril metil cetona 4.9 que se desconecta, mediante una
operación basada en una reacción SEAr, al cloruro de acetilo 4.14 y al isobutilbenceno 4.10.
COOH IGF
Ibuprofeno
CN AGF CN
OH
IGF
OO
SEArCl+
4.4 4.13
4.144.10 4.9
Esquema 4.9
4.5.1.3b. Síntesis de ibuprofeno mediante cianohidr ina
La síntesis del ibuprofeno se inicia con la reacción de acilación Friedel-Crafts del
isobutilbenceno 4.10 con el cloruro de acetilo 4.14 (esquema 4.10). Esta reacción
proporciona la aril metil cetona 4.9 que se convierte en la cianohidrina 4.13 mediante
reacción con NaCN. Finalmente, la hidrólisis del grupo nitrilo y la hidrogenolisis
concomitante del grupo hidroxilo, por reacción de 4.13 con HI acuoso y fósforo, conduce al
ibuprofeno.
Esquema 4.10
Las reacciones que explican la transformación del compuesto 4.13 en ibuprofeno se
indican en el esquema 4.11. Muy probablemente, la cianohidrina 4.13, por hidrólisis ácida
del grupo nitrilo y reacción de sustitución nucleofílica del hidroxilo bencílico, se transforma
en el yodoácido 4.15 el cual, ya sea mediante hidrogenolisis directa del enlace C-I, o por
hidrogenación del metilenoácido 4.16, se convierte en ibuprofeno.

Síntesis de antiinflamatorios
34
Esquema 4.11
En el esquema 4.11 se indica la generación de hidrógeno molecular por reacción entre
HI y el fósforo. En primer lugar (reacción 1), el ácido yodhídrico se disocia para formar yodo
e hidrógeno molecular. Esta reacción se hace irreversible porque el yodo molecular
reacciona con el fósforo en medio acuoso para generar el ácido hipofosforoso. La suma de
las dos reacciones proporciona la reacción 3, en la cual la oxidación del fósforo por reacción
con agua forma ácido hipofosforoso e hidrógeno molecular:
Esquema 4.12
La ecuación global ajustada para la transformación de la cianohidrina 4.13 en
ibuprofeno, por reacción con HI y fósforo es la siguiente:
COOH
Ibuprofeno
CN
OH
4.13
+ HI + 2 P + 5 H2O + 2 H3PO2 + NH4I
Esquema 4.13
4.5.2. Síntesis de flurbiprofeno
El flurbiprofeno se administra como racemato y se emplea en el tratamiento del dolor y
de la artritis. Muy a menudo es uno de los componentes de las pastillas para la tos, como
las tabletas Strepsils. La actividad antiinflamatoria del flurbiprofeno se debe al enantiómero
S. El enantiómero R carece de actividad antiinflamatoria y se ha demostrado que no inhibe
ninguna de las dos COX. El enantiómero R se bioconvierte en el S, aunque de manera muy
poco eficiente ya que sólo el 1.5% del R se biotransforma en S.
Aunque el enantiómero R no inhibe las COX sin embargo ha demostrado ser un potente
reductor de los niveles de β-amiloide, el principal constituyente de las placas amiloides que
se observan en los enfermos de Alzheimer. El enantiómero R, denominado tarenflurbil,
también está en siendo estudiado en fase clínica como fármaco para el tratamiento del
cáncer metastático de próstata.

Tema 4. Inflamación
35
4.5.2.a. Análisis retrosintético
El analisis retrosintético del flurbiprofeno se inicia con la desconexión del sistema
bifenílico (esquema 4.14). Esta operación está basada en una reacción de acoplamiento
arilo-arilo catalizada por paladio y genera el fragmento nucleofílico 4.17 (Y=metal o
metaloide) y el fragmento electrofílico 4.18 (X=halógeno). Una operación de intercambio de
grupo funcional convierte el dihaloácido 4.18 en el nitroácido 4.19. La última operación del
análisis retrosintético desconecta la parte de acido propiónico del anillo aromático. Esta
desconexión se basa en una reacción SNAr y genera el sintón nucleofílico 4.20 y el sintón
electrofílico 4.21 (X=halógeno).
Esquema 4.14
4.5.2.b. Síntesis
Para la síntesis del flurbiprofeno se elige como material de partida el 2,4-
difluoronitrobenceno 4.21 (esquema 4.15).7 La reacción SNAr entre este compuesto y el
anión derivado del metilmalonato de dietilo 4.22, que se emplea como equivalente sintético
del sintón aniónico 4.20, proporciona el diéster 4.23. La hidrólisis ácida del diéster, seguida
de descarboxilación in situ, conduce al nitroácido 4.19, que se transforma en el anilinoácido
4.24 mediante hidrogenación. El dihaloácido 4.18, necesario para la proyectada reacción de
acoplamiento bifenílico, se sintetiza a partir del anilinoácido 4.23 mediante reacción de
Sandmeyer vía la correspondiente sal de arildiazonio. El flurbiprofeno se obtiene mediante
reacción de acoplamiento de tipo Suzuki entre el dihaloácido 4.18 y el tetrafenoilborato
sódico (NaBPh4) en presencia de paladio depositado sobre carbono.8
7 G. Lu, R. Franzen, X. J. Yu, Y. J. Xu. Chin. Chem. Lett. 2006, 17, 461-464. 8 G. Lu, R. Franzen, Q. Zhang, Y. Xu. Tetrahedron Lett. 2005, 46, 4255-4259.

Síntesis de antiinflamatorios
36
COOH
F
O2N4.18
COOEt
FO2N
F
+
4.21
EtOOC NaOH, DMF
23ºC
COOEt
FO2N
COOEt
HOAc, H2SO4, H2O, reflujo,
(87% 2 pasos)
H2, Pd/C
23ºC (98%)
COOH
F
H2N
NaNO2, 40% HBr
CuBr, H2O (83%)
COOH
FBr
NaBPh4, Na2CO3, H2O0.05 mol% de Pd/C al 5%,
reflujo al aire durante 1 h (98%)
COOH
F
Flurbiprofeno
4.22 4.23
4.24
4.18
Esquema 4.15
4.5.2.c. Cuestiones
1) ¿Por qué la reacción SNAr del anión del metilmalonato de dietilo 4.22 sobre el 2,4-
difluoronitrobenceno es regioselectiva? ¿Por qué no se sustituye el átomo de flúor en orto
con respecto al grupo nitro?
2) Explique mecanísticamente la conversión de la arilamina 4.24 en el bromoarilo 4.25.
3) La reacción ajustada para el acoplamiento de Suzuki entre el compuesto 4.18 y el
NaBPh4 es la siguiente:
Esquema 4.16
La reacción anterior se explica mediante la intervención de cuatro ciclos catalíticos. Con
estos datos proponga un mecanismo que explique la formación del flurbiprofeno mediante la
reacción de Suzuki.
4.5.3. Síntesis de naproxeno
El naproxeno se patentó en 1967 por los laboratorios Syntex (Patente GB 1211134) y su
uso médico se aprobó en 1972. Este fármaco antiinflamatorio no esteroideo (AINE) se
receta en el tratamiento del dolor leve a moderado, la fiebre, la inflamación y la rigidez
provocados por afecciones como la osteoartritis, la artritis reumatoide, la artritis psoriásica,
la espondilitis anquilosante, la tendinitis, la bursitis, y en el tratamiento de la dismenorrea

Tema 4. Inflamación
37
primaria y los calambres menstruales. El naproxeno también está disponible como sal
sódica, que se absorbe más rápidamente que el naproxeno en el tracto gastrointestinal.
4.5.3.1a. Análisis retrosintético
La retrosíntesis del naproxeno se inicia con la interconversión del grupo funcional
carboxilo en éster (esquema 4.17). Esta operación genera el naproxenato de alquilo 4.25
que por escisión del grupo metilo, basada en una reacción SN2, conduce al compuesto 4.26.
El siguiente paso retrosintético convierte el éster 4.26 en la naftilmetil cetona 4.27. En el
esquema 4.16 a esta operación se ha indicado como migración 1,2 de grupo carbonilo. La
última desconexión escinde el grupo acetilo y conduce al cloruro de acetilo 4.14 y al 2-
metoxinaftaleno 4.28.
Esquema 4.17
4.5.3.1b. Síntesis
La primera síntesis a gran escala del naproxeno se llevó a cabo por la empresa Syntex
en 1970 y permitía la producción de 500 kg de este fármaco.9 La síntesis del naproxeno se
inicia con la obtención de metoxinaftil metil cetona 4.24 por reacción SEAr entre el 2-
metoxinaftaleno 4.28 y el cloruro de acetilo (esquema 4.18). El paso clave de la síntesis es
la migración 1,2 del grupo carbonilo cetónico, con aumento concomitante de su estado de
oxidación. Esta conversión se consigue mediante reacción de la cetona 4.24 con morfolina y
azufre a reflujo. El proceso proporciona la tiomorfolida 4.30 que por hidrólisis ácida se
convierte en el ácido carboxílico 4.31. La esterificación de Fischer del ácido 4.31, seguida
de enolización con NaH y alquilación con yoduro de metilo, proporciona el naproxenato de
metilo racémico (+/-)-4.25. La saponificación de la función éster permite la obtención del
naproxeno racémico (+/-)-4.32. La resolución óptica del racemato (+/-)-4.32 se lleva a cabo
con el alcaloide (-)-cinconidina 4.33. Así, la reacción de (+/-)-4.32 con el alcaloide genera las
correspondientes sales diastereoisoméricas, de las cuales la de estructura 4.34 cristaliza en
el seno de la reacción. La separación de esta sal mediante filtración, seguida de tratamiento
ácido, proporciona el (S)-naproxeno.
9 I. T. Harrison, B. Lewis, Peter, P. Nelson, W. Rokks, A. Roszkowski, A. Tomolonis, J. H. Fried. J. Med. Chem. 1970, 13, 203-205.

Síntesis de antiinflamatorios
38
Esquema 4.18
La transposición 1,2 de carbonilo en la cetona 4.27, por reacción con morfolina y azufre,
se conoce como reacción de Willgerodt-Kindler. El mecanismo de la primera parte de este
proceso, que es la formación de una tioamida, se indica en el esquema 4.19. El proceso
comienza con la formación de la enamina 4.36 por reacción de la morfolina con la aril metil
cetona 4.27. A continuación, la enamina nucleofílica ataca al azufre y genera la betaína 4.37
que se transforma en el α-morfolino-tioaldehído 4.38 por migración intramolecular de
hidruro. El subsiguiente ataque nucleofílico intramolecular del nitrógeno al doble enlace C=S
forma el aziridinio 4.39 el cual, por pérdida de protón, se transforma en la tioenamina 4.40.
Este compuesto se convierte en el tioamida 4.30 mediante tautomería tioenólica.

Tema 4. Inflamación
39
Esquema 4.19
4.5.3.2a. Análisis retrosintético de naproxeno medi ante acoplamiento organometálico
En el esquema 4.20 se indica un segundo análisis retrosintético para el naproxeno. Así,
la desconexión de la parte de propionato sobre el éster 4.25 conduce al fragmento
nucleofílico 4.41 (Y=metal) y al fragmento electrofílico 4.42 (X=halógeno). El compuesto
organometálico 4.41 derivará del compuesto halogenado 4.43 (X=halógeno) que se
obtendrá del 2-naftol 4.45 mediante halogenación SEAr seguida de metilación fenólica.
MeOO
OH
MeOO
ORIGF C-C
MeO
O
OR
MeO
X
HO
Naproxeno 4.25 4.41
4.434.45
Y
X
+
HO
X IGFC-X
IGF
SEAr
4.42
4.44 Esquema 4.20
4.5.3.2b. Síntesis de naproxeno mediante acoplamien to organometálico
Los inconvenientes de la síntesis industrial indicada en el esquema 4.18 son varios. El
primero de ellos es que la reacción de acilación Friedel-Crafts no es regioselectiva y
produce también el regioisómero de acilación en la posición 1. El segundo es la formación

Síntesis de antiinflamatorios
40
de cantidades estequiométricas de hidróxido de aluminio. El tercero es el empleo de
disolventes poco apropiados a escala industrial como el nitrobenceno, que se utiliza como
disolvente en la reacción de acilación. El cuarto es la utilización de reactivos peligrosos en
grandes cantidades, como el yoduro de metilo y el hidruro sódico.
A fin de evitar los inconvenientes asociados a la síntesis del naproxeno, descrita en el
esquema 4.18, durante los años 1972-1975, la empresa Syntex aplicó la secuencia sintética
que se describe en el esquema 4.21.10 En esta síntesis el compuesto de partida es el �-
naftol 4.45, que por reacción con bromo molecular proporciona el dibromonaftol 4.46. La
reacción con hidrogenosulfito sódico provoca la eliminación reductiva regioselectiva del
bromo en C-1 y proporciona el bromonaftol 4.44, que se convierte en el 2-bromo-6-
metoxinaftaleno 4.34 por metilación con cloruro de metilo en medio básico. Este compuesto
se transforma en el reactivo de Grignard 4.41, que por transmetalación con ZnCl2 y reacción
con 2-bromopropanoato de etilo proporciona el éster racémico (+/-)-4.47. La hidrólisis del
éster seguida de resolución con cinconidina permiten la obtención del (S)-naproxeno.
HO HO
BrBr2
HO
Br 4.46
NaHSO3
O
OEt
MeO
Br
4.41 4.43
4.45
Br
4.42
4.44
CH3Cl, NaOH(85-90% desde 4.45)
Mg
MeO
MgBr
ZnCl2
MeOO
OEt
(+/-)-4.47
NaOH, H2O
MeOO
OH
(+/-)-4.32
1. Resolución con (-)-cinconidina2. HCl ac.
MeOO
OH
Naproxeno
Br
Esquema 4.21
La aplicación industrial de la secuencia sintética del esquema 4.21 tampoco está exenta
de inconvenientes. El primero de ellos es el empleo de cantidades estequiométricas de
ZnCl2, que se requieren para generar el reactivo organometálico de tipo naftilzinc (no
dibujado en el esquema 4.21). La utilización del ZnCl2 genera grandes cantidades de
hidróxido de zinc como subproducto. El segundo inconveniente está relacionado con el
acoplamiento del reactivo organometálico con el 2-bromopropanoato de etilo, que transcurre
10 P. J. Harrington, E. Lodewijk. Org. Process Res. Dev. 1997, 1, 72-76.

Tema 4. Inflamación
41
con bajos rendimientos. Además, en esta reacción se forma también el 2-metoxinaftaleno,
como subproducto de reducción, y el dímero binaftílico, como consecuencia de una vía
secundaria que procede mediante acoplamiento radicalario del reactivo organometálico.
En los años 1976-1993 la producción de naproxeno por Syntex se llevó a cabo mediante
la secuencia de reacciones que se indica en el esquema 4.22. La principal novedad de esta
secuencia, en relación con la descrita en el esquema 4.21, estriba en el acoplamiento con el
reactivo organometálico, que se lleva a cabo sobre la sal cloromagnésica 4.48 derivada del
ácido 2-bromopropanoico.
Esquema 4.22
En la síntesis del esquema anterior no necesita ZnCl2 para la reacción de acoplamiento,
y por tanto no generan residuos de zinc, y además el subproducto 2-metoxinaftaleno y el
dímero binaftílico se forman en mucha menor proporción.
Otra mejora asociada al esquema anterior está relacionada con el paso de resolución
óptica del racemato (+/-)-4.32. En esta secuencia la cinconidina se sustituye por una amina
quiral preparada mediante aminación reductiva de la glucosa.
Otra secuencia sintética que desarrolló la empresa Syntex para la fabricación de
naproxeno se detalla en el esquema 4.23. En esta secuencia se recurre a la utilización de
(S)-lactato de etilo 4.49, compuesto quiral que es accesible en grandes cantidades a precios
relativamente moderados. Así, el lactato de etilo 4.49, mediante mesilación, saponificación y
cloración, se convierte en el cloruro de ácido 4.50. El acoplamiento de este compuesto con
el reactivo de Grignard 4.41 proporciona la naftilcetona quiral 4.51. Cuando este compuesto
se trata con el 2,2-dimetilpropan-1,2-diol se obtiene el acetal 4.52 que experimenta una
reacción de hidrólisis estereoespefíca, por calentamiento en presencia de la resina IRC-50S,
y se convierte en el éster quiral 4.53. La hidrólisis ácida del éster conduce al naproxeno.

