
UNIVERSIDAD DE GUAYAQUIL FACULTAD DE …repositorio.ug.edu.ec/bitstream/redug/8178/1/BCIEQ-T-0095...
93
UNIVERSIDAD DE GUAYAQUIL FACULTAD DE CENCIAS QUIMICAS UNIDAD DE TITULACIÓN PROYECTO TEMA: EVALUACIÓN DE LA NORMA ISO 9001 E ISO 15189 EN EL INSTITUTO DE BIOMEDICINA DE LA UNIVERSIDAD CATÓLICA DE SANTIAGO DE GUAYAQUIL, 2014 TRABAJO DE TITULACION PRESENTADO COMO REQUISITO PREVIO PARA OPTAR AL TÍTULO DE QUÍMICA Y FARMACEUTICA. AUTOR VELIZ GARCIA, LEYLA LAIZ TUTOR ACADÉMICO Q.F. PATRICIA JIMENEZ GUAYAQUIL, ECUADOR 2015
Transcript of UNIVERSIDAD DE GUAYAQUIL FACULTAD DE …repositorio.ug.edu.ec/bitstream/redug/8178/1/BCIEQ-T-0095...

UNIVERSIDAD DE GUAYAQUIL FACULTAD DE CENCIAS QUIMICAS
UNIDAD DE TITULACIÓN PROYECTO
TEMA:
EVALUACIÓN DE LA NORMA ISO 9001 E ISO 15189 EN EL INSTITUTO DE
BIOMEDICINA DE LA UNIVERSIDAD CATÓLICA DE SANTIAGO DE
GUAYAQUIL, 2014
TRABAJO DE TITULACION PRESENTADO COMO REQUISITO PREVIO
PARA OPTAR AL TÍTULO DE QUÍMICA Y FARMACEUTICA.
AUTOR
VELIZ GARCIA, LEYLA LAIZ
TUTOR ACADÉMICO
Q.F. PATRICIA JIMENEZ
GUAYAQUIL, ECUADOR
2015

ii

iii
CERTIFICADO DEL TUTOR
En calidad de tutora del trabajo de titulación, Certifico: que he asesorado,
guiado y revisado el trabajo de titulación en la modalidad de Proyecto de
Investigación, cuyo título es “Evaluación de la norma ISO 9001 e ISO 15189 en el Instituto de Biomedicina de la Universidad Católica de Santiago de Guayaquil, 2014”, presentado por la Srta. Leyla Laíz Véliz
García con cédula de ciudadanía #0930784269, previo a la obtención del título
de Química y Farmacéutica.
Este trabajo ha sido aprobado en su totalidad y se adjunta el informe de Anti-
plagio del programa URKUND. Lo certifico.
En honor a la verdad,
____________________
QF. Patricia Jiménez Granizo. Mg.
TUTOR DE TESIS
Guayaquil, Febrero del 2015

iv
CERTIFICADO DEL TUTOR INFORME DE ANTI-PLAGIO DEL PROGRAMA URKUND.
Por la presente certifico que el Proyecto de Investigación cuyo tema es
“Evaluación de la norma ISO 9001 e ISO 15189 en el Instituto de Biomedicina de la Universidad Católica de Santiago de Guayaquil, 2014”,
correspondiente a la Srta. Leyla Laíz Véliz García, con C.I.: 0930784269, ha
sido subido al programa URKUND y revisado, arrojando un porcentaje de
similitud del 3% el cual se encuentra dentro de los límites aceptables.

v
CARTA DE AUTORÍA DEL TRABAJO DE TITULACIÓN.
Yo, LEYLA LAIZ VELIZ GARCIA autor de este trabajo declaro ante las
autoridades de la Facultad de Ciencias Químicas de la Universidad de
Guayaquil, que la responsabilidad del contenido de este TRABAJO DE
TITULACIÓN, me corresponde exclusivamente; y el patrimonio intelectual de la
misma a la Facultad de Ciencias Químicas de la Universidad de Guayaquil.
Declaro también que todo el material escrito me pertenece, salvo el que está
debidamente referenciado en el texto. Además ratifico que este trabajo no ha
sido parcial ni totalmente presentado para la obtención de un título, ni en la
universidad nacional, ni en una extranjera.
Guayaquil, 2 de febrero del 2015.
Leyla Laíz Véliz García
CI: 0930784269

vi
DEDICATORIA
Esta investigación está dedicada a mi hijo Matías, el amor de mi vida, mi
pequeño milagro y bendición eres el mayor motivo para seguir adelante.
Agradezco a mis padres y mis hermanos que estuvieron conmigo apoyándome
en cada etapa de mi vida.

vii
AGRADECIMIENTO
A todas las personas y amigos por su colaboración incondicional en esta
investigación.
Al instituto de biomedicina por las facilidades que me brindaron para poder
llevar a cabo este trabajo.
Blgo. Gustavo Escobar Valdivieso, Coordinador Administrativo/ Jefe de
Laboratorio del Instituto de biomedicina de la Facultad de Ciencias Médicas de
la Universidad Católica Santiago de Guayaquil
QF. Cecibel Ramírez Morán Química Farmacéutica, miembro del instituto de
Biomedicina de la Universidad Católica Santiago de Guayaquil.
QF Patricia Jiménez docente y tutora de la Facultad De Ciencias Químicas.

