
ciqa.repositorioinstitucional.mx · tesis “estudio de los factores que afectan la polimerizaciÓn...
113
TESIS “ESTUDIO DE LOS FACTORES QUE AFECTAN LA POLIMERIZACIÓN RADICÁLICA CONTROLADA POR NITRÓXIDOS EN EMULSIÓN” Presentada por: ANDRES CANO VALDEZ Para obtener el grado de: MAESTRO EN TECNOLOGÍA DE POLÍMEROS Asesor: Dr. Enrique Saldívar Guerra Saltillo, Coahuila. Agosto 2011 - -- - - CENTRO DE INVESTIGACIÓN EN QUÍMICA APLICADA r:- r-
-
Upload
nguyendung -
Category
Documents
-
view
216 -
download
0
Transcript of ciqa.repositorioinstitucional.mx · tesis “estudio de los factores que afectan la polimerizaciÓn...

TESIS
“ESTUDIO DE LOS FACTORES QUE AFECTAN LA
POLIMERIZACIÓN RADICÁLICA CONTROLADA
POR NITRÓXIDOS EN EMULSIÓN”
Presentada por:
ANDRES CANO VALDEZ
Para obtener el grado de:
MAESTRO EN TECNOLOGÍA DE POLÍMEROS
Asesor:
Dr. Enrique Saldívar Guerra
Saltillo, Coahuila. Agosto 2011
- -- - -CENTRO DE INVESTIGACIÓN EN QUÍMICA APLICADA
r:- r-

DEDICATORIA
A mis padres Rubén Cano Caballero y Silvia Valdez Patiño por apoyarme en
todo momento y enseñarme a ser un hombre de bien, así como a mis
hermanas Carolina Cano Valdez y Estefania Cano Valdez por brindarme su
apoyo y comprensión.
A la familia de mi esposa Velazquez Garcia por apoyarnos y guiarnos en
todo momento.
A mi esposa Areli Irasu Velazquez Garcia por toda su compresión, apoyo y
amor incondicional.
Te amo Princesa.

AGRADECIMIENTOS
Al Consejo Nacional de Ciencia y Tecnología (CONACYT) y al Centro de
Investigación en Química Aplicada (CIQA) por el apoyo en el proyecto para obtener mi
grado de Maestro en Tecnología en Polímeros.
A mi asesor el Dr. Enrique Saldívar Guerra por su apoyo y confianza durante la
realización de esta tesis y su asesoramiento en la escritura de tesis.
A mis sinodales, al Dr. Enrique Jiménez, al Dr. Jorge Herrera y al Dr. Raúl
Guillermo López Campos por su cooperación en la revisión del documento y sugerencias.
Al Dr. Enrique Jiménez y a la I.Q. Areli Irasu Velazquez Garcia por su apoyo con
material de laboratorio. A la M.C. Hened Saade y la M.C. Gladys Cortez por su ayuda en el
laboratorio.
A mi grupo de trabajo, M.C. Almendra Ordáz, Ing. Ricardo Mendoza, M.C. Roberto
Blanco, M.C. Omar García, M.C. David Contreras y M.C. Enrique García por su ayuda y
consejos.
Al Dr. Ramiro Guerrero y a su grupo de trabajo, Dr. Francisco Javier Enriquez,
M.C. Hortensia Maldonado Textle por su apoyo en la parte experimental y caracterización
de ésta tesis.
Al Dr. Ramón Díaz de León y a su grupo de trabajo, Ing. Jose Alejandro Díaz y
M.C. Beatriz Reyes por su ayuda en la parte experimental de la tesis.
Al Dr. Enrique Elizalde y al M.C. Jorge Félix por su ayuda en la caracterización de
ésta tesis. Y a la M.C. Gladys de los Santos por su apoyo y consejos como tutora.

i
CONTENIDO
Índice de figuras…………………………………………………………………………..iv
Índice de tablas…………………………………………………………………………..vii
Resumen…………………………………………………………………………………..ix
1. Introducción……………………………………………………………………….1
2. Antecedentes……………………………………………………………………....2
2.1 Polimerización radicálica controlada…………………………………………2
2.1.1 NMRP (Nitroxide Mediated Radical Polymerization)………………...4
2.1.1.1 Proceso Bimolecular……………………………………………….6
2.1.1.2 Proceso Unimolecular……………………………………………...7
2.2 Polimerización radicálica en medios dispersos……………………………….9
2.2.1 Suspensión……………………………………………………………10
2.2.2 Emulsión……………………………………………………………...11
2.2.3 Otros………………………………………………………………….13
2.3 NMRP en emulsión ab initio…………………………………………………14
2.4 Problemas en NMRP en emulsión…………………………………………...16
2.5 Posibles soluciones…………………………………………………………...16
2.5.2 Reparto de nitróxido velocidad de agitación y presión..……………….17
2.6 Modelos matemáticos para polimerización radicálica en emulsión………….19
2.7 Modelos matemáticos para polimerización radicálica en medios
heterogéneos mediada por nitróxidos………………………………………..21
2.8 Justificación…………………………………………………………………..24
3. Hipótesis y Objetivos…………………………………………………………….26
3.1 Hipótesis……………………………………………………………………...26
3.2 Objetivo………………………………………………………………………26
4. Parte experimental………………………………………………………………..27
4.1 Reactivos y Materiales……………………………………………………….27
4.2 Equipo………………………………………………………………………..27
4.2.1 Reparto de nitróxidos………………………………………………...27

ii
4.2.2 Polimerización de estireno medidada por nitróxidos
en emulsión ab initio………………………………………………...28
4.3 Caracterización………………………………………………………………29
4.3.1 Cuantificación del nitróxido en los experimentos del
reparto………….……………………………………………………..29
4.3.2 Polimerización………………………………………………………..29
4.4 Metodología………………………………………………………………….29
4.4.1 Reparto de nitróxidos………………………………………………...29
4.4.1.1 Cálculo del coeficiente de extinción molar………………………29
4.4.1.2 Cálculo del coeficiente de reparto………………………………..30
4.4.2 Polimerización de estireno mediada por nitróxidos
en emulsión ab initio…………………………………………………31
4.4.3 Caracterización……………………………………………………….33
4.4.3.1 Cuantificación del nitróxido en los experimentos
de reparto…………………………………………………………33
4.4.3.2 Polimerización……………………………………………………33
5. Resultados y Discusión…………………………………………………………..34
5.1 Coeficientes de reparto a 25, 90, 120 y 135 °C para el
OH-TEMPO………………………………………………………………….34
5.1.1 Cálculo de los coeficientes de extinción molar en estireno
y agua desionizada…………………………………………………...34
5.1.2 Cálculo de los coeficientes de reparto………………………………..37
5.2 Coeficientes de reparto a 25, 90, 120 y 135 °C para el
OXO-TEMPO……………………………………………………………….39
5.2.1 Cálculo de los coeficientes de extinción molar en estireno
y agua desionizada…………………………………………………...39
5.2.2 Cálculo de los coeficientes de reparto………………………………..41
5.3 Coeficientes de reparto a 25, 90, 120 y 135 °C para el
TEMPO………………………………………………………………………43
5.3.1 Cálculo de los coeficientes de extinción molar en estireno
y agua desionizada………..………………………………………….43

iii
5.3.2 Cálculo de los coeficientes de reparto……………………………….44
5.4 Coeficientes de reparto a 25, 90, 120 y 135 °C para el
TIPNO……………………………………………………………………….46
5.4.1 Cálculo de los coeficientes de extinción molar en estireno
y agua desionizada…………………………………………………..46
5.4.2 Cálculo de los coeficientes de reparto………………………………..48
5.5 Comparación de los coeficientes de reparto del OH-TEMPO,
OXO-TEMPO, TEMPO y TIPNO a diferentes temperaturas………………49
5.6 Polimerización de estireno mediada por nitróxidos en emulsión
ab initio………………………………………………………………………53
5.6.1 Efecto de la presión a nivel alto y bajo de agitación
en la conversión, Dp, Np, Mn y PDI. Comparación
de los tratamientos R1(+)(-) con R3(+)(+), R4(-)(-)
con R2(-)(+)….……………………………………………………….54
5.6.2 Efecto de la velocidad de agitación a nivel alto y
bajo de presión en la conversión, Dp, Np, Mn y PDI.
Comparación de los tratamientos R2(-)(+) con
R3(+)(+) y R4 (-)(-) con R1(+)(-).…………………………………...59
5.6.3 Análisis estadístico…………………………………………………...65
5.6.4 Comparación de los cuatro nitróxidos utilizados…………………….72
5.7 Introducción de los términos del nitróxido a un modelo
matemático simplificado para polimerización en emulsión…………………76
6. Conclusiones……………………………………………………………………..93
7. Referencias……………………………………………………………………….94
ANEXO……………………………………………………………………………..100

iv
ÍNDICE DE FIGURAS
2.1 Mecanismo de una polimerización controlada/viviente……………………………….5
2.2 Esquema de un sistema bimolecular de una polimerización
mediada por nitróxidos. ………………………………..…………………………….7
2.3 Esquema de un sistema unimolecular de una polimerización
mediada por nitróxidos. ………………………………..…………………………….8
4.1 Sistema utilizado para los experimentos de reparto………………………………….28
4.2 Sistema utilizado para las polimerizaciones………………………………..………..28
5.1 Curva de calibración para el cálculo del coeficiente de extinción
molar para el OH-TEMPO en estireno. ………………………………..…………...35
5.2 Curva de calibración para el cálculo del coeficiente de extinción
molar para el OH-TEMPO en agua desionizada. ………………………………..….36
5.3 Gráfica comparativa de los experimentos de reparto a diferentes
temperaturas para el cálculo de los coeficientes de reparto del
OH-TEMPO. ………………………………..………………………………..……...37
5.4 Curva de calibración para el cálculo del coeficiente de extinción
molar para el OXO-TEMPO en estireno. ………………………………..………….39
5.5 Curva de calibración para el cálculo del coeficiente de extinción
molar para el OXO-TEMPO en agua desionizada. …………………………………40

v
5.6 Gráfica comparativa de los experimentos de reparto a diferentes
temperaturas para el cálculo de los coeficientes de reparto del
OXO-TEMPO. ………………………………..………………………………..……41
5.7 Curva de calibración para el cálculo del coeficiente de extinción
molar para el TEMPO en estireno. ………………………………..………………...43
5.8 Curva de calibración para el cálculo del coeficiente de extinción
molar para el TEMPO en agua desionizada. ………………………………..………44
5.9 Gráfica comparativa de los experimentos de reparto a diferentes
temperaturas para el cálculo de los coeficientes de reparto del
TEMPO. ………………………………..………………………………..…………..45
5.10 Curva de calibración para el cálculo del coeficiente de extinción
molar para el TIPNO en estireno. ………………………………..…………………46
5.11 Curva de calibración para el cálculo del coeficiente de extinción
molar para el TIPNO en agua desionizada. ………………………………..………..47
5.12 Gráfica comparativa de los experimentos de reparto a diferentes
Temperaturas para el cálculo de los coeficientes de reparto del
TIPNO. ………………………………..………………………………..…………...48
5.13 Gráficas de los coeficientes de reparto para el OH-TEMPO,
OXO-TEMPO, TEMPO y TIPNO a 25, 90, 120, y 135°C. ………………………...51
5.14 Efecto de la presión a nivel alto (350 rpm) y bajo (250 rpm)
de agitación sobre la conversión final (X). Comparación de los
tratamientos R1(+)(-) con R3(+)(+) y R4(-)(-) con R2(-)(+).………………………..55

vi
5.15 Efecto de la presión a nivel alto (350 rpm) y bajo (250 rpm)
de agitación sobre el Np y Dp. Comparación de los tratamientos
R1(+)(-) con R3(+)(+) y R4(-)(-) con R2(-)(+).………………………………..…….56
5.16 Efecto de la presión a nivel alto (350 rpm) y bajo (250 rpm)
de agitación sobre el Mn y PDI. Comparación de los tratamientos
R1(+)(-) con R3(+)(+) y R4(-)(-) con R2(-)(+).……………………………………...57
5.17 Gráfica comparativa de la distribución de peso molecular
final del diseño experimental. ………………………………..……………………..58
5.18 Efecto de la velocidad de agitación a nivel alto (200 psia)
y bajo (100 psia) de presión sobre la conversión final (X).
Comparación de los tratamientos R2(-)(+) con R3(+)(+) y
R4 (-)(-) con R1(+)(-).………………………………..………………………………60
5.19 Efecto de la velocidad de agitación a nivel alto (200 psia)
y bajo (100 psia) de presión sobre el Np y Dp. Comparación de
los tratamientos R2(-)(+) con R3(+)(+) y R4 (-)(-) con R1(+)(-).…………………...61
5.20 Efecto de la velocidad de agitación a nivel alto (200 psia)
y bajo (100 psia) de presión sobre el Mn y PDI. Comparación
de los tratamientos R2(-)(+) con R3(+)(+) y R4 (-)(-) con R1(+)(-).………………..62
5.21 Gráfica comparativa de la distribución de peso molecular final
de las pruebas R3 (+)(+), R5, R6, R7 a 125°C y R8 a 90°C). ……………………...75
5.22 Espectro de resonancia magnética nuclear obtenido de la prueba
R3 (+)(+) (PS-OH-TEMPO). ………………………………..………………………76

vii
ÍNDICE DE TABLAS
2.1 Modelos matemáticos y variables obtenidas en polimerización
en emulsión…………………………………………………………………………..20
4.1 Cantidades empleadas en cada uno de los experimentos de
reparto………………………………………………………………………………..30
4.2 Niveles de agitación (RPM) y presión (P) utilizados con
St/OH-TEMPO………………………………………………………………………31
4.3 Reacciones de polimerización de estireno utilizando el mejor
tratamiento (R3 (+)(-)) con OXO-TEMPO, TEMPO y TIPNO…………………….32
4.4 Composición inicial de la prueba R1 (+)(-) del diseño experimental………………..32
5.1 Coeficientes de reparto y ajustes lineales de los datos experimentales
obtenidos del OH-TEMPO………………………………………………………….38
5.2 Coeficientes de reparto y ajustes lineales de los datos experimentales
obtenidos del OXO-TEMPO………………………………………………………...42
5.3 Coeficientes de reparto y ajustes lineales de los datos experimentales
obtenidos del TEMPO………………………………………………………………..46
5.4 Coeficientes de reparto y ajustes lineales de los datos experimentales
obtenidos del TIPNO………………………………………………………………..49
5.5 Coeficientes de reparto calculados a diferentes temperaturas para
el OH-TEMPO, OXO-TEMPO, TEMPO y TIPNO………………………………50

viii
5.6 Ajustes polinomiales para la interpolación de los coeficientes de
reparto………………………………………………………………………………..51
5.7 Porciento mol de los nitróxidos en cada una de las fases a diferentes
temperaturas…………………………………………………………………………52
5.8 Resumen de resultados de las polimerizaciones del diseño experimental…………...54
5.9 Peso molecular promedio peso, peso molecular promedio número
e índice de polidispersidad en e l polímero a diferente velocidad
de agitación y presión………………………………………………………………..58
5.10 Peso molecular número teórico, peso molecular número experimental
y eficiencia aparente del nitróxido, para cada uno de los tratamientos…………...64
5.11 Efecto de las variables de velocidad de agitación y presión en las variables
de respuesta del diseño experimental………………………………………………………...71
5.12 Resumen de resultados de las polimerizaciones con OH-TEMPO,
OXO-TEMPO, TEMPO y TIPNO…………………………………………………...72
5.13 Peso molecular número teórico, peso molecular número experimental
y eficiencia aparente del iniciador, para cada uno de las pruebas…………………...73
5.14 Peso molecular promedio peso, peso molecular promedio número
e índice de polidispersidad en el polímero para las pruebas R3(+)(+),
R5, R6, R7 y R8……………………………………………………………………...75

ix
RESUMEN
En esta investigación se presenta el cálculo de los coeficientes de reparto para los
nitróxidos OH-TEMPO, OXO-TEMPO, TEMPO y TIPNO a diferentes temperaturas 25,
90, 120 y 135 °C. Los valores obtenidos para el OH-TEMPO fueron 0.87, 9.9, 12.8 y
14.5 %mol/%mol, para el OXO-TEMPO fueron 28, 160, 91 y 77 %mol/%mol, para el
TEMPO fueron 719, 911, 683 y 635 %mol/%mol y finalmente para el TIPNO fueron
707, 943, 710 y 612 %mol/%mol. Se observó que a pesar de que el OH-TEMPO tiene un
mayor carácter hidrófilo que los otros tres, éste presenta un porcentaje mol muy bajo en
la fase acuosa y posiblemente no es suficiente para atrapar los radicales primarios en el
agua, lo cual provoca un bajo control y estabilidad coloidal. Posteriormente se realizó un
diseño experimental para observar el efecto de la velocidad de agitación y presión en las
variables de respuesta de la NMRP en emulsión. El mejor tratamiento fue el que se
realizó con los niveles altos de velocidad de agitación y presión R3(+)(+), el cual
presentó un buen control y estabilidad coloidal. También el tratamiento R1(+)(-) presentó
buenos resultados, sin embargo se optó por las condiciones del tratamiento R3(+)(+) para
utilizarlas en la polimerización con OXO-TEMPO, TEMPO y TIPNO, con el fin de
comparar los cuatro nitróxidos y observar el efecto del reparto del nitróxido en el control
y estabilidad coloidal. Los resultados mostraron que el OH-TEMPO fue el nitróxido que
controló de mejor manera la polimerización y con éste se obtuvo una mejor estabilidad
coloidal. Sin embargo, posiblemente un nitróxido más hidrófilo controle mejor ya que
éste podrá atrapar los radicales primarios y por ende incrementar el número de partículas
y disminuir el tamaño de partícula.

1
1. INTRODUCCIÓN
La polimerización radicálica ha sido una importante área tecnológica por setenta
años debido a su impacto en la sociedad. Aproximadamente un 50 % de todos los
polímeros que utilizamos hoy en día (plásticos, elastómeros, pinturas, fibras, etc.) se
producen por un mecanismo de radicales libres. Sin embargo, en los últimos 15 años la
química de polimerización por radicales libres ha experimentado una renovación debido al
descubrimiento de nuevas técnicas de polimerización por radicales libres denominadas
controladas/vivientes (CRP: Controlled Radical Polymerization o Polimerización
Radicálica Controlada). Estas nuevas técnicas combinan la robustez de la química
tradicional de polimerización de radicales libres (tolerancia a impurezas y versatilidad en
cuanto al tipo de monómeros y el tipo de procesos de polimerización que pueden ser
utilizados) con la posibilidad de diseñar y producir polímeros con pesos moleculares y
estructuras que no pueden ser obtenidas mediante un proceso tradicional por radicales
libres.
Las polimerizaciones controladas/vivientes en medios acuosos presentan retos y
problemas que aun no se han resuelto, por un lado se tiene el problema del control de la
estabilidad coloidal debido a los fenómenos de superhinchamiento y de Ostwald ripening,
los cuales han sido poco estudiados.1 Posibles soluciones para estos problemas pueden
provenir del estudio del reparto de los nitróxidos entre la fase acuosa y orgánica, por ser un
fenómeno de importancia en estos sistemas.
Es por eso que en este trabajo se tratará de entender de una mejor manera el reparto
del nitróxido entre la fase acuosa y orgánica, para posteriormente realizar una comparación
del control y estabilidad coloidal de cada uno de los nitróxidos considerados en ésta
investigación, en un proceso de polimerización en emulsión ab initio.

2
2. ANTECEDENTES
2.1 Polimerización Radicálica Controlada
El término de polimerización viviente fue definido por primera vez por Szwarc2
como el crecimiento de una cadena que no experimenta reacciones de terminación y/o
transferencia; además, si se adiciona más monómero las cadenas seguirán creciendo, esto
debido al control de los grupos terminales (polimerizaciones aniónica, catiónica y
coordinación), sin embargo no todas las polimerizaciones vivientes poseen un buen control
de los principales parámetros (índice de polidispersidad (PDI), peso molecular (MW),
conversión).1
Para poder controlar dichos parámetros, el iniciador debe consumirse al inicio
de la polimerización rápidamente y el intercambio entre las diferentes especies debe ser
muy rápido(cuando hay un equilibrio entre especies vivas y durmientes).3
Más tarde se encontró que existían polimerizaciones no totalmente vivientes que se
denominaron “controladas” o pseudovivientes, en las cuales se tiene un control del peso
molecular y de la distribución de pesos moleculares (MWD) pero que aún siguen
experimentando en mucho menor grado reacciones de terminación como en el mecanismo
de radicales libres convencional.4
Finalmente se utilizó el término viviente/controlada para
este tipo de polimerizaciones, sin embargo cabe mencionar que las polimerizaciones
pueden ser vivientes pero no controladas, esto quiere decir que el índice de polidispersidad
aumenta, e incluso se tienen distribuciones de pesos moleculares bimodales, también puede
ocurrir el caso opuesto en el que existe un buen control pero la polimerización no es
viviente.5
Una polimerización bien controlada provee:
1. La conversión en escala logarítmica varía linealmente con el tiempo, si la
reacción es de primer orden con respecto a la concentración de monómero.
2. La evolución del peso molecular es lineal con la conversión.

3
3. La polidispersidad (Mw/Mn) debe de disminuir con la conversión para sistemas
con iniciación e intercambio rápido de las especies.
Una iniciación rápida es un prerrequisito para la síntesis de polímeros con grados de
polimerización predeterminados por la relación de monómero/iniciador ([M]/[I]0). En
sistemas sin intercambio y sin desactivación de las cadenas, el índice de polidispersidad es
superior a 1.351, como resultado de una iniciación lenta. Una iniciación lenta lleva a una
polimerización más lenta, usualmente acompañada de periodos de inducción y de pesos
moleculares mayores a los predichos. El control de los pesos moleculares y
polidispersidades puede ser afectado también por la lenta velocidad de propagación y la
poca homogeneidad del sistema. La poca solubilidad del iniciador (el solvente depende del
sistema: masa, suspensión, emulsión, etc.) o que el polímero no sea soluble en su
monómero también pueden llevar a altas polidispersidades.1
Igualmente es importante mencionar el efecto del radical persistente, el cual hace
referencia a cualquier radical cuya concentración aumenta durante la reacción en lugar de
disminuir, un ejemplo muy sencillo es tomar dos radicales que se encuentran en una
reacción X• y Y
•, se esperaría que las reacciones que ocurrieran fueran X-X, Y-Y y X-Y,
sin embargo la característica de los radicales persistentes (en nitróxidos y en sistemas
ATRP), es que no reaccionan entre ellos mismos, esto debido a que a ciertas temperaturas
estos radicales prefieren por completo la terminación cruzada, por lo tanto al no reaccionar
X-X, las únicas reacciones que se llevan a cabo son Y-Y y X-Y, esto hace que la
concentración del radical X• aumente durante la reacción. Cuando radicales estables, como
los nitróxidos o especies intermedias controladoras (ATRP) son generados en conjunto con
los radicales primarios (iniciador), y radicales monómericos, el acoplamiento cruzado es
mucho más rápido que el acoplamiento entre las mismas especies de radicales. A las
especies nuevas se les llama cadenas durmientes, debido a que pueden ser activadas
nuevamente.1
En la actualidad existen varios mecanismos químicos de radicales libres que
presentan carácter viviente, la mayoría de ellos requieren de la adición de uno o más

4
compuestos a la polimerización (agente o sistema controlador) los cuales se encargan de
darle el carácter viviente. Los mecanismos más importantes son los siguientes:
1. Polimerización radicálica de transferencia de átomo (Atom Transfer Radical
Polymerization o ATRP)6
2. Polimerización vía transferencia – adición – fragmentación reversible
(Reversible Addition Fragmentation Transfer o RAFT)7,8
3. Polimerización radicálica controlada por nitróxidos (Nitroxide Mediated Radical
Polymerization o NMRP)9-12
De estos tres mecanismos, la polimerización radicálica controlada por nitróxidos es
la que presenta mayores posibilidades de comercialización a corto plazo, esto debido a que
los agentes controladores (nitróxidos) son fáciles de conseguir comercialmente y se pueden
usar sin violar patentes a diferencia de los componentes para ATRP o RAFT.
2.1.1 NMRP (“Nitroxide Mediated Radical Polymerization”)
En este mecanismo químico, la terminación reversible de las cadenas en crecimiento
es el paso clave para reducir la concentración de radicales terminales propagantes. También
asumiendo que no existen reacciones laterales tales como la del nitróxido con el monómero
vinílico, la concentración de cadenas activas es extremadamente baja, esto quiere decir que
el nitróxido (N•) no funciona como iniciador, por lo tanto no puede reaccionar con el
monómero y solo se encarga de atrapar los diferentes radicales (primarios (R•),
monómericos (M•) o cadenas activas de polímero (P•)), es por eso que se busca que el
iniciador se descomponga lo más rápido posible para asegurar que todas las cadenas se
inicien al mismo tiempo y que éstas tengan en un extremo el nitróxido para controlar el
crecimiento de la cadena, es por esto que se reduce la probabilidad de terminación
irreversible (combinación o desproporción). Algo muy importante es que las cadenas deben
de ser iniciadas solo a partir de las especies deseadas (iniciador), debido a que este tipo de
polimerización se lleva a cabo a temperaturas mayores a los 120 °C, esto para que el
nitróxido funcione como controlador.

