
2. FÓRMULA QUÍMICA ESTRUCTURAL 3. DESCRIPCIÓN · ... químicamente designado como...
29
MONOGRAFÍA DEPACON ® Inyección de Valproato de sodio Lista No. 1564 SOLID 1000603902V9.0 Revisión marzo 2017 Monografía V13.03.17 Página 1 de 29 1. NOMBRE DEL PRODUCTO DEPACON ® Inyección de Valproato de sodio 2. FÓRMULA QUÍMICA ESTRUCTURAL 3. DESCRIPCIÓN El valproato de sodio es la sal de sodio del ácido valproico y está químicamente designado como 2-propilpentanoato de sodio. El valproato de sodio tiene un peso molecular de 166.2. Se presenta como un polvo delicuescente blanco, cristalino e inodoro. Cada mL de valproato de sodio contiene el equivalente a 100 mg de ácido valproico, 0.4 mg de edetato disódico y agua para inyección para dar volumen. El pH se ajusta a 7.6 con hidróxido de sodio y/o ácido clorhídrico. La solución es clara e incolora. 4. CLASIFICACIÓN FARMACOLÓGICA Y TERAPÉUTICA Anticonvulsivante Código-ATC: N03AG01. 5. INDICACIONES Y USOS Depacon ® está indicado como una alternativa intravenosa en pacientes para quienes la administración oral de productos de valproato no es factible temporalmente en las siguientes condiciones: Depacon ® está indicado como monoterapia o tratamiento adjunto en pacientes con crisis parciales complejas que ocurren aisladamente o en asociación con otro tipo de convulsiones. Depacon ® está indicado como monoterapia o tratamiento adjunto en pacientes con crisis de ausencias simples o complejas, y como tratamiento adjunto en pacientes con múltiples tipos de convulsiones que incluyen crisis de ausencias. Ausencia simple se define como un bloqueo momentáneo del sensorio o pérdida de la conciencia acompañada por ciertas descargas epilépticas generalizadas sin otros signos clínicos detectables. Ausencias complejas es el término que se usa cuando están presentes otros signos.
-
Upload
trinhduong -
Category
Documents
-
view
219 -
download
0
Transcript of 2. FÓRMULA QUÍMICA ESTRUCTURAL 3. DESCRIPCIÓN · ... químicamente designado como...

MONOGRAFÍA DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902V9.0 Revisión marzo 2017
Monografía V13.03.17 Página 1 de 29
1. NOMBRE DEL PRODUCTO
DEPACON® Inyección de Valproato de sodio 2. FÓRMULA QUÍMICA ESTRUCTURAL
3. DESCRIPCIÓN El valproato de sodio es la sal de sodio del ácido valproico y está químicamente designado como 2-propilpentanoato de sodio. El valproato de sodio tiene un peso molecular de 166.2. Se presenta como un polvo delicuescente blanco, cristalino e inodoro. Cada mL de valproato de sodio contiene el equivalente a 100 mg de ácido valproico, 0.4 mg de edetato disódico y agua para inyección para dar volumen. El pH se ajusta a 7.6 con hidróxido de sodio y/o ácido clorhídrico. La solución es clara e incolora.
4. CLASIFICACIÓN FARMACOLÓGICA Y TERAPÉUTICA Anticonvulsivante Código-ATC: N03AG01. 5. INDICACIONES Y USOS Depacon® está indicado como una alternativa intravenosa en pacientes para quienes la administración oral de productos de valproato no es factible temporalmente en las siguientes condiciones: Depacon® está indicado como monoterapia o tratamiento adjunto en pacientes con crisis parciales complejas que ocurren aisladamente o en asociación con otro tipo de convulsiones. Depacon® está indicado como monoterapia o tratamiento adjunto en pacientes con crisis de ausencias simples o complejas, y como tratamiento adjunto en pacientes con múltiples tipos de convulsiones que incluyen crisis de ausencias. Ausencia simple se define como un bloqueo momentáneo del sensorio o pérdida de la conciencia acompañada por ciertas descargas epilépticas generalizadas sin otros signos clínicos detectables. Ausencias complejas es el término que se usa cuando están presentes otros signos.

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 2 de 29
Limitaciones importantes: Debido al riesgo para el feto de coeficiente intelectual disminuido, defectos del tubo neural y otras malformaciones congénitas mayores, lo que puede ocurrir muy temprano en el embarazo, el valproato no debe administrarse a una mujer en edad fértil a menos que el fármaco sea esencial para el manejo de su condición médica. Ver ADVERTENCIAS Y PRECAUCIONES en declaración relacionada con disfunción hepática fatal.
6. POSOLOGÍA Y MODO DE ADMINISTRACIÓN
General Depacon® es para uso intravenoso exclusivamente. Está indicado como monoterapia y terapia adjunta en crisis parcial compleja en adultos y pacientes pediátricos desde los 10 años de edad en adelante; y en crisis de ausencia complejas y simples. El uso de Depacon® por periodos de más de 14 días no ha sido estudiado. Los pacientes debieran cambiar a productos de valproato oral tan rápido como sea clínicamente factible. Se debe administrar Depacon® como una infusión de 60 minutos (pero no más de 20 mg/min) con la misma frecuencia que los productos orales, sin embargo, la supervisión de la concentración de plasma y cambio en la dosis si fuera necesario. Los productos médicos parenterales deben ser inspeccionados visualmente en busca de material o partículas y decoloración antes de su administración, cuando la solución y el recipiente lo permitan. Lineamientos para la administración La infusión rápida se asocia con el aumento de efectos secundarios. No se ha estudiado la infusión en menos de 60 minutos o mayor de 20 mg/min en pacientes con epilepsia (ver REACCIONES ADVERSAS). La inyección de valproato debe ser administrada por la vía intravenosa como una infusión de 60 minutos. Debe ser diluida con al menos 50 mL de un diluyente compatible. Se debe descartar lo que sobre en el frasco. Exposición Inicial al Valproato de Sodio Las siguientes recomendaciones sobre la dosis se obtuvieron de estudios utilizando productos orales de valproato de sodio. Crisis Parciales Complejas Para adultos y niños de 10 años o mayores. Monoterapia (Tratamiento Inicial) Depacon® no ha sido estudiado sistemáticamente como terapia inicial. Los pacientes deben iniciar una terapia con 10 a 15 mg/Kg/ al día. La dosis debe ser incrementada de 5 a 10 mg/Kg/ por semana para lograr la respuesta clínica deseada, la cual se obtiene generalmente con dosis por debajo de 60 mg/Kg/ al día. Si no se ha obtenido una respuesta óptima, se deben medir los niveles plasmáticos para determinar si se encuentran dentro del rango aceptado (50-100 mcg/mL). No se pueden hacer recomendaciones respecto a la seguridad de valproato para uso de dosis por encima de 60 mg/Kg al día. La probabilidad de trombocitopenia aumenta significativamente en concentraciones plasmáticas de valproato por encima de 110 mcg/mL en mujeres y de 135 mcg/mL en hombres. Los beneficios de mejorar el control de las convulsiones con dosis más altas deben ser evaluados en cuanto a la posibilidad de mayor incidencia de reacciones adversas. (Ver ADVERTENCIAS Y PRECAUCIONES – Trombocitopenia) Conversión a Monoterapia Iniciar con 10 a 15 mg/Kg al día, incrementando 5 a 10 mg/Kg por semana para lograr la respuesta clínica óptima, la cual se obtiene generalmente con dosis por debajo de 60 mg/Kg al día; si no se obtiene la respuesta óptima, se deben medir los niveles plasmáticos para determinar si se encuentran dentro del rango aceptado (50-100 mcg/mL). No hay

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 3 de 29
recomendaciones respecto a la seguridad de usar el valproato en dosis > 60 mg/Kg al día. Dosis de fármacos antiepilépticos concomitantes pueden ser comúnmente reducidas en un 25% cada dos semanas. La reducción debe empezar al iniciar el uso del divalproato de sodio; o retrasarse una o dos semanas si hay la sospecha de que con la reducción ocurran convulsiones. La rapidez y la duración de la retirada de los medicamentos antiepilépticos concomitantes pueden ser altamente variables, y se debe monitorear estrechamente a los pacientes durante este periodo en cuanto al aumento en la frecuencia de las convulsiones. Tratamiento Adjunto Depacon® debe agregarse al régimen del paciente en 10 a 15 mg/Kg por día, incrementando 5 a 10 mg/Kg por semana para lograr la respuesta clínica deseada, la cual se obtiene generalmente con dosis por debajo de 60 mg/Kg al día; si no se obtiene la respuesta óptima, se deben medir los niveles plasmáticos para determinar si se encuentran dentro del rango aceptado (50-100 mcg/mL). No se pueden hacer recomendaciones sobre la seguridad del uso de valproato en dosis por encima de 60 mg/Kg al día. Si la dosis diaria total excede los 250 mg, se debe administrar en dosis divididas. En un estudio de tratamiento adjunto de convulsiones parciales complejas en pacientes que recibían ya sea carbamazepina o fenitoína, además del valproato de sodio, no se requirió ajustar la dosis de los dos primeros. Sin embargo, ya que el valproato tiene interacciones con estos y otros medicamentos antiepilépticos concomitantes, se recomienda determinar las concentraciones plasmáticas de medicamentos antiepilépticos concomitantes desde el inicio del tratamiento (ver INTERACCIONES MEDICAMENTOSAS). Crisis de Ausencia Simples y Complejas La dosis inicial que se recomienda es de 15 mg/Kg al día, aumentando 5 a 10 mg/Kg al día cada semana hasta controlar las convulsiones o hasta que los efectos colaterales eviten seguirlo aumentando. La dosis máxima recomendada es de 60 mg/Kg al día. Si la dosis diaria excede 250 mg, se debe administrar en dosis divididas. No se ha establecido una buena correlación entre la dosis diaria, las concentraciones séricas y el efecto terapéutico. Sin embargo, las concentraciones séricas terapéuticas en la mayoría de los pacientes con crisis de ausencia estarán en el rango de 50 a 100 mcg/mL. Algunos pacientes se controlarán con concentraciones menores y otros con mayores (ver FARMACOLOGÍA). Conforme aumenta la dosis de valproato de sodio, se pueden afectar las concentraciones de fenobarbital y fenitoína (ver INTERACCIONES). Los otros antiepilépticos no se deben suspender abruptamente en los pacientes en quienes el fármaco se administre para prevenir convulsiones mayores, debido a la fuerte posibilidad de precipitar status epilepticus, con hipoxia y riesgo de vida. En niñas, adolescentes, mujeres en edad fértil y mujeres embarazadas El valproato de sodio debe ser iniciado y supervisado por un especialista con experiencia en el tratamiento de la epilepsia. El tratamiento solo se debe iniciar si otros tratamientos son ineficaces o no se toleran y el riesgo-beneficio debe ser cuidadosamente reconsiderado en revisiones regulares del tratamiento. De preferencia valproato de sodio debe ser prescrito como monoterapia y en la dosis eficaz más baja, de ser posible en una formulación de liberación prolongada para evitar concentraciones plasmáticas muy elevadas. La dosis diaria debe dividirse en al menos dos dosis individuales. Tratamiento de Reemplazo Cuando se cambia desde los productos orales de valproato, la dosis diaria total de valproato de sodio inyectable debe ser equivalente al producto oral (ver FARMACOLOGÍA) y debe ser administrado en infusión durante 60 minutos (pero no a más de 20 mg/min), con la misma frecuencia que los productos por la vía oral, aunque puede ser necesario el monitoreo de las concentraciones plasmáticas y el ajuste de la dosis. El monitoreo debe ser estrecho en aquellos pacientes que reciben dosis cercana a la máxima recomendada (60 mg/Kg al día), en particular los que no reciben fármacos inductores enzimáticos. Si la dosis total excede 250 mg, se debe dar en régimen dividido. Sin embargo, la

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 4 de 29
equivalencia mostrada entre valproato de sodio inyectable y en formas orales (divalproato de sodio) en el estado estable, sólo se evaluó en un régimen de cada seis horas. Se desconoce qué sucede si la inyección de valproato de sodio se da con menor frecuencia (2 a 3 veces al día), con niveles por debajo de los que resultan de la dosis oral dada en el mismo régimen; por tanto, si se administra valproato de sodio inyectable 2 ó 3 veces al día, se requerirá un monitoreo cuidadoso de los niveles plasmáticos. Advertencias para la Dosificación en General Dosificación en Pacientes Ancianos Debido al incremento en la depuración del valproato no unido y la sensibilidad al incremento de somnolencia en ancianos, la dosis inicial debiera ser reducida en estos pacientes. La dosis debe aumentarse más despacio y con monitoreo regular en consumo de líquidos y sólidos nutricionales, deshidratación, somnolencia, y otros efectos adversos. La reducción en la dosis o la descontinuación de valproato debe ser considerada en pacientes de bajo consumo de alimentos y fluidos y en pacientes con somnolencia excesiva. La dosis terapéutica última se debe lograr en base a la tolerancia y respuesta clínica (ver PRECAUCIONES Y ADVERTENCIAS - Somnolencia en ancianos – Propiedades farmacológicas - Ancianos). Eventos Adversos Relacionados Con La Dosis La frecuencia de eventos adversos (particularmente enzimas hepáticas elevadas y trombocitopenia) pueden estar relacionadas con la dosis. La probabilidad de trombocitopenia aparenta incrementar significativamente a concentraciones totales de valproato de ≥ 110 mcg/mL (mujeres) ó ≥ 135 mcg/mL (hombres) (ver ADVERTENCIAS Y PRECAUCIONES - TROMBOCITOPENIA). El beneficio en mejora de efectos terapéuticos con dosis altas debe ser considerado contra la posibilidad de una mayor incidencia de reacciones adversas. Compatibilidad y estabilidad El valproato de sodio en inyección es físicamente compatible y químicamente estable con las siguientes soluciones parenterales durante por lo menos 24 horas, siempre que se conserve en envases de cloruro de polivinilo (PVC) o vidrio a temperatura ambiente controlada de 15 a 30ºC (59 a 86ºF).
Dextrosa (5%) Inyección, USP
Cloruro de Sodio (0.9%) Inyección, USP
Lactato de Ringer Inyección, USP
Dosificación en pacientes que toman Rufinamida: Los pacientes estabilizados con rufinamida antes de ser prescrito valproato deben comenzar la terapia con valproato a dosis bajas y se valora a una dosis clínicamente efectiva.
7. CONTRAINDICACIONES
La inyección de valproato de sodio no debe ser administrado en pacientes con enfermedad o disfunción hepática significativa (Ver ADVERTENCIAS Y PRECAUCIONES – Hepatotoxicidad). El valproato de sodio esta contraindicado en pacientes que se conoce que tienen desórdenes mitocondriales causados por mutaciones en el ADN mitocondrial polimerasa gamma (POLG; por ejemplo síndrome de Alpers-Huttenlocher) y niños menores de dos años de edad que se sospecha que tienen un desorden relacionado con POLG (Ver ADVERTENCIAS Y PRECAUCIONES – Hepatotoxicidad).

