
“COLECCIÓN” UNIDAD FARMACOLOGIA …farmacus.com.co/wp-content/uploads/2016/06/5... ·...
47
“COLECCIÓN” FARMACOLOGIA SIMPLIFICADA PRINCIPIOS DE FARMACOLOGIA BIOFARMACIA Y FARMACOCINETICA: BIODISPONIBILIDAD Y BIOEQUIVALENCIA DE FARMACOS Jorge Luis Maya Benavides Q F, MSc www.farmacus.com.co UNIDAD 5
Transcript of “COLECCIÓN” UNIDAD FARMACOLOGIA …farmacus.com.co/wp-content/uploads/2016/06/5... ·...

“COLECCIÓN”
FARMACOLOGIA SIMPLIFICADA
PRINCIPIOS DE FARMACOLOGIA
BIOFARMACIA Y FARMACOCINETICA:
BIODISPONIBILIDAD Y
BIOEQUIVALENCIA
DE FARMACOS
Jorge Luis Maya Benavides Q F, MSc
www.farmacus.com.co
UNIDAD
5

CONTENIDO
Unidad 5
5.1. Definiciones
5.2. Biodisponibilidad
5.3. Factores condicionantes de la Biodisponibilidad
5.4. Cuando se realizan los estudios de Bioequivalencia
5.5. Fases y requisitos de los estudios de Bioequivalencia
5.6. Marco regulatorio en Colombia
5.7. Recomendaciones
5.8. Los medicamentos Genéricos
5.8.1 Garantía de calidad, eficacia y seguridad de los
medicamentos genéricos
5.9. Ventana terapéutica
Este tema no constituye en modo alguno un sistema de diagnostico y mucho menos de recomendación
terapéutica, por lo tanto no se debe utilizar esta información para auto medicarse, solo es con fines
académicos

Unidad 5
BIODISPONIBILIDAD
Y
BIOEQUIVALENCIA

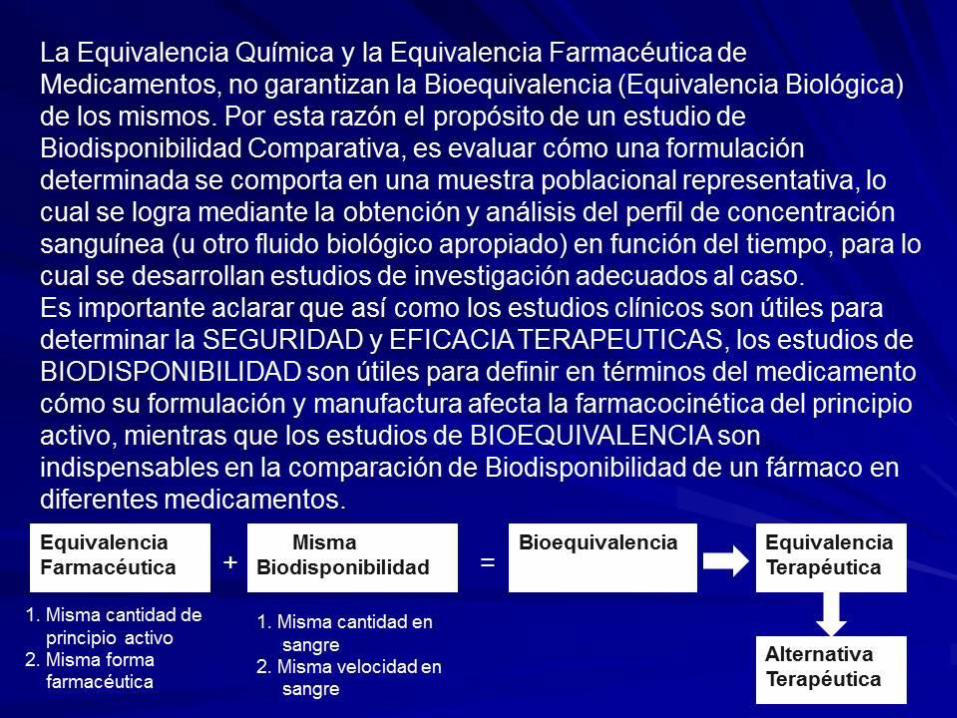
5.1. DEFINICIONES (Resolución 1400 agosto 24 de 2001)
BIODISPONIBILIDAD:
Es la medida de velocidad y de la cantidad que del ingrediente
terapéuticamente activo o fármaco contenido en un medicamento, alcanza la
circulación general.
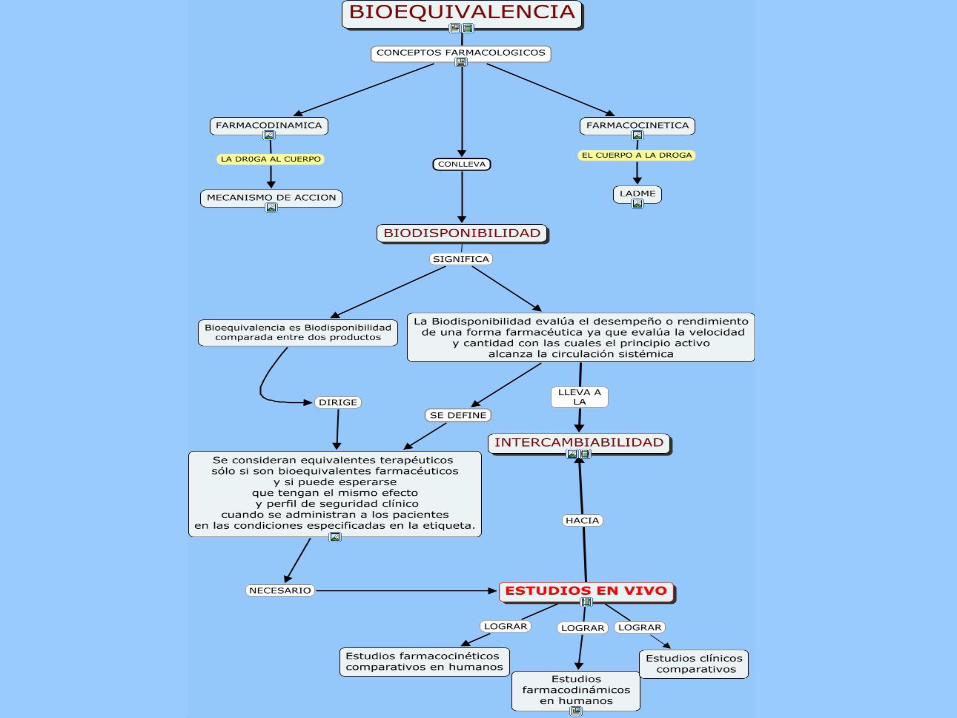
BIOEQUIVALENCIA:
Dos productos farmacéuticos son equivalentes si ellos son farmacéuticamente
equivalentes y su Biodisponibilidad luego de su administración en la misma dosis
molar son similares en tal grado que puede esperarse que sus efectos sean
esencialmente los mismos. Los estudios de Bioequivalencia son estudios de
Biodisponibilidad comparativa entre dos o mas medicamentos, del mismo fármaco y
evaluados bajo el mismo diseño experimental. Los estudios de Bioequivalencia se
utilizan como herramienta para demostrar que un medicamento genérico tiene la
misma eficacia terapéutica que el medicamento innovador de referencia.
“En la practica los estudios de Bioequivalencia son el método mas apropiado para
demostrar la equivalencia terapéutica entre dos medicamentos y en la actualidad
es la vara de medida para la autorización de los medicamentos genéricos en
numerosos países, si se demuestra la Bioequivalencia, entonces existe la
posibilidad de la intercambiabilidad” ¿Que tan similares deben ser?...
Es arbitrario pero por consenso se acepta que no difieran en mas del 20%. (Abad Santos, F; Martínez Sancho, E; Gálvez Múgica, M A: Estudios de bioequivalencia: análisis y
aspectos metodológicos. Cap. 4 en El ensayo clínico en España. García, A; Gandía, L. Serie científica.
Farmaindustria. Madrid, 2001).


