
AROILPIRROLHETEROARIL Y METANOLES UTILES · PDF fileel sistema nervioso periférico o...
32
19 OFICINA ESPAÑOLA DE PATENTES Y MARCAS ESPAÑA 11 Número de publicación: 2 286 328 51 Int. Cl.: A61K 31/4439 (2006.01) A61K 31/4178 (2006.01) A61P 25/00 (2006.01) A61P 25/08 (2006.01) A61P 25/02 (2006.01) A61P 29/00 (2006.01) C07D 401/06 (2006.01) C07D 409/14 (2006.01) C07D 403/06 (2006.01) 12 TRADUCCIÓN DE PATENTE EUROPEA T3 86 Número de solicitud europea: 02799923 .4 86 Fecha de presentación : 10.12.2002 87 Número de publicación de la solicitud: 1458386 87 Fecha de publicación de la solicitud: 22.09.2004 54 Título: Aroilpirrolheteroaril y metanoles útiles para tratar trastornos del sistema nervioso central. 30 Prioridad: 27.12.2001 US 343768 P 45 Fecha de publicación de la mención BOPI: 01.12.2007 45 Fecha de la publicación del folleto de la patente: 01.12.2007 73 Titular/es: Ortho-McNeil Pharmaceutical, Inc. U.S. Route nº 202, P.O. Box 300 Raritan, New Jersey 08869-0602, US 72 Inventor/es: Carson, John, R.; Codd, Ellen, E. y Pitis, Philip, M. 74 Agente: Ungría López, Javier Aviso: En el plazo de nueve meses a contar desde la fecha de publicación en el Boletín europeo de patentes, de la mención de concesión de la patente europea, cualquier persona podrá oponerse ante la Oficina Europea de Patentes a la patente concedida. La oposición deberá formularse por escrito y estar motivada; sólo se considerará como formulada una vez que se haya realizado el pago de la tasa de oposición (art. 99.1 del Convenio sobre concesión de Patentes Europeas). ES 2 286 328 T3 Venta de fascículos: Oficina Española de Patentes y Marcas. Pº de la Castellana, 75 – 28071 Madrid
Transcript of AROILPIRROLHETEROARIL Y METANOLES UTILES · PDF fileel sistema nervioso periférico o...

19© OFICINA ESPAÑOLA DEPATENTES Y MARCAS
ESPAÑA
11© Número de publicación: 2 286 32851© Int. Cl.:
A61K 31/4439 (2006.01)
A61K 31/4178 (2006.01)
A61P 25/00 (2006.01)
A61P 25/08 (2006.01)
A61P 25/02 (2006.01)
A61P 29/00 (2006.01)
C07D 401/06 (2006.01)
C07D 409/14 (2006.01)
C07D 403/06 (2006.01)
12© TRADUCCIÓN DE PATENTE EUROPEA T3
86© Número de solicitud europea: 02799923 .486© Fecha de presentación : 10.12.200287© Número de publicación de la solicitud: 145838687© Fecha de publicación de la solicitud: 22.09.2004
54© Título: Aroilpirrolheteroaril y metanoles útiles para tratar trastornos del sistema nervioso central.
30© Prioridad: 27.12.2001 US 343768 P
45© Fecha de publicación de la mención BOPI:01.12.2007
45© Fecha de la publicación del folleto de la patente:01.12.2007
73© Titular/es: Ortho-McNeil Pharmaceutical, Inc.U.S. Route nº 202, P.O. Box 300Raritan, New Jersey 08869-0602, US
72© Inventor/es: Carson, John, R.;Codd, Ellen, E. yPitis, Philip, M.
74© Agente: Ungría López, Javier
Aviso: En el plazo de nueve meses a contar desde la fecha de publicación en el Boletín europeo de patentes, dela mención de concesión de la patente europea, cualquier persona podrá oponerse ante la Oficina Europeade Patentes a la patente concedida. La oposición deberá formularse por escrito y estar motivada; sólo seconsiderará como formulada una vez que se haya realizado el pago de la tasa de oposición (art. 99.1 delConvenio sobre concesión de Patentes Europeas).E
S2
286
328
T3
Venta de fascículos: Oficina Española de Patentes y Marcas. Pº de la Castellana, 75 – 28071 Madrid

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
DESCRIPCIÓN
Aroilpirrolheteroaril y metanoles útiles para tratar trastornos del sistema nervioso central.
Campo de la invención
Esta invención se relaciona con compuestos útiles como agentes para tratar o modular un trastorno del sistemanervioso central. Más concretamente, esta invención se relaciona con compuestos de aroilpirrolhetero-arilmetanona ymetanol útiles como agentes para tratar o modular un trastorno del sistema nervioso central.
Antecedentes de la invención
Las condiciones agrupadas bajo el término “trastorno del sistema nervioso central” constituyen un área de continuanecesidad médica. Dichas condiciones incluyen aquellos trastornos asociados a dolor neuropático, dolor inflamatorio,dolor relacionado con la inflamación o epilepsia.
Se ha sugerido que los canales del sodio tienen un papel en (y que los bloqueantes de los canales del sodio sonútiles en el tratamiento de) muchos trastornos del sistema nervioso central (Madge, D., Sodium Chanels: RecentDevelopments and Therapeutic Potential, Annual Reports in Medicinal Chemistry, 1998, 33, 51-60, 56). La mayoríade los compuestos estudiados hasta la fecha muestran algún potencial en más de uno de estos trastornos; se hanidentificado muy pocos compuestos con actividad anticonvulsivante, analgésica o neuroprotectora selectiva (Madge,D., p. 56).
En los últimos años, se ha desarrollado un entendimiento mucho mejor de los canales del sodio y de fármacos queinteraccionan con ellos (Anger, T., Madge, D., Mulla M. y Riddall, D., Medicinal Chemistry of Neuronal Voltage-Gated Sodium Channel Blockers, Journal of Medicinal Chemistry, 2001, 44(2), 115-137). Ha quedado claro que unaserie de fármacos que tienen un mecanismo de acción desconocido realmente actúan modulando la conductanciade los canales del sodio, incluyendo anestésicos locales, antiarrítmicos de la clase I y anticonvulsivantes (Anger,T. y col., p. 123). Los bloqueantes de los canales del sodio neuronales han encontrado aplicación con su uso enel tratamiento de la epilepsia (fenitoína y carbamazepina, usados desde hace mucho tiempo como anticonvulsivantes,pero sin un claro entendimiento de su mecanismo de acción), la neuroprotección (como resultado del ictus isquémico yde otros traumatismos cerebrales), en la prevención de la neurodegeneración (tal como en el tratamiento de la esclerosislateral amiotrófica, primariamente, por bloqueo de los canales del sodio) y en la reducción del dolor neuropático(como resultado de neuralgia del trigémino, de neuropatía diabética, de neuralgia post-herpética, dolor por neuroma ysíndrome de miembros fantasma) (Anger, T. y col., pp. 124, 126, 129).
El dolor neuropático y otros síndromes de dolor asociados a condiciones crónicas y debilitantes están todos asocia-dos a cambios en la excitabilidad neuronal (Brau, M.E. y col., Effect of drugs used for neuropathic pain managementon tetrodotoxin-resistant Na(+) currents in rat sensory neurons, Anesthesiology, 2001, Enero, 94(1), 137-44; SiddallP.J. y Loeser J.D., Pain following spinal cord injury, Spinal Cord, 2001, Febrero, 39(2), 63-73; Kontinen V.K. y col.,Electrophysiologic evidence for increased endogenous gabanergic but not glycinergic inhibitory tone in the rat spinalnerve ligation model of neuropathy, Anesthesiology, 2001, Febrero, 94(2), 333-9).
Diversos fármacos antiepilépticos (FAE) que estabilizan la excitabilidad neuronal son efectivos en el dolor neu-ropático (Johannessen C.U., Mechanisms of action of valproate: a commentatory, Neurochem. Int., 2000, Agosto-Septiembre, 37(2-3), 101-110, y Magnus, L., Non- epileptic uses of gabapentin, Epilepsia, 1999, 40 Supl. 6, S66-72;Nadin Attal y col., Effects of Gabapentin on the Different Components of Peripheral and Central Neuropathic PainSyndromes: A Pilot Study, Fr. Eur. Neurol., 1998, 40(4), 191-200.
En particular, el dolor neuropático se define como el dolor causado por procesamiento somatosensorial aberrante enel sistema nervioso periférico o central e incluye el dolor neuropático resultante de condiciones crónicas o debilitantes(tales como la neuropatía periférica diabética dolorosa, la neuralgia post-herpética, la neuralgia del trigémino, eldolor post-ictus, el dolor asociado a la esclerosis múltiple, el dolor asociado a neuropatías (tal como en la neuropatíaidiopática o post-traumática y la mononeuritis), el dolor neuropático asociado al VIH, el dolor neuropático asociado alcáncer, el dolor neuropático asociado al túnel carpiano, el dolor asociado a lesiones de la médula espinal, el síndromedel dolor regional complejo, el dolor neuropático asociado a fibromialgia, el dolor lumbar y cervical, la distrofiasimpática refleja, el síndrome de los miembros fantasma y otros síndromes de dolor asociados a condiciones crónicasy debilitantes), el dolor mantenido simpáticamente o el dolor asociado al dolor de cabeza en racimo y en migraña;el dolor asociado al cáncer, la fibromialgia, trastornos de la espalda o el dolor de cabeza en migraña o crónico, laadiposis dolorosa y el dolor por quemadura, condiciones dolorosas centrales después de un ictus, lesiones talámicas oesclerosis múltiple o el dolor resultante de lesión del sistema nervioso periférico o central (tras amputación, paraplejía,herpes o como resultado de la polineuropatía diabética).
Se observa un aumento en la expresión o en la actividad de los canales del sodio en varios modelos animalesde dolor inflamatorio. La expresión de ARNm α-SNS y la corriente de sodio resistente a tetrodotoxina en pequeñasneuronas GRD aumentaron tras inyección de carragenina en la superficie plantar del pie posterior de la rata (TanakaM., NeuroReport, 1998, 9, 967-972). De forma similar, la inducción de inflamación crónica mediante la inyección deAdyuvante Completo de Freund iba seguida del desarrollo de hipersensibilidad térmica inflamatoria y de un aumento
2

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
en la tinción de los canales del sodio (Gould H.J. y col., Brain Res., 1998, 802(1), 69-74; Gould H.J. y col., Brain res.,1999, 824, 296-299). El antisentido (pero no el sentido o el sentido alterado) para el canal del sodio PN3 previno eldesarrollo de la hiperalgesia refleja por flexión mecánica tras la administración del agente inflamatorio PGE2 (KhasarS.G. y col., Neurosci. Lrs., 1998, 256, 17-20).
Referencias de patentes describen compuestos como moduladores o antagonistas de los canales del sodio para usoen el tratamiento o la modulación de trastornos del sistema nervioso central en una serie de modelos in vitro e in vivo.
La Patente EE.UU. 6.288.278 describe derivados de 3-amino-3-arilpropan-1-ol como bloqueantes de los canalesdel sodio en un ensayo de unión a BTX (S.W. Postma y W.A. Catterall, Mol. Pharmacol., 1984, 25, 219-227) y métodospara uso como agentes anestésicos locales, anti-arrítmicos, antieméticos y nootrópicos (neurotrópicos) y como agentespara el tratamiento/terapia de enfermedades cardiovasculares, de la incontinencia urinaria, de la diarrea, del prurito,de la dependencia del alcohol o de fármacos y de la inflamación.
La Patente EE.UU. 6.288.123 describe compuestos de guanidina disubstituidos como moduladores o inhibidoresde la liberación de neurotransmisores tales como el glutamato por las células neuronales isquémicas bloqueando loscanales presinápticos del calcio y/o del sodio en un ensayo de inhibición de la liberación de glutamato, en un ensayode inhibición de la captación de 45Ca a través de los canales presinápticos del calcio, en un ensayo de inhibición de lacaptación de 45Ca a través de los canales del calcio de tipo L (sensibles a dihidropiridina), en un ensayo de inhibiciónde la captación de [14C]-guanidinio a través de los canales del sodio activados por voltaje neuronal de Tipo II y en unmodelo in vivo anticonvulsivante/ataques audiogénicos en ratones D6A/2 y métodos para uso en el tratamiento y/o laprofilaxis de condiciones neurológicas tales como la epilepsia, condiciones y enfermedades neurodegenerativas (talescomo la enfermedad de Parkinson, la enfermedad de Huntington, la Esclerosis Lateral Amiotrófica, la enfermedadde Alzheimer, el Síndrome de Down, la enfermedad de Korsakoff, la atrofia olivopontocerebelar, la demencia y laceguera inducidas por el VIH o la demencia multiinfarto) y muerte de las células nerviosas (como resultado de hipoxia,de hipoglicemia, de isquemia cerebral o de la médula espinal, de traumatismo cerebral o de la médula espinal o deisquemia cerebral global (como resultado de ictus, ataque cardíaco, ahogamiento o envenenamiento por monóxido decarbono)); para uso en el tratamiento de la hipertensión, de arritmias cardíacas o de la angina de pecho, de trastornosendocrinos (tales como la acromegalia y la diabetes insípida) y del dolor crónico (incluyendo el uso como anestésicolocal); y para uso en el tratamiento de enfermedades en las cuales la patofisiología del trastorno implica una secrecióncelular excesiva o de algún otro modo inapropiada (v.g., hipersecretora) (v.g., secreción de una substancia endógenatal como una catecolamina, una hormona o un factor de crecimiento).
La Patente EE.UU. 6.281.211 describe compuestos semicarbazida como bloqueantes de los canales del sodioen un ensayo electrofisiológico con neuronas del hipocampo disociadas, en un ensayo de membranas del cerebroanterior de rata dependientes del voltaje neuronal, en células HEK-293 que expresan establemente canales de sodiohSkM1, en un ensayo [3H]BTX-B y en un modelo de ataques inducidos por electroshock máximo (ESM) en ratonesy métodos para tratar, prevenir o mejorar la pérdida de neuronas (asociada a ictus, isquemia global y focal, traumadel SNC, hipoglicemia, cirugía y trauma de la médula espinal), para el tratamiento o la prevención de condicionesneurodegenerativas (tales como la enfermedad de Alzheimer, la esclerosis lateral amiotrófica (ELA), la enfermedad deParkinson, la ansiedad, las convulsiones, el glaucoma, el dolor de cabeza en migraña y el espasmo muscular), comodepresores antimaníacos, como anestésicos locales, como antiarrítmicos, como anticonvulsivantes, como agentes parael tratamiento o la prevención de la neuropatía diabética y para el tratamiento del dolor (dolor agudo, crónico yquirúrgico, dolor neuropático y dolor de cabeza en migraña).
La Patente EE.UU. 6.265.405 describe derivados de 5-aminotriazina como bloqueantes de los canales del sodioen un ensayo de clamp de voltaje con células enteras (canal de Na+ de cerebro humano recombinante de tipo IIAexpresado en células de ovario de hámster Chino), como anticonvulsivantes en un modelo ESM y una prueba de in-fusión de pentilenotetrazol en ratones, como agentes para tratar la hiperalgesia aguda y la inflamación en un modelode carragenina en pie de rata, como un agente neuroprotector en un modelo de neurotoxicidad inducida por MPTPpara la enfermedad de Parkinson y modelos para tratar la epilepsia (incluyendo los ataques parciales simples, los ata-ques parciales complejos, los ataques generalizados secundarios y los ataques generalizados (incluyendo también losataques de ausencia, los ataques mioclónicos, los ataques clónicos, los ataques tónicos, los ataques tónico-clónicos ylos ataques atónicos)), el trastorno bipolar (alternativamente conocido como depresión maníaca, incluyendo el Tipo Io II) y la depresión unipolar; para tratar o prevenir el dolor agudo (dolor musculoesquelético, post-operatorio y qui-rúrgico), el dolor crónico (dolor inflamatorio (por artritis reumatoide y osteoartritis), dolor neuropático (por neuralgiapost-herpética, neuralgia del trigémino y dolor simpáticamente mantenido) y dolor asociado al cáncer, fibromialgiay dolor asociado a migraña); para tratar el tinnitus, los trastornos funcionales del intestino (dispepsia no ulcerativa,dolor de pecho no cardíaco y síndrome del intestino irritable) y enfermedades neurodegenerativas (enfermedad deAlzheimer, ELA, enfermedad de las neuronas motoras, enfermedad de Parkinson, esclerosis muscular, degeneraciónmacular y glaucoma), para la neuroprotección (tratamiento de la neurodegeneración tras un ictus, un paro cardíaco,un bypass pulmonar, una lesión traumática del cerebro y una lesión de la médula espinal) y para prevenir o reducirla dependencia/tolerancia/tolerancia inversa a un agente inductor de dependencia (tal como opioides, depresores delSNC, psicoestimulantes y nicotina).
La Patente EE.UU. 6.262.078 describe derivados de fenoximetilpiperidina como bloqueantes de los canales del so-dio en un ensayo de nervio vago de rata in vitro (Kourtney y Stricharz, Local Anesthetics, Springer-Verlag, New York,1987) y como agentes para el tratamiento del dolor neuropático en un modelo de alodinia mecánica en rata in vivo
3