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 5 de 29
La inyección de valproato está contraindicada en pacientes de quienes se tiene hipersensibilidad conocida al medicamento. (Ver ADVERTENCIAS Y PRECAUCIONES – Reacciones de hipersensibilidad multiorgánica). La inyección de valproato de sodio está contraindicado en pacientes con trastornos conocidos del ciclo de la urea (ver ADVERTENCIAS Y PRECAUCIONES- Desordenes del ciclo de la urea). Valproato está contraindicado en pacientes con porfiria.
8. ADVERTENCIAS Y PRECAUCIONES
Hepatotoxicidad Han ocurrido fatalidades en personas con insuficiencia hepática que recibieron ácido valproico. Estos incidentes usualmente, sucedieron durante los primeros seis meses de tratamiento. Hepatotoxicidad seria o fatal, puede estar precedida por síntomas inespecíficos; malestar, debilidad, letargia, edema facial, anorexia y vómito. En pacientes con epilepsia podría también ocurrir pérdida del control de las convulsiones. Los pacientes deben ser monitoreados detenidamente si aparecen estos síntomas. Se requiere control estrecho mediante pruebas de función hepática, antes de usar valproato y luego a intervalos frecuentes, principalmente los seis primeros meses. Sin embargo, los médicos no deberían confiar totalmente en la bioquímica sérica dado que las pruebas podrían no ser anormales en todas las instancias, pero deberían también considerar cuidadosamente los resultados en el ínterin de la historia clínica y el examen físico. Se deberá tener precaución cuando se administre productos con valproato a pacientes con antecedentes de hepatopatía. Los pacientes que reciben múltiples anticonvulsivantes, los niños, aquellos con trastornos metabólicos congénitos, aquéllos con severos trastornos convulsivos acompañados de retardo mental y aquellos con enfermedad cerebral orgánica, pueden constituir un grupo de particular riesgo. La experiencia ha indicado que los niños menores de dos años están expuestos a un riesgo considerablemente mayor de hepatotoxicidad fatal, especialmente si reúnen las condiciones mencionadas anteriormente. Cuando se utilice en estos pacientes divalproato de sodio, deberá emplearse con extrema precaución y como agente único. Los beneficios terapéuticos deberán ser evaluados frente a los riesgos. No se ha estudiado el uso de valproato de sodio en niños menores de dos años. Por encima de este grupo de edad, la experiencia con productos de valproato en epilepsia ha indicado que la incidencia de hepatotoxicidad fatal baja considerablemente en grupos de pacientes progresivamente mayores. El medicamento deberá suspenderse de inmediato ante la sospecha o evidencia de disfunción hepática significativa o sospechada. En algunos casos, la disfunción hepática ha progresado a pesar de haberse interrumpido la administración del medicamento. (Ver CONTRAINDICACIONES). Valproato de sodio está contraindicado en pacientes que se conoce que tienen desórdenes mitocondriales causados por mutaciones en el ADN mitocondrial polimerasa gamma (POLG; por ejemplo síndrome de Alpers-Huttenlocher) y niños menores de dos años de edad que se sospecha que tienen un desorden relacionado con POLG (ver CONTRAINDICACIONES). Valproato induce la insuficiencia hepática aguda y se han reportado muertes relacionadas con el hígado en pacientes con síndromes neurometabólicos hereditarios causados por mutaciones en el gen del ADN mitocondrial polimerasa gamma (POLG) (por ejemplo síndrome de Alpers-Huttenlocher) a un ritmo mayor que aquellos que no tienen estos síndromes. La mayor parte de los casos reportados de insuficiencia hepática en los pacientes con estos síndromes han sido identificados en niños y adolescentes. Desórdenes relacionados con POLG se debe sospechar en pacientes con historia familiar o síntomas sugestivos de desórdenes relacionados con POLG, incluyendo pero no limitado a encefalopatía inexplicable, epilepsia refractaria (focal,

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 6 de 29
mioclónica), estado epiléptico como presentación, atrasos en el desarrollo, regresión psicomotora, neuropatía axonal sensoriomotora, ataxia cerebelar miopática, oftalmoplegía, o migraña complicada con aura occipital. El ensayo de mutación POLG debe ser realizado de acuerdo a la práctica clínica actual para el diagnóstico de la evaluación de dichos desórdenes. Las mutaciones A467T y W748S están presentes en aproximadamente 2/3 de los pacientes con trastornos relacionados con POLG-autosómicos recesivos. En pacientes mayores de dos años de edad que tienen sospecha clínica de tener una enfermedad mitocondrial hereditaria, valproato de sodio debe ser utilizado únicamente después que otros anticonvulsivantes han fallado. Este grupo de mayor edad debe ser estrechamente monitorizado durante el tratamiento con valproato de sodio por el desarrollo de una lesión de hígado aguda con asesorías clínicas regulares y control de pruebas de función hepática. El fármaco debe suspenderse inmediatamente en presencia de disfunción hepática significativa, presunta o aparente. En algunos casos, la disfunción hepática ha progresado a pesar de la interrupción del fármaco. Defectos del Nacimiento: El Valproato puede causar daño fetal cuando se administra a una mujer embarazada. Los datos del registro del embarazo muestran que el uso de valproato materna puede causar defectos del tubo neural y otras anomalías estructurales (por ejemplo, defectos craneofaciales, malformaciones cardiovasculares, hipospadias, malformaciones de las extremidades). La tasa de malformaciones congénitas en los bebés nacidos de madres que usan valproato es de aproximadamente cuatro veces mayor que la tasa entre los bebés nacidos de madres epilépticas utilizando otras monoterapias contra las convulsiones. La evidencia sugiere que los suplementos de ácido fólico antes de la concepción y durante el primer trimestre del embarazo reduce el riesgo de defectos congénitos del tubo neural en la población general. Pancreatitis Se han reportado casos de vida o muerte con pancreatitis tanto en adultos como en niños en tratamiento de valproato. Algunos de estos casos han sido descritos como hemorragias de rápida progresión desde los síntomas iniciales hasta la muerte. Algunos casos han ocurrido poco después del uso inicial así como después de varios años de uso. El índice basado en los casos excede lo que se esperaba en la población en general, y ha habido casos en los cuales la pancreatitis fue recurrente luego de retomar el tratamiento con valproato. En estudios clínicos, hubo dos casos de pancreatitis sin etiología alternativa en 2416 pacientes, representando experiencia en 1044 pacientes-años. Los pacientes y tutores deben ser advertidos que el dolor abdominal, nausea, vómitos, y /o anorexia pueden ser síntomas de pancreatitis que requieren pronta evaluación médica. Cuando la pancreatitis se diagnostica, el valproato debe ser descontinuado. El tratamiento alternativo para la condición médica subyacente deberá iniciarse según indicación clínica. Desorden de Ciclo de Urea (UCD, por sus siglas en inglés) El valproato de sodio está contraindicado en pacientes con trastornos del ciclo de la urea conocidos (UCD) La encefalopatía hiperamonémica, algunas veces fatal, se ha reportado seguido al inicio del tratamiento de valproato en pacientes con desórdenes del ciclo de urea, un grupo de anomalías genéticas no comunes, particularmente déficit de ornitina transcarbamilasa. Antes del inicio del tratamiento de valproato, se debe considerar una evaluación de UCD en los siguientes pacientes: 1) aquellos con un historial de encefalopatía o coma inexplicable, encefalopatía asociada con carga de proteína, embarazo relacionado o encefalopatía postparto, retardo mental inexplicable, o historial de elevaciones de amoníaco o glutamina en plasma. 2) aquellos con vómitos cíclicos y letargia, irritabilidad extrema episódica, ataxia, bajo BUN, pérdida de proteínas. 3) aquellos con historia familiar de UCD o historia familiar de muertes inexplicadas de infantes (particularmente varones). 4) aquellos con otros signos o síntomas de UCD. Pacientes que desarrollan síntomas de encefalopatía hiperamonémica inexplicable mientras recibían terapia de valproato deberían

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 7 de 29
recibir tratamiento rápido (incluyendo la suspensión de terapia con valproato) y ser evaluados por desorden del ciclo de la urea subyacente. (Ver CONTRAINDICACIONES y ADVERTENCIAS - Hiperamonemia y encefalopatía asociada al uso concomitante de topiramato). Comportamiento e ideas suicidas Se ha reportado un aumento en el riesgo de pensamientos o comportamiento suicida en pacientes que toman fármacos antiepilépticos (FAE) para cualquier indicación. El mayor riesgo de pensamientos o comportamiento suicida con FAE se observó tan sólo una semana después de comenzar el tratamiento con medicamentos antiepilépticos y persistió en la duración del tratamiento evaluado. El riesgo relativo para pensamientos o comportamiento suicida fue superior en los estudios clínicos para la epilepsia que en los estudios clínicos para trastornos psiquiátricos u otras condiciones, pero la diferencia de riesgo absoluto fue similar para las indicaciones de epilepsia e indicaciones psiquiátricas. Los pacientes tratados con FAE para cualquier indicación deben ser monitoreados por la aparición o empeoramiento de depresión, pensamiento o comportamiento suicida, y/o cualquier cambio inusual en el estado de humor o comportamiento. Cualquiera que considere la prescripción de valproato de sodio en inyección o cualquier otro FAE debe sopesar el riesgo de los pensamientos o comportamientos suicidas con el riesgo de enfermedad no tratada. La epilepsia y muchas otras enfermedades para las cuales se prescriben los FAE son en sí asociadas con morbilidad y un incremento en el riesgo de pensamientos y comportamientos suicidas. Si los pensamientos y comportamientos suicidas surgen durante el tratamiento, el médico necesita considerar si el surgimiento de estos síntomas en cualquier paciente dado puede estar relacionado con la enfermedad que está siendo tratada. Los pacientes, sus cuidadores, y sus familias deben ser informados sobre los FAE que incrementan el riesgo de pensamientos y comportamientos suicidas y deben ser advertidos de la necesidad de estar alertas por el surgimiento o empeoramiento de los signos y síntomas de depresión, cualquier cambio inusual en el estado de ánimo o en el comportamiento, o el surgimiento de ideas suicidas, comportamiento suicida o pensamientos para autodañarse. Los comportamientos de preocupación deben reportarse inmediatamente a los profesionales en atención médica. Interacción con antibióticos Carbapenems Los antibióticos carbapenems (ertapenem, imipenem, meropenem) pueden reducir las concentraciones del ácido valproico en suero a niveles subterapéuticos, resultando en pérdida del control de la crisis. Las concentraciones de ácido valproico en suero deben monitorearse frecuentemente después de iniciar la terapia con carbapenem. Debe considerarse una terapia alterna antibacteriana o anticonvulsiva si las concentraciones del ácido valproico en suero bajan significativamente o se deteriora el control de la crisis (ver INTERACCIONES MEDICAMENTOSAS - Antibióticos carbapenémicos). Somnolencia en el Adulto Mayor En un ensayo multicéntrico doble-ciego de valproato en pacientes ancianos con demencia (media de edad=83 años), las dosis se incrementaron en 125 mg/día a una dosis objetivo de 20 mg /Kg/día. Una significativa mayor proporción de pacientes con valproato padecieron somnolencia en comparación con los de placebo, y aunque no estadísticamente significativo, hubo una mayor proporción de pacientes con deshidratación. La descontinuación por somnolencia también fue significativamente superior con la de placebo. En algunos pacientes con somnolencia (aproximadamente la mitad), hubo una reducción asociada de consumo nutricional y pérdida de peso. Hubo una tendencia de los pacientes que experimentaron estos acontecimientos de tener menor concentración basal de albúmina, remoción menor de valproato, y un mayor BUN. En pacientes ancianos, la dosis debiera aumentarse más lentamente y con monitoreo regular de consumo nutricional y de fluidos, somnolencia, deshidratación, y otros eventos adversos. La reducción de la dosis o descontinuación del valproato debiera considerarse en pacientes con bajo consumo de alimentos y fluidos, y en pacientes con somnolencia excesiva (ver DOSIS Y ADMINISTRACIÓN). Trombocitopenia Le frecuencia de efectos adversos (particularmente elevaciones de las enzimas hepáticas y trombocitopenia) podrían estar relacionadas con la dosis. En un estudio clínico de divalproato de sodio como monoterapia en pacientes con

