
Metodolog ía Sint ética Aplicada a la S íntesis de F … · Inhibidores de la Transcriptasa...
85
Metodolog Metodologí a Sint a Sinté tica Aplicada tica Aplicada a la S a la Sí ntesis de F ntesis de Fá rmacos rmacos Miguel Carda Miguel Carda Má ster en Qu ster en Quí mica Aplicada y Farmacol mica Aplicada y Farmacológica gica Universidad Jaume I Universidad Jaume I NH 2 HOOC MeO HN N N N N Ph3C (PriO)2B Br Br Síntesis de losartan
Transcript of Metodolog ía Sint ética Aplicada a la S íntesis de F … · Inhibidores de la Transcriptasa...

MetodologMetodolog íía Sinta Sint éética Aplicada tica Aplicada a la Sa la Sííntesis de Fntesis de F áármacosrmacos
Miguel CardaMiguel CardaMMááster en Quster en Qu íímica Aplicada y Farmacolmica Aplicada y Farmacol óógicagica
Universidad Jaume IUniversidad Jaume I
NH2
HOOCMeO
HN
N
NN
NPh3C
(PriO)2B
Br
Br
Síntesis de losartan


Tema 6 Enfermedades víricas: síntesis de
antivirales
Máster en Química Aplicada y Farmacológica
Universidad Jaume I


Tema 6. Enfermedades víricas: síntesis de antivirales
6.1. Los virus 1
6.2. Ácidos nucleicos virales 1
6.2.1. Virus con ADN bicatenario 2
6.2.2. Virus con ADN monocatenario 2
6.2.3. Virus con ARN 2
6.2.3.a Virus con ARN bicatenario 3
6.2.3.b. Virus con ARN monocatenario positivo 3
6.2.3.c. Virus con ARN monocatenario negativo 3
6.2.3.d. Virus con ARN monocatenario y bicatenario retrotranscrito 3
6.3. Morfologia de los virus 4
6.4. Transmisión 6 6.5. Origen evolutivo 7
6.6. Propiedades de vida 7 6.7. Genoma 7 6.8. Ciclo de replicación de los virus 9
6.8.1. Fijación o adsorción 9
6.8.2. Penetración 9
6.8.3. Eclipse 10
6.8.4. Multiplicación 11
6.8.5. Ensamblaje 11 6.8.6. Liberación de los nuevos virus 12
6.8.6.a. Infección persistente 12
6.9. Efectos en la célula huésped 13
6.10. Virus y enfermedades humanas 13
6.11. Epidemiología 13
6.11.1. Epidemias y pandemias 14
6.12. Virus y cáncer 15 6.13. Respuesta inmune del huésped 16
6.14. Vacunas 17
6.15. El virus de la inmunodeficiencia humana (VIH) 18
6.15.1. Proteínas reguladoras 22
6.15.1.a. Rev 22
6.15.1.b. Tat y Rev: acción conjunta 22
6.15.2. Proteínas accesorias 22
6.15.2.1. Vif: incremento en infectividad y protección del genoma viral 22
6.15.2.2. Vpu 22
6.15.2.2.a. Degradación de la proteína CD4 23

6.15.2.2.c. Desprendimiento de viriones de la membrana celular 23
6.15.3. Ciclo de replicación 23
6.16. Fármacos antivirales 26
6.16.1. Inhibidores de la Transcriptasa Reversa Análogos
de los Nucleósidos (ITRN) 28
6.16.2. Inhibidores de la Transcriptasa Reversa NO Análogos
de los Nucleósidos (ITRNN) 28
6.17. Síntesis de antivirales 31
6.17.1. Aciclovir 31
6.17.1.a. Análisis retrosintético 32
6.17.1.b. Síntesis 33
6.17.1.c. Cuestiones 32 6.17.2. Azidotimidina (Zidovudina, AZT) 33
6.17.2.a. Análisis retrosintético 35
6.17.2.b. Síntesis 35
6.17.2.c. Cuestiones 36
6.17.3. Ribavirina 37
6.17.3.a. Análisis retrosintético 37
6.17.3.b. Síntesis 37
6.17.3.c. Cuestiones 38
6.17.4. Síntesis de adefovir dipivoxilo 37
6.17.4.a. Síntesis 38
6.17.4.b. Síntesis 38
6.17.4.c. Cuestiones 39
6.17.5. Síntesis de telbivudina 40 6.17.5.a. Análisis retrosintético 40 6.17.5.b. Síntesis 40 6.17.5.c. Cuestiones 41
6.17.6. Síntesis de clevudina 42 6.17.6.a. Análisis retrosintético 42 6.17.6.b. Síntesis 42 6.17.6.c. Cuestiones 43
6.17.7. Nevirapina 44
6.17.7.a. Análisis retrosintético 48
6.17.7.b. Síntesis 49
6.17.7.c. Cuestiones 49
6.17.8. Síntesis de efavirenz 49
6.17.8.a. Análisis retrosintético 50
6.17.8.b. Síntesis 51

6.17.8.c. Cuestiones 53
6.17.9. Síntesis de delavirdina (Rescriptor®) 54
6.17.9.a. Análisis retrosintético 54
6.17.9.b. Síntesis 55
6.17.9.c. Cuestiones 56
6.17.10. Síntesis de raltegravir (Isentress®) 58
6.17.10.a. Análisis retrosintético 62
6.17.10.b. Síntesis 63
6.17.10.c. Cuestiones 64
6.17.11. Síntesis de vicriviroc 65
6.17.11.a. Análisis retrosintético 68
6.17.11.b. Síntesis 69
6.17.11.c. Cuestiones 70
6.17.12. Síntesis de plecoranil 71
6.17.12.a. Análisis retrosintético 73
6.17.12.b. Síntesis 74
6.17.12.c. Cuestiones 75
6.17.13. Síntesis de arbidol 75
6.17.12.a. Análisis retrosintético 75
6.17.12.b. Síntesis 76
6.17.12.c. Cuestiones 77


Tema 6. Enfermedades víricas 1
6.1. Los virus
En biología, un virus, del latín virus (toxina o veneno), es una entidad infecciosa microscópica que sólo puede multiplicarse dentro de las células de otros organismos. Los virus son fragmentos de ácido nucleico (ADN o ARN), capaces de multiplicarse en una célula y pasar a otras para iniciar un nuevo ciclo de replicación. Los ácidos nucleicos son muy vulnerables en el medio extracelular y, antes de abandonar la célula que han infectado, se rodean de una cubierta de estructura proteica denominada cápside. Algunos virus rodean a la cápside con una envoltura lipídica denominada peplo.
Los virus infectan todos los tipos de organismos, desde animales y plantas hasta bacterias y arqueas. El primer virus conocido, el virus del mosaico del tabaco, fue descubierto por Martinus Beijerinck en 1899. Actualmente se han descrito más de 5.000, si bien algunos autores opinan que podrían existir millones de tipos diferentes. Los virus se hallan en casi todos los ecosistemas de la Tierra y son el tipo de entidad biológica más abundante.
Figura 6.1. Virus del mosaico del tabaco
6.2. Ácidos nucleicos virales
Los ácidos nucleicos de los virus contienen la información específica y el potencial para llevar a cabo reacciones biológicas en el seno de la célula infectada. Existen cuatro tipos de ácidos nucleicos virales:
1) ADN de cadena sencilla o ssADN (del inglés single stranded DNA).
2) ADN de cadena doble o dsADN (del inglés double stranded DNA).
3) ARN de cadena sencilla o ssARN (del inglés single stranded RNA).
4) ARN de cadena doble dsARN (del inglés double stranded RNA).
La replicación del genoma de la mayoría de los virus ADN se produce en el núcleo de la célula infectada. Si la célula tiene el receptor adecuado en su superficie el virus accede a ella por fusión con la membrana celular o por endocitosis. La mayoría de virus ADN son completamente dependientes de la maquinaria de síntesis de ADN y ARN de la célula hospedadora.
Los virus se pueden clasificar según su forma de replicación que está basada a su vez en el tipo de ácido nucleico que contienen.

Síntesis de antivirales 2
6.2.1. Virus con ADN bicatenario
Este tipo de virus se replica usando una ADN polimerasa de la célula hospedadora y, por lo tanto, son altamente dependientes del ciclo celular. Para que pueda realizarse la infección, y la producción de progenie del virus, se requiere que la célula esté en la fase de replicación, que es cuando las polimerasas de la célula están activas. En la figura 6.2 se indica esquemáticamente la estructura de un virus con ADN bicatenario.
Figura 6.2. Virus con ADN bicatenario
6.2.2. Virus con ADN monocatenario
Este tipo de virus posee en su material genético ADN de cadena sencilla y se replica usando una ADN polimerasa dependiente del ADN, al igual que los virus con ADN bicatenario.Para que exista la replicación de esta clase de virus, es necesario que el ADN de cadena simple se convierta en ADN de cadena doble en las células infectadas.
6.2.3. Virus con ARN
Los virus que contienen ARN se replican en el citoplasma y no el núcleo de la célula infectada. Los virus ARN se pueden clasificar en cuatro grupos según su modo de replicación.
Figura 6.3. Imagen de un virus con ARN (flavivirus)

Tema 6. Enfermedades víricas 3
6.2.3.a Virus con ARN bicatenario
Los virus con ARN bicatenario se replican en el citoplasma y no dependen de las polimerasas de la célula hospedadora como lo hacen los virus ADN, pues incluyen estas enzimas en el virión. La traducción suele ser monocistrónica, lo que significa que cada uno de los segmentos codifica una sola proteína, a diferencia de otros virus que exhiben una traducción más compleja. Una característica particular de estos virus es su capacidad para llevar a cabo la transcripción de los segmentos de ARN bicatenarios bajo las condiciones apropiadas dentro de la cápside.
6.2.3.b. Virus con ARN monocatenario positivo
En virología un ARN viral de sentido positivo es el que puede ser directamente traducido a las proteínas virales deseadas. Así, el genoma ARN viral es idéntico al ARNm viral y puede ser inmediatamente traducido usando la maquinaria de traducción de la célula huésped. La replicación tiene lugar principalmente en el citoplasma. Una de las proteínas codificadas por los ARN positivos es la ARN replicasa, que es una ARN polimerasa que copia el ARN viral sin necesidad de pasar por una cadena de ADN intermedia.
6.2.3.c. Virus con ARN monocatenario negativo
El ARN de sentido negativo no puede ser traducido directamente a una proteína, por lo que tiene que ser convertido en un ARNm de sentido positivo. Los virus ARN de sentido negativo utilizan una ARN polimerasa o transcriptasa para formar ARN de sentido positivo. Esto significa que el virus debe aportar la enzima ARN polimerasa puesto que ésta es dependiente del ARN. Una vez fabricada la molécula de ARN de sentido positivo ésta actúa como un ARNm viral traduciéndose en proteínas por los ribosomas del huésped. Las proteínas resultantes se dedican directamente a la producción de los elementos de los nuevos viriones, tales como las proteínas de la cápside y la ARN replicasa, que se encarga de la producción de nuevas moléculas de ARN de sentido negativo.
6.2.3.d. Virus con ARN monocatenario y bicatenario retrotranscrito
Un virus que contiene ARN monocatenario retrotranscrito (o virus ssRNA-RT) es un virus con ARN de cadena sencilla en su genoma que se replica en la célula hospedadora mediante transcripción inversa, es decir, mediante la formación de ADN a partir del molde ARN. Estos virus usan transcriptasa reversa codificada viralmente, utilizando una ADN polimerasa dependiente del ARN para producir ADN a partir del genoma de ARN viral. Este ADN a menudo se integra en el genoma del huésped, como en el caso de los retrovirus y seudovirus, donde es replicado y transcrito por el huésped. El virus de la inmunodeficiencia humana (VIH) pertenece a esta clase de virus.

Síntesis de antivirales 4
Figura 6.4. Virus VIH
6.3. Morfologia de los virus
Los virus presentan una amplia diversidad de formas y tamaños llamadas «morfologías». Son unas 100 veces más pequeños que las bacterias. La mayoría de los virus estudiados tienen un diámetro de entre 10 y 300 nanómetros. Algunos filovirus tienen un tamaño total de hasta 1.400 nm, sin embargo, sólo miden unos 80 nm de diámetro.
La mayoría de los virus no pueden ser observados con un microscopio óptico, de manera que se utilizan microscopios electrónicos de barrido y de transmisión para visualizarlos. Para aumentar el contraste entre los virus y el trasfondo se utilizan tinciones densas en electrones, que son soluciones de sales de metales pesados, como wolframio, que dispersan los electrones en las regiones cubiertas por la tinción.
Una partícula vírica completa, conocida como virión, consiste en un ácido nucleico rodeado por una capa de protección proteica llamada cápside que encierra y protege al genoma viral de la acción de nucleasas y otros factores adversos del medio exterior. Además, en los virus desnudos carentes de envoltura, la cápside es la encargada de establecer, a través de alguna de sus proteínas, la unión con la célula que será parasitada por el virus. Asimismo, las proteínas de la cápside contienen los determinantes antigénicos contra los que el sistema inmune del huésped elaborará la respuesta de anticuerpos en defensa del organismo. Las cápsides virales pueden presentar dos tipos básicos de estructura:
a) Cápside icosaédrica, en la que los capsómeros, que suelen ser de varios tipos, se ajustan formando un icosaedro regular (es decir, 20 caras triangulares y 12 vértices), dejando un hueco central donde se sitúa el ácido nucleico fuertemente apelotonado. Cuando este tipo de virus se observan al microscopio se aprecian de forma aproximadamente esférica.
La mayoría de virus que infectan los animales son icosaédricos o casi esféricos. Un ejemplo de virus icosaédricos lo constituyen los adenovirus, entre los que se encuentran los virus de los resfriados y la faringitis.

Tema 6. Enfermedades víricas 5
Figura 6.5. Representaciones de virus icosaédricos
b) Cápside helicoidal, que se compone de un único tipo de capsómero apilado alrededor de un eje central formando una estructura helicoidal que puede tener una cavidad central o un tubo hueco. Esta formación produce viriones en forma de barra o de hilo, que pueden ser cortos y muy rígidos, o largos y muy flexibles. El material genético es normalmente ARN monocatenario, pero a veces también puede ser ADN monocatenario. El ácido nucleico queda unido a la hélice proteica por interacciones entre la carga negativa de aquél con la carga positiva de las proteínas. En general, la longitud de una cápside helicoidal está en relación con la longitud del ácido nucleico que contiene. El diámetro de la cápside depende del tamaño y de la distribución de los capsómeros. El conocido virus del mosaico del tabaco es un ejemplo de virus helicoidal (figura 6.6).
Figura 6.6. Representaciones de virus helicoidales
c) Un tercer tipo de virus, denominados complejos, se caracterizan por poseer una cabeza de estructura icosaédrica que alberga el ácido nucleico, una cola de estructura helicoidal que constituye un cilindro hueco, un collar de capsómeros entre la cabeza y la cola y una placa basal al final de la cola, con unos puntos de anclaje que sirven para fijar el virus a la membrana celular. De la placa salen también unas fibras proteicas que ayudan a la fijación del virus sobre la célula hospedadora. Entre esta clase de virus se encuentran los denominados bacteriófagos, los virus que infectan a las bacterias.

Síntesis de antivirales 6
Figura 6.7. Representaciones de bacteriófagos
6.4. Transmisión
Los virus se diseminan de muchas maneras diferentes y cada tipo de virus tiene un método distinto de transmisión. Entre estos se encuentran los vectores de transmisión, que son otros organismos que los transmiten entre portadores. Los virus vegetales se propagan frecuentemente por insectos que se alimentan de su savia, como los áfidos, mientras que los virus animales se suelen propagar por medio de insectos hematófagos. Otros virus, como el virus de la gripe (rinovirus), no precisan de vectores propagándose por el aire a través de los estornudos y la tos. Los norovirus tampoco necesitan de vectores de propagación y se transmiten por vía fecal-oral, o a través de las manos, alimentos y agua contaminados. Los rotavirus se diseminan, muy a menudo, por contacto directo con niños infectados. El VIH es uno de los muchos virus que se transmiten por contacto sexual o por exposición con sangre infectada.
No todos los virus provocan enfermedades, ya que muchos virus se reproducen sin causar ningún daño al organismo infectado. Algunos virus, como el VIH pueden producir infecciones permanentes o crónicas porque el virus continúa replicándose en el cuerpo, evadiendo los mecanismos de defensa del huésped. En los animales, sin embargo, es frecuente que las infecciones víricas produzcan una respuesta inmunitaria que confiere una inmunidad permanente a la infección. Los microorganismos como las bacterias también tienen defensas contra las infecciones víricas, conocidas como sistemas de restricción-modificación. Los antibióticos no tienen efecto sobre los virus, pero se han desarrollado medicamentos antivirales para tratar infecciones potencialmente mortales.
Los avances en la caracterización de los virus a nivel molecular sugieren que los virus coevolucionan con sus organismos hospedadores, debido a que los virus son parásitos intracelulares extremos y, por lo tanto, requieren de la supervivencia del hospedador para poder asegurar su propia supervivencia. Es interesante notar que cuando un virus se replica en su hospedador natural, tiende a no causar enfermedad en el mismo o a causar una enfermedad leve y autolimitada en la mayoría de los casos. Varios de los virus conocidos producen enfermedades severas sólo cuando infectan organismos diferentes a sus hospedadores naturales. Lo anterior sugiere que buena parte de los virus asociados con la producción de enfermedades son virus que están en proceso de adaptación a un nuevo tipo de hospedador y que, una vez lograda dicha adaptación, la estrategia del virus consiste en perpetuarse y propagarse sin afectar al organismo hospedador.