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
(Kim y Chung, Pain, 1992, 50: 355-363), en un modelo de alodinia fría aguda y crónica, mononeuropatía unilateral ylesión por constricción crónica en rata in vivo (Benent y Xie, Pain, 1988, 33: 87-107), en un modelo de hiperalgesiamecánica en rata in vivo (Bennet y Xie, Pain, 1988, 33:87-107) y en un modelo de hiperalgesia térmica en rata in vivoy métodos para tratar neuropatías periféricas (neuralgia del trigémino, neuralgia post-herpética, neuropatía diabética,neuralgia glosofaríngea, radiculopatías lumbares y cervicales, distrofia simpática refleja y causalgia), neuropatía se-cundaria a infiltración metastática, adiposis dolorosa y dolor por quemadura y condiciones dolorosas centrales tras unictus, lesiones talámicas y esclerosis múltiple.
La Patente EE.UU. 6.255.307 describe una clase de derivados de fenilpirazina como bloqueantes de los canalesdel sodio y métodos para tratar la epilepsia (incluyendo los ataques parciales simples, los ataques parciales complejos,los ataques generalizados secundarios y los ataques generalizados (incluyendo también los ataques de ausencia, losataques mioclónicos, los ataques clónicos, los ataques tónicos, los ataques tónico-clónicos y los ataques atónicos)), eltrastorno bipolar (alternativamente conocido como depresión maníaca, incluyendo el Tipo I o II) y la depresión uni-polar; para tratar o prevenir el dolor agudo (dolor musculoesquelético, post-operatorio y quirúrgico), el dolor crónico(dolor inflamatorio (por artritis reumatoide y osteoartritis), dolor neuropático (por neuralgia post-herpética, neuralgiadel trigémino y dolor simpáticamente mantenido) y dolor asociado al cáncer, fibromialgia y dolor asociado a migra-ña); para tratar el tinnitus, los trastornos funcionales del intestino (dispepsia no ulcerativa, dolor de pecho no cardíacoy síndrome del intestino irritable) y enfermedades neurodegenerativas (enfermedad de Alzheimer, ELA, enfermedadde las neuronas motoras, enfermedad de Parkinson, esclerosis muscular, degeneración macular y glaucoma), para laneuroprotección (tratamiento de la neurodegeneración tras un ictus, un paro cardíaco, un bypass pulmonar, una lesióntraumática del cerebro y una lesión de la médula espinal) y para prevenir o reducir la dependencia/tolerancia/toleranciainversa a un agente inductor de dependencia (tal como opioides, depresores del SNC, psicoestimulantes y nicotina).
La Patente EE.UU. 6.169.116 describe tetrahidronaftalenaminas como bloqueantes de los canales del sodio en unensayo de inhibición de glutamato inducida por veratridina en hipocampo de rata (modificación de M.J. Leach y col.,Epilepsia, 1986, 27, 490-497, y Stroke, 1993, 24, 1063-1067, usando glutamato exógeno) y en un ensayo de unión averatridina (J.B. Brown, Journal of Neuroscience, 1986, 6, 2064-2070) como agentes para reducir la lesión neuronalinducida por isquemia y los consiguientes síntomas en un modelo de oclusión de la arteria cerebral media (ACM) derata (A. Tamura y col., J. Cereb. Blood Flow Metabol., 1981, 1, 53-60; A. Sauter y M. Rudin, Stroke, 1986, 17, 1228-1234) y métodos para el tratamiento de cualquier condición clínica que conlleve un componente de anoxia, hipoxiao isquemia cerebral (lesión isquémica de la materia gris y blanca) como resultado de un ictus, de una hemorragiasubaracnoidea, de una lesión/traumatismo del cerebro y de la médula espinal, de una alta presión intracraneal, dedemencia multiinfarto o de demencia vascular, como resultado de cualquier procedimiento quirúrgico potencialmenteasociado a anoxia, hipoxia y/o isquemia cerebral (bypass cardíaco, operaciones sobre los vasos extracerebrales), comoresultado de cualquier patología, trastorno o condición clínica que conlleve liberación de glutamato en su etiología(incluyendo trastornos psiquiátricos (tales como esquizofrenia, depresión, ansiedad, ataques de pánico, déficit de aten-ción y trastornos cognitivos o evitación social), condiciones hormonales (tales como un exceso de secreción de GH(como en la diabetes mellitus, la angiopatía o la acromegalia) o de LH (como en la hipertrofia prostática y el síndromemenopáusico) o secreción de corticosterona en el estrés)), lesión cerebral inducida metabólicamente (hipoglicemia, hi-perglicinemia no cetósica (encefalopatía por glicina), deficiencia en la sulfito oxidasa o encefalopatía hepática asociadaa fallo hepático), emesis, espasticidad, tinnitus, dolor (como resultado del cáncer o de la artritis) y abuso y retirada defármacos (como resultado del uso de etanol, de opiáceos (incluyendo sintéticos con efectos de tipo opiáceo), cocaí-na, anfetamina, barbiturato y otros sedantes y benzodiazepinas)), como resultado de cualquier patología que conllevelesión neuronal (incluyendo trastornos neurodegenerativos tales como las enfermedades de Alzheimer, de Huntingtono de Parkinson, la neurodegeneración inducida por virus (incluyendo el VIH), la ELA, la parálisis supranuclear, laatrofia olivopontocerebelar (AOPC) y las acciones de neurotoxinas exógenas ambientales.
La Patente EE.UU. 6.172.085 describe compuestos éter cíclicos como bloqueantes de los canales del sodio enun modelo de unión a fracciones del córtex cerebral de rata y métodos para tratar enfermedades y trastornos delsistema nervioso central (SNC) tales como la isquemia del SNC, el traumatismo del SNC (traumatismo del cerebro,lesión de la médula espinal o lesión de latigazo), la epilepsia, enfermedades neurodegenerativas (ELA, enfermedad deAlzheimer, corea de Huntington, enfermedad de Parkinson o neuropatía diabética), la demencia vascular (demenciamultiinfarto o enfermedad de Binswanger), la psicosis maníaco-depresiva, la depresión, la esquizofrenia, el dolorcrónico, la neuralgia del trigémino, la migraña y el edema cerebral.
La Patente EE.UU. 6.051.583 describe derivados de 2,3,3a,4,9,9a-hexahidro-8-hidroxi-1H-benz[f]indol substitui-dos como bloqueantes de los canales del sodio en un ensayo de unión a BTX (S.W. Postma y W.A. Catterall, Mol.Pharmacol., 1984, 25, 219-227) y en experimentos de clamp de parches (W.A. Catterall, Trends Pharmacol. Sci., 1987,8, 57-65), como anticonvulsivantes en un modelo de ESM en ratón (M.A. Rogawski y R.J. Porter, Pharmacol. Rev.,1990, 42, 223-286), como agentes neuroprotectores en un ensayo de inhibición de glutamato inducida por veratridina(S. Villanueva, P. Frenz, Y. Dragnic y F. Orrego, Brain Res., 1988, 461, 377-380) y en un modelo de MCAO en rata (U.Pschorn y A.J. Carter, J. Stroke Cerebrovascular Diseases, 1996, 6, 93-99) y métodos para tratar enfermedades neuro-degenerativas (resultantes de arritmia, espasmo e isquemia cardíaca y cerebral, hipoglicemia, hipoxia, anoxia, traumadel cerebro, edema cerebral, ictus y asfixia perinatal) y las asociadas a la epilepsia, a la esclerosis lateral amiotrófica,a la enfermedad de Huntington, a la enfermedad de Alzheimer, a la enfermedad de Parkinson, a la ciclofrenia, a lahipotonía, al infarto cardíaco, a trastornos del ritmo cardíaco, a la angina de pecho, al dolor (dolor nociceptor, dolorneuropático, dolor resultante de lesión del sistema nervioso periférico o central (tras amputación, paraplejía, herpes oen la polineuropatía diabética) y al dolor causado por trastornos funcionales (migraña y dolor de espalda)).
4

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
La solicitud PCT WO 01/23570 describe ácidos nucleicos y proteínas variantes con ayustamiento de la subunidadβ1A de los canales de sodio abiertos por voltaje como útiles en el tratamiento del dolor neuropático. La solicitud PCTWO 00/61231 describe el uso de antagonistas de los canales del sodio para tratar enfermedades mediadas o exacerba-das por apoptosis neuronal sensorial: en particular, estados dolorosos (tales como el dolor crónico) tras una agresiónnerviosa asociada a daños tisulares (debidos a lesión o infección), enfermedades neurodegenerativas (tales como laesclerosis múltiple y la enfermedad de Parkinson) e inflamación. La solicitud PCT WO 00/02865 describe el uso deagentes farmacéuticos en el bloqueo de la actividad de los canales de sodio sensibles a voltaje para tratar los dañosneuronales resultantes de sucesos agudos, tales como isquemia o hipoxia, o de enfermedades neurodegenerativas talescomo la enfermedad de Alzheimer, la enfermedad de Parkinson, la enfermedad de Huntington o la esclerosis lateralamiotrófica. La solicitud PCT WO 00/48584 describe derivados de aroilaminoacilpirrol para uso en el tratamiento deldolor neuropático.
Resumen de la invención
La presente invención proporciona compuestos aroilpirrolheteroarilmetanona y metanol como agentes para trataro modular un trastorno del sistema nervioso central seleccionados entre la Fórmula (I) o la Fórmula (II):
donde
A es seleccionado entre el grupo consistente en:
arilo (eventualmente substituido con 1 a 4 substituyentes independientemente seleccionado entre el grupo consis-tente en halógeno, alquilo C1−8, alcoxi C1−8, hidroxi, hidroxialquilo C1−8, hidroxialcoxi C1−8, (halo)1−3alquilo(C1−8)y (halo)1−3alcoxi(C1−8)) y
heteroarilo (eventualmente substituido sobre 1 a 4 miembros del anillo de átomos de carbono disponibles con unsubstituyente seleccionado entre el grupo consistente en halógeno, alquilo C1−8, alcoxi C1−8, hidroxi, hidroxial-quilo C1−8, hidroxialcoxi C1−8, (halo)1−3alquilo(C1−8) y (halo)1−3alcoxi(C1−8); y eventualmente substituido sobrelos miembros del anillo de átomos de nitrógeno disponibles con un substituyente seleccionado entre el grupoconsistente en alquilo C1−8, hidroxialquilo C1−8 y (halo)1−3-alquilo(C1−8));
B es seleccionado entre heteroarilo eventualmente substituido sobre 1 a 4 miembros del anillo de átomos de carbo-no disponibles con un substituyente seleccionado entre el grupo consistente en halógeno, alquilo C1−8, alcoxi C1−8,hidroxi, hidroxialquilo C1−8, hidroxialcoxi C1−8, (halo)1−3alquilo(C1−8) y (halo)1−3alcoxi(C1−8); y eventualmentesubstituido sobre los miembros del anillo de átomos de nitrógeno disponibles con un substituyente seleccionadoentre el grupo consistente en alquilo C1−8, alquenilo C2−8, hidroxialquilo C1−8, (halo)1−3alquilo(C1−8) y óxido;
Z es seleccionado entre el grupo consistente en oxo e hidroxi;
R1 es seleccionado entre el grupo consistente en:
alquilo C1−8 {donde el alquilo está eventualmente substituido sobre un carbono terminal con un substituyenteseleccionado entre el grupo consistente en alcoxi C1−8, -C(O)-(substituido con un substituyente seleccionado entreel grupo consistente en H, OH, alquilo C1−8, alcoxi C1−8, NH2, -NHalquilo(C1−8), -N(alquilo(C1−8))2), -NHC(O)-(substituido con un substituyente seleccionado entre el grupo consistente en H, OH, alquilo C1−8, alcoxi C1−8,NH2, -NHalquilo(C1−8), -N(alquilo(C1−8))2), -OC(O)-(substituido con un substituyente seleccionado entre el grupoconsistente en H, OH, alquilo C1−8, alcoxi C1−8, NH2, -NHalquilo(C1−8), -N(alquilo(C1−8))2), NH2, -NHalquilo(C1−8), -N(alquilo(C1−8))2, -Salquilo(C1−8), -SO2alquilo(C1−8), ciano, (halo)1−3, hidroxi y nitro),
cicloalquilo y arilo {donde el cicloalquilo y el arilo están eventualmente substituidos con 1 a 4 substituyentesindependientemente seleccionados entre el grupo consistente en ciano, halo, hidroxi y nitro; y donde el cicloalquiloy el arilo están eventualmente substituidos con un substituyente seleccionado entre el grupo consistente en alquiloC1−8, (donde el alquilo está eventualmente substituido sobre un carbono terminal con un substituyente seleccionadoentre el grupo consistente en NH2, -NHalquilo(C1−8), -N(alquilo(C1−8))2, ciano, (halo)1−3, hidroxi y nitro), alcoxiC1−8, NH2, -NHalquilo(C1−8) y -N(alquilo(C1−8))2);
5

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
R2 y R3 son independientemente seleccionados entre el grupo consistente en hidrógeno, alquilo C1−8 y halógeno;
y sus sales de adición de ácido, sales de amonio cuaternario y N-óxidos farmacéuticamente aceptables, siendo losgrupos heteroarilo para A y B como se define en la reivindicación 1.
Las realizaciones de la presente invención incluyen una composición farmacéutica consistente en un soporte far-macéuticamente aceptable y un compuesto seleccionado entre la Fórmula (I) o la Fórmula (II).
Descripción detallada de la invención
Las realizaciones de la presente invención incluyen compuestos seleccionados entre la Fórmula (I) o la Fórmu-la (II) donde A es seleccionado entre arilo eventualmente substituido con 1 a 4 substituyentes independientementeseleccionados entre el grupo consistente en halógeno, alquilo C1−4, alcoxi C1−4, hidroxi, hidroxialquilo C1−4, hidro-xialcoxi C1−4, (halo)1−3alquilo(C1−4) y (halo)1−3alcoxi(C1−4), donde el arilo es seleccionado entre un anillo monocíclicoaromático que tiene seis miembros o un anillo bicíclico aromático que tiene diez miembros.
Preferiblemente, A es seleccionado entre el grupo consistente en fenilo y naftalenilo eventualmente substituido con1 a 4 substituyentes como se ha descrito previamente.
Más preferiblemente, A es seleccionado entre fenilo eventualmente substituido con 1 a 4 substituyentes como seha descrito previamente.
Preferiblemente, los substituyentes de arilo de A son independientemente seleccionados entre el grupo consistenteen halógeno, alquilo C1−4 e hidroxi.
Más preferiblemente, los substituyentes de arilo de A son independientemente seleccionados entre el grupo con-sistente en cloro, flúor, metilo e hidroxi.
Más preferiblemente, los substituyentes de arilo de A son independientemente seleccionados entre el grupo con-sistente en cloro, flúor y metilo.
Las realizaciones de la presente invención incluyen compuestos seleccionados entre la Fórmula (I) o la Fórmula(II) donde A es seleccionado entre el grupo consistente en heteroarilo eventualmente substituido sobre 1 a 4 miembrosdel anillo de átomos de carbono disponibles con un substituyente seleccionado entre el grupo consistente en halógeno,alquilo C1−4, alcoxi C1−4, hidroxi, hidroxialquilo C1−4, hidroxialcoxi C1−4, (halo)1−3alquilo(C1−4) y (halo)1−3alcoxi(C1−4);y eventualmente substituido sobre los miembros del anillo de átomos de nitrógeno disponibles con un substituyenteseleccionado entre el grupo consistente en alquilo C1−4, hidroxialquilo C1−4 y (halo)1−3alquilo(C1−4)).
El heteroarilo de A es seleccionado entre el grupo consistente en furilo, tienilo, pirrolilo, oxazolilo, tiazolilo,imidazolilo, pirazolilo, isoxazolilo, isotiazolilo, triazolilo, piridinilo, piridazinilo, pirimidinilo, pirazinilo, quinoliniloe isoquinolinilo eventualmente substituido sobre 1 a 4 miembros del anillo de átomos de carbono disponibles con unsubstituyente como se ha descrito previamente; y eventualmente substituido sobre los miembros del anillo de átomosde nitrógeno disponibles con un substituyente como se ha descrito previamente.
Más preferiblemente, el heteroarilo de A es seleccionado entre el grupo consistente en tienilo, piridinilo, quinoliniloe isoquinolinilo eventualmente substituido sobre 1 a 4 miembros del anillo de átomos de carbono disponibles con unsubstituyente como se ha descrito previamente y eventualmente substituido sobre los miembros del anillo de átomosde nitrógeno disponibles con un substituyente como se ha descrito previamente.
Más preferiblemente, el heteroarilo de A es seleccionado entre el grupo consistente en tienilo y piridinilo eventual-mente substituido sobre 1 a 4 miembros del anillo de átomos de carbono disponibles con un substituyente como se hadescrito previamente y eventualmente substituido sobre los miembros del anillo de átomos de nitrógeno disponiblescon un substituyente como se ha descrito previamente.
Preferiblemente, los substituyentes del heteroarilo de A eventualmente substituido sobre 1 a 4 miembros del anillode átomos de carbono disponibles son independientemente seleccionados entre el grupo consistente en halógeno yalquilo C1−4 y los del eventualmente substituido sobre los miembros del anillo de átomos de nitrógeno disponibles sonseleccionados entre alquilo C1−4.
Más preferiblemente, los substituyentes del heteroarilo de A eventualmente substituido sobre 1 a 4 miembros delanillo de átomos de carbono disponibles son independientemente seleccionados entre el grupo consistente en cloro,flúor y metilo y los del eventualmente substituido sobre los miembros del anillo de átomos de nitrógeno disponiblesson seleccionados entre metilo.
Las realizaciones de la presente invención incluyen compuestos seleccionados entre la Fórmula (I) o la Fórmula (II)donde B es heteroarilo eventualmente substituido sobre 1 a 4 miembros del anillo de átomos de carbono disponiblescon un substituyente seleccionado entre el grupo consistente en halógeno, alquilo C1−4, alcoxi C1−4, hidroxi, hidro-xialquilo C1−4, hidroxialcoxi C1−4, (halo)1−3alquilo(C1−4) y (halo)1−3alcoxi(C1−4); y eventualmente substituido sobre los
6