5.1.DEFINICIONES (Resolución 1400 agosto 24 de 2001) EQUIVALENCIA QUIMICA:
Medicamentos que contienen el mismo principio activo o fármaco en la misma sal, ester o
derivado químico y son idénticos en potencia o concentración y forma de dosificación.
EQUIVALENCIA FARMACEUTICA:
Medicamentos que contienen el mismo principio activo o fármaco en la misma sal, ester o
derivado químico y son idénticos en potencia o concentración, forma de dosificación y vía de
administración. Los medicamentos equivalentes químicos son equivalentes farmacéuticos y
como aquellos pueden diferir en su excipiente pero deben cumplir los mismos estándares de
potencia, calidad, pureza e identidad, también pueden diferir en características como color,
sabor, forma, marcas de configuración, preservantes, empaques, tiempo de vida útil y dentro
de ciertos limites, en el etiquetado.
Sin embargo, “la equivalencia farmacéutica no necesariamente implica equivalencia
terapéutica”: diferencias en los excipientes, en el proceso de elaboración, u otras, pueden
determinar disparidades en el comportamiento de los productos (OMS).
Ejemplo : Metoclopramida 0,2% gotas marca A y Metoclopramida 0,2% gotas marca B.
EQUIVALENTES TERAPÉUTICOS:
Son medicamentos que contienen el mismo fármaco terapéuticamente activo, producen el
mismo efecto terapéutico y presentan la misma potencialidad de efectos adversos. Los
medicamentos equivalentes terapéuticos pueden diferir en ciertas características tales como
color, marcado, sabor, configuración, agentes de preservación, empaque y fecha de
expiración. Los medicamentos Equivalentes terapéuticos deben ser productos (1) Seguros y
Efectivos, (2) Bioequivalentes, (3) adecuadamente etiquetados y (4) manufacturados en
cumplimiento con las buenas Prácticas de Manufactura Vigentes.
Ejemplo: Lansoprazol 30mg/día = Pantoprazol 40mg/día = Rabeprazol 20mg/día

5.1. DEFINICIONES (Resolución 1400 agosto 24 de 2001) ALTERNATIVA FARMACÉUTICA
Medicamentos que contienen la misma sustancia o principio terapéuticamente activo, pero en
forma de diferente derivado (sal, éster o complejos) o diferentes formas de dosificación y
potencia o concentración dentro de una línea de producto de un mismo fabricante, son
alternativas farmacéuticas poseyendo la misma vía de administración, la misma indicación
Terapéutica y la misma posología.
Ejemplo1: suspensiones de palmitato de cloranfenicol y suspensión de estearato de
cloranfenicol
Ejemplo 2: cápsulas de 250 mg de ampicilina con cápsulas de 500 mg de ampicilina.
Ejemplo 3: Amoxicilina 500 mg cápsulas y Amoxicilina 500 mg / 5 ml suspensión.
Ejemplo 4: Diclofenaco sódico 75 mg comprimidos y Diclofenaco potásico 50 mg comprimidos
ALTERNATIVA TERAPÉUTICA
Medicamentos que contienen diferentes principios activos que son indicados para el mismo
objetivo terapéutico o clínico. En las alternativas terapéuticas, los componentes o principios
activos o fármacos pertenecen a la misma clase farmacológica, comparten la misma
indicación y se espera que tengan el mismo efecto terapéutico cuando se administran a los
pacientes en la condición indicada para su empleo.
Se espera que los efectos terapéuticos sean similares cuando se administren en dosis
terapéuticas equivalentes.
Ejemplo 1: Ketorolac 10 mg comprimidos y Diclofenaco 50 mg comprimidos.
Ejemplo 2: Pantoprazol 20 mg cápsulas y Omeprazol 10 mg comprimidos
.

5.2 BIODISPONIBILIDAD
Las formas farmacéuticas solidas deben liberar su principio activo en el
organismo de modo que éste pueda ser absorbido en óptimas condiciones
y llegar, por este mecanismo, al sitio de acción. Se ha demostrado que las
diferencias en formulación y método de manufactura de un Medicamento,
inciden en la variabilidad de la respuesta alcanzada con el mismo, medida
esta variabilidad en términos del inicio, la duración y la intensidad de la
respuesta obtenida.
De este modo, una inyección intravenosa no involucra proceso de absorción
y la dosis administrada llega en forma casi instantánea y completa(100%) a
la circulación sistémica. Por este motivo, la inyección intravenosa se emplea
corrientemente como punto de referencia para evaluar la Biodisponibilidad
absoluta de un fármaco en una forma farmacéutica particular.
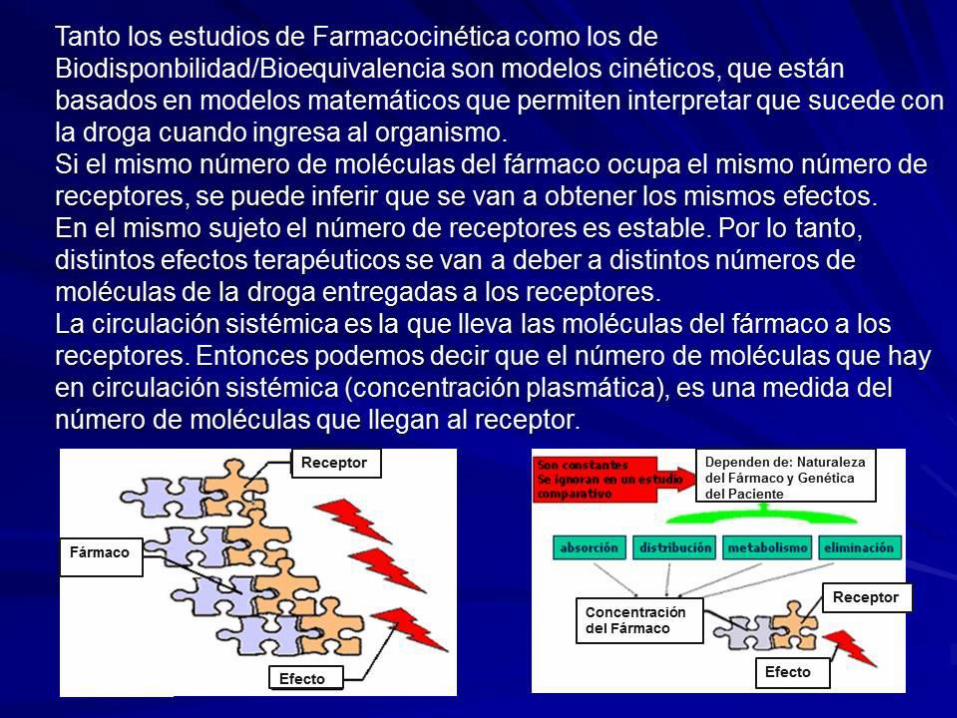

La distribución, el metabolismo y la eliminación dependen de la naturaleza
química del fármaco y de la genética del paciente. Cuando se hace un
estudio comparativo estas tres variables son constantes en un mismo
paciente. Entonces en un estudio comparativo de dos productos con el
mismo principio activo, la diferencia entre estos dos productos sólo va estar
dado por diferencias en la absorción y esta depende de la naturaleza
química del fármaco y de las características del producto farmacéutico o
diferencias en la formulación. Entonces:
Las diferencias de absorción, metabolismo, distribución y eliminación entre
distintas personas aumentan la variabilidad en la concentración plasmática
La concentración plasmática de una droga determina el número de
moléculas que llega al receptor y por ende su efecto terapéutico.
La concentración plasmática está gobernada por la absorción, distribución,
metabolismo y eliminación de la droga..
Dependen de: Naturaleza de
la Formulación
Concentración
del Fármaco
Receptor
Efecto
Zanen, P. Bioequivalence and generics medicines.
www.math.umbc.edu/caoli/bioequ.pdf.Acceso
23.06.04