Tema 6. Enfermedades víricas 7
6.5. Origen evolutivo
El origen evolutivo de los virus aún es incierto. Se piensa que algunos podrían haber evolucionado a partir de plásmidos (fragmentos de ADN que se mueven entre las células), mientras que otros podrían haberse originado desde bacterias. Desde el punto de vista de la evolución de otras especies, los virus son un medio importante de transferencia horizontal de genes, la cual incrementa la diversidad genética.
Los virus son organismos parásitos que infectan células y producen viriones para difundir sus genes. La mayoría de las proteínas virales no tienen homólogos en las células modernas, en contradicción con la visión tradicional de los virus como los «ladrones de genes celulares». Esto sugiere que los genes virales básicamente tienen su origen durante la replicación de los genomas virales y/o fueron reclutados de linajes celulares ahora extintos. Algunas proteínas virales específicas están presentes en virus que infectan a los miembros de los tres dominios de la vida, lo que sugiere que los virus son en realidad muy antiguos. En particular, los análisis estructurales de proteínas de la cápside han revelado que al menos dos tipos de viriones se originaron de manera independiente antes que el último antepasado universal celular, conocido con el acrónimo LUCA (del inglés Last Universal Common Ancestor).
Se pueden encontrar virus dondequiera que haya organismos vivos, y probablemente existen desde la aparición de las primeras células. Su origen es incierto, puesto que no fosilizan, de manera que sólo se puede especular a partir de diferentes técnicas y ensayos de biología molecular. Estas técnicas dependen de la disponibilidad de ADN o ARN vírico antiguo, pero desgraciadamente la mayoría de virus que han sido preservados y almacenados en laboratorios tienen menos de 90 años.
6.6. Propiedades de vida
Existen opiniones dispares sobre si los virus son una forma de vida o estructuras orgánicas que interactúan con los seres vivos. Por ello algunos autores se refieren a ellos como organismos al límite de la vida. Por una parte, los virus se asemejan a los organismos que tienen genes y evolucionan por selección natural, y se reproducen creando múltiples copias de sí mismos para autoensamblarse. Sin embargo, carecen de estructura celular, lo que es considerado como la unidad básica de la vida. Además, los virus no tienen un metabolismo propio, y necesitan una célula hospedadora para crear nuevos metabolitos. Por tanto, no se pueden reproducir en el exterior de una célula hospedadora, aunque bacterias como Rickettsia y Chlamydia son considerados organismos vivos a pesar de tener la misma limitación.
6.7. Genoma
Las distintas especies de virus contienen una diversidad genómica superior a la de los reinos de plantas, animales o bacterias. Hay millones de diferentes tipos de virus y únicamente alrededor de 5.000 de ellos han sido descritos detalladamente. Un virus tiene un genoma compuesto de ADN o de ARN, y recibe respectivamente el nombre de «virus ADN» y «virus ARN». La gran mayoría de virus utilizan el ARN. Los virus de las plantas tienden a tener ARN monocatenario y los bacteriófagos tienden a tener ADN bicatenario.
Los genomas víricos pueden ser circulares, como los polyomaviridae o lineales, como los adenoviridae. El tipo de ácido nucleico es irrelevante para la forma del genoma. En los virus

Síntesis de antivirales 8
ARN, el genoma está a menudo dividido en partes separadas dentro del virión y se le califica de «segmentado». Cada segmento suele codificar una proteína y los segmentos suelen estar reunidos en una cápside.
En los virus ARN o en los virus ADN monocatenarios, las cadenas pueden ser o bien positivas (cadenas plus) o negativas (cadenas minus), que son complementarias con el ARN mensajero (ARNm) vírico.
El ARN viral positivo es idéntico al ARNm viral y por tanto puede ser traducido inmediatamente por la célula huésped.
El ARN viral negativo es complementario del ARNm y por tanto debe ser convertido en ARN positivo por una ARN polimerasa antes de ser traducido, siendo la nomenclatura del ADN similar a la del ARN viral negativo.
El tamaño del genoma varía mucho entre especies. Los genomas víricos más pequeños sólo codifican cuatro proteínas y pesan unos 106 daltons. Los más grandes pesan unos 108 daltons y codifican más de un centenar de proteínas. Los virus ARN suelen tener genomas más pequeños que los virus ADN debido a una tasa de error más alta a la hora de replicarse. Así, los virus ARN tienen un límite superior de tamaño por encima del cual los errores en la replicación del genoma hacen que el virus sea inofensivo o, incluso, incompetente. Para compensar esto, los virus ARN a menudo inician un proceso de segmentación que escinde el genoma en moléculas más pequeñas, reduciendo así las posibilidades de error. En cambio, los virus ADN tienen genomas mayores gracias a la elevada fidelidad de sus enzimas de replicación.
Los virus sufren cambios genéticos por diversos mecanismos como:
a) El proceso de deriva genética. Este mecanismo provoca la mutación de bases individuales del ADN o del ARN. La mayoría de estas mutaciones puntuales son imperceptibles pues la proteína codificada por el gen no cambia, pero aún así, puede conferir ventajas evolutivas como la resistencia a los medicamentos antivíricos.
b) El cambio antigénico, que se produce cuando hay un cambio significativo en el genoma del virus como consecuencia de una recombinación genética. Cuando esto se produce, por ejemplo en los virus de la gripe, pueden resultar pandemias. Los virus ARN suelen existir como quasiespecies o en enjambres de virus de la misma especie pero con secuencias de nucleósidos del genoma ligeramente diferentes. Estos grupos son un objetivo destacado por la selección natural.
c) La recombinación genética, que es el proceso por el cual una cadena de ADN es escindida y luego unida al extremo de una molécula de ADN diferente. Este proceso puede tener lugar cuando diferentes virus infectan las mismas células al mismo tiempo. La recombinación es común en los virus ARN y ADN. Los genomas segmentados ofrecen ventajas evolutivas ya que diferentes cepas de un virus con el genoma segmentado pueden intercambiar y combinar genes, produciendo virus progenénicos (o descendientes) con características únicas, lo que se conoce como «sexo vírico».

Tema 6. Enfermedades víricas 9
6.8. Ciclo de replicación de los virus
El ciclo reproductivo de los virus consta generalmente de las siguientes fases:
1) Fijación
2) Penetración en la célula
3) Eclipse
4) Multiplicación
5) Ensamblaje
6) Liberación
6.8.1. Fijación o adsorción
El primer paso en la infección viral es la fijación del virus sobre la membrana de la célula. Este proceso tiene lugar mediante adhesión a receptores superficiales de la célula diana de los ligandos virales, que son proteínas de la cápside o glucoproteínas de las espículas. Algunos bacteriófagos también son capaces de adherirse a los flagelos, a las vellosidades (pili ) o a las cápsulas presentes en la superficie de la bacteria hospedadora. Para que esto suceda la bacteria debe contener el factor sexual "F" o ciertas colicinas (factores de resistencia contra agentes antimicrobianos). Los bacteriófagos filamentosos con ADN de cadena sencilla se adhieren a las puntas de estos pili , mientras que los bacteriófagos esféricos de ARN se adhieren a los costados de éstos.
La especificidad de unión proteína-cápside se determina por la variedad de hospedadores de los virus. Por ejemplo, el VIH sólo infecta linfocitos T humanos, pues su proteína de superficie, gp120, solo puede interactuar con la CD4 y con receptores de la superficie del linfocito T.
El mecanismo de fijación ha evolucionado para favorecer a los virus que sólo pueden infectar células en las que se pueden replicar.
6.8.2. Penetración
Después de la adhesión del virus se produce la penetración, introduciéndose el virión en la célula huésped por endocitosis mediada por receptores, o por fusión de membrana.
Los virus complejos, como los bacteriófagos, producen una rotura en la membrana celular del hospedador en uno de los puntos de anclaje gracias a la presencia de enzimas hidrolíticas en las proteínas de la cápside. Después de la rotura, el tubo central inyecta el ácido nucleico vírico, quedando la cápside vacía en el exterior de la célula diana y el ácido nucleico libre en el citoplasma de la célula hospedadora. La presencia de cápsides en la superficie celular es un buen indicio de que se ha sufrido una infección vírica.

Síntesis de antivirales 10
Figura 6.8. Penetración de ADN en una célula de E. coli infectada por un macrófago
Otros virus sin envoltura lipídica se introducen en la célula con cápside, lo cual puede realizarse de dos maneras:
a) Por penetración directa después de la fijación. En este caso se produce la apertura de una brecha en la membrana celular que va seguida de la introducción del material vírico en el citoplasma de la célula huésped.
b) Por endocitosis. En esta forma de penetración la membrana celular forma una invaginación en torno al virus, llegando éste a formar una vesícula que penetra en la célula. Una vez formada la vesícula, el virus abre una brecha en la membrana de la misma con ayuda de algunas enzimas hidrolíticas que él mismo transporta, difundiéndose así en el citoplasma.
Los virus con envoltura lipídica burlan la barrera de la membrana celular porque su cubierta lipídica se funde con la membrana, ya que son de la misma naturaleza. Esta fusión de membranas puede realizarse en dos lugares distintos:
c) Fusión en la superficie celular, con penetración directa del virión en el citoplasma.
d) Fusión con un lisosoma, mediante formación de una vesícula por endocitosis a la que se une un lisosoma para digerir la partícula introducida. A continuación, la cubierta lipídica del virus se funde con la membrana del lisosoma y el virión se difunde dentro del citoplasma.
6.8.3. Eclipse
Según la duración de la fase de eclipse, se suelen distinguir dos modalidades de ciclo infeccioso de un virus:
a) Ciclo ordinario o lítico, en el cual el ácido nucleico vírico inicia inmediatamente la transcripción de su mensaje genético en los ARN necesarios para su multiplicación. Este tipo de ciclo es el más extendido en la naturaleza.
b) Ciclo lisogénico, en el cual el ADN vírico se cierra por sus extremos generando un ADN circular. Este ADN se inserta en el ADN bacteriano en un lugar específico, en el cual la secuencia de nucleótidos bacterianos es semejante a la región del ADN vírico. La bacteria prosigue sus funciones vitales sin que el virus realice ninguna acción, y cuando el ADN

Tema 6. Enfermedades víricas 11
bacteriano se duplica también lo hace el ADN vírico, de manera que el genoma del virus pasa a las dos bacterias hijas. La multiplicación bacteriana puede seguir durante generaciones sin que el virus se manifieste. Pero ante una alteración de las condiciones ambientales, el ADN vírico se separa del ADN bacteriano y prosigue entonces las restantes fases de ciclo infeccioso, produciendo la muerte de la bacteria y nuevos ejemplares del virus
Algunos virus que infectan células animales siguen también el ciclo lisogénico, como los papilomavirus de las verrugas y algunos retrovirus que producen algunos tipos de cáncer.
En el caso de los retrovirus conviene recordar que el ácido nucleico es ARN monocatenario, por lo que la transcriptasa reversa ha de copiar el genoma vírico en forma de ADN antes de que pueda insertarse en el ADN celular.
6.8.4. Multiplicación
La multiplicación del virus consta de:
a) La replicación de su material genético.
b) La transcripción de su mensaje en una molécula de ARN.
c) La traducción del mensaje para producir proteínas víricas, tanto las que formarán parte de la cápside, como las proteínas enzimáticas necesarias para el ensamblaje de las piezas del virión y para algunas de las funciones anteriores. Los ribosomas y la mayor parte de las enzimas que los ácidos nucleicos víricos utilizan en estos procesos son los de la célula infectada.
Los virus con ADN realizan la replicación del material genético de la misma manera que las células.
Los virus con ADN monocatenario, previamente a la replicación, sintetizan una cadena de ADN complementario para formar la doble hélice.
Los virus con ARN replican el material genético sin necesidad de pasar por ADN, actuando cada cadena de ARN como molde para la síntesis de su complementaria.
Los retrovirus constituyen una excepción a lo dicho anteriormente, ya que su ARN sintetiza un ADN bicatenario, que será el que posteriormente realice la síntesis de nuevos ejemplares de ARN vírico.
6.8.5. Ensamblaje
Posteriormente a la multiplicación tiene lugar el ensamblaje de las piezas para construir nuevos viriones. En muchos virus, como el del mosaico del tabaco, el ensamblaje es automático y depende de la concentración salina del medio. En otros virus, el ensamblaje necesita de la intervención de enzimas codificadas en el ácido nucleico del virus.
Los virus pueden autoensamblarse en un proceso similar a la cristalización, ya que las partículas virales, al igual que los cristales, constituyen estructuras que se encuentran en un estado de mínima energía libre. Sin embargo, el genoma viral también puede especificar ciertos factores morfogenéticos que no contribuyen directamente a formar la estructura del virión, pero son necesarios para el proceso de ensamblaje.
Tras el ensamblaje de partículas víricas, a menudo se produce una modificación postraduccional de las proteínas víricas. En virus como el VIH, esta modificación, a veces

Síntesis de antivirales 12
llamada maduración, se produce después de que el virus haya sido liberado de la célula huésped. 6.8.6. Liberación de los nuevos virus
Después de la multiplicación del virus tiene lugar la salida de los nuevos viriones, que saldrán con capacidad de infectar nuevas células. Las principales modalidades de liberación dan nombre a nuevas variantes del ciclo vital de los virus, que se explican a continuación.
6.8.6.a. Infección persistente
En el proceso de infección persistente los nuevos virus no esperan a la muerte de la célula hospedadora para abandonarla, sino que van saliendo de la célula al mismo tiempo que se van produciendo, de manera que la célula puede seguir viva produciendo nuevas partículas víricas. La liberación puede hacerse de dos maneras:
a) Los virus sin envoltura lipoproteica salen directamente, sin arrastrar ningún resto de la membrana plasmática abriendo una brecha en la membrana o aprovechando los mecanismos de exocitosis o de salida de sustancias al exterior de la célula.
b) Los virus con envoltura lipoproteica salen por gemación, rodeándose de una porción de membrana plasmática que acaba separándose de la célula y constituye la cubierta.
En la figura 6.9 se indican esquemáticamente las fases del ciclo de replicación de un bacteriófago.
1. Adsorción viral
2. Penetración y liberacióndel ácido nucleico
3. Integración del ácidonucleico viral
4. Producción deARNm viral
5. Producción de proteinasvirales de la cápside
7. Lisis celular yliberación de nuevosviriones
6. Autoensamblaje delas cápsides
Figura 6.9. Ciclo de replicación de un bacteriófago

Tema 6. Enfermedades víricas 13
6.9. Efectos en la célula hospedadora
La mayoría de infecciones víricas acaban provocando la muerte de la célula hospedadora, entre cuyas causas están la lisis de la célula, las alteraciones de la membrana superficial de la célula y la apoptosis. A menudo, la muerte de la célula es causada por el paro de sus actividades normales debido a la acción de proteínas específicas del virus.
Algunos virus no causan cambios aparentes en la célula infectada. Este es el caso de los virus del herpes simple, incluyendo el virus de Epstein-Barr (que causa mononucleosis infecciosa) y el virus de la varicela zoster (que causa la varicela), que pueden existir de manera relativamente inofensiva en un organismo, permaneciendo en un estado durmiente dentro del cuerpo humano. Las infecciones latentes de varicela pueden provocar en la etapa adulta del ser humano la enfermedad del herpes zóster. Sin embargo, otros virus son altamente dañinos para el organismo hospedador, como los papilomavirus, que son una causa demostrada de cáncer.
6.10. Virus y enfermedades humanas
Las enfermedades humanas provocadas por virus incluyen, entre otras, el resfriado, la gripe, la varicela, el herpes simple, el ébola, el VIH, la hepatitis B, la gripe aviar y el SARS (Severe Acute Respiratory Syndrome).
La capacidad relativa de los virus de provocar enfermedades se describe en términos de «virulencia». Los virus tienen diferentes mecanismos mediante los cuales causan enfermedades a un organismo, que dependen en gran medida en la especie de virus. Los mecanismos a nivel celular incluyen principalmente la lisis de la célula, es decir, la ruptura y posterior muerte de la célula.
Algunos virus pueden causar infecciones permanentes o crónicas, en las cuales los virus continúan replicándose en el cuerpo a pesar de los mecanismos de defensa del hospedador. Este es el proceso habitual en las infecciones provocadas por el virus de la hepatitis B y de la hepatitis C. Los enfermos crónicos son conocidos como portadores, pues sirven de reservorio de los virus infecciosos. En poblaciones con una proporción elevada de portadores, se dice que la enfermedad es endémica. Algunos virus pueden mutar dentro de las células hospedadoras, reforzando sus defensas contra diversos antivirales, proceso conocido como mutación.
6.11. Epidemiología
La epidemiología viral es la rama de la ciencia médica que estudia la transmisión y el control de infecciones víricas en los humanos. La transmisión de virus puede ser vertical (de madre a hijo) u horizontal (de una persona a otra). Ejemplos de transmisión vertical incluyen el virus de la hepatitis B o el VIH, en cual el bebé ya nace infectado con el virus. Otro ejemplo más raro es el virus de la varicela zóster que, normalmente, causa infecciones relativamente leves en los humanos, pero puede resultar fatal para los fetos y los bebés recién nacidos.
La transmisión horizontal es el mecanismo de contagio de virus más extendido. La transmisión puede ser por intercambio de sangre o por el cambio de fluidos en la actividad sexual (infecciones por VIH, hepatitis B y hepatitis C), por el intercambio de saliva (infecciones por virus de Epstein-Barr), por alimentos o agua contaminados (infecciones por norovirus), por la respiración de virus en forma de aerosol (infecciones por el virus de la gripe) o por insectos vectores como los mosquitos (infecciones por dengue o malaria).

Síntesis de antivirales 14
La tasa y la velocidad de la transmisión de infecciones víricas dependen de factores como la densidad de población, el número de individuos susceptibles (los que no son inmunes), la calidad del sistema sanitario y el tiempo.
La epidemiología se utiliza para romper la cadena de infecciones en poblaciones durante brotes de enfermedades víricas, utilizándose medidas de control basadas en el conocimiento del modo de transmisión del virus.
Una vez identificado el virus se puede romper la cadena de infecciones por medio de vacunas. Cuando no se puede contar con vacunas pueden resultar eficientes el saneamiento y la desinfección. A menudo se aíslan las personas o animales infectados del resto de la comunidad y los que han estado expuestos al virus son puestos en cuarentena. Para controlar el brote de fiebre aftosa en bovinos británicos en 2001, se sacrificaron miles de cabezas de ganado.
La mayoría de infecciones víricas de los humanos y otros animales tienen un periodo de incubación durante el cual la infección no causa ningún síntoma. Los períodos de incubación de las enfermedades víricas van desde unos días hasta semanas. Tras el periodo de incubación hay un «periodo de comunicabilidad», un tiempo durante el cual el individuo o animal infectado es contagioso y puede infectar a otra persona o animal. Este periodo también es conocido en muchas infecciones, y el conocimiento de la longitud de ambos periodos es importante en el control de brotes. Cuando un brote causa una proporción inusualmente elevada de infecciones en una población, comunidad o región, se le llama epidemia. Si un brote se extiende en todo el mundo se le llama pandemia.
6.11.1. Epidemias y pandemias
Una pandemia es una epidemia global. Las poblaciones amerindias fueron devastadas por enfermedades contagiosas, especialmente la viruela, llevada a América por los colonos europeos. La pandemia de gripe de 1918, a menudo llamada gripe española, fue una pandemia de gripe de categoría 5 provocada por un virus de la gripe A inusualmente grave y mortal. Las víctimas a menudo eran adultos jóvenes sanos, en contraste con la mayoría de brotes de gripe, que afectan predominantemente niños, ancianos o pacientes débiles. La pandemia de gripe española duró de 1918 a 1919. Las estimaciones más recientes sugieren que podrían haber muerto hasta 100 millones de personas, un 5% de la población mundial en 1918.
La mayoría de investigadores creen que el VIH se originó en el África subsahariana durante el siglo XX. Actualmente es una pandemia, con un número estimado de 38,6 millones de enfermos en todo el mundo. El Programa Conjunto de las Naciones Unidas sobre el VIH/SIDA (UNAIDS) y la Organización Mundial de la Salud (OMS) estiman que el sida ha matado a más de 25 millones de personas desde que la enfermedad fue reconocida por primera vez el 5 de junio de 1981, siendo una de las epidemias más destructivas de la historia. En 2007 hubo 2,7 millones de infecciones con VIH.
Algunos patógenos víricos muy letales son miembros de la familia de los Filoviridae. Los Filovirus son virus similares a filamentos que causan la fiebre hemorrágica vírica, e incluyen el Ébola y los virus de Marburg. El virus de Marburg atrajo la atención de la prensa en abril de 2005 por un brote en Angola. El brote, que comenzó en 2004 y se extendió en 2005, fue la peor epidemia del mundo de cualquier tipo de fiebre hemorrágica vírica.

