
7.LIPIDOS
89
7 LÍPIDOS 7.1 ESTRUCTURA DE LOS LÍPIDOS Introducción. Los lípidos constituyen un grupo importante y heterogéneo de com- puestos que se caracterizan por compartir las siguientes propiedades: I. Solubilidad. Estos compuestos se ca- racterizan por ser insolubles en agua y sol- ventes polares y solubles en solventes no polares, también llamados solventes orgáni- cos o solventes de grasas (cloroformo, ace- tona, éter, tetracloruro de carbono, hexano, etc.). Estos solventes son útiles para extraer- los de células y tejidos. II. Estructura Química. Estos compues- tos se caracterizan por poseer cuando menos una función éster y por hidrólisis dan alco- holes y ácidos grasos. Pero existen algunas excepciones. III. Asimilación. Los lípidos son asimila- bles por los organismos vivos y en esto se diferencian de los aceites minerales. Existen algunas excepciones a estas pro- piedades, ya que algunos lípidos son relati- vamente solubles en solventes polares (metanol, etanol y otros). Existen lípidos que en lugar de la función éster (Rj-CO-O- CH2-R2) poseen la función amida (RpCO- NH-CH2-R2). 7.2 FUNCIÓN DE LOS LÍPIDOS Los lípidos son compuestos que desempe- ñan varias funciones en todos los seres vivos las cuales se pueden dividir en 2 tipos. 7.2.1 Estructurales Los lípidos son componentes fundamentales de todos los organismos, constituyen en pro- medio el 10% del peso de todos los seres vi- vos, son un componente importante de todas las membranas celulares y subcelu lares (mi- tocondriai, nuclear, vacuolar, lisosomal, etc.) El tejido adiposo desempeña importan- tes funciones de relleno, amortiguadoras y de sostén; actúan como aislante térmico y lubricante y morfogenético sexual y como tejido conectivo. 7.2.2 Energéticas La función más importante de los lípidos es la energética; se ha calculado que en prome- dio el 40% de las calorías que utiliza el orga- nismo proviene de los lípidos. Obviamente el tejido adiposo tiene como una de sus funcio-
-
Upload
jorge-ordonez -
Category
Documents
-
view
29 -
download
1
Transcript of 7.LIPIDOS

7 LÍPIDOS
7.1 ESTRUCTURA DE LOS LÍPIDOS
Introducción. Los lípidos constituyen un grupo importante y heterogéneo de compuestos que se caracterizan por compartir las siguientes propiedades:
I. Solubilidad. Estos compuestos se caracterizan por ser insolubles en agua y solventes polares y solubles en solventes no polares, también llamados solventes orgánicos o solventes de grasas (cloroformo, acetona, éter, tetracloruro de carbono, hexano, etc.). Estos solventes son útiles para extraerlos de células y tejidos.
II. Estructura Química. Estos compuestos se caracterizan por poseer cuando menos una función éster y por hidrólisis dan alcoholes y ácidos grasos. Pero existen algunas excepciones.
III. Asimilación. Los lípidos son asimilables por los organismos vivos y en esto se diferencian de los aceites minerales.
Existen algunas excepciones a estas propiedades, ya que algunos lípidos son relativamente solubles en solventes polares (metanol, etanol y otros). Existen lípidos que en lugar de la función éster (Rj-CO-O-CH2-R2) poseen la función amida (RpCO-NH-CH2-R2).
7.2 FUNCIÓN DE LOS LÍPIDOS
Los lípidos son compuestos que desempeñan varias funciones en todos los seres vivos las cuales se pueden dividir en 2 tipos.
7.2.1 Estructurales
Los lípidos son componentes fundamentales de todos los organismos, constituyen en promedio el 10% del peso de todos los seres vivos, son un componente importante de todas las membranas celulares y subcelu lares (mi-tocondriai, nuclear, vacuolar, lisosomal, etc.) El tejido adiposo desempeña importantes funciones de relleno, amortiguadoras y de sostén; actúan como aislante térmico y lubricante y morfogenético sexual y como tejido conectivo.
7.2.2 Energéticas
La función más importante de los lípidos es la energética; se ha calculado que en promedio el 40% de las calorías que utiliza el organismo proviene de los lípidos. Obviamente el tejido adiposo tiene como una de sus funcio-

Figura 7.2. Formación de inositol-1,4, 5-trifosfato.
cebadas, liberación de serotonina, agregación plaquetaria, secreción de insulina, secreción de adrenalina, contracción de músculo liso, transducción visual en los fotorrecepto-res de invertebrados y otras.
Como se puede apreciar, varios tipos de señales extracelülares se convierten en muchas señales intracelulares. En la misma forma que la adenilato ciclasa actúa como un transductor de mensajes y un estímulo externo se convierte en una cascada de reacciones intracelulares, que dependiendo del tipo de célula da lugar a diferentes respuestas, así también los productos de hidrólisis del fosfatidil inositol, 4, 5-difosfato por acción de la fosfolipasa "C" o fosfoinositidasa, enzima que se encuentra en la membrana celular, al interactuar con una hormona como la serotonina se activa y produce el 1,4, 5-tri-fosfoinositol (IP3) y el diacilglicerol (DAG) que actúan como mensajeros intracelulares.
7.5.1.4 Cardiolipinas
Este tipo de glicerofosfolípido está constituido por 2 moléculas de ácido fosfatídico unidas por una molécula de glicerina. Estas moléculas tienen importancia porque son constituyentes de la membrana interna de las mitocondrias.
7.5.1.5 Plasmalo'genos
Este es un tipo especial de glicerofosfolípi-dos que se caracterizan por tener una estructura química muy similar a todos los demás glicerofosfolípidos ya mencionados, la única diferencia es que el C-1 en lugar de estar presente un ácido graso se encuentra un aldehido graso en forma de enol, unido en forma de éter enólico (fíg. 7.3).
Los plasmalógenos pueden tener en la posición X colina, etanolamina o serina.
Existe un fosfolípido con estructura similar a los plasmalógenos que se ha descrito como un factor activante de las plaquetas aún a concentraciones tan bajas como 0.1 nm produce agregación de plaquetas y dilatación de vasos sanguíneos (fig. 7.4).
7.5.2 Esfingolípidos
7.5.2.1 Esfíngofosfolípidos
Los más abundantes de este tipo de fosfo-lipidos son las estingomieYmas. üstos compuestos por hidrólisis total dan: esfingosina + ácido graso + H3PO4 + colina.
La esfingosina es un aminoalcohol alifáti-co de 18 carbonos insaturado, que se obtiene a partir de la palmitoil CoA y serina (fig. 7.5).
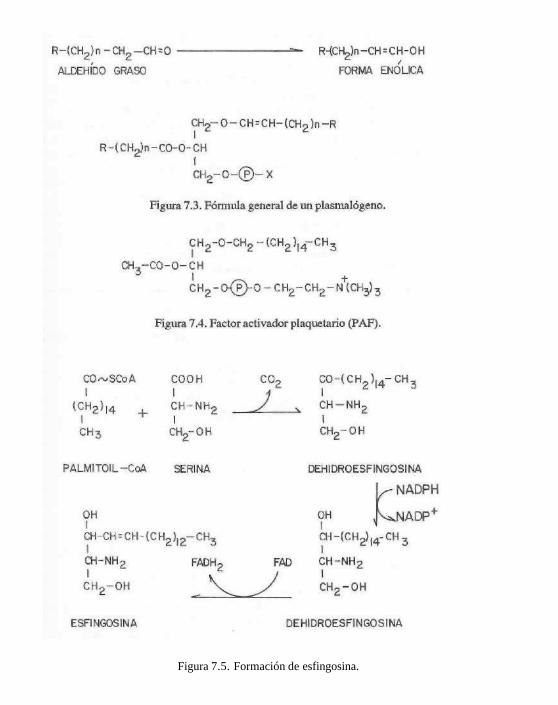
Figura 7.5. Formación de esfingosina.

En los enfingolípidos los ácidos grasos no están esterificados con la esfingosina a pesar de que posee grupos alcohólicos, el ácido graso se une al grupo amina y forma una amida (Fig. 7.6).
El ácido fosfórico se une al grupo alcohólico de la ceramida formando un fosfoéster y el mismo ácido fosfórico se une al grupo alcohólico de la colina para formar la molécula de esfingomielina. (fig. 7.7). La colina se une a la ceramida en forma activa como CDP-colina.
Las esfingomielinas son compuestos íntimamente relacionados con el sistema nervioso. Son los principales componentes de las vainas de mielina que existen en las fibras nerviosas mielínicas. Su función es actuar como aislante de las corrientes bioe-
léctricas y como material de reserva para la biosíntesis de neurotrasmisores.
7.5.2.2 Glucolípidos
Son lípidos que se caracterizan por poseer en su molécula glúcidos, en forma de mono-sacáridos u oligosacáridos. Los más abundantes son los esfingolípidos: cerebrósidos y gangliósidos, que tienen como alcohol la esfingosina. Se han aislado y caracterizado glucolípidos que tienen como alcohol glice-rina, su estructura y función son similares a los esfingolípidos, ya que por el carácter polar de los glúcidos las moléculas resultantes son, al igual que los fosfolípidos, sustancias antipáticas.

7.5.2.2.1 Cerebrósidos
Este tipo de lípidos complejos como su nombre lo indica se encuentra preferentemente en el sistema nervioso, en la sustancia blanca del cerebro y en las vainas de mieiina y en menor cantidad en las membranas celulares.
En forma general se puede decir que un cerebrósido es una molécula de ceramida (N-acil-esfingosina) unida a un monosacári-do, generalmente, galactosa (fig. 7.8). Otra característica estructural es que generalmente el ácido que esta unido al grupo amino es de 24 carbonos (C24: Lignocérico; C24-2-hi-droxi: Cerebrónico; C24A14 : Nervónico y C24A14-2 hidroxi: Hidroxinervónico).
Acompañando a estos glucolfpidos se han aislado e identificado algunos glucocerebró-sidos o sea, cerebrósidos que en lugar de galactosa contienen glucosa, aunque en cantidades pequeñas. También se han aislado glucoesfingolípidos de estructura más compleja, que en lugar de tener un monosa-cárido presentan varios monosacáridos y algunos derivados de ellos como el ácido N-acetil-neuramínico o ácido siálico. Este compuesto deriva del ácido fosfoenolpirúvi-
co (3c) y de la N-acetil manosamina. Se ci-cliza entre el C-2 y el C-6 formando la siguiente estructura de la figura 7.9.
Estos glucolípidos de estructura compleja, algunos aún no completamente caracterizados, pueden tener azufre en su molécula, debido a la esterificación de ácido sulfúrico con los grupos alcohólicos de los monosacáridos y oligosacáridos. Este tipo de compuestos se denominan gangliósidos y sulfolípidos.
7.5.2.2 Gangliósidos
Los gangliósidos son un grupo de glucoesfingolípidos, con una estructura similar a los cerebrósidos, pero en lugar de un monosacá-rido tiene un oligosacárido, casi siempre ramificado y cuando menos un azúcar ácido (ácido siálico) (fig. 7.10).
Se han aislado y caracterizado muchos gangliósidos que difieren en el número, tipo y unión de los azúcares y ácidos siálicos. Los gangliósidos se representan por la letra "G", y como subíndice otra letra "M", "D", o "T" que indica el número de ácidos siálicos que tiene en su molécula.

Figura 7.10. Representación simplificada del gangliósido GMi. Cer = ceramida; Glu = glucosa; Gal = galactosa; NAcGal = N-acetil-galactosamina; NANA = ácido N-acetilneuramínico.
Los gangliósidos se encuentran en altas concentraciones en el sistema nervioso, en especial en la materia gris, donde llega a existir en proporciones hasta del 6% de los lípidos totales. Los gangliósidos no son moléculas inertes, tienen un intercambio meta-bólico muy intenso ya que existen glicosilhidrolasas que eliminan los azúcares terminales. Estas enzimas son muy específicas y se encuentran en los lisosomas. Existe una enfermedad genética denominada enfermedad de Tay-Sachs en la que falta la enzima -N-Acetimexosaminidasa que elimina el residuo final de la N-acetilgalactosamina en el gangliósido GM2- Esto produce un incremento en la concentración de este gangliósido, varias veces arriba de lo normal, en el
cerebro de los niños. Los síntomas son progresivos e irreversibles: debilidad, retardo psicomotor, alteraciones visuales que llevan a la ceguera, demencia y muerte entre los 2-3 años (ver adelante).
Recientemente se ha descubierto que los gangliósidos desempeñan funciones muy importantes en nuestro organismo. Se ha comprobado que varias toxinas bacterianas (cólera, tétanos, botulismo y difteria) se unen selectivamente a los gangliósidos de la membrana. Si se adiciona el gangliósido específico se bloquea el sitio de unión y la toxina se vuelve inocua. Aparentemente los gangliósidos también actúan como receptores de los interferones, agentes biológicos con actividad antiviral.

7.5.2.2.3 Sulfolípidos
Estos lípidos complejos tienen una estructura química similar a los cerebrósidos y gangliósidos con la única diferencia que tienen moléculas de ácido sulfúrico esterifica-das con los grupos alcohólicos de los azúcares. Se considera que sus propiedades y funciones deben ser similares aunque aún no se conocen con precisión. Se sabe que son especialmente abundantes en las membranas de los tilacoides, organelos membranosos que se encuentran dentro del cloroplasto, y que semejan las crestas de las mitocondrias ya que también son invaginaciones de la membrana interna. Estas membranas contienen en su interior las enzimas responsables del transporte de electrones y de biosíntesis del ATP por acción de la luz solar. Estas membranas al igual que las membranas internas de las mitocondrias se caracterizan por ser impermeables a la mayor parte de iones y moléculas. Químicamente se caracterizan por poseer la misma cantidad de lípidos y proteínas. La composición de lípidos es la siguiente: 40% de glucolípidos, 10% de fosfolípidos y 4% de sulfolípidos.
7.5.3 Lipoproteínas
Los lípidos se asocian con proteínas y constituyen las lipoproteínas, que es la forma como se transportan los lípidos en la sangre, ya que por ser sustancias no polares hidrofóbi-cas no pueden circular libres.
Las proteínas que forman parte de las lipoproteínas son proteínas glubulares y los componentes lipidíeos, que son hidrofóbi-cos, se localizan en el interior de la macro-molécula en la zona hidrofóbica; los grupos polares de los lípidos complejos (fosfolípidos y glucolípidos) están orientados hacia la región polar externa. Esta disposición espacial o conformación asegura su estabilidad en medio acuoso.
Dependiendo de su composición cuali y cuantitativa, las lipoproteínas se subclasifi-cacn en varios grupos que se caracterizan por el tipo y proporción de lípidos y proteínas. En base a su composición presentan diferentes propiedades que permiten su separación y cuantificación como son: densidad, velocidad de sedimentación (Sf) y movilidad electroforética. Las propiedades y composición química cuali y cuantitativa de las diferentes lipoproteínas que se encuentran en el plasma humano, se resumen en la Tabla 7.6.
La densidad de las lipoproteínas es inversamente proporcional a su proporción de lípidos. Cuando las lipoproteínas se centrifugan en una solución de NaCl con una densidad de 1.063, algunas de ellas tienden a "flotar", o sea aparecen en la superficie de la solución. Esta propiedad de flotar en un medio isopícnico se expresa cuantitativamente como unidades Svedberg de flotación (Sf).
La hipercolesterolemia se caracteriza por una elevada proporción de lipoproteínas LDL (Low Density Lipoprotein) o en español LBD (Lipoproteínas de Baja Densidad).
Las apoproteínas de las lipoproteínas, son heterogéneas, se han aislado y caracterizado varios tipos que se designan como A-1, A-2, A-4, B-48, B-100, C y E, que pueden identificarse por pruebas inmunoquímicas.
Las lipoproteínas de baja densidad (LBD o LDL) poseen exclusivamente las apoproteínas B-100, C y E. Las de alta densidad (LAD o HDL) poseen principalmente apoproteínas A-l y A-2 y porciones pequeñas de C, D y E. Las de muy baja densidad (LMBD o VLDL) y los quilomicrones tienen principalmente apoproteínas: A- l , A-2, A-4 y B-48.
7.5.4 Grupo Misceláneo
Pertenecen a este grupo de compuestos una abundante serie de sustancias que poseen algunas características de los lípidos, como la
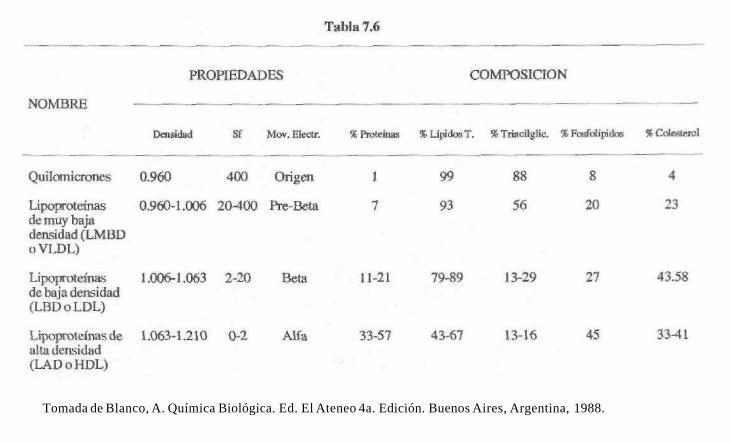
Tomada de Blanco, A. Química Biológica. Ed. El Ateneo 4a. Edición. Buenos Aires, Argentina, 1988.

insolubilidad en agua, pero carecen de otras como el tener una función éster. Debido a esto se encuentran como fracción insaponi-ficable. Algunas de estas sustancias derivan de lípídos y también se les conoce como derivados de lípidos. Sin embargo, existen algunas que no derivan de ninguno de los lípidos conocidos, razón por la cual se les conoce también como sustancias asociadas a lípidos. Este conjunto heterogéneo de sustancias a pesar de que se encuentran en pequeñísimas cantidades tienen importantes funciones y se pueden dividir para su estudio en los siguientes grupos: a) Terpenos y terpenoides; b) Esteróles y esteroides; c) Ei-cosanoides (Prostaglandinas, tromboxanos y leucotrienos)
7.5.4.1 Terpenos y Terpenoides
Los terpenos constituyen un grupo muy abundante e importante de sustancias que se encuentran universaImente distribuidos en la naturaleza y que realmente derivan de una simple unidad de 5C que se repite. Esta molécula es un hidrocarburo insaturado que se denomina isopreno. El isopreno como tal no existe libre en la naturaleza, su equivalente
es el llamado isopreno activo que se denomina isopentenil-pirofosfato o su isómero, el dimetil- alil-pirofosfato, que derivan de la acetil. CoA vía ácido mevalónico.
Biosíntesis del Isopreno activo. La síntesis del isopreno activo, y, por lo
tanto, la biosíntesis de todos los terpenos y terpenoides se inicia a partir de acetil-CoA, la cual se condensa con otra molécula de acetil CoA para formar primero, la acetoacetil-CoA; ésta se condensa con otra acetil-CoA para formar el 3-hidroxi-3-metil-glutaril CoA, precursor del ácido mevalónico.
Estas moléculas se condensan cabeza-cola, o cabeza-cabeza para formar hidrocarburos políméricos, que se denominan terpenos,
y según el número de moléculas de isopreno que los formen se clasifican como: monoter-penos (2(0$) -* 10C); Sesquiterpenos (3(C5) - 15C), diterpenos (4(C5) -» 20C): tríterpetios (6(C5) -* 30C); politerpenos (n(Cs) -* (5C)n). Los terpenos son hidrocarburos. Si existe algún grupo funcional, o si el número de carbonos ya no es múltiplo de 5, los compuestos se denominan terpenoides; algunos ejemplos son:

Como puede apreciarse, por sus nombres, los terpenos y terpenoides de bajo peso molecular se caracterizan por ser los principales componentes de los aceites esenciales responsables de las fragancias naturales de flores, frutas, hojas y tallos. La función que desempeñan estas moléculas no está bien definida. Pueden servir para atraer insectos, indispensables para la fecundación; debido a su alto contenido de carbono e hidrógeno son material energético de reserva. Los terpenos y terpe-
7.5.4.2 Esteróles y Esteroides
Estos compuestos aunque se revisan por separado por su importancia particular, pertenecen al grupo anterior, ya que se ha demostrado que todos los carbonos del colesterol provienen de la acetil-CoA. Los trabajos iniciales fueron realizados por Kon-rad Bloch (1940) y posteriormente se han aclarado y aislado todas las enzimas que participan en su biosíntesis.
El colesterol y todos los esteróles y compuestos que de ellos derivan como son: vitaminas D, hormonas sexuales y adreno-corticales, ácidos bilares, glucósidos cardiacos, saponinas, etc. se caracterizan por tener una estructura básica común: el ciclopenta-
noides de mayor peso molecular desempeñan importantes funciones como reguladores me-tabólicos (vitaminas liposolubles), además de actuar como pigmentos y participar en procesos fotoactivadores. Los terpenos de elevado P.M., como el hule se forma como una cubierta protectora cuando las hojas y tallos son lesionados. Cuando el caucho o hule natural se trata con azufre en caliente (vulcanización), las largas cadenas moleculares se unen por enlace azufrados y el compuesto es más fuerte, compacto, rígido y resistente a solventes.
noperhidrofenantreno y se denominan esteroides.
La molécula del ciclopentanoperhidrofe-nantreno está formada por la estructura del fenantreno totalmente hidrogenado (perhi-
l dro) unida al ciclopentano (Figura 7.11). ; Los carbonos 5, 8, 9, 10, 13 y 14 presen
tan centros de asimetría. Si se considera que i la molécula es plana puede existir isóme-í ros geométricos cis/trans. Los compuestos
de interés biológico sólo presentan isómeros en el C-5. En el caso del 5 a-androsta-
: no el H unido al C-5 está por debajo del plano y esto se indica con una línea punteada. En cambio el C-19, que está unido al
r C-10 está por arriba del plano. El H del C-5, puede estar en posición P o cis, con res-

CICLOPENTANOPERHIDROFENANTRENO
Figura 7.11 Estructura del ciclopentano perhidrofenantreno.
pecto al C-19, o 5a, que indica posición trans.
en realidad la verdadera estructura de los esferoides no es plana, adoptan la conforn-mación de silla en los anillos A, B y C que aparentemente es la más estable.
En los esteróles existe un grupo -OH en el C-3 en posición p o ecuatorial y en el C-17 existe una cadena hidrocarbonada en posición ecuatorial.
El esterol más abundante e importante en los organismos animales es el colesterol, el cual es materia prima en la biosíntesis de las
hormonas sexuales masculinas y femeninas, las hormonas adenocorticales, los ácidos cólicos y los ácidos biliares, todos estos compuestos desempeñan importantísimas funciones. Los vegetales no tienen colesterol; el esterol más abundante es el ergosterol, que tiene importancia por ser provitamina D cuando se expone a la luz solar.
A continuación se muestran las estructuras químicas de un miembro de cada grupo, indicando en cada caso el nombre del compuesto, el grupo al que pertenece y las principales características estructurales del grupo.

nes más importantes la de actuar como material de reserva energética.
7.2.3 De transporte
Sirven también como un eficaz vehículo de sustancias liposolubles como vitaminas y hormonas y en esa forma regulan la actividad metabólica. Son además importantes en el transporte de materiales alimenticios como lipoproteínas.
7.3 CLASIFICACIÓN
Para facilitar su estudio los lípidos se clasifican en 3 grupos.
I. Lípidos simples. Son aquellos que están cosntituídos únicamente por alcoholes y ácidos grasos.
II. Lípidos Complejos. Son aquellos que por hidrólisis total dan alcoholes, ácidos grasos y otra molécula diferente.
m. Grupo Misceláneo. Se incluyen en este grupo una serie de sustancias que tienen algunas características de lípidos, pero que no poseen ácidos grasos, por lo tanto a diferencia de los lípidos simples y complejos no son capaces de formar jabones por hidrólisis alcalina y por ello a este conjunto de sustancias se le conoce como "fracción insaponificable".
Los lípidos simples a su vez se subclasifi-can en los siguientes grupos, dependiendo del tipo de alcohol.
a) Acilgliceroles. Son aquellos lípidos que tienen como alcohol a la glicerina o gli-cerol (propanotriol).
b)Ceras. Se caracterizan por poseer un alcohol monohidroxílico alifático y de elevado peso molecular.
Acilgliceroles. (triglicéridos). Son lípidos constituidos por glicerol y
ácidos grasos. Estructura del glicerol. Es un alcohol po
livalente. Químicamente es el propanotriol,
posee tres carbonos y estos se designan con números arábigos o letras griegas.
l o a CH2-OH
2o p CH-OH
3 o a' CH2-OH
Fórmula lineal
7.3.1. Estructura de Ácidos Grasos
Los ácidos grasos que existen en los seres vivos constituyendo los lípidos simples y complejos son generalmente monocarboxílicos y de cadena lineal saturada o insaturada. Sin embargo, existen en ciertos organismos (microorganismos, semillas) ácidos grasos cíclicos, ramificados y con funciones alcohólicas.
Los ácidos carboxílicos se pueden considerar como productos de oxidación de los hidrocarburos:
°2 °2 °2 R-CHJ-CHJ - R-CHJ-CHJ-OH - R-CHj-CH-O - R-CHj-COOH
Hidrocarburo Alcohol Aldehido Acido
Nomenclatura. Los ácidos grasos se denominan generalmente en dos formas; el nombre sistémico se forma con el nombre del hidrocarburo del que proviene más el sufijo oico.^ El nombre común o trivial termina en "ico" y el nombre generalmente está relacionado con la fuente natural de la cual proviene. (Tabla 7.1) Los carbonos de los ácidos grasos se numeran a partir del grupo carboxilo.
4 3 2 1 R-CH2-CH2-CH2-COOH
También se acostumbra utilizar letras griegas. La letra alfa (a) para el carbono, anexo al carboxilo (C2); (P) para C3 y así sucesivamente. Se designa como co (omega)