Síntesis de antiinflamatorios
42
Esquema 4.23
4.5.3.2c. Cuestiones
1) Proponga un mecanismo que explique la transformación del acetal 4.52 en el éster 4.53.
4.5.4. Síntesis de indoprofeno
El indoprofeno es un fármaco AINE que se recetaba para el tratamiento del dolor
asociado a enfermedades oncológicas, osteoartritis y artritis reumatoide. En los años 80 del
siglo pasado fue retirado del mercado por reportes de graves reacciones gastrointestinales y
de casos de cáncer en ratas de laboratorio. Sin embargo, un estudio publicado en el año
2004 ha demostrado que el indoprofeno incrementa la producción de una proteína clave
para la supervivencia de las células nerviosas afectadas por la atrofia muscular espinal, una
enfermedad mortal en niños.11
4.5.4.a. Análisis retrosintético
La retrosíntesis del indoprofeno se inicia con la desconexión del sistema de
isoindolinona que se obtendrá mediante la cicloadición entre el ftalaldehído 4.54 y el 2-(4-
aminofenil)propanoato de alquilo 4.55 (esquema 4.24). Este compuesto derivará del 2-(4-
nitrofenil)propanoato de alquilo 4.55 que se obtendrá mediante la reacción entre el
nitrobenceno 4.57 y un equivalente sintético del sintón aniónico 4.58.
11 M. R. Lunn, D. E. Root, A. M. Martino, S. P. Flaherty, B. P. Kelley, D. D. Coovert, A. H. Burghes, N. T. Man, G. E. Morris, J. Zhou, E. J. Androphy, C. J. Sumner, B. R. Stockwell. Chem Biol. 2004, 11, 1489-1493.

Tema 4. Inflamación
43
Esquema 4.24
4.5.4.b. Síntesis
La síntesis del indoprofeno que se indica en el esquema 4.25 está basada en la
preparación de este compuesto publicada por el grupo de N. J. Lawrence.12 La síntesis se
inicia con la adición del anión derivado del 2-cloropropionato de etilo al nitrobenceno
(esquema 4.25). Después de la hidrólisis ácida de la mezcla de reacción se obtiene el 2-(4-
nitrofenil)propanoato de etilo 4.56. La reducción del grupo nitro proporciona el 2-(4-
aminoenil)propanoato de etilo 4.55. Por último, la reacción entre 4.55 y el ftalaldehído 4.54,
en presencia de 2-mercaptoetanol y de 1,2,3-H-benzotriazol en acetonitrilo,13 proporciona el
etil éster del indoprofeno 4.60 que por saponificación conduce al indoprofeno.
Esquema 4.25
4.5.4.c. Cuestiones
1) Proponga un mecanismo que explique la formación del compuesto 4.56.
12 N. J. Lawrence, J. Liddle, S. M. Bushell, D. A. Jackson. J. Org. Chem. 2002, 67, 457-464. 13 I. Takahashi, T. Kawakami, M. Kimino, E. Hiano, S. Kamimura, T. Tamura, H. Kitajima, M. Hatanaka, H. Uchida, A. Nomura, M. Tanaka. Heterocycles 2001, 54, 635-638. Para mecanismos relacionados con esta reacción véase: P. Zuman. Chem. Rev. 2004, 104, 3217.3238.

Síntesis de antiinflamatorios
44
4.5.5. Síntesis de indometacina
La indometacina es un derivado del indol que se emplea como antiinflamatorio no
esteroideo (AINE). Este fármaco se prescribe para el alivio del dolor, la fiebre y la
inflamación en pacientes con osteoartritis, artritis reumatoide, dolor muscular,
espondiloartropatías, osteítis deformante, dismenorrea, bursitis, tendinitis, dolor de cabeza,
neuralgia y, por sus efectos antipiréticos, para el alivio de la fiebre en pacientes con cáncer
maligno.
La capacidad inhibitoria de la indometacina es más acusada en COX-1 que en COX-2.
La mayor inhibición de COX-1 explica los efectos secundarios de este fármaco, como las
gastritis y la nefritis.
4.5.5.a. Análisis retrosintético
La retrosíntesis de la indometacina se inicia con la desconexión del grupo p-
clorobenzoilo y el intercambio de la función de ácido carboxílico por éster (esquema 4.26).
En esta doble operación se genera el indol 4.61 y el cloruro de p-clorobenzoilo 4.62. La
siguiente operación retrosintética se lleva a cabo sobre el núcleo de indol y se ha
denominado con el acrónimo SIF, de Síntesis del Indol de Fischer. Esta metodología
general remite a la fenilhidrazona 4.63 como precursora del indol 4.61. La fenilhidrazona se
obtendrá mediante condensación entre el levulinato 4.64 y la fenilhidracina 4.65.
Esquema 4.26
4.5.5.b. Síntesis
La síntesis de la indometacina se inicia con la preparación de la p-metoxifenilhidrazona
4.63, lo que se consigue por condensación de la p-metoxifenilhidracina 4.65 con el
levulinato de metilo 4.64 (esquema 4.27). El tratamiento de la hidrazona 4.63 con ácido
clorhídrico en etanol proporciona el indol 4.61. Este compuesto se convierte en el t-butiléster
4.66 mediante saponificación y esterificación con diciclohexilcarbodimida (DCC) y t-butanol.
La indometacina se obtiene mediante N-acilación del nitrógeno indólico con cloruro de p-
clorobenzoilo, lo que proporciona el indoléster 4.68, seguida de termólisis del grupo t-
butiléster en este último compuesto.

Tema 4. Inflamación
45
Esquema 4.27
4.5.5.c. Cuestiones
1) Explique mecanísticamente la formación del indol 4.61.
2) ¿Por qué se cambia el éster de metilo en 4.61 por éster de t-butilo? ¿Qué podría ocurrir si
no se efectuase este cambio?
3) Explique mecanísticamente la formación del t-butiléster por reacción de 4.49 con DCC,
tBuOH y ZnCl2.
4) La conversión del grupo t-butilo en el t-butiléster 4.68 se lleva a cabo sin utilización de
ningún ácido, simplemente por calentamiento a 210ºC. Explique mecanísticamente la
conversión del compuesto 4.68 en indometacina.
En el esquema 4.28 se describe una síntesis mejorada de indometacina que evita las
etapas de saponificación-esterificación-eliminación del éster. Así, la p-metoxifenilhidracina
4.65 se convierte en la arilhidrazona 4.69 por reacción con acetaldehído. La N-acilación de
4.69 con cloruro de p-clorobenzoilo proporciona el compuesto 4.70 que se hidroliza a la N-
acilhidracina 4.71. La condensación de 4.71 con ácido levulínico genera la hidrazona 4.72.
La reacción de condensación del indol de Fischer sobre la hidrazona 4.72. proporciona la
indometacina.
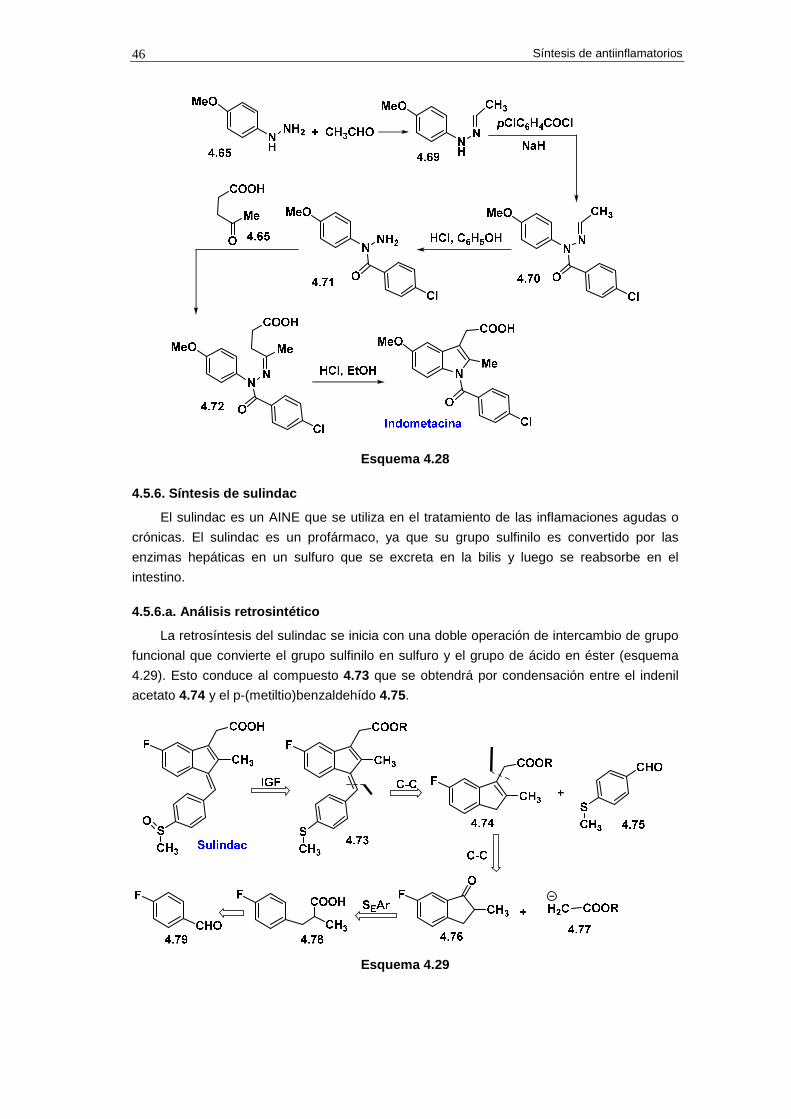
Síntesis de antiinflamatorios
46
Esquema 4.28 4.5.6. Síntesis de sulindac
El sulindac es un AINE que se utiliza en el tratamiento de las inflamaciones agudas o
crónicas. El sulindac es un profármaco, ya que su grupo sulfinilo es convertido por las
enzimas hepáticas en un sulfuro que se excreta en la bilis y luego se reabsorbe en el
intestino.
4.5.6.a. Análisis retrosintético
La retrosíntesis del sulindac se inicia con una doble operación de intercambio de grupo
funcional que convierte el grupo sulfinilo en sulfuro y el grupo de ácido en éster (esquema
4.29). Esto conduce al compuesto 4.73 que se obtendrá por condensación entre el indenil
acetato 4.74 y el p-(metiltio)benzaldehído 4.75.
Esquema 4.29

Tema 4. Inflamación
47
El compuesto 4.74 se sintetizará por condensación entre la indenona 4.76 y el anión
4.77, la base conjugada de un acetato de alquilo. La indenona 4.76 se obtendrá mediante
reacción SEAr intramolecular en el ácido 4.78 el cual derivará del p-fluorobenzaldehído 4.79.
4.5.6.b. Síntesis
La síntesis del sulindac se inicia con la condensación de tipo Claisen-Schmidt entre el
p-fluorobenzaldehído 4.79 y el anhídrido propiónico 4.80 (esquema 4.30).14 Esta reacción
proporciona el ácido conjugado 4.81 que por hidrogenación se convierte en el ácido 4.78. El
tratamiento de este compuesto con ácido polifosfórico (PPA) provoca la reacción SEAr
intramolecular con la consiguiente formación de la indenona 4.76. Este compuesto se hace
reaccionar con el enolato lítico derivado del acetato de etilo. El producto resultante de la
reacción proporciona el indenil acetato 4.74 mediante deshidratación con H2SO4 en AcOH.
El compuesto 4.74 se somete a una reacción de condensación de tipo Claisen-Schmidt
viníloga mediante enolización con metóxido de sodio en metanol seguida de reacción con el
p-(metiltio)benzaldehído 4.75.15 Este proceso conduce al compuesto 4.73, el cual por
hidrólisis ácida y oxidación con NaIO4 se convierte en el sulindac.
Esquema 4.30
4.5.6.c. Cuestiones
1) Proponga un mecanismo para la reacción del esquema 4.31.
Esquema 4.31
14 Z-G. Wang, L.Chen, J. Chen, J-F. Zheng, W. Gao, Z. Zeng, H. Zhou, X-k. Zhang, P-Q, Huang, Y. Su. Eur. J. Med. Chem. 2013, 62, 632-648. 15 Patente US2015/266842

Síntesis de antiinflamatorios
48
4.5.7. Síntesis de etodolaco
El etodolaco se receta para el tratamiento del dolor y la inflamación provocada por la
osteoartritis y la artritis reumatoide. Es unas 10 veces más selectivo por la COX-2 que por la
COX-1.
4.5.7.a. Análisis retrosintético
La retrosíntesis del etodolaco se inicia con la desconexión del centro cuaternario en el
anillo dihidropiránico. Esta operación conduce al indol 4.82 y al 3-oxopentanoato de alquilo
4.83 (esquema 4.32). El indol se obtendrá mediante síntesis del indol del Fischer entre la (2-
etilfenil)hidracina 4.84 y el 4-hidroxibutanal 4.85.
Esquema 4.32
4.5.7.b. Síntesis
La (2-etilfenil)hidracina 4.84 necesaria para la síntesis del etodolaco se obtiene a partir
de la 2-etilanilina 4.86 por conversión en la correspondiente sal de 2-etildiazonio seguida de
reducción con SnCl2 (esquema 4.33).16 La reacción de síntesis del indol de Fischer entre la
hidracina 4.84 y el 4-hidroxibutanal 4.85 proporciona el indol 4.82.17 Cuando este compuesto
se hace reaccionar con 3-oxopentanoato de etilo en presencia de ácido p-toluensulfónico se
obtiene el compuesto tricíclico 4.87. La saponificación de este compuesto proporciona el
etodolaco.
NH
OCOOEt
Etodolaco
NH
OH
COOEtO
CHO
OH
4.82
4.83
4.84
4.85NH2
NH
NH2
4.86
1) NaNO2, HCl2) SnCl2
HCl
TsOH
KOH
NH
OCOOH
4.87
Esquema 4.33
4.5.7.c. Cuestiones
1) Proponga un mecanismo para la formación del compuesto 4.87.
16 C. A. Demerson, L. G. Humber, T. Dobson, I. L. Jirkovsky. US Patent 1976/3.393.178. 17 Para una síntesis del indol 4.82 que emplea dihidrofurano como sustituto de 4-hidroxibutanal 4.85 véase: M. C. Sekharayya, G. J. Narayana, S. Nigam, G. Madhusudhan. Indian J. Chem. B. 2012, 51B, 1763-1766.