Tema 6. Enfermedades víricas 15
En 2009 surgió en México una supuesta pandemia de Influenzavirus A (H1N1), conocida como Virus H1N1/09 Pandémico. El origen de la infección es una variante de la cepa H1N1, con material genético proveniente de una cepa aviaria, dos cepas porcinas y una humana que sufrió una mutación y dio un salto entre especies (o heterocontagio) de los cerdos a los humanos, contagiándose posteriormente de persona a persona. La pandemia fue clasificada, según la OMS, de Nivel 6. Aproximadamente, unas 14.000 personas han muerto en todo el mundo a causa de esta enfermedad.
6.12. Virus y cáncer
Los virus son una causa establecida de cáncer en los humanos y otras especies. Los virus que producen cáncer pueden provenir de muchas familias, tanto de virus ADN como de virus ARN, y no únicamente del oncovirus, un término obsoleto para referirse a los retrovirus. El desarrollo del cáncer puede deberse a gran cantidad de factores como la debilidad inmunitaria del huésped y mutaciones en éste. Los virus más importantes asociados con cánceres humanos son el papilomavirus humano, el virus de la hepatitis B, el virus de Epstein-Barr, y el virus T-linfotrópico humano. El más reciente descubrimiento de un virus que causa cáncer es el poliomavirus (Merkel cell polyomavirus) que es la causa de un raro cáncer de piel denominado carcinoma de células de Merkel. Los virus de la hepatitis pueden causar una infección crónica que provoca cáncer de hígado. La infección con virus T-linfotrópico humano puede causar paraparesia espástica tropical y leucemia de linfocitos T del adulto. Los papilomavirus humanos son una causa establecida de cáncer de cérvix, piel, ano y pene. Dentro de los Herpesviridae, el human herpesvirus 8 causa sarcoma de Kaposi y linfoma de las cavidades corporales, y el virus de Epstein-Barr causa linfoma de Burkitt, enfermedad de Hodgkin, trastorno linfoproliferativo de los linfocitos B y carcinoma nasofaríngeo. El Merkel cell polyomavirus está estrechamente relacionado con el SV40 y con los poliomavirus del ratón que han sido usados como modelos de animales para los virus del cáncer desde hace 50 años.
Figura 6.10. Infección de una célula por virus asociado a cáncer

Síntesis de antivirales 16
6.13. Respuesta inmune del hospedador
La primera línea de defensa del organismo contra los virus es el sistema inmunitario innato, que incluye a las células y a los mecanismos que defienden al organismo de la infección de una forma no específica. Así, las células del sistema innato reconocen y responden a los agentes patógenos de una manera genérica, pero, a diferencia del sistema inmune adaptativo, no confieren protección de larga duración o inmunidad.
Muchos virus tienen una estrategia de replicación que implica ARN bicatenario (dsARN). Cuando tales virus infectan a una célula y liberan sus moléculas de ARN, inmediatamente una proteína compleja denominada DICER se une al ARN y lo corta en pedazos más pequeños.1 La protección celular se pone en marcha mediante la activación de una vía bioquímica denominada complejo RISC que degrada el ARNm viral. Los rotavirus inhiben este mecanismo evitando desnudarse completamente dentro de la célula de manera que el dsARN genómico continúa protegido en el interior del núcleo del virión, liberando los nuevos ARNm producidos a través de los poros de la cápside.
Figura 6.11. Imagen de dos rotavirus: el derecho está cubierto por anticuerpos que impiden la adhesión a la célula huésped
Cuando el sistema inmunitario adaptativo de un vertebrado encuentra un virus, produce anticuerpos específicos que se unen al virus y lo hacen no infeccioso, lo que se denomina inmunidad humoral.
Existen dos tipos de anticuerpos antivirales. El primero se denomina IgM (Inmunoglobulina M) y es altamente eficaz para neutralizar los virus, pero sólo es producido por las células del sistema inmune durante unas pocas semanas. El segundo, denominado IgG (Inmunoglobulina G), se produce indefinidamente.
La presencia de IgM en la sangre del huésped se utiliza para determinar una infección aguda, mientras que el IgG indica una infección en el pasado. Los dos tipos de anticuerpos se analizan cuando se llevan a cabo las pruebas de inmunidad.
Una segunda línea de defensa de los vertebrados frente a los virus se denomina inmunidad celular. Las células del organismo muestran cortos fragmentos de sus proteínas en la superficie celular. Si un linfocito T reconoce en una célula un fragmento sospechoso de ser viral, destruye
1 Para una visualización animada del modo de actuación de DICER véase: http://www.youtube.com/watch?v=RdxtoTRkHSk

Tema 6. Enfermedades víricas 17
dicha célula y, a continuación, se produce una proliferación de los linfocitos T específicos para ese virus.
Los interferones, que son glicoproteínas, impiden la replicación de una amplia variedad de virus de tipo ssARN y ssADN. Los interferones son secretados por las células infectadas estimulando, en las células no infectadas, la producción de proteínas que inhiben la replicación de diferentes tipos de virus.
Figura 6.12. Interferon beta-05
No todas las infecciones por virus producen una respuesta inmune protectora como la que se acaba de comentar. El VIH evade al sistema inmunológico por el cambio constante de la secuencia de aminoácidos de las proteínas en la superficie del virión. Estos persistentes virus eluden el control mediante el secuestro y bloqueo de la presentación antigénica, resistencia a las citoquinas, evasión a las actividades de los linfocitos T, inactivación de la apoptosis y mediante el cambio antigénico. Otros virus, denominados virus neurotróficos, se propagan en el sistema neural, donde el sistema inmunológico es incapaz de llegar a ellos.
6.14. Vacunas
La vacunación es una forma barata y eficaz en la prevención de las infecciones causadas por los virus. Las vacunas se han utilizado para prevenir las enfermedades virales desde mucho antes del descubrimiento de los virus. Su uso ha dado lugar a una espectacular disminución de la morbilidad (enfermedad) y mortalidad (muerte) asociada a infecciones virales como poliomielitis, sarampión, paperas y rubeola.
En la actualidad se dispone de vacunas para prevenir más de trece infecciones virales en los seres humanos y algunas otras se utilizan para prevenir infecciones virales en animales. La vacunación provoca la infección en condiciones controladas, estimulando el sistema inmune del individuo vacunado para que produzca un arsenal de anticuerpos y células inmunes con capacidad para destruir o neutralizar cualquiera otra invasión por parte del mismo agente infeccioso.
Las vacunas pueden consistir en virus vivos atenuados, en virus muertos, o contener sólo las proteínas virales (antígenos).

Síntesis de antivirales 18
Las vacunas vivas contienen formas debilitadas del virus que causa la enfermedad. Las vacunas vivas pueden ser peligrosas cuando se administran a las personas inmunodeficientes, puesto que en estas personas incluso el virus debilitado puede causar la enfermedad original. Sin embargo, la vacuna contra el virus de la fiebre amarilla, obtenida de una cepa atenuada denominada 17D, es posiblemente una de las vacunas más seguras y eficaces fabricadas hasta la fecha.
La biotecnología y las técnicas de ingeniería genética se utilizan para producir vacunas de subunidades. Estas vacunas usan sólo la cápside de proteínas del virus. La vacuna de la hepatitis B es un ejemplo de este tipo de vacuna. Las vacunas de subunidades son seguras para pacientes inmunodeficientes, ya que no pueden causar la enfermedad.
6.15. El virus de la inmunodeficiencia humana (VIH)
El virus de la inmunodeficiencia humana (VIH) fue descubierto por Luc Montagnier en Francia en 1983. La estructura del virus es esférica, con un diámetro de aproximadamente 100 nanómetros (véase la figura 6.13). Su parte exterior contiene una membrana, que originalmente pertenecía a la célula de donde el virus emergió, en la que se encuentra una proteína del virus, la gp41, o glicoproteína transmembrana. Conectada a la gp41 está la gp120. Estos antígenos proteicos de la envoltura exterior se acoplan de forma específica con proteínas de la membrana de las células infectables, especialmente de los linfocitos T4.
Figura 6.13. Virus VIH
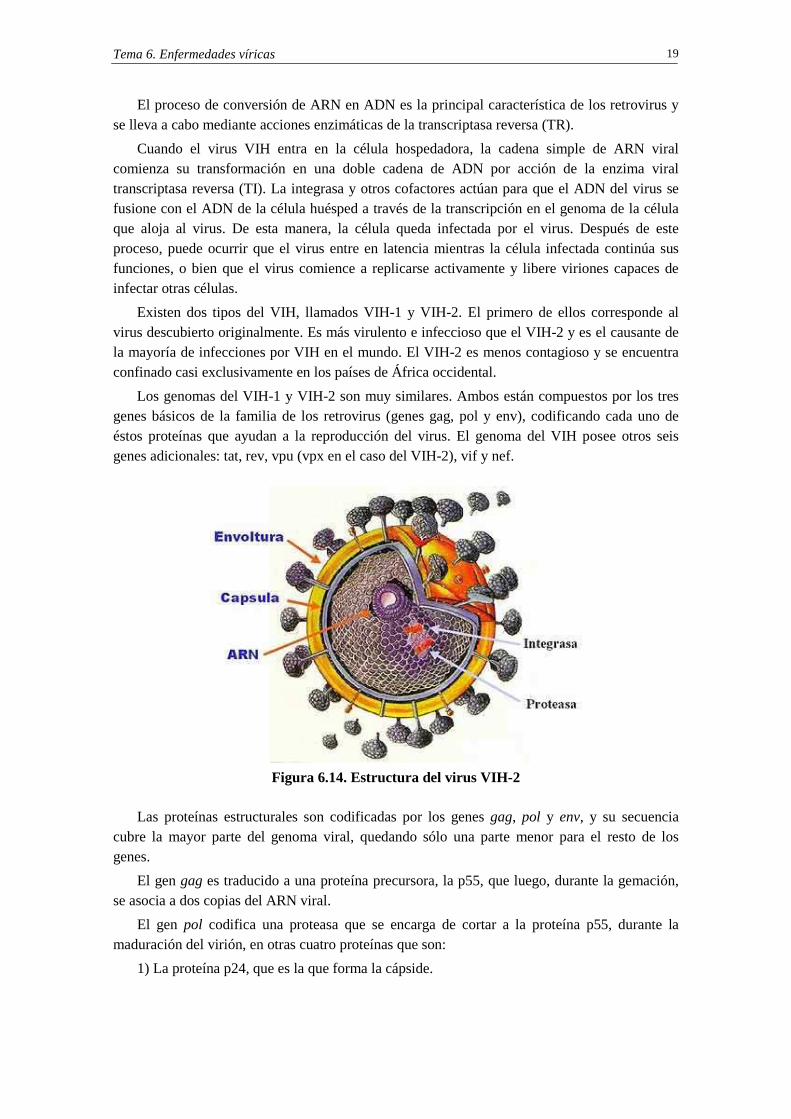
En el núcleo del virus se encuentra la cápside, compuesta por la proteína p24. En su interior está el ARN que contiene varios genes, cada uno de los cuales codifica las diversas proteínas que el VIH necesita para reproducirse. Dentro de la cápside también se encuentran las transcriptasas reversas (TR, también denominas transcriptasas inversas) y las integrasas.
El genoma del virus está contenido en una cadena de ARN monocatenario (ssARN) , que debe copiarse provisionalmente a ADN para poder multiplicarse e integrarse en el genoma de la célula que infecta.
Tema 6. Enfermedades víricas 19
El proceso de conversión de ARN en ADN es la principal característica de los retrovirus y se lleva a cabo mediante acciones enzimáticas de la transcriptasa reversa (TR).
Cuando el virus VIH entra en la célula hospedadora, la cadena simple de ARN viral comienza su transformación en una doble cadena de ADN por acción de la enzima viral transcriptasa reversa (TI). La integrasa y otros cofactores actúan para que el ADN del virus se fusione con el ADN de la célula huésped a través de la transcripción en el genoma de la célula que aloja al virus. De esta manera, la célula queda infectada por el virus. Después de este proceso, puede ocurrir que el virus entre en latencia mientras la célula infectada continúa sus funciones, o bien que el virus comience a replicarse activamente y libere viriones capaces de infectar otras células.
Existen dos tipos del VIH, llamados VIH-1 y VIH-2. El primero de ellos corresponde al virus descubierto originalmente. Es más virulento e infeccioso que el VIH-2 y es el causante de la mayoría de infecciones por VIH en el mundo. El VIH-2 es menos contagioso y se encuentra confinado casi exclusivamente en los países de África occidental.
Los genomas del VIH-1 y VIH-2 son muy similares. Ambos están compuestos por los tres genes básicos de la familia de los retrovirus (genes gag, pol y env), codificando cada uno de éstos proteínas que ayudan a la reproducción del virus. El genoma del VIH posee otros seis genes adicionales: tat, rev, vpu (vpx en el caso del VIH-2), vif y nef.
Figura 6.14. Estructura del virus VIH-2
Las proteínas estructurales son codificadas por los genes gag, pol y env, y su secuencia cubre la mayor parte del genoma viral, quedando sólo una parte menor para el resto de los genes.
El gen gag es traducido a una proteína precursora, la p55, que luego, durante la gemación, se asocia a dos copias del ARN viral.
El gen pol codifica una proteasa que se encarga de cortar a la proteína p55, durante la maduración del virión, en otras cuatro proteínas que son:
1) La proteína p24, que es la que forma la cápside.

Síntesis de antivirales 20
2) La proteína p17, que constituye la matriz, situada bajo la envoltura, a la que estabiliza.
3) Las proteínas p6 y p7 (ó p9), que son las que forman la nucleocápside. La región de la p55 correspondiente al polipéptido p6 es responsable de la incorporación de la proteína accesoria Vpr (producto de la traducción del gen vpr) al virión en formación y de la interacción con la membrana de la célula que hace posible la gemación. La p7 (ó p9) es responsable del reconocimiento y la incorporación del ARN al virión y además interviene en la transcripción inversa.
Dentro de la cápside coexisten dos copias idénticas del ARN viral junto con las enzimas necesarias para la multiplicación del virus: una transcriptasa reversa, una integrasa, una proteasa y una ARNasa. Todos estos enzimas se producen a partir de la proteína Pol.
a) La transcriptasa reversa (p50), que es una ADN-polimerasa y tiene como función la síntesis del ADN de doble cadena del provirus, usando como patrón la cadena singular del ARN viral. Así, una vez formada la primera cadena de ADN, complementaria del ARN viral, la ARNasa lo separa de él, lo que permite a la transcriptasa inversa ejecutar la síntesis de la segunda cadena de ADN tomando como molde la primera que se formó. Así pues, en la síntesis de la primera cadena la actividad de la transcriptasa reversa es ARN-dependiente, pero en la síntesis de la segunda cadena es ADN-dependiente.
Existen múltiples fármacos que ejercen su acción inhibiendo la actividad de la transcriptasa reversa.
b) La integrasa (p31), que realiza la inserción del ADN proviral en el genoma de la célula hospedadora. No se requiere ATP para su actividad y debe cumplir sucesivamente tres funciones:
.- Actividad exonucleasa, cortando dos núcleótidos del extremo 3' de cada una de las dos cadenas del ADN proviral.
.- Actividad endonucleasa (de doble cadena), cortando el ADN de la célula huésped en el punto de integración.
.- Actividad ligasa, uniendo el ADN proviral en el ADN celular mediante un sólo enlace covalente en cada extremo.
En la actualidad existe un fármaco comercializado contra la actividad de la integrasa denominado raltegravir.
Figura 6.15. Imágenes de la integrasa de VIH

Tema 6. Enfermedades víricas 21
c) La proteasa (p10), que es una aspartil-proteasa que actúa cortando las piezas de las proteínas Gag, Pol y Gag-Pol. Una parte de los fármacos empleados contra el VIH son inhibidores de su función.
d) La ARNasa (p15), que separa la cadena de ARN de la de ADN durante el proceso de la transcripción inversa.
La envoltura del VIH consta de una bicapa lipídica, lo mismo que cualquier membrana biológica, y sus componentes estructurales básicos proceden de la membrana plasmática de la célula parasitada. La envoltura del VIH porta regularmente espaciadas 72 espículas, que son complejos proteicos integrados en la membrana formados por proteínas virales codificadas por el gen env. Cada espícula está formada por una pieza de la proteína gp41, integrada en la membrana, y una cabeza externa formada por la proteína gp120, esencial para el acoplamiento con el exterior de las células que van a ser infectadas por el VIH.
Cuando no se produce la interacción con receptores de la superficie celular, la gp41 está cubierta por la gp120, lo que la hace indetectable por el sistema inmunológico. Una vez que la gp120 se ha unido con receptores CD4 y otros correceptores de la célula hospedador, la gp120 cambia su conformación y permite la exposición de la gp41, que asiste en la fusión del virus con la célula hospedadora.
Glicoproteína gp120
Glicoproteína gp41
Membrana viral
ARN
Transcriptasa
reversa
Figura 6.16. Virus VIH mostrando las proteínas transmembrana gp120 y gp41
Entre los dos componentes de las espículas existe una unión no covalente. Las proteínas gp41 y gp120 se sintetizan como una sola poliproteína, gp160, con la información del gen env

Síntesis de antivirales 22
antes de que sea cortada por una proteasa de la célula. La proteína Env existe como trímero en la superficie de los viriones y las células infectadas.
Los fármacos inhibidores de la fusión funcionan contra la proteína gp41, para evitar su unión a los linfocitos.
6.15.1. Proteínas reguladoras
La proteína Tat existe en dos formas, una larga, de 101 aminoácidos de longitud, y otra más corta, de sólo 72. La segunda se produce cuando en fase temprana se produce una edición completa del ARNm viral, la primera cuando en una fase más tardía sólo se realiza una edición parcial. La proteína Tat (por transactivator) promueve activamente la producción de nuevos viriones. Así, se une a una región de 59 nucleótidos situada en el extremo 5' del ARN viral llamada TAR (Transactivator Active Region) y actúa como un transactivador, algo excepcional, puesto que éstos suelen unirse al ADN, no al ARN. En cuanto este extremo inicial del genoma viral ha sido transcrito desde el ADN proviral, la proteína Tat se une a él y promueve su elongación favoreciendo la transcripción del resto de la cadena.
6.15.1.a. Rev
La proteína Rev regula la expresión del ARN viral controlando el ritmo de exportación del ARNm.
6.15.1.b. Tat y Rev: acción conjunta
La acción sinergística de Tat y Rev incrementa fuertemente la expresión de proteínas virales. Los papeles que Tat y Rev desempeñan en la regulación transcripcional del VIH-1 y en la expresión de proteínas estructurales, respectivamente, hacen que Tat y Rev sean esenciales para el ciclo de vida del VIH. Sus funciones facilitan la expresión de proteínas virales en dos etapas. Después de la integración del ADN proviral y de su transcripción en un nivel basal, solamente los ARNm de 2 KB se transportan al citoplasma, lo que permite la síntesis de Tat, Rev y Nef. Una vez sintetizadas, las proteínas Tat y Rev son transportadas al núcleo, donde actúan aumentando la transcripción del ADN del provirus (Tat) y el transporte de todos los ARNm virales al citoplasma (Rev).
6.15.2. Proteínas accesorias
6.15.2.1. Vif: incremento en infectividad y protección del genoma viral
Vif es una proteína de 193 aminoácidos que está presente en bajos niveles dentro de los viriones donde interactúa con ARN genómico viral. La proteína Vif se emplea en la eliminación del factor ApoBEC3G, una desaminasa de citidinas que convierte la citosina en uracilo, y emplea como sustrato el ADN de cadena sencilla.
6.15.2.2. Vpu
La Vpu es una proteína de 81 aminoácidos que es insertada en las membranas vía su terminal nitrogenado. La Vpu se acumula en el aparato de Golgi y en los endosomas celulares. La Vpu es única en VIH-1 y no hay homólogos en lentivirus relacionados como el VIH-2 y el VIS. A la VPU se le han atribuido dos actividades que se explican a continuación.