5
En el caso de ciertos monómeros vinílicos se puede presentar la autoiniciación térmica a lo
largo de la polimerización, lo cual provoca distribuciones de pesos no deseadas e incluso
bimodales. Otra cosa de suma importancia es que el crecimiento debe ser en una manera
pseudo-viviente, esto quiere decir que las cadenas crecen en forma intermitente, cuando de
una cadena polimérica durmiente (P-N) se desprende el nitróxido (N•) dicha cadena se
activa y es capaz de crecer. Esto depende de la concentración de N• en el sistema, si hay
una baja concentración la cadena crecerá considerablemente y si hay mucha concentración
crecerá poco, al acoplarse nuevamente el nitróxido a la cadena ésta pasará a cadena o
especie durmiente. Además, todas las cadenas en crecimiento (caso ideal) deben de tener
durante toda la polimerización una unidad de nitróxido para que se lleve a cabo el
crecimiento controlado de las mismas.
También se debe de tomar en cuenta la identidad del radical persistente (o
controlador), el cual es crítico para el proceso de polimerización.13-15
Entre los radicales y
controladores más utilizados están los nitróxidos y las alcoxiaminas (nitróxido+iniciador),
estos últimos desarrollados como radicales controladores por Solomon, Rizzardo y Moad.16
Iniciación
II 2
1PMI
Propagación
Figura 2.1. Mecanismo de una polimerización controlada/viviente.
En la Figura 2.1 se muestra el mecanismo general de una polimerización
controlada/viviente, donde la especie X• representa el radical controlador, que en este caso
puede ser un nitróxido o la parte controladora de la alcoxiamina. Se pueden observar del

6
lado derecho del equilibrio las especies durmientes o de terminación reversible y del lado
izquierdo las especies en estado de propagación o desacopladas. Existen dos formas de
llevar a cabo la iniciación, la primera es en un sistema bimolecular, en el cual el nitróxido y
el iniciador se adicionan por separado, y el segundo es en un sistema unimolecular, que
consta de una alcoxiamina (nitróxido+iniciador) la cual sirve como iniciador y controlador
al iniciar la polimerización.17
2.1.1.1 Proceso Bimolecular
Este proceso fue desarrollado por el grupo de Georges en XEROX, en un artículo
reporta la preparación de poliestireno de baja polidispersidad por medio de polimerización
mediada por nitróxidos y posteriormente la síntesis de un copolímero en bloques.18
En esta
polimerización se adicionó por separado el iniciador y el controlador, es por eso que se
habla de un sistema bimolecular, en este tipo de sistema se tiene que definir la relación
molar nitróxido/iniciador (N/I), ya que ésta es importante para tener un buen control de la
polimerización. En la Figura 2.2. se aprecia el mecanismo de dicha polimerización, en la
cual al inicio se tienen los tres principales componentes ( iniciador, monómero y nitróxido);
primero el iniciador se rompe homolíticamene para dar lugar a dos moléculas de radicales,
después el iniciador reacciona con el monómero, pero inmediatamente un radical nitróxido
se une al oligómero formando la especie durmiente, por lo tanto cada vez que el nitróxido
se desprende la cadena polimérica crece de forma intermitente, agregándose 1 o 2 unidades
monoméricas. Por último, cuando el monómero se consume y se baja la temperatura, la
mayoría de las cadenas poliméricas tienen ancladas una molécula de nitróxido, por lo tanto
si se agrega más monómero y se eleva nuevamente la temperatura por arriba de los 120°C,
la cadena polimérica seguirá creciendo. Cabe recalcar que los nitróxidos a bajas
temperaturas actúan como inhibidores, sin embargo al trabajar a elevadas temperaturas (>
120°C) éstos actúan como controladores en la polimerización. El trabajo de Georges reveló
muchos de los aspectos fundamentales de las polimerizaciones controladas-vivientes,
trabajos posteriores a éste confirmaron estos aspectos. Es importante recalcar que en este
tipo de polimerizaciones se pueden trabajar con una amplia gama de iniciadores como en la
polimerización radicálica convencional.19

7
Figura 2.2. Esquema de un sistema bimolecular de una polimerización mediada por nitróxidos.
El nitróxido se encarga de reducir la concentración de cadenas en propagación,
generando las especies durmientes que crecen en forma intermitente y controlada, y aunque
existe terminación bimolecular, ésta ocurre en mucho menor grado que en radicálica
convencional y es por ello que estas polimerizaciones no son consideradas como 100%
vivientes.20
2.1.1.2 Proceso Unimolecular
El reconocimiento de la naturaleza pobremente definida de las especies iniciantes en
el proceso bimolecular provocó el desarrollo de un sistema iniciado y controlado por una
sola molécula. Tomando el concepto de los iniciadores bien definidos para las
0 -(}--0) • © • 0-9 Inciador l\1onómero Nitróxido
j 120 ºC
. ··-9 Polímero en crecimiento Nitróxido
Polímero durmiente

8
polimerizaciones vivientes como la aniónica y catiónica, se sintetizaron iniciadores
unimoleculares para la polimerización mediada por nitróxidos. En la Figura 2.3, se puede
ver el mecanismo de un proceso unimolecular, el cual tiene como única diferencia al del
proceso bimolecular la etapa de iniciación, ya que en este tipo de proceso la alcoxiamina se
disocia al alcanzar la temperatura de reacción, generando el radical iniciador y el radical
nitróxido, de aquí en adelante la polimerización se lleva a cabo igual que en el caso
anterior.21
Figura 2.3. Esquema de un sistema unimolecular de una polimerización mediada por nitróxidos.
La estructura de estos iniciadores se basó en la funcionalidad de la alcoxiamina que
está presente en el extremo de la cadena en crecimiento en su etapa durmiente. El enlace
carbono-oxígeno de la alcoxiamina es térmicamente inestable y se descompone con la
presencia de calor, generando una cadena activa en crecimiento. La ventaja de este sistema
unimolecular es que se tiene un mejor control sobre la polimerización. Debido a que se
conoce el número de los sitios de iniciación de la polimerización, el peso molecular puede
Radicales
120 ºC -Alcoxiamina
o :Monómero
Polímero durmiente

9
ser controlado con precisión. También se puede funcionalizar el iniciador unimolecular
para permitir la introducción controlada de grupos funcionales al final de las cadenas del
polímero.
2.2 Polimerización Radicálica en Medios Dispersos
En la actualidad existen diversos procesos de polimerización de gran importancia
industrial que se llevan en medios dispersos acuosos. Los procesos de polimerización
heterogéneos más conocidos van desde la polimerización en suspensión y en emulsión
hasta los procesos para obtener polímeros con determinada tacticidad a partir de
catalizadores Ziegler Natta o metalocenos. Lo que caracteriza a estos procesos es que se
trabaja en un sistema heterogéneo el cual en la mayoría de los sistemas consta de una fase
acuosa que por lo general se encuentra en mayor cantidad, y una fase orgánica en menor
proporción. Según el sistema en que se esté trabajando, además del iniciador o agentes de
transferencia como convencionalmente se utiliza, se adicionan otros componentes a la
receta, por ejemplo tensoactivos, agentes de suspensión, agente hidrófobo etc. El propósito
de estos componentes es ayudar a la estabilidad coloidal del sistema, ya que para alguno de
estos procesos mantener la identidad de las partículas es vital. Existen ventajas y
desventajas de utilizar los procesos antes mencionados, las cuales se abordarán más
adelante en el capitulo.
Un aspecto importante en todos estos casos es el papel que tiene el reparto
termodinámico del monómero, pues determina la concentración de monómero y otros
reactivos en cada una de las fases presentes. Es por eso que se pueden presentar problemas
debido a que el monómero no se encuentra en el sito de reacción resultando en velocidades
bajas. A continuación se hará una breve descripción de los procesos de polimerización
heterogéneos más utilizados.

10
2.2.1 Suspensión
Como antes se mencionó, este sistema heterogéneo acuoso consta por lo general de
una fase acuosa, una fase orgánica en la cual se disuelve el iniciador y finalmente se
adiciona el agente de suspensión el cual puede ser por lo general de tres tipos: polímeros
solubles en agua, polvos inorgánicos finamente divididos y estabilizadores mixtos. Estos
estabilizadores tienen como tarea la disminución de la tensión interfacial entre las gotas de
monómero y agua. Las moléculas del estabilizador se adsorben en la superficie de las gotas
de monómero para generar una barrera que evita la coalescencia que se puede presentar por
la agitación del sistema. Es posible controlar la distribución de tamaños de partícula por
medio del agente de suspensión y la agitación.
Existen tres tipos de polimerización en suspensión bien definidos; 1) polimerización
en perlas, aquí el polímero es soluble en el monómero y las gotas de monómero pasan por
una etapa pegajosa hasta finalmente formar partículas rígidas; 2) en la polimerización en
polvo el polímero no es soluble en su monómero, por lo tanto precipita y forma partículas
porosas; 3) finalmente en masa-suspensión se inicia la polimerización en masa y a
determinada conversión se pasa a un reactor para trabajar en suspensión. El tamaño
promedio de partícula que se obtiene es de 50 a 2000 micras.
Entre las ventajas de la polimerización en suspensión, está la fácil remoción del
calor debido a la baja viscosidad del sistema, también la fácil separación del polímero en
forma de perlas, lo que facilita el manejo posterior en un proceso de extrusión o inyección y
finalmente está el bajo nivel de impurezas del polímero. Las desventajas de este proceso
son: el pobre control de la estabilidad de las gotas, el control del tamaño de partícula es de
manera empírica, se desperdicia volumen del reactor con el agua por lo que se tiene una
baja productividad, y un punto importante es la necesidad de tratar el agua después de
utilizarla en este proceso.22

11
2.2.2 Emulsión
En este tipo de polimerización se tienen los mismos componentes generales de un
proceso heterogéneo pero con la adición de un tensoactivo, que se encarga de la estabilidad
coloidal; al igual que en suspensión el monómero es escasamente soluble en agua y a
diferencia de suspensión el iniciador es soluble en la fase acuosa. Algo importante es la
presencia de dos o tres fases en el sistema (fase acuosa, fase partículas y posiblemente gotas
de monómero). El tamaño promedio de partícula es de 20 a 500 nm.24
Como ya se mencionó, uno de los componentes más importantes es el tensoactivo
que posee un carácter anfífilo, esto quiere decir que tiene un parte hidrófoba y una
hidrófila. Los tensoactivos se pueden caracterizar de diferentes formas, sin embargo la más
común es por la carga de su grupo hidrófilo: tensoactivos aniónicos (más usados),
tensoactivos catiónicos y tensoactivos anfóteros o zwitteriónicos son aquellos que poseen
las dos cargas (+,-). Se deben de tomar en cuenta dos puntos importantes para la formación
de una emulsión, el primero es trabajar en o sobre la concentración micelar crítica (CMC).
La CMC es la concentración de tensoactivo necesaria para saturar la fase acuosa y formar
micelas. Otra forma de describir la CMC es como la concentración a la cual la tensión
interfacial ya no decrece y se mantiene constante. El segundo es la importancia del punto
Krafft (Tk) del tensoactivo, el cual se define como la temperatura a la que la solubilidad
iguala la CMC; en caso de que se estuviera trabajando por debajo de esta temperatura no
existiría formación de micelas.
Desde un enfoque mecanístico Harkins104
fue el precursor y posteriormente Smith y
Ewarth105
aportaron la parte matemática del proceso, finalmente Gardon106
definió los tres
intervalos de la polimerización en emulsión. El primer intervalo abarca la nucleación de
partículas (incremento en el número de partículas), en las cuales se lleva a cabo la
polimerización. Para que el iniciador pueda llegar hasta estos sitios de reacción es necesario
que éste se descomponga en la fase acuosa generando radicales libres los cuales reaccionan
con el poco monómero soluble en la fase acuosa hasta convertirse en cadenas oligoméricas
con suficiente fuerza hidrófoba para introducirse a una micela. La velocidad de

12
polimerización está dada por la suma de las velocidades de todas las partículas en
crecimiento, por lo que al haber más partículas la velocidad de polimerización se
incrementa en gran magnitud. Este intervalo termina cuando la concentración de
tensoactivo disminuye por debajo de su CMC (fin de la nucleación). Es importante recalcar
que los radicales oligoméricos prefieren entrar a las partículas debido a la gran área
superficial total que presentan a diferencia de las gotas de monómero.
El segundo intervalo se caracteriza por que el número de partículas se mantiene
constante, así como también la velocidad de polimerización. Aquí las gotas de monómero
sirven como reservas para las partículas que están polimerizando y así, mientras existan
gotas de monómero, la concentración de éste se mantendrá constante. El intervalo termina
cuando las gotas de monómero se agotan en el sistema.
Por último, al comienzo del tercer intervalo, debido a que las gotas de monómero se
han agotado, la concentración de monómero dentro de las partículas decrece y por
consiguiente la velocidad de polimerización también. Dentro de las partículas comienza a
aumentar la viscosidad debido a que la relación polímero/monómero aumenta y es posible
que pueda presentarse el efecto gel o Trommsdorff.
Otra característica de este proceso es que se puede formar una gran variedad de
tipos de sistema dentro de las emulsiones, como lo son: emulsiones normales aceite/agua,
emulsiones inversas agua/aceite y sistemas Winsor. Como ventajas de este proceso se tiene,
al igual que en suspensión, una fácil remoción del calor, y es posible alcanzar velocidades
de polimerización y pesos moleculares más altos que en masa y suspensión.
La ventaja más grande de este proceso es el poder utilizar el producto como látex,
por ejemplo en la industria de adhesivos o pinturas. Por otro lado, las desventajas de este
proceso también son importantes como lo es la baja productividad debido al pobre
contenido de sólidos obtenido esto comparado con la polimerización en masa; también el
agua de este proceso es contaminada por los componentes del sistema, por lo cual es
necesario tratarla antes de volver a usarla o desecharla y finalmente la mayor desventaja en

13
el caso de que se necesite el polímero seco es el requerimiento de varios procesos de
separación. A pesar de lo antes mencionado este proceso es utilizado industrialmente para
producir adhesivos, pinturas y algunos copolímeros.25-28
2.2.3 Otros
En 1962, Higuchi y Misra107
propusieron la formación de emulsiones en las que no
se presentaba la difusión molecular mediante la adición de un tercer componente, el cual
debe de ser soluble en la fase dispersa. Una miniemulsión es un sistema en heterofase que a
diferencia de emulsión está compuesto solo por gotas pequeñas de monómero estabilizadas
con un poco de tensoactivo y agente hidrófobo el cual se encarga de contrarrestar la presión
osmótica y por lo tanto suprimir la difusión del monómero; es importante mencionar que
aquí la agitación juega un papel muy importante en la polimerización. Una característica
muy particular de este proceso es que la distribución de tamaños de gotas es relativamente
cerrada y por lo tanto no existe una alta difusión de las gotas pequeñas a las grandes
(Ostwald ripening). Las miniemulsiones se preparan al someter a un alto esfuerzo de corte
un sistema compuesto por agua, aceite, tensoactivo y agente hidrófobo. Los tamaños
promedio de partículas caen en el rango de 50-500 nm. El tiempo de estabilidad de una
miniemulsión está dado en días hasta meses a diferencia de las emulsiones que en cuestión
de horas pierden su estabilidad. Una de las principales ventajas es para el caso de
copolímeros(reactor batch), es que la composición final del copolímero será la misma en
todas las partículas; sin embargo existen limitantes como lo es el modo de operación, es
necesario trabajar en reactores batch ya que el agregar monómero rompería el equilibrio del
sistema.29-32
En 1943, Schulman encontró que si a una emulsión se le agregaba un alcohol de
bajo peso molecular se formaba espontáneamente un sistema transparente. Las
microemulsiones son dispersiones coloidales, termodinámicamente estables, translúcidas
formadas por una fase dispersa y una fase continua (agua). Para poder generar una
microemulsión es necesario realizar un diagrama de fases monómero/tensoactivo/agua para
encontrar la región en la que se forman éstas. Su estabilidad viene dada por la energía libre

14
de Gibbs, la cual nos dice que si el valor es negativo, el proceso se llevará de manera
espontánea sin necesidad de suministrar energía. Esto quiere decir que el valor entrópico
del sistema es demasiado grande a diferencia del valor entálpico, por lo tanto al ser mucho
mayor la entropía, el valor de la energía libre de Gibbs será menor de cero.
STHG
G = energía libre de Gibbs
H diferencia de entalpías
S diferencia de entropías
T = temperatura
El uso excesivo de tensoactivo y el bajo contenido de sólidos son variables
importantes a considerar en la polimerización por lotes, sin embargo existen trabajos
recientes en donde se reporta que trabajar en un régimen en semicontinuo reduce en gran
cantidad el tensoactivo, permitiendo un contenido más alto de sólidos.108,109
A diferencia de la técnica de emulsión aquí la polimerización consta de dos
intervalos, esto debido a que no se presentan gotas de monómero en el sistema por lo que
no existe el segundo intervalo. Una de las ventajas de este tipo de polimerización es que se
pueden fabricar partículas funcionalizadas de tamaños “nano”, estas nano-partículas son
muy utilizadas en la industria farmacéutica como liberadores de medicamentos, entre otras
aplicaciones. Sin embargo, la baja concentración de sólidos obtenidos por polimerización
en microemulsión y los altos costos de procesos de separación, hace de ésta una opción
casi exclusivamente para el desarrollo de productos de especialidad.33-37
2.3 NMRP en Emulsión ab initio
El llevar a cabo una polimerización controlada por nitróxidos en emulsión ab initio
presenta un gran reto, debido a que se presentan problemas significativos con el control y la
estabilidad coloidal. En emulsión lo más importante es la formación de las partículas de
polímero, las cuales se forman durante la etapa de nucleación a baja conversión. Para poder

15
tener una polimerización en emulsión controlada por nitróxidos ideal, es de vital
importancia evitar la autoiniciación térmica en las gotas de monómero, ya que como se está
trabajando a temperaturas muy altas en algunos sistemas se presenta esté fenómeno, dando
como resultado un mal control de la polimerización. Otro factor importante es la difusión
del nitróxido a través de la fase acuosa a las partículas, ya que si éste no se encuentra en el
sitio de reacción, la polimerización no será controlada. La concentración de nitróxido
dentro de las partículas está dada por el reparto de éste entre la fase acuosa y la orgánica,
por lo que mientras la concentración de nitróxidos sea suficientemente alta dentro de las
partículas, la polimerización será controlada.38
Los primeros intentos de una polimerización en emulsión ab initio fueron en general
malos en cierto grado, esto debido a los problemas latentes asociados a la nucleación y el
reparto.39-41
Se han reportados varios trabajos de la polimerización de estireno utilizando
KPS (persulfato de potasio) y una gran variedad de nitróxidos de la familia del TEMPO (2,
2, 2, 6-Tetrametilpiperidina 1-oxil), algunos de estos fueron OH-TEMPO, nitróxido de di-
tert-butilo, 4-carboxi-TEMPO, 4-benzoiloxi-TEMPO y amino TEMPO.40,41
Los resultados
obtenidos fueron bajas conversiones y una pobre estabilidad coloidal de la emulsión,
excepto con el amino-TEMPO y el acetoxi-TEMPO41
, utilizando SDS (dodecilsulfato de
sodio) como tensoactivo. En estos casos se obtuvieron látexes con una conversión
intermedia y una buena estabilidad coloidal, señales de un buen control de la
polimerización. El éxito relativo de estos nitróxidos puede ser atribuido al reparto de éstos
entre la fase acuosa y la orgánica. También se ha reportado la polimerización ab initio en
emulsión de estireno a 135 oC, utilizando una alcoxiamina de TEMPO/KPS soluble en
agua y SDBS (dodecil-benzil sulfonato de sodio ) como tensoactivo. Los resultados fueron
buenos mostrando un buen control de la polimerización pero una mala estabilidad
coloidal.42
También se han reportado trabajos utilizando SG1 (nitróxido N-tert-butil-N-[1-
dietilfóforo-(2, 2-dimetilpropilo)]) a 90 oC en uno de los cuales se obtuvieron conversiones
demasiado bajas, un pobre control de la estabilidad y coagulación.43

16
2.4 Problemas en NMRP en Emulsión
Uno de los problemas más comunes que se presentan en este tipo de
polimerizaciones es la polimerización de las gotas de monómero, lo cual se le atribuye a la
autoiniciación térmica del monómero. En este estudio se trabajará con el estireno (está
reportado que a partir de los 100 °C se autopolimeriza), por lo tanto al estar en un sistema
en emulsión por arriba de los 120 °C se favorece este fenómeno; otro problema que se
presenta es que el nitróxido no se encuentra en el sitio de reacción debido a la repartición
de éste (dado por su carácter hidrófobo), por ende no se tiene control de la polimerización.
El fenómeno de Ostwald ripening se presenta con mayor incidencia que en una
polimerización en emulsión convencional, esto gracias a la preferencia de difusión de
monómero de las gotas pequeñas a las grandes, ya que éstas son termodinámicamente más
estables debido a su menor área superficial. Finalmente se tiene el fenómeno del
superhinchamiento de las partículas de polímero, que se debe a moléculas de bajo peso
molecular u oligómeros.38,44,45,51-60
2.5 Posibles Soluciones
Recientemente (2008) Cunningham et al.46
utilizaron dos nitróxidos, TEMPO y 4-
estearoil-TEMPO, el cual es extremadamente hidrófobo, en una polimerización en
emulsión ab initio de estireno (KPS/SDBS/135oC). En las polimerizaciones en las que solo
se utilizó TEMPO, se presentó coagulación de un 25%. El coágulo estaba formado por una
mezcla de partículas pequeñas (≈500 nm) y grandes (>1μm). Sin embargo, se tuvo un buen
control de la polimerización con un índice de polidispersidad (PDI) de 1.19, además la
distribución de pesos moleculares de las partículas del coágulo y de la fase dispersa fueron
idénticas. Cuando se utilizaron ambos nitróxidos en una relación de 1.33 (4-estearoil-
TEMPO/TEMPO), se formaron partículas con un diámetro de 45 nm sin la presencia de
coágulos y el control de la polimerización fue excelente. Esto debido a que el TEMPO se
difunde de las gotas de monómero hacia las micelas donde la polimerización ocurre, pero el
4-estearoil-TEMPO es demasiado hidrófobo como para difundirse a través de la fase acuosa
y por lo tanto permanece en las gotas. Si la concentración de éste es suficientemente alta

17
dentro de las gotas de monómero puede actuar como inhibidor y prevenir la polimerización
de las gotas. Con este trabajo se comprueba que el mayor de los problemas es la
autoiniciación térmica de las gotas de monómero.
En 2006 Charleaux et al. desarrollaron una polimerización controlada por nitróxidos
en un sistema en emulsión ab initio, la cual se basa en el principio de autoensamblaje
dentro de las micelas hechas de un copolímero dibloque anfífilo (SG1-poli acrilato de
sodio) que se forma in situ durante la polimerización.47,48
Esta macromolécula sirve
también como un macroiniciador, sin embargo es necesario usarse bajo condiciones de pH
básicas para obtener un buen control, ya que el SG1 se descompone a pH bajos.39,49,50
Aquí
no es necesario el uso de tensoactivos, solo del monómero, agua y el macroiniciador, ya
que el copolímero dibloque provee estabilidad electrostática, así se evitó la coagulación.
Todas las pruebas fueron llevadas a cabo a 120 oC y el pH >7, el diámetro promedio fue de
450 nm, con un índice de polidispersidad de 1.2.
2.5.2 Reparto de Nitróxido, Velocidad de agitación y Presión
Para calcular el coeficiente de reparto de algún soluto entre dos solventes, es
necesario calcular primero el coeficiente de extinción molar. El cual nos dice qué tan fuerte
una sustancia absorbe la luz a una longitud de onda, por unidad de masa o por
concentración molar. De acuerdo a la ley de Lambert-Beer, el coeficiente de extinción
molar es proporcional a la conductividad del soluto absorbente.
Para el cálculo de los coeficientes de extinción molar de cada una de las fases, es
necesario hacer una curva de calibración en donde la pendiente que se presenta esta curva
es igual al coeficiente de extinción molar ( ), el cual es una relación inversa con la
concentración del nitróxido en dicha fase. A continuación se presenta la ley de Lambert-
Beer para el cálculo de la concentración:
Alcee
I
I
0
1 (2.1)
I0, I1 = intensidades entrante y saliente respectivamente

