
Malalties hemorràgiques per inhibidors adquirits de la coagulació · 2011-10-08 · Hemofilia...
45
Malalties hemorràgiques per inhibidors adquirits de la coagulació Dra. Carme Altisent Unitat d’Hemofília. Hospital Vall d’Hebron
Transcript of Malalties hemorràgiques per inhibidors adquirits de la coagulació · 2011-10-08 · Hemofilia...

Malalties hemorràgiques per inhibidors adquirits de la coagulació
Dra. Carme AltisentUnitat d’Hemofília. Hospital Vall d’Hebron
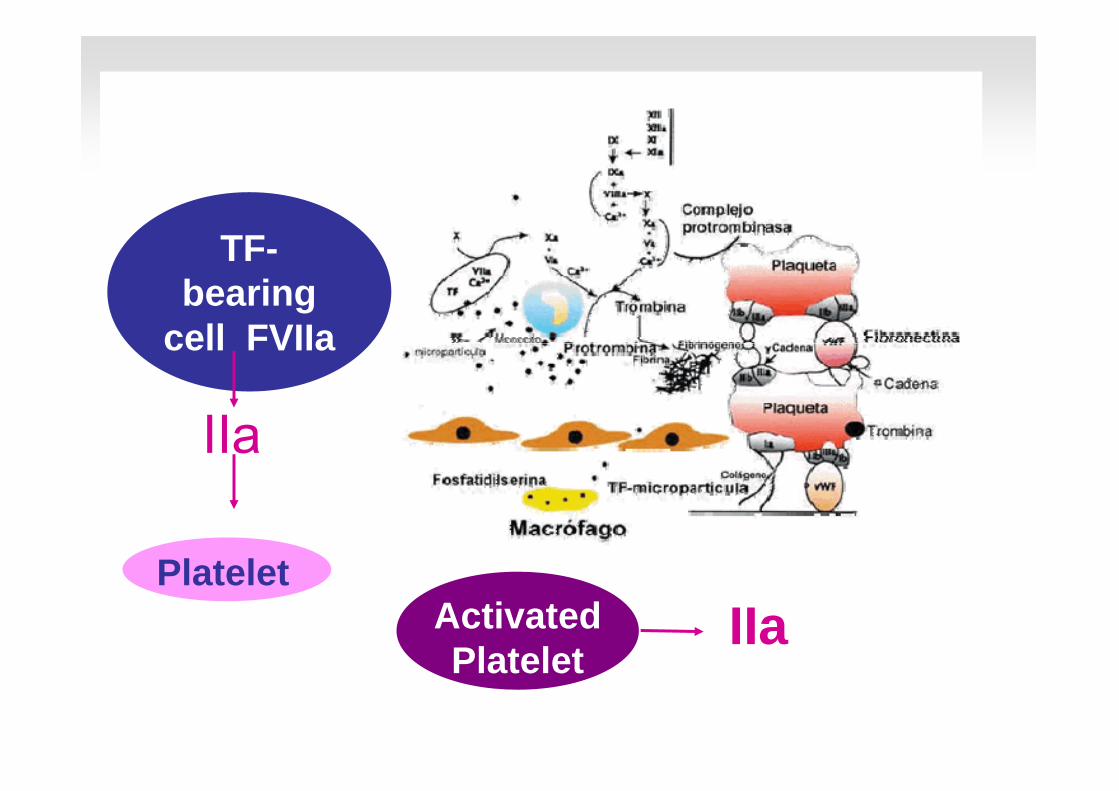
TF-bearing
cell FVIIa
IIa
PlateletActivated Platelet
IIa

Trastorn hemorràgic d’origen autoimmune degut a anticossos específics capaços d’inhibir l’acció dels factors de la coagulació.
El seu diagnòstic sovint és complex
L’hemofília adquirida és la malaltia més freqüent
Concepte

AloanticossosEn malalts amb coagulopaties després de rebre tractament substitutiu
AutoanticossosEn malalts sense coagulopatia, de forma espontània o associats a d’altres malalties
NaturalsEn el 20% dels donants sans
Classificació

Incidència anual: 1,3 casos per mil·lió d’habitantsNens <16 anys: 0,045
Adults >85 anys: 14,7
InfradiagnosticadaBaix títol
Assimptomàtics
Epidemiologia
Collins PW et al. Br J Hematol. 2009; 148: 183-194.

Fisiopatologia
Anticossos policlonals IgG : anti-IgG1 i -IgG4
No fixen complement
Especificitat de l’epítop FVIII: A2 o C2
Títol >10 UB en el 90% de casos

Combinació de factors genètics i ambientals associada a l’envelliment
del sistema immune
Canvis en la població de cèl·lules T reguladores
Limfòcits T amb receptors específics d’hiperesposta (Vβ2, Vβ5, Vβ9)
Polimorfismes de CTLA-4 (-318 C / T, +49 A / G i CT60 A / G
Polimorfismes del gen del factor VIII i de l’HLA
Fisiopatologia
Oldenburg J et al. Haemophilia. 2010; 16 (supl 3): S41-S45.
Tiede A et al. Ann Hematol. 2010; 89 (6): 607-6012.