Tema 6. Enfermedades víricas 23
6.15.2.2.a. Degradación de la proteína CD4
En ausencia de la Vpu, la proteína CD4 interactúa con la proteína viral gp160 recién sintetizada para formar un complejo insoluble, el cual retiene gp120 dentro de la célula. La región citoplásmica de la Vpu se puede unir con CD4 y con la proteína β-TrCP lo que induce la ubiquitinizacion de CD4 y su subsiguiente degradación por el proteasoma, incrementando así la expresión de gp120 en la superficie celular.
6.15.2.2.b. Desprendimiento de viriones de la membrana celular
Esta actividad depende de la región transmembranal de la Vpu. En ausencia de Vpu, los viriones se acumulan en la superficie celular en un estado parcialmente desprendido. La expresión de la Vpu tiene como resultado la liberación de viriones de la membrana celular. Este efecto no está restringido solamente al VIH-1, puesto que la Vpu también facilita el desprendimiento de otros virus no relacionados. El mecanismo mediante el que actúa la Vpu no está claro aunque se ha sugerido que la Vpu facilita la fluidez de la membrana celular por medio de un canal de cationes y también que la Vpu causa disrupción de interacciones entre proteínas del VIH y de la superficie celular; lo que previene la endocitosis de viriones recientemente desprendidos de la célula.
6.15.3. Ciclo de replicación
Las células que el VIH invade son esencialmente los linfocitos T CD4+, pero también en menor medida los monocitos/macrófagos, las células dendríticas, las células de Langerhans y las células de microglía del cerebro. La replicación viral tiene lugar en tejidos diversos, como por ejemplo de ganglios linfáticos, intestino, cerebro, timo, etc. Los órganos linfoides, sobre todo los ganglios linfáticos, constituyen la principal sede de su replicación. El virus está presente en numerosos líquidos del organismo, en particular la sangre y las secreciones genitales.
La replicación del virus se desarrolla en las siguientes etapas:2
1) Fijación. La fijación representa la primera etapa en la invasión de una célula. Se basa en el reconocimiento mutuo y acoplamiento de proteínas de la envoltura del virión, las gp120 y gp41, y los receptores de la célula blanca, los CD4. Este reconocimiento no es posible sin ayuda de correceptores propios de las células susceptibles de ser invadidas, que en el caso de los macrófagos son los CCR5 y en el caso de los LT4, los CXCR4, que interactúan con la proteína superficial. Los macrófagos y los LT4 tienen en común el receptor CD4. Este reconocimiento es condición obligada para que el virus llegue a penetrar en la célula y continuar con el proceso de infección.
2 En las siguientes páginas web se puede visualizar el proceso de replicación del virus VIH: http://www.youtube.com/watch?v=36UDFKEpc2E http://www.youtube.com/watch?v=hdgNnXLY8LU

Síntesis de antivirales 24
Figura 6.17. Acoplamiento de la proteína gp120 a un receptor CD4 de linfocito
2) Penetración. Una vez reconocido el virión por los receptores de superficie se vacía dentro de la célula, fusionándose la envoltura lipídica del virión con la membrana plasmática de la célula. Los dos ARN mensajeros, que forman el genoma viral, y sus proteínas asociadas pasan al citoplasma de la célula hospedadora protegidos por la cápside y las nucleocápsides.
3) Eliminación. En el tercer paso se produce la eliminación de las cubiertas proteicas: cápside y nucleocápsides. Una vez dentro de la célula hospedadora, el ARN vírico es liberado en el citoplasma y listo para ser procesado.
4) Transcripción inversa. El cuarto paso es el de la transcripción inversa del ARN vírico para formar ADNc (ADN complementario, monocatenario). Cada una de las dos moléculas de ARN llega desde el virión asociada a una molécula de transcriptasa reversa que es la encargada de traducir el ARN en ADNc. Las moléculas de ADNc se asocian para formar una molécula de ADN, que es la forma en la que guarda la información la célula eucariota. El paso siguiente es la integración del genoma vírico en el genoma de la célula huésped. Para ello el ADN viral penetra en el núcleo y se inserta en el ADN celular con ayuda de una integrasa, que procede del virión infectante.
La transcripción del ADN vírico se lleva a cabo mediante la maquinaria bioquímica que contiene la célula hospedadora. El resultado de la transcripción es un ARNm que está constituido por una sucesión de intrones (partes no informativas) y exones (partes informativas). El ARNm viríco debe ser procesado mediante cortes y reempalmes antes de que la información que contiene pueda servir para fabricar las proteínas víricas.

Tema 6. Enfermedades víricas 25
Figura 6.18. Ciclo de replicación del VIH
5) Salida del ARNm. En el quinto paso el ARNm sale, a través de las nucleoporinas, del núcleo de la célula huesped y pasa al citoplasma.
6) Traducción del ARNm. Una vez en el citoplasma el ARNm proporciona la información para su traducción en proteínas. Esta operación se realiza usando el sistema de la célula hospedadora. El resultado de la traducción no consiste inmediatamente en proteínas víricas funcionales, sino en poliproteínas que deben ser cortadas en fragmentos. En este punto entran en acción las proteasas específicas del VIH que cortan las poliproteínas producto de la traducción dando lugar a las proteínas constitutivas del virus. Las proteínas víricas fabricadas se ensamblan, junto con ARN provirales, para formar los componentes internos de la estructura del virión que constituyen la cápside y su contenido.
7) Gemación. El último paso del proceso infeccioso es la gemación. Los nucleoides víricos se aproximan a la membrana plasmática y se hacen envolver en una verruga que termina por desprenderse, formando un nuevo virión o partícula infectante. En cada célula infectada se

Síntesis de antivirales 26
ensamblan varios miles de nuevos viriones, aunque muchos son incompletos y no pueden infectar.
Figura 6.19. Imágenes de la transcriptasa reversa mostrando la molécula de ARN y de ADN transcrito
El VIH sólo se puede transmitir a través del contacto entre fluidos corporales que poseen una alta concentración viral. El virus no se transmite de manera casual. No se han reportado casos en los que abrazos, besos secos o saludos con las manos hayan sido causantes de infección. El virus ha sido aislado en la saliva, las lágrimas y la orina, el semen, el líquido preseminal, los fluidos vaginales, el líquido amniótico, la leche materna, el líquido cefalorraquídeo y la sangre, entre otros fluidos corporales humanos.
6.16. Fármacos antivirales
El primer agente antiviral verdaderamente eficiente fue el aciclovir, que se empleó como tratamiento profiláctico del herpes genital y cutáneo, y también en el tratamiento de las lesiones causadas por el Herpes zoster.
Durante los últimos veinte años, el desarrollo de fármacos antivirales ha continuado aumentado impulsado por la epidemia del sida. Los medicamentos antivirales son a menudo análogos de nucleósidos (falsos nucleósidos, los bloques de construcción de los ácidos nucleicos) que los virus incorporan a sus genomas durante la replicación. Los análogos de nucleósidos consiguen detener el ciclo de vida del virus porque las nuevas cadenas de ADN sintetizadas son defectuosas debido a que los análogos de nucleósidos carecen de los grupos hidroxilos que, junto con los grupos fosfato, forman los enlaces de la columna vertebral de la molécula de ADN.
Ejemplos de análogos de nucleósidos son el aciclovir, que se emplea en el tratamiento del virus del herpes y la lamivudina, que se utiliza en el tratamiento de las infecciones de VIH y

Tema 6. Enfermedades víricas 27
hepatitis B. El aciclovir es uno de los fármacos antivirales más antiguos y más frecuentemente prescritos.
Aciclovir
N
N
NH2
O
O
SHO
Lamivudina
NH
N
O
OHOO
N3
H3C
Azidotimidina (AZT)
NHOO
HO HO
N
N
O
NH2
Ribavirina
NH
NN
N
O
NH2O
HO
Figura 6.20. Estructuras de fármacos antivirales
La hepatitis C es causada por un virus ARN. En el 80% de las personas infectadas, la enfermedad es crónica y sin tratamiento continúan siendo infecciosas para el resto de sus vidas. El tratamiento actual de la hepatitis C emplea el fármaco ribavirina, un análogo de nucleósido, en combinación con interferón. Actualmente se está desarrollando una estrategia similar con lamivudina para el tratamiento de los portadores crónicos de hepatitis B. Otros fármacos antivirales en uso tienen como objetivo diferentes etapas del ciclo replicativo viral.
El VIH depende de una enzima proteolítica denominada proteasa VIH-1 para ser plenamente infeccioso. Existe una clase de medicamentos, denominados inhibidores de la proteasa, que han sido diseñados para inactivar esta enzima.
El sida, provocado por el VIH, tiene un tratamiento antiviral de zidovudina (azidotimidina o AZT). La zidovudina es un potente inhibidor de la transcriptasa reversa (TR), enzima esencial en el proceso de replicación del VIH.
Figura 6.21. Modo de acción de los inhibidores de la transcriptasa reversa
Desgraciadamente, los efectos del AZT no son duraderos y, en algunos casos, son inútiles porque la zidovudina no tiene ningún efecto sobre el ADN vírico (provirus), ya que sólo inhibe la formación de éste pero no su expresión en las células huésped.
Por otro lado, el consumo prolongado de zidovudina puede provocar mutaciones del VIH, haciendo que el virus se haga resistente al tratamiento.

Síntesis de antivirales 28
6.16.1. Inhibidores de la Transcriptasa Reversa Análogos de Nucleósidos (ITRN)
El tratamiento actual de la infección por VIH se basa en una combinación de fármacos que incluye un inhibidor de la transcriptasa reversa análogo de nucleósido (ITRN), un inhibidor de la transcriptasa reversa no análogo de nucleósido (ITRNN) y un inhibidor de proteasa (IP). De entre los fármacos ITRN destacan la azidotimidina (AZT), la didanosina (Videx®, zalcitabina®), la lamivudina (3TC) y la estavudina (Zerit®).
Al grupo de medicamentos anteriores se le ha añadido la emtricitabina (Emtriba®) y el abacavir (Ziagenavir®). Este compuesto es un análogo de guanosina con una excelente actividad antiviral in vitro. Desafortunadamente el abacavir produce una reacción de hipersensibilidad hasta en el 3% de los casos y no es activo en contra de virus que son resistentes a AZT o 3TC.
Figura 6.22. Inhibidores de la transcriptasa reversa análogos de nucleósidos (ITRN)
6.16.2. Inhibidores de la Transcriptasa Reversa No Análogos de Nucleósidos (ITRNN)
Los inhibidores de la transcriptasa reversa no análogos de nucleósidos (ITRNN) también inhiben la mencionada enzima pero, aunque son muy potentes, originan una rápida resistencia, incluso con la aparición de una sola mutación. En el grupo de los ITRNN cabe mencionar la nevirapina (Viramune®), el efavirenz (Stocrin®) y la delavirdina (Rescriptor®).
N
HN
NN
H3C O
Nevirapina
NH
O
F3C
Cl
O
Efavirenz
NH
N
N NHN
O
SH3C
O O
HN
H3CCH3
Delavirdina
Figura 6.23. Inhibidores de la transcriptasa reversa no análogos de nucleósidos (ITRNN)

Tema 6. Enfermedades víricas 29
En la figura 6.24 se indica esquemáticamente el modo de acción de los ITRNN (en inglés NNRTI).
Figura 6.24. Modo de acción de los fármacos ITRNN
6.16.3. Inhibidores de Proteasa (IP)
En su proceso de replicación el virus VIH produce largas cadenas de proteínas que necesitan ser fragmentadas en trozos más pequeños para dar lugar a las proteínas y a las enzimas que se empleará el virus en su proceso de reproducción. La fragmentación de las cadenas más largas se lleva a cabo por una proteasa y sus inhibidores impiden dicha fragmentación. De este modo, las proteínas no cortadas originan a copias defectuosas del virus VIH que, si bien pueden destruir la célula que infectaron, ya no pueden infectar más células.
Al producir nuevos virus defectuosos se logra parar la propagación del virus dentro del organismo, consiguiéndose una cronificación de la infección del VIH. Como el organismo del individuo infectado contiene menos virus, son también menos las células CD4 infectadas, con lo que el paciente puede combatir mejor las infecciones y vivir durante más tiempo.
Los inhibidores de la proteasa (IP) no pueden eliminar completamente el VIH del organismo. Sin embargo, con este tipo de medicamentos, se ha observado que pueden reducir la cantidad del VIH hasta en un 99% aunque algunos sigan latentes dentro de las células infectadas.
Entre los fármacos inhibidores de proteasa se encuentran el indinavir (Crixivan®), ritonavir (Norvir®), saquinavir (Invirase®, Fortovase®), nelfinavir (Viracept®), fosamprenavir calcio (Lexiva®, Telxir®), tripanavir (Aptivus®), darunavir (Prezista®) y atazanavir sulfato (Reayataz®).

Síntesis de antivirales 30
Figura 6.25. Estructuras de fármacos inhibidores de proteasa (IP) del VIH

Tema 6. Enfermedades víricas 31
6.17. Síntesis de antivirales
6.17.1. Aciclovir
El año 1962 la compañia Burroughs Wellcome & Company, actualmente GlaxoSmithKline, inició una línea de investigación en drogas antivirales. En 1974 Howard Schaeffer y Lilia Beauchamp, investigadores de Burroughs Wellcome & Co, descubrieron el aciclovir. Las pruebas clínicas se iniciaron en 1977 y en el año 1982 se comercializó el aciclovir para uso tópico. El aciclovir significó el comienzo de una nueva era en la terapia antiviral ya que es extremadamente selectivo y posee un bajo nivel de citotoxicidad. La farmacóloga Gertrude Belle Elion y George H. Hitchings recibieron el Premio Nobel de Medicina en 1988 debido, en parte, al desarrollo de este compuesto.
El aciclovir es un fármaco antiviral que se usa en el tratamiento de las infecciones producidas por el virus herpes humano (VHH), entre las que se incluyen el herpes genital, el herpes bucal, el herpes zóster, la varicela y la mononucleosis infecciosa. Este fármaco impide la replicación viral disminuyendo la extensión y duración de la enfermedad. El aciclovir es un análogo de la guanosina cuyo anillo de ribosa ha sido reemplazado por una cadena abierta.
Figura 6.26. Estructuras de la guanosina y del aciclovir
El aciclovir se considera una prodroga ya que son sus metabolitos las sustancias antivirales activas. Los virus sensibles al aciclovir lo convierten selectivamente, por intervención de una timidina quinasa, en una forma monofosfatada A continuación, el monofosfato, por acción de quinasas celulares, es convertido, primero en difosfato y luego en trifosfato, que es el metabolito activo capaz de inhibir la síntesis de ADN viral (véase el esquema 6.1).
Esquema 6.1. Fosforilación del aciclovir
Uno de los mecanismos de acción de los análogos de nucleósido antivirales se inicia con la penetración de fármaco en la célula. A continuación, el fármaco es fosforilado por quinasas virales, lo que permite su incorporación a la cadena de ADN viral que se está replicando. La incorporación del análogo provoca la terminación de la cadena de ADN viral (véase el esquema

Síntesis de antivirales 32
6.2). El efecto final es la detención de la replicación del virus. Este efecto es muy selectivo en los virus herpes simple tipos 1 y 2 (VHH 1 y 2), virus varicela-zóster (VZV), virus de Epstein-Barr (VEB) y virus herpes humano 6 (VHH6).
Esquema 6.2
La baja toxicidad del fármaco se explica por su escasa afinidad hacia las polimerasas celulares y a que su fosforilación ocurre sólo en células infectadas.
6.17.1.a. Análisis retrosintético
En el esquema 6.3 se indica el análisis retrosintético para el aciclovir que se inicia con la desconexión, basada en una reacción SN2, de la cadena lateral oxigenada. Esta operación genera el sintón aniónico nucleofílico 6.1 y el sintón catiónico electrofílico 6.2. El precursor del equivalente sintético del sintón aniónico 6.1 será la guanina protegida 6.3 mientras que el equivalente sintético del sintón catiónico 6.2 será el haloéter 6.4 (X=halógeno).
Esquema 6.3
6.17.1.b. Síntesis
El compuesto de partida para la síntesis del aciclovir es la guanina 6.5 (esquema 6.4).3 La reacción de este compuesto con bis(trimetilsilil)amina proporciona la guanina protegida 6.3 que reacciona con el 1-benzoiloxi-2-clorometoxietano 6.4 para dar lugar, después de la hidrólisis, al compuesto 6.7. La eliminación del grupo benzoilo, por reacción con una disolución metanólica de amoníaco, permite la obtención del aciclovir.
3 (a) Patente US 4,294,831. (b) Patente US 4,294,831,

Tema 6. Enfermedades víricas 33
Esquema 6.4
El compuesto 6.4 se prepara mediante la secuencia de reacciones que se indica a
continuación:
Esquema 6.5
6.17.1.c. Cuestiones
1) La reacción SN2 entre la guanina protegida 6.3 sobre el cloroéter 6.4 se produce de forma regioselectiva, siendo el grupo NH del anillo imidazólico el que provoca el desplazamiento nucleofílico del cloro ¿por qué el grupo NH del anillo imidazólico es más nucleofílico que el grupo NH del anillo de pirimidinona?
Esquema 6.6
2) Explique mecanísticamente las reacciones implicadas en la síntesis del compuesto 6.4 a partir de benzonitrilo.
6.17.2. Azidotimidina (Zidovudina, AZT)
La azidotimidina, también denominada Zidovudina o AZT, se aprobó en 1987 para el tratamiento de personas infectadas con el VIH. Se comercializa bajo el nombre de Retrovir® y Retrovis®, y es un ingrediente en el Combivir®, Epzicom® y Trizivir®. El AZT fue sintetizado en 1964 por Jerome Horwitz en el Wayne State University School of Medicine como agente anticáncer. El AZT es un análogo de la timidina.