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
miembros del anillo de átomos de nitrógeno disponibles con un substituyente seleccionado entre el grupo consistenteen alquilo C1−4, alquenilo C2−4, hidroxialquilo C1−4, (halo)1−3alquilo(C1−4) y óxido.
El heteroarilo de B es seleccionado entre el grupo consistente en furilo, tienilo, pirrolilo, oxazolilo, tiazolilo,imidazolilo, pirazolilo, isoxazolilo, isotiazolilo, triazolilo, piridinilo, piridazinilo, pirimidinilo, pirazinilo, quinolinilo,isoquinolinilo y quinoxalinilo eventualmente substituido sobre 1 a 4 miembros del anillo de átomos de carbono dis-ponibles con un substituyente como se ha descrito previamente y eventualmente substituido sobre los miembros delanillo de átomos de nitrógeno disponibles con un substituyente como se ha descrito previamente.
Más preferiblemente, el heteroarilo de B es seleccionado entre el grupo consistente en furilo, tienilo, pirrolilo, imi-dazolilo, piridinilo, quinolinilo e isoquinolinilo eventualmente substituido sobre 1 a 4 miembros del anillo de átomosde carbono disponibles con un substituyente como se ha descrito previamente y eventualmente substituido sobre losmiembros del anillo de átomos de nitrógeno disponibles con un substituyente como se ha descrito previamente.
Más preferiblemente, el heteroarilo de B es seleccionado entre el grupo consistente en tienilo, imidazolilo y piridi-nilo eventualmente substituido sobre 1 a 4 miembros del anillo de átomos de carbono disponibles con un substituyentecomo se ha descrito previamente y eventualmente substituido sobre los miembros del anillo de átomos de nitrógenodisponibles con un substituyente como se ha descrito previamente.
Preferiblemente, los substituyentes de B eventualmente substituidos sobre 1 a 4 miembros del anillo de átomos decarbono disponibles son independientemente seleccionados entre el grupo consistente en halógeno y alquilo C1−4 y loseventualmente substituidos sobre un miembro del anillo de átomos de nitrógeno son seleccionados entre óxido.
Más preferiblemente, los substituyentes de B eventualmente substituidos sobre 1 a 4 miembros del anillo deátomos de carbono disponibles son independientemente seleccionados entre el grupo consistente en cloro y meti-lo y los eventualmente substituidos sobre un miembro del anillo de átomos de nitrógeno son seleccionados entreóxido.
Las realizaciones de la presente invención incluyen aquellos compuestos en los que R1 es seleccionado entre elgrupo consistente en:
alquilo C1−4 {donde el alquilo está eventualmente substituido sobre un carbono terminal con un substituyenteseleccionado entre el grupo consistente en alcoxi C1−4, -C(O)-(substituido con un substituyente seleccionado entreel grupo consistente en H, OH, alquilo C1−4, alcoxi C1−4, NH2, -NHalquilo(C1−4), -N(alquilo(C1−4))2), -NHC(O)-(substituido con un substituyente seleccionado entre el grupo consistente en H, OH, alquilo C1−4, alcoxi C1−4,NH2, -NHalquilo(C1−4), -N(alquilo(C1−4))2), -OC(O)-(substituido con un substituyente seleccionado entre el grupoconsistente en H, OH, alquilo C1−4, alcoxi C1−4, NH2, -NHalquilo(C1−4), -N(alquilo(C1−4))2), NH2, -NHalquilo(C1−4), -N(alquilo(C1−4))2, -Salquilo(C1−4), -SO2alquilo(C1−4), ciano, (halo)1−3, hidroxi y nitro),
cicloalquilo y arilo {donde el cicloalquilo y el arilo están eventualmente substituidos con 1 a 4 substituyentesindependientemente seleccionados entre el grupo consistente en ciano, halo, hidroxi y nitro; y donde el cicloalquiloy el arilo están eventualmente substituidos con un substituyente seleccionado entre el grupo consistente en alquiloC1−4, (donde el alquilo está eventualmente substituido sobre un carbono terminal con un substituyente seleccionadoentre el grupo consistente en NH2, -NHalquilo(C1−4), -N(alquilo(C1−4))2, ciano, (halo)1−3, hidroxi y nitro), alcoxiC1−4, NH2, -NHalquilo(C1−4) y -N(alquilo(C1−4))2).
Preferiblemente, R1 es seleccionado entre alquilo C1−4 eventualmente substituido en un carbono terminal con unsubstituyente como se ha descrito previamente.
Más preferiblemente, R1 es seleccionado entre alquilo C1−4.
Preferiblemente, el substituyente eventual en el carbono terminal del alquilo C1−4 es seleccionado entre el grupoconsistente en alcoxi C1−4, NH2, -NHalquilo(C1−4), -N(alquilo(C1−4))2, ciano, (halo)1−3, hidroxi y nitro.
Las realizaciones de la presente invención incluyen aquellos compuestos en los que, preferiblemente, R2 y R3
son independientemente seleccionados entre el grupo consistente en hidrógeno, alquilo C1−4 y halógeno. Más prefe-riblemente, R2 y R3 son independientemente seleccionados entre el grupo consistente en hidrógeno y alquilo C1−4.Más preferiblemente, R2 y R3 son independientemente seleccionados entre el grupo consistente en hidrógeno ymetilo.
7

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
Ejemplifica la invención un compuesto seleccionado entre la Fórmula (I):
TABLA 1
donde A, B, Z, R2 y R3 son independientemente seleccionados entre
y sus sales de adición de ácido, sales de amonio cuaternario y N-óxidos farmacéuticamente aceptables.
8

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
Ejemplifica la invención un compuesto seleccionado entre la Fórmula (II):
TABLA 2
donde B, A, R2 y R3 son independientemente seleccionados entre
y sus sales de adición de ácido, sales de amonio cuaternario y N-óxidos farmacéuticamente aceptables.
Una realización de la presente invención incluye una composición farmacéutica consistente en un soporte farma-céuticamente aceptable y un compuesto seleccionado entre la Fórmula (I) o la Fórmula (II).
Para preparar las composiciones farmacéuticas de esta invención, se mezclan íntimamente uno o más compuestosseleccionados entre la Fórmula (I) o la Fórmula (II) con un soporte farmacéutico según las técnicas de composiciónfarmacéuticas convencionales, cuyo soporte puede adoptar una amplia variedad de formas dependiendo de la formade preparación deseada para administración (v.g., oral, tópica, por supositorios o parenteral).
Al preparar las composiciones en forma de dosificación oral, se puede emplear cualquiera de los medios farmacéu-ticos habituales. Así, para preparaciones orales líquidas, tales como suspensiones, elixires y soluciones, como soportesy aditivos adecuados se incluyen agua, glicoles, aceites, alcoholes, agentes saborizantes, conservantes, agentes colo-rantes y similares. Para preparaciones orales sólidas, tales como polvos, cápsulas y tabletas, o para preparacionestópicas, tales como cremas, como soportes y aditivos adecuados se incluyen almidones, azúcares, diluyentes, agentesgranulantes, lubricantes, ligantes, agentes desintegrantes y similares.
Por su facilidad de administración, las tabletas y las cápsulas representan la forma unitaria de dosificación oral másventajosa, en cuyo caso se usan obviamente soportes farmacéuticos sólidos. Si se desea, las tabletas pueden ser dotadasde un revestimiento de azúcar o de un revestimiento entérico por técnicas estándar. Se pueden preparar supositorios, encuyo caso se podría usar manteca de cacao como soporte. Para los parenterales, el soporte consistirá normalmente enagua estéril, aunque se pueden incluir otros ingredientes, por ejemplo con fines tales como añadir solubilidad o comoconservación. También se pueden preparar suspensiones inyectables, en cuyo caso se pueden emplear soportes líquidosapropiados, agentes suspensores y similares. Para los agentes tópicos, se emplearían diluyentes, agentes granulantes,lubricantes y agentes desintegrantes adecuados para ayudar a la dispersión y absorción.
9

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
Las presentes composiciones farmacéuticas pueden contener, por unidad de dosificación (por ejemplo, tableta,cápsula, polvo, inyección, cucharadita, supositorio y similares), de aproximadamente 0,001 mg a aproximadamente500 mg del ingrediente activo.
Los términos usados en la descripción de la invención son comúnmente usados y conocidos por los expertos en latécnica. Tal como se usan aquí, las siguientes abreviaturas tienen los significados indicados:
DCE 1,2-Dicloroetano
Et2O Éter dietílico
EtOH Etanol
h Hora
K2CO3 Carbonato de potasio
MeOH Metanol
min minuto
MTBE Éter metil-t-butílico
2-ProOH 2-Propanol
Métodos sintéticos generales
Se pueden sintetizar compuestos representativos de la presente invención según los métodos sintéticos generalesdescritos a continuación y se ilustran más particularmente en los esquemas que siguen. Como los esquemas son unailustración, no se ha de considerar que la invención está limitada por las reacciones químicas y condiciones expresadas.La preparación de los diversos materiales de partida usados en los esquemas está dentro de los conocimientos de laspersonas versadas en la técnica.
El Esquema A ejemplifica la preparación de compuestos objeto en los que A está en la posición 2 del pirrol y Bestá en la posición 4 y Z es oxo. Alternativamente, variando los materiales de partida y usando las mismas condicionesseñaladas en el Esquema A, se pueden preparar compuestos objeto en los que B está en la posición 2 del pirrol y Aestá en la posición 4 y Z es oxo.
Haciendo referencia al Esquema A, en la primera etapa se acila un Compuesto pirrólico A1 simple con un Com-puesto de cloruro de aroílo apropiadamente substituido A2 (A-C(O)Cl) para producir un Compuesto aroilpirrólico A3.Esta acilación puede ser llevada a cabo calentando simplemente el Compuesto cloruro de aroílo A2 y el Compuestopirrólico A1 en un solvente aprótico. La temperatura de la acilación variará dependiendo de la velocidad de reaccióndeseada y de los substituyentes del Compuesto pirrólico A1. Preferiblemente, la acilación es llevada a cabo a unatemperatura de aproximadamente 50ºC a aproximadamente 250ºC.
A continuación, se acila el Compuesto aroilpirrólico A3 en la posición 4 en una reacción de Friedel-Crafts conun compuesto cloruro de heteroarilmetanona (B-C(Z)Cl, donde Z es oxo), el Compuesto A4, para producir el pro-ducto deseado, el compuesto A5. Se lleva preferiblemente a cabo la reacción de Friedel-Crafts a una temperatura deaproximadamente 0ºC a aproximadamente 100ºC. Como catalizadores ácidos de Lewis de Friedel-Crafts adecuados,se incluyen cloruro de aluminio, cloruro de zinc, BF3 o TiCl4. Como solventes adecuados, se incluyen cloruro demetileno, 1,2-dicloroetano, tetracloruro de carbono, nitrometano, nitrobenceno, diclorobenceno o cloroformo.
10

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
Esquema A
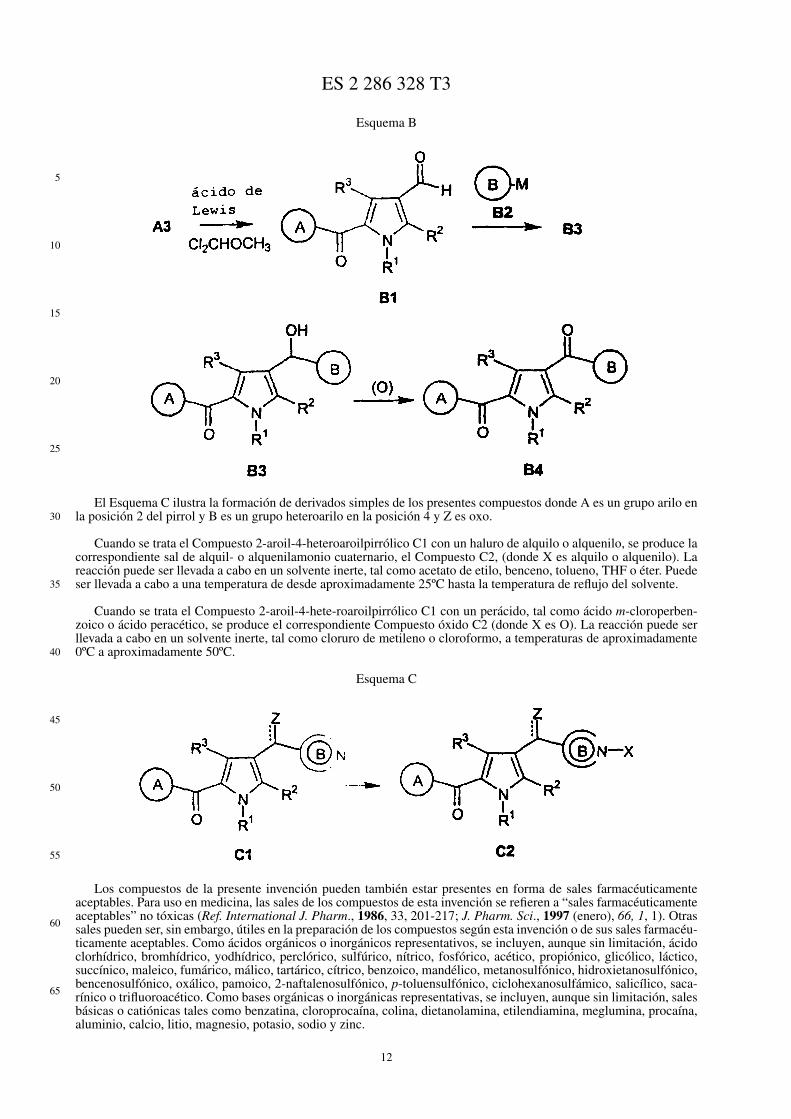
Se ilustra en el Esquema B una ruta alternativa a la preparación de compuestos donde A es un grupo arilo oheteroarilo en la posición 2 del pirrol y B es un grupo heteroarilo en la posición 4 y Z es oxo.
Se somete el Compuesto 2-aroilpirrólico A3 (donde A es un grupo arilo y Z es oxo) a formilación de Friedel-Crafts usando 1,1-diclorometil metil éter y un catalizador ácido de Lewis de Friedel-Crafts adecuado, tal como clo-ruro de aluminio, cloruro de zinc, BF3 o TiCl4, para obtener el Compuesto 2-aroil-4-pirrol-2-carboxaldehído B1. Selleva a cabo la reacción de Friedel-Crafts a una temperatura de desde aproximadamente -40ºC hasta aproximada-mente 50ºC. Como solventes adecuados, se incluyen cloruro de metileno, 1,2-dicloroetano, tetracloruro de carbono,nitrometano, nitrobenceno, diclorobenceno, cloroformo o sus mezclas. Se hizo entonces que el aldehído reaccionaracon el Compuesto B-metálico B2 (donde B es un grupo heteroarilo) para obtener el Compuesto carbonilo B3. Co-mo compuestos heteroarilometálicos adecuados, se incluyen organolitio, organomagnesio (Grignard) y compuestosde organozinc. Dichos compuestos heteroarilometálicos pueden ser preparados por intercambio metal-halógeno entreun compuesto halo heteroaromático y un organometálico simple, tal como n-butil-litio, bromuro de etilmagnesio odietilzinc. Son solvente preferidos para este procedimiento de dos etapas solventes etéreos, tales como éter dietílico oTHF. La reacción de intercambio metal-halógeno puede ser llevada a cabo a temperaturas de desde aproximadamente-78ºC hasta aproximadamente 25ºC. La adición del compuesto heteroarilmetálico al aldehído puede ser llevada a caboa una temperatura de desde aproximadamente -78ºC hasta aproximadamente 50ºC.
Se oxidó entonces el Compuesto carbinol B3 al correspondiente Compuesto cetónico B4 usando un oxidanteadecuado (O), tal como dióxido de manganeso, trióxido de cromo o permanganato de potasio. Se realizó la oxidacióncon dióxido de manganeso agitando en un solvente halocarbonado o hidrocarbonado. Cuando el material de partidaheterocíclico deseado lleva una funcionalidad ácida o sensible, se puede realizar la secuencia global con un grupoprotector, tal como tritilo o bencilo, sobre el heterociclo. Se puede retirar entonces el grupo protector después de laadición organometálica por hidrogenolisis o tratamiento ácido.
11
5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
Esquema B
El Esquema C ilustra la formación de derivados simples de los presentes compuestos donde A es un grupo arilo enla posición 2 del pirrol y B es un grupo heteroarilo en la posición 4 y Z es oxo.
Cuando se trata el Compuesto 2-aroil-4-heteroaroilpirrólico C1 con un haluro de alquilo o alquenilo, se produce lacorrespondiente sal de alquil- o alquenilamonio cuaternario, el Compuesto C2, (donde X es alquilo o alquenilo). Lareacción puede ser llevada a cabo en un solvente inerte, tal como acetato de etilo, benceno, tolueno, THF o éter. Puedeser llevada a cabo a una temperatura de desde aproximadamente 25ºC hasta la temperatura de reflujo del solvente.
Cuando se trata el Compuesto 2-aroil-4-hete-roaroilpirrólico C1 con un perácido, tal como ácido m-cloroperben-zoico o ácido peracético, se produce el correspondiente Compuesto óxido C2 (donde X es O). La reacción puede serllevada a cabo en un solvente inerte, tal como cloruro de metileno o cloroformo, a temperaturas de aproximadamente0ºC a aproximadamente 50ºC.
Esquema C
Los compuestos de la presente invención pueden también estar presentes en forma de sales farmacéuticamenteaceptables. Para uso en medicina, las sales de los compuestos de esta invención se refieren a “sales farmacéuticamenteaceptables” no tóxicas (Ref. International J. Pharm., 1986, 33, 201-217; J. Pharm. Sci., 1997 (enero), 66, 1, 1). Otrassales pueden ser, sin embargo, útiles en la preparación de los compuestos según esta invención o de sus sales farmacéu-ticamente aceptables. Como ácidos orgánicos o inorgánicos representativos, se incluyen, aunque sin limitación, ácidoclorhídrico, bromhídrico, yodhídrico, perclórico, sulfúrico, nítrico, fosfórico, acético, propiónico, glicólico, láctico,succínico, maleico, fumárico, málico, tartárico, cítrico, benzoico, mandélico, metanosulfónico, hidroxietanosulfónico,bencenosulfónico, oxálico, pamoico, 2-naftalenosulfónico, p-toluensulfónico, ciclohexanosulfámico, salicílico, saca-rínico o trifluoroacético. Como bases orgánicas o inorgánicas representativas, se incluyen, aunque sin limitación, salesbásicas o catiónicas tales como benzatina, cloroprocaína, colina, dietanolamina, etilendiamina, meglumina, procaína,aluminio, calcio, litio, magnesio, potasio, sodio y zinc.
12