Estudi TTPA allargat
Excloure contaminació amb heparina
Demostrar la presència d’un anticoagulant circulant
Excloure la presència d’un anticoagulant lúpic
Documentar els nivells baixos de factor
Quantificar l’inhibidor
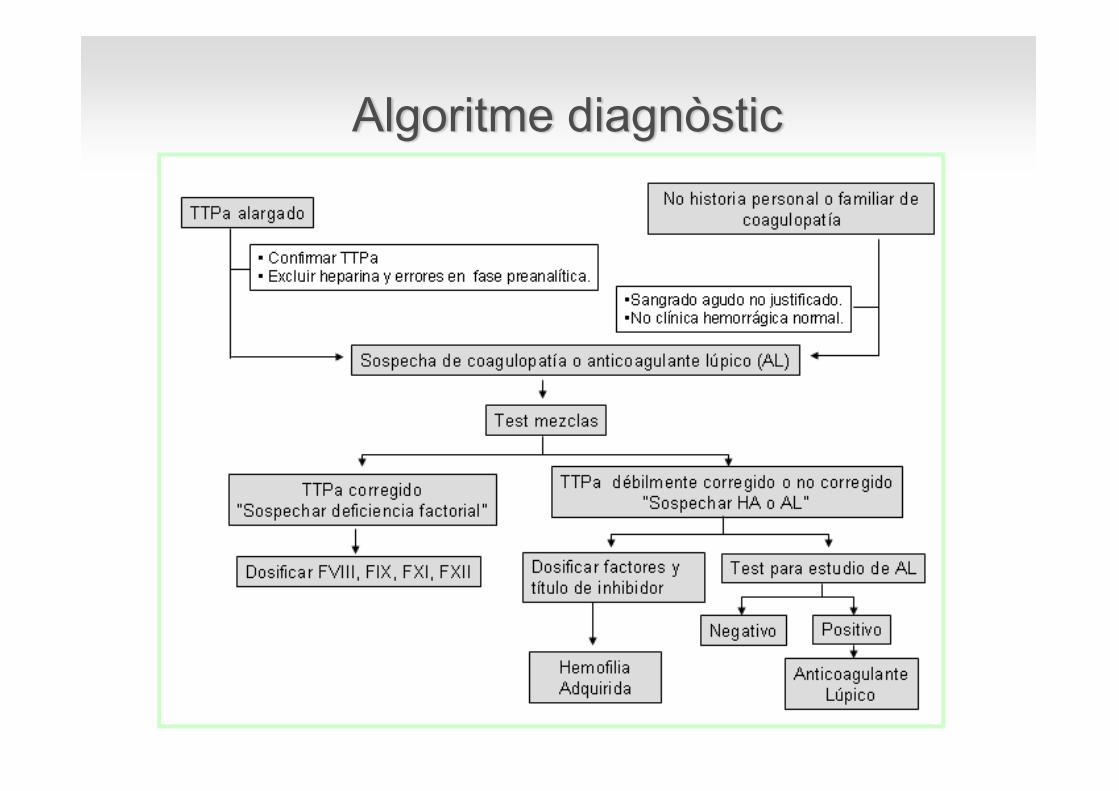
Algoritme diagnòstic
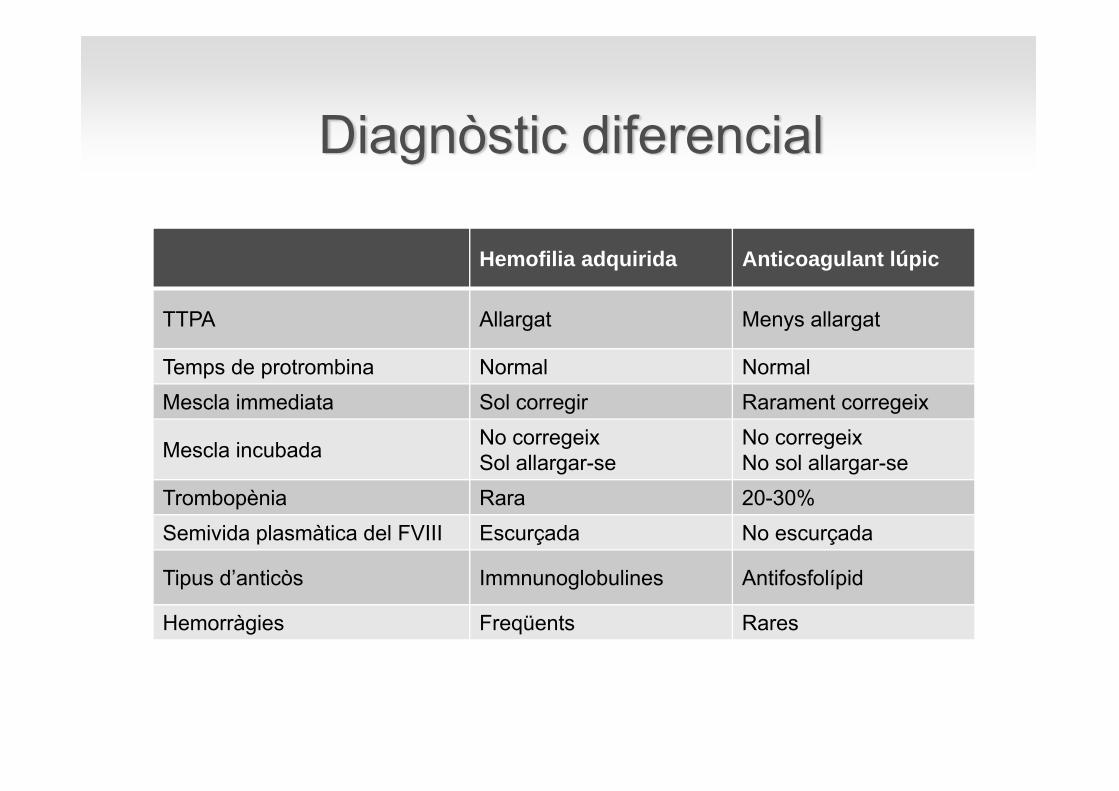
Hemofilia adquirida Anticoagulant lúpic
TTPA Allargat Menys allargat
Temps de protrombina Normal Normal Mescla immediata Sol corregir Rarament corregeix
Mescla incubada No corregeixSol allargar-se
No corregeixNo sol allargar-se
Trombopènia Rara 20-30%Semivida plasmàtica del FVIII Escurçada No escurçada
Tipus d’anticòs Immnunoglobulines Antifosfolípid
Hemorràgies Freqüents Rares
Diagnòstic diferencial
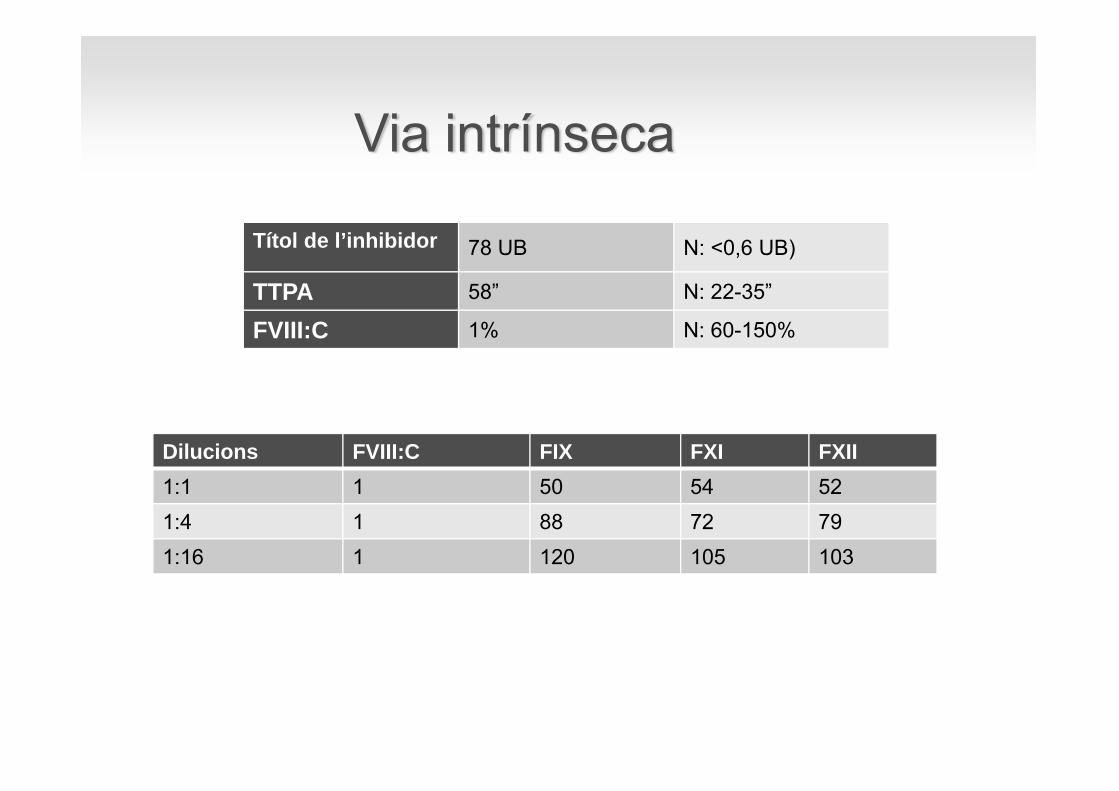
Dilucions FVIII:C FIX FXI FXII1:1 1 50 54 521:4 1 88 72 791:16 1 120 105 103
Via intrínseca
Títol de l’inhibidor 78 UB N: <0,6 UB)
TTPA 58” N: 22-35”
FVIII:C 1% N: 60-150%
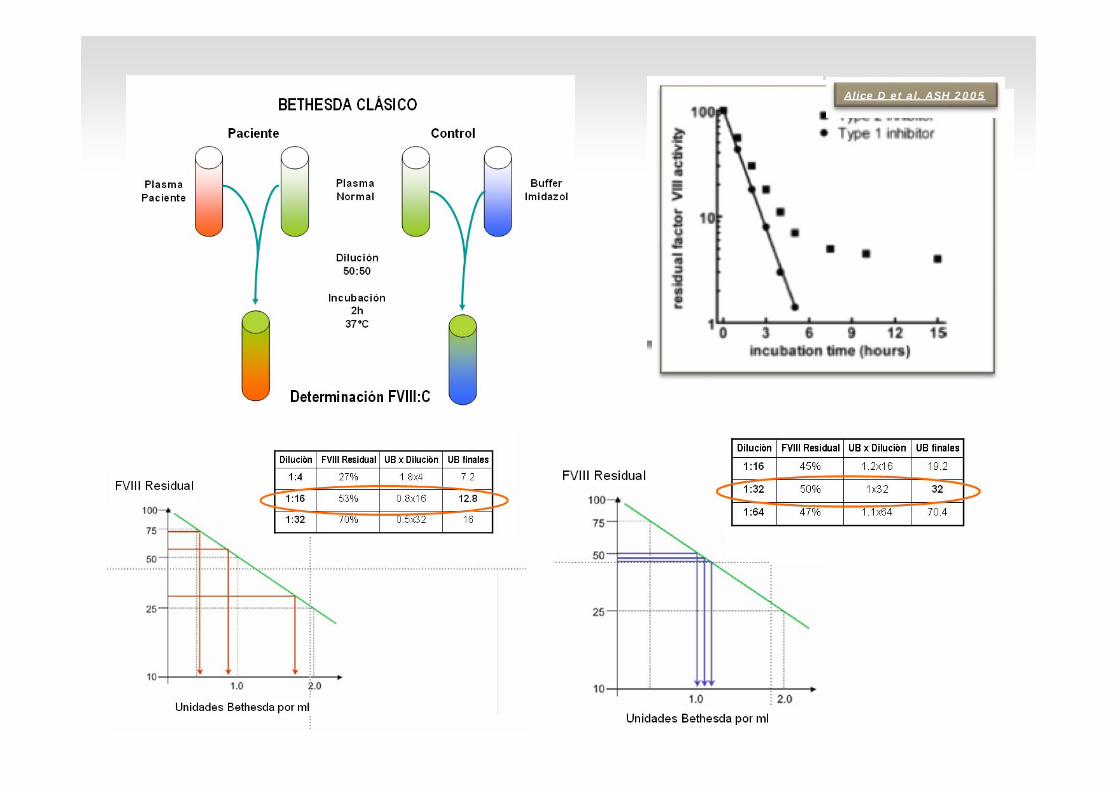
Alice D et a l. ASH 2 0 0 5
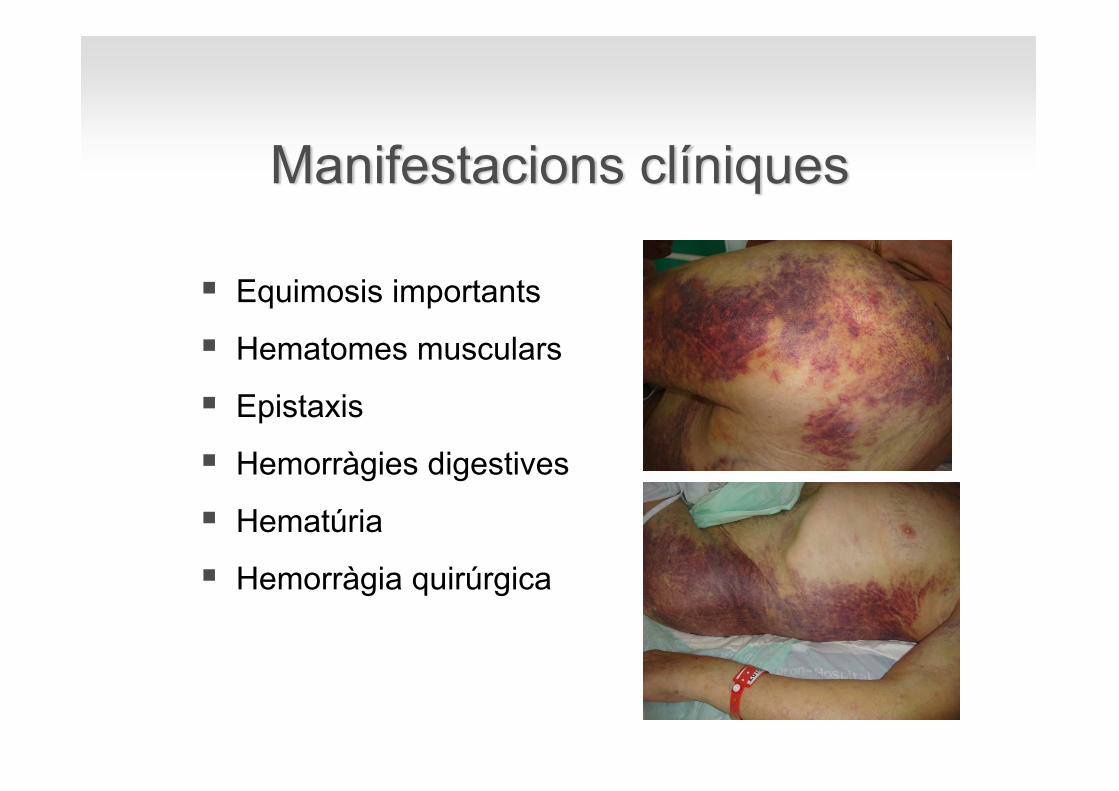
Equimosis importants
Hematomes musculars
Epistaxis
Hemorràgies digestives
Hematúria
Hemorràgia quirúrgica
Manifestacions clíniques
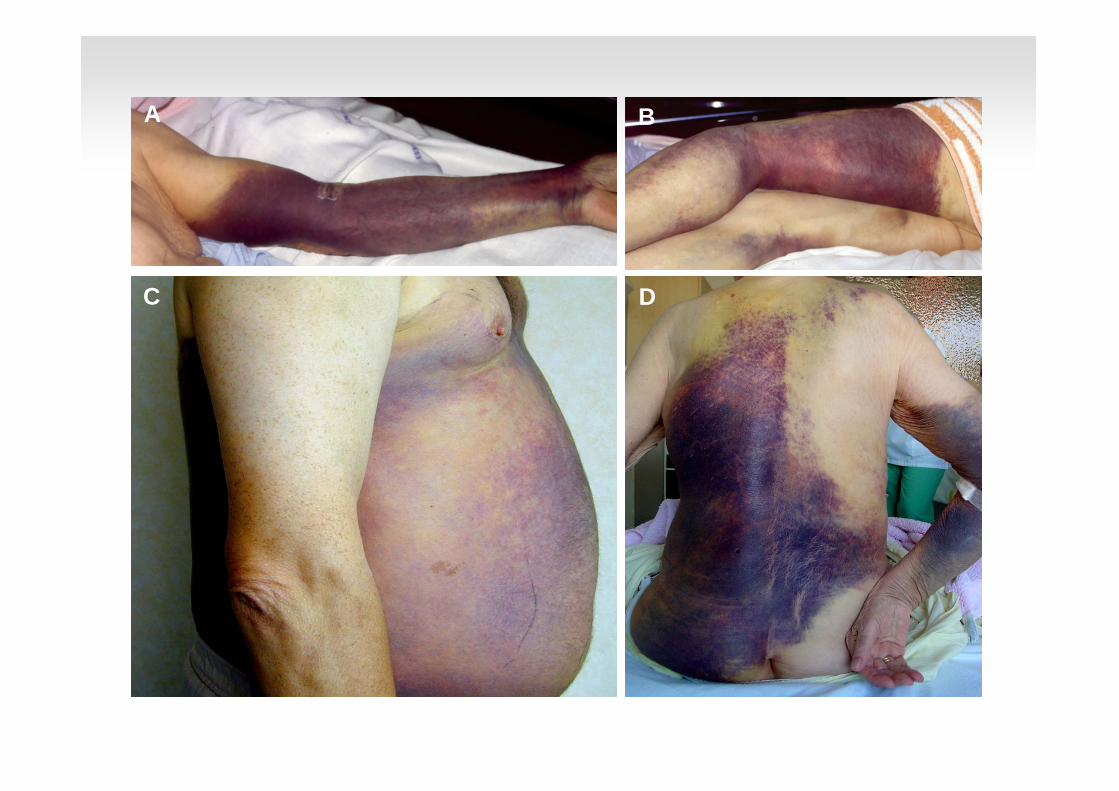
C
A B
D
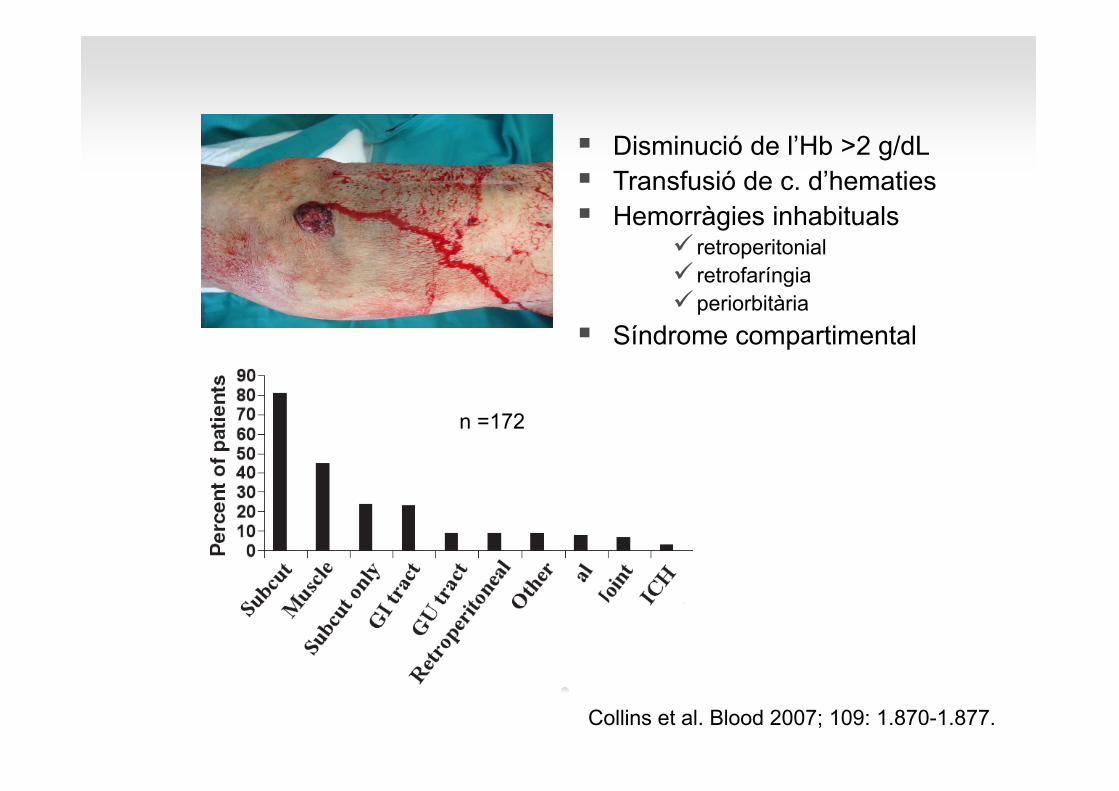
Disminució de l’Hb >2 g/dLTransfusió de c. d’hematiesHemorràgies inhabituals
retroperitonialretrofaríngiaperiorbitària
Síndrome compartimental
Collins et al. Blood 2007; 109: 1.870-1.877.
n =172
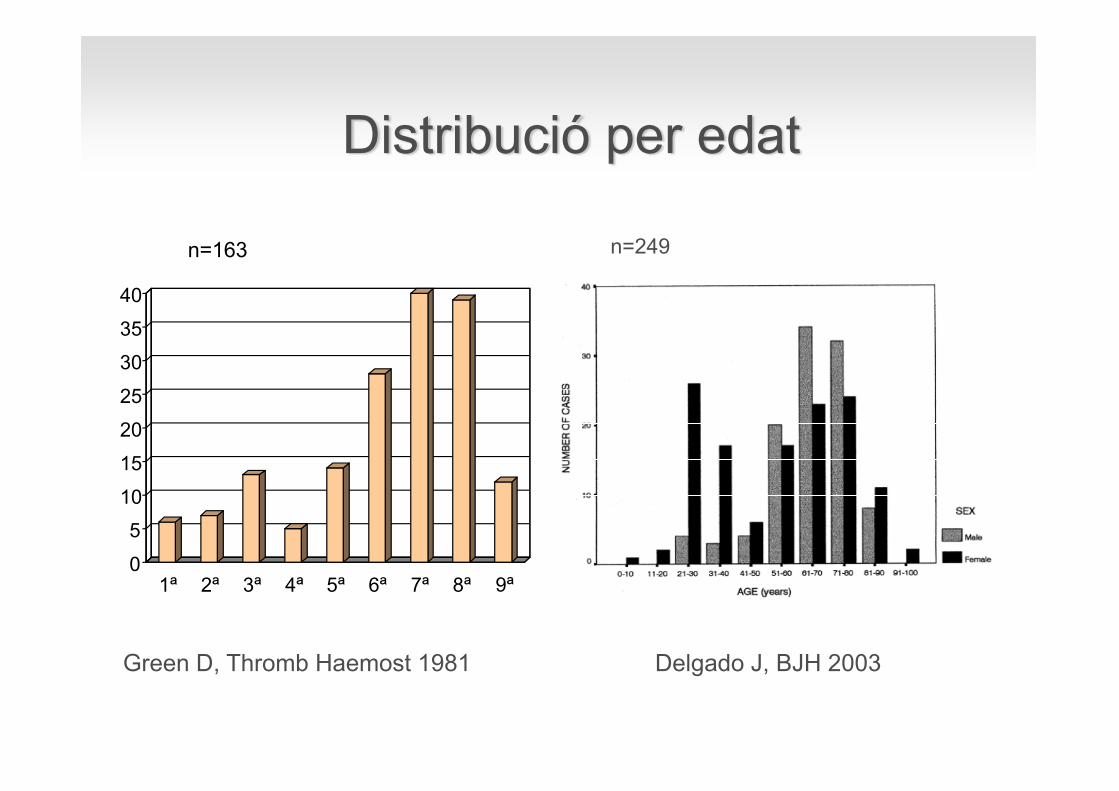
0
5
10
15
20
25
30
35
40
1ª 2ª 3ª 4ª 5ª 6ª 7ª 8ª 9ª
Green D, Thromb Haemost 1981
n=163
Delgado J, BJH 2003
n=249
Distribució per edat