18
A = absorbancia
l = longitud atravesada por la luz en el medio o longitud de la celda (1 cm)
c = concentración del absorbente en el medio (mol/L)
= coeficiente de extinción molar (L/ mol*cm)
Por lo tanto la absorbancia se puede expresar como:
lcA (2.2)
Ahora tomando en cuenta que la celda de cuarzo del espectrofotómetro UV-visible
es de una longitud de 1 cm la ecuación se reduce a:
l
Ac
* (2.3)
Es importante señalar que esta ecuación es válida solo para concentraciones del absorbente
que se comportan de manera lineal, por lo general en curvas de calibración muy amplias la
linealidad se pierde. Debido a que la concentración del nitróxido es relativamente baja en
las dos fases es posible aplicar esta ley.61
En 2001 Cunningham et al. presentaron el primer estudio del reparto de nitróxidos
en miniemulsión, para tener un mejor entendimiento del proceso a escala industrial.
Debido a los problemas antes mencionados es importante entender de una mejor manera el
reparto de los nitróxidos en sistemas heterogéneos y las posibles influencias que este
reparto puede tener en las reacciones de la fase acuosa u orgánica. En este artículo se
midieron los coeficientes de reparto de tres nitróxidos comunes: TEMPO, OH-TEMPO y
NH-TEMPO, trabajando a temperaturas de 20, 90, 120 y 135 oC. Estos nitróxidos también
fueron escogidos debido a sus diferentes grados de solubilidad en agua. Se obtuvo una
mejor conversión con el OH-TEMPO así como también un mejor control de la
polimerización. Los coeficientes de reparto fueron obtenidos por espectrometría de UV-
visible. El coeficiente de reparto del TEMPO (711 - 652 mol% / mol%-1
) fue mucho mayor
en todos los casos que el del mas hidrófilo OH-TEMPO (2.9 - 14.3 mol% / mol%-1
).50
En la literatura solo existe un artículo relacionado con el cálculo de los coeficientes
de reparto de nitróxidos en sistemas de polimerización heterogéneos.50

19
Otra posible solucion para los problemas en la NMRP en emulsión, es el incremento
en la presión del sistema ya que esto podría ayudar a mejorar la estabilidad del sistema, esto
debido a que algunos nitróxidos poseen bajos puntos de sublimación y al trabajar a altas
temperaturas es muy probable tener al nitróxido en fase vapor, por lo tanto esto contribuye
a que la polimerización no sea controlada. En la literatura no existe ningún reporte de
NMRP en emulsión donde se estudie la presión.
Otro factor importante es la velocidad de agitación del sistema, ya que con esta
variable es posible ayudar a la difusión de las especies y además se puede incrementar la
nucleación de partículas y por ende se incrementa el número de partículas del sistema. Con
esta variable es posible ajustar hasta cierto punto también el tamaño de partícula. En la
literatura no existe ningún reporte donde se estudie la velocidad de agitación en la NMRP
en emulsión.
Cabe recalcar que éstas variables nunca han sido de importancia en las
investigaciones relacionadas con la NMRP en emulsión, ya que casi todas se centran en
desarrollar nuevos nitróxidos, los cuales son demasiado difíciles de sintetizar y sobretodo
extremadamente costosos.
2.6 Modelos matemáticos para polimerización radicálica en emulsión
En la literatura existe un gran número de artículos en los cuales se modelan sistemas
de polimerización heterogéneos (suspensión, emulsión, etc.), sin embargo para el caso de la
polimerización radicálica mediada por nitróxidos en emulsión, es casi nulo.
Como bien se sabe, la polimerización en emulsión es un proceso heterogéneo en el
cual se involucra el transporte de los monómeros, radicales libres y otras especies entre la
fase acuosa y orgánica. Comparada con las demás polimerizaciones heterogéneas como
suspensión o precipitación, la polimerización en emulsión es el sistema más complicado. La
velocidad de polimerización en la fase orgánica no solo es controlada por el reparto del

20
monómero, sino también por la nucleación de partículas y la absorción y desorción de
radicales. La estabilidad de las partículas se ve afectadas por el tipo, cantidad y fuerza
iónica del tensoactivo. Todos estos factores hacen que el modelamiento de este sistema sea
muy complicado. Otras dificultades en el modelamiento de este sistema, es la resolución de
las ecuaciones ordinarias diferenciales no lineales (integro-diferenciales) acopladas con
ecuaciones algebraicas.62
En la Tabla 2.1 se presentan los modelos en emulsión para homo/co-polimerización
más representativos.
Tabla 2.1. Modelos matemáticos y variables obtenidas en polimerización en emulsión.
Grupos Avances importantes y resultados Sistema
Ballard et al.63
Modelo de co-polimerización para sistemas 0-1 St/BD
Broadhead et al.64
Modelo de co-polimerización para un reactor
continuo, PSD. St/BD
Congalidis et al.65
PSD, modelo de co-polimerización MMA, St
Dougherty66
Co-polimerización, MWD y PSD St, MMA
Dubé et al.67
Ramificación de cadena larga, moldelo de co-
polimerización, PSD y MWD BD/AN
Forcada y Asua68,69
Unidades por cadena y PSD St/MMA
Giannetti et al.70
Reviews en PSD St/MMA
Giannetti71
Modelo de co-polimerización St/AN
Guillot72
Aspectos termodinámicos en co-polimerización St/AN
Hamielec et al.73,74
Ramificación, entrecruzamiento, PSD y MWD St/AN
Lichti et al.75
Reviews en PSD y MWD St/AN
Lin et al.76
Composición azeotrópica, modelo de co-
polimerización St/AN
Mead y Poehelein77,78
Modelo de co-polimerización y PSD St/MA, St/AN
Min y Ray79,80,81
Modelo de homo-polimerización PSD y MWD MMA
Nomura et al.82,83
Modelo de co-polimerización y PSD MMA/St
Penlidis et al.84,85,86
Modelo para un reactor continuo, batch, PSD,
MWD, modelamiento en estado estacionario y
dinámico en un reactor batch, semi-batch y
continuo, review de modelos de co-
polimerización
PVC, VAc,
VAc/PVC
Rawlings87 Modelo de homo-polimerización, PSD, MWD en
un reactor continuo MMA, St, VAc
Richards et al.88
Mejora al modelo de Congalidis et al. modelo de
co-polimerización asumiendo no nucleación MMA, MMA/St

21
micelar, MWD y PSD
Rawlings y Ray89,90
Mejora al modelo de Min y Ray, PSD, MWD,
operación en un reactor continuo MMA
Saldivar et al.91
Versión más reciente del modelo Min y Ray,
PSD, MWD y modelo para co-polimerización
MMA/St, St/BD,
St/α-metil St
Morbidelli et al.92,93
Modelamiento de la distribución de peso
molecular y modelo para copolimerización AN/St/VAc
Urretabizkaia et al.94,95
Modelo de terpolimerización MMA/BA/VAc
St: estireno, BD: butadieno, MMA: metil. meta-acrilato, AN: acrilonitrílo, VAc: Alcohol vinílico, PVC: policloruro de
polivinilo, BA: butil acrilato PSD: distribución de tamaño de partícula, MWD: distribución de peso molecular,
Como se puede ver existen un gran número de artículos en la literatura en los que se
estudian diferentes variables presentes en emulsión. Sin embargo, para la polimerización
radicálica mediada por nitróxidos en emulsión, no existe un modelo para ésta. La mayoría
de los estudios de modelamiento para nitróxidos son para sistemas homogéneos o en
miniemulsión y microemulsión.
2.7 Modelos matemáticos para polimerización radicálica en medios hetrogéneos
mediada por nitróxidos
Actualmente no existen en la literatura reportes sobre el modelamiento de la
polimerización en emulsión mediada por nitróxidos. Los únicos reportes que existen son en
miniemulsión y microemulsión. A continuación se presentan los artículos más
sobresalientes en este campo.
En 2005 Zetterlund et al.96
reportaron la modificación del modelo de Storti y
Morbidelli92,93
para la polimerización en miniemulsión mediada por nitróxidos de estireno,
suponiendo iniciación solamente en la fase orgánica a 125 °C. Para poder resolver las
ecuaciones del balance del nitróxido supusieron los valores para el coeficiente de reparto
del nitróxido de 98, 2.2 y 0.2. Esto se hizo con el fin de simular un nitróxido muy hidrófobo
como el TEMPO y uno muy hidrófilo como el OH-TEMPO. Tomando en cuenta que la
iniciación se lleva solo en la fase orgánica, encontraron que la cinética es independiente del
coeficiente de reparto del nitróxido en estado estacionario. En la etapa de pre-equilibrio, la
concentración de radicales propagantes aumenta y la concentración de radicales nitróxidos

22
disminuye con el decremento del coeficiente de reparto. Al ser más hidrófilo el nitróxido, la
concentración de éste disminuye en la fase orgánica y por lo tanto el numero de radicales
propagantes aumenta, por lo tanto la terminación bimolecular aumenta perdiéndose el
carácter viviente de la polimerización.
En 2006 Zetterlund et al.97
modificó las ecuaciones de Smith-Ewart para determinar
el efecto de compartimentación en la polimerización mediada por nitróxidos en sistemas
dispersos. Estudió el sistema estireno/TEMPO a 125 °C, además tomó en cuenta la
iniciación térmica del monómero en el modelo. Se encontró que al ir disminuyendo el
diámetro de partícula la velocidad de polimerización disminuye, debido a que solo se
encuentra un radical propagante y un radical nitróxido dentro de la partícula. Esto ocasiona
un incremento en el carácter viviente de la polimerización, así cada cadena polimérica
tendrá en un extremo una molécula de nitróxido. Por lo tanto, es posible calcular el número
exacto de unidades monoméricas en la cadena y por ende el peso molecular teórico es igual
al experimental, dando como resultado un carácter viviente/controlado excelente. El efecto
de compartimentación se presenta a tamaños menores de 110 nm.
En 2009 Zetterlund et al.98
agregó los términos de la compartimentación al modelo
antes mencionado. El efecto de compartimentación se compone de dos efectos, el efecto de
segregación se refiere a dos especies localizadas en partículas diferentes, las cuales no
pueden reaccionar entre sí. Por lo tanto, este efecto puede aplicarse a los radicales
propagantes, ya que al disminuir el tamaño de partícula la terminación bimolecular se
reduce en gran medida. También este efecto lleva a incrementar el carácter viviente de la
polimerización y por lo tanto una alta funcionalidad terminal de las cadenas. Por otra parte,
el efecto de confinamiento propone que dos especies reaccionan mucho más rápido en una
partícula pequeña que en una partícula grande, sin embargo al estar presente una molécula
del nitróxido la velocidad de polimerización disminuye considerablemente, pero el control
en la polimerización es muy bueno, lo cual se puede ver en la distribución de pesos
moleculares cercana a uno. Existe una competición de estos dos efectos en la
polimerización, ya que mientras el efecto de segregación tiende a incrementar la velocidad
de polimerización, el efecto de confinamiento la disminuye. Por lo tanto el resultado final

23
de la velocidad depende de la magnitud de estos dos efectos. El sistema que se estudió fue
estireno con TEMPO y TIPNO a 125 °C, la constante de equilibrio es mucho más grande
para el TIPNO que para el TEMPO, lo cual resulta en grandes diferencias en los efectos de
compartimentación en los dos sistemas. En los resultados se aprecia que el efecto de
confinamiento comienza a influenciar la velocidad de polimerización para tamaños de
partícula menores a 55 nm para el TIPNO, a diferencia del TEMPO en donde a partir de los
70 nm se presenta este efecto. El efecto de segregación se presenta en mayor medida en el
TIPNO; debido a su fuerte carácter hidrófobo la concentración de radicales es mucho
mayor en la fase dispersa, esto hace que se reduzca en gran medida la terminación
bimolecular y por lo tanto aumenta el carácter viviente/controlado de la polimerización.
Finalmente, se tienen dos regiones en las cuales se puede estar operando tomando en cuenta
estos efectos, la primera región es cuando el efecto de segregación es más fuerte que el
efecto de confinamiento, por lo tanto la velocidad de polimerización aumenta, la
distribución de pesos moleculares es muy amplia, sin embargo el carácter viviente es mayor
que en masa. En la segunda región donde el efecto de confinamiento es mayor que el efecto
de segregación, la velocidad de polimerización es menor, la distribución de pesos
moleculares es estrecha y el carácter viviente es mucho mayor que en masa. La constante
de equilibrio del nitróxido nos indica en qué región estamos trabajando, en este estudio para
el TIPNO se tiene una constante de equilibrio muy alta, por lo tanto se trabaja en la primera
región. Pero también se tienen que tener partículas mayores a 15 nm y menores a 100 nm
para asegurar un buen carácter viviente/controlado. Para el caso del TEMPO se tiene una
constante de equilibrio muy baja, por lo tanto se requiere tamaños mayores a los 55 nm y
menores a 100 nm para estar cerca de la primera región.
En 2010 Zetterlund99
estudió de forma más profunda los efectos de confinamiento y
de segregación, así como también el efecto del reparto del nitróxido. Propone que bajo
ciertas circunstancias, es posible obtener una mejora en el control y el carácter viviente.
Propone principalmente trabajar con un tamaño crítico de partícula en donde el efecto de
segregación se presente en mayor medida, por lo tanto así se asegurará un buen control y
carácter viviente, sin embargo esto es para sistemas como miniemulsión o microemulsión
en donde se tienen tamaños de partícula lo suficientemente pequeños, por eso es difícil

24
aplicarlo en emulsión. El efecto de segregación tiene una fuerte influencia en la terminación
y el efecto de confinamiento en la desactivación de las cadenas poliméricas (especies
durmientes). Por otra parte, si el nitróxido fuese muy hidrófilo es posible que se presente
una desorción de radicales nitróxido de las partículas provocando una pérdida del control y
el carácter viviente. Aquí se aprecia de forma latente que la compartimentación viene dada
por el coeficiente de reparto del nitróxido, ya que si el radical nitróxido es lo
suficientemente hidrófilo, se pierde el control de la polimerización.
2.8 JUSTIFICACIÓN
En el grupo del Dr. Enrique Saldivar Guerra en CIQA, actualmente la investigación
se ha enfocado a desarrollar procesos de polimerización controlada/viviente en emulsión y
otros sistemas110
, dando como resultado técnicas ingeniosas para lograr polimerizaciones de
este tipo, sin embargo no ha existido un esfuerzo para estudiar a profundidad en forma
conjunta los mecanismos y variables que intervienen en la estabilidad coloidal de una
emulsión controlada/viviente. Los intentos más exitosos reportados hasta el momento
recurren a métodos sofisticados y difíciles de realizar, o útiles sólo para ciertos sistemas.
En la polimerización radicálica mediada por nitróxidos en emulsión se presentan
principalmente tres fenómenos: superhinchamiento, Ostwald ripening y el reparto del
nitróxido, los cuales afectan la estabilidad, control y carácter viviente del látex. Este último
fenómeno se puede entender por medio de la estimación de los coeficientes de reparto, sin
embargo esto presenta retos cuando se habla de polimerización en emulsión. El principal de
ellos es la alta temperatura en la que se opera, debido a esto la mayoría de los componentes
de la emulsión se encuentran en un equilibrio liquido-vapor. Por lo tanto, la pérdida de
estabilidad, control y carácter viviente es inevitable. En los reportes de la literatura, se
trabaja por lo general a presiones bajas y no se enfatiza la importancia de manejar presiones
más altas. Trabajar a presiones altas podría ayudar a mejorar el control y la estabilidad del
látex. Además otra variable importante en la difusión del sistema es la velocidad de
agitación, por lo tanto es importante estudiar en conjunto estas dos variables. Otro
problema importante, es la toma de muestras del sistema, ya que al muestrear se va

25
perdiendo la homogeneidad de la emulsión. Estos problemas se tratarán de resolver para
poder determinar estos coeficientes y entender de una mejor manera el proceso.
También el cálculo de los coeficientes de reparto ayudará en un futuro en la
simulación de la homo/co-polimerización de diferentes monómeros. Como se mencionó
anteriormente, no existe el modelamiento para un sistema en emulsión con nitróxidos.
De acuerdo al planteamiento anterior, sería científicamente novedoso saber cómo el
reparto del nitróxido afecta la distribución de peso molecular, el tamaño de partícula, índice
de polidispersidad y estabilidad coloidal en la polimerización del estireno en emulsión. Se
seleccionaron los nitróxidos: OH-TEMPO, OXO-TEMPO, TEMPO y TIPNO, debido la
gran diferencia del carácter hidrófilo o hidrófobo que presentan. Por lo tanto, se estudiará
desde un nitróxido (OH-TEMPO) relativamente hidrófilo hasta uno muy hidrófobo
(TIPNO). Como iniciador se utilizará el KPS el cual está reportado que funciona bien en
este tipo de sistema50
Para estabilizar el sistema se eligió el SDBS, el cual puede soportar
altas temperaturas sin hidrolizarse o descomponerse.100

26
3. HIPÓTESIS Y OBJETIVOS
3.1 HIPÓTESIS
1. El mejor nitróxido será el OH-TEMPO en la polimerización radicálica
controlada por nitróxidos de estireno en emulsión.
2. Al incrementar la presión del sistema se favorecerá el control de y estabilidad
coloidal del látex.
3. Al incrementar la velocidad de agitación hasta cierto punto se favorecerá el
control y estabilidad coloidal del látex.
3.2 OBJETIVOS
1. Estudiar la termodinámica de reparto de los nitróxidos OH-TEMPO, OXO-TEMPO,
TEMPO y TIPNO en un sistema que contenga agua y monómero.
2. Determinar el efecto de la velocidad de agitación y presión, sobre la distribución de
peso molecular, tamaño de partícula, índice de polidispersidad y estabilidad coloidal
en la polimerización en emulsión mediada por nitróxidos de estireno.
3. Posteriormente realizar una comparación en control y estabilidad coloidal de los
nitróxidos en conjunto con los coeficientes de reparto estimados, en una
polimerización mediada por nitróxidos en emulsión ab initio.
4. Introducción de los términos del nitróxido a un modelo matemático simplificado
para polimerización en emulsión.

27
4. PARTE EXPERIMENTAL
4.1 Reactivos y materiales
Todas las sustancias utilizadas fueron grado reactivo. El persulfato de potasio (KPS)
(K2S2O8) (99.99 % de pureza), el dodecil bencen sulfonato de sodio (SDBS)
(C18H30NaO3S) (98.50 %), la hidroquinona (C6H8O2) (99.00 %), el tetrahidrofurano grado
HPLC (THF) (99.99 %), el 4-hidroxi Tetrametilpiperidina-1-oxil (OH-TEMPO) (99.99 %),
el 4-oxo-2,2,6,6-Tetrametilpiperidina-1-oxil (OXO-TEMPO) (99.99 %), el 2,2,6,6-
Tetrametilpiperidina-1-oxil (TEMPO), el 2,2,5-Trimetíl-4-fenil-3-azahexano (TIPNO)
(99.99 %) y el estireno (St) (C8H8) (99.00 %) fueron suministrados por Sigma-Aldrich. El
agua destilada desionizada que se utilizó se obtuvo de un sistema de dos columnas de
intercambio iónico de Cole Parmer. El nitrógeno fue grado UAP.
4.2 Equipo
4.2.1 Reparto de nitróxidos
Para llevar a cabo los experimentos de reparto de cada uno de los nitróxidos se
utilizó un reactor Parr enchaquetado de vidrio serie 5100 de baja presión (150 psia), de un
volumen de 600 mL y equipado con 6 puertos en la tapa del reactor. En uno de los puertos
se colocó un manómetro, en otros dos puertos se colocaron tubos buzos a diferentes alturas
para tomar las muestras de cada una de las fases, en otro se instaló un sello de seguridad
para liberar presión, en otro se instaló un termopozo donde se colocó un termopar, y en el
último puerto se instaló una línea para introducir el gas nitrógeno. El sistema de agitación
constaba de una varilla de acero inoxidable con doble propela de 6 álabes a 45 ° de
inclinación y con un sello magnético, dicho sistema era activado por un motor mecánico
que iba conectado a un controlador Parr modelo 4835. Para mantener la temperatura
deseada en el sistema de reparto, se hizo circular aceite a través de la chaqueta del reactor
por medio de un baño de temperatura constante Fisher Scientific modelo 3013H. El sistema
empleado se muestra en la Figura 4.1.

28
Figura 4.1. Sistema utilizado para los experimentos de reparto.
. 4.2.2 Polimerización de estireno mediada por nitróxidos en emulsión ab initio
Para llevar a cabo la reacción de polimerización mediada por nitróxidos en emulsión
ab initio del estireno se utilizó un reactor Parr de acero inoxidable de alta presión (2000
psia), con un volumen de 1 L y equipado con 6 puertos en la tapa del reactor. La
configuración de los puertos fue casi la misma que en los estudios de reparto, la única
diferencia es que se removió uno de los tubos buzos y se tapó. El sistema de agitación fue
exactamente el mismo, usado en los experimentos de reparto. Para mantener la temperatura
deseada en el sistema de reacción se utilizó una resistencia eléctrica conectada a un
controlador Parr modelo 4848. El sistema empleado se muestra en la Figura 4.2.
Figura 4.2. Sistema utilizado para las polimerizaciones.

29
4.3 Caracterización
4.3.1 Cuantificación del nitróxido en los experimentos del reparto.
Para la cuantificación del nitróxido en cada una de las fases se utilizó un
espectrofotómetro de UV-visible Hewlett Packard 8452A.
4.3.2 Polimerización
Para el estudio del látex y polímero obtenido se emplearon los siguientes equipos
analíticos:
1) Dispersor de Luz – Malvern Nano z-sizer S90
2) Cromatógrafo de Permeación en Gel (GPC) – Hewlett Packard modelo
1110 equipado con un detector de índice de refracción. El equipo se calibró
con estándares de poliestireno (Polyscience).
3) Resonancia Magnética Nuclear (RMN) 1H – Espectrofotómetro FT-NMR
Jeol Eclipse de 300 MHz.
4.4. Metodología
4.4.1 Reparto de nitróxidos
4.4.1.1 Cálculo del coeficiente de extinción molar
Antes de calcular los coeficientes de extinción molar fue necesario identificar la
banda de absorción de cada uno de los nitróxidos en las dos fases (St/H20); posteriormente
se realizaron curvas de calibración para cada nitróxido en las dos fases. El intervalo que se
trabajó en las curvas de calibración de la fase orgánica y acuosa de cada uno de los
nitróxidos fue de 0.01–0.5 de absorción.