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
Cuando los compuestos según esta invención tienen al menos un centro quiral, pueden existir consiguientementecomo enantiómeros. Cuando los compuestos poseen dos o más centros quirales, pueden adicionalmente existir comodiastereómeros. Cuando los procedimientos para la preparación de los compuestos según la invención dan lugar a unamezcla de estereoisómeros, estos isómeros pueden ser separados por técnicas convencionales, tales como cromatogra-fía preparatoria. Los compuestos pueden ser preparados en forma racémica, o se pueden preparar los enantiómerosindividuales por síntesis enantioespecífica o por resolución. Los compuestos pueden ser, por ejemplo, resueltos en suscomponentes enantioméricos por técnicas estándar, tales como la formación de pares diastereoméricos por formaciónde sal con un ácido ópticamente activo, tal como ácido (-)-di-p-toluoil-d-tartárico y/o ácido (+)-di-p-toluoil-l-tartárico,seguido de cristalización fraccionada y regeneración de la base libre. Los compuestos pueden ser también resueltospor formación de ésteres o amidas diastereoméricos, seguido de separación cromatográfica y eliminación del auxiliarquiral. Alternativamente, los compuestos pueden ser resueltos usando una columna de HPLC quiral. Hay que entenderque todos esos isómeros y sus mezclas quedan abarcados dentro del alcance de la presente invención.
Durante cualquiera de los procedimientos para la preparación de los compuestos de la presente invención, puedeser necesario y/o deseable proteger grupos sensibles o reactivos sobre cualquiera de las moléculas implicadas. Sepuede conseguir esto por medio de grupos protectores convencionales, tales como los descritos en Protective Groupsin Organic Chemistry, ed. J.F.W. Mc-Omie, Plenum Press, 1973, y en T.W. Greene & P.G.M. Wuts, Protective Groupsin Organic Synthesis, John Wiley & Sons, 1991. Se pueden eliminar los grupos protectores en una etapa posteriorconveniente usando métodos bien conocidos en la técnica.
Más aún, algunas de las formas cristalinas para los compuestos pueden existir como polimorfos, y como tales sepretende que queden incluidas en la presente invención. Además, algunos de los compuestos pueden formar solva-tos con agua (es decir, hidratos) o solventes orgánicos comunes, y se pretende también que dichos solvatos quedenabarcados dentro del alcance de esta invención.
Métodos sintéticos específicos
Se pueden preparar compuestos específicos representativos de esta invención según los siguientes ejemplos, ofre-cidos a modo de ilustración. Además, se designan ejemplos específicamente utilizados para preparar intermediariospara la posterior síntesis de los compuestos de la invención mediante “Procedimiento”. No se ha hecho ningún intentode optimizar los rendimientos obtenidos en cualquiera de las reacciones. Un experto en la técnica sabrá cómo aumen-tar dichos rendimientos por variaciones rutinarias en los tiempos de reacción, las temperaturas, los solventes y/o losreactivos.
Ejemplo 1
[5-(4-Clorobenzoil)-1-metil-1H-pirrol-3-il]piridin-4-il-metanona (Comp. 1)
Se calentó una solución de 17,5 g (0,08 moles) de (4-clorofenil)-(1-metil-1H-pirrol-2-il)metanona, 14,4 g (0,088moles) de cloruro de isonicotinoílo y 26,6 g de cloruro de aluminio (0,2 moles) en 280 ml de DCE a reflujo durante 16h. Después de enfriar, se repartió la mezcla entre CH2Cl2 y una solución diluida de NaOH. Se secó la capa orgánicay se evaporó el solvente a vacío. Se trituró el residuo con éter. Se recristalizó el sólido resultante con 2-PrOH paraobtener 7 g (27%) del compuesto del título, p.f. 174-175ºC. ES-MS m/z = 325 (M++H). 1H RMN (300 MHz, CDCl3)δ 4,1 (s, 3H), 7,1 (s, 1H), 7,3 (dd, 2H), 7,42 (dd, 2H), 7,45 (s, 1H), 7,8 dd, 2H), 8,8 (dd, 2H). Análisis calculado para:C18H13ClN2O2: C, 66,57; H, 4,03; N, 8,63. Encontrado: C, 66,34; H, 3,94; N, 8,53.
Ejemplo 2
(5-Benzoil-1-metil-1H-pirrol-3-il)piridin-4-ilmetanona (Comp. 2)
Siguiendo el protocolo del Ejemplo 1 y empleando (1-metil-1H-pirrol-2-il)fenilmetanona en lugar de (4-clorofenil)-(1-metil-1H-pirrol-2-il)metanona, se obtuvo el compuesto del título en forma bruta y se purificó por cromatografíainstantánea (acetona al 20% en hexano): p.f. 127-129ºC. ES-MS m/z = 290 (M++H). 1H RMN (300 MHz, DMSO-d6)δ 4,05 (s, 3H), 7,0 (s, 1H), 7,6 (m, 5H), 7,8 (m, 4H), 7,95 (s, 1H).
Ejemplo 3
Clorhidrato de [1-metil-5-(4-metilbenzoil)-1H-pirrol-3-il]piridin-4-ilmetanona (Comp. 3)
Siguiendo el protocolo del Ejemplo 1 y empleando (1-metil-1H-pirrol-2-il)-p-tolilmetanona en lugar de (4-clorofe-nil)-(1-metil-1H-pirrol-2-il)metanona, se obtuvo el compuesto del título en forma bruta, se trató con HCl 3N paraobtener la sal clorhidrato y se recristalizó con MeOH/EtOH: p.f. 134-136ºC. ES-MS m/z = 305 (M++H). 1H RMN(300 MHz, DMSO-d6) δ 2,44 (s, 3H), 4,0 (s, 3H), 7,03 (s, 1H), 7,4 (dd, 2H), 7,75 (dd, 2H), 8,0 (dd, 2H), 8,05 (s, 1H),8,95 (dd, 2H). Análisis calculado para: C19H16N2O2.HCl: C, 66,96; H, 5,03; N, 8,22. Encontrado: C, 67,2; H, 4,99; N,8,0.
13

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
Ejemplo 4
Clorhidrato de [1-metil-5-(tiofeno-2-carbonil)-1H-pirrol-3-il]piridin-4-ilmetanona (Comp. 4)
Siguiendo el protocolo del Ejemplo 1 y empleando (1-metil-1H-pirrol-2-il)tiofen-2-ilmetanona en lugar de (4-clorofenil)-(1-metil-1H-pirrol-2-il)metanona, se obtuvo el compuesto del título en forma bruta, se trató con HCl 3Npara obtener la sal clorhidrato y se purificó por cromatografía instantánea (MeOH al 10% en CH2Cl2): p.f. 243-245ºC.ES-MS m/z = 297 (M++H). 1H RMN (300 MHz, DMSO-d6) δ 4,0 (s, 3H), 7,3 (t, 1H), 7,4 (s, 1H), 7,9 (m, 3H), 8,02 (s,1H), 8,1 (d, 1H), 8,95 (dd, 2H). Análisis calculado para: C16H12N2O2S.HCl: C, 57,74; H, 3,94; N, 8,42. Encontrado:C, 57,61; H, 3,99; N, 8,21.
Ejemplo 5
Clorhidrato de [5-(4-clorobenzoil)-1-metil-1H-pirrol-3-il]piridin-3-ilmetanona hidrato [2:1:1] (Comp. 5)
Siguiendo el protocolo del Ejemplo 1 y empleando clorhidrato de cloruro de nicotinoílo en lugar de clorhidrato decloruro de isonicotinoílo, se obtuvo el compuesto del título en forma bruta y se purificó por cromatografía instantánea(acetona al 30% en hexano). Se obtuvo la sal clorhidrato a partir de THF/Et2O/HCl: p.f. 138-140ºC. ES-MS m/z = 325(M++H). 1H RMN (300 MHz, DMSO-d6) δ 4,05 (s, 3H), 5,0 (s amplio, 2H), 7,1 (s, 1H), 7,62 (dd, 2H), 7,72 (m, 1H),7,82 (dd, 2H), 8,1 (s, 1H), 8,35 (m, 3H), 8,9 (m, 1H), 9,07 (s, 1H). Análisis calculado para: C18H13ClN2O2.0,5 HCl.0,5H2O: C, 66,96; H, 5,03; N, 8,22. Encontrado: C, 67,2; H, 4,99; N, 8,0.
Ejemplo 6
[5-(4-Clorobenzoil)-1-metil-1H-pirrol-3-il]piridin-2-il-metanona (Comp. 6)
Siguiendo el protocolo del Ejemplo 1 y empleando clorhidrato de cloruro de picolinoílo en lugar de clorhidratode cloruro de isonicotinoílo, se obtuvo el compuesto del título con un rendimiento del 36%, se recristalizó dos vecescon 2-PrOH y una vez con EtOAc: p.f. 138-139ºC. ES-MS m/z = 325 (M++H). 1H RMN (300 MHz, CDCl3) δ 4,1 (s,3H), 7,5 (dd, 2H), 7,55 (m, 2H), 7,84 (dd, 2H), 7,9 (t, 1H), 8,1 (d, 1H), 8,35 (s, 1H), 8,7 (d, 1H). Análisis calculadopara: C18H13ClN2O2: C, 66,57; H, 4,03; N, 8,63. Encontrado: C, 67,2; H, 4,99; N, 8,0. Encontrado: C, 66,2; H, 4,11;N, 8,55.
Procedimiento 1
(1-Metil-1H-pirrol-2-il)piridin-3-ilmetanona
Se calentó una mezcla de 25 g (0,14 moles) de clorhidrato de cloruro de nicotinoílo y 10,4 ml (0,14 moles) deN-metilpirrol a reflujo en 200 ml de tolueno seco mientras se burbujeaba una corriente de nitrógeno lentamente através de la mezcla de reacción. Después de someter a reflujo durante la noche, se enfrió la mezcla de reacción y sefiltró el sólido. Se convirtió el sólido en la base libre repartiendo entre Et2O/NaOH 3N. Se lavaron los orgánicos conagua y salmuera y se secaron (K2CO3). Se cromatografió el residuo sobre sílice (CH2Cl2:MeOH:NH4OH 90:10:1) paraobtener 8,9 g de (2-piridinil)-(1-metil-1H-pirrol-2-il)metanona (34%) como una goma. CI-MS m/z = 188 (M++H). 1HRMN (300 MHz, CDCl3) δ 9,0 (ar, 1H), 8,7 (ar, 1H), 8,1 (ar, 1H), 7,4 (ar, 1H), 6,7 (ar, 1H), 6,2 (ar, 1H), 4,0 (s, 3H).
Ejemplo 7
Clorhidrato de (4-fluorofenil)-[1-metil-5-(piridina-3-carbonil)-1H-pirrol-3-il]metanona (Comp. 7)
Se agitó una solución de 3,33 g (0,015 moles) de clorhidrato de (1-metil-1H-pirrol-2-il)piridin-3-ilmetanona, 1,94ml (0,0165 moles) de cloruro de 4-fluorobenzoílo y 5 g de cloruro de aluminio (0,037 moles) en 50 ml de DCE a25ºC durante 16 h. Se repartió la mezcla entre CH2Cl2 y una solución diluida de NaOH. Se secó la capa orgánica yse evaporó el solvente a vacío. Se sometió el residuo a cromatografía instantánea con acetona al 35% en hexano, paraobtener 2,14 (46% de rendimiento) de la base libre. Se obtuvo la sal clorhidrato del compuesto del título a partir deEt2O/HCl: p.f. 220-223ºC. ES-MS m/z = 309 (M++H). 1H RMN (300 MHz, DMSO-d6) δ 4,0 (s, 3H), 6,2 (s amplio,2H), 7,16 (s, 1H), 7,35 (t, 2H), 7,85 (m, 1H), 7,95 (m, 2H), 8,1 (s, 1H), 8,4 (d, 1H), 8,95 (d, 1H), 9,05 (s, 1H), 10,5 (samplio, 1H). análisis calculado para: C18H13FN2O2.HCl: C, 62,71; H, 4,09; N, 8,13. Encontrado: C, 63,08; H, 4,23; N,8,11.
Ejemplo 8
[1-Metil-5-(piridina-3-carbonil)-1H-pirrol-3-il]naftalen-2-ilmetanona (Comp. 8)
Siguiendo el protocolo del Ejemplo 7 y empleando clorhidrato de cloruro de 2-naftoílo en lugar de cloruro de 4-fluorobenzoílo y calentando a 55ºC, se obtuvo el compuesto del título y se recristalizó con EtOAc: p.f. 132-133ºC. ES-MS m/z = 341 (M++H). 1H RMN (300 MHz, CDCl3) δ 4,1 (s, 3H), 7,25 (s, 1H), 7,45 (m, 1H), 7,56 (m, 3H), 7,9 (m,4H), 8,15 (d, 1H), 8,35 (s, 1H), 8,8 (d, 1H), 9,05 (s, 1H). Análisis calculado para: C22H16N2O2: C, 77,63; H, 4,74; N,8,23. Encontrado: C, 77,14; H, 4,55; N, 8,02.
14