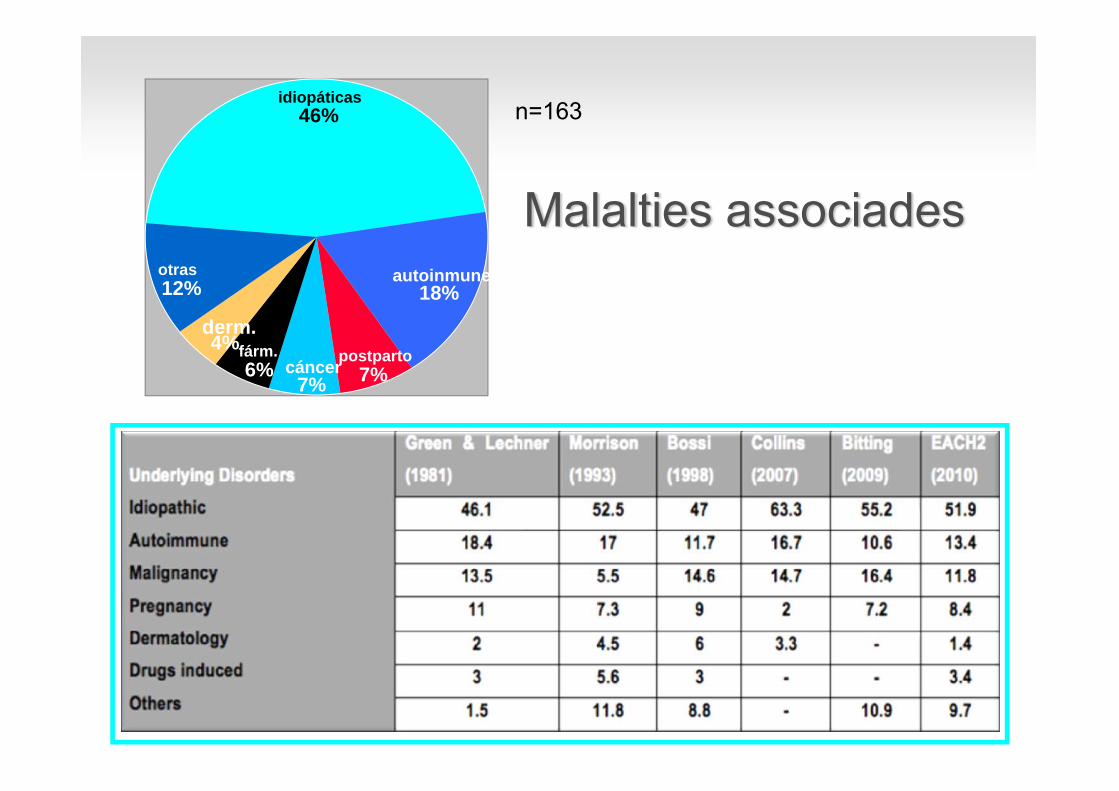
Malalties associades
idiopáticas46%
postparto7%cáncer
7%
fárm.6%
derm.4%
otras12% autoinmunes
18%
n=163