30
4.4.1.2 Cálculo del coeficiente de reparto
El coeficiente de reparto es la relación de la concentración del nitróxido en la fase
orgánica entre la concentración del nitróxido en la fase acuosa, el cual se escribe como:
ac
org
NN
NK (4.1)
Se calcularon los coeficientes de reparto de cada uno de los nitróxidos a las
temperaturas de 25, 90, 120 y 135°C. La mezcla St/H2O para los experimentos de reparto
se presenta en la tabla 4.1.
Tabla 4.1. Cantidades empleadas en cada uno de los experimentos de reparto.
Peso (g) Volumen (L) % peso
Fase Orgánica
(Estireno) 62.5 0.0569 25
Fase Acuosa
(Agua desionizada) 187.5 0.1875 75
Total 250 0.2444 100
Se trabajó con 0.1, 0.15, 0.2 y 0.25 g de cada uno de los nitróxidos para cada
temperatura. Para evitar la autoiniciación del estireno se agrego 0.1 g de hidroquinona
como inhibidor. Ya preparadas las mezclas de St/H2O junto con el nitróxido y la
hidroquinona, se procedió a colocarlas en el reactor y el sistema se desgasificó durante 30
minutos con gas nitrógeno para mantener una atmósfera inerte, después se presurizó a 140
psia y se comenzó a pasar aceite por la chaqueta del reactor a la temperatura deseada. Cabe
recalcar que se hicieron pruebas a una presión más baja (50 psia) sin embargo se observó
una incongruencia en el coeficiente de reparto. Una vez alcanzada la temperatura deseada
se encendió el controlador de la agitación y se fijó en 700 rpm para asegurar un buen
mezclado de las dos fases y también aumentar la velocidad de difusión del nitróxido para
alcanzar el equilibrio. Se realizaron varias pruebas preliminares para determinar el tiempo
mínimo para alcanzar el equilibrio y se observó que con 90 minutos era más que suficiente
para alcanzarlo. Una vez alcanzado el tiempo, se detuvo la agitación y el sistema se dejó
31
reposar durante 30 minutos para que las fases se separaran. Finalmente, a la temperatura
deseada se tomaron 5 mL de muestra de cada una de las fases en viales de 10 mL
previamente enfriados para evitar la evaporación de cualquier componente. Las muestras se
extrajeron por la presión del sistema por dos válvulas conectadas a dos tubos buzos. Las
muestras se taparon y se dejaron reposar durante 3 días aproximadamente para su posterior
cuantificación por UV-visible.
4.4.2 Polimerización de estireno mediada por nitróxidos en emulsión ab initio
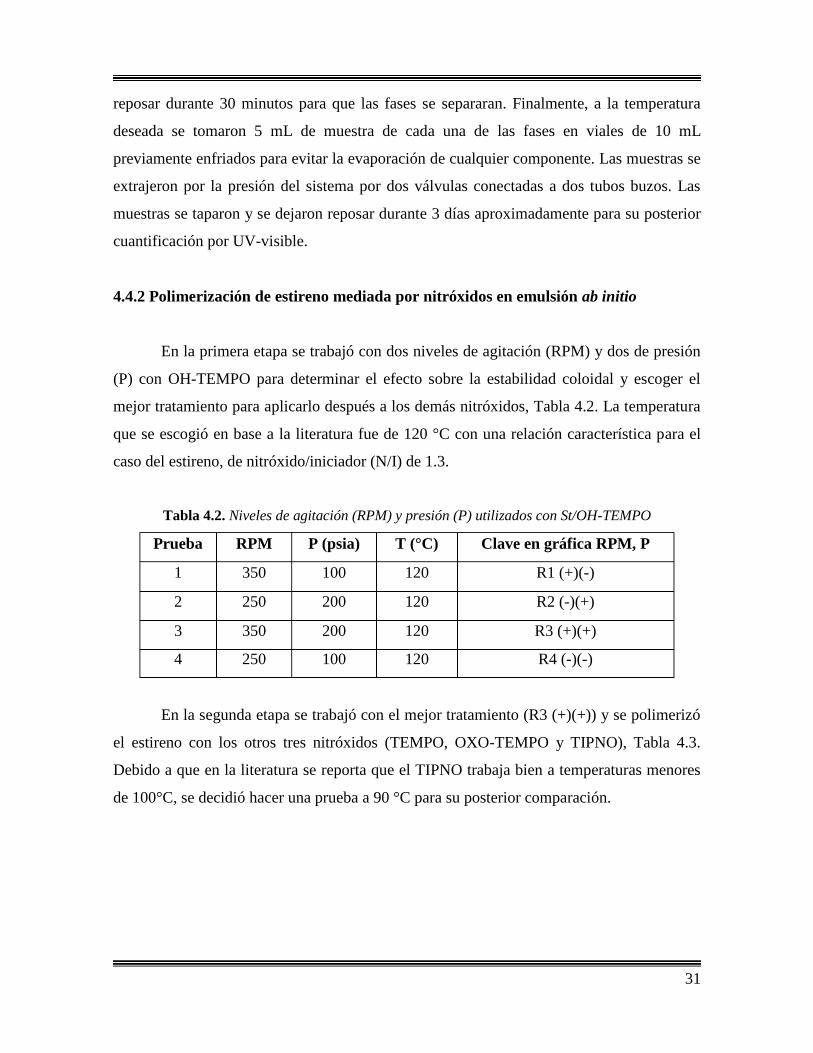
En la primera etapa se trabajó con dos niveles de agitación (RPM) y dos de presión
(P) con OH-TEMPO para determinar el efecto sobre la estabilidad coloidal y escoger el
mejor tratamiento para aplicarlo después a los demás nitróxidos, Tabla 4.2. La temperatura
que se escogió en base a la literatura fue de 120 °C con una relación característica para el
caso del estireno, de nitróxido/iniciador (N/I) de 1.3.
Tabla 4.2. Niveles de agitación (RPM) y presión (P) utilizados con St/OH-TEMPO
Prueba RPM P (psia) T (°C) Clave en gráfica RPM, P
1 350 100 120 R1 (+)(-)
2 250 200 120 R2 (-)(+)
3 350 200 120 R3 (+)(+)
4 250 100 120 R4 (-)(-)
En la segunda etapa se trabajó con el mejor tratamiento (R3 (+)(+)) y se polimerizó
el estireno con los otros tres nitróxidos (TEMPO, OXO-TEMPO y TIPNO), Tabla 4.3.
Debido a que en la literatura se reporta que el TIPNO trabaja bien a temperaturas menores
de 100°C, se decidió hacer una prueba a 90 °C para su posterior comparación.
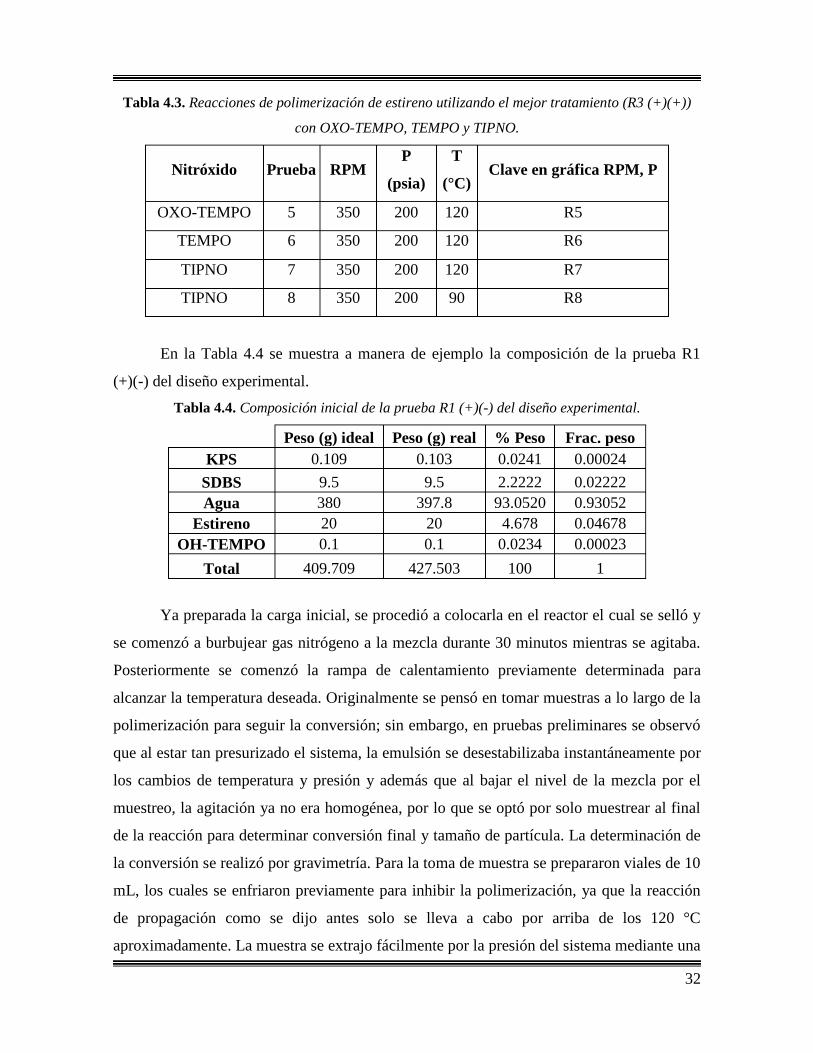
32
Tabla 4.3. Reacciones de polimerización de estireno utilizando el mejor tratamiento (R3 (+)(+))
con OXO-TEMPO, TEMPO y TIPNO.
Nitróxido Prueba RPM P
(psia)
T
(°C) Clave en gráfica RPM, P
OXO-TEMPO 5 350 200 120 R5
TEMPO 6 350 200 120 R6
TIPNO 7 350 200 120 R7
TIPNO 8 350 200 90 R8
En la Tabla 4.4 se muestra a manera de ejemplo la composición de la prueba R1
(+)(-) del diseño experimental.
Tabla 4.4. Composición inicial de la prueba R1 (+)(-) del diseño experimental.
Peso (g) ideal Peso (g) real % Peso Frac. peso
KPS 0.109 0.103 0.0241 0.00024
SDBS 9.5 9.5 2.2222 0.02222
Agua 380 397.8 93.0520 0.93052
Estireno 20 20 4.678 0.04678
OH-TEMPO 0.1 0.1 0.0234 0.00023
Total 409.709 427.503 100 1
Ya preparada la carga inicial, se procedió a colocarla en el reactor el cual se selló y
se comenzó a burbujear gas nitrógeno a la mezcla durante 30 minutos mientras se agitaba.
Posteriormente se comenzó la rampa de calentamiento previamente determinada para
alcanzar la temperatura deseada. Originalmente se pensó en tomar muestras a lo largo de la
polimerización para seguir la conversión; sin embargo, en pruebas preliminares se observó
que al estar tan presurizado el sistema, la emulsión se desestabilizaba instantáneamente por
los cambios de temperatura y presión y además que al bajar el nivel de la mezcla por el
muestreo, la agitación ya no era homogénea, por lo que se optó por solo muestrear al final
de la reacción para determinar conversión final y tamaño de partícula. La determinación de
la conversión se realizó por gravimetría. Para la toma de muestra se prepararon viales de 10
mL, los cuales se enfriaron previamente para inhibir la polimerización, ya que la reacción
de propagación como se dijo antes solo se lleva a cabo por arriba de los 120 °C
aproximadamente. La muestra se extrajo fácilmente por la presión del sistema mediante una

33
válvula conectada a un tubo buzo. Se tomaron aproximadamente 20 g de muestra, que se
repartió en 5 viales, registrando el peso exacto de cada uno de ellos. Las muestras se
congelaron y enseguida se colocaron en un liofilizador Labconco para ser secadas a vacío.
4.4.3 Caracterización
4.4.3.1 Cuantificación del nitróxido en los experimentos de reparto.
La concentración del nitróxido en cada una de las fases se midió a 25 °C. Primero se
corrieron los blancos de cada una de las fases, posteriormente cada una de las muestras se
inspeccionó para ver si no se presentaba algún precipitado del nitróxido y por último se
analizaron por espectrofotometría de UV-visible.
4.4.3.2 Polimerización
1) Tamaño de partícula- Dispersor de Luz – El tamaño de partícula se midió
a 25 °C. Se realizó una dilución del látex al 0.1% peso de polímero en agua
filtrada. El diámetro obtenido mediante esta técnica es el diámetro de
intensidad, Dz.
2) Distribución de pesos moleculares (MWD) y peso molecular promedio
del polímero. Para eliminar el tensoactivo del polímero, éste se disolvió en
cloroformo grado reactivo y se precipitó en metanol grado reactivo (4
veces). Se prepararon muestras a una concentración de 1 mg/mL en THF
grado HPLC empleándose éste mismo como fase móvil.
3) Espectro de RMN H1 – Se prepararon muestras del polímero con una
concentración de 15 mg/mL en cloroformo deuterado (CDCl3). Las pruebas
fueron analizadas con 16 scans de resolución.

34
4) Peso de la fase separada (PFS) – Se dejó reposar la emulsión durante una
semana y posteriormente por medio de un embudo de separación, se extrajo
la fase nadante y se cuantificó. Este parámetro nos indica que tan estable es
la emulsión.

35
5. RESULTADOS Y DISCUSIÓN
5.1 Coeficientes de reparto a 25, 90, 120 y 135 °C para el OH-TEMPO
5.1.1 Cálculo de los coeficientes de extinción molar en Estireno y agua desionizada.
En la Figura 5.1., se muestra la absorción contra la concentración del nitróxido en
la fase orgánica (estireno). Para poder calcular el coeficiente de extinción molar se debe
realizar un ajuste lineal a los datos experimentales. Como se puede observar en la figura, el
valor de R2 es muy cercano a 1, lo cual nos indica que las mediciones fueron confiables. El
valor de la longitud de onda en que el OH-TEMPO en estireno absorbe la luz es 464 nm.
Figura 5.1. Curva de calibración para el cálculo del coeficiente de extinción molar para el OH-
TEMPO en Estireno.
Tomando en cuenta que el valor de la intersección con el eje “y” (absorción) es
muy cercano a cero, se puede despreciar éste valor y nos queda la siguiente ecuación:
cA 74.10
Donde la pendiente es igual al coeficiente de extinción molar multiplicado por la
longitud de la celda de cuarzo (1 cm), por lo tanto el valor del coeficiente de extinción
A = 10.74*c + 0.0152
R² = 0.9975
0
0.1
0.2
0.3
0.4
0 0.005 0.01 0.015 0.02 0.025 0.03 0.035
Ab
sorc
ión
[OH-TEMPO]org (mol/L)

36
molar y la ecuación que se utilizará para calcular la concentración de las muestras de la fase
orgánica en los experimentos de reparto son:
ελ= 10.74 L/mol
(5.1)
En la curva de calibración del OH-TEMPO en agua desionizada, se observa de igual
manera una buena confiabilidad de los datos experimentales, dando como resultado un
ajuste lineal casi perfecto, Figura 5.2.
Figura 5.2. Curva de calibración para el cálculo del coeficiente de extinción molar para el OH-
TEMPO en agua desionizada.
La longitud de onda que se observó para el OH-TEMPO en agua desionizada fue
430 nm, este corrimiento de la longitud de onda es debido a las interacciones que tiene el
soluto con el solvente.
A = 13.044*c + 0.0083
R² = 0.9976
0
0.1
0.2
0 0.002 0.004 0.006 0.008 0.01 0.012 0.014
Ab
sorc
ión
[OH-TEMPO]ac (mol/L)
74.10
Ac

37
De igual manera que en la fase orgánica, se desprecia el valor de la intersección con
el eje “y”, y se obtiene el coeficiente de extinción molar para la fase acuosa, quedando la
ecuación:
ελ= 13.0044 L/mol
(5.2)
5.1.2 Cálculo de los coeficientes de reparto
Para calcular la concentración del nitróxido en las fases orgánica y acuosa se
utilizaron las ecuaciones 5.1 y 5.2 respectivamente. En la Figura 5.3 se muestra la
concentración del nitróxido en la fase orgánica ([OH-TEMPO]org) contra la concentración
del nitróxido en la fase acuosa ([OH-TEMPO]ac), donde se observa cómo cambia la
concentración en cada una de las fases a diferentes temperaturas.
Figura 5.3. Gráfica comparativa de los experimentos de reparto a diferentes temperaturas para el
cálculo de los coeficientes de reparto del OH-TEMPO.
Como se muestra en la figura anterior, el ajuste de los datos experimentales es
relativamente bueno, por lo tanto al cumplirse la linealidad de estos y su intersección con el
0
0.005
0.01
0.015
0.02
0.025
0.03
0.035
0 0.002 0.004 0.006 0.008 0.01 0.012 0.014 0.016
[OH
-TE
MP
O] o
rg (
mol/
L)
[OH-TEMPO]ac (mol/L)
135 °C
120 °C
90 °C
25 °C
044.13
Ac

38
origen, es posible calcular los coeficientes de reparto. En caso de que el ajuste fuera bueno
pero estuviera muy alejado del origen, no sería posible aplicar la ley de Lambert-Beer.
En el capítulo anterior se presentó la ecuación para el cálculo de los coeficientes de
reparto (ec. 4.4), por lo tanto la pendiente de estas líneas representa el coeficiente de reparto
de cada temperatura, en la Tabla 5.1 se presentan los ajustes lineales, el valor de R2 y los
coeficientes de reparto a las diferentes temperaturas.
Tabla 5.1. Coeficientes de reparto y ajustes lineales de los datos experimentales obtenidos del
OH-TEMPO.
Temperatura
(°C) Ajuste lineal R
2 KN
(%mol / %mol)
KNa
(%mol / %mol)
25 y = 0.8688x - 0.0005 0.9788 0.87 2.9
90 y = 8.3487x - 0.0049 0.9928 9.887 11.0
120 y = 10.099x + 0.0014 0.8904 12.799 13.8
135 y = 14.623x - 0.0032 0.9626 14.523 14.3
y= concentración del nitróxido en la fase orgánica.
x=concentración del nitróxido en la fase acuosa.
KNa= coeficientes de reparto para el OH-TEMPO, Cunningham et al.50
Al comparar los valores obtenidos con los de Cunningham et al.50
se observaron
valores similares entre los coeficientes, por lo que se comprueba una vez más la
confiabilidad de los datos experimentales. Debido a las características hidrófilas que posee
el OH-TEMPO, sus coeficientes de reparto son relativamente bajos, sin embargo al
incrementar la temperatura el coeficiente de reparto aumenta. Esto quiere decir que al ir
incrementando la temperatura del sistema, la fase orgánica comienza a incrementar su
solubilidad y así puede disolver mayor cantidad del nitróxido.
Al igual que la temperatura, la presión es un factor importante en el reparto del
nitróxido, esto se correlaciona con el “flash point”o punto de inflamación, el cual es
demasiado bajo para algunos nitróxidos. El punto de inflamación se define como la
temperatura mínima a la cual un líquido emite un vapor, en concentración suficiente como
para formar con el aire una mezcla inflamable cerca de la superficie del líquido, dentro de

39
un recipiente especificado, según procedimientos de prueba e instrumentos apropiados. El
peligro relativo aumenta a medida que baja el punto de inflamación. Cuando se lo calienta a
su punto de inflamación ( o sobre ese punto) cualquier liquido combustible producirá
vapores inflamables. El OH-TEMPO es un nitróxido que no posee punto de inflamación,
debido a que éste se encuentra en estado sólido y es por eso que sí presenta temperatura de
fusión (70 °C). Como se está trabajando a temperaturas relativamente altas es posible tener
en fase vapor un porcentaje del nitróxido en el sistema, es por eso que se necesitó aumentar
la presión aun más que la calculada para el vapor de agua.
5.2 Coeficientes de reparto a 25, 90, 120 y 135 °C para el OXO-TEMPO
5.2.1 Cálculo de los coeficientes de extinción molar en Estireno y agua desionizada.
En la siguiente figura se muestra la absorción contra la concentración del nitróxido
en la fase orgánica. De igual manera que con el nitróxido anterior se realizó el ajuste lineal
a los datos experimentales para calcular el coeficiente de extinción molar.
Figura 5.4. Curva de calibración para el cálculo del coeficiente de extinción molar para el OXO-
TEMPO en Estireno.
El valor de la longitud de onda en que el OXO-TEMPO en Estireno absorbe la luz
es 438 nm. La ecuación resultante que queda para el cálculo de la concentración del
nitróxido en la fase orgánica y el coeficiente de extinción molar son:
A = 6.3868*c + 0.0082
R² = 0.9999
0
0.1
0.2
0.3
0 0.005 0.01 0.015 0.02 0.025 0.03 0.035
Ab
sorc
ión
[OXO-TEMPO]org (mol/L)
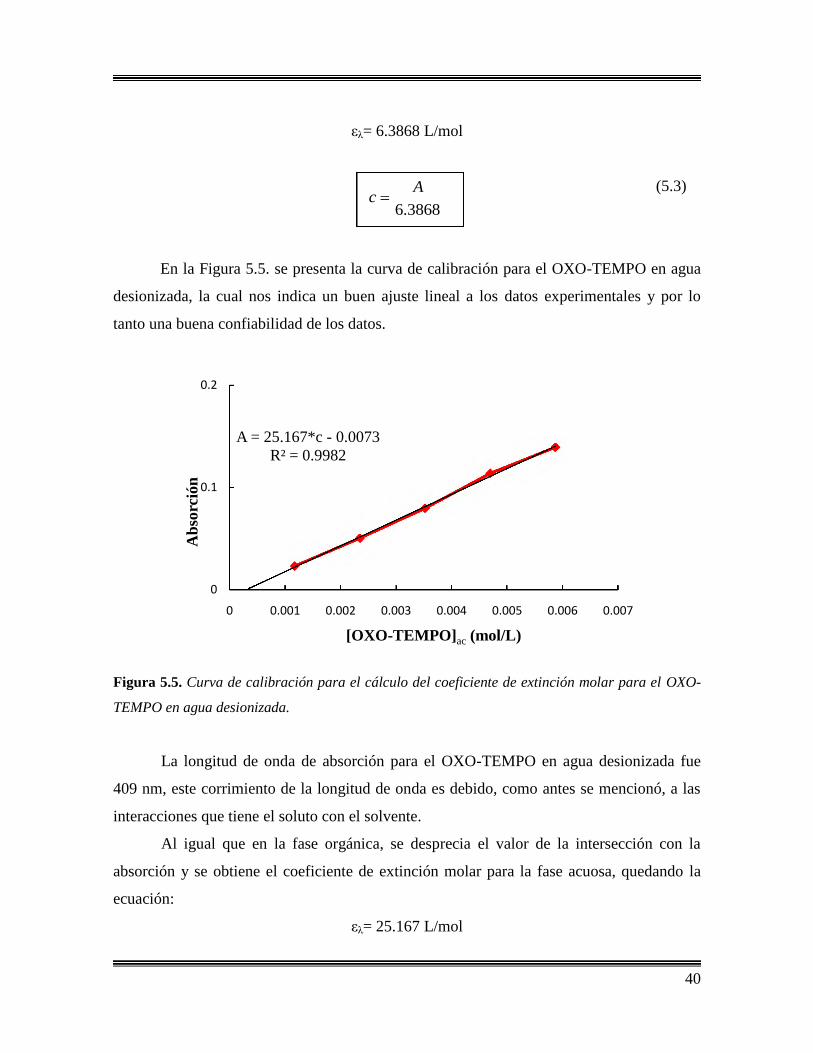
40
ελ= 6.3868 L/mol
(5.3)
En la Figura 5.5. se presenta la curva de calibración para el OXO-TEMPO en agua
desionizada, la cual nos indica un buen ajuste lineal a los datos experimentales y por lo
tanto una buena confiabilidad de los datos.
Figura 5.5. Curva de calibración para el cálculo del coeficiente de extinción molar para el OXO-
TEMPO en agua desionizada.
La longitud de onda de absorción para el OXO-TEMPO en agua desionizada fue
409 nm, este corrimiento de la longitud de onda es debido, como antes se mencionó, a las
interacciones que tiene el soluto con el solvente.
Al igual que en la fase orgánica, se desprecia el valor de la intersección con la
absorción y se obtiene el coeficiente de extinción molar para la fase acuosa, quedando la
ecuación:
ελ= 25.167 L/mol
A = 25.167*c - 0.0073
R² = 0.9982
0
0.1
0.2
0 0.001 0.002 0.003 0.004 0.005 0.006 0.007
Ab
sorc
ión
[OXO-TEMPO]ac (mol/L)
3868.6
Ac

41
(5.4)
5.2.2 Cálculo de los coeficientes de reparto
Se utilizaron las ecuaciones 5.3 y 5.4 para calcular las concentraciones en cada una
de las fases. En la Figura 5.6 se muestra la concentración del nitróxido en la fase orgánica
([OXO-TEMPO]org) contra la concentración del nitróxido en la fase acuosa (OXO-
TEMPO]ac) y se observa cómo cambia la concentración en cada una de las fases a
diferentes temperaturas.
Figura 5.6. Gráfica comparativa de los experimentos de partición a diferentes temperaturas para
el cálculo de los coeficientes de reparto del OXO-TEMPO.
A diferencia del OH-TEMPO, el OXO-TEMPO es menos hidrófilo y es por eso que
no presenta el mismo comportamiento ascendente en los coeficientes de reparto. Como se
puede apreciar en la Tabla 5.2., el coeficiente de reparto presenta un máximo a 90°C;
después de esta temperatura, el coeficiente de reparto disminuye debido a varios
fenómenos; el primero es que el comportamiento del coeficiente de reparto no es lineal,
esto hace que el equilibrio se desplace hacia la fase acuosa; el segundo fenómeno es debido
0
0.001
0.002
0.003
0.004
0.005
0.006
0.007
0 0.00005 0.0001 0.00015 0.0002
[OX
O-T
EM
PO
] org
(m
ol/
L)
[OXO-TEMPO]ac (mol/L)
135 °C
120
90 °C
25 °C
25.167
Ac

42
a que la solubilidad en el agua aumenta en gran medida a partir de 90 °C, más adelante en
la discusión de las polimerizaciones se ahondará en éste fenómeno. Es posible que el
máximo del coeficiente de reparto en el OH-TEMPO se alcance a una temperatura más alta,
sin embargo es impráctico trabajar a temperaturas mayores a 135 °C. El tercer fenómeno es
el incremento en la entropía del sistema, ya que al estar presurizado a una temperatura
relativamente alta, el número de choques entre las moléculas aumenta, desplazando más
soluto hacia la fase que se encuentra en mayor cantidad.
Tabla 5.2. Coeficientes de reparto y ajustes lineales de los datos experimentales obtenidos del
OXO-TEMPO.
Temperatura
(°C) Ajuste lineal R
2 KN
(%mol / %mol)
25 y = 27.598x - 0.0001 0.9984 27.598
90 y = 160.34x - 0.0032 0.9575 160.34
120 y = 90.81x - 0.00011 0.9513 90.81
135 y = 77.32x - 0.0005 0.9925 77.32
y= concentración del nitróxido en la fase orgánica.
x=concentración del nitróxido en la fase acuosa.
Algo que es muy importante mencionar, es que éste nitróxido y el TEMPO poseen
puntos de inflamación bajos. Para el OXO-TEMPO y el TEMPO se tienen los valores de 30
°C y 65 °C respectivamente, por lo tanto se solucionó con un aumento en la presión del
sistema. Además algunos de los nitróxidos poseen muy bajas temperaturas de sublimación
lo que ocasiona que al incrementar la temperatura, el nitróxido comienza a vaporizarse por
arriba de su punto de sublimación, hasta quedar en la tapa del reactor, por lo que disminuye
la concentración del nitróxido en todo el sistema (St/H2O), lo cual ocasiona que al tomar
las muestras, la concentración en éstas sea muy baja. Como antes se mencionó, para no
tener el nitróxido en fase vapor en el sistema, se utilizó para todos los experimentos de
reparto, una presión más alta que la del vapor del agua, la cual fue de 140 psia.