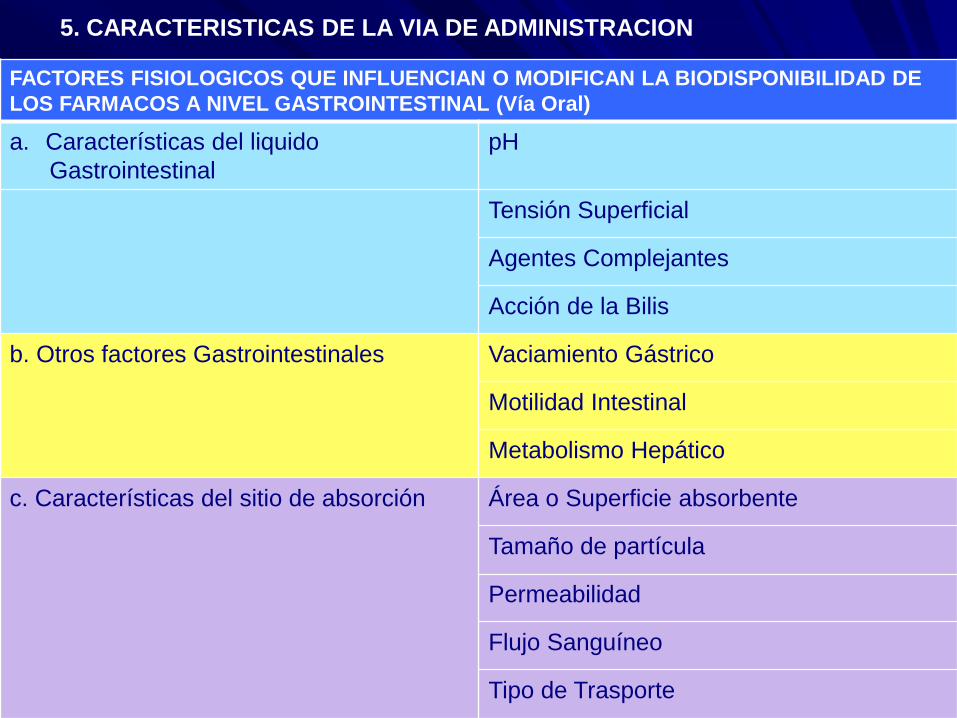
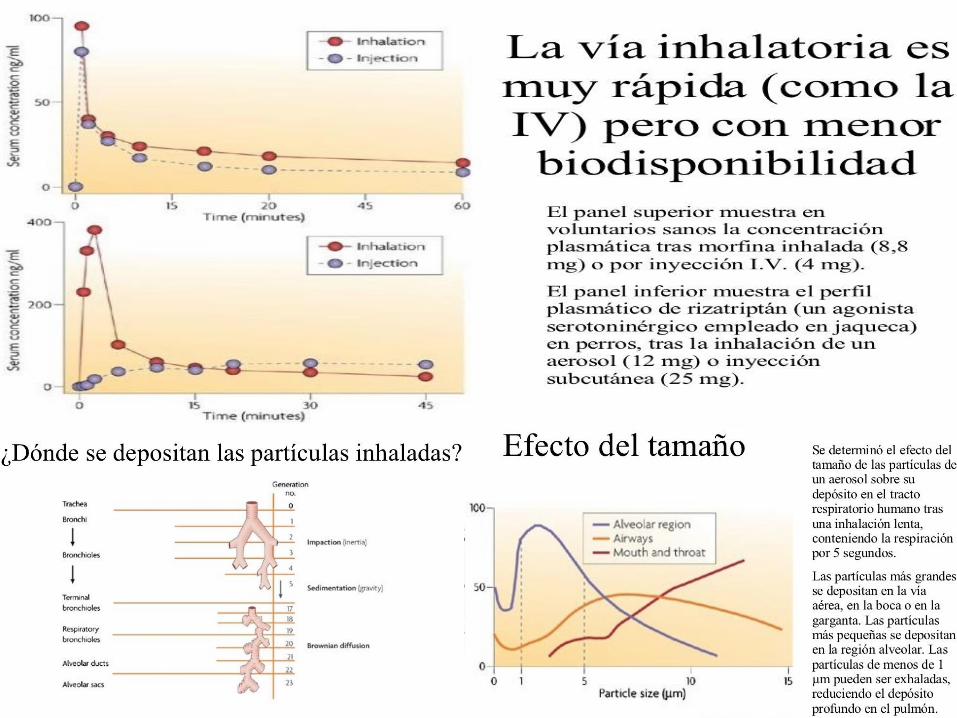
5. CARACTERISTICAS DE LA VIA DE ADMINISTRACION
FACTORES FISIOLOGICOS QUE INFLUENCIAN O MODIFICAN LA BIODISPONIBILIDAD DE
LOS FARMACOS A NIVEL GASTROINTESTINAL (Vía Oral)
a. Características del liquido
Gastrointestinal
pH
Tensión Superficial
Agentes Complejantes
Acción de la Bilis
b. Otros factores Gastrointestinales Vaciamiento Gástrico
Motilidad Intestinal
Metabolismo Hepático
c. Características del sitio de absorción Área o Superficie absorbente
Tamaño de partícula
Permeabilidad
Flujo Sanguíneo
Tipo de Trasporte


1.FÍSICO: dureza, densidad, conductividad eléctrica a térmica.
2.FÍSICO-QUÍMICO: absorción, estabilidad, punto de fusión.
3.QUÍMICO: reactividad, estabilidad, solubilidad, superficie específica.
4.TECNOLÓGICO: piezoelectricidad, magnetismo, refracción, reflexión y
absorción de la luz.
5.FARMACOLÓGICO: Biodisponibilidad, inefectividad, tolerabilidad y
toxicidad.
Ejemplos de polimorfismo en fármacos. Aspirina, Amoxicilina, Diclofenaco,, Nifedipina
(*) POLIMORFISMO FARMACÉUTICO
Polimorfismo farmacéutico es la capacidad de los principios activos para
adoptar diferentes configuraciones espaciales y que tienen su origen en
las condiciones fisicoquímicas especificas en las que se realiza la
síntesis en el laboratorio, en tal sentido que Fármacos químicamente
idénticos pueden dar compuestos físicamente diferentes y
consecuentemente, las características que derivan de su estructura en
estado sólido también difieren desde el punto de vista físico,
fisicoquímico, químico, tecnológico y farmacológico, a saber:

Medicamento “A” Forma Farmacéutica: comprimidos
1. Principio activo: Amlodipino 5 mg
2. Excipientes:
Celulosa de magnesio
Celulosa microcristalina
Cera carnauba
Croscaramelosa sódica NF
Lactosa monohidrato
Hidroxipropilcelulosa
Estearato de magnesio
Laurilsulfato de sodio
Opadry amarillo
Medicamento “B” Forma Farmacéutica: comprimidos
1. Principio activo: Amlodipino 5 mg
2. Excipientes:
Almidón glicolato sódico
Estearato de magnesio
Lactosa anhidra
Celulosa microcristalina PH 102
Dióxido de silicio
Polivinilpirrolidona K 30
Opadry 285 G28725
Dióxido de titanio
Talco
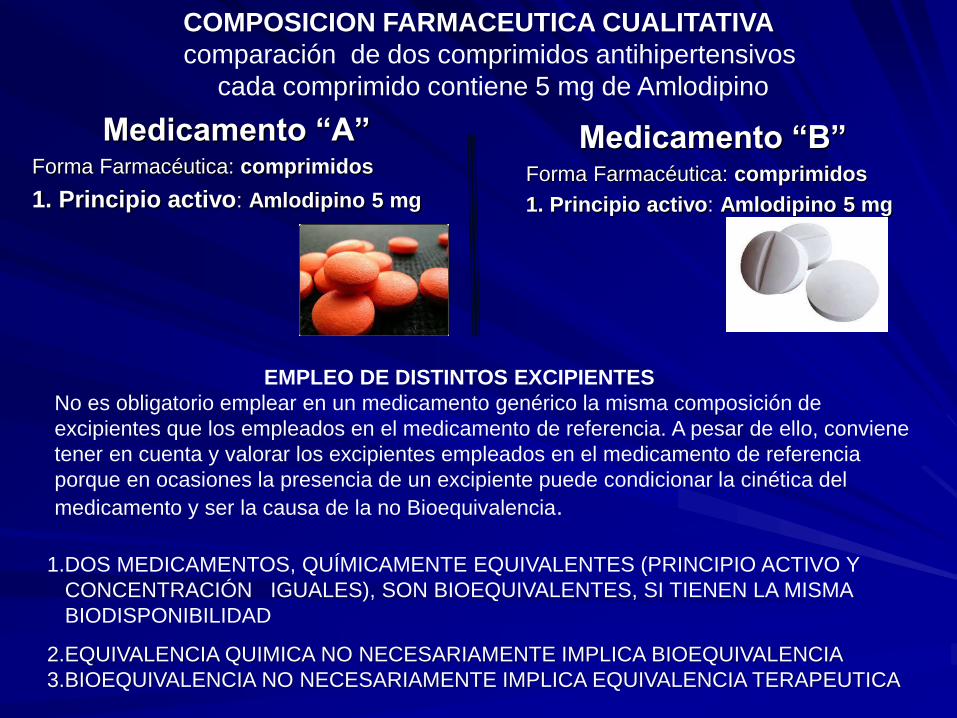
COMPOSICION FARMACEUTICA CUALITATIVA
comparación de dos comprimidos antihipertensivos
cada comprimido contiene 5 mg de Amlodipino
(anexo lista de excipientes)
PROCESO DE FABRICACION (FARMACOTECNIA)
Aunque dos medicamentos tengan los mismos principios activos, en la misma concentración y en
la misma forma farmacéutica, por el hecho de cambiar el proceso de fabricación se podría
modificar la disolución o la absorción del medicamento (Biodisponibilidad)
Medicamento “A” Forma Farmacéutica: comprimidos
1. Principio activo: Amlodipino 5 mg
Medicamento “B” Forma Farmacéutica: comprimidos
1. Principio activo: Amlodipino 5 mg
COMPOSICION FARMACEUTICA CUALITATIVA
comparación de dos comprimidos antihipertensivos
cada comprimido contiene 5 mg de Amlodipino
2.EQUIVALENCIA QUIMICA NO NECESARIAMENTE IMPLICA BIOEQUIVALENCIA
3.BIOEQUIVALENCIA NO NECESARIAMENTE IMPLICA EQUIVALENCIA TERAPEUTICA
1.DOS MEDICAMENTOS, QUÍMICAMENTE EQUIVALENTES (PRINCIPIO ACTIVO Y
CONCENTRACIÓN IGUALES), SON BIOEQUIVALENTES, SI TIENEN LA MISMA
BIODISPONIBILIDAD
EMPLEO DE DISTINTOS EXCIPIENTES
No es obligatorio emplear en un medicamento genérico la misma composición de
excipientes que los empleados en el medicamento de referencia. A pesar de ello, conviene
tener en cuenta y valorar los excipientes empleados en el medicamento de referencia
porque en ocasiones la presencia de un excipiente puede condicionar la cinética del
medicamento y ser la causa de la no Bioequivalencia.

