
FACULTAD DE MEDICINA CENTRO UNIVERSITARIO...
115
FACULTAD DE MEDICINA CENTRO UNIVERSITARIO DE INVESTIGACIONES BIOMÉDICAS EFECTO CONCOMITANTE DEL ÓXIDO NÍTRICO Y EL CIANURO DE SODIO EN EL SENO CAROTÍDEO IN VIVO SOBRE EL REFLEJO HIPERGLUCÉMICO EN RATAS NORMALES Y DIABÉTICAS Tesis Que para obtener el Grado de Doctor en Ciencias Fisiológicas Presenta: M. en C. Héctor Rafael Tejeda Chávez Asesores: Biol. Elena Roces de Álvarez-Buylla D. en C. Sergio Adrián Montero Cruz Colima, Col., Enero 2008.
Transcript of FACULTAD DE MEDICINA CENTRO UNIVERSITARIO...

FACULTAD DE MEDICINA
CENTRO UNIVERSITARIO DE INVESTIGACIONES BIOMÉDICAS
EFECTO CONCOMITANTE DEL ÓXIDO NÍTRICO Y EL CIANURO DE SODIO EN EL SENO CAROTÍDEO IN VIVO SOBRE EL REFLEJO HIPERGLUCÉMICO
EN RATAS NORMALES Y DIABÉTICAS
Tesis
Que para obtener el Grado de Doctor en Ciencias Fisiológicas
Presenta:
M. en C. Héctor Rafael Tejeda Chávez
Asesores: Biol. Elena Roces de Álvarez-Buylla
D. en C. Sergio Adrián Montero Cruz
Colima, Col., Enero 2008.


FACULTAD DE MEDICINA
CENTRO UNIVERSITARIO DE INVESTIGACIONES BIOMÉDICAS
EFECTO CONCOMITANTE DEL ÓXIDO NÍTRICO Y EL CIANURO DE SODIO EN EL SENO CAROTÍDEO IN VIVO SOBRE EL REFLEJO HIPERGLUCÉMICO
EN RATAS NORMALES Y DIABÉTICAS
Tesis
Que para obtener el Grado de Doctor en Ciencias Fisiológicas
Presenta:
M. C. Héctor Rafael Tejeda Chávez
Asesores: Biol. Elena Roces de Álvarez-Buylla
D. en C. Sergio Adrián Montero Cruz
Colima, Col., Enero 2008.

Este trabajo de tesis fue realizado en el laboratorio de
neuroendocrinología del Centro Universitario de Investigaciones
Biomédicas de la Universidad de Colima, con la beca del Consejo
Nacional de Ciencia y Tecnología (CONACYT). Registro: 171469.

DEDICATORIA
Dedico este trabajo:
Especialmente:
A mis Hijos y Esposa.
A mis Padres.
A mis Hermanos y Hermanas.
A mis Sobrinos.
A mis Hermanos Fraternos.
Con cariño y respeto a toda la humanidad que le pueda ser de utilidad este Conocimiento.
“Nuestra recompensa se encuentra en el esfuerzo y no en el resultado. Un esfuerzo total es una victoria completa”.
Mahatma Gandhi

AGRADECIMIENTOS
Expreso mi sincero agradecimiento:
A la Universidad de Colima, en particular al Centro Universitario de Investigaciones Biomédicas (CUIB), por aceptarme como alumno de su
reconocido programa académico en Ciencias Fisiológicas
Al Dr. Ramón Álvarez-Buylla de Aldana, a quien considero como uno de los
científicos más prominentes que ha tenido la comunidad científica en México, y a
quien debo me haya sembrado la curiosidad de explorar lo desconocido en el
campo del saber científico, cuando aún era un estudiante de medicina.
A la Biol. Elena Roces de Álvarez-Buylla y al Dr. Sergio Adrián Montero Cruz, asesores de este trabajo de Tesis, por abrirme las puertas del laboratorio
de neuroendocrinología y guiarme en el camino de la investigación científica; por
su invaluable ayuda, sus consejos, su apoyo permanente durante el proceso de
mi formación en el doctorado, y muchas otras cosas que quedan grabadas tanto
en mis pensamientos como en mis sentimientos.
Al Dr. Sergio Adrián Montero Cruz, por ser un apreciable amigo y por
compartir conmigo su vasto conocimiento con gran generosidad.
A todos mis Maestros que integran el Cuerpo Académico de este Centro Universitario, que me enseñaron a comprender los diversos mecanismos
implicados en los procesos biológicos estudiados por ellos; muy especialmente a
mis sinodales: Dra. Irene Díaz Reval, Dr. Miguel Huerta Viera, Dra. Xóchitl
Angélica Rosio Trujillo Trujillo y Dr. José Clemente Vásquez Jiménez, que con sus
aportaciones y comentarios han contribuido a enriquecer de manera importante la
estructura de esta Tesis.

Al Dr. Eliseo Portilla de Buen por sus aportaciones y a la Dra. Caridad Áurea Leal Cortés por el apoyo técnico para la cuantificación de los nitritos en la
sangre encefálica.
A todo el personal técnico y administrativo del CUIB, por su apoyo
constante y su amistad.
Al CONACyT por la beca recibida para la realización de mi doctorado, sin
este apoyo me hubiera sido imposible continuar estudiando.

ÍNDICE
Página Índice de tablas y figuras 3
Abreviaturas 6
Resumen 8
Abstract 9
Introducción 10
Antecedentes - La homeostasis de la glucosa y los quimiorreceptores.
- El óxido nítrico y los quimiorreceptores.
- El óxido nítrico y la homeostasis de la glucosa.
- El óxido nítrico en la diabetes.
14
14
26
34
36
Justificación 41
Planteamiento del problema 42
Hipótesis 42
Objetivos - General.
- Específicos.
42
43
Metodología - Animales y técnicas quirúrgicas generales.
- Estimulación de los RSCC.
- Obtención de sangre y procedimientos bioquímicos.
- Determinación de los niveles de glucosa en el plasma.
- Determinación de los niveles de nitritos en el plasma. Método de
Griess.
- Reactivos de Griess
- Fármacos utilizados.
- Ratas diabéticas.
- Protocolo experimental.
44
45
46
47
48
50
50
52
53

2
- Análisis estadístico. 54
Resultados - Control 1. Inyección de sol. sal. por dos ocasiones en el seno
carotídeo aislado circulatoriamente aislado.
- Control 2. Estimulación de los RSCC con NaCN seguida de una
inyección de sol. sal. en el seno carotídeo circulatoriamente aislado.
- Experimental 1. Estimulación de los RSCC con NaCN en el seno
carotídeo circulatoriamente aislado seguida de un inhibidor de la
NOS (L-NAME).
- Experimental 2. Estimulación de los RSCC con NaCN en el seno
carotídeo circulatoriamente aislado seguida de un donador del NO
(NPS).
- Comparación de la retención de glucosa cerebral entre los grupos
control 2 y experimentales 1 y 2.
55
58
61
65
69
Discusión 71
Conclusiones 83
Perspectivas 85
Referencias 86

3
ÍNDICE DE TABLAS Y FIGURAS
Página Tabla 1. Protocolo para la determinación de nitritos. 51
Tabla 2. Concentración de glucosa plasmática (mg/dL) en el grupo
control 1.
55
Tabla 3. Retención de glucosa cerebral (mg/dL) en el grupo control
1.
56
Tabla 4. Concentración de nitritos (nmol/mL) en la sangre venosa
cerebral en el grupo control 1.
56
Tabla 5. Concentración de glucosa plasmática (mg/dL) en el grupo
control 2.
59
Tabla 6. Retención de glucosa cerebral (mg/dL) en el grupo control
2.
59
Tabla 7. Concentración de nitritos (nmol/mL) en la sangre venosa
cerebral en el grupo control 2.
61
Tabla 8. Concentración de glucosa plasmática (mg/dL) en el grupo
experimental 1.
63
Tabla 9. Retención de glucosa cerebral (mg/dL) en el grupo
experimental 1.
63
Tabla 10. Concentración de nitritos (nmol/mL) en la sangre venosa
cerebral en el grupo experimental 1.
65
Tabla 11. Concentración de glucosa plasmática (mg/dL) en el grupo
experimental 2.
66
Tabla 12. Retención de glucosa cerebral (mg/dL) en el grupo
experimental 2.
67
Tabla 13. Concentración de nitritos (nmol/mL) en la sangre venosa
cerebral en el grupo experimental 2.
67
Figura 1. Microfotografía de la morfología de las células glómicas en
una rebanada delgada del CC de rata.
20

4
Figura 2. Cambios en la concentración de la glucosa sanguínea y
en la retención de glucosa cerebral en las ratas
anestesiadas.
22
Figura 3. Vías nerviosas desde los receptores del CC al hipotálamo. 23
Figura 4. Velocidad de infusión de glucosa en la vena cava durante
el estado hipoglucémico producido por la insulina (1
mU/k/min) en perros controles con CC intactos (●) y en
perros con extirpación de ambos CC (ο).
24
Figura 5. Respuesta secretora de las células glómicas del CC a
diferentes concentraciones de glucosa en condiciones de
hipoxia y normoxia.
25
Figura 6. Cadena respiratoria (CR) en la mitocondria. 28
Figura 7. Esquema hipotético de un modelo de membrana de las
células del CC sensibles al oxígeno.
31
Figura 8. Síntesis, difusión y esfera de acción del NO en el sistema
nervioso.
33
Figura 9. Secuencia de mediciones del cuerpo carotídeo perfundido
de gato.
35
Figura 10. Expresión de la NOSe y NADPH-Diaforasa en el cuerpo
carotídeo de ratas control en condiciones de normoxia o
hipoxia, y en ratas diabéticas durante la normoxia.
39
Figura 11. Esquema que muestra el procedimiento quirúrgico en la
rata.
46
Figura 12. Concentración de glucosa en el plasma y retención de
glucosa cerebral en las ratas normales (A) y diabéticas (B)
del grupo control 1.
57
Figura 13. Concentración de nitritos en la sangre venosa cerebral en
ratas normales (A) y diabéticas (B) del grupo control 1.
58
Figura 14. Concentración de glucosa en los plasmas arterial y
venoso, y retención de glucosa cerebral en ratas normales
(A) y diabéticas (B) del grupo control 2.
60

5
Figura 15. Concentración de nitritos en la sangre venosa cerebral en
ratas normales (A) y diabéticas (B) del grupo control 2.
62
Figura 16. Concentración de glucosa en los plasmas arterial y
venoso, y retención de glucosa cerebral en las ratas
normales (A) y diabéticas (B) del grupo experimental 1.
64
Figura 17. Concentración de nitritos en la sangre venosa cerebral en
las ratas normales (A) y diabéticas (B) del grupo
experimental 1.
65
Figura 18. Concentración de glucosa en los plasmas arterial y
venoso, y retención de glucosa cerebral en las ratas
normales (A) y diabéticas (B) del grupo experimental 2.
68
Figura 19. Concentración de nitritos en la sangre venosa cerebral en
las ratas normales (A) y diabéticas (B) del grupo
experimental 2.
69
Figura 20. Comparación de las retenciones de glucosa cerebral en
las ratas normales y diabéticas entre los grupos: control 2,
experimental 1 y 2.
70
Figura 21 Esquema hipotético de un modelo de la membrana de las
células del CC sensibles al oxígeno.
74

6
ABREVIATURAS aa Aorta abdominal i.v. intravenosa AC Arteria carótida K+ Ión potasio acc Arteria carótida común LC Locus coeruleus ace Arteria carótida externa LCR Liquido cefalorraquídeo. Aci ACh
Arteria carótida interna Acetilcolina
L-NAME N-nitro-L-arginina metil éster
ADN Ácido desoxiribonucleíco L-NMMA N-monometil-L-arginina ADP Difosfato de adenosina m Metros af Arteria femoral. ME Membrana externa al Arteria lingual. mg Miligramos ATP Trifosfato de adenosina MI Membrana interna Ca2+ Ión calcio. min Minuto CC Cuerpo carotídeo mL Mililitro Cit-c Citocromo oxidasa c mM Milimolar CO2 Bióxido de carbono mmHg Milímetros de mercurio CP Carótida primitiva mmol Milimol CR Cadena respiratoria mU Miliunidades Cu Cobre Na+ Ión sodio dL Decilitro. NaCN Cianuro de sodio D. O. Densidad óptica NAD Nicotinamida-adenina
dinucleótido EDTA Ácido etilen diamino
tetraacético NADH Nicotinamida-adenina
dinucleótido reducido E.E. Error estándar NaOH Hidróxido de sodio EIM Espacio intermembranal
mitocondrial NED N-1-nafiletilendiamina
FAD Dinucleótido flavina adenina NH4Cl Cloruro de amonio FADH Dinucliótido flavina adenina
reducido Nm
Nmol Nanómetros Nanomol
fC Femtocoulombio NO Óxido nítrico g Gramo NO2 Nitritos GCS Ganglio cervical superior NOS Sintasa de óxido nítrico GLUT Proteína transportadora de
glucosa NOSe Sintasa de óxido nítrico
endotelial GMPc Guanosin monofasfato
cíclico. NOSi Sintasa de óxido nítrico
inducible GP Ganglio petroso NOSmt Sintasa de óxido nítrico
mitocondrial H+ Ión hidrógeno NOSn Sintasa de óxido nítrico
neuronal i.p. Intraperitoneal NPS Nitroprusiato de sodio NPV Núcleo paraventricular SCA Seno carotídeo aislado

7
NSC Nervio del seno carotídeo SH Sulfhidrilo NSO Núcleo supraóptico SNC Sistema nervioso central NTS Núcleo del tracto solitario STZ Estreptozotocina O2 Oxígeno SY Seno yugular OONO- Peroxinitrito t tiempo p Probabilidad V3 Tercer ventrículo pCO2 Presión parcial de bióxido
de carbono V4 Cuarto ventrículo
pO2 Presión parcial de oxígeno µL Microlitro RCC Receptores del cuerpo
carotídeo µmol Micromol
rpm Revoluciones por minuto µm Micrómetros RSCC Receptores del seno cuerpo
carotídeo ∆ψ Potencial de membrana
mitocondrial sec Segundos ∆pH Gradiente electroquímico SC Seno carotídeo

8
RESUMEN
Se ha postulado la existencia de glucorreceptores en diversos sitios del
organismo, particularmente en el SNC, y en los cuerpos carotídeos (CC). El óxido
nítrico (NO) modula la descarga quimiosensora en los receptores seno-cuerpo
carotídeos (RSCC) inhibiendo en forma reversible el consumo de O2 y bloqueando la
citocromo-c oxidasa (cit-c) mitocondrial. Se sabe que los desórdenes metabólicos
observados en la diabetes reducen la utilización de glucosa, afectando la actividad
neuronal y la respuesta a la hipoxia. En esta tesis, se analiza el efecto concomitante
del NO y el cianuro de sodio (NaCN) en el seno carotídeo vascularmente aislado in
vivo (SCA), sobre la respuesta hiperglucemiante con retención de glucosa por el
cerebro en ratas normales y diabéticas. Los experimentos se realizaron en ratas
Sprague-Dawley macho de 250 a 300 g de peso, anestesiadas con pentobarbital
sódico, según el siguiente protocolo: a) control 1, infusión simultánea de solución
salina (sol.sal.) en el SCA; b) control 2, infusión de NaCN y sol.sal. en el SCA; c)
experimental 1: infusión de NaCN y L-NAME (N-nitro-L-arginina metil éster), un
inhibidor de la NOS (sintasa de óxido nítrico), en el SCA; d) experimental 2: infusión
de NaCN y NPS (nitroprusiato de sodio), donador de NO, en el SCA. Se valoraron
los niveles de glucosa y de nitritos en la sangre venosa cerebral. En el experimental
2 el NO reforzó el estímulo anóxico, e incrementó la glucosa arterial con retención de
glucosa cerebral (reflejo hiperglucémico), así como los niveles de nitritos en el SNC.
En las ratas diabéticas, el NO falló para incrementar el reflejo hiperglucémico descrito
pero sí aumentó los niveles de nitritos. El L-NAME y el NPS mostraron efectos
opuestos sobre las variables estudiadas. El primero redujo la retención de glucosa
cerebral y los niveles de nitritos, mientras que el segundo los incrementó, tanto en
ratas normales como diabéticas. Las ratas diabéticas siempre mostraron niveles
inferiores de nitritos en comparación con las ratas normales, antes y después de la
estimulación RSCC. Se concluye que el NO, igual que el NaCN, inhibe la cadena
respiratoria mitocondrial y aumenta el reflejo hiperglucémico únicamente en las ratas
normales.

9
ABSTRACT
It has been postulated the existence of glucoreceptors in various sites of the
organism, particularly in the CNS and in the carotid bodies (CBs). Chemoreceptors of
the CBs (RSCC) have been found to increase afferent nerve activity to regulate
respiratory performance during hypoxia or hypoglycemia, causing glucose retention
by the brain. In the CBs, nitric oxide (NO) modulates chemosensory responses,
inhibiting the mitochondrial cytochrome c oxidase and oxygen consumption. The
metabolic disorders observed in diabetes mellitus are known to reduce glucose
utilization, affecting neuronal activity and the hypoxic sensitivity. The aim of this
thesis was to study the concomitant effects of NO and cyanide (NaCN), infused into
the carotid sinus temporarily isolated from the systemic circulation in vivo (SCA), on:
glucose retention by the brain and on nitrite levels in brain blood in normal and
diabetic rats. The experiments were carried out in male Sprague-Dawley rats
weighing 250-300 g, anesthetized with sodium pentobarbital, according to the
following protocol: a) control 1, saline was injected into the SCA; b) control 2, saline.
and NaCN injections were made into the SCA; c) experimental 1, NaCN and N-nitro-
L-arginine methyl éster (L-NAME- NO-synthase inhibitor) injections were made into
the SCA; d) experimental 2, NaCN and sodium nitroprusside (NPS- NO donor)
injections into the SCA. Glucose retention by the brain and nitrites in CNS blood
were assayed. In control 2 experiments, NO improves RSCC effect, increasing
arterial glucose levels and glucose retention by the brain (hyperglycemic reflex), as
well as nitrite levels in CNS. Opposite effects were obtained on the variables studied
when L-NAME or NPS were applied before NaCN infusion. In the first case, a
significant reduction in glucose retention by the brain and in nitrite levels in cerebral
blood were observed; while in the second, the values significantly increased, both, in
normal and diabetic rats. Before RSCC stimulation was performed or afterwards,
diabetic rats always showed lower nitrite levels in relation to normal rats. It is
concluded that NO, as NaCN, inhibits respiratory chain in the mitochondria, to
increase the hyperglycemic reflex only in normal rats.

10
INTRODUCCIÓN
La glucosa es un substrato esencial para el metabolismo celular, y en forma
especial para las células del sistema nervioso central (SNC), cuya actividad depende
de la disponibilidad de este carbohidrato. En los mamíferos, la neurotransmisión y en
general todas las funciones cerebrales, demandan un suministro continuo de energía
de substratos metabólicos (glucosa) que mantienen, entre otros, el potencial de
membrana. En efecto, los cambios en la utilización de la glucosa cerebral
constituyen un índice de la actividad neuronal. La barrera hemato-encefálica
condiciona el paso de substratos energéticos al cerebro, y en particular de la glucosa
(Lund-Andersen, 1979), por lo que es importante conocer los mecanismos y factores
precisos que regulan dicho transporte (Fray, Forsyth, Boutelle y Fillenz, 1996);
aunque sabemos que en el SNC las proteínas transportadoras de glucosa (GLUT 1 y
GLUT 3 principalmente) juegan un papel importante en este proceso.
La homeostasis de la glucosa se regula dentro de márgenes estrechos, tanto
por mecanismos nerviosos (Álvarez-Buylla y Carrasco-Zanini, 1960; Niijima, 1982;
Frizzell, Jones, Davis, Biggers, Myers, Connolly, Neal, Jaspan, Cherrington, 1993;
Burcelín, Dolci y Thorens, 2000; García, Millán, Balmaceda-Aguilera, Castro, Pastor,
Montecinos, Reinicke, Zuniga, Vera, Onate, Nualart, 2003; Uyama, Geerts y
Reynaert, 2004), como hormonales (Frohman, 1983; Heimberg, De Vos, Moens,
Quartier, Bouwers, Pipeleers, Van Schaftingen, Madsen y Schuit, 1996; Álvarez-
Buylla, Álvarez-Buylla, Mendoza, Montero y Álvarez-Buylla, 1997). La detección de
estados de hiper o hipoglucemia constituye el primer paso de dicha regulación, y
estas alteraciones en el nivel glucémico se acompañan, entonces, por reacciones
compensadoras (contrarregulación) que tienen por objeto regresar los niveles de
glucosa en la sangre a las concentraciones fisiológicas. Los desórdenes metabólicos
que alteran el suministro o la utilización de la glucosa, pueden afectar la actividad
neuronal, como ocurre en la diabetes mellitus tipo 1 (insulino-dependiente). Es de
esperar que existan sensores a la glucosa en distintas regiones del organismo

11
(Hevener, Bergman y Donovan, 2000) y particularmente en el SNC (Levin, Dun-
Meynell y Routh 1999; Penicaud, Leloup, Lorsignol, Alquier y Guillaod, 2002). El
papel fisiológico que juegan las distintas regiones glucosensibles no está bien
definido, y ninguna de ellas por sí sola, puede explicar las respuestas de
contrarregulación a la hiper o hipoglucemia (Koyama, Coker, Stone, Lacy, Jabbour,
Williams y Wasserman, 2000). Álvarez-Buylla y colaboradores demuestran la
participación del SNC en la homeostasis de la glucosa (Álvarez-Buylla, 1960;
Álvarez-Buylla, Segura y de Álvarez-Buylla, 1961a; 1961b; Goldraij y Álvarez-Buylla,
1971; Álvarez-Buylla, 1973; Álvarez-Buylla y de Álvarez-Buylla, 1975; Álvarez-Buylla
y Bencosme, 1981; Álvarez-Buylla, Rojas, de Álvarez-Buylla y Faria, 1986; Guarner y
Álvarez-Buylla, 1991; Álvarez-Buylla, Huberman, Montero, Lemus, Valles, de
Álvarez-Buylla, 2003). Aunque el hígado, la vena porta y el páncreas ejercen un
control periférico para mantener estables los niveles de glucosa en la sangre, los
cuerpos carotídeos (CC) que ocupan una posición estratégica en el inicio de la
circulación cerebral, así como sus características fisiológicas únicas, como son: una
rica vascularización (flujo sanguíneo de 2000 mL/min/100 g de tejido) y una tasa
metabólica muy elevada, llevaron al Dr. Ramón Álvarez-Buylla a postular su
participación en esta función vital para el organismo a nivel central. Las neuronas de
los CC son particularmente vulnerables a la falta simultánea de oxígeno y glucosa,
por lo que tendrían una función de especial importancia en la homeostasis cerebral
(Álvarez-Buylla y de Álvarez-Buylla, 1988, Álvarez-Buylla y Roces de Álvarez-Buylla,
1994). Durante la hipoxia crónica se presenta una reacción homeostática de
adaptación que tiende a compensar la hipoxia con hiperventilación (Álvarez-Buylla,
1951; Eyzaguirre y Zapata, 1984) e hiperglucemia (Álvarez-Buylla y Álvarez-Buylla,
1988, 1994). Este proceso homeostático compromete tanto a componentes
periféricos como centrales.
Experimentos posteriores in vitro, comprueban que los CC detectan cambios en
la glucemia del medio de cultivo (Pardal y López-Barneo, 2002). En el mismo
sentido, Koyama y col. en el 2000 plantean que los CC juegan un papel importante
en la respuesta contrarregulatoria después de una hipoglucemia insulínica ligera en

12
perros. Los receptores del seno-cuerpo carotídeo (RSCC) generan señales
quimiorreceptoras (Álvarez-Buylla, 1951, 1952; Eyzaguirre y Zapata, 1984) que
viajan por el nervio del seno carotídeo (NSC) hasta el ganglio cervical superior, rama
del nervio glosofaríngeo (IX par), para hacer su primer relevo en el núcleo del tracto
solitario (NTS) y mandar las proyecciones a centros superiores como el hipotálamo
(Ricardo y Koh, 1978; Housley, Martin-Body, Dawson y Sinclair, 1987; Finley y Katz,
1992). Se desconocen aún los mecanismos de la quimiotransducción en el CC, así
como los blancos precisos en las vías aferentes y efectoras de la regulación
glucémica.
El óxido nítrico (NO), que se sintetiza a partir de la L-arginina por la sintasa de
NO (NOS), es un mensajero biológico en el SNC y otros tejidos, que participa en
funciones fisiológicas con implicaciones homeostáticas (Wu y Morris, 1998). El NO
interviene en el transporte de glucosa en el músculo esquelético y durante el ejercicio
crónico se estimula la expresión de la NOS (Roberts, Barnard, Scheck y Balon,
1997). En experimentos in vitro el NO modula, también, la descarga quimiosensora
en el CC (Di Giulio, Grilli, De Lutiis, Di Natale, Sabatino y Felaco, 1998; Prabhakar,
Fields, Baker y Fletcher, 2001). Los inhibidores de la NOS incrementan la actividad
en el nervio del seno carotídeo en forma dosis-dependiente (Buerk y Lahiri, 2000).
Es decir, el NO en el CC actuaría mimetizando la respuesta al oxígeno e inhibiendo
la actividad quimiosensora. Sin embargo, algunos autores encuentran un papel
excitatorio del NO cuando el CC está en estado de normoxia, aumentando la
descarga quimiosensitiva; efecto mediado probablemente por una alteración en el
transporte de electrones y la fosforilación oxidativa en la cadena respiratoria (CR)
mitocondrial (Iturriaga, 2001). Se sabe que la mitocondria contiene una isoforma de
la NOS (probablemente, variante de la NOSn) denominada NOSm, capaz de
estimular la producción NO, en cantidad suficiente para regular su propia respiración
(Ghafourifar y Richter, 1997; Giulivi, Poderoso, Boveris, 1998; Iturriaga 2001).
La diabetes representa una serie de alteraciones de los mecanismos
homeostáticos de los niveles de glucosa en la sangre con alteración en la respuesta

13
ventilatoria a la hipoxia e hipercapnia. La insulina que es la hormona más importante
en el transporte de la glucosa a nivel periférico, participa también en la vasodilatación
y en la liberación del NO (Felaco, Grilli, De Lutiis, Patruno, Libertini, Taccardi, Di
Napoli, Di Giulio, Barbacane y Conti, 2001); es decir, la diabetes tiene semejanzas
con los estados de hipoxia crónica, pero existen controversias sobre la influencia de
la diabetes (hiperglucemia) en los niveles de NO en la sangre sistémica y en el SNC
(Kino, Yamato, Aomine, 2004). Con estos antecedentes se pensó que las ratas
diabéticas podrían representar un modelo que permitiera ahondar en el estudio de
las vías quimiorreceptoras del CC y la participación del NO en la homeostasis de la
glucosa.
En esta tesis de doctorado, se analiza el efecto concomitante del NO y el
cianuro de sodio (NaCN) en el seno carotídeo vascularmente aislado in vivo, en la
respuesta hiperglucemiante con retención de glucosa por el cerebro en ratas
normales y diabéticas.