Registre internacional multicèntric, prospectiu sobre hemofília adquirida
Periode: 2003-2009
90 hospitals d’11 països europeus
AlemàniaAustriaFrançaEspanyaHolandaHongria
ItàliaPortugalRegne UnitSuèciaSuïssa
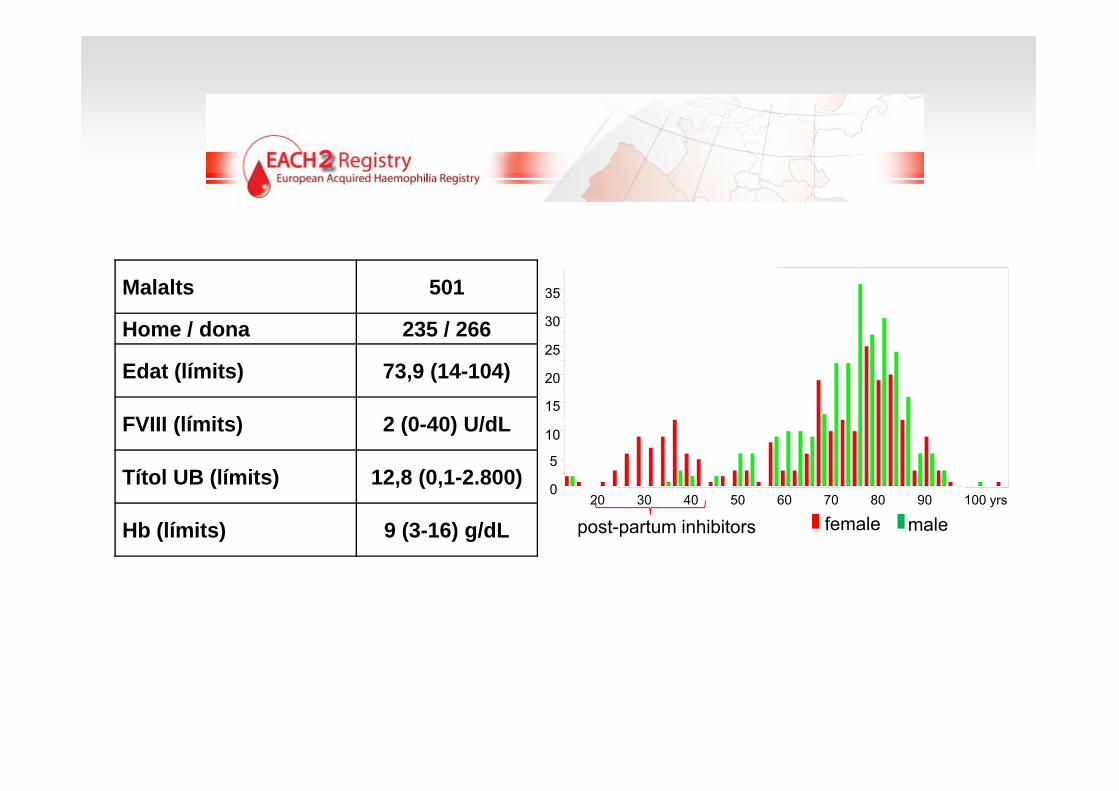
Malalts 501
Home / dona 235 / 266
Edat (límits) 73,9 (14-104)
FVIII (límits) 2 (0-40) U/dL
Títol UB (límits) 12,8 (0,1-2.800)
Hb (límits) 9 (3-16) g/dL male
0
5
10
15
20
25
30
35
20 30 40 50 60 70 80 90 100 yrs
femalepost-partum inhibitors