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 8 de 29
epilepsia, 34/126 pacientes (27%) recibiendo aproximadamente 50 mg/Kg/ al día en promedio, obtuvieron al menos un valor de plaquetas ≤75 x 109/L. Aproximadamente la mitad de estos pacientes habían interrumpido el tratamiento, con un retorno del conteo de plaquetas a la normalidad. En el resto de los pacientes, el conteo de las plaquetas se normalizó con la continuación del tratamiento. En este estudio, la probabilidad de trombocitopenia pareció aumentar significativamente a concentraciones totales de valproato de ≥ 110 mcg/mL (mujeres) ó ≥ 135 mcg/mL (hombres). El beneficio terapéutico, el cual podría acompañar las dosis más altas, deberá por lo tanto, sopesar la posibilidad de una mayor incidencia en efectos adversos. Convulsiones Post-traumáticas Un estudio fue realizado para evaluar el efecto del valproato IV en la prevención de convulsiones post-traumáticas en pacientes con agudas lesiones en la cabeza. Los pacientes fueron asignados al azar para recibir valproato IV durante una semana (seguido de valproato oral durante uno o seis meses por asignación de tratamiento aleatorio) o fenitoína IV suministrada durante una semana (seguido por placebo). En este estudio, la incidencia de muerte demostró ser más alta en los dos grupos asignados con tratamiento de valproato, con respecto al índice del grupo asignado con tratamiento de fenitoína IV (13% contra 8.5%, respectivamente). Muchos de estos pacientes se encontraban en estado crítico con múltiple y/o lesiones graves, y la evaluación de las causas de la muerte, no precisamente sugirió estar relacionada con un medicamento en específico. Además, en la ausencia de un control de placebo concurrente durante la semana inicial de terapia intravenosa, es imposible determinar si el índice de mortalidad en pacientes tratados con valproato, fue mayor o menor que la que se esperaba en un grupo similar no tratado con valproato, o si la tasa vista en el grupo de pacientes tratados con fenitoína IV fue menor de la que se podría esperar. No obstante, hasta obtener mayor información, parece ser prudente no utilizar valproato de sodio en paciente con trauma agudo en la cabeza para la profilaxis de convulsiones post-traumáticas. Niñas, adolescentes y mujeres en edad fértil y embarazo: Valproato de sodio no debe ser utilizado en niñas, adolescentes, mujeres en edad fértil y mujeres embarazadas a no ser que los tratamientos alternativos sean ineficaces o no tolerados debido a su alto potencial teratogénico y riesgo de trastornos del desarrollo en infantes expuestos dentro del útero al valproato. Los beneficios y los riesgos deben ser cuidadosamente reconsiderados con revisiones regulares al tratamiento, en la pubertad y urgentemente cuando una mujer en edad fértil que esté siendo tratada con valproato de sodio planee un embarazo o si queda embarazada. Las mujeres en edad fértil deben utilizar un método anticonceptivo eficaz durante el tratamiento y estar informadas de los riesgos asociados con el uso de valproato de sodio durante el embarazo. La terapia de valproato solo debe continuarse después una nueva evaluación de los beneficios y los riesgos del tratamiento con valproato para el paciente, realizada por un médico con experiencia en el tratamiento de epilepsia. Las mujeres que planean quedar embarazadas, deben ser asesoradas sobre los riesgos y beneficios del uso de valproato relativos durante el embarazo y las opciones terapéuticas alternativas que deben ser considerados para estos pacientes. Para evitar crisis mayores, valproato no debe interrumpirse bruscamente, ya que esto puede precipitar el estado epiléptico con el resultado de la hipoxia y la amenaza a la vida de la madre y del feto. La evidencia sugiere que los suplementos de ácido fólico antes de la concepción durante el primer trimestre del embarazo reducen el riesgo de defectos congénitos del tubo neural y den la población general. No se sabe si el riesgo de defectos del tubo neural o disminuye el coeficiente intelectual de los hijos de mujeres que reciben valproato se reduce por la administración de suplementos de ácido fólico. Suplementos de ácido fólico en la dieta, tanto antes de la concepción y durante el embarazo debe ser recomendado rutinariamente para los pacientes que usan valproato. Disfunción Hepática Ver CONTRAINDICACIONES y ADVERTENCIAS – Hepatotoxicidad

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 9 de 29
Hiperamonemia La hiperamonemia ha sido reportada en asociación al tratamiento con valproato y puede estar presente a pesar de las pruebas de función hepática normal. En pacientes que desarrollan inexplicables episodios de letargo y vómitos o cambios en su estado mental, se debiera considerar encefalopatía hiperamonémica y la medición del nivel de amoníaco. También debe considerarse la hiperamonemia en pacientes que presentan hipotermia (ver PRECAUCIONES – Hipotermia). Si el amonio aumenta, la terapia de valproato debe ser descontinuada. Intervenciones apropiadas para el tratamiento de hiperamonemia deben ser iniciadas, y los pacientes deben someterse a investigación subyacentes de trastornos del ciclo de la urea (véase CONTRAINDICACIONES y ADVERTENCIAS — Desorden del Ciclo de la Urea – Elevaciones asintomáticas de amoníaco son más frecuentes y, cuando están presentes, se requiere especial monitoreo de los niveles de plasma de amoníaco. Si la elevación persiste, se debe considerar la descontinuación del valproato. Hiperamonemia y Encefalopatía Asociadas al Uso de Topiramato Concomitante La administración de topiramato concomitante y ácido valproico ha sido asociada con hiperamonemia con o sin encefalopatía en pacientes que han tolerado cualquiera de estos fármacos en forma aislada. Los síntomas clínicos de encefalopatía hiperamonemica suelen incluir alteraciones agudas en el nivel de conciencia y/o funciones cognitivas con letargo o vómitos. La hipotermia también puede ser una manifestación de hiperamonemia (ver Hipotermia). En la mayoría de los casos, los síntomas y signos disminuyen con la descontinuación de cualquiera de los medicamentos. Este evento adverso no se debe a una interacción farmacocinética. Se desconoce si la monoterapia de topiramato esta asociada con hiperamonemia. Los pacientes con errores innatos del metabolismo o actividad mitocondrial hepática reducida, pueden estar en mayor riesgo de padecer de hiperamonemia con o sin encefalopatía. Aunque no se ha estudiado, una interacción de topiramato y ácido valproico puede exacerbar los defectos existentes o desenmascarar deficiencias en personas susceptibles. En las personas que desarrollan letargo, vómitos inexplicables, o cambios en el estado mental, la encefalopatía hiperamonémica debe ser considerado medir el nivel de amoníaco (ver CONTRAINDICACIONES y ADVERTENCIAS - Desorden del Ciclo de Urea y PRECAUCIONES-Hiperamonemia). Hipotermia La hipotermia, definida como una baja no intencional en la temperatura del cuerpo a <35°C (95°F), se ha reportado en asociación con la terapia con valproato, tanto en combinación como en la ausencia de hiperamonemia. Esta reacción adversa también puede ocurrir en pacientes usando topiramato concomitantemente con valproato después de iniciar el tratamiento con topiramato o después de incrementar la dosis diaria de topiramato (ver INTERACCIONES MEDICAMENTOSAS – Topiramato). Debe considerarse la interrupción del valproato en pacientes que desarrollan hipotermia, la cual puede manifestarse por una variedad de anormalidades clínicas incluyendo letargo, confusión, coma y alteraciones significativas en otros sistemas de órganos principales como el sistema cardiovascular y el sistema respiratorio. El manejo clínico y la evaluación deben incluir el análisis de los niveles de amoníaco en sangre. Atrofia cerebral Ha habido reportes post mercadeo de atrofia cerebral reversible e irreversible y atrofia cerebelar temporal asociada con el uso de productos conteniendo valproato. En algunos casos, los pacientes se recuperaron con secuelas permanentes (ver sección REACCIONES ADVERSAS). Las funciones motoras y cognitivas de pacientes tomando valproato deben ser controlados rutinariamente y el fármaco debe ser descontinuado en presencia de signos sospechosos o aparentes de atrofia cerebral. Reportes de atrofia cerebral con varias formas de problemas neurológicos incluyendo atrasos en el desarrollo y deterioro psicomotor han sido reportados también en niños que fueron expuestos a productos conteniendo valproato in útero (ver sección de EMBARAZO Y LACTANCIA).

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 10 de 29
Disminución del IQ exposición en el útero: El Valproato puede causar disminución de las puntuaciones de IQ siguientes exposición en el útero. Estudios epidemiológicos publicados han indicado que los niños expuestos al valproato en el útero tienen menores resultados de las pruebas cognitivas que los niños expuestos en el útero a cualquier otro medicamento antiepiléptico o no drogas antiepilépticas. El mayor de estos estudios es un estudio de cohorte prospectivo realizado en los Estados Unidos y el Reino Unido, que encontró que los niños con exposición prenatal al valproato (n=62) tuvieron menor coeficiente intelectual a los 6 años (97 [ 95% IC 94 – 101]) que los niños con exposición prenatal a los otros tratamientos de monoterapia de fármaco antiepiléptico evaluados: Lamotrigina (108 [95% C.I. 105–110]), carbamazepina (105 [95% C.I. 102–108]), y fenitoína (108 [95% C.I. 104–112]). No se sabe cuándo se producen durante el embarazo efectos cognitivos en los niños expuesto al valproato. Debido a que las mujeres de este estudio fueron expuestos a fármacos antiepilépticos durante el embarazo, si el riesgo para la disminución IQ se relaciona con un periodo de tiempo determinado durante el embarazo no puede ser evaluada. A pesar de todos los estudios disponibles tienen limitaciones metodológicas, el peso de la evidencia apoya la conclusión de que la exposición valproato en el útero puede causar una disminución del índice de inteligencia en los niños. En estudios con animales, las crías con la exposición prenatal al valproato tenían malformaciones similares a los observados en los seres humanos y demostraron déficits neuroconductuales. Las mujeres con epilepsia que están embarazadas o que planean quedar embarazadas no deben ser tratadas con valproato a menos que otros tratamientos han fallado en proporcionar un control adecuado de los síntomas o son de otra manera inaceptable. En estas mujeres, los beneficios del tratamiento con valproato durante el embarazo pueden aun ser mayores que los riesgos. General Debido a reportes de trombocitopenia (ver ADVERTENCIAS- Trombocitopenia), la inhibición de la segunda fase de la agregación plaquetaria, y anormalidades en parámetros de la coagulación (por ejemplo, fibrinógeno bajo), se recomienda realizar pruebas de coagulación y recuentos plaquetarios antes de iniciar el tratamiento y a intervalos regulares durante el mismo. En los pacientes que reciben valproato de sodio se recomienda controlar el recuento de plaquetas y los parámetros de coagulación antes de ser sometidos a procedimientos quirúrgicos. En un estudio clínico de monoterapia con divalproato de sodio en pacientes con epilepsia, 34/126 pacientes (27%) recibiendo aproximadamente 50 mg/Kg al día en promedio, hubo al menos un valor de plaquetas de ≤ 75 x 109/L. Aproximadamente la mitad de estos pacientes habían interrumpido el tratamiento, con el regreso del número de plaquetas a la normalidad. En el resto de los pacientes, se normalizó el conteo de plaquetas con la continuación del tratamiento. En este estudio, la probabilidad de trombocitopenia pareció aumentar significativamente a un total de concentraciones de valproato de ≥ 110 mcg/mL (mujeres) ó ≥ 135 mcg/mL (hombres). La evidencia de hemorragia, hematomas o un desorden de homeostasis/coagulación es una indicación para la reducción de la dosis o de descontinuación del tratamiento. Ya que el valproato de sodio puede interactuar simultáneamente con medicamentos concomitantemente administrados que son capaces de inducción enzimática, se recomienda durante el inicio de la terapia, determinaciones periódicas de las concentraciones plasmáticas de valproato y de los fármacos concomitantes, (ver INTERACCIONES MEDICAMENTOSAS). Valproato es parcialmente eliminado en la orina como un cetometabolito que puede conducir a una falsa interpretación de la prueba de cetona en la orina. Ha habido informes de la función tiroidea alterada relacionada con valproato. La importancia clínica de éstos se desconoce. Hay estudios in vitro que sugieren que el valproato estimula la replicación del virus VIH y CMV bajo ciertas condiciones experimentales. La consecuencia clínica, si la hay, no se conoce. Además, la relevancia de estos hallazgos in vitro es incierta para los pacientes que reciben terapia antirretroviral máximamente supresora. Sin embargo, estos datos deben tenerse en cuenta al interpretar los resultados de supervisión periódica de la carga viral en pacientes infectados por VIH en tratamiento con valproato o pacientes infectados con CMV en observación clínica.