14
ANTECEDENTES
Homeostasis de la glucosa y los quimiorreceptores
La homeostasis de la glucosa en los mamíferos; es decir, su regulación en el
medio interno, es un proceso controlado por múltiples factores. El organismo de los
mamíferos ha desarrollado mecanismos que mantienen las concentraciones de
glucosa dentro de márgenes estrechos, con sistemas sofisticados capaces de iniciar
reacciones compensadoras (contrarregulación) ante las desviaciones fisiológicas
(Koyama y col., 2000; La Fleur, 2003). Dentro de estas fronteras bioquímicas, está
presente un ritmo diario de las concentraciones de glucosa en el plasma, relacionado
con un reloj biológico localizado en estructuras superiores como el núcleo
supraquiasmático (La Fleur, 2003; Sankaran y Subramanian, 2006). El cerebro es un
órgano especialmente vulnerable y depende de un suministro eficiente de glucosa y
otros substratos energéticos desde la sangre (Almeida, Cidad, Delgado-Esteban,
Fernández, García-Nogales y Bolaños, 2005); la neurotransmisión, y el resto de las
funciones encefálicas, requieren un suministro constante de substratos metabólicos
capaces de mantener activo el potencial de membrana en los axones (Hawkins,
1989), por lo que, los cambios en la utilización de glucosa cerebral constituyen un
índice de la actividad neuronal (Sokoloff, 1977). Durante el desarrollo postnatal
temprano, el cerebro requiere, también, grandes cantidades de substratos
energéticos para apoyar el crecimiento de la neuroglia y otros tejidos; en este periodo
crítico se consume más de la mitad de la totalidad energética disponible para todo el
organismo tanto en el humano como en la rata (Gibbons, 1998). Y aquí surge la
pregunta de cómo es posible que el cerebro, adulto o en formación, compita con
tanto éxito por las fuentes energéticas. Este trabajo va dirigido a contestar, en parte,
esta incógnita.
Diversas estructuras, especializadas en detectar cambios en los niveles de
glucosa, participan en el control de la homeostasis energética y en las funciones

15
neuroendocrinas. Se han encontrado sensores a la glucosa en el páncreas
(Heimberg y col., 1996; Hatakeyama, Kishimoto, Nemoto, Kasai y Takahashi, 2006),
en el hígado (Adachi, Kobashi y Funahashi, 1995), en el SNC (Levin, 2002; Pocai,
Obici. Schwartz y Rosetti, 2005), en la vena porta (Donovan, Halter y Bergman, 1991;
Hevener y col., 2000) y en los CC (Álvarez-Buylla y de Álvarez-Buylla, 1988; Koyama
y col., 2000; Pardal y López Barneo, 2002). Las células glucosa-excitables se
despolarizan para aumentar su frecuencia de descarga cuando suben los niveles de
glucosa, y por el contrario, se hiperpolarizan disminuyendo su frecuencia de
descarga cuando disminuyen los niveles de glucosa; concurrentemente, la
concentración de K+ baja durante la hiperglucemia y sube durante la hipoglucemia.
Estos receptores se regulan por medio de una combinación de glucocinasa y
apertura o cierre de los canales de K+ sensibles a trifosfato de adenosina (ATP)
(KATP) derivado del metabolismo de la propia glucosa, aunque en los CC no se han
encontrado canales KATP. Cuando los niveles de glucosa bajan en forma crítica, este
carbohidrato adquiere la primacía para estimular a los detectores de glucosa, con
objeto de activar los mecanismos contrarreguladores que restauran el suministro vital
de este substrato (Levin, 2002). El glucógeno, cuyas concentraciones en el cerebro
son pequeñas (2-3 µmol/g en la rata), se encuentra principalmente en los astrocitos,
en contacto con los capilares sanguíneos (Kacem, Lacombe, Seylaz, Bonvento,
1998) y los espacios sinápticos (Grosche, Matyash, Moller, Verkhratsky,
Reichenbach, Kettenmann, 1999) por lo que son las primeras células del SNC en
captar la glucosa. Este hecho aunado a la característica de que el glucógeno no está
supeditado al ATP para iniciar su metabolismo (Clarke y Sokoloff, 1998), convierte al
glucógeno astrocitario en la reserva energética cerebral más importante para el
metabolismo del SNC. El conocimiento de los factores que regulan la utilización de
la glucosa en los astrocitos es relevante para entender el metabolismo energético
neuronal. Como ocurre con la glucosa, los niveles de glucógeno en el cerebro
cambian ante las variaciones de las concentraciones de glucosa plasmática, así
como durante las maniobras experimentales (Goldberg y O’Toole, 1969; Dienel y
Cruz, 2006). El papel del glucógeno no sólo está supeditado a ser una reserva de

16
carbohidratos durante la hipoglucemia, sino también bajo condiciones de equilibrio,
por lo que su degradación y síntesis son continuas.
A diferencia de otros tejidos, que tienen la capacidad de utilizar nutrientes
energéticos diversos para su metabolismo celular, el cerebro; es decir, las neuronas
en su estado normal, dependen casi exclusivamente de la glucosa como substrato
oxidativo, por lo que las únicas diferencias arterio-venosas positivas encontradas en
el cerebro humano, son para la glucosa y el oxígeno. La glucosa, en su metabolismo
oxidativo, utiliza casi la totalidad del consumo del oxígeno cerebral para proporcionar
una energía equivalente a 0.25 kcal/min (Sokoloff, 1960). La glucopenia o hipoxemia
cerebral evocan, en pocos segundos, una disminución de la actividad
electroencefalográfica con pérdida de la conciencia. La hipoxia o hipoglucemia
focales también producen cambios neuronales severos, con alteración de la
homeostasis iónica y la liberación anormal de neurotransmisores (Prabhakar y
Jacono, 2005; Levin, 2002). El único substrato capaz de substituir a la glucosa en
casos de una hipoglucemia severa, es la manosa, pero su efecto para restaurar las
funciones cerebrales tiene lugar a través de mecanismos que elevan los niveles de
glucosa en la sangre (Sloviter y Kamimoto, 1970). En los procesos de adaptación a
la hipoxia, generalmente aumenta la utilización de glucosa por vías que no involucran
el metabolismo aeróbico mitocondrial. Este fenómeno depende de la sobre-
expresión de los transportadores de la glucosa y de las enzimas glucolíticas (Beitner-
Johnson, Leibold y Millhorn, 1998). Aunque la insulina es un factor primordial para
aumentar la utilización de los sustratos energéticos en los tejidos periféricos, su
participación en la regulación del metabolismo cerebral es dudosa (Baskin, Figlewicz,
Woods, Porte y Dorsa, 1987; Hasselbach, Knudsen, Videbaek, Pinborg, Schmidt,
Holm y Paulson, 1999; Cheng, Reinhardt, Lee, Joncas, Patel y Bondy, 2000); se llega
a esta conclusión por el hecho de que tanto la cantidad de insulina sintetizada en el
cerebro, como la insulina circulante que cruza la barrera hematoencefálica, son muy
pequeñas (Coker, Studelska, Harmon, Burke y O’Malley, 1990; Reinhardt y Bondy,
1994). Aunque la barrera hematoencefálica condiciona el paso de los substratos
energéticos al cerebro, en particular de la glucosa (Lund-Andersen, 1979),

17
desconocemos en forma detallada los mecanismos y factores que regulan dicho
transporte (Fray y col., 1996). El paso a través de las membranas biológicas se lleva
a cabo por proteínas de membrana específicas, que en los mamíferos son
principalmente de dos clases, cotransportadoras Na+/glucosa y facilitadoras del
transporte de glucosa (GLUT) (Mueckler, 1994; Gould y Holman, 1993; Ibberson,
Uldry y Thorens, 1999). La glucosa atraviesa la barrera hematoencefálica por las
proteínas GLUT 1 y GLUT 3, aunque en los últimos años se han descubierto algunas
otras que también participan en el transporte de este carbohidrato (Ngarmukos,
Bauer y Kumagai, 2001; Komori, Morikawa, Tamura, Doi, Nanjo y Senba, 2005). Las
GLUT se encuentran en mayor proporción en el endotelio de los capilares cerebrales
y son capaces de transportar dos a tres veces más glucosa que la que normalmente
metaboliza el cerebro; actúan en forma estereoespecífica y son insulina-
independientes (Kalaria, Gravina, Schmidley, Perry y Harik, 1988). Los trastornos
metabólicos que alteran la expresión de estas proteínas, y por lo tanto, el suministro
de glucosa o su utilización por el SNC afectan significativamente la actividad
neuronal, como ocurre en el retardo mental, las convulsiones y la diabetes tipo 1 o
insulino-dependiente.
Canonn, Newton, Bright, Menkin y Moore (1929) encontraron la primera
evidencia de la participación del sistema nervioso simpático en la regulación de la
glucemia; demostraron que la secreción de insulina por hipoglucemia sistémica
produce un aumento de la actividad simpática, con la presencia de una glucopenia
local en el sistema nervioso autónomo. Estas investigaciones tienen el antecedente
de los trabajos de Claudio Bernard, que demuestran alteraciones en el metabolismo
de la glucosa después de producir lesiones en distintos puntos del SNC (Bernard,
1857). En años posteriores, utilizando microinyecciones de glucosa en el cerebro, se
propuso la existencia de “glucorreceptores” en el SNC, principalmente en el
hipotálamo ventromedial y lateral (Marshall y Mayer, 1956; Shimazu, 1981; Oomura,
1984; Levin, 2002). Estos últimos experimentos llevaron al concepto de que el
cerebro, por sí mismo, es un “detector” de la glucemia ambiental, y es capaz de
poner en marcha los mecanismos glucorreguladores necesarios ante variaciones no

18
fisiológicas. Muchos de los trabajos que involucran al SNC en la detección
glucémica se basan en experimentos que alteran el comportamiento normal, como es
el caso de las lesiones cerebrales, la estimulación eléctrica o la administración
directa en el SNC de análogos de la glucosa o de la glucosa misma (Benzo, 1982;
Cane, Artal y Bergman, 1986). En realidad estos procedimientos no proporcionan
una idea clara del papel que juega el cerebro para detectar de manera cuantitativa
los cambios en la glucemia y, lo que es más importante, para cuantificar los procesos
glucorregulatorios ante una hipoglucemia in vivo.
Lo descrito hasta ahora, indica que una disminución y/o un incremento de los
niveles de glucosa en el plasma inicia una respuesta neuroendocrina compleja que
corrige los cambios y preserva la función cerebral (Auer, 1986; Martin, Lloyd y
Cowan, 1994), pero desconocemos como y donde se detectan estos cambios en los
niveles de glucosa (Cane et al, 1986; Álvarez-Buylla y de Álvarez-Buylla, 1988;
Koyama y col., 2000). Álvarez-Buylla y col. proponen un nuevo papel para las
células del CC (células tipo I o glómicas) como “glucosa-detectoras”, y consideran
que los quimio-barorreceptores carotídeos intervienen en la integración de la
información de las dos variables más importantes para el metabolismo cerebral,
glucosa y oxígeno en experimentos in vivo (Álvarez-Buylla y de Álvarez-Buylla, 1988,
Álvarez-Buylla y Roces de Álvarez-Buylla, 1994; Álvarez-Buylla y col., 1997).
Los quimiorreceptores carotídeos localizados en los CC, son pequeños órganos
situados de forma bilateral en la bifurcación de ambas carótidas primitivas, y ayudan
a mantener la composición química de la sangre controlando la ventilación (Nurse,
2005). Su situación estratégica en la entrada del árbol arterial central los convierte
en estructuras ideales para llevar a cabo la retroalimentación mencionada, su rica
vascularización e inervación les permite detectar cambios mínimos en las variables
metabólicas (De Castro, 1928). Los receptores carotídeos, cuyos cuerpos
neuronales se encuentran en el ganglio petroso (McDonnald y Mitchell, 1981)
mandan su información sensora a través del nervio del seno carotídeo (NSC), rama
del nervio glosofaríngeo (González, Almara, Obeso, Rigual, 1994). Durante los

19
últimos años se han realizado intensas investigaciones para estudiar los mecanismos
de transducción por medio de los cuales los CC son capaces de percibir los cambios
en la pO2, pCO2 y pH; así como los mecanismos que se encargan de traducir el
potencial de receptor en un incremento de las descargas aferentes. Dichos estudios
llegan a la conclusión que las células quimiorreceptoras propiamente dichas, para
todos los estímulos, son las células tipo I o células glómicas (López-Barneo, Ortega-
Saenz, Piruat, García-Fernández, 2006; López-Barneo, Del Toro, Levitsky, Chiara,
Ortega-Sáenz, 2004; Nurse, 2005). Las células glómicas se encuentran formando
cúmulos, donde se realizan las sinapsis químicas y eléctricas en forma recíproca.
Estas células están en estrecha relación con las células tipo II, sustentaculares (glia-
semejantes) (McDonald y Mitchell, 1975) inervadas por fibras aferentes que se
activan ante los estados de hipoxia o hipercapnia, liberando dopamina y otros
transmisores como acetilcolina (ACh) y ATP (Acker y Starlinger, 1984; Fitzgerald,
Shirahata, Chang y Balbir, 2000; López Barneo, Pardal y Ortega Sáenz, 2001; Nurse,
2005) para estimular las fibras sensitivas aferentes (Ortega-Sáenz, Pardal,
Castellano, López Barneo, 2000) (Figura 1). Este tipo de células neurosecretoras
también se encuentran en el pulmón y en las células cromafines de las glándulas
adrenales, donde detectan cambios en la pO2, liberando catecolaminas ante la caída
de la pO2 (Wyatt y Peers, 1995; Zinker, Namdaran, Wilson, Lacy y Wasserman,
1994). El consumo de O2 del CC es de 1.3 mL/100 g -1/min (González y col., 1994),
esto daría una cantidad de ATP de 3.5 mmol kg -1 min -1, y la utilización de glucosa
del CC in vitro es aproximadamente de 120 µmol kg -1 min -1. Aunque sólo el 20 %
del CC está formado por células glómicas, éstas utilizan el 90 % de la glucosa. Por
lo tanto la producción de ATP por el metabolismo oxidativo en las células tipo 1 del
CC estaría en el rango de de 2.6 mmol kg -1 min-1. Los CC están formados por un
tejido neuroepitelial, con características similares a las glándulas endocrinas, que
posee mecanismos para detectar cambios en los niveles de oxígeno (Lawson, 1980;
Paulding, Schnell, Bauer, Striet, Nash, Kuznetsova, Czyzyk-Krzeska, 2002), pero
desconocemos los mecanismos precisos de la quimiotransducción (Bianchi, Cacchio,
Artese, Ferrero, Rapi, Grilli, Felaco, Di Giulio, 2003). Se sabe que las células tipo I
dopaminérgicas en los CC son las transductoras del estímulo hipóxico que en sus

20
vias de señalización comprenden el cierre de canales de K+ de distintos tipos,
facilitando la despolarización de la membrana, con entrada de Ca2+ extracelular y
liberación del neurotransmisor (Prabhakar, 2000). Durante la hipoxia o durante la
hipercapnia, se observa una co-liberación de ACh y ATP en cultivos de largo plazo
de células de CC tanto en las células tipo I como en las tipo II (Nurse y Fearon,
2002). Estos experimentos demuestran la existencia de mecanismos autocrinos y
paracrinos interactuando en el CC para liberar los distintos transmisores (Fearon y
col., 2001). La utilización de este tipo de cultivos ayuda a resolver controversias
sobre la biología del desarrollo, la fisiología y la bioquímica de las células
quimiorreceptores, así como su respuesta a la hipoxia
Figura 1. Microfotografía de las células glómicas en una rebanada delgada del CC de rata. (A) Pocas horas después del corte; baja resolución. (B) Agregados de células glómicas (glomérulos). Las células individuales pueden verse claramente (flecha). (C) Rebanada del CC teñida con anticuerpos anti-tirosina hidroxilasa. Note la apariencia típica de las células del glomus con núcleo grande y organización en glomérulos. Modificado de Pardal, Ludewig, García-Hirschfeld y López-Barneo y Pardal, 2000.
Citando al Dr. Álvarez-Buylla “las zonas reflexogénicas, cardioaórtica y
senocarotídea, ocupan posiciones estratégicas, en la iniciación de las circulaciones

21
sistémica y cerebral. Los baro y quimiorreceptores del seno-cuerpo carotídeo
proporcionan al cerebro la información sancional sobre diversas variables
fisiológicas, como son la presión arterial, los niveles de O2, de CO2, de pH y de
osmolaridad (Álvarez-Buylla, 1951, 1952; Eyzaguirre y Zapata, 1984) y también, de
los niveles de glucosa “ (Álvarez-Buylla y de Álvarez-Buylla, 1988; Álvarez-Buylla y
Roces de Álvarez-Buylla, 1994; Koyama y col., 2000; Pardal y López Barneo, 2002).
En efecto, en preparaciones de CC in vivo aislados de las circulaciones sistémica y
cerebral, en ratas anestesiadas con pentobarbital, la perfusión del seno carotídeo
con sangre cuyas concentraciones de glucosa son bajas (2.7 mM), aumenta la
glucosa en la sangre arterial (reflejo hiperglucémico), así como la captación de
glucosa cerebral; por el contrario, la perfusión del seno carotídeo con sangre rica en
glucosa (16.7 mM), no aumenta de forma significativa la glucosa arterial, pero si
disminuye significativamente la captación de glucosa cerebral y como consecuencia
aumenta la concentración de glucosa venosa (Álvarez-Buylla y col., 1988) (Fig. 2).
La estimulación baro-quimiorreceptora por oclusión de la carótida primitiva
(hipoxia) también produce un reflejo hiperglucémico, que no desaparece después de
la denervación de las fibras barorreceptoras ipsi- y contralaterales de los senos
carotídeos. En estos experimentos aumenta la concentración de glucosa en la vena
suprahepática indicando que el hígado participa en el mecanismo efector del reflejo
hiperglucémico (Álvarez-Buylla y col., 1997). Las vías efectoras de los reflejos
descritos no están bien aclaradas pero se sabe que participan la hipófisis, las
adrenales (Álvarez-Buylla y col., 1997) y la vasopresina (Montero, Yarkov, Álvarez-
Buylla, 2000; Yarkov, Montero, Lemus, Roces de Álvarez-Buylla y Álvarez-Buylla,
2001; Montero, Yarkov, Lemus, Mendoza, Valles, de Álvarez-Buylla y Álvarez-Buylla,
2003).
El CC madura después del nacimiento adquiriendo características funcionales y
estructurales en respuesta a los cambios ambientales; es decir, a los cambios en la

22
Figura 2. Cambios en la concentración de la glucosa sanguínea y en la retención de glucosa cerebral en ratas anestesiadas con perfusión de sangre rica o pobre en glucosa en el seno carotídeo circulatoriamente aislado. La perfusión de sangre con alta glucosa, provoca una disminución en la retención de glucosa cerebral (panel superior). Por el contrario, cuando se perfunde el seno carotídeo con sangre pobre en glucosa, se observa un aumento en las glucemias arterial y venosa, así como en la retención de glucosa cerebral (panel inferior). Tomado de: Álvarez-Buylla y de Álvarez-Buylla, 1988.
pO2, y probablemente, a los niveles de glucosa en la circulación local (Calder, Kumar
y Hanson, 1997; Fitzgerald, Shirahata, Chang, Balbir, 2006). Las poblaciones que
viven en localidades cuya altitud es mayor de 3000 m sobre el nivel del mar,
proporcionan un modelo experimental útil para estudiar los procesos de adaptación y
homeostasis de la glucosa ante este tipo de estrés a la hipoxia crónica (Sarkar,
Banerjee, Selvamurthy, 2003; Wilson, Roy y Lahiri, 2005). En estas condiciones, los
CC adquieren características especiales que se deben a las alteraciones en la
expresión genética de las proteínas encargadas de regular el crecimiento de las
células del glomus, fenómeno que depende de la sobre-regulación de las proteínas
23
GLUT y de las enzimas que participan en la degradación de la glucosa (Bunn y
Poyton, 1996; Behrooz y Ismail-Beigi, 1999; Semenza, 1999).
La información aferente proveniente de los quimiorreceptores del CC viaja por el
NSC y por el nervio glosofaríngeo hasta la médula dorso-medial en el núcleo del
tracto solitario (NTS) como primer relevo, aunque algunas de las fibras terminan en el
área postrema y en el núcleo del trigémino, para dirigirse después al hipotálamo
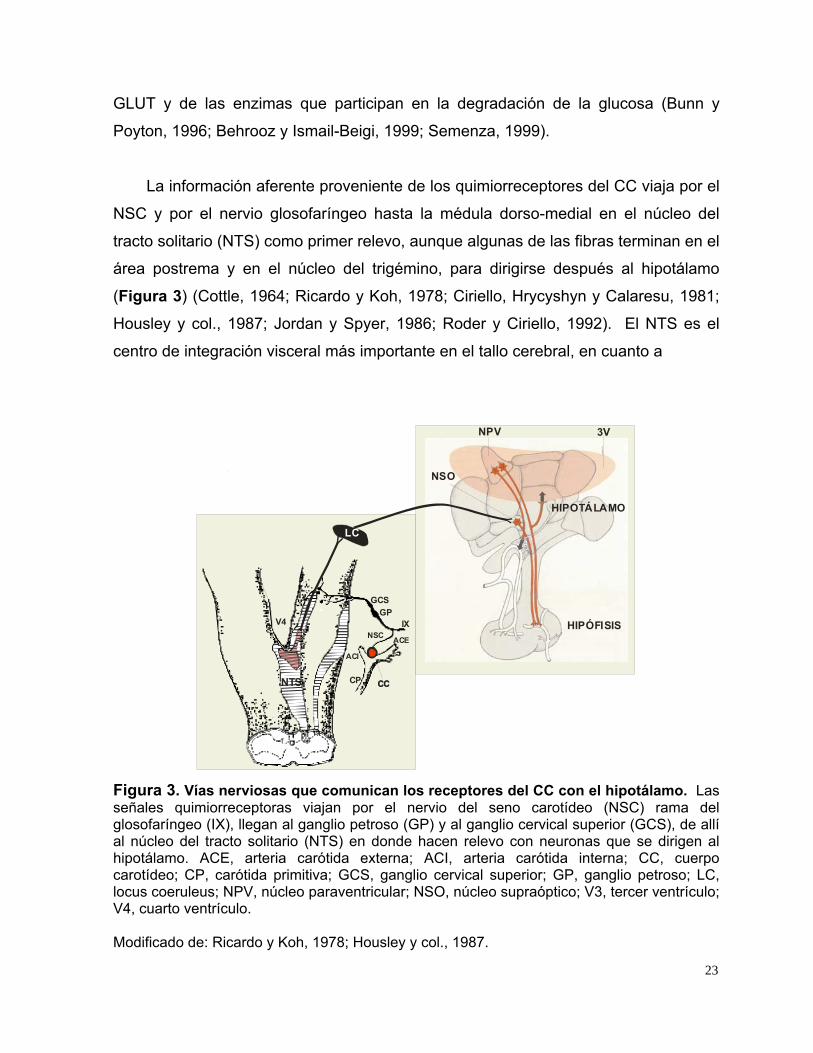
(Figura 3) (Cottle, 1964; Ricardo y Koh, 1978; Ciriello, Hrycyshyn y Calaresu, 1981;
Housley y col., 1987; Jordan y Spyer, 1986; Roder y Ciriello, 1992). El NTS es el
centro de integración visceral más importante en el tallo cerebral, en cuanto a
LCV
NTS
GCSGP
IXNSC
CC
V
HIPÓFISIS
HIPOTÁLAMO
NSO
CP
3VNPV
CC
ACI
ACE
V4
Figura 3. Vías nerviosas que comunican los receptores del CC con el hipotálamo. Las señales quimiorreceptoras viajan por el nervio del seno carotídeo (NSC) rama del glosofaríngeo (IX), llegan al ganglio petroso (GP) y al ganglio cervical superior (GCS), de allí al núcleo del tracto solitario (NTS) en donde hacen relevo con neuronas que se dirigen al hipotálamo. ACE, arteria carótida externa; ACI, arteria carótida interna; CC, cuerpo carotídeo; CP, carótida primitiva; GCS, ganglio cervical superior; GP, ganglio petroso; LC, locus coeruleus; NPV, núcleo paraventricular; NSO, núcleo supraóptico; V3, tercer ventrículo; V4, cuarto ventrículo. Modificado de: Ricardo y Koh, 1978; Housley y col., 1987.
24
funciones cardiovasculares y respiratorias se refiere (Daly, Ward, Wood, 1986;
McRitchie y Törk, 1994); y es importte señalar que el reflejo quimiorreceptor arterial
produce alteraciones en ambos efectores en forma simultánea (Davidson, Goldner y
McCloskey, 1976). Haciendo una analogía con lo que ocurre en el asta dorsal de la
médula espinal, se sugiere que el NTS modula las señales aferentes que le llegan
por mecanismos presinápticos en las propias terminales, o postsinápticos en los
cuerpos neuronales (Jordan y Spyer, 1986; Housley y Sinclair, 1988).
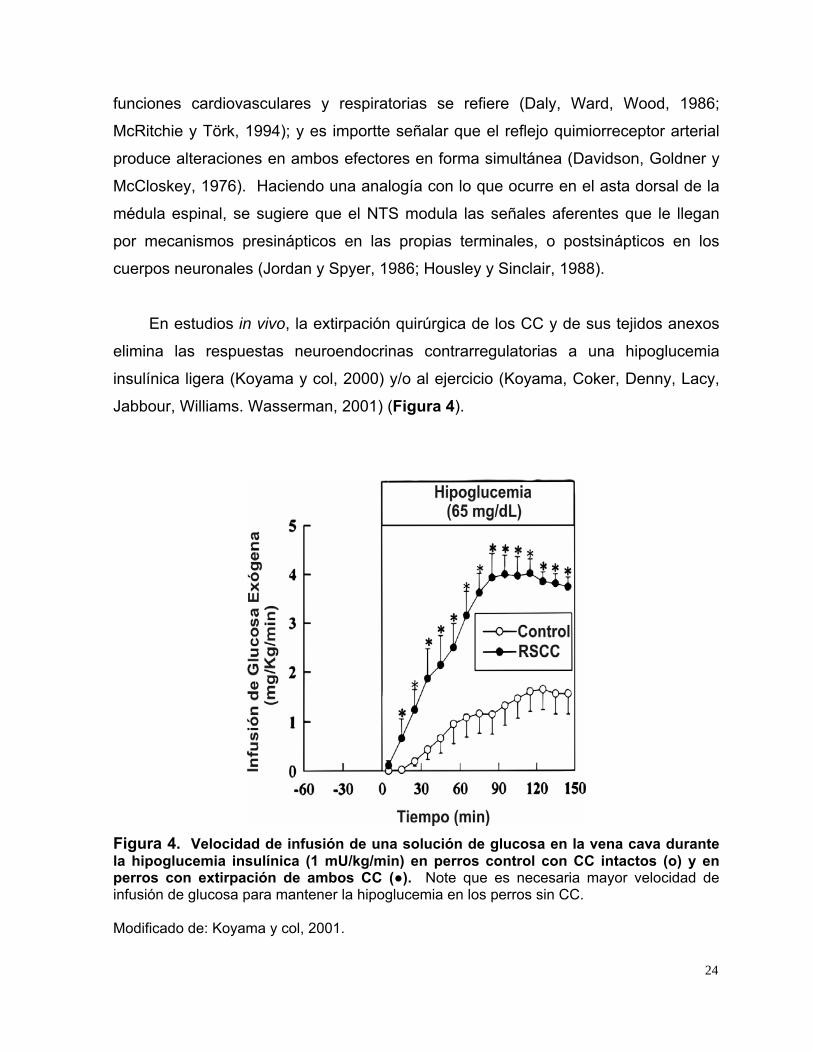
En estudios in vivo, la extirpación quirúrgica de los CC y de sus tejidos anexos
elimina las respuestas neuroendocrinas contrarregulatorias a una hipoglucemia
insulínica ligera (Koyama y col, 2000) y/o al ejercicio (Koyama, Coker, Denny, Lacy,
Jabbour, Williams. Wasserman, 2001) (Figura 4).
Figura 4. Velocidad de infusión de una solución de glucosa en la vena cava durante la hipoglucemia insulínica (1 mU/kg/min) en perros control con CC intactos (o) y en perros con extirpación de ambos CC (●). Note que es necesaria mayor velocidad de infusión de glucosa para mantener la hipoglucemia en los perros sin CC. Modificado de: Koyama y col, 2001.