7.5.4.3 Eicosanoides
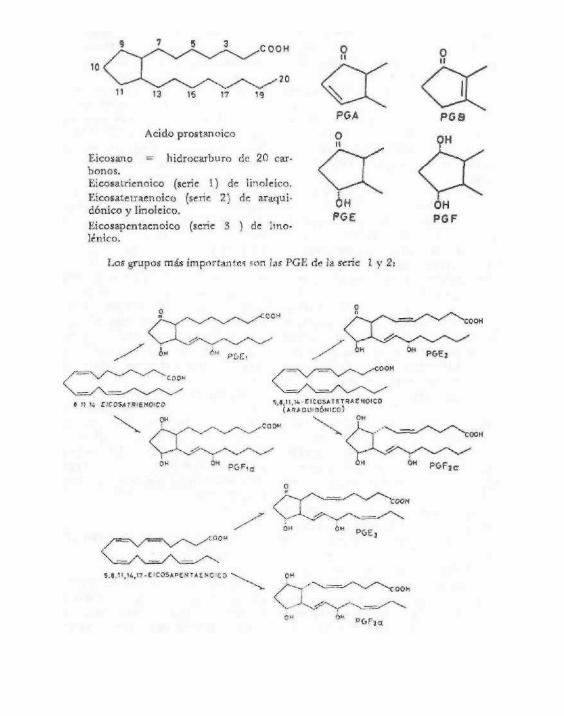
Son un grupo de sustancias bioactivas que modulan la función celular y que derivan de los ácidos grasos poliinsaturados de 20 carbonos (C20: eicosano). Este grupo incluye las prostaglandinas (PG), los tromboxanos (TX) y los leucotrienos (LT). Recientemente se han agregado algunos trihidroxideriva-dos del ácido araquidónico llamados lipoxinas.
7.5.4.3.1 Prostaglandinas
Constituyen un importante grupo de compuestos relacionados estructuralmente con los ácidos grasos, en especial, con el ácido C2oA5,8,l 1,14 - araquidónico.
Estas moléculas se descubrieron y reportaron desde 1930 por Kurzrok y Lieb, quienes observaron que el semen humano era capaz de producir contracciones en el útero "in vitro". Su descubrimiento pasó inadvertido hasta 1960, cuando Von Euler y Bergs-trom lograron aislar y comprobar su actividad biológica. En un principio se consideraban como secreciones características de las glándulas prostáticas y vesículas seminales. Sin embargo, se ha demostrado que se sintetizan en todas las células de mamíferos, excepto en los eritrocitos, y se han encontrado en gran cantidad en una alga marina Plexaura homomalla, utilizada desde hace tiempo como abortivo.
Aunque las prostaglandinas fueron los primeros compuestos descubiertos que derivan de un ácido graso de 20C (eicosanoico) poliinsaturado, se han descubierto otros compuestos relacionados con pequeñas variaciones en su estructura, aunque con diferencias en su biosíntesis y en su función. Estas sustancias se han denominado Hormonas eicosanoides y comprenden además de las prostaglandinas (PG), a los tromboxanos (TX), las prostaciclinas (PGI) y los leucotrienos
(LT). Todas estas sustancias se caracterizan por tener una gran actividad biológica, actúan a concentraciones pequeñísimas del orden de los nanogramos o picogramos, tienen una vida media muy corta y su efecto se observa en la célula que los produce o en células vecinas, por lo que el nombre de hormona no es adecuado a menos que se aclare que son hormonas locales o autacoi-des; (tienen acción autocrina o paracrina).
Los ácidos grasos precursores de estas moléculas son de 20C generalmente insatu-rados; el más abundante es el araquidónico C20: A5-8'11-14. Estos ácidos se encuentran en los fosfolípidos de la membrana en posición P (C2) y se liberan por acción de la fos-folipasa Á2. El ácido araquidónico puede servir como materia prima para la biosíntesis de prostaglandinas, tromboxanos, prosta-ciclinas o leucotrienos.

La biosíntesis de prostaglandinas, trom-boxanos y prostaciclinas se inicia por acción de una enzima denominada ciclooxigenasa, que convierte al ácido araquidónico en un endoperóxido cíclico, que es la prostaglan-dina G2 o PGG2. Este compuesto por acción de una peroxidasa se convierte en un hidro-xiderivado que se denomina prostaglandina H2 (PGH2). Este compuesto sirve como materia prima para la síntesis de prostaglandinas, tromboxanos o prostaciclinas, por una serie de reacciones que son específicas. John Vane demostró que la aspirina (ácido acetilsalicílico) inhibe a la ciclooxigenasa componente de la prostaglandina sintetasa.
Las prostaglandinas se pueden considerar como hídroxiácidos insaturados con un ci-clopentano o ciclopenteno. El nombre se forma con las letras PG, seguida de una tercera letra (que va de la A a la I) y de un número en forma de subíndice, que indica el número de dobles enlaces presentes en la molécula. La letra indica si son derivados del ciclopentano o ciclopenteno y el grado de oxidación, si presenta grupos hidróxilo o cetona. Las más importantes son las PGE que tienen un grupo cetona en el C-9, mientras que las PGF tiene un grupo hidróxilo.
7.5.4.3.2 Tromboxanos
Los tromboxanos (TX) son compuestos que se caracterizan por poseer en su estructura un anillo etéreo de 6 eslabones con propiedades fisiológicas diferentes y en ocasiones antagónicas con las prostaglandinas y prostaciclinas.
Las prostaglandinas H2 (PGH2) por acción de la prostaciclina sintetasa se convierte en prostaciclina, la cual también se conoce como prostaglandina I2 (PGI2). Se caracterizan por presentar además del ciclopentano otra estructura heterocíclica derivada del fu-rano que se forma al reaccionar un oxígeno del endoperóxido (PGH2) con los C-6 y C-9.
7.5.4.3.2 Leucotrienos
Los leucotrienos como su nombre lo dice son eicosanoides que se caracterizan por presentar tres dobles enlaces en su molécula por lo menos en los primeros que se aislaron de leucocitos. Su biosíntesis es diferente de las prostaglandinas, prostaciclinas y tromboxanos. La biosíntesis se inicia por acción de la lipooxigenasa sobre el ácido araquidónico que lo convierte en el ácido 5-hidroperóxido-eicostetraenoico (5-HETE), y éste se convierte luego en el leucotrieno A2 (LTA2). Este compuesto por adición de agua se convierte en el leucotrieno B4. El mismo LTA2 por adición de glutatión al C-6 da el leucotrieno C4 (LTC4) y éste por hidrólisis parcial pierde el ácido glutámico y da el leucotrieno D4 (LTD4). Cuando este compuesto pierde la molécula de glicina se forma el leucotrieno E4 (LTE4), el cual sólo tiene la cisteína en el C-6. La mezcla de estos tres últimos compuestos LTC4, LTD4 y LTE4) constituyen la que antes se conocía como SRS-A (sustancia de reacción lenta de la anafilaxia) de gran importancia en reacciones inmunitarias.
Los eicosanoides se caracterizan por ser moléculas muy activas, de acción pasajera y muy variada y en ocasiones antagónicas, lo cual ha dificultado su empleo terapéutico por ejemplo la PGF2 que un poderoso broncocons-trictor; en cambio la PGE2 produce dilatación de bronquios y de vasos sanguíneos cardiacos, renales, mesentéricos y musculares. Existen algunas acciones comunes a todas las prostaglandinas. Otro aspecto importante es que estas moléculas afectan casi todos los sistemas y aparatos del organismo (respiratorio, circulatorio, digestivo, reproductor, endocrino, nervioso, etc.). Por todo lo anterior, se presenta en forma muy resumida y parcial las principales acciones fisiológicas de los eicosanoides.
Sistema Nervioso. Algunas prostaglandinas (PGF) favorecen la liberación de neutro-transmisores, otras (PGE2) la inhiben.

Aparato Digestivo. Las PGE y PGI inhiben las secreciones digestivas gástricas y tienen un efecto estimulante sobre la secreción de moco, por lo que se tienen justificadas esperanzas de su utilidad terapéutica en el tratamiento de la úlcera. Las PGE incrementan la peristalsis del estómago e intestino, al estimular la contracción de la musculatura lisa longitudinal e inhibir a la circular. Las PGF estimulan tanto las circulares como longitudinales, y la PGI2 inhibe a ambas fibras.
Aparato Reproductor. Las prostaglandi-nas participan en todas las etapas de la reproducción; participan en la ovulación, en la ruptura del folículo, tienen acción luteolítica y aparentemente inician el trabajo de parto al estimular la contracción de la musculatura uterina. La oxitocina aparentemente participa en la 2a. etapa del parto.
Aparato Respiratorio. Las PGD2, PGF, el TXA2 y los leucotrienos LTC4 LTD4 y LTE4 (SRS-A) sonbroncoconstrictores. Los leucotrienos son 1000 veces más protentes que la histamina como broncoconstrictores y se considera que están muy relacionados con el asma. Las PGE son potentes bronco-dilatadores.
Aparato Circulatorio. Las PGE y PGI son vasodilatadores e hipotensores, mientras que el TXA2 es vasoconstrictor. Las PGI2 inhiben la agregación plaquetaria, mientras que el TXA2 estimula la agregación de plaquetas.
Inflamación, fiebre, dolor y respuestas, inmunitarias anormales. Los leucotrienos y las PG juegan un papel importante en los procesos de inflamación, fiebre y dolor. Incrementan la movilidad de leucocitos poli-morfonucleados e incrementan la permeabilidad celular y de esta forma contribuyen al edema. Se ha demostrado que la sensación de dolor y la hipertermia también están asociadas a las prostaglandinas. Esto explica el efecto antiinflamatorio, antitérmico y analgésico de la aspirina y una serie de
fármacos que inhiben la ciclooxigenasa y, por lo tanto, la biosíntesis de PG, TX y PGI. Los corticosteroides también son poderosos agentes antiinflamatorios que inhiben la fos-folipasa A2 y por lo tanto bloquean la biosíntesis de los eicosanoides en general.
Aparentemente las reacciones de hiper-sensibilidad y crisis asmática están relacionadas con la SRS-A (LTC4, LTD4 y LTE4) y la posibilidad de interferir farmacológicamente en la producción de estas sustancias tiene un futuro promisorio.
El mecanismo de acción de los eicosanoides aun se desconoce. Se ha propuesto que se unen a receptores específicos y modifican los niveles de AMPc o de Ca++, también se ha propuesto que actúan como segundos mensajeros, en respuesta a diferentes hormonas y que los cambios fisiológicos que producen son característicos de la célula y no de la hormona. Sin embargo, todas estas hipótesis requieren ser confirmadas o modificadas, por esto el estudio de éstas moléculas es actualmente una de las áreas de investigación más promisorias y apasionantes.
Las lipoxinas se forman por oxidación secuencial de ácido araquidónico mediada por la 15- y la 5-lipoxigenasa. Son reguladores intracelulares específicos; la lipoxi-na A estimula la formación de superóxidos en los neutrófilos, la quimiotaxis, produce vasoconstricción y activa la proteinci-nasa C.
7.6 DIGESTIÓN Y ABSORCIÓN DE LÍPIDOS
Las grasas y aceites alimenticios son mezclas de triglicéridos mixtos. En países industrializados se consumen entre 80 y 150 gramos diarios por persona. La capacidad de absorción de grasa del intestino delgado normal es considerable, casi 95% de los triglicéridos ingeridos.

A partir de las primeras observaciones anatómicas y funcionales de los vasos linfáticos intestinales, por Gaspare Aselli a principios del Siglo XVII, y de la aportación de Claude Bernard, en 1856, se demostró que los lípidos de la dieta alcanzaban el sistema linfático, la cuestión central en la digestión de los lípidos quedó circunscrita a cómo los lípidos de la luz intestinal atravesaban la pared del intestino para llegar a la linfa. A comienzos del presente siglo dos hipótesis contrapuestas eran intensamente debatidas. Immanuel Munk, en su "teoría particulada", sostenía que la mayor parte de los triacilgli-ceroles de la dieta eran absorbidos intactos en forma de una fina emulsión, y así transportados a través de la mucosa intestinal hacia los vasos linfáticos. Contrariamente, la "teoría lipolítica" de E.F. Pflüger, afirmaba que los triacilglicéridos eran hidrolizados totalmente a ácidos grasos y glicerol, y que estos últimos eran los compuestos absorbidos. En 1936, Verzar y McDougall demostraron que la degradación de los triglicéridos por acción de la lipasa pancreática y su emulsificación con las sales biliares, eran procesos esenciales para la absorción de lípidos. Este último trabajo, sumado a las aportaciones de Frazer, Bollman y otros autores, apoyaron definitivamente la "Teoría Lipolítica" de Pflüger.
Aunque debe convenirse que la digestión de los lípidos es un proceso dinámico que ocurre en todo el tubo digestivo, pueden diferenciarse fases caracterizadas por el lugar anatómico donde se realizan y por las enzimas hidrolíticas que intervienen.
7.6.1 Digestión gástrica. Lipasa lingual y lipasa gástrica
Como respuesta a la ingestión oral de grasas, a la masticación y a estímulos nerviosos, un grupo de glándulas serosas localizadas por debajo de las papilas circun
valadas de la lengua, segregan a la boca la lipasa lingual (conocida también como lipasa de la saliva).
Se sabe que la lipasa de la lengua es una glucoproteína hidrofóbica, escasamente soluble en agua, que muestra especificidad para la hidrólisis de triacilglicéridos. Se trata de una verdadera lipasa y no de una esterasa en general, ya que actúa sobre sustratos en forma de agregados insoluoles. La enzima es parcialmente inhibible por sales biliares y muestra un pH óptimo ácido. (Herrera E, 1991).
La lipasa de la lengua es más activa con los triacilglicéridos de ácidos grasos de cadena corta que con los de cadena larga. Presenta especificidad por los enlaces éster primarios de los triacilglicéridos (posición a y a ' o 1 y 3) y no hidroliza los enlaces en posición P (2), ni los enlaces éster de fosfo-glicéridos y del colesterol.
La acción de la lipasa lingual determina el comienzo de la digestión de los lípidos de la dieta en el estómago. Las contracciones del estómago producen los movimientos suficientes para favorecer la emulsificación de los lípidos, aunque también parecen participar otros agentes presentes en el propio alimento, como ciertos péptidos, polisacáridos complejos y fosfolípidos. De todos modos, la emulsificación en el estómago es más bien grosera y no puede calificarse de verdadera emulsión. Los ácidos grasos liberados por la lipasa lingual son hidrofílicos y solubles en medio acuoso, tanto en su estado ionizado como en el no ionizado, de manera que pueden escapar de las gotitas de grasa y estar en disposición de ser absorbidos. Se ha demostrado que la mucosa gástrica absorbe pasivamente ácidos grasos de cadena corta o media, los cuales son liberados a la circulación portal.
En resumen, puede considerarse que la lipasa de la lengua actúa en el estómago, hi-drolizando los enlaces a (1) de los triacilglicéridos, y permite la absorción ahí mismo de los ácidos grasos de cadena corta

o media, pero también es importante su acción facilitadora sobre la hidrólisis de los acilglicéridos restantes que ocurre en el intestino delgado.
El estómago produce una lipasa gástrica capaz de hidrolizar triacilgliceroles de ácidos grasos de cadena corta. A esta enzima también se le conoce como tributirasa por su especificidad para separar ácidos grasos de cuatro átomos de carbono (ác. butírico). La acción lipolítica de esta enzima es poco importante.
7.6.2 Digestión intestinal
El contenido del estómago ó quimo, es vaciado al intestino delgado a través de la válvula pilórica. El contenido alcalino de las secreciones pancreáticas y biliar neutraliza el ácido del quimo y hace variar el pH de éste a la alcalinidad. Este cambio en el pH es necesario para la actividad de las enzimas contenidas en los jugos pancreático e intestinal.
7.6.2.1 Secreción biliar
La absorción de los primeros ácidos grasos de cadena larga en el duodeno, así como otros factores (pH, ciertos aminoácidos etc.), desencadenan la secreción de colecis-tocinina por la mucosa intestinal. Esta hormona induce a su vez la secreción de la bilis (por contracción de la vesícula biliar y relajación del esfínter de Oddi) y la del jugo pancreático. Es a la altura de la ampolla de Vater donde las gotitas de grasa se encuentran y mezclan con estas secreciones.
Las sales biliares tienen una considerable capacidad para disminuir la tensión superficial del agua. Esto les permite emulsionar las grasas en el intestino y disolver los ácidos grasos y los jabones insolubles en agua. La presencia de la bilis en el intestino es un coadyuvante importante para llevar a cabo
la digestión y absorción de las grasas, así como la de las vitaminas liposolubles A, D, E y K. Cuando está alterada la digestión de las grasas, también son mal digeridos otros alimentos, pues la grasa cubre las partículas de alimento y evita que las enzimas actúen sobre ellas. En estas condiciones, la actividad de las bacterias intestinales provoca considerable putrefacción y producción de gases.
7.6.2.2 Secreción pancreática
La secreción pancreática al intestino contiene al menos tres actividades enzimáticas distintas: lipasa pancreática, fosfolipasa A2 y colesterol esterasa, y una coenzima, la colipasa.
La actividad de la lipasa es cuantitativamente la más importante de las tres y, de hecho, la actividad en el intestino es de 100 a 1,000 veces superior a la necesaria para la hidrólisis completa de los triacilgliceroles en el intestino delgado superior, tras una ingesta habitual. La actividad de la fosfolipasa A2 es bastante menor y la hidrólisis de los fosfolípidos requiere un trayecto en la luz intestinal más dilatado en el tiempo y en el espacio, llegando a ocurrir incluso en el íleo.
La lipasa pancreática degrada el enlace éster primario de triacilgliceroles; actúa en la interfase aceite-agua, de las gotitas finamente emulsificadas de los lípidos, formadas por la agitación mecánica en el intestino en presencia de los productos de la actividad de la lipasa lingual, sales biliares, colipasa, fosfolípidos y fosfolipasa A2. La fosfolipasa A2 y la colipasa son secretadas en forma de zimógenos y requieren activación por hidrólisis tríptica de enlaces peptídicos específicos. El Ca2+ es necesario para la actividad de la fosfolipasa A2. Una hidrólisis limitada del enlace éster en la posición 2 del fosfolípidos por la fosfolipasa A2 fija la lipasa a la interfase del sustrato y acelera la hidrólisis del triacilglicerol. La colipasa se une a la in-

terfase sal biliar-triacilglicerol-agua proporcionando un anclaje de elevada afinidad para la lipasa. La hidrólisis completa de los triacilgliceroles produce glicerol y ácidos grasos. La lipasa pancreática es virtualmen-te específica para la hidrólisis de enlaces éster primarios, es decir, en las posiciones 1 y 3 de los triacilgliceroles. Durante la digestión de grasas, la fase acuosa o "fase mice-lar" contiene una mezcla de micelas en forma de disco y liposomas de sales biliares saturadas con productos lipolíticos.
A causa de la dificultad para la hidrólisis del enlace éster secundario en el triacilglice-
rol, es probable que la digestión de estos lí-pidos avance hacia la remoción de los ácidos grasos terminales para producir 2 monoacil-gliceroles. Puesto que este último ácido graso está unido por un enlace éster secundario, su remoción necesita isomerización a un enlace éster primario. Este proceso es relativamente lento; como consecuencia, los 2-monoacilgliceroles son los productos principales de la digestión de triacilgliceroles y menos de una cuarta parte de los triacilgliceroles ingeridos es degradado en forma completa a glicerol y ácidos grasos (Fig.7.12).
Figura 7.12. Digestión y absorción de los triacilgliceroles, AG, ácidos grasos de cadena larga.
Los esteres de colesterol son degradados por una hidrolasa específica, en la luz intestinal, la colesterol esterasa que cataliza la hidrólisis de los esteres de colesterol, los cuales son así absorbidos en el intestino en una forma libre no esterificada.
La fosfolipasa pancreática, al igual que la co-lipasa, se segrega en forma de precursor no activo (proenzima). Al llegar al duodeno se activa por hidrólisis tríptica de un enlace Arg-Gli en la región proximal al NH2 terminal.
La enzima activa cataliza la hidrólisis de los ácidos grasos esterificados en la posición
P(2) de una variedad de fosfoglicéridos y no tiene efecto por ejemplo sobre los esfingolí-pidos. Los productos de su acción son 1-acil-2-lisofosfoglicéridos y ácidos grasos. La enzima tiene un requerimiento absoluto de iones Ca2+ que parecen asegurar la fijación y estabilización del complejo enzima-sustrato y las sales biliares son esenciales para la acción de la enzima, ya que aseguran la adecuada presentación física del sustrato.
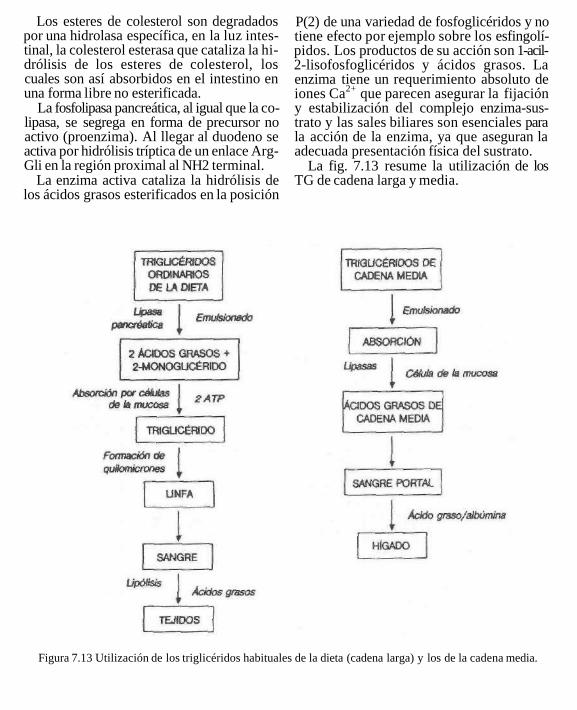
La fig. 7.13 resume la utilización de los TG de cadena larga y media.
Figura 7.13 Utilización de los triglicéridos habituales de la dieta (cadena larga) y los de la cadena media.

7.6.3 Absorción intestinal
7.6.3.1 Absorción micelar
Los 2-monoacilgliceroles, ácidos grasos y cantidades pequeñas de 1-monoacilglicero-les, dejan la fase de aceite de la emulsión de lípidos y difunden entre las micelas mixtas y liposomas consistentes en sales biliares, fos-fatidilcolina y colesterol, proporcionadas por la bilis. Debido a que las micelas son solubles permiten que los productos de la digestión sean transportados a través del medio acuoso de la luz intestinal hasta el borde en cepillo de las células de la mucosa donde son absorbidos al interior del epitelio intestinal. Las sales biliares pasan al íleon, donde la mayor parte son absorbidas penetrando a la circulación enterohepática. Los fosfolí pidos de origen alimentario y biliar son hidrolizados por la fosfolipasa A2 de la secreción pancreática hasta ácidos grasos y lisofosfolí pidos, los cuales también son absorbidos desde las micelas. Los esteres de colesterol son hidrolizados por la colesterol esterasa del jugo pancreático y el colesterol libre junto con la mayor parte del colesterol biliar es absorbido a través del borde en cepillo después de su transporte en las micelas. Normalmente se absorbe más del 95% de los lípidos de la alimentación.
En el interior de la célula intestinal, los 1-monoacilgliceroles son hidrolizados aún más para producir glicerol libre y ácidos grasos por una lipasa, que es distinta de la li-pasa pancreática. Los 2-monoacilgliceroles pueden ser convertidos de nuevo a triacilgli-ceroles a través de la vía del monoacilglice-rol (fig. 7.12). La utilización de ácidos grasos para una nueva síntesis de triacilgli-ceroles requiere primero su activación.
Es probable que la síntesis de triacilglice-roles procede en la mucosa intestinal en una forma semejante a la que se lleva a cabo en otros tejidos. Los fosfolípidos, junto con gran parte del colesterol, absorbidos, tam
bién son reacilados con Acil-CoA para regenerar fosfolípidos y esteres de colesterol.
El glicerol libre no es reutilizado sino que pasa directamente a la vena porta. Sin embargo, el glicerol liberado dentro de las células de la pared intestinal puede ser reutilizado en la síntesis de triacilgliceroles por activación con ATP y glicerocinasa para dar glicerol-3-fosfato. Por lo tanto, todos los ácidos grasos de cadena larga absorbidos en las células de la mucosa de la pared intestinal, son utilizados en la formación de novo de triacilgliceroles.
Los triacilgliceroles sintetizados en la mucosa intestinal no son transportados por la sangre venosa portal. La mayor parte de los lípidos, incluyendo fosfolípidos, esteres de colesterol, colesterol libre y vitaminas lipo-solubles, generan quilomicrones que le dan el aspecto lechoso al quilo, el cual es recolectado por los vasos linfáticos de la región abdominal y llevado a la circulación general por el conducto torácico.
7.6.3.2 Síntesis intestinal de quilomicrones
Los quilomicrones (QM) son los principales transportadores de triacilgliceroles con ácidos grasos de cadena larga de origen exó-geno. Los triacilglicéridos ingeridos se hi-drolizan en la luz del intestino delgado principalmente. Dentro de las células intestinales a nivel del yeyuno nuevos triglicéri-dos se sintetizan en el retículo endoplásmico liso. En la fracción microsomal de los ente-rocitos se encuentran también las enzimas responsables de la síntesis de fosfolípidos y de colesterol. La síntesis de las proteínas está asociada con los ribosomas del retículo endoplásmico rugoso. Los quilomicrones formados en el retículo se transfieren al aparato de Golgi y se descargan desde las células hasta la linfa por fusión de éstas vesículas con la membrana plasmática por el proceso de pinocítosis inversa o exocitosis.

