







Idiomas
Páginas
Jurídico


Máster en Descubrimiento de Fármacos
Registro de un medicamento
06.07.2016
Juan Domingo Sánchez Cebrián
- Técnico Registros UCB Pharma, S.A.
- Profesor Asociado en el Dpto. de Química Orgánica y Farmacéutica de la Facultad de Farmacia de la U.C.M.

DEFINICIONES

DEFINICIONES 3
¿QUÉ ES UNA SUSTANCIA ACTIVA?
De acuerdo al Real Decreto Legislativo 1/2015, de 24 de julio, por el que se aprueba el texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios, una sustancia activa es: “Toda sustancia o mezcla de sustancias destinadas a la fabricación de un medicamento y que, al ser utilizadas en su producción, se convierten en un componente activo de dicho medicamento destinado a ejercer una acción farmacológica, inmunológica o metabólica con el fin de restaurar, corregir o modificar las funciones fisiológicas, o de establecer un diagnóstico.”

DEFINICIONES 4
¿QUÉ ES UN MEDICAMENTO?
De acuerdo al Real Decreto Legislativo 1/2015, de 24 de julio, por el que se aprueba el texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios, un medicamento es: “Toda sustancia o combinación de sustancias que se presente como poseedora de propiedades para el tratamiento o prevención de enfermedades en seres humanos, o que pueda usarse, o administrarse a seres humanos con el fin de restaurar, corregir o modificar las funciones fisiológicas ejerciendo una acción farmacológica, inmunológica o metabólica, o de establecer un diagnóstico médico.”

CLASIFICACION DE LAS SUSTANCIAS ACTIVAS

TIPOS DE SUSTANCIAS ACTIVAS 6
MEDICAMENTOS INNOVADORES O DE REFERENCIA
Moléculas pequeñas = sustancias con estructura química pequeña. - Son naturales o sintéticas. - Su modo de acción en muchas ocasiones se desconoce en gran medida. - Alta frecuencia de fallos en los ensayos clínicos. - Efectos adversos y toxicidad inesperada.

TIPOS DE MEDICAMENTOS 7
MEDICAMENTOS INNOVADORES O DE REFERENCIA
Moléculas grandes = sustancias de origen biológico - Su modo de acción se conoce. - Baja frecuencia de fallos en los ensayos clínicos. - Reducción de los efectos adversos y toxicidad inesperada - Son sustancias obtenidas a partir de organismos vivos, como bacterias y levaduras. - Pueden tener estructura de moléculas “relativamente” pequeñas, por ejemplo
la insulina o eritropoyetina, o pueden ser moléculas complejas como los anticuerpos monoclonales.

TIPOS DE MEDICAMENTOS 8
MEDICAMENTOS GENÉRICOS Y BIOSIMILARES
Medicamentos Genéricos - Medicamento que tenga la misma composición cualitativa y cuantitativa
en principios activos y la misma forma farmacéutica, y cuya bioequivalencia con el medicamento de referencia haya sido demostrada por estudios adecuados de biodisponibilidad (R.D. 1345/2007).
Medicamentos Biosimilares - Medicamento biológico que es similar a otro medicamento biológico,
que ya está autorizado para su uso (R.D. 1345/2007).

TIPOS DE MEDICAMENTOS 9
TERAPIAS AVANZADAS
Los medicamentos de terapia avanzada son medicamentos de uso humano basados en genes (terapia génica), células (terapia celular) o tejidos (ingeniería tisular) e incluyen productos de origen autólogo, alogénico o xenogénico. Constituyen nuevas estrategias terapéuticas y su desarrollo contribuirá a ofrecer oportunidades para algunas enfermedades que hasta el momento carecen de tratamientos eficaces (R.D. 1345/2007).
TERAPIA GÉNICA Es un medicamento biológico que incluye un principio activo que contiene un ácido nucleico recombinante, o está constituido por él, que se utiliza en seres humanos, o administrado a los mismos, con objeto de regular, reparar, sustituir, añadir o eliminar una secuencia génica.

TIPOS DE MEDICAMENTOS 10
TERAPIAS AVANZADAS
TERAPIA CELULAR Es un medicamento biológico que contiene células o tejidos, o está constituido por ellos, que han sido objeto de manipulación sustancial de modo que se hayan alterado sus características biológicas, funciones fisiológicas o propiedades estructurales pertinentes para el uso clínico previsto, o por células o tejidos que no se pretende destinar a la misma función esencial en el receptor y en el donante. INGENIERÍA DE TEJIDOS Son medicamentos que contiene o está formado por células o tejidos manipulados por ingeniería, que tiene tales propiedades, que se emplea o se administra a las personas para regenerar, restaurar o reemplazar un tejido humano.