Tema 4. Inflamación
49
4.5.8. Síntesis de diclofenaco
El diclofenaco y sus sales como el diclofenaco sódico (nombre comercial Voltaren®) son
fármacos antiinflamatorios que posee actividades analgésicas y antipiréticas y están
indicados por vía oral e intramuscular para el tratamiento de enfermedades reumáticas
agudas, artritis reumatoidea, espondilitis anquilosante, artrosis, lumbalgia, gota en fase
aguda, inflamación postraumática y postoperatoria, cólico renal y biliar, migraña aguda, y
como profilaxis para dolor postoperatorio y dismenorrea.
El mecanismo exacto de acción del diclofenaco no está totalmente aclarado, pero se
cree que su acción antiinflamatoria y analgésica se basa en el bloqueo de la síntesis de
prostaglandinas mediante la inhibición de las ciclooxigenasas. Parece ser que el diclofenaco
también inhibe las funciones de la lipooxigenasa, por lo que reduce la formación de
leucotrienos (sustancias inflamatorias), y la producción de la fosfolipasa A2. Estas acciones
adicionales explican su alta efectividad.
La preferencia del diclofenaco por COX-2 es unas 10 veces mayor que por COX-1, lo
que explica su relativamente baja incidencia de efectos negativos gastrointestinales, en
comparación con los mostrados por la indometacina y el ácido acetilsalicílico. No obstante,
el principal efecto secundario del diclofenaco es la formación de úlceras gástricas debido a
la inhibición de COX-1, lo que provoca una menor producción de prostaglandinas en el
epitelio del estómago, haciéndolo mucho más vulnerable a la corrosión por los ácidos
gástricos.
A pesar de que el consumo de inhibidores selectivos de COX-2, como los coxibes, ha
provocado paros cardiacos en algunos pacientes, el consumo de diclofenaco, que inhibe
mayoritariamente COX-2, no provoca ningún síntoma de afectación cardíaca.
4.5.8.a. Análisis retrosintético
La retrosíntesis del diclofenaco se inicia con una operación de reconexión (esquema
4.34). Así la reconexión de las funciones amida y ácido conduce a la lactama 4.88 que se
obtendrá mediante reacción SEAr intramolecular sobre la halogenoamida 4.89 (X=halógeno).
La escisión del enlace amida conduce a la 2,6-dicloro-N-fenilanilina 4.90 que se sintetizará
mediante acoplamiento de la 2,6-dicloroanilina 4.91 con el halogenobenceno 4.92
(X=halógeno).
Esquema 4.34

Síntesis de antiinflamatorios
50
4.5.8.b. Síntesis
La síntesis del diclofenaco se inicia con el acoplamiento de Ullman entre la N-(2,6-
diclorofenil)acetamida 4.93 y el bromobenceno 4.92 (esquema 4.35).18 Esta reacción
proporciona el compuesto 4.94 cuya saponificación conduce a la 2,6-dicloro-N-fenilanilina
4.90. La acilación de este compuesto con cloruro de 2-cloroacetilo permite la obtención de la
cloroacetamida 4.89 que se convierte en la lactama 4.88 mediante reacción con AlCl3 a
160ºC. La saponificación de este compuesto proporciona el diclofenaco.
Esquema 4.35
4.5.8.c. Cuestiones
1) En el esquema 4.36 se indica una secuencia sintética para la obtención de la N-(2,6-
diclorofenil)acetamida 4.93. El proceso se inicia con la obtención de la p-bromoanilina 4.95
por monobromación de la anilina con N-bromosuccinimida en polietilenglicol-400.19 La
cloración de 4.95 proporciona la 4-bromo-2,6-dicloroanilina 4.96.20 Cuando este compuesto
se trata con HBr en AcOH, en presencia de anilina para capturar el Br2 generado en la
reacción,20 se obtiene la 2,6-dicloroanilina 4.97. La acetilación de este compuesto conduce a
la N-(2,6-diclorofenil)acetamida 4.93.
NH2
Cl
Cl
NH2
4.93
NH
Cl
ClO
CH3
4.95Anilina
NBS
PEG-400NH2
Br
NH2
Cl
Cl
Br
4.96
4.97
Ac2O
NaClO3
HCl
HBr, AcOH
anilina
Esquema 4.36
18 P. Moser, A. Sallmann, I. Wiesenbergt. J. Med. Chem. 1990, 33, 2358-2368. 19 K. Venkateswarlu, K. Suneel, B. Das, K. N. Reddy, T. S. Reddy. Synth. Commun. 2009, 39, 215-219. 20 H. Y. Choi, D. Y. Chi. J. Am. Chem. Soc. 2001, 123, 9202-9203.
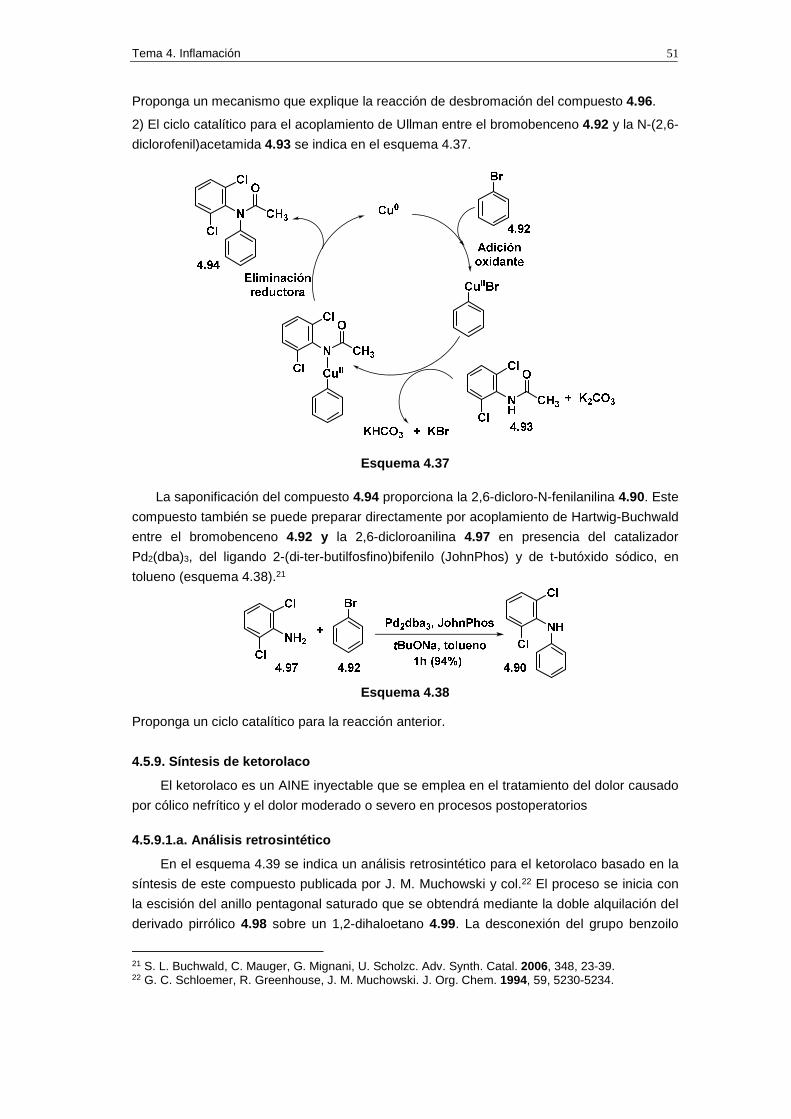
Tema 4. Inflamación
51
Proponga un mecanismo que explique la reacción de desbromación del compuesto 4.96.
2) El ciclo catalítico para el acoplamiento de Ullman entre el bromobenceno 4.92 y la N-(2,6-
diclorofenil)acetamida 4.93 se indica en el esquema 4.37.
Esquema 4.37
La saponificación del compuesto 4.94 proporciona la 2,6-dicloro-N-fenilanilina 4.90. Este
compuesto también se puede preparar directamente por acoplamiento de Hartwig-Buchwald
entre el bromobenceno 4.92 y la 2,6-dicloroanilina 4.97 en presencia del catalizador
Pd2(dba)3, del ligando 2-(di-ter-butilfosfino)bifenilo (JohnPhos) y de t-butóxido sódico, en
tolueno (esquema 4.38).21
Esquema 4.38
Proponga un ciclo catalítico para la reacción anterior.
4.5.9. Síntesis de ketorolaco
El ketorolaco es un AINE inyectable que se emplea en el tratamiento del dolor causado
por cólico nefrítico y el dolor moderado o severo en procesos postoperatorios
4.5.9.1.a. Análisis retrosintético
En el esquema 4.39 se indica un análisis retrosintético para el ketorolaco basado en la
síntesis de este compuesto publicada por J. M. Muchowski y col.22 El proceso se inicia con
la escisión del anillo pentagonal saturado que se obtendrá mediante la doble alquilación del
derivado pirrólico 4.98 sobre un 1,2-dihaloetano 4.99. La desconexión del grupo benzoilo
21 S. L. Buchwald, C. Mauger, G. Mignani, U. Scholzc. Adv. Synth. Catal. 2006, 348, 23-39. 22 G. C. Schloemer, R. Greenhouse, J. M. Muchowski. J. Org. Chem. 1994, 59, 5230-5234.

Síntesis de antiinflamatorios
52
conduce al pirrolil-acetato 4.100 que se obtendrá mediante la C-alquilación del pirrol con un
haloacetato 4.101.
Esquema 4.39
4.5.9.1.b. Síntesis
La preparación del ketorolaco se inicia con la C-alquilación del pirrol. Este proceso se
lleva a cabo del siguiente modo. En primer lugar se trata el pirrol con un exceso de MeMgCl
en THF. Esto genera el cloruro de pirrolilmagnesio 4.102 indicado en el esquema 4.40.
Después de agitación a temperatura ambiente, se adiciona el bromoacetato de isopropilo
4.101 a -10ºC, se deja subir la temperatura hasta temperatura ambiente y se agita durante
unas 2 h. El intermedio 4.103 experimenta el desplazamiento nucleofílico intramolecular del
bromuro obteniéndose, después del procesado de la reacción, el 2-pirrolilacetato de
isopropilo 4.100. La benzoilación de 4.100 se lleva a cabo mediante reacción de tipo
Vilsmeier-Hack con el reactivo generado al tratar la N,N-dimetilbenzamida con dicloruro de
oxalilo. Este proceso conduce al compuesto 4.98. Las reacciones de ciclación sobre
compuestos del tipo 4.98 con 1,2-dihaloetanos no funcionan bien, obteniéndose en muchos
casos el producto ciclopropánico. Para evitar esta reacción indeseable, el compuesto 4.98
se somete a un proceso de carbometoxilación por reacción con LDA (3 equivalentes)
seguido de adición de ClCOOMe. Esto permite la obtención del (5-benzoil-2-pirrolil)malonato
de metilo e isopropilo 4.104. Cuando este compuesto se trata a reflujo de 1,2-dicloroetano,
en presencia de K2CO3 y de bromuro de tetra-n-butilamonio, se obtiene el producto de
ciclación 4.105. La saponificación de este compuesto con NaOH 1 N en metanol seguida de
acidificación-descarboxilación con HCl proporciona el ketorolaco.
Esquema 4.40

Tema 4. Inflamación
53
4.5.9.1.c. Cuestiones
1) La benzoilación de 4.100 por reacción con N,N-dimetilbenzamida y dicloruro de oxalilo se
indica en el esquema 4.41.
Esquema 4.41
La reacción de benzoilación se lleva a cabo del siguiente modo. El dicloruro de oxalilo
se añade a una disolución de N,N-dimetilbenzamida en éter. Esto provoca la aparición de un
precipitado con la consiguiente evolución de un gas. Se elimina el disolvente y el exceso de
dicloruro de oxalilo y el residuo se disuelve en diclorometano. Luego se añade el 2-
pirrolilacetato de isopropilo 4.100 disuelto en diclorometano y se agita a temperatura
ambiente durante 40 horas. El procesado de la reacción proporciona el (5-benzoilpirrol-2-
il)acetato de isopropilo 4.98. Con estos datos proponga un mecanismo para la reacción
indicada en el esquema 4.41.
4.5.9.2. Síntesis enantioselectiva de ketorolaco
El grupo de P. S. Baran ha publicado una síntesis enantioselectiva de (S)-kerotolaco
que emplea como reacción clave un acoplamiento oxidante intramolecular del anillo pirrólico
sobre un enolato (acoplamiento Csp2-Csp2) y un auxiliar quiral para conseguir la
enantioselectividad.23 La secuencia sintética se dibuja en el esquema 4.42 y se inicia con la
obtención del ácido 4-(pirrol-1-il)butanoico 4.107 por ionización del pirrol con hidruro
potásico y reacción subsiguiente con la valerolactona 4.106.24 El ácido 4.107 se convierte
en un anhídrido mixto, por reacción con trietilamina y cloroformiato de metilo, el cual se trata
con la sultama quiral (sal lítica) 4.108 para dar lugar al compuesto 4.109. La reacción clave
de ciclación oxidante se ensaya sobre 4.109 con múltiples agentes oxidantes de CuII, FeIII,
AgI, AgII, TiIV, MnIII y CeIV. Al final la ciclación se consigue generando el enolato lítico
derivado de 4.109 con LiHMDS y Et3N y oxidando el correspondiente enolato con
hexafluorofosfato de ferroceno 4.110 (oxidante de FeIII). En el proceso de ciclación oxidante
se genera el compuesto 4.111 con una relación diastereoisomérica de 4.5:1. La
benzoilación de 4.111 conduce al compuesto 4.112 el cual se convierte en (S)-ketorolaco
mediante eliminación del auxiliar quiral con hidroperóxido de tetrabutilamonio (TBAH) en
1,2-dimetoxietano (DME).
23 P. S. Baran, J. M. Richter, D. W. Lin. Angew, Chem. Int. Ed. 2005, 44, 609-612. 24 J-H. Li, J. K. Snyder. J. Org. Chem. 1993, 58, 516-519.

Síntesis de antiinflamatorios
54
Esquema 4.42
4.5.10. Síntesis de zomepiraco
El zomepiraco es un AINE que se recetaba para el tratamiento del dolor suave o
severo. El fármaco es metabolizado por la UDP-glucuronil-transferasa a un glucurónido
reactivo que se une de manera irreversible a la albúmina del plasma provocando
anafilaxis.25 Por esta razón fue retirado del mercado USA en 1983.
4.5.10.a. Análisis retrosintético
El análisis retrosintético del zomepiraco se inicia con la desconexión del grupo p-
metoxibenzoilo (esquema 4.43). Esta operación conduce al imidazol 4.113 que por adición
del grupo funcional carboxialquilo se convierte en el compuesto 4.114. La desconexión del
anillo de imidazol, de la manera indicada en el esquema 4.43 origina la metilamina, una
haloacetona 4.115 y el 3-oxopentanodioato de dialquilo 4.116.
Esquema 4.43
25 La anafilaxis es una reacción inmunitaria grave, que compromete a todo el cuerpo, y se produce como resultado de reacciones inmunológicas a alimentos, fármacos o picaduras de insectos.