viii
ÍNDICE
PAG
CERTIFICADO DEL TRIBUNAL .................................... ¡Error! Marcador no definido. CERTIFICADO DEL TUTOR ........................................................................................ iii CERTIFICADO DEL TUTOR ........................................................................................ iv CARTA DE AUTORÍA DEL TRABAJO DE TITULACIÓN. ............................................. v DEDICATORIA ............................................................................................................ vi AGRADECIMIENTO ................................................................................................... vii ÍNDICE ........................................................................................................................viii ÍNDICE DE TABLAS ..................................................................................................... x ÍNDICE DE GRÁFICOS ............................................................................................... xi RESUMEN .................................................................................................................. xii ABSTRACT .................................................................................................................xiii 1. EL PROBLEMA .........................................................................................................1
1.1. PLANTEAMIENTO DEL PROBLEMA .................................................................1
1.1.1 FORMULACION DEL PROBLEMA………………………………………..….2 1.2. OBJETIVOS........................................................................................................3
1.2.1 OBJETIVO GENERAL ......................................................................................3
1.2.2. OBJETIVOS ESPECÍFICOS ............................................................................3
1.3. JUSTIFICACIÓN E IMPORTANCIA ...................................................................4
2. MARCO TEORICO ................................................................................................... 5
2.1. INTRODUCCIÓN………………………………………………………….………5
2.2. ANTECEDENTES…………………………………………………………………6
2.3. DEFINICIONES……………………………………………………………………8
2.3.1. CALIDAD……………………………………………………………………...8
2.3.2. ENFOQUE MEDIANTE PROCESOS……………………………………..9
2.3.3. PUNTOS DE DEMING……………………………………………………...10
2.3.4. CONTROL DE CALIDAD…………………………………………………..12
2.3.5. ASEGURAMIENTO DE LA CALIDAD…………………………………..12
2.3.6. GESTION DE LA CALIDAD……………………………………………….12
2.3.7. SISTEMA DE GESTION DE LA CALIDAD………………………………13
2.3.8. POLÍTICAS DE CALIDAD………………………………………………....14
2.3.9. PLANIFICACIÓN DE LA CALIDAD………………………………………14
2.3.10. REQUISITO………………………………………………………………….14
2.4. NORMAS ISO …………………………………………………………………..15
2.4.1. EVOLUCIÓN DE LAS NORMAS ISO 9000……………………………..15
2.4.2. NORMA ISO 9001:2008……………………………………………………16

ix
2.4.3. NORMA ISO 15189:2012………………………………………………18
2.4.3.1. COMPONENTES DEL SISTEMA DE GESTION DE CALIDAD.
(ISO 15189)……………………………………………………18
2.4.4. EVOLUCIÓN DE LA ACREDITACIÓN DE LOS
LABORATORIOS………………………………………………….….19
2.4.4.1. ACREDITACION EN UN LABORATORIO………..……….20
2.4.4.2. PREPARACIONES Y CONSIDERACIONES DE
ACREDITACION EN BASE A LA NORMA ISO 15189…..21
2.4.4.3. DIFERENCIA ENTRE UNA ACREDITACIÓN Y
CERTIFICACIÓN……………………………………………..22
2.4.4.4. VENTAJA DE UNA CERTIFICACIÓN………………………22
2.4.4.5. ¿QUE ORGANISMOS SE ENCARGAN DE LA
NORMALIZACIÓN?..............................................................23
2.4.4.6. QUIEN SE ENCARGA DE EVALUAR LA ISO 9001 E ISO
15189…………………………………………………………...24
2.5. ANTECEDENTE DEL INSTITUTO DE BIOMEDICINA ………….…..…25
2.5.1. MISIÓN………………………………………………………….25
2.5.2. VISIÓN………………………………………………………….26
2.5.3. PLAN NACIONAL DEL BUEN VIVIR………………..……….26
3. METODOLOGIA…………………………………………………………...27
3.1. GENERALIDADES…………………………………………………...27
3.2. DISENO DE LA INVESTIGACION………………………………….27
3.3. PROCEDIEMIENTO DE LA INVESTIGACION……………………28
3.4INSTRUMENTOS DE INVESTIGACION…………………………….29
3.5 LUGAR DE ESTUDIO…………………………………………………30
3.6 CONFIDENCIALIDAD…………………………………………………30
3.7 RESULTADOS………………………………………………………….31
4. PROPUESTA……………………………………………………………………….36
4.1 CONCLUSION………………………………………………………….37
4.2 RECOMENDACIÓN……………………………………………………38
5. BIBLIOGRAFIA…………..……………………………………………….39
ANEXO…………………………………………………………………………………41