25
Varios años después de los experimentos de Álvarez-Buylla (1981), Obeso, Almaraz,
González (1986) y Pardal y López-Barneo (2002), trabajando en preparaciones del
CC in vitro, llegan a la misma conclusión. Utilizando rebanadas finas de tejido que
mantienen la estructura del CC, así como su sensibilidad a la hipoxia, demuestran
que la falta de glucosa (glucopenia) en el medio induce una actividad secretora
semejante a la producida por la hipoxia (Pardal y López-Barneo, 2002); así mismo, la
estimulación hipóxica induce la activación simpática con la secreción de
catecolaminas (Figura 5). Las células del glomus carotídeo, al ser estimuladas por
Figura 5. Respuesta secretora de las células glómicas del CC in vitro a diferentes concentraciones de glucosa en condiciones de hipoxia y normoxia. (A) Aumento de la respuesta secretora durante la hipoxia (PO2 25 mmHg) a 0 mM de glucosa, y reducción de la secreción al aumentar la concentración de glucosa (5 mM). (B) Índice de secreción durante las 3 respuestas en A; 1,837, 5,816 y 1,729 fC/min. (C) Modulación de la actividad secretora al aumentar los niveles de glucosa en el baño (5-10 mM) en una célula glómica expuesta a tensión normal de oxígeno (PO2 = 90 mmHg). (D) Curva que expresa la secreción máxima de catecolaminas en respuesta a la concentración de glucosa. Modificado de Pardal y López Barneo, 2002.

26
la hipoxia, liberan catecolaminas y otros transmisores como la sustancia P, la
metencefalina, la ACh, etc. (Ureña, Fernández-Chacón, Benot, Álvarez de Toledo y
López-Barneo, 1994).
Óxido Nítrico (NO) y quimiorreceptores
El NO es un radical libre, gaseoso, liposoluble, membrana-permeante, de vida
media muy corta, reactivo y muy difusible (Fernández-Álvarez, Abudara y Morales,
1999), que participa como un mensajero biológico en diversas funciones fisiológicas,
tales como la regulación de la presión sanguínea, la transmisión sináptica, la
supresión de patógenos (Lowenstein, Dinerman y Snyder, 1994), así como en los
procesos de señalización intra- e intercelular en diversas funciones como en la
homeostasis de la glucosa (Shankar, Zhu, Ladd, Henry, Shen y Baron, 1998;
Bradley, Kingwell y McConell, 1999; Higaki, Hirshman, Fujii y Goodyear, 2001).
Interviene, también, en eventos fisiopatológicos, activando cadenas metabólicas
intracelulares a través de la síntesis de GMPc (Boucher, Moali y Tenu, 1999; Wang y
Robinson, 1997; Di Giulio y col., 1998; Fernández-Álvarez y col., 1999; Prabhakar y
col., 2001). Asimismo, en los últimos años numerosos reportes sugieren que el NO
participa en la modulación de la respiración durante la hipoxia (Prabhakar, 1999). La
vida media del NO en solución buffer a pH fisiológico y en condiciones de normoxia
es de 30 s, con una distancia de difusión radial de 540 µm; en la sangre, estos
valores disminuyen a 1 s y 100 µm respectivamente (Kelm y Schrader, 1988).
El NO fue encontrado originalmente en el endotelio vascular por lo que en un
principio se denominó como “factor de la relajación endotelial”; se sintetiza en
diversos tipos de células, como neuronas, músculo liso y macrófagos, entre otros
(Moncada, Palmer y Higgs, 1991). La biosíntesis del NO se lleva a cabo a partir de
la L-arginina y el NADPH, generando citrulina como coproducto, con la participación
de la sintasa del NO (NOS) y la diaforasa de NADPH como cofactores. Las
isoformas de la NOS más conocidas en los mamíferos son la NOSi (inducible),

27
sintetizada en respuesta a estímulos inmunológicos o inflamatorios, la NOSn
(neuronal), la NOSe (endotelial), y por último la NOSmt (mitocondrial) que parece ser
una modificación de la NOSn (Saavedra-Molina, Ramírez-Emiliano, Clemente-
Guerrero, Pérez-Vázquez, Aguilera-Aguirre, Gonzalez-Hernández, 2003). Aunque
las NOS varían ligeramente en su modo de expresión y regulación, todas las
isoformas catalizan la misma reacción, pero sólo la NOSn y NOSe son
Ca2+/calmodulina dependientes (Lowenstein y col., 1994; Boucher y col., 1999). La
NOSe y la NOSn son sensibles a distintas concentraciones de O2, sugiriendo que las
condiciones fisiopatológicas que cursan con una disminución de O2 en los tejidos,
producen un decremento en la producción de NO (Rengasamy y Johns, 1993).
El CC es altamente excitable por la inhibición de la fosforilación oxidativa, hecho
que convierte a la CR mitocondrial como organelo fundamental en la detección de
oxígeno a través de cambios en el metabolismo energético. Al parecer todos los
estímulos que actúan sobre los quimiorreceptores lo harían del mismo modo, esto es,
inhibiendo los canales de potasio, como los canales semejantes a TASK (canales de
potasio de doble poro rectificadores entrantes tardíos sensibles a acidez, por sus
siglas en inglés) denominados canales KB, localizado en la membrana de las células
tipo 1 del glomus. Estos canales son inhibidos por hipoxia o acidosis, despolarizando
a la célula e iniciando la actividad eléctrica y la entrada del calcio dependiente de
voltaje que estrimula la neurosecreción de las células tipo 1 del glomus carotídeo
para excitar las terminales nerviosas aferentes quimiorreceptoras. Los canales KB
son sensibles a inhibidores de la fosforilación oxidativa mitocondrial, lo que los hace
sensibles a los cambios de pH y oxígeno. Se ha señalado que la integridad de la
mitocondria es necesaria para la detección de oxígeno y pH; el hecho de que estas
células no respondan a la hipoxia cuando se inhibe la función mitocondrial, sugiere
una fuerte vinculación entre el metabolismo energético y la sensibilidad al oxígeno en
las células glómicas tipo 1 (Varas, Wyatt, Buckler, 2007). La CR mitocondrial
transporta los electrones provenientes del metabolismo oxidativo hacia un receptor
final, el O2, y acopla este proceso al bombeo de protones hacia el espacio
intermembranal, proceso que es responsable de la generación del potencial

28
electroquímico transmembranal necesario para la síntesis del ATP y la homeostasis
iónica en la mitocondria. En la CR se encuentran diversos grupos prostéticos con los
que se une el NO producido por la NOSmt para alterar su actividad (figura 6). El
cianuro, igual que el NO, es un inhibidor específico de la cit-c, que disminuye la
Figura 6. Cadena respiratoria (CR) en la mitocondria. Mitocondria formada por las membranas externa (ME) e interna (MI), la matriz y el espacio intermembranal (EIM). Los complejos I, II, III IV y V de la CR, la coencima Q y la citocromo oxidasa c (cit-c) se encuentran en la MI, el complejo V (ATP sintasa) se encuentra en el EIM; su lugar en la CR es de acuerdo a sus potenciales redox (-250 mV hasta +250 mV). Al oxidarse el NADH o el FADH2 ingresan electrones, pasan por los complejos hasta llegar al complejo IV donde se reduce el O2 a H2O. Se expulsan los protones y se establece el potencial trasmembranal (∆ψ, negativo adentro) con un gradiente electroquímico a través de la membrana. Los protones reingresan a la matriz por la ATP sintasa. El ∆ψ es la fuerza motriz de los cationes como el Ca2+. El Ca2+ ionizado en la mitocondria se mantiene bajo, por la formación de depósitos insolubles [Ca3 (PO4] o por intercambio con otros cationes como H+ o Na+. El NO compite con el O2 en el complejo IV y el consumo de O2 de manera reversible. Cuando el NO inhibe el consumo de O2 disminuye el ∆ψ y el ∆pH.
Tomado de Ghafourifar y Cadenas, 2005.

29
velocidad de degradación del NO en una solución. La concentración inhibitoria
media (IC50) del cianuro para el consumo de O2 es de 13.2 ± 1.8 µM. Una
concentración de 100 µM de SNAP (donador de NO) atenúa la IC50 del cianuro de
potasio (KCN). En cambio, el L-NAME aumenta la IC50 del NaCN hasta 59.6 ± 0.9
µM en el consumo de O2 (Leavesley, Prabhakaran, Borowitz y Isom, 2007). Estos
datos indican que el KCN activa la producción de NO mitocondrial y de este modo
potencia el bloqueo de la cit-c. La unión del O2, del NO o del KCN a la cit-c depende
del estado redox de la enzima, es decir, del estado reducido del núcleo de la enzima
(Fe2+/CuB+), del estado oxidado (Fea3
3+/CuB2+) o del estado parcialmente reducido
(Fea33+/CuB
+). El O2 y el NO se unen a la la cit-c en el estado reducido, mientras que
el KCN se une a la cit-c en los tres estadios, reducido, oxidado o parcialmente
reducido (Leavesley y col., 2007). El NO oxida la cit-c en presencia de NADH y
FADH con la formación de nitrosilo en milisegundos (ms), pero en presencia de
NaCN disminuye el tiempo de reducción del NO a nitrosilo. Es decir, el cianuro
compite con el NO por el sitio de unión a la cit-c mitocondrial, provocando un
aumento en la concentración de NO en las células (Staubauer, Giuffrè, Brunori, Sarti,
1998). Brown y Cooper (1994) señalan que el NO inhibe en forma reversible el
consumo de O2 bloqueando la cit-c mitocondrial in vivo. Aunque el NO mitocondrial
interfiere con la respiración, la síntesis de ATP y la regulación del potencial de
membrana mitocondrial (Okada, Takehara, Yabuki, Yoshioka, Yasuda, Inoue,
Utsumi, 1996; Ghafourifar, Bringold, Klein, Richter, 2001), la disminución del ATP no
contribuye a la inhibición persistente de la respiración provocada por el NO
(Clementi, Brown, Feelisch, Moncada, 1988). Por sus acciones sobre la CR, el NO
debe intervenir en los efectos citotóxicos sobre el SNC y otros tejidos (Cleete,
Cooper, Darley-Usmar, Moncada y Schapira, 1994), y puede causar una inhibición
persistente en la CR debido a la formación generalizada de peroxinitritos (OONO-)
(Beckman, Beckman, Chen, Marshall y Freeman, 1990) y/o nitroso-tioles (Clementi y
col., 1988) en el complejo I de la CR.
Por estos antecedentes, el papel fisiológico del NO en la regulación del
metabolismo energético mitocondrial de la célula es cada vez más evidente, pero

30
falta definir cómo se llevan a cabo esta función. Las acciones del NO sobre la CR
parecen depender de muchos factores entre los que tenemos la propia concentración
de NO en función de la regulación de las NOS (Ghafourifar y Richter, 1997;
Saavedra-Molina y col., 2003).
El NO es capaz de reaccionar en forma rápida y reversible con el Fe2+ del grupo
hemo de distintas metaloproteínas (Radi, 1996), mecanismo que activaría a la
guanilatociclasa citosólica (GCS) (Arnold, Aldred y Murad, 1977) e inhibiría a todas
las NOS (autoregulación) (Rogers e Ignarro, 1992). Una vez que el NO activa a la
CGS, se incrementa la concentración de GMPc intracelular a partir de GMP
(Schmidt, Schrammel, Koesling y Mayer 2001). El NO reacciona también con los
grupos hemo de la oxihemoglobina y mioglobina (“secuestradores” del NO) (Gillespie
y Sheng, 1989). Se han caracterizado tres genes diferentes que codifican para las
tres isoformas de NOS (Valdés, Mosqueira, Rey, Del Río e Iturriaga, 2002), y gran
número de trabajos sugieren que la biosíntesis del NO constituye un factor clave en
las respuestas fisiopatológicas del cerebro a los estados de hipoxia-isquemia, es
decir, de hipoglucemia (Bolaños y Almeida, 1999; Beltrán, Mathur, Duchen,
Erusalimsky y Moncada, 2000).
En la neurotransmisión y en el sistema cardiovascular las NOSn y NOSe en los
CC actúan como moduladores (Boucher y col., 1999; Kline y Prabhakar, 2000;
Valdés y col., 2002) (Figura 7). Utilizando técnicas inmunohistoquímicas se detecta
actividad NOS-positiva en las neuronas de la periferia del CC, así como en las
arterias y arteriolas intraglómicas, que desaparece después de la sección del NSC,
indicando su origen sensorial (Prabhakar, Kumar, Chang, Agani y Haxhiu, 1993;
Chugh, Katayama, Mokashi, Debout, Ray y Lahiri, 1994; Hohler, Mayer, Kummer,
1994). La NOS también se localiza en la vecindad del CC, tanto en las neuronas
preganglionares simpáticas como en las postganglionares parasimpáticas (Hohler y
col., 1994). Las hipótesis actuales sobre la función detectora de los
quimiorreceptores del CC en respuesta a la hipoxia, a la hipercapnia o a la acidez de
la sangre arterial, indican que las células dopaminérgicas del glomus son las

31
transductoras del estímulo químico inhibiendo los canales de K+; con la
despolarización consecuente, la entrada de Ca2+ y liberación del neurotransmisor
(González y col., 1994; López-Barneo, 1996; Prabhakar, 2000; Prabhakar y Overholt,
2000). Aunque se ha propuesto la presencia de una proteína, ya sea asociada a la
membrana, al grupo hemo, o al canal de K+, para identificar los detectores al oxígeno
(Nurse y Fearon, 2002) no hay evidencias al respecto. En el CC se han encontrado,
también, otros transmisores, como la ACh, la adenosina y el ATP, que generan
también la descarga quimiosensitiva en las terminales nerviosas de las neuronas
aferentes ante la hipoxia (Fitzgerald y col., 2000).
Figura 7. Síntesis, difusión y esfera de acción del NO en el sistema nervioso. En la parte central se representa la sinapsis glutamatérgica que establece contacto sináptico con un elemento postsináptico que contiene NOS. El neurotransmisor glutamato interactúa con el receptor NMDA para generar una corriente entrante de Ca2+ al elemento postsináptico. El aumento del Ca2+ intracelular promueve la síntesis de NO por activación de la NOS Ca2+/calmodulina dependiente. La línea punteada representa la esfera de acción del NO. El NO que difunde desde la postsinapsis podría actuar en cualquiera de los elementos celulares vecinos siempre que contengan la GC. GC, guanilato ciclasa; NMDA, N-metil-D-aspartato; NO, óxido nítrico; NOS, sintasa de NO. Modificado de: Fernández-Álvarez y col. 1999; Denicola, Souza, Radi y Lissi, 1996.

32
La administración de NO o sus donadores como el precursor L-arginina, reducen
la respuesta quimiosensitiva a la hipoxia en el CC del gato in vitro (Chugh,
Katayama, Mokashi, Bebout, Ray, Lahiri, 1994; Iturriaga, Alcayaga y Rey, 1998;
Iturriaga, Mosqueira y Villanueva, 2000; Valdés y col., 2002). Otro donador del NO,
como es el nitroprusiato de sodio (NPS, reduce en forma reversible las respuestas
quimiosensitivas del CC inducidas con una dosis única de NaCN o de nicotina en el
CC del gato (Alcayaga, Iturriaga, Ramírez, Readi, Quezada y Salinas, 1997). En
cambio, la inhibición de la NOS con la consecuente disminución en la producción de
NO, incrementa la frecuencia de las descargas quimiosensitivas carotídeas en el CC
in situ (Iturriaga y col., 1998) e in vitro (Wang, Stensaas, Bredt, Dinger y Fidone,
1994; Wang, Stensaas, Dinger y Fidone, 1995). En experimentos farmacológicos o
de supresión genética, se demuestra la participación de la NOSn y NOSe en la
producción de NO en el CC, tanto in vitro (Kline, Yang, Huang y Prabhakar, 1998)
como in vivo (Gozal, Torres, Gozal y Littwin., 1996), comprobando, de manera
indirecta, la participación de dichas isoformas de la NOS sobre la respuesta
ventilatoria inducida por la hipoxia histotóxica (NaCN).
Un inhibidor específico de la NOSn, como es la s-metil-L-tíocitrulina no modifica
la respuesta ventilatoria inducida por el NaCN en las ratas, en tanto que un inhibidor
no específico como el L-NAME aumenta significativamente la respuesta ventilatoria
(Gozal y col., 1996). Sin embargo, en ratones mutantes carentes de NOSn o NOSe,
la deficiencia de NOSn origina mayor respuesta ventilatoria a la hipoxia histotóxica y
a la hiperoxia cuando se compara con los controles silvestres; mientras que la
respuesta al NaCN y/o a la hiperoxia en los ratones desprovistos de NOSe no está
clara (Kline y col., 1998). En estos estudios no se hicieron registros en el NSC para
verificar las respuestas quimiosensitivas del CC. Buerk y Lahiri (2000) demuestran
una reducción del NO en los CC aislados de gatos que no altera la frecuencia de las
descargas en el NSC, después de perfundir con L-NAME (50 µM) durante 10 min. Al
aumentar la dosis de L-NAME hasta 250 µM, los niveles de NO disminuyen a los 22
min aumentando la frecuencia de las descargas en el NSC. Con dosis mayores de

33
L-NAME (500 µM) desaparece el NO en el tejido carotídeo, y decrece la actividad en
el NSC (Figura 8).
El nitroprusiato de sodio (NPS), donador de NO, incrementa el GMPc intracelular
en las células del glomus carotídeo y en las arteriolas del CC, para producir
hiperventilación (Wang, Stensaas, de Vente, Dinger y Fidone, 1991; Knowles y
Moncada, 1992). Todos estos experimentos demuestran la presencia de un control
nitroxidérgico en el CC, tanto sobre la actividad neuronal como sobre el flujo
sanguíneo.
Figura 8. Relación entre el nivel de NO en el cuerpo carotídeo perfundido de gato y la actividad eléctrica en el NSC. Al disminuir el la concentración de óxido nítrico (NO) en el tejido (panel inferior), por la perfusión de concentraciones crecientes de L-NAME, se incrementan las descargas en el nervio del seno carotídeo (ND) (panel superior). En el t = 50 min el NSC fue incapaz de sostener la actividad máxima.
Modificado de: Buerk y Lahiri, 2000.
En preparaciones del CC in vitro perfundidas con donadores y/o inhibidores de
la NOS a una presión constante, para evitar efectos vasculares (Iturriaga y col.,
2000), se demuestra que el NO sintetizado por la NOSe regula el flujo sanguíneo y

34
los cambios subsecuentes en la PO2 tisular del CC; mientras que el NO sintetizado
por la NOSn en las terminales de las fibras sensitivas “C” y en las neuronas
parasimpáticas, regula la excitabilidad de las células glómicas (Prabhakar y col.,
1993; Chugh y col., 1994; Wang y col., 1994, 1995; Lahiri y Buerk, 1998; Prabhakar,
1999; Valdés y col., 2002). Al inhibir la NOS con L-NAME se incrementa tanto la
respuesta basal en el NSC, como las respuestas a la hipoxia tisular producidas por el
NaCN y la dopamina en gatos anestesiados (Iturriaga y col., 1998), mientras que el
NPS incrementa la respuesta basal, pero reduce sólo la inhibición obtenida por
dopamina pero no por hiperoxia. Es posible que el NO, por ser una molécula ubicua,
module la quimiorrecepción del CC en diferentes blancos utilizando varios
mecanismos (Iturriaga y col., 1998; Prabhakar, 1999; Valdés y col., 2002).
En resumen, el NO produce: vasodilatación en el CC (Chugh y col., 1994; Wang
y col., 1994; Lahiri y Buerk, 1998), inhibición retrógrada de la excitabilidad de las
células del glomus (Wang y col., 1994), inhibición de la actividad de los canales de
Ca2+ de las células del glomus, modulación de la excitabilidad de las neuronas del
ganglio petroso (Alcayaga y col., 1997) e inhibición del metabolismo mitocondrial
(Iturriaga y col., 2000).
Óxido nítrico y la homeostasis de la glucosa
Es indudable que el SNC monitorea y regula los requerimientos energéticos del
organismo; pero queda por determinar el mecanismo por medio del cual el cerebro
realiza esta función detectora de los niveles de glucosa, y la forma en que dicha
información se utiliza para regular la homeostasis energética bajo condiciones
fisiológicas. Datos anteriores sugieren que el SNC participa en la patogénesis de
algunas formas de resistencia a la insulina (Shankar y col., 1998), pero será
importante estudiar si el NO participa en la homeostasis de la glucosa en el SNC en
condiciones fisiológicas. El NO podría ser el mediador potencial del transporte de
glucosa en el SNC, como ocurre en el músculo esquelético (Figura 9), tanto en los

35
estados de reposo como después del ejercicio (Balon y Nadler, 1997). El NO induce
la expresión de los transportadores a la glucosa, como son el GLUT1, GLUT3 y
GLUT4 (Etgen, Fryburg y Gibbs, 1997; Higaki y col., 2001), pero no se ha estudiado
el papel que juega dentro del SNC.
Vasodilatación
glucosainsulina
IRS IRS
PKC
Akt
insulina
SS
SS SS
PI3K
NO
NOSe
glucosainsulina
P P
+
+
++
Akt GLU
T4
GLU
T1
Miocito
Intersticio
Capilar
Sensibilidad a la Insulina
Figura 9. Modelo hipotético de la regulación que ejerce el NO sobre la acción de la insulina en la homeostasis de la glucosa a nivel del músculo esquelético. Akt, proteína kinasa serina-dependiente; GLUT1 y 4, proteína transportadora de glucosa;.IRS, substrato receptor de la insulina; NO, óxido nítrico; NOSe, sintasa de óxido nítrico endotelial; p, fosfato; PI3K, fosfatidilinositol 3-kinasa; PKC, proteína kinasa C. Modificado de Dallaire y Marette, 2004.
La inhibición de los niveles del NO, por infusión intracerebroventricular de L-
NAME, incrementa los niveles de glucosa en el plasma por encima de los
encontrados en grupos controles (Shankar y col., 1998). Observaciones recientes
sugieren que el NO es la molécula señalizadora que modula en forma fisiológica el
metabolismo de la glucosa, tanto en los astrocitos como en las neuronas (Almeida y
col., 2005); en los astrocitos el NO aumenta la glucólisis a través de un mecanismo
independiente de la glucogenolisis (Almeida, Cidad y Bolaños, 2002). Sin embargo,

36
la inhibición de la NOS no altera la utilización de glucosa cerebral, por lo que algunos
autores concluyen que el NO no participa en el metabolismo energético del cerebro
de la rata (Takahashi, Cook, Jehle, Kennedy y Sokoloff, 1995).
Trabajos anteriores de este laboratorio demuestran que el NO en el SNC
participa en el reflejo hiperglucemiante con retención de glucosa por el cerebro
después de estimular los RSCC con NaCN. En efecto, los donadores del NO
(nitroglicerina y NPS) inyectados en la cisterna magna, incrementan la retención de
glucosa por el cerebro, y por el contrario, un inhibidor del NO como el L-NAME
disminuye estos reflejos (Cadenas, 2003).
El óxido nítrico en la diabetes
La diabetes mellitus representa un grupo de enfermedades metabólicas
caracterizadas por presentar hiperglucemia con alteración en los mecanismos
homeostáticos, que desencadenan una serie de procesos patogénicos como la
destrucción autoinmune de las células beta del páncreas con la consecuente
disminución en la secreción de insulina, y anormalidades que dan como resultado un
aumento en la resistencia a la acción de la insulina (Fauci, Braunwald, Isselbacher,
1998). La fisiopatología de la diabetes se caracteriza por angiopatía, hipoxia tisular,
reducción en la respuesta ventilatoria a la hipoxia e hipercapnia. Además, la
diabetes se acompaña de aterosclerosis, neoformación de la red capilar, reducción
en el flujo sanguíneo de órganos, que induce una reducción en el suplemento de O2
al CC, hipertensión arterial y anomalías en el metabolismo de las lipoproteínas (The
Expert Committee on the Diagnosis and Classification of Diabetes Mellitus, 2003).
Las complicaciones agudas de la enfermedad son, entre otras, hiperglucemia con
cetoacidosis y el síndrome hiperosmolar sin cetoacidosis, o estados de hipoglucemia
que pueden ocurrir por el tratamiento con insulina o los hipoglucemiantes orales. Las
complicaciones crónicas, secundarias a la diabetes mellitus, incluyen: retinopatía con
pérdida potencial de la visión, nefropatía hasta insuficiencia renal, neuropatía

37
periférica con ulceración en las extremidades, disfunción gastrointestinal,
genitourinaria, cardiovascular y/o sexual. Existen múltiples evidencias que asocian a
la disfunción vascular con la diabetes mellitus.
La Organización Mundial de la Salud considera 4 tipos de diabetes:
1. La diabetes mellitus tipo 1, con destrucción de las células beta del
páncreas y deficiencia absoluta de insulina.
2. La diabetes mellitus tipo 2, con resistencia a la insulina y deficiencia
relativa de la misma hasta su falta total.
3. La diabetes gestacional, con intolerancia a la glucosa que se inicia o se
reconoce por primera vez durante el embarazo.
4. Otros tipos específicos de diabetes secundarias: a) defecto genético de la
función de las células ß; b) defecto genético de la acción de la insulina; c)
pancreatitis; d) trauma/pancreatectomía; e) drogas o inducción química
(pentamidina, ácido nicotínico, glucocorticoides, hormona tiroidea); f)
agonistas beta adrenérgicos (tiazidas); g) endocrinopatías como la
acromegalia, el síndrome de Cushing, el hipertiroidismo, el glucagonoma
y el feocromocitoma; h) infecciones (rubéola congénita, citomegalovirus).
La patogénesis de la diabetes se ha estudiado profusamente, pero la etiología
de este desorden metabólico permanece sin aclarar. La diabetes se caracteriza por
presentar una función anomal de las células beta con disminución de la secreción de
insulina (Polonsky, Sturis y Bell, 1996) o resistencia a la acción de la insulina
(principalmente en el músculo esquelético y en el tejido adiposo) (Yki-Jarvinen,
1994); se presenta también una disfunción hepática con sobreproducción de glucosa
(Garvey, Revers, Kolterman, Rubenstein y Olefsky, 1985). En todos los casos, la
atención de los investigadores se centra en las vías metabólicas de los órganos
involucrados. Es notorio que durante este estado diabético, los niveles de glucosa
en ayuno son elevados pero relativamente constantes dentro de márgenes
estrechos; hecho que sugiere la presencia de mecanismos reguladores “hacia

38
arriba”, es decir para un nivel alto de glucosa (DeFronzo, 1988). Este alto grado de
regulación en coordinación con el flujo de glucosa hepático, indica también, la
presencia de un sistema de control centralizado y complejo. La utilización de ratas
diabéticas como modelos para entender el proceso de esta enfermedad, permitirá
estudiar los cambios patofisiológicos que ocurren, y en última instancia poder
acceder a su tratamiento.
La administración sistémica de altas dosis de un competidor de la NOS como la
N-monomethyl-L-arginina (L-NMMA), induce una resistencia muy notoria a la insulina
en las ratas sanas, datos que apoyan la existencia de mecanismos centrales que
participan en la patogénesis de la diabetes, particularmente en algunas formas de
resistencia a la insulina (Shankar y col. 1998), es decir, en estos casos el NO a nivel
central influye sobre las vías efectoras para cambiar el nivel de la glucorregulación a
la alta (Shankar y col., 1998). Trabajos recientes encuentran que en los estados de
hipoxia, se presenta un aumento en la frecuencia ventilatoria como una reacción
homeostática de las células periféricas a los requerimientos de O2 y probablemente
de glucosa (Lahiri, Di Giulio y Roy, 2002). La disminución en la respuesta ventilatoria
a la hipoxia en los pacientes diabéticos, podría correlacionarse también con la
hiperglucemia, y la consecuente reducción en la conductividad del NSC, por la
neuropatía de las fibras sensitivas aferentes y motoras efectoras (Bianchi y col.,
2003). Existen controversias sobre la influencia de la diabetes (hiperglucemia) en los
niveles de NO en la sangre sistémica y en el SNC (Kino y col., 2004). Algunos
investigadores, utilizando técnicas inmunohistoquímicas, encuentran que en el CC in
vitro de ratas diabéticas aumenta la expresión de la NOSe y del NO (Bianchi y col.,
2003) (Figura 10A), hechos que asocian a la falta de sensibilidad de los CC a la
hipoxia por un incremento en la NOS, mecanismo que piensan juega un papel en la
adaptación del CC a la hipoxia. Otros autores, en cambio, indican que durante la
hiperglucemia se inhibe la producción de NOSmt (Brodsky, Gao, Li, Goligorsky,
2002) (Figura 10B). El NO disminuye durante la diabetes como consecuencia de
una reducción en los niveles de L-arginina (Pieper y Dondlinger, 1997; Kino y col.,
2004; Lei, Venkatakrishnan, Yu, Kazlauskas, 2007).