Agents bypass
Complex protrombínic activat
Origen plasmàtic
Vida mitja de 12 h
No hi ha una prova de control
Possible resposta mnemònica
Risc de trombosis
Cost elevat
FVIIar
Origen recombinant
Vida mitja de 2-3 h
No hi ha una prova de control
Sense resposta mnemònica
Menor risc de trombosis
Cost elevat
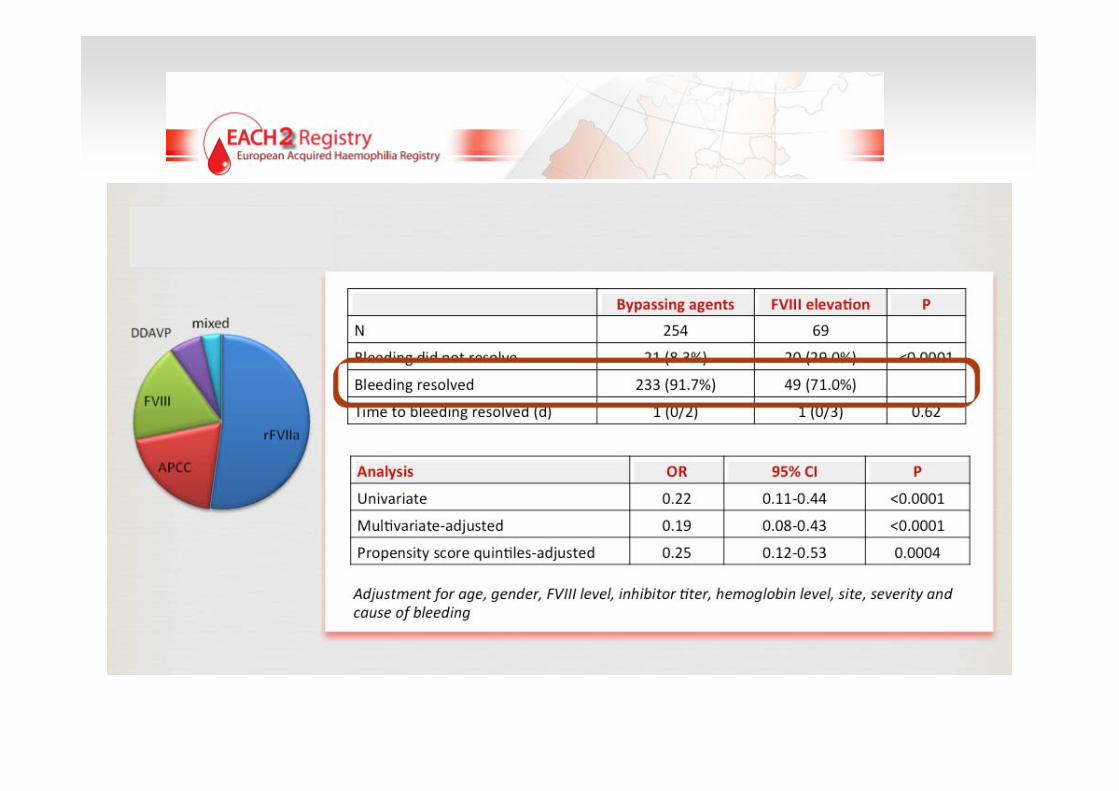
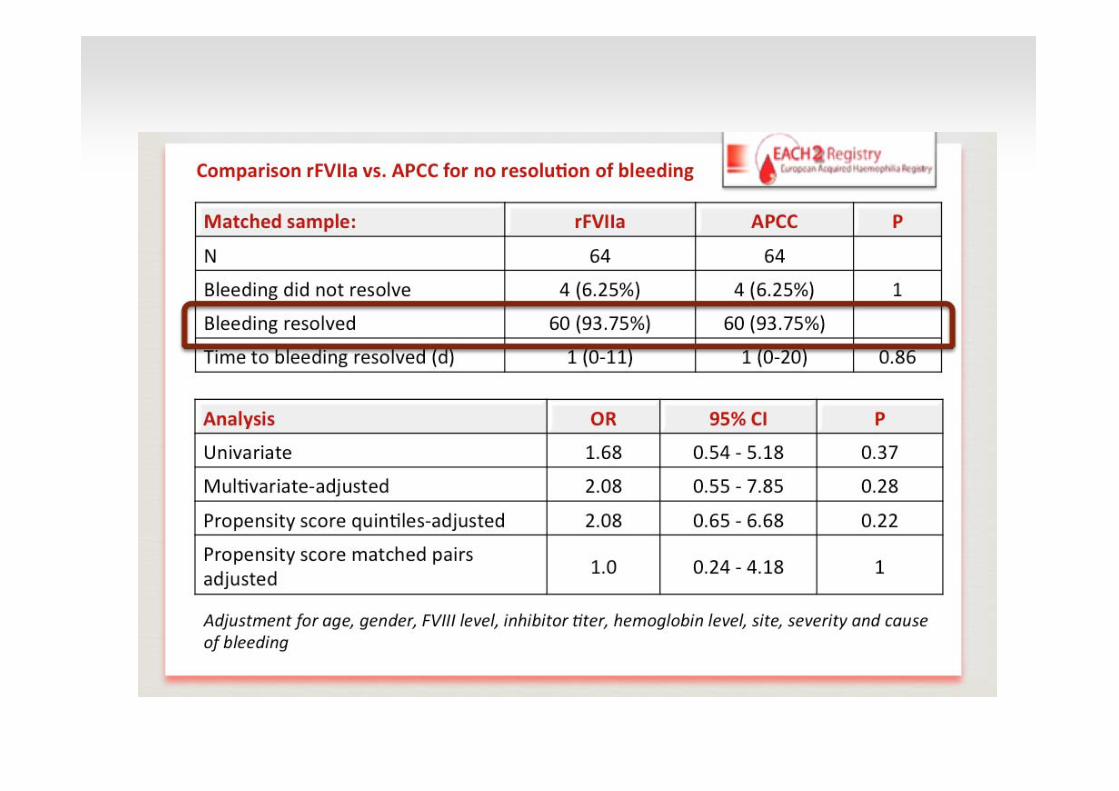

3 Thromboembolic events38 patients
(67 Bleedings)
10 Thromboembolic events 151 patients
(207 bleedings)
Deaths due to bleeding 3.0 % Myocardial infarction 1.4 % Stroke 0.2 % Venous thromboembolism 1.0 %
N o significant association of death or severe adverse events with a specific hemostatic therapy
aPCC rFVIIa


CPADosi: 50-100 UI/Kg cada 8-12 hDosi máxima: 200 UI/Kg dia
FVIIarDosi: 90 µg/Kg cada 2-3 h
Agents bypass