Tabla 7.1 Nomenclatura sistémica y trivial de algunos ácidos grasos.
al último carbono de la cadena, independientemente del número de carbonos.
© y (3 a CH3-CH2--CH2-CH2-CH2-COOH
Una forma muy simplificada de representar un ácido graso es indicar el número de carbonos, dos puntos y otro número que indica el número de dobles enlaces (tablas 7.2,7.3 y 7.4). Si el ácido graso es insaturado se debe indicar la posición de la(s) dobíe(s) ligadura(s), colocando entre paréntesis, el o los números de los carbonos en los que empieza el doble enlace. También se utiliza la letra griega (A) seguido del número o números de los carbonos donde empieza la doble ligadura, en forma de supraíndice (A9). La mayor parte de los ácidos grasos que existen en la naturaleza tienen número par de carbonos. Esto se debe a que estas moléculas se sintetizan y degradan por la adición o separación de unidades de dos carbonos.
7.3.3 Propiedades de los Ácidos Grasos
Los ácidos grasos son moléculas antipáticas tienen un grupo polar (carboxilo) y un grupo no polar (la cadena hidrocarbonada). Los de bajo peso molecular (C2-C8) son solubles en agua; a medida que aumenta el peso molecular disminuye la solubilidad en agua y se incrementa la solubilidad en solventes no
polares. El punto de fusión también aumenta con el peso molecular. Los de bajo peso molecular (C2-Cg) son líquidos a temperatura ambiente, los de mayor peso molecular son sólidos. El grado de insaturación disminuye el punto de fusión.
Los ácidos grasos que poseen dobles enlaces presentan un tipo de isomería, que se denomina isomería geométrica o cis-trans ejemplo:
La mayor parte de los ácidos grasos que existen en la naturaleza son CIS.
7.3.4 Ácidos grasos esenciales
Los ácidos grasos poliinsaturados (linoleico, linolénico) no pueden ser sintetizados por los organismos superiores y deben de ser administrados en la dieta, ya que constituyen del 10 al 20% de los acilgliceroles y fosfolí-pidos. Por esto se les conoce como ácidos grasos esenciales (o vitamina F), su carencia produce alteraciones fisiológicas que pueden llegar a producir la muerte. Este tipo

El enlazamiento de carbohidratos al componente proteico ocurre en el retículo endo-plásmico liso pero en mayor proporción en el aparato de Golgi. Los triacilgliceroles con ácidos grasos de cadena larga se transportan en los quilomicrones por vía linfática y entran al torrente sanguíneo por el conducto torácico. Los triglicéridos con ácidos grasos de cadena corta y media pasan directamente a la vena porta en donde se unen a la albúmina para su transporte al hígado. Los quilomicrones que se encuentran en la linfa, llamadas partículas primarias o partículas nacientes, son más ricos en fosfolípidos y pobres en proteínas que los quilomicrones de la sangre, partículas secundarias ó partículas maduras. Los QM nacientes contienen Apo A-l y Apo B, pero son pobres en Apo C y Apo E. Los QM nacientes obtienen sus ApoC y E de las HDL en la sangre. La Apo B-48 es una apo-lipoproteína estructural e indispensable para la formación de los quilomicrones.
Catabolismo
La depuración sanguínea de los quilomicrones marcados es rápida; el tiempo medio
de degradación es de unos cuantos minutos. Cuando se administran intravenosamente quilomicrones con ácidos grasos marcados, casi el 80% de la marca aparece en el tejido adiposo, corazón y músculo y aproximadamente 20% en hígado. El catabolismo de los quilomicrones tiene lugar en dos etapas. En la primera etapa, los quilomicrones se adhieren a las células endoteliales de los capilares y sus triglicéridos se hidrolizan por la lipoproteínlipasa (LPL), un proceso que requiere la presencia de Apo C-II. Parte de los fosfolípidos se hidrolizan por la misma enzima. Conjuntamente con la hidrólisis de los triglicéridos, los compuestos superficiales de los quilomicrones, como son Apo C, fosfolípidos y colesterol no esterificado, se transfieren a las HDL (fig. 7.14). Las partículas producidas en esta primera etapa se denominan remanentes de quilomicrón. Estos remanentes en una segunda etapa son eliminados de la circulación por el hígado al enlazarse a los receptores hepáticos de alta afinidad para los Apo E. Tan pronto como se enlazan a los receptores, los remanentes se internalizan y sus componentes se hidrolizan para su posterior reutilización.
Figura 7.14. Metabolismo de los quilomicrones y de los remanentes de quilomicrones. TG, triglicéridos; CE, esteres de colesterol.

Metabolismo de las VLDL (LMBD) Síntesis
Las VLDL son transportadoras de trigli-céridos endógenos. Son sintetizados continuamente en el hígado y en el intestino y transportan en menor proporción triglicéri-dos de origen exógeno. La secuencia de eventos para la formación de las VLDL es parecida a la descrita para los quilomicrones excepto que poseen Apo B-100. En la zona de transición entre los retículos endoplásmi-cos rugoso y liso, sitio de la síntesis de los triglicéridos, pueden observarse partículas osmiofílicas de 300-800 A de diámetro. Estas partículas se transportan al aparato de Golgi y hacia la membrana plasmática en vesículas secretoras membranosas; las VLDL se descargan luego hacia la circulación o hacia la linfa por fusión de estos dos elementos. Los esteres de colesterol de las VLDL derivan de la transferencia de esteres de colesterol de las HDL a las VLDL, a cambio de triglicéridos.
Catabolismo
Las VLDL son degradadas en 2 etapas. Los triglicéridos de las VLDL son hidroliza-dos por la lipoprotein lipasa. Conjuntamente con la hidrólisis de los triglicéridos de las VLDL los fosfolípidos, el colesterol no este-rificado y las Apo C son transferidos de las VLDL a las HDL. En esta primera etapa las VLDL pierden gran parte de sus triglicéridos, colesterol y fosfolípidos y casi todas las Apo C. El producto principal del catabolismo de las VLDL es una lipoproteína de densidad intermedia (IDL) conocida también como remanente de VLDL. Dos destinos posibles esperan a la IDL. Pueden ser captadas de manera directa por el hígado a través del receptor para IDL (Apo B-100 y Apo E) o convertirse en LDL. Al parecer la mayor parte de las LDL son formadas a partir de las
VLDL, pero existen evidencias de producción directa en el hígado, la conversión de las IDL (ricas en Apo B) en LDL representa la segunda etapa del catabolismo de las VLDL (ver fig. 7.15).
Figura 7.15 Metabolismo de las VLDL. Conversión de las VLDL en IDL y LDL. TG, triglicéridos; CE,
esteres de colesterol.
Metabolismo de las LDL Síntesis. Las LDL se forman casi exclusi
vamente en la circulación a partir de las VLDL vía IDL, pero también hay evidencias experimentales que apoyan su producción directa por el hígado. (Fig. 7.16).
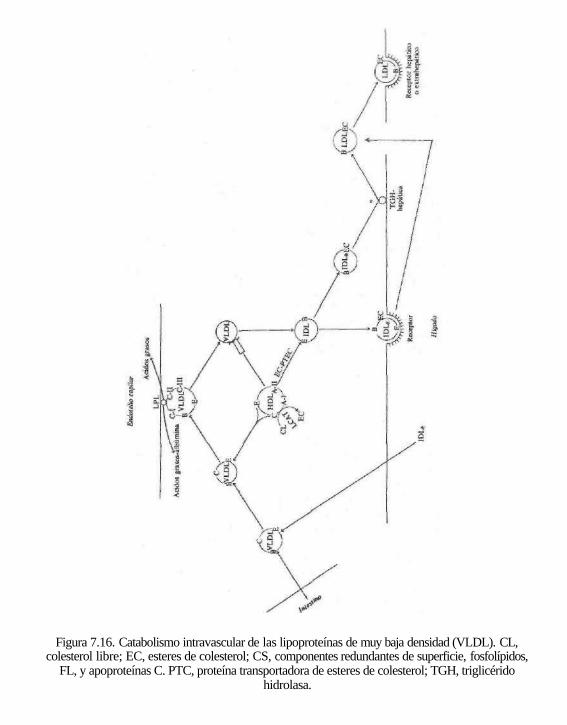
Figura 7.16. Catabolismo intravascular de las lipoproteínas de muy baja densidad (VLDL). CL, colesterol libre; EC, esteres de colesterol; CS, componentes redundantes de superficie, fosfolípidos,
FL, y apoproteínas C. PTC, proteína transportadora de esteres de colesterol; TGH, triglicérido hidrolasa.

Catabolismo
La vida media de las LDL en sujetos normales es de 2.5 a 3.5 días. Las LDL son degradadas tanto en el hígado como en tejidos extrahepáticos (fibroblastos, linfocitos, ce-lulas de músculo liso arterial, etc.). Las LDL se enlazan a los receptores para Apo B-100 y Apo E y, después de su internalización por endocitosis, la Apo B y los esteres de colesterol se hidrolizan por enzimas lisosomales.
Parte del colesterol se excreta de las células y el resto se transfiere al citoplasma, donde es reesterificado. La degradación de las LDL por la vía de los receptores LDL de alta afinidad es mucho más eficiente que su degradación por los macrófagos. Sin embargo, la degradación de las LDL vía sistema retículo endotelial adquiere importancia a medida que los niveles de las LDL en el plasma se incrementan. Se estima que entre el 33 y el 66% de las LDL circulantes se degradan por el sistema de los receptores LDL de los hepatoritos y de los tejidos extrahepáticos, y el resto por las células de Kupffer en el hígado y por los demás macrófagos del sistema retículoendotelial. Dado que las LDL transportan las dos terceras partes del colesterol circulante, estas partículas son las principales proveedoras de colesterol al hígado y a los demás tejidos del organismo.
La mayor parte de los esteres de colesterol de las LDL representan colesterol reciclado: el colesterol libre de las VLDL de origen hepático es transferido a las HDL para su este-rificación por la LCAT (lecitina colesterol a cil transferasa) y su transferencia en forma esterificada a las IDL por la PTEC (Proteina de transferencia de esteres de colesterol) las IDL a su vez, se degradan a LDL, entregando parte de su colesterol el hígado. El colesterol esterificado que se acumula en los tejidos extrahepáticos y en los macrófagos se hidroliza a colesterol libre por la colesterol hidrolasa, y el colesterol libre se secreta hacia los compartimientos intravasculares e
intersticiales, donde se incorpora a las HDL que lo transportan al hígado después de su esterificación por la LCAT.
Metabolismo de las HDL
Las lipoproteínas de alta densidad han despertado gran interés, por la relación inversa que existe entre su concentración y el riesgo de desarrollar aterosclerosis y sus complicaciones, ya que no se lleva a cabo en un solo órgano, los componentes de las HDL son aportados por varios tejidos ó por las lipoproteínas circulantes (figura 7.17). Las HDL se constituyen principalmente por Apo A-I, fosfolípidos y colesterol. La Apo A-l sintetizada en el hígado e intestino, es secretada en asociación a las lipoproteínas ricas en TG (QM y VLDL). Durante la hidrólisis de los TG de estas lipoproteínas por la LPL ocurre un fenómeno de intercambio de componentes con las HDL. Los QM y VLDL ceden la Apo A-l a las HDL y reciben apoproteínas C II, C III y Apo E de las HDL. Los fosfolípidos y TG son transferidos al núcleo de las HDL y los esteres de colesterol pasan a formar parte del núcleo de los remanentes de QM y VLDL. El paso de estas moléculas lipídicas, de caráctei hidro-fóbico entre las lipoproteínas mencionadas requiere de la proteína de transferencia de esteres de colesterol (PTEC). Esta proteína transportadora puede ser sintetizada y secretada por los macrófagos, que de esta manera facilitan el "transporte en reversa" del colesterol.
Las HDL, después de la asociación de la Apo A-I y los fosfolípidos, adquieren un aspecto discoide denominándose "HDL discoides o nacientes". En estrecha unión con estas HDL discoides, se encuentra la enzima LCAT, sintetizada y secretada por el hígado. Los sustratos de la LCAT son la fosfatidil-colina y el colesterol libre, éste último llega a la membrana de las HDL discoides procedente de la membrana de las células o de
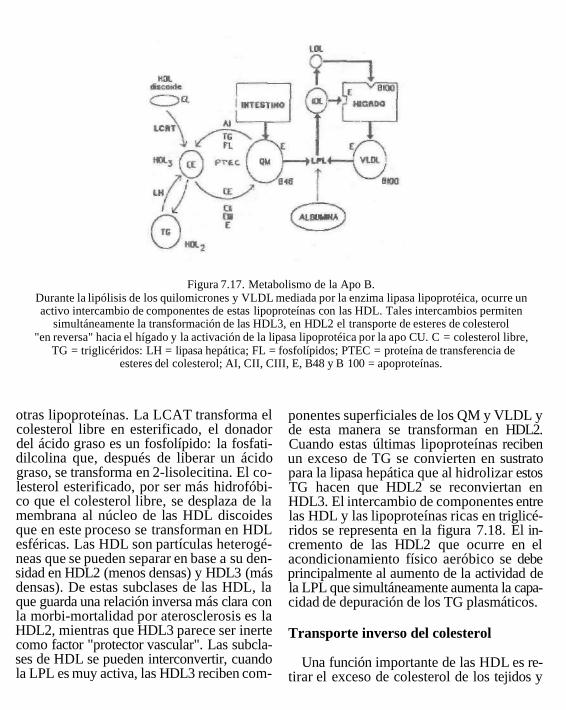
Figura 7.17. Metabolismo de la Apo B. Durante la lipólisis de los quilomicrones y VLDL mediada por la enzima lipasa lipoprotéica, ocurre un activo intercambio de componentes de estas lipoproteínas con las HDL. Tales intercambios permiten
simultáneamente la transformación de las HDL3, en HDL2 el transporte de esteres de colesterol "en reversa" hacia el hígado y la activación de la lipasa lipoprotéica por la apo CU. C = colesterol libre,
TG = triglicéridos: LH = lipasa hepática; FL = fosfolípidos; PTEC = proteína de transferencia de esteres del colesterol; AI, CII, CIII, E, B48 y B 100 = apoproteínas.
otras lipoproteínas. La LCAT transforma el colesterol libre en esterificado, el donador del ácido graso es un fosfolípido: la fosfati-dilcolina que, después de liberar un ácido graso, se transforma en 2-lisolecitina. El colesterol esterificado, por ser más hidrofóbi-co que el colesterol libre, se desplaza de la membrana al núcleo de las HDL discoides que en este proceso se transforman en HDL esféricas. Las HDL son partículas heterogéneas que se pueden separar en base a su densidad en HDL2 (menos densas) y HDL3 (más densas). De estas subclases de las HDL, la que guarda una relación inversa más clara con la morbi-mortalidad por aterosclerosis es la HDL2, mientras que HDL3 parece ser inerte como factor "protector vascular". Las subclases de HDL se pueden interconvertir, cuando la LPL es muy activa, las HDL3 reciben com
ponentes superficiales de los QM y VLDL y de esta manera se transforman en HDL2. Cuando estas últimas lipoproteínas reciben un exceso de TG se convierten en sustrato para la lipasa hepática que al hidrolizar estos TG hacen que HDL2 se reconviertan en HDL3. El intercambio de componentes entre las HDL y las lipoproteínas ricas en triglicéridos se representa en la figura 7.18. El incremento de las HDL2 que ocurre en el acondicionamiento físico aeróbico se debe principalmente al aumento de la actividad de la LPL que simultáneamente aumenta la capacidad de depuración de los TG plasmáticos.
Transporte inverso del colesterol
Una función importante de las HDL es retirar el exceso de colesterol de los tejidos y
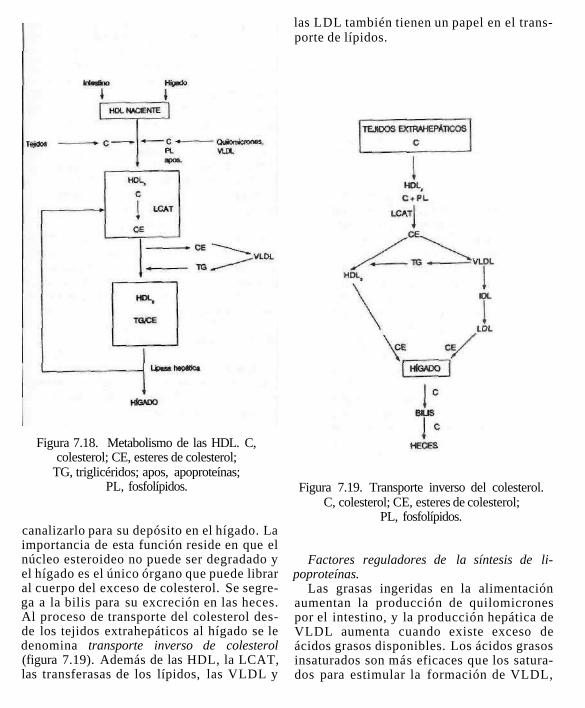
Figura 7.18. Metabolismo de las HDL. C, colesterol; CE, esteres de colesterol;
TG, triglicéridos; apos, apoproteínas; PL, fosfolípidos.
canalizarlo para su depósito en el hígado. La importancia de esta función reside en que el núcleo esteroideo no puede ser degradado y el hígado es el único órgano que puede librar al cuerpo del exceso de colesterol. Se segrega a la bilis para su excreción en las heces. Al proceso de transporte del colesterol desde los tejidos extrahepáticos al hígado se le denomina transporte inverso de colesterol (figura 7.19). Además de las HDL, la LCAT, las transferasas de los lípidos, las VLDL y
las LDL también tienen un papel en el transporte de lípidos.
Figura 7.19. Transporte inverso del colesterol. C, colesterol; CE, esteres de colesterol;
PL, fosfolípidos.
Factores reguladores de la síntesis de li-poproteínas.
Las grasas ingeridas en la alimentación aumentan la producción de quilomicrones por el intestino, y la producción hepática de VLDL aumenta cuando existe exceso de ácidos grasos disponibles. Los ácidos grasos insaturados son más eficaces que los saturados para estimular la formación de VLDL,

al tiempo que los ácidos grasos de cadena larga producen mayor efecto que los de cadena corta o mediana. Los carbohidratos de la dieta también aumentan la producción de las VLDL al aumentar la síntesis de los ácidos grasos y estimular la liberación de insulina. El alcohol también incrementa la producción de las VLDL al hacer que un mayor número de ácidos grasos esté disponible para la síntesis hepática de los triglicé-ridos.
La acción del colesterol en la formación de las lipoproteínas es complicada y no se comprende en su totalidad. En experimentos con animales, una dieta rica en colesterol altera el tipo de partículas formada. En algunas especies, el hígado produce un tipo de VLDL modificada rica en esteres de colesterol. Por el contrario, las LDL, lipoproteínas que suelen estar presentes en la circulación, se elevan en los seres humanos cuando la alimentación es rica en colesterol. Las partículas LDL que se acumulan no parecen tener una estructura ni composición anómala, y no se sabe si el aumento de LDL que se presenta en los seres humanos como respuesta a la dieta con alto contenido en colesterol es consecuencia de algún cambio producido en la formación de lipoproteínas.
7.6.3.3 Síndrome de absorción intestinal deficiente de lípidos
El síndrome se caracteriza por la falta de digestión y absorción de grasas a nivel de mucosa intestinal. El cuadro clínico se manifiesta principalmente por evacuaciones diarreicas con grasa macroscópica (esteato-rrea), baja de peso, distensión abdominal y flatulencia. El síndrome de malabsorción de grasas es multicausal y puede deberse a cuatro causas principales:
1. Alteraciones digestivas: cáncer pancreático, pancreatitis crónica, gastrectomía, insuficiencia hepática crónica.
2. Alteraciones en la absorción: síndrome de intestino corto o resección intestinal.
3. Alteraciones linfáticas: linfomas, enteritis postirradiación, conexión anormal entre órganos (quiluria, quilotórax).
4. Alteraciones de causa incierta: enfermedad de Addison, hipertiroidismo, administración de medicamentos como neomicina y colchicina.
La absorción defectuosa de grasas puede alterar también la utilización de otros nutrimentos como las vitaminas liposolubles (A, D, E y K). En cirugía de vesícula biliar debe prevenirse la falta subsecuente de vitaminas K y el consiguiente sangrado.
Las pruebas de laboratorio que ayudan al diagnóstico son el tiempo de protrombina, que aumenta en casos de esteatorrea, la prueba microscópica de grasa en heces y por rayos X la determinación de edad ósea, la cual se retrasa en casos crónicos.
Modelo clínico: esteatorrea
Niña de 10 años de edad, sin antecedentes familiares de esteatorrea, nivel socioeconómico medio, producto de parto eutócico con peso de 3000 g al nacer.
Inicial el padecimiento hace dos años con retraso en el crecimiento (talla y peso) y cuadros diarreicos frecuentes. A la exploración física se reportan los siguientes valores: peso 24 kg; talla 110 cm; FC 85x min; FR 26x min; temperatura 36.5°C; TA 90/80.
A su ingreso se encuentra a la paciente consciente, por debajo de los porcentiles de peso y talla, y signos característicos de avitaminosis A y D.
Abdomen globoso y doloroso, perista lsis aumentada.
El laboratorio reporta los siguientes resultados:
Tinción de lugol y sudan III: +++
cuantificación de ácidos grasos (TÉCNICA DEVANDEKAMER) 13 g/24 hs (normal: hasta 6 g / 24 hs).

7.7 TRANSPORTE Y DISTRIBUCIÓN DELÍPIDOS
Los triacilgliceroles (antes triglicéridos) es la principal forma de almacenamiento de energía en los seres humanos. Los adipoci-tos del tejido adiposo (10% del peso corporal) se dedican exclusivamente a almacenar la grasa. En la abundancia alimenticia, una de las estrategias fundamentales del organismo es convertir el exceso de calorías de los carbohidratos en grasas. La obesidad se trata de una hiperplasia y una hipertrofia del tejido adiposo; la presencia de un número excesivo de adipocitos induce al cuerpo a sintetizar más triacigliceroles para poder llenarlos.
Cuando se presenta un estado de estrés u otra forma de déficit de energía, se liberan ácidos grasos libres (AGL) que unidos a la albúmina plasmática llegan al hígado u otros tejidos donde son oxidados. Así, los triacilgliceroles del tejido adiposo se encuentran en equilibrio dinámico presentando lipólisis y reesterificación constantemente. Los triacilgliceroles son sintetizados en hígado, intestino y tejido adiposo (ver figura 7.20).
7.7.1 Papel del hígado en el transporte y distribución de lípidos
Como se resume en la figura 7.21, el hígado es el principal receptor de los productos de la lipólisis, el glicerol y los ácidos grasos libres. El glicerol es inmediatamente transformado en glicerol-3-fosfato, mediante la glicerocinasa presente en el hígado. El gli-cerol-3-fosfato puede ser esterificado en el hígado con las acil-CoA provenientes de la sangre o sintetizados en el propio órgano. Se forman triacilgliceroles que, junto con los procedentes de las lipoproteínas circulantes, son temporalmente acumulados para su posterior utilización, sin embargo, el hígado no es un reservorio de grasa. Normalmente
contiene 5 % de lípidos, y cuando se supera esta cantidad se presenta el llamado hígado graso. En este caso, el contenido lipídico llega a ser hasta de 25-30% y las gotas de grasa llegan a reemplazar el citoplasma de la célula hepática. El daño del tejido puede terminar en el desarrollo de cirrosis hepática, como ocurre en el alcoholismo crónico.
El hígado regula los niveles de colesterol plasmático a través de biosíntesis de coleste-rol y su conversión en ácidos biliares. Los triacilgliceroles formados se utilizan para la formación de VLDL o son hidrolizados a glicerol y ácidos grasos libres por acción de lipasas hepáticas, distintas a las del tejido adiposo y otros tejidos. Una de ellas es la triacilglicérido hidrolasa hepática y otra es la lipasa lisosómica que hidroliza los triglicéridos que llegan a los lisosomas por auto-fagocitosis celular (proceso facilitado por glucagon e inhibido por insulina)
7.7.2 Movilización y depósito del tejido adiposo
Para salir del adipocito, el triacilglicérido debe ser hidrolizado a glicerol y ácidos grasos, proceso conocido como lipólisis. El glicerol producido no es utilizado por los adipocitos por la baja actividad de glicerocinasa y pasa a la sangre donde es transportado al hígado (que contiene glicerocinasa) y transformado en glucosa por gluconeo-génesis. En la síntesis de triacilgliceroles por tejido adiposo se utiliza más bien el a-glicerofosfato proveniente de glucosa-6-fos-fato.
El ayuno, la estimulación del sistema nervioso simpático o la liberación hormonal (adrenalina, ACTH, hormona del crecimiento o glucagon) estimulan la liberación de AGL por parte del tejido adiposo. Estas hormonas activan la triacilglicerol lipasa {lipasa sensible a hormonas) de los adipocitos, acción mediada por AMPc(fig. 7.22).