39
Se postula que el CC podría jugar un papel importante en la regulación de la
glucosa in vivo (Koyama y col., 2000). El incremento en la NOS sería el responsable
de la reducción en la respuesta ventilatoria en el estado diabético, así como en la
adaptación respiratoria durante la hipoxia crónica. La hipoxia aguda inhibe
normalmente la actividad enzimática, mientras que la hipoxia crónica estimula varias
enzimas entre ellas la NOS, aumentando la producción del NO, que representaría un
mecanismo de defensa homeostática (Bianchi y col., 2003).
Figura 10. A) Expresión de la NOSe en el cuerpo carotídeo de ratas control en condiciones de normoxia o hipoxia, y en ratas diabéticas durante la normoxia. B) Concentración de NO (fluorescencia) en ratas normales (ZL) y diabéticas (ZDF) en condiciones control y después de la estimulación hipóxica (A23187). A) Modificado de: Bianchi y col., 2003. B) Modificado de: Brodsky y col., 2000.
La estimulación de la NOS en el CC de las ratas diabéticas puede
correlacionarse también con los estados de hipoxia crónica en otros órganos debido
a la aterosclerosis, al incremento de la matriz extracelular y a otros factores que
aumentan la distancia de difusión entre la sangre y la mitocondria (Popel, Goldman,
Vadapalli, 2003). Bianchi y col. (2003) consideran que la diabetes afecta a los
quimiorreceptores por una modificación en la respuesta quimiotransductora, como
Fluorescencia
F luorescencia
Control A23187
B
Control DiabetesHipoxia
%NOS
10
20
30
40
50
A

40
resultado del incremento en la producción de radicales libres y enzimas oxidativas
que dañan potencialmente la membrana celular. Para estos autores, la correlación
entre diabetes e hipoxia crónica estaría relacionada con una disminución en la pO2
sanguínea, hecho que ocurre en ambas situaciones.

41
JUSTIFICACIÓN
La homeostasis de la glucosa es esencial para la vida. El número de personas
con trastornos en los mecanismos reguladores que controlan la homeostasis
glucémica es muy grande, y se preveé una tendencia de aumento en el número de
pacientes diabéticos durante los próximos años a nivel mundial. Como no
conocemos con precisión los mecanismos que regulan el metabolismo energético en
el medio interno, y en particular en el SNC, es difícil que podamos implementar los
enfoques terapéuticos para controlar la diabetes, en particular, los tipos 1 y 2.
Además de la existencia de neuronas capaces de detectar cambios en los niveles de
glucosa en diversas áreas cerebrales como en el hipotálamo, en los últimos años se
presentan evidencias que apoyan la presencia de glucorreceptores en los CC, que
juegan un papel importante en la glucorregulación in vivo, participando en las
respuestas contrarregulatorias a la hipoglucemia.
La insulina producida por las células beta del páncreas participa en: el
transporte de la glucosa a nivel periférico, y con ello en la regulación energética; en
la reabsorción de sodio; en la regulación vasomotora y en la liberación de NO.
Deben existir mecanismos en el SNC, que regulen el aporte de glucosa a nivel
central, es decir, que participen en el mantenimiento de los niveles apropiados de
este energético fundamental para la bioquímica cerebral. El estado diabético
representa un paradigma experimental que nos permitirá dilucidar los cambios que
ocurren a nivel central ante una falta de oxígeno en los RSCC in vivo, de manera
concomitante con inhibidores y/o donadores del NO.

42
PLANTEAMIENTO DEL PROBLEMA
Las zonas reflexogénicas seno-carotídeas participan en la homeostasis de la
glucosa, en particular en la captación de glucosa por el SNC. Sin embargo, se
desconoce el o los mecanismos para llevar a cabo dicha regulación, en especial a
nivel del CC. La diabetes implica una serie de alteraciones en los procesos
homeostáticos semejantes a los que se presentan en un estado de hipoxia crónica
con alteración de la producción de óxido nítrico (NO). En esta tesis se plantea
dilucidar el efecto concomitante del NO y el cianuro de sodio (NaCN) en el seno
carotídeo aislado in vivo sobre el reflejo hiperglucémico por estimulación RSCC y los
niveles de nitritos en la sangre procedente del cerebro en ratas normales y
diabéticas.
HIPÓTESIS
El NO en el cuerpo carotídeo aislado de la circulación in vivo, en condiciones de
anoxia histotóxica (por estimulación de los quimiorreceptores del CC con NaCN),
interviene de manera diferencial para modular el metabolismo de la glucosa en ratas
normales y/o diabéticas.
OBJETIVOS
General
- Estudiar el efecto del NO en el cuerpo carotídeo sobre el reflejo
hiperglucemiante inducido por la anoxia histotóxica después de la inyección de
NaCN en el seno carotídeo aislado in vivo de ratas normales y diabéticas.

43
Específicos
- Determinar las concentraciones de nitritos en la sangre venosa del seno yugular
(proveniente del SNC), y las concentraciones de glucosa en la sangre arterial y
venosa encefálica, antes y después de inyectar sol. sal. en el seno carotídeo
circulatoriamente aislado en ratas normales y diabéticas.
- Determinar los niveles de nitritos en la sangre venosa del seno yugular y la
concentración de glucosa en la sangre arterial y venosa encefálica, antes y
después de estimular los RSCC con NaCN en el seno carotídeo
circulatoriamente aislado en ratas normales y diabéticas.
- Evaluar el efecto del L-NAME (inhibidor de la NOS) en el seno carotídeo
circulatoriamente aislado, sobre el reflejo hiperglucemiante en ratas normales y
diabéticas, antes y después de estimular los RSCC con NaCN; cuantificando los
niveles de nitritos en la sangre venosa del seno yugular y la concentración de
glucosa en la sangre arterial y venosa encefálica.
- Evaluar el efecto del NPS (donador de NO) en el seno carotídeo aislado, sobre
el reflejo hiperglucemiante en ratas normales y diabéticas, antes y después de
estimular los RSCC con NaCN; cuantificando los niveles de nitritos en la sangre
venosa del seno yugular y la concentración de glucosa en la sangre arterial y
venosa encefálica.

44
METODOLOGÍA
Animales y técnicas quirúrgicas generales
Los experimentos se realizaron en ratas Sprague-Dawley macho, de 250 a 300
g de peso corporal con ayuno de 12 horas y libre acceso al agua con glucosa al 10%;
experimentos anteriores mostraron que bajo estas condiciones los niveles de glucosa
se mantienen más estables en las ratas anestesiadas (Álvarez-Buylla y de Álvarez-
Buylla, 1988; Álvarez-Buylla y Roces de Álvarez-Buylla, 1994).
Todos los procedimientos experimentales se realizaron de acuerdo con la guía
de los Institutos Nacionales de Salud de los Estados Unidos (Guide for Care and Use
of Laboratory Animals, 1996). Los animales se anestesiaron con pentobarbital
sódico (3 mg/100 g, Anestesal, Pfizer), por vía intraperitoneal (i.p.). El nivel de
anestesia se mantuvo constante durante todo el experimento por goteo i.p. continuo
del anestésico diluido en sol sal. (0.063 mg/min). La profundidad de la anestesia se
vigiló periódicamente examinando el reflejo flexor de la pata al dolor y el reflejo
palpebral. Las ratas se mantuvieron con respiración artificial utilizando un respirador
Palmer conectado a una cánula endotraqueal (por traqueostomía), con frecuencia de
40 respiraciones/min y presión positiva hasta evitar los movimientos respiratorios
espontáneos de la rata. Se mantuvo la temperatura corporal a 37°C por medio de
una lámpara (Álvarez-Buylla y de Álvarez-Buylla, 1988). Las variables respiratorias,
basadas en los valores de pH, pO2 y pCO2 en la sangre arterial obtenida 10 min
antes de las manipulaciones experimentales y 16 min después, se mantuvieron
dentro de los límites normales (Ueki, Linn y Hossman, 1988). La rata se colocó
sobre la mesa de operaciones (Álvarez-Buylla, Quintanar-Stephano, Quintanar-
Stephano, Álvarez-Buylla, 1991) en decúbito dorsal. Se realizó una incisión por la
línea media en la cara ventral del cuello, desde el extremo cefálico del esternón
hasta 2 mm de la base del maxilar, para disecar la traquea e introducir la cánula
endotraqueal.

45
Estimulación de los RSCC
Para estimular los RSCC con NaCN e inyectar los fármacos utilizados, el seno
carotídeo izquierdo se aisló temporalmente de la circulación cefálica y general,
utilizando la técnica de Álvarez-Buylla y de Álvarez-Buylla (1988) (Figura 11). El
método fue el siguiente: disección roma para separar los músculos esternohioideo,
homohioideo y esternomastoideo hasta llegar a la carótida común o primitiva
izquierda, que se disecó en un tramo de 2-3 cm partiendo de la bifurcación carotídea
en dirección caudal. Las arterias: lingual, carótida externa y carótida interna
ipsilaterales se disecaron con la ayuda de ganchillos de vidrio sin interrumpir la
circulación en dichos vasos. La arteria lingual se puncionó con una aguja del número
20, para introducir un tubo de polietileno (Clay Adams PE-20) cuya punta se introdujo
hasta la carótida externa a 5 mm de la bifurcación de la carótida común. Este catéter
se utilizó para la inyección del NaCN durante 4-5 s, y para la extracción posterior de
la sangre que baña al seno carotídeo (6-7 s). Se pasó un hilo en forma de asa por
debajo de la carótida común (2 cm posterior a la bifurcación) y otro hilo igual en la
carótida interna (1.5 cm anterior a la bifurcación); estas ligaduras se utilizaron en
forma temporal durante el tiempo que duraron la administración de los fármacos. La
carótida interna (cerca del foramen yugular) y la carótida externa (por encima de la
arteria lingual) ipsilaterales se ocluyeron temporalmente (10-15 s) durante las
inyecciones de NaCN para evitar su paso a la circulación cefálica y/o circulación
general. Para producir isquemia cerebral sería necesario ocluir estos vasos por más
de 5 min (Wu, Fujihara, Yao, Qi, Li, Shimoji y Baba, 2003). Las arterias faríngea y
occipital izquierdas se ligaron en forma permanente. La estimulación de los RSCC
se realizó por una inyección lenta de 5 µg/100 g de NaCN en 0.1 mL de solución
salina (sol. sal.) a través de una aguja del No. 27 para evitar la estimulación
barorreceptora (Álvarez-Buylla, 1954). Con objeto de bloquear o aumentar los
efectos de la estimulación de los RSCC, inmediatamente después de terminar la
estimulación de estos receptores, se inyectaron un inhibidor de la NOS (L-NAME) o
un donador del NO (NPS) en el seno carotídeo circulatoriamente aislado.

46
Figura 11. Esquema que muestra el procedimiento quirúrgico en la rata. Cateterización de la arterial femoral y la vena yugular para la obtención de muestras de sangre arterial y venosa; estimulación de los receptores seno-cuerpo carotídeo aislados circulatoriamente. aa, aorta abdominal; acc, arteria carótida común; ace, arteria carótida externa; aci, arteria carótida interna; af, arteria femoral; al, arteria lingual; NSC, nervio del seno carotídeo; RCC, receptores del cuerpo carotídeo; sc, seno carotídeo; sy, seno yugular; y tc, tronco celiaco. Modificado de: Montero y col., 2000.
Obtención de sangre y procedimientos bioquímicos
Con la ayuda de ganchillos de vidrio, se disecó la vena yugular externa derecha
hasta llegar hasta el seno venoso yugular, para posteriormente cateterizarlo con un
tubo de silastic (Dow Corning 602-155). Después de incidir la región inguinal en la

47
cara interna y media del muslo izquierda, en sentido transversal al pliegue, hasta el
tercio superior, se visualizó el paquete vásculo-nervioso, se disecó la arteria femoral
para cateterizarla con un tubo de polietileno (PE-20 Clay Adams) hasta la aorta
abdominal. Las cateterizaciones de los vasos se realizaron con cánulas previamente
heparinizadas sin interrumpir su circulación normal (Álvarez-Buylla y Bencosme,
1981), verificando al final de cada experimento la posición correcta de los catéteres
(Figura 11).
Todas las medidas de gases (O2 y CO2) en sangre y pH en trabajos similares
determinados anteriormente en un analizador Micro 13 (Instrumentation Laboratory)
en mmHg y valores absolutos, respectivamente. se han encontrado dentro del rango
de los estándares comerciales (Ueki y col., 1988; Guarner y Álvarez-Buylla, 1991;
Montero, Mendoza, Valles, Lemus, Álvarez-Buylla, de Álvarez-Buylla, 2006). Por lo
que no hay cambios en estas variables por la cirugía o por las maniobras
experimentales.
Determinación de los niveles de glucosa en el plasma
Después de lavar por tres veces, con su propia sangre, los catéteres descritos,
se colectaron las muestras de sangre arterial y venosa de acuerdo al protocolo
experimental. Se tomaron 4 muestras de sangre arterial (arteria femoral) y 4
muestras de sangre venosa (seno yugular), durante un período de 20 min, para la
determinación de las concentraciones de glucosa en el plasma; la muestra t = -4 min
correspondió a la basal, y las muestras t = 4 min, t = 8 min y t =16 min
correspondieron a los tiempos experimentales, después de la estimulación de los
RSCC con NaCN simultáneamente con las inyecciones de sol. sal, L-NAME o NPS
en el seno carotídeo circulatoriamente aislado en el t = 0 min. En cada tiempo se
colectaron 0.15 mL de sangre arterial y 0.15 mL de sangre venosa. Para compensar
la pérdida de líquido, después de obtener cada par de muestras, las ratas recibieron
una inyección de 0.3 mL de sol. sal. Las muestras se mantuvieron en refrigeración

48
hasta su centrifugación. La sangre se centrifugó durante 5 min a una velocidad de
3000 rpm, utilizando una centrífuga refrigerada (Beckman T J-6). Como los niveles
de glucosa en la sangre de la aorta abdominal son iguales a los de la carótida, y los
valores de flujo sanguíneo no cambiaron después de la estimulación de los RSCC, la
retención de glucosa cerebral se calculó a partir de las diferencias arterio-venosas (a-
v) de glucosa entre las concentraciones de glucosa en la aorta abdominal y los del
seno yugular en µmol/mL (Álvarez-Buylla y col., 2003).
La concentración de glucosa en el plasma se midió por el método de la glucosa-
oxidasa (Analizador de glucosa Beckman 2) en µmol/mL, en muestras de 10 µL de
plasma. La glucosa se determinó a partir de la depleción de O2 en una solución de
glucosa-oxidasa saturada con O2; el consumo de O2 es directamente proporcional a
la concentración de glucosa en la muestra.
Determinación de los niveles de nitritos en el plasma. Método de Griess.
El reactivo de Griess consiste en una sulfanilamida y bicloruro de N-1-
nafiletilendiamina (NED), bajo condiciones ácidas (ácido fosfórico). Este sistema
detecta los nitritos (NO2-) en una gran variedad de líquidos biológicos y
experimentales tales como el plasma, suero, LCR, saliva, sudor, orina y medios de
cultivo celular, entre otros. La sensibilidad del método para los nitritos depende del
medio utilizado para su medición. El límite de detección es de 2.5 µM (125 pmol) de
nitritos (en agua desionizada ultrapura) (Granger, Taintor, Boockvar y Hibbs, 1996).
Para evitar la contaminación en las muestras de plasma con hemoglobina y
proteínas (requisito para esta técnica), la extracción de sangre se hizo lentamente, a
goteo. Para la determinación de los niveles de nitritos en el plasma se tomaron dos
muestras de sangre venosa encefálica de 1 mL cada una, a los tiempos t = -4 min
(control) y t = 8 min (experimental), restituyendo el volumen con sol. sal. después de
la extracción. El volumen total de sangre colectado fue de 3.2 mL aproximadamente,

49
menor del 15 % del volumen total, por lo que no produjo alteraciones hemodinámicas
significativas (Álvarez-Buylla y col., 1988). La muestra de sangre total para valorar
nitritos, se centrifugó a 3000 rpm en una centrífuga refrigerada (Beckman T-J 6)
durante 10 min para obtener el plasma. A 400 mL de plasma se les agregaron 400
mL de ácido perclórico frío al 20% para desnaturalizar las proteínas, se mezcló en el
vórtex durante 30 s y se centrifugó a 3000 rpm en una centrífuga refrigerada
(Beckman T-J 6) durante 10 min. Una vez obtenido el sobrenadante, se agregaron 5
µL de naranja de metilo al 0.05% y K2CO3 (3 M) gota a gota, para neutralizar, al
mismo tiempo que la mezcla se agitaba en el vórtex suavemente hasta que el color
rosado se tornó en amarillo claro, indicando un pH de 7.4. La muestra se conservó a
-70 ºC en el ultracongelador.
Protocolo para determinar la concentración de nitritos
Se preparó una curva de referencia con la solución de nitritos antes de cada
experimento con el fin de comparar la concentración de nitritos en las muestras
experimentales. Se preparó la solución de nitrato reductasa (5 µL = 50 mU) en el
buffer usado para las muestras experimentales añadiendo 20 µL en cada muestra
experimental o de estudio. Se agregaron 20 µL de NADPH (25 µM) y EDTA (10 mM)
y se incubó durante 30 min a temperatura ambiente agregando 20 µL de glutámico
deshidrogenasa (5 µL = 100 mU). Posteriormente, se agregaron 20 µL de NH4Cl
(100mM) y 20 µL del ácido alfa-cetoglutárico (4 mM), que se incubó durante 10 min
a temperatura ambiente. Una vez transcurrido este tiempo, se agregaron 60 µL de
ácido 5-sulfasalicílico al 6%, se agitó cada 5 min durante 30 min, y se centrifugó a
10,000 rpm en una centrifuga refrigerada (Sorvall Instruments RC5C) durante 15 min.
Se extrajo el sobrenadante en alícuotas de 200 µL, agregando 50 µL de NH4Cl al 30
% y 30 µL de NaOH al 5 % para completar un volumen de 280 µL, para incorporar,
después, el reactivo de Griess con el mismo volumen (280 µL). Se incubó durante
20 min a temperatura ambiente protegido de la luz hasta desarrollar un color
magenta casi de manera inmediata. Se midió la absorbancia en todas las muestras

50
en un lapso de 40 min (lector de ELISA - Spectronic 21 D) con un filtro entre 520-550
nm (Tabla 1). Se determinó la absorbancia promedio de cada muestra experimental
calculando su concentración por comparación con la curva del estándar de los
nitritos. La concentración de nitritos se calculó a partir de la curva estándar corrida a
la par de los problemas. La curva se diseñó de acuerdo a los valores máximos y
mínimos esperados en las muestras problema con el fin de que todas las muestras
entraran en el rango de la curva. Una vez obtenida la curva de referencia con los
estándares de nitritos, se graficó el valor promedio de la absorbancia de cada
concentración del estándar de nitrito como función de ¨Y¨ contra la concentración de
nitritos como función de ¨X¨.
Reactivos de Griess
- Solución A:
Naphthyl-ethylen diamina 0.1%, disuelto en agua bidestilada.
- Solución B:
H3PO4 al 5 % (ácido fosfórico) disuelto en agua bidestilada.
- Sulfanilamida al 1% en ácido fosfórico se disuelve y se afora en el ácido
fosfórico al 5%.
- El reactivo de Griess se preparó mezclando las soluciones A y B 1:1, la mezcla
dura 12 horas a una temperatura de 4 °C, o más tiempo a -20 °C.
- La curva estándar se hizo con el NaNO2 100 µM en agua bidestilada.
Fármacos utilizados
• Pentobarbital sódico (Anestesal, Pfizer), 3 mg/100 g.
• Solución salina al 0.9 % (Pisa), 0.1 mL.
• Cianuro de sodio (NaCN, Sigma), 5 µg/100 g en 0.1 mL de sol. sal.
(Álvarez-Buylla y col., 1988).

51
• N-nitro-L-arginina metil éster (L-NAME, Sigma), 250 µM en 0.1mL de sol.
sal. (Kadekaro, Terrell, Liu, Gestl, Bui, Summy-Long, 1998).
• Nitroprusiato de Sodio. (NPS, Sigma) 250 µg/100 g en 0.1mL de sol. sal.
(Iturriaga y col., 2000).
• Estreptozotocina. (Sigma) 50 mg/kg en 0.5 mL de sol. sal. i.p. (Szkudelski,
2001; Días, Porawski, Alonso, Marroni, Collado y González-Gallego, 2005).
TABLA 1. Protocolo para la determinación de nitritos.
BR (µL)
S1 S2 S3-7 P1 (µL)
Pn
1) Muestra o estándar - 200 1’) Agua 280 √ 2) Nitrato reductasa (5µl = 50mU) - 20 3) NADPH 25µM, EDTA 10mM EDTA - 20 4) Incubar a temperatura ambiente (30 min) - √ 5) Glutámico deshidrogenasa (5µl =100 mU) - 20 6) NH4CL 100 mM - 20
7) Ácido α-cetoglutárico 4 mM - 20 8) Incubar a temperatura ambiente (10 min) - √ 9) Ácido 5-sulfosalicílico al 6% - 60 10) Agitar cada 5 min durante 30 min - √ 11) Centrifugar a 10,000 rpm/15 min - √ 12) Alícuota de sobrenadante - 200 13) NH4Cl al 30% - 50 14) NaOH al 5% - 30 15) Reactivo de Griess 280 280 16) Incubar a temperatura ambiente (20 min) √ √ 17) Leer D. O. a 550 nm √ √
BR, agua con reactivo de Griess; D.O., densidad óptica en el espectrofotómetro; min, minutos; Pn; número de muestras experimentales; S, muestras estándar de la 1 a la 7.