PROBLEMAS DE
BIODISPONIBILIDAD
Aminofilina
Ampicilina
Cabamazepina
Cloranfenicol
Cloroquina
Clorpromazina
Dihidroergotamina
Ergotamina
Eritromicina
Furosemida
Glibenclamida
Medicamentos con problemas potenciales conocidos
de Biodisponibildad y de Estabilidad Fuente: Managing Drug Supply (MSH 1997), pag.273
PROBLEMAS DE
BIODISPONIBILIDAD
Nitrofurantoina
Estrógenos
Fenitoina
Prednisolona
Quinidina
Rifampicina
Espironolactona
Griseofulvina
Sulfato Ferroso
Levodopa
Metrotexato
Metildopa
PROBLEMAS DE
ESTABILIDAD
Comprimidos de
acido acetil salicílico
Comprimidos de amoxicilina
Comprimidos de Ampicilina
Comprimidos de Penicilina V
Comprimidos de Retinol
Paracetamol Liquido
Suspensión de Penicilina V
Inyección de Ergometrina
Inyección de Metilergometrina

5.4. CUANDO SE REALIZAN LOS ESTUDIOS DE BIOEQUIVALENCIA
Un estudio de Bioequivalencia compara cómo dos fármacos con el mismo
principio activo liberan el mismo a la circulación sistémica.
Además, la absorción depende solamente de las características del
fármaco y del paciente.
Entonces, en realidad el medicamento es sólo responsable de liberar el
fármaco en el organismo para que ésta se absorba.
Un estudio de bioequivalencia compara cómo dos medicamentos, idénticos
desde el punto de vista de su composición y administración, liberan el
fármaco y la dejan en disponibilidad de ser absorbida por el organismo.
Por lo tanto, si el fármaco en el medicamento esta solubilizada, no tiene
sentido realizar estudios de Bioequivalencia.
Si el fármaco es muy soluble, no es necesario realizar esta
experimentación "in-vivo" (en voluntarios sanos), sólo se realizan
evaluaciones “in-vitro” (en ensayos de laboratorio).
Fármaco 1= muy soluble, muy permeable
Fármaco 2= poco soluble, poco permeable
In-Vitro In-Vitro/
In-Vivo
1 2

Actualmente no se exige estudios de Bioequivalencia para formas parenterales
(inyectables) que se den en solución, porque el fármaco ya ingresa solubilizada al
organismo. Es importante aclarar que en un producto liofilizado o un polvo estéril,
no es necesario constatar Bioequivalencia porque si bien el fármaco está en estado
sólido se debe solubilizar para inyectar al organismo.
Tampoco se realizan estudios de Bioequivalencia para productos orales que se
administran en solución. Ejemplos de esto pueden ser los jarabes o soluciones, o
polvos para reconstituir que una vez reconstituidos, se solubilizan completamente.
En el caso de productos de acción local, no se exigen estudios de Bioequivalencia
porque la droga para ser efectiva no ingresa a la circulación sistémica, pero si se
exigen para los parches transdermicos y supositorios, ya que actúan
sistémicamente.
Se exigen estudios de Bioequivalencia para formas orales sólidas, por ejemplo
comprimidos, cápsulas o suspensiones orales y para los productos orales de
liberación sostenida o retardada.
También para otras formas farmacéuticas, por ejemplo polvos inhalatorios o
aerosoles inhalatorios, donde el fármaco ingresa al organismo en forma sólida y
que actúan por absorción sistémica.
Si bien en los casos mencionados anteriormente, se exigen estudios de
Bioequivalencia teniendo en cuenta las características de solubilidad y
permeabilidad del fármaco, en algunas situaciones es suficiente con probar la
Bioequivalencia "in-vitro".

5.5.FASES Y REQUISITOS DE LOS ESTUDIOS DE BIOEQUIVALENCIA
La guía Note for Guidance on the investigation of bioavailability and bioequivalence
(CPMP/EWP/QWP/1401/98), publicada el 14 de diciembre de 2000 por el Comité
de Especialidades Farmacéuticas de Uso Humano de la Agencia Europea de
Evaluación de Medicamentos (EMEA), trata de definir, para productos con un efecto
sistémico, los estudios de Biodisponibilidad y Bioequivalencia, así como establecer
criterios para su diseño, su procedimiento y su evaluación a nivel de toda la Unión
Europea.

5.5.FASES Y REQUISITOS DE LOS ESTUDIOS DE BIOEQUIVALENCIA
1. PERSONAS PARTICIPANTES
Las personas participantes en el estudio de Bioequivalencia se deben escoger
cuidadosamente con el fin de minimizar al máximo la variabilidad de los resultados
permitiendo a su vez que se puedan detectar diferencias entre los medicamentos
que se comparan.
El tipo de participantes requeridos para los estudios de Bioequivalencia son
voluntarios sanos de ambos sexos, con edades comprendidas entre 18 y 50-55
años, y con un peso que no supere un ± 10-15% del peso ideal para su altura y
sexo. Además, se prefiere escoger participantes no fumadores, sin historia previa
de alcoholismo ni toma de drogas, factores que podrían interferir en los resultados
obtenidos del estudio. También para determinados fármacos es interesante prestar
atención a si los participantes son o no personas con un fenotipo/genotipo
metabólico característico.
En general, suelen ser de 12 a 24 los participantes en el estudio, siempre
dependiendo de la variabilidad cinética del fármaco. Estas personas deben firmar
específicamente su consentimiento a participar en el estudio y se les asegura la
confidencialidad de los datos y la protección con una póliza de seguros por los
posibles daños que se les pueda causar

5.5.FASES Y REQUISITOS DE LOS ESTUDIOS DE BIOEQUIVALENCIA
2. FORMULACIÓN DE REFERENCIA
La elección de la formulación de referencia con la que se va a comparar la
formulación genérica debe tener un perfil de seguridad establecido por su uso
clínico continuado.
3. TIPO DE DISEÑO
Aprobación por un Comité Ético de Investigación Clínica (CEIC), además de la
autorización del Ministerio de Sanidad.
El estudio debe respetar los principios básicos de la Declaración de Helsinki.
Cumplir las normas de Buena Práctica Clínica y las normas de Buena Práctica de
Laboratorio para asegurar que los datos resultantes del estudio son fiables.
El tipo de diseño suele ser aleatorio, cruzado y abierto:
Las condiciones en las que se realiza el estudio deben estar debidamente
estandarizadas para todas las personas, con el fin de minimizar las variaciones
debidas a otros factores distintos de la toma de los medicamentos que se
comparan. De este modo, se controla que la administración de los fármacos se
realiza en ayunas de por lo menos la noche anterior para alimentos, que el volumen
de agua ingerido con el fármaco es el mismo siempre (unos 100-240 ml) y se
controlan las condiciones de alimentos, bebida, ejercicio, postura y la no toma de
otros medicamentos