43
5.3 Coeficientes de reparto a 25, 90, 120 y 135 °C para el TEMPO
5.3.1 Cálculo de los coeficientes de extinción molar en Estireno y agua desionizada.
En la Figura 5.7. se observa la absorción contra la concentración del nitróxido en la
fase orgánica. La longitud de onda en la que el TEMPO en estireno absorbe la luz es 466
nm.
Figura 5.7. Curva de calibración para el cálculo del coeficiente de extinción molar para el
TEMPO en Estireno.
De igual manera que en los casos anteriores se calculó el coeficiente de extinción
molar, por lo tanto la ecuación de la concentración queda:
ελ= 10.562 L/mol
(5.5)
A = 10.562*c - 0.0033
R² = 0.9997
0
0.1
0.2
0.3
0.4
0.5
0 0.01 0.02 0.03 0.04 0.05
Ab
sorc
ión
[TEMPO]org (mol/L)
10.562
Ac
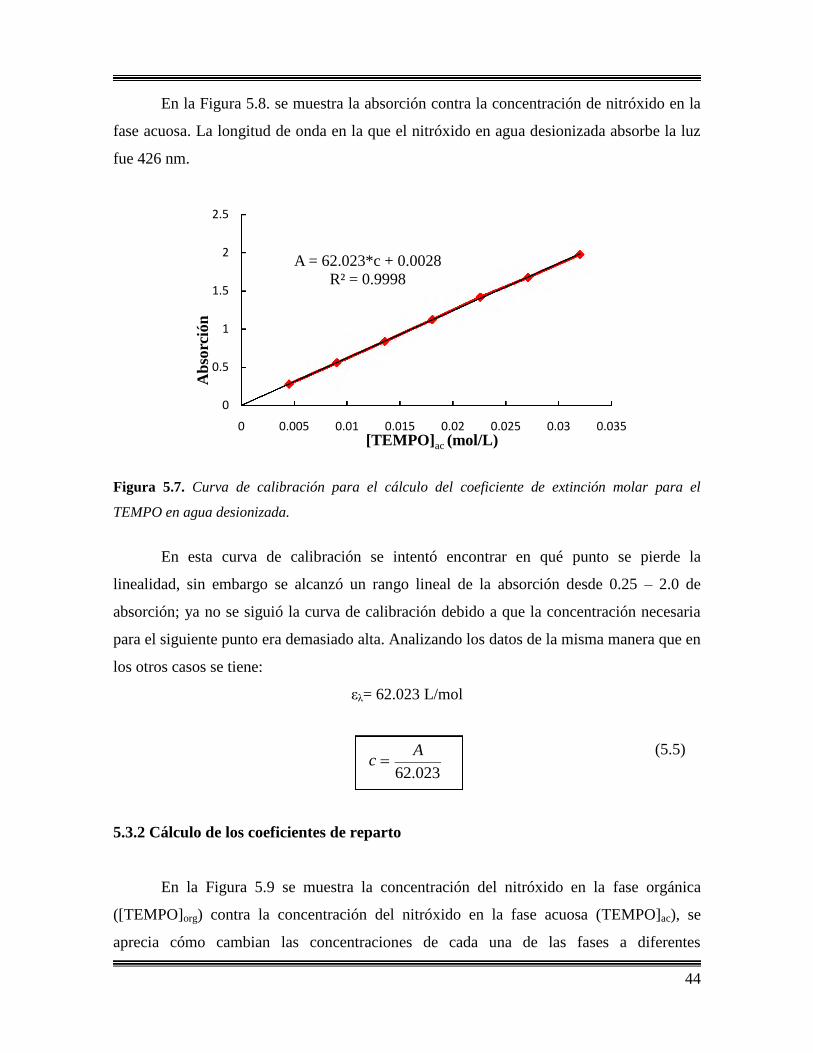
44
En la Figura 5.8. se muestra la absorción contra la concentración de nitróxido en la
fase acuosa. La longitud de onda en la que el nitróxido en agua desionizada absorbe la luz
fue 426 nm.
Figura 5.7. Curva de calibración para el cálculo del coeficiente de extinción molar para el
TEMPO en agua desionizada.
En esta curva de calibración se intentó encontrar en qué punto se pierde la
linealidad, sin embargo se alcanzó un rango lineal de la absorción desde 0.25 – 2.0 de
absorción; ya no se siguió la curva de calibración debido a que la concentración necesaria
para el siguiente punto era demasiado alta. Analizando los datos de la misma manera que en
los otros casos se tiene:
ελ= 62.023 L/mol
(5.5)
5.3.2 Cálculo de los coeficientes de reparto
En la Figura 5.9 se muestra la concentración del nitróxido en la fase orgánica
([TEMPO]org) contra la concentración del nitróxido en la fase acuosa (TEMPO]ac), se
aprecia cómo cambian las concentraciones de cada una de las fases a diferentes
A = 62.023*c + 0.0028
R² = 0.9998
0
0.5
1
1.5
2
2.5
0 0.005 0.01 0.015 0.02 0.025 0.03 0.035
Ab
sorc
ión
[TEMPO]ac (mol/L)
62.023
Ac
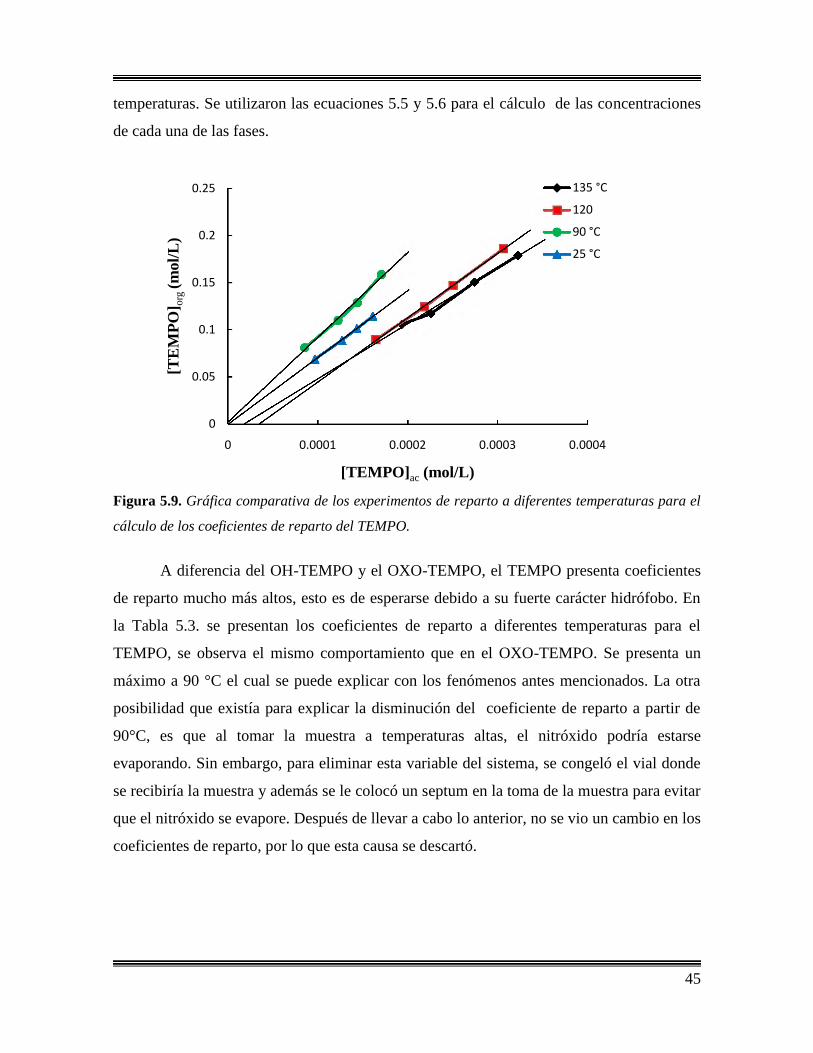
45
temperaturas. Se utilizaron las ecuaciones 5.5 y 5.6 para el cálculo de las concentraciones
de cada una de las fases.
Figura 5.9. Gráfica comparativa de los experimentos de reparto a diferentes temperaturas para el
cálculo de los coeficientes de reparto del TEMPO.
A diferencia del OH-TEMPO y el OXO-TEMPO, el TEMPO presenta coeficientes
de reparto mucho más altos, esto es de esperarse debido a su fuerte carácter hidrófobo. En
la Tabla 5.3. se presentan los coeficientes de reparto a diferentes temperaturas para el
TEMPO, se observa el mismo comportamiento que en el OXO-TEMPO. Se presenta un
máximo a 90 °C el cual se puede explicar con los fenómenos antes mencionados. La otra
posibilidad que existía para explicar la disminución del coeficiente de reparto a partir de
90°C, es que al tomar la muestra a temperaturas altas, el nitróxido podría estarse
evaporando. Sin embargo, para eliminar esta variable del sistema, se congeló el vial donde
se recibiría la muestra y además se le colocó un septum en la toma de la muestra para evitar
que el nitróxido se evapore. Después de llevar a cabo lo anterior, no se vio un cambio en los
coeficientes de reparto, por lo que esta causa se descartó.
0
0.05
0.1
0.15
0.2
0.25
0 0.0001 0.0002 0.0003 0.0004
[TE
MP
O] o
rg (
mol/
L)
[TEMPO]ac (mol/L)
135 °C
120
90 °C
25 °C

46
Tabla 5.3. Coeficientes de reparto y ajustes lineales de los datos experimentales obtenidos.
Temperatura
(°C) Ajuste lineal R
2 KN
(%mol / %mol)
25 y = 719.24x - 0.0004 0.9889 719.24
90 y = 911.3x + 0.0002 0.9836 911.3
120 y = 682.63x - 0.0236 0.9994 682.63
135 y = 634.74x - 0.0103 0.9905 634.74
y= concentración del nitróxido en la fase orgánica.
x=concentración del nitróxido en la fase acuosa.
5.4 Coeficientes de reparto a 25, 90, 120 y 135 °C para el TIPNO
5.4.1 Cálculo de los coeficientes de extinción molar en Estireno y agua desionizada.
En la siguiente figura se muestra la absorción contra la concentración del nitróxido
en la fase orgánica. La longitud de onda observada para el TIPNO en estireno fue 444 nm.
Figura 5.10. Curva de calibración para el cálculo del coeficiente de extinción molar para el
TIPNO en Estireno.
De igual manera que en los casos anteriores se cálculo el coeficiente de extinción
molar, la ecuación queda:
ελ= 2.1989 L/mol
A = 2.1989*c + 0.0434
R² = 0.9975
0
0.02
0.04
0.06
0.08
0.1
0 0.005 0.01 0.015 0.02 0.025
Ab
sorc
ión
[TIPNO]org (mol/L)

47
(5.7)
En la Figura 5.11. se muestra la absorción contra la concentración del nitróxido en
la fase acuosa. La longitud de onda en que el TIPNO en agua desionizada absorbe la luz es
426 nm.
Figura 5.11. Curva de calibración para el cálculo del coeficiente de extinción molar para el
TIPNO en agua desionizada.
De igual manera que en los casos anteriores se calculó el coeficiente de extinción
molar, la ecuación queda:
ελ= 62.023 L/mol
(5.8)
y = 28.52x + 0.0334
R² = 0.9981
0
0.2
0.4
0.6
0.8
0 0.005 0.01 0.015 0.02 0.025
Ab
sorc
ión
[TIPNO]aq (mol/L)
2.1989
Ac
62.023
Ac

48
5.4.2 Cálculo de los coeficientes de reparto
Para el cálculo de las concentraciones en cada una de las fases se utilizaron las
ecuaciones 5.7 y 5.8. En la Figura 5.12 se muestra la concentración del nitróxido en la fase
orgánica ([TIPNO]org) contra la concentración del nitróxido en la fase acuosa (TIPNO]ac),
se aprecia claramente un cambio en las pendientes de cada una de las temperaturas.
Figura 5.12. Gráfica comparativa de los experimentos de reparto a diferentes temperaturas para el
cálculo de los coeficientes de reparto del TIPNO.
En la Tabla 5.4 se puede observar cómo varían los coeficientes de reparto en
función de la temperatura; al igual que los nitróxidos anteriores, se presenta un máximo a
90 °C. Algo importante de mencionar, es que el valor máximo del coeficiente de reparto,
nos da una idea del fuerte carácter hidrófobo. De igual manera se corrobora el
comportamiento que se presentó con el OXO-TEMPO y TEMPO.
0
0.05
0.1
0.15
0.2
0.25
0.3
0.35
0.4
0 0.0001 0.0002 0.0003 0.0004
[TIP
NO
] org
(m
ol/
L)
[TIPNO]ac (mol/L)
135 °C
120
90 °C
25 °C

49
Tabla 5.4. Coeficientes de reparto y ajustes lineales de los datos experimentales obtenidos.
Temperatura
(°C) Ajuste lineal R
2 KN
(%mol / %mol)
25 y = 707.03x + 0.0008 0.984 707.03
90 y = 943.02x - 0.0001 0.9971 943.02
120 y = 710.36x - 0.0004 0.9993 710.36
135 y = 612.32x - 0.0009 0.9909 612.32
y= concentración del nitróxido en la fase orgánica.
x=concentración del nitróxido en la fase acuosa.
El punto de inflamación del TIPNO es de 96 °C, por lo cual no se presentaron los
mismos problemas que en el caso del OXO-TEMPO y el TEMPO, dándole una ventaja al
igual que el OH-TEMPO. Sin embargo la ventaja de estos nitróxidos viene restringida por
su incapacidad de controlar en el medio de polimerización, el cual se comparará más
adelante.
5.5 Comparación de los coeficientes de reparto del OH-TEMPO, OXO-TEMPO,
TEMPO y TIPNO a diferentes temperaturas.
Al tomar en cuenta las características de cada uno de los nitróxidos y compararlas
con las necesidades de la polimerizacion en emulsión, se puede especular que el OH-
TEMPO puede controlar de una mejor manera, debido a que el iniciador es soluble en agua
y por lo tanto éste podrá ser atrapado por el nitróxido para iniciar la polimerización
controlada en la fase acuosa, sin embargo esto se verá restringido por los problemas del
superhinchamiento y Ostwald ripening1. Por otra parte, el relativo carácter hidrófilo del
OH-TEMPO puede dificultar la difusión hacia las gotas de monómero, donde se lleva a
cabo la auto-iniciación térmica del estireno y donde es importante tener una alta
concentración de nitróxido para controlar ahí la polimerización, sin embargo este fenómeno
se ve contrarrestado un poco por las unidades monoméricas que se van uniendo a la cadena.
Es posible que la ausencia de nitróxido en la fase acuosa ocasione que la longitud de las
cadenas poliméricas durmientes que entran a micelas sea mucho mayor, provocando una
desviación importante del peso molecular número experimental. Sin embargo no hay que

50
descartar los demás nitróxidos ya que al ser más hidrófobos podrían controlar de una mejor
manera el problema de la auto-iniciación térmica del monómero.
A continuación se presenta una tabla comparativa de los nitróxidos estudiados a las
diferentes temperaturas, Tabla 5.5. Se aprecia claramente que el más hidrófobo de los
nitróxidos es el TIPNO, su valor más alto de coeficiente de reparto fue 931.09 %mol /
%mol a una temperatura de 90 °C, por otra parte el nitróxido más hidrófilo fue el OH-
TEMPO con un coeficiente de reparto de 11.0 %mol / %mol a 90 °C. Los resultados
concordaron con los esperado OH-TEMPO > OXO-TEMPO > TEMPO > TIPNO en
carácter hidrófilo. También es importante mencionar el aumento en la solubilidad del agua,
la cual se dispara a partir de los 90 °C provocando un descenso en el coeficiente de reparto
de los nitróxidos más hidrófobos. El aumento de la solubilidad del agua repercute
directamente en la polimerización, ya que al no conocerse la CMC de los tensoactivos a
temperaturas mayores de 70 °C, hace imposible determinar la concentración necesaria para
formar micelas.
Tabla 5.5. Coeficientes de reparto calculados a diferentes temperaturas para el OH-TEMPO,
OXO-TEMPO, TEMPO y TIPNO.
KN (%mol / %mol)
T (°C) OH-TEMPO OXO-TEMPO TEMPO TIPNO
25 0.87 27.6 719.24 707.03
90 8.34 160.3 911.3 943.02
120 10.1 90.8 682.63 710.36
135 14.62 77.3 634.74 612.32
En la Figura 5.13 se muestra las gráficas de los coeficientes de reparto de cada uno
de los nitróxidos a las diferentes temperaturas para una mejor apreciación de la tendencia
que presentan y en la Tabla 5.6 se muestran los ajustes polinomiales para la interpolación
de algún valor.
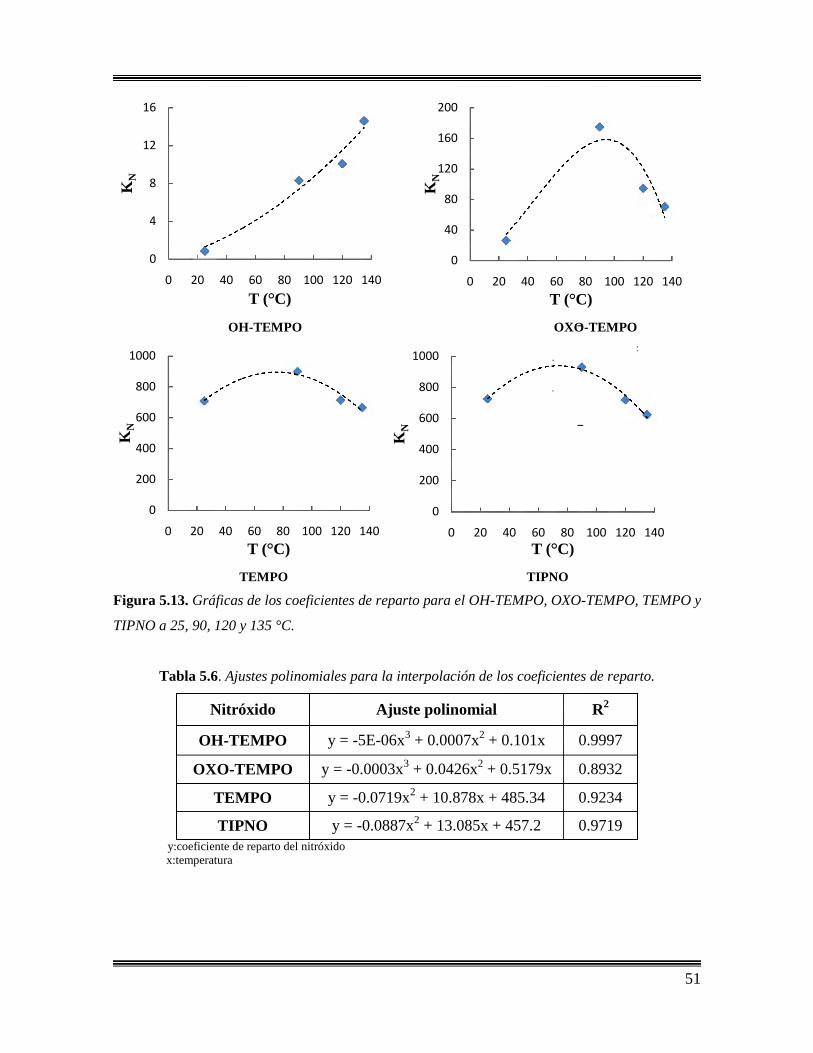
51
OH-TEMPO OXO-TEMPO
TEMPO TIPNO
Figura 5.13. Gráficas de los coeficientes de reparto para el OH-TEMPO, OXO-TEMPO, TEMPO y
TIPNO a 25, 90, 120 y 135 °C.
Tabla 5.6. Ajustes polinomiales para la interpolación de los coeficientes de reparto.
Nitróxido Ajuste polinomial R2
OH-TEMPO y = -5E-06x3 + 0.0007x
2 + 0.101x 0.9997
OXO-TEMPO y = -0.0003x3 + 0.0426x
2 + 0.5179x 0.8932
TEMPO y = -0.0719x2 + 10.878x + 485.34 0.9234
TIPNO y = -0.0887x2 + 13.085x + 457.2 0.9719
y:coeficiente de reparto del nitróxido
x:temperatura
0
40
80
120
160
200
0 20 40 60 80 100 120 140
KN
T (°C)
0
200
400
600
800
1000
0 20 40 60 80 100 120 140
KN
T (°C)
0
200
400
600
800
1000
0 20 40 60 80 100 120 140
KN
T (°C)
0
4
8
12
16
0 20 40 60 80 100 120 140
KN
T (°C)
_______ !/
,,.,,,'
-- -------·-.--
, , ,,'. /
• , , ,
, • , , ,
, , , , ,
, ,
, , ,
• ... -- ........ ,
---------•--,' ... - -
' ' ' ' \ • '-
- -. - +-,
\
\ ~

52
Para entender de una mejor manera lo que representa el coeficiente de reparto, en la
Tabla 5.7 se presenta el porciento mol en cada una de las fases de los nitróxidos a las
diferentes temperaturas.
Tabla 5.7. Porciento mol de los nitróxidos en cada una de las fases a diferentes temperaturas.
OH-TEMPO OXO-TEMPO TEMPO TIPNO
T (°C)
% mol N en St
% mol N en H20
% mol N en St
% mol N en H20
% mol N en St
% mol N en H20
% mol N en St
% mol N en H20
25 46.5241 53.4759 96.5035 3.4965 99.8612 0.1388 99.8588 0.1412
90 89.2934 10.7066 99.3800 0.6200 99.8904 0.1096 99.8941 0.1059
120 90.9910 9.0090 98.9107 1.0893 99.8537 0.1463 99.8594 0.1406
135 93.5980 6.4020 98.7229 1.2771 99.8427 0.1573 99.8370 0.1630 % mol N en St= porciento mol del nitróxido en la fase orgánica.
% mol N en H2O= porciento mol en la fase acuosa.
Para el caso del OH-TEMPO, en la tabla anterior se puede apreciar como el porcentaje
mol del nitróxido en la fase acuosa disminuye y en qué medida aumenta en la fase orgánica.
Por otra parte, el coeficiente de reparto del nitróxido sirve para estimar la
concentración que se tendrá en cada una de las fases al inicio de la polimerización, sin
embargo este coeficiente de reparto varía en el transcurso de ésta. Además se tiene un
coeficiente de alcoxiamina, esto debido a que el nitróxido atrapará una molécula de
iniciador al principio, pero al agregarse unidades de monómero, la alcoxiamina irá ganando
carácter hidrófobo hasta nuclear una micela. Estos coeficientes de reparto se pueden
calcular para cualquier tiempo con la introducción de los términos del nitróxido a un
modelo de balance de población en emulsión ya existente, más adelante se presentarán las
ecuaciones modificadas y en un futuro cercano se llevará a cabo la programación de éstas,
para obtener los resultados. De igual manera se podrá obtener más información de este
modelo, como la cinética, número de partículas y distribución de pesos moleculares
completa, entre otras.