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 11 de 29
Pacientes con una subyacente deficiencia de carnitina palmitoiltransferasa (CPT por sus siglas en inglés) tipo II deben ser advertidos sobre un riesgo mayor de rabdomiolisis al tomar valproato. Reacción de Hipersensibilidad Multiorgánica Raramente se ha reportado reacciones de hipersensibilidad multiorgánica en asociación temporal cercana después del inicio de la terapia con valproato en pacientes adultos y pediátricos (el tiempo promedio para la detección es de 21 días; rango de 1 a 40). Aunque ha habido un número limitado de reportes, muchos de estos casos resultaron en hospitalización y una muerte como mínimo. Los signos y síntomas de este trastorno fueron diversos; sin embargo, los pacientes normalmente, aunque no exclusivamente, presentaron cuadros de fiebre y erupción asociados con la participación de otros órganos. Otras manifestaciones asociadas pueden incluir linfadenopatía, hepatitis, anormalidades en la prueba de función hepática, anormalidades hematológicas (por ejemplo: eosinofilia, trombocitopenia, neutropenia), prurito, nefritis, oliguria, síndrome hepato-renal, artralgia astenia. Debido a que el desorden es variable en su expresión, otros síntomas del sistema orgánico y signos, no observados aquí pueden ocurrir. Si esta reacción se sospecha, valproato debería ser interrumpido y comenzar un tratamiento alternativo. Aunque la existencia del cruce de sensibilidad con otros medicamentos que producen este síndrome no está clara, la experiencia entre fármacos asociados con la hipersensibilidad multiorgánica podría indicar una posibilidad. La reacción cutánea con eosinofilia y síntomas sistémicos (DRESS), también conocido como Hipersensibilidad multiorgánica, se ha reportado en pacientes que toman valproato. DRESS puede ser fatal o potencialmente mortal. DRESS por lo general, aunque no exclusivamente, se presentan con fiebre, erupción cutánea y/ o linfadenopatia, en asociación con afectación de otros órganos y sistemas, tales como hepatitis, nefritis, anormalidades hematológicas, miocarditis, o miositis a veces se asemeja a una infección viral aguda. La eosinofilia está a menudo presente. Debido a que este trastorno es variable en su expresión, otros sistemas de órganos no citados aquí pueden estar involucrados. Es importante tener en cuenta que las manifestaciones tempranas de hipersensibilidad, tales como fiebre o linfadenopatía, puede ser incluso cuando la erupción no es evidente. Si tales signos o síntomas están presentes, el paciente debe ser evaluado inmediatamente. El Valproato debe interrumpirse y no se reanudara si no se puede establecer una etiología alternativa para los signos y síntomas. Información para Pacientes Los pacientes y tutores deben ser advertidos de que el dolor abdominal, náuseas, vómitos y/o anorexia podrían ser síntomas de pancreatitis y, por lo tanto, requieren mayor evaluación médica con prontitud. Pacientes y tutores deben ser informados de los signos y síntomas asociados con la encefalopatía hiperamonémica (Ver PRECAUCIONES Y ADVERTENCIAS - Hiperamonemia) y deben ser informados al prescriptor si cualquiera de estos síntomas ocurre. Efectos Sobre La Capacidad Para Conducir y Utilización de Máquinas Dado que el valproato de sodio podría producir depresión del SNC, especialmente cuando se combina con otros depresores del mismo (por ej., alcohol), se aconsejará a los pacientes evitar actividades riesgosas, tales como conducir automóviles y operar maquinaria peligrosa hasta asegurarse de que el medicamento no les provoque somnolencia. Datos De Seguridad Preclínica Carcinogénesis Se administró ácido valproico a ratas Sprague Dawley y ratones ICR (HA/ICR) en dosis de 80 y 170 mg/Kg/día (aproximadamente de 10 al 50% de la dosis humana máximas en un régimen de mg/m2) durante dos años. A pesar de haberse observado una variedad de neoplasmas en ambas especies, los hallazgos principales fueron un incremento estadísticamente significativo en la incidencia de fibrosarcomas subcutáneos en las ratas macho que recibieron altas dosis de ácido valproico y una tendencia de codepencencia estadísticamente significativa en los ratones machos con adenomas pulmonares benignos que recibieron ácido valproico.

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 12 de 29
Se desconoce el significado de estos hallazgos para los seres humanos. Mutagénesis El valproato no produjo mutaciones en la prueba de Ames in vitro, tampoco produjo efectos letales dominantes en ratones ni aumentó la frecuencia de aberraciones cromosómicas en un estudio citogenético in vivo, en ratas. En niños epilépticos que recibían valproato de sodio, se observó un incremento en la frecuencia de intercambio de cromátidas hermanas (ICH), pero no se observó en los adultos. Hay algunas evidencias de que el incremento en la frecuencia de ICH puede estar asociado con la epilepsia. Se desconoce el significado biológico de un aumento en la frecuencia de ICH. Fertilidad Estudios de toxicidad crónica en ratas y perros, jóvenes y adultos, mostraron que el valproato reduce la espermatogénesis y produce atrofia testicular en dosis de 400 mg/Kg al día o más, o de 150 mg/Kg/día o más, respectivamente. Tales dosis son equivalentes a 1.4 veces la dosis diaria máxima en humanos con base a mg/m2, respectivamente. Estudios de fertilidad del segmento I, en ratas, han mostrado que dosis de hasta 350 mg/Kg/día (la dosis diaria máxima humana con base en mg/m2), durante 60 días no afecta la fertilidad. Uso en Pediatría La experiencia con valproato de sodio oral ha indicado que los niños menores de dos años están expuestos a un riesgo considerablemente mayor de hepatotoxicidad fatal, especialmente si reúnen las condiciones mencionadas (ver ADVERTENCIAS Y PRECAUCIONES - Hepatotoxicidad). No se ha estudiado la seguridad de la inyección de valproato en individuos de menos de dos años de edad. Por tanto, si se toma la decisión de utilizarlo en ellos, deberá hacerlo con extrema precaución y como único agente. Se deben tomar en cuenta los beneficios de la inyección contra los riesgos. Por arriba de dos años de edad, la experiencia en la epilepsia ha señalado que la incidencia de hepatotoxicidad fatal disminuye considerablemente en grupos de pacientes progresivamente de mayor edad. Los niños pequeños, en especial aquéllos que reciben fármacos inductores enzimáticos, requerirán mayores dosis de mantenimiento para obtener las concentraciones de ácido valproico totales y libres deseadas. La variabilidad en los límites de la fracción libre limita la utilidad clínica del monitoreo de la concentración plasmática total de ácido valproico. La interpretación de las concentraciones de ácido valproico en niños debe considerar los factores que afectan el metabolismo hepático y la unión a proteínas plasmáticas. No se identificaron aspectos importantes respecto a la seguridad en estudios clínicos con 24 pacientes entre 2 y 17 años de edad, que recibieron valproato de sodio inyectable. La toxicología básica y las manifestaciones patológicas del valproato de sodio en ratas recién nacidas y jóvenes (4 y 14 días de edad, respectivamente) son similares a las observadas en ratas adultas jóvenes. Sin embargo, adicionalmente se han encontrado alteraciones renales en las jóvenes y alteraciones renales con displasia retinal en las recién nacidas, con dosis de 240 mg/Kg/día (equivalente a la dosis máxima diaria en humanos con base en mg/m2). Tales anomalías no se observaron con 90 mg/Kg/día, 40% de la dosis en humanos con mg/m2. Uso en Geriatría Los estudios clínicos prospectivos doble-ciego, llevados a cabo para el tratamiento de la manía asociada con el trastorno bipolar, no incorporaron pacientes de más de 65 años. En un estudio basado en una revisión de casos de 583 pacientes, 72 de ellos (12%) eran mayores de 65 años. Un mayor porcentaje de estos últimos pacientes presentó lesión accidental, infección, dolor, somnolencia y temblor. La suspensión del valproato fue ocasionalmente asociada con los 2 últimos episodios. No se sabe con exactitud si dichos episodios constituyen un riesgo adicional o si son consecuencia de una enfermedad clínica preexistente y del empleo de medicaciones concomitantes entre estos pacientes. Un estudio con pacientes ancianos con demencia reveló somnolencia relacionada al medicamento y descontinuación por somnolencia (ver ADVERTENCIAS-Somnolencia en Ancianos). La dosis inicial debiera reducirse en estos pacientes o su descontinuación debiera ser considerada en pacientes con somnolencia excesiva (ver DOSIS Y ADMINISTRACIÓN).

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 13 de 29
No se identificaron aspectos importantes de seguridad en 19 pacientes > 65 años de edad que recibieron valproato de sodio en estudios clínicos. Convulsiones Agravadas: Al igual que con otros fármacos antiepilépticos, algunos pacientes pueden experimentar, en lugar de una mejora, un empeoramiento reversible de la frecuencia de la convulsión y la gravedad (incluyendo estado epiléptico), o la aparición de nuevos tipos de convulsiones con valproato. En caso de convulsiones agravadas, se debe recomendar a los pacientes consultar a su médico inmediatamente. Información relacionada con excipientes Este producto medicinal contiene 3.5 mmol (141.5 mg) de hidróxido de sodio por dosis (correspondiente a 81.33 mg de sodio). Esto debe ser considerado en pacientes con una dieta controlada de sodio.
9. INTERACCIONES MEDICAMENTOSAS
Efectos de la coadministración de fármacos sobre la depuración del Valproato. Los medicamentos que afectan el nivel de expresión de las enzimas hepáticas, particularmente aquellas que elevan los niveles de las glucuronosiltransferasas (como ritonavir), pueden aumentar la eliminación del valproato. Por ej., la fenitoína, la carbamazepina y el fenobarbital (o la primidona) pueden duplicar la depuración del valproato. Por lo tanto, los pacientes bajo monoterapia generalmente presentarán vidas medias más largas y concentraciones más elevadas que los pacientes que reciben politerapia con medicamentos antiepilépticos. Por el contrario, los inhibidores de las isoenzimas del citocromo P450, por ej. antidepresivos, ejercen poco efecto sobre la depuración del valproato debido a que la oxidación mediada por los microsomas del citocromo P450 es una vía metabólica secundaria de relativamente poca importancia en comparación con la glucuronización y la beta-oxidación. Debido a estas variaciones en la depuración del valproato, siempre que se agreguen o se suspendan inductores enzimáticos, se deberá intensificar el monitoreo de las concentraciones de valproato y fármacos concomitantes. La siguiente lista proporciona información sobre el potencial de influencia de varias medicaciones de prescripción corriente sobre la farmacocinética del valproato. Esta lista no está completa y no podría estarlo nunca ya que continuamente se está informando de nuevas interacciones. Medicamentos Por Los Cuales Importantes Interacciones Potenciales Han Sido Observados Aspirina: Un estudio que comprendió la administración de aspirina a dosis antipiréticas (11 a 16 mg/Kg) con valproato en niños (n = 6), reveló sacar una disminución en la unión a la proteína e inhibición del metabolismo del valproato. La fracción libre de valproato se cuadriplicó en presencia de la aspirina en comparación con el valproato sólo. La vía de la β-oxidación que comprende el 2-E-ácido valproico, 3-OH-ácido valproico y 3-ceto ácido valproico, disminuyó del 25% de los metabolitos totales excretados con valproato a sólo 8.3% en presencia de aspirina. Se deberá observar con precaución al medicar valproato y aspirina. Antibióticos Carbapenémicos: Una reducción clínicamente significativa en la concentración de ácido valproico en suero se ha reportado en pacientes recibiendo antibióticos carbapenems (ertapenem, imipenem, meropenem) y puede resultar en pérdida del control de la crisis. El mecanismo de esta interacción no ha sido bien entendido. Las concentraciones de ácido valproico en suero deben monitorearse frecuentemente después de iniciar la terapia con carbapenem. Debe considerarse una terapia alterna antibacteriana o anticonvulsiva si las concentraciones del ácido valproico en suero bajan significativamente o se deteriora el control de la crisis (ver ADVERTENCIAS Y PRECAUCIONES - Interacciones con antibióticos carbapenémicos).