Síntesis de antivirales 34
NH
N
O
OHOO
N3
H3C
Azidotimidina (AZT)
NH
N
O
OHO
O
OH
H3C
Timidina Figura 6.27. Estructuras de AZT y de la timidina
En febrero de 1985, Samuel Broder, Hiroaki Mitsuya y Robert Yarchoan, tres cientificos del National Cancer Institute (NCI), en colaboración con Janet Rideout y otros cientificos del Burroughs Wellcome (ahora GlaxoSmithKline) comenzaron a investigar la potencialidad del AZT como fármaco contra el sida. En ese mismo año los laboratorios Burroughs Wellcome Co. presentaron una patente sobre el AZT.
El 20 de marzo de 1987, la Food and Drug Administration (FDA) dio su aprobación al empleo del AZT como fármaco para su uso contra el VIH. En un principio este fármaco era administrado en elevadas dosis (unos 400 mg cada cuatro horas), lo que provocaba anemia, entre otros efectos secundarios. En los tratamientos actuales se suelen recetar dosis más bajas de AZT (por ejemplo, 300 mg) dos veces al día.
Desde 1996, el AZT, al igual que otros medicamentos antirretrovirales, se utiliza como parte de la terapia antirretroviral de gran actividad (TARAA, en inglés HAART de Highly Active Antriretroviral Therapy), en combinación con otros fármacos, con el fin de evitar la mutación del VIH resistente al AZT. El virus incorpora el AZT a su genoma durante la replicación, deteniéndose su ciclo de vida porque las nuevas cadenas de ADN viral son defectuosas.
En la figura 6.28 se indica esquemáticamente el modo de acción de los análogos de nucleósidos. En esta figura se representa a la transcriptasa reversa, la enzima que convierte a la hebra de ARN en una hebra de ADN, con un círculo de color verde.
Figura 6.28. Representación del modo de acción de los análogos de nucleósidos

Tema 6. Enfermedades víricas 35
La zona de unión del cebador (primer) se indica con un círculo de color azul (P). La zona de unión del análogo de nucleósido con un círculo de color blanco (N). La plantilla de ARN viral está coloreada en azul y la hebra de (-)-ADN en granate. El análogo de nucleósido fosforilado (dibujado en color verde) se incorpora en la cadena de ADN creciente pero la elongación de esta cadena queda bloqueada por la ausencia de grupo hidroxilo en la posición 3 o, como en el caso de AZT, por la presencia del grupo azida en esta posición.
6.17.2.a. Análisis retrosintético
El análisis retrosintético del AZT (esquema 6.7) se inicia con la desconexión de la función azida, que se instalará mediante reacción SN2 del anión azida sobre el compuesto 6.8 (X=grupo saliente), que procederá del compuesto 6.9. Este intermedio se obtendrá del nucleósido timidina 6.10.
NH
N
O
OHO
O
N3
H3C
Azidotimidina (AZT)
SN2
NH
N
O
OPO
O
H3C
X
N3
+6.8
NH
N
O
OPO
O
H3C
OH
6.9
IGF
NH
N
O
OHO
O
H3C
OH 6.10
Esquema 6.7
6.17.2.b. Síntesis El compuesto de partida para la síntesis del AZT es el nucleósido desoxitimidina 6.10
(esquema 6.8).4 La tritilación selectiva del hidroxilo primario, por reacción con Ph3CCl (cloruro de tritilo) en piridina, proporciona la 5-O-tritiltimidina 6.11, que se convierte en el mesilato 6.12. Cuando el compuesto 6.12 se trata con hidróxido sódico acuoso en etanol, a temperatura ambiente durante toda la noche, se obtiene el producto de ciclación 6.13 como resultado del desplazamiento nucleofílico intramolecular de la agrupación mesilato. El calentamiento de 6.13 a reflujo de hidróxido sódico acuoso proporciona el compuesto 6.9, que se convierte en el azido derivado 6.7 mediante mesilación del hidroxilo y desplazamiento SN2 con azida de litio. La escisión del tritil éter, mediante reacción de 6.7 con cloruro de hidrógeno clorofórmico, proporciona el AZT.5
4 (a) Fox, J. J.; Miller, N. C. J. Org. Chem. 1963, 28, 941-936. (b) Lin, T-S.; Prusoff. W. H. J. Med. Chem. 1978, 21, 109-112. 5 Horwitz, J.; Chua, J. Noel, M. J. Org. Chem. 1964, 29, 2076-2078.

Síntesis de antivirales 36
Esquema 6.8
6.17.2.c. Cuestiones
1) ¿Por qué se produce la tritilación selectiva del hidroxilo primario de la timidina en la reacción con cloruro de tritilo (Ph3CCl) en piridina?
Esquema 6.9
2) Explique mecanísticamente la conversión de 6.13 en 6.9.
NH
N
O
OTrO
O
H3C
OH
6.9
N
N
O
TrO
OO
CH3
6.13
NaOH ac.
reflujo, 5 h
Esquema 6.10

Tema 6. Enfermedades víricas 37
3) Explique mecanísticamente la reacción de destritilación que se emplea en la conversión de 6.7 en AZT.
Esquema 6.11
6.17.3. Ribavirina
La ribavirina, también conocida como virazole, es un nucleósido sintético que inhibe in vitro el crecimiento de virus tanto ADN como ARN, tales como mixovirus, paramixovirus, arenavirus, bunyavirus, virus del herpes, adenovirus y poxvirus.
En las células infectadas la ribavirina sufre un proceso de fosforilación provocado por enzimas tisulares como la adenosina-quinasa. La ribavirina monofosfato inhibe la síntesis de guanosína monofosfato, reduciendo sus niveles intracelulares. Por otro lado, la ribavirina trifosfato inhibe la enzima mRNA-guanililtransferasa inhibiendo la síntesis de ARN mensajero vírico y también de la ARN polimerasa. A altas concentraciones la ribavirina inhibe in vitro la transcriptasa inversa del VIH.
6.17.3.a. Análisis retrosintético
El análisis retrosintético de la ribavirina implica la desconexión del enlace C-N del glucósido (esquema 6.12). Esta operación genera el sintón catiónico 6.15 y el sintón aniónico 6.14.
NHOO
HOHO
N
N
O
NH2
Ribavirina
N
HOO
HO HO
N
N
O
NH2
6.14
6.15
Esquema 6.12
6.16.3.b. Síntesis
Para la síntesis de la ribavirina se eligen como materiales de partida el 1,2,4-triazol-3-carboxilato de metilo 6.166 y la 1,2,3,5-tetra-O-acetyl-β-D-ribofuranosa 6.17 (esquema 6.13).7 El calentamiento de estos dos compuestos a 160-165ºC en presencia de fosfato de bis(p-
6 de Keczer1, A. A.; R. Masjedizadeh1, M. R.; Wu, S-Y.; Lara-Jaime, T.; Comstock1, K.; Charles Dvorak, C.; Liu, T-Y.; Berger, W. J. Label. Compd. Radiopharm. 2006, 49, 1223-1236. 7 Witkowski, J. T.; Robins, R. K.; Sidwell, R. W.; Simon, L. N. J. Med. Chem. 1972, 15, 1150-1154.

Síntesis de antivirales 38
nitrofenilo) proporciona el análogo de nucleósido 6.18. La ribavirina se obtiene mediante desacetilación de 6.18 por reacción con amoníaco metanólico.
NHOO
HOHO
N
N
O
NH2
Ribavirina
OAcAcOO
AcO OAc
NH
N
N
O
OMe
(pNO2C6H4O)2P(O)OH
160-165ºC (78%)
+
NAcO
O
AcO OAc
N
N
O
OMe
NH3, MeOH
25ºC, 18h (90%)
6.16
6.176.18
Esquema 6.13
6.17.3.c. Cuestiones
Proponga un mecanismo que explique la formación del compuesto 6.18.
Esquema 6.14
6.17.4. Síntesis de adefovir dipivoxilo
La hepatitis B es una infección que afecta a más de 400 millones de personas. Esta enfermedad puede provocar cirrosis, bloqueo de la función del hígado y carcinoma hepatocelular. El adefovir dipivoxilo (Hepsera®) fue el primer análogo de nucleósido aprobado por la FDA para el tratamiento de la hepatitis crónica tipo B. Este fármaco actúa bloqueando la replicación viral.
6.17.4.a. Análisis retrosintético
En el esquema 6.15 se indican los tres principales bloques de construcción en la síntesis del adefovir dipivoxilo. Estos son la adenina 6.19, el carbonato de etileno 6.20 y el p-toluensulfoniloximetanofosfato de dietilo 6.21.

Tema 6. Enfermedades víricas 39
Esquema 6.15
6.17.4.b. Síntesis
La síntesis del antiviral se inicia con la reacción de la adenina 6.19 con carbonato de etileno 6.20 en DMF en presencia de NaOH (esquema 6.16). La reacción de la adenina con el carbonato de etileno proporciona la (2-hidroxietil)adenina 6.22 (esquema 6.16).8 La alquilación de 6.22 con el p-toluensulfoniloximetanofosfonato de dietilo 6.21 en DMF, en presencia de t-butóxido de sodio, conduce al fosfonato 6.23. La hidrólisis del fosfonato se lleva a cabo por reacción con TMSBr en acetonitrilo. El correspondiente ácido alquilfosfónico (intermedio 6.24) se convierte en adefovir dipivoxilo por reacción con pivalato de clorometilo 6.25.
Esquema 6.16
6.17.4.c. Cuestiones
1) Explique mecanísticamente la formación del compuesto 6.22.
Esquema 6.17
8 Yu, R. H.; Schultze, L. M.; Rohloff, J. C.; Dudzinski, P. W.; Kelly,D. E. Org. Process Res. Dev. 1999, 3, 53.

Síntesis de antivirales 40
6.17.5. Síntesis de telbivudina
La telbivudina, denominada Tyzeka® en Estados Unidos y Sebivo® in Suiza, es un fármaco empleado en el tratamiento de la hepatitis crónica tipo B. La resistencia a este fármaco afecta, después de dos años de tratamiento, al 2.5% de los pacientes, lo cual es un porcentaje mucho menor que el que presentan otro antivirales como el adefovir dipivoxilo. La telvibudina es fosforilada por las células humanas al correspondiente trifosfato. Este compuesto inhibe a la polimerasa del virus de la hepatitis B pero no a las polimerasas humanas.
6.17.5.a. Análisis retrosintético
La desconexión de la base pirimidínica conduce a la timina 6.26 y al carbohidrato 6.27 que por adición de grupo funcional hidroxilo se convierte en 6.58 cuya estructura remite a la del carbohidrato L-arabinosa 6.59 (esquema 6.18).
Esquema 6.18
6.17.5.b. Síntesis
La síntesis de la telbivudina comienza con la reacción de glicosidación de la L-arabinosa 6.29 por reacción con metanol en presencia de ácido sulfúrico y sulfato cálcico (esquema 6.19).9 El subsiguiente tratamiento con cloruro de benzoilo proporciona el furanósido 6.30. La acetolisis de este compuesto conduce a la mezcla de acetatos anoméricos 6.28. Esta mezcla se condensa directamente con la sililtimina para dar lugar al nucleósido benzoilado 6.31 el cual, mediante metanolisis, se convierte en el nucleósido 6.32. Para conseguir la desoxigenación reductiva de la posición 2´-OH se opera del siguiente modo. El nucleósido 6.32 se silila selectivamente en los hidroxilos 3´ y 5´-OH mediante reacción con 1,3-dicloro-1,1,3,3-tetraisopropildisiloxano en piridina. El producto de esta reacción, compuesto 6.33, se somete a la desoxigenación de Barton-McCombie. Así, la reacción de 6.33 con O-fenil clorotionoformiato (PhOC(S)Cl) conduce al tionocarbonato 6.34 que se trata con Bu3SnH en presencia de azobis-isobutironitrilo (AIBN), como iniciador radicalario, en tolueno a reflujo. El resultado de esta reacción es el compuesto 6.35 que por desililación con fluoruro de tetra-n-butilamonio se convierte en la telbivudina.
9 (a) Czernecki, S.; Le Diguarher, T. Synthesis, 1991, 683. (b) Genu-Dellac, C.; Gosselin, G.; Imbach, J.-L. Tetrahedron Lett., 1991, 32, 79.

Tema 6. Enfermedades víricas 41
Esquema 6.19
6.17.5.c. Cuestiones
1) Explique la exclusiva formación del anómero beta 6.31 en la reacción entre la timina sililada y la mezcla de acetatos anoméricos 6.28.
N
NH
O
O
BzO
BzOO
H3C
6.28
O
BzO
BzOOAc
OBz
(Me3Si)2NH
TMSCl, SnCl4
CH3CN, reflujo (60%)
OBz6.31
+
NH
NH
O
O
H3C
6.26
Esquema 6.20
2) Explique mecanísticamente el proceso de desoxigenación radicalaria reductiva que convierte 6.34 en 6.35.

Síntesis de antivirales 42
Esquema 6.21
6.17.6. Síntesis de clevudina
La clevudina (Levovir®) es un inhibidor de polimerasa que se emplea en el tratamiento de la hepatititis B. Estructuralmente es el enantiómero fluorado de la desoxitimidina.
Figura 6.29. Estructuras de la clevudina y de la desoxitimidina
6.17.6.a. Análisis retrosintético
La desconexión de la base pirimidiníca conduce a la timina y al fluorocarbohidrato 6.36 cuya estructura remite a la del carbohidrato L-arabinosa 6.29 (esquema 6.22).
Esquema 6.22
6.17.6.b. Síntesis
La síntesis de la clevudina se inicia con la acetilación de la L-arabinosa por reacción con HBr del 30% en AcOH/Ac2O. Esta reacción proporciona el bromoglicósido 6.38 con un 57% de rendimiento después de la cristalización en éter etílico (esquema 6.123).10 Cuando el
10 Sznaidman, M. L.; Almond, M. R.; Pesyan, A. Nucleosides Nucleotides Nucleic Acids, 2002, 21, 155-63.

Tema 6. Enfermedades víricas 43
bromoazúcar se trata con zinc en polvo, en presencia de CuSO4 y NaOAc en AcOH/H2O, se obtiene el L-arabinal 6.39. La reacción de 6.39 con Selectflúor (FTEDA-BF4) a reflujo en una mezcla de nitrometano/H2O proporciona el correspondiente fluoroazúcar diacetilado el cual, mediante metanolisis básica, forma el compuesto 6.37. El tratamiento de este compuesto con metanol en presencia de ácido sulfúrico lleva al furanósido 6.40 que por benzoilación conduce a la correspondiente mezcla de benzoatos anoméricos, de la cual se separa el anómero alfa mediante cromatografía. Este anómero se transforma en el bromoglicósido 6.36 por reacción con HBr del 30% en AcOH/CH2Cl2. El bromoglicósido 6.36 se disuelve en cloroformo y se condensa con la timina sililada 6.41, preparada por reacción de la timina con bis(trimetilsilil)amina a reflujo en presencia de sulfato amónico. El producto de la reacción de condensación es la 3,5-di-O-benzoilclevudina 6.42 que se obtiene en un 42% de rendimiento después de la recristalización de etanol. La clevudina se consigue por desbenzoilación de 6.42 con n-butilamina a reflujo de metanol.
Esquema 6.23
6.17.6.c. Cuestiones 1) Explique mecanísticamente la reacción de formación del L-arabinal 6.39.
Esquema 6.24
2) El 1-clorometil-4-fluoro-1,4-diazoniabiciclo[2.2.2]octano bis(tetrafluoroborato), se conoce comercialmente como Selectfluor® y se emplea como reactivo dador de fluoro electrofílico. La

Síntesis de antivirales 44
síntesis del Selectfluor® se indica en el esquema 6.25 y se inicia con la alquilación del diazabiciclo[2.2.2]octano (DABCO) con diclorometano. El producto de la reacción, compuesto 6.43, se trata con tetrafluoroborato sódico mediante una resina de intercambio iónico para reemplazar el anión cloruro por el anión tetrafluoroborato. Por último, la sal 6.74 se convierte en Selectfluor® por reacción con fluoro elemental y tetrafluoroborato sódico.
Selectfluor®
N
N
DABCO
CH2Cl2, 40ºC
N
N
ClCl
NaBF4
CH3CN, 20ºC N
N
ClBF4
F2, NaBF4
CH3CN, -35ºC N
N
Cl
F
BF4
BF46.43 6.44
Esquema 6.25
Explique mecanísticamente la formación de 6.37 por reacción de 6.39 con Selectfluor® en nitrometano acuoso seguida de metanolisis básica.
Esquema 6.26
6.17.7. Síntesis de nevirapina
La nevirapina, comercializada con el nombre de Viramune® por Boehringer Ingelheim, es un inhibidor de la transcriptasa reversa (TR) no análogo de los nucleósidos (ITRNN) que se emplea en el tratamiento de la infección por VIH tipo 1.
En la figura 6.30 se representa la estructura de la transcriptasa reversa de VIH-1. La enzima tiene dos dominios: el p66 (el coloreado en la figura 6.30) y el p51 (de color gris en la figura 6.30).
El dominio polimerasa muestra una forma que se parece a la de la mano derecha. La parte de los dedos está colorada en magenta en la figura 6.30, la de la palma está coloreada en morado y la parte del dedo pulgar en azul. La subunidad p66 también contiene el domino de conexión (coloreado en naranja en la figura 6.30) y el dominio RNasa-H (coloreado en amarillo).11
En el dominio polimerasa el ssARN viral se transcribe a una doble hélice ARN-ADN. A continuación, el sitio ARnasa separa la cadena de ARN de la de ADN. Luego el sitio polimerasa completa la hebra que queda de ADN formando el dsADN.
11 Esposito, F.; Corona, A.; Tramontano, E. Mol. Biol. Inter. 2012, doi:10.1155/2012/586401

Tema 6. Enfermedades víricas 45
Figura 6.30. Representación de la transcriptasa reversa
La zona catalítica de la parte de polimerasa está situada en los subdominios de la palma, de los dedos y del pulgar. En la zona de la palma se encuentran tres residuos de ácido aspártico (Asp-110, Asp-185 y Asp-186) que son claves en la actividad catalítica de este dominio. El dominio RNasa-H está localizado en la zona C-terminal de la subunidad p66, a una distancia de 60Å de la zona de actividad polimerasa.
En la figura 6.31 se representa el dominio p66 de la transcriptasa reversa. Se puede apreciar su forma de mano (véase la parte derecha de la figura). Los dedos de la mano se indican en azul, la palma en rosa y el dedo pulgar en verde. El sitio activo son los átomos de color rojo, donde se produce la elongación del ADN (la zona de la palma de la mano). En amarillo se representa la colocación de un inhibidor de la transcriptasa reversa.12
Figura 6.31. Representación del dominio polimerasa de la transcriptasa reversa
12 Béthune, M-P. Antiviral Res. 2010, 85, 75-90.