TIPOS DE MEDICAMENTOS 11
MEDICAMENTOS HUÉRFANOS Y ENFERMEDADES RARAS
MEDICAMENTO HUÉRFANO Es un medicamento que se puede utilizar para diagnosticar, prevenir o tratar una enfermedad rara. ENFERMEDAD RARA Aquella que afecta a menos de 5 sujetos por cada 10.000 habitantes. VENTAJAS
• Exclusividad comercial durante 10 años. • Asesoramiento científico gratuito. • Acceso al P.Centralizado de registro, y evaluación rápida (fast-track). • Exención de tasas. • Investigación subvencionada por la Unión Europea.
OBLIGACIÓN • El fármaco tiene que estar disponible en toda la Unión Europea.

ANTECEDENTES

ANTECEDENTES 13
JARABE DE SULFANILAMIDA
En 1937, en E.E.U.U., 105 personas fallecieron por consumir un jarabe de sulfanilamida, con sabor a frambuesa. Dicho jarabe, estaba constituido en un 70% por etilenglicol, un anticongelante.
En 1938, se publica la “Federal Food, Drugs and Cosmetic Act”

ANTECEDENTES 14
TALIDOMIDA
En Europa entre 1957 y 1963, se comercializó el principio activo Talidomida. Se utilizaba como sedante y tratamiento de la hiperémesis gravídica (evitar náuseas durante los 3 primeros meses/embarazo) Durante el tiempo que se usó, se observaron unos 12.000 casos de niños con focomegalia (malformación que se caracterizaba por acortamiento de las extremidades), de mujeres que durante el embarazo habían tomado Talidomida.
En E.E.U.U., la FDA se negó a autorizar el medicamento, a no ser que se incluyeran más estudio clínicos que avalaran la seguridad del medicamento.
Necesidad de límites más estrictos y mayor número de datos de seguridad para la autorización de nuevos medicamentos. Nacimiento de la Farmacovigilancia.

AGENCIAS EVALUADORAS

AGENCIAS EVALUADORAS 16
FUNCIONES
Las funciones comunes de todos los órganos reguladores son:
REGISTRO DE MEDICAMENTOS
ASESORÍA CIENTÍFICA
FARMACOVIGILANCIA
AGENCIAS DE REFERENCIA • AEMPS (Agencia Española de Medicamentos y Productos Sanitarios) http://www.aemps.gob.es/ • Agencias Nacionales en todos los países europeos • EMA (European Medicine Agency) http://www.ema.europa.eu/ema • FDA (Foods and Drugs Administration) http://www.fda.gov/default.htm

AGENCIAS EVALUADORAS 17
FDA vs EMA
FDA EMA
Creación en 1938 Creación en 1995
No tiene una única sede Sede Central en Londres ¿Por cuánto tiempo tras el Brexit?
Tiene asesores internos Asesores procedentes de las diferentes Agencias nacionales
Evalúa y autoriza el medicamento Evalúa el medicamento, pero la autorización la concede la Comisión Europea.