5.5.FASES Y REQUISITOS DE LOS ESTUDIOS DE BIOEQUIVALENCIA
4. REALIZACIÓN DEL ESTUDIO
Después de la administración del fármaco, se realizan extracciones de sangre a
distintos tiempos (según la semivida de eliminación del fármaco) y se determinan
las concentraciones del principio activo en sangre. Durante el estudio se
controlarán en todo momento las constantes vitales de las personas y se evaluará
la posible aparición de efectos indeseados.
La determinación de las concentraciones en sangre permite calcular los parámetros
cinéticos de Biodisponibilidad que nos van a permitir determinar la Bioequivalencia
entre el fármaco genérico y el de referencia.
Se asume que las concentraciones del fármaco en sangre son representativas de
las concentraciones del fármaco en el lugar donde ejerce la acción.
Los parámetros cinéticos de Biodisponiblidad que se determinan más
frecuentemente para evaluar la Bioequivalencia entre dos formulaciones son los
siguientes:
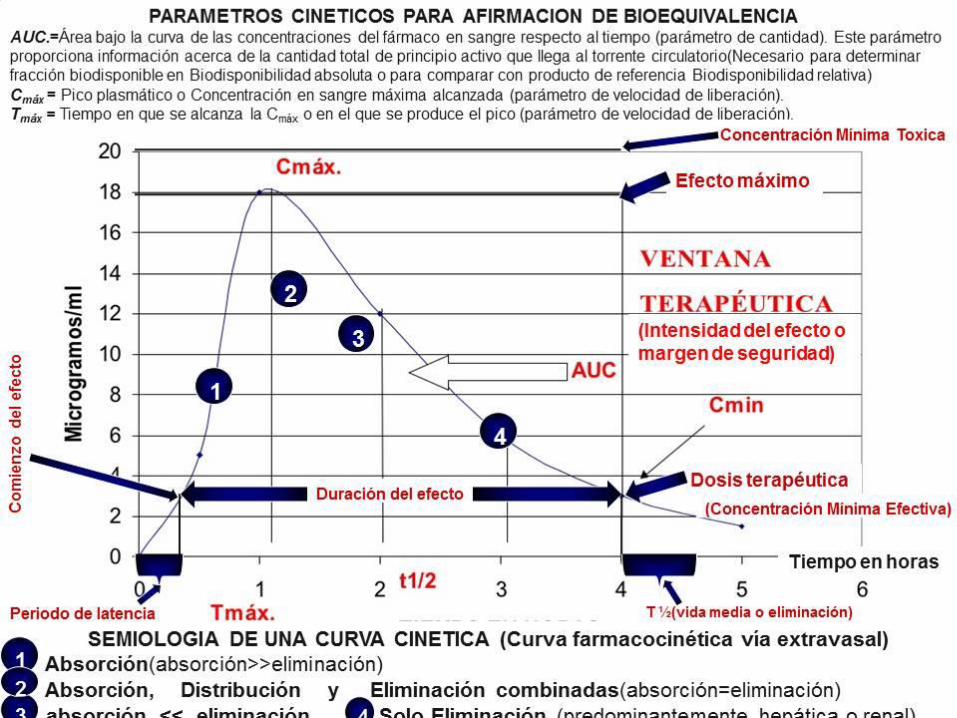
AUC. Área bajo la curva de las concentraciones del fármaco en sangre respecto al
tiempo (parámetro de cantidad). Este parámetro proporciona información acerca de
la cantidad total de principio activo que llega al torrente circulatorio.
Cmáx. Concentración en sangre máxima alcanzada (parámetro de velocidad).
Tmáx. Tiempo en que se alcanza la Cmáx (parámetro de velocidad).
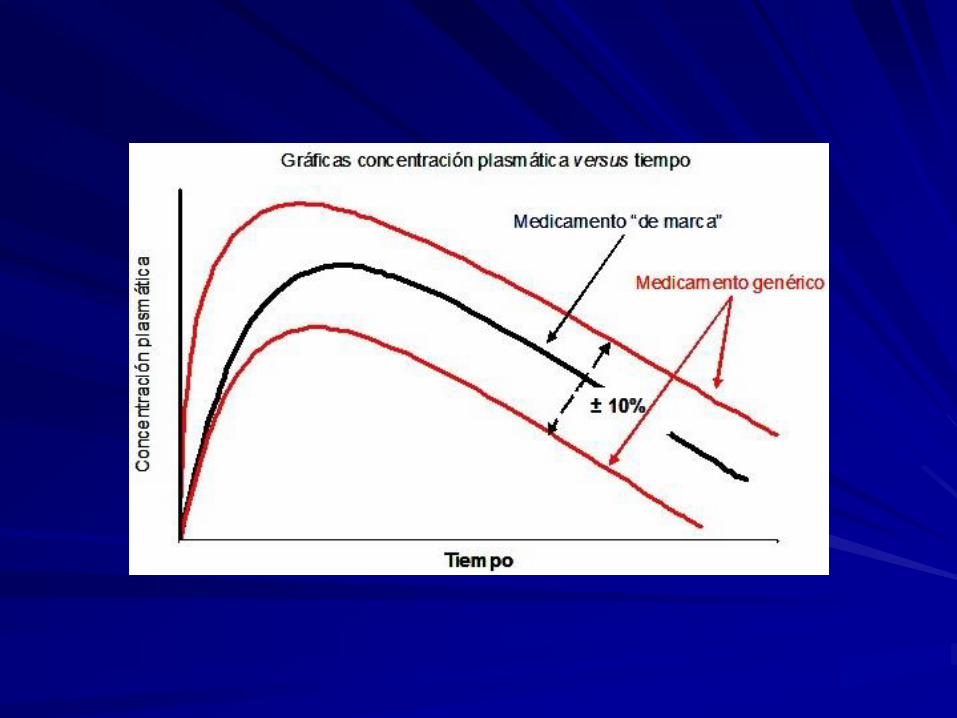
La bioequivalencia es, en general, el método más apropiado para constatar la
equivalencia terapéutica entre dos medicamentos.

5.5.FASES Y REQUISITOS DE LOS ESTUDIOS DE BIOEQUIVALENCIA
5. VALORACIÓN DE LOS RESULTADOS OBTENIDOS
Se aceptan como Bioequivalentes dos especialidades con la misma composición
cualitativa y cuantitativa, cuyas diferencias en los parámetros farmacinéticos
exigidos se encuentren:
Entre un ± 20% (80-120%) para el parámetro farmacocinético AUC.
Entre un ± 30% (70-130%) para los parámetros farmacocinéticos Cmáx y Tmáx.
Obviamente, estos rangos no son aceptados para fármacos con un estrecho
margen terapéutico en los que estas variaciones porcentuales tendrían una
repercusión clínica, ya sea de eficacia o de seguridad.