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 14 de 29
Anticonceptivos hormonales que contienen estrógenos - Los anticonceptivos hormonales que contienen estrógeno pueden incrementar la depuración de valproato, lo que puede resultar en una disminución de la concentración de valproato y potencialmente mayor frecuencia de convulsiones. Los prescriptores deben monitorizar las concentraciones séricas de valproato y la respuesta clínica cuando se añade o se interrumpe productos que contienen estrógenos,
preferiblemente durante los intervalos del ciclo anticonceptivo hormonal. Felbamato: Un estudio sobre la administración de 1200 mg/día de felbamato en combinación con valproato a pacientes con epilepsia (n = 10) reveló un aumento en la unión de proteínas en una concentración media de valproato de 35% (de 86 a 115 mcg/mL) en comparación con sólo valproato. El incremento de la dosis de felbamato a 2400 mg/día, incrementó la concentración media de valproato a 133 mcg/mL (otro 16% más de incremento). Una disminución de la dosis de valproato puede ser necesaria cuando se inicia un tratamiento con felbamato. Rifampicina: Un estudio que investigó la administración de una dosis única de valproato (7 mg/Kg) 36 horas después durante 5 noches de administración diaria de rifampicina (600 mg), reveló un 40% de aumento en la depuración oral del valproato. La administración de valproato y rifampicina podrá requerir el ajuste de la dosis de valproato.
Inhibidores de la proteasa: Los inhibidores de la proteasa tales como lopinavir, ritonavir disminuyen el nivel de
plasma del valproato cuando son coadministrados.
Colestiramina: La colestiramina puede conducir a una disminución del nivel plasmático de valproato cuando son coadministrados. Medicamentos para los que no se ha observado ninguna interacción o probablemente interacción clínica irrelevante. Antiácidos: Un estudio durante el cual se administró 500 mg de valproato con antiácidos (Malox, Trisogel, y Titralac – 160 mEq) no reveló ningún efecto sobre el grado de absorción del valproato. Clorpromazina: Un estudio en el que se administraron de 100 a 300 mg/día de clorpromazina a pacientes esquizofrénicos que ya recibían valproato (200 mg 2 veces al día), reveló un 15% de aumento en los niveles plasmáticos mínimos de valproato. Haloperidol: En un estudio en el que se administraron de 6 a 10 mg/día de haloperidol a pacientes esquizofrénicos que ya recibían valproato (200 mg 2 veces al día), no se registraron cambios significativos en los niveles plasmáticos mínimos de valproato. Cimetidina y Ranitidina: Cimetidina y ranitidina no afectan la depuración de valproato. Efectos de Valproato en Otros Medicamentos Se ha comprobado que el Valproato es un inhibidor débil de algunos isoenzimas P450, epóxido hidrolasas, y glucuroniltransferasas. La siguiente lista provee información sobre el potencial de influencia de la coadministración de valproato sobre la farmacocinética o farmacodinamia de varios medicamentos comúnmente prescritos. Esta lista no está completa, ya que continuamente se reportan nuevas interacciones. Medicamentos en Los Cuales se Han Observado Una Importante Interacción Potencial de Valproato. Amitriptilina/Nortriptilina: La administración de una dosis oral única de 50 mg de amitriptilina en 15 voluntarios sanos (10 hombres y 5 mujeres) que recibían valproato (500 mg 2 veces al día), produjo una disminución del 21% en la

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 15 de 29
depuración de la amitriptilina y del 34% en la depuración neto de la nortriptilina. Se han obtenido pocos informes previos de comercialización del uso simultáneo de valproato y amitriptilina que resultaran en un aumento en el nivel de amitriptilina. El uso simultáneo de valproato y amitriptilina casi no ha sido asociado a toxicidad. Se debe considerar el monitoreo de los niveles de amitripilina en pacientes que toman valproato concomitantemente con amitriptilina. En presencia de valproato, se debe considerar la disminución de la dosis de amitriptilina/nortriptilina. Carbamazepina/Carbamazepina-10, 11-epóxido (CBZ-E): La administración de valproato y de CBZ, reduce un 17% los niveles séricos de CBZ e incrementa hasta 45% los de CBZ-E. Clonazepam: El empleo concomitante de ácido valproico y clonazepam podría inducir estados de ausencia en pacientes con antecedentes de crisis de ausencia. Diazepam: El valproato desplaza al diazepam de sus sitios de unión a la albúmina plasmática e inhibe su metabolismo. La administración de valproato (1500 mg diarios) aumentó la fracción libre del diazepam (10 mg) en un 90% en voluntarios sanos (n = 6). La depuración plasmática y el volumen de distribución del diazepam libre, se redujeron en un 25% y 20%, respectivamente en presencia del valproato. La vida media de eliminación del diazepam no se vio alterada con el agregado de valproato. Etosuximida: El valproato inhibe el metabolismo de la etosuximida. La administración de una sola dosis de 500 mg de etosuximida con valproato (800 a 1600 mg/día) a voluntarios sanos (n = 6), se vio acompañada por un incremento del 25% en la vida media de eliminación de la etosuximida y una disminución del 15% en su depuración total en comparación con sólo etosuximida. Los pacientes que reciben valproato y etosuximida, especialmente junto con otros anticonvulsivantes, deberán ser controlados para detectar alteraciones en las concentraciones séricas de ambos medicamentos. Lamotrigina: En un estudio fijo de diez voluntarios sanos, la eliminación de lamotrigina aumentó de 26 a 70 horas administrado con valproato (a 165% aumento). Se debe disminuir la dosis de lamotrigina cuando se administra con valproato. Reacciones adversas en la piel (tales como el síndrome de Stevens-Johnson y necrosis tóxica epidérmica) han sido reportadas con la administración concomitante de lamotrigina y valproato. Ver inserto del empaque de lamotrigina para detalles en dosis de lamotrigina con valproato concomitante. Fenobarbital: El valproato demostró inhibir el metabolismo del fenobarbital. La administración concomitante de valproato (250 mg dos veces al día por 14 días) con fenobarbital en individuos normales (n = 6) resultó en un 50% de aumento de la vida media y un 30% en disminución de la depuración plasmático de fenobarbital (60 mg única dosis). La fracción de la dosis de fenobarbital excretada sin modificar aumentó un 50% en presencia de valproato. También se ha informado que la combinación de valproato y fenobarbital produce depresión del SNC con o sin elevaciones significativas en los niveles séricos de ambos fármacos. Todos los pacientes que reciben tratamiento concomitante con barbitúricos deberán controlarse estrechamente para detectar signos de toxicidad neurológica. Se deberán obtener las concentraciones séricas de los barbitúricos, si fuera posible, y luego disminuir la dosificación del barbitúrico en caso de ser necesario. Fenitoína: El valproato desplaza a la fenitoína de sus sitios de unión a la albúmina plasmática e inhibe su metabolismo hepático. La administración de valproato (400 mg tres veces por día) con fenitoína (250 mg) en voluntarios sanos (n = 7) fue asociada con un incremento del 60% en la fracción libre de la fenitoína. La depuración plasmática total y el volumen de distribución aparente de la fenitoína, aumentaron un 30% en presencia de valproato. Hubo informes de crisis convulsivas importantes con la combinación de valproato y fenitoína en pacientes con epilepsia. La dosis de fenitoína deberá ajustarse según la situación clínica.

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 16 de 29
Antiepilépticos con efecto de inductores enzimáticos (incluyendo fenitoína, fenobarbital, carbamazepina) reducen las concentraciones séricas de Valproato. Las dosis deben ser ajustadas de acuerdo a la respuesta clínica de los niveles de sangre en caso de terapia combinada. Primidona: La primidona se metaboliza en un barbitúrico y, por lo tanto, también puede estar involucrado en una interacción similar con valproato como fenobarbital. Propofol: Una interacción clínicamente significativa entre Valproato y Propofol puede llevar a un incremento en el nivel sanguíneo de Propofol. Por lo tanto, cuando se coadminstra con Valproato, la dosis de propofol debe ser reducida. Nimodipino: El tratamiento concomitante de nimodipino con ácido valproico puede incrementar las concentraciones plasmaticas del nimodipino en un 50%. Tolbutamida: Los experimentos in vitro revelaron que la fracción libre de la tolbutamida se vio incrementada de 20% a 50% cuando se le agregó a las muestras de plasma recolectadas de pacientes tratados con valproato. Se desconoce su relevancia clínica. Topiramato y acetazolamida: La administración concomitante de valproato y topiramato o acetazolamida ha sido asociada con encefalopatía y/o hiperamonemia. Los pacientes tratados con esos dos medicamentos deben ser monitorizados cuidadosamente por la aparición de signos y síntomas de encefalopatía hiperamonémica.
La administración concomitante de topiramato con ácido valproico se ha asociado con la hipotermia en los pacientes que han tolerado cualquiera de los medicamentos solos. Los niveles de amoníaco en la sangre se debe medir en pacientes cuando se reporta hipotermia (ver PRECAUCIONES - Hipotermia e Hiperamonemia).
Warfarina - En un estudio in vitro, el valproato aumentó la fracción libre de la warfarina hasta un 32.6%. Se desconoce la relevancia terapéutica de este dato; sin embargo, se deberá controlar los parámetros de coagulación cuando se administre valproato a pacientes que reciben anticoagulantes. Zidovudina: En 6 pacientes HIV-seropositivo, la depuración de la zidovudina (100 mg c/8 hrs) disminuyó en un 38% después de la administración de valproato (250 ó 500 mg c/8 hrs); la vida media de la zidovudina no se vio alterada. Quetiapina: La co-administración de quetiapina y valproato puede incrementar el riesgo de neutropenia/leucopenia Medicamentos para los cuales no se ha observado interacción ni probable interacción clínica sin importancia. Acetaminofen: El valproato no afectó ninguno de los parámetros farmacocinéticos del acetaminofen cuando se administró concomitantemente a 3 pacientes epilépticos. Clozapina: En pacientes psicóticos (n = 11) no se observó interacción cuando se administró valproato concomitante con clozapina. Litio: La administración de valproato concomitante (500 mg dos veces por día) y carbonato de litio (300 mg tres veces por día) a voluntarios hombres sanos (n = 16), no afectó la cinética del litio en el estado estable. Lorazepam: La administración concomitante de valproato (500 mg dos veces al día) y lorazepam (1 mg dos veces al día) en hombres voluntarios sanos (n = 9), se acompañó de un descenso del 17% en la depuración plasmática de lorazepam.

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 17 de 29
Olanzapina- El ácido valproico puede disminuir la concentración plasmática de olanzapina. Rufinamida: El ácido valproico puede provocar un aumento del nivel plasmático de la rufinamida. Este aumento depende de la concentración del ácido valproico. Se debe tener precaución, especialmente en los niños, dado que este efecto es mayor en esta población. Con base a un análisis farmacocinético de la población, el aclaramiento de rufinamida se redujo con el valproato. Las
concentraciones de rufinamida se incrementaron en ˂16% a 70 %, dependiendo de la concentración de valproato (con
los aumentos más grandes que se observan en paciente pediátricos a dosis altas o concentraciones de valproato). Los pacientes estabilizados con rufinamida antes de ser prescrito valproato deben comenzar la terapia con valproato a dosis bajas, y se valora a una dosis clínicamente eficaz. Del mismo modo, los pacientes tratados con valproato deberían comenzar en una dosis de rufinamida más baja de 10 mg/kg por día (pacientes pediátricos) o 400 mg por día (adultos).
10. EMBARAZO Y LACTANCIA
Categoría D en embarazo para las indicaciones de epilepsia. Valproato de sodio no debe utilizarse en niñas, adolescentes, mujeres en edad fértil y en mujeres embarazadas a menos que otros tratamientos sean ineficaces o no se toleren. Las mujeres en edad fértil tienen que utilizar un método anticonceptivo eficaz durante el tratamiento. En las mujeres que estén planeando embarazarse deben hacer todos los esfuerzos posibles para cambiar a alternativas de tratamiento adecuadas antes de la concepción, si es posible
Riesgo de exposición en el embarazo relacionado con el valproato
Tanto valproato en monoterapia como valproato en politerapia o terapia combinada están asociados con resultados
anormales de embarazo. Los datos disponibles sugieren que la politerapia antiepiléptica que incluye valproato se asocia
con un mayor riesgo de malformaciones congénitas comparado con el valproato en monoterapia.
Malformaciones congénitas
Los datos obtenidos a partir de un metaanálisis (incluyendo los registros y estudios de cohorte) ha demostrado que
10.73% de los niños de mujeres epilépticas que fueron expuestos al valproato en monoterapia durante el embarazo
sufren de malformaciones congénitas (95% IC: 8.16 -13.29). Este es un riesgo elevado de malformaciones mayores que
para la población en general, para los cuales el riesgo es de aproximadamente 2-3%. El riesgo depende de la dosis,
pero no se puede establecer una dosis de umbral menor de la cual no existe ningún riesgo.
Los datos disponibles indican un aumento en la incidencia de malformaciones mayores y menores. Los tipos más
comunes de las malformaciones incluyen defectos del tubo neural, dismorfias faciales, labio leporino y paladar hendido,
craneoestenosis, defectos cardiacos, renales y urogenitales, defectos en las extremidades (incluidos aplasia bilateral del
radio), y múltiples anomalías que involucran a varios sistemas del organismo.
Trastornos del desarrollo
Los datos han demostrado que la exposición al valproato en el útero puede tener efectos adversos en el desarrollo
mental y físico de los niños expuestos. El riesgo parece ser dependiente de la dosis, pero una dosis de umbral menor
de la cual no existe el riesgo, no puede ser establecida basada en los datos disponibles. El período gestacional exacto
de riesgo para estos efectos es incierto y no se puede excluir la posibilidad de riesgo durante todo el embarazo.