Tema 4. Inflamación
55
4.5.10.b. Síntesis
La síntesis del zomepiraco se inicia con la preparación del imidazol funcionalizado
4.114 (esquema 4.44). La obtención de este compuesto se consigue mezclando en primer
lugar la metilamina acuosa con el 3-oxopentanoato de dietilo 4.116. Esto provoca la
aparición de un precipitado (que debe ser la sal amónica 4.117), el cual, después de la
adición de la cloroacetona 4.115, proporciona el imidazol 4.114.26 La saponificación de los
grupos éster seguida de esterificación regioselectiva conduce al ácido 4.118, que se
convierte en el imidazol 4.113 por descarboxilación. Este compuesto proporciona el
zomepiraco por benzoilación de Vilsmeier-Hack con N,N-dimetil-p-clorobenzoilacetamida y
POCl3, seguida de saponificación.
Esquema 4.44
4.5.10.c. Cuestiones
1) Explique mecanísticamente la formación del imidazol 4.114.
4.5.11.a. Síntesis de piroxicam
El piroxicam es un fármaco AINE indicado para el alivio de los síntomas de la artritis
reumatoide, la osteoartritis, el dolor menstrual primario y el dolor postoperatorio. El
piroxicam es un inhibidor no selectivo de la ciclooxigenasa por lo que posee propiedades
tanto analgésicas como antiinflamatorias por inhibición de la síntesis de prostaglandinas.
4.5.11.a. Análisis retrosintético
La retrosíntesis del piroxicam se inicia con la escisión del enlace amida (esquema 4.45).
Esta operación genera el compuesto 4.119 y la 2-aminopiridina 4.120. A continuación, la
eliminación del grupo metilo en 4.119 origina la benzotiazina 4.121. La operación clave del
análisis retrosintético es la que convierte la benzotiazina 4.121 en la benzoisotiazolona
4.122. Esta operación indicada como CA (Contracción de Anillo) será, en el sentido
sintético, una operación de expansión de anillo y convertirá el anillo de 5 eslabones de
4.122 en el de 6 eslabones de 4.121. La desconexión de la parte de acetato en el
compuesto 4.122 conduce a la o-sulbobenzimida 4.123 y al halogenoacetato 4.124. La o-
sulfobenzimida 4.123 se obtendrá a partir del ácido 2-sulfobenzoico 4.125.
26 J. R. Carson, S. Wong. J. Med. Chem. 1973, 16, 172-174.
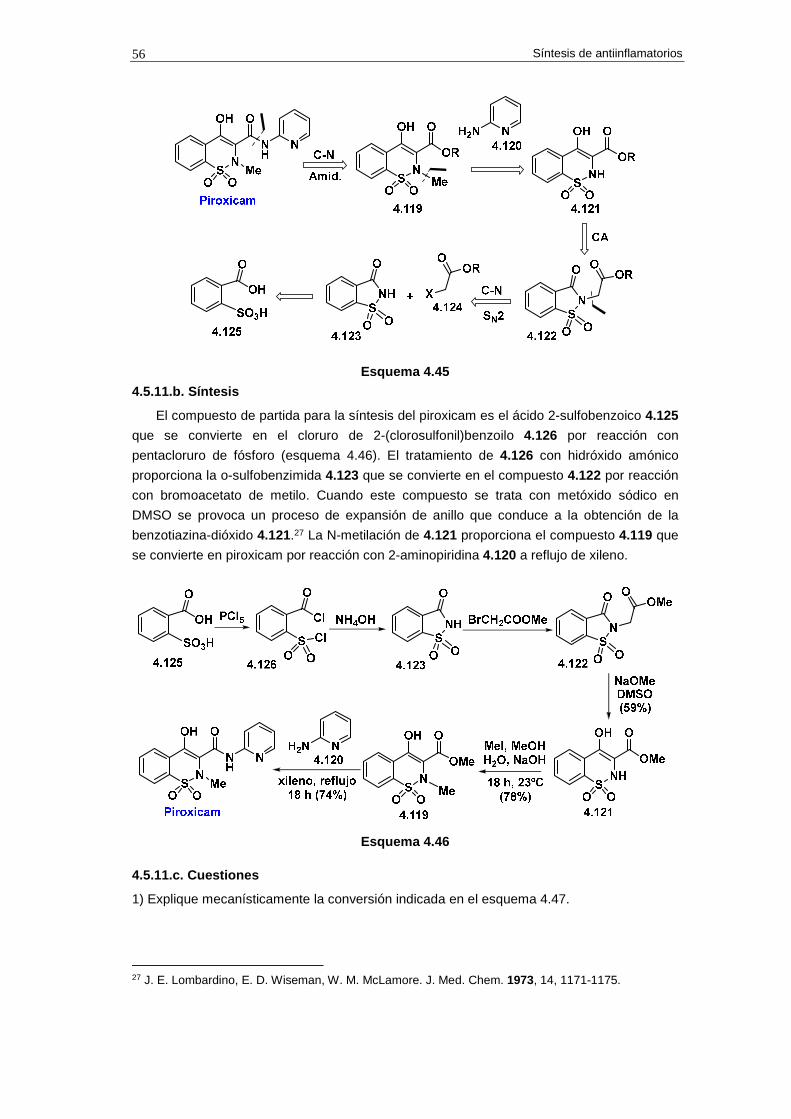
Síntesis de antiinflamatorios
56
Esquema 4.45
4.5.11.b. Síntesis
El compuesto de partida para la síntesis del piroxicam es el ácido 2-sulfobenzoico 4.125
que se convierte en el cloruro de 2-(clorosulfonil)benzoilo 4.126 por reacción con
pentacloruro de fósforo (esquema 4.46). El tratamiento de 4.126 con hidróxido amónico
proporciona la o-sulfobenzimida 4.123 que se convierte en el compuesto 4.122 por reacción
con bromoacetato de metilo. Cuando este compuesto se trata con metóxido sódico en
DMSO se provoca un proceso de expansión de anillo que conduce a la obtención de la
benzotiazina-dióxido 4.121.27 La N-metilación de 4.121 proporciona el compuesto 4.119 que
se convierte en piroxicam por reacción con 2-aminopiridina 4.120 a reflujo de xileno.
Esquema 4.46
4.5.11.c. Cuestiones
1) Explique mecanísticamente la conversión indicada en el esquema 4.47.
27 J. E. Lombardino, E. D. Wiseman, W. M. McLamore. J. Med. Chem. 1973, 14, 1171-1175.

Tema 4. Inflamación
57
Esquema 4.47
4.5.12. Síntesis de fenilbutazona
La fenilbutazona es un fármaco AINE que actúa mediante inhibición reversible de las
ciclooxigenasas. Se emplea en el tratamiento de la artritis reumatoide y otras poliartritis y
espondilitis anquilosantes. Su uso es limitado en humanos debido a sus efectos adversos
severos tales como la supresión de los glóbulos blancos y la anemia aplásica. Al igual que
otros AINEs, la fenilbutazona no debe ser administrada en pacientes con úlcera gástrica,
trombocitopenia, trastornos de la coagulación, insuficiencia cardíaca, insuficiencia hepática
o insuficiencia renal, hipertensión arterial grave ni síndrome de Sjögren.28
4.5.12.a. Análisis retrosintético
El análisis retrosintético de la fenilbutazona se inicia con la desconexión de la cadena de
butilo (esquema 4.48). Esta operación genera la 1,2-difenilpirazolidin-3,5-diona 4.127 que se
sintetizará mediante reacción entre la 1,2-difenilhidrazina 4.128 y el derivado de ácido
malónico 4.129.
Esquema 4.48
4.5.12.b. Síntesis
La síntesis de la fenilbutazona se indica en el esquema 4.49 y se inicia con la obtención
de la 1,2-difenilpirazolidin-3,5-diona 4.127 por reacción entre la 1,2-difenilhidrazina 4.128 y
el malonato de dietilo 4.129 (esquema 4.49).29 Esta condensación se lleva a cabo mediante
adición lenta de malonato de dietilo 4.129 a una mezcla que contiene la 1,2-difenilhidrazina
4.128 e hidruro sódico en clorobenceno. Luego se calienta a reflujo durante 4 horas y luego
5 horas más con destilación del etanol formado en la reacción. La reacción SN2 del anión
derivado de 4.127 con bromuro de butilo conduce a la fenilbutazona.
28 El síndrome Sjögren es una enfermedad autoinmune sistémica que se caracteriza por afectar principalmente a las glándulas exocrinas y que conduce a la aparición de sequedad. Las glándulas exocrinas son las encargadas de producir líquidos como la saliva, las lágrimas, las secreciones mucosas de la laringe y de la tráquea y las secreciones vaginales. 29 J. L. Vennerstrom, T.J. Holmes Jr. J. Med. Chem. 1987, 30, 563-567.

Síntesis de antiinflamatorios
58
Esquema 4.49
4.5.13. Síntesis de ácido flufenámico
El ácido flufenámico es un fármaco antiinflamatorio no esteroide (AINE) con
propiedades antiinflamatorias, analgésicas, antipiréticas y antiplaquetario. Se utiliza
principalmente en el tratamiento de trastornos musculoesqueléticos. Se administra por vía
oral o por vía tópica.
4.5.13.a. Análisis retrosintético
La retrosintesis del ácido flufenámico se indica en el esquema 4.50 y se basa en la
escisión del enlace C-N, lo que conduce a la 3-(trifluorometil)anilina 4.130 y al ácido 2-
halobenzoico 4.131.
Esquema 4.50
4.5.13.b. Síntesis
El ácido flufenámico se obtiene mediante acoplamiento de Ullman de la 3-
(trifluorometil)anilina 4.130 con el ácido 2-clorobenzoico 4.131. La reacción se lleva a cabo
calentando a reflujo de DMF, durante 2 horas, una mezcla de los dos compuestos anteriores
en presencia de cobre y carbonato potásico.30
Esquema 4.51
El ácido flufenámico también se ha preparado mediante acoplamiento Hartwig-
Buchwald de la 3-(trifluorometil)anilina 4.130 con el 2-bromobenzoato de metilo 4.132
seguida de saponificación (véase el esquema 4.52).31
30 R. F. Pellón, R. Carrasco, T. Márquez, T. Mamposo. Tetrahedron Lett. 1997, 38, 5107-5110. 31 (a) A. O. Adeniji, B. M. Twenter, M. C. Byrns, Y. Jin, M. Chen, J. D. Winkler, T. M. Penning. Bioorg. Med. Chem. Lett. 2011, 21, 1464-1468. (b) A. O. Adeniji, B. M. Twenter, M. C. Byrns, J. Yin, J. D. Winkler, T. M. Penning. J. Med. Chem. 2012, 55, 2311-2323

Tema 4. Inflamación
59
Esquema 4.52
4.5.13.c. Cuestiones
1) Proponga ciclos catalíoticos para las reacciones de acoplamiento de Ullman y Hartwig-
Buchwald de los esquemas 4.51 y 4.52.
4.5.14. Síntesis de tolmetina
La tolmetina es un medicamento AINE que se receta en el tratamiento del dolor leve o
moderado asociado a osteoartritis y a la artritis reumatoide. La tolmetina no ha demostrado
ser efectiva en el alivio de los síntomas de la gota.
Aunque el mecanismo de acción aún no está totalmente dilucidado, se sabe que la
tolmetina disminuye las concentraciones plasmáticas de la prostaglandina E, por lo que se
piensa que esa acción inhibitoria sobre las prostaglandinas es la responsable de sus efectos
antiinflamatorios.
4.5.14.a. Análisis retrosintético
El análisis retrosintético de la tolmetina se indica en el esquema 4.53 y comienza con la
desconexión de la parte de 4-metilbenzoilo. Esta operación, basada en una reacción SEAr,
conduce al pirrol 4.134 y al cloruro de 4-metilbenzoilo 4.135. La adición del grupo funcional
carbonilo en el pirrol 4.134 forma el pirrol-oxoacetato 4.136, que por desconexión de la parte
de oxoéster proporciona el N-metilpirrol 4.137 y el cloruro de oxalato 4.138.
Esquema 4.53
4.5.14.b. Síntesis
La síntesis de la tolmetina se inicia con la reacción SEAr del N-metilpirrol 4.137 con el
dicloruro de oxalilo 4.139 (esquema 4.54). Esta reacción forma el cloruro de ácido 4.140 que
es hidrolizado al ácido carboxílico 4.141.32 La reducción del carbonilo cetónico de 4.141, por
32 L. A. Reddy, S. Chakraborty, R. Swapna, D. Bhalerao, et al. Org. Process Res. Dev. 2010, 14, 362-368.

Síntesis de antiinflamatorios
60
reacción con hidrazina en presencia de KOH acuosa, proporciona el ácido pirrolacético
4.142, que es convertido en el metiléster 4.136 por reacción con sulfato de dimetilo en
diclorometano. La reacción SEAr de 4.136 con el cloruro de 4-metilbenzoilo 4.135, mediante
calentamiento en xileno a 145ºC durante 24 horas, proporciona el éster de tolmetina 4.143
que por saponificación se convierte en tolmetina.
Esquema 4.54
4.5.14.c. Cuestiones
1) El cetoácido 4.141 se convierte en el ácido pirrolacético 4.142 por reacción con hidrazina
en presencia de KOH acuosa (reducción de Wolff-Kishner). En el esquema 4.55 se indica la
reacción ajustada de este proceso (no se ha tenido en cuenta en este esquema que la
función de ácido carboxílico se ionizará al carboxilato potásico por reacción con KOH).
Esquema 4.55
Proponga un mecanismo que explique la reacción anterior.
2) Recientemente se ha publicado un método que permite efectuar reacciones de acilación
Friedel-Crafts sobre pirroles, bajo catálisis con 1,5-diazabiciclo[4.3.0]non-5-eno (DBN), que
actúa como catalizador nucleofílico (esquema 4.56).33
Esquema 4.56
33 J. E. Taylor, M. D. Jones, J. M. J. Williams, S. D. Bull. Org. Lett. 2010, 12, 5740-5743.

Tema 4. Inflamación
61
Proponga un mecanismo que explique la actividad catalítica del DBN en la anterior reacción
de acilación
4.5.15. Síntesis de oxaprocina
La oxaprocina se usa para aliviar el dolor, la inflamación y la rigidez causada por la
osteoartritis y la artritis reumatoide. También se usa para aliviar el dolor, la sensibilidad, la
hinchazón y la rigidez causada por la artritis reumatoide juvenil en los niños a partir de 6
años de edad. 4.5.15.a. Análisis retrosintético
La oxaprocina contiene en su estructura un anillo de oxazol trisustituido y su análisis
retrosintético se basa en una operación de CicloDeshidratación (CDH), lo que conduce a la
benzoina 4.98, al amoníaco y al ácido succínico 4.99 (esquema 4.57). Como se verá en la
sección de síntesis, el equivalente sintético del ácido succínico será el anhidrido succínico
4.100.
Esquema 4.57
4.5.15.b. Síntesis
La oxaprocina se obtiene del siguiente modo (esquema 4.58). Una mezcla formada por
benzoina 4.144, anhidrido succínico 4.146, y piridina se calienta a 90-95ºC durante 1.5
horas. Luego se añade ácido acético glacial y acetato amónico y se vuelve a calentar a 90-
95ºC durante 2 horas. Luego se flitra, se añade agua destilada al filtrado y se calienta de
nuevo a 90-95ºC. Después de enfriar, se filtra y el residuo sólido se lava con AcOH-H2O
(2:1). El sólido se mezcla con agua y la papilla se vuelve a filtrar y lavar con agua, lo que
proporciona la oxaprocina como un sólido blanco con un 67% de rendimiento.34
N
O
COOH
Oxaprocina
O
OH
O
O
O4.144 4.146
+
1) Piridina, 90-95ºC2) NH4OAc, AcOH, 90-95ºC
Esquema 4.58
4.5.15.c. Cuestiones
1) Explique mecanísticamente la formación de la oxaprocina indicada en el esquema 4.58.
34 John Wyeth and Brother Limited Patent: US4190584A1, 1980.

Síntesis de antiinflamatorios
62
El grupo de Y. Du y K. Zhao ha publicado un método que permite conseguir la síntesis
de oxazoles sustituidos. La aplicación de esta metodología a la preparación de la
oxaprocina se indica en el esquema 4.59.35 La secuencia se inicia con la conversión de la
1,2-difeniletanona 4.147 en la enamida 4.148. La reacción de ciclación sobre este
compuesto se consigue mediante calentamiento a reflujo de dicloroetano (DCE) en
presencia de diacetato de fenilyodonio y BF3·Et2O. Estas condiciones permiten la obtención
del isoxazol 4.149 que por saponificación proporciona la oxaprocina.
Esquema 4.59
El mecanismo que explica la formación del anillo de oxazol mediante reacción de 4.148
con PhI(OAc)2 (especie de yodo trivalente) se indica en el esquema 4.60. El proceso se
inicia con el ataque nucleofílico de la enamida 4.148 al PhI(OAc)2 lo que forma el intermedio
imínico 4.149, que se convierte en la yodoenamida 4.150 por pérdida de AcOH. La reacción
intramolecular de este compuesto forma el intermedio cíclico de 6 eslabones 4.152 que por
eliminación reductiva forma el oxazol 4.149 y yodobenceno.
Esquema 4.60
35 Y. Zheng, X. Li, C. Ren, D. Zhang-Negrerie, Y. Du, K. Zhao. J. Org. Chem. 2012, 77, 10353-10361.