53
5.6 Polimerización de estireno mediada por nitróxidos en emulsión ab initio
Como antes se mencionó, en una polimerización en emulsión mediada por
nitróxidos, la estabilidad se ve comprometida por los problemas de superhinchamiento y
Ostwald ripening. Por lo tanto, estas polimerizaciones serán pruebas exploratorias para
identificar variables importantes y entender el rol del reparto en el control y la estabilidad,
por lo cual no se pretende tener altas conversiones o un control perfecto en las
polimerizaciones. Al realizar los experimentos de reparto se observó que la presión es un
factor importante en el sistema de reparto, debido a las altas temperaturas que se trabaja es
posible que el nitróxido no esté en el sitio de reacción. Estas variables no son tomadas en
cuenta en los trabajos reportados en la literatura, por lo que se trabaja a presiones
relativamente bajas48,50
y a velocidades de agitación estándar.47,48
También hay que tomar
en cuenta, que la mayoría de los nitróxidos son muy volátiles y para contrarrestar esto es
necesario un aumento de la presión.
En la Tabla 5.8 se presenta un resumen de los resultados obtenidos en la conversión
final (X%), tamaño de partícula (Dp), número de partícula (Np), peso molecular en número
(Mn), peso molecular en peso (Mw), índice de polidispersidad (PDI= Mw/Mn) y porcentaje
de la fase separada (PFS) en cada una de las polimerizaciones. Para asegurar la
confiabilidad del método se realizaron 2 repeticiones para el tratamiento R1 (+)(-), las
cuales presentaron gran reproducibilidad en la conversión, tamaño de partícula y número de
partícula. Para calcular la densidad en número de partículas, Np, se utilizó la siguiente
ecuación:
(5.9)
Donde:
Pp = Concentración del polímero.
ρp = Densidad del polímero.
Dp = Diámetro de partícula.
3**
*6
p
p
pD
PN

54
Tabla 5.8. Resumen de resultados de las polimerizaciones del diseño experimental.
Diseño experimental con OH-TEMPO
Tiempo (min)
X (%)
Dp (nm)
Np/ mL agua
Mn g/mol
Mw g/mol
PDI RPM P
(psia) T
(°C) PFS (g)
R1 (+)(-)
240 10.4502 485 8.77E+10 16,439 23,028 1.4 350 100 120 3.8
R1a
(+)(-) 240 10.9834 499 8.51E+10 - - - 350 100 120 3.1
R1b
(+)(-) 240 9.5934 450 9.01E+10 - - - 350 100 120 4
R2 (-)(+)
240 13.7049 949 1.54E+10 9,772 29,079 2.98 250 200 120 10.1
R3 (+)(+)
240 17.7699 510 1.28E+11 25,100 34,007 1.36 350 200 120 1.9
R4 (-)(-)
240 15.4272 297 5.64E+11 15,925 273, 635 17.18 250 100 120 14.6
PFS-peso de la fase separada
5.6.1 Efecto de la presión a nivel alto y bajo de agitación en la conversión, Dp, Np, Mn y
PDI. Comparación de los tratamientos R1(+)(-) con R3(+)(+), R4 (-)(-) con R2(-)(+).
Conversión final
En la figura 5.14 se comparan las conversiones finales (X) para el nivel alto y bajo
de agitación para los dos niveles de la presión (R1(+)(-) con R3 (+)(+) y R4(-)(-) con R2(-
)(+)). Se observa una relación directamente proporcional de la conversión con la presión al
nivel alto de la agitación. Como se dijo antes, al trabajar a temperaturas relativamente altas,
se tiene algunos de los componentes en un equilibrio líquido vapor. Por lo tanto al
aumentar la presión, el sistema se vuelve más estable y esto hace que los componentes se
encuentren en la zona de reacción. El nitróxido, en lugar de condensarse en la tapa del
reactor, se mantiene disuelto en la mezcla de reacción, de igual manera el agua al estarse
evaporando a presiones bajas, arrastra más nitróxido disminuyendo la concentración de
éste; al aumentar la presión se solucionan estos problemas.
Para el caso de la conversión a nivel bajo de agitación, se observa una relación
inversa con la presión, este fenómeno se podrá explicar de mejor manera más delante con
las gráficas de Mn y PDI.

55
50 100 150 200 2500
5
10
15
20
X (
%)
Presión (psia)
Figura 5.14. Efecto de la presión a nivel alto (350 rpm) y bajo (250 rpm) de agitación sobre la
conversión final (X). Comparación de los tratamientos R1(+)(-) con R3(+)(+) y R4(-)(-) con R2
(-)(+).
Número y tamaño de partícula
En la Figura 5.15 se comparan el Np y Dp para el nivel alto y bajo de agitación a los
dos niveles de presión R1(+)(-) con R3 (+)(+) y R4 (-)(-) con R2 (-)(+). Como se puede
apreciar en el nivel alto de agitación, el tamaño y número de partículas se incrementa con la
presión. A pesar de ser un poco más grandes las partículas a presión alta, el número de
partículas es mayor en el nivel alto de la presión que en el bajo. Analizando la fórmula de
Np se puede deducir que aunque el tamaño promedio es mayor en el nivel alto, la
concentración de polímero es mayor en éste, por lo tanto se tiene mayor número de
partículas. Así, la presión ayuda a la estabilidad del polímero para mantener una mayor
concentración de éste en el látex.
Nivel alto de velocidad de
agitación
Nivel bajo de velocidad de
agitación
---x Rl (+X-) y R3 (+X+) a 350 rpm __._ X R4 (-)(-) y R2 (-X+) a 250 rpm

56
Np a nivel alto de velocidad de
agitación
Np a nivel bajo de
velocidad de agitación
Dp a nivel alto de
velocidad de agitación
Dp a nivel bajo de velocidad de
agitación
En el caso del nivel bajo de la agitación, se presenta una relación directa tamaño de
partícula con la presión, sin embargo una relación inversa del número de partículas con la
presión. Esto se debe a que en la prueba R4 (-)(-) se presentó una trimodalidad en el tamaño
de partícula, donde las poblaciones fueron 117 nm, 240 y 5438 nm, pero las áreas que
predominaron en mayor porcentaje fueron las poblaciones más pequeñas, dando como
resultado un tamaño aparente menor, por lo tanto para esta prueba el número de partículas
fue muy alto, debido a la relación inversa que existe con el Dp (ec. 5.9), a diferencia de las
demás pruebas en las que se presentó solo una población en los escaneos del dispersor de
luz. Es probable que la baja velocidad de difusión de las especies y la baja presión del
sistema provoquen esta trimodalidad en el tamaño de partícula.
50 100 150 200 2500.00E+000
2.00E+011
4.00E+011
6.00E+011
8.00E+011
Presión (psia)
Np
/ m
L d
e a
gu
a
0
200
400
600
800
1000
Dp
(nm
)
Figura 5.15. Efecto de la presión a nivel alto (350 rpm) y bajo (250 rpm) de agitación sobre el Np
y Dp. Comparación de los tratamientos R1(+)(-) con R3(+)(+) y R4(-)(-) con R2(-)(+).
Peso molecular y PDI
En la Figura 5.16 se comparan el Mn y PDI para el nivel alto de agitación a los dos
niveles de presión R1(+)(-) con R3 (+)(+) y R4 (-)(-) con R2 (-)(+).
---DpR4 (-X-) y R2 (-X+) a 250 rpm -a-NpR4 (-X-) y R2 (-X+) a 250 rpm
___.... Dp Rt (+X-) y R3 (+) (+) a 350 rpm ___.... Np Rl (+X-) y R3 (+) (+) a 350 rpm
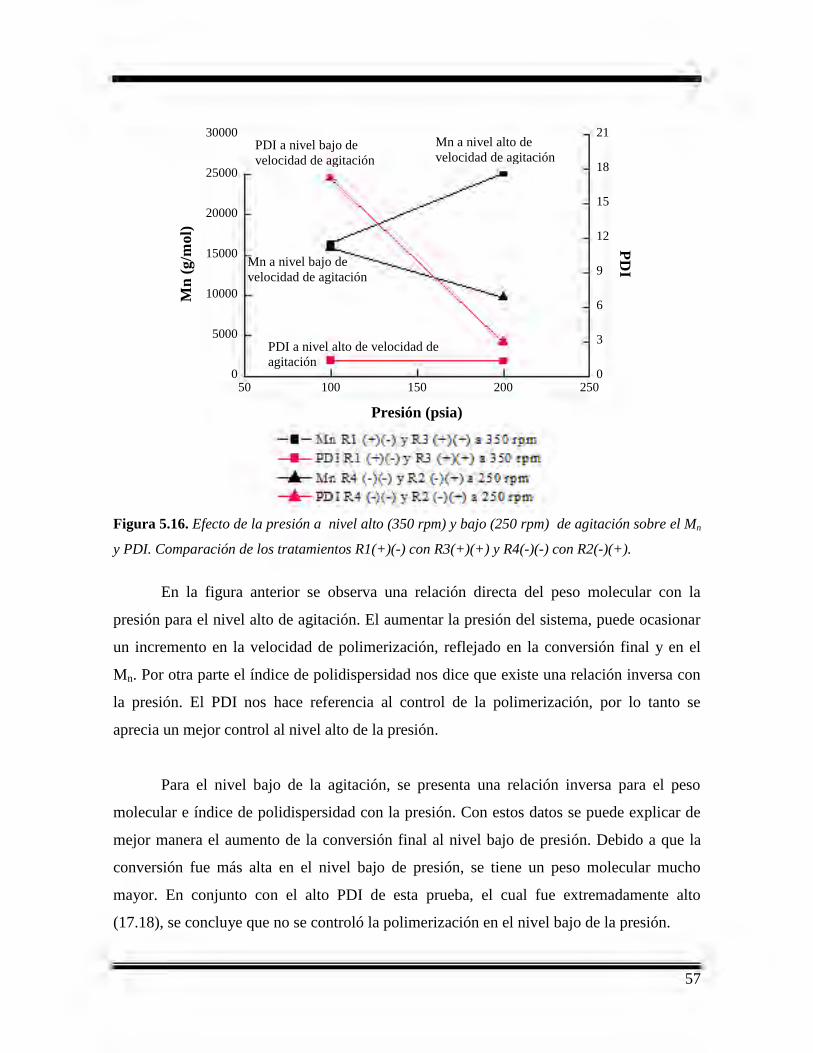
57
Mn a nivel bajo de
velocidad de agitación
PDI a nivel alto de velocidad de
agitación
50 100 150 200 2500
5000
10000
15000
20000
25000
30000
Presión (psia)
Mn
(g
/mo
l)
0
3
6
9
12
15
18
21
PD
I
Figura 5.16. Efecto de la presión a nivel alto (350 rpm) y bajo (250 rpm) de agitación sobre el Mn
y PDI. Comparación de los tratamientos R1(+)(-) con R3(+)(+) y R4(-)(-) con R2(-)(+).
En la figura anterior se observa una relación directa del peso molecular con la
presión para el nivel alto de agitación. El aumentar la presión del sistema, puede ocasionar
un incremento en la velocidad de polimerización, reflejado en la conversión final y en el
Mn. Por otra parte el índice de polidispersidad nos dice que existe una relación inversa con
la presión. El PDI nos hace referencia al control de la polimerización, por lo tanto se
aprecia un mejor control al nivel alto de la presión.
Para el nivel bajo de la agitación, se presenta una relación inversa para el peso
molecular e índice de polidispersidad con la presión. Con estos datos se puede explicar de
mejor manera el aumento de la conversión final al nivel bajo de presión. Debido a que la
conversión fue más alta en el nivel bajo de presión, se tiene un peso molecular mucho
mayor. En conjunto con el alto PDI de esta prueba, el cual fue extremadamente alto
(17.18), se concluye que no se controló la polimerización en el nivel bajo de la presión.
Mn a nivel alto de
velocidad de agitación PDI a nivel bajo de
velocidad de agitación
-•- Mn Rl ( +) (-) y R3 ( +) (+) a 3S0 rpm ---- PDI Rl ( +) (-) y R3 ( +X+) • 3; 0 rpm __._ Mn R4 ( -) (-) y R2 (-) (+ ) • 2 ; 0 rpm __._ PDI R4 ( -) (-) y R2 ( ·) (+) • 2 ; 0 rpm

58
A continuación se presenta la distribución de pesos moleculares final, para cada uno
de los tratamientos, Figura 5.17. De igual manera se pueden apreciar los valores de pesos
moleculares e índices de polidispersidad en la Tabla 5.8.
Figura 5.17. Gráfica comparativa de la distribución de peso molecular final del diseño
experimental.
Tabla 5.9. Peso molecular promedio peso, peso molecular promedio número e índice de
polidispersidad en el polímero a diferente velocidad de agitación y presión.
Prueba Mn (g/mol) Mw (g/mol) PDI
R1 (+)(-) 16,439 23,028 1.4
R2 (-)(+) 9,772 29,079 2.98
R3 (+)(+) 25,100 34,007 1.36
R4 (-)(-) 15,925 273,635 17.18
En la gráfica anterior se aprecia de una mejor manera el grado de control de cada
una de las polimerizaciones. Se observa una gran pérdida del control de la polimerización
en el tratamiento R4 (-)(-) con la trimodalidad de la distribución de peso molecular, esto
debido a la ausencia del nitróxido en la zona de reacción. Por lo tanto, al trabajar a
presiones bajas la vaporización parcial del agua y del nitróxido ocasiona la pérdida del
control de la polimerización, así como también de la estabilidad del látex.
0
0.2
0.4
0.6
0.8
1
1.2
3 3.5 4 4.5 5 5.5 6 6.5 7
dw
/dlo
gM
log M
R1 (+)(-)
R2 (-)(+)
R3 (+)(+)
R4 (-)(-)

59
Algo importante de mencionar, es que en todas las pruebas se presentó una
separación de fases parcial o total de la emulsión. Esto debido a los problemas que se
presentan en la NMRP en emulsión, sin embargo se pudo disminuir la separación de fases
en gran medida con el tratamiento R3 (+)(+), este aspecto se cuantificó pesando la fase
separada o nadante de la emulsión.
5.6.2 Efecto de la velocidad de agitación a nivel alto y bajo de presión en la
conversión, Dp, Np, Mn y PDI. Comparación de los tratamientos R2(-)(+) con R3(+)(+)
y R4 (-)(-) con R1(+)(-).
Conversión final
En la Figura 5.18 se muestra como varía la conversión final (X) en función de la
agitación, para el nivel alto (200 psia) y bajo (100 psia) de la presión, R2(-)(+) con
R3(+)(+) y R4 (-)(-) con R1(+)(-).
Analizando el nivel alto de la presión, se observa una relación directa de la
conversión final con la velocidad de agitación. El aumento en la velocidad de agitación
tiene varios objetivos positivos en una polimerización heterogénea, el primero es el
aumento del esfuerzo de corte para la disminución del tamaño de partícula, este efecto
viene restringido por un máximo en la agitación, ya que si se pasa de este límite las
partículas comienzan a coalescer o aglomerarse. El segundo, es la homogenización de las
variables del sistema como la transferencia de calor y de masa, por lo tanto la difusión del
monómero a las partículas en crecimiento se incrementa en gran medida102
. Por último, el
incremento de la velocidad de agitación ayuda a la suspensión del polímero en el medio
continuo, lo cual evita que el polímero se precipite durante la polimerización.
Por otra parte, se observa una relación inversa de la conversión final con la
velocidad de agitación en el nivel bajo de la presión. Como se dijo anteriormente, el que la
conversión sea mayor a baja agitación, es debido a la pérdida de control de la prueba R4 (-

60
)(-), donde lo más probable es que se llevó una polimerización radicálica convencional, lo
cual se puede corroborar con el peso molecular número e índice de polidispersidad.
200 250 300 350 4000
5
10
15
20X
(%
)
Velocidad de agitación (RPM)
Figura 5.18. Efecto de la velocidad de agitación a nivel alto (200 psia) y bajo (100 psia) de
presión sobre la conversión final (X). Comparación de los tratamientos R2(-)(+) con R3(+)(+) y R4
(-)(-) con R1(+)(-).
Número y tamaño de partícula
En la Figura 5.19 se muestra como varía el Np y Dp en función de la agitación, para
el nivel alto (200 psia) y bajo (100 psia) de la presión, R2(-)(+) con R3(+)(+) y R4 (-)(-)
con R1(+)(-). En la figura se aprecia una relación de Np directa con la velocidad de
agitación y una relación inversa para el Dp a nivel alto de presión. Al incrementar la
velocidad de agitación del sistema se incrementa la velocidad de difusión de las especies
provocando un aumento en la nucleación de micelas, lo cual da como resultado un mayor
número de partículas y por ende un diámetro menor.
En el nivel bajo de la presión, se aprecia una relación inversa de Np con la velocidad
de agitación y una relación directa de Dp con ésta. Este comportamiento anómalo se debe a
Nivel alto de presión
Nivel bajo de presión
--- X R2 (-X+) y R3 (+)(+) a 200 psia ---T-- X R4 (-X-) y Rl (+)(-) a 100 psia

61
Dp a Nivel alto de presión
Np a Nivel alto de presión
Np a Nivel bajo de presión
la trimodalidad del tamaño de partícula antes mencionado, el cual afecta de manera
importante el número de partículas.
200 250 300 350 400
0.00E+000
1.00E+011
2.00E+011
3.00E+011
4.00E+011
5.00E+011
6.00E+011
Velocidad de agitación (rpm)
Np
/ m
L a
gua
0
200
400
600
800
1000D
p (n
m)
Figura 5.19. Efecto de la velocidad de agitación a nivel alto (200 psia) y bajo (100 psia) de
presión sobre el Np y Dp. Comparación de los tratamientos R2(-)(+) con R3(+)(+) y R4 (-)(-) con
R1(+)(-).
Peso molecular y PDI
En la Figura 5.20 se muestra la comparación del Mn y PDI a diferente velocidad de
agitación para el nivel alto (200 psia) y bajo (100 psia) de la presión, R2(-)(+) con R3(+)(+)
y R4 (-)(-) con R1(+)(-).
Dp a Nivel bajo de presión
- • - Np R2 (-X+) y R3 (+X+) a 200 psia --DpR2 (-X+) y R3 (+X+) a 200 psia --4---- Np R4 (-X-) y RI (+X-) a 100 psi a --4- DpR4 (-X-)y RI (+X-) a I00 psia

62
Mn a Nivel bajo de presión
Mn a Nivel alto de presión PDI a Nivel bajo de presión
PDI a Nivel alto de presión
200 250 300 350 4000
5000
10000
15000
20000
25000
30000
Velocidad de agitación (rpm)
Mn
(g
/mo
l)
0
2
4
6
8
10
12
14
16
18
20
PD
I
Figura 5.20. Efecto de la velocidad de agitación a nivel alto (200 psia) y bajo (100 psia) de
presión sobre el Mn y PDI. Comparación de los tratamientos R2(-)(+) con R3(+)(+) y R4 (-)(-) con
R1(+)(-).
Se observa una relación directa del Mn con la velocidad de agitación en el nivel alto
de la presión. Al incrementar la velocidad en que el monómero se difunde de las gotas
hacia las partículas, la velocidad de polimerización aumenta y por lo tanto el Mn también.
De forma lógica se presenta una relación inversa del PDI con la velocidad de agitación en
el nivel alto de la presión, ya que al incrementar la homogeneidad del sistema debido a los
niveles altos de la presión y velocidad de agitación, el nitróxido puede cumplir con su papel
de controlador de forma más eficiente que en los demás casos.
Por otra parte, se observa una relación directa del Mn con la velocidad de agitación
en el nivel bajo de la presión, sin embargo hay que mencionar que la prueba R4 (-)(-)
presentó una trimodalidad en la distribución de peso molecular, lo cual indica que a niveles
bajos de presión y velocidad de agitación, el control y la estabilidad del sistema se pierde.
-•- MnR2 (-X+) y R3 (+X+) a 200 psia -- PDI R2 (-X+) y R3 (+X+) a 200 psia -4- Mn R4 (-)(-) y RI (+X-) a IOO psia -4- PDIR4 (-X-) y RI (+X-) a IOO psia

63
De cualquier manera los resultados obtenidos en la prueba R1 (+)(-) son razonablemente
buenos, ya que se tiene un buen control y estabilidad coloidal. La prueba está en la relación
inversa del PDI con la velocidad de agitación en el nivel bajo de la presión.
En la gráfica anterior se comprueba que el mejor tratamiento de los cuatro es el R3
(+)(+), ya que al compararlo en niveles bajos y altos de presión y velocidad de agitación, es
el que presenta los mejores resultados en cuanto a control y estabilidad coloidal. De
cualquier manera, se puede comparar de otra forma la calidad del control de cada una de las
pruebas, con el cálculo del peso molecular número teórico por medio de la siguiente
ecuación.111
NMI
T
n MwMwXN
MMwM **
0
0 (5.10)
Donde:
MwI = peso molecular del iniciador
MwN = peso molecular del nitróxido
MwM = peso molecular del monómero
0M = moles iniciales de monómero
0N = moles iniciales de nitróxido
X = conversión final
Posteriormente, para comparar los pesos moleculares se definirá a “f” como una
eficiencia aparente del nitróxido, lo cual nos puede dar indicios del control de la
polimerización.
E
n
T
n
M
Mf (5.11)
Donde:
T
nM = peso molecular número teórico
E
nM = peso molecular número experimental

64
En la Tabla 5.10 se presenta el valor de “f” el cual nos indica aproximadamente la
porción del nitróxido que está formando cadenas de polímero. Como se puede apreciar los
valores de “f” son muy bajos, esto lo explica Matyjaszewski102
como una iniciación
ineficiente debido a que el nitróxido no se encuentra unido a la cadena polimérica formando
la especie durmiente, esto ocasiona que exista en gran medida terminación bimolecular por
lo cual aumenta demasiado el peso molecular, estos sistemas se caracterizan por una
iniciación lenta e intercambio lento de las especies. Esto también es debido a que la
alcoxiamína formada en la fase acuosa no alcanza el suficiente carácter hidrófobo para
nuclear micelas y por lo tanto existe una disminución en el número de partículas.
Tabla 5.10. Peso molecular número teórico, peso molecular número experimental y eficiencia
aparente del nitróxido, para cada uno de los tratamientos.
Prueba MnT
MnE
f
R1 (+)(-) 4045 16,439 0.2461
R2 (-)(+) 5029 9,772 0.51475
R3 (+)(+) 6435 25,100 0.2564
R4 (-)(-) 5723 15,925 0.3594 Mn
T= peso molecular número téorico
MnE= peso molecular número experimental
f= eficiencia aparente del nitróxido
Es posible incrementar la eficiencia del nitróxido utilizando una alcoxiamina como
iniciador (sistema unimolecular), donde no se tiene que calcular la relación
nitróxido/iniciador. Por lo tanto, al tener exactamente una molécula de nitróxido e iniciador
el número de cadenas no se verá tan afectado por la terminación bimolecular.
A pesar de que el tratamiento R3 (+)(+) presenta una baja eficiencia aparente, hay
que tomar en cuenta que los tratamientos que presentaron mayor eficiencia, fueron en los
que el peso molecular y el índice de polidispersidad mostraron un bajo control de la
polimerización. Por lo tanto, las condiciones del tratamiento R3 (+)(+) serán utilizadas para
las polimerizaciones con los demás nitróxidos.