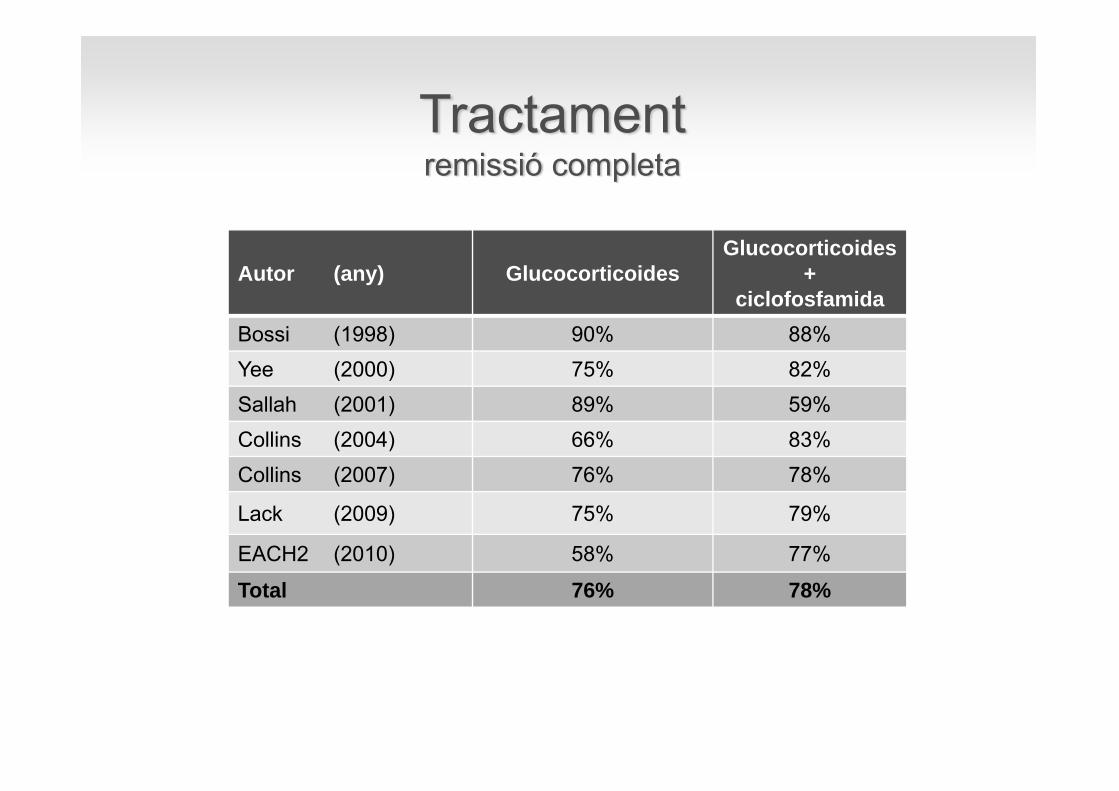
Autor (any) GlucocorticoidesGlucocorticoides
+ciclofosfamida
Bossi (1998) 90% 88%Yee (2000) 75% 82%Sallah (2001) 89% 59%Collins (2004) 66% 83%Collins (2007) 76% 78%
Lack (2009) 75% 79%
EACH2 (2010) 58% 77%
Total 76% 78%
Tractamentremissió completa
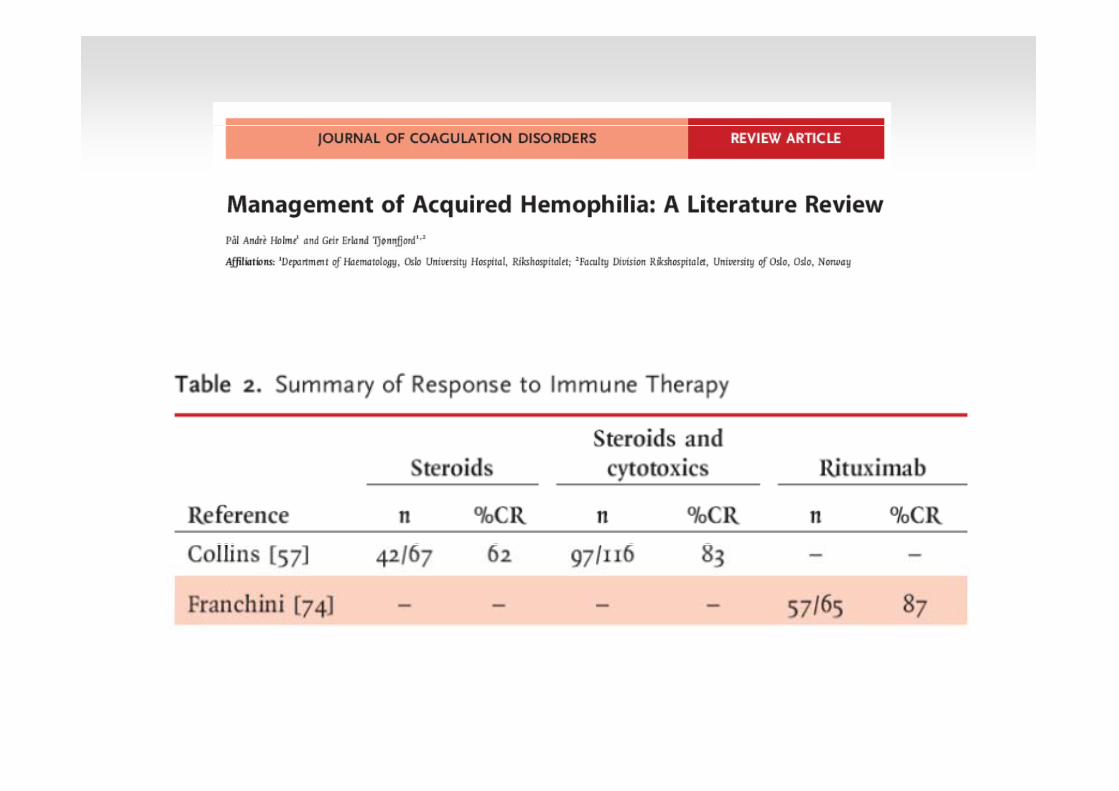

Dosis recomanades
Glucocorticoides: 1-2 mg/kg/d (6 setmanes; 30% de resposta)
Ciclofosfamida sola o amb glucocorticoides (60-100% de resposta)
• 1-2 mg/kg/d
• 10-12 mg/kg/d iv dos dies i continuar amb 1-2 mg/kg/d
Rituximab: 375 mg/m2/set durant 4 setmanes
Ciclosporina: 200-300 mg/d
Azatioprina: 2mg/kg/d durant 6 setmanes
Vincristina: 1 mg/m2 (dosi única)
Immunosupresssió
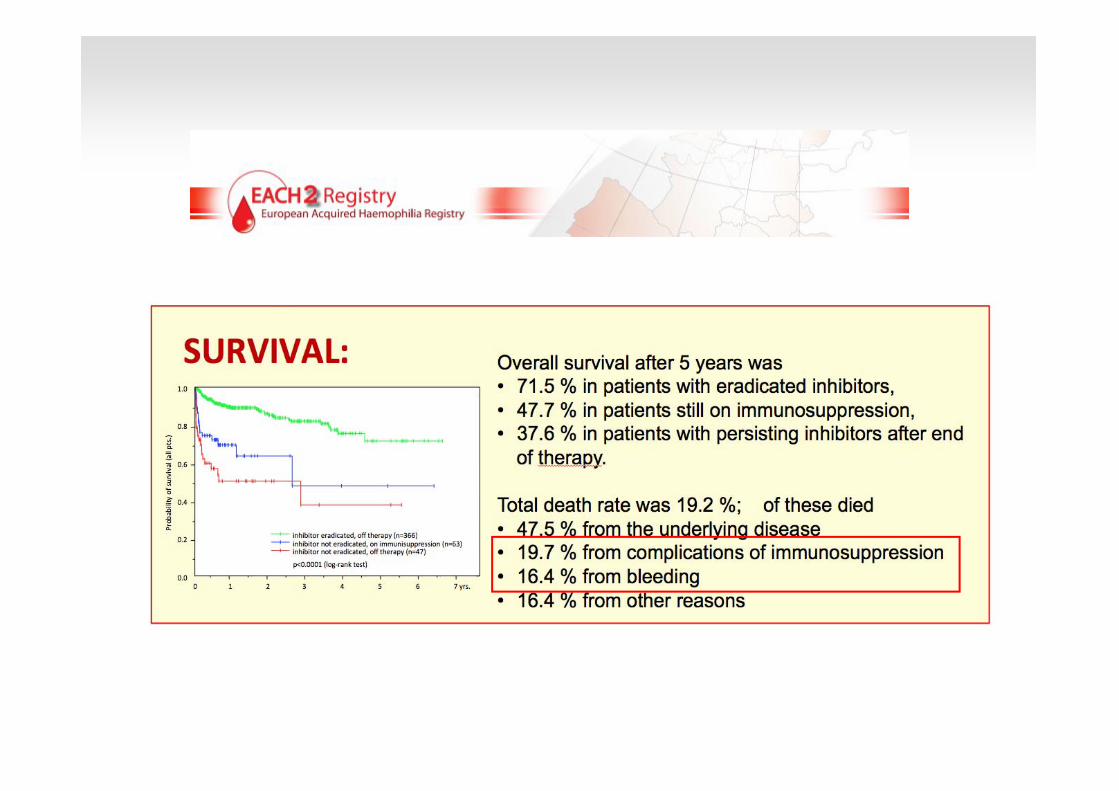
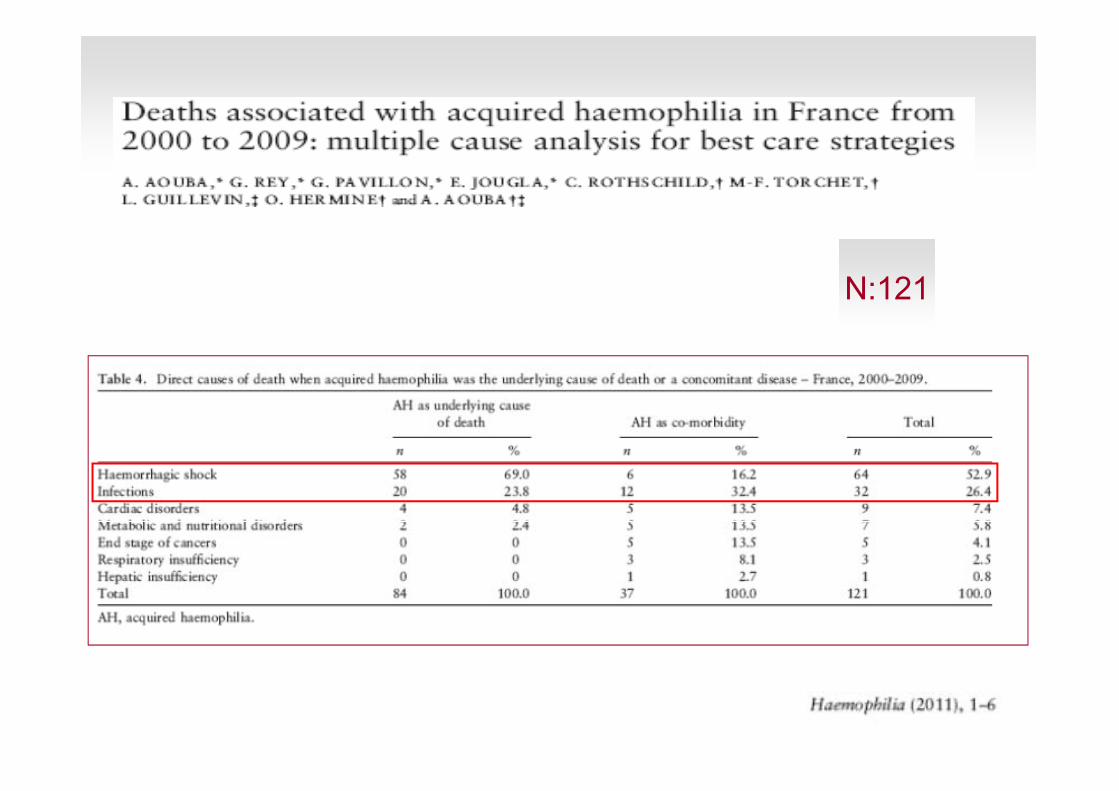
N:121
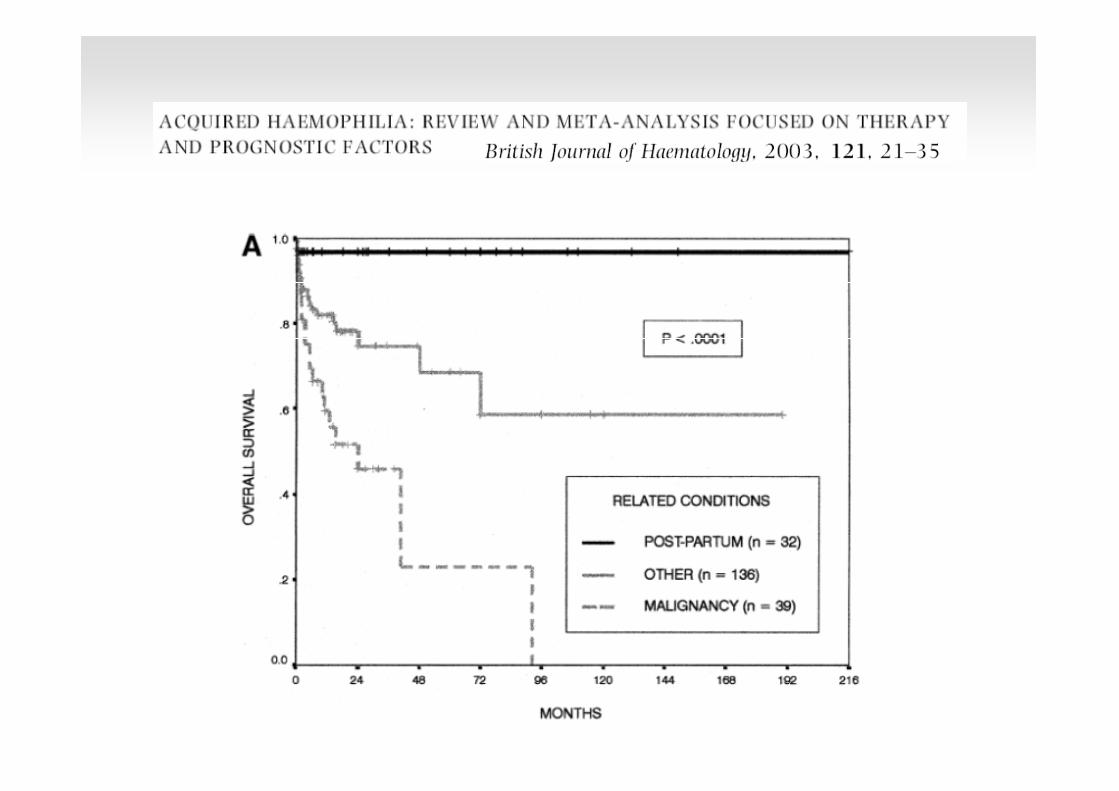