Tema 4. Inflamación
63
4.5.16. Síntesis de nimesulida
La nimesulida es un antiinflamatorio relativamente COX-2 selectivo, con efectos
analgésicos y antipiréticos. Se receta en el tratamiento del dolor agudo, de la osteoartritis y
la dismenorrea. Debido al potencial riesgo de hepatotoxicidad, la nimesulida ha sido retirada
del mercado en muchos países excepto en Colombia, Uruguay, Chile y Mexico. Aunque la
nimesulida nunca fue aprobada por la FDA, en la actualidad está disponible en más de 50
países como Francia, Portugal, Grecia, Suiza, Bélgica, Brasil e India. En 2002 la EMEA
(Agencia Europea de Medicamentos) revisó el perfil beneficio/riesgo de la nimesulida y tomó
la decisión de suspenderlo temporalmente a partir de marzo de 2002, debido a su potencial
hepatotoxicidad. Más tarde se demostró que la ocurrencia de reacciones adversas
hepáticas debidas a la nimesulida era similar a la de otros AINEs.36
4.5.16.a. Análisis retrosintético
La retrosíntesis de la nimesulida se inicia con la desconexión del grupo nitro (esquema
4.61). Esta operación genera el compuesto 4.153 que por escisión del grupo
metanosulfonilo conduce a la 2-fenoxianilina 4.154. La interconversión del grupo amino en
nitro en 4.154 lleva al 1-nitro-2-fenoxibenceno 4.155 que se obtendrá a partir del 1-halo-2-
nitrobenceno 4.156 (X=halógeno) mediante reacción SNAr con fenol.
NimesulidaNO2
O
NHS
H3C
O O
SEArO
NHS
H3C
O O
O
NH2
N-S
O
NO2
SNAr
NO2
X
4.153
4.1544.1554.156
IGF
Esquema 4.61
4.5.16.b. Síntesis
El compuesto de partida para la síntesis de la nimesulida es el 1-cloro-2-nitrobenceno
4.156 que se convierte en el 1-nitro-2-fenoxibenceno 4.155 por reacción con fosfato de
trifenilo en presencia de KOH a reflujo de DMF (esquema 4.62).37 La reducción del grupo
nitro a amina, por reacción con Fe(0), proporciona la 2-fenoxianilina 4.154. Cuando este
compuesto se trata con cloruro de metanosulfonilo en diclorometano, en presencia de
trietilamina, se obtiene el producto de monomesilación 4.153 contaminado con un 5% del
producto de dimesilación 4.157. La separación de estos compuestos se lleva a cabo
mediante disolución de la mezcla en diclorometano seguida de extracción en medio básico
acuoso (NaOH) de la base conjugada del compuesto 4.153 y acidificación de la fase acuosa
36 G. Traversa, C. Bianchi, R. Da Cas, I. Abraha, F. Menniti-Ippolito, M. Venegoni. Brit. Med. J. 2003, 327, 18-22. 37 K. Prasad, M. L. Sharma, S. Kanwar, R. Rathee, S. D. Sharma. J. Sci. Ind. Res. 2005, 64, 756-760.

Síntesis de antiinflamatorios
64
básica hasta pH=2. La reextracción de la fase acuosa ácida con diclorometano proporciona
el compuesto 4.153 puro, el cual, por nitración SEAr se convierte en la nimesulida. En esta
reacción también se forma un pequeño porcentaje del producto de dinitración [N-(2,4-dinitro-
6-fenoxifenil)metanosulfonamida], que se separa mediante cristalización de metanol de la
mezcla de reacción.
Esquema 4.62
4.5.16.c. Cuestiones
1) Explique mecanísticamente la siguiente reacción:
Esquema 4.63
4.5.17. Síntesis de tenidap
El tenidap es un fármaco antireumático con actividad antiinflamatoria y analgésica que
actúa mediante inhibición de las ciclooxigenasas.38 Además de estas propiedades, el
tenidap también provoca una disminución de la proteína reactiva C (CRP, del inglés C
Reactive Protein) y de la velocidad de sedimentación de eritrocitos (ESR, del inglés
Erythrocyte Sedimentation Rate), lo que diferencia a este fármaco de la mayoría de los
AINEs.39
4.5.17.a. Análisis retrosintético
El análisis retrosintético del tenidap se inicia con la conversión de la parte de urea en
carbamato, lo que conduce al compuesto 4.158. Este compuesto está en equilibrio con su
forma carbonílica 4.159, cuya relación 1,5-dicarbonílica permite su desconexión al
carboxilato de oxoindol 4.160 y al derivado del ácido tiofeno-2-carboxílico 4.161. La escisión
38 P. F. Moore, D. L. Larson, I. G. Otterness, A. Weissman, S. B. Kadin, F. J. Sweeney, J. D. Eskra, A. Nagahisa, M. Sakakibara, T. J. Carty. Inflamm Res. 1996, 45, 54-61. 39 J. M. G. Canvin, R. Madhok. Ann. Rheum. Dis. 1996, 55, 79-82

Tema 4. Inflamación
65
de la función carbamato en 4.160 origina la 5-cloroindolin-2-ona 4.162 que se obtendrá del
2-(2-bromo-5-clorofenil)acetonitrilo 4.163.
Esquema 4.64
4.5.17.b. Síntesis
La síntesis del tenidap se inicia con la conversión del 2-(2-bromo-5-clorofenil)acetonitrilo
4.163 en la 5-cloroindolin-2-ona 4.162 (esquema 4.65). Recientemente se ha publicado un
método de ciclación que permite conseguir esta transformación mediante reacción con CuI
(3 mol%), KI (19 mol%), N-acetilglicina (6 mol%, que actúa como ligando del cobre) y NaOH
(3 equiv) en t-butanol. La mezcla de reacción se calienta a 100ºC durante 18 horas y
proporciona la 5-cloroindolin-2-ona 4.162 con un 57% de rendimiento.40
Esquema 4.65
El tratamiento de 4.162 con cloroformiato de fenilo en THF en presencia de trietilamina
conduce a la formación del 5-cloro-1-fenoxicarbonil-2-(fenoxicarboniloxi)-indol 4.164.41 La
reacción de este compuesto con carbonato amónico proporciona el 5-cloro-1-fenoxicarbonil-
2-oxoindol 4.160. Cuando este oxoindol se trata con el cloruro del ácido tiofeno-2-carboxílico
40 J-C. Hsieh, A-Y Cheng, J-H. Fu, T-W Kang. Org. Biomol. Chem. 2012, 10, 6404-6409. 41 M. Porcs-Makkay, G. Simig. Org. Process Res. Dev. 2000, 4, 10-16

Síntesis de antiinflamatorios
66
4.161, en DMF en presencia de DMAP, se obtiene el compuesto 4.158 que se convierte en
tenidap mediante calentamiento con carbonato amónico en DMF.
El ciclo catalítico que explica la conversión del bromonitrilo 4.163 en el oxindol 4.162 se
indica en el esquema 4.66 y se inicia con la formación del complejo de 4.165 (Cu(II)). Este
intermedio es atacado nucleofílicamente en el triple enlace por el anión hidróxido, lo que
genera el intermedio azaenólico 4.166, que se equilibra con el complejo amídico 4.167. La
inserción oxidante intramolecular forma el intermedio cíclico 4.168 (Cu(III)), que experimenta
un proceso de eliminación reductora proporcionando el oxindol 4.162 y regenerando el
complejo de Cu(I).
Esquema 4.66
4.5.17.c. Cuestiones
1) Proponga una secuencia sintética que permita la conversión del 1-bromo-4-cloro-2-
metilbenceno en el 2-(2-bromo-5-clorofenil)acetonitrilo 4.163.
4.5.18. Síntesis de benzidamina
La benzidamina es un antagonista de las aminas vasoactivas. Actúa estabilizando las
membranas celulares y lisosómicas e inhibiendo las prostaglandinas que intervienen en los
procesos inflamatorios. Se emplea en el tratamiento del dolor y/o inflamación de origen
traumático, muscular, reumático o vascular. Este fármaco se suele emplear mediante vía
tópica.
4.5.18.a. Análisis retrosintético
La retrosíntesis de la benzidamina se inicia con la desconexión de la cadena de N,N-
dimetilpropilo (esquema 4.67). Esta operación genera el 1-bencil-1H-indazol-3-ol 4.169 y la
3-halo-N,N-dimetilpropanamina 4.170. La desconexión del grupo bencilo en el compuesto

Tema 4. Inflamación
67
4.169 conduce al 1H-indazol-3-ol 4.171 que se obtendrá del ácido 2-hidrazinobenzoico
4.172.
Esquema 4.67
4.5.18.b. Síntesis
La síntesis de la benzidamina se indica en el esquema 4.68 y se inicia con la
ciclodeshidratación del ácido 2-hidrazinobenzoico 4.172. Esta reacción proporciona el 1H-
indazol-3-ol 4.171 que por N-bencilación se convierte en el compuesto 4.169.42 La O-
alquilación de 4.169 se lleva a cabo mediante calentamiento de la sal sódica derivada de
este compuesto, en xileno en presencia de un exceso de 3-cloro-N,N-dimetilpropanamina
4.170 durante 6 horas, lo que permite la obtención de la benzidamina con un 74% de
rendimiento.43
Esquema 4.68
4.5.19. Síntesis de celecoxib (celebrex)
El desarrollo del rofecoxib, el etoricoxib, el celecoxib y el valdecoxib (véanse sus
estructuras en la figura 4.36) confirmó que los inhibidores selectivos de COX-2 podían
actuar como fármacos antiinflamatorios sin provocar toxicidad gastrointestinal ni renal.
Desafortunadamente, algunos inhibidores selectivos de COX-2, como el rofecoxib y el
valdecoxib, alteran el balance natural de la vía modulada por las enzimas COX. Así, los
efectos vasodilatadores y anti-agregantes de la prostaciclica PGI2 se ven disminuidos al
reducirse los niveles de esta prostaciclina, aumentándose al mismo tiempo la
42 T. M. Sokolenko, L. M. Yagupolskii. Chem. Heterocyc. Compd. 2011, 46, 1335-1343. 43 Aziende Chimiche Riunute Angelini Francesco A.C.R.A.F. S.p.A. Patente: US4749794 A1, 1988.

Síntesis de antiinflamatorios
68
vasoconstricción debido al aumento de los niveles del prototrombótico tromboxano A2
(TAX2). Estos dos efectos son los que explican los graves efectos secundarios, como el
aumento de la presión arterial y los infartos de miocardio, asociados a la administración de
rofecoxib y valdecoxib.
Figura 4.36
El celecoxib es un inhibidor altamente selectivo del enzima COX-2, teniendo una
selectividad 20 veces mayor por COX-2 que por COX-1. Está indicado para el alivio de
dolores agudos por traumatismos, dolor en pacientes con osteoartritis y dolores de la
menstruación. También reduce el número de pólipos en el colon y recto en pacientes con
poliposis adenomatosa hereditaria.
El celecoxib accede al centro catalítico del enzima COX-2 intercalándose en la
membrana lipídica y difundiéndose desde allí hasta el centro activo hidrofóbico del enzima.
Otros fármacos antiinflamatorios pueden seguir vías de acceso diferentes a la que sigue el
celecoxib.
En la parte de la izquierda de la figura 4.37 se indica esquemáticamente el camino que
sigue el celecoxib desde la zona extracelular hasta el centro activo de la enzima COX-2. En
la parte de la derecha de la figura 4.37 se representa la posición del celecoxib en el centro
activo de la enzima COX-2.
Acceso del celecoxib al centro activo de COX-2 Celec oxib en el centro activo de COX-2
CelecoxibCOX-2
Figura 4.37. Acceso del celecoxib al centro activo de COX-2
En la figura 4.38 se comparan esquemáticamente los centros activos de las enzimas
COX-1 y COX-2. El celecoxib no puede encajar en el centro activo de COX-1 pero si pueda
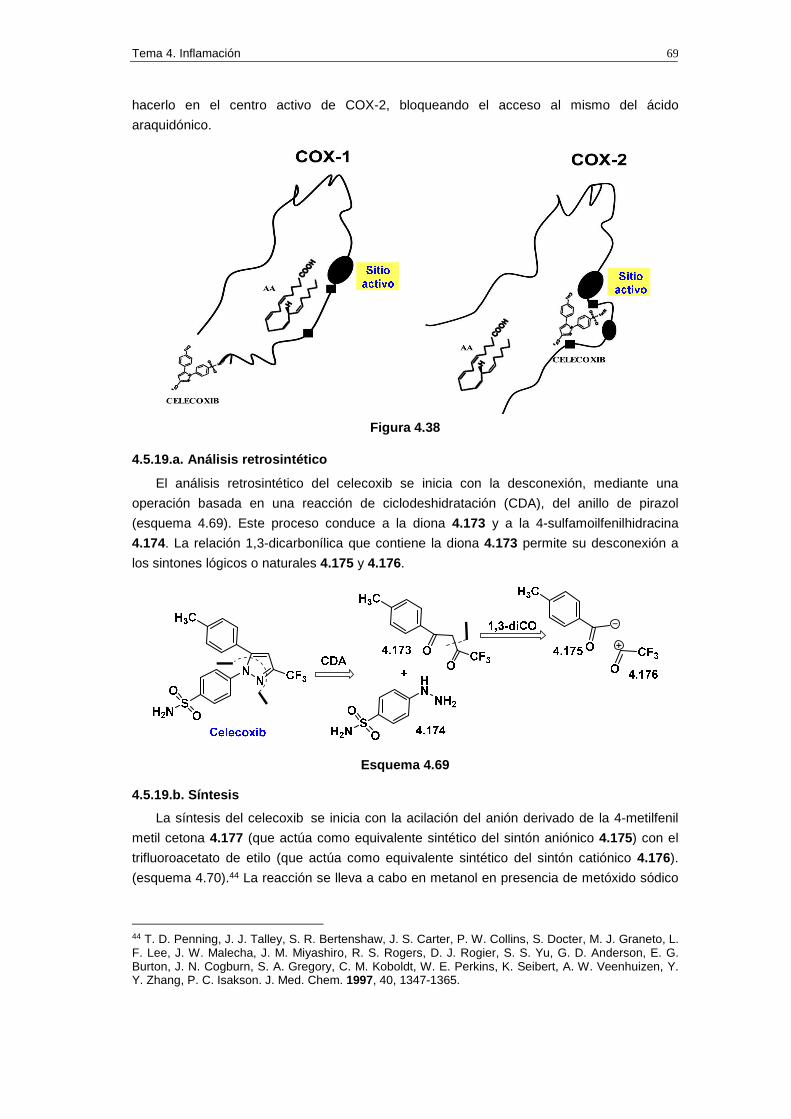
Tema 4. Inflamación
69
hacerlo en el centro activo de COX-2, bloqueando el acceso al mismo del ácido
araquidónico.
Figura 4.38
4.5.19.a. Análisis retrosintético
El análisis retrosintético del celecoxib se inicia con la desconexión, mediante una
operación basada en una reacción de ciclodeshidratación (CDA), del anillo de pirazol
(esquema 4.69). Este proceso conduce a la diona 4.173 y a la 4-sulfamoilfenilhidracina
4.174. La relación 1,3-dicarbonílica que contiene la diona 4.173 permite su desconexión a
los sintones lógicos o naturales 4.175 y 4.176.
Esquema 4.69
4.5.19.b. Síntesis
La síntesis del celecoxib se inicia con la acilación del anión derivado de la 4-metilfenil
metil cetona 4.177 (que actúa como equivalente sintético del sintón aniónico 4.175) con el
trifluoroacetato de etilo (que actúa como equivalente sintético del sintón catiónico 4.176).
(esquema 4.70).44 La reacción se lleva a cabo en metanol en presencia de metóxido sódico
44 T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Carter, P. W. Collins, S. Docter, M. J. Graneto, L. F. Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J. Rogier, S. S. Yu, G. D. Anderson, E. G. Burton, J. N. Cogburn, S. A. Gregory, C. M. Koboldt, W. E. Perkins, K. Seibert, A. W. Veenhuizen, Y. Y. Zhang, P. C. Isakson. J. Med. Chem. 1997, 40, 1347-1365.