x
ÍNDICE DE TABLAS
Tabla 1 ............................................................................................................. 31
Tabla 1 porcentaje de cumplimiento del instituto de la norma ISO 9001:2008 ....... 31
Tabla 2 .............................................................................................................. 33 Tabla 2 porcentaje de cumplimiento del instituto de la norma ISO 15189:2012 ..... 33
Tabla 4………………………………………………………………………..………………..34 Tabla 4 cuestionario de preguntas para el personal del Instituto De Biomedicina….34

xi
ÍNDICE DE GRÁFICOS
Grafica 1 ..................................................................................................................... 32 Grafica 1. Porcentaje de cumplimiento del instituto de la norma ISO 9001:2008 ... 32
Grafica 2 ..................................................................................................................... 33 Grafica 1. Porcentaje de cumplimiento del instituto de la norma ISO 15189:2012 . 33
Grafica 3……………………………………………………………………………………….35 Grafica 3. Encuesta realizada al personal del laboratorio
del instituto de biomedicina…………………………………………………35

xii
RESUMEN
Las exigencias actuales de los organismos certificadores y acreditadores
nacionales como el “Servicio de Acreditación Ecuatoriano y el Instituto
Ecuatoriano de Normalización” demandan que los laboratorios de diagnóstico
aseguren la calidad de sus resultados mediante la implementación de normas
internacionales que normalicen sus procesos. El objetivo de este estudio tiene
como finalidad evaluar el cumplimiento de la norma ISO 9001:2008 e ISO
15189:2012 en el Instituto De Biomedicina de La Universidad Católica De
Santiago De Guayaquil. Para el estudio se recurrió a métodos
observacionales, descriptivos y prospectivos se efectuaron listas de verificación
en base a las normas ISO 9001:2008 e ISO 15189:2012 para analizar el estado
actual de la documentación respectiva y así ver el grado de cumplimiento bajo
las normas. Se efectuaron preguntas dirigidas al personal que labora para ver
sus conocimientos en lo que compete al tema de la investigación. De acuerdo
con los requisitos de la norma la norma ISO 9001:2008 el Instituto de
Biomedicina de la Universidad Católica de Santiago de Guayaquil cuenta con
una implementación del 80.45% de cumplimiento, su otra norma también
analizada en el proceso de investigación la Norma ISO 15189:2012, cuenta con
el 84.86% de cumplimiento. Por medio del análisis realizado se determinó que
es de vital importancia la completa implementación de las normas para que el
Instituto pueda ajustarse a las exigencias establecidas por parte de los
organismos. Es recomendable planes de capacitación continua y participación
del personal administrativo y técnico del instituto de biomedicina.
Palabras claves: Normas ISO 9001:2008 e ISO 15189:2012, Acreditación, certificación, laboratorios.

xiii
ABSTRACT
Current requirements of the certification bodies and national accreditors as
"Ecuadorian Accreditation Service and the Ecuadorian Institute of
Standardization" claim that diagnostic laboratories ensure the quality of their
results by implementing international standards to standardize its processes.
The objective of this study aims to assess compliance with the ISO 9001: 2008
and ISO 15189: 2012 at the Institute of Biomedicine of the Catholic University of
Santiago de Guayaquil. For the study relied on observational, descriptive and
prospective methods checklists were made based on the ISO 9001: 2008 and
ISO 15189: 2012 to analyze the current state of the relevant documentation and
thus see the degree of compliance under the rules . Questions to the staff
working for their knowledge in what concerns the subject of the investigation
were made. In accordance with the requirements of EN ISO 9001: 2008 the
Institute of Biomedicine at the Catholic University of Santiago de Guayaquil has
an implementation of 80.45% compliance, his other standard also analyzed in
the research process ISO 15189: 2012, has 84.86% compliance. Through
analysis determined that it is vital the full implementation of standards for the
Institute to meet the standards set by the agencies. It is recommended plans for
continuous training and participation of administrative and technical staff of the
Institute of Biomedicine.
Keywords: ISO Standards 9001:2008 and ISO 15189:2012 accreditation,
certification, laboratories.

1
1. El PROBLEMA
1.1. Planteamiento del problema
El Laboratorio de Biomedicina inició sus actividades en el año 1993 como
Laboratorio de Investigación en enfermedades tropicales que ofrecía servicios
de Laboratorio de Análisis con técnicas moleculares de enfermedades
infecciosas.
El Instituto de Biomedicina siendo un Laboratorio de Investigación y
docencia que pertenece a la Universidad Católica de Santiago de Guayaquil se
acoge a ciertas exigencias de la Educación Superior, por esta razón resulta de
vital importancia certificar y posteriormente acreditar cada uno de los
Laboratorios que forman parte de la institución.
Debido a la exigencia por parte de los organismos de acreditación, el
Instituto de Biomedicina de la Universidad Católica de Santiago de Guayaquil
se ha visto en la necesidad de establecer un sistema de gestión de la calidad,
para así brindar resultados de calidad.
Es por ello que para poder certificar y acreditar se deben evaluar el estado
actual de la Institución bajo las normas ISO