DESARROLLO DE UN MEDICAMENTO
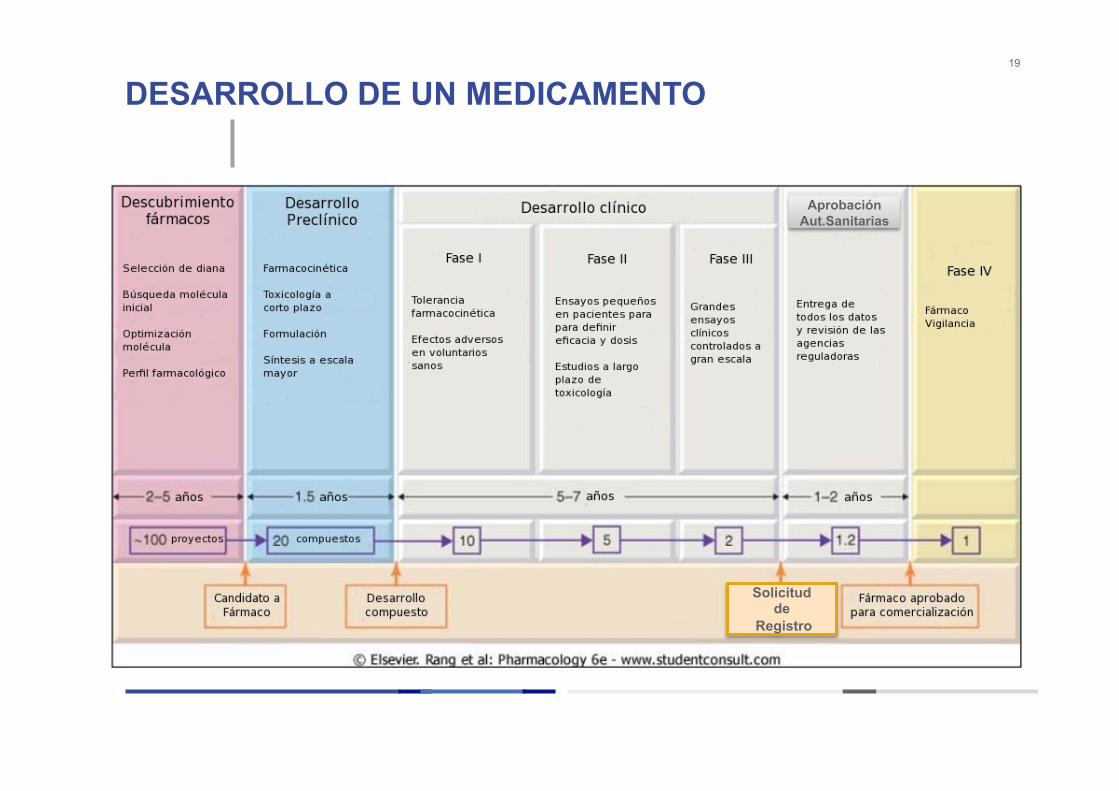
DESARROLLO DE UN MEDICAMENTO 19
Aprobación Aut.Sanitarias
Solicitud de
Registro

DESARROLLO DE UN MEDICAMENTO 20
Esquema
INVESTIGACIÓN
ACCESO
COMERCIALIZACIÓN
DESARROLLO PRECLÍNICO CLÍNICO/GALÉNICO
AUTORIZACIÓN DEL MEDICAMENTO ORIGINAL
PATENTE
AUTORIZACIÓN DE GENÉRICOS Comercialización
de genéricos
MARCA(S)
PERIODOS DE PROTECCIÓN
Protección efectiva frente a
genérico (DATOS Y PATENTE)
EFG2 EFG1

CONCEPTO DE PATENTE Y PROTECCIÓN DE DATOS 21
PATENTE
Derecho que protege al titular a impedir el uso y/o explotación de su invención por un tercero sin su consentimiento. La patente viene reglada por nuestra Ley 11/1986, de 20 de marzo, de Patentes de Invención y Modelos de Utilidad, que adaptó la normativa comunitaria sobre patentes a nuestro ordenamiento jurídico e introdujo el reconocimiento de la patente de producto, siendo el principio del fin de los medicamentos copia (medicamentos esencialmente similares que no han demostrado bioequivalencia, autorizados con la normativa anterior).
Según esta norma, en su artículo 49 se indica: “La patente tiene una duración de veinte años improrrogables, contados a partir de la fecha de presentación de la solicitud y produce sus efectos desde el día en que se pública la mención de que ha sido concedida”.

CONCEPTO DE PATENTE Y PROTECCIÓN DE DATOS 22
PROTECCIÓN DE DATOS
Es un periodo que otorga la Autoridad Sanitaria para proteger y fomentar el I+D de la industria farmacéutica.
Este periodo se fija en 10 años, independiente de las patentes, más uno adicional si durante los primeros 8 años del período de 10 años, el titular de la autorización de comercialización del medicamento de referencia obtiene una autorización para una o varias indicaciones terapéuticas nuevas y, durante la evaluación científica previa a su autorización, se establece que dichas indicaciones aportarán un beneficio clínico significativo en comparación con las terapias existentes (Cláusula Bolar).

DESARROLLO DE UN MEDICAMENTO 23
CICLO DE VIDA
CALIDAD (Fabricación) SEGURIDAD (Estudios en animales) EFICACIA Y SEGURIDAD (EECC en humanos)
DOSSIER DE REGISTRO
EVALUACIÓN AASS
I+D
Asesoría Científica
AUTORIZACIÓN DE COMERCIALIZACIÓN
SOLICITUD DE MODIFICACIONES, REVALIDACIONES,
EXTENSIONES DE LÍNEA, VARIOS…
EVALUACIÓN AASS
Mantenimiento actualizado del dossier de registro

¿QUÉ SE OBTIENE CON LA AUTORIZACIÓN DE REGISTRO DE UN MEDICAMENTO?