Síntesis de antiinflamatorios
70
y proporciona la 1,3-dicetona 4.173. El celecoxib se obtiene por condensación de este
compuesto con el clorhidrato de la 4-sulfamoilfenildracina 4.174.
Esquema 4.70
4.5.19.c. Cuestiones
1) Explique mecanísticamente la formación del celecoxib por condensación entre la 1,3-
dicetona 4.173 y el clorhidrato de la 4-sulfamoilfenildracina 4.174.
2) La reacción de condensación entre la 1,3-dicetona 4.173 y el clorhidrato de la 4-
sulfamoilfenilhidracina 4.174 conduce a una mezcla formada por celecoxib y un
regioisómero. ¿Cuál es la estructura de este regioisómero?
4.5.20. Síntesis de etoricoxib
El etoricoxib es un inhibidor selectivo de COX-2, siendo su afinidad hacia COX-2 unas
106 veces mayor que hacia COX-1. El etoricoxib se receta para el tratamiento de la artritis
reumatoide, artritis psoriásica, osteoartritis, espondilosis anquilosante, dolor lumbar crónico,
dolor agudo y gota.
El etoricoxib no inhibe la síntesis de las prostaglandinas gástricas ni afecta la función
plaquetaria. En los estudios realizados en más 3.000 pacientes no se observaron
diferencias significativas entre el etoricoxib, el placebo y otros AINES en lo que se refiere a
la incidencia de episodios cardiovasculares trombóticos. Sin embargo, en comparación con
el naproxeno (500 mg dos veces al día), el etoricoxib provocó una mayor incidencia de
episodios cardiovasculares trombóticos, debido probablemente que, a diferencia de los
inhibidores de la COX-1, el etoricoxib inhibe la formación de la prostaciclina sin inhibir la
formación del tromboxano plaquetario.
4.5.20.a. Análisis retrosintético
El análisis retrosintético del etoricoxib comienza con la desconexión del anillo piridínico
trisustituido, que se generará mediante reacción de ciclodeshidratación intramolecular en la
dienaminocetona 4.178 (esquema 4.71). La siguiente operación retrosintética desconecta el
grupo amino, en forma de amoníaco, y genera el compuesto 4.179. Esta operación se ha
denominado AD-E (Adición-Eliminación) porque en el sentido sintético el amoníaco se
adicionará de forma conjugada a un sistema de tipo aceptor Michael, como el que contiene
el compuesto 4.179 (X grupo electrón-atrayente), y a continuación se producirá la

Tema 4. Inflamación
71
eliminación de Y (grupo saliente). La escisión del sistema aceptor Michael en el sustrato
4.179 conduce al sintón catiónico 4.180 y al sintón aniónico 4.181. Una operación de adición
de grupo funcional sobre el ácido conjugado de 4.181 proporciona el cetoéster 4.182, que
se desconecta en el sistema 1,3-dicarbonílico para generar los sintones naturales 4.183 y
4.184.
N
N
Cl
Me
SO2Me
Etoricoxib
CDA
N
Cl
Me
SO2Me
O
N
Cl
Me
SO2MeX
Y ONH2
4.178NH3
AD-E
4.179
N
Cl
Me
SO2Me
X
Y
O
C-C
4.180
4.181
N Me
SO2Me
O
4.182
ROOC
N Me
SO2Me
O
4.184
ROOC
1,3-diCO
4.183+
AGF
Esquema 4.71
4.5.19.b. Síntesis
Para la síntesis del etoricoxib se elige como compuesto de partida la sulfonilfenil metil
cetona 4.185 (esquema 4.72). Esta cetona se convierte en el ácido 4.186 mediante reacción
de Willgerodt-Kindler seguida de hidrólisis (para el mecanismo de esta reacción véase el
esquema 4.19).45 La adición del dianión derivado del ácido 4.186 al 6-metilnicotinato de
metilo 4.187, en THF a 50ºC, proporciona directamente la cetona 4.188. Cuando este
compuesto se trata con la sal de vinilamidinio 4.189 (equivalente sintético del sintón
catiónico 4.181), se genera el compuesto 4.190, el cual, por tratamiento ácido, se convierte
en 4.191. La reacción de este intermedio con hidróxido amónico acuoso a reflujo
proporciona el etoricoxib.
45 I. W. Davies, J-F. Marcoux, E. G. Corley, M. Journet, D-W. Cai, M. Palucki, J. Wu, R. D. Larsen, K. Rossen, P. J. Pye, L. DiMichele, P. Dormer, P. J. Reider. J. Org. Chem. 2000, 65, 8415-8420.

Síntesis de antiinflamatorios
72
Esquema 4.72
La síntesis de sal de vinilamidinio 4.189 se indica en el esquema 4.73.46
Esquema 4.73
4.5.20.c. Cuestiones
1) Explique mecanísticamente la formación del etoricoxib a partir del compuesto 4.188.
2) Proponga un mecanismo que explique la formación del compuesto 4.192 en la reacción 1
del esquema 4.73.
46 M. Palucki, Z. Lin, Y. Sun. Org. Process Res. Dev. 2005, 9, 141-148.

Tema 4. Inflamación
73
4.5.21. Síntesis de rofecoxib (vioxx)
El rofecoxib es un inhibidor altamente selectivo de COX-2. En septiembre de 2004 la
compañía Merck retiró el rofecoxib del mercado ante la posibilidad de que el medicamento
estuviese asociado a un incremento del riesgo de infartos y derrames cerebrales en
pacientes que tomaban rofecoxib por largo tiempo o en dosis muy elevadas. Aunque los
graves efectos secundarios asociados al rofecoxib han obligado a su retirada como fármaco
anti-inflamatorio, se han descubierto recientemente otras potenciales aplicaciones
terapéuticas del mismo, en particular en el campo de la lucha contra el cáncer. Por ejemplo,
el rofecoxib actúa como un sensibilizador a los efectos de los fármacos citostáticos en
modelos in vitro de cáncer gástrico, como consecuencia de la inhibición de los genes MRP1
y GST-pi asociados con el fenómeno de multirresistencia a fármacos.47 También se ha
demostrado que el rofecoxib produce una disminución en la carcinogénesis cólica,
farmacológicamente inducida en ratas.48
4.5.21.1a. Análisis retrosintético
En el esquema 4.74 se indica un análisis retrosintético para el rofecoxib. El proceso de
desconexión se inicia con la escisión del sistema carbonílico α,β-insaturado. Esta operación
forma el cetoéster 4.193, que por desconexión de la función éster proporciona el sintón
catiónico no natural 4.194 y el sintón aniónico natural 4.195.
Esquema 4.74
4.5.21.1b. Síntesis
Para la síntesis del rofecoxib se elige como compuesto de partida la 4-tiometil-
acetofenona 4.196, que se obtiene mediante acilación Friedel-Crafts del tioanisol. La
oxidación de 4.196 con oxono (2KHSO5.KHSO4
.K2SO4, componente activo
monoperoxosulfato potásico KHSO5) proporciona la sulfona 4.197 (esquema 4.75).49 Este
compuesto se convierte en la bromocetona 4.198 mediante bromación con bromo molecular
en presencia de tricloruro de aluminio. El rofexocib se obtiene por reacción de la
bromocetona 4.198 (equivalente sintético del sintón catiónico 4.194) con ácido fenilacético
4.199 en presencia de Et3N y DBU (1,8-diazabiciclo[5.4.0]undec-7-eno).
47 F. S. Zhu, X. M. Chen, Z. G. Huang, Z. R. Wang, D. W. Zhang, X. Zhang. J. Digestive Dis. 2010, 11, 34.42. 48 F. Noguera, I. Amengual, J. M. Morón, A. Plaza, J. A. Martínez-Córcoles, C. Tortajada, J. J. Pujol Rev. Esp. Enferm. Dig. 2005, 97, 405-415. 49 Y. Ducharme, J. Y. Gauthier, Y. Leblanc, Z. Wang, S. Léger, M. Théren. Patente: WO95/18799.

Síntesis de antiinflamatorios
74
Esquema 4.75
4.5.21.1c. Cuestiones
1) Proponga un mecanismo que explique la formación del rofecoxib mediante reacción de la
bromocetona 4.198 con el ácido fenilacético 4.199 en presencia de Et3N y DBU (1,8-
diazabiciclo[5.4.0]undec-7-eno).
4.5.21.2a. Análisis retrosintético de rofexocib med iante acoplamiento sp2-sp2
En el esquema 4.76 se indica un análisis retrosintético alternativo para el rofecoxib. El
proceso de retrosíntesis se inicia con la desconexión del grupo fenilsulfona. Esta operación
se basa en una reacción de acoplamiento sp2-sp2 y conduce al fragmento nucleofílico 4.200
(Y=metal o metaloide) y al fragmento electrofílico 4.201 (X=halógeno).
Esquema 4.76
El intercambio de grupo funcional en el compuesto 4.201 proporciona el ácido
feniltetrónico 4.202 cuya forma cetónica es la cetolactona 4.203. La desconexión del
sistema 1,3-dicarbonílico de 4.203 conduce al éster de ácido glicólico 4.204 que por escisión

Tema 4. Inflamación
75
de la función éster proporciona el sintón catiónico no natural 4.205 y el sintón aniónico
natural 4.206.
4.5.21.2b. Síntesis de rofexocib mediante acoplamie nto sp2-sp2
La síntesis alternativa del rofecoxib se indica en el esquema 4.77 y comienza con la O-
alquilación del ácido fenilacético 4.199 con bromoacetato de etilo 4.207 (equivalente
sintético del sintón catiónico no natural 4.205).50 Esta reacción se lleva a cabo en presencia
de trietilamina y proporciona el diéster 4.204. La condensación Michael intramolecular de
4.204, inducida por t-butóxido de potasio, permite la obtención del ácido 2-feniltretrónico
4.202. Para llevar a cabo la proyectada reacción de acoplamiento sp2-sp2 se necesita activar
la posición C-3 del ácido tetrónico 4.202, lo que se consigue mediante una secuencia
sintética en dos etapas. En la primera de ellas el ácido tetrónico 4.202 se convierte en el
triflato 4.208 por reacción con Tf2O y diisopropil etil amina (DIPEA). En la segunda etapa se
provoca la sustitución formal del grupo triflato por bromuro mediante reacción de 4.208 con
bromuro de litio en acetona. La bromolactona resultante, compuesto 4.201, se somete a la
reacción de acoplamiento de Suzuki con el ácido 4-tiometilfenilborónico 4.209 en tolueno, en
presencia de Pd(PPh3)4 y de carbonato potásico acuoso. En estas condiciones se obtiene el
producto de acoplamiento 4.210 que se convierte en rofecoxib mediante oxidación de la
función sulfuro a sulfona con oxono.
Esquema 4.77
4.5.21.c.2. Cuestiones
1) Explique mecanísticamente la conversión del triflato 4.208 en el bromuro 4.201.
50 R. Desmond, U. Dolling, B. Marcune, R. Tiller, D. Tschaen. Patente: WO 96/08482.

Síntesis de antiinflamatorios
76
2) Proponga un ciclo catalítico para la formación del compuesto 4.210 mediante reacción de
acoplamiento de Suzuki entre el ácido borónico 4.209 y el bromuro 4.201.
3) El compuesto 4.202 se puede englobar dentro de la familia de los denominados ácidos
tetrónicos, cuyo miembro más simple es el propio ácido tetrónico (4-hidroxifuran-2(5H)-ona,
figura 4.39). De la familia de los ácidos tetrónicos forman parte moléculas con importante
actividad biológica como el ácido ascórbico (vitamina C, figura 4.39).
Figura 4.39
En la tabla 4.3 se compara la fuerza ácida de varios compuestos orgánicos
Tabla 4.3
3a) Explique por qué el acido ascórbico es un ácido más fuerte, en su primera ionización
que los ácidos benzoico y acético y que el fenol.

Tema 4. Inflamación
77
3b) Explique por qué el ácido ascórbico es un ácido más débil, en su segunda ionización,
que la 2-hidroxicliclohex-2-enona.
4.5.22. Síntesis de lumiracoxib
El lumiracoxib es un inhibidor selectivo de COX-2 producido por Novartis y
comercializado por primera vez en Brasil, en 2005, con el nombre de Prexige®. Se receta
para el tratamiento de la osteoartritis y del dolor agudo.
La estructura del lumiracoxib difiere de los otros coxibes y se encuadra dentro de la
familia de los ácidos aril-acéticos. De hecho es el único inhibidor selectivo de COX-2 que es
ácido, siendo su estructura muy similar a la del diclofenaco, que muestra una preferencia de
inhibición de COX-2 unas 10 veces mayor que por COX-1 (veáse la figura 4.40 para la
comparación de las estructuras de estos dos compuestos). El lumiracoxib se enlaza a COX-
2 en un sitio de unión diferente del de los coxibes, presentando la mayor selectiva hacia
COX-2 que ninguno de los AINEs.51
Figura 4.40
4.5.22.1a. Análisis retrosintético
La retrosíntesis del lumiracoxib se inicia con una estrategia de reconexión de los grupos
funcionales amina y ácido carboxílico en grupo funcional amida (esquema 4.78). Esta
operación genera la lactama 4.211 que se obtendrá del cloruro de ácido 4.212 mediante una
reacción SEAr intramolecular.
Esquema 4.78
51 S. Tacconelli, M. L. Capone, P. Patrignani. Curr Pharm. Des. 2004, 10, 589-601.

Síntesis de antiinflamatorios
78
La desconexión del enlace amídico en 4.212 conduce a la diarilamina 4.213 que por
escisión del anillo p-metilfenílico origina la 2-cloro-6-fluoroanilina 4.214 y el 1-halo-4-
metilbenceno 4.215. En el sentido sintético la conexión de estos dos fragmentos se llevará a
cabo mediante una reacción de acoplamiento catalizada por un metal de transición.
4.5.22.1b. Síntesis
La síntesis del lumiracoxib se inicia con la reacción de acoplamiento entre 2-cloro-6-
fluoroanilina 4.214 y el 1-bromo-4-metilbenceno 4.215 (esquema 4.79). Esta reacción se
lleva a cabo en presencia de Pd2(dba)3, tristributilfosfina y t-butóxido de sodio en tolueno y
proporciona la anilina 4.213.52 La amidación de este compuesto con cloruro de cloroacetilo
conduce a la cloroacetilamida 4.212 que se convierte en la lactama 4.211 mediante reacción
SEAr intramolecular. La hidrólisis del anillo lactámico permite la obtención del lumiracoxib.
Lumiracoxib
F
NH
Cl
CH3
COOH
F
N
Cl
CH3
O
F
N
Cl
CH3
O
ClF
NH
Cl
CH3
F
NH2
Cl
CH3
Br
+
4.211
4.2124.213
4.2144.215
NaOtBu, Pd2(dba)3
(n-Bu3)P, tolueno
ClCl
O
neat, 90ºC
AlCl3, neat
160-170ºCNaOH, EtOH
H2O, reflujo
Esquema 4.79
4.5.22.1c. Cuestiones
1) Proponga un ciclo catalítico para la formación del compuesto 4.213.
4.5.22.2a. Análisis retrosintético de lumiracoxib m ediante homologación
El segundo análisis retrosintético del lumiracoxib se inicia con la desconexión del enlace
Csp2-N tal y como se indica en el esquema 4.80. Esta escisión conduce a la 2-cloro-6-
fluoroanilina 4.214 y al ácido 2-(2-halo-5-metilfenil)acético 4.216 que derivará del ácido 2-
halo-5-metilbenzoico 4.217 mediante homologación.
52 R. A. Fujimoto, L. W. Mcquire, B. B. Mugrage, J. H. Van Duzer, H. Xu, Patente: WO9911605 A1 1999.

Tema 4. Inflamación
79
Lumiracoxib
F
NH
Cl
CH3
COOHF
NH2
Cl
Csp2-N
4.216
4.214
CH3
OHOX
+
CH3
COOH
X
4.217
Homologación
Esquema 4.80
4.5.22.2b. Síntesis de lumiracoxib mediante homolog ación
El ácido 2-yodo-5-metilbenzoico 4.217 se reduce al alcohol bencílico 4.218 el cual se
convierte en el bromuro de bencilo 4.219 mediante reacción con HBr (esquema 4.81).53
Esquema 4.81
El desplazamiento SN2 del bromuro con cianuro conduce al nitrilo 4.220 que por
hidrólisis proporciona el ácido 2-(2-yodo-5-metilfenil)acético 4.216. Este compuesto se
convierte en la amida 4.222, vía el cloruro de ácido 4.221. La amida se somete a la reacción
de acoplamiento con 2-cloro-6-fluoroanilina 4.214 en presencia de Cu, Cu2I2 y K2CO3 en
xileno a reflujo. En estas condiciones se obtiene la diarilamina 4.223 que se transforma en el
lumiracoxib mediante hidrólisis de la función amida.
4.5.22.2c. Cuestiones
1) Explique mecanísticamente la formación de 4.223.
53 Novartis AG Patente: US6291523 B1, 2001.