5.5.FASES Y REQUISITOS DE LOS ESTUDIOS DE BIOEQUIVALENCIA
6. CONCLUSIONES DE UN ESTUDIO DE BIOEQUIVALENCIA
De los resultados obtenidos en un estudio de Bioequivalencia se pueden extraer
tres conclusiones:
Fármacos genérico y original son iguales en cantidad absorbida y velocidad de
absorción. Este resultado implica que la especialidad genérica y la original son
bioequivalentes.
Fármacos genérico y original son iguales en cantidad absorbida, pero no en
velocidad de absorción. Este resultado no implica necesariamente la no
Bioequivalencia, pero se tiene que estudiar muy bien la situación dependiendo del
fármaco del que se trate.
Fármacos genérico y original no son iguales en cantidad. Si el resultado es de
infrabiodisponibilidad, eso significa que los fármacos genérico y original son no
Bioequivalentes. Si el resultado es de supradisponibilidad, eso implica que genérico
y original son no Bioequivalentes.
En cualquier caso, la calificación última de los estudios de Bioequivalente o no
Bioequivalente de un Genérico con respecto al de referencia para su posterior
comercialización en Colombia es ejercido por el Instituto de Vigilancia de
Medicamentos y Alimentos (INVIMA)
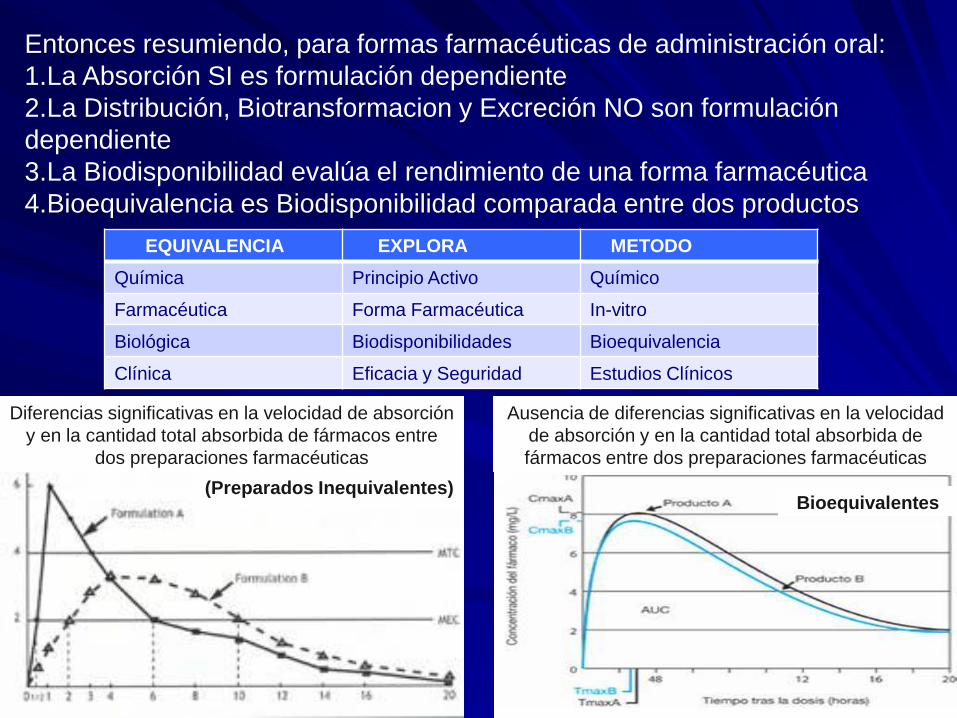
Entonces resumiendo, para formas farmacéuticas de administración oral:
1.La Absorción SI es formulación dependiente
2.La Distribución, Biotransformacion y Excreción NO son formulación
dependiente
3.La Biodisponibilidad evalúa el rendimiento de una forma farmacéutica
4.Bioequivalencia es Biodisponibilidad comparada entre dos productos
Ausencia de diferencias significativas en la velocidad
de absorción y en la cantidad total absorbida de
fármacos entre dos preparaciones farmacéuticas
Diferencias significativas en la velocidad de absorción
y en la cantidad total absorbida de fármacos entre
dos preparaciones farmacéuticas
(Preparados Inequivalentes) Bioequivalentes
EQUIVALENCIA EXPLORA METODO
Química Principio Activo Químico
Farmacéutica Forma Farmacéutica In-vitro
Biológica Biodisponibilidades Bioequivalencia
Clínica Eficacia y Seguridad Estudios Clínicos
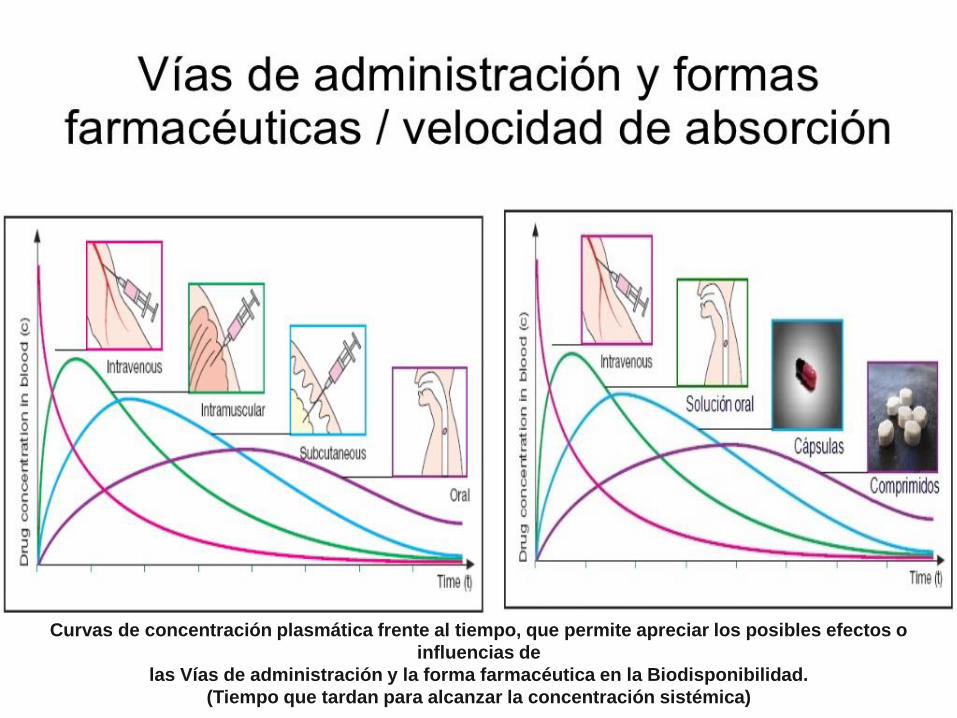
FARMACOCINETICA
Curvas de concentración plasmática frente al tiempo, que permite apreciar los posibles efectos o
influencias de
las Vías de administración y la forma farmacéutica en la Biodisponibilidad.
(Tiempo que tardan para alcanzar la concentración sistémica)

5.6. MARCO REGULATORIO EN COLOMBIA
RESOLUCION 1400 DE 2001
Por el cual se establece la Guía de Biodisponibilidad y de Bioequivalencia
de Medicamentos que trata el Decreto 677 de 1995.
PROPOSITO DE LA GUIA.
Diseño y la evaluación de los estudios de Biodisponibilidad y de Bioequivalencia
de medicamentos que contienen fármacos de acción sistémica, o sea aquellos
que requieren de un proceso de absorción y cuya respuesta terapéutica, está
íntimamente relacionada con los niveles sanguíneos del mismo.
Se exigirá́ la presentación de los estudios de Biodisponibilidad absoluta para los
medicamentos que pertenezcan a los grupos farmacológicos aquí ́relacionados,
además de los contemplados en la Guía de Biodisponibilidad acogida mediante la
presente resolución así:́
a) Antineoplásicos;
b) Anticoagulantes;
c) Antiarrítmicos;
d) Anticonvulsivantes;
e) Antiparkinsonianos;
f) Digitálicos;
g) Inmunosupresores;
h) Teofilina y sus sales;
i) Antirretrovirales; .

5.6. MARCO REGULATORIO EN COLOMBIA
RESOLUCION 1400 DE 2001
Por el cual se establece la Guía de Biodisponibilidad y de Bioequivalencia
de Medicamentos que trata el Decreto 677 de 1995.
Se exigirán estudios de Bioequivalencia para los medicamentos que se
comercializan en Colombia bajo denominación genérica o de marca, cuando el
productor interesado solicite la certificación de intercambiabilidad con el innovador
en el mercado.
Se exigirá́ Bioequivalencia “in vivo” a los siguientes grupos farmacológicos:
a) Anticonvulsivantes;
b) Inmunosupresores;
Y demás Medicamentos definidos por el INVIMA cuando lo considere pertinente
por sus características de alto riesgo, tales como, toxicidad, margen terapéutico
estrecho y comportamiento farmacocinética, previo concepto y sustentación
científica de la Comisión Revisora, Sala Especializada de Medicamentos.

5.6. MARCO REGULATORIO EN COLOMBIA
RESOLUCION 1400 DE 2001 CRISTERIOS BIOFARMACEUTICOS PARA NO EXIGIR ESTUDIOS DE
BIOEQUIVALENCIA.
En determinadas situaciones se puede asegurar que un medicamento genérico es
bioequivalente con un medicamento original sin la necesidad de tener que realizar
estudios de Bioequivalencia en humanos.
De acuerdo con las recomendaciones de la OMS informe de 1996 se consideran
exentos de estudios de Bioequivalencia a:
A. Los productos destinados a ser administrados parenteralmente (por ejemplo:
vía intravenosa, intramuscular o intratecal) en solución acuosa que contengan
el o los mismos fármacos en la misma concentración.
B. Soluciones para administración por vía oral que contengan el o los mismos
fármacos en la misma concentración y no contengan un excipiente que se
conozca o se sospeche afecta el tránsito gastrointestinal o la absorción del
principio activo.
C. Medicamentos presentados como gases o vapores.
D. Polvos y/o granulados para reconstitución como solución, cuando cumpla con
lo anotado en A y B.
E. Productos otológicos u oftalmológicos que contengan el o los mismos fármacos
en la misma concentración y esencialmente los mismos excipientes.