EVALUACIÓN Y AUTORIZACIÓN. TIPOS DE PROCEDIMIENTO 25
OBJETIVOS DEL PROCEDIMIENTO DE AUTORIZACIÓN
De acuerdo al art. 9 y siguientes, del Real Decreto Legislativo 1/2015, de 24 de julio, por el que se aprueba el texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios, los objetivos del procedimiento de autorización son:
• Comprobar que el medicamento alcanza los requisitos de calidad establecidos.
• Comprobar que el medicamento es seguro, no produciendo en condiciones normales de autorización efectos tóxicos o indeseables desproporcionados al beneficio que procura.
• Comprobar que es eficaz en las indicaciones terapéuticas aprobadas.
• Está correctamente identificado y va acompañado de la información precisa para su utilización.
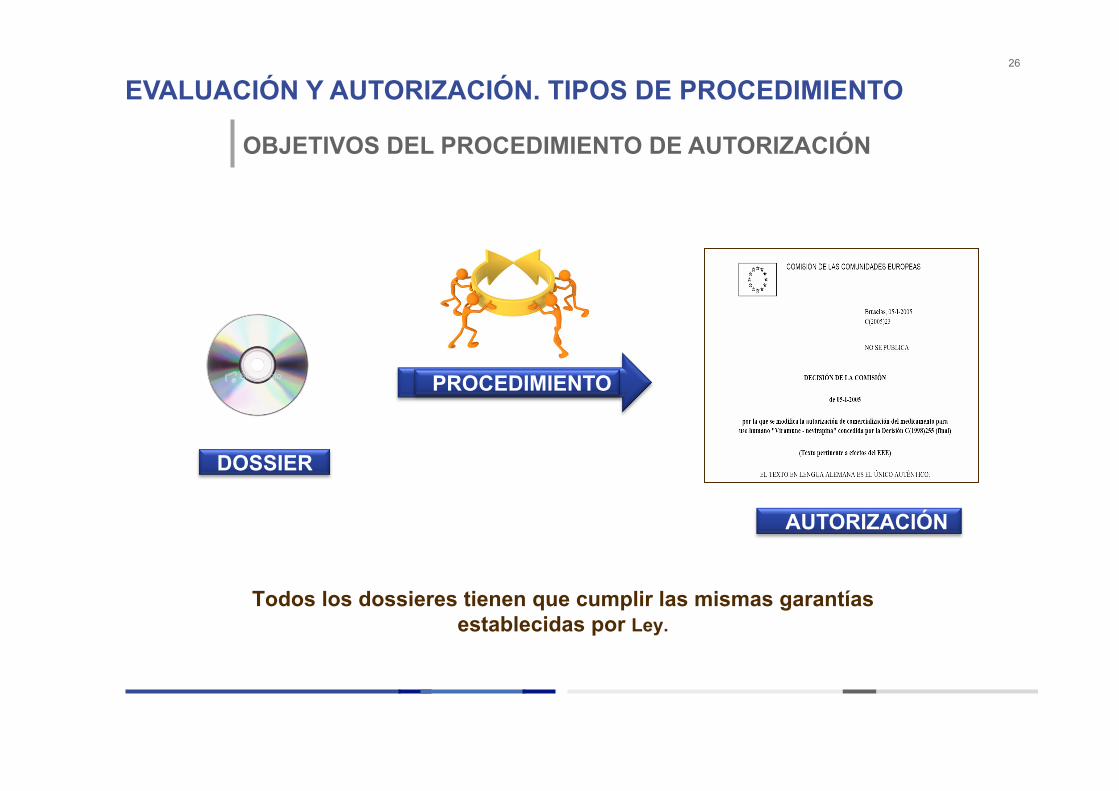
EVALUACIÓN Y AUTORIZACIÓN. TIPOS DE PROCEDIMIENTO 26
OBJETIVOS DEL PROCEDIMIENTO DE AUTORIZACIÓN
DOSSIER
AUTORIZACIÓN
PROCEDIMIENTO
Todos los dossieres tienen que cumplir las mismas garantías establecidas por Ley.