Figura 7.20. Metabolismo lipídico en las células del tejido adiposo. Reproducción modificada, con autorización, de: N. V. Bhagavan Biochemistry. A. comprehensive review, pág. 681 J. B. Lippincott Co., 1974.
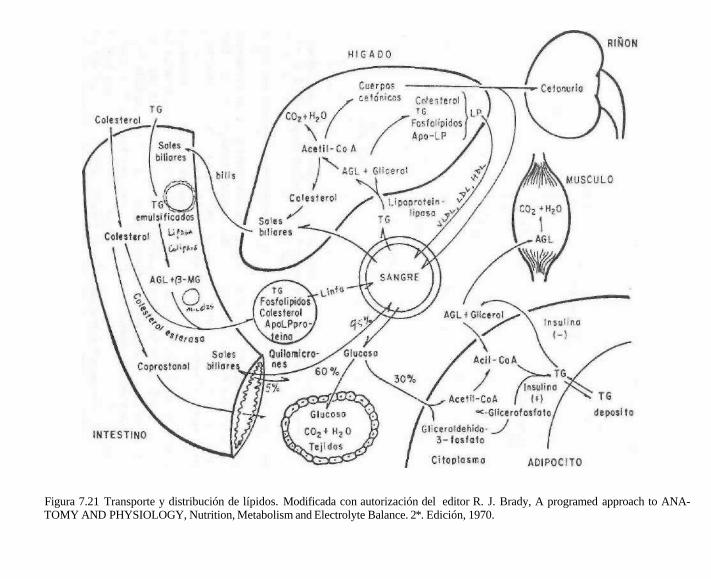
Figura 7.21 Transporte y distribución de lípidos. Modificada con autorización del editor R. J. Brady, A programed approach to ANA-TOMY AND PHYSIOLOGY, Nutrition, Metabolism and Electrolyte Balance. 2*. Edición, 1970.

de ácidos están relacionados con las prosta-glandinas, tromboxanos y leucotrienos, reguladores de gran importancia fisiológica.
La reactividad de los ácidos grasos depende fundamentalmente de su grupo funcional, el carboxilo, el cual puede disociarse y modificar el pH del medio. Sin embargo, todos los ácidos orgánicos son ácidos débiles y, a excepción del acético y algo el butírico, los demás son muy poco solubles en agua y esto disminuye su carácter ácido.
Los ácidos grasos pueden formar sales al reaccionar con álcalis. Estas sales se conocen como jabones y se caracterizan por ser potentes agentes tensoactivos negativos, y esto tiene aplicaciones prácticas y fisiológicas.
Los ácidos grasos pueden reaccionar con alcoholes para formar e'steres, con aminas para formar amidas y con otros ácidos para formar anhídridos. Él grupo carboxílico se puede reducir a aldehido, alcohol o hidrocarburo según la magnitud de la reducción.
Los ácidos grasos no saturados pueden presentar reactividad en la que se involucran los dobles enlaces, como en las reacciones de oxidación y de adición de hidrógeno, o halógenos (I, Br, Cl o F).
7.4 LÍPIDOS SIMPLES
Son compuestos constituidos por alcoholes y ácidos grasos, y se subclasifican en:
7.4.1 Acilgliceroles (triglicéridos)
Estos lípidos están constituidos por glice-rol o glicerina (propanotriol).
El glicerol posee 3 grupos alcohólicos, que se designan con números arábigos o letras griegas. Según el número de grupos alcohólicos esterificados los acilgliceroles, se denominan como monoacilgliceroles, dia-cilgliceroles y triacilgliceroles.
lo CH2-OH C H J - O - C H ^ R J CH2-OH
I I I 2o CH-OH CH-OH CH-O-CO-CH^R,
I i I 3o CH2-OH CH2-OH CH^O-CO-CHJ-RJ
Glicerol Monoacilglicerol Diacilglicerol
CRj-O-CCHCH^) 14-CH3 CH2-0-CO-CH2-Rj
I I CH-0-CO-(CH2)14-CH3 CH-O-CO-CRj-I^ I I CH2-0-CO-(CH2) 14-CH3 CH-J -O-CO-CH^RJ Tripalmitoiglicerol Heterotriacilglicerol (Homotriacilglicerol)
Si los ácidos grasos que esterifican al glicerol son iguales los di o tri acilgliceroles se denominan homoacilgliceroles, si los ácidos grasos son diferentes se denominan heteroa-cilgliceroles.
Si se observa la estructura de un L-monoa-cilglicerol, se comprueba que el carbono 2 es asimétrico y existen 2 esteroisomeros D y L.
CH^O-CO-R, CH^O-CO-CHj^
I I H-C-OH HO-C-H
I I CHj-OH Cf^-OH
D-monoacilglicerol L-monoacilglicerol
Los di y triacilgliceroles mixtos pueden presentar isomería D y L. Sin embargo, todos los productos naturales pertenecen a la serie L.
Todas las grasas y aceites naturales, ya sean de origen animal o vegetal se pueden considerar como mezclas de triglicéridos mixtos con pequeñísimas cantidades de mono y diacilgliceroles, ácidos grasos libres y sustancias liposolubles de estructura química diversa que son las responsables del color, olor y sabor (pigmentos y aromas) y además de su actividad biológica (hormonas esteroidales, vitaminas y prostaglandinas).
El término grasa se aplica a los triacilgliceroles que son sólidos a temperatura ambiente. Si son líquidos se denominan

Figura 7.22. Acción de la lipasa sensible a hormonas en el tejido adiposo. Reproducida, con autorización, de: R. Montgomery y cois., Biochemistry. A case-oriented approach, 4a edición, pág. 421. St. Louis,
1983, C. V. Mosby Co.
Esta lipasa sensible a hormonas cataliza la hidrólisis de trigHcéridos y diglicéridos. La monoacilglicerol lipasa cataliza la última etapa del proceso hasta glicerol y ácidos grasos.
Los glucocorticoides no tienen efecto li-político directo sino que facilitan la acción de otras hormonas movilizadoras de grasas. La tiroxina y la hormona del crecimiento actúan con más lentitud.
Los niveles elevados de glucosa e insulina en la sangre estimulan la acumulación de triacilgliceroles en el tejido adiposo. Cuando esto ocurre, disminuye el contenido de AMPc de los adipocitos. Las metilxantinas
(cafeína y teofilina) también favorecen la movilización de grasas del tejido adiposo, por inhibición de la fosfodiesterasa que inactiva al AMPc.
En la diabetes mellitus existe un transporte inadecuado de glucosa, por lo que falta el glicerofosfato y disminuye la lipogénesis (adelgazamiento). Además, la falta de insulina (antilipolítica) favorecería la lipólisis y el flujo de AGL a la sangre. Los AGL unidos a albúmina se elevan y llegan al hígado donde se ve favorecida la presentación de hígado graso.
En casos de hipoglucemia, los ácidos grasos proporcionan energía para todos los teji-

dos excepto cerebro; eritrocitos, hígado y médula suprarrenal. Aunque como tal los ácidos grasos no proporcionan glucosa para el cerebro, la acetil-CoA estimula la piruvato carboxilasa y con ello, la gluconeogénesis.
piruvato carboxilasa
Alanina -* Piruvato— r- OAA -* PEP -» glucosa CO2
7.7.3 Hiperlipoproteinemias
Hiperlipidemia es el aumento de una o más fracciones lipídicas con un incremento simultáneo de lipoproteínas. Las hiperlipi-demias primarias son enfermedades "sui ge-neris" o sea, trastornos del metabolismo lipídico que no dependen de otro enfermedad aparente básica. A diferencia de ellas, las secundarias son consecuencia de un padecimiento existente, y como síntoma de otra enfermedad subyacente cambian durante el transcurso de ella. La clasificación de las hiperlipidemias, que se manifiestan sólo en presencia de ciertos factores provocadores (obesidad, por ejemplo), en primarias o secundarias, en un poco al azar, para ciertos autores son primarias y para otros secundarias.
Se habla de hiperlipidemia, cuando una o más fracciones se encuetran aumentadas. Puesto que en el diagnóstico usual se siguen cuantificando sólo los componentes químicos, no hay necesidad de substituir al termino hiperlipidemia por hiperlipoproteinemia. (*hiperlipidemias: G. Hartmann 6 H. Stahe-lin. Ed. Manual Moderno 1987.
Clasificación fenotipica de las hiperlipidemias
La sugirieron por primera vez Fredrick-son y Loes, y fue modificada un poco por un grupo de trabajo de la OMS y sirve hasta la fecha como medio de referencia. Aun así
hay que tener en cuenta que se basa en los patrones electroforéticos de las lipoproteínas del suero en ayuno, y describe su fenotipo. La ventaja de la clasificación de Fredrickson consiste en que se logra mediante electrofo-resis de LP y también, en grandes rasgos, con otros métodos de separación (ultracen-trífuga, métodos de precipitación), e incluso a través de la sencilla determinación del co-lesterol y los triglicéridos.
Aún se justifican los términos hipertrigli-ceridemia, hipercolesterolemia, si se trata de describir un hallazgo sin fijarse en un tipo, o antes de clasificarlo. Se habla de "hiperlipidemia" o sencillamente "lipemia", si se desea hacer constar un enturbiamiento de suero por lípidos y por último "hiperlipoproteinemia" que es la forma más correcta debido a que tanto el colesterol como los triglicéridos circulan en el plasma en diferentes lipoproteínas en combinación con fosfolípidos y con proteínas.
Hiperlipoproteinemia tipo I (hipertrigli-ceridemia inducible por grasas, hiperlipemia exógena).
La deficiencia de la enzima lipasa lipo-proteica (LPL) resulta en una menor capacidad para depurar QM. En casi todos los casos hasta ahora observados se ha hallado también una activación restringida de esta lipasa por la heparina.
La herencia tiene lugar por vía recesiva autosómica. La enfermedad se manifiesta en una edad precoz de la vida. En los heteroci-gotos se observa una ligera disminución de la actividad de la LPL.
En el cuadro clínico destacan en primer lugar los cólicos abdominales, que a menudo se diagnostican erróneamente de abdomen agudo. El acumulo de grasa en el sistema retículo endotelial provoca hepa-toesplenomegalia. Es posible que los cólicos abdominales sean consecuencia de la tensión de la cápsula hepática, otra causa posible serían las microembolias de grasa como

consecuencia de la considerable lipemia. También podría contribuir a los dolores abdominales la pancreatitis secundaria a menudo exis tente . Además , después de comidas ricas en grasas se observan leucocitosis (neutrofilia) y fiebre ligera.
Después de una alimentación rica en grasas aparecen xantomas cutáneos (predominantemente en glúteos) como consecuencia de la hiperlipemia. La aterosclerosis no es importante. Se observa con frecuencia lipemia retiniana. En la biopsia se encuentran células espumosas en órganos ricos en tejido reticu-loendotelial (p. ej., en la médula ósea, bazo e hígado). Los enfermos no muestran ningún trastorno de la tolerancia a la glucosa.
Diagnóstico. El diagnóstico de deficiencia familiar de LPL lo sugiere el hallazgo de plasma lipémico en un individuo joven que ha estado en ayunas al menos por 12 horas. Cuando se recolecta este plasma en presencia de EDTA tiene una apariencia característica después de refrigerado toda la noche a 42C. En el extremo superior del tubo aparece una capa de crema blanca. Debajo de esta capa el plasma es claro. El diagnóstico de deficiencia familiar de LPL se establece por el hallazgo de un patrón tipo I en la electro-foresis de lipoproteínas. Se confirma por la demostración de que no aumentan los niveles de LPL después de la inyección de heparina.
Tratamiento, Cuando el paciente recibe una dieta sin grasas, todos los signos y síntomas son reversibles. Debe hacerse cualquier intento para mantener el nivel plasmático de TG en ayunas por abajo de 1000 mg/100 mi para evitar la pancreatitis. Se ha demostrado empíricamente que en los pacientes adultos afectados la ingestión de grasas debe ser menor de 20 g al día para prevenir la hiperlipemia sintomática. Se han empleado TG de cadena mediana para ayudar a lograr una ingestión calórica normal, ya que éstos normalmente no son incorporados a los QM. La dieta debería complementarse con vitaminas liposolubles.
Hiperlipoproteinemia Tipo l ia (sinónimos: Hiperbetalipoproteinemia, hipercoles-terolemia).
En esta hiperlipidemia hay un aumento aislado de LDL y, con ello, del colesterol; los triglicéridos están normales. Solo una pequeña parte de las hiperlipidemias Tipo lia pertenece a la forma familiar.
La forma familiar homocigota depende de la ausencia o un defecto de receptores para las LDL. La heterocigota, mucho más frecuente, tiene la mitad del número de receptores funcionalmente intactos. Se desconoce la génesis de los casos esporádicos que son más frecuentes, y de los que se presentan en una hiperlidemia familiar combinada.
En 20 a 50% de todas las hiperlipidemias se encuentra un tipo Ha. La forma heterocigota se presenta con una frecuencia de 1:500 a 1:1,000. La homocigota familiar es muy rara en Europa central (con una frecuencia de 1:5 x 106 a 1:106). Existen grandes variaciones en todo el mundo, quizá en parte debido a consanguidad. Hay frecuencias anormales en Líbano y Sudáfrica, donde se observa en Transval un caso de homocigotos por 30,000 habitantes. El Tipo Ha se manifiesta desde la niñez.
El diagnóstico del fenotipo Ha se basa siempre en los valores elevados del colesterol. En la electroforesis se encuentra como característica un gradiente beta pequeño y una banda prebeta poco desarrollada. El aumento del colesterol afecta con exclusividad a la fracción LDL. Las LDL están normales o ligeramente bajas. La presentación clínica orienta el diagnóstico. Se puede confirmar el tipo familiar estudiando a todo el linaje por cultivo de fibroblastos en donde se examina la ausencia de receptores.
Son característicos el arco lipoide y xante-lasmas a menudo grotescos en pacientes ancianos, así como xantomas patognomónicos en el tendón de Aquiles, el dorso de la mano y,con menos frecuencia, en las rodillas y codos. Son duros y tuberosos, circundados por

el tendón y móviles bajo la piel. Los pacientes con lia por lo regular, tienen peso normal o en ocasiones incluso bajo.
De todas las hiperlipoproteinemias el tipo lia tiene el mayor riesgo de aterosclerosis. Entre los homocigotos 60 a 80% desarrollan una coronariopatía aterosclerótica que se manifiesta antes de los 20 años; pocos llegan a los 40.
Hiperlipoproteinemia Tipo Ilb (sinónimo: hiperlipidemia combinada).
La elevación moderada de los niveles de colesterol y TG en la mayoría de los pacientes con aumento simultáneo de LDL y VLDL y suero poco turbio es lo que caracteriza a este trastorno hereditario.
Es poco clara la patogenia de la hiperlipidemia Ilb. Se supone se debe a un aumento de la producción de las VLDL y de la apo-proteína B. Una parte de los pacientes pertenecen a familias con hiperl ipidemias combinadas, la mayoría son casos debidos a alimentación hipercalórica. De todas las hiperlipidemias primarias representa entre 10-30%. Es rara en niños.
Son característicos el aumento moderado del colesterol y TG. Los valores promedio de colesterol van de 300 a 340 mg/dl y de TG de 217 a 345 mg/dl. El suero puede estar claro o poco turbio. La electroforesis debe mostrar una banda prebeta bien marcada. En la mitad de los pacientes Tipo Ilb está alterada la tolerancia a la glucosa, y en un tercio valores elevados de ácido úrico.
En más de la mitad de los casos existe arco lipoide, pero sólo en menores de 50 años tiene importancia diagnóstica. Los xantomas y xantelasmas son raros. En menos del 5 % de los pacientes hay xantomas tuberosos. Un signo frecuente es la obesidad moderada.
El riesgo de aterosclerosis, ante todo de una enfermedad coronaria, es muy elevado. En pacientes tipo Ilb la frecuencia de coronariopatía manifiesta o de una enfermedad oclusiva arterial periférica llega a 25 % en la
tercera década, 59% en la cuarta y 62% en la quinta.
Hiperlipoproteinemia Tipo III. Disbeta-lipoproteinemia familiar.
Este es un trastorno hereditario en el que están elevadas las concentraciones plasmáticas de colesterol y TG debido a la acumulación en el plasma de moléculas similares a los residuos derivados del catabolismo parcial de la VLDL y de los QM por la acción de la LPL. En los sujetos normales, las moléculas residuales de los QM son captados rápidamente por el hígado, y por lo tanto son poco detectables en el plasma. Una fracción de los residuos de VLDL también es captada por el hígado, y el resto se convierte en LDL. En pacientes con disbetalipoproteine-mia familiar se bloquea la captación hepática de remanentes de QM y VLDL, y se acumulan estas lipoproteínas en grandes cantidades en el plasma y en los tejidos, produciendo xantomas y aterosclerosis.
La mutación responsable de esta enfermedad abarca el gen que codifica la Apo E, una proteína que se encuentra normalmente en los remanentes de QM y VLDL. Esta proteína se une a los receptores hepáticos y por lo tanto media la captación de los remanentes por este órgano.
Manifestaciones clínicas. Característicamente los individuos afectados no manifiestan hiperlipidemia ni otra manifestación clínica de la enfermedad hasta después de los 20 años de edad. Una característica única del trastorno es la existencia de dos tipos de xantomas cutáneos. Estos son el xantoma estriado palmar que aparece como una mancha anaranjada o amarilla en los pliegues palmares o digitales, y el xantoma tuberoso ó tuberoeruptivo, que son xantomas cutáneos bulbosos cuyo tamaño puede variar. Estos xantomas tuberosos se localizan en forma característica sobre codos y rodillas. También se presentan xantelasmas, pero no son característicos de este trastorno.

También es característica la aterosclerosis grave y fulminante que afecta arterias coronarias, carótidas internas y aorta abdominal. Las secuelas clínicas incluyen infartos al miocardio prematuros, claudicación intermitente y gangrena de las extremidades inferiores. Los pacientes que presentan estas manifestaciones clínicas frecuentemente tienen hi-potiroidismo, obesidad, o diabetes mellitus.
Diagnóstico. El hallazgo de xantomas palmares o tuberosos en un paciente con aumento de los niveles plasmáticos de colesterol y TG sugiere el diagnóstico. En aproximadamente 80% de pacientes sintomáticos aparecen estos xantomas. También sugiere el diagnóstico una elevación moderada de las concentraciones plasmáticas de colesterol y TG, de tal forma que las concentraciones absolutas de estas dos sustancias son aproximadamente iguales (aproximadamente 300 mg/ml). Sin embargo, esto no siempre es regla exacta y segura, ya que cuando la enfermedad se exacerba en forma grave tienden a elevarse predominante los TG.
El diagnóstico se confirma por el hallazgo de la llamada banda beta ancha en la electro-foresis de lipoproteínas (patrón tipo 3). Esto se produce por la existencia de residuos moleculares que migran entre las lipoproteínas P y las pre-p y producen una mancha notoria en esta región del electroforograma.
Tratamiento. Debe buscarse insistentemente hipotiroidismo oculto y si hay debe indicarse L-tiroxina. Además debe intentarse controlar la obesidad y la diabetes mellitus por medio de una dieta y tratamiento con insulina. Si estas medidas no tienen éxito, los pacientes deben tratarse con clofibrato. Los pacientes afectados casi siempre muestran reducción espectacular y constante de los niveles de lípidos plasmáticos cuando se tratan con este medicamento.
Hiperlipoproteinemia Tipo IV. (Sinónimos: hiperlipidemia endógena, hipertrigli-ceridemia idiopática, hiperprebeta lipopro-teinemia).
Es un trastorno autosómico dominante común en el que aumenta la concentración plasmática de VLDL, lo que produce hiper-trigliceridemia. No se conoce el defecto bioquímico; en la mayoría de los pacientes depende de una producción elevada de VLDL y rara vez de una disminución en su catabolismo. El que se transmita como rasgo autosómico dominante, implica mutación en un solo gen. Sin embargo, no se ha identificado la naturaleza del gen mutante ni el mecanismo por el cual se produce la hipertrigliceri-demia. Es probable que este trastorno sea genéticamente heterogéneo; esto es, que el fenotipo de hipertrigliceridemia puede resultar de mutaciones diferentes.
En algunos pacientes afectados aumenta la velocidad de producción de VLDL, especialmente cuando ingieren dietas altas en carbohidratos. Sin embargo, muchos de estos pacientes tienen obesidad y diabetes mellitus. Cuando la velocidad de producción de VLDL está aumentada debido a obesidad o diabetes mellitus, son incapaces de aumentar el catabolismo en forma proporcional y se produce la hipertrigliceridemia. No obstante, la actividad de la lipasa lipoproteica aumenta normalmente después de la administración de heparina, y no se han identificado anormalidades en la estructura de las lipoproteínas.
En las formas masivas se presentan a veces xantomas eruptivos que desaparecen después, según el contenido de TG. Los xan-telasmas son extraordinarios, con frecuencia se nota un arco lipoide prematuro. Las hiperli-pidemias masivas tipo IV pueden provocar hepatomegalia por esteatosis y son causa de pancreatitis aguda. Con el tipo IV se combinan diabetes, alteración de la tolerancia a la glucosa, obesidad y gota. La gota primaria muestra una gran frecuencia de hiperlipide-mias tipo IV (hasta 80%). A diferencia de conceptos antiguos, sólo una pequeña parte de esta hiperlipidemia es sensible a los carbohidratos. Al parecer la más frecuente es

por alcohol, sin abuso. La abstinencia estricta por una semana permite comprobarlo.
Diagnóstico. El hallazgo de una elevación moderada en el nivel plasmático de triglicé-ridos, junto con un nivel normal de colesterol, aumenta la posibilidad de hipertrigliceride-mia familiar. En la mayoría de los pacientes cuando se examina el plasma es claro o levemente opaco. La electroforesis del plasma revela aumento de la fracción prebeta.
El riesgo de morbilidad coronaria y enfermedad oclusiva arterial periférica es menor que en hiperlipidemias tipo II, pero definitivamente más alto que en normolipidemias; no se cuenta con datos exactos, ya que en muy pocos estudios se clasifican las hiperlipidemias. Por otro lado, entre los pacientes con enfermedad coronaria hay con relativa frecuencia hiperlipidemias tipo IV.
Hiperlipoproteinemia Tipo V (Sinónimos: hipertrigliceridemia mixta, hiperlipoproteinemia endógena y exógena, síndrome de quilomicronemia).
En esta hiperlipidemia está alterada la clarificación de las grasas. La herencia de esta enfermedad es confusa; se cree que se debe a un desorden multifactorial de varios genes; también se cree que este padecimiento lo causa la deficiencia familiar de apoproteí-na C-II, cofactor esencial de la lipoprotein lipasa.
La turbiedad lechosa del suero en ayunas indica elevación del contenido de lipoproteí-nas especialmente ricas en grasas como son los QM y los VLDL; el aumento de estas fracciones hace que se eleven los otros elementos que constituyen estas partículas como el colesterol y triglicéridos.
Al parecer la principal causa en una pequeña parte de los pacientes es una deficiencia hereditaria de lipoproteinlipasa. En la mayor parte de los casos cabe suponer una hiperproducción genética de VLDL combinada con un trastorno adquirido del catabolismo de quilomicrones y VLDL. Este
último se acompaña a menudo de consumo de alcohol, hiperinsulinemia, tolerancia a la glucosa alterada, obesidad y, en casos excepcionales, dislipidemia.
La frecuencia es de 1 a 5% de todas las hiperlipidemias primarias. Son más comunes en familias con hiperlipidemia mixta y en las de tipo IV. Solo se presenta en personas de más de 20 años. Más frecuente en pacientes con xantomas eruptivos y pancreatitis.
Los pacientes con esta hiperlipidemia presentan xantomas eruptivos, hepatomegalia, dolores abdominales y pancreatitis. En 20% de los casos hay lipemia retiniana. El suero muy lipémico falsea una serie de pruebas de laboratorio. Existen trabajos que describen una frecuencia aproximada de 40% de enfermedades coronarias y oclusivas arteriales periféricas.
Hipolipoproteinemias
Se habla de hipolipidemia, cuando el colesterol total es menor de 180 mg/dl en una persona mayor de 40 años, 160 mg/dl en los de 20 años y en niños de 140 mg/dl. Respecto a los triglicéridos los límites deben ser muy bajos, ya que tan solo dejar de comer por poco tiempo puede causar valores de unos 50 mg/dl; sólo cuando son más bajos se puede hablar de hipotrigliceridemia.
Las hipolipidemias pueden deberse a trastornos congénitos del metabolismo de las li-poproteínas; se habla entonces de formas primarias, que son poco comunes. Con más frecuencia, las hipopolipidemias son secundarias a muy diversas enfermedades.
Hipoalfalipoproteinemia primaria
Se han descrito varios trastornos primarios del metabolismo de C-HDL y Apo A-I que se caracterizan por disminución de la concentración plasmática de las HDL y sus componentes. Dentro de estos padecimientos están la hipoalfalipoproteinemia familiar,