52
Con las dosis mencionadas se produjo un efecto óptimo en estudios previos por
los autores citados.
Ratas diabéticas
Los animales diabéticos se pueden obtener por varios métodos (Álvarez-Buylla
y Carrasco-Zanini, 1960): la extirpación total del páncreas, la inyección i.p. o i.v. de
aloxano, y la inyección i.p. o i.v. de estreptozotocina (STZ). En este estudio se utilizó
la última opción por ser la menos traumática y menos tóxica para el organismo. La
inducción de la diabetes experimental en la rata utilizando fármacos que destruyen
selectivamente las células beta es un método muy conveniente y simple de llevar a
cabo, pero es necesario comprender los cambios que ocurren en las células beta del
páncreas, así como en todo el organismo, después de utilizar fármacos
diabetogénicos como el aloxano (2, 4, 5, 6-tetraoxipirimidina) o la estreptozotocina
(STZ) que tienen una acción citotóxica en los islotes del páncreas (Szkudelski,
Kandulkska, Okulicz, 1998). La acción tóxica del aloxano es la suma de varios
procesos oxidantes de grupos–SH esenciales, con inhibición de la glucocinasa y la
generación de radicales libres con alteraciones intracelulares en la homeostasis del
calcio. La STZ (2-deoxy-2-[3-(methyl1-3-nitrosoureido)-D-glucopiranosa] se sintetiza
por el Streptomycetes achromogenes, y se utiliza para inducir tanto la diabetes
mellitus tipo 1 (insulino-dependiente-IDDM) y como la tipo 2 (no insulino-
dependiente-NIDDM), según el momento de la inyección, la dosis empleada o su
frecuencia de inyección. Una dosis i.v. de STZ (40 a 60 mg/kg) en ratas adultas
puede inducir diabetes tipo 1 (Ganda, Rossini y Like, 1976), pero es posible utilizar
dosis mayores (Szkudelski, 2001). La STZ también se puede administrar por vía i.p.
en una dosis similar o mayor (Katsumata, Katsumata, Katsumata, 1992). Cuando se
inyectan 50 mg/kg de STZ por vía i.v. en ratas, 2 semanas después, la glucosa
sanguínea alcanza valores de 15 mmol/L (Szkudelski, 2001), y podemos considerar,
entonces, que la rata presenta diabetes. La STZ altera primero la oxidación de la
glucosa para ser captada por las células (Bedoya, Solano y Lucas, 1996), para

53
después disminuir la biosíntesis y sedcreción de insulina (Nukatsuka, Yoshimura,
Nishida, Kawada, 1990). Las células beta del páncreas captan la STZ a través del
transportador GLUT2, por lo que cuando se reduce la expresión de este
transportador, se previene la acción diabetogénica de la STZ (Thulesen, Orskov,
Holst, Poulsen, 1997). Este fármaco ocasiona cambios intracelulares en las células
beta, como fragmentación y alcalinización del DNA, hasta producir su muerte (Elsner,
Guldbakke, Tiedge, Munday, Lenzen, 2000).
Para verificar el establecimiento del estado diabético en el grupo de ratas que
recibieron la inyección de una dosis única de STZ (50 mg/kg peso, i.p., preparada
diariamente en una solución de citrato de sodio 10 mmol/L a pH 4.5), se realizó un
seguimiento de los niveles de de glucosa en la sangre total en ayuno durante 3 días
después de la inyección, tomando la muestra sanguínea de la vena de la cola. La
valoración de los niveles de glucosa, en estos casos, se realizó mediante una tira
reactiva (Accu-Chek). El principio de esta prueba consiste en la conversión, por la
enzima glucosa deshidrogenasa, de la glucosa en gluconolactona. La
gluconolactona crea una corriente eléctrica proporcional a la concentración de
glucosa en la sangre, que se lee en el medidor. Un nivel mayor de 14 mmol/L de
glucosa se consideró indicativo de diabetes tipo 1 de acuerdo a la literatura
anteriormente citada.
Protocolo experimental
Las ratas se dividieron al azar en 4 grupos, de 10 ratas cada uno; a su vez,
estos grupos se dividieron en dos subgrupos: ratas normales que recibieron una
inyección i.p. del vehículo para diluir la STZ (0.5 mL de citrato de sodio-pH 4.5), y
ratas diabéticas, que recibieron una inyección i.p. de STZ (50 mg/kg/0.5 mL de citrato
de sodio-pH 4.5) para alcanzar una concentración de glucosa en el plasma de 14
mmol/L o mayor. Los grupos se denominaron como:

54
- Control 1: a) dos inyecciones consecutivas de sol. sal. (100 µL) en el seno
carotídeo circulatoriamente aislado (15-20 s) en las ratas normales (n = 5); b)
mismas inyecciones que en “a” en las ratas diabéticas (n = 5).
- Control 2: a) estimulación de los RSCC con NaCN (5 µg/100 g/100 µL de sol.
sal.) en el seno carotídeo circulatoriamente aislado, seguida de una inyección
de sol. sal. (100 µL) en el mismo lugar (15-20 s) en las ratas normales (n = 5);
b) mismo procedimiento que en “a” en las ratas diabéticas (n = 5).
- Experimental 1: a) estimulación de los RSCC con NaCN (5 µg/100 g/100 µL
de sol. sal.) en el seno carotídeo circulatoriamente aislado, seguida de una
inyección de L-NAME (250 mM/100 µL de sol. sal.) en el mismo lugar (15-20
s) en las ratas normales (n = 5); b) mismo procedimiento que en “a” en las
ratas diabéticas (n = 5).
- Experimental 2: a) estimulación de los RSCC con NaCN (5 µg/100 g/100 µL
de sol. sal.) en el seno carotídeo circulatoriamente aislado, seguida de una
inyección de NPS (250 µg/100 g/100 µL de sol. sal.) en bolo en el mismo
lugar (15-20 s), en las ratas normales (n = 5); b) mismo procedimiento que en
“a” en las ratas diabéticas (n = 5).
Análisis estadístico
Las comparaciones estadísticas se realizaron utilizando la prueba t de Student
comparando los valores experimentales con sus propias basales, y la prueba de
ANOVA con la prueba de Scheffè para la comparación múltiple. El nivel de
significancia se fijó en p < 0.05. En todas las gráficas los valores son medias
aritméticas y las líneas verticales representan los errores estándar. Las
probabilidades (valor de p) entre los datos reportados con respecto a su basal, se
calcularon como muestras pareadas.

55
RESULTADOS
Control 1. Inyección de sol. sal. por dos ocasiones en el seno carotídeo
circulatoriamente aislado
En condiciones basales, la concentración media de glucosa en la sangre arterial
en los subgrupos de ratas normales y diabéticas, fue de 126 ± 1 mg/dL y 456 ± 19
mg/dL, respectivamente; la concentración media de glucosa en la sangre venosa fue
de 115 ± 1 mg/dL en las ratas normales, y de 437 ± 16 mg/dL en las ratas diabéticas.
La retención de glucosa cerebral media fue de 11 ± 1 mg/dL en las ratas normales y
de 19 ± 4 mg/dL en las ratas diabéticas. Cuando se inyectó la sol. sal. (100 µL), en
el seno carotídeo circulatoriamente aislado, por dos ocasiones consecutivas, tanto en
las ratas normales como en las ratas diabéticas, no se observaron cambios
significativos en las concentraciones de glucosa arterial y venosa, ni en la retención
de glucosa cerebral en los tiempos estudiados (t = 4 min, t = 8 min, t = 16 min) (n = 5)
(tablas 2 y 3, figura 12A y B).
Tabla 2. Concentración de glucosa plasmática (mg/dL) en el grupo control 1.
Tiempo (min)
Ratas normales
(n=5)
Ratas diabéticas
(n=5)
Arterial
Venosa
Arterial
Venosa
-4
126 ± 1
115 ± 1
456 ± 19
437 ± 16
4 133 ± 2 122 ± 2 453 ± 22 436 ± 19
8 135 ± 2 123 ± 2 456 ± 23 438 ± 19
16 135 ± 2 122 ± 2 458 ± 24 438 ± 19
Se inyectó sol. sal. (100 µL) en dos ocasiones consecutivas en el seno carotídeo circulatoriamente aislado (t = 0). Los datos se expresan como medias aritméticas ± EE (t de Student).

56
Tabla 3. Retención de glucosa cerebral (mg/dL) en el grupo control 1.
Tiempo (min)
Ratas normales
(n=5)
Ratas diabéticas
(n=5)
-4
11 ± 1
19 ± 4
4 11 ± 1 17 ± 3
8 12 ± 1 18 ± 5
16 13 ± 1 20 ± 6
Se inyectó sol. sal. (100 µL) en dos ocasiones consecutivas en el seno carotídeo circulatoriamente aislado (t = 0). Los datos se expresan como medias ± EE (t de Student).
Con respecto a los niveles basales de nitritos en la sangre venosa cerebral, los
valores promedio observados en el subgrupo de las ratas normales fue de 0.6957 ±
0.089 nmol/mL, mientras que en el subgrupo de las ratas diabéticas fue de 0.1987 ±
0.062 nmol/mL. Después de las inyecciones de sol. sal. en el seno carotídeo
circulatoriamente aislado, no se observaron variaciones significativas (tabla 4 y figura 13A y 13 B).
Tabla 4. Concentración de nitritos (nmol/mL) en la sangre venosa cerebral en el grupo control 1.
Tiempo (min)
Ratas normales
(n=5)
Ratas diabéticas
(n=5)
-4
0.6957 ± 0.089
0.1987 ± 0.062
8 0.6892 ± 0.093 0.1782 ± 0.047
Se inyectó sol. sal. (100 µL) en dos ocasiones consecutivas en el seno carotídeo circulatoriamente aislado (T = 0). Los datos se expresan como medias ± EE (t de Student).

57
Figura 12. Concentración de glucosa en el plasma y retención de glucosa cerebral en las ratas normales (A) y diabéticas (B) del grupo control 1. El tiempo cero (flecha) corresponde al momento de la inyección de 100 µL de solución salina (sol. sal.) por dos ocasiones de forma consecutiva en el seno carotídeo circulatoriamente aislado (SCA). Los valores se expresan en medias aritméticas ± E.E. (t de Student).
-4 0 4 8 12 160
40
80
300
400
500
B
Ret
enci
ón d
egl
ucos
a (m
g/dL
)
Glu
cem
ia(m
g/dL
)
M in
-4 0 4 8 12 160
40
80
120
180
240
300
A Arterial Venosa- sol. sal. (100 µL)
- sol. sal. (100 µL) en el SCA
Glu
cem
ia
(mg/
dL)
Ret
enci
ón d
egl
ucos
a (m
g/dL
)

58
Figura 13. Concentración de nitritos en la sangre venosa cerebral en ratas normales (A) y diabéticas (B) del grupo control 1. La flecha indica el tiempo cero en el cual se inyectaron 100 µL de solución salina (sol. sal.) por dos ocasiones de forma consecutiva en el seno carotídeo circulatoriamente aislado (SCA). Los valores se expresan en medias aritméticas ± E. E. (t de Student). Control 2. Estimulación de los RSCC con NaCN seguida de una inyección de sol. sal. en el seno carotídeo circulatoriamente aislado
Cuando se estimularon los RSCC con NaCN (5 µg/100 g/100 µL de sol. sal.) la
concentración media de glucosa en la sangre arterial en los subgrupos de ratas
normales y diabéticas, antes de la estimulación fue de 129 ± 1 mg/dL y 447 ± 17
mg/dL, respectivamente; la concentración media de glucosa en la sangre venosa fue
de 118 ± 3 mg/dL en las ratas normales, y de 430 ± 18 mg/dL en las ratas diabéticas.
La retención de glucosa cerebral media fue de 17 ± 2 mg/dL en las ratas normales y
de 17 ± 3 mg/dL en las ratas diabéticas. Después de la inyección de NaCN en el
seno carotídeo circulatoriamente aislado, se observaron cambios significativos en las
concentraciones de glucosa arterial y en la retención de glucosa cerebral tanto en las
ratas normales, como en las diabéticas. En las ratas normales, la glucosa arterial y
la retención de glucosa cerebral aumentaron de manera paulatina en todos los
tiempos estudiados (t = 4, t = 8 y t = 16) hasta llegar a 284 ± 5 mg/dL (t = 8, p =
0,0
0,2
0,4
0,6
0,8
1,0
1,2
Min
A
80-4
- sol. sal. (100 µL)- sol. sal. (100 µL) en el SCA
Con
cent
raci
ón d
e ni
trito
s(n
mol
/mL)
0,0
0,2
0,4
0,6
0,8
1,0
1,2
80-4
B
- sol. sal. (100 µL)- sol. sal. (100 µL) en el SCA
Min

59
0.00046) y 72 ± 4 mg/dL (t = 8 min, p = 0.016) respectivamente. En las ratas
diabéticas, la concentración de glucosa arterial también aumentó, llegando hasta 500
mg/dL ± 10 mg/dL en el t = 8 min (p = 0.0087), pero en la retención de glucosa
cerebral solo se observó un incremento significativo en el t = 16 min (p = 0.0086)
(tablas 5 y 6, figura 14A y 14B).
Tabla 5. Concentración de glucosa plasmática (mg/dL) en el grupo control 2.
Tiempo
Ratas normales
Ratas diabéticas
Arterial
Venosa
Arterial
Venosa
-4
129 ± 1
118 ± 3
447 ± 17
430 ± 18
4 151 ± 3** 127 ± 4 486 ± 13** 449 ± 20
8 217 ± 6** 166 ± 4 500 ± 10** 456 ± 18
16 284 ± 5*** 212 ± 3 503 ± 16* 460 ± 16
Se inyectó NaCN (5 µg/100 g/100 µL) y sol. sal. (100 µL) en forma consecutiva en el seno carotídeo circulatoriamente aislado (t = 0). Los datos se expresan como medias aritméticas ± E. E. * p < 0.05, ** p < 0.01, *** p < 0.001 (t de Student).
Tabla 6. Retención de glucosa cerebral (mg/dL) en el grupo control 2.
Tiempo (min)
Ratas normales
(n=5)
Ratas diabéticas
(n=5)
-4
17 ± 2
17 ± 3
4 29 ± 2* 37 ± 8
8 51 ± 3* 44 ± 11
16 72 ± 4* 43 ± 7**
Se inyectó NaCN (5 µg/100 g/100 µL) y sol. sal. (100 µL) de forma consecutiva en el seno carotídeo circulatoriamente aislado (t = 0). Los datos se expresan como medias aritméticas ± E. E. * p < 0.05, ** p < 0.01 (t de Student).

60
Figura 14. Concentración de glucosa en el plasma arterial y venoso, y retención de glucosa cerebral en ratas normales (A) y diabéticas (B) del grupo control 2. La flecha indica el tiempo cero en el cual se inyectaron el NaCN (5 µg/100 g/100 µL) y la sol. sal. (100 µL) de forma consecutiva en el seno carotídeo circulatoriamente aislado (SCA). Note el aumento paulatino en las concentraciones de glucosa arterial y venosa (reflejo hiperglucémico) y la retención de glucosa en el tiempo. Los valores se expresan en medias aritméticas ± E. E. * p< 0.05, p < 0.01, p < 0.001 (t de Student contra sus propias basales).
-4 0 4 8 12 160
40
80
120
180
240
300
*
A
**
**
***
**
Arterial Venosa
- NaCN (5 µg/100 g/100 µL)- sol. sal. (100 µL) en el SCA
Ret
enci
ón d
egl
ucos
a (m
g/dL
)G
luce
mia
(mg/
dL)
-4 0 4 8 12 160
40
80
300
360
420
480
540 *B
*
****
Min
Ret
enci
ón d
egl
ucos
a (m
g/dL
) G
luce
mia
(m
g/dL
)

61
Con respecto a los niveles de nitritos en la sangre venosa cerebral, los valores
promedio encontrados en el subgrupo de las ratas normales fue de 0.6928 nmol/mL,
mientras que en el subgrupo de las ratas diabéticas fue de 0.1844 ± 0.041 nmol/mL.
Después de la estimulación con NaCN los nitritos en la sangre venosa cerebral
aumentaron significativamente en el t = 8 min hasta 1.4347 ± 0.17 (p = 0.019 t de
Student). Cuando comparamos este último valor con el obtenido en las ratas
normales del grupo 1 se observó un aumento significativo (p = 0.010) después de la
inyección de NaCN (N = 5); es decir, la estimulación anóxica elevó los niveles de
nitritos en la sangre procedente del cerebro (tabla 7, figura 13A y 15A).
Tabla 7. Concentración de nitritos (nmol/mL) en sangre venosa cerebral en el grupo control 2.
Tiempo (min)
Ratas normales
(n=5)
Ratas Diabéticas
(n=5)
-4
0.6928 ± 0.075
0.1844 ± 0.041
8 1.4347 ± 0.17** 1.1650 ± 0.223*
Se inyectó el NaCN (5 µg/100 g/100 µL) y la sol. sal. (100 µL), de forma consecutiva, en el seno carotídeo circulatoriamente aislado (t = 0). Los datos se expresan como medias aritméticas ± E. E. * p < 0.05; ** p < 0.01 (t de Student con su propia basal).
Experimental 1. Estimulación de los RSCC con NaCN seguida de un inhibidor
de la NOS (L-NAME) en el seno carotídeo circulatoriamente aislado
La estimulación de los RSCC con NACN (5 µg/100 g en 100 µL de sol.sal.)
seguida de una inyección de un inhibidor de la NOS (L-NAME) (250 µM/100 µL) dio
como resultado la abolición del reflejo hiperglucemiante en todos los tiempos
estudiados en las ratas normales, por el contrario, en las ratas diabéticas se observó
un aumento significativo, desde una basal de 334 ± 19 mg/dL hasta alcanzar niveles
de 361 ± 19 mg/dL en el t = 4 min y 376 ± 19 mg /dL en el t = 8 min (p = 0.0086 y p =

62
0.0078, respectivamente) (tabla 8). La retención de glucosa por el cerebro no varió
significativamente durante el experimento (tabla 9). La concentración media de
glucosa basal en la sangre arterial en los subgrupos de ratas normales y diabéticas,
fue de 125 ± 5 mg/dL y 334 ± 19 mg/dL, respectivamente, mientras que la
concentración media de glucosa en la sangre venosa fue de 117 ± 4 mg/dL en las
ratas normales, y de 324 ± 21 mg/dL en las ratas diabéticas. La retención de glucosa
cerebral no varió significativamente durante el experimento, la basal media fue de 8 ±
3 mg/dL en las ratas normales y de 11 ± 4 mg/dL en las ratas diabéticas (n = 5)
(tablas 8 y 9, fig. 16A y 16B).
Figura 15. Concentración de nitritos en la sangre venosa cerebral en ratas normales (A) y diabéticas (B) del grupo control 2. La flecha indica el tiempo cero [inyección del NaCN - 5 µg/100 g/100 µL y de la sol. sal. - 100 µL, de forma consecutiva en el seno carotídeo circulatoriamente aislado (SCA)]. Los valores se expresan en medias aritméticas ± E. E. (t de Student).
Con respecto a los niveles de nitritos en la sangre venosa cerebral, los valores
basales promedio encontrados en el subgrupo de las ratas normales fueron de
0.6390 ± 0.015 nmol/mL; después de la estimulación seguida por la infusión de un
inhibidor de la NOS, se observó un decremento significativo hasta 0.3011 ± 0.040
nmol/mL (p = 0.044), mientras que en el subgrupo de las ratas diabéticas fue de
0.3414 ± 0.103 nmol/mL en el t = -4 min y 0.1485 ± 0.097 nmol/mL en el t = 8 min
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
1,8
Con
cent
raci
ón d
e ni
trito
s(n
mol
/mL)
A
80-4
Min
**
- NaCN (5 µg/100 g/100 µL)- sol. sal. (100 µL)
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
1,8
B
*
80-4
- NaCN (5 µg/100 g/100 µL)- sol. sal. (100 µL)
Min

63
(tabla 10, figura 17A y B). Al comparar el valor obtenido en las ratas normales
(control 2), después de la estimulación anóxica, con el valor obtenido en este grupo
en que se inyectó L-NAME después de una estimulación semejante (n = 5), se
registró un descenso significativo (p = 0.015) (tablas 7 y 10, figuras 15A y 17A).
Tabla 8. Concentración de glucosa plasmática (mg/dL) en el grupo experimental 1.
Tiempo
Ratas normales
Ratas diabéticas
Arterial
Venosa
Arterial
Venosa
-4
125 ± 5
117 ± 4
334 ± 19
324 ± 21
4 135 ± 7 122 ± 7 361 ± 19** 348 ± 19
8 141 ± 9 123 ± 10 376 ± 19** 357 ± 14
16 174 ± 20 141 ± 12 392 ± 35 366 ± 37
Se inyectó NaCN (5 µg/100 g/100 µL) y L-NAME (250 µM/100 µL) de forma consecutiva en el seno carotídeo circulatoriamente aislado (t = 0). Los datos se expresan como medias ± E.E., ** p < 0.01 (t de Student).
Tabla 9. Retención de glucosa cerebral (mg/dL) en el grupo experimental 1.
Tiempo (min)
Ratas normales
(n=5)
Ratas diabéticas
(n=5)
-4
8 ± 3
11 ± 4
4 13 ± 3 14 ± 5
8 18 ± 3 19 ± 6
16 33 ± 10 26 ± 4
Se inyectó NaCN (5 µg/100 g/100 µL) y L-NAME (250 µM/100 µL) de forma consecutiva en el seno carotídeo circulatoriamente aislado (t = 0). Los datos se expresan como medias aritméticas ± E. E. (t de Student).

64
Figura 16. Concentración de glucosa en el plasma arterial y venoso, y retención de glucosa cerebral en las ratas normales (A) y diabéticas (B) del grupo experimental 1. La flecha indica el tiempo cero, en el cual se inyectaron el NaCN (5 µg/100 g/100 µL) y el L-NAME (250 µM/100 µL) de forma consecutiva en el seno carotídeo circulatoriamente aislado (SCA). Los valores se expresan en medias aritméticas ± E. E. ** p < 0.001 (t de Student).
-4 0 4 8 12 160
40
80
120
180
240
300
A - NaCN (5 µg/100 g/100 µL)- L-NAME (250 µM/100 µL) en el SCA
Ret
enci
ón d
e g
luco
sa (m
g/dL
)
Arterial Venosa
Glu
cem
ia(m
g/dL
)
-4 0 4 8 12 160
40
80
300
350
400
450B
****
Ret
enci
ón d
egl
ucos
a (m
g/dL
) G
luce
mia
(m
g/dL
)
M in

65
Tabla 10. Concentración de nitritos (nmol/mL) en la sangre venosa cerebral en el grupo experimental 1.
Tiempo (min)
Ratas normales
(n=5)
Ratas diabéticas
(n=5)
-4
0.6390 ± 0.015
0.3414 ± 0.103
8 0.3011 ± 0.040* 0.1485 ± 0.097*
Se inyectó NaCN (5 µg/100 g/100 µL) y L-NAME (250 µM/100 µL) de forma consecutiva en el seno carotídeo circulatoriamente aislado (t = 0). Los datos se expresan como medias aritméticas ± E. E. * p < 0.05 (t de Student con su propia basal).
Figura 17. Concentración de nitritos en la sangre venosa cerebral en las ratas normales (A) y diabéticas (B) del grupo experimental 1. La flecha indica el tiempo cero, en el cual se inyectaron el NaCN (5 µg/100 g/100 µL) y el L-NAME (250 µM/100 µL) de forma consecutiva en el seno carotídeo circulatoriamente aislado (SCA). Los valores se expresan en medias aritméticas ± E. E. * p < 0.05 (t de Student). Experimental 2: Estimulación de los RSCC con NaCN seguida de un donador
del NO (NPS) en el seno carotídeo circulatoriamente aislado.
La estimulación de los RSCC con NaCN (5 µg/100 g en 100 µL de sol. sal.),
seguida de una inyección de NPS (donador de NO) (250 µg/100 g/100 µL) en el
0,0
0,2
0,4
0,6
0,8
1,0
A
Min
-40-4
*
- NaCN (5 µg/100 g/100 µL)- L-NAME (250 µM/100 µL)
Con
cent
raci
ón d
e ni
trito
s(n
mol
/mL)
-4 0 40,0
0,2
0,4
0,6
0,8
1,0
*
- NaCN (5 µg/100 g/100 µL)- L-NAME (250 µM/100 µL)
B
Min

66
SCA, dio como resultado un incremento en el reflejo hiperglucemiante con retención
de glucosa por el cerebro cuando este grupo experimental se comparó con el grupo
control 2 (sólo estimulación de los RSCC) en los tiempos t = 8 min (p = 0.015,
ANOVA) y t = 16 min (p = 0.033, ANOVA) en las ratas normales (figura 19). En
efecto, las concentraciones de glucosa en la sangre arterial subieron desde 126 ± 1
Tabla 11. Concentración de glucosa plasmática (mg/dL) en el grupo experimental 2.
Tiempo
Ratas normales
Ratas diabéticas
Arterial
Venosa
Arterial
Venosa
-4
126 ± 1
110 ± 1
410 ± 22
395 ± 21
4 169 ± 2*** 127 ± 5 447 ± 25* 418 ± 27
8 236 ± 11*** 154 ± 7 488 ± 18** 449 ± 22
16 303 ± 5*** 196 ± 10 473 ± 28 446 ± 34
Se inyectó NaCN (5 µg/100 g/100 µL) y NPS (250 µg/100 g/100 µL) de forma consecutiva en el seno carotídeo circulatoriamente aislado (t = 0). Los datos se expresan como medias aritméticas ± E. E. ** p < 0.01, *** p < 0.001 (t de Student).
mg/dL hasta 303 ± 5 mg/dL en el t = 16 min (p = 0.00024); en la sangre venosa se
observó un aumento desde 110 ± 1 hasta 196 ± 10 mg/dL en el t = 16 (p = 0.0035, t
de Student). Por el contrario, en las ratas diabéticas no se observó dicho
incremento, pero la concentración de glucosa arterial después de la estimulación
anóxica aumentó significativamente desde 410 ± 22 mg/dL hasta 488 ± 18 mg/dL en
el t = 8 min con respecto a sus valores basales (p = 0.0017, t de Student). La
retención de glucosa por el cerebro en las ratas normales, también aumentó
significativamente en todos los tiempos estudiados, desde 16 ± 2 mg/dL hasta 107 ±
4 mg /dL en el t = 16 min (p = 0.0015, t de Student) (n = 5) (tablas 11 y 12, figura 18A y B).

67
Tabla 12. Retención de glucosa cerebral (mg/dL) en el grupo experimental 2.
Tiempo (min)
Ratas normales
(n=5)
Ratas diabéticas
(n=5)
-4
16 ± 2
15 ± 1
4 42 ± 5* 29 ± 7
8 82 ± 8** 39 ± 7*
16 107 ± 4*** 27 ± 7
Se inyectó NaCN (5 µg/100 g/100 µL) y NPS (250 µg/100 g/100 µL) de forma consecutiva en el seno carotídeo circulatoriamente aislado (t = 0). Los datos se expresan como medias aritméticas ± E.E. * p < 0.05, ** p < 0.01, *** p<0.001 (t de Student).
Tabla 13. Concentración de nitritos (nmol/mL) en la sangre venosa cerebral en el grupo experimental 2.
Tiempo (min)
Ratas normales
(n=5)
Ratas diabéticas
(n=5)
-4
0.947 ± 0.304
0.3078 ± 0.083
8 1.850 ± 1.160* 1.2713 ± 0.305**
Se inyectó NaCN (5 µg/100 g/100 µL) y NPS (250 µg/100 g/100 µL) de forma consecutiva en el seno carotídeo circulatoriamente aislado (t = 0). Los datos se expresan como medias aritméticas ± E. E. * p < 0.05, ** p < 0.01 (t de Student).
Con respecto a los niveles de nitritos en la sangre venosa procedente del
cerebro los resultados obtenidos fueron los siguientes: el promedio basal en las ratas
normales fue de 0.947 ± 0.304 nmol/mL, mientras que en el grupo de las ratas
diabéticas fue de 0.3078 ± 0.083 nmol/mL; después de la estimulación RSCC y el
NPS en el SCA los valores aumentaron significativamente hasta 1.850 ± 0.160
nmol/mL (p = 0.035) y 1.213 ± 0.305 nmol/mL (p = 0.023), respectívamente (tabla 13, figura 19A y B).