DOCUMENTACIÓN DE REGISTRO 27
REGISTRO DE UN MEDICAMENTO
DESARROLLO REGISTRO COMERCIALIZACIÓN MANTENIMIENTO
CALIDAD (Fabricación) SEGURIDAD (Estudios en animales) EFICACIA Y SEGURIDAD (EECC en humanos)
DOSSIER DE REGISTRO
AUTORIZACIÓN DE COMERCIALIZACIÓN
EVALUACIÓN AASS

DOCUMENTACIÓN DE REGISTRO 28
REGISTRO DE UN MEDICAMENTO Ficha Técnica
Prospecto
Se autoriza toda la documentación de CALIDAD, PRECLINICA y CLÍNICA presentada en el dossier
Datos administrativos
CALIDAD SEGURIDAD EFICACIA

NUEVOS REGISTROS

EVALUACIÓN Y AUTORIZACIÓN 30
TIPOS DE PROCEDIMIENTO DE REGISTRO
• PROCEDIMIENTO CENTRALIZADO
• PROCEDIMIENTO RECONOCIMIENTO MUTUO/DESCENTRALIZADO
• PROCEDIMIENTO NACIONAL

EVALUACIÓN Y AUTORIZACIÓN 31
PROCEDIMIENTO CENTRALIZADO
¿Qué medicamentos de uso humano pueden acogerse al mismo? 1) Los medicamentos designados como medicamentos huérfanos de conformidad
con el Reglamento (CE) no 141/2000 del Parlamento Europeo y del Consejo de 16 de diciembre de 1999.
2) Medicamentos de uso humano desarrollados por medio de uno de los siguientes procesos biotecnológicos:
• Técnica del ADN recombinante.
• Expresión controlada de codificación de genes para las proteínas biológicamente activas en procariotas y eucariotas, incluidas las células de mamífero transformadas.
• Métodos del hibridoma y del anticuerpo monoclonal.

EVALUACIÓN Y AUTORIZACIÓN 32
PROCEDIMIENTO CENTRALIZADO
3) Medicamentos de uso humano que contengan una sustancia activa nueva, cuya indicación terapéutica sea el tratamiento de alguna de las enfermedades siguientes:
• Síndrome de inmunodeficiencia adquirida • Cáncer • Trastornos neurodegenerativos • Diabetes
y con efectos a partir de 20 de mayo de 2008:
• Enfermedades autoinmunes y otras disfunciones inmunes, • Enfermedades víricas.

EVALUACIÓN Y AUTORIZACIÓN 33
PROCEDIMIENTO CENTRALIZADO – ACTORES PRINCIPALES
Industria Estados Miembros
EMA CHMP
Comisión Europea
EMA: European Medicines Agency CHMP: Committee for Human Medicinal Products OPINIÓN FINAL
COORDINA ACTIVIDADES
EMITE LA DECISIÓN, AUTORIZACIÓN

EVALUACIÓN Y AUTORIZACIÓN 34
P. CENTRALIZADO – FASE DE EVALUACIÓN
Recepción del borrador del Informe de Evaluación inicial del ponente/coponente del CHMP, EMA y solicitante
Comienzo del procedimiento
Sesión plenaria del CHMP:
CHMP adopta la lista de preguntas y la recomendación provisional para el solicitante
Día 1
Día 80
Día 120
…
Parada reloj
Hasta 6 meses para responder preguntas
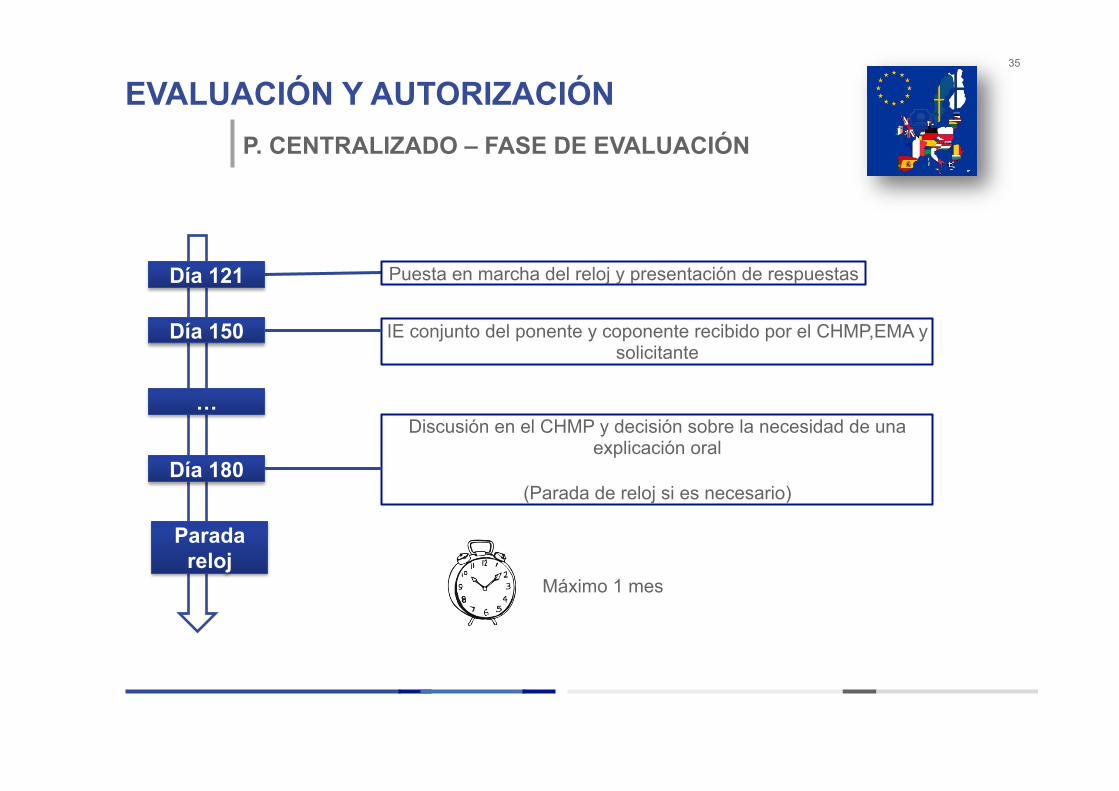
EVALUACIÓN Y AUTORIZACIÓN 35
P. CENTRALIZADO – FASE DE EVALUACIÓN
IE conjunto del ponente y coponente recibido por el CHMP,EMA y solicitante
Puesta en marcha del reloj y presentación de respuestas
Discusión en el CHMP y decisión sobre la necesidad de una explicación oral
(Parada de reloj si es necesario)
Día 121
Día 150
Día 180
…
Parada reloj
Máximo 1 mes