la enfermedad de Tangier, la deficiencia familiar de la enzima lecitina colesterol acil transferasa la enfermedad de "ojos de pescado", la deficiencia familiar de HDL con xan-tomas planos, la deficiencia familiar de Apo A-I y C-III y el síndrome de opacificación corneal asociado a ausencia de Apo A-I. Además, existen enfermedades caracterizadas por la modificación de la secuencia de aminoácidos de la Apo A-I, como la llamada Apo A-I Milán, que pueden condicionar un cambio de la tasa de depuración de esta molécula reduciendo su vida media y, consecuentemente su concentración en el plasma. De estos padecimientos, la hipoalfalipopro-teinemia familiar es la más frecuente y se caracteriza por:
1. Niveles bajos de C-HDL y Apo A-I; 2. Ausencia de enfermedades u otros factores que pudieran explicar la disminución de C-HDL; 3. Existencia de un defecto similar en parientes de primer grado; 4. Los individuos afectados tienen un riesgo elevado de experimentar eventos cardíacos prematuros. Los estudios de longevidad demuestran que la esperanza de vida de los sujetos afectados se acorta y el gene aberrante se transmite en forma autosómica dominante. El defecto metabólico se desconoce, existiendo la posibilidad de disminución de la síntesis o aumento de la tasa de depuración de las HDL. Es factible que el uso de otros marcadores del metabolismo de las HDL más específicos como la Apo A-I permita aclarar la naturaleza bioquímica del padecimiento. Es posible que se trate de un grupo heterogéneo de trastornos y que exista más de un defecto metabólico con expresión clínica idéntica. No se han podido identificar alteraciones de la secuencia de aminoácidos o del metabolismo de la Apo A-I. Los estudios de DNA recombinantte, utilizando la técnica del "punteado" de Southern han permitido demostrar el polimorfismo de los genes de la Apo A-I. J. Ordovas en un estudio reciente de los "fragmentos de restricción" del DNA
(RFLPs) , encont ró que el haplot ipo Pstl+/Sac I-se asocia fuertemente a la hi-poalfalipoproteinemia familiar.
Con excepción de la hipoalfalipoproteine-mia familiar que es relativamente frecuente y cuyo mecanismo de transmisión es autosó-mico dominante, los otros tipos son muy raros y se transmiten en forma recesiva. La enfermedad de Tangier se caracteriza por la acumulación de colesteril esteres en el sistema retículo endotelial y otros tejidos y la ausencia o deficiencia severa de HDL en el plasma. El estado heterocigótico generalmente es asintomático pero los niveles de HDL y Apo A-II se reducen un 50%. En el homocigoto la concentración plasmática de Apo A-I es menor al 1 % y la de Apo A-II es de 5 a 7 % de los valores normales correspondientes. El defecto genético, es el catabolismo acelerado de la Apo A-I. En esta entidad existe una mayor tendencia a la ate-roesclerosis, aunque no en forma acelerada ya que en series grandes de pacientes con enfermedad de Tangier no se encontró evidencia de enfermedad vascular antes de los 35 años. Es posible que esto se deba a los niveles bajos de LDL, que se asocian a tal padecimiento. Otra de las manifestaciones clásicas de esta enfermedad resultan del depósito de esteres de colesterol en diveros tej idos, incluyendo principalmente a las tonsilas que adquieren un color anaranjado, el bazo (esplenomegalia), nervios periféricos (neuropatía sensorial y motora) y córnea.
La deficiencia familiar de LCAT es un padecimiento sumamente raro, en el que las manifestaciones clínicas y el perfil lipopro-teico resulta de la incapacidad para esterifi-car el colesterol. Las HDL en este trastorno tienen una morfología discoide y están enriquecidas en Apo E. Clínicamente los pacientes presentan opacidades corneales, anemia con eritrocitos en "blanco de tiro", proteinuria y progresión a insuficiencia renal y ateroesclerosis prematura; las placas

de ateroma contiene menor proporción de colesterol que otros procesos ateromatosos.
Hipoalfalipoproteinemía secundaria
Existen múltiples factores "secundarios" capaces de reducir los niveles de HDL en el plasma incluyendo obesidad, tabaquismo, dieta hipercalórica rica en carbohidratos, diabetes mellitus, progestágenos, probucol y beta-bloqueadores. La hipertrigliceridemia generalmente se asocia a valores bajos de C-HDL, que se normalizan al tratar la hipertrig l icer idemia . Es to puede deberse a l intercambio de colesterol por triglicéridos entre las lipoproteínas ricas en TG y las HDL. la reducción de C-HDL observada en la hipertrigliceridemia es un posible nexo indirecto entre los TG y la aterosclerosis. Como se ha mencionado, es indispensable identificar y tratar la hipoalfalipoproteine-mia primaria y secundaria por la fuerte correlación inversa exis ten te entre los componentes de dichas lipoproteínas y la aterosclerosis.
Hiperlipoproteinemias secundarias
Las hiperlipidemias secundarias se presentan como consecuencia de ciertas enfermedades que no afectan en primer lugar el metabolismo de las grasas, dependen de la influencia de factores tóxicos y medicamentos. Es muy importante definir estas formas de hiperlipidemia, ya que desaparecen cuando mejora la enfermedad primaria o al retirar el tóxico.
1. Diabetes mellitus: La diabetes se acompaña de una excesiva movilización de ácidos grasos de los depósitos, lo cual condiciona un aumento de los lípidos totales en particular triglicéridos. Durante los episodios de aci dosis y coma diabético se observa una reducción en los niveles de colesterol y LDL. Al normalizarse el episodio estos vuelven a sus niveles normales, pudiendo
permanecer ligeramente elevados. Cuando al paciente no está bien controlado el patrón electroforético muestra un cuadro compatible con el tipo IV (60-80%) pero también son frecuentes el Ha (10 a 20%) y Ilb (10%) y el tipo V en menor proporción.
2. Síndrome nefrótico: La hiperlipidemia es parte integral de este síndrome. Los valores del colesterol son, por lo regular, entre 300 y 600 mg/dl y los triglicéridos de 300 a 1,000 mg/dl. Se han observado todos los fenotipos, pero la Ilb con mayor frecuencia.
Las alteraciones de las lipoproteínas dependen, al parecer, de la pérdida de proteína por riñon lo que condiciona hipoalbumine-mia y baja presión oncótica del suero y una "excesiva" producción hepática de proteínas para tratar de compensar. Es posible que se estimule también por compensación, la síntesis hepática de TG y lipoproteínas en espe-c i a l V L D L , p e r o a l m i s m o t i e m p o disminuye la actividad de la LPL y la lipasa hepática. Son típicos los valores muy bajos de HDL, ya que estas LP se pierden, en parte, por la proteinuria y lipiduria. En la orina también hay fragmentos de apo-proteínas.
3. Alcoholismo: La estimulación de la síntesis de TG, VLDL y HDL en el hígado, es un efecto fisiológico del alcohol. Cabe resaltar dos particularidades: (1) El problema no es por abuso; si hay hiperlipidemia, el consumo usual de alcohol puede provocar hiperlipidemia en individuos sensibles por cantidades regulares ó grandes de alcohol. (2) Al parecer, la hiperlipidemia depende del caso (paciente) que se trate. En el alcoholismo crónico (100 g de etanol al día o más) solo 10 a 25 % de los pacientes desarrollan hiperlipidemias que corresponden al tipo IV y con menor frecuencias Tipos V ó Ilb.
El diagnóstico de hiperlipidemia etílica se confirma con la prueba de abstinencia, en la cual deben de normalizarse los lípidos en el transcurso de una semana.

7.8 METABOLISMO DE LÍPIDOS
El metabolismo de lípidos no es tan homogéneo como el de carbohidratos en virtud de la gran diversidad de tales sustancias. Sin embargo, existe una interrelación importante a nivel de acetil-CoA como vía de entrada común al ciclo de Krebs.
A pesar de la ventaja numérica en rendimiento calórico de las grasas (9 Kcal/g) respecto a carbohidratos (5 Kcal/g), el organismo utiliza preferentemente carbohidratos como combustible primario. El uso excesivo de grasas con fines energéticos acarrea graves problemas como la cetosis por ejemplo. Los fosfolípidos y esteróles realizan funciones distintas a la producción de energía.
7.8.1 Oxidación de ácidos grasos
Antecedentes históricos de la oxidación de ácidos grasos.
Los conocimientos actuales sobre los detalles de la oxidación de los ácidos grasos se empezaron a desarrollar a principios de 1950. Sin embargo, en 1905, F. Knoop reportó una serie de experimentos que indicaron que los ácidos grasos eran oxidados por separación simultánea de dos átomos de carbono. Alimentó conejos con ácidos grasos en los que el grupo metilo fue sustituido por un grupo fenilo. Si el ácido graso modificado tenía número par de átomos de carbono (por ejemplo, QH5-CH2 (CH2)2-COOH), el metabolito primario era el ácido fenil acético (C6H5-CH2-COOH), excretado por la orina en forma de su conjugado con glicina, el ácido fenilacetúrico (C6H5-CH2-CO-NHCH2-COOH). Cuando alimentó conejos con el fenil derivado de un ácido graso con número impar de átomos de carbono (por ejemplo, QHs-CHHCHab-COOH, encontró ácido benzoico (C<3H5- COOH), excretado en la orina como su conjugado con la glicina, el ácido hipúrico (C6H5-CO-NH-
CH2-COOH). Knoop postuló que los ácidos grasos eran oxidados en el carbono p (de aquí el nombre de p-oxidación) y degradados a ácido acético y un ácido graso con dos átomos de carbono menos:
R-CH2-CH2-COOH • R-CO-CHj-COOH R-COOH+CH3-COOH
El siguiente paso experimental importante fue la demostración en 1944 por Luis Le-loir de que los ácidos grasos podían ser oxidados en un sistema libre de células. A continuación Albert Lehninger demostró que el proceso ocurría en la mitocondria de células hepáticas y, aparentemente, involucraba un "acetato activo". Los experimentos de Fritz Lipmann probaron que la coenzima A estaba involucrada en la formación del "acetato activo".
Acetato + ATP + CoA • "Acetato Activo"
Posteriormente, en 1951, Feodor Lynen trabajando en Munich con levaduras, demostró que el "acetato activo" era la acetil CoA. En base a esto varios laboratorios concibieron la idea de que la CoA podía jugar un papel en la activación de los ácidos grasos para la p-oxidación y, en 1954, fue desarrollado el modelo básico de la p-oxidación tal como se conoce ahora.
Las enzimas degradativas conocidas como "oxidasa de ácidos grasos" se encuentran en la matriz mitocondrial adyacente a la cadena respiratoria. En el proceso se forman NADH y FADH2 que se acoplan a la fosforilación de ADP a ATP. La acetil-CoA que se forma al final, puede pasar al ciclo de Krebs donde termina de oxidarse y producir más energía. Así, la oxidación es eminentemente intramitocondrial.
El rendimiento de ATP producido en la oxidación del ácido hexanoico (caproico) es de 44 moléculas de ATP (la glucosa produce

38 moléculas); es decir, se producen más moles de ATP por mol de C 0 2 que en la oxidación de carbohidratos.
7.8.1.1 p-oxidación de ácidos grasos saturados con número par de átomos de carbono
En la figura 7.23 se muestra un resumen de la P-oxidación de un ácido graso saturado de cadena par de átomos de carbono. En la primera reacción, la acil-CoA es deshidrogenada para producir el a-p (ó 2-3) trans-enoil-CoA y FADH2. Esta enoil-CoA es posteriormente hidratada estereoespecíficamente para producir 3-L-hidroxiacil-CoA. El grupo hi-droxilo es oxidado por NAD+ , y una deshi-drogenasa para producir p-cetoacil-CoA y NADH. El paso final en la secuencia involucra una tiolisis para formar acetil-CoA y una acil CoA dos átomos de carbono más corta que el sustrato inicial. Esta acil-CoA puede llevar a cabo otra secuencia de oxidación para producir FADH2 NADH, acil-CoA. Los pasos enzimáticos son repetidos hasta que en la última secuencia de reacciones, la buti-ril CoA (CH3-CH2-CH2-CO-C0A) es degradada a dos moléculas de acetil-CoA.
Balance energético de la p-oxidación
Las reacciones para la oxidación completa de palmitil-CoA se presenta en la fig. 7.23. Cada uno de los productos es posteriormente oxidada directamente por la cadena respiratoria o por el ciclo de Krebs y luego la cadena respiratoria. Cuando estas reacciones totales son sumadas, al ser oxidada una molécula de palmitil-CoA se producen 16C02+131ATP+146H20 y CoA. Sin embargo, la formación de palmitil-CoA requiere hidrólisis de dos enlaces de alta energía, CoA y consumo de una molécula de H2Ó durante la hidrólisis del PPi producido por la reacción de la tiocinasa. Así, el rendi
miento neto son 129 moléculas de ATP y 145 de agua.
El ATP producido durante la oxidación del ácido palmítico puede ser comparado con el que se produce al oxidar la glucosa. Ambos sustratos son las principales fuentes de energía en nuestro organismo. Puesto que el palmitato tiene 16 carbonos y la glucosa solo 6, la comparación deberá hacerse entre una molécula de palmitato y 2 2/3 de glucosa. Como se vio en el capítulo de metabolismo de carbohidratos, se producen 38 ATP a partir de la oxidación completa de la glucosa. La producción a partir de 2 2/3 moléculas de glucosa es por lo tanto de 101 ATP. De esta manera, la oxidación del palmitato produce 28 ATP adicionales. Por lo tanto, el palmitato es una molécula más eficiente que la glucosa en el almacenamiento de energía. Por otro lado, los lípidos están menos hidratados que los carbohidratos por lo cual ocupan menos espacio. Estos dos factores son probablemente la explicación de que sea la grasa la principal forma de almacenamiento de energía. Desde el punto de vista químico la diferencia en la energía producida durante la oxidación de la glucosa y la del palmitato, es que este último está casi completamente en estado reducido, mientras que la glucosa está parcialmente oxidada con 6 oxígenos en su molécula.
7.8.1.2 p-oxidación de ácidos grasos saturados con número impar de átomos de carbono
La oxidación de estos ácidos grasos se lleva a cabo también por p-oxidación, y al completarse el último ciclo, la reacción de tiolisis no produce dos moléculas de acetil-CoA sino una de acetil-CoA y otra de pro-pionil-CoA:
o o II II
CH3-C-SC0A+ CH3-CH2-C-SCoA (Acetil-CoA) (Propionil-CoA)
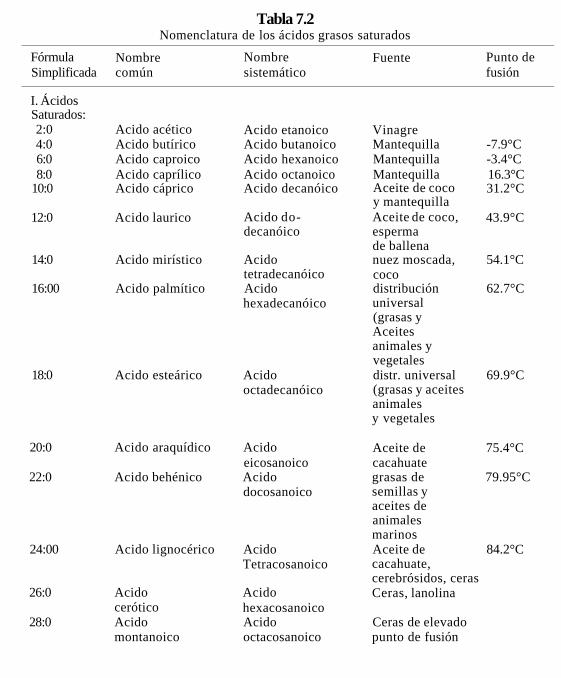
Tabla 7.2 Nomenclatura de los ácidos grasos saturados
Fórmula Simplificada
I. Ácidos Saturados: 2:0 4:0 6:0 8:0
10:0
12:0
14:0
16:00
18:0
20:0
22:0
24:00
26:0
28:0
Nombre común
Acido acético Acido butírico Acido caproico Acido caprílico Acido cáprico
Acido laurico
Acido mirístico
Acido palmítico
Acido esteárico
Acido araquídico
Acido behénico
Acido lignocérico
Acido cerótico Acido montanoico
Nombre sistemático
Acido etanoico Acido butanoico Acido hexanoico Acido octanoico Acido decanóico
Acido do-decanóico
Acido tetradecanóico Acido hexadecanóico
Acido octadecanóico
Acido eicosanoico Acido docosanoico
Acido Tetracosanoico
Acido hexacosanoico Acido octacosanoico
Fuente
Vinagre Mantequilla Mantequilla Mantequilla Aceite de coco y mantequilla Aceite de coco, esperma de ballena nuez moscada, coco distribución universal (grasas y Aceites animales y vegetales distr. universal (grasas y aceites animales y vegetales
Aceite de cacahuate grasas de semillas y aceites de animales marinos Aceite de cacahuate, cerebrósidos, ceras Ceras, lanolina
Ceras de elevado punto de fusión
Punto de fusión
-7.9°C -3.4°C 16.3°C 31.2°C
43.9°C
54.1°C
62.7°C
69.9°C
75.4°C
79.95°C
84.2°C

Figura 7.23. Oxidación del ácido palmítico.
La acetil-CoA es sustrato para la citrato 1) La propionil-CoA se carboxila trans-sintetasa, por lo que puede entrar en el ciclo formándose en metil-malonil-CoA, proceso de Krebs, no así la propionil-CoA que pre- catalizado por la enzima propionil carboxi-senta un metabolismo diferente que puede lasa: describirse de la siguiente manera:

2) Mediante la actividad de la enzima me-til-malonil mutasa, la metil-malonil-CoA se isomeriza a succinil-CoA:
3) La succinil-CoA, por acción de la enzima succinil tiocinasa puede perder la coenzima-A (CoA-SH) y transformarse en ácido succínico libre:
El ácido succínico libre puede incorporarse al ciclo de Krebs. El metabolismo de la propionil-CoA constituye una ruta metabó-lica secundaria.
7.8.1.3 Oxidación de ácidos grasos insaturados
Los ácidos grasos insaturados utilizan también como ruta degradativa la p-oxidación, pero requieren la participación, de dos enzimas adicionales, una isomerasa y una epimerasa.
Supongamos que un ácido graso natural A3:4 monoinsaturado, que como sabemos posee una configuración cis, penetra en la mitocondria para ser oxidado (fig. 7.24).
Este compuesto no es sustrato para la primera oxidación que cataliza la enzima acil-CoA deshidrogenasa, ni tampoco puede sufrir la hidratación, puesto que la enoil trans hidratasa actúa exclusivamente sobre el doble enlace A2:3. Por lo tanto, la oxidación no se llevaría a cabo de no existir la enzima adicional, descubierta por Stoffel, llamada Cis A3:4, trans A2:3 enoil-CoA isomerasa, o sencillamente isomerasa. Esta enzima cataliza el desplazamiento reversible del doble enlace de cis A3:4 a trans A2:3 (fig. 7.25).
Una vez realizada la isomerización, el derivado trans A2:3 enoil- CoA puede proseguir las etapas de la P-oxidación, puesto que se ha convertido en sustrato normal de la enoil hidratasa (ver figura 7.23).
En el caso de los ácidos grasos poliinsatura-dos suele ser necesaria una segunda enzima adicional. Tomemos como ejemplo para la descripción del proceso, la oxidación del ácido linoleico, que posee 18 átomos de carbono y dos dobles enlaces cis A9-12 (fig. 7.26).
El linoleico puede experimentar 3 ciclos normales de oxidación en los que pierde 6 átomos de carbono, obteniéndose el derivado cis A3' 6-enoil CoA (fig. 7.27).
Llegando a este punto la isomerasa descrita anteriormente transforma el isómero cis A 3 4 en el isómero trans A2:3 y continúa la oxidación; se desprenden dos porciones di-
carbonadas (acetil-CoA) más hasta obtener el derivado Cis A3:4 enoil-CoA.
Este derivado puede ser hidratado por la enoil hidra tasa, pero puesto que el doble enlace tiene configuración Cis A2:3, se obtiene el isómero D-3 hidroxi-acil-CoA, que no es sustrato para la enzima de la siguiente etapa L-3 hidroxiacil-CoA deshidrogenasa, con lo que la oxidación quedaría interrumpida a este nivel de no existir la segunda enzima adicional D-3 hidroxiacil-CoA epimerasa, que transforma el isómero D en isómero L.
El isómero L. puede proseguir la secuencia oxida ti va en forma normal (ver figura 7.23), hasta alcanzar el siguiente doble enlace sobre el que actuará la isomerasa o la epimerasa según la posición que ocupe; si se sitúa en posición cis A3:4, respecto al carbono carbo-xílico, actuará la isomerasa; y si su localiza-ción es cis A 2 3 actuará la epimerasa. De este
Figura 7.27. Oxidación de Cis-A ' enoil-CoA

modo, mediante la actividad combinada de ambas enzimas, los ácidos grasos insatura-dos son oxidados completamente.
7.8.1.4 a-oxidación y enfermedad de Refsum
Aunque la p-oxidación de alguno ácidos grasos. En una dieta normal, se ingieren pequeñas cantidades de fitol, un componente de la clorofila. Como se muestra en la figura 7.28, este alcohol de cadena larga es oxidado a ácido titánico, el cual es el componente dietético más importante en la grasa de rumiantes y en derivados lácteos. La ingesta diaria estimada de ácido fitánico es de 50-100 mg. Debido a la presencia de un grupo metilo en el carbono 3, el ácido fitánico no es sustrato para la acil-CoA deshidrogenasa (primera enzima de la p-oxidación). Este problema es resuelto por otra enzima mito-condrial que hidroxila el carbono a del ácido fitánico. El intermediario hidroxilado es descarboxilado para producir ácido pristáni-co y CO2. El ácido pristánico no está sustituido en el carbono 3 y puede ser oxidado por la acil-CoA deshidrogenasa y el resto de las enzimas normales de la p-oxidación para producir propionil-CoA y acil-CoA. La cual puede ser degradada por medio de cuatro ciclos de p-oxidación, produciendo acetil-CoA y propionil-CoA. La secuencia final de reacciones produce acetil CoA e isobutiril-CoA, la cual puede ser convertida en succinil-CoA y metabolizada en el ciclo de Krebs.
Nuestro conocimiento actual de cómo el humano metaboliza el fitol y el ácido fitánico es en gran parte resultado de los estudios de Daniel Steinberg y colaboradores acerca de la enfermedad de Refsum, un raro síndrome hereditario caracterizado por trastornos neuro-lógicos (temblores, marcha inestable, disminución del campo visual y visión nocturna deficiente). Probablemente los síntomas se deben a la acumulación de ácido fitánico en
el sistema nervioso. En estos pacientes, del 5 al 30% de los ácidos grasos plasmáticos (20-100 mg/100 mi) y aproximadamente el 50% de los ácidos grasos hepáticos son ácido fitánico; en contraste con la cantidad normal de ácido fitánico en el plasma que está por abajo del 1% (0.3 mg/100 mi). La enfermedad se conoce ahora como síndrome de almacenamiento de ácido fitánico. Los estudios bioquímicos han demostrado que esta acumulación se debe a una deficiencia en la <x-oxidac ión de ác ido f i tánico a ác ido pristánico. Lo más probable es que el defecto metabólico esté en la a-hidroxilación del ácido fitánico, puesto que individuos con este síndrome son capaces de oxidar fitol a ácido fitánico y ácido pristánico a CO2 y H2O. Cuando los pacientes son diagnosticados, los síntomas de la enfermedad pueden disminuir por un régimen dietético estricto con alimentos libres de ácido fitánico.
7.8.1.5 co-oxidación
Se ha observado en microsomas hepáticos de rata una ruta metabólica menor para la oxidación de ácidos grasos (oxidasa de función mixta) que involucra la oxidación del metilo terminal (carbono) de ácidos grasos por medio de NADPH y oxígeno molecular. Esta ruta metabólica, no es de importancia cuantitativa en la oxidación de ácidos grasos de cadena larga. Sin embargo, puede ser importante en el metabolismo de ácidos grasos de cadena corta (C6 a C10) (fig. 7.29).
7.8.1.6 Papel de la carnitina en el transporte mitocondrial de ácidos grasos
La entrada de ácidos grasos o sus acil-CoA derivados a la mitocondria requiere de un mecanismo de transporte, ya que la mitocondria es impermeable a estos compuestos. La molécula acarreadora es la carnitina (car-
Figura 7.29 Productos de la co-oxidación.

ne=músculo). Asimismo, la acetil-CoA producida en mitocondrias por degradación de carbohidratos, ácidos grasos y aminoácidos, no pueden pasar a través de la mitocondria y requieren un mecanismo de transporte similar (fig. 7.30).
La carnitina está muy distribuida, pero es particularmente abundante en músculo, de aquí la capacidad de este tejido para oxidar ácidos grasos. La carnitina también estimula la oxidación de ácidos grasos de cadena larga por la mitocondria.
Modelo clínico: deficiencia de carnitina
Una chica de 19 años acudió al hospital debido a que se fatigaba fácilmente y tema poca tolerancia al ejercicio. El examen neu-rológico reveló debilidad muscular en las
extremidades. Se realizaron biopsias musculares encontrándose al examen microscópico que el músculo estaba lleno de vacuolas lipídicas, principalmente triacilglicéridos; el contenido de carnitina era 6 veces inferior a lo normal.
Cuestionario:
1. ¿Cuál es la principal función intracelu-lar de la carnitina?
2. ¿Estará alterada la p-oxidación en esta paciente?
3. ¿Estará afectada la oxidación de piru-vato (proveniente de glucosa) en esta paciente?
4. ¿Cómo se explicaría la acumulación de triacilgliceroles en músculo?
Figura 7.30 Sistema de transporte de ácidos grasos.