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
Procedimiento 2
(6-Cloropiridin-3-il)-(1-metil-1H-pirrol-2-il)metanona
Siguiendo el protocolo del Ejemplo 1 y empleando clorhidrato de cloruro de 5-cloronicotinoílo en lugar de clor-hidrato de cloruro de nicotinoílo, se obtuvo el compuesto del título. ES-MS m/z = 221 (M++H). 1H RMN (300 MHz,CDCl3) δ 4,1 (s, 3H), 6,2 (m, 1H), 6,75 (m, 1H), 7,0 (s, 1H), 7,45 (d, 1H), 8,05 (d, 1H), 8,8 (s, 1H).
Ejemplo 9
(4-Clorofenil)-[5-(6-cloropiridina-3-carbonil)-1-metil-1H-pirrol-3-il]metanona (Comp. 9)
Siguiendo el protocolo del Ejemplo 8 y empleando clorhidrato de (6-cloropiridin-3-il)-(1-metil-1H-pirrol-2-il)metanona en lugar de clorhidrato de (1-metil-1H-pirrol-2-il)piridin-3-ilmetanona y cloruro de 4-cloro-benzoílo enlugar de cloruro de 4-fluorobenzoílo, se obtuvo el compuesto del título y se recristalizó con MTBE: p.f. 150-152ºC.ES-MS m/z = 360 (M++H). 1H RMN (300 MHz, CDCl3) δ 4,1 (s, 3H), 7,2 (s, 1H), 7,5 (m, 3H), 7,56 (s, 1H), 7,8 (dd,2H), 8,05 (d, 1H), 8,8 (s, 1H). Análisis calculado para C18H12Cl2N2O2: C, 60,19; H, 3,37; N, 7,8. Encontrado: C, 59,9;H, 3,34; N, 7,71.
Procedimiento 3
Clorhidrato de (1-metil-1H-pirrol-2-il)piridin-3-ilmetanona
Siguiendo el protocolo del Ejemplo 1 y empleando clorhidrato de cloruro de picolinoílo en lugar de clorhidrato decloruro de nicotinoílo, se obtuvo el compuesto del título en un rendimiento del 38%. ES-MS m/z = 187 (M++H). 1HRMN (300 MHz, CDCl3) δ 4,05 (s, 3H), 6,2 (m, 1H), 6,95 (m, 1H), 7,32 (m, 1H), 7,42 (m, 1H), 7,86 (m, 1H), 7,95 (d,1H), 8,7 (d, 1H).
Ejemplo 10
(4-Fluorofenil)-[1-metil-5-(piridina-2-carbonil)-1H-pirrol-3-il]metanona (Comp. 10)
Siguiendo el protocolo del Ejemplo 7 y empleando clorhidrato de (1-metil-1H-pirrol-2-il)piridin-2-ilmetanona enlugar de clorhidrato de (1-metil-1H-pirrol-2-il)piridin-3-ilmetanona, se obtuvo el compuesto del título en un rendi-miento del 56%. Se recristalizó con 2-PrOH: p.f. 159-161ºC. ES-MS m/z = 309 (M++H). 1H RMN (300 MHz, CDCl3)δ 4,1 (s, 3H), 7,2 (m, 2H), 7,5 (m, 2H), 7,9 (m, 4H), 8,05 (d, 1H), 8,7 (d, 1H). Análisis calculado para C18H13FN2O2:C, 70,12; H, 4,25; N, 9,09. Encontrado: C, 70,01; H, 4,15; N, 8,88.
Procedimiento 4
Clorhidrato de (1-metil-1H-pirrol-2-il)piridin-4-ilmetanona
Siguiendo el procedimiento del Ejemplo 1 y empleando clorhidrato de cloruro de isonicotinoílo en lugar de clor-hidrato de cloruro de nicotinoílo, se obtuvo el compuesto del título. ES-MS m/z = 187 (M++H). 1H RMN (300 MHz,CDCl3) δ 4,05 (s, 3H), 6,2 (m, 1H), 6,7 (m, 1H), 7,0 (s, 1H), 7,6 (dd, 2H), 8,75 (dd, 2H).
Ejemplo 11
(4-Fluorofenil)-[1-metil-5-(piridina-4-carbonil)-1H-pirrol-3-il]metanona (Comp. 11)
Siguiendo el protocolo del Ejemplo 7 y empleando clorhidrato de (1-metil-1H-pirrol-2-il)piridin-4-ilmetanona enlugar de clorhidrato de (1-metil-1H-pirrol-2-il)piridin-3-ilmetanona, se obtuvo el compuesto del título y se recristalizócon EtOAc: p.f. 155-7ºC. ES-MS m/z = 309 (M++H). 1H RMN (300 MHz, CDCl3) δ 4,1 (s, 3H), 7,2 (m, 3H), 7,5 (s,1H), 7,6 (dd, 2H), 7,8 (dd, 2H), 8,8 (dd, 2H).
Procedimiento 5
5-(4-Clorobenzoil)-1-metil-1H-pirrol-3-carboxaldehído
A una mezcla de 5,05 g (23 mmol) de (4-clorofenil)-(1-metil-1H-pirrol-2-il)metanona y 6,11 g (46 mmol) de AlCl3en 42 ml de 1,2-dicloroetano y 42 ml de nitrometano a -20ºC bajo argón, se le añadieron 2,27 ml (25,3 mmol) de éter1,1-diclorodimetílico. Se agitó la mezcla a -20ºC durante una hora y a 25ºC durante 16 h. Se vertió en hielo/HCl yse extrajo con CH2Cl2. Se secó la fase orgánica (MgSO4) y se concentró. Se recristalizó el residuo con EtOAc paraobtener 2,5 g (43%) del compuesto del título como un sólido marrón: CI-MS m/z = 248 (M++H). 1H RMN (300 MHz,CDCl3) δ 4,1 (s, 3H), 7,1 (s, 1H), 7,45 (dd, 2H), 7,5 (s, 1H), 7,8 (dd, 2H), 9,8 (s, 1H).
15

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
Ejemplo 12
(4-Clorofenil)-[4-(hidroxipiridin-4-ilmetil)-1-metil-1H-pirrol-2-il]metanona (Comp. 12)
Se enfrió una solución de 1,36 g (8,64 mmol) de 4-bromopiridina en 12 ml de éter a -40ºC bajo argón y se añadierongota a gota 5,4 ml (8,64 mmol) de n-butil-litio 1,6 M en hexano. Se añadió gota a gota una solución de 1,07 g (4,3mmol) de 5-(4-clorobenzoil)-1-metil-1H-pirrol-3-carboxaldehído (obtenido por el Procedimiento 5) en 15 ml de THF(exotermia). Se agitó la reacción durante 5 min y se vertió en agua y se extrajo con CH2Cl2. Se secó la fase orgánica(MgSO4) y se concentró. Se sometió el residuo a cromatografía instantánea (acetona 50%/hexano). Se concentró laestela de mancha y se recristalizó con EtOAc, para obtener 0,74 g (52% de rendimiento) del compuesto del título comoun sólido blanco: p.f. 145-146ºC. ES-MS m/z = 327 (M++H). 1H RMN (300 MHz, CDCl3) δ 2,55 (s, 1H), 4,0 (s, 3H),5,8 (s, 1H), 6,56 (s, 1H), 6,8 (s, 1H), 7,3 (dd, 2H), 7,4 (dd, 2H), 7,75 (dd, 2H), 8,55 (dd, 2H). Análisis calculado paraC18H15ClN2O2: C, 66,16; H, 4,63; N, 8,57. Encontrado: C, 65,8; H, 4,65; N, 8,63.
Procedimiento 6
(4-Clorofenil)-{4-[hidroxi(1-tritil-1H-imidazol-4-il)metil]-1-metil-1H-pirrol-2-il}metanona
A 4,4 g (0,01 moles) de 4-yodo-1-tritilimidazol en 80 ml de CH2Cl2 se añadieron gota a gota 3,3 ml de bromurode etilmagnesio 3,0M en Et2O. Después de agitar durante 1 h, se añadió gota a gota una solución de 1,0 g (0,004moles) de 5-(4-clorobenzoil)-1-metil-1H-pirrol-3-carboxaldehído en 10 ml de CH2Cl2. Se agitó la mezcla de reacciónresultante durante 2,5 h, después de lo cual se vertió en agua. Se filtró el sólido a través de celita. Se separaron losorgánicos del filtrado, se lavaron con agua y salmuera y se secaron (Na2SO4). Se evaporó el solvente a vacío, paraobtener 3,1 g de (4-clorofenil)-{4-[hidroxi(1-tritil-1H-imidazol-4-il)metil]-1-metil-1H-pirrol-2-il}metanona: CI-MSm/z = 559 (M++H). 1H RMN (CDCl3) δ 7,7 (m, 5H), 7,4-7,0 (m, 16H), 6,5 (m, 1H), 3,75 (s, 3H).
Procedimiento 7
(4-Clorofenil)-[1-metil-4-(1-tritil-1H-imidazol-4-carbonil)-1H-pirrol-2-il]metanona
Se agitó una solución de 2,2 g de (4-clorofenil)-{4-[hidroxi(1-tritil-1H-imidazol-4-il)metil]-1-metil-1H-pirrol-2-il}metanona en 50 ml de CH2Cl2 con 2,5 g de dióxido de manganeso activado durante la noche. Se filtró la reaccióny se evaporó el filtrado a vacío, para obtener 2,0 g de (4-clorofenil)-[1-metil-4-(1-tritil-1H-imidazol-4-carbonil)-1H-pirrol-2-il]metanona. CI-MS m/z = 556 (M++H).
Ejemplo 13
(4-Clorofenil)-[4-(3H-imidazol-4-carbonil)-1-metil-1H-pirrol-2-il]metanona (Comp. 13)
Se agitó una muestra de 2,0 g de 1-[5-(4-clorobenzoil)-1-metil-1H-pirrol-3-il]-2-(1-tritil-1H-imidazol-4-il)etanonaen 50 ml de MeOH y 35 ml de HCl 2N durante 5 h. Se evaporó el solvente a vacío y se pasó el residuo resultante através de Biotage Flash 40L (gel de sílice: CH2Cl2:MeOH), para obtener 0,14 g de (4-clorofenil)-[4-(3H-imidazol-4-carbonil)-1-metil-1H-pirrol-2-il]-metanona: p.f. 198-200ºC. CI-MS m/z = 314 (M++H). 1H RMN (CD3CN) δ 8,4 (ar,1H), 7,9-7,8 (ar, 4H), 7,7 (ar, 1H), 7,6 (ar, 3H), 7,5 (ar, 1H), 4,0 (s, 3H), 2,0 (m, 2H).
Ejemplo 14
Trifluorometanosulfonato de 4-[[5-(4-clorobenzoil)-1-metil-1H-pirrol-3-il]carbonil]-1-metilpiridinio hidrato [5:1](Comp. 14)
Se agitó una mezcla de 0,64 g (2 mmol) de [5-(4-clorobenzoil)-1-metil-1H-pirrol-3-il]piridin-4-ilmetanona y 0,22ml (2 mmol) de triflato de metilo durante 16 h en 20 ml de CH2Cl2. Se evaporó el solvente y se recristalizó el residuocon EtOAc, para obtener 0,76 g (77% de rendimiento) del compuesto del título como un sólido blanco: p.f. 113-114ºC.ES-MS m/z = 339 (M++H). 1H RMN (300 MHz, CDCl3) δ 4,0 (s, 3H), 4,55 (s, 3H), 7,32 (dd, 2H), 7,8 (m, 3H), 8,2(dd, 2H), 9,0 (dd, 2H). Análisis calculado para: C19H16ClN2O2.CF3O3S.0,2 H2O: C, 48,77; H, 3,35; N, 5,68; KF, 0,73.Encontrado: C, 48,91; H, 3,5; N, 5,57; KF, 1,03.
Ejemplo 15
(4-Clorofenil)-[1-metil-4-(1-oxipiridina-4-carbonil)-1H-pirrol-2-il]metanona (Comp. 15)
Una solución de 324 mg (10 mmol) de [5-(4-clorobenzoil)-1-metil-1H-pirrol-3-il]piridin-4-ilmetanona y 300 mg(> 10 mmol) de ácido m-cloroperbenzoico en 5 ml de CHCl3 durante 3 h. Se lavó la solución con NaOH diluido, sesecó (Na2SO4) y se concentró. Se recristalizó el residuo con EtOAc para obtener 260 mg (rendimiento del 76%) delcompuesto del título como un sólido blanco: p.f. 207-208ºC. ES-MS m/z = 341 (M++H). 1H RMN (300 MHz, CDCl3)δ 4,1 (s, 3H), 7,1 (s, 1H), 7,5 (dd, 2H), 7,53 (s, 1H), 7,7 (dd, 2H), 7,8 (dd, 2H), 8,23 (dd, 2H).
16

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
Ejemplo 16
(4-Clorofenil)-{4-[hidroxi(1-oxipiridin-4-il)metil]-1-metil-1H-pirrol-2-il}metanona (Comp. 16)
Siguiendo el protocolo del Ejemplo 15 y empleando (4-clorofenil)-[4-(hidroxipiridin-4-ilmetil)-1-metil-1H-pirrol-2-il]metanona en lugar de [5-(4-clorobenzoil)-1-metil-1H-pirrol-3-il]piridin-4-ilmetanona, se obtuvo el compuesto deltítulo y se recristalizó con CHCl3, para obtener el compuesto del título con un rendimiento del 50%. ES-MS m/z = 344(M++H). 1H RMN (300 MHz, DMSO-d6) δ 3,95 (s, 3H), 5,58 (d, 1H), 5,9 (d, 1H), 6,6 (s, 1H), 7,15 (s, 1H), 7,4 (dd,2H), 7,56 (dd, 2H), 7,7 (dd, 2H), 8,1 (dd, 2H).
Procedimiento 8
Piridin-3-il(1,3,5-trimetil-1H-pirrol-2-il)metanona
Siguiendo el protocolo para el Ejemplo 1 y empleando 1,2,4-trimetilpirrol en lugar de N-metilpirrol, se obtuvo elcompuesto del título: p.f. 195-197ºC. CI-MS m/z = 215 (M++H). 1H RMN (CDCl3) δ 8,95-8,85 (ar, 2H), 8,7 (ar, 1H),8,1 (ar, 1H), 5,9 (ar, 1H), 3,8 (s, 3H), 2,3 (s, 3H), 1,7 (s, 3H).
Ejemplo 17
(4-Fluorofenil)-[1,2,4-trimetil-5-(piridina-3-carbonil)-1H-pirrol-3-il]metanona (Comp. 17)
Siguiendo el protocolo del Ejemplo 7 y empleando clorhidrato de piridin-3-il(1,3,5-trimetil-1H-pirrol-2-il)meta-nona en lugar de clorhidrato de (1-metil-1H-pirrol-2-il)piridin-3-ilmetanona, se obtuvo el compuesto del título. P.f.85-88ºC. CI-MS m/z = 337 (M++H). 1H RMN (DMSO-d6) δ 8,85-8,75 (ar, 2H), 8,3 (ar, 1H), 7,8 (ar, 3H), 7,35 (ar,2H), 3,7 (s, 3H), 2,2 (s, 3H), 1,6 (s, 3H).
Procedimiento 9
(4-Clorofenil)-[4-(hidroxipiridin-4-ilmetil)-1,3,5-trimetil-1H-pirrol-2-il]metanona
Mediante el método del Ejemplo 12, usando 5-(4-clorobenzoil)-1,2,4-trimetil-1H-pirrol-3-carbaldehído en lugarde 2-(4-clorobenzoil)-1-metil-1H-pirrol-3-carboxaldehído, se obtuvo el compuesto del título en un rendimiento del47%. ES-MS m/z = 355 (M++H).
Ejemplo 18
(4-Clorofenil)-[4-(hidroxipiridin-4-ilmetil)-1,3,5-trimetil-1H-pirrol-2-il]metanona (Comp. 18)
Mediante el método del Procedimiento 7, substituyente con (4-clorofenil)-[4-(hidroxipiridin-4-ilmetil)-1,3,5-tri-metil-1H-pirrol-2-il]metanona la 5-[[2-(4-clorobenzoil)-1-metil-1H-pirrol-4-il]carbonil]-3-(trifenmetil)-1H-imidazo-lio, se preparó el compuesto del título. El tratamiento con HCl etéreo dio un 35% de rendimiento de la sal clorhidrato.P.f. 174-176ºC. ES-MS m/z = 353 (M++H). 1H RMN (CDCl3) δ 8,9 (ar, 2H), 8 (ar, 2H), 7,7 (ar, 2H), 7,5 (ar, 2H), 3,75(s, 3H), 2,3 (s, 3H), 2,65 (s, 3H).
Ejemplo 19
(4-Clorofenil)-[4-(2-cloropiridina-4-carbonil)-1-metil-1H-pirrol-2-il]metanona (Comp. 19)
Se sometió a reflujo una solución de 3,6 g (0,010 moles) de 4-[2-(4-clorobenzoil)-1-metil-1H-pirrol-4-il]-1-óxido-4-piridinilmetanol en 32 ml de POCl3 durante 4 h. Después de enfriar, se evaporó el solvente a vacío. Se recogió elresiduo en CH2Cl2, se lavó con NaOH 3N, agua y salmuera y se secó (Na2SO4). Se recogió el residuo en 15 ml deEtOH y 15 ml de tolueno y se eliminó por reflujo el agua de la mezcla azeotrópica con una trampa Dean-Stark durante2 h. Se evaporó el solvente a vacío. Se suspendió el residuo en EtOH caliente y se filtró el sólido. Se disolvió el sólidoen MeOH con CH2Cl2 al 10% y se añadió HCl etéreo. Se filtró el sólido para obtener un rendimiento del 24% delcompuesto del título. P.f. 182-184ºC. ES-MS m/z = 359. 1H RMN (DMSO-d6) δ 8,6 (ar, 1H), 8,1 (ar, 1H), 7,9-7,7 (ar,6H), 7,1 (ar, 1H), 4,0 (s, 3H).
Ejemplos biológicos
Los compuestos de la presente invención son útiles como agentes para tratar o modular un trastorno del sistemanervioso central. Los siguientes ejemplos biológicos demuestran el uso de los presentes compuestos para tratar omodular un trastorno del sistema nervioso central.
Los métodos incluyen un método para tratar o modular un trastorno del sistema nervioso central donde el trastornodel sistema nervioso central incluyen, sin limitación, el dolor neuropático, el dolor crónico (incluyendo el dolor crónicocausado por inflamación o una condición relacionado con la inflamación, la osteoartritis o la artritis reumatoide),el dolor, condiciones neurológicas (incluyendo la epilepsia y el trastorno bipolar), enfermedades cardiovasculares y
17