EVALUACIÓN Y AUTORIZACIÓN 36
P. CENTRALIZADO – FASE DE EVALUACIÓN
Borrador inicial de FT,PP y etiquetado en inglés para enviar al ponente, EMA y solicitante
Puesta en marcha del reloj de nuevo y explicación oral
Decisión de la Comisión - AUTORIZACIÓN
Día 181
Día 181-210
Día 255
Día 210
Día 277
Día 215
…
Adopción de la Opinión del CHMP e Informe de Evaluación
El solicitante proporciona FT,PP y etiquetado en los idiomas oficiales
La EMA prepara la Opinión en todos los idiomas y envía copias finales a la Comisión

EVALUACIÓN Y AUTORIZACIÓN 37
PROCEDIMIENTO CENTRALIZADO
¿Qué se obtiene tras la autorización por el Procedimiento Centralizado?
• Se autoriza el medicamento. Autorización válida en todos los Estados Miembros de la UE.
• Un nombre común/única marca en todos los Estados Miembros de la UE • Una FT (Anexo I) común en todos los estados miembros de la U.E. • Unas condiciones de autorización. • Un prospecto y un etiquetado (Anexo III) común para todos los países de la U.E.

EVALUACIÓN Y AUTORIZACIÓN 38
PROCEDIMIENTO CENTRALIZADO
BLUE BOX: Espacio destinado para requisitos locales (específico de cada país)