Discusión:
Los músculos de esta paciente, deficientes en camitina, no podrían transportar eficientemente grupos acil-CoA formados dentro y fuera de la mitocondria. Como la oxidación de ácidos grasos es la principal fuente de energía del músculo, los síntomas musculares y la intolerancia al ejercicio se explican por la incapacidad para obtener energía a partir dé la oxidación de ácidos grasos.
En contraste con la oxidación de ácidos grasos no sería de esperar algún defecto en la oxidación de la glucosa en esta paciente. Al igual que los ácidos grasos, la glucosa se convierte en acetil-CoA antes de su oxidación en el ciclo de Krebs. La glucosa entra a la mitocondria como piruvato, y éste se convierte en acetil-CoA; por lo tanto, este paso no requiere de camitina y no se afectará la oxidación de la glucosa!. De hecho, a fin de reemplazar la energía que proviene ordina
riamente de la oxidación de ácidos grasos, probablemente más cantidad de glucosa se oxida en el ciclo de Krebs.
Es probable que se acumulen triacilglice-roles en los músculos de la paciente debido a que no hay alteración en la activación de ácidos grasos. Sin embargo, estos acil-CoA no pueden entrar a la mitocondria para oxidarse y se depositan en forma de triacilgliceroles.
7.8.2 Biosíntesis de ácidos grasos
El mecanismo es diferente a la oxidación, aunque al principio se pensó que sería el mecanismo inverso, como en la glucogénesis. Los ácidos grasos pueden sintetizarse dentro o fuera de la mitocondria. A diferencia de la extramitocondrial, en la vía mitocondrial no interviene la malonil-CoA y una de las reducciones utiliza NADH en lugar de NADPH(fig.7.31).
Figura 7.31. Biosíntesis de ácidos grasos.

La fuente de acetil CoA para la síntesis extramitocondrial de ácidos grasos es acetil-CoA producida dentro de la mitocondria y transportada por el sistema carnitina (Fig. 7.32).
7.8.2.1 Regulación de la biosíntesis de ácidos grasos
Si bien la membrana mitocondrial es impermeable a acetil-CoA, es permeable al ci-trato. Una vez en el citoplasma, el proceso se invierte y el citrato se transforma en oxa-lacetato (OAA) y acetil-CoA por medio de la enzima citrato liasa.
El citrato se convierte en factor estimulante de la acetil-CoA carboxilasa que forma malonil-CoA y sustrato de la citrato liasa formadora de acetil-CoA.
Queda así claro que una dieta alta en carbohidratos conduce a aumento de ácidos grasos por aumentar la acetil-CoA. Por el contrario, una dieta grasa inhibe la biosínte
sis de ácidos grasos por inhibir la acetil-CoA carboxilasa y la citrato liasa (fig. 7.33).
Además, una dieta no grasa aumenta la actividad de la sintetasa multienzimática; la inanición o una dieta alta en grasa disminuye esa actividad.
Como el ciclo de Krebs produce CoA-SH, el funcionamiento de dicho ciclo modifica la disponibilidad de CoA-SH y también el equilibrio síntesis-degradación de ácidos grasos.
Al parecer, la vía intramitocondrial permite sólo alargar cadenas preexistentes de ácidos grasos y no síntesis de novo. La vía intramitocondrial opera sólo en condiciones anaeróbicas. El sistema ACP parece ser diferente en los dos tipos de sintetasa. El principal producto final de la vía extramitocondrial es palmitato libre.
Los ácidos grasos insaturados; linoleico, linolénico y araquidónico, no pueden ser sintetizados por el organismo y se requieren, como las vitaminas, en la dieta. Se conocen como ácidos grasos esenciales.
Figura 7.32. Sistema carnitina de transporte de acetilos y acilos a través de la membrana mitocondrial. Reproducida con autorización de: N.V. Bhagavan, Biochemistry. A comprehensive review,
pág. 647, J. B. Lippincott Coa., 1974.

Figura 7.33.Reproducida con autorización de: J. M. Orten y O. W. Neuhaus, Human Biochemistry, 10a,. edición, pág. 300, St. Louis, 1982, C. V. Mosby Co.
7.8.3 Regulación del metabolismo de ácidos grasos
El mantenimiento del equilibrio entre el catabolismo y anabolismo de los ácidos grasos en el hígado involucra efectos intra-celulares (metabolitos) así como efectos extracelulares (hormonas y dieta). Estas influencias dependen obviamente de las relaciones metabólicas con otras sustancias (carbohidratos y proteínas), del suministro de energía de la células hepática y del movimiento de lípidos entre el hígado y otros tejidos. En su relación con otros tejidos, el hígado juega un papel análogo a su participación en la homeostasis de glucosa, ya que contribuye significativamente con proteínas, fosfolípidos, colesterol y algunos lípidos de las lipoproteí-nas plasmáticas, y es el principal consumidor de triacilgliceroles de los quilomicrones y de los ácidos grasos libres plasmáticos.
El principal factor involucrado en el control de la (3-oxidación, es la disponibilidad de sustrato (figura 7.34). Los ácidos grasos hepáticos pueden originarse de 3 fuentes: 1) La captación de ácidos grasos no esterifica-dos (AGL) provenientes del tejido adiposo; 2) La captación de ácidos grasos liberados
por medio de la actividad de la lipoproteinli-pasa sobre los quilomicrones (esta fuente depende principalmente del nivel de la grasa dietética; y 3) Los ácidos grasos liberados por la actividad de la lipasa intracelular sobre los triacilgliceroles hepáticos. Como en el caso de la lipasa del tejido adiposo, la actividad de la lipasa hepática es incrementada por aquellas hormonas que promueven el aumento del nivel de AMPc a través de la estimulación del sistema adenilciclasa. De esta manera, en el hígado el glucagon es el principal promotor de la producción intracelular de ácidos grasos (fig. 7.34).
Un segundo factor es el contenido energético de la células (Figura 7.35). La P-oxida-ción depende del flujo de electrones a través de la cadena respiratoria y el acoplamiento con la fosforilación oxidativa; de este modo, la velocidad de utilización de ácidos grasos con fines oxidativos depende de las concentraciones relativas de ADP y ATP. Cuando los niveles de ADP son altos (reservas energéticas disminuidas), la tendencia es hacia la síntesis de ATP con flujo de electrones desde las deshidrogenasas de la P-oxida-ción. Cuando, por otro lado, los niveles de ATP están elevados, la tendencia anterior se
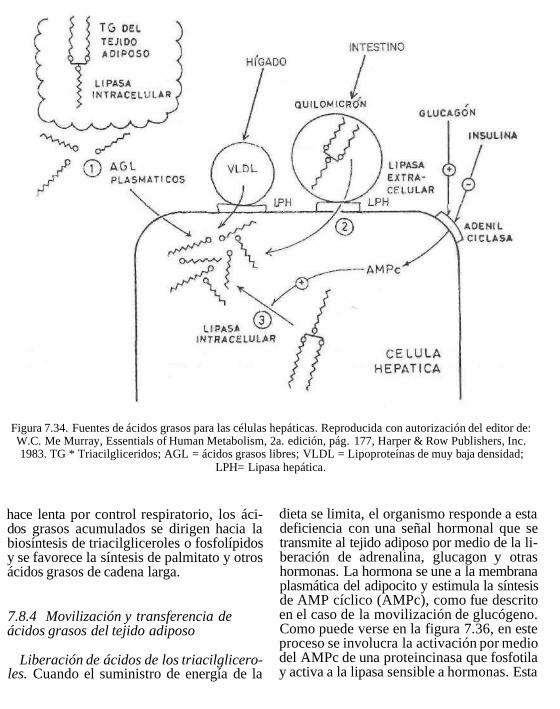
Figura 7.34. Fuentes de ácidos grasos para las células hepáticas. Reproducida con autorización del editor de: W.C. Me Murray, Essentials of Human Metabolism, 2a. edición, pág. 177, Harper & Row Publishers, Inc. 1983. TG * Triacilgliceridos; AGL = ácidos grasos libres; VLDL = Lipoproteínas de muy baja densidad;
LPH= Lipasa hepática.
hace lenta por control respiratorio, los ácidos grasos acumulados se dirigen hacia la biosíntesis de triacilgliceroles o fosfolípidos y se favorece la síntesis de palmitato y otros ácidos grasos de cadena larga.
7.8.4 Movilización y transferencia de ácidos grasos del tejido adiposo
Liberación de ácidos de los triacilgliceroles. Cuando el suministro de energía de la
dieta se limita, el organismo responde a esta deficiencia con una señal hormonal que se transmite al tejido adiposo por medio de la liberación de adrenalina, glucagon y otras hormonas. La hormona se une a la membrana plasmática del adipocito y estimula la síntesis de AMP cíclico (AMPc), como fue descrito en el caso de la movilización de glucógeno. Como puede verse en la figura 7.36, en este proceso se involucra la activación por medio del AMPc de una proteincinasa que fosfotila y activa a la lipasa sensible a hormonas. Esta

Tabla 7.3 Nomenclatura de los ácidos grasos insaturados
Fórmula simplificada
CnA10
CieA9
CisA9
CigA9
C22A13
CigA9-12
Cl8A9.12.15
C2oA5'8>n.i4
Nombre común
Acido unde-cilénico. Acido palmitoleico Acido oleico
Acido elaídico
Acido erúcico
Acido linolei-co.
Acido linolénico
Acido araquidóníco
Nombre sistemático
Ac.Al° undecenoico AcA9
hexadecenoico AcA9 (Cis) octadecenoico AcA9
(Trans) octadecenoico. AcA1 3
docosenoco
AcA9»!2
octadecadi-enoico. Ac_A9,12;15 octadecatri-enoico. Ac>A5,8,lI,W eicosatetraenoico
Fuente
Antifúngico
Grasas y aceites Muy abundante
Aceite de colza, mostaza alhelí Aceite de algodón, linaza, cártamo. Ac. linaza, cártamo.
Lecitinas, cefalinas
Punto de fusión
0.5°C
13.4°C
33.8°C
-5.0°C
-10°C
-49.5°C
aceites. En forma general se puede decir que el estado físico depende de la composición química de los ácidos grasos. En los aceites predominan los ácidos grasas de bajo peso molecular e insaturados y generalmente son de origen vegetal. Por el contrario, las grasas o mantecas tienen mayor proporción de ácidos grasos de elevado PM y con alto índice de saturación. Los aceites vegetales se pueden convertir en grasas por hidrogena-ción e incluso dependiendo de la magnitud de la hidrogenación se obtienen grasas con diferente punto de fusión.
Las grasas y aceites son materiales muy ricos en energía. La oxidación de lg de grasa produce 9 kcal, debido a esto su principal función es la de actuar como material energético de reserva. La composición y natura
leza de las grasas varían según la especie animal, pero incluso pueden variar en el mismo animal según su localización. La grasa que rodea y sostiene a los ríñones es sólida con una elevada proporción de ácidos grasos saturados y de elevado peso molecular. Por el contrario las grasas que van a ser metabolizadas son blandas o líquidas según la temperatura corporal, lo cual facilita su utilización.
7.4.2 Ceras y céridos
Los céridos son esteres de ácidos grasos y alcoholes monohidroxílicos, alifáticos de elevado peso molecular. Su principal característica es que son fuertemente hidrofóbi-
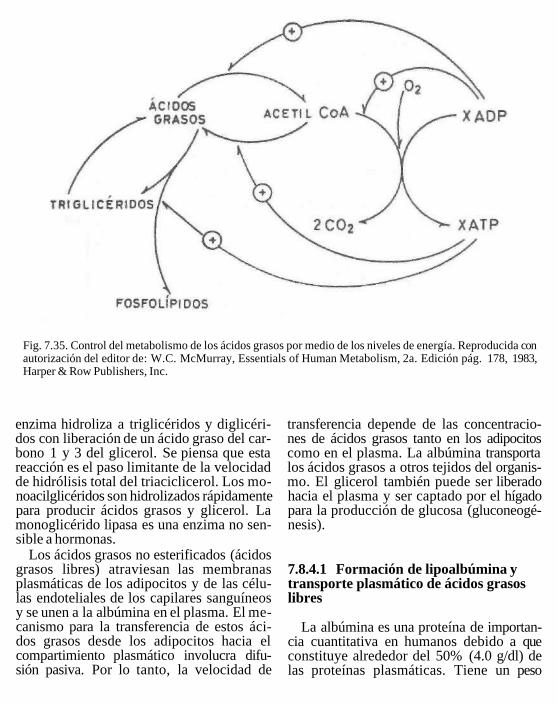
Fig. 7.35. Control del metabolismo de los ácidos grasos por medio de los niveles de energía. Reproducida con autorización del editor de: W.C. McMurray, Essentials of Human Metabolism, 2a. Edición pág. 178, 1983, Harper & Row Publishers, Inc.
enzima hidroliza a triglicéridos y diglicéri-dos con liberación de un ácido graso del carbono 1 y 3 del glicerol. Se piensa que esta reacción es el paso limitante de la velocidad de hidrólisis total del triaciclicerol. Los mo-noacilglicéridos son hidrolizados rápidamente para producir ácidos grasos y glicerol. La monoglicérido lipasa es una enzima no sensible a hormonas.
Los ácidos grasos no esterificados (ácidos grasos libres) atraviesan las membranas plasmáticas de los adipocitos y de las células endoteliales de los capilares sanguíneos y se unen a la albúmina en el plasma. El mecanismo para la transferencia de estos ácidos grasos desde los adipocitos hacia el compartimiento plasmático involucra difusión pasiva. Por lo tanto, la velocidad de
transferencia depende de las concentraciones de ácidos grasos tanto en los adipocitos como en el plasma. La albúmina transporta los ácidos grasos a otros tejidos del organismo. El glicerol también puede ser liberado hacia el plasma y ser captado por el hígado para la producción de glucosa (gluconeogé-nesis).
7.8.4.1 Formación de lipoalbúmina y transporte plasmático de ácidos grasos libres
La albúmina es una proteína de importancia cuantitativa en humanos debido a que constituye alrededor del 50% (4.0 g/dl) de las proteínas plasmáticas. Tiene un peso
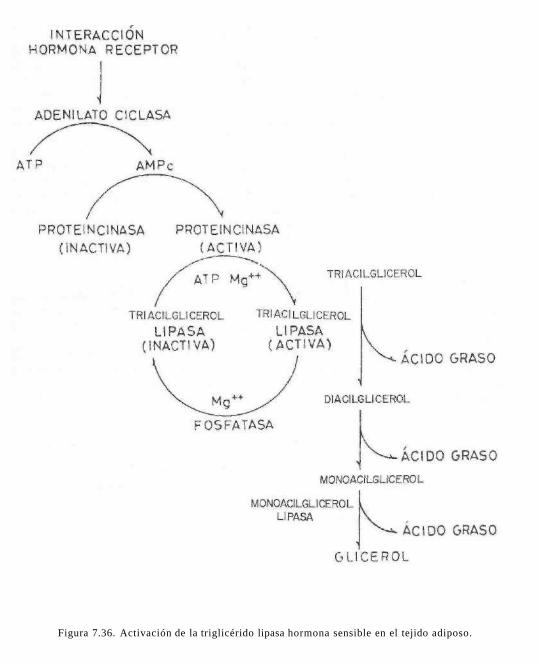
Figura 7.36. Activación de la triglicérido lipasa hormona sensible en el tejido adiposo.

molecular de 66,200 y está formada por una sola cadena polipeptídica que posee 17 puentes disulfuro. Cada molécula tiene la capacidad de unirse a 6 ácidos grasos; las dos primeras se unen más estrechamente (Ka ~ 108M) que las otras cuatro (Ka ~ 106M). Parece tener dos o tres sitios primarios de unión, localizados en regiones apola-res dadas por residuos de aminoácidos hidrofóbicos. La unión entre la molécula de albúmina y los ácidos grasos ocurre a través de interacciones físico químicas, esto es, no se forman enlaces covalentes.
La longitud de la cadena del ácido graso determina la afinidad de unión entre este y la albúmina (por ejemplo, el estearato se une más fácilmente que el palmitato). Por otro lado, los ácidos grasos monoinsaturados se unen con mayor afinidad que los saturados (así por ejemplo, el oleato se une más fácilmente que el estearato). Sin embargo el linoleato se une con menor facilidad que el estearato. La vida media de los ácidos grasos unidos a la albúmina es de 1 a 2 minutos.
Además de proporcionar un complejo hi-drosoluble para el transporte eficiente a través de la circulación, la albúmina facilita la captación rápida de ácidos grasos hacia los tejidos periféricos, actuando como un reser-
vorio, que permite una concentración mucho más alta de ácidos grasos cerca de la superficie celular de la que se obtendría si estuvieran en solución acuosa.
Por medio del complejo lipoalbúmina, son acarreados los ácidos grasos hacia los tejidos deficientes de energía, pasando del compartimiento plasmático al intracelular por un proceso de difusión. La cantidad de ácidos grasos utilizada por un tejido depende de las concentraciones relativas tanto en el plasma como en las células de dicho tejido. El músculo cardíaco y esquelético así como el hígado utilizan ácidos grasos como principal fuente de energía removiendo por lo tanto grandes cantidades de la circulación.
7.8.5 Cetogénesis
Como consecuencia de la acumulación normal de acetil-CoA se produce la condensación para dar acetoacetil-CoA. En el hígado, la acetoacetil-CoA se transforma en acetoa-cetato por medio de una tiolasa, reacción irreversible en este órgano, pero no así en otros tejidos. El acetoacetato en hígado se reduce a P-OH-butirato y una parte se descarboxila espontáneamente en acetona (fig. 7.37).
Figura 7.37. Formación de cuerpos cetónicos.

Estos tres compuestos pasan del hígado a la sangre que los lleva a otros tejidos donde ingresan al ciclo de Krebs y se oxidan completamente.
La enzima clave en el catabolismo de cuerpos cetónicos es la P-cetoácido-CoA transferasa (tioforasa) que no existe en el hígado. Los tejidos que tienen esta enzima (como el músculo, cerebro, corazón, riñon y testículos) si pueden metabolizar los cuerpos cetónicos (Figura 7.38).
La formación de cuerpos cetónicos en hígado a partir de acetil-CoA o acetoacetil-CoA incluye el beta-hidroxi-beta-metil-glutaril-CoA (HMG-CoA) como intermediario obligado (fig. 7.39).
Debe notarse que la HMG-CoA es también el punto de partida para la síntesis de colesterol. Sin embargo, la colesterogénesis no se modifica durante la cetogénesis.
Los cuerpos cetónicos son transportados por la sangre a tejidos extrahepáticos. En estos tejidos, el beta-hidroxi-butirato se convierte en acetoacetato y éste se reactiva a
acetoacetil-CoA por dos mecanismos que se muestran en la fig. 7.40.
El acetoacetil-CoA se oxida a CO7 y H2O en ríñones, músculo, corazón, cerebro y testículos por medio de la p-ceto tiolasa que produce 2 acetil-CoA.
En condiciones normales, la concentración de cuerpos cetónicos es baja (1 mg/100 mi sangre) y la eliminación urinaria es menor de 1 mg/24 horas. Cuando aumenta el metabolismo de las grasas y disminuye el de carbohidratos, la concentración sanguínea alcanza niveles anormales (cetonemiá), se eliminan los cuerpos cetónicos en orina (ce-tonurid) y se instala el cuadro clínico llamado cetosis.
La cetosis es causada por (1) producción aumentada de cuerpos cetónicos por el hígado (principal órgano formador) o (2) por utilización disminuida en tejidos extrahepáticos. Se presenta en la diabetes grave, inanición, anestesia o por dieta mal balanceada; alta en grasa y baja en carbohidratos. Como los cuerpos cetónicos son ácidos que agotan
Figura 7.38. Formación y utilización de cuerpos cetónicos. Reproducida con autorización de; N. V. Bhagavan, Biochemistry. A comprehensive review, pág. 683, J. B. Lippincott, Co., 1974.

Figura 7.40. Utilización tisular de cuerpos cetónicos.
la reserva alcalina se presenta la cetoácido-sis metabolica. Sin embargo, debe apreciarse que los cuerpos cetónicos representan una fuente muy importante de acetil-CoA para la producción celular de ATP. A diferencia de la glucosa o los ácidos grasos, que sólo forman acetil-CoA después de largas secuencias metabólicas, pudiendo además ser metabolizados a otros productos como glucógeno o triglicéridos, los cuerpos cetónicos forman acetil-CoA por medio de unos cuantos pasos (figura 7.41). Por otro lado, la producción de acetil-CoA es la única ruta metabolica disponible para los cuerpos ce-
tónicos en las células. Por lo anterior, pueden considerarse como la forma más útil en que la "energía metabolica" circula en la sangre.
7.8.5.1 Regulación de la cetogénesis
Cuando baja la concentración plasmática de glucosa se origina una disminución en la secreción de insulina y un aumento en la secreción de glucagon. El resultado de estos cambios es un estímulo de la lipolisis, con la consecuente liberación de AGL del tejido

Figura 7.41. Oxidación de cuerpos cetónicos en el músculo. Reproducida con autorización del editor de W.C. McMurray, Essentials of Human Metabolism, 2a. edición, pág. 195,1983,
Harper «fe Row Publishers, Inc.
adiposo. Estos son captados por el h ígado; en donde pueden ser reesterificados y secretados en forma de lipoproteínas de muy baja densidad o catabolizados a acetil-CoA y después a cuerpos cetónicos. No se conoce con exactitud qué es lo que determina que los ácidos grasos se degraden en lugar de dar origen a la síntesis de triglicéridos. Sin embargo, se piensa que el estímulo se origina en el hígado y se ubica a nivel de la entrada de los ácidos grasos a la mitocondria mediada por la carnitina.
Cuando el nivel de cuerpos cetónicos aumenta se estimula la liberación de insulina y de esta manera disminuye la liberación de AGL del tejido adiposo (efecto antilipolíti-co). En otras palabras, cuando los cuerpos cetónicos se están produciendo a una velocidad que excede la de su utilización, son capaces de reducir el suministro hepático de sus precursores (autorregulación). Esto con
serva el depósito de triglicéridos y minimiza los cuerpos cetónicos y en consecuencia la pérdida de combustible por la orina. Además evita la cetoácidosis típica de una ceto-génesis no controlada (fíg. 7.42).
No obstante la gran asociación de los cuerpos cetónicos con la diabetes, no deben ser considerados como entidades químicas patológicas; ya que son importantes combustibles metabólicos (representan la transformación de un combustible, incapaz de entrar a las células del cerebro, como son los ácidos grasos, en un material hidrosoluble que puede abandonar los capilares y difundir fácilmente hacia las células). Es su sobreproducción no controlada la que es patológica.
Los cuerpos cetónicos son capaces de suministrar mucha de la energía requerida por el encéfalo durante la inanición y son oxidados preferentemente a la glucosa y los ácidos grasos por otros tejidos.
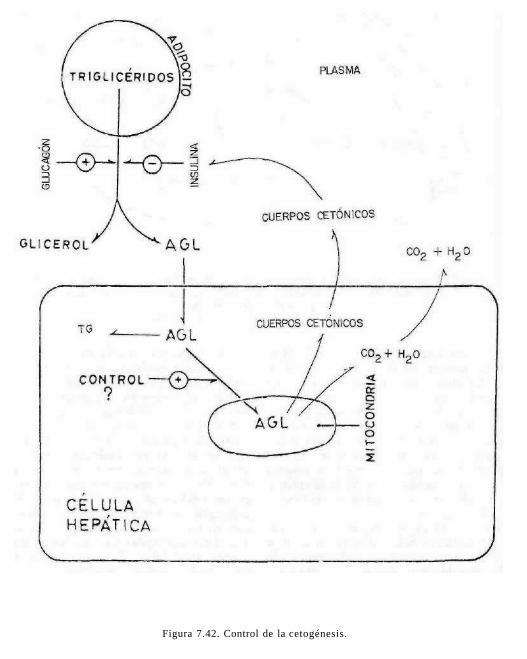
Figura 7.42. Control de la cetogénesis.
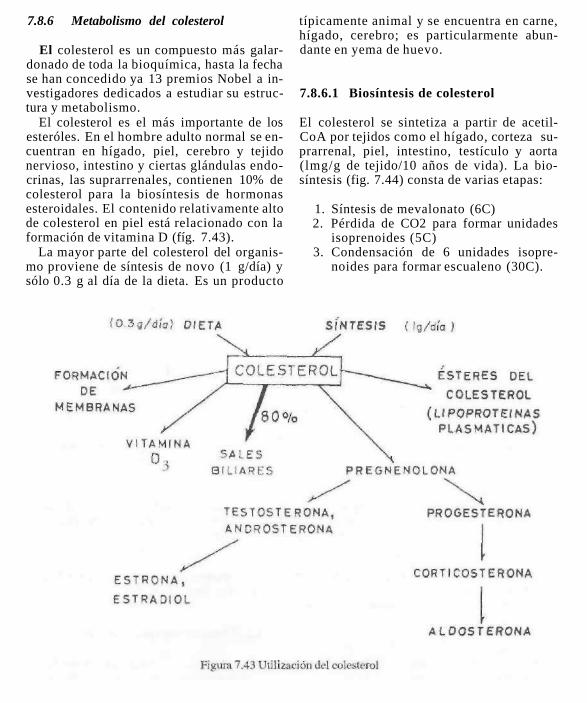
7.8.6 Metabolismo del colesterol
El colesterol es un compuesto más galardonado de toda la bioquímica, hasta la fecha se han concedido ya 13 premios Nobel a investigadores dedicados a estudiar su estructura y metabolismo.
El colesterol es el más importante de los esteróles. En el hombre adulto normal se encuentran en hígado, piel, cerebro y tejido nervioso, intestino y ciertas glándulas endocrinas, las suprarrenales, contienen 10% de colesterol para la biosíntesis de hormonas esteroidales. El contenido relativamente alto de colesterol en piel está relacionado con la formación de vitamina D (fíg. 7.43).
La mayor parte del colesterol del organismo proviene de síntesis de novo (1 g/día) y sólo 0.3 g al día de la dieta. Es un producto
típicamente animal y se encuentra en carne, hígado, cerebro; es particularmente abundante en yema de huevo.
7.8.6.1 Biosíntesis de colesterol
El colesterol se sintetiza a partir de acetil-CoA por tejidos como el hígado, corteza suprarrenal, piel, intestino, testículo y aorta (lmg/g de tejido/10 años de vida). La biosíntesis (fig. 7.44) consta de varias etapas:
1. Síntesis de mevalonato (6C) 2. Pérdida de CO2 para formar unidades
isoprenoides (5C) 3. Condensación de 6 unidades isopre
noides para formar escualeno (30C).