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 18 de 29
Los estudios en niños en edad preescolar expuestos en el útero a valproato muestran que hasta un 30-40% sufren de
retrasos en su desarrollo temprano, tal como hablar y caminar más tarde, menor capacidad intelectual, malas aptitudes
del lenguaje (hablar y entender) y problemas de la memoria. El coeficiente intelectual (CI) medido en niños en edad escolar (6 años de edad) con un historial de exposición al valproato en el útero fue en promedio 7 a 10 puntos menos que aquellos niños expuestos a otros antiepilépticos. A pesar de que el papel de los factores de confusión no puede excluirse, existe evidencia en los niños expuestos al valproato de que el riesgo de deterioro intelectual puede ser independiente del coeficiente intelectual (CI) de la madre. Existen datos limitados sobre los resultados a largo plazo. Los datos disponibles muestran que los niños expuestos al valproato en el útero están en mayor riesgo de trastorno del espectro autista (aproximadamente tres veces) y autismo infantil (alrededor de cinco veces) en comparación con la población general. Datos limitados sugieren que los niños expuestos al valproato en el útero pueden ser más propensos a desarrollar síntomas del trastorno por déficit de atención/hiperactividad (TDAH). Niñas, adolescentes y mujeres en edad fértil Si una mujer quiere planificar un embarazo:
Durante el embarazo, las convulsiones tónico clónicas maternas y estado epiléptico con hipoxia pueden llevar a un riesgo particular de muerte para la madre y el feto.
En las mujeres que estén planeando quedar embarazadas o que ya están embarazadas, se debe volver a evaluar la terapia con valproato
En las mujeres que estén planeando quedar embarazadas deben hacer todos los esfuerzos posibles para cambiar a alternativas de tratamiento adecuadas antes de la concepción, si es posible.
La terapia con valproato no debe ser descontinuada sin una nueva evaluación de los beneficios y los riesgos del tratamiento con valproato para el paciente, realizada por un médico con experiencia en el tratamiento de epilepsia. Si basado en una evaluación cuidadosa de los riesgos y los beneficios se continúa con el tratamiento con valproato durante el embarazo, se recomienda lo siguiente: Utilizar la menor dosis eficaz y dividir la dosis diaria de valproato en varias dosis pequeñas a tomarse durante todo
el día. El uso de una formulación de liberación prolongada puede ser preferible a otras formulaciones de tratamiento a fin de evitar concentraciones plasmáticas muy elevadas.
Los suplementos de ácido fólico antes del embarazo pueden disminuir el riesgo de defectos del tubo neural que es común para todos los embarazos. Sin embargo, la evidencia disponible no sugiere que evite los defectos de nacimiento o malformaciones debido a la exposición del valproato.
Instituir un control prenatal especializado para detectar la posible aparición de defectos del tubo neural y otras malformaciones.
Informar al pediatra y a cualquier otro profesional de la salud que esté a cargo del control del bebé luego del nacimiento, para que lo vigilen estrechamente a fin de detectar posibles trastornos del desarrollo y se establezcan en forma oportuna las medidas más adecuadas según cada caso.
Riesgo en el neonato
- Muy raramente se han reportado casos de síndrome hemorrágico en los recién nacidos cuyas madres han tomado valproato durante el embarazo. Este síndrome hemorrágico se relaciona con la trombocitopenia, hipofibrinogenemia y/o a la disminución de otros factores de la coagulación. También se ha reportado afibrinogenemia y puede ser mortal. Sin embargo, este síndrome debe distinguirse de la disminución de los factores de la vitamina K inducidos por el fenobarbital y por los inductores enzimáticos. Por lo tanto, el recuento de plaquetas, el nivel plasmático del fibrinógeno, las pruebas de coagulación y los factores de coagulación deben ser investigados en los recién nacidos.
- Se han reportado casos de hipoglucemia en recién nacidos cuyas madres han tomado valproato durante el tercer

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 19 de 29
trimestre de su embarazo. - Se han reportado casos de hipotiroidismo en los recién nacidos cuyas madres han tomado valproato durante el
embarazo. - Puede ocurrir el Síndrome de abstinencia (por ejemplo, en particular, agitación, irritabilidad, hiperexcitabilidad,
nerviosismo, hiperquinesia, trastornos de tonicidad, temblor, convulsiones y trastornos en la alimentación) en los recién nacidos cuyas madres han tomado valproato durante el último trimestre de su embarazo.
Lactancia materna El valproato se excreta en la leche materna con una concentración que oscila desde 1% a 10% de los niveles séricos maternos. Se han demostrado trastornos hematológicos en los recién nacidos y los infantes amamantados de las mujeres tratadas. Se debe tomar una decisión si se va a interrumpir la lactancia materna o si se descontinúa o se abstiene de la terapia de valproato de sodio teniendo en cuenta el beneficio de la lactancia materna para el niño y el beneficio de la terapia para la mujer. Fertilidad Se ha reportado amenorrea, ovarios poliquísticos e incremento en los niveles de testosterona en las mujeres utilizando valproato. La administración de valproato también puede dañar la fertilidad en los hombres. Informes de caso indican que las disfunciones de la fertilidad son reversibles después de la descontinuación del tratamiento.
11. REACCIONES ADVERSAS
Los eventos adversos que induce la inyección de valproato de sodio incluyen a los observados en sus presentaciones orales. Lo siguiente describe la experiencia específicamente con la inyección de valproato de sodio. La forma inyectable, por lo general, ha sido bien tolerada en estudios clínicos realizados en 111 hombres voluntarios sanos y en 352 pacientes epilépticos, en dosis de 125 a 6,000 mg (dosis total/diaria). Un total de 2% de pacientes descontinuaron el tratamiento con valproato de sodio inyectable debido a eventos adversos, siendo los más comunes (2 casos cada uno) náusea/vómito y amilasa elevada. Otros que llevaron a la suspensión del uso del fármaco fueron: alucinaciones, neumonía, cefalea, reacción en el sitio de inyección y marcha anormal. El mareo y el dolor en el sitio de inyección fueron observados más frecuentemente a tasas de infusión de 100 mg/min que a tasas de hasta 33 mg/min. A velocidad de 200 mg/min, el mareo y la percepción del sabor ocurrieron con mayor frecuencia que a 100 mg/min. La velocidad máxima de infusión estudiada fue de 200 mg/min. Los eventos adversos informados por al menos 0.5% de todos los pacientes en los estudios clínicos con valproato de sodio inyectable, se muestran en resumen en la tabla 1.
Tabla 1
Eventos adversos reportados durante estudios con valproato de sodio inyectable
Sistema corporal/Evento n = 463
General
Dolor torácico 1.7%
Cefalea 4.3%
Inflamación en el sitio de inyección 0.6%
Dolor en el sitio de inyección 2.6%
Reacción en el sitio de inyección 2.4%
Dolor (inespecífico) 1.3%
Cardiovascular
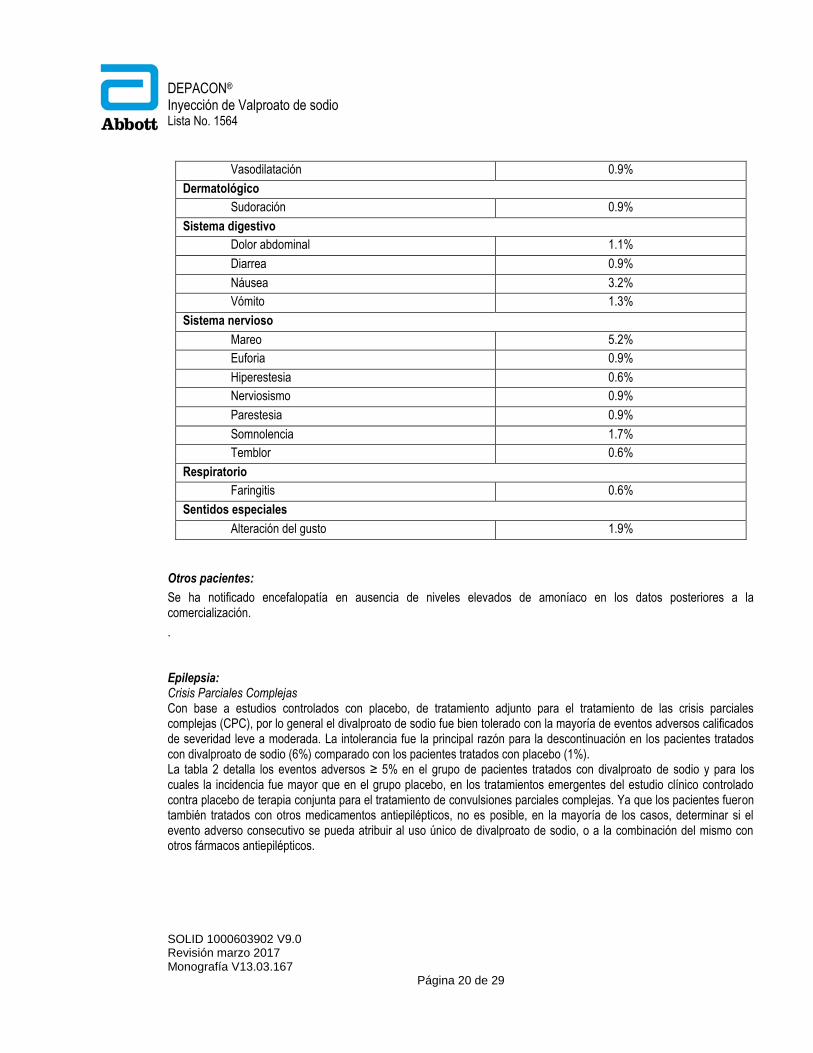
DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 20 de 29
Vasodilatación 0.9%
Dermatológico
Sudoración 0.9%
Sistema digestivo
Dolor abdominal 1.1%
Diarrea 0.9%
Náusea 3.2%
Vómito 1.3%
Sistema nervioso
Mareo 5.2%
Euforia 0.9%
Hiperestesia 0.6%
Nerviosismo 0.9%
Parestesia 0.9%
Somnolencia 1.7%
Temblor 0.6%
Respiratorio
Faringitis 0.6%
Sentidos especiales
Alteración del gusto 1.9%
Otros pacientes:
Se ha notificado encefalopatía en ausencia de niveles elevados de amoníaco en los datos posteriores a la comercialización.
. Epilepsia: Crisis Parciales Complejas Con base a estudios controlados con placebo, de tratamiento adjunto para el tratamiento de las crisis parciales complejas (CPC), por lo general el divalproato de sodio fue bien tolerado con la mayoría de eventos adversos calificados de severidad leve a moderada. La intolerancia fue la principal razón para la descontinuación en los pacientes tratados con divalproato de sodio (6%) comparado con los pacientes tratados con placebo (1%). La tabla 2 detalla los eventos adversos ≥ 5% en el grupo de pacientes tratados con divalproato de sodio y para los cuales la incidencia fue mayor que en el grupo placebo, en los tratamientos emergentes del estudio clínico controlado contra placebo de terapia conjunta para el tratamiento de convulsiones parciales complejas. Ya que los pacientes fueron también tratados con otros medicamentos antiepilépticos, no es posible, en la mayoría de los casos, determinar si el evento adverso consecutivo se pueda atribuir al uso único de divalproato de sodio, o a la combinación del mismo con otros fármacos antiepilépticos.