5.6. MARCO REGULATORIO EN COLOMBIA
RESOLUCION 1400 DE 2001
F. Productos para empleo tópico, líquidos, que contengan el o los mismos
fármacos y esencialmente los mismos excipientes.
G. Formas farmacéuticas de aplicación tópica (crema, pomada, gel, loción, etc.),
de uso externo, sólidas, que contengan el o los mismos fármacos y
esencialmente los mismos excipientes.
H. Productos destinados a ser utilizados por inhalación o aerosoles nasales que
sean administrados con o sin esencialmente el mismo dispositivo, sean
preparados como soluciones acuosas y que contengan el o los mismos
fármacos y esencialmente los mismos excipientes en concentraciones
comparables.
I. Productos que contienen principios activos de administración oral que no deban
absorberse.
NOTA: Se considera “esencialmente los mismos excipientes” a excipientes del
mismo tipo en cuanto a que posean la misma función en la formulación
(dispersante, aglomerante, espesante, etc.), aunque no se trate de la misma
molécula y que los excipientes que contengan no afecten el transito
gastrointestinal, la absorción o la estabilidad del principio activo.
Para los medicamentos cuyo efecto se ejerce a nivel local sin absorción sistémica
(administración dérmica, nasal, vaginal, colirios, gotas oticas, etc.) no se pueden
realizar estudios de Bioequivalencia. En estos casos se realizan estudios clínicos
comparativos para demostrar la Bioequivalencia.

5.7. RECOMENDACIONES
Aunque nacional e internacionalmente se acepte que existen formas
farmacéuticas que por sus características, no requieren estudios de
Bioequivalencia (Res. 1400 de 2001) (OMS, 1996), de acuerdo a las
características de solubilidad y permeabilidad (absorción), puede existir
excepciones(FDA, 2000,“Dispensa de estudios de biodisponibilidad y bioequivalencia in vivo para
formas farmacéuticas orales sólidas de liberación inmediata”)
CLASIFICACION BIOFARMACEUTICA
Clase 1: alta solubilidad y alta permeabilidad
Clase 2: baja solubilidad y alta permeabilidad
Clase 3: alta solubilidad y baja permeabilidad
Clase 4: baja solubilidad y baja permeabilidad (Kasim, NA, et al: Molecular proprierties of WHO essential drugs and provisional Biopharmaceutical
Classification. Molecular Pharmaceutics 2004: 1(1) 85-96
En este documento se establece que la demostración de Bioequivalencia
puede no ser necesaria para medicamentos que contienen principios
activos comprendidos en la Clase 1 (alta solubilidad y alta permeabilidad).

El test de disolución in vitro tiene que ser optimizado para que sea
sensible a los cambios en las variables críticas de manufactura, dentro de
un rango de valores establecidos durante el proceso de elaboración. Esto
asegura que las variaciones entre lotes no resulten Bioinequivalentes
Dayami Carrión Recio. Rev Cubana Farm 1999; 33: 137-42.
“Cuando los países carecen de los medios para aplicar plenamente el
estándar (de Bioequivalencia),se recomienda que se aplique
gradualmente”. Consultation of Experts on Bioequivalence of Pharmaceutical Products. Caracas,
Venezuela, Enero 13-15 de 1999. Documento OMS-OPS, Junio de 1999.
Es fácil comprender que la implementación de un programa de
Biodisponibilidad/Bioequivalencia es un proceso gradual y progresivo,
de acuerdo a las características de cada país.
Esta es la razón por la cual el control de calidad sugerido por la
farmacopea para formas de dosificación oral no asegura en muchos
casos la Bioequivalencia.
Por ello en este tipo de fármacos el estudio de disolución in vitro no
puede sustituir al estudio de Bioequivalencia, hasta tanto no sea
relacionado con datos in vivo.

En la reunión de caracas de 1999 se recomendó el abordaje de la
problemática de la Bioequivalencia, teniendo en cuenta el concepto de
RIESGO SANITARIO. En algunos países para la selección de principios
activos que deben ser sometidos a estudios de Bioequivalencia, se
tienen en cuenta dos aspectos:
1. El riesgo sanitario
2. Fármacos en los que se les exige Bioequivalencia y cada tipo de riesgo se le asignó un puntaje de acuerdo a su gravedad
(alto: 3 puntos; intermedio: 2 puntos; bajo: 1 punto). Las categorías adoptadas fueron las siguientes
a) Riesgo sanitario alto: complicaciones graves o RAMS graves
b) Riesgo sanitario intermedio: complicaciones y RAMS no graves
c) Riesgo sanitario bajo: complicaciones y RAMS menores
La aplicación de este modelo dio como resultado una categorización de
los fármacos, que se vio plasmada en un listado. Allí, naturalmente,
figuran en primer lugar aquellas drogas con ventana terapéutica estrecha
(igual o menor que 2).

Según el riesgo sanitario de cada fármaco, la equivalencia química
(principios activos “idénticos”, igual concentración y forma
farmacéutica similar) no es suficiente y se requiere Bioequivalencia
para disminuir al mínimo la probabilidad de alteraciones en las
respuestas terapéuticas por cambios debidos a factores
condicionantes de la Biodisponibilidad
Seria un error pensar que todos los medicamentos químicamente
equivalentes y con similar forma farmacéutica tienen la misma
Biodisponibilidad, eficacia y seguridad independientemente de su
fabricante.
Teniendo en cuenta que la problemática de la Bioequivalencia es
dinámica y adaptable a la evolución del conocimiento científico, a las
necesidades sanitarias y la normatividad de cada país esto no
constituye un sistema cerrado sino abierto a modificaciones y nuevas
incorporaciones.
Aun existen detalles mejorables de la normatividad y el impacto de
los estudios de Bioequivalencia sobre los hábitos de prescripción, el
mercado farmacéutico en Colombia y la accesibilidad a
medicamentos genéricos eficaces y seguros.

FARMACOS CON RIESGO SANITARIO
RIESGO ALTO RIESGO INTERMEDIO
Carbamazepina Azatioprina
Ciclosporina Biperideno HCl
Fenitoina sodica Carbidopa
Litio carbonato Etambutol HCl
Procainamida Etinilestradiol
Tolbutamida Etoposido
Valproato sodico Flucitosina
Verapamilo sodico Fludrocortisona
Etosuximida Levodopa
Insulina Levonorgestrel
Quinidina Mefloquina HCl
Warfarina Metotrexato sodico
Propranolol
Rifampicina
Salbutamol
Atenolol
Diazepam
Metildopa

5.8. LOS MEDICAMENTOS GENERICOS La exigencia de demostración de la intercambiabilidad de medicamentos genéricos en relación con los innovadores comenzó en el mundo desarrollado en la década de 1970, cuando se constataron problemas de Bioequivalencia. Luego a principios de la primera década de este siglo, impulsado por un grupo de trabajo de la Organización Panamericana de la Salud (OPS) se comenzó a legislar sobre esta materia.
¿POR QUÉ SON MÁS BARATOS LOS MEDICAMENTOS GENÉRICOS?
1. No hay investigación preclínica ni clínica por lo que no deben soportar los importantes gastos en I+D. 2. No hay recursos para demostrar la seguridad y eficacia ya que son las mismas que el original y en muchas ocasiones no asume costos de investigación de Bioequivalencia. 3.Se dedican pocos recursos a la promoción farmacéutica 4. Los procesos de registro legal son más simples mas rápidos y menos costosos.
En ningún caso, UN MEDICAMENTO GENERICO como consecuencia de
calidad, eficacia y/o seguridad debe ser inferiores respecto al
medicamento innovador o de referencia.
Sólo en un mercado de medicamentos de calidad incuestionable, se
puede elegir por precio porque la intercambiabilidad está
garantizada.