Figura 7.44 Biosíntesis de colesterol. Reproducida con modificaciones, de Martin D. W. Jr. y cois., Harper's Review of Biochemistry, 20a. edición pág. 251, 1985, con la gentil autorización de Lange Medical
Publications, Los Altos, California
4. Conversión de escualeno lineal en la- concentraciones de ácidos grasos saturados nosterol (cíclico). de cadena larga y colesterol.
5. Transformación de lanosterol en colesterol (27C).
La enzima clave en la regulación de la 7.8.6.2 Eliminación de colesterol biosíntesis de colesterol es la P-hidroxi-P -metil-glutaril-CoA reductasa (HMG-CoA Debido a nuestra incapacidad para oxidar el reductasa); la enzima es inhibida por altas colesterol hasta C0 2 , el organismo humano
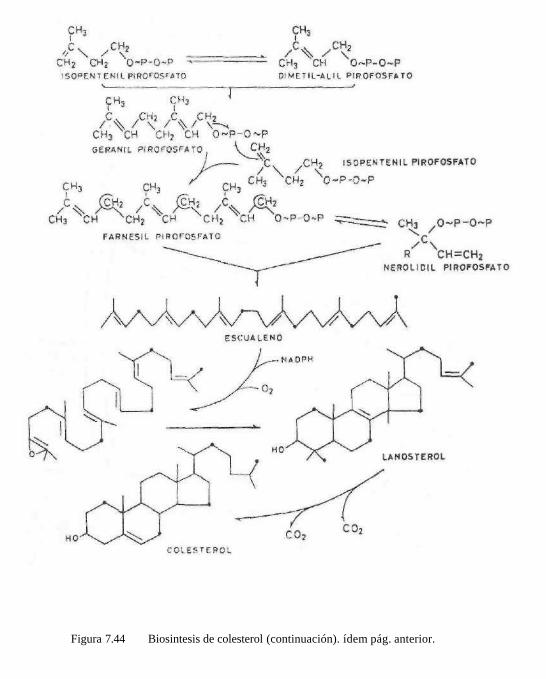
Figura 7.44 Biosintesis de colesterol (continuación). ídem pág. anterior.

Tabla 7.4 Nomenclatura de los ácidos grasos hidroxilados y cíclicos
Fórmula simplificada
C24 2-hidroxi
C24A14
2-hidroxi
C24A14
CisA9
12-hidroxi
Ci8 Acido chaulmúgrico
nombre común
Acido cerebrónico
Acido hidroxi-nervónico
Acido nervónico
Acido ricinoleico
Acido chaulmúgrico
Nombre sistemático
Acido 2-hidroxi-tetracosanoico
AcA14
2-hidroxite-tracosanoico
AcA14
tetracosenoico
AcA9
12-hidroxiocta-decenoico.
Acido ciclopen-ten 1-tridecanoico
Fuente
Cerebrona, esfinogolípidos
Fosfolípidos, esfingolípidos
Fosfolípidos, esfingolípidos
Esfingolípidos, aceite de ricino
(Tratamiento de la lepra)
Punto de fusión
-100°C
68.5°C
eos, por lo que su principal función es proteger a las estructuras del organismo de la des-hidratacíón, fundamentalmente las que están expuestas al sol o que requieren lubricación o imperrneabilización como la piel, pelos y plumas de aves y la superficie de hojas y frutas. Las abejas usan la cera para construir sus panales. Algunos animales utilizan las ceras como material energético de reserva.
Las ceras tanto animales como vegetales son mezclas de céridos, alcoholes superiores, ácidos grasos libres e hidrocarburos de elevado peso molecular. La estructura química de un cérido presente en la cera de abeja es la siguiente:
CH3-(CH2)28-CH2-O-C0-(CH2)14-CH3
7.5 LIPIDOS COMPLEJOS
Son lípidos que en su estructura poseen además de un alcohol y ácidos grasos una o
varias moléculas diferentes, y según el tipo de heteromoléculas de denominan: Fosfolípidos, Glucolípidos, Sulfolípidos y Lipopro-teínas.
Fosfolípidos. Estos lípidos se caracterizan por poseer en su molécula una molécula de ácido fosfórico en forma de éster o de fosfo-diéster. Los fosfolípidos pueden ser glicero-fosfolípidos, si poseen como alcohol a la glicerina o esfingofosfolípidos cuando el alcohol es esfingosina.
7.5.1 Glicerofosfolípidos o fosfoacilgliceroles
Estos son los lípidos complejos más abundantes. Existen en algunos tejidos en proporciones muy elevadas como en el sistema nervioso. Son además los principales componentes de las membranas celulares. Estos compuestos se subclasifican en varios grupos en base a su estructura química. Todos
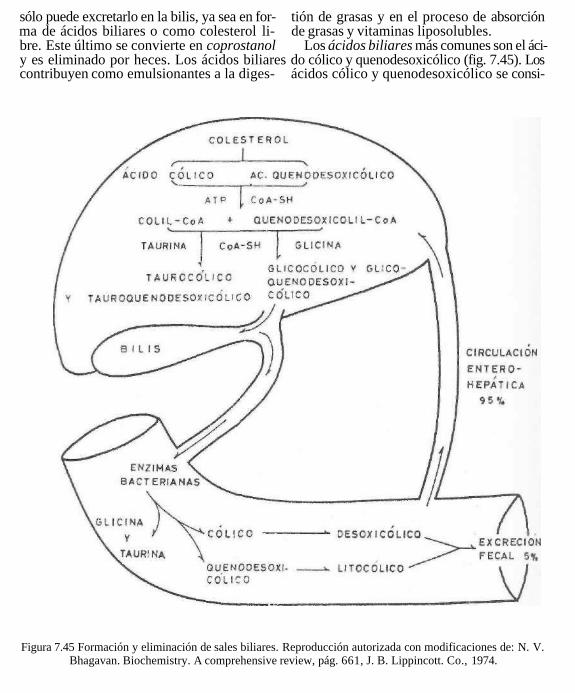
sólo puede excretarlo en la bilis, ya sea en for- tión de grasas y en el proceso de absorción ma de ácidos biliares o como colesterol li- de grasas y vitaminas liposolubles. bre. Este último se convierte en coprostanol Los ácidos biliares más comunes son el áci-y es eliminado por heces. Los ácidos biliares do cólico y quenodesoxicólico (fig. 7.45). Los contribuyen como emulsionantes a la diges- ácidos cólico y quenodesoxicólico se consi-
Figura 7.45 Formación y eliminación de sales biliares. Reproducción autorizada con modificaciones de: N. V. Bhagavan. Biochemistry. A comprehensive review, pág. 661, J. B. Lippincott. Co., 1974.

deran primarios por sintetizarse directamente en el hígado a partir de colesterol. La enzima clave del proceso es la 7-a-hidroxilasa que es inhibida por los ácidos biliares y activada al incrementarse el colesterol de la dieta.
El grupo carboxilo de estos ácidos se encuentra generalmente unido a glicina o taurina dando lugar a los ácidos glicocólico, taurocólico, glicoquenodesoxicólico y tau-roquenodesoxicólico. Cuando estos ácidos sufren modificaciones químicas por acción de las bacterias intestinales, se transforman en los ácidos biliares secundarios; desoxicó-lico y litocólico.
Como la cantidad de ácidos biliares sintetizados por el hígado es insuficiente para satisfacer las necesidades fisiológicas, éstos son reabsorbidos mediante un proceso activo dependiente de ATP y Na+ a nivel del íleo, y transportados de nuevo al hígado unidos a la albúmina. Este continuo reciclaje de ácidos biliares recibe el nombre de circulación enterohepática, a través de la cual regresan al hígado más del 95% de los ácidos biliares. La circulación enterohepática es muy activa, realiza de 4 a 12 ciclos al día.
Los ácidos biliares no absorbidos son eliminados por heces y aunque el porcentaje es bajo (1-5%), constituye la vía principal de eliminación de colesterol por el organismo; la excreción en forma de hormonas esteroi-dales o sus derivados es de menor cuantía.
El colesterol sanguíneo puede disminuir consumiendo grasas con ácidos grasos po-liinsaturados (cártamo, maíz, algodón), mientras los saturados lo aumentan (mantequilla, coco) probablemente los ácidos grasos poliinsaturados actúan:
a) estimulando su excreción en intestino. b) estimulando su oxidación a sales biliares c) acelerando el metabolismo de esteres
de colesterol d) cambiando la distribución en los tejidos
7.8.6.3 Fármacos que disminuyen los niveles de colesterol sanguíneo
Clofibrato. Aunque el mecanismo es desconocido, este fármaco inhibe la síntesis hepática de colesterol. También disminuye las VLDL. Es útil en las hiperlipoproteínemias tipo III, IV y V. Efectos colaterales son aumento de sensibilidad a cumarínicos y a veces miositis y debilidad muscular (fig. 7.46).
C| -< \ / > - 0 - C - C O O - E t
CH 3
Figura 7.46. Estructura del clofibrato.
D.tiroxina. Este fármaco acelera el cata-boliso del colesterol y de la LBD. Se usa en lugar de isómero L en virtud del menor efecto calorigénico. Efectos secundarios son aumento de sensibilidad a cumarínicos, insuficiencia cardíaca y curvas de tolerancia a la glucosa anormales (fig. 7.47)
Fig. 7.47. Estructura de la D-tiroxina.
Colestiramina. Es una resina de intercambio iónico; impide la reabsorción intestinal de sales biliares y con ello aumenta el catabolismo del colesterol. Es útil en la hiperli-poproteinemia tipo II. Efectos colaterales son náusea y estreñimiento, acidosis hiper-clorémica y falta de absorción de vitaminas A , D , E y K .

Otros fármacos utilizados son el ácido ni-cotínico que interfiere con la movilización de ácidos grasos y los estrógenos que disminuyen las LDL (p y aumentan las VLDL (pre-P); los andrógenos muestran efecto contrario. Recientemente se han utilizado los ácidos quenodesoxicólico y ursodesoxi-cólico para disminuir los niveles de coleste-rol plasmático, ya que inhiben la HMG-CoA reductasa. Otros agentes inhibidores de la reductasa utilizados son la mevastatina y lo-vastatina, que reducen las concentraciones de colesterol unido a LDL con pocos efectos adversos.
Xantomatosís
Los xantomas son acumulación de lípidos (sobre todo esteres de colesterol) en forma de tumores grasos múltiples en piel e hiper-colesterolemia grave y aterosclerosis prematura (ver también hiperlipoproteinemias).
La enfermedad de Hans-Schüler-Chris-tian (xantomatosís idiopática crónica) es una lipidosis de colesterol asociada con diabetes insípida y depósitos de lípido en hígado, bazo y huesos planos del cráneo.
7.8.7 Metabolismo de fosfolípidos
En la bilis, los fosfolípidos mantienen en solución al colesterol. En pulmones, las leci-tinas son componentes del surfactante.
Los fosfolípidos (FL) pueden sintetizarse a partir de un diglicérido intermediario común en la síntesis de triacilgliceroles que reacciona con CDP-etanolamina, o bien, a partir de ácido fosfatídico y CTP en una reacción común en la formación de fosfatidil-inositol y cardiolipinas. (fig. 7.48 y 7.49).
La degradación de fosfolípidos se lleva a cabo por fosfolipasas. En el humano es la fosfolipasa A2 que hidroliza en posición p (2). El lisofosfolípido resultante es hidroli-zado luego por una lisofosfolipasa, en posi
ción a (1) y una esterasa lo degrada finalmente a a-glicerofosfato (fig. 7.50).
Las lisolecitinas son poderosos detergentes y agentes hemolíticos. El veneno de serpiente contiene fosfolipasa A dependiente de Ca++, de aquí que su mordedura produzca hemolisis masiva.
7.8.7.1 Síntesis de esfingomielinas, cerebrósidos y gangliósidos
Las esfingomielinas son fosfolípidos que no contienen glicerol ni enlace tipo éster, característicos de los demás lípidos. La molécula precursora es la esfingosina formada a partir de palmitil-CoA y serina. Esta molécula es a su vez precursora de esfingomielinas, cerebrósidos, gangliósidos y sulfátidos.
Los gangliósidos son glucolípidos que, al igual que las glucoproteínas, parecen ser sintetizados en las membranas del retículo endoplásmico. De aquí son transportados al aparato de Golgi y eventualmente al exterior donde se unen a la superficie externa de la membrana celular.
Cada uno de los pasos biosintéticos indicados en la figura 7.51, son catalizados por glicosil transferasas, de las cuales depende la secuencia oligosacárida del gangliósido así como de la concentración relativa de los azúcares existentes en el medio.
Un aspecto notable del metabolismo de los glucolípidos es la existencia de enfermedades por almacenamiento causadas por deficiencia en las enzimas degradativas conocidas como esfingolipidosis y gangliosidosis.
7.8.7.2 Esfingolipidosis y gangliosidosis
Normalmente estas moléculas se localizan en cerebro. Cuando faltan las enzimas degradativas, se acumulan estas sustancias y dan lugar a una serie de síntomas y signos característicos de las enfermedades (Tabla 7.7).

D H A P DIHIDROXIACETONAFOSFATO
N A D NICOTINADENINDINUCLEOTIDO
N A D P NICOTINADENINDINUCLEOTIDO FOSFATO
Figura 7.48. Formación de ácido fosfatídico. Modificado con permiso de J. M. Orten y O. W. Neuhaus, Human Biochemistry, 10a. edición, pág. 300, St. Louis, 1982, C. V. Mosby Co.
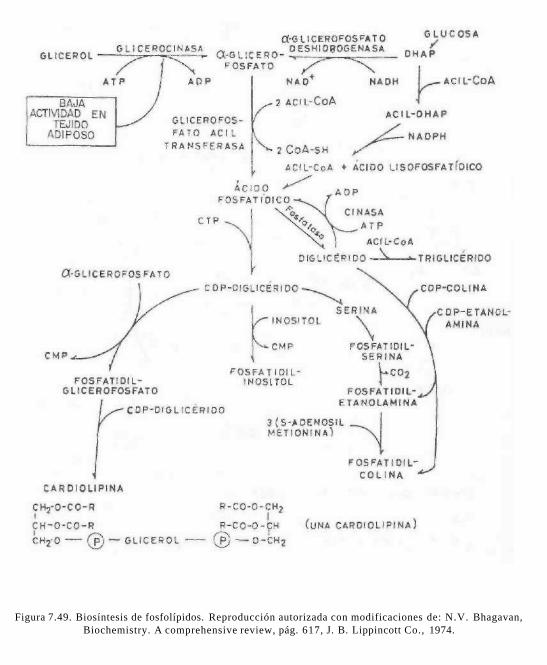
Figura 7.49. Biosíntesis de fosfolípidos. Reproducción autorizada con modificaciones de: N.V. Bhagavan, Biochemistry. A comprehensive review, pág. 617, J. B. Lippincott Co., 1974.
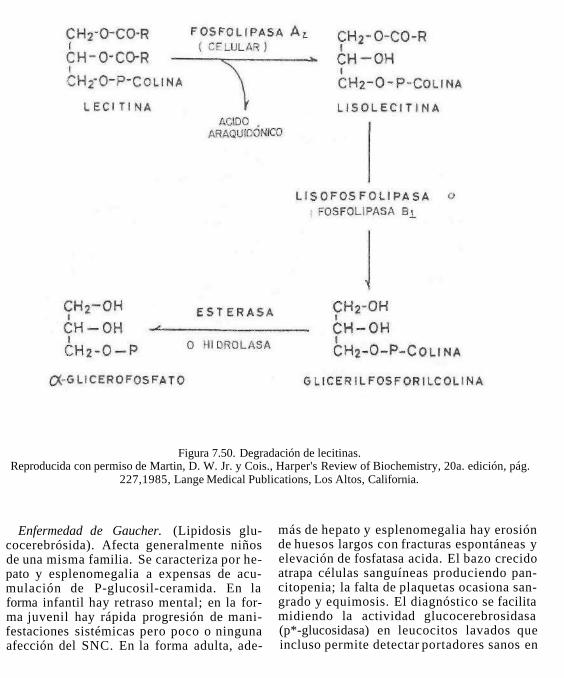
Figura 7.50. Degradación de lecitinas. Reproducida con permiso de Martin, D. W. Jr. y Cois., Harper's Review of Biochemistry, 20a. edición, pág.
227,1985, Lange Medical Publications, Los Altos, California.
Enfermedad de Gaucher. (Lipidosis glu-cocerebrósida). Afecta generalmente niños de una misma familia. Se caracteriza por he-pato y esplenomegalia a expensas de acumulación de P-glucosil-ceramida. En la forma infantil hay retraso mental; en la forma juvenil hay rápida progresión de manifestaciones sistémicas pero poco o ninguna afección del SNC. En la forma adulta, ade
más de hepato y esplenomegalia hay erosión de huesos largos con fracturas espontáneas y elevación de fosfatasa acida. El bazo crecido atrapa células sanguíneas produciendo pan-citopenia; la falta de plaquetas ocasiona sangrado y equimosis. El diagnóstico se facilita midiendo la actividad glucocerebrosidasa (p*-glucosidasa) en leucocitos lavados que incluso permite detectar portadores sanos en
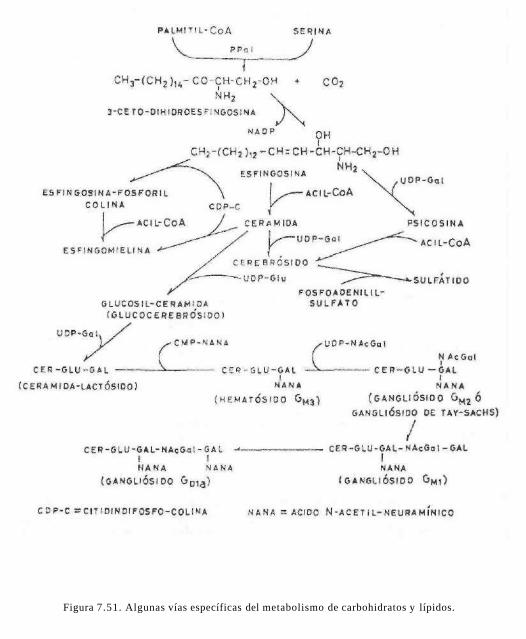
Figura 7.51. Algunas vías específicas del metabolismo de carbohidratos y lípidos.

Tabla 7.7 Enzimas alteradas en las esfingolipidosis y gangliosidosis
Tomada de: Handbook of Neurochemistry, Vol. 7. Abel Lajtha Ed. 1972, con modificaciones.

estudios prenupciales, o bien en cultivo de células amnióticas para predecir si el feto ha heredado el defecto.
Enfermedad de Niemann-Pick (Lipidosis esfingomielínica). También hay hepato y es-plenomegalia por depósitos de fosfolípidos, sobretodo lecitinas y esfingomielinas. Ocurre en la infancia y causa la muerte en unos cuantos meses con daño cerebral profundo. Se observa color amarillo-oliva en la piel y mancha rojo-cereza en la mácula. La mayoría de los pacientes tienen ancestros judíos; sin embargo, no se restringe la enfermedad a estas familias. El diagnóstico se hace por determinación de esfingomielinasa en leucocitos lavados y sonicados, o en cultivo de células amnióticas por amniocentesis.
Enfermedad de Tay-Sachs (Idiocia amau-rótica infantil o gangliosidosis-GM2). Es la más frecuente de las gangliosidosis. Se presenta precozmente en la infancia con daño cerebral y ceguera por acumulación del gan-gliósido en cerebro y tejido nervioso. También hay mancha rojo-cereza en la mácula. El diagnóstico se hace por determinación flu-rorométrica de hexosaminidasa sérica. Desde 1887 a la fecha se han descrito 500 casos.
Variante de Tay-Sachs o enfermedad de Sandhojf (Lipodistrofia globósida). La primera descripción se hizo en 1968 y se han presentado hasta la fecha una docena de casos. Las manifestaciones neurológicas semejan el Tay-Sachs.
Gangliosidosis generalizada (Gangliosidosis GM1). Hay degeneración cerebral progresiva, hepatomegalia y deformaciones esqueléticas.
Enfermedad de Krabbe (Leucodistrofia globoide). Existe deterioro mental progresivo, desmielinización y acumulación de células globoides en la adventicia de vasos sanguíneos pequeños y en la sustancia gris cerebral. La muerte ocurre generalmente a los 10 meses.
Enfermedad de Fabry. (Lipidosis cerami-da trihexósido). Es la más frecuente de las
esfingolipidosis. Está ligada al cromosoma X. Los hombres presentan lesiones cutáneas (pápulas púrpuras en escroto), dolor en extremidades y paresias de nervios craneales. La afección cardíaca, renal y nervios periféricos hace que la causa principal de muerte sea insuficiencia renal, cardíaca o cerebral. El diagnóstico se hace por determinación de actividad ceramida trihexosidasa en biopsia renal o intestinal.
Leucodistrofia metacromática. (Lipidosis sulfátida). Se caracteriza por desmielinización, parálisis progresiva y demencia cerebral con metacromasia, lo cual significa que ciertos colorantes catiónicos cambian su valor de absorción a longitudes de onda más cortas. Así, el azul de toluidina aparece rojo o rosado. En la forma infantil hay defecto en la locomoción, debilidad, ataxia, hipotonía y parálisis seguida por dificultad al hablar y tragar, y atrofia óptica. En la forma adulta hay trastornos psicológicos seguidos por demencia progresiva. Es importante distinguir esta enfermedad de la esclerosis múltiple que también afecta la sustancia blanca pero tiene etiología viral o autoinmune.
Lipidosis ceramida kctósida. Sólo hay tres casos descritos en la literatura. Se produce hepato y esplenomegalia, daño cerebral progresivo y deterioro neurológico.
Enfermedad de Farber (lipogranulomato-sis). Se manifiesta por ronquera, dermatitis, deformación del esqueleto y retraso psico-motor. Es mortal al inicio de la vida. Se llegan a encontrar nodulos subcutáneos, hepato y esplenomegalia.
MODELO CLÍNICO: Enfermedad de Gaucher
Una mujer de 34 años fue admitida en el hospital debido a que presentaba fácilmente equimosis y sangrado excesivo. El examen abdominal reveló una masa firme en cuadrante superior izquierdo que fue juzgada

como esplenomegalia. También estaba presente un aumento de tamaño del hígado. El examen sanguíneo reveló pancitopenia. Las pruebas de coagulación indicaron un tiempo de sangrado prolongado como única anormalidad.
7.8.7.3 Tratamiento de las enfermedades lisosomales
Existe actualmente enorme interés en la posibilidad de terapia de reemplazo enzimá-tico en las enfermedades por deficiencia li-sosomal. Se ha demostrado que las células pueden incorporar enzimas en cultivo de tejidos. Parece que en el proceso de pinocito-sis la membrana externa es englobada para formar vacuolas pinocíticas que luego se fusionan con lisosomas. Las hidrolasas lisosomales degradan luego los polisacáridos en un proceso que es aparentemente esencial para la función normal de la célula. La pino-citosis también proporciona un medio de incorporación de material enzimático del medio externo y la esperanza de una posible terapia de reemplazo. El problema surge porque la inyección de enzimas en el torrente sanguíneo puede conducir a respuestas alérgicas. La alternativa consiste en el mi-croencapsulamiento de las enzimas en "fantasmas" de eritrocitos del propio paciente. En el caso de las enfermedades de Gaucher y Fabry, se espera que el tratamiento de lactantes y niños pequeños pueda prevenir el daño cerebral. Sin embargo, en el Tay-Sachs, el sitio primario de acumulación del gangliósido GM2 s o n l ° s ganglios y células guales del encéfalo. Debido a la barrera he-matoencefálica y la severidad del daño parece poco probable que la enfermedad pueda tratarse con éxito.
El enfoque usado actualmente consiste en identificar portadores de defectos genéticos altamente indeseables y ofrecer consejo genético. Si ambos padres son portadores, el
riesgo de tener un hijo con Tay-Sachs es de uno en cuatro. En un grupo de 32 mujeres con antecedentes de un hijo con la enfermedad, el estado genético del feto fue establecido por amniocentesis. Como lo predicho, 8 de los 32 tienen la enfermedad. En este caso, 7 de las mujeres escogieron el aborto; en el octavo caso, el niño nació con el defecto.
7.8.8 Metabolismo de eicosanoides
Los eicosanoides son compuestos bioacti-vos que modulan la función celular. En la actualidad tienen un enorme interés bioquímico y farmacológico. El término fue introducido por Corey en 1979 en virtud de derivar de ácidos grasos poliinsaturados de 20 carbonos (eicosano, hidrocarburo de 20 carbonos).
Los precursores comunes de los eicosanoides son tres ácidos grasos insaturados de 20 carbonos; el 8,11,14-eicosatrienoico (diho-mo-y-linolénico), el 5, 8, 11, 14-eicosate-traenoico (araquidónico) y el ácido 5, 8, 11, 14,17- eicosapentaenoico. Los dos primeros derivan del ácido linoleico y pertenecen a la clase omega-6. El ácido eicosapentaenoico, de la clase omega-3, deriva del ácido linolé-nico.
Cuando se activa la fosfolipasa A2 celular (fig. 7.50) se libera principalmente ácido araquidónico a partir de los fosfolípidos de membrana. De aquí que el grueso de los eicosanoides sintetizados por el hombre derive del ácido araquidónico. Además, la enzima central de la vía biosintética de pros-taglandinas, la ciclooxigenasa, utiliza el ácido araquidónico con mayor eficacia.
Los principales eicosanoides son prosta-glandinas (PG), tromboxanos (TX), hidro-peróxidos de ácidos grasos (HPETE), hidróxidos de ácidos grasos (HETE), epóxi-dos de ácidos grasos (EET), dihidr óxidos de ácidos grasos (diHETE), leucotrienos (LT) y lipoxinas (fig. 7.52).