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
otros trastornos (incluyendo los trastornos funcionales del intestino), trastornos psicóticos, trastornos del movimiento,trastornos de ansiedad o trastornos neurodegenerativos.
El dolor neuropático incluye, sin limitación, el dolor neuropático resultante de condiciones crónicas o debilitantes(tales como la neuropatía periférica diabética dolorosa, la neuralgia post-herpética, la neuralgia del trigémino, eldolor post-ictus, el dolor asociado a la esclerosis múltiple, el dolor asociado a neuropatías (tal como en la neuropatíaidiopática o post-traumática y la mononeuritis), el dolor neuropático asociado al VIH, el dolor neuropático asociado alcáncer, el dolor neuropático asociado al túnel carpiano, el dolor asociado a lesiones de la médula espinal, el síndromedel dolor regional complejo, el dolor neuropático asociado a fibromialgia, el dolor lumbar y cervical, la distrofiasimpática refleja, el síndrome de los miembros fantasma y otros síndromes de dolor asociados a condiciones crónicasy debilitantes), el dolor mantenido simpáticamente o el dolor asociado al dolor de cabeza en racimo y en migraña;el dolor asociado al cáncer, la fibromialgia, trastornos de la espalda o el dolor de cabeza en migraña y crónico, laadiposis dolorosa y el dolor por quemadura, condiciones dolorosas centrales después de un ictus, lesiones talámicas oesclerosis múltiple o el dolor resultante de lesión del sistema nervioso periférico o central (tras amputación, paraplejía,herpes o como resultado de la polineuropatía diabética).
El dolor crónico incluye, sin limitación, el dolor crónico causado por inflamación o una condición relacionada conla inflamación, osteoartritis o artritis reumatoide o como secuela de una enfermedad, lesión aguda o traumatismo eincluye el dolor de la espalda superior o dolor de la espalda inferior (resultante de una enfermedad espinal sistemática,regional o primaria (tal como la radiculopatía), inflamación o una condición relacionada con la inflamación), el doloróseo (debido a osteoartritis, osteoporosis, metástasis óseas o razones desconocidas), el dolor pélvico el dolor asociadoa lesiones de la médula espinal, el dolor cardíaco de pecho, el dolor no cardíaco de pecho, el dolor post-ictus central,el dolor miofascial, el dolor del cáncer, el dolor del SIDA, el dolor de células falciformes, el dolor geriátrico o eldolor causado por dolor de cabeza (tal como el crónico o la migraña), la neuralgia del trigémino, el síndrome dela articulación temporomandibular, el síndrome de la fibromialgia, la osteoartritis, la artritis reumatoide, la gota, lafibrositis o síndromes de la salida torácica e incluye el uso de los presentes compuestos como anestésico local para sutratamiento.
El dolor incluye, sin limitación, el dolor de mediación central, el dolor de mediación periférica, el dolor causadopor lesión de tejidos estructurales, el dolor causado por lesión de tejidos blandos o el dolor causado por una enferme-dad progresiva, el dolor agudo (causado por una lesión aguda, un traumatismo, una enfermedad, lesiones de medicinadeportiva, el síndrome del túnel carpiano, quemaduras, torceduras y tensiones musculoesqueléticas, tensión musculo-tendinosa, síndromes de dolor cervicobraquial, dispepsia, úlcera gástrica, úlcera duodenal, dismenorrea, endometriosiso cirugía (tal como cirugía a corazón abierto o de bypass), dolor post-operatorio, dolor por cálculos renales, dolor dela vesícula biliar, dolor por cálculos biliares, dolor obstétrico, dolor reumatológico o dolor dental, e incluye el uso delos presentes compuestos como anestésico local para su tratamiento.
Las condiciones neurológicas incluyen, sin limitación, condiciones tales como la ansiedad, las convulsiones, laciclofrenia, la hipotonía, la epilepsia (incluyendo los ataques parciales simples, los ataques parciales complejos, losataques generalizados secundarios y los ataques generalizados (incluyendo además los ataques de ausencia, los ataquesmioclónicos, los ataques clónicos, los ataques tónicos, los ataques tónico-clónicos y los ataques atónicos)), el trastornobipolar (tal como el trastorno bipolar de tipo I, el trastorno bipolar de tipo II, el trastorno ciclotímico, el ciclo rápido, elciclo ultradiano, la depresión bipolar, la manía aguda, la manía, la manía mixta, la hipomanía y los episodios asociadoscon el trastorno bipolar) o la depresión unipolar.
Las enfermedades cardiovasculares y otros trastornos incluyen, aunque sin limitación, las arritmias (incluyendola arritmia cardíaca, el infarto cardíaco o la angina de pecho), la hipertensión, trastornos endocrinos (tales como laacromegalia o la diabetes insípida), el tinnitus, el espasmo muscular, la incontinencia urinaria, la diarrea, el prurito,los trastornos funcionales del intestino (tales como la dispepsia no ulcerativa, el dolor no cardíaco de pecho o elsíndrome del intestino irritable), la esclerosis muscular, la degeneración macular o el glaucoma, enfermedades en lascuales la patofisiología del trastorno implica una secreción celular excesiva o hipersecretora o de algún otro modoinapropiada de una substancia endógena (tal como una catecolamina, una hormona o un factor de crecimiento) o laemesis.
Como trastornos psicóticos se incluyen, sin limitación, la esquizofrenia (incluyendo la esquizofrenia paranoide, laesquizofrenia hebefrénica, la esquizofrenia catatónica, la esquizofrenia indiferenciada, la depresión post-esquizofréni-ca, la esquizofrenia residual, la esquizofrenia simple o la esquizofrenia no especificada), el trastorno esquizofrenifor-me, el trastorno esquizoafectivo, el trastorno psicótico breve, el trastorno psicótico compartido, el trastorno psicóticodebido a una condición médica general, el trastorno psicótico inducido por substancias o un trastorno psicótico noespecificado de algún otro modo.
Los trastornos del movimiento incluyen, sin limitación, el temblor esencial benigno, el temblor en la enfermedadde Parkinson, el temblor del Parkinsonismo, otros temblores esenciales o del Parkinsonismo no relacionados (talescomo temblores centrales o temblores no clásicos (incluyendo temblor en reposo de cabeza/miembros, temblor ciné-tico simple, temblor de intención, temblor ortostático, temblor fisiológico aumentado, temblor psicogénico, temblorcerebelar, temblor rubral o temblores asociados a la postura, posición, voz o tarea) o temblores inducidos por fár-macos y trastornos del movimiento (tales como temblor postural, distonía aguda, corea, acatisia, disquinesia tardíao síndromes de tipo Parkinson)), el síndrome de las piernas inquietas, el síndrome de los brazos inquietos, la corea
18

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
en la enfermedad de Huntington, temblores asociados a la esclerosis múltiple o al síndrome de Gilles de la Touret-te, los espasmos post-lesión de la médula espinal, los espasmos post-anóxicos, la distonía por torsión idiopática, ladistonía por torsión focal, el mioclono, la atetosis, los trastornos de movimiento paroxístico (tales como la distoníaparoxística, la ataxia paroxística o los temblores paroxísticos) o los movimientos anormales (tales como la enfermedadde Wilson).
Los trastornos de la ansiedad incluyen, sin limitación, el trastorno de ansiedad generalizada, los trastornos de pá-nico (tales como la agorafobia, los trastornos de pánico sin agorafobia, la ansiedad anticipatoria, los ataques de pánicodel sueño recurrentes, los síntomas angustiantes (tales como la dispnea, la taquicardia, las palpitaciones, los doloresde cabeza, el mareo, las parestesias, la sofocación, la sensación de ahogo, la náusea o el meteorismo) o las sensacio-nes de muerte inminente); los trastornos del control de los impulsos (tales como el trastorno obsesivo-compulsivo,la bulimia, el discontrol episódico, la tricotilomanía, el juego compulsivo y la cleptomanía); los trastornos fóbicos(tales como por fobia social, fobia social global, fobia social específica, fobia simple, agorafobia, apifobia, tropofobia,astrapofobia, triscaidecafobia, blenofobia, talasofobia, claustrofobia, esfecsofobia, cinofobia, esciofobia, decidofo-bia, eletrofobia, escolionofobia, eremofobia, pirofobia, gamofobia, pnigerofobia, ofidiofobia, odinofobia, nictofobia,oclofobia, musofobia, queraunofobia, quetagelofobia, cacorrafiofobia, hidrofobia, ginofobia, gatofobia, gefirofobia,acrofobia o amatofobia); el trastorno de estrés post-traumático, estados disociativos (tales como amnesia, sonambu-lismo, trastorno de identidad disociativa o despersonalización), los estados de ansiedad pre-quirúrgica, los estados deansiedad post-quirúrgica u otras condiciones de ansiedad inducida médicas o psiquiátricas (tales como la ansiedadresultante de una lesión traumática del cerebro, los trastornos de dolor crónico u otras condiciones de enfermedadcrónicas).
Los trastornos neurodegenerativos incluyen, sin limitación, los trastornos neurodegenerativos agudos (tales comolos asociados a una agresión abrupta resultante de lesión aguda (tal como trauma cerebral, trauma cerebral focal,lesión cerebral difusa, lesión de la médula espinal, lesiones intracraneales (incluyendo lesiones por contusión, pe-netración, corte, compresión o laceración), lesiones intravertebrales (incluyendo lesiones por contusión, penetración,corte, compresión o laceración) o el síndrome del niño agitado por latigazo), la isquemia anóxica, la isquemia hipóxi-ca, la isquemia hipoglicémica (donde la isquemia es el resultado de insuficiencia cerebrovascular, isquemia o infartocerebral (originado por edema, oclusión embólica, oclusión trombótica, reperfusión tras isquemia aguda, lesión pe-rinatal hipóxica-isquémica o por asfixia, arritmia cardíaca, isquemia, espasmo o paro o hemorragia intracraneal (talcomo hemorragia epidural, subdural, subaracnoidea o intracerebral)), ahogamiento o envenenamiento por monóxidode carbono) o su combinación resultante en muerte o compromiso de las células neuronales); trastornos neurodegene-rativos crónicos (tales como los asociados a muerte o compromiso progresivos de las células neuronales a lo largo deun período de tiempo (incluyendo la enfermedad de Alzheimer, la enfermedad de Pick, la enfermedad de cuerpos deLewy difusos, la parálisis supranuclear progresiva (tal como el síndrome de Steel-Richardson), la degeneración mul-tisistémica (tal como el síndrome de Shy-Drager), condiciones epilépticas crónicas asociadas a neurodegeneración,enfermedades de las neuronas motoras (tales como la esclerosis lateral amiotrófica (ELA)), la esclerosis múltiple, lasataxias degenerativas, la degeneración basal cortical, el complejo ELA-Parkinson-Demencia de Guam, la demencia yla ceguera inducidas por el VIH, la panencefalitis esclerosante subaguda, la enfermedad de Huntington, la enferme-dad de Parkinson, el Síndrome de Down, la enfermedad de Korsakoffs, las sinucleinopatías (tales como la atrofia demúltiples sistemas), la afasia progresiva primaria, estriadonigral, la enfermedad de Machado-Joseph/ataxia espinoce-rebelar de tipo 3 y la atrofia o degeneración olivopontocerebelar, la enfermedad de Gilles De La Tourette, la parálisisbulbar y pseudobulbar, la atrofia muscular espinal y espinobulbar (tal como la enfermedad de Kennedy), la esclerosislateral primaria, la paraplejía espástica familiar, la enfermedad de Werdnig-Hoffmann, la enfermedad de Kugelberg-Welander, la enfermedad de Tay-Sach, la enfermedad de Sandhoff, la enfermedad espástica familiar, la enfermedad deWohlfart-Kugelberg-Welander, la paraparesia espástica, la leucoencefalopatía multifocal progresiva, la disautonomíafamiliar (tal como el síndrome de Riley-Day) o las enfermedades priónicas (tal como la enfermedad de Creutzfeldt-Jakob, la enfermedad de Gerstmann-Sträussler-Scheinker, la enfermedad de Kuru o el insomnio familiar fatal)); otrostrastornos neurodegenerativos agudos o crónicos asociados a pérdida de memoria (como resultado de la demenciarelacionada con la edad, de la demencia vascular, de la demencia multiinfarto, de la enfermedad de la materia blancadifusa (tal como la enfermedad de Biswanger), la demencia de origen endocrino o metabólico, la demencia de traumade la cabeza o de lesión cerebral difusa, la demencia pugilística o la demencia de lóbulos frontales); u otros trastor-nos neurodegenerativos agudos o crónicos asociados a lesión neuronal (tales como los asociados a lesión por agentesquímicos, tóxicos, infecciosos y por radiación del sistema nervioso, lesión durante el desarrollo fetal, premadurez enel momento del nacimiento, isquemia anóxica, lesión de origen hepático, glicémico, urémico, electrolítico y endocri-no, lesión de origen psiquiátrico (como resultado de psicopatología, depresión o ansiedad), lesión por enfermedadesperiféricas y plexopatía (tal como parálisis de plexos); y lesión por neuropatía (como resultado de neuropatías mul-tifocales, sensoriales, motoras, sensoriales-motoras, autonómicas, sensoriales-autonómicas o desmielinizantes (talescomo el síndrome de Gillain-Barré o la polirradiculoneuropatía desmielinizante inflamatoria crónica), por neuropa-tías que se originan por infecciones, inflamación, trastornos inmunes, dependencia/tolerancia/tolerancia inversa a unagente inductor de dependencia (tal como el alcohol, los opioides, los depresores del SNC, los psicoestimulantes o lanicotina), tratamientos farmacológicos, toxinas, traumatismo (tal como traumatismos por compresión, aplastamiento,laceración o segmentación), trastornos metabólicos (tales como endocrinos o paraneoplásicos), enfermedad de Char-cot-Marie-Tooth (tal como la de tipo 1a, 1b, 2, 4a o ligada a 1-X), ataxia de Fried-reich, leucodistrofia metacromática,enfermedad de Refsum, adrenomieloneuropatía, ataxia-telangiectasia, Déjerine-Sottas (tipo A o B), síndrome de Lam-bert-Eaton o trastornos de los nervios craneales), por neuropatías periféricas (tales como la neuralgia del trigémino, laneuralgia post-herpética, la neuropatía diabética, la neuralgia glosofaríngea, las radiculopatías lumbares y cervicales,la distrofia simpática refleja y la causalgia) o por neuropatía secundaria a infiltración metastática); pérdida neuronal
19

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
(asociada a ictus, isquemia global y focal, traumatismo del SNC, hipoglicemia, cirugía y traumatismo de la médulaespinal), e incluye el uso de los presentes compuestos como agentes neuroprotectores (tratamiento de la neurodegene-ración tras un ictus, un paro cardíaco, bypass pulmonar, lesión traumática del cerebro y lesión traumática de la médulaespinal).
Ejemplo biológico 1
Ensayo de rastreo para antagonistas de los canales del sodio abiertos por voltaje
Ensayo de potencial de membrana fluorométrico
Se demostró la actividad de los presentes compuestos como agentes moduladores de los canales del sodio por elensayo de potencial de membrana fluorométrico en células SK-N-SH como se describe a continuación.
Procedimiento
Se mantuvieron células de neuroblastoma SK-N-SH en un medio de propagación que contenía medio de Eaglemodificado y un 10% (v/v) de suero bovino fetal y se incubaron a 37ºC en una atmósfera de CO2 al 5%. Sólo se usaronlas células con menos de 20 pases en cada ensayo. Todos los fármacos fueron preparados en DMSO y la concentraciónfinal de solvente era menor del 0,1% (v/v). La veratridina, una toxina que promueve selectivamente la activaciónpersistente de los canales de sodio abiertos por voltaje, sirvió como estímulo. Veinticuatro horas antes de la prueba, seplaquearon 1 x 105 células/pocillo en placas de 96 pocillos específicamente diseñadas para uso en un lector de placasde imagen fluorescente (FLIPR) (Molecular Devices, Sunnyvale, CA).
Antes del rastreo, se cargaron monocapas de células con 100 µl/pocillo de un tinte sensible a voltaje patentado(Molecular Devices, Sunnyvale, CA) durante 30 min a 37ºC. Tras este período de incubación, se estimularon las célulascon veratridina 5 µM en presencia o ausencia de los compuestos investigados (preparados a una concentración finalde 10 µM en DMSO). Una muestra que contenía veratridina 5 µM y tetrodotoxina 10 µM respondían de la actividadinespecífica. Se monitorizó la fluorescencia por medio del FLIPR (Molecular Devices) desde los 10 segundos previos ala adición del fármaco (50 µl) hasta los 170 segundos posteriores a la adición del fármaco a 25ºC. Durante la activaciónde los canales del sodio, las células se despolarizan y el tinte se asocia a las membranas celulares, dando lugar a unaumento en la fluorescencia.
Análisis
Tal como se muestra en la Tabla 3, se calculó el % de inhibición de la fluorescencia (% IF) en la membrana por uncompuesto según la fórmula:
% IF = 100 X [1-[(FCE-FNE)/(FCC-FNE)]],
donde FCE se define como la Fluorescencia del Compuesto de Ensayo, FCC es la Fluorescencia del CompuestoControl (donde se usó veratridina 5 µM como control) y FNE es la Fluorescencia No Específica.
(Tabla pasa a página siguiente)
20