65
5.6.3 Análisis estadístico
Para determinar si las variables del diseño experimental tenían algún efecto en las
variables de respuesta, para esto se realizó un análisis de varianza (ANOVA) y para
determinar el porcentaje del efecto sobre la variable de respuesta se realizó una regresión en
el modo stepwise con el software Minitab. A continuación se presenta la nomenclatura
utilizada para el modelo estadístico.
x1= velocidad de agitación
x2= presión
Conversión final
Análisis de varianza para X(%) vs x1
Como se observa no existe efecto de la
variable x1 en la conversión final. El
valor calculado (F) es muy similar al de
tablas (P).
Donde:
DF= grados de libertad
SS= suma de cuadrados
MS= promedio de cuadrados
F= valor calculado
P= valor de tablas
Fuente DF SS MS F P
x1 1 7.5 7.5 0.68 0.455
Error 4 43.8 11.0
Total 5 51.3

66
Análisis de varianza para X(%) vs x2
Se observa un efecto de la variable x2 en
la conversión final. El valor calculado (F)
es mayor que el de tablas (P).
Regresión modo Stepwise de X(%) vs x1, x2
valor de alfa de entrada: 0.15 valor de alfa de salida: 0.15
Con éste valor nos damos una idea del porcentaje del efecto
que tiene la variable x2 sobre la conversión final.
Tamaño de partícula
Análisis de varianza para Dp vs x1
Se observa que no existe efecto de la
variable x1 sobre el tamaño de partícula.
El valor calculado (F) es menor al de
tablas (P).
Fuente DF SS MS F P
x2 1 22.67 22.67 3.17 0.150
Error 4 28.64 7.16
Total 5 51.31
Step 1
Constant 13.68
x2 2.1
T-Value 1.78
P-Value 0.150
S 2.68
R-Sq 44.19
R-Sq(adj) 30.24
C-p 1.3
PRESS 69.2714
R-Sq(pred) 0.00
Fuente DF SS MS F P
x1 1 25025 25025 0.47 0.532
Error 4 214594 53648
Total 5 239619

67
Análisis de varianza para Dp vs x2
Existe un efecto de la variable x2 sobre el
tamaño de partícula. El valor calculado (F)
es mayor que el de tablas (P).
Regresión modo Stepwise de Dp vs x1, x2
valor de alfa de entrada: 0.15 valor de alfa de salida: 0.15
Se corrobora que solamente la variable x2 tiene efecto sobre el
tamaño de partícula.
Número de partícula
Análisis de varianza para Np vs x1
Se observa que no existe un efecto
de la variable x1 en el número de
partículas. El valor de F es muy
pequeño.
Fuente DF SS MS F P
x2 1 117414 117414 3.84 0.121
Error 4 122205 30551
Total 5 239619
Step 1
Constant 581.1
x2 148
T-Value 1.96
P-Value 0.121
S 175
R-Sq 49.00
R-Sq(adj) 36.25
C-p 1.1
PRESS 431388
R-Sq(pred) 0.00
Fuente DF SS MS F P
x1 1 4.914E+22 4.914E+22 1.30 0.319
Error 4 1.517E+23 3.793E+22
Total 5 2.009E+23

68
Análisis de varianza para Np vs x2
No existe un efecto de la variable
x2 en el número de partículas. El
valor de F es similar al de tablas
(P).
Debido a que no existe efecto de ninguna de las dos variables en el número de
partícula, la regresión no se puede hacer ya que no hay porcentaje de efecto que observar.
Peso molecular
Análisis de varianza para Mn vs x1
Podría considerarse que existe un
pequeño efecto de x1 en el peso
molecular. Ya que el valor de F es
un poco más grande que P.
Análisis de varianza para Mn vs x2
No existe efecto de la variable x2 en
el peso molecular. El valor de F es
extremadamente pequeño.
Fuente DF SS MS F P
x2 1 2.431E+22 2.431E+22 0.55 0.499
Error 4 1.765E+23 4.414E+22
Total 5 2.009E+23
Fuente DF SS MS F P
x1 1 44171544 44171544 2.35 0.200
Error 4 75189395 18797349
Total 5 119360939
Fuente DF SS MS F P
x2 1 1689000 1689000 0.06 0.822
Error 4 117671939 29417985
Total 5 119360939

69
Regresión modo Stepwise de Mn vs x1, x2
valor de alfa de entrada: 0.25 valor de alfa de salida: 0.25
Se puede apreciar que solo la variable x1 tiene efecto sobre el
peso molecular y que el tamaño de este efecto es muy pequeño,
esto concuerda con el análisis de varianza.
Indice de polidispersidad
Análisis de varianza para PDI vs x1
Existe efecto de la variable x1 en el índice
de polidispersidad. El valor de F es mayor
que P.
Análisis de varianza para PDI vs x2
No existe efecto de la variable x2 en el
índice de polidispersidad. El valor de F es
muy pequeño.
Step 1
Constant 15726
x1 2878
T-Value 1.53
P-Value 0.200
S 4336
R-Sq 37.01
R-Sq(adj) 21.26
C-p 1.4
PRESS 1.76E+08
R-Sq(pred) 0.00
Fuente DF SS MS F P
x1 1 100.8 100.8 4.00 0.116
Error 4 100.8 25.2
Total 5 201.6
Fuente DF SS MS F P
x2 1 13.5 13.5 0.29 0.620
Error 4 188.1 47.0
Total 5 201.6

70
Regresión modo Stepwise de PDI vs x1, x2
valor de alfa de entrada: 0.15 valor de alfa de salida: 0.15
Se aprecia claramente que solo la variable x1 es la que efecto
sobre el índice de polidispersidad, con un porcentaje
considerable.
Peso de la fase separada
Análisis de varianza para PFS vs x1
Se observa el efecto de la variable x1
muy marcado en el peso de la fase
separada. El valor de F es muy grande
en comparación de P.
Análisis de varianza para PFS vs x2
No existe efecto de la variable x2 en el
peso de la fase separada. El valor de F es
muy pequeño.
Step 1
Constant 5.732
x1 -4.3
T-Value -2.00
P-Value 0.116
S 5.02
R-Sq 50.00
R-Sq(adj) 37.49
C-p 3.0
PRESS 403.285
R-Sq(pred) 0.00
Fuente DF SS MS F P
x1 1 107.40 107.40 32.91 0.005
Error 4 13.05 3.26
Total 5 120.45
Fuente DF SS MS F P
x2 1 0.4 0.4 0.01 0.913
Error 4 120.1 30.0
Total 5 120.5

71
Regresión modo Stepwise de PDI vs x1, x2
valor de alfa de entrada: 0.15 valor de alfa de salida: 0.15
Se observa claramente que existe un efecto muy marcado de la
variable x1 en el peso de la fase separada y el porcentaje de éste
es extremadamente grande.
Tabla 5.11. Efecto de las variables de velocidad de agitación y presión en las variables de
respuesta del diseño experimental.
Variable de respuesta Efecto de la
Vel. de agitación
Efecto de la
presión
Conversión final No Si (+)
Tamaño de partícula No Si (+)
Número de partícula No No
Peso molecular Si (+) No
Indice de polidispersidad Si (-) No
Peso de la fase separada Si (-) No
(+) Efecto directo-al incrementar, la variable respuesta aumenta
(-) Efecto inverso-al incrementar, la variable respuesta disminuye
En la tabla anterior se presentan los efectos en cada una de las variables del sistema
y también si este efecto es directo o inverso. Se observa que para el caso de la conversión
final y tamaño de partícula el efecto de la presión es directo. Para el caso del peso
Step 1
Constant 7.863
x1 -4.49
T-Value -5.74
P-Value 0.005
S 1.81
R-Sq 89.16
R-Sq(adj) 86.45
C-p 18.1
PRESS 45.7044
R-Sq(pred) 62.06

72
molecular el efecto es directo con la velocidad de agitación y para el índice de
polidispersidad y peso de la fase separada el efecto es inverso.
5.6.4 Comparación de los cuatro nitróxidos utilizados
Tomando las condiciones del mejor tratamiento 200 psia y 350 rpm (R3 (+)(+) con
OH-TEMPO), se polimerizó el estireno con los otros tres nitróxidos (OXO-TEMPO,
TEMPO y TIPNO), con el fin de compararlos y concluir cuál de los cuatro es el que
controla de mejor manera, además se realizó una polimerización extra para el TIPNO, el
cual se tiene reportes que trabaja bien a temperaturas menores de los 100 °C.103
En la Tabla
5.12 se presenta un resumen de los resultados obtenidos en la conversión final (X%),
tamaño de partícula (Dp), número de partícula (Np), peso molecular en número (Mn), peso
molecular en peso (Mw), índice de polidispersidad (PDI= Mw/Mn) y porcentaje de la fase
separada (PFS) en cada una de las polimerizaciones. La relación utilizada de N/I fue la
misma para todas las polimerizaciones, al igual que las cantidades de los demás reactivos.
Esto se hizo con el fin de estudiar la influencia del reparto de cada uno de los nitróxidos en
la polimerización.
Tabla 5.12. Resumen de resultados de las polimerizaciones con OH-TEMPO, OXO-TEMPO,
TEMPO y TIPNO.
Tiempo
(min)
X
(%)
Dp
(nm)
Np/ mL
agua
Mn
(g/mol)
Mw
(g/mol) PDI
T
(°C)
PFS
(g)
OH-
TEMPO
R3
(+)(+) 240 17.7699 510 1.28E+11 25,100 34,007 1.36 120 1.9
OXO-
TEMPO R5 240 17.1282 429 2.08E+11 30,469 49,242 1.61 120 4.9
TEMPO R6 240 21.4846 526 1.41E+11 35,155 57,348 1.63 120 2.4
TIPNO R7 240 23.9085 517 1.66E+11 52,251 81,932 1.57 120 1.2
TIPNO R8 240 54.9429 137 2.05E+13 264,321 553,419 2.09 90 0.2

73
Conversión final
La iniciación de una polimerización en emulsión se lleva a cabo en la fase acuosa,
por lo tanto se requiere que el nitróxido sea parcialmente soluble en el agua para que éste
pueda controlar desde el inicio de la polimerización, sin embargo si el nitróxido es
demasiado hidrófilo, éste no controlara la autoiniciación térmica del estireno en las gotas y
micelas hinchadas de monómero. Por lo tanto se requiere un equilibrio cargado hacia la
fase orgánica para controlar la polimerización aquí y una cantidad menor para la
polimerización en la fase acuosa.
En la Tabla 5.12 se observa una conversión razonablemente alta para el TIPNO a
90°C R8, sin embargo al analizar los valores de “f” presentados en la Tabla 5.13, se
concluye que en esta polimerización no hubo control, ya que valores cercanos a cero de “f”
concuerdan con una polimerización radicálica convencional, además del alto peso
molecular y la pobre estabilidad coloidal. Sin embargo, en donde se presentó una mayor
conversión fue en la R7 con el TIPNO a 120 °C, donde se observa un buen control de la
polimerización a pesar de tener un bajo valor de “f”. De cualquier manera se observa que el
OH-TEMPO, OXO-TEMPO y TEMPO presentan una mayor eficiencia que el TIPNO
Tabla 5.13. Peso molecular número teórico, peso molecular número experimental y eficiencia
aparente del iniciador, para cada uno de las pruebas.
Prueba MnT
MnE
f
R3 (+)(+) OH-TEMPO 6435 25,100 0.2564
R5 OXO-TEMPO 6052 30,469 0.1987
R6 TEMPO 7680 35,155 0.2185
R7 TIPNO 9152 52,251 0.1752
R8TIPNO 20129 264,321 0.0762 Mn
T= peso molecular número téorico
MnE= peso molecular número experimental
f= eficiencia aparente del iniciador
Como se puede apreciar la eficiencia aparente del nitróxido está en función del
carácter hidrófilo de éste. A pesar de que el OH-TEMPO presenta un coeficiente de reparto

74
mucho menor que los demás nitróxidos, no olvidemos que aun así el porcentaje mol
presente en la fase acuosa es demasiado bajo, por lo tanto se podría esperar que un
nitróxido más hidrófilo pudiera controlar de mucha mejor manera, sin embargo la síntesis
de un nitróxido con estas características es difícil, complicado y sobre todo costoso.
Número y tamaño de partícula
Como se aprecia en la tabla 5.12, los valores de tamaño de partícula son muy
parecidos entre sí para las pruebas R3 (+)(+), R5, R6 y R7 sin embargo para R8 a 90°C se
presenta un tamaño de partícula característico de una polimerización en emulsión
convencional, además el número de partículas presente en esta prueba es demasiado alto
como resultado del tamaño de partícula, como anteriormente se dijo. Esto quiere decir que
el nitróxido no cumplió con su papel de controlador debido a la baja temperatura de
reacción, que ocasionó que el nitróxido no se reactivara después de formar a la especie
durmiente. El nitróxido atrapa todos los radicales del sistema hasta que cada molécula de
nitróxido se acople con un radical, sin embargo al no presentarse el efecto del radical
persistente y además de que por cada molécula de iniciador salen dos radicales primarios,
el iniciador en exceso podría polimerizar de manera convencional ocasionando la pérdida
del control, sin embargo la prueba R7 con TIPNO a 120°C presenta valores más
congruentes con una polimerización controlada por nitróxidos en emulsión.
Peso molecular y PDI
En la figura 5.21 se presenta una gráfica comparativa de la distribución de peso
molecular completa final de las pruebas R3 (+)(+), R5, R6, R7 y R8. Como se puede
apreciar en la gráfica todas las pruebas presentaron una distribución de peso molecular

75
unimodal, lo cual indica un buen control de las polimerizaciones, a excepción de la prueba
R8 la cual presenta un muy alto peso molecular y una nula eficiencia del nitróxido.
Figura 5.21. Gráfica comparativa de la distribución de peso molecular final de las pruebas R3
(+)(+), R5, R6, R7 a 120°C y R8 a 90°C).
En la Tabla 5.14 se aprecia que el índice de polidispersidad más bajo se presenta
con el OH-TEMPO, como resultado del buen control de la polimerización. De cualquier
manera los demás nitróxidos presentan un aceptable control de la polimerización.
Tabla 5.14. Peso molecular promedio peso, peso molecular promedio número e índice de
polidispersidad en el polímero para las pruebas R3(+)(+), R5, R6, R7 y R8.
Prueba Mn (g/mol) Mw (g/mol) PDI
R3 (+)(+) OH-TEMPO 25,100 34,007 1.36
R5 OXO-TEMPO 30,469 49,242 1.61
R6 TEMPO 35,155 57,348 1.63
R7 TIPNO 52,251 81,932 1.57
R8TIPNO 264,321 553,419 2.09
Al tomar en cuenta la estabilidad de los látex finales, se observa que las emulsiones
más estables hasta el momento han sido la R7 TIPNO y R6 TEMPO. En el látex R3 OH-
TEMPO se presentó un poco mas de separación que en estas pruebas y en la prueba R5
0
0.2
0.4
0.6
0.8
1
1.2
3 4 5 6 7
dw
/dlo
gM
log M
R3(+)(+) OH-TEMPO 125°C
R5 OXO-TEMPO 125°C
R6 TEMPO 125°C
R7 TIPNO 125°C
R8 TIPNO 90°C

76
Hg
OXO-TEMPO se presentó una separación casi total de la emulsión. El látex de la prueba
R8 TIPNO es estable, sin embargo sus resultados no son comparables debido a que se llevo
a cabo una polimerización radicálica convencional. Por último, en la Figura 5.22 se
presenta el espectro de resonancia magnética nuclear 1H para el polímero obtenido en la
prueba R3 (+)(+), con la que se obtuvo los mejores resultados.
Figura 5.22. Espectro de resonancia magnética nuclear obtenido de la prueba R3 (+)(+) (PS-OH-
TEMPO).
En el espectro anterior se puede observar el hidrógeno alfa al oxigeno (Hg) del OH-
TEMPO, el cual nos indica que el nitróxido está unido químicamente al polímero.
5.7 Introducción de los términos del nitróxido a un modelo matemático simplificado
para polimerización en emulsión
El modelo está basado ampliamente en el modelo de Saldívar et al.91
, pero se ha
modificado para incluir los términos del nitróxido. Asimismo, el esquema cinético utilizado
es un subconjunto del descrito en ese modelo. Las reacciones excluidas son todas aquellas
que sólo tienen influencia en la distribución de pesos moleculares y que no afectan
Hg
He
7 .0 6 .0 5.0 • . o PP'Ol
Hi Hi
0-k_OH ·vHj Hi Hi
HfyHb
3 .0 2.0
Hd
Hm
1.0 o

77
esencialmente a la cinética ni a la distribución de tamaños de partícula (DTP). En
consecuencia, el mecanismo cinético incluye las siguientes reacciones.
Mecanismo cinético
Fase acuosa:
- Iniciación
- Propagación
- Terminación
Fase partículas:
- Propagación
- Terminación por acoplamiento y por desproporción
- Terminación por inhibición
- Transferencia de cadena al monómero y a agente de transferencia
- Reiniciación después de transferencia
Este modelo se simplificó para la función de distribución de tipos de partículas, que
elimina la dependencia del tamaño (utiliza un tamaño promedio de partícula), pero no
elimina la dependencia del número de radicales. También se desprecian los términos de
coagulación. El corazón del modelo es la ecuación que describe el número de partículas y
las demás ecuaciones son complementarias, por lo tanto el modelo resultante es:
Función de distribución
Fn(t) = Número de partículas por litro de agua con n radicales al tiempo t
La ecuación de balance de población correspondiente es:

78
c
i
nnpdiipw
c
i
nnpciipw
nn
c
i
c
j
tijji
A
w
c
i
niniwnn
c
i
iwAwn
FFNPkvVnFFnNkpV
FnnFnnkppvN
V
nFdFndVFFeeVNdt
VdF
1
1
1
1
2
1 1
1
11
1
0
1
)1()1)(2(2
)1(
(5.12)
n>1
Con condiciones iniciales:
@ t=0 Fn(t)=0
El primer término de la ecuación 5.12 hace referencia al fenómeno de nucleación
micelar, el segundo se refiere a la desorción y absorción de radicales, el tercero se refiere a
la reacción de terminación de dos radicales. Por último tenemos los términos de
acoplamiento y desacoplamiento del nitróxido con un radical propagante los cuales son
nuevos en el modelo. Lo que se describe con estos términos es la pérdida o ganancia de un
radical propagante dentro de una partícula con “n” radicales. La explicación más detallada
de cada uno de los términos, así como también las demás ecuaciones del sistema, se pueden
ver en Saldivar et al.91
En el anexo de la tesis se puede ver que representa cada una de las
variables del modelo.
Para el caso particular de F1(t) hay que añadir el término de nucleación homgénea,
quedando:
c
i
pdiipw
c
i
pciipw
c
i
c
j
tijji
A
wc
i
iiw
c
i
iwA
c
i
mmiaqAw
FFNPkvVFFNkpV
FkppvN
VFdFdV
FFeeVNHomNuceeVNdt
VdF
1
10
1
12
3
1 11
12
10
1
0
1
01
2
62
2 (5.13)

79
Donde la nucleación homogénea se expresa:
c
i
c
j
cr
l
cr
crrw
r
w
l
jitcijw
aqA
c
i
c
jwjw
cr
ipijw
aqA
c
i
c
jwjw
cr
ipijw
aqA
PPppkVN
MPpkVN
MPpkVNHomNuc
1 1
1
1
1
1
1 1
1
1 1
1
(5.13a)
Y el número total de partículas FT es:
0i
iT FF (5.14)
Asumiendo Ff = 0 para un valor de f finito, entonces:
fT F...FFFF 321 (5.15)
fT F...FFFF 210 (5.16)
La ecuación para FT es:
HomNuceeVNdt
VdF c
i
mmiaqA
wT
1
0 (5.17)
Notar que si se considera que sólo radicales poliméricos acuosos con longitud
mayor a j = je se permite que entren a partículas y micelas, entonces emi = em0 = 0.
HomNuceeVNdt
dFV
c
i
mmiaqAT
w
1
0 (5.18)
Por otro lado, adimensionalizando las ecuaciones (5.17), (5.13) y (5.12) se tiene:
Si Vw = cte y haciendo que FT sea:

80
lessf
TT
M
FF
dim_
(5.19)
HomNuceeVNdt
FdMV
c
i
mmiaqA
T
lessfw
1
0dim_ (5.20)
lessfw
c
i
mmi
lessfw
aqAT
MV
HomNucee
MV
VN
dt
Fd
dim_1
0
dim_
(5.17b)
Similarmente en la ecuación (5.13), haciendo que F1 sea:
lessfM
FF
dim_
11 (5.21)
c
i
pdiip
c
i
pciip
c
i
c
j
tijji
A
c
i
ii
c
i
iA
lessfw
c
i
mmi
lessfw
aqA
FFNPkvFFNkp
FkppvN
FdFd
FFeeNMV
HomNucee
MV
VN
dt
Fd
1
10
1
12
3
1 11
12
10
1
0
dim_1
0
dim_
1
2
62
12
(5.13b)
Así mismo, para la ecuación (5.12) se tiene:
lessf
nn
M
FF
dim_
donde n ≥ 1 (5.22)
c
i
nnpdiip
c
i
nnpciip
nn
c
i
c
j
tijji
A
c
i
nininn
c
i
iA
n
FFNPkvFnFnNkp
FnnFnnkppvN
FndFndFFeeNdt
Fd
1
1
1
1
2
1 1
1
11
1
0
1
)1()1)(2(2
1
)1(
(5.12b)
Crecimiento de partículas:

81
1111
/][)/1(n
npj
c
j
piji
c
in
nA FMkpnFNdt
dm (5.23)
Balances de las especies
Iniciador
Ik
dt
dId (5.24)
Monómero j (Mj) y Unidades Monoméricas en Cadena (Hj)
pj
c
i
piji
n
n
A
wwjwi
c
i
pijaq
j][MkpnF
N
V][M][PkV
dt
dM
111 (5.25)
j = 1,..,c, con c = número de monómeros
pj
c
i
piji
n
n
A
w
wjwi
c
i
pijaq
jMkpnF
N
VMPkV
dt
dH][][][
111
(5.26)
Surfactante
0
dt
dS (5.27)
Nitróxido
ww
c
i
ciiww
c
i
diiw
ww
c
i
ciiww
c
i
diiw
aq
p
c
i
ciip
on
n
A
wp
c
i
diip
on
npw
NPkpPNk
NRkpRNk
V
NkpnFN
VPNkvFVV
dt
dN
11
11
00
(5.28)

82
Coeficiente de reparto del nitróxido
aqwppTotal VNVNNN
Total
(5.29)
Donde:
0n
nwpp FVVVTotal
(5.30)
Vp= volumen de una partícula
Vw= volumen de agua
Sustituyendo 5.30 en 5.29 queda:
aqw
n
nwppTotal VNFVVNN
0
(5.31)
El coeficiente de reparto se define como:
w
p
NN
NK
(5.32)
Despejando para cada una de las concentraciones:
aq
n
nwpN
Totalw
VFVVK
NN
0
(5.33)
N
aq
n
nwp
Totalp
K
VFVV
NN
0
(5.34)
Sustituyendo:

83
aq
n
nwpN
Total
wi
c
i
ciiwwi
c
i
diiw
aq
n
nwpN
Totalw
c
i
ciiww
c
i
diiw
aq
N
aq
n
nwp
Totalc
i
ciip
on
n
A
w
pi
c
i
diip
on
npw
VFVVK
NPkpPNk
VFVVK
NRkpRNk
V
K
VFVV
NkpnF
N
VPNkvFVV
dt
dN
0
11
0
11
0
11
(5.35)
Haciendo la consideración de cuasi-equilibrio del nitróxido y sobre todo para
nitróxidos muy hidrofóbicos, los términos del agua se pueden despreciar en el balance del
nitróxido.
N
aq
n
nwp
Totalc
i
ciip
on
n
A
w
pipi
c
i
diip
on
npw
K
VFVV
NkpnF
N
V
PNPNkvFVVdt
dN
0
1
1
(5.35b)
Notar que N=NTotal
En la ecuación anterior los términos de piPN y
piPN se explican mas
delante con el balance de las especies durmientes.
Balance para especies durmientes totales de tipo i:
widiwiwci
pipidi
N
aq
n
nwp
Totalcii
n
n
A
wi
PNkPNk
PNPNk
K
VFVV
NkpnF
N
V
dt
PdN
0
0
(5.36)
i= 1,…, c

84
El primer término de la ecuación hace referencia al radical de tipo “i” que reacciona
con el nitróxido para formar N-Pi dentro de partículas, el segundo representa la reacción
inversa y el ultimo representa la reacción del nitróxido con un radical primario en la fase
acuosa.
Haciendo la consideración del cuasi-equilibrio se desprecian los términos del agua
en el balance total.
pipidi
N
aq
n
nwp
Totalcii
n
n
A
wi PNPNk
K
VFVV
NkpnF
N
V
dt
PdN
0
0
(5.37)
Tomando en cuenta que la especie alcoxiamina dependiendo de su longitud de
cadena se puede encontrar en cualquiera de las fases, es necesario definir el equilibrio
termodinámico de la alcoxiamina. Para evitar el balance para cada especie por longitud de
cadena, se definé una longitud crítica la cual nos dice que a partir de esta longitud la
alcoxiamina se encontrará en la fase orgánica.
iii PNPNPN
(5.38)
iPN = moles totales de alcoxiamina
iPN = moles de alcoxiamina que se encuentra en partículas (> que longitud crítica)
iPN = moles de alcoxiamina que se encuentra repartida (< que longitud crítica)
Reparto termodinámico de
iPN
wiwpipi PNVPNVPNTotal
(5.39)
Sustituyendo el volumen total de partículas:
wiwpi
n
nwpi PNVPNFVVPN
0
(5.40)

85
Definiendo un coeficiente de reparto para la alcoxiamina:
wi
pi
PNPN
PNK
(5.41)
Despejando para wiPN y sustituyendo en la ecuación 5.40 anterior:
PN
pi
wpi
n
nwpiK
PNVPNFVVPN
0
(5.42)
Despejando para piPN :
PN
w
n
nwp
i
pi
K
VFVV
PNPN
0
(5.43)
Como piPN es la concentración de alcoxiamina que se encuentra solamente en
partículas, se calcula de la siguiente manera:
ipip PNPNVTotal
(5.44)
Despejando:
Totalp
i
piV
PNPN
(5.45)
Sustituyendo el volumen total de partículas:
0n
nwp
i
pi
FVV
PNPN
(5.46)
Ahora tomando las ecuaciones de piPN y
piPN y sustituyendo en las
ecuaciones 5.35 y 5.37 queda:

86
000
0
n
nwp
i
PN
w
n
nwp
i
di
aq
n
nwpN
Total
cii
n
n
A
wi
FVV
PN
K
VFVV
PNk
VFVVK
NkpnF
N
V
dt
PdN
(5.37b)
(5.35c)
Definiendo una relación para resolver el sistema:
i
iPN
PN
PN
(5.47)
Ahora bien si:
iii PNPNPN
(5.38)
Con el parámetro ajustable y la ecuación anterior, se despejó para
iPN y
iPN
queda:
PNii PNPN
1
(5.38a)
PN
PNii PNPN
1 (5.38b)
Sustituyendo en el balance del nitróxido y de alcoxiamína (5.35c y 5.37b) queda:
000
0
11
n
nwp
PNi
PN
w
n
nwp
PN
PNi
di
aq
n
nwpN
Totalcii
n
n
A
wi
FVV
PN
K
VFVV
PN
k
VFVVK
NkpnF
N
V
dt
PdN
(5.37c)
N
aq
n
nwp
Totalc
i
ciip
on
n
A
w
n
nwp
i
PN
w
n
nwp
ic
i
diip
on
npw
K
VFVV
NkpnF
N
V
FVV
PN
K
VFVV
PNkvFVV
dt
dN
0
1
00
1