68
Figura 18. Concentración de glucosa en el plasma arterial y en el venoso, y retención de glucosa cerebral en las ratas normales (A) y diabéticas (B) del grupo experimental 2. La flecha indica el tiempo cero en el cual se inyectó el NaCN (5 µg/100 g/100 µL) y el NPS (250 µg/100 g/100 µL) de forma consecutiva en el seno carotídeo circulatoriamente aislado (SCA). Los valores se expresan en medias aritméticas ± E. E. * p < 0.05, ** p < 0.01, *** p< 0.001 (t de Student).
-4 0 4 8 12 160
40
80
120
180
240
300
A
***
*
***
***
***
**
- NaCN (5 µg/100 g/100 µL)- NPS (250 µg/100 g/100 µL) en el SCA
Ret
enci
ón d
egl
ucos
a (m
g/dL
)
Arterial Venosa
Glu
cem
ia(m
g/dL
)
-4 0 4 8 12 160
40
80
300
360
420
480
540B
Ret
enci
ón d
egl
ucos
a (m
g/dL
)G
luce
mia
(mg/
dL)
Min
*
***

69
Figura 19. Concentración de nitritos en la sangre venosa cerebral en ratas normales (A) y diabéticas (B) del grupo experimental 2. La flecha indica el tiempo cero en el cual se inyectó el NaCN (5 µg/100 g/100 µL) y el NPS (250 µg/100 g/100 µL) de forma consecutiva en el seno carotídeo circulatoriamente aislado (SCA). Los valores se expresan en medias aritméticas ± E. E. * p < 0.05 (t de Student).
Comparación de la retención de glucosa cerebral entre los grupos control 2 y
experimentales 1 y 2.
Al comparar los dos grupos experimentales con el control 2, en el que solo se
produjo una anoxia histotóxica en el SCA (comparación múltiple por ANOVA, con la
prueba de Scheffè), no se observaron diferencias significativas en el t = 4 min (p =
0.073 y 0.197 respectívamente), pero la comparación en los t = 8 min y t = 16 min si
fue significativa (p = 0.09 y p = 0.015 respectivamente). Cuando se aplicó un
inhibidor de la NOS en el SCA, se observó una clara inhibición del efecto de
retención de glucosa cerebral después de la estimulación RSCC solo en las ratas
normales. Por el contrario, la infusión del NPS en el SCA después de la estimulación
con NaCN incrementó el efecto de la retención de glucosa por el cerebro en las ratas
normales anestesiadas, mientras que en las ratas diabéticas anestesiadas los
0,0
0,4
0,8
1,2
1,6
2,0
2,4
2,8
3,2
B
*
80-4Min
0,0
0,4
0,8
1,2
1,6
2,0
2,4
2,8
3,2
A
*
80-4
Con
cent
raci
ón d
e ni
trito
s(n
mol
/mL)
MIn

70
valores se mantuvieron sin cambio (figura 20A y B). La comparación entre los dos
grupos experimentales en las ratas normales dio diferencias significativas en todos
los tiempos estudiados (p = 0.003, p = 0.000 y p = 0.000).
Figura 20. Comparación de las retenciones de glucosa cerebral en las ratas normales (A) y diabéticas (B) después del NaCN (5 µg/100 g/100 µL) en el SCA (C2), o después de la misma estimulación de los RSCC seguida por L-NAME (250 µM/100 µL) (E1), o después de la misma estimulación de los RSCC seguida por NPS (250 µg/100 g/100 µL) (E2). Los valores se expresan como medias aritméticas ± E. E. * + p < 0.05; ** ++ p < 0.01, +++ p < 0.001 (ANOVA); *se comparan las curvas entre E1 y E2 contra C2; + se comparan las curvas E1 y E2 entre sí.
0
20
40
60
80
100
120
++
+++
+++
A
*
*
*
**
1684-4
Ret
enci
ón d
e gl
ucos
a (m
g/dL
)
Min
0
20
40
60
80
100
120
B C2 E1 E2
1684-4Min

71
DISCUSIÓN
En este trabajo se presentan evidencias sobre el efecto del NO en el SCA
modificando el reflejo hiperglucémico con retención de glucosa por la estimulación de
los RSCC in situ en ratas normales y diabéticas. La estimulación de los receptores
seno-cuerpo carotídeo (RSCC) aumentó los niveles de glucosa en las sangres arterial
y venosa en las ratas anestesiadas normales y diabéticas por STZ. Esta maniobra
motivó, también, un incremento en los niveles de nitritos en la sangre venosa
procedente del encéfalo. Cuando se valoró la respuesta a la infusión de un inhibidor
de la NOS (L-NAME) o un donador del NO (NPS) en el SCA, se encontró que el
primero inhibió significativamente los efectos de la estimulación de los RSCC, en
tanto que el donador del NO los incrementó significativamente. Por lo que respecta a
las variables glucémicas, estos efectos o no se observaron, o disminuyeron
significativamente en las ratas diabéticas.
Las ratas diabéticas por STZ constituyen un modelo experimental bien
caracterizado para estudiar la diabetes tipos I y 2, valorando tanto el metabolismo
como las funciones vasculares de los animales (Wu y Meininger, 1995; Ozcelikay,
Tay, Guner, Tasyaran, Yildizoglu-Ari y Altan, 2000). El CC está formado por tejido
neuroepitelial y exhibe algunas características semejantes a los tejidos de las
glándulas endocrinas (Lawson, 1980; Paulding y col., 2002), precisamente, una de las
propiedades de la diabetes mellitus es la disfunción endotelial, que resulta en una
producción deficiente de NO (DeVriese, Verbeuren, Van de Voorde, Lamiere y
Vanhoutte, 2000). Estos hechos indicarían que este radical libre diatómico interviene
en la diabetes insulino-dependiente (Welsh y Sandler, 1994) incrementando la
sensibilidad a la insulina y mejorando, por lo tanto, la homeostasis de la glucosa
(Kohli, Meininger, Hayness, Yan, Self y Wu, 2004).
La presencia de peroxinitritos, formados espontáneamente cuando coexisten el
NO y el superóxido, llevó a suponer que estos compuestos tenían un efecto

72
coadyuvante en la destrucción de las células beta del páncreas durante las
inyecciones de STZ (Yamamoto, Uchigata y Okamoto, 1981). Si bién, en
determinadas condiciones experimentales, el NO se encuentra involucrado en los
efectos de la STZ, cuando se utiliza en una sola dosis, como en este trabajo, este
fármaco no participa de manera directa en la destrucción de las células beta del
páncreas para inducir la diabetes (Flodström, Tyrberg, Eizirik y Sandler, 1999;
Papaccio, Pisanti, Latronico, Ammendola y Galdieri, 2000); pero no se debe descartar
que en un proceso citotóxico como este, sean varias estructuras las que participen en
la destrucción de los islotes de Langerhans. La metodología empleada para obtener
las ratas diabéticas fue adecuada, y como se puede observar en todas las tablas del
presente trabajo, los niveles de glucosa basales, en ayuno, en las ratas diabéticas,
fueron significativamente mayores que los de las ratas normales (p = 0.0029). El
valor promedio de los niveles de glucosa venosa para las ratas diabéticas
anestesiadas fue de 437 ± 16 mg/dL, mientras que en las ratas normales
anestesiadas fue de 114 ± 1 mg/dL. Los valores de la glucemia venosa obtenidos en
las ratas diabéticas de este estudio se encuentran por encima de los reportados en
trabajos anteriores (274 ± 12 mg/dL). Esta diferencia puede deberse a la cepa de
ratas utilizadas y al tiempo que transcurre entre la inyección i.p. de STZ y el desarrollo
experimental en los estudios consultados (ratas Wistar, 7 días) (Rajasekaran,
Sivagnanam y Subramanian, 2005). Los procedimientos quirúrgicos generales
empleados en estos experimentos no motivaron cambios significativos en los niveles
de pO2 pCO2 y pH en la sangre arterial (Montero, 2006), por lo que los efectos del
NaCN en el SCA se deben atribuir a la estimulación de los quimiorreceptores del CC
(Eyzaguirre y Zapata, 1984) ya que la concentración utilizada de NaCN no estimula a
los barorreceptores (Álvarez-Buylla, 1954).
El NaCN provocó una anoxia histotóxica en las células tipo 1 o glómicas del CC,
es decir, simuló un efecto hipóxico, sin disminuir los niveles de O2. A diferencia de
experimentos anteriores in vitro en que se induce una hipoxia directamente en la
solución de perfusión; en estos casos, la disminución de O2 bloquea los canales de K+
sensibles al O2, provocando la despolarización de la membrana de las células del CC

73
para activar a los canales de Ca2+ dependientes de voltaje (aumenta el Ca2+
intracelular de manera directa); el Ca2+q es utilizado para liberar ell transmisor por
exocitosis con la consiguiente excitación de las fibras del NSC (Sato, Ikeda, Yoshizaki
y Koyano, 1991; Stea y Nurse, 1991; Acker, 1994). Normalmente, la hipoxia aguda
inhibe la actividad enzimática, y por el contrario, la hipoxia crónica la estimula,
constituyendo este proceso un mecanismo de defensa homeostático (Schmidt, 2002).
Cabe mencionar que en los experimentos aquí presentados, el animal tiene
respiración asistida, es decir, está ventilado con una pO2 tisular adecuada, por lo que
se piensa que en estas condiciones no entran en juego los mecanismos antes
citados.
El NO y el cianuro comparten vías de acción similares para inhibir la actividad
de la cit-c, y compiten por el O2 para regular la respiración mitocondrial (Brown,
2001). Es posible, entonces, que los mecanismos en los que interviene el NO como
neurotransmisor, participen en la homeostasis energética en forma semejante a lo
sugerido para el O2 (Clementi y col., 1999). En el CC, una vez que el NO difunde
hacia las células del glomus, inhibe a la cit-c y bloquea la CR alterando el potencial
electroquímico mitocondrial para liberar Ca2+ al citoplasma y estimular la migración
de las vesículas que almacenan la dopamina para su exocitosis (López-Barneo y
col,, 2001) (Figura 21). Trabajos anteriores sugieren que existe una interacción NO-
O2 como parte de un mecanismo de inhibición que modula la quimiorrecepción
carotídea. Además de su efecto inhibidor, el NO tiene un efecto dual que depende
de la presión parcial de O2. Durante la hipoxia el NO es un modulador inhibitorio,
mientras que durante la normoxia aumenta la actividad quimiorreceptora (Iturriaga,
2001).
Debido a la alta difusión y degradación del NO, los efectos biológicos que
desencadena este neurotransmisor son difíciles de interpretar, además, las
concentraciones utilizadas en experimentos in vitro varían considerablemente de las
que se pueden encontrar in vivo (Kelm, 1999; Schulz, Kelm y Heusch, 2004) como es
el caso de los experimentos aquí presentados.

74
Figura 21. Esquema hipotético de un modelo de la membrana de las células del CC
sensibles al oxígeno.
Por su capacidad de enlace con los compuestos hemo que compiten por el O2, el
NO puede mimetizar las respuestas de los CC a cambios en la pO2 (Katayama,
Chugh, Mokashi, Ray, Bebout y Lahiri, 1994), y durante la hipoxia crónica la expresión
de la NOS depende del nivel de hipoxia o de glucopenia, así como del tiempo real de
la exposición a dichos niveles de O2 o de glucosa ya sea en la sangre o en el medio
de cultivo (Schmidt, 2002).
La detección de O2 es de vital importancia para la sobrevida celular debido al
papel central del O2 como aceptor de electrones en la CR de la mitocondria, haciendo
posible la síntesis de ATP por medio de la fosforilación oxidativa, por lo que la falta de
O2 participa en forma crítica en la patogénesis de las principales causas de
mortalidad, como el infarto cerebral, el infarto de miocardio o la enfermedad pulmonar
crónica. La hipoxia aguda desencadena rapidamente ajustes contrarreculatorios
respiratorios y cardiovasculares (en la escala de segundos a un min). Respuestas
agudas que dependen de la modulación en los canales iónicos regulados por O2, que
se expresan preferencialmente en células específicas como los quimiorreceptores del
CC. La exposición de los CC a inhibidores del transporte de electrones (NaCN) que
Fibraaferente
Célula delglomus
Dopamina
[Ca ]2+
SNC
Núcleo
Mitocondria
ON

75
impiden la fosforilación oxidativa, aumenta la actividad en el NSC (López-Barneo y
col, 2001). El hecho fundamental para considerar la hipótesis mitocondrial como
primordial en la detección de los cambios de O2 es que la hipoxia reduce la actividad
de la cit-c en la mitocondria, con liberación de Ca2+ y secreción de neurotransmisores
(Chandel y Schumacker, 2000). El cianuro actúa a nivel mitocondrial de manera
semejante al NO, con la diferencia de que la concentración de NO en condiciones
normales se encuentra limitada a las concentraciones de la NOS, mientras que el
cianuro se administra en concentraciones tóxicas para producir un bloqueo total en la
CR que impide la transferencia de electrones y la salida de H+; al irse acumulando los
H+ en la matriz mitocondrial, baja el potencial de membrana mitocondrial y el potencial
electroquímico para iniciar la liberación de Ca2+ evitando su recaptura (Ghafourifar y
col., 2001). Las células glómicas in vitro responden a la hipoxia aumentando la
producción de NO, por un incremento en la producción de NOSe que se activa por la
entrada de Ca2+ (Ye, Tipoe, Fung, Fung, 2002); la presencia del NO en las
mitocondrias sugiere que la regulación hipóxica es a través de esta molécula (Di
Giulio, Grilli, Ciocca, Macri, Daniele, Sabatino, Cacchio, De Lutis, Da Porto, Di Natale
y Felaco, 2000; Yamamoto y col., 2006).
Aunque en las células del glomus carotídeo no se ha demostrado la presencia de
canales sensibles a potasio TASK-semejantes (modulados por ATP), las células 1 del
CC expresan el canal de potasio (KB) con propiedades similares a los canales TASK
(Williams y Buckler, 2004), por lo que su modulación por medio de neurotransmisores
tiene una influencia marcada sobre estas células y abre la posibilidad de que la
señalización del NO esté ligado a estos canales (Varas y col., 2007). Canales
semejantes son los reponsables de la función sensora de las células
quimiorreceptores en el locus coeruleus y en neuronas serotoninérgicas del núcleo
rafé (Washburn, Bayles, Guyenet, 2003).
En el grupo control 1, en el que las ratas normales y/o diabéticas se mantuvieron
en las condiciones experimentales, sin estimular los RSCC, no se observaron
cambios significativos en las variables estudiadas; los niveles de la glucosa y de los

76
nitritos permanecieron estables. Por el contrario, en el grupo control 2, cuando las
ratas recibieron una infusión de sol.sal. en el SCA después de estimular los RSCC
con NaCN, tanto las concentraciones de la glucosa arterial y venosa, como las
diferencias a-v de glucosa cerebral (retención de glucosa por cerebro) se elevaron
significativamente en ambos subgrupos de ratas. Estos resultados, confirmaron la
participación de los RSCC en la homeostasis de la glucosa (Álvarez-Buylla y de
Álvarez-Buylla, 1990; Montero, 1998; Montero y col., 2000; Cadenas, 2003), aunque,
en las ratas diabéticas el efecto sobre la retención de glucosa cerebral fue
significativamente menor y se presentó con mayor latencia. El comportamiento
observado en las ratas diabéticas recapitula un punto toral en el estudio de la
diabetes mellitus insulina-dependiente con el síndrome de resistencia a la insulina
(DeFronzo, 1988); los datos de esta tesis indican que las vías nitroxidérgicas
centrales podrían estar regulando y manteniendo la homeostasis de la glucosa en los
niveles de euglucemia, por lo que las alteraciones en dichas vías participarían en la
etiopatogénesis de ciertas formas de resistencia a la insulina, de manera semejante a
lo reportado por Dallaire y Marette (2002) en el sentido de que el NO es el eslavón
perdido entre la obesidad y el desarrollo de la resistencia a la insulina en la diabetes.
Por consiguiente, se puede considerar que el daño en las vías nitroxidérgicas
aferentes desde el cuerpo carotídeo pudiera estar relacionado con alteraciones en las
vías nitroxidérgicas eferentes, estrechamente vinculadas con alteraciones en el
metabolismo de la glucosa (Shankar y col., 1998). Es probable que el nivel tan alto
de glucosa arterial en la sangre que baña a los CC modifique su estructura,
disminuyendo la sensibilidad a la hipoxia histotóxica que representa la inyección de
NaCN. Aunque el descenso de glucosa activa a la NOSmt (Horn, Wolf, Duffy, Weiss,
Keilhoff y MacVicar, 2002), la hiperglucemia baja los niveles de NO en las
mitocondrias (Brodsky y col., 2002). Estos resultados no están relacionados con el
desarrollo de una neuropatía de las vías nerviosas aferentes y efectoras, ya que la
duración del estado diabético fue de solo 3 o 4 días y son necesarios más de 30 para
que aparezca este daño (Ferlito y Gallina, 1998; Dobretsov, Romanovsky y Stimers,
2007). La estimulación de los RSCC, por sí sola, elevó significativamente los niveles
de nitritos en el plasma de las ratas normales y diabéticas, indicando que el estímulo

77
a los RSCC con NaCN activa la producción del NO, no sólo en condiciones de
normoglucemia (Cadenas, 2005; Montero y col., 2006), sino también en condiciones
de hiperglucemia.
Durante la hipoxia intermitente, la NOSi contribuye para prevenir el daño por
isquemia/reperfusión; es decir, a diferencia de lo que ocurre con la hiperglucemia,
durante la isquemia se produce un aumento compensatorio de los nitritos en el tejido
cerebral (Bolanos y Almeida, 1999). Iturriaga y col. (2000) y otros (Tuzgen, Tanriover,
Uzan, Tureci, Tanriverdi, Gumustas, y Kuday, 2003) encuentran una reducción en la
producción del NO sólo en el inicio de la hipoxia (antes de 1 min). Ante la hipoxia
central y/o periférica en las ratas, se produce la activación de la expresión de genes
NOSn, con aumento de las proteínas NOS en las neuronas periféricas y centrales,
para aumentar la generación del NO (Prabhakar y col., 1996; Shibata, Araki, Hamada,
Sasaki, Shimazu y Fukuuchi, 1996). Es probable que el incremento de los nitritos que
observamos en nuestras ratas normales y diabéticas anestesiadas después de la
estimulación de los RSCC sea indicativo de este efecto homeostático.
Como ya se señaló, la L-arginina representa el substrato fisiológico responsable
de la síntesis del NO catalizada por la NOS (Pieper, 1998), y es interesante observar
que tanto en las ratas como en los humanos diabéticos, los niveles de L-arginina se
encuentran por debajo de los encontrados en los organismos normales (Ardawi, 1995;
Pieper y Dondlinger, 1997). En este trabajo, los niveles de nitritos en la sangre
procedente del SNC fueron siempre menores en las ratas diabéticas que en las ratas
normales en condiciones basales, es decir, antes de producir la hipoxia histotóxica en
el SCA. El descenso en los niveles de los nitritos podría correlacionarse, también,
con el aumento de la distancia de difusión entre la sangre y las mitocondrias que tiene
lugar en la diabetes (Habeck, Huckstorf, Behm, 1988), o con el daño que sufren los
propios CC (Lei y col., 2007). Sin embargo, la explicación más plausible es que la
hiperglucemia y/o la diabetes producen una disminución en la actividad de la NOS y
consecuentemente una disminución del NO, por un aumento de la PKA que provoca

78
la translocación de la proteína de choque térmico necesaria para la actividad de la
NOS (Lei y col., 2007).
Estimulación de los RSCC simultáneamente con la infusión de un inhibidor de la
NOS (L-NAME) en el seno carotídeo circulatoriamente aislado
La infusión de un inhibidor de la NOS como es el L-NAME en el SCA
inmediatamente después de la estimulación de los RSCC (experimental 1) abolió el
reflejo hiperglucemiante observado en las ratas normales anestesiadas después de la
estimulación de los RSCC. Tanto los niveles de glucosa en la sangre como la
retención de glucosa por el cerebro permanecieron sin cambios significativos. Es
interesante observar, que en las ratas diabéticas anestesiadas de este mismo grupo
sí hubo una hiperglucemia arterial significativa (con respecto a sus valores basales)
pero fue menos importante y se presentó con una latencia mayor que lo encontrado
para las ratas sanas. Esta respuesta es controversial y puede explicarse por el
incremento en la expresión de diversas NOS con aumento en los niveles de NO
observado in vitro en los CC de las ratas diabéticas (Bianchi y col., 2003). Es posible
que la dosis de L-NAME, utilizada en este trabajo, fuera insuficiente para inhibir la
gran concentración de la NOS reportada en las ratas diabéticas en el estudio anterior.
Será interesante estudiar en el futuro un grupo de ratas diabéticas, utilizando
concentraciones mayores a 250 µM de L-NAME para abolir el reflejo hiperglucémico,
caracterizando y cuantificando las NOS en los CCs extirpados al final de los
diferentes protocolos experimentales.
El L-NAME en el SCA inhibió el reflejo hiperglucemiante en las ratas normales,
efecto que se puede explicar por la reducción en los niveles de NO a nivel
mitocondrial inducido por el L-NAME (Tong, Wang y Cheng, 1997). Se sabe que el
L-NAME aumenta la amplitud de la respuesta a la hipoxia en las células del glomus
in vitro (Valdés y col., 2003), y no cambia la respuesta secretora de las células del
glomus a la hipoxia (Ortega-Sáenz, García-Fernández, Pardal, Álvarez y López-

79
Barneo, 2003). Es decir, el NaCN necesita de la presencia concomitante del NO
para realizar su efecto tóxico. El bloqueo de la NOS con L-NAME (20 mg/kg, i.p.)
incrementa los niveles plasmáticos de insulina y glucosa arterial, sugiriendo la
participación del NO en la homeostasis de la glucosa en forma independiente a la
secreción de insulina, por un efecto compensador del NO, más prominente en esta
función homeostática (Tong y col., 1997). La relación NO/O2 en la mitocondria debe
jugar un papel fisiológico crucial en la sensibilidad al oxígeno, y en la homeostasis de
la glucosa en las ratas (Tong y col., 1997). Los resultados de esta tesis en relación
con los efectos del L-NAME en el CC difieren de los encontrados por Buerk y Lahiri
(2000) en preparaciones in vitro, donde no encuentran hiperglucemia cuando
aumentan las descargas en el NSC después de la perfusión con L-NAME en
condiciones de hipoxia; es posible que en estas condiciones, la falta de O2 (20%)
produzca alteraciones en la homeostasis celular. En cambio, en los experimentos
realizados in vivo, en condiciones fisiológicas, y con una pO2 normal, el NaCN
generador de la hipoxia histotóxica, inhibió la CR mitocondrial mediante un bloqueo
selectivo de la citocromo c oxidasa (cit-c) (Leavesley y col., 2007).
Los nitritos en la sangre venosa cerebral disminuyeron en ambos subgrupos de
ratas, indicando que el L-NAME inhibe la producción del NO, tanto durante la
normoglucemia como durante la hiperglucemia, como se ha reportado en trabajos
previos de este laboratorio y otros (Valdés y col., 2003; Cadenas y col., 2003; Montero
y col., 2006).
Estimulación de los RSCC simultáneamente con la infusión de un donador del
NO (NPS) en el seno carotídeo circulatoriamente aislado
En la segunda serie experimental, se hizo la estimulación de los RSCC
acompañada de una infusión de un donador de NO (NPS) en el SCA de ratas
normales y diabéticas. Se observó una respuesta del reflejo hiperglucemiante mucho
más robusta con respecto a lo observado en las ratas normales con estimulación de

80
los RSCC sin el donador; es decir, las concentraciones de glucosa arterial fueron
mayores en el primer caso que en el segundo. El NO aumentó la actividad
quimiorreceptora ante un estímulo hipóxico a los RSCC reforzando el reflejo
hiperglucemiante con aumento en la retención de glucosa cerebral en las ratas
normales. Es posible que las concentraciones de NO liberado por el NPS alteren la
cadena de transporte de electrones y la fosforilación oxidativa en las mitocondrias de
las células del glomus (Iturriaga y col, 2000). Este aumento en la respuesta al NO
podría deberse al aumento de Ca2+ intracelular a través de la interacción del NO
sobre la GC soluble y el GMPc para activar a las fosfolipasas, y la formación de
bifosfato de inositol (IP2) y trifosfato de inositol (IP3) con la consiguiente liberación de
Ca2+ desde el retículo endoplásmico (Koesling y Friebe, 2000), para favorecer la
exocitosis y la liberación de dopamina u otros neurotransmisores de las células
glómicas (Horn y col., 2002). Otra explicación posible para el efecto sinérgico del NO
con el cianuro, observado unicamente en las ratas normales, sería el bloqueo de la
cit-c por el NO en la cadena CR (Leist, Single, Naumann, Fava, Simon, Kuhnle y
Nicotera, 1999), con el desarrollo de un déficit de ATP (Brookes, Bolanos y Heales,
1999), sufrimiento metabólico de las células y su despolarización. Como hemos visto,
el aumento en la concentración de Ca2+ intracelular para la liberación del
neurotransmisor puede provenir del exterior a través del NO aportado por el NPS, en
respuesta a la despolarización por el estímulo anóxico, y de las mitocondrias. Es
probable que el bloqueo de la CR provoque un predominio de la glucólisis anaeróbica
con la producción de ácido láctico, acidosis y el cierre de los canales de K+ (Buckler y
Vaughan-Jones, 1994). Aunque se conoce el papel del NaCN para inhibir
selectivamente la actividad de la cit-c (Ikegaya, Iwase, Hatanaka, Sakurada, Yoshida
y Takatori, 2001), su papel exacto en la quimiotransducción sensorial y
particularmente en la homeostasis de la glucosa estaría por definirse. No obstante, la
presencia necesaria del NO en el CC para que los quimiorreceptores respondan al
estímulo anóxico del cianuro en las ratas normales, en las ratas diabéticas este efecto
se enmascara por la hiperglucemia con un probable aumento en la producción de
ATP que compensa el sufrimiento metabólico durante la anoxia y disminuye la
actividad quimiorreceptora. Los resultados observados en los niveles de nitritos en el

81
plasma venoso cerebral después del NaCN y el NPS en el SCA en las ratas normales
y diabéticas, sugieren que el efecto del NO en las ratas normales es sinérgico al
efecto anóxico sobre la captación de glucosa por el cerebro.
Los resultados encontrados in vivo difieren con lo observado in vitro por otros
investigadores utilizando como donador del NO a la S-nitro-N-acetilpenicilamina
(SNAP) (Chugh y col, 1994; Wang y col., 1994; Alcayaga y col., 1997). El SNAP
redujo la descarga basal quimiosensora en el CC de gatos perfundidos in vitro
durante la hipoxia. La diferencia más importante entre estos dos tipos de
experimentos sería la presencia, en condiciones normales de oxigenación, de los
vasos sanguíneos y el tejido de músculo liso, que participan en la irrigación de los
tejidos con la aportación de hormonas, metabolitos, y nutrientes diversos, que
modifican las respuestas en el caso de los experimentos in vivo (Batrakova, Miller, Li,
Alakhov, Kabanov y Elmquist, 2001). Durante la normoxia la inyección de donadores
del NO incrementa la descarga quimiosensora en relación a la dosis, mostrando un
efecto dual del NO en el CC dependiente de los niveles de la pO2 (Iturriaga,
Villanueva, Mosqueira, 2000). El NPS en el seno carotídeo después de la
estimulación de los RSCC produjo incrementos en la glucemia arterial y en la
captación de glucosa por el cerebro mayores que los incrementos encontrados
únicamente con la estimulación de los RSCC tanto en las ratas normales como en
las diabéticas (control 2) (p = 0.015, t = 8 min). En trabajos anteriores de este
laboratorio el NPS infundido en la cisterna magna sin estimulación de los RSCC
produjo, también, indujo un incremento en la captación de glucosa cerebral
(Cadenas, 2003; Montero y col., 2006).
En las ratas diabéticas el NPS no reforzó el reflejo hiperglucemiante con
aumento en la retención de glucosa inducido por el NaCN, indicando, como ya se
dijo, que en condiciones de hiperglucemia la inhibición de la NOS disminuye la
síntesis de NO (Brodsky y col., 2002). La inhibición de la NOS por la hiperglucemia,
evita el desarrollo de la hipertrofia de los CC inducida por la anoxia (Lahiri, Mokashi,
Shirahata, Andronikou, 1990; Prabhakar y col., 2001). No debemos descartar el

82
efecto del NO sobre el aumento en la retención de glucosa cerebral a través de la
relajación vascular, que causaría un aumento en el flujo sanguíneo cerebral (Bolanos
y Almeida, 1999); si bién, este mecanismo no es tan claro en el cerebro como en el
músculo esquelético (Higaki y col., 2001). Con objeto de discriminar los efectos
sobre la musculatura lisa vascular, en experimentos futuros será interesante estudiar,
el reflejo hiperglucémico utilizando un inhibidor específico de la NOSn.