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
TABLA 3
Los resultados del ensayo de potencial de membrana fluorométrico sugieren que los compuestos de la presenteinvención pueden ser efectivos en el tratamiento o la modulación de un trastorno del sistema nervioso central.
Para tratar o modular un trastorno del sistema nervioso central, se puede emplear un compuesto seleccionado entrela Fórmula (I) o la Fórmula (II) a una dosificación diaria en el rango de aproximadamente 30 a aproximadamente4.000 mg, normalmente en 2 a 4 dosis divididas, para un humano adulto medio. Una dosis unitaria contendría de apro-ximadamente 10 a aproximadamente 500 mg del ingrediente activo. Más en general, para mamíferos, el tratamientoconsistiría en la administración diaria de aproximadamente 0,001 mg/kg a aproximadamente 500 mg/kg.
En particular, una composición farmacéutica de la presente invención consistente en un soporte farmacéuticamenteaceptable y un compuesto seleccionado entre la Fórmula (I) o la Fórmula (II) administrada oralmente puede serespecialmente adecuada para uso en el tratamiento o la modulación de un trastorno del sistema nervioso central.
Ejemplo biológico 2
Ensayo de rastreo para antagonistas de los canales del sodio abiertos por voltaje
Ensayo de flujo de [14C]guanidinio
Se demostró la actividad de los presentes compuestos como agentes moduladores de los canales del sodio por elensayo de flujo de [14C]guanidinio en células SK-N-SH como se describe a continuación.
Procedimiento
Se mantuvieron células de neuroblastoma SK-N-SH en un medio de propagación que consistía en medio de Eaglemodificado y un 10% (v/v) de suero bovino fetal y se incubaron a 37ºC en una atmósfera de CO2 al 5%. Sólo se usaronlas células con menos de 20 pases en cada ensayo. Todos los fármacos fueron preparados en DMSO y la concentraciónfinal de solvente era menor del 0,1% (v/v). La veratridina, una toxina que promueve selectivamente la activaciónpersistente de los canales de sodio abiertos por voltaje, sirvió como estímulo.
21

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
Se plaquearon células SK-N-SH a 1 x 105 células/pocillo en placas de microcentelleo Cytostar-T de 96 pocillosen medio de propagación y se realizaron los experimentos 6-7 días después del plaqueo. Antes del ensayo, se lavaronlas células SK-N-SH dos veces con tampón de ensayo que contenía HEPES 50 mM (pH 7,45), cloruro de potasio 5,4mM, cloruro de magnesio 0,8 mM, cloruro de colina 130 mM, glucosa 5,5 mM y 0,1 mg/ml de seroalbúmina bovina.A continuación, se dejó que las placas se equilibraran durante 10 min a 37ºC en tampón de ensayo. Después de esteperíodo de incubación, se estimularon las células con veratridina 100 µM en presencia o ausencia de los compuestosinvestigados preparados en una placa de 96 pocillos (preparados a una concentración final de 10 µM en DMSO). Sepreparó la placa de dosificación de 96 pocillos mezclando 25 µl de una solución de fármaco/tampón de ensayo porpocillo con 25 µl de 0,7 µCi/ml de clorhidrato de [14C]guanidina en tampón de ensayo. Se añadieron entonces alícuotasde 50 µl de la placa de dosificación a las células. Una muestra que contenía veratridina 100 µM y tetrodotoxina 10µM respondía de la actividad no específica. Se incubaron las placas durante 1 h a temperatura ambiente y se contó laradiactividad en un contador de centelleo 1450 Microbeta.
Análisis
Tal como se muestra en la Tabla 4, el % de inhibición del flujo de guanidinio (% IFG) por un compuesto estudiadofue calculado según la fórmula:
% IFG = 100 X [1-[(CCE-CNS)/(CCC-CNS)]],
donde CCE se define como las Cuentas de Compuesto de Ensayo (por minuto), CCC son las Cuentas del CompuestoControl (por minuto; se usó veratridina 100 µM como control) y CNS son las Cuentas No específicas (por minuto).
TABLA 4
Los resultados del ensayo del flujo de guanidinio sugieren que los compuestos de la presente invención pueden serefectivos en el tratamiento o la modulación de un trastorno del sistema nervioso central.
Para tratar o modular un trastorno del sistema nervioso central, se puede emplear un compuesto seleccionado entrela Fórmula (I) o la Fórmula (II) a una dosificación diaria en el rango de aproximadamente 30 a aproximadamente4.000 mg, normalmente en 2 a 4 dosis divididas, para un humano adulto medio. Una dosis unitaria contendría de apro-ximadamente 10 a aproximadamente 500 mg del ingrediente activo. Más en general, para mamíferos, el tratamientoconsistiría en la administración diaria de aproximadamente 0,001 mg/kg a aproximadamente 500 mg/kg.
En particular, una composición farmacéutica de la presente invención consistente en un soporte farmacéuticamenteaceptable y un compuesto seleccionado entre la Fórmula (I) o la Fórmula (II) administrada oralmente puede serespecialmente adecuada para uso en el tratamiento o la modulación de un trastorno del sistema nervioso central.
22

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
Ejemplo biológico 3
Procedimiento de estudio en un modelo anticonvulsivante en ratón
Se determinó la actividad de los presentes compuestos como anticonvulsivantes usando una “prueba de electros-hock máximo” (ESM) en ratón estándar. En esta prueba, la actividad venía indicada por un bloque del ataque extensortóxico, como describen Swinyard y col. (J. Pharmacol. Exptl. Therap., 1952, 106, 319). Se da una descripción másreciente de rastreo de fármacos anticonvulsivantes actual en Swinyard y col., en Epilepsia, 1978, 19, 409.
En la Tabla 5 se muestra la actividad anticonvulsivante de los presentes compuestos y se evaluó según el métodode Swinyard (1952). Las abreviaturas usadas en la tabla tienen los siguientes significados: mpk se refiere a la dosisadministrada oralmente en “mg por kg” y DE50 se refiere a la “Dosis Efectiva del 50%” oralmente administrada enmpk. Los valores marcados por un * representan el número de ratones protegidos/número de ratones estudiados; porlo tanto, un valor de 3/3 representa que el número de ratones protegidos se correspondía con el número de ratonesestudiados a la dosis indicada.
TABLA 5
Los resultados del modelo de ESM en ratón sugieren que los compuestos de la presente invención pueden serefectivos en el tratamiento de la epilepsia o en la modulación de sus síntomas.
Para tratar la epilepsia o modular sus síntomas, un compuesto seleccionado entre la Fórmula (I) o la Fórmula (II) nodebe tener A seleccionado entre 2-nafta-lenilcarbonilo unido al anillo de pirrol en la posición 4 o, independientemente,no debe tener B seleccionado entre 2-tienilcarbonilo, 2-piridinilcarbonilo o 4-piridinilcar-bonilo unido al anillo depirrol en la posición 2.
Para tratar la epilepsia o modular sus síntomas, un compuesto seleccionado entre la Fórmula (I) o la Fórmula (II)puede ser empleado a una dosificación diaria en el rango de aproximadamente 30 a aproximadamente 4.000 mg, nor-malmente en 2 a 4 dosis divididas, para un humano adulto medio. Una dosis unitaria contendría de aproximadamente10 a aproximadamente 500 mg del ingrediente activo. Más en general, para mamíferos, el tratamiento consistiría en laadministración diaria de aproximadamente 0,001 mg/kg a aproximadamente 50 mg/kg.
Un compuesto seleccionado entre la Fórmula (I) o la Fórmula (II) puede ser usado en el tratamiento de la epilepsiao en la modulación de sus síntomas de forma similar a la usada para la fenitoína. Los aspectos médicos del tratamientode la epilepsia están descritos en L.S. Goodman y col., en “The Pharmacological Basis of Therapeutics”, 5ª Ed.,páginas 201 a 226, Macmillan (1975).
En particular, una composición farmacéutica de la presente invención consistente en un soporte farmacéuticamenteaceptable y un compuesto seleccionado entre la Fórmula (I) o la Fórmula (II) administrada oralmente puede serespecialmente adecuada para uso en el tratamiento de la epilepsia o en la modulación de sus síntomas.
Ejemplo biológico 4
Procedimiento para estudiar el efecto antialodínico
El procedimiento usado para detectar el efecto antialodínico de un compuesto de la presente invención, para elque existe una buena correlación con la eficacia humana para el tratamiento del dolor, es el procedimiento para la
23

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
medición de la alodinia encontrada en el Modelo de Chung (Kim S.H. y Chung J.M., An Experimental Model forPeripheral Neuropathy Produced by Segmental Spinal Nerve Ligation in the Rat, Pain, 1992, 50, 355-363). El efectoantialodínico de la composición de la presente invención en el Modelo de Chung se expresa en % de EMP (EfectoMáximo Posible).
Se anestesiaron ratas Sprague-Dawley machos de 200 g de peso aproximadamente cada una con isoflurano. Seexpuso el nervio espinal a nivel de L5 a través de una incisión justo a la izquierda de la línea media dorsal y seligó apretadamente con seda 6-0. A diversos tiempos tras la cirugía, los animales fueron estudiados en cuanto a laalodinia mecánica con pelos de von Frey (monofilamentos que están calibrados para doblarse bajo una cierta cantidadde presión, que varía entre 0,41 y 15,1 g). Para calcular el umbral de retirada del pie (URP), se midió la alodiniatáctil registrando la presión a la que se retiraba el pie afectado de estímulos graduados según el procedimiento de S.R.Chaplan, J.W. Pogrel, T.L. Yaksh, Role of Voltage-Dependent Calcium Channel Subtypes in Experimental TactileAllodynia, J. Pharmacol. Exp. Ther., 1994, 269, 1117-1123. Las ratas normales pueden soportar al menos 15 g depresión sin responder. Las ratas operadas, sin embargo, pueden responder a tan sólo 0,25 g de presión. Se consideróque la cirugía había tenido éxito si el animal respondía con un URP de menos de 4 g de presión aplicada al pie afectado.Se incluyeron las ratas en el estudio sólo si no exhibían disfunción motora (v.g., arrastre o caída del pie) y su URPestaba por debajo de 39,2 mN (equivalente a 4,0 g). Se usó el URP para calcular el % de efecto máximo posible (%(EMP) según la fórmula:
% EMP = 100 X [(URP-UC)/(CO-UC)],
donde URP se define como el Umbral de Retirada del Pie, UC es el Umbral Control y CO es el Corte (definido como15 g).
El simulacro de operación consistía en una cirugía similar; se visualizó el nervio espinal sin ligarlo. Estos animalestambién fueron estudiados en cuanto a la alodinia mecánica y no mostraron respuesta a más de 15 g de fuerza aplicadaal pie ipsilateral.
Se estudiaron compuestos seleccionados entre la Fórmula (I) y la Fórmula (II) en cuanto a su actividad en el trata-miento o modulación del dolor neuropático disolviéndolos o suspendiéndolos en agua o en hidroxipropilmetilcelulosa,respectivamente. Se dejaron en ayunas animales post-operatorios de entre 14 y 42 días durante la noche antes de ladosificación. Los animales fueron dosificados oralmente y los volúmenes de dosificación fueron calculados sobre unabase de 4 ml/kg.
La Tabla 6 muestra los resultados bien como la DE50, bien como el % de EMP, en el Modelo de Chung paraciertos compuestos seleccionados entre la Fórmula (I) y la Fórmula (II). Las abreviaturas usadas en la tabla tienen lossiguientes significados: IA se refiere a “inactivo a la dosis de rastreo”, DE50 se refiere a la “Dosis Efectiva del 50%”oralmente administrada en mg por kg (mpk).
TABLA 6
24

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
Los resultados del estudio del “Modelo de Chung” sugieren que los compuestos antialodínicos de la presenteinvención pueden ser efectivos en el tratamiento o la modulación del dolor neuropático.
Para tratar o modular el dolor neuropático, un compuesto seleccionado entre la Fórmula (I) o la Fórmula (II) nodebe tener B seleccionado entre 2-tienilcar-bonilo unido al anillo de pirrol en la posición 2 o 1H-imidazol-5-ilcarbonilounido al anillo de pirrol en la posición 4.
Para tratar o modular el dolor neuropático, un compuesto seleccionado entre la Fórmula (I) o la Fórmula (II) puedeser empleado a una dosificación diaria en el rango de aproximadamente 30 a aproximadamente 4.000 mg, normalmenteen 2 a 4 dosis divididas, para un humano adulto medio. Una dosis unitaria contendría de aproximadamente 10 aaproximadamente 500 mg del ingrediente activo. Más en general, para mamíferos, el tratamiento consistiría en laadministración diaria de aproximadamente 0,001 mg/kg a aproximadamente 500 mg/kg.
En particular, una composición farmacéutica de la presente invención consistente en un soporte farmacéuticamenteaceptable y un compuesto seleccionado entre la Fórmula (I) o la Fórmula (II) administrada oralmente puede serespecialmente adecuada para uso en el tratamiento o en la modulación del dolor neuropático.
Ejemplo biológico 5
Procedimiento para estudiar el efecto hiperalgésico
El procedimiento usado para detectar la actividad antihiperalgésica de un compuesto de la presente invención, parael cual existe una buena correlación con la eficacia humana, es la Prueba de la Hiperalgesia del Pie por Carragenina(según se describe en Sammons y col., Brain Research, 2000, 876, 48-54).
Animales
Se albergan ratones CD-1 machos (Charles River Laboratories) de 22-30 g de peso cada uno en el momento de laprueba en un ambiente libre de virus y de clima controlado durante al menos 7 días antes del ensayo. Disponen dealimento y agua ad libitum hasta el momento de la prueba.
Dosificación de los animales
Los ratones de ensayo son inmunizados por inyección de un irritante (v.g., 0,02 ml de una solución de carragenina al1,0% en solución salina al 0,9%) subcutáneamente en el tejido subplantar de uno de los pies posteriores para estimularuna reacción inflamatoria aguda. Se evalúa a continuación la respuesta del animal tras la inyección de carragenina enun momento posterior fijado y se verifica que es hiperalgésica en relación a una respuesta basal inmediatamente antesde la inyección de carragenina.
Se dosificaron los ratones oralmente con el Compuesto 1 disuelto en una suspensión de hidroxipropil-beta-ciclo-dextrina al 20% en agua destilada o con vehículo solo en un momento fijado tras la inyección de carragenina. Elvolumen de dosificación era de 10 ml/kg. Se evaluaron luego las latencias de respuesta en momentos fijados despuésde la dosificación oral para valorar la reversión de la hiperalgesia causada por el tratamiento con Compuesto 1 encomparación con el tratamiento con vehículo.
Medición de la hiperalgesia
Se valora la hiperalgesia por medición de una respuesta a un estímulo térmico. La medición de la hiperalgesia tér-mica es realizada con un aparato de placa caliente de laboratorio estándar, cuya temperatura superficial es determinadacon precisión y mantenida de modo uniforme. Alternativamente, se evalúa la hiperalgesia con un aparato Hargreavescomercializado que eleva selectivamente la temperatura de un pie individual (Dirig y col., J. Neurosci. Methods, 1997,76, 183). Se define una respuesta como cualquier agitación, lamido o doblamiento del pie tratado. Con cualquiera de
25

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
los aparatos, se define la hiperalgesia como una latencia reducida a la respuesta en comparación con la latencia basalregistrada antes de la inyección de carragenina y se ve el efecto antihiperalgésico del compuesto de ensayo como unarestauración (parcial) de la latencia hacia la normalidad (Dirig y col., J. Phrmacol. Expt. Therap., 1998, 285, 1031).
Se expresa la reversión de la hiperalgesia producida por la intervención terapéutica como el porcentaje de recu-peración (% R), es decir, el porcentaje de la reversión total posible, teniendo en cuenta las diferencias individualesen las latencias de respuesta basal y la gravedad de la hiperalgesia post-carragenina valorada antes de la intervenciónterapéutica.
El % de Recuperación (% R) por un compuesto estudiado fue calculado según la fórmula:
% R = 100 X [(LPF-LPC)/(LB-LPC)],
donde LPF se define como la Latencia Post-Fármaco, LPC es la Latencia Post-Carragenina y LB es la Latencia Basal.
Los resultados del Modelo del Pie con Carragenina para el Compuesto 1 oralmente administrado a 30 mg por kgdemuestran un % de Recuperación del 84% en comparación con el % de Recuperación de un control de vehículo del12% cuando se valoró cada grupo a los 90 minutos de la dosificación oral.
Los resultados del Modelo del Pie con Carragenina sugieren que los compuestos de la presente invención puedenser efectivos como agentes antihiperalgésicos en el tratamiento o la modulación del dolor asociado a inflamación o aun trastorno relacionado con la inflamación.
Para tratar o modular la inflamación o un trastorno relacionado con la inflamación, se puede emplear un compuestoseleccionado entre la Fórmula (I) o la Fórmula (II) a una dosificación diaria en el rango de aproximadamente 30 aaproximadamente 4.000 mg, normalmente en 2 a 4 dosis divididas, para un humano adulto medio. Una dosis unitariacontendría de aproximadamente 10 a aproximadamente 500 mg del ingrediente activo. Más en general, para mamífe-ros, el tratamiento consistiría en la administración diaria de aproximadamente 0,001 mg/kg a aproximadamente 500mg/kg.
En particular, una composición farmacéutica de la presente invención consistente en un soporte farmacéuticamenteaceptable y un compuesto seleccionado entre la Fórmula (I) o la Fórmula (II) administrada oralmente puede serespecialmente adecuada para uso en el tratamiento o en la modulación de la inflamación o de un trastorno relacionadocon la inflamación.
Ejemplo biológico 6
Procedimiento para estudiar la respuesta antiinflamatoria
El procedimiento usado para detectar la actividad antiinflamatoria de un compuesto de la presente invención, parael que existe una buena correlación con la eficacia humana, es el Modelo de Edema del Pie por Carragenina (descritoen Levy, L, Carrageenan Paw Edema in the Mouse, Life Sci., 1969, 8, 11, 601-6).
Animales
Se albergan ratas Sprague-Dawley machos (Charles River Laboratories) de 175-200 g en un ambiente libre de virusy de clima controlado durante al menos 5 días antes del ensayo.
Procedimiento
Se prepara carragenina en solución salina (solución de carragenina al 0,5% en solución salina al 0,9%) y se inyectaen un volumen de 0,1 ml en la almohadilla plantar de la rata. Se administra el compuesto de ensayo oralmente porsondaje 30-60 minutos antes de la inoculación en la almohadilla plantar. Se sumergen los pies en un ordenador deedema pletismógrafo de mercurio (Buxco Electronics) y se registra el desplazamiento del pie en el tiempo 0 y endiversos tiempos tras la inoculación de carragenina. Se comparan las ratas tratadas con compuesto de ensayo con loscontroles de vehículo en cuanto a cualquier inhibición de la hinchazón del pie inducida por carragenina.
Se calculó el % de inhibición (% I) por un compuesto estudiado según la fórmula:
% I = 100 - [100 X (CVPT/CVPV)],
donde CVPT se define como el Cambio medio en el Volumen del Pie en el grupo Tratado y CVPV es el Cambio medioen el Volumen del Pie en el grupo del Vehículo.
26