Síntesis de antiinflamatorios
80
4.6. Migraña
Se denomina migraña al dolor pulsátil intenso o sensación de latido en la cabeza,
generalmente de un solo lado. Los episodios migrañosos pueden durar horas o días y venir
acompañados de náuseas, vómitos, sensibilidad extrema a la luz o al ruido y en algunos
casos a los olores y al tacto. Pueden provocar visión borrosa y sensación de vértigo seguida
algunas veces por desmayo.
La migraña se produce como consecuencia de un trastorno neurovascular que
involucra la vasodilatación local de los vasos sanguíneos intracraneales y extracerebrales y
la estimulación simultánea de la vía del dolor nervioso sensorial del trigémino circundante,
que provoca en última instancia el dolor de cabeza. La activación del sistema
trigeminovascular causa la liberación de diversos vasodilatadores, especialmente el péptido
relacionado con el gen de la calcitonina (CGRP del inglés Calcitonin Gene-Related Peptide)
que induce la respuesta al dolor. Los episodios migrañosos también van acompañados de
una disminución de los niveles de determinados neurotransmisores, como la serotonina.54
4.6.1. Factores desencadenantes de la migraña
Los factores más comunes que pueden dar lugar a fenómenos migrañosos son:
a) Los cambios hormonales en las mujeres que originan fluctuaciones de los niveles de
estrógenos, lo que explica que muchas mujeres presenten dolores de cabeza
inmediatamente antes o durante la menstruación. Otras mujeres también desarrollan
migrañas durante el embarazo o la menopausia.
b) Determinados alimentos como quesos curados y los alimentos procesados o muy
salados. Los aditivos alimentarios como el aspartamo, utilizado como edulcorante y el
glutamato monosódico, empleado como conservante, pueden provocar migrañas.
c) Bebidas alcohólicas, particularmente el vino, y las bebidas con alto contenido en cafeína.
d) El estrés en el trabajo o en el hogar.
e) Los estímulos sensoriales como las luces muy brillantes y el brillo del sol pueden
provocar las migrañas, al igual que los sonidos fuertes. Los olores muy penetrantes, como el
de algunos perfumes, disolventes de pintura, etc también pueden originar migrañas.
f) Esfuerzos físicos intensos.
g) Cambios en el patrón de sueño-vigilia, como el insomnio o el jet lag.
h) Cambios bruscos del tiempo, como descensos acusados de la presión barométrica.
i) Determinados fármacos, como los anticonceptivos orales y los vasodilatadores
(nitroglicerina).
4.6.2. Factores de riesgo
Los factores que favorecen la propensión a padecer migrañas son:
a) Componentes genéticos o hereditarios. Un gran porcentaje de personas que sufren
migraña tienen antecedentes familiares de ataques de migraña. Si uno o ambos padres
tienen migrañas, es muy probable que sus hijos la padezcan también.
54 M. Aggarwal, V. Puri, S. Puri. Ann. Neurosci. 2012, 19, 88–94.

Tema 4. Inflamación
81
b) Edad. Las migrañas pueden comenzar a cualquier edad, aunque la mayoría de las
personas experimentan su primera migraña durante la adolescencia.
c) Sexo. Las mujeres son tres veces más propensas que los hombres a tener migrañas. Sin
embargo, durante la infancia la migraña afecta más a los niños que a las niñas aunque en la
pubertad los episodios migrañosos afectan más a las adolescentes.
d) Los cambios hormonales. En el apartado anterior ya se ha indicado que los cambios
hormonales son un factor favorecedor de las migrañas. Muchas mujeres sufren migrañas
poco antes o durante la menstruación, durante el embarazo o la menopausia. En general,
las migrañas mejoran después de la menopausia.
4.6.3. Etapas del proceso migrañoso
a) Aura . Los síntomas de la migraña se denominan aura y pueden aparecer antes del
dolor de cabeza o junto con él. Entre estos síntomas se incluyen la visión de luces o puntos
destellantes, pérdida de visión, sensación de hormigueo en una parte de la cara, el brazo o
la pierna, percepción de ruidos, dificultad para hablar, movimientos espasmódicos o difíciles
de controlar, debilidad en una extremidad (migraña hemipléjica). Cada uno de estos
síntomas generalmente comienza en forma gradual, empeora durante pocos minutos y dura
entre 20 y 60 minutos.
b) Pródromo . Uno o dos días antes de la migraña, se pueden notar cambios sutiles
que avisan del ataque migrañoso, como estreñimiento, cambios de humor (desde depresión
hasta euforia), antojos de alimentos, rigidez en el cuello, aumento de la sed y la frecuencia
de las micciones, bostezos frecuentes, hiperactividad, irritabilidad
c) Crisis (cefalea ). Las migrañas sin tratar generalmente duran entre 4 y 72 horas. La
frecuencia con la cual aparecen las migrañas varía según la persona pudiendo ser poco
frecuentes o aparecer varias veces al mes. El episodio migrañoso puede cursar con dolor en
un lado o en ambos lados de la cabeza, dolor punzante, sensibilidad a la luz, a los sonidos y
a veces a los olores, náuseas y vómitos, visión borrosa y mareo, que a veces va seguido de
desmayos.
d) Pósdromo . La fase final de la migraña se denomina pósdromo. En esta fase
algunas personas tienen sensación de agotamiento aunque otras han manifestado sentirse
eufóricas. Durante el pósdromo también se puede tener sensación de confusión.
4.7. Fármacos contra la migraña
Se han propuesto diversas teorías para explicar el origen de la migraña. Una de ellas,
denominada teoría vascular, explica que la migraña se produce como consecuencia de una
vasoconstricción intracerebral seguida de una fase posterior de vasodilatación extracerebral.
Otra teoría basa su hipótesis en una depresión cortical en la que se observa un fuerte
desequilibrio iónico con flujo sanguíneo reducido. Para la hipótesis inflamatoria la migraña
se origina como consecuencia de la activación de las meninges y los vasos extracraneales
provocada por la liberación de neuropéptidos y otros agentes inflamatorios en las
terminaciones nerviosas.
Un neurotransmisor que juega un papel clave en los procesos migrañosos es la
serotonina. Las meninges y los vasos cerebrales están inervadas por el nervio trigémino y

Síntesis de antiinflamatorios
82
las raíces cervicales superiores. Las neuronas del nervio trigémino contienen receptores del
tipo 5HT1D, mientras que los receptores de los vasos son del tipo 5HT1B. Estos receptores
presinápticos intervienen en la contracción vascular craneal y su estimulación disminuye la
liberación de serotonina. Cuando se inicia el episodio migrañoso se produce una
disminución del flujo sanguíneo cerebral, lo que provoca la activación del sistema
noradrenérgico con la consiguiente liberación de serotonina y dilatación de los vasos
sanguíneos. Esto activa las terminaciones nerviosas del trigémino y provoca dolor en la
corteza cerebral. Los triptanos son agonistas de los receptores 5HT1B/D y producen
vasoconstricción al reducir la liberación de serotonina. De esta manera contrarrestan el
efecto vasodilatador producido por la serotonina y alivian los dolores asociados a la
migraña.54
En la figura 4.41 se indican las estructuras de fármacos de tipo triptano empleados en
el tratamiento de la migraña.
Figura 4.41
4.8. Síntesis de triptanos
4.8.1. Síntesis de sumatriptan
El sumatriptan es un agonista selectivo del receptor 5-HT1D y su unión al receptor
reduce la liberación de serotonina, y también la de otros neuropéptidos vasoactivos,
provocando vasoconstricción selectiva de los vasos craneales de la circulación carotídea
dilatados e inflamados. El sumatriptan se emplea en el tratamiento de ataques agudos de
migraña.
4.8.1.a. Análisis retrosintético
La retrosítesis del sumatriptan se inicia con la desconexión del grupo dimetilamino, lo
que genera el indol 4.224 (X= grupo saliente, esquema 4.82). El sistema indólico de 4.224
se obtendrá mediante Síntesis del Indol de Fischer (SIF) en la hidrazona funcionalizada
4.225. Este compuesto se sintetizará mediante condensación entre la arilhidrazina 4.226 y
el aldehído 4.227. La arilhidrazina derivará de la anilina 4.228 que, a su vez, se obtendrá del
(4-bromometil)nitrobenceno 4.229.

Tema 4. Inflamación
83
Esquema 4.82
4.8.1.b. Síntesis
En el esquema 4.83 se indica la síntesis del sumatriptan que se inicia con la conversión
del (4-bromometil)nitrobenceno 4.229 en el ácido (4-nitrofenil)metanosulfónico 4.230 por
reacción con sulfito sódico.55 La reacción de este compuesto con PCl5 proporciona el cloruro
de ácido 4.231 que se convierte en la N-metil-1-(4-nitrofenil)metanosulfonamida 4.232 por
reacción con metilamina.
Esquema 4.83
La reducción de 4.232 con hidrógeno molecular en presencia de Ni-Raney conduce a la
N-metil-1-(4-aminofenil)metanosulfonamida 4.228. Cuando este compuesto se trata con
ácido nitroso se genera la correspondiente sal de arildiazonio que se reduce in situ con
SnCl2 para proporcionar la N-metil-1-(4-hidracinofenil)metanosulfonamida 4.233. La
55 C. Bhanja, S. Chakroborty, S. Jena. Asian J. Pharm. Anal. Med. Chem. 2014, 2, 168-175.

Síntesis de antiinflamatorios
84
reacción del indol de Fischer sobre este compuesto se lleva a cabo empleando el
dihidrofurano como equivalente sintético del aldehído 4.227 que aparece en el análisis
retrosintético. Esta reacción proporciona el indol 4.234, que se convierte en sumatriptan por
mesilación y reacción con dimetilamina.
Una síntesis alternativa del sumatriptan basada en una reacción de Japp-Klingemann
se indica en el esquema 4.84.56 El proceso se inició con la protección del grupo sulfonamido
de la N-metil-1-(4-nitrofenil)metanosulfonamida 4.232 por reacción con cloroformiato de etilo
en presencia de carbonato potásico. Esta reacción de protección resultó ser necesaria
porque la proyectada reacción de Japp-Klingemann no funcionó sobre un sustrato de tipo N-
metilsulfonamida. El producto de carbometoxilación 4.235 se convirtió en la amina 4.236 por
reducción del grupo nitro. La reacción de Japp-Klingemann se llevó a cabo del siguiente
modo: el compuesto 4.235 se convirtió en la sal de diazonio 4.237 por reacción con NaNO2
en HCl acuoso a -4ºC. Después de 15 minutos se añadió el 2-(3-dimetilaminopropil)-3-
oxobutanoato de etilo 4.238 y acetato sódico. Luego se calentó a 50ºC durante 30 minutos,
manteniendo el pH de la reacción entre 3-4 mediante adición de NaOAc. Después de
enfriar, se filtró la reacción y el filtrado se extrajo con diclorometano. La evaporación del
disolvente proporcionó la hidrazona 4.239. La reacción del indol de Fischer sobre 4.239 se
llevó a cabo disolviendo este compuesto en AcOH glacial seguido de adición de HCl gas y
agitando la mezcla de reacción a temperatura ambiente durante 24 horas. En estas
condiciones se obtuvo el indol 4.240. La saponificación de este compuesto con KOH acuoso
en metanol condujo al ácido 4.241 que se convirtió en sumatriptan por descarboxilación
mediante calentamiento a 200ºC en quinolina en presencia de Cu.
Esquema 4.84
56 B. Pete, I. Bitter, K. Harsanyi, L. Töke. Heterocycles 2000, 53, 665-673.

Tema 4. Inflamación
85
4.8.1.c. Cuestiones
1) Explique mecanísticamente la reacción del indol de Fischer que convierte la hidracina
4.233 en el indol 4.234 por reacción con dihidrofurano (esquema 4.85).
Esquema 4.85
2) Proponga un mecanismo para la reacción de Japp-Klingemann del esquema 4.85.
Esquema 4.85
4.8.2. Síntesis de eletriptan
El eletriptan es un agonista de los receptores 5-HT1B y 5-HT1D. Produce
vasoconstricción y se receta en el tratamiento de los ataques agudos de migraña.
4.8.2.1a. Análisis retrosintético
La retrosíntesis del eletriptan se inicia una adición del grupo funcional doble enlace al
sistema de etil fenil sulfona lo que conduce al compuesto 4.242 (esquema 4.86). La
desconexión del sistema de fenil vinil sulfona, basada en una reacción de acoplamiento
Csp2-Csp2, origina la propia fenil vinil sulfona 4.243 y el indol 4.244. Una adición del grupo
funcional carbonilo en 4.244 proporciona el acilbromoindol 4.245 que se desconecta al
cloruro de ácido 4.246 y al bromoindol 4.247.
NH
SPh
O O
Eletriptan
N
H
AGF
NH
SPh
O O
N
H
sp2-sp2C-C
NH
BrN
H
SPh
O O
+
AGFNH
BrN
HO
NH
Br N
HO
Cl
+
4.242
4.244
4.243
4.2454.247
4.246
Esquema 4.86
4.8.2.1b. Síntesis
El cloruro de ácido 4.246 que aparece en el análisis retrosintético remite al aminoácido
(R)-D-prolina. En la síntesis del eletriptan indicada en el esquema 4.87 no se emplea el

Síntesis de antiinflamatorios
86
cloruro de ácido 4.246 derivado de la N-metil-D-prolina, sino el cloruro de ácido derivado de
la D-prolina N-Cbz protegida, actuando el grupo Cbz (benciloxicarbonilo) como forma latente
del grupo metilo. De acuerdo con este planteamiento, la síntesis del eletriptan se inicia con
la reacción de acilación del bromoindol 4.247 lo que se consigue mediante formación de la
sal bromomagnésica 4.248 seguida de reacción con el cloruro de ácido 4.249.57 El producto
de acilación, compuesto 4.250, se convierte en el derivado N-metilpirrolidínico 4.244
mediante reducción con LiAlH4. En esta reacción el grupo carbonilo se reduce a metileno y
el grupo Cbz (COOCH2Ph) se convierte en metilo. El grupo de vinilsulfona se instala
mediante reacción de Heck del bromoindol 4.244 con fenil vinil sulfona catalizada por
Pd(OAc)2 en presencia de Et3N y de (o-tolil)3P como ligando del paladio. El producto de la
reacción de acoplamiento, compuesto 4.242 se convierte en eletriptan mediante
hidrogenación.
Esquema 4.87
En el esquema 4.88 se dibuja el ciclo catalítico de la reacción de Heck, que se inicia
con la etapa de adición oxidante por reacción del bromoindol 4.247 con el complejo de
paladio(0) simbolizado en el esquema como L-Pd(0)-L. Esta reacción forma el complejo II
que experimenta una reacción de carbopaladación con la fenil vinil sulfona 4.243. El
intermedio III resultante de este proceso forma el producto de acoplamiento 4.242 y el
hidruro de paladio III por reacción de β-eliminación de hidruro. El hidruro III por eliminación
reductora regenera el catalizador de paladio(0) y forma HBr, que es neutralizado por la
trietilamina.
57 J. E. Macor, M. J. Wythes. US Patent 5.545.644, 1996. Véase también N. Baumann, I. R. Baxendale, S. V. Ley, N. Nikbin. Beilstein J.Org. Chem. 2011, 7, 422-495.