5.8.1.GARANTIA DE LA CALIDAD, EFICACIA Y SEGURIDAD DE LOS MEDICAMENTOS GENÉRICOS
Los prescriptores y el público pueden estar seguros que si
la FDA declara que un medicamento genérico es terapéuticamente
equivalente al original, ambos productos proporcionarán los mismos
efectos clínicos esperados. Jane E. Henney (Commissioner of Food and Drugs); JAMA 1999
Aproximadamente, el 50 % de los medicamentos que se prescriben en
Estados Unidos son genéricos.
Todos los productos que fueron aprobados por la FDA cumpliendo las
especificaciones de la USP han mostrado ser Bioequivalentes. Jerome P. Skelly. 2005 President, American Association of Pharmaceutical Scientist . Drug Discovery
Today; 2005, 10: 682-685
“La calidad de un medicamento se mide por la capacidad de ejercer el efecto
terapéutico que de él se espera. Esa capacidad es determinada por las
propiedades que influencian en los resultados, como su identidad, pureza,
contenido o potencia, las propiedades químicas, físicas y biológicas o de su
proceso de fabricación Estrategia de los medicamentos genericos de los paises de la subregion andina (Julio 2012)

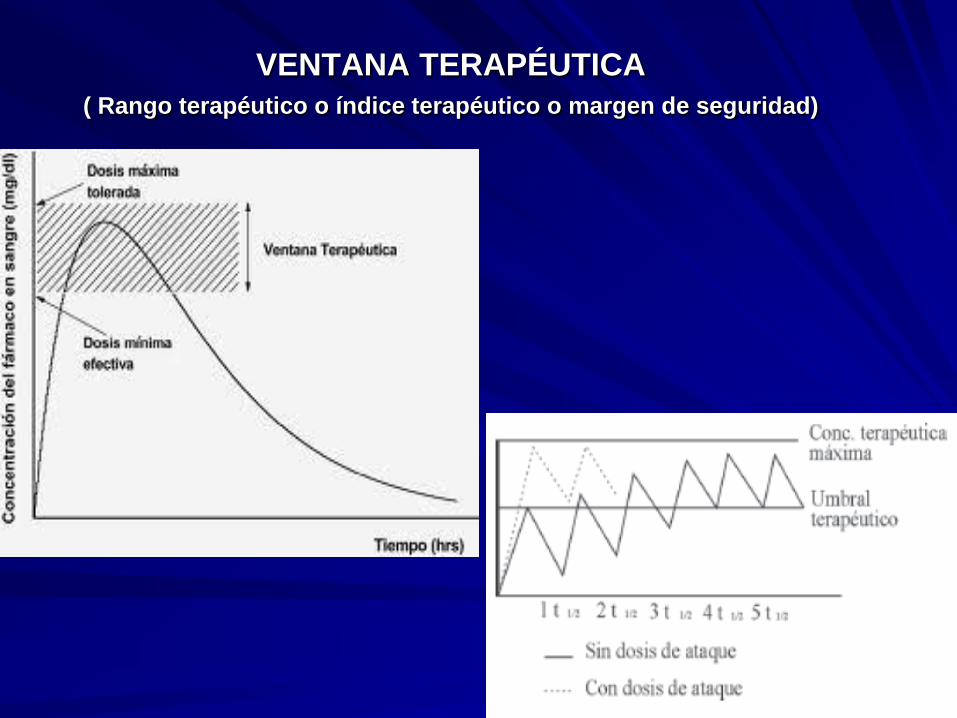
5.9. VENTANA TERAPÉUTICA
( Rango terapéutico o índice terapéutico o margen de seguridad)
La ventana terapéutica es el rango en el cual se puede utilizar un fármaco sin
provocar efectos tóxicos o letales en el organismo vivo.
se establece bajo un margen superior, en el cual se corre el riesgo que durante el
uso de un fármaco, éste cause un efecto tóxico; y un rango mínimo bajo el cual, el
uso del fármaco tendrá un efecto ineficaz en el individuo.
es un valor que se establece matemáticamente como la relación entre la dosis con
la cual se produce un daño en el 50% de la población (DT50 o DL50) y la dosis con
la cual se obtiene un efecto terapéutico en el 50% de la población muestra (DE50)
de tal forma que se obtiene un valor numérico. En este contexto, si el valor
obtenido de la razón llamada Índice Terapéutico es cercano a "1" se corre un
riesgo muy alto de producir un efecto tóxico en el uso del fármaco, entonces, a
medida que el valor obtenido se aleja de la unidad la seguridad relativa, es decir, la
seguridad en el uso de cierto fármaco, es mayor, de tal forma que su uso no
causaría tan rápidamente un efecto tóxico, por lo cual el efecto terapéutico es más
seguro, y la dosis a usar pueden ser más amplias
Cociente entre la Concentración Máxima Efectiva (No tóxica) y la
Concentración Mínima Efectiva. Se estima que debe realizarse el
seguimiento de dosificación de los pacientes que reciben principios activos
con una Ventana Terapéutica igual o menor que 2 (dos)
Ej. digoxina, litio, teofilina, fenitoina y warfarina
VENTANA TERAPÉUTICA
( Rango terapéutico o índice terapéutico o margen de seguridad)
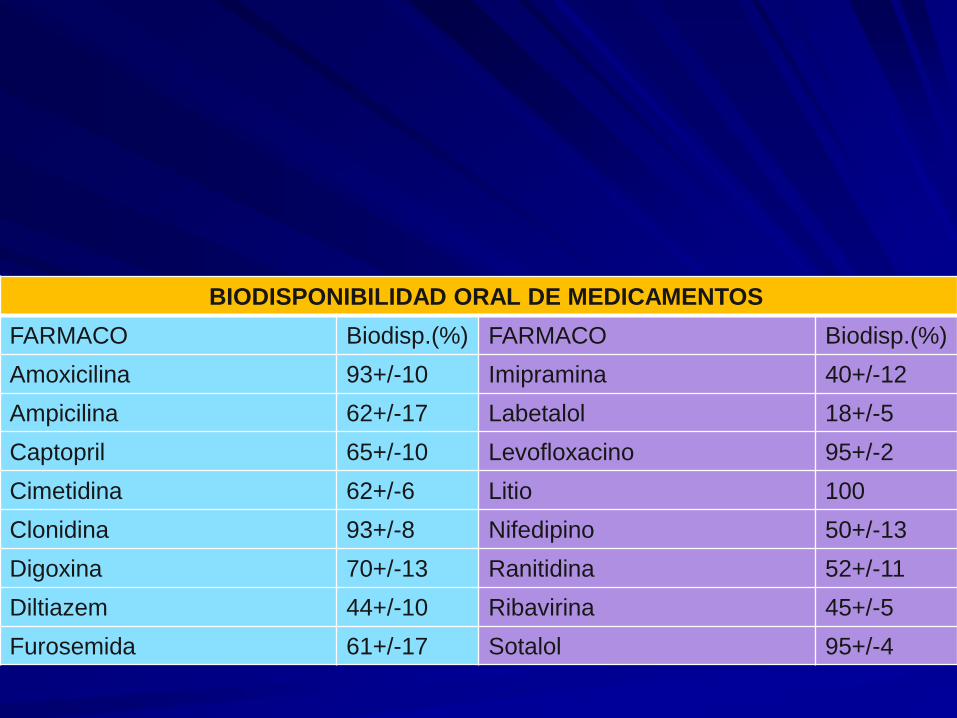
BIODISPONIBILIDAD ORAL DE MEDICAMENTOS
FARMACO Biodisp.(%) FARMACO Biodisp.(%)
Amoxicilina 93+/-10 Imipramina 40+/-12
Ampicilina 62+/-17 Labetalol 18+/-5
Captopril 65+/-10 Levofloxacino 95+/-2
Cimetidina 62+/-6 Litio 100
Clonidina 93+/-8 Nifedipino 50+/-13
Digoxina 70+/-13 Ranitidina 52+/-11
Diltiazem 44+/-10 Ribavirina 45+/-5
Furosemida 61+/-17 Sotalol 95+/-4

BIBLIOGRAFIA
Brunton, L; Lazo, J; Parker, K. Goodman & Gilman. Las bases
farmacológicas de la terapéutica. Mc Graw Hill. 11 edición, 2007
Helman, José. Farmacotecnia. Teoría y practica. Editorial continental.
1982
Resolución 1400 (agosto 24) de 2001
Resolución 1890 de 2001(modifica la resolución 1400 de 2001)
Note for Guidance on the investigacion of bioavailability and
bioequivalence (CPMP/EW/QWP/1401/98).EMEA,2000
http://apps.who.int/gb/archive/pdf_files/WHA54/sa5417.pdf
NOTA: Recuerda que las leyes y las interpretaciones cambian.
Esta presentación que estas leyendo solo refleja lo que opine el día de su publicación.
Esta presentación no tiene finalidad comercial. Algunas imágenes publicadas han sido extraídas de
internet y son etiquetadas para reutilización, por lo que damos nuestros mas sinceros
agradecimientos a las fuentes:
Wikipedia; Public Domain; Good Free Photos; Free Stock Photos.
En todo caso no es nuestra intención atribuirse la autoría de ellas, solo se utilizan para crear un
concepto grafico asociado al tema.
www.farmacus.com.co [email protected] [email protected]
Síguenos en twitter: @GRUPOFARMACUS Búscanos en Facebook: farmacus.grupodecapacitacion


