J Thromb Haemost. 2008; 6 (4): 565-568
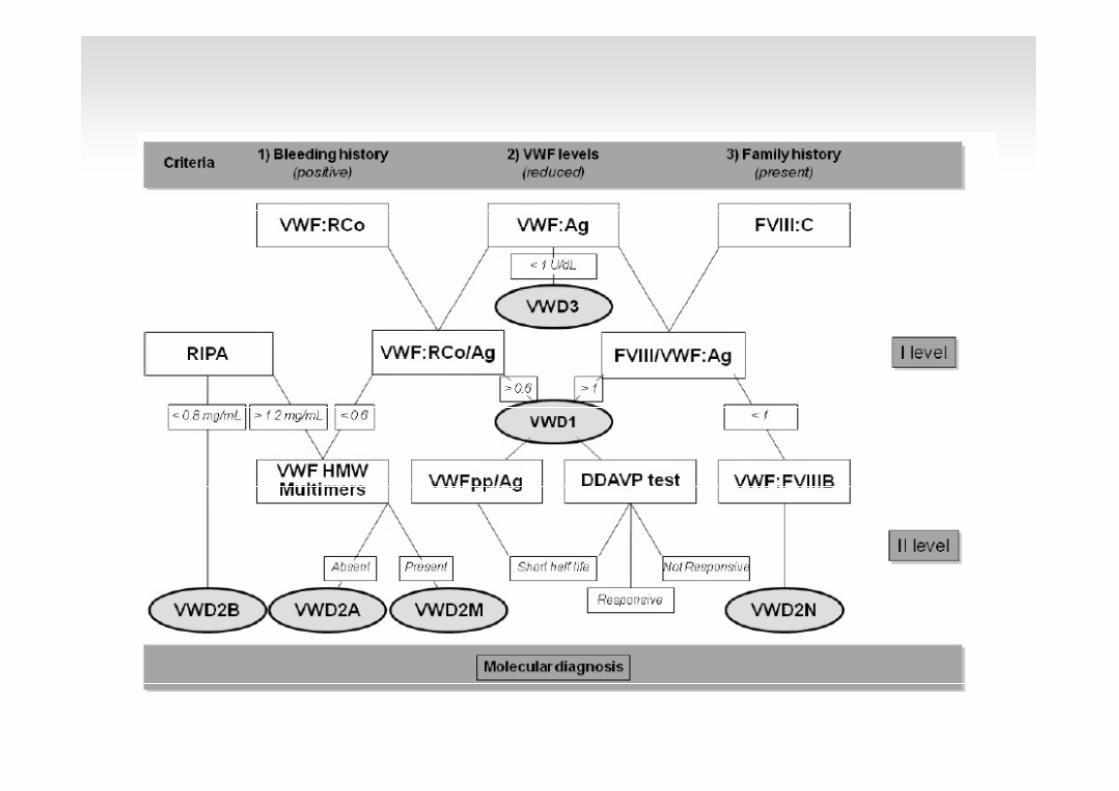
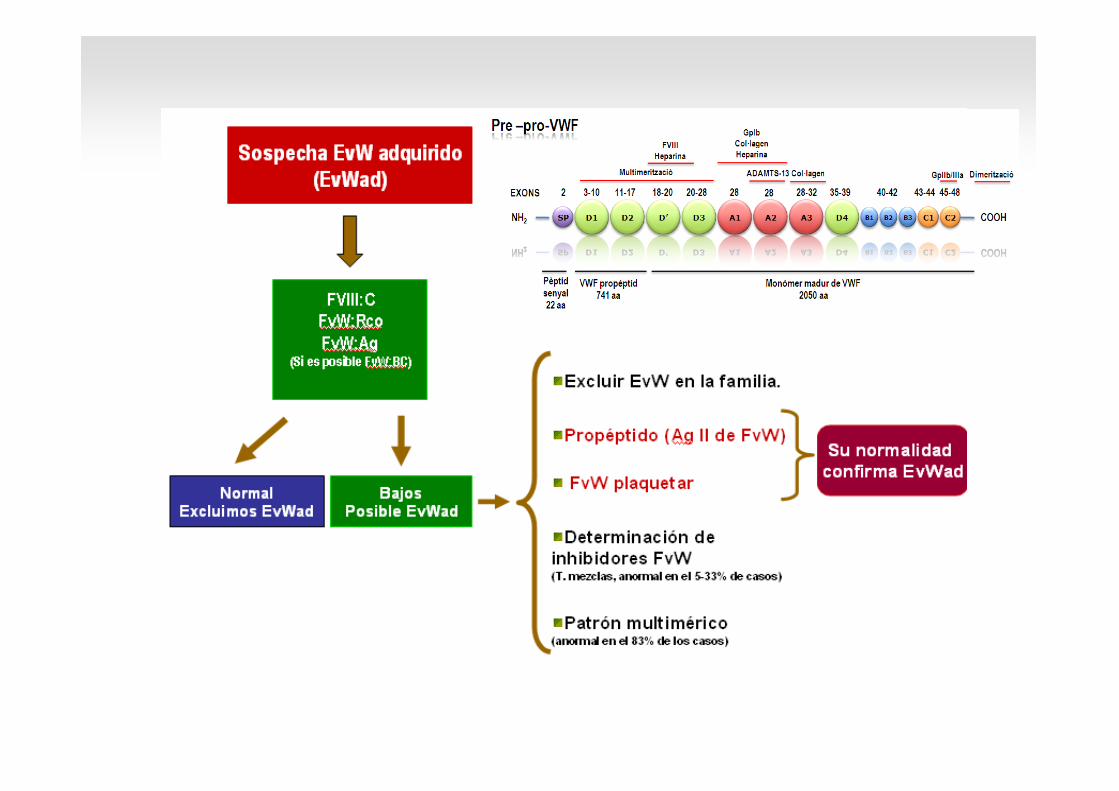