87
(5.35d)
Balances de especies en fase acuosa
Radicales primarios en fase acuosa
wc
i
diiwaqww
c
i
ciiwaq
ww
w
taqmoaq
n
onwwiw
c
i
riaqd
RNkvVNRkpV
PRkVeVeFVMRkVIfkdt
dR
11
11
2
(5.48)
wc
i
diiwaqww
c
i
ciiwaqw RNkvVNRkpV
dt
RdN
11
(5.49)
Método de pseudohomopolímero
Aplicando el método del pseudohomopolímero se obtienen las siguientes
ecuaciones:
wjwwj PpP (5.50)
c
jwjw PP
1
(5.51)
0l
w
l
jwj PP (5.52)
N
aq
n
nwp
Totalc
i
ciip
on
n
A
w
n
nwp
PNi
PN
w
n
nwp
PN
PNic
i
diip
on
npw
K
VFVV
NkpnF
N
V
FVV
PN
K
VFVV
PN
kvFVVdt
dN
0
1
00
1
11

88
w
wj
jwP
Pp (5.53)
c
jwj
wi
iw
PN
PNv
1
(5.54)
Radicales poliméricos de longitud “1” fase acuosa (P1
w)
(5.55)
wc
i
diiwaqww
c
i
ciiwaq
wPNkvVNPkpV
dt
PdN 1
1
1
1
1
(5.56)
Radicales monoméricos fase Acuosa (P0
w)
wc
i
diiwaq
ww
c
i
ciiwaqww
c
j
jwiw
w
t
c
i
aq
wjw
c
j
iw
w
p
c
i
aq
c
i
wiwmmaq
w
c
i
iwm
n
n
n
w
c
jpijtr
c
i
i
n
n
A
ww
PNkvV
NPkpVPPppkV
MPpkVPMpkaV
PpkFrVMpknFN
V
dt
dP
ij
ijmi
piji
0
1
0
1
0
11
0
111
0
0
11111
0
)(
4
(5.57)
wc
i
diiwaqww
c
i
ciiwaq
ww
c
j
w
tjwiw
c
i
aq
c
i
wiwmmaq
c
i
wmiw
n
n
n
ww
c
jwjiw
w
p
c
i
aq
wwi
c
j
jw
w
p
c
i
aqw
c
iwi
w
paqw
PNkvVNPkpV
PPkppVPMpkaV
PkpFrVPMpkV
PMpkVRMkVdt
dP
ijmi
piij
jii
1
1
1
1
1
111
1
1
1
1
1
11
0
111
1
)(
4

89
wc
i
diiwaqww
c
i
ciiwaq
wPNkvVNPkpV
dt
PdN 0
1
0
1
0
(5.58)
Radicales poliméricos de longitud “l” fase Acuosa ( lP W)
wlc
i
diiwaq
ww
lc
i
ciiwaqww
l
jwiw
c
j
w
t
c
i
aq
c
i
w
l
iwmmaq
c
i
w
l
iwm
n
n
n
w
w
l
wj
c
j
iw
w
p
c
i
aq
lc
i
c
jwijw
w
paq
l
w
PNkvV
NPkpVPPppkV
PMpkaVPpkFrV
PMpkVPMpkVdt
dP
ij
mipi
ijji
1
111
111
11
1
1 1
)(4
(5.59)
wlc
i
diiwaqww
lc
i
ciiwaq
wl
PNkvVNPkpVdt
PdN
11
(5.60)
Nota: para l > 1
Solución para radicales en fase acuosa (QSSA)
D
PNkvVPMkpVVRMk
P
w
c
i
diiwaqw
c
iwi
w
pji
c
j
jwaqaqw
c
iwi
w
pi
W
1
1
0
1 111
(5.61)
donde:
wc
i
ciiwaqw
c
i
c
j
w
tjwiwaq
c
i
c
j
c
i
c
i
iwmm
n
aqnwim
n
wwjiw
w
p
NkpVPkppV
MpkaVFpkrVMpkD
ij
mipiij
11 1
1 1 1 11
4
(5.62)
Para wlP con QSSA se obtiene:

90
D
PNkvVMPpkV
P
lc
i
diiwaq
c
i
c
jwiw
l
jw
w
paq
w
lji
11 1
1
(5.63)
Un balance general sobre radicales totales, se obtiene al sumar las ecuaciones de
wR , wP0, wP1
y wlP , donde l = 1,cr-1. El resultado es:
1
01
1
01
1 111 1
2
1 1
1
1
1
0
1
1
01
42
cr
l
w
l
w
c
i
diiw
cr
l
w
l
ww
c
i
ciiwaq
c
j
c
jpijtri
n
n
A
wc
i
c
j
wiwjw
w
taq
c
i
c
jwjw
cr
iw
w
paq
c
i
cr
l
w
l
iwmmaq
wmmRmaq
n
n
w
cr
l
lc
i
iwmwmpR
n
wdw
PNRNkvPRNkpV
MpknFN
VPppkV
MPpkVPMpkaV
RMkaVFPpkRkrVIfkdt
dP
jiij
ijmi
pi
(5.64)
Recordando que:
1
0
cr
l
wl
ww PRP (5.65)
1
0
cr
l
wl
ww PNRNPN
(5.66)
ww
c
i
ciiww
c
i
diiwaq
c
i
c
jpijtri
n
n
A
w
c
i
c
j
wiwjw
w
tijaq
c
i
c
jwjw
cr
iw
w
paq
w
c
i
iwmmaqw
c
i
iwm
n
n
n
wdw
PNkpPNkvVMpknFN
V
PppkVMPpkV
PMpkaVPpkFrVIfkdt
dP
ji
ij
mipi
111 11
1 1
2
1 1
1
111
42
(5.67)
Con QSSA esta ecuación se aproxima a cero y resultará una sola ecuación
algebraica no lineal en wP ; sin embargo, todavía depende de wcrP 1, que a su vez

91
depende de wP0, wR y wP . Para escribir todo en términos de wP , conviene escribir
wR y wP0 en términos de wP solamente:
Tomando QSSA para 0dt
dRw y despejando wR queda:
)4
(2
01 111
110
NkpVPppkVpkFrV
MpkaVMkVRNkvVIfkR
c
i
ciiwaq
c
j
c
i
wjwiw
w
tijaq
c
i
iwmpi
n
n
n
w
c
i
iwmmaqwi
c
i
riaqw
c
i
diiwaqdw mi
(5.68)
Sin embargo, si primero se asume cuasi-equilibrio para wRN , entonces
0
dt
RdN w por lo tanto se eliminan los términos que involucran al nitróxido, quedando:
( (5.69)
y haciendo QSSA para wP0 queda:
0
0
dt
Pd w (5.69)
Entonces:
)
4(
1 11 1
111111
0
c
i
c
j
w
w
tjwiwaq
c
i
c
jwjiw
w
paq
c
i
iwmm
c
i
aqmiw
n
n
nw
c
jpijtr
c
i
i
c
n
n
A
ww
PkppVMpkV
MpkaVkprFVMpknFN
VP
ijij
mipiji
(5.68)
Así, resulta una sola ecuación algebraica no lineal en wP ; y una vez que se
resuelve, se obtienen de manera explícita (hacia atrás) wP0, wR , wlP , etc, donde l =
)
4(2
1 1
1111
c
j
c
i
wjwiw
w
taq
c
i
iwm
n
n
n
w
c
i
iwmmaqwi
c
i
riaqdw
PppkV
pkFrVMpkaVMkVIfkR
ij
pimi
/
/
/

92
cr-1, 2, 3... Por lo tanto aplicando el cuasi-equilibrio en 5.56, 5.58 y 5.60 se desprecian los
términos del nitróxido de las ec 5.61, 5.63, 5.67 y 5.68. De esta manera se considera que el
nitróxido no afecta en la cinética en la fase acuosa.
Sin embargo, si se quiere ser más riguroso se pueden conservar las ecuaciones
diferenciales 5.56, 5.58 y 5.60 las cuales se tendrían que resolver simultáneamente con las
ecuaciones algebraicas completas (incluyendo los términos del nitróxido).

93
6. CONCLUSIONES
1. La presión es un factor que afecta en gran medida al reparto termodinámico del
nitróxido. Esto se comprobó con el cálculo de un coeficiente de reparto a presión
relativamente baja y otro a una presión alta, donde se observó un notable
incremento en el coeficiente de reparto.
2. A pesar de que el OH-TEMPO presentó coeficientes de reparto mucho más
pequeños que los demás nitróxidos, el porcentaje mol en la fase acuosa de éste
nitróxido es considerablemente baja.
3. El mejor tratamiento fe el realizado con los niveles altos de velocidad de agitación y
presión R3 (+)(+), ya que presenta un buen control de la polimerización, buena
estabilidad coloidal, un PDI aceptable y una mayor eficiencia del nitróxido
aparente.
4. Comparando las polimerizaciones llevadas a cabo con el OH-TEMPO, OXO-
TEMPO, TEMPO y TIPNO, se concluye que con el OH-TEMPO se obtuvo un
mejor control sobre la polimerización.
5. Es muy probable que una gran parte de la alcoxiamina primaria formada en la fase
acuosa no alcance el carácter hidrófobo necesario para nuclear las micelas, lo cual
provoca una disminución en el número de partículas y por ende un aumento en el
diámetro de partícula.

94
7. REFERENCIAS
1. K. Matyjaszewski, T. P. Davis, Handbook of Radical Polymerization, John Wiley
and Sons (2002).
2. M. Szwarc, Nature 178, 1168 (1956).
3. O. W. Webster, Science 251, 887 (1991).
4. K. Matyjaszewski and A. H. E. Muller, Polym. Prepr. (Am. Chem. Soc., Div.
Polym. Chem.) 38(1), 6 (1997).
5. K. Matyjaszewski and P. Sigwalt, Polym. Int. 35, 1 (1994).
6. K. Matyjaszewski, Controlled / Living Radical Polymerization: State of the Art in
2005. Chapter 1 in Controlled / Living Radical Polymerization, Ed. By K.
Matyjaszewski, ACS Symposium Series 944, ACS Washington D.C. (2006).
7. Y. Chiefari, K. Chong, F. Ercole, J. Krstina, J. Jeffery, T.P.T. Le, R. T. A.
Mayadunne, G. F. Meijs, C. L.Moad, G. Moad, E. Rizzardo, S. H. Thang,
Macromolecules, 31, 5559, (1998).
8. Y. K. Chong, T. P. T. Le, G. Moad, E. Rizzardo, S. H. Thang, Macromolecules, 32,
2071, (1999).
9. T. Fukuda, A. Goto, K. Ohno, Macromol. Rapid Commun. 21, 151, (2000).
10. H. Fischer, J. Polym. Sci. A: Polym. Chem. 37, 1885, (1999).
11. C. J. Hawker, A. W. Bosman, E. Harth, Chem. Rev. 101, 3661 37-42, (2001).
12. H. Fischer, Chem. Rev. 101, 3581, (2001).
13. E. D. Leon-Saenz, G. Morales, R. Guerrero-santos, and Y. Gnanou, Macromol.
Chem. Phys. 201, 74 (2000).
14. B. Yamada, Y. Nobukane, and Y. Miura, Polym. Bull. 41, 539 (1998).
15. 17. M. Steenbock, M. Klapper, and K. Mullen, Macromol. Chem. Phys. 199, 763
(1998).
16. D. H. Solomon, E. Rizzardo, and P. Cacioli, U.S. Patent 4,581,429 (1986).
17. G. Moad, E. Rizzardo, and D. H. Solomon, Macromolecules 15, 909 (1982).
18. M. K. Georges, R.P.N. Veregin, P. M. Kazmaier, and G. K. Hamer,
Macromolecules 26, 2987 (1993).

95
19. K. Matyjaszewski, S. Gaynor, D. Greszta, D. Mardare, and T. Shigemoto, J. Phys.
Org. Chem. 8, 306 (1995).
20. P. G. Odell, R.P.N. Veregin, L. M. Michalak, and M. K. Georges, Macromolecules
30, 2232 (1997).
21. C. J. Hawker, J. Am. Chem. Soc. 116, 11185 (1994).
22. Vivaldo-Lima, E.; Wood, P. E.; Hamielec, A. E.; Penlidis, A. Ind. Eng. Chem. Res.
36, 939, (1997).
23. Peter A. Lovell, Mohamed S. El-Aasser. Emulsion Polymerization and Emulsion
Polymer, John Wiley and Sons Inc. Capitulo 2, (1997)..
24. Emulsion Polymerization. A mechanistic approach. Robert G. Gilbert. Academic
Press Inc. Capitulo 1, (1995).
25. P. Y. Chow, L. M. Gan, Adv. Polym. Sci. 175, 257, (2005).
26. Gardon, J. L.; J. Polym. Sci. 6 (A-1) , p. 643, (1968),
27. Landfester, K.; Annu. Rev. Mater. Res. 36:231–79, (2006).
28. Chern, C. S,; Chen, T. J.; Colloids Surf A 138:65–74, (1998).
29. Durand, A. ; Marie, E. ; Rotureau, E.; Leonard, M. ; Dellacherie, E. ; Langmuir
20:6956–63, (2004).
30. Kim, N.; Sudol, E. D.; Dimonie V. L.; El-Aasser M. S.; Macromolecules 37:3180–
87, (2004).
31. Landfester, K.; Pawelzik, U.; Antonietti, M.; Polymer 46:9892–98,(2005).
32. O´Donnell, J.; Kaler, E. W. Macromol. Rapid Commun. 28, 1445–1454, (2007),
33. Texter, F. Yan, Soft Matter 2, 109, (2006).
34. Liu, S.; Hermanson, k. D.; Kaler, W.; Macromolecules 39, 4345, (2006).
35. Zheng, C.; He, W.; Liu, W.; Li, J.; Li, J. F.; Macromol. Rapid Commun. 27, 1229.
(2006).
36. Trevino, M.; Lopez, R. G.; Peralta, R. D.; Becerra, F.; Mendizabal, E.; Puig, J. E.;
Polym. Bull. 42, 411, (1999).
37. Zetterlund, Per B.; Kagawa, Y.; Okubo M.; Chem. Rev., 108 (9), pp 3747–3794,
(2008).
38. Lansalot, M.; Farcet, C.; Charleux, B.; Vairon, J. P.; Pirri, R.; Tordo, P. In
Controlled/LiVing Radical Polymerization: Progress in ATRP, NMP and RAFT;

96
Matyjaszewski, K., Ed.; ACS Symposium Series 768; American Chemical Society:
Washington, DC, p 138, (2000).
39. Marestin, C.; Noel, C.; Guyot, A.; Claverie, J. Macromolecules 31, 4041, (1998).
40. Cao, J.; He, J.; Li, C.; Yang, Y. Polym. J 33, 75, . (2001).
41. Szkurhan, A. R.; Georges, M. K. Macromolecules 37, 4776, (2004).
42. Simms, R. W.; Hoidas, M. D.; Cunningham, M. F. Macromolecules 41, 1076,
(2008).
43. Jung, K. J. Polym. Sci. 46, 6983 (2008).
44. Cunningham, M. F.; Prog. Polym. Sci. 33, 365–398, (2008).
45. Maehata, H.; Liu, X.; Cunningham, M.; Keoshkerian, B. Macromol. Rapid Commun
29, 479, . (2008).
46. Delaittre, G.; Nicolas, J.; Lefay, C.; Save, M.; Charleux, B. Chem. Commun. 5, 614,
(2005).
47. Delaittre, G.; Nicolas, J.; Lefay, C.; Save, M.; Charleux, B. Soft Matter 2, 223,
(2006).
48. Farcet, C.; Lansalot, M.; Charleux, B.; Pirri, R.; Vairon, J. P. Macromolecules 33,
8559, (2000).
49. Nicolas, J.; Charleux, B.; Guerret, O.; Magnet, S. Macromolecules 37, 4453,
(2004).
50. Ma, J. W.; Cunningham, M. F.; McAuley, K. B.; Keoshkerian, B.; Georges, M. K.;
J. Polym. Sci., Part A: Polym. Chem 39, 1081, . (2001).
51. Zetterlund, Per.; Macromol. Theory Simul. 19, 11–23, (2010).
52. M. N. Alam, P. B. Zetterlund, M. Okubo, J. Polym. Sci., Part A: Polym. Chem. 45,
4995, (2007).
53. M. N. Alam, P. B. Zetterlund, M. Okubo, Polymer 50, 1632, (2009).
54. M. N. Alam, P. B. Zetterlund, M. Okubo, Polymer 49, 3428, (2008).
55. M. N. Alam, P. B. Zetterlund, M. Okubo, Polymer 49, 883. (2007).
56. M. Lin, J. C. C. Hsu, M. F. Cunningham, J. Polym. Sci., Part A: Polym. Chem. 44,
5974, (2006).
57. P. B. Zetterlund, T. Nakamura, M. Okubo, Macromolecules 40, 8663, (2007).
58. H. Tobita, Macromol. Theory Simul.,16, 810, (2007).

97
59. H. Tobita, F. Yanase, Macromol. Theory Simul. 16, 476, (2007).
60. M. Wulkow, Macromol. React. Eng. 2, 461, (2008).
61. Jentoft F.C. Diffuse Reflectance IR and UV-vis Spectroscopy Fritz-Haber-Institut
der Max-Planck-Gesellschaft. (2004)
62. Gao J.; Penlidis A. Prog. Polym. Sci. 27 403-535 (2002).
63. Ballard M.J.; Gilbert R.G.; Napper D.H. J. Polym Sci. 19, 533, (1981).
64. Broadhead T.O.; Hamielec A.E.; MacGregor J.F. Makromol. Chem Suppl. 10/11,
105-28, (1985).
65. Congalidis J.P; Richards J.R; Bilbert R.G. ACS Symp. Ser. 404, 360-78, (1989).
66. Dougherty E.P. J. Appl. Polym. Sci. 32, 3051-78, (1986).
67. Dubé M.A.; Penlidis A.; Mutha R.K.; Cluett W.R. Ind. Engng. Chem Res. 35, 4434-
48, (1996).
68. Forcada J.; Asua J.M. J. Polym. Sci. Chem. 28, 987-1009, (1990).
69. Forcada J.; Asua J.M. J. Polym. Sci. Chem. 29, 1231-42, (1991).
70. Giannetti E.; Storti G.; Morbidelli M. J. Polym. Sci. Chem. 26, 2307-43, (1988).
71. Giannetti E. Macromol. 22, 2094-102, (1989).
72. Guillot J.; New York: Huthig & Wepd. 147-64, (1986).
73. Hamielec A.E.; MacGregor J.F; Penlidis A. Macromol. Symp. 10/11, 521-70,
(1987).
74. Hamielec A.E.; MacGregor J.F; Penlidis A. Polym. React. Eng. 21-7, (1983).
75. Lichti G.; Gilbert R.G.; Napper D.H. New York: Academic Press. 94-144, (1982).
76. Lin C.; Ku H.C.; Chiu W.Y. J. Appl. Sci. 26, 1327, (1981).
77. Mead R.N.; Poehlein G.W.; Int Engng. Chem. Res. 28, 51, (1988).
78. Mead R.N.; Poehlein G.W.; J. Appl. Sci. 238, 105-22, (1989).
79. Min K.W., Ray W.H. J. Macromol. Sci. 11, 177-255, (1974).
80. Min K.W., Ray W.H. J. Appl. Sci. 22, 89-112, (1978).
81. Min K.W., Ray W.H. J. Macromol. Sci. 11, 177-255, (1974).
82. Nomura M.; Kubo M.; Fujita K. J. Appl. Sci. 28, 2767-76, (1983).
83. Nomura M.; Fujita K. Makromol. Chem. Suppl. 10, 25-42, (1985).
84. Penlidis A.; Hamielec A.E.; MacGregor A.E. J Vinyl Technol. 6, 134-42, (1984).

98
85. Penlidis A.; Hamielec A.E.; MacGregor A.E. Polym. Porc. Engng. 3, 185-218,
(1985).
86. Penlidis A.; Hamielec A.E.; MacGregor A.E. AIChE. J. 31, 881-9, (1985).
87. Rawlings J.B. Phd Thesis. University of Wisconsin, (1985).
88. Richards J.R.; Cangalidis J.P; Gilbert R.G. J. Appl. Polym. Sci. 37, 2727-78, (1989).
89. Rawlings J.B.; Ray W.H. Polym Engng Sci. 28, 237-56, (1988).
90. Rawlings J.B.; Ray W.H. Polym Engng Sci. 28, 257-74, (1988).
91. Saldivar E.; Dafnitois P.; Ray W.H. J. Macromol. Sci. 38, 207-325, (1998).
92. Storti G.; Morbidelli M.; Carra S. Comput. Appl. Polym. Sci. II ACS Symp. Ser. 404,
79-402, (1989).
93. Storti G.; Polotti G.; Cociani M.; Morbidelli M. J. Polym. Sci. Polym. Chem. 30,
731-50, (1992).
94. Urretabizkaia A.; Asua J.M. J. Polym. Sci. Chem. 32, 1761-78, (1994).
95. Urretabizkaia A.; Arzamendi A.; Unzue M.J.; Asua J.M. J. Polym. Sci. Chem. 32,
1779-88, (1994).
96. Zetterlund P.; Okubo M. Macromol. Theory Simul. 14, 415–420, (2005).
97. Zetterlund P.; Okubo M. Macromol. 39, 8959–8967, (2006).
98. Zetterlund P.; Okubo M. Macromol. Theory Simul. 18, 277–286, (2009).
99. Zetterlund P. Macromol. Theory Simul. 19, 11-23, (2010).
100. Lin M.; Hsu J.; Cunningham M. J. Polym. Sci. Part A Polym. Chem. 44, 5974-
5986, (2006).
101. Lagrille O.; Cameron N.R.; Lovell P.A.; Blanchard G. Goeta; Koch. J. Polym.
Sci. 44, 1926-1940, (2006).
102. Sajjadi S.; Macromol. Rapid Commun. 25, 882-887, (2004).
103. Braslau R.; O´Bryan G.; Nilsen A.; Henise J.; Thongpaisanwong T.; Murphy E.;
Mueller L.; Ruehl J.; Template for Synlett and Synthesis. 1-14 (2006).
104. Harkins W. D.; A general Theory of Mechanism of Emulsion Polymerization. 69,
(1947).
105. Smith W. ;Ewart R. Journal of Chem Phys. 16, No 6 (1948).
106. Gardon L. Jornal of Polym Sci. 6, 623-641, (1968).

99
108. Ledezma R.; Elizalde L.; Pérez-Carrillo L.; Mendizábal E.; Puig J. E.; López R. G.
Journal of Poly Sci Part A: Poly Chem. 5, 8, 1463, (2007).
109. Sajjadi S. Division of Engineering, King´s College London. (2007).
110. Kendall J.; Canelas D.; Young J.; DeSimone J. Chem Rev. 99, 543-563, (1999).
111. Nicolas J.; Charleux B.; Guerret O.; Magnet S. Macromol. 37, 4453-4463, (2004).

100
ANEXO
Fn(t) Número de partículas por litro de agua con n radicales al tiempo t, (s)
n Número de radicales
NA Número de Avogadro
Vw Volumen de agua en el reactor, (L)
ei, e0 Tasa de entrada de radicales de longitud i y de radicales primarios a
partículas, respectivamente, (mol/s)
di Tasa de desorción de radicales de partículas, (1/s)
v Volumen de partícula, (L)
pi, pj Fracción de radicales de tipo i y j (unidad monomérica activa tipo i y j)
ktij Constante cinética de terminación entre radicales de tipo i y j, (L/mol*s)
pip Fracción de radicales de tipo i dentro de partículas
vi Fracción de alcoxiamína con unidad monomérica tipo i dentro de partículas
kci, kdi Constante de activación y desactivación de la cadena polimérica con el
nitróxido
pN Concentración de nitróxido en partículas, (mol/L)
pi NP Concentración de alcoxiamína en partículas (mol/L)
Vaq Volumen de la fase acuosa
kw
pij Constante cinética de propagación de radical tipo i con monómero j en la
fase acuosa, (L/mol*s)
kw
tcij Constante cinética de terminación de radical tipo i con monómero j en la fase
acuosa, (L/mol*s)
wcrP 1 Concentración de polímero con longitud cr-1 en la fase acuosa, (mol/L)
cr Longitud crítica de radical para precipitación
wjM Concentración de monómero j en la fase acuosa, (mol/L)
pjM
Concentración de monómero j en la fase partículas, (mol/L)
wiP
Concentración de polímero con unidad monomérica terminal de tipo i en la
fase acuosa, (mol/L)
c Número de monómeros

101
wlP Concentración de polímero con longitud l en la fase acuosa, (mol/L)
wrP Concentración de polímero con longitud r en la fase acuosa, (mol/L)
m Masa de partícula, (g)
I Moles de inciador
kd Constante cinética de descomposición del iniciador, (1/s)
f Eficiencia del inciador
S Moles de tensoactivo
Hj Unidades monómericas en cadena
N Moles de nitróxido
wR Concentración de radicales primarios en la fase acuosa, (mol/L)
wNR Concentración de alcoxiamína primaria, (mol/L)
VpTotal Volumen de partículas total (L)
KN Coeficiente de reparto del nitróxido


