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 21 de 29
Tabla 2 Eventos adversos informados por ≥ 5% de pacientes tratados con divalproato de sodio durante estudios
controlados acerca del tratamiento conjunto para convulsiones parciales complejas
Sistemas corporales/eventos DVP* (%) (n = 77)
Placebo (%) (n = 70)
General
Cefalea 31 21
Astenia 27 7
Fiebre 6 4
Sistema gastrointestinal
Náusea 48 14
Vómito 27 7
Dolor abdominal 23 6
Diarrea 13 6
Anorexia 12 0
Dispepsia 8 4
Constipación 5 1
Sistema nervioso
Somnolencia 27 11
Temblor 25 6
Mareo 25 13
Diplopía 16 9
Ambliopía/visión borrosa 12 9
Ataxia 8 1
Nistagmo 8 1
Labilidad emocional 6 4
Pensamiento anormal 6 0
Amnesia 5 1
Sistema respiratorio
Síndrome gripal 12 9
Infección 12 6
Bronquitis 5 1
Rinitis 5 4
Otros
Alopecia 6 1
Pérdida de peso 6 0
* DVP = Divalproato de sodio
En la tabla 3 se listan eventos adversos que surgen durante el tratamiento y que fueron informados en ≥ 5% de pacientes en el grupo con la dosis más alta de divalproato de sodio y en quienes la incidencia fue mayor para el grupo con la dosis más baja, en estudios controlados como monoterapia de convulsiones parciales complejas. Ya que durante la primera parte del estudio, los pacientes habían sido titulados en retirada de otro fármaco antiepiléptico, no es posible
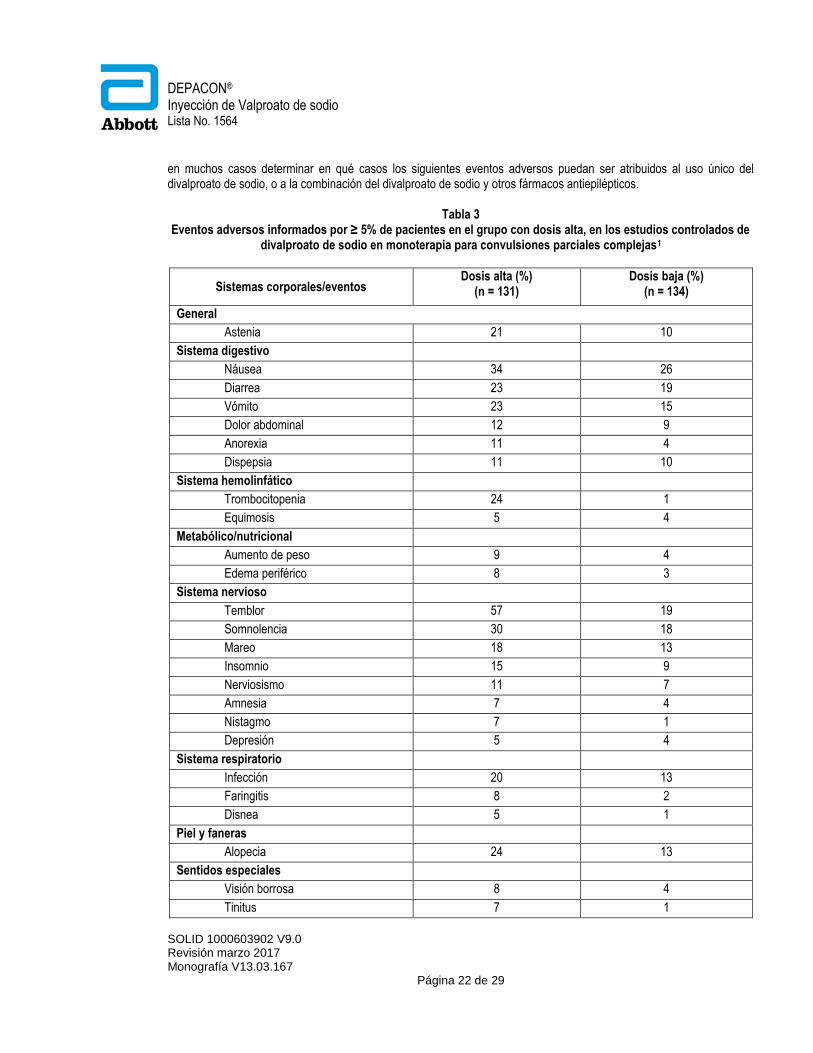
DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 22 de 29
en muchos casos determinar en qué casos los siguientes eventos adversos puedan ser atribuidos al uso único del divalproato de sodio, o a la combinación del divalproato de sodio y otros fármacos antiepilépticos.
Tabla 3 Eventos adversos informados por ≥ 5% de pacientes en el grupo con dosis alta, en los estudios controlados de
divalproato de sodio en monoterapia para convulsiones parciales complejas1
Sistemas corporales/eventos Dosis alta (%)
(n = 131) Dosis baja (%)
(n = 134)
General
Astenia 21 10
Sistema digestivo
Náusea 34 26
Diarrea 23 19
Vómito 23 15
Dolor abdominal 12 9
Anorexia 11 4
Dispepsia 11 10
Sistema hemolinfático
Trombocitopenia 24 1
Equimosis 5 4
Metabólico/nutricional
Aumento de peso 9 4
Edema periférico 8 3
Sistema nervioso
Temblor 57 19
Somnolencia 30 18
Mareo 18 13
Insomnio 15 9
Nerviosismo 11 7
Amnesia 7 4
Nistagmo 7 1
Depresión 5 4
Sistema respiratorio
Infección 20 13
Faringitis 8 2
Disnea 5 1
Piel y faneras
Alopecia 24 13
Sentidos especiales
Visión borrosa 8 4
Tinitus 7 1

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 23 de 29
1Dolor de cabeza fue el único evento secundario que ocurrió en ≥ 5% de pacientes en el grupo con dosis alta y una incidencia igual o mayor en el grupo con dosis baja.
Los siguientes eventos secundarios adicionales fueron informados en proporción > 1% y < 5% en 358 pacientes tratados con divalproato de sodio, en los estudios controlados, de convulsiones parciales complejas: Cuerpo total: Dolor de espalda, dolor torácico y malestar. Sistema Cardiovascular: Taquicardia, hipertensión y palpitaciones. Sistema Digestivo: Hiperfagia, flatulencia, hematemesis, eructos, pancreatitis y abscesos periodontales. Sistema Hemolinfático: Petequias. Metabólico/nutricional: Aumento de TGO y TGP. Sistema Musculoesquelético: Mialgia, contracciones, artralgias, calambres en las piernas y miastenia. Sistema Nervioso: Ansiedad, confusión, marcha anormal, parestesia, hipertonía, incoordinación, sueños anormales y trastornos de personalidad. Sistema Respiratorio: Sinusitis, aumento de tos, neumonía y epistaxis. Piel y anexos: Salpullido, prurito y piel seca. Sentidos especiales: Alteración del gusto, visión anormal, sordera y otitis media. Sistema urogenital: Incontinencia urinaria, vaginitis, dismenorrea, amenorrea y frecuencia urinaria. Otra población de pacientes Son listados a continuación, los eventos adversos que han sido reportados con todas las dosis de valproato en los estudios clínicos de epilepsia, reportes espontáneos y otras fuentes. Gastrointestinal Unos de los efectos secundarios más comunes que se reportan al iniciar la terapia son: náusea, vómito e indigestión. Estos efectos son transitorios y rara vez se requiere suspender el tratamiento. La diarrea, calambres abdominales, constipación, desórdenes gingivales (principalmente hiperplasia gingival), anorexia con algo de pérdida de peso, hiperorexia con aumento de peso también han sido reportados. El uso de una forma de liberación retardada oral de divalproato de sodio puede reducir los efectos gastrointestinales en algunos pacientes con terapia oral. Se ha reportado obesidad en casos raros en la experiencia post mercadeo. Efectos SNC Se han notado efectos sedantes en pacientes que sólo reciben valproato, pero esto ocurre más a menudo en pacientes que reciben terapia combinada. La sedación usualmente desaparece con la reducción de otro medicamento antiepiléptico. Temblor (puede tener relación a la dosis), alucinaciones, ataxia, cefalea, nistagmo, diplopía, asterixis, “manchas ante los ojos”, disartria, mareo, confusión, hipoestesia, vértigo, incoordinación, discapacidad en la memoria, desorden cognitivo y trastornos extrapiramidales incluyendo parkinsonismo han sido reportados con el uso del valproato. Extraños casos de coma han ocurrido en pacientes recibiendo solo valproato o en combinación con fenobarbital. En raras instancias se ha desarrollado luego de la introducción de valproato monoterapia, sin evidencia de disfunción hepática o niveles inapropiadamente altos de valproato plasmático, encefalopatía con o sin fiebre. La mayoría de los pacientes se recuperaron con mejoría notable de los síntomas, al discontinuar el fármaco, aunque ha habido fatalidades en pacientes con encefalopatía hiperamonémica, particularmente en pacientes con desórdenes del ciclo de la urea (ver ADVERTENCIAS y PRECAUCIONES – Desórdenes del Ciclo de la Urea - Hiperamonemia y encefalopatía asociada al uso concomitante de topiramato e Hiperamonemia). Además, se han notificado casos de encefalopatía en ausencia de niveles elevados de amoníaco. Convulsiones agravadas (frecuencia: poco frecuentes) también han sido reportados en pacientes con epilepsia y tratados con monoterapias de valproato. Ha habido reportes post mercadeo de atrofia cerebral reversible e irreversible y atrofia cerebelar temporal asociada con el uso de productos conteniendo valproato. En algunos casos, los pacientes se recuperaron con secuelas permanentes (ver sección ADVERTENCIAS Y PRECAUCIONES – Atrofia cerebral). Atrofia cerebral vista en niños expuestos a

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 24 de 29
valproato in útero condujo a varias formas de eventos neurológicos, incluyendo atrasos en el desarrollo y deterioro psicomotor. Se han reportado también malformaciones congénitas y trastornos del desarrollo (ver sección de EMBARAZO Y LACTANCIA). Dermatológico Pérdida del cabello transitoria, desórdenes del cabello (como textura anormal, cambio en el color, crecimiento anormal), erupción cutánea, fotosensibilidad, prurito generalizado, eritema multiforme, y síndrome de Stevens-Johnson. Se han reportado casos raros de necrólisis epidérmica tóxica incluyendo un caso fatal en un infante de seis meses de edad medicado con valproato y otros medicamentos concomitantes. Un caso adicional de necrólisis epidérmica tóxica que resultó fatal, se informó en un paciente con SIDA de 35 años de edad tomando varios medicamentos concomitantes y con un historial de múltiples reacciones a medicamentos cutáneos. Las reacciones adversas cutáneas se han informado en la administración concomitante de lamotrigina y valproato (ver INTERACCIONES MEDICAMENTOSAS - Lamotrigina). Trastornos de la uña y el lecho ungueal se han reportado también en el marco de la experiencia post mercadeo. Psiquiátricos Se ha reportado alteración emocional, depresión, psicosis, agresividad, hiperactividad psicomotora, hostilidad, agitación, alteración en la atención, comportamiento anormal, desorden de aprendizaje y deterioro del comportamiento. Musculoesquelético Debilidad Se han recibido reportes de reducción de la masa ósea, lo que potencialmente causaría osteoporosis y osteopenia, durante la terapia a largo plazo con medicaciones anticonvulsivos, incluyendo valproato. Algunos estudios han indicado que el suplemento con calcio y vitamina D puede ser de beneficio para los pacientes que están con una terapia crónica con valproato. Hematológico Trombocitopenia e inhibición de la segunda fase de agregación plaquetaria, el cual se ve reflejado en la alteración del tiempo de sangrado, petequias, moretones, hematoma, epistaxis y hemorragia franca (ver PRECAUCIONES- General e INTERACCIONES MEDICAMENTOSAS - Warfarina). Linfocitosis relativa, macrocitosis, hipofibrinogenemia, leucopenia, eosinofilia, anemia incluyendo macrocítica con o sin deficiencia de folato, supresión de médula ósea, pancitopenia, anemia aplásica, porfiria aguda intermitente y agranulocitosis. Hepáticos Es frecuente observar ligeras elevaciones de las transaminasas (por ej. TGO y TGP) y de la LDH, las que parecen ser dosis-dependientes. Ocasionalmente, los resultados de las pruebas de laboratorio incluyen también aumentos en la bilirrubina sérica y alteraciones en otras pruebas de la función hepática. Estos resultados pueden ser reflejo de hepatotoxicidad potencialmente severa (ver ADVERTENCIAS - Hepatotoxicidad). Endocrino Menstruación irregular, amenorrea secundaria, crecimiento de los senos, galactorrea e hinchazón de la glándula parótida, incluyendo hipotiroidismo. Hiperandrogenismo (hirsutismo, virilismo, acné, alopecia parcheada masculina y/o incremento de andrógenos). Pruebas de función tiroidea anormal (ver PRECAUCIONES - General). Ha habido casos muy raros de enfermedad del ovario poliquístico. No se ha establecido una relación de causa y efecto. Pancreáticos Se han comunicado episodios de pancreatitis aguda, incluyendo casos fatales (ver PRECAUCIONES - Pancreatitis).

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 25 de 29
Metabólicos Hiperamonemia (ver PRECAUCIONES- Hiperamonemia), hiponatremia y secreción inadecuada de hormona antidiurética. Se han registrado casos raros de síndrome de Fanconi, principalmente en niños. Se ha informado de concentraciones disminuidas de carnitina, aunque se desconoce su relevancia clínica. Se ha informado de hiperglicinemia, la que fue asociada con la muerte de un paciente con hiperglicinemia no-cetósica preexistente. Resistencia a la insulina y dislipidemia han sido también reportados en el marco de la experiencia post-comercialización. Genitourinario Enuresis, fallo renal, nefritis tubulointersticial e infección del tracto urinario. Reproducción Se ha reportado infertilidad masculina incluyendo azoospermia, análisis anormales de esperma, disminución de la cuenta de esperma, anormalidad en la morfología de espermatozoides, aspermia, y disminución en la movilidad de espermatozoides. Sentidos Especiales Se han comunicado casos reversibles e irreversibles de pérdida de la audición; sin embargo, no se ha establecido una relación de causa y efecto. También se ha reportado dolor de oído. Neoplasmas benignos, malignos e inespecíficos (incluyendo quistes y pólipos) Síndrome mielodisplásico Desórdenes respiratorios, torácicos y mediastinales Efusión pleural Otros Reacciones alérgicas, anafilaxis, edema de las extremidades, lupus eritematoso, rabdomiólisis, deficiencia de biotina/ deficiencia de biotinidasa, dolor de huesos, aumento de tos, neumonía, otitis media, bradicardia, vasculitis cutánea, fiebre, e hipotermia. Manía Aunque valproato de sodio I.V. no ha sido evaluado para la seguridad y eficacia en el tratamiento de episodios maníacos asociados con el trastorno bipolar, las siguientes reacciones adversas no detalladas anteriormente, fueron reportadas por el 1% o más, de pacientes de dos estudios clínicos controlados con placebo y tabletas de divalproato de sodio.
General: Escalofríos, dolor de cuello y rigidez de cuello. Sistema Cardiovascular: Hipotensión, hipotensión postural, vasodilatación. Sistema Digestivo: incontinencia fecal, gastroenteritis, glositis. Sistema Musculoesquelético: Artrosis Sistema Nervioso: Agitación, reacción catatónica, hipocinesia, aumento de reflejos, discinesia tardía, vértigo. Piel y Anexos: Furunculosis, erupción maculopopular, seborrea. Sentidos Especiales: Conjuntivitis, ojos secos, dolor de ojos. Urogenitales: Disuria
Otros pacientes: Se han notificado trastornos extrapiramidales y encefalopatía en ausencia de niveles elevados de amoniaco en los datos posteriores a la comercialización.