Autoanticossos específics
Autoanticossos inespecífics i formació d’immunocomplexos que depuren el FvW a través de cèl·lules amb receptors Fc del sistema retículo-endotelial
Absorció pels clons de cèl·lules malignes
Augment de la degradació proteolítica
Síndrome de von Willebrand adquiridapatogènia

Mieloma múltiple
Limfoma
Leucèmia limfàtica crònica
Waldenström
Síndromes mieloproliferatives
Càncertumor de Wilmsadenocarcinomacarcinoma cèl·lules escamoseshepatocarcinoma
AngiodisplàsiaLupusMalformacions cardíaques
Federici AB. Haemophilia 1998; 4: 654-660
Síndrome de von Willebrand adquiridamalalties associades
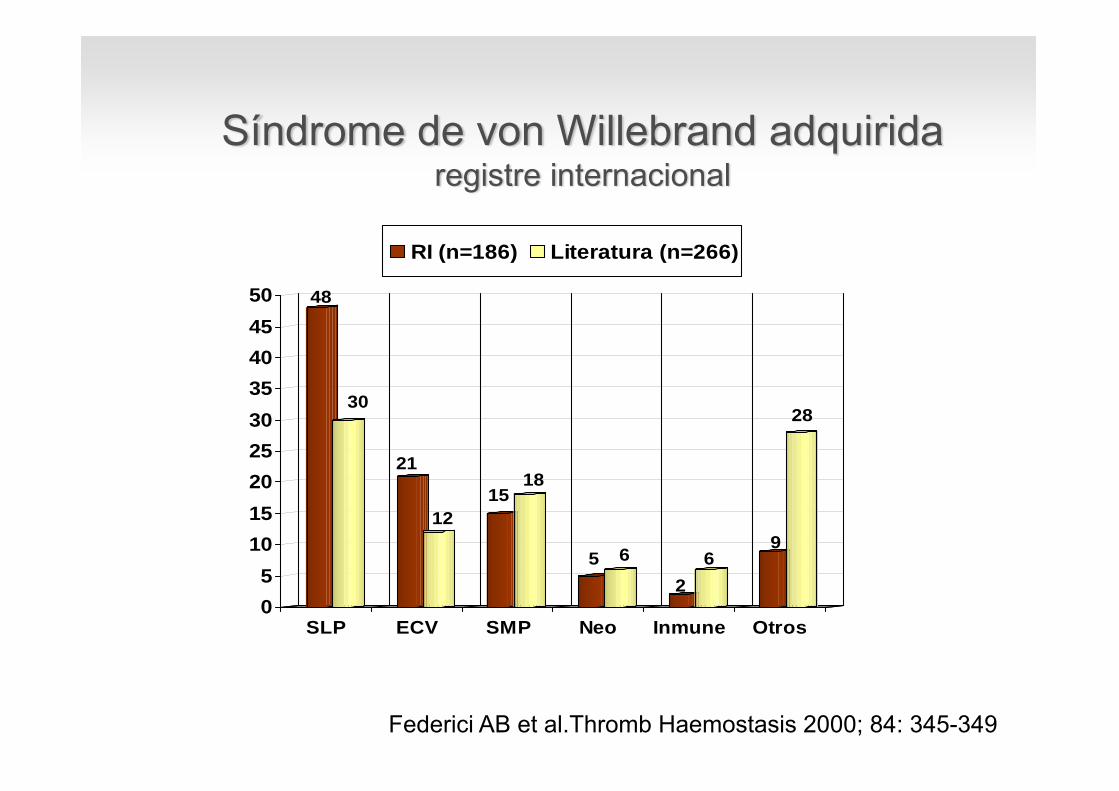
48
30
21
1215
18
5 6
26
9
28
05
101520253035404550
SLP ECV SMP Neo Inmune Otros
RI (n=186) Literatura (n=266)
Federici AB et al.Thromb Haemostasis 2000; 84: 345-349
Síndrome de von Willebrand adquiridaregistre internacional

Hemorràgies (70-80%)lleus o moderades com a la malaltia congènita tipus 1 i 2muco-cutàniesquirúrgiques
Alteració del laboratori (20-30%)
Síndrome de von Willebrand adquiridamanifestacions clíniques
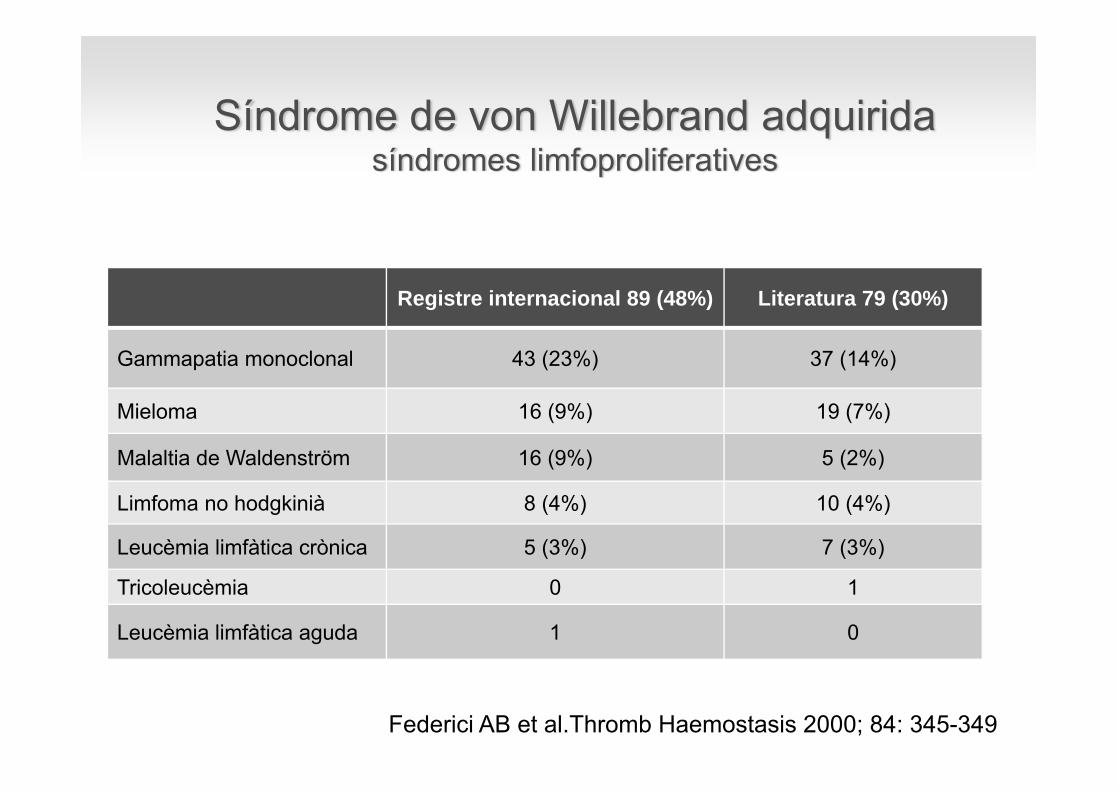
Registre internacional 89 (48%) Literatura 79 (30%)
Gammapatia monoclonal 43 (23%) 37 (14%)
Mieloma 16 (9%) 19 (7%)
Malaltia de Waldenström 16 (9%) 5 (2%)
Limfoma no hodgkinià 8 (4%) 10 (4%)
Leucèmia limfàtica crònica 5 (3%) 7 (3%)
Tricoleucèmia 0 1
Leucèmia limfàtica aguda 1 0
Síndrome de von Willebrand adquiridasíndromes limfoproliferatives
Federici AB et al.Thromb Haemostasis 2000; 84: 345-349

Registre internacional 29 (15%) Literatura 48 (18%)
Trombocitèmia essencial 21 (11%) 17 (6%)
Policitèmia vera 1 9 (3%)
Leucèmia mieloide crònica 5 (3%) 22 (8%)
Mielofibrosi 2 (1%) 0
Síndrome de von Willebrand adquiridasíndromes mieloproliferatives
Federici AB et al.Thromb Haemostasis 2000; 84: 345-349

Fibrinògen i factors II, V, VII, IX, XI i XIII
FII i FV: exposició a trombina bovina
FX: amiloïdosi sistèmica
FXI: trombosi amb dèficit de FXII
Fibrinògen: no són útils els agents bypass
Inhibidors contra altres factors

Els inhibidors adquirits de la coagulació són causa d’hemorràgies potencialment molt greus
Un estudi bàsic d’hemostàsia permet orientar el diagnòstic
El tractament és complicat i cal derivar els malalts a centres de referència
No hi ha evidència científica sobre el tractament òptim. Les guies terapèutiques es basen en l’experiència de casos publicats, en estudis retrospectius i en l’opinió d’experts
Conclusió















![Coagulació i anticoagulació: buscant l'equilibri al ...Coagulació i anticoagulació: buscant l'equilibri al sistema cardiovascular Pablo García de Frutos . sang 1 1 f. [MD] [ZOA]](https://static.fdocuments.ec/doc/165x107/5f02bfa27e708231d405d155/coagulaci-i-anticoagulaci-buscant-lequilibri-al-coagulaci-i-anticoagulaci.jpg)