83
CONCLUSIONES
En base a los resultados obtenidos en las ratas normales y diabéticas en las que
se valoró el efecto del NO en el CC, inyectando donadores o inhibidores de este
neurotransmisor en el SCA, sobre el reflejo hiperglucemiante con retención de
glucosa cerebral después de la estimulación de los RSCC, se concluye lo siguiente:
1. La inyección de sol. sal. en el SCA sin la estimulación de los RSCC en las
ratas normales y diabéticas no produjo el reflejo hiperglucemiante ni aumentó
la retención de glucosa por el cerebro, tampoco se observaron modificaciones
en los niveles de nitritos.
2. La estimulación de los RSCC seguida de la inyección de sol. sal. en el SCA
produjo el reflejo hiperglucemiante con aumento en la retención de glucosa
cerebral y un incremento en los niveles de nitritos en la sangre venosa
encefálica, tanto en las ratas normales como en las diabéticas. Sin embargo,
el aumento en la retención de glucosa cerebral en las ratas diabéticas sólo se
observó en el t = 16 min.
3. La estimulación de los RSCC seguida de la inyección de L-NAME en el SCA
inhibió el reflejo hiperglucemiante y el aumento en la retención de glucosa
cerebral, los niveles de nitritos en la sangre venosa encefálica en las ratas
normales disminuyeron. En las ratas diabéticas el L-NAME produjo los
mismos efectos que en las ratas normales sin afectar el reflejo
hiperglucemiante que permaneció sin cambios..
4. La estimulación de los RSCC seguida de la inyección de NPS en el SCA
incrementó el reflejo hiperglucemiante con aumento en la retención de glucosa
cerebral sólo en las ratas normales. En las ratas diabéticas el NPS no reforzó
el reflejo hiperglucemiante ni el aumento en la retención de glucosa cerebra,

84
que sólo se observó en el t = 8 min. En ambas ratas se encontró un aumento
en los niveles de nitritos en la sangre venosa encefálica.
5. Los niveles de nitritos en la sangre procedente del SNC fueron siempre
menores en las ratas diabéticas que en las ratas normales, aún en
condiciones basales, es decir, antes de inducir la hipoxia histotóxica en el
SCA.
Estos resultados aportan evidencias en relación al estudio de la
quimiotransducción y la homeostasis de la glucosa. Es probable que la CR
mitocondrial tenga un papel central en este proceso ante el bloqueo selectivo de la cit-
c por el NO y el NaCN.

85
PERSPECTIVAS
Es cada vez más evidente que el NO sintetizado en el SNC juega papeles
fisiológicos importantes en la homeostasis de la glucosa. A la luz de la relación que
existe entre las concentraciones de glucosa en la sangre, la actividad
quimiorreceptora y los niveles de NO, se podrá ampliar el conocimiento sobre el
mecanismo por medio del cual el SNC es capaz de captar la glucosa para mantener
su metabolismo. El hecho de que la mitocondria tenga NO para regular su propia
respiración (Ghafourifar y Richter, 1997) sugiere que esta molécula debe ser
importante en el metabolismo energético. En este sentido se propone un modelo
hipotético de la quimiotransducción. En próximos trabajos, será importante estudiar:
a) si en la sangre procedente del cerebro, después de reforzar el reflejo
hiperglucemiante con NPS, se detecta la presencia de insulina en ratas normales y
diabéticas tipos 1 y 2; b) si el cociente mitocondrial NO/O2 es o no crucial para regular
la sensibilidad al oxígeno o a la glucosa en la propia función mitocondrial, y
profundizar en el estudio de estos mecanismos para entender la participación del NO
mitocondrial en la homeostasis energética, en particular del SNC en ratas normales y
diabéticas; c) estudiar el tejido de los CCs de las ratas normales y/o diabéticas
después de las manipulaciones aquí descritas, para analizar la expresión de la
NOSmt; d) será interesante estudiar un grupo de ratas diabéticas sometidas a los
protocolos aquí presentados, perfundiendo el SCA con concentraciones de L-NAME
mayores a 250 µM para abolir el reflejo hiperglucémico, caracterizando y
cuantificando las NOS a través de la técnica de reacción en cadena de la polimerasa
con retrotranscripción (RT-PCR) en los CCs extirpados al finalizar los experimentos.
En el futuro, la disponibilidad de herramientas bioquímicas específicas para
determinar los niveles del NO y su participación en el metabolismo de los mamíferos
in vivo, nos permitirá desarrollar estrategias terapéuticas más adecuadas.

86
REFERENCIAS
● Acker H. Cellular oxygen sensor. Ann N Y Akd Sci. 1994; 718:3-10.
● Acker H, Starlinger H. Adenosine triphosphate content in the cat carotid body under different arterial O2 and CO2 conditions. Neurosci Lett. 1984;
50:175-179.
● Adachi A, Kobashi M, Funahashi M. Glucose-responsive neurons in the brainstem. Obes Res. 1995; 5:735S-740S.
● Alcayaga J, Iturriaga R, Ramirez J, Readi R, Quezada C, and Salinas S. Cat carotid body chemosensory response to non-hypoxic stimuli is inhibited by sodium nitroprusside both in situ and in vitro. Brain Res. 1997;
767:384–387.
● Almeida A, Cidad P, Bolaños JP. Nitric oxide accounts for an increased glycolytic rate in activated astrocytes through a glycogenolysis-independent mechanism. Brain Res. 2002; 945:131-134.
● Almeida A, Cidad P, Delgado-Esteban M, Fernández E, García-Nogales P,
Bolaños JP. Inhibition of mitochondrial respiration by nitric oxide: its role in glucose metabolism and neuroprotection. J Neurosci Res. 2005; 79:166-
171.
● Álvarez-Buylla, R. Actividad quimiorreceptora del glomus carotídeo. Arch
Inst Cardiol. 1951; XXI:724-739.
● Álvarez-Buylla, R. Estudio de la actividad quimiorreceptora del seno carotídeo. Ciencia. 1952; XII: 129-140.
● Álvarez-Buylla, R. Disociación de las actividades quimiorreceptoras y barorreceptoras en gatos. Arch Inst Nac Card. 1954; 24:6-37.
● Álvarez-Buylla, R. Importancia fisiológica de las zonas refelxigénicas del seno carotídeo. Gac. Med. Mex. 1960; 8:671-677.
● Álvarez-Buylla R. Efecto reflejo de la insulina y regulación de la glucemia. Ciencia. 1973; 28:143-148.

87
● Älvarez-Buylla R. Glucose influence on the carotid sinus chemoreceptor function. En: Arterial Chemoreceptors. Leicester University Press. 1981; 327-
333.
● Álvarez-Buylla R, Álvarez-Buylla E, Mendoza H, Montero SA, Álvarez-Buylla A.
Pituitary and adrenals are required for hyperglycemic reflex initiated by stimulation of CBR with cyanide. Am J Physiol. 1997; 272:R392-399.
● Álvarez-Buylla R, Bencosme SA. Reflex hypoglycemia initiated by insulin. Acta Physiol Lat Amer. 1981; 31:5-11.
● Álvarez-Buylla R, Carrasco-Zanini J. A conditioned reflex which reproduces the hypoglycemic effect of insulin. Acta Physiol Lat Am. 1960; 10:153-158.
● Álvarez-Buylla R, de Álvarez-Buylla ER. Hypoglycemic conditioned reflex in rats: preliminary study of its mechanism. J Comparative and Physiol
Psychol. 1975; 88:155-160.
● Álvarez-Buylla R, de Álvarez-Buylla ER. Carotid sinus receptors participate in glucose homeostasis. Respir Physiol. 1988; 72:347-359.
● Álvarez-Buylla R, de Álvarez-Buylla ER. Carotid sinus receptors participate in glucose homeostasis. En: Arterial Chemoreceptors. Springer Verlag. 1990
● Álvarez-Buylla R, Huberman A, Montero S, Lemus M, Valles V, de Álvarez-
Buylla ER. Induction of brain glucose uptake by a factor secreted into cerebrospinal fluid. Brain Res. 2003; 994:124-133.
● Álvarez-Buylla R, Quintanar-StephaON A, Quintanar-StephaON JL, Álvarez-
Buylla de ER. Removal of the unfragmented pituitary gland (hypophysectomy) in the rat. Bol Inst Invest Bioméd. 1991; 39:33-38.
● Álvarez-Buylla R, Roces de Álvarez-Buylla E. Changes in blood glucose concentration in the carotid body-sinus modify brain glucose retention. Brain Res. 1994; 654:167-170.
● Álvarez-Buylla R, Rojas M, de Álvarez-Buylla ER, Faria N. Effects of intracisternal glucose or insulin injections on glucose homeostasis in cat. Diabetes. 1986; 35:826-831.
● Álvarez-Buylla R, Segura ET, de Álvarez-Buylla ER. A study of the afferent path of the hypoglycemic reflex of insulin. Acta Physiol Lat Am. 1961a;

88
11:43-50.
● Álvarez-Buylla R, Segura ET, de Álvarez-Buylla ER. Participation of the hipophysis in the conditioned reflex which reproduces the hypoglycemic effect of insulin. Acta Physiol Lat Am. 1961b; 11:113-119.
● Ardawi MS. Diabetes mellitus. En: Amino Acid Metabolism and Therapy in
Health and Nutricional Disease. Cynober, L.A. ed. CRC Press. 1995: pp. 287-
298.
● Arnold WP, Aldred R, Murad F. Cigarette smoke activates guanylate cyclase and increases guanosine 3', 5'-monophosphate in tissues. Science. 1977;
198:934-936.
● Auer RN. Progress review: hypoglycemic brain damage. Stroke. 1986;
17:699-708.
● Balon TW y Nadler JL. Evidence that nitric oxide increase glucose transport in skeletal muscle. J Appl Physiol. 1997; 82:359-363.
● Baskin DG, Figlewicz DP, Woods SC, Porte D Jr, Dorsa DM. Insulin in the brain. Annu Rev Physiol. 1987; 49:335-347.
● Batrakova EV, Miller DW, Li S, Alakhov VY, Kabanov AV, Elmquist WF.
Pluronic P85 enhances the delivery of digoxin to the brain: in vitro and in vivo studies. J Pharmacol Exp Ther. 2001; 296:551-7.
● Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. 1990;
87:1620-1624.
● Bedoya FJ, Solano F, Lucas M. N-monomethyl-arginine and nicotinamide prevent streptozotocin-induced double strand DNA break formation in pancreatic rat islets. Experientia. 1996; 52:344-347.
● Behrooz A, Ismail-Beigi F. Stimulation of Glucose Transport by Hypoxia: Signals and Mechanisms. News Physiol Sci. 1999; 14:105-110.
● Beitner-Johnson D, Leibold J, Millhorn DE. Hypoxia regulates the cAMP- and Ca2+/calmodulin signaling systems in PC12 cells. Biochem Biophys Res
Commun. 1998; 242:61-66.

89
● Benzo CA. The hypothalamus and blood glucose regulation. Life Sci.
1982; 32:2509-2515.
● Bernard C. Lecons Sur le Systéme Nerveux. París. 1857. ● Bianchi G, Cacchio M, Artese L, Ferrero G, Rapino C, Grilli A, Felaco M, Di
Giulio C. Carotid body nitric oxide activity in spontaneously diabetic BB rat. Adv Exp Med Biol. 2003; 536:359-366.
● Bolanos JP, Almeida A. Roles of nitric oxide in brain hypoxia-ischemia. Biochim Biophys Acta. 1999; 1411:415-436.
● Boucher J. L, Moali C, Tenu J. P. Nitric oxide biosynthesis, nitric oxide synthase inhibitors and arginase competition for L-arginine utilization. Cell Mol Life Sci. 1999; 55:1015-1028.
● Bradley SJ, Kingwell BA, McConell GK. Nitric oxide synthase inhibition reduces leg glucose uptake but not blood flow during dynamic exercise in humans. Diabetes. 1999; 48:1815-1821.
● Brodsky SV, Gao S, Li H, Goligorsky MS. Hyperglycemic switch from mitochondrial nitric oxide to superoxide production in endothelial cells. Amer J Physiol. 2002; 283:H2130-H2139.
● Brookes, PS, Bolanos JP, Heales SJ. The assumption that nitric oxide inhibits mitochondrial ATP synthesis is correct. FEBS Lett. 1999; 446:261-
263.
● Brown GC. Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim Biophys Acta. 2001; 1504:46-
57.
● Brown GC, Cooper CE. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994; 356:295-298.
● Buckler KJ, Vaughan-Jones RD. Role of intracellular pH and [Ca2+]i in acid chemoreception in type-1 cells of the carotid body. En: Arterial
Chemoreception: Cell to System. Plenum Press, New Cork. 1994: pp. 41-55,
● Buerk DG, Lahiri S. Evidence that nitric oxide plays a role in O2 sensing from tissue NO and pO2 measurements in cat carotid body. Adv Exp Med

90
Biol. 2000; 475:337-347.
● Bunn HF, Poyton RO. Oxygen sensing and molecular adaptation to hypoxia. Physiol Rev. 1996; 76:839-885.
● Burcelin R, Dolci W, Thorens B. Glucose sensing by the hepatoportal sensor is GLUT2-dependent: in vivo analysis in GLUT2-null mice. Diabetes.
2000; 49:1643-1648.
● Cadenas, JL. (2003). Papel del óxido nítrico en la retención de glucosa cerebral post-estimulación de los receptores carotídeos con cianuro de sodio en ratas. Tesis de Maestría, CUIB, Universidad de Colima.
● Cadenas JL. (2006). Captación de glucosa cerebral post-estimulación de los receptores carotídeos con NaCN en ratas: participación del óxido nítrico. Tesis de Doctorado, CUIB, Universidad de Colima.
● Calder NA, Kumar P, Hanson MA. Development of carotid chemoreceptor dynamic and steady-state sensitivity to CO2 in the newborn lamb. J
Physiol. 1997; 503:187-194.
● Cane P, Artal R, Bergman RN. Putative hypothalamic glucoreceptors play NO essential role in the response to moderate hypoglycemia. Diabetes.
1986; 35:268-77.
● Cannon WB, Newton HF, Bright EM, Menkin V, Moore RM. Some aspects of the physiology of animals surviving complete exclusion of sympathetic nerve impulses. Am J Physiol. 1929; 79: 466-480.
● Chandel NS, Schumacker PT. Cellular oxygen sensing by mitochondria: old questions, new insight. J Appl Physiol. 2000; 88:1880-1889.
● Cheng CM, Reinhardt RR, Lee WH, Joncas G, Patel SC, Bondy CA. Insulin-like growth factor 1 regulates developing brain glucose metabolism. Proc
Natl Acad Sci U S A. 2000; 97:10236-41.
● Chugh DK, Katayama M, Mokashi A, Bebout DE, Ray DK, Lahiri S. Nitric oxide-related inhibition of carotid chemosensory nerve activity in the cat. Respir Physiol. 1994; 97:147–156.
● Ciriello J, Hrycyshyn AW, Calaresu FR. Glossopharyngeal and vagal afferent projections to the brain stem of the cat: a horseradish peroxidase study. J

91
Auton Nerv Syst. 1981; 4:63-79.
● Clarke D, Sokoloff. Circulation and Energy Metabolims of the Brain en: Basic Neurochimestry Mollecular. En: Cellular and Medical Aspecs. 6ta ed.
Lippin cott/raven 1999: pp. 237-669.
● Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH.
Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett. 1994; 345:50-54.
● Clementi E, Brown GC, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci U S A.
1998; 95:7631-636.
● Coker GT 3rd, Studelska D, Harmon S, Burke W, O'Malley KL. Analysis of tyrosine hydroxylase and insulin transcripts in human neuroendocrine tissues. Brain Res Mol Brain Res. 1990; 8:93-98.
● Cottle MK. The generation studies of primary afferents of IX and cranial nerves in the cat. 1964; 122:329-343.
● Pilon G, Dallaire P, Marette A. Inhibition of inducible nitric-oxide synthase by activators of AMP-activated protein kinase: a new mechanism of action of insulin-sensitizing drugs. J Biol Chem. 2004; 279:20767-74.
● Dallaire P, Marette A. Obesity-linked Insulin Resistance: Is Nitric Oxide the Missing Link ?. Canadian J of Diabetes. 2004; 28:59-66.
● Daly MB, Ward J, Wood LM. Modification by lung inflation of the vascular responses from the carotid body chemoreceptors and other receptors in dogs. J Physiol. 1986; 378:13-30.
● Davidson NS, Goldner S, McCloskey DI. Respiratory modulation of barareceptor and chemoreceptor reflexes affecting heart rate and cardiac vagal efferent nerve activity. J Physiol. 1976; 259:523-530.
● De Castro F. Sur la structure et l'innervation du sinus carotidien de l'homme et des mammiféres. nouveaux faits sur l'innervation et la fonction du glomus caroticum. Estudes anatomiques et physiologiques. Trab Lab

92
Invest Biol. Univ. Madrid. 1928; 25:331-380.
● DeFronzo RA. Lilly lecture 1987. The triumvirate: beta-cell, muscle, liver. A collusion responsible for NIDDM. Diabetes. 1988; 37:667-687.
● Denicola A, Souza JM, Radi R, Lissi E. Nitric oxide diffusion in membranes determined by fluorescence quenching. Arch Biochem Biophys. 1996;
328:208-212.
● De Vriese AS, Verbeuren TJ, Van de Voorde J, Lamiere HN, Vanhoutte PM.
Endothelial dysfunction in diabetes mellitus. Br. J. Pharmacol. 2000;
130:963-974.
● Dias AS, Porawski M, Alonso M, Marroni N, Collado PS, Gonzalez-Gallego J.
Quercetin decreases oxidative stress, NF-kappaB activation, and iNOS overexpression in liver of streptozotocin-induced diabetic rats. J Nutr.
2005; 135:2299-2304.
● Dienel GA, Cruz NF. Astrocyte activation in working brain: energy supplied by minor substrates. Neurochem Int. 2006; 48:586-595.
● Di Giulio C, Grilli A, Ciocca I, Macri MA, Daniele F, Sabatino G, Cacchio M, De
Lutiis MA, Da Porto R, Di Natale F, Felaco M. Carotid body NO-CO interaction and chronic hypoxia. Adv Exp Med Biol. 2000; 475:685-690.
● Di Giulio C, Grilli A, De Lutiis MA, Di Natale F, Sabatino G, Felaco M. Does chronic hypoxia increase rat carotid body nitric oxide ?. Comp Biochem
Physiol A Mol Integr Physiol. 1998; 120:243-247.
● Dobretsov M, Romanovsky D, Stimers J. Early diabetic neuropathy: Triggers and mechanisms. World J Gastroenterol. 2007; 13:175-191.
● Donovan CM, Halter JB, Bergman RN. Importance of hepatic glucoreceptors in sympathoadrenal response to hypoglycemia. Diabetes. 1991; 40:155-
158.
● Elsner M, Guldbakke B, Tiedge M, Munday R, Lenzen S. Relative importance of transport and alkylation for pancreatic beta-cell toxicity of streptozotocin. Diabetologia. 2000; 43:1528-1533.
● Etgen GJ Jr, Fryburg DA, Gibbs EM. Nitric oxide stimulates skeletal muscle glucose transport through a calcium/contraction and phosphatidylinositol-

93
3 kinase-independent pathway. Diabetes. 1997; 46:1915-1919.
● Eyzaguirre C, Zapata P. Perspectives in carotid body research. J Appl
Physiol. 1984; 57:931-957.
● Fauci AS, Braunwald E, Isselbacher KJ. Diabetes mellitus. En: Principios de
Medicina Interna. 2 nd ed. Mc. Graw-Hill. Interamericana. 1998: pp. 2341-2365.
● Felaco M, Grilli A, De Lutiis MA, Patruno A, Libertini N, Taccardi AA, Di Napoli
P, Di Giulio C, Barbacane R, Conti P. Endothelial nitric oxide synthase (eNOS) expression and localization in healthy and diabetic rat hearts. Ann
Clin Lab Sci. 2001; 31:179-186.
● Fernández-Álvarez A, Abudara V, Morales FR. El Óxido Nítrico como Neurotransmisor y Neuromodulador. Act de Fisiol. 1999; 5:39-77.
● Ferlito, S, Gallina M. Nitrite plasma levels in type 1 and 2 diabetics with and without complications. Panminerva Med. 1998; 40:304-308.
● Finley JC, Katz DM. The central organization of carotid body afferent projections to the brainstem of the rat. Brain Res. 1992; 572:108-116.
● Fitzgerald RS, Shirahata M, Wang HY. Acetylcholine is released from in vitro cat carotid bodies during hypoxic stimulation. Adv Exp Med Biol.
2000; 475:485-494.
● Fitzgerald RS, Shirahata M, Chang I, Balbir A. Modulators of cat carotid body chemotransduction. Adv Exp Med Biol. 2006; 580:307-311.
● Flodström M, Tyrberg B, Eizirik DL, Sandler S. Reduced sensitivity of inducible nitric oxide synthase-deficient mice to multiple low-dose streptozotocin-induced diabetes. Diabetes. 1999; 48:706-713.
● Fray AE, Forsyth RJ, Boutelle MG, Fillenz M. The mechanisms controlling physiologically stimulated changes in rat brain glucose and lactate: a microdialysis study. J Physiol. 1996; 496:49-57.
● Frizzell RT, Jones EM, Davis SN, Biggers DW, Myers SR, Connolly CC, Neal
DW, Jaspan JB, Cherrington AD. Counterregulation during hypoglycemia is directed by widespread brain regions. Diabetes. 1993; 42:1253-1261.
● Frohman LA. CNS peptides and glucoregulation. Annu Rev Physiol. 1983;
45:95-107.

94
● Ganda OP, Rossini AA, Like AA. Studies on streptozotocin diabetes. Diabetes. 1976; 25:595-603.
● Garcia MA, Millan C, Balmaceda-Aguilera C, Castro T, Pastor P, Montecinos H,
Reinicke K, Zuniga F, Vera JC, Onate SA, Nualart F. Hypothalamic ependymal-glial cells express the glucose transporter GLUT2, a protein involved in glucose sensing. J Neurochem. 2003; 86:709-24.
● Garvey WT, Revers RR, Kolterman OG, Rubenstein AH, Olefsky JM.
Modulation of insulin secretion by insulin and glucose in type II diabetes mellitus. J Clin Endocrinol Metab. 1985; 60:559-568.
● Ghafourifar P, Bringold U, Klein SD, Richter C. Mitochondrial nitric oxide synthase, oxidative stress and apoptosis. Biol Signals Recept. 2001; 10:57-
65.
● Ghafourifar P, Cadenas E. Mitochondrial nitric oxide synthase. TRENDS in
Pharmacol Sci. 2005; 26:190-195.
● Ghafourifar P, Richter C. Nitric oxide synthase activity in mitochondria. FEBS Lett. 1997; 418:291-6.
● Gibbons RD, Lavigne JV. Emergence of childhood psychiatric disorders: a multivariate probit analysis. Stat Med. 1998; 17:2487-2499.
● Gillespie JS, Sheng H. A comparison of haemoglobin and erythrocytes as inhibitors of smooth muscle relaxation by the NANC transmitter in the BRP and rat anococcygeus and by EDRF in the rabbit aortic strip. Br J
Pharmacol. 1989; 98:445-450.
● Giulivi C, Poderoso JJ, Boveris A. Production of nitric oxide by mitochondria. J Biol Chem. 1998; 273:11038-11043.
● Goldberg ND, O'Toole AG. The properties of glycogen synthetase and regulation of glycogen biosynthesis in rat brain. J Biol Chem. 1969;
244:3053-3061.
● Goldraij A, Álvarez-Buylla R. Eosinopenic response to glucose. Acta Physiol
Lat Am. 1971; 21:40-47.
● González C, Almaraz L, Obeso A, Rigual R. Carotid body chemoreceptors: from natural stimulai to sensory discharges. Physiol Rev. 1994; 74:829-

95
898.
● Gould GW, Holman GD. The glucose transporter family: structure, function and tissue-specific expression. Biochem J. 1993; 295:329-341.
● Gozal D, Torres JE, Gozal YM, Littwin SM. Effect of nitric oxide synthase inhibition on cardiorespiratory responses in the conscious rat. J Appl
Physiol. 1996; 81:2068-2077.
● Granger DL, Taintor RR, Boockvar KS, Hibbs JB. Determination of nitrate and nitrite in biological samples using bacterial nitrate reductase coupled with the Griess reaction. Methods Enzymol. 1996; 7:78-85.
● Grosche J, Matyash V, Moller T, Verkhratsky A, Reichenbach A, Kettenmann H.
Microdomains for neuron-glia interaction: parallel fiber signaling to Bergmann glial cells. Nat Neurosci. 1999; 2:139-143.
● Guarner V, Álvarez-Buylla R. Changes in brain glucose retention produced by the stimulation of an insulin-sensitive reflexogenic zone in rats. J Auton
Nerv Syst. 1991; 34:89-94.
● Guide for Care and Use of Laboratory Animals. Nat. Res. Concil. 1996, Ed
Nat. Acad Press.
● Habeck JO, Huckstorf C, Behm R. The paraganglia within the carotid bifurcation regions of young and old spontaneously hypertensive rats (SHR) after exposure to chronic hypobaric hypoxia. I. The carotid bodies. Anat Anz. 1988; 165:45-54.
● Hasselbalch SG, Knudsen GM, Videbaek C, Pinborg LH, Schmidt JF, Holm S,
Paulson OB. NO effect of insulin on glucose blood-brain barrier transport and cerebral metabolism in humans. Diabetes. 1999; 48:1915-1921.
● Hatakeyama H, Kishimoto T, Nemoto T, Kasai H, Takahashi N. Rapid glucose sensing by protein kinase A for insulin exocytosis in mouse pancreatic islets. J Physiol. 2006; 570:271-282.
● Hawkins RA. Brain energy metabolism and function. Eur J Clin Invest.
1989; 19:313-314.
● Heimberg H, De Vos A, Moens K, Quartier E, Bouwens L, Pipeleers D, Van
Schaftingen E, Madsen O, Schuit F. The glucose sensor protein glucokinase