Síntesis de antivirales 46
La nevirapina fue descubierta por el equipo de Hargrave en los laboratorios de Boehringer Ingelheim Pharmaceuticals, Inc. y cubierta por la patente 5,366,972 de los Estados Unidos y sus correspondientes en otros países. Este fármaco fue el primero de los ITRNN aprobados por la Administración de Drogas y Alimentos de los Estados Unidos (FDA). El visto bueno de esta agencia estadounidense fue concedido el 21 de junio de 1996 para el tratamiento de adultos seropositivos al VIH, y el 11 de septiembre de 1998 fue aprobado su empleo en niños infectados por VIH. En 1997 fue aprobado su uso en Europa.
La nevirapina pertenece a la familia de los inhibidores de la transcriptasa reversa no nucleósidos (ITRNN). La nevirapina es ineficaz en contra del VIH tipo 2, debido a la distinta estructura de la transcriptasa reversa de esta variedad del virus, que le confiere una resistencia inherente a los ITRNN. Si la reproducción del virus no es suficientemente suprimida por la acción de la nevirapina, las posibilidades de una mutación resistente a este fármaco se incrementan. Las mutaciones más frecuentes que son capaces de desarrollar los virus de inmunodeficiencia humana a la nevirapina son las conocidas como Y181C y K103N, que también son típicas en pacientes que han sido tratados con otros ITRNN. Desafortunadamente, cuando el VIH ha desarrollado resistencia a un medicamento de la familia de los ITRNN es muy probable que también haya desarrollado resistencia a otros ITRNN, como la delavirdina y el efavirenz. En la figura 6.32 se esquematiza el modo de acción de un ITRNN.
Figura 6.32. Modo de acción de los fármacos ITRNN
En el proceso de retrotranscripción el ARN viral de una única hebra (ssARN) es convertido en ADN de doble hebra (dsADN). Cada partícula de VIH contiene dos copias de (+)-ssARN de 9,7 kb. Cerca del extremo 5´ del genoma viral existe una secuencia de 18 nucleótidos denominada sitio de unión del cebador (en inglés primer binding site PBS), que es complementaria con el extremo 3´ del ADN humano denominado tARNLys3. La secuencia de retrotranscripción consta de los siguientes pasos (véase la figura 6.33).
1) El tARNLys3 celular es hibridizado a PBS cerca del extremo 5´ de la cadena de (-)-ARN viral (en naranja en la figura 6.33) y la polimerasa asociada a la transcriptasa reversa inicia la síntesis de la cadena (-)-ADN (en azul en la figura 6.33) empleando el genoma del ARN viral como plantilla.
2) La RNasa-H hidroliza una parte del ARN viral y la cadena híbrida de (-)-ADN queda liberada para unirse a la secuencia complementaria del extremo 3´ de uno de los dos ssARN virales.

Tema 6. Enfermedades víricas 47
3) La unión a la región R del extremo 3´ del genoma de ssARN inicia la síntesis de la cadena (-)-ADN.
4) Mientras tiene lugar la síntesis de ADN (mediada por la transcripta reversa), la RNasa-H rompe la cadena de ARN viral en numerosos puntos, dejando intactos dos secuencias específicas ricas en purinas, denominadas cPPT y 3´PPT (del inglés PolPurine Tracts) que son resistentes a la RNasa-H.
5) Se inicia la síntesis de la cadena (+)-ADN (en verde) utilizando como cebadores los segmentos PPT y en dirección 5´.
6) La RNasa-H hidroliza los segmentos PPT y corta también el segmento híbrido tARN-ADN, liberando la secuencia PBS de la hebra (+)-ADN
7) La transferencia de la secuencia PBS de la cadena (+)-ADN permite la anelación con el sitio complementario del extremo 3´ de la cadena (-)-ADN.
8) Se produce una síntesis bidireccional, lo que permite completar el dsADN viral.
9) Finalmente, se produce una ruptura específica de los cebadores PPT exponiéndose la secuencia de integración para facilitar la inserción del dsADN viral en el cromosoma de la célula hospedadora.
Iniciación de la síntesis
de (-)-ADN
Transferencia de (-)-ADN
Síntesis de (-)-ADN
Ruptura por RNasa-Hy selección de PPT
Síntesis de (+)-ADN y
eliminación de tARN y PPT
Transferencia de (+)-ADNy síntesis bidireccional
Figura 6.33. Representación del proceso de retrotranscripción
En la figura 6.34 se indica la colocación de la nevirapina en la transcriptasa reversa. Este fármaco ejerce un efecto de cuña, impidiendo el completo cierre de la zona bisagra de la

Síntesis de antivirales 48
enzima, lo que tiene como consecuencia la colocación de la zona de agarre y la triada catalítica (compuesta por unidades de ácido aspártico), demasiado alejadas para que puedan ejercer su función catalítica.13
Zona deagarre
Nevirapina
Triada deaspártico
A
Figura 6.34. Colocación de la nevirapina en el centro activo de la transcriptasa reversa
6.17.7.a. Análisis retrosintético
La retrosíntesis de la nevirapina se inicia con la desconexión del anillo de diazepinona que se preparará mediante reacción SNAr intramolecular en la nicotinamida 6.45 (X=halógeno, esquema 6.27). Una nueva desconexión, basada esta vez en una reacción SNuAr intermolecular, desconecta la función ciclopropilamina y proporciona la amida 6.46 (X=halógeno). Por último, la desconexión del enlace amida conduce a la aminopiridina 6.48 y al derivado del ácido nicotínico 6.49 (X=halógeno).
Esquema 6.27
13 Ivetac, A.; McCammon. J. A. J. Mol. Biol. 2009, 388, 644-658.

Tema 6. Enfermedades víricas 49
6.17.7.b. Síntesis
La síntesis de la nevirapina se inicia con la reacción de amidación entre la 2-cloro-4-metil-piridin-3-amina 6.48 y el cloruro del ácido 2-cloronicotínico 6.49 (esquema 6.28).14 La amidación proporciona la nicotinamida 6.46, que por reacción SNAr con la ciclopropilamina conduce a la amida 6.45. La nevirapina se obtiene mediante reacción SNAr intramolecular por calentamiento del compuesto 6.45 en diglime (bis(2-metoxietil)éter) a 160ºC en presencia de hidruro sódico.
Esquema 6.28
6.17.7.c. Cuestiones
¿Por qué la reacción SNAr de la ciclopropilamina sobre la amida 6.46 se produce sobre el átomo de cloro del anillo de nicotinamida y no sobre el átomo de cloro del anillo de piridina?
6.17.8. Síntesis de efavirenz
El efavirenz se emplea como parte de la terapia antirretroviral altamente activa (TARAA) en el tratamiento de la infección por virus de inmunodeficiencia humana tipo 1 (VIH-1).
El efavirenz fue aprobado por la FDA de los Estados Unidos el 21 de septiembre de 1998, convirtiéndose en el decimocuarto medicamento antirretroviral aprobado en ese país.
El efavirenz, al igual que los otros ITRNN, se adhiere a la transcriptasa inversa y evita que el ARN del VIH se transforme en ADN. De esta manera, el virus no puede insertar su información genética en las células sanas impidiéndose su reproducción. El fármaco es ineficaz contra el VIH tipo 2, porque la transcriptasa inversa de este tipo del virus posee una estructura distinta a la del VIH-1, lo que le confiere una resistencia intrínseca a los medicamentos de la familia de los ITRNN.
14 Hargrave, K. D.; Proudfoot, J. J. R.; Grozinger, K. G.; Cullen, E.; Kapadia, S. R.; Patel, U. R.; Fuchs, V. U.; Mauldin, S. C.; Vitous, J.; Behnke, M. L.; Klunder, J. M.; Pal, K.; Skiles, J. W.; McNeil, D. W.; Rose, J. M.; Chow, G. C.; Skoog, M. T.; Wu. J. C.; Schmidt, G.; Engel, W. W.; Eberlein, W. G.; Saboe, T. D.; Campbell, S. J.; Rosenthal, A. S.; Adams, J. J. Med. Chem. 1991, 34, 2231-2241.
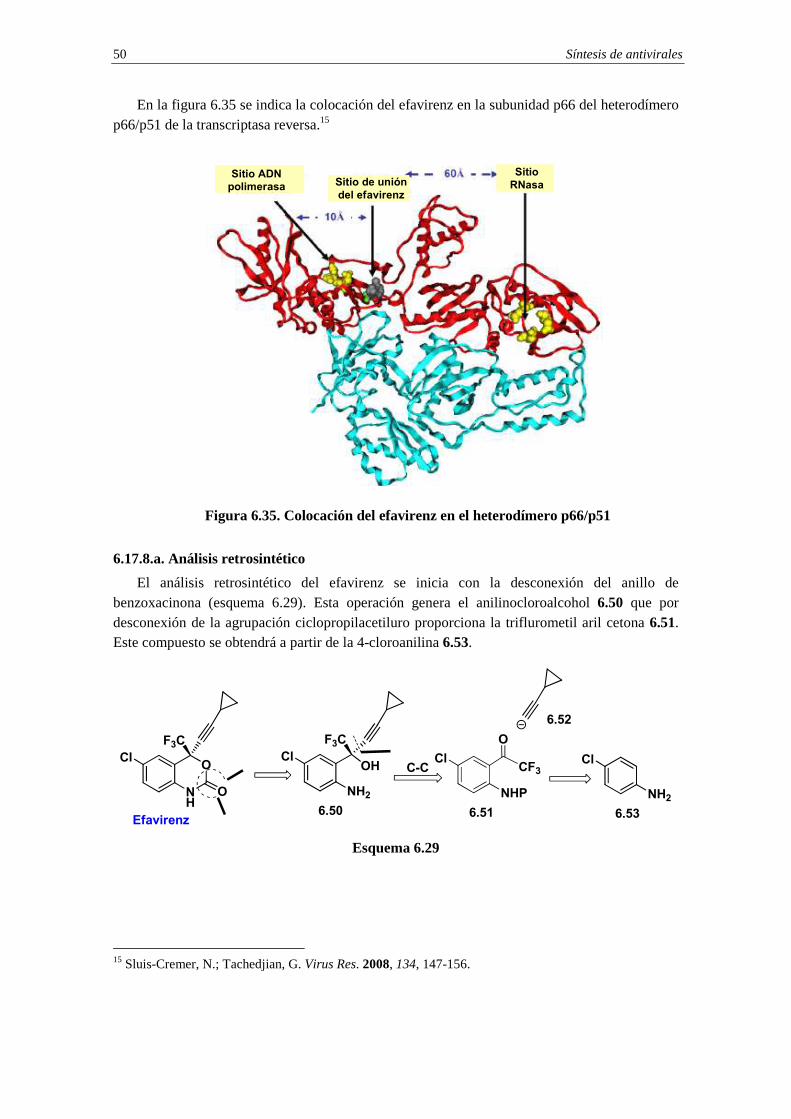
Síntesis de antivirales 50
En la figura 6.35 se indica la colocación del efavirenz en la subunidad p66 del heterodímero p66/p51 de la transcriptasa reversa.15
Sitio ADN
polimerasa Sitio de unión
del efavirenz
Sitio
RNasa
Figura 6.35. Colocación del efavirenz en el heterodímero p66/p51
6.17.8.a. Análisis retrosintético
El análisis retrosintético del efavirenz se inicia con la desconexión del anillo de benzoxacinona (esquema 6.29). Esta operación genera el anilinocloroalcohol 6.50 que por desconexión de la agrupación ciclopropilacetiluro proporciona la triflurometil aril cetona 6.51. Este compuesto se obtendrá a partir de la 4-cloroanilina 6.53.
NH
O
F3C
Cl
O
Efavirenz
NH2
OH
F3C
Cl
NHP
O
CF3Cl
C-C
NH2
Cl
6.50 6.51
6.52
6.53
Esquema 6.29
15 Sluis-Cremer, N.; Tachedjian, G. Virus Res. 2008, 134, 147-156.

Tema 6. Enfermedades víricas 51
6.17.8.b. Síntesis
El compuesto de partida para la síntesis del efavirenz es la p-cloroanilina 6.53. La reacción de este compuesto con el cloruro de pivaloilo 6.53 proporciona la amida 6.55 (esquema 6.30). Cuando la amida se trata con 2 equivalentes de n-BuLi se genera el dianión lítico 6.56 que reacciona con trifluoroacetato de etilo para dar lugar a la amidocetona 6.51. La hidrólisis ácida de la función amida genera el cloridrato hidratado 6.57 que se transforma en la aminocetona 6.58 mediante reacción con acetato sódico.
Esquema 6.30
Antes de la instalación del estereocentro cuaternario se procede a la protección del grupo amino, lo que se consigue mediante la reacción de 6.58 con el alcohol p-metoxibencílico en presencia de cantidades catalíticas de ácido p-toluensulfónico. Esta reacción proporciona la

Síntesis de antivirales 52
trifluorocetona 6.59, el sustrato sobre el que se llevará a cabo la instalación asimétrica del grupo ciclopropilacetileno.16
La adición enantioselectiva de ciclopropilacetileno se consigue del siguiente modo: se añade una disolución de n-butil-litio (mínimo 4 equivalentes) y tetrametiletilendiamina (TMEDA), en metil t-butil éter (MTBE), a una disolución de (1R,2S)-N-propilidinilnorefedrina 6.60 (2 equivalentes) y ciclopropilacetileno 6.61 (2 equivalentes) a -10ºC. La mezcla resultante se deja calentar hasta 0ºC. En estas condiciones se genera el complejo quiral responsable de la inducción asimétrica (véase la estructura I del esquema 6.19. Nota: en esta estructura el éter que se coordina con el litio es THF en lugar de MTBE).
La disolución que contiene el complejo quiral se enfría a -50ºC y se le añade la trifluorocetona 6.59 disuelta en THF. Después de la agitación durante 1 hora a -50ºC se añade ácido cítrico acuoso a la mezcla de reacción. La fase orgánica se extrae con tolueno y el producto se cristaliza de hexano. El alcohol 6.62 se obtiene con un rendimiento químico del 93% y un exceso enantioselectivo del 99.5%.
La eliminación del grupo p-metoxibencilo del compuesto 6.62 se consigue en dos pasos. En primer lugar la reacción con diclorodicianoquinona (DDQ) forma el aminal 6.63. A continuación, este compuesto se convierte en el aminoalcohol 6.50 por reacción con metóxido sódico en metanol. A la reacción se le añade borohidruro sódico para transformar in situ el p-metoxibenzaldehído, generado como consecuencia de la metanolisis básica del aminal, en alcohol p-metoxibencílico. Si no se añade NaBH4, la mezcla de aminoalcohol 6.50 y p-metoxibenzaldehído resulta difícil de separar. El efavirenz se obtiene por reacción del aminoalcohol 6.50 con fosgeno.
La enantioselectividad de la reacción de alquinilación se explica del siguiente modo. La desprotonación de la (1R,2S)-N-propilidinilnorefedrina 6.60 y del cicloprilacetileno 6.61 genera el agregado mixto I , que presenta simetría C2 (esquema 6.31).17 El intercambio del ligando THF por la trifluorometilcetona 6.59 dispone el escenario para la adición intramolecular del ciclopropilacetiluro (complejo II del esquema 6.31).
El ataque a la cara Si de la cetona se produce mediante la intervención de estado de transición III , que está favorecido sobre el estado de transición IV (ataque a la cara Re) porque en éste las interacciones estéricas desfavorables son más desestabilizantes debido a la fuerte interacción estérica del voluminoso grupo arilo con el grupo metilo y el grupo fenilo de la parte de propilidinilnorefedrina.
16 Pierce, M. E. ; Parsons, R. L.; Radesca, L. A.; Lo, Y. S.; Silverman, S.; Moore, J Islam, J. R. Q.; Choudhury, A.; Fortunak, J. M. D.; Nguyen, D.; Luo, C.; Morgan, S. J.; Davis, W. P.; Confalone, P. N.; Chen, C.; Tillyer, R. D.; Frey, L.; Tan,L.; Xu, F.; Zhao, D.; Thompson, A. S.; Corley, E. G.; Grabowski, E. J. J.; Reamer, R.; Reider, P. J. J. Org. Chem. 1998, 63, 8536-8543. 17 Thompson, A.; Corley, E. G.; Huntington, M. F.; Grabowski, E. J. J.; Remenar, J. F.; Collum, D. B. J. Am. Chem. Soc. 1998, 120, 2028-2038.

Tema 6. Enfermedades víricas 53
Esquema 6.31 6.17.8.c. Cuestiones
1) ¿Por qué se convierte la p-cloroanilina 6.53 en la amida del ácido piválico 6.55? ¿Por qué no se convierte la p-cloroanilina 6.53 en otra amida, por ejemplo en la amida de ácido acético? 2) Explique mecanísticamente la conversión del compuesto 6.62 en el aminal 6.63 mediante reacción con DDQ.