EVALUACIÓN Y AUTORIZACIÓN 39
PROCEDIMIENTO RECONOCIMIENTO MUTUO
Se utiliza cuando ya existe una primera autorización nacional en un Estado Miembro (EM Referencia) y el Titular de la primera Autorización de Comercialización quiere comercializar el mismo medicamento en otros Estados Miembros Concernidos (EMC) VENTAJAS: Evaluación coordinada entre todos los EMCs ayudados por el Informe de Evaluación del EMR. CALENDARIO: 90 días tras recibir una solicitud válida en todos los EMCs
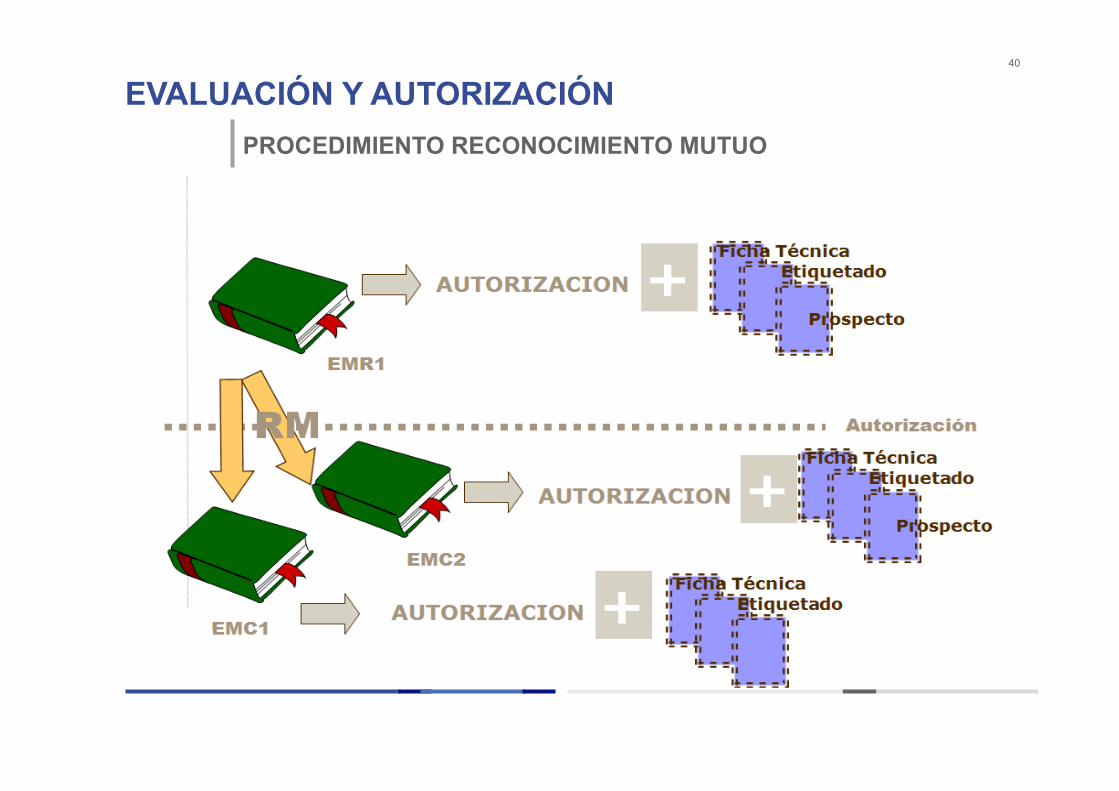
EVALUACIÓN Y AUTORIZACIÓN 40
PROCEDIMIENTO RECONOCIMIENTO MUTUO

EVALUACIÓN Y AUTORIZACIÓN 41
PROCEDIMIENTO DESCENTRALIZADO
Evaluación coordinada entre todos los EMCs ayudados por el Informe de Evaluación del EMR.
No tiene una primera Autorización de Comercialización CALENDARIO: 210 días
• Fase I de Evaluación (día 0-119) • Fase II de Evaluación (día 120-210 =90) RM • Autorización de Comercialización Nacional: 30 días

EVALUACIÓN Y AUTORIZACIÓN 42
PROCEDIMIENTO DESCENTRALIZADO

EVALUACIÓN Y AUTORIZACIÓN 43
PROC. RECONOCIMIENTO MUTUO Y DESCENTRALIZADO
Reconocimiento Mutuo Descentralizado
Es necesario que el medicamento esté aprobado en un país europeo
No es necesario que el medicamento se encuentre autorizado en un país europeo
El EMR es el país donde se encuentra autorizado el medicamento
El solicitante elige el estado miembro que quiere que actúe como EMR.
Los EMCs, donde se quiere obtener la autorización de comercialización, deben reconocer la Autorización ya otorgada en el EMR.
El informe de evaluación es elaborado por el EMR.

EVALUACIÓN Y AUTORIZACIÓN 44
PROC. RECONOCIMIENTO MUTUO Y DESCENTRALIZADO
¿Qué se autoriza por el Procedimiento de RM/Descentralizado?
• Se autoriza el medicamento en los países elegidos por la Cía. • Un nombre que puede ser diferente en cada país • Una FT (Anexo I) para todos los países donde está autorizado. • Un prospecto y un etiquetado (Anexo III) común para todos los países donde se
encuentra autorizado el medicamento, aunque con los requerimientos locales diferentes, a negociar con las A.A.S.S. Nacionales.