96
is expressed in glucagon-producing alpha-cells. Proc Natl Acad Sci U S A.
1996; 93:7036-7041.
● Hevener AL, Bergman RN, Donovan CM. Portal vein afferents are critical for the sympathoadrenal response to hypoglycemia. Diabetes. 2000; 49:8-12.
● Higaki Y, Hirshman MF, Fujii, N, Goodyear LJ. Nitric oxide increases glucose uptake through a mechanism that is distinct from the insulin and contraction pathways in rat skeletal muscle. Diabetes. 2001; 50:241-247.
● Hohler B, Mayer B, Kummer W. Nitric oxide synthase in the rat carotid body and carotid sinus. Cell Tissue Res. 1994; 276:559-564.
● Horn TF, Wolf G, Duffy S, Weiss S, Keilhoff G, MacVicar B. Nitric oxide promotes intracellular calcium release from mitochondria in striatal neurons. FASEB J. 2002; 161611-161622.
● Housley GD, Martin-Body RL, Dawson NJ, Sinclair JD. Brain stem projections of the glossopharyngeal nerve and its carotid sinus branch in the rat. Neurosci. 1987; 22:237-250.
● Housley GD, Sinclair JD. Localization by kainic acid lesions of neurones transmitting the carotid chemoreceptor stimulus for respiration in rat. J
Physiol. 1988; 406:99-114.
● Ibberson M, Uldry M, Thorens B. GLUTX1, a novel mammalian glucose transporter expressed in the central nervous system and insulin-sensitive tissues. J Biol Chem. 2000; 275:4607-4612.
● Ikeyaga H, Iwase H, Hatanaka K, Sakurada K, Yoshida K, Takatori T.
Diagnosis of cyanide intoxication by measurement of cytochrome c oxidase activity. Toxicol Lett. 2001; 119:117-123.
● Iturriaga R. Nitric oxide and carotid body chemoreception. Biol. Res. 2001;
34: 135-139.
● Iturriaga R, Alcayaga J, Rey S. Sodium nitroprusside blocks the cat carotid chemosensory inhibition induced by dopamine, but not that by hyperoxia. Brain Res. 1998; 799:26–34.
● Iturriaga R, Mosqueira M y Villanueva S. Effects of nitric oxide gas on cat carotid body chemosensory response to hypoxia. Brain Res. 2000; 855:

97
282-286.
● Iturriaga R, Villanueva S, Mosqueira M. Dual effects of nitric oxide on cat carotid body chemoreception. J Appl Physiol. 2000; 89:1005-1012.
● Jordan D, Spyer KM. Brain integration of cardiovascular and pulmonary afferent activity. En: Progress in Brain Research. Ed. Elsevier Science
Publishers. 1986; 67:295-314.
● Kacem K, Lacombe P, Seylaz J, Bonvento G. Structural organization of the perivascular astrocyte endfeet and their relationship with the endothelial glucose transporter: a confocal microscopy study. Glia. 1998; 23:1-10.
● Kadekaro M, Terrell ML, Liu H, Gestl S, Bui V, Summy-Long JY. Effects of L-NAME on cerebral metabolic, vasopressin, oxytocin, and blood pressure responses in hemorrhaged rats. Am J Physiol. 1998; 274:R1070-R1077.
● Kalaria RN, Gravina SA, Schmidley JW, Perry G, Harik SI. The glucose transporter of the human brain and blood-brain barrier. Ann Neurol. 1988;
24:757-764. ● Katayama M, Chugh DK, Mokashi A, Ray, D, Bebout DE, Lahiri S. NO mimics
O2 in the carotid body chemoreception. En: Arterial Chemoreception: Cell to
System. Plenum Press. 1994: pp. 225-226.
● Katsumata K, Katsumata K Jr, Katsumata Y. Protective effect of diltiazem hydrochloride on the occurrence of alloxan- or streptozotocin-induced diabetes in rats. Horm Metab Res. 1992; 24:508-510.
● Kelm M. Nitric oxide metabolism and breakdown. Biochim Biophys Acta.
1999; 1411:273-289.
● Kelm M, Schrader J. Nitric oxide release from the isolated guinea pig heart. Eur J Pharmacol. 1988; 155:317-321.
● Kino M, Yamato T, Aomine M. Simultaneous measurement of nitric oxide, blood glucose, and monoamines in the hippocampus of diabetic rat: an in vivo microdialysis study. Neurochem Int. 2004; 44 :65-73
● Kline DD, Prabhakar NR. Peripheral chemosensitivity in mutant mice deficient in nitric oxide synthase. Adv Exp Med Biol. 2000; 475:571-579.
● Kline DD, Yang T, Huang PL, Prabhakar NR. Altered respiratory responses

98
to hypoxia in mutant mice deficient in neuronal nitric oxide synthase. J
Physiol. 1998; 511:273-287.
● Knowles RG, Moncada S. Nitric oxide as a signal in blood vessels. Trends
Biochem Sci. 1992; 17:399-402.
● Koesling D, Frieber A. Structure-Function Relationships in NO-Sensitive Guanylyl Cyclase. En: Nitric Oxide. Biology and Pathobiology. Acad Press,
New York. 2000.
● Kohli R, Meininger CJ, Haynes TE, Yan W, Self JT, Wu G. Dietary L-arginine supplementation enhances endothelial nitric oxide synthesis in streptozotocin-induced diabetic rats. J Nutr. 2004; 134:600-608.
● Komori T, Morikawa Y, Tamura S, Doi A, Nanjo K, Senba E. Subcellular localization of glucose transporter 4 in the hypothalamic arcuate nucleus of ob/ob mice under basal conditions. Brain Res. 2005; 1049:34-42.
● Koyama Y, Coker RH, Denny JC, Lacy DB, Jabbour K, Williams PE,
Wasserman DH. Role of carotid bodies in control of the neuroendocrine response to exercise. Am J Physiol Endocr Metab. 2001; 281:E742-748.
● Koyama Y, Coker RH, Stone EE, Lacy DB, Jabbour K, Williams PE, Wasserman
DH. Evidence that carotid bodies play an important role in glucoregulation In Vivo. Diabetes. 2000; 49:1434-1442.
● La Fleur SE. Daily rhythms in glucose metabolism: suprachiasmatic nucleus output to peripheral tissue. J Neuroendocrinol. 2003; 15:315-322.
● Lahiri S, Buerk DG. Vascular and metabolic effects of nitric oxide synthase inhibition evaluated by tissue pO2 measurements in carotid body. Adv Exp
Med Biol. 1998; 454:455–460.
● Lahiri S, Di Giulio C, Roy A. Lessons from chronic intermittent and sustained hypoxia at high altitudes. Respir Physiol Neurobiol. 2002;
130:223-233.
● Lahiri S, Mokashi A, Shirahata M, Andronikou S. Chemical respiratory control in chronically hyperoxic cats. Respir Physiol. 1990; 82:201-215.
● Lawson W. The neuroendocrine nature of the glomus cells: an experimental, ultrastructural, and histochemical tissue culture study.

99
Laryngoscope. 1980; 90:120-144.
● Leavesley HB, Li L, Prabhakaran K, Borowitz JL, Isom GE. Interaction of cyanide and nitric oxide with cytochrome C oxidase: implications for acute cyanide toxicity. Toxicol Sci. 2008; 101:101-11.
● Lei H, Venkatakrishnan A, Yu S, Kazlauskas A. Protein kinase A-dependent translocation of Hsp90 alpha impairs endothelial nitric-oxide synthase activity in high glucose and diabetes. J Biol Chem. 2007; 282:9364-71.
● Leist, M, Single B, Naumann H, Fav F, Simon B, Kuhnle S, Nicotera P.
Inhibition of mitochondrial ATP generation by nitric oxide switches apoptosis to necrosis. Ex Cell Res. 1999; 249:396-403.
● Levin BE, Dun-Meynell AA, Routh VH. Brain glucose sensing and body energy homeostasis: role in obesity and diabetes. Amer J Physiol Regul
Integ Comp Physiol. 1999; 276:R1223-R1231.
● Levin BE. Glucosensing neurons: the metabolic sensors of the brain?. Diabetes Nutr Metab. 2002; 15:274-280.
● López-Barneo J. Oxygen-sensing by ion channels and the regulation of cellular functions. Trends Neurosci. 1996; 19:435-440.
● López-Barneo J, del Toro R, Levitsky KL, Chiara MD, Ortega-Saenz P.
Regulation of oxygen sensing by ion channels. J Appl Physiol. 2004;
96:1187-1195.
● López-Barneo J, Ortega-Saenz P, Piruat JI, Garcia-Fernandez M. Oxygen-sensing by ion channels and mitochondrial function in carotid body glomus cells. Novartis Found Symp. 2006; 272:54-64.
● López-Barneo J, Pardal R, Ortega-Sáenz P. Cellular mechanism of oxygen sensing. Ann Rev Physiol. 2001; 63:259-287.
● Lowenstein CJ, Dinerman JL, Snyder SH. Nitric Oxide: a physiologic messenger. Ann Intern Med. 1994; 120:227-237.
● Lund-Andersen H. Transport of glucose from blood to brain. Physiol Rev.
1979; 59:305-352.
● Martin RL, Lloyd HG, Cowan AI. The early events of oxygen and glucose deprivation: setting the scene for neuronal death ?. Trends Neurosci. 1994;

100
17:251-257.
● Marshall NB, Mayer J. Specificity of gold thioglucose for ventromedial hypothalamic lesions and hyperphagia. Nature. 1956; 178:1399-1400.
● McDonald DM, Mitchell RA. The innervation of glomus cells, ganglion cells and blood vessels in the rat carotid body. A quantitative ultraestructural analysis. 1975; J Neurocytol. 4:177-230.
● McDonald DM, Mitchell RA. The neural pathway involved in "efferent inhibition" of chemoreceptors in the cat carotid body. J Comp Neurol.
1981; 201:457-476.
● McRitchie DA, Törk I. Distribution of substance P-like immunoreactive neurons and terminals throughout the nucleus of the solitary tract in the human brainstem. J Comp Neurol. 1994; 343:83-101.
● Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology and pharmacology. Pharmacol Rev. 1991; 43:109-142.
● Montero, S. (1998). Mediadores efectores en la respuesta hiperglucemiante a la estimulación de los quimiorreceptores del cuerpo carotídeo con cianuro en ratas. Tesis de Doctorado, CUIB. Universidad de Colima, Colima.
● Montero S, Mendoza H, Valles V, Lemus M, Alvarez-Buylla R, de Alvarez-Buylla
ER. Arginine-vasopressin mediates central and peripheral glucose regulation in response to carotid body receptor stimulation with Na-cyanide. J Appl Physiol. 2006; 100:1902-1909.
● Montero SA, Yarkov A, Álvarez-Buylla R. Carotid chemoreceptors participation in brain glucose regulation. Role of arginine-vasopressin.
Adv Exp Med Biol. 2000; 475:749-760.
● Montero SA, Yarkov A, Lemus M, de Álvarez-Buylla ER, Álvarez-Buylla R.
Carotid chemoreceptor reflex modulation by arginine-vasopressin microinjected into the nucleus tractus solitarius in rats. Arch Med Res.
2006; 37:709-716.
● Montero SA, Yarkov A, Lemus M, Mendoza H, Valles V, de Álvarez-Buylla ER,
Álvarez-Buylla R. Enhancing effect of vasopressin on the hyperglycemic response to carotid body chemoreceptor stimulation. Role of metabolic

101
variables. Adv Exp Med Biol. 2003; 536:95-107.
● Mueckler M. Facilitative glucose transporters. Eur J Biochem. 1994; 219:
713-725.
● Ngarmukos C, Baur EL, Kumagai AK. Co-localization of GLUT1 and GLUT4 in the blood-brain barrier of the rat ventromedial hypothalamus. Brain Res.
2001; 900:1-8.
● Niijima A. Glucose-sensitive afferent nerve fibres in the hepatic branch of the vagus nerve in the guinea-pig. J Physiol. 1982; 332:315-323.
● Nukatsuka M, Yoshimura Y, Nishida M, Kawada J. Importance of the concentration of ATP in rat pancreatic beta cells in the mechanism of streptozotocin-induced cytotoxicity. J Endocrinol. 1990; 127:161-165.
● Nurse CA. Neurotransmission and neuromodulation in the chemosensory carotid body. Auton Neurosci. 2005; 120:1-9.
● Nurse CA, Fearon IM. Carotid body chemoreceptors in dissociated cell culture. Microsc Res Tech. 2002; 59:249-255.
● Okada S, Takehara Y, Yabuki M, Yoshioka T, Yasuda T, Inoue M, Utsumi K.
Nitric oxide, a physiological modulator of mitochondrial function. Physiol
Chem Phys Med NMR. 1996; 28:69-82.
● Oomura Y, Yoshimatsu H. Neural network of glucose monitoring system. J
Auton Nerv Syst. 1984; 10:359-372.
● Ortega-Sáenz P, García-Fernández M, Pardal R, Alvarez E, López-Barneo J.
Studies on glomus cell sensitivity to hypoxia in carotid body slices. Adv
Exp Med Biol. 2003; 536: 65-73
● Ortega-Saenz P, Pardal R, Castellano A, López-Barneo J. Collapse of conductance is prevented by a glutamate residue conserved in voltage-dependent K (+) channels. J Gen Physiol. 2000; 116:181-90.
● Ozcelikay AT, Tay A, Guner S, Tasyaran V, Yildizoglu-Ari N, Dincer UD Altan
VM. Reversal effects of L-arginine treatment on blood pressure and vascular responsiveness of streptozotocin-diabetic rats. Pharmacol Res.
2000; 41:201-209.
● Papacio G, Pisanti FA, Latronico MV, Ammendola E, Galdieri M. Multiple low-

102
dose and single high-dose treatments with streptozotocin do not generate nitric oxide. J Cell Biochem. 2000; 77:82-91.
● Pardal R, Ludewig U, García-Hirschfeld J, López-Barneo J. Secretory responses of intact glomus cells in thin slices of rat carotid body to hypoxia and tetraethylammonium. Proc Natl Acad Sci USA. 2000; 97:2361-
2366.
● Pardal R, López-Barneo J. Low glucose-sensing cells in the carotid body. Nat Neurosci. 2002; 5:197-198.
● Paulding WR, Schnell PO, Bauer AL, Striet JB, Nash JA, Kuznetsova AV,
Czyzyk-Krzeska MF. Regulation of gene expression for neurotransmitters during adaptation to hypoxia in oxygen-sensitive neuroendocrine cells. Microsc Res Tech. 2002; 59:178-187.
● Penicaud L, Leloup C, Lorsignol A, Alquier T, Guillaod E. Brain glucose sensing mechanism and glucose homeostasis. Curr Opin Clin Nutr Metab
Care. 2002; 5:539-543.
● Pieper GM. Review of alterations in endotelial nitric oxide production in diabetes: protective role of arginine on endothelial dysfunction. Hypertension. 1998; 31:1047-1060.
● Pieper GM, Dondlinger LA. Plasma and vascular tissue arginine are decreased in diabetes: acute arginine supplementation restores endothelium dependent relaxation by augmenting cGMP production. J
Pharmacol Exp Ther. 1997; 283:684-691.
● Pocai A, Obici S, Schwartz GJ, Rossetti L. A brain-liver circuit regulates glucose homeostasis. Cell Metab. 2005; 1:53-61.
● Polonsky KS, Sturis J, Bell GI. Seminars in Medicine of the Beth Israel Hospital, Boston. non-insulin-dependent diabetes mellitus - a genetically programmed failure of the beta- cell to compensate for insulin resistance.
N Engl J Med. 1996; 334:777-783.
● Popel AS, Goldman D, Vadapalli A. Modelling of oxygen diffusion from the blood vessels to intracellular organelles. Adv Exp Med Biol. 2003; 530:485-
495.

103
● Prabhakar NR. NO and CO as second messengers in oxygen sensing in the carotid body. Respir Physiol. 1999; 115:161-168.
● Prabhakar NR. Oxygen sensing by the carotid body chemoreceptors. J
Appl Physiol. 2000; 88:2287-2295.
● Prabhakar NR, Fields RD, Baker T, Fletcher EC. Intermittent hypoxia: cell to system. Am J Physiol Lung Cell Mol Physiol. 2001; 281:L524-L528.
● Prabhakar NR, Jacono FJ. Cellular and molecular mechanisms associated with carotid body adaptations to chronic hypoxia. High Alt Med Biol. 2005;
6:112-120.
● Prabhakar NR, Kumar GK, Chang CH, Agani FH, Haxhiu MA. Nitric oxide in the sensory function of the carotid body. Brain Res. 1993; 625:16–22.
● Prabhakar NR, Overholt JL. Cellular mechanisms of oxygen sensing at the carotid body: heme proteins and ion channels. Respir Physiol. 2000;
122:209-221.
● Radi R. Reactions of nitric oxide with metalloproteins. Chem Res Toxicol.
1996; 9:828-835.
● Rajasekaran S, Sivagnanam K, Subramanian S. Mineral content of aloe vera leaf gel and their role on streptozotocin-inuced diabetic rat. Pharmacol
Rev. 2005; 57:90-96.
● Reinhardt RR, Bondy CA. Insulin-like growth factors cross the blood-brain barrier. Endocrinology. 1994; 135:1753-1761.
● Rengasamy A, Johns RA. Inhibition of nitric oxide synthase by a superoxide generating system. J Pharmacol Exp Ther. 1993; 267:1024-
1027.
● Ricardo JA, Koh ET. Anatomical evidence of direct projections from the nucleus of the solitary tract to the hypothalamus, amygdala, and other forebrain structures in the rat. Brain Res. 1978; 153:1-26.
● Roberts CK, Barnard RJ, Scheck SH, Balon TW. Exercise-stimulated glucose transport in skeletal muscle is nitric oxide dependent. Am J Physiol. 1997;
273:E220-E225.
● Roder S, Ciriello J. Caudal ventrolateral medullary projections to the

104
nucleus of the solitary tract in the cat. Neurosci Lett. 1992; 134:161-164.
● Rogers NE, Ignarro LJ. Constitutive nitric oxide synthase from cerebellum is reversibly inhibited by nitric oxide formed from L-arginine. Biochem
Biophys Res Commun. 1992; 189:242-249.
● Saavedra-Molina A, Ramírez-Emiliano J, Clemente-Guerrero M, Pérez-Vázquez
V, Aguilera-Aguirre L, Gonzalez-Hernández JC. Mitochondrial nitric oxide inhibits ATP synthesis. Effect of free calcium in rat heart. Amino Acids.
2003; 24:95-102.
● Sankaran M, Subramanian P. Modulation of biochemical circadian rhythms during long-term melatonin treatment in rats. Singapore Med J. 2006;
47:42-47.
● Sarkar S, Banerjee PK, Selvamurthy W. High altitude hypoxia: an intrincate interplay of oxygen responsive macroevents and micromolecules. Moll
Cell Biochem. 2003; 253:280-305.
● Sato M, Ikeda K, Yoshizaki K, Koyano H. Response of cytosolic calcium to anoxia and cyanide in cultured glomus cells of newborn rabbit carotid body. Brain Res. 1991; 551: 327-330.
● Schmidt W. Effects of intermittent exposure to high altitude on blood volume and erythropoyetic activity. High Alt Med Biol. 2002; 3:167-176.
● Schmidt K, Schrammel A, Koesling D, Mayer B. Molecular mechanisms involved in the synergistic activation of soluble guanylyl cyclase by YC-1 and nitric oxide in endothelial cells. Mol Pharmacol. 2001; 59:220-224.
● Schulz R, Kelm M, Heusch G. Nitric oxide in myocardial ischemia/reperfusion injury. Cardiovasc Res. 2004; 61:401-413.
● Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol. 1999; 15:551-578.
● Shankar R, Zhu JS, Ladd B, Henry D, Shen HQ, Baron AD. Central nervous system nitric oxide synthase activity regulates insulin secretion and insulin action. J Clin Invest. 1998; 102:1403-1412.
● Shibata M, Araki N, Hamada J, Sasaki T, Shimazu K, Fukuuchi Y. Brain nitrite production during global ischemia and reperfusion: an in vivo

105
microdialysis study. Brain Res. 1996; 734:86-90.
● Shimazu T. Central nervous system regulation of liver and adipose tissue metabolism. Diabetologia. 1981; 20:343-356.
● Sloviter HA, Kamimoto T. The isolated, persed rat brain preparation metabolizes mannose but not maltose. J Neurochem. 1970; 17:1109-11.
● Sokoloff L. Quantitative measurements of cerebral blood flow in man.
Methods Med Res. 1960; 8:253-61.
● Sokoloff L. Relation between physiological function and energy metabolism in the central nervous system. J Neurochem. 1977; 29:13-26.
● Stea A, Nurse CA. Whole cell and perforated patch recordings from =2-sensitive rat carotid body cells grown in short and long term culture. Pflüg
Arch. 1991; 418:92-101.
● Stubauer G, Giuffre A, Brunori M, Sarti P. Cytochrome c oxidase does not catalyze the anaerobic reduction of NO. Biochem Biophys Res Commun.
1998; 245:459-465.
● Szkudelski T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol Res. 2001; 50:537-546.
● Szkudelski T, Kandulska K, Okulicz M. Alloxan in vivo does not only exert deleterious effects on pancreatic B cells. Physiol Res. 1998; 47:343-346.
● Takahashi S, Cook M, Jehle J, Kennedy C, Sokoloff L. Lack of effects of inhibition of nitric oxide synthesis on local glucose utilization in the rat brain. J Neurochem. 1995; 65, 414-419.
● The Expert Committe on the Diagnosis and Classification of Diabetes Mellitus.
Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 2003; 26:S5-S20.
● Thulesen J, Orskov C, Holst JJ, Poulsen SS. Short-term insulin treatment prevents the diabetogenic action of streptozotocin in rats. Endocrinology.
1997; 138:62-68.
● Tong Y-CH, Wang Ch-J, Cheng J-T. The role of nitric oxide in the control of plasma glucose concentration in spontaneously hypertensive rats.
Neurosc Lett. 1997; 233:93-96.

106
● Tuzgen S, Tanriover N, Uzan M, Tureci E, Tanriverdi T, Gumustas K, Kuday C.
Nitric oxide levels in rat cortex, hippocampus, cerebellum, and brainstem after impact acceleration head injury. Neurol Res. 2003; 25:31-34.
● Ueki M, Linn F, Hossmann KA. Functional activation of cerebral blood flow and metabolism before and after global ischemia of rat brain. J Cereb
Blood Flow Metab. 1988; 8:486-494.
● Ureña J, Fernandez-Chacón R, Benot AR, Álvarez de Toledo GA, López-Barneo
J. Hypoxia induces voltage-dependent Ca2+ entry and quantal dopamine secretion in carotid body glomus cells. Proc Natl Acad Sci USA. 1994;
91:10208-10211.
● Uyama N, Geerts A, Reynaert H. Neural connections between the hypothalamus and the liver. Anat Rec A Discov Mol Cell Evol Biol. 2004;
280:808-820.
● Valdés V, Mosqueira M, Rey S, Del Río R, Iturriaga R. Inhibitory effects of NO on carotid body: contribution of neural and endothelial nitric oxide synthase isoforms. Am J Physiol Lung Cell Mol Physiol. 2002; 284: L57-L68.
● Varas R, Wyatt CN, Buckler KJ. Modulation of TASK-like background potassium channels in rat arterial chemoreceptor cells by intracellular ATP and other nucleotides. J Physiol. 2007; 583:521-36.
● Wang X, Robinson PJ. Cyclic GMP-dependent protein kinase and cellular signaling in the nervous system. J Neurochem. 1997; 68:443-456.
● Wang ZZ, Stensaas LJ, Bredt DS, Dinger B, and Fidone SJ. Localization and actions of nitric oxide in the cat carotid body. Neuroscience. 1994; 60:275–
286.
● Wang ZZ, Stensaas LJ, de Vente J, Dinger B, Fidone SJ.
Immunocytochemical localization of cAMP and cGMP in cells of the rat carotid body following natural and pharmacological stimulation.
Histochemistry. 1991; 96:523-530.
● Wang ZZ, Stensaas LJ, Dinger BG y Fidone SJ. Nitric oxide mediates chemoreceptor inhibition in the cat carotid body. Neuroscience. 1995;
65:217-229.

107
● Washburn CP, Bayliss DA, Guyenet PG. Cardiorespiratory neurons of the rat ventrolateral medulla contain TASK-1 and TASK-3 channel mRNA.
Respir Physiol Neurobiol. 2003; 138:19-35.
● Welsh N, Sandler S. Protective action by hemin against interleukin-1 beta induced inhibition of rat pancreatic islet function. Mol Cell Endocrinol.
1994; 103:109-114.
● Williams BA, Buckler KJ. Biophysical properties and metabolic regulation of a TASK-like potassium channel in rat carotid body type 1 cells. Am J
Physiol Lung Cell Mol Physiol. 2004; 286:L221-30.
● Wilson DF, Roy A, Lahiri S. Immediate and long-term responses of the carotid body to high altitude. High Alt Med Biol. 2005; 6:97-111.
● Wu C, Fujihara H, Yao J, Qi S, Li H, Shimoji K, Baba H. Different expression patterns of Bcl-2, Bcl-xl, and Bax proteins after sublethal forebrain ischemia in C57Black/Crj6 mouse striatum. Stroke. 2003; 34:1803-1808.
● Wu G, Meininger CJ. Impaired arginine metabolism and nitric oxide synthesis in coronary endotelial cells of the spontaneously diabetic BB rat. Am J Physiol Heart Circ Physiol. 1995; 260:1312-1318.
● Wu G, Morris SM Jr. Arginine metabolism: nitric oxide and beyond. Biochem J. 1998; 336:1-17.
● Wyatt CN, Peers C. Ca2+-activated K+ channels in isolated type I cells of the neonatal rat carotid body. J Physiol. 1995; 483:559-565.
● Yamamoto H, Uchigata Y, Okamoto H. Streptozotocin and alloxan induce DNA strand breaks and poly (ADP-ribose) synthetase in pancreatic islets. Nature. 1981; 294:284-286.
● Yarkov A, Montero S, Lemus M, Roces de Álvarez-Buylla E, Álvarez-Buylla R.
Arginine-vasopressin in nucleus of the tractus solitarius induces hyperglycemia and brain glucose retention. Brain Res. 2001; 902:212-222.
● Ye JS, Tipoe GL, Fung PC, Fung ML. Augmentation of hypoxia-induced nitric oxide generation in the rat carotid body adapted to chronic hypoxia: an involvement of constitutive and inducible nitric oxide synthases.
Pflugers Arch. 2002; 444:178-85.

108
● Yki-Jarvinen H. Pathogenesis of non-insulin-dependent diabetes mellitus.
Lancet. 1994; 343:91-95.


