Síntesis de antivirales 54
Esquema 6.32
3) Proponga un mecanismo que explique la conversión del aminal 6.63 en el aminoalcohol 6.50.
Esquema 6.33
6.17.9. Síntesis de delavirdina (Rescriptor®)
La delavirdina (Rescriptor®) fue el segundo inhibidor de la transcriptasa reversa no análogo de nucleósido (ITRNN) aprobado por la FDA, en abril de 1.997, para uso en combinación con otros agentes antirretrovirales en el tratamiento de la infección VIH/SIDA. Al igual que otros ITRNN, no se debe emplear como droga única (monoterapia), para evitar la aparición de resistencia al fármaco.
In vitro, la delavirdina inhibe selectivamente al VIH-1 (pero no al VIH-2) de manera no competitiva, pero puede ser inactivo frente al grupo O del VIH-1. Es inactiva frente a otras polimerasas de ADN humano, por lo que, in vitro, presenta escasa toxicidad celular y en cultivos celulares ha demostrado un efecto aditivo o sinérgico con AZT, ddI, ddC, 3TC e inhibidores de la proteasa. La delavirdine se une directamente a la transcriptasa reversa y bloquea las actividades de las polimerasas ARN yt ADN-dependientes.
6.17.9.a. Análisis retrosintético
El análisis retrosintético de la delavirdina se inicia con la desconexión de la función metanosulfonamida que se obtendrá de la amina 6.65 mediante metanosulfonilación (esquema 6.34). El intercambio del grupo funcional amino por nitro conduce al nitrocompuesto 6.66 que por escisión del enlace amida origina el ácido 6-nitroindol-2-carboxílico 6.68 y la piridilpiperazina 6.67. Una operación IGF sobre la parte de isopropilamina de 6.67 genera la imina 6.69 que se desconecta a la aminopiridilpiperazina 6.70. El intercambio del grupo amino por nitro conduce a la nitropiridilpiperazina 6.71 que se obtendrá mediante reacción SNAr entre la piperazina 6.72 y la 2-halo-3-nitropiridina 6.73.

Tema 6. Enfermedades víricas 55
Esquema 6.34
6.17.9.b. Síntesis La síntesis de la delavirdina se inicia con la reacción SNAr entre la piperazina 6.72 y la 2-
cloro-3-nitropiridina 6.73 (esquema 6.35).18 La síntesis de 6.71 se lleva a cabo mediante reacción de la 2-cloro-3-nitropiperidina 6.73 con 10 equivalentes de piperazina 6.72 en presencia de diisopropiletilamina. La reacción proporciona un 60% de 1-(3-nitro-2-piridil)piperazina 6.71 junto con un 15% del producto de di-sustitución 1,4-bis-(3-nitro-2-piridil)piperazina. El producto 6.71 se obtiene puro mediante cromatografía sobre alúmina. La N-Boc protección de 6.71, mediante reacción con bicarbonato de di-t-butilo, conduce a la N-Boc-nitropiridilpiperazina 6.74. La reducción del grupo nitro a amino, con hidrógeno molecular en presencia de Pd/C proporciona la N-Boc-aminopiridilpiperazina 6.75. Este compuesto se somete a aminación reductiva con acetona en presencia de NaBH3CN. El producto resultante de la reacción anterior se trata con ácido trifluoroacético, lo que conduce a la isopropilaminopiridilpiperazina 6.67. El acoplamiento de este compuesto con el ácido 6-nitroindol-2-carboxílico 6.68 se lleva a cabo en presencia de 1,1´-carbonildiimidazol (CDI) o en presencia del clorhidrato de 1-etil-3-(3-dimetilamino)propilcarbodiimida (EDCI), lo que permite la obtención de la amida 6.66. La reducción del grupo nitro proporciona el compuesto 6.65 el 18 (a) Patente US 5,563,142. (b) Romero, D. L.; Merge,, R. A.; Biles, C.; Norman Berrios-Pena, et al. J. Med. Chem. 1994, 37, 999-1014. (d) Pinna, G. A.; Loriga, G.; Murineddu, G.; Grella, G,: Mura, M.; Vargiu, L.; Murgioni, C.; La Colla, P. Chem. Pharm. Bull. 2001, 49, 1406-1411.

Síntesis de antivirales 56
cual, mediante sulfonilación con cloruro de metanosulfonilo en piridina-diclorometano, proporciona la delavirdina, que se transforma en el mesilato de delavirdina (la forma en la que se comercializa el fármaco) mediante reacción con ácido metanosulfónico.
Esquema 6.35
6.17.9.c. Cuestiones
1) ¿Por qué se protege el grupo amino de 6.71?
2) La reacción ajustada para el acoplamiento amídico en presencia de 1,1´-carbonildiimidazol (CDI) se da en el esquema 6.36. Proponga un mecanismo para esta reacción.

Tema 6. Enfermedades víricas 57
Esquema 6.36
3) El clorhidrato de 1-etil-3-(3-dimetilamino)propilcarbodiimida (EDCI) se emplea en el acoplamiento peptídico en presencia de cantidades catalíticas de N,N-dimetilaminopiridina (DMAP). La ventaja que ofrece la EDCI es que la urea resultante del acoplamiento es soluble en agua, con lo que resulta muy fácil su separación de la amida que se forma en la reacción.
En el esquema 6.37 se indica la reacción ajustada para el proceso de amidación con EDCI. Proponga un mecanismo para esta reacción.
Esquema 6.37 4) En el esquema 6.38 se indica una síntesis del ácido 6-nitroindol-2-carboxílico 6.68. El proceso se inicia con la reacción de Japp-Klingemann sobre la 4-nitroanilina 6.76, que se convierte en la hidrazona 6.79 mediante transformación de 6.76 en la sal 4-nitrofenildiazonio 6.77 seguida de reacción con 2-metil-3-oxobutanoato de etilo en medio básico.19
Esquema 6.38
Proponga un mecanismo que explique la formación de la hidrazona 6.79.
5) Explique mecanísticamente la formación del ácido 6-nitroindol-2-carboxílico 6.68 por reacción de la hidrazina 6.79 con ácido polifosfórico (PPA) seguida de saponificación.
19 Tois, J.; Franzén, R.; Aitio, O.; Huikko, K.;Taskinen, J. Tetrahedron Lett. 2000, 41, 2443-2446.

Síntesis de antivirales 58
6.17.10. Síntesis de raltegravir (Isentress®)
El raltegravir en un fármaco antirretroviral producido por la compañía farmacéutica Merck & Co. Fue aprobado por la FDA en octubre de 2007 y se emplea en el tratamiento de la infección por VIH (véase su estructura en la figura 6.35).
Figura 6.35
El raltegravir actúa inhibiendo la integrasa viral e impidiendo la fusión del ADN del virus con el ADN de la célula hospedadora. De momento, el raltegravir sólo está aprobado para pacientes con VIH resistente al resto de fármacos antirretrovirales de alta eficacia (TARGA). Se recomienda utilizarlo en politerapia, dado que se prevé la aparición de resistencias si se utiliza como único fármaco.
Cuando una célula es infectada por el virus VIH el ssARN se convierte en dsADN. Una copia de este ADN viral es insertada en el ADN de la célula hospedadora (véase la figura 6.36).
Figura 6.36. Representación del modo de acción de un inhibidor de integrasa
La inserción del ADN viral la lleva a cabo la enzima viral denominada integrasa. En primer
lugar la integrasa forma un complejo con el ADN viral. Este complejo entra en el núcleo de la célula infectada y allí la integrasa elimina dos nucleótidos desde cada final 3´ del ADN viral. En el esquema 6.39 se representa la reacción de corte del extremo 3´ del ADN viral provocado por la integrasa. Tres residuos ácidos del centro activo de esta enzima se coordinan con dos cationes

Tema 6. Enfermedades víricas 59
Mg2+ que quedan expuestos sobre la superficie de la enzima. Esta colocación facilita su cooordinación con la parte de fosfato del extremo 3´ del ADN viral (representado en azul en el esquema 6.39), que experimenta un proceso de hidrólisis, escindiéndose el dinucleótido del dicho extremo 3´.20
Esquema 6.39
A continuación, el ADN viral es integrado covalentemente en el ADN de la célula hospedadora. En el esquema 6.40 se representa la unión covalente del ADN viral con el ADN de la célula infectada provocada por el ataque nucleofílico SN2 del grupo hidroxilo 3´ del ADN viral sobre el fosfato de la cadena de ADN de la célula hospedadora.
Esquema 6.40
El inhibidor de integrasa se coordina con los dos átomos metálicos del centro activo bloqueando la unión al ADN de la célula infectada. En el esquema 6.41 se representa esta inhibición.
20 Kawasuji, T.; Fuji, N.; Yoshinaga, T.; Sato, A.; Fujiwara, T.; Kiyama, R. Bioorg. Med. Chem. 2006, 14, 8420-8429.

Síntesis de antivirales 60
Esquema 6.41
En la figura 6.37 se muestra la colocación del raltegravir bloqueando el extremo 3´ del ADN viral e impidiendo la unión de éste al centro activo de la integrasa. Se puede apreciar la interacción entre los átomos de oxígeno quelantes del raltegravir (en rojo) con los cationes magnesio (en verde) del centro activo.21
Figura 6.37. Colocación del raltegravir entre el sitio catalítico de VIH-1 y el extremo 3´ del ADN viral
La integrasa es una proteína dimérica. Cada monómero contiene un centro catalítico, un dominio C-terminal y uno N-terminal (véase la figura 6.38 para la estructura de la integrasa de VIH-1).22 La estructura del dímero tiene forma de Y provocada por la presencia de seis hélices alfa que unen, en cada uno de los monómeros, el sitio catalítico con el dominio C-terminal. Una pequeña diferencia en una de las hélices alfa, que tiene lugar en la treonina-210, actúa de pivote flexible y permite orientar las dos hélices en un ángulo de 90º. Esta flexibilidad juega un papel clave en el proceso de integración del ADN.
21 http://www.proteopedia.org/wiki/index.php/Retroviral_Integrase. 22 http://maptest.rutgers.edu/drupal/?q=node/436.

Tema 6. Enfermedades víricas 61
Hélices alfa
Dominios C-terminales
Dominioscatalíticos
Figura 6.38. Estructura de la integrasa de VIH-1
A lo largo del dímero que constituye la integrasa se encuentra una banda de residuos
cargados positivamente que van desde Lys-159 en un monómero a Lys-219 en el otro. Esta banda se extiende desde el final del sitio catalítico de un monómero al domino C-terminal del otro monómero (véase la figura 6.39).
Figura 6.39. Estructura de la integrasa de VIH-1 con indicación de los aminoácidos clave

Síntesis de antivirales 62
Debido a la carga positiva, se cree que esta es la zona de estabilización del ADN, lo que permite el corte de los dos nucleótidos del extremo 3´ y la transferencia del ADN viral al ADN de la célula hospedadora.
En la figura 6.40 se indica la estructura tridimensional del sitio catalítico de la integrasa de VIH-1. La estructura del raltegravir se indica en esferas de color amarillo. Los residuos activos del centro catalítico (Asp-64, Asp-116, Glu-152) se muestran en blanco. En rojo se muestran los residuos (Glu-92, Glu-138, Gly-140, Gln-148 y Asn-155) cuyas mutaciones provocan resistencia a raltegravir.23
Figura 6.40. Colocación del raltegravir en el sitio catalítico de VIH-1
6.17.10.a. Análisis retrosintético
El análisis retrosintético del raltegravir se indica en el esquema 6.41 y se inicia con la escisión del enlace de la parte de amida de (4-fluorofenil)metanamina. Este proceso forma la estructura 6.80, en la cual el grupo hidroxilo del anillo de 5-hidroxipirimidin-4-ona se indica en su forma protegida (en esta operación retrosintética no se ha dibujado la estructura de la (4-fluorofenil)metanamina que surge del proceso de desconexión). La segunda operación retrosintética se encarga de escindir el otro enlace amidíco, lo que conduce al compuesto 6.81 (de nuevo en esta operación retrosintética no se ha dibujado la estructura del ácido 5-metil-1,3,4-oxadiazol-2-carboxílico que surge del proceso de escisión del enlace amídico). La tercera operación del análisis retrosintético se encarga de desconectar el anillo de 5-hidroxipirimidin-4-ona. Esta operación no es evidente y baste ahora decir que este anillo se obtendrá por condensación entre la hidroxiacetimidamina 6.82 y el acetilendicarboxilato 6.83. El compuesto 6.78 se sintetizará a partir del ácido 2-amino-2-metilpropanoico 6.84.
23 http://hivdb.stanford.edu/pages/3DStructures/in.html.

Tema 6. Enfermedades víricas 63
Esquema 6.41
6.17.10.b. Síntesis
La síntesis del raltegravir se inicia con la protección del grupo amino del ácido 2-amino-2-metilpropanoico 6.84, por reacción con cloroformiato de bencilo y carbonato de potasio, seguida de tratamiento con cloroformiato de etilo en presencia de amoniaco. Esta secuencia conduce a la amida 6.85 (esquema 6.42).24 La deshidratación de este compuesto con anhidrido trifluoroacético y trietilamina conduce al nitrilo 6.86 que se convierte en la hidroxiacetimidamina 6.82 por reacción con hidroxilamina en presencia de KOH. El anillo de 5-hidroxipirimidin-4-ona se obtiene en dos pasos. Así, la reacción de 6.82 con acetilendicarboxilato de dimetilo (DMADC) proporciona el compuesto 6.87, que se convierte en la 5-hidroxipirimidin-4-ona 6.88 por calentamiento a reflujo de xileno. La benzoilación del hidroxilo conduce al compuesto 6.89, el cual, mediante reacción con hidruro de lito y sulfato de dimetilo proporciona el producto de N-metilación 6.90. La eliminación del grupo benciloxicarbonilo, mediante hidrogenolisis en medio ácido, conduce al clorhidrato 6.81. La reacción de este compuesto con el cloruro de ácido 6.91, en presencia de trietilamina, proporciona la amida 6.80 que se convierte en el raltegravir mediante reacción con (4-fluorofenil)metanamina 6.92. El tratamiento del raltegravir con KOH permite la obtención de su sal potásica (raltegravir potasio).
24 Summa, V.; Petrocchi, A.; Bonelli, F.; Crescenzi, B.; Donghi, M.; Ferrara, M.; Fiore, F.; Gardelli, C.; González Paz, O.; Hazuda, D. J.; Jones, P.; Kinzel, O.; Laufer, R.; Monteagudo, E.; Muraglia, E.; Nizi, E.; Orvieto, F.; Pace, P.; Pescatore, G.; Scarpelli, R.; Stillmock, K.; Witmer, M. V.; Rowley, M. J. Med. Chem. 2008, 51, 5843-5855.

Síntesis de antivirales 64
N
NHN
OH F
O
Me
HN
Me Me
O
NN
O
Me
O
Raltegravir
N
NOBz
O
Me
HN
Me Me
O
NN
O
Me
O
OMe
N
HNOH
O
CbzHN
Me Me O
OMe NH2
NOH
CbzHN
Me Me
COOHH2N
Me Me
6.80
6.87 6.82
6.84
CbzHN
Me Me
O
NH2
1) CbzCl, K2CO3
2) ClCOOEt, NH3, H2O
6.85
CNCbzHN
Me Me
(CF3CO)2O, Et3N
6.86
NH2OH·HClKOH
CHCl3
N
NOBz
O
Me
H3N
Me Me O
OMe
6.81
N
HNOBz
O
CbzHN
Me Me O
OMe
6.88
LiH, Me2SO4
N
NOBz
O
CbzHN
Me Me O
OMe
6.89
Me
dioxano
H2, 10% Pd/C, HCl
ONN
OMe Cl6.91
Et3N
ClH2N
F
MeOH
6.92
KOH N
NHN
OK F
O
Me
HN
Me Me
O
NN
O
Me
O
Raltegravir Potasio
CH3CN
DMADC
NH2
N
CbzHN
Me Me
O COOMe
COOMe
piridina(PhCO)2O
Xileno
reflujo
6.90
Esquema 6.42
6.17.10.c. Cuestiones
1) La reacción ajustada para la conversión de la amida 6.85 en el nitrilo 6.86 se indica en el esquema 6.43.
Esquema 6.43
Proponga un mecanismo que explique la reacción anterior.

Tema 6. Enfermedades víricas 65
2) Explique mecanísticamente la reacción indicada en el esquema 6.44.
Esquema 6.44
3) La síntesis del cloruro del ácido 5-metil-1,3,4-oxadiazol-2-carboxílico 6.91 se lleva a cabo a partir del 5-metil-1H-tetrazol 6.93, tal y como se indica en el esquema 6.45.25
Esquema 6.45
Explique mecanísticamente la formación del 5-metil-1,3,4-oxadiazol-2-carboxílato de etilo 6.95.
6.17.11. Síntesis de vicriviroc
La gran mayoría de los compuestos anti-VIH permiten la entrada del virus en la célula, actuando a continuación sobre las células ya infectadas mediante inhibición de la transcriptasa reversa o de otras proteasas. Una vía de acción muy diferente a la anterior se centra en impedir la entrada del virus en la célula. Este es el modo de acción del vicriviroc, cuya estructura se indica en la figura 6.41.
Figura 6.41
El virus VIH se une a los macrófagos o a las células T con la ayuda de las proteínas gp120 y gp41. Estas moléculas se asocian entre sí de forma no covalente en la membrana del virus, formando un heterotrímero gp120/gp41. La fusión comienza con la unión de la proteína gp120 a la glicoproteína CD4, que está situada en la superficie de la membrana de la célula hospedadora. Para que el proceso de fusión pueda continuar, la proteína gp120 debe unirse a unos
25 Wang, Z.; Wang, M.; Yao, X.; Li, Y.; Qiao, W.; Geng, Y.; Liu, Y.; Wang, Q. Eur. J. Med. Chem. 2012, 50, 361-369.

Síntesis de antivirales 66
determinados correceptores ubicados en la membrana de la célula hospedadora. En el caso de los virus VIH-1 estos correceptores son las proteínas transmembrana denominadas CCR5 y CXCR4 (véase la parte A de figura 6.42). Así, después de la unión de la proteína gp120 a CD4 aquélla cambia su conformación, lo que provoca la formación del sitio de unión del correceptor en la gp120. Una vez ocurrido este cambio, gp120 se une al correceptor y la proteína gp41 experimenta un cambio conformacional que provoca la inserción de su dominio hidrofóbico N-terminal (conocido como dominio de fusión) en la membrana de la célula hospedadora. La inserción de la gp41 hace que esta proteína atraiga hacia sí a dos colas triméricas, denominadas HR1 y HR2, formando una hélice sextuple que provoca la aproximación de las membranas del virus y de la célula y, finalmente, la fusión, con el subsiguiente vertido en la célula hospedadora del genoma viral (véase la parte B de la figura 6.42).
Virus
Sitio de unión
del correceptor
Correceptor
(CCR5 o CXCR4) Célula
Membranacelular
A B
Fusión virus-célula
Figura 6.42. Representación del proceso de fusión virus-célula hospedadora
Los correceptores CCR5 y CXCR4 son receptores transmembrana acoplados a proteína G. Los virus VIH se pueden clasificar según se unan a uno u otro correceptor. Los denominados virus R5, o virus M-trópicos, son los que se unen al correceptor CCR5 de los macrofágos, mientras que los virus X4, o virus T-trópicos, son los que se unen al correceptor CXCR4 de las células T. También existen virus duales, capaces de unirse a ambos correceptores. La selectividad hacia uno u otro correceptor depende del bucle V3, una región estructuralmente flexible y altamente variable, compuesta por 35 aminoácidos y ubicada en la proteína gp120.
En la figura 6.43 se representa con más detalle la unión de la proteína gp120 de un virus R5 (M-trópico) y de un virus X4 (T-trópico) a su correceptor. Se puede apreciar el bucle V3 que marca la selectividad hacia uno u otro correceptor.