Tema 4. Inflamación
87
adición oxidante
PdBr
L
L
carbopaladación
H Pd
L
L
Br
-eliminación de hidruro
HBr
I
III
L LPd(0)eliminación reductora
Et3N
NH
RSPh
O O SPh
O O4.242
4.243
NH
Br
N
H
4.244
NH
R
II
PdL L
Br
II
IV
NH
RSPh
O O
Et3NHBr
II
II
Esquema 4.88
La reacción de Heck es E-estereoselectiva. Esto se explica porque la reacción de
carbopaladiación transcurre mediante adición sin al doble y porque, a continuación, la b-
eliminación de hidruro tiene lugar mediante eliminación sin. Son posibles dos estados de
transición en la eliminación, tal y como se dibuja en el esquema 4.89. El estado de
transición que conduce al producto Z está más desfavorecido, porque coloca al grupo
indolilo (Ar en el esquema) en interacción estérica con el grupo fenilsulfona. Por el contrario,
el estado de transición que conduce al producto E está más favorecido porque coloca al
grupo indolilo en una posición anti con respecto al grupo fenilsulfona.
Esquema 4.89
4.8.2.2a. Análisis retrosintético del eletriptan me diante síntesis del indol de Fischer
En el esquema 4.90 se indica un análisis retrosintético del eletriptan basado en una
reacción de síntesis del indol de Fischer, lo que conduce a la arilhidracina 4.251 y al
aldehído (o su equivalente sintético) 4.252.

Síntesis de antiinflamatorios
88
Esquema 4.90
4.8.2.2b. Síntesis
a) Síntesis del equivalente sintético del aldehído 4.252
Como equivalente sintético del aldehído 4.252 se empleó el acetal 4.260 cuya síntesis
se indica en el esquema 4.91. El compuesto de partida para la síntesis del acetal fue el
ácido N-metil-4-aminobutírico (en su forma de clorhidrato, compuesto 4.253).58 La reacción
de este compuesto con cloroformiato de bencilo, en presencia de KOH, proporcionó el
compuesto N-Cbz protegido 4.254. Cuando este compuesto se trató con N-
metilhidroxilamina, en presencia de carbonil diimidazol (CDI) y trietilamina, se obtuvo la
amida de Weinreb 4.255 que por tratamiento con el reactivo de Grignard 4.256 condujo a la
cetona 4.257. Cuando este compuesto se hizo reaccionar con hidrógeno molecular, a
presión y bajo catálisis con Pd/C, se provocó la N-Cbz desprotección, la formación
intramolecular de la correspondiente imina y su subsiguiente hidrogenación, lo que condujo
a la obtención de la pirrolidina 4.258 en su forma racémica. Este producto era aceitoso pero
la reacción con ácido fumárico proporcionó la sal cristalina 4.259. La resolución de esta sal
se consiguió con ácido L-dibenzoiltartárico, lo que daba lugar a la precipitación de la
pirrrolidina de configuración R en forma de la sal 4.260 (93% de exceso enantiomérico en la
pirrolidina).
Esquema 4.90
58 C. A. Ishcroft, P. Hellier, A. Pettman, S. Watkinson. Org. Process Res. Dev. 2011, 15, 98-103.

Tema 4. Inflamación
89
b) Pasos finales
Los pasos finales en la síntesis del eletriptan se indican en el esquema 4.91. Para la
síntesis de la fenilhidracina se eligió como compuesto de partida el 1-(2-bromoetil)-4-
nitrobenceno 4.261 (esquema 4.91). Cuando este compuesto se calentó con fenilsulfinato
de sodio en isopropanol se obtuvo el 1-nitro-4-(2-(fenilsulfonil)etil)benceno 4.262. La
hidrogenación de este compuesto proporcionó la 4-(2-(fenilsulfonil)etil)anilina 4.263. El
tratamiento de este compuesto con ácido nitroso, seguido de la reducción de la
correspondiente sal de diazonio con SnCl2 proporcionó la hidracina 4.251 pero con tan solo
el 13% de rendimiento. Este resultado se mejoró cuando la reducción se llevaba a cabo con
ácido ascórbico (vitamina C). Así, a una disolución de la anilina 4.263 en acetonitrilo se le
añadía H2SO4 y NaNO2 a -4ºC. Después de 11 h a -4ºC, se añadía el ácido ascórbico y se
agitaba luego 16 horas a temperatura ambiente. En estas condiciones se obtenía la
hidracina en su forma de oxalilhidrazida (véase la estructura 4.264 del esquema 4.91). Este
compuesto no era aceitoso pero su podía obtener en su forma cristalina como la sal cálcica
4.265. Cuando este compuesto se trataba con ácido se descomponía para generar in situ la
correspondiente hidracina. De esta forma, el eletriptan se obtuvo añadiendo H2SO4 acuoso
a una mezcla de 4.265 y de 4.260 en acetonitrilo y calentando luego a 80ºC durante 16
horas. La adición de KOH acuoso y extracción con acetato de etilo proporcionaba el
eletriptan.
4.265
NH
SPh
O O
N
H
CH3
NH2
SPh
O O
NaNO2, H2SO4
H2O, CH3CN
ácido ascórbico
NH
SPh
O O
NH
Eletriptan
4.264
4.263NO2
Br
4.261iPrOH
PhSO2Na
NO2
H2, Pd/CS
Ph
O O
4.262
O
O
OH
KOH, CaCl2H2O, CH3CN
NH
SPh
O O
NH
O
O
O
2
Ca2+
H2O, CH3CN
H2SO4
4.260
Esquema 4.91
4.8.2.3b. Cuestiones
1) Proponga una secuencia sintética para la obtención del reactivo de Grignard 4.256 a
partir del ácido acrílico (CH2=CHCOOH).
2) Explique mecanísticamente la formación del eletriptan por reacción de 4.265 con 4.260
en presencia de ácido sulfúrico acuoso.

Síntesis de antiinflamatorios
90
4.8.3. Síntesis de zolmitriptan
El zolmitriptán se emplea en el tratamiento del dolor agudo causado por la migraña. Es
un agonista de los receptores 5-HT1B y 5-HT1D. El principal efecto secundario del fármaco, al
igual que el de los restantes triptanos, es la vasoconstricción de las arterias coronarias, por
lo que no se debe emplear en personas con cardiopatía isquémica, angina de pecho o que
hayan sufrido infarto agudo de miocardio.
4.8.3.a. Análisis retrosintético
La retrosíntesis del zolmitriptan comienza con una operación de intercambio del grupo
dimetilamino por un grupo X (X=grupo saliente, esquema 4.92) lo que conduce al
compuesto 4.266. La siguiente operación del análisis retrosintético se efectúa sobre el anillo
de indol que se obtendrá mediante síntesis del indol de Fischer entre la hidracina 4.267 y el
aldehído funcionalizado 4.268. La hidracina derivará de la anilina 4.269 y esta del
nitrocompuesto 4.270. El anillo de oxazolidinona de este compuesto se obtendrá por
ciclocondensación del aminoalcohol 4.271 el cual se obtendrá a partir del aminoácido L-
fenilalanina 4.272.
NH
NHN
O
H
O
Zolmitriptan
H2N
HO
H
O
NH
XHN
O
H
O
SIF
NH
NH2
HN
O
H
O
X
OH
+IGF
NH2
HN
O
H
O
IGF
NO2
HN
O
H
ONO2
H2N
HO
H
4.266
4.267
4.2714.270
4.269 4.268
4.272
IGF
Esquema 4.92
4.8.3.b. Síntesis
La síntesis del zolmitriptan se indica en el esquema 4.93 y se inicia con la nitración
SEAr de la L-fenilalanina 4.272. Esta reacción proporciona la 4-nitro-L-fenilalanina 4.273, la
cual, por esterificación con cloruro de tionilo en metanol seguida de reducción, se convierte
en el aminoalcohol 4.271.59 El tratamiento de este compuesto con fosgeno en presencia de
KOH lleva a la obtención de la oxazolidinona 4.270. La reducción del grupo nitro de 4.270
conduce a la anilina 4.269 que se convierte en la hidracina 4.267 (en su forma de
clorhidrato) por reacción con ácido nitroso seguida de reducción in situ de la
correspondiente sal de arildiazonio con SnCl2. La reacción del indol de Fischer se lleva a
cabo entre el clorhidrato de la arilhidracida 4.267 y el dimetilacetal del 4-clorobutanal
(Cl(CH2)3CH(OMe)2) mediante calentamiento a reflujo en una mezcla de etanol/agua. Esta
59 B. Xu, Z. Huang, C. Liu, Z. Cai, W. Pan, P. Cao, X. Hao, G. Liang. Bioorg. Med. Chem. 2009, 17, 3118-3125.

Tema 4. Inflamación
91
reacción proporciona la indolil-etilamina 4.274. El zolmitriptan se obtiene mediante la
amidación reductiva de 4.274 con formaldehido y cianoborohidruro sódico.60
NH
NHN
O
H
O
Zolmitriptan
H2N
HO
H
O
NH
NH2HN
O
H
ONH
NH2
HN
O
H
O
NH2
HN
O
H
O
NO2
HN
O
H
O
NO2
H2N
HO
H
4.2744.267
4.271
4.2704.269
4.272
H2N
HO
H
O NO24.273
1) SOCl2, MeOH2) NaBH4, EtOH
COCl2KOH, H2OH2, Pd/C
NaNO2, HClluego SnCl2
Cl(CH2)3CH(OMe)2
EtOH, H2O
H2SO4
HNO3
Na(BH3)CN, HCHO
HCl
Esquema 4.93
4.8.3.c. Cuestiones
1) Explique mecanísticamente indicada en el esquema 4.94. en la que se forma el
compuesto 4.274 mediante síntesis del indol de Fischer.
Esquema 4.94
4.8.4. Síntesis de naratriptan
El naratriptan es un agonista de los receptores 5-HT1B/1D empleado en el tratamiento de
la migraña.
4.8.4.a. Análisis retrosintético
La retrosíntesis del naratriptan comienza con la desconexión del anillo de N-
metilpiperidina que se instalará por condensación del sulfonil-indol 4.275 con la 4-N-
metilpirrolidona 4.276 (esquema 4.95). El compuesto 4.275 se obtendrá mediante reacción
de Heck entre la N-metilvinilsulfonamida 2.477 y el bromoindol 4.247.
60 R. C. Glen, G. R. Martin, A. P. Hill, R. M. Hyde, P. M. Woollard, J. A. Salmon, J. Buckingham, A. D. Robertson. J. Med. Chem. 1995, 38, 3566-3580.

Síntesis de antiinflamatorios
92
Esquema 4.95
4.8.4.b. Síntesis
La síntesis del naratriptan comienza con la reacción de Heck entre la N-
metilvinilsulfonamida 2.477 y el bromoindol 4.247 (esquema 4.96). Esta reacción
proporciona el compuesto 4.278 que por hidrogenación se convierte en la indolil-
sulfonamida 4.275.61 La condensación de este compuesto con la 4-N-metilpirrolidona 4.276
proporciona el derivado 4.279 que se transforma en naratriptan por hidrogenación.62
Esquema 4.96
4.8.4.c. Cuestiones
1) El compuesto 4.279 se obtiene del siguiente modo: lentejas de KOH se agitan en metanol
entre 25-45ºC hasta su completa disolución. Luego se añade el compuesto 4.275 y a
continuación la 4-N-metilpirrolidinona y la mezcla de reacción se calienta a reflujo 5 horas.
Luego se añade agua con lo que se produce la precipitación del compuesto 4.279, que se
obtiene por flitración. Proponga un mecanismo para la formación del compuesto 4.279.
4.8.5. Síntesis de frovatriptan
El frovatriptan es un agonista selectivo de los receptores 5-HT1B/1D que intervienen en la
contracción vascular craneal. Se emplea en el tratamiento de los dolores producidos por la
migraña.
4.8.5.a. Análisis retrosintético
La retrosíntesis del frovatriptan se inicia con el intercambio del grupo formamido por el
grupo ciano lo que conduce al compuesto 4.280 (esquema 4.97). La siguiente operación
61 Patente WO 2009/118753,A2 62 U. S. Kumar, V. R. Sankar, S. B. Kumar, M. P. Prabhu, S. M. Rao. Org. Process Res. Dev. 2009, 13, 468-470.

Tema 4. Inflamación
93
desconecta el anillo de indol, que se obtendrá mediante síntesis del indol de Fischer entre la
hidracina 4.281 y la aminocetona (o su equivalente sintético) 4.282.
Esquema 4.97
4.8.5.b. Síntesis
El compuesto de partida para la síntesis del frovatriptan es la ciclohexano-1,4-diona
4.283 (esquema 4.98). La monocetalización de este compuesto, mediante reacción con 2,2-
dimetilpropano-1,3-diol, en presencia de ácido sulfúrico acuoso, proporciona la cetona
4.284.63 La aminación reductiva de este compuesto con metilamina conduce al aminoacetal
4.285.64 Cuando este compuesto se sometió a la síntesis del indol de Fischer, por reacción
con el 4-hidracinilbenzonitrilo 4.281, se obtuvo el indol racémico (+/-)-4.280. La resolución
se llevó a cabo con ácido L-piroglutámico y proporcionó la sal 4.286 (94% de exceso
enantiomérico en el indol). El frovatriptan se obtuvo por reacción de esta sal con BF3·AcOH
en AcOH acuoso.
NH
NC
HN
NH
NC
NH2
(+/-)-4.280
4.281
O
O
OH OH
H2SO4, H2O
O
OO
MeNH2 ac, EtOHluego H2, Pd/C
HN
OO
HCl, H2O, 80-90ºC
ácido L-piroglutámicoMeOH reflujo
luego AcOH, 0ºCNH
NC
HN
.NHO
COOH
H2O, 95ºC NH
HN
Frovatriptan
4.283 4.284 4.285
4.286 (94% ee)
BF3·AcOH, AcOH H2N
O
Esquema 4.98
En el esquema 4.99 se dibuja una síntesis para el 4-hidracinilbenzonitrilo 4.281 a partir
de la anilina. Así, la monobromación de este compuesto, por reacción con N-
63 J. H. Babler, K. P. Spina. Synth. Commun. 1984, 14, 39-44. 64 Patente US6359146B1.

Síntesis de antiinflamatorios
94
bromosuccinimida en polietilenglicol, proporciona la 4-bromoanilina 4.287.65 Para la
cianación de la 4-bromoanilina se empleó cianuro de zinc en presencia de Pd/C como
catalizador en N,N-dimetilacetamida (DMA) a 110ºC. Como ligando del paladio se emplea
el difenilfosfinoferroceno (dppf) y también se añade formiato de zinc dihidratado. El cianuro
de zinc se emplea, en lugar de otras fuentes de cianuro como el cianuro sódico o el cianuro
potásico, en que aquél es un compuesto mucho más covalente que éstos, que suelen
formar complejos no reactivos de cianuro con el paladio. El formiato de zinc se añade para
favorecer la reducción de complejos de Pd(II) en complejos de Pd(0) y así facilitar la
regeneración del catalizador.66 La 4-bromoanilina 4.287 se convierte en el 4-
hidracinilbenzonitrilo 4.281 mediante reacción con ácido nitroso seguido de reducción in situ
de la sal de arildiazonio 4.289 con SnCl2.67
NH
NC
NH2
4.281
NH24.281
NBS, PEG
NH2
Br
4.287
Pd/C, dppf, Zn(CN)2
Zn(HCHO)2, DMANH2
NC
4.288
NaNO2, HClN2 Cl
NC
SnCl2
4.289
Esquema 4.99
65 K. Venkateswarlu , K. Suneel, B. Das, K. N. Reddy, T. S. Reddy. Synth. Commun. 2009, 39, 215-219. 66 H. Yu, R. N. Richey, W. D. Miller, J. Xu, S. A. May. J. Org. Chem. 2011, 76, 665–668. 67 N. Chandna, S. Kumar, P. Kaushik, D. Kaushik, S. K. Roy, G. K. Gupta, S. M. Jachak, J. K. Kapoor, P. K. Sharma, Bioorg. Med. Chem. 2013, 21, 4581–4590.


