EJEMPLO PRÁCTICO 45
ASTROCETAM UCB
1) Descentralizado: Austria & Alemania. Austria se retira del proceso, solo se autoriza en Alemania.
¿TIPO DE PROCEDIMIENTO DE REGISTRO?
¿Centralizado? ¿RM? ¿Descentralizado?
2) Reconocimiento Mutuo: es necesario que el medicamento esté aprobado en un país europeo* Austria República Checa *Alemania Grecia España Hungría Portugal Eslovaquia

EVALUACIÓN Y AUTORIZACIÓN 46
PROCEDIMIENTO NACIONAL

EJEMPLO PRÁCTICO 47
YIGO
¿TIPO DE PROCEDIMIENTO DE REGISTRO QUE PODRIAMOS HABER UTILIZADO?
¿Centralizado? ¿RM? ¿Descentralizado?
No puede ser centralizado
No puede ser descentralizado
Debería ser un RM porque ya está registrado en un país
FINALMENTE SE REALIZÓ UN REGISTRO NACIONAL
Situación inicial: Autorizado en Alemania

EJEMPLO PRÁCTICO 48
Rupertiracetam
¿TIPO DE PROCEDIMIENTO DE REGISTRO?
¿Centralizado? ¿RM? ¿Descentralizado?
1) Antecedentes Descentralizado: Austria & Alemania. Austria se retira del proceso, solo se autoriza en Alemania.
2) Reconocimiento Mutuo (es necesario que el medicamento esté aprobado en un país europeo) Austria República Checa Alemania (EMR) Grecia España Hungría Portugal Eslovaquia
3) Centralizado Sólo si se quisiera comercializar en todos los países de la EU
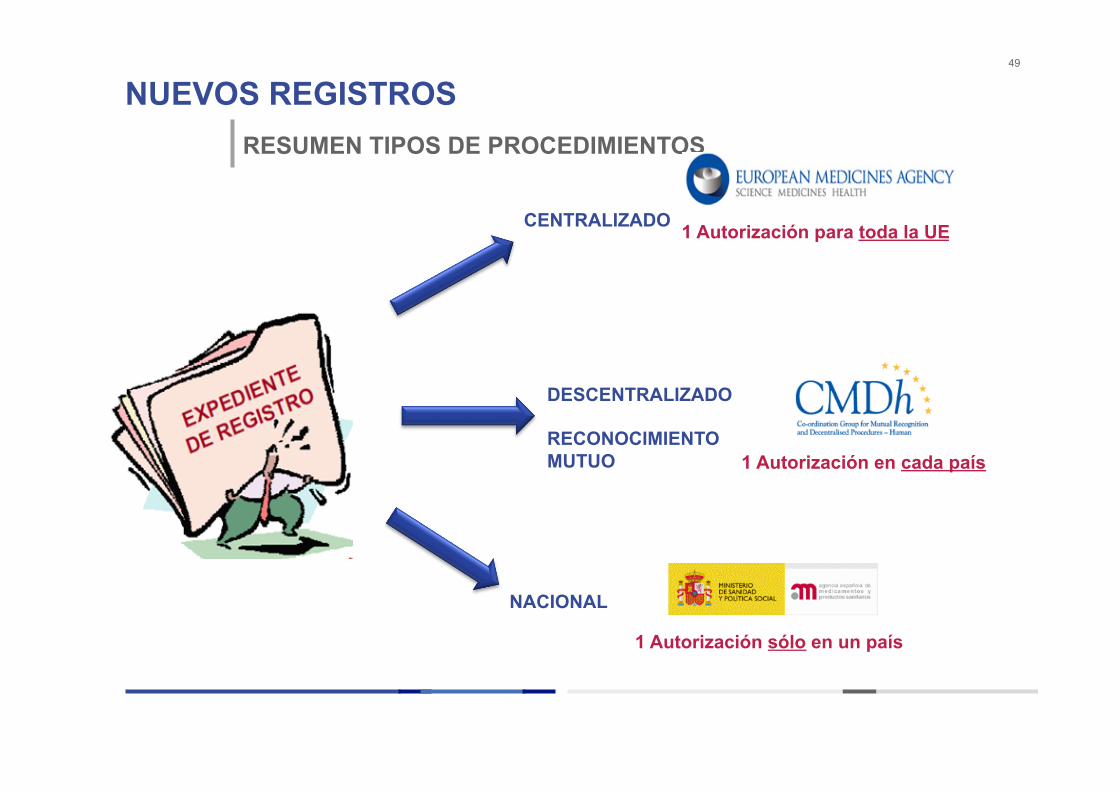
NUEVOS REGISTROS 49
RESUMEN TIPOS DE PROCEDIMIENTOS
CENTRALIZADO
DESCENTRALIZADO RECONOCIMIENTO MUTUO
NACIONAL
1 Autorización para toda la UE
1 Autorización en cada país
1 Autorización sólo en un país