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
Las ratas tratadas con Compuesto 1 a una dosis de 100 mg/kg, p.o., exhibían una inhibición del 35% en la hinchazóndel pie inducida por carragenina a las 2 horas de la administración de carragenina. Los resultados del Modelo de Edemadel Pie sugieren que los compuestos de la presente invención pueden ser efectivos como agentes antiinflamatorios enel tratamiento o la modulación de la inflamación o de un trastorno relacionado con la inflamación.
Para tratar o modular la inflamación o un trastorno relacionado con la inflamación, se puede emplear un compuestoseleccionado entre la Fórmula (I) o la Fórmula (II) a una dosificación diaria en el rango de aproximadamente 30 aaproximadamente 4.000 mg, normalmente en 2 a 4 dosis divididas, para un humano adulto medio. Una dosis unitariacontendría de aproximadamente 10 a aproximadamente 500 mg del ingrediente activo. Más en general, para mamí-feros, el tratamiento consistiría en la administración diaria de aproximadamente 0,001 mg/kg a aproximadamente500 mg/kg.
En particular, una composición farmacéutica de la presente invención consistente en un soporte farmacéuticamenteaceptable y un compuesto seleccionado entre la Fórmula (I) o la Fórmula (II) administrada oralmente puede serespecialmente adecuada para uso en el tratamiento o en la modulación de la inflamación o de un trastorno relacionadocon la inflamación.
Aunque la anterior descripción enseña los principios de la presente invención, facilitando ejemplos con fines deilustración, se entenderá que la práctica de la invención abarca todas las variaciones, adaptaciones y/o modificacioneshabituales que entren dentro del alcance de las siguientes reivindicaciones.
27

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
REIVINDICACIONES
1. Un compuesto seleccionado entre la Fórmula (I) o la Fórmula (II):
donde
A es seleccionado entre el grupo consistente en:
arilo (eventualmente substituido con 1 a 4 substituyentes independientemente seleccionado entre el grupo con-sistente en halógeno, alquilo C1−8, alcoxi C1−8, hidroxi, hidroxialquilo C1−8, hidroxialcoxi C1−8, (halo)1−3alquilo(C1−8) y (halo)1−3alcoxi(C1−8)) y
heteroarilo seleccionado entre furilo, tienilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, isoxazolilo,isotiazolilo, triazolilo, piridinilo, piridazinilo, pirimidinilo, pirazinilo, quinolinilo e isoquinolinilo (estando di-cho heteroarilo eventualmente substituido sobre 1 a 4 miembros del anillo de átomos de carbono disponiblescon un substituyente seleccionado entre el grupo consistente en halógeno, alquilo C1−8, alcoxi C1−8, hidroxi,hidroxialquilo C1−8, hidroxialcoxi C1−8, (halo)1−3alquilo(C1−8) y (halo)1−3-alcoxi(C1−8); y eventualmente substi-tuido sobre los miembros del anillo de átomos de nitrógeno disponibles con un substituyente seleccionado entreel grupo consistente en alquilo C1−8, hidroxialquilo C1−8 y (halo)1−3alquilo(C1−8));
B es heteroarilo seleccionado entre furilo, tienilo, pirrolilo, oxazolilo, tiazolilo, imidazolilo, pirazolilo, isoxazolilo,isotiazolilo, triazolilo, piridinilo, piridazinilo, pirimidinilo, pirazinilo, quinolinilo, isoquinolinilo y quinoxalinilo(estando dicho heteroarilo eventualmente substituido sobre 1 a 4 miembros del anillo de átomos de carbono dispo-nibles con un substituyente seleccionado entre el grupo consistente en halógeno, alquilo C1−8, alcoxi C1−8, hidroxi,hidroxialquilo C1−8, hidroxialcoxi C1−8, (halo)1−3alquilo(C1−8) y (halo)1−3alcoxi(C1−8); y eventualmente substituidosobre los miembros del anillo de átomos de nitrógeno disponibles con un substituyente seleccionado entre el grupoconsistente en alquilo C1−8, alquenilo C2−8, hidroxialquilo C1−8, (halo)1−3alquilo(C1−8) y óxido);
Z es seleccionado entre el grupo consistente en oxo e hidroxi;
R1 es seleccionado entre el grupo consistente en:
alquilo C1−8 {donde el alquilo está eventualmente substituido sobre un carbono terminal con un substituyenteseleccionado entre el grupo consistente en alcoxi C1−8, -C(O)-(substituido con un substituyente seleccionadoentre el grupo consistente en H, OH, alquilo C1−8, alcoxi C1−8, NH2, -NHalquilo(C1−8), -N(alquilo(C1−8))2),-NHC(O)-(substituido con un substituyente seleccionado entre el grupo consistente en H, OH, alquilo C1−8, al-coxi C1−8, NH2, -NHalquilo(C1−8), -N(alquilo(C1−8))2), -OC(O)-(substituido con un substituyente seleccionadoentre el grupo consistente en H, OH, alquilo C1−8, alcoxi C1−8, NH2, -NHalquilo(C1−8), -N(alquilo(C1−8))2), NH2,-NHalquilo(C1−8), -N(alquilo(C1−8))2, -Sal-quilo(C1−8), -SO2alquilo(C1−8), ciano, (halo)1−3, hidroxi y nitro),
cicloalquilo y arilo {donde el cicloalquilo y el arilo están eventualmente substituidos con 1 a 4 substituyentesindependientemente seleccionados entre el grupo consistente en ciano, halo, hidroxi y nitro; y donde el cicloal-quilo y el arilo están eventualmente substituidos con un substituyente seleccionado entre el grupo consistente enalquilo C1−8, (donde el alquilo está eventualmente substituido sobre un carbono terminal con un substituyenteseleccionado entre el grupo consistente en NH2, -NHalquilo(C1−8), -N(alquilo(C1−8))2, ciano, (halo)1−3, hidroxiy nitro), alcoxi C1−8, NH2, -NHalquilo(C1−8) y -N(alquilo(C1−8))2);
R2 y R3 son independientemente seleccionados entre el grupo consistente en hidrógeno, alquilo C1−8 y halógeno;
y sus sales de adición de ácido, sales de amonio cuaternario y N-óxidos farmacéuticamente aceptables.
2. El compuesto de la reivindicación 1, donde A es seleccionado entre arilo eventualmente substituido con 1 a4 substituyentes independientemente seleccionados entre el grupo consistente en halógeno, alquilo C1−4, alcoxi C1−4,hidroxi, hidroxialquilo C1−4, hidroxialcoxi C1−4, (halo)1−3alquilo(C1−4) y (halo)1−3alcoxi(C1−4), donde el arilo es selec-cionado entre un anillo monocíclico aromático de seis miembros o un anillo bicíclico aromático de diez miembros.
28

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
3. El compuesto de la reivindicación 2, donde el arilo de A es seleccionado entre el grupo consistente en fenilo ynaftalenilo eventualmente substituido con 1 a 4 substituyentes.
4. El compuesto de la reivindicación 3, donde el arilo de A es fenilo eventualmente substituido con 1 a 4 substitu-yentes.
5. El compuesto de la reivindicación 2, donde los substituyentes de arilo de A son independientemente selecciona-dos entre el grupo consistente en halógeno, alquilo C1−4 e hidroxi.
6. El compuesto de la reivindicación 5, donde los substituyentes de arilo de A son independientemente selecciona-dos entre el grupo consistente en cloro, flúor, metilo e hidroxi.
7. El compuesto de la reivindicación 6, donde los substituyentes de arilo de A son independientemente selecciona-dos entre el grupo consistente en cloro, flúor y metilo.
8. El compuesto de la reivindicación 1, donde A es seleccionado entre heteroarilo eventualmente substituido sobre1 a 4 miembros del anillo de átomos de carbono disponibles con un substituyente seleccionado entre el grupo consis-tente en halógeno, alquilo C1−4, alcoxi C1−4, hidroxi, hidroxialquilo C1−4, hidroxialcoxi C1−4, (halo)1−3-alquilo(C1−4) y(halo)1−3alcoxi(C1−4); y eventualmente substituido sobre los miembros del anillo de átomos de nitrógeno disponiblescon un substituyente seleccionado entre el grupo consistente en alquilo C1−4, hidroxialquilo C1−4 y (halo)1−3alquilo(C1−4)).
9. El compuesto de la reivindicación 8, donde el heteroarilo de A es seleccionado entre el grupo consistente entienilo, piridinilo, quinolinilo e isoquinolinilo eventualmente substituido sobre 1 a 4 miembros del anillo de átomosde carbono disponibles con un substituyente y eventualmente substituido sobre los miembros del anillo de átomos denitrógeno disponibles con un substituyente.
10. El compuesto de la reivindicación 9, donde el heteroarilo de A es seleccionado entre el grupo consistente entienilo y piridinilo eventualmente substituido sobre 1 a 4 miembros del anillo de átomos de carbono disponibles conun substituyente y eventualmente substituido sobre los miembros del anillo de átomos de nitrógeno disponibles conun substituyente.
11. El compuesto de la reivindicación 8, donde los substituyentes de heteroarilo de A eventualmente substituidossobre 1 a 4 miembros del anillo de átomos de carbono disponibles son independientemente seleccionados entre elgrupo consistente en halógeno y alquilo C1−4 y los eventualmente substituidos sobre miembros del anillo de átomos denitrógeno disponibles son seleccionados entre alquilo C1−4.
12. El compuesto de la reivindicación 11, donde los substituyentes de heteroarilo de A eventualmente substituidossobre 1 a 4 miembros del anillo de átomos de carbono disponibles son independientemente seleccionados entre elgrupo consistente en cloro, flúor y metilo y los eventualmente substituidos sobre miembros del anillo de átomos denitrógeno disponibles son seleccionados entre metilo.
13. El compuesto de la reivindicación 1, donde B es heteroarilo eventualmente substituido sobre 1 a 4 miembrosdel anillo de átomos de carbono disponibles con un substituyente seleccionado entre el grupo consistente en halógeno,alquilo C1−4, alcoxi C1−4, hidroxi, hidroxialquilo C1−4, hidroxialcoxi C1−4, (halo)1−3alquilo(C1−4) y (halo)1−3alcoxi(C1−4);y eventualmente substituido sobre los miembros del anillo de átomos de nitrógeno disponibles con un substituyenteseleccionado entre el grupo consistente en alquilo C1−4, alquenilo C2−4, hidroxialquilo C1−4, (halo)1−3alquilo(C1−4) yóxido.
14. El compuesto de la reivindicación 13, donde el heteroarilo de B es seleccionado entre el grupo consistenteen furilo, tienilo, pirrolilo, imidazolilo, piridinilo, quinolinilo e isoquinolinilo eventualmente substituido sobre 1 a 4miembros del anillo de átomos de carbono disponibles con un substituyente como se ha definido en la reivindicación13 y eventualmente substituido sobre los miembros del anillo de átomos de nitrógeno disponibles con un substituyentecomo se ha definido en la reivindicación 13.
15. El compuesto de la reivindicación 14, donde el heteroarilo de B es seleccionado entre el grupo consistente entienilo, imidazolilo y piridinilo eventualmente substituido sobre 1 a 4 miembros del anillo de átomos de carbono dis-ponibles con un substituyente como se ha definido en la reivindicación 13 y eventualmente substituido sobre los miem-bros del anillo de átomos de nitrógeno disponibles con un substituyente como se ha definido en la reivindica-ción 13.
16. El compuesto de la reivindicación 13, donde los substituyentes de B eventualmente substituidos sobre 1 a 4miembros del anillo de átomos de carbono disponibles son independientemente seleccionados entre el grupo consis-tente en halógeno y alquilo C1−4 y los eventualmente substituidos sobre un miembro del anillo de átomos de nitrógenoson seleccionados entre óxido.
17. El compuesto de la reivindicación 16, donde los substituyentes de B eventualmente substituidos sobre 1 a 4miembros del anillo de átomos de carbono disponibles son independientemente seleccionados entre el grupo consis-
29

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
tente en cloro y metilo y los eventualmente substituidos sobre un miembro del anillo de átomos de nitrógeno sonseleccionados entre óxido.
18. El compuesto de la reivindicación 1, donde R1 es seleccionado entre el grupo consistente en:
alquilo C1−4 {donde el alquilo está eventualmente substituido sobre un carbono terminal con un substituyenteseleccionado entre el grupo consistente en alcoxi C1−4, -C(O)-(substituido con un substituyente seleccionado entreel grupo consistente en H, OH, alquilo C1−4, alcoxi C1−4, NH2, -NHalquilo(C1−4), -N(alquilo(C1−4))2), -NHC(O)-(substituido con un substituyente seleccionado entre el grupo consistente en H, OH, alquilo C1−4, alcoxi C1−4,NH2, -NHalquilo(C1−4), -N(alquilo(C1−4))2), -OC(O)-(substituido con un substituyente seleccionado entre el grupoconsistente en H, OH, alquilo C1−4, alcoxi C1−4, NH2, -NHalquilo(C1−4), -N(alquilo(C1−4))2), NH2, -NHalquilo(C1−4), -N(alquilo(C1−4))2, -Salquilo(C1−4), -SO2alquilo(C1−4), ciano, (halo)1−3, hidroxi y nitro),
cicloalquilo y arilo {donde el cicloalquilo y el arilo están eventualmente substituidos con 1 a 4 substituyentesindependientemente seleccionados entre el grupo consistente en ciano, halo, hidroxi y nitro; y donde el cicloalquiloy el arilo están eventualmente substituidos con un substituyente seleccionado entre el grupo consistente en alquiloC1−4, (donde el alquilo está eventualmente substituido sobre un carbono terminal con un substituyente seleccionadoentre el grupo consistente en NH2, -NHalquilo(C1−4), -N(alquilo(C1−4))2, ciano, (halo)1−3, hidroxi y nitro), alcoxiC1−4, NH2, -NHalquilo(C1−4) y -N(alquilo(C1−4))2).
19. El compuesto de la reivindicación 18, donde R1 es seleccionado entre alquilo C1−4 eventualmente substituidoen un carbono terminal con un substituyente.
20. El compuesto de la reivindicación 18, donde R1 es seleccionado entre alquilo C1−4.
21. El compuesto de la reivindicación 19, donde el substituyente eventual en el carbono terminal del alquilo C1−4es seleccionado entre el grupo consistente en alcoxi C1−4, NH2, -NHalquilo(C1−4), -N(alquilo(C1−4))2, ciano, (halo)1−3,hidroxi y nitro.
22. El compuesto de la reivindicación 1, donde R2 y R3 son independientemente seleccionados entre el grupoconsistente en hidrógeno, alquilo C1−4 y halógeno.
23. El compuesto de la reivindicación 22, donde R2 y R3 son independientemente seleccionados entre el grupoconsistente en hidrógeno y alquilo C1−4.
24. El compuesto de la reivindicación 23, donde R2 y R3 son independientemente seleccionados entre el grupoconsistente en hidrógeno y metilo.
25. El compuesto de la reivindicación 1, donde el compuesto es seleccionado entre la Fórmula (I):
30

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
donde A, B, Z, R2 y R3 son independientemente seleccionados entre el grupo consistente en:
26. El compuesto de la reivindicación 1, donde el compuesto es seleccionado entre la Fórmula (II):
donde B, A, R2 y R3 son independientemente seleccionados entre el grupo consistente en:
31

5
10
15
20
25
30
35
40
45
50
55
60
65
ES 2 286 328 T3
27. El compuesto de la reivindicación 1, seleccionado entre el grupo consistente en:
[5-(4-clorobenzoil)-1-metil-1H-pirrol-3-il]-piridin-4-ilmetanona,
clorhidrato de (4-fluorofenil)-[1-metil-5-(piridina-3-carbonil)-1H-pirrol-3-il]metanona y
(4-clorofenil)-{4-[hidroxi(1-oxipiridin-4-il)metil]-1-metil-1H-pirrol-2-il}metanona.
28. Una composición farmacéutica consistente en un compuesto de la reivindicación 1 y un soporte farmacéutica-mente aceptable.
29. Una composición farmacéutica preparada mezclando un compuesto de la reivindicación 1 y un soporte farma-céuticamente aceptable.
30. Un procedimiento para preparar una composición farmacéutica consistente en mezclar un compuesto de lareivindicación 1 y un soporte farmacéuticamente aceptable.
32


