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 26 de 29
Migraña A pesar de que el valproato de sodio I.V. no ha sido evaluado para la seguridad y eficacia en tratamientos profilácticos de cefalea migrañosa, los efectos negativos que no se mencionaron anteriormente fueron reportados por un 1% o más de pacientes de dos estudios clínicos con control de placebo de divalproato de sodio en tabletas.
General: Edema facial Sistema Digestivo: Boca seca, estomatitis. Urogenital: Cistitis, metrorragia, hemorragia vaginal.
Otros pacientes:
Se ha notificado encefalopatía en ausencia de niveles elevados de amoníaco en los datos posteriores a la comercialización.
Reporte de sospecha de reacciones adversas
Es importante reportar cualquier sospecha de reacción adversa después de la autorización del medicamento. Esto
permite el control continuo del equilibrio entre el beneficio/riesgo del medicamento. Se solicita a los profesionales de
salud que reporten sobre cualquier sospecha de reacciones adversas a través del sistema nacional de informes.
12. SOBREDOSIS
La sobredosis con valproato puede producir somnolencia, bloqueo cardiaco, hipotensión y shock/colapso circulatorio y coma profundo. Se han informado casos fatales; sin embargo, los pacientes se han recuperado de niveles de valproato tan altos como 2,120 mcg/mL. La presencia del contenido de sodio en las formulaciones de valproato pueden causar hipernantremia cuando se toma una sobredosis. En los casos de sobredosificación, la fracción del fármaco no unido a las proteínas es alta, y con la hemodiálisis o la hemodiálisis tándem junto con la hemoperfusión, se puede eliminar gran parte del mismo. Se deberá aplicar medidas generales de apoyo prestando particular atención al mantenimiento de una adecuada diuresis. Naloxona puede revertir los efectos depresores de la sobredosis de valproato sobre el SNC. Debido a que naloxona teóricamente también podría revertir los efectos antiepilépticos del valproato, deberá emplearse con precaución en pacientes con epilepsia. 13. FARMACOLOGÍA Farmacodinamia El Valproato de sodio existe como ion valproato en la sangre. El mecanismo mediante el cual el valproato ejerce sus efectos terapéuticos, no se ha establecido. Se ha sugerido que su actividad en la epilepsia está relacionada con el incremento de concentraciones de ácido gammaaminobutírico (GABA) en el cerebro. Farmacocinética — Absorción/Biodisponibilidad Se espera que dosis equivalentes de valproato I.V. y de productos orales de valproato, resulten en valores equivalentes de Cmax, Cmin y de exposición sistémica total al ion de valproato. Sin embargo, la proporción de la absorción de ion

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 27 de 29
valproato podría variar con la formulación utilizada. Bajo las condiciones de estado estable alcanzado con el uso crónico en el tratamiento de la epilepsia, estas diferencias deberían ser de importancia clínica menor. La administración de divalproato de sodio en tabletas y valproato I.V. (infusión para una hora), en dosis de 250 mg, cada 6 horas, durante 4 días en 18 voluntarios masculinos sanos, resultó en ABC, Cmax, Cmín equivalentes en estado estable, al igual que después de la primera dosis. El Tmax después del valproato de sodio I.V. ocurrió al final de la primera hora de infusión, mientras que el Tmax posterior a la dosis oral de valproato semisódico ocurrió a las 4 horas aproximadamente. Debido a que la cinética del valproato no unido es lineal, se puede asumir la bioequivalencia entre valproato semisódico y divalproato de sodio, hasta las dosis máximas recomendadas de 60 mg/Kg/día. También fueron equivalentes el ABC y la Cmax resultantes de la administración de valproato I.V. 500 mg como una sola infusión durante 1 hora y una sola dosis de 500 mg de ácido valproico en jarabe a 17 adultos masculinos sanos. Los pacientes mantenidos con dosis diarias de 750 mg ó 4,250 mg de ácido valproico (en dosis divididas cada 6 horas) como divalproato sódico oral solo (n = 24), o con otro fármaco antiepiléptico estabilizador [carbamazepina, (n = 15); fenitoína, (n = 11) o fenobarbital, (n =1)] mostraron niveles plasmáticos comparables de ácido valproico cuando se cambió del divalproato de sodio oral al valproato I.V. (infusión de 1 hora). Farmacocinética – Distribución Unión de Proteínas La unión del valproato a las proteínas plasmáticas depende de la concentración y la fracción libre aumenta alrededor del 10% a 40 mcg/mL hasta el 18.5% a 130 mcg/mL. La unión del valproato a las proteínas se ve reducida en los ancianos, en pacientes con hepatopatías crónicas, pacientes con insuficiencia renal y en presencia de otros medicamentos (por ej. aspirina). Por el contrario, el valproato puede desplazar a ciertos medicamentos que se unen a las proteínas (por ej. fenitoína, carbamazepina, warfarina y tolbutamida). (Ver INTERACCIONES MEDICAMENTOSAS para más información detallada en interacciones de farmacocinética del valproato y otros fármacos). Distribución en el SNC. Las concentraciones de valproato en el líquido cerebroespinal (CSF, por sus siglas en inglés) son cercanas a las concentraciones libres en plasma (cerca de 10% de la concentración total). Farmacocinética – Metabolismo El valproato se metaboliza casi totalmente en el hígado. En pacientes adultos, bajo monoterapia, 30 a 50% de una dosis administrada aparece en la orina como un conjugado glucorónico. La otra vía metabólica principal es la β-oxidación mitocondrial, que típicamente representa más del 40% de la dosis. Usualmente, menos de 15 a 20% de la dosis se elimina por otros mecanismos oxidativos y menos del 3% se elimina como tal, por la orina. La relación entre la dosis y la concentración total de valproato es no lineal; la concentración no aumenta proporcionalmente con la dosis; en vez de ello, incrementa en menor extensión, debido a la saturación de la unión a las proteínas plasmáticas. La cinética del fármaco libre es lineal. Farmacocinética – Eliminación La depuración plasmática promedioy el volumen de distribución para el total de valproato son de 0.56 L/h/1.73 m2 y 11 L/1.73 m2 , respectivamente. La vida media terminal para el valproato en monoterapia después de 60 minutos de infusión intravenosa de 1,000 mg fue de 16 ± 3.0 horas. Los datos representados se pueden aplicar principalmente a los pacientes que no están tomando fármacos que afecten los sistemas hepáticos de enzimas metabolizadoras. Por ejemplo, los pacientes que toman antiepilépticos inductores de enzimas (carbamazepina, fenitoína, fenobarbital) pueden aclarar el valproato más rápidamente. Debido a esos cambios en la depuración del valproato, se debe intensificar el monitoreo de las concentraciones de los antiepilépticos, siempre que se introduzca o se retire un antiepiléptico concomitante.

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 28 de 29
Poblaciones Especiales Neonatos Menores de dos meses de edad tienen una habilidad muy reducida para eliminar el valproato, en relación a los niños mayores y los adultos. Esta es una consecuencia de depuración reducido (quizá debido al retraso en el desarrollo de glucuronosiltransferasa y otros sistemas enzimáticos implicados en la eliminación del valproato) y de aumento en el volumen de distribución (en parte por disminución de unión a proteínas plasmáticas). Por ejemplo, en un estudio, la vida media en niños menores de 10 días de edad fue de 10 a 67 horas, comparada con 7 a 13 horas en niños mayores de dos meses. Ancianos La capacidad de los ancianos (entre 68 y 89 años) para eliminar el valproato es menor respecto a los adultos jóvenes (edad: 22 a 26 años). La depuración intrínseca se reduce por 39%; la fracción libre de valproato se incrementó por 44%. Por tanto, la dosis inicial debe reducirse en el anciano (véase DOSIS Y ADMINISTRACIÓN). Niños Los pacientes pediátricos (por ejemplo, entre tres meses y 10 años) tienen 50% más depuración expresado en peso (es decir, mL/min/Kg), que los adultos. Después de los 10 años, los niños tienen parámetros farmacocinéticos que se aproximan a los de los adultos. Género No hay diferencias en la depuración del fármaco libre (no unido) ajustado por área de superficie corporal entre hombres y mujeres (4.8 ± 0.17 y 4.7 ± 0.07 L/h por 1.73 m2, respectivamente). Etnia No se han realizado estudios en las etnias sobre resultados en la cinética del valproato. Enfermedad Renal Se ha informado de una ligera reducción (27%) de la eliminación del valproato no unido, en pacientes con insuficiencia renal (depuración de creatinina < 10 mL/min); sin embargo, la hemodiálisis normalmente reduce las concentraciones de valproato cerca de un 20%. Por tanto, parece no ser necesario ajustar la dosis en pacientes con insuficiencia renal. En estos pacientes, la unión a proteínas está sustancialmente reducida; por consiguiente, el monitoreo de las concentraciones totales puede llevar a errores. Enfermedades Hepáticas (Ver CONTRAINDICACIONES y ADVERTENCIAS - Hepatotoxicidad) La enfermedad hepática impide la capacidad de eliminar el valproato. En un estudio, la depuración del valproato libre se redujo en 50% en 7 pacientes con cirrosis y en 16% en 4 pacientes con hepatitis aguda, en comparación con 6 individuos sanos. En tal estudio, la vida media del valproato aumentó de 12 a 18 horas. La enfermedad hepática se asocia con la disminución de las concentraciones de albúmina y de mayores fracciones de valproato libre (no unido) (2 a 2.6 veces mayor). Por consiguiente, el monitoreo de las concentraciones totales puede inducir a error, puesto que las concentraciones libres pueden estar sustancialmente elevadas en pacientes con enfermedad hepática mientras que las concentraciones totales pueden parecer normales. Niveles Plasmáticos y Efecto Clínico Las relaciones entre concentraciones plasmáticas y respuesta clínica no están bien documentadas. Un factor que contribuye es la concentración no lineal, dependiente de la unión de proteínas del valproato, el cual afecta la depuración del fármaco. De manera que, el monitoreo del valproato sérico total no proporciona un índice confiable de la fracción bioactiva del valproato.

DEPACON® Inyección de Valproato de sodio Lista No. 1564
SOLID 1000603902 V9.0 Revisión marzo 2017 Monografía V13.03.167
Página 29 de 29
Por ejemplo, debido a que la unión del valproato a las proteínas plasmáticas es dependiente de la concentración, la fracción libre aumenta cerca de 10 a 40 mcg/mL hasta 18.5% al llegar a 130 mcg/mL. Ocurren fracciones libres mayores a las esperadas en los ancianos, en los pacientes hiperlipidémicos y en aquellos con insuficiencia renal y hepática. Epilepsia En la epilepsia, el rango terapéutico comúnmente se considera entre 50 a 100 mcg/mL de valproato total, si bien algunos pacientes pueden ser controlados con menores o mayores concentraciones plasmáticas que las referidas. Las dosis equivalentes de valproato y divalproato de sodio producen niveles plasmáticos equivalentes al ion valproato (ver FARMACOLOGÍA CLINICA - Farmacocinética). 14. COMPATIBILIDAD Y ESTABILIDAD El valproato de sodio en inyección es físicamente compatible y químicamente estable con las siguientes soluciones parenterales durante por lo menos 24 horas, siempre que se conserve en envases de cloruro de polivinilo (PVC) o vidrio a temperatura ambiente controlada de 15 a 30ºC (59 a 86ºF).
Dextrosa (5%) Inyección, USP
Cloruro de Sodio (0.9%) Inyección, USP
Lactato de Ringer Inyección, USP Almacenar a temperatura no mayor a 30ºC. No contiene preservantes. La porción no utilizada en el recipiente debe desecharse.
15. PRESENTACIÓN La inyección de Valproato de sodio, equivalente a 100 mg de ácido valproico por mL, es una solución clara, sin color, viene en viales de 5mL única dosis, disponible en caja de 10 viales. 16. REFERENCIAS
I. Abbott Laboratories II. FDA
III. PRAC (Comité para la Evaluación de Riesgos en Farmacovigilancia Europeo). Agreement of the co-ordination group for mutual recognition and decentralized procedures for human use, pursuant to article 107k (1) and (2) of directive 2001/83/EC for/ EMA /CMDH / 654826 /2014.
Fecha de Revisión: Marzo 2017


