Tema 6. Enfermedades víricas 67
Virus R5 (M-trópico) Virus X4 (T-trópico)
ReceptorCD4
ReceptorCD4
CorreceptorCXCR4
CorreceptorCCR5
Bucle V1/V2
Bucle V3
Lámina puente
Bucle V3
Figura 6.43. Representación de la unión gp120-correceptor
El empleo de un correceptor u otro depende del curso de la infección. En los estadíos tempranos de la enfermedad por VIH el 90% de los pacientes presentan virus R5. Sin embargo, después de unos cinco años de sufrir la infección, un 50% de los pacientes tienen cantidades apreciables de virus X4. Estos cambios de correceptor aceleran el progreso de la enfermedad.
El vicriviroc es un inhibidor antagonista alostérico no-competitivo de CCR5. Se une a un pequeño bolsillo hidrofóbico que se encuentra ubicado entre dos hélices transmembrana, cerca de la superficie extracelular de CCR5. La unión a este bolsillo provoca un cambio conformacional del segmento extracelular de CCR5 impidiendo de este modo la unión de gp120 a la célula hospedadora.
Las interacciones que se establecen entre el vicriviroc y el correceptor CCR5 se detallan en la figura 6.44.26 El grupo trifluorometilo del vicriviroc establece una fuerte interacción hidrofóbica con el residuo de isoleucina I-198 de la quinta hélice transmembrana (TM5). Otras interacciones importantes son el puente salino que se establece entre el átomo de nitrógeno terciario positivamente cargado del vicriviroc y el glutámico E-283 de la séptima hélice transmembrana (TM7), la interacción π-stacking del anillo de pirimidina con el triptófano W-86 de TM2, la interacción π-stacking del anillo aromático con el triptófano W-248 de TM6, la
26 Kondru, R.; Zhang, J.; Ji, C.; Mirzadegan, T.; Rotstein, D.; Sankuratri, S.; Dioszegi, M. Mol. Pharmacol, 2008, 73, 789-800.

Síntesis de antivirales 68
interacción por puente de hidrógeno de la tirosina Y-108 (TM3) y la interacción hidrofóbica de la tirosina Y-251 de TM6 con el anillo de piperazina.
Puente salino
-Stacking aromáticoInteracciones hidrofóbicas
Puente de hidrógeno
Figura 6.44. Interacciones del vicriviroc con el correceptor CCR5
6.17.11.a. Análisis retrosintético
El análisis retrosintético del vicriviroc se inicia con la desconexión del enlace amídico. Esta operación genera el compuesto 6.96 y el ácido 4,6-dimetilpirimidin-5-carboxílico (esquema 6.46). La siguiente operación retrosintética se encarga de adicionar dos grupos carbonilo sobre el anillo de metilpiperazina, lo que conduce a la piperazinodiona 6.98 (en este paso también se ha incluido un grupo protector en el átomo de nitrógeno del anillo de piperidina). En la siguiente operación retrosintética se lleva a cabo una doble desconexión sobre el anillo de piperazinodiona, lo que conduce al amidoéster 6.99 y a la 4-amino-4-metilpiperidina protegida 6.100. La escisión del enlace amidíco en el compuesto 6.99 forma el aminoéster 6.101 cuya ruptura del enlace C-N origina el alcohol 6.102 y el aminoéster 6.103. Esta desconexión, basada en una reacción SN2, se produce sobre un carbono estereogénico, por lo que derivado activado el alcohol 6.102, y su derivado activado que es el que debe participar en el proceso SN2, deben ser de configuraciones opuestas a la de la aminoéster 6.101.
El alcohol 6.102 se obtendrá de la metoxicetona 6.104 mediante un proceso de reducción enantioselectiva. Este último compuesto se obtendrá de la 4-trifluorometilfenil metil cetona 6.105.

Tema 6. Enfermedades víricas 69
Esquema 6.46
6.17.11.b. Síntesis
La síntesis del vicriviroc se inicia con la bromación de la 4-trifluorometilfenil metil cetona 6.105 que se lleva a cabo mediante reacción con bromo molecular (esquema 6.47).27 La bromocetona 6.106 se convierte en la metoxicetona 6.104 mediante reacción con metanol en presencia de trifluoruro de boro y óxido de plata. Para la reducción enantioselectiva de la metoxicetona 6.104 se aplica el método de Corey-Bakshi-Shibata empleando el complejo BH3·SMe2 como agente reductor en presencia de la oxazaborilidina quiral derivada de L-prolina.28 El alcohol resultante de esta reducción, compuesto 6.102, se convierte en el mesilato 6.107, el cual se somete a la reacción SN2 con alaninato de metilo en acetonitrilo a reflujo en presencia de carbonato potásico. Este proceso permite la obtención del aminoéster 6.100 que se convirte en la clorometilamida 6.99 por reacción con cloruro de cloroacetilo. El tratamiento de 6.99 con la 4-amino-4-metilpiperidina N-Boc protegida 6.101 proporciona la piperazinodiona 6.98. La N-Boc desprotección con TFA seguida de reducción de las funciones amida con borano permite la obtención de la piperazina 6.96 la cual, por reacción de amidación con el ácido 4,6-dimetilpirimidin-5-carboxílico 6.97 se convierte en el vicriviroc.
27 Feng, D-Z.; Song, Y-L.; Jiang, X-H.; Che, L.; Long, T-Q. Org. Biomol. Chem. 2007, 5, 2690-2697. 28 (a) E. J. Corey, S. Shibata, R. K. Bakshi. J. Org. Chem. 1988, 53, 2861-2863. (b) E. J. Corey, K. Bakshi, S. Shibata, C. Chen, V. K. Singh. J. Am. Chem. Soc. 1987, 109, 7925-7926. Para el mecanismo de esta reacción véase la síntesis de sertralina en el tema 3.

Síntesis de antivirales 70
Esquema 6.47
6.17.11.c. Cuestiones
1) Dibuje el ciclo catalítico y el modelo estereoquímico para la reacción indicada en el esquema 6.48.
Esquema 6.48

Tema 6. Enfermedades víricas 71
2) La síntesis de la 4-amino-4metilpiperidina N-Boc protegida 6.100 se indica en el esquema 6.49. El compuesto de partida es el etil éster del ácido isonipecótico (compuesto 6.108). La N-Boc protección conduce al compuesto 6.109.29 La enolización de este compuesto con LDA, seguida de C-alquilación del correspondiente enolato lítico con yoduro de metilo, proporciona el éster 6.110.30 La saponificación conduce al ácido 6.111 que se somete a reacción de transposición de Curtius. Para ello se convierte en primer lugar en el anhidrido mixto 6.112 el cual, por reacción con azida sódica, conduce a la azida de acilo 6.113. Cuando este compuesto se calienta en tolueno a 90ºC experimenta el proceso de transposición de Curtius y se convierte en el isocianato 6.114. Finalmente, la hidrólisis básica del isocianato proporciona la 4-amino-4-metilpiperidina N-Boc protegida 6.100.
Esquema 6.49
Explique mecanísticamente la reacción de transposición de Curtius que convierte la azida de acilo 6.113 en el isocianato 6.114.
6.17.12. Síntesis de pleconaril
Los Enterovirus constituyen un amplio grupo de virus causantes de importantes infecciones humanas. La familia Picornaviridae está constituida por los géneros Poliovirus, Echovirus, Coxsackievirus A y B, Enterovirus y Rhinovirus, los cuales presentan más de 200 serotipos diferentes.
Las infecciones causadas por Enterovirus van desde cuadros benignos y autolimitados, como la rinitis y el catarro común, hasta procesos graves como la sepsis neonatal, la meningitis linfocitaria, la miocarditis y las infecciones crónicas y debilitantes en pacientes inmunodeprimidos.
29 Jansen, M.; Rabe, H.; Strehle, A.; Dieler, S.; Debus, F.; Dannhardt, G.; Akabas, M. H.; Lüddens, H. J. Med. Chem. 2008, 51, 4430-4448. 30 Jiang, X-H.; Song, Y-L.; Long, Y-Q. Biorg. Med. Chem. Lett. 2004, 14, 3675-3678.

Síntesis de antivirales 72
Los estudios de cristalización y difracción con rayos X han permitido conocer con gran exactitud la estructura tridimensional de la cápside de los Enterovirus y establecer la localización en ella de las diferentes proteínas estructurales. La cápside de los Enterovirus está formada por 60 protómeros, cada uno de los cuales contiene una copia de cada una de las proteínas estructurales designadas como VP1, VP2, VP3 y VP4. La unión de las proteínas VP1 y VP3 da lugar a la formación de un canal estructural que es utilizado por el virus para unirse al receptor celular. En el interior de este canal se localiza una cavidad hidrofóbica llamada bolsillo de fijación, una estructura prácticamente constante en todos los Enterovirus estudiados con sólo pequeñas variaciones en su longitud y carga hidrofóbica entre los diferentes enterovirus.
A partir del conocimiento de la existencia de esta zona de fijación se han ido probando diferentes compuestos químicos, como la rodamina, los flavonoides, las chalconas, algunas aralquilamino-piridinas, oxazolinil-isoxazoles, piridazinaminas y fenoxil-imidazoles, capaces de unirse a ella y, por lo tanto, de disminuir o eliminar la fijación de los Enterovirus al receptor celular.
Se han establecido diferentes hipótesis para explicar la acción de los anteriores compuestos.
1) Una de ellas establece que la liberación del RNA viral de Enterovirus necesita de un cierto grado de flexibilidad en la cápside. Así, la unión de estos compuestos al bolsillo de fijación reduciría esta flexibilidad, dando lugar a estructuras virales mucho más rígidas y menos adaptables al proceso de infección celular.
2) Otra hipótesis indica que el bolsillo de fijación y su cavidad interna actúan como conducto iónico, permitiendo la entrada de iones al interior del Enterovirus. De acuerdo con esta hipótesis, los compuestos que se unen al bolsillo de fijación actuarían como bloqueantes físicos del flujo iónico, alterando el pH del entorno bloqueando el proceso de descapsidación.
El pleconaril (véase la estructura en la figura 6.45) pertenece a la serie de compuestos oxazolinil-isoxazoles con demostrada actividad frente a la mayoría de Enterovirus.
Figura 6.45
Su estructura se adapta perfectamente al bolsillo de fijación de los Enterovirus, bloqueando la fijación del virus a la célula e impidiendo su descapsidación. Como el bolsillo de fijación existente en la cápside de la mayoría de los Enterovirus tiene una estructura altamente conservada, el pleconaril muestra un potente y amplio espectro de actividad que abarca a la casi totalidad de los miembros de esta familia de virus.
En la figura 6.46 se representa en la parte A la estructura de Enterovirus (virus eicosaédrico) mostrando en color rojo un corte en la zona del bolsillo de fijación. En la parte b dela figura 6.46 se observa aumentado este corte y en la parte c de la figura se indica el bolsillo de fijación.

Tema 6. Enfermedades víricas 73
Figura 6.46
El pleconaril se une al bolsillo de fijación mediante interacciones hidrofóbicas con los 17 aminoácidos que se indican en la parte de la izquierda de la figura 6.47.31 La integración del plecoranil provoca un cambio conformacional en la cápside del virus, lo que impide la unión al receptor celular ICAM-1 (empleado por más del 90% de los rinovirus) aumentando la rigidez de la cápside, lo que inhibe la descapsidación del virión y la liberación del ARN dentro de la célula hospedadora.
Figura 6.47. Interacciones del plecoranil con los aminoácidos del bolsillo de fijación
6.17.12.a. Análisis retrosintético
En el esquema 6.50 se indica el análisis retrosintético del plecoranil.
Esquema 6.50
31 Ledford, R. M.; Patel, N. R.; Demenczuk, T. M.; Watanyar, A.; Herbertz, T.; Collett, M. S.; Pevear, D. C. J. Virol. 2004, 78, 3663-3674.

Síntesis de antivirales 74
El proceso se inicia con la desconexión del anillo de oxadiazol, que se obtendrá mediante reacción de ciclodeshidratación sobre la amidoxima 6.115 que, a su vez, derivará del nitrilo 6.112. Por último, la desconexión del enlace C-O, basada en una reacción SN2, conduce al isoxazol 6.118 y al 4-hidroxi-3,5-dimetilbenzonitrilo 6.117.
6.17.12.b. Síntesis
Para la síntesis del pleconaril se elige como compuesto de partida el 3,5-dimetilisoxazol 6.119 (esquema 6.51). La desprotonación de este compuesto, mediante reacción con BuLi, seguida de C-alquilación con el 1-bromo-2-cloroetano, proporciona el 5-(3-cloropropil)-3-metilisoxazol 6.118.32 La reacción SN2 del 4-hidroxi-3,5-dimetilbenzonitrilo 6.117 con el isoxazol 6.118 se lleva a cabo en N-metilpirrolidona (NMP) en presencia de yoduro potásico y cabonato potásico y proporciona el compuesto 6.112. La reacción con clorhidrato de hidroxilamina, en etanol, en presencia de carbonato potásico, conduce a la amidoxima 6.115.33 El tratamiento de este compuesto con anhidrido trifluoroacético en piridina proporciona el plecoranil.34
Esquema 6.51
32 Diana, G. D.; Cutcliffe, D.; Volkots, D. L.; Mallamo, J. P.; Bailey, T. R.; Vescio, N.; Oglesby, R. C.; Nitz, T. J.; Wetzel, J.; Giranda, V.; Pevear, D. C.; Dutko, F. J. J. Med. Chem. 1993, 36, 3240-3250. 33 Diana, G. D.; Volkots, D. L.; Nitz, T. J.; Bailey, T. R.; Long, M. A.; Vescio, N.; Aldous, S.; Pevear, D. C.; Dutko, F. J. J. Med. Chem. 1994, 37, 2421-2436. 34 Diana, G. D.; Rudewicz, P.; Pevear, D. C.; Nitz, T. J.; Aldous, S. A.; Aldous, D. J.; Robinson, D. T.; Draper, T.; Dutko, F. J.; Aldi, C.; Gendron, G.; Oglesby, R. C.; Volkots, D. L.; Reurnan, M.; R.; Bailey, T. R.; Czerniak, R.; Block, T.; Roland, R.; Oppermann, J. J. Med. Chem. 1996, 38, 1355-1371.

Tema 6. Enfermedades víricas 75
6.17.12.c. Cuestiones
1) Explique mecanísticamente la formación del 5-(3-cloropropil)-3-metilisoxazol 6.118 a partir del 3,5-dimetilisoxazol 6.119. ¿Por qué se produce la C-alquilación en el metilo en la posición en C-3?
2) En el esquema 6.52 se indica una síntesis para el 4-hidroxi-3,5-dimetilbenzonitrilo 6.117.35
Esquema 6.52
Explique mecanísticamente la formación de 6.117 por reacción del 4-bromo-2,6-dimetilfenol 6.125 con cianuro cuproso.
6.17.12. Síntesis de arbidol
El arbidol es un fármaco antiviral, comercializado en Rusia y China, que se emplea en el tratamiento de la gripe.36 El arbidol inhibe la entrada del virus en la célula hospedadora y estimula la respuesta inmune.37 Algunos estudios han demostrado que el arbidol es más efectivo en infecciones por virus ARN que por virus ADN.38
7.17.12.a. Análisis retrosintético
El análisis retrosintético del arbidol se indica en el esquema 6.53 y se inicia con la desconexión del fragmento de dimetilamino metilo. Esta operación conduce al compuesto 6.126 el cual, por escisión del grupo tiofenilo, origina el indol dihalogenado 6.127 (X=halógeno) que se obtendrá a partir hidroxi-indol 6.129. Este compuesto se sintetizará mediante reacción de Nenitzescu de la 1,4-benzoquinona 6.130 con el acetilacetato de etilo 6.131 y la metilamina 6.132.
35 Wang, W.; Wu, W.; Shen, Z.; Chen, L. J. Label. Compd. Radiopharm. 2011, 54, 371-373. 36 Leneva, I. A.; Russell, R. J.; Boriskin, Y. S.; Hay, A. J. Antiviral Res. 2009, 81, 132-140. 37 Boriskin, Y. S.; Leneva, I. A.; Pécheur, E. I.; Polyak, S. Curr. Med. Chem. 2008, 15, 997-1005. 38 Shi, L.; Xiong, H.; He, J.; Deng, H.; Li, Q.; Zhong, Q.; Hou, W.; Cheng, L.; Xiao, H.; Yang, Z. Arch. Virol.2007, 152, 1447-1455.

Síntesis de antivirales 76
Esquema 6.53
7.17.12.b. Síntesis
La síntesis del arbidol se inicia con la reacción de Nenitzescu. Así, la metilamina se calienta con acetilacetato de etilo, a reflujo de tolueno en presencia de TsOH y la enamina resultante se hace reaccionar con 1,4-benzoquinona en nitrometano,39 lo que proporciona el hidroxi-indol 6.129 (esquema 6.54).
N
COOEtHO
Br
CH3
S
Arbidol
N
COOEtHO
Br
CH3
S
N
COOEtAcO
Br
CH3
Br N
COOEtHO
CH3
CH3
O
O
O
COOEt
CH3+ CH3NH2
6.126
6.128
6.129
6.130
6.131
6.132
TsOH
CH3NO2 H3CHN
COOEt
CH3
6.133
ClCH2CH2Cl, reflujo
CH3COCl
piri-acetonaN
COOEtAcO
CH3
CH3Br2
6.1346.135
SH
KOH, MeOH
HCHO
Me2NH
AcOH
3h, rt (96%)
CCl4, 2h(87%)
(60%)
Me2N
Esquema 6.54
39 Pawlak, J. M.; Khau, V. V.; Hutchison, D. R.; Martinelli, M. J. J. Org. Chem. 1996, 61, 9055-9059.

Tema 6. Enfermedades víricas 77
La acetilación del hidroxilo fenólico conduce al compuesto 6.134 que se convierte en el derivado dibromado 6.135 mediante reacción con bromo molecular.40 El tratamiento de 6.135 con tiofenol, en presencia de KOH, permite la obtención del compuesto 6.126 que se convierte en el arbidol por reacción de Mannich con formaldehído y dimetilamina.
7.17.12.c. Cuestiones
1) Explique mecanísticamente la reacción de Nenitzescu que permite la obtención del hidroxi-indol 6.129.
Esquema 6.55
2) Explique mecanísticamente la reacción indicada en el esquema 6.56.
Esquema 6.56
40 (a) Chai, H.; Zhao, Y.; Zhao, C.; Gong, P. Biorg. Med. Chem. 2006, 14, 911-917 (b) Boriskin, Y. S.; Leneva, I. A. ; Pécheur, E.-I. ; Polyak, S. J. Curr. Med. Chem. 2008, 15, 997-1005.


