ellos se caracterizan por poseer una estructura básica. Todos los glicerofosfolípidos se pueden considerar derivados de un ácido L-a-fosfatídico, que posee un ácido graso saturado en el C-l y un ácido graso insatura-do en el C-2. La fórmula general de los fos-foglicerolípidos se puede resumir en el siguiente cuadro: Ácidos Fosfatídicos Los ácidos fosfatídi-
cos son los precursores de todos los fosfo-
Tabla7.5. Nomenclatura de fosfoglicerolípidos
Nombre Naturaleza de R
Ac. fosfatídico
Fosfatidilcolinas (Lecitinas)
Fosfatidiletanolaminas (Cefalinas)
Fosfatidilserinas (Cefalinas)
Fosfatidil inositoles
Fosfatidilgliceroles
Fosfatidil fosfatídicos (Cardiolipinas)
-H
-0-CH2-CH2-N+(CH3)3 colina
-O-CH2-CH2-NH 3 etanolamina
-O-CH2-CH-COO"

OH
Tromboxano (TXA,)
Lipoxina (lipoxina A)
Figura 7.52. Clases de eicosanoides. Se ha ilustrado cada clase con un compuesto representativo.

No obstante que los eicosanoides poseen actividades biológicas diferentes, presentan una serie de características comunes como son:
1. Se forman a partir de ácidos grasos. 2. Se comportan como autacoides, es de
cir, ejercen sus efectos biológicos en las células cercanas a las que los producen.
3. Actúan como biorreguladores en la formación de nucleótidos cíclicos y en la movilización del calcio.
La liberación del ácido araquidónico desde sus reservas esterificadas es una etapa clave, puesto que la biosíntesis de eicosanoides está limitada por la disponibilidad de araquidónico libre. En los fosfolípidos de membranas, el ácido araquidónico se encuentra principalmente en la posición 2. Por lo tanto, existen varias rutas para la liberación de este ácido (fig. 7.53).
Las enzimas liberadoras del ácido araquidónico son estimuladas por numerosos factores. La hormona TSH activa la fosfolipasa A2 en tiroides, la ACTH en corteza suprarrenal y la LH en el ovario. En las plaquetas se ha descrito un sistema de activación tanto de fosfolipasa A2 como de la fosfolipasa C. Además, situaciones patológicas como el estrés, algunas alergias y otras, pueden incrementar la síntesis de prostaglandinas, tromboxanos y leucotrienos.
Existen tres vías principales de utilización de ácido araquidónico en la síntesis de eicosanoides; la ruta de la ciclooxigenasa, que origina las prostaglandinas y tromboxanos; la ruta de la lipooxigenasa, que produce leucotrienos. HETE y lipoxinas; y la ruta de cito-cromo P450 que da lugar a epóxidos, que luego se convierte en HETE o diHETE (fig. 7.54). Habitualmente, una célula sintetiza fundamentalmente uno de estos productos. Por ejemplo, las células endoteliales forman prostaglandinas; los neutrón los, productos de la lipooxigenasa; las plaquetas, productos de lipo y ciclooxigenasa.
7.8.8.1 Prostaglandinas y prostaciclinas
La necesidad de prostaglandinas es una de las principales razones para que los ácidos grasos poliinsaturados omega-6 sean esenciales en la dieta humana. El precursor más importante de prostaglandinas es el ácido graso esencial linoleico.
Las reacciones de síntesis están catalizadas por la prostaglandina endoperóxido sin-tetasa {prostaglandina sintetasa), la cual posee dos actividades enzimáticas: una actividad ciclooxigenasa (o dioxigenasa) que transforma al ácido araquidónido en PGG2, y una actividad hidroxiperoxidasa que transforma a este último en PGH2 (fig. 7.55). Las dos actividades necesitan la presencia de un grupo hemo.
Una propiedad muy interesante de la ciclooxigenasa es la de autoinactivarse después de funcionar durante 15-30 segundos, es decir, es una "enzima suicida". Los ácidos ei-cosapentaenoicos procedentes de la dieta o del ácido linolénico son de una potencia terapéutica excelente debida a su acción inhibidora de la actividad ciclooxigenasa. La aportación a la dieta del ácido eicosapentaenoico abre una vía terapéutica en el campo de las enfermedades tromboembólicas, la ate-rosclerosis, la inflamación y la autoinmu-nidad. En los esquimales, cuya alimentación es muy rica en pescado que aporta mucho eicosapentaenoico, se ha observado menor incidencia de enfermedades tromboembólicas.
Los antiinflamatorios no esteroidales son todos inhibidores competitivos de la ciclooxigenasa, en particular aspirina e indometa-cina. Los efectos de la aspirina sobre la inflamación, el dolor, la activación plaque-taria, la fiebre y la mucosa gástrica, se han explicado por inhibición de la biosíntesis de eicosanoides cíclicos.
Los antiinflamatorios esteroides (hidro-cortisona, prednisona y betametasona) parecen actuar por bloqueo de la liberación de

Figura 7.53. Rutas de liberación de ácido araquidónico.
Figura 7.54. Biosíntesis de eicosanoides. Para mayor explicación ver texto.
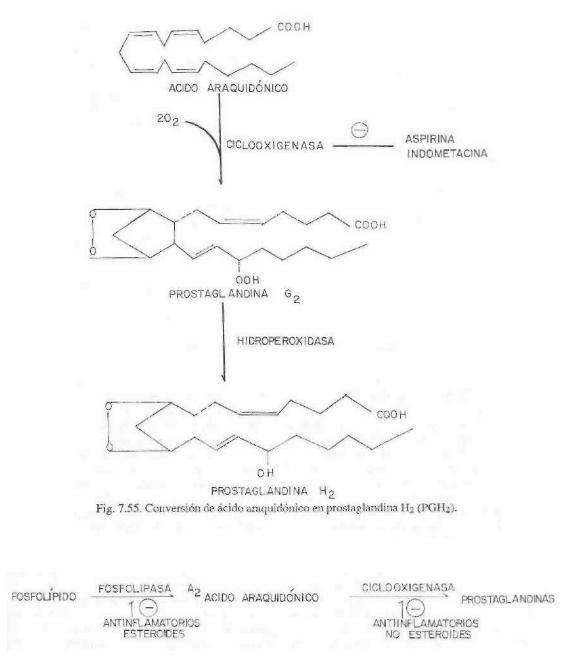

Figura 7.56. Principales prostaglandinas y derivados.
ácido araquidónico al inhibir la actividad fosfolipasa A2.
Las PGG2 y PGH2 son los intermediarios precursores en la síntesis de las otras prostaglandinas. Sólo cinco de ellas se encuentran en abundancia en el organismo. Se conocen como prostaglandinas primarias y todas pertenecen a la serie 2: PGD2 , PGE2, PGF2a. En vasos sanguíneos se produce principalmente PGI2 (también llamada prostacicli-ná). En corazón, la PGE2, PGF2CX y la PGI2 se producen en cantidades iguales. El trom-boxano A2 (TXA2) es el principal derivado de prostaglandinas formado en las plaquetas.
La PGI2 (prostaciclina), principal pros-taglandina del endotelio vascular, es un vasodilatador, especialmente de arterias coronarias, y también previene la agregación de las plaquetas. En cambio, el tromboxano
A2 tiene efectos contrarios a los de la prostaciclina, ya que contrae las arterias y desencadena la agregación plaquetaria.
Las prostaglandinas se encuentran entre las sustancias biológicas más potentes descubiertas hasta la fecha. Tienen una potencial utilidad terapéutica en el tratamiento de la hipertensión, el alivio del asma bronquial y la congestión nasal, como anticonceptivos y en la curación de la úlcera péptica.
Muchas de las acciones de las prostaglandinas están mediadas por el sistema adenila-to ciclasa-AMP cíclico-proteincinasa A. No obstante, una misma PG puede tener efectos contrarios en células diferentes. Por ejemplo, la PGE! activa la adenilato ciclasa en muchos tejidos, pero la inhibe en adipocitos, impidiendo la lipólisis. En cambio, en útero estimula la contracción, incrementando la concentración intracelular de calcio.

7.8.8.2 Tromboxanos
El término tromboxano deriva del hecho que estos compuestos tienen capacidad de formar trombos. La enzima que forma el TXA2 a partir de PGH2 es la tromboxano sintetasa. Debido al potente poder de agregación plaquetaria de los tromboxanos, la búsqueda de inhibidores específicos de su síntesis es objetivo primordial de investigación. El TXA2 es de vida media muy corta (30-40 segundos); se hidroliza espontáneamente a TXB2, que es inactivo.
El ácido eicosapentaenoico, ya mencionado, da origen a las PGI3 y los TX3. La PGI3
es tan potente como antiagregante plaqueta-rio como la PGI2, pero el TXA3 es un agre-gador plaquetario más débil que el TXA2; por lo tanto, el equilibrio está desplazado contra la agregación.
Así, la regulación de la hemostasia sanguínea va a depender del equilibrio entre la síntesis de PGI2 y de TXA2. Varias enfermedades se han relacionado con un desequilibrio en estos dos eicosanoides, entre las que destacan los procesos tromboembólicos y la aterosclerosis. En las lesiones ateromatosas en humanos existe disminución en la biosín-tesis de PGI2 y activación plaquetaria con aumento en la síntesis de TXA2, lo que predispone a tromboembolias e incluso al infarto de miocardio.
En pacientes diabéticos disminuye la actividad prostaciclina sintetasa y aumento en la biosíntesis de tromboxanos, con alta predisposición a aterosclerosis y trombosis. La insulina restaura este desequilibrio y mejora las lesiones vasculares en el hombre.
7.8.8.3 Leucotrienos y otros derivados
Los leucotrienos deben su nombre por haber sido aislados de leucocitos y poseer en su estructura tres dobles ligaduras conjugadas (tríenos). Se sintetizan a partir del 5-
HPETE de la ruta de la 5-lipooxigenasa. A diferencia de la ciclooxigenasa, que está unida a la membrana, las lipooxigenasas son enzimas citosólicas. En las células animales existen tres lipooxigenasas que insertan oxígeno en las posiciones 5, 12 y 15 del ácido araquidónico (5 - , 12- y 15-lipooxigenasa), pero sólo la 5-lipooxigenasa forma leucotrienos.
Células diferentes poseen lipooxigenasas diferentes: los neutrófilos son ricos en 5-lipooxigenasa, las plaquetas en 12-lipooxige-nasa y los eosinófilosen 15-lipooxigenasa. Los productos hidroperóxidos derivados (HPETE) no son hormonas, sino intermediarios altamente reactivos que se convierten en sus alcoholes análogos (HETE) o en leucotrienos.
En plaquetas, islotes endocrinos pancreáticos y células glomerulares predomina el 12-HPETE. Este es un inhibidor de la agregación plaquetaria, y pacientes con trastornos mieloproliferativos, que carecen de actividad 12-lipooxigenasa, tienen una alta incidencia de procesos hemorrágicos.
En reticulocitos, eosinófilos y linfocitos T, los principales productos de la 15-lipooxigenasa son el 15-HPETE y el 15-HETE; este último tiene efecto inhibidor sobre las 5 y 12-lipooxigenasas (fig. 7.57).
En neutrófilos, mastocitos, queratinoci-tos, pulmón, bazo, cerebro y corazón la 5-lipooxigenasa cataliza la formación de leucotrienos, después del intermediario 5-HPETE. A diferencia de otras lipooxigenasas, la 5-lipooxigenasa requiere calcio para su actividad.
El primero en ser sintetizado es el LTA4, que a su vez es metabolizado a LTB4 o en los leucotrienos LTC4 o LTD4 . La conversión del leucotrieno A4 en los leucotrienos C4, D4 y E4 requiere de glutatión reducido y peptidasas específicas (fig. 7.58).
El LTB4 es un potente agente quimiotácti-co de los leucocitos, mientras que los leucotrienos LTC4, y LTE4 muestran otro tipo de

Figura 7.57. Vía de la lipooxigenasa.
acciones como aumento de la permeabilidad vascular (edema) y la constricción de arterias y bronquios. Las acciones biológicas de los leucotrienos C4, D4 y E4 abarcan lo que durante décadas se ha denominado sustancia de reacción lenta de la anafilaxia (SRS-A), estos LT son hasta 1000 veces más potentes que la histamina como broncocons-trictores.
En conjunto, los leucotrienos parecen ser mediadores importantes en trastornos que involucran reacciones inflamatorias o de hi-persensibilidad inmediata como el asma bronquial y otras manifestaciones alérgicas.
Así pues, los eicosanoides, sintetizados por todas las células del organismo, tienen una enorme repercusión clínica (alergia, inflamación, procesos tromboembólicos) y farmacológica. Actualmente se están utilizando estas sustancias, p. ej. la prostacicli-na, como agente vasodilatador e inhibidor de la agregación plaquetaria.
7.8.9 Obesidad
Trastorno mas común del metabolismo de los lípidos.
La anormalidad mas común del metabolismo lipídico es la OBESIDAD. Toda discusión sobre obesidad sólo se puede realizar en base a la ley de conservación de la energía; cuando la ingestión de alimento es mayor que la emisión de energía en forma de calor o trabajo, aumenta la grasa corporal. Así, la obesidad es el resultado de ingerir mas calorías de las que se necesitan para los requerimientos energéticos del organismo.
En el origen de la obesidad son importantes las pequeñas cantidades adicionales de comida que se toman diariamente. Es decir, no son las comidas voluminosas aisladas las que conducen a la gordura, sino las cantidades de energía en "pequeneces" por el simple gozo de comer.
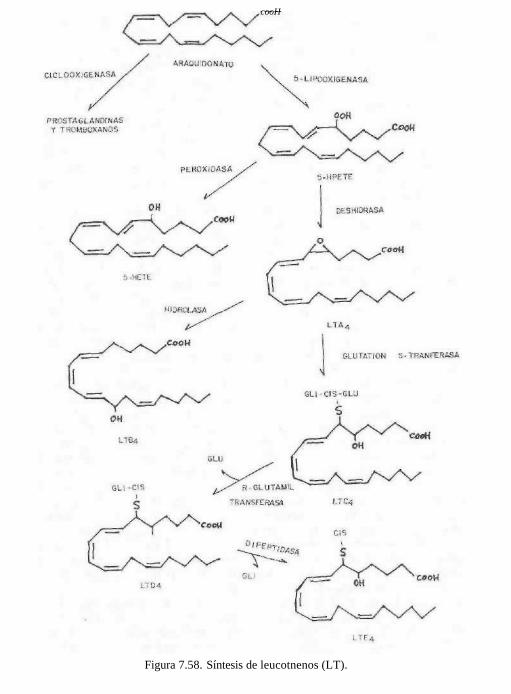
cooH
Figura 7.58. Síntesis de leucotnenos (LT).

No se pueden pasar por alto los factores psíquicos. Aunque la expresión "grasa de aflicción" exprese que algunas personas comen mas por pena o dolor, no hay que olvidar que a muchas personas les ocurre lo contrario cuando sufren alteraciones psíquicas, y adelgazan visiblemente.
Los trastornos endocrinos también pueden causar obesidad. En los portadores de adenomas insulares, la obesidad está relacionada con la secreción excesiva de insulina que aumenta la síntesis de grasas. Pero solamente son obesos los que por el peligro de hipoglicemia toman muchos carbohidratos y por acción de la hormona los acumulan como grasa.
En cuanto a la obesidad parcial del tronco y cara de luna llena en el síndrome de Cus-hing debido a superproducción de glucocor-ticoides, podemos indicar que es producida por la desintegración proteínica que conduce a gluconeogénesis y lipogénesis. A este respecto, algunos autores han propuesto que la obesidad madura (40-50 años) es en realidad un síndrome de Cushing no tumoral por exceso de ACTH y P-endorfinas pituitarias liberadas por estímulos estresantes. El exceso de ACTH estimula la liberación de gluco-cor t ico ides que conduce a desgas te proteínico y gluconeogénesis. La P-endorfi-na estimula la liberación insulina que promueve la lipogénesis. En otras palabras, la glucosa obtenida por gluconeogénesis de proteínas se transforma en ácidos grasos, los cuales se almacenan en tejido adiposo como triglicéridos. Así, el individuo transforma sus propias proteínas en grasas por la acción de estas dos hormonas. Esto hace que el individuo se vuelva gordo sin sobrealimentarse. En muchos maduros obesos hay además sobrealimentación lo que contribuye al círculo vicioso cuando el stress genera glucosa, insulina y comida, lo que genera mas glucosa, insulina y comida.
Debe señalarse que la gluconeogénesis y lipogénesis benignas son concomitantes
normales del proceso de envejecimiento. Por ejemplo, un hombre de 25 años y 70 kg de peso tiene 14% de tejido adiposo y 86% de masa corporal magra. A los 40 años tiene 22% de adiposo y 78% magro; a los 55 tendrá 25% de adiposo y 75% de peso magro. Así, es necesario perder peso al envejecer para mantener la misma proporción de grasa corporal de los jóvenes.
El depósito excesivo de grasa está generalmente asociado con aumento en la incidencia de diabetes mellitus y enfermedades coronarias.
Recientes estudios han definido dos tipos de anormalidades aparentemente no relacionadas, en individuos obesos. Primero, los niveles de fibrinógeno plasmático se elevan y la actividad fibrinolítica disminuye pro-porcionalmente al aumento de obesidad; ambas anormalidades se corrigen al reducir de peso. Este proceso en obesos es relevante en el desarrollo de trombosis intravascu-lar, factor importante en la patogénesis de enfermedades coronarias. El segundo grupo incluye anormalidades en el metabolismo de grasas y carbohidratos. Un individuo obeso segrega cantidades excesivas de insulina a fin de mantener su glucosa sanguínea en niveles normales, es decir, hay una resistencia a la insulina interpretada como disminución de receptores celulares a la insulina en respuesta a elevadas cantidades de la hormona. Estudios "in vitro" muestran que el tejido adiposo se vuelve progresivamente mas resistente a la insulina a medida que el volumen de lípidos depositados aumenta; la sensibilidad a la insulina (aumento del número de receptores) vuelve a lo normal cuando se reduce el tamaño de la célula adiposa.
Finalmente, hay que mencionar que con un exceso de peso de 8 kg (a los 45-50 años) la mortalidad es del 18%. Con exceso de 18 kg el aumento es ya del 45% y con un exceso de peso de 40 kg del 116%.

Modelo clínico: Obesidad
Una paciente de 19 años acudió al médico por tener un sobrepeso de 30 kg, principalmente a expensas de depósito de triglicéri-dos en tejido adiposo. La historia clínica reveló que su dieta en verdad era baja en grasas, pero su ingestión calórica dependía de carbohidratos (dulces, galletas, bebidas acuosas y cerveza).
Cuestionario:
1. ¿Cómo es posible formar cantidades excesivas de triglicéridos en el cuerpo si la
dieta contiene principalmente carbohidratos?
2. ¿Cómo llega al citoplasma la acetil-CoA generada dentro de la mitocondria para ser utilizada en la síntesis de ácidos grasos?
3. ¿Por qué se requiere bicarbonato en la síntesis de ácidos grasos?
4. ¿Cuál es la enzima limitante en la bio-síntesis de novo de ácidos grasos?
5. Si esta paciente fuera deficiente en biotina, ¿interferiría esta deficiencia con la biosíntesis de ácidos grasos?

glicerolípidos. Todos son L-a-fosfatídicos y la diferencia entre ellos se debe a la naturaleza de los ácidos grasos. Existen en pequeña proporción en productos naturales ya que son productos intermedios en la biosíntesis de otros fosfoglicerolípidos.
7.5.1.1 Lecitinas o Fosfatidilcolinas
Estos compuestos se encuentran ampliamente distribuidos en los organismos donde desempeñan múltiples e importantes funciones. Se encuentran en las membranas celulares, y debido a su carácter anfipático se ordenan en forma radial y contribuyen al potencial de membrana.
La solubilidad de estas sustancias y, en general, de todos los fosfoglicerolípidos es especial, debido a su carácter anfipático. En sistema difásicos se ordenan en base a su polaridad. En agua forman capas monomole-culares ordenadas y si se aumenta la concentración forman núcelas, en las cuales, al igual que los jabones quedan expuestas las cabezas hidrofílicas polares y las cadenas hidrofóbicas se unen formando una zona hidrofóbica común interna (Figura 7.1).
Las fosfatidilcolinas también desempeñan un importante papel en el transporte de lípi-
dos, son constituyentes de los quilomicrones y lipoproteínas.
7.5.1.3 Fosfatidil inositoles
Estos lípidos se caracterizan por poseer en su molécula en lugar de una base nitrogenada un polialcohol cíclico denominado inositol, del cual existen 9 isómeros, según la posición de los grupos alcohólicos, el más abundante en la naturaleza y el que forma parte de los inositolípidos es el mioinosotol, que es la forma mesosimétrica y ópticamente inactiva.
Recientemente se ha encontrado que un inositolípido que posee 3 grupos fosfato en lugar de uno, denominado fosfatodilinositol, 4,5-difosfato (figura 7.2).
Este compuesto se encuentra en las membranas celulares y se hidroliza aparentemente como respuesta a varios compuestos:hormo-nas o intermediarios metabólicos, dando el inositol 1,4,5-trifosfato y el diacil glicerol que pueden actuar como "segundos mensajeros" iniciando distintas funciones en la célula. Algunas de las funciones que pueden ser disparadas por el inositol, 1,4,5-trifosfato (IP3) son: glucogenolisis en las células hepáticas, secreción de histamina por las células
Figura 7.1 Estructura y distribución esquemática de la disposición de las moléculas de fosfolípidos.













![Lipidos Y Lipidos Simples [Modo De Compatibilidad]](https://static.fdocuments.ec/doc/165x107/557d1d50d8b42a6e4f8b4d75/lipidos-y-lipidos-simples-modo-de-compatibilidad.jpg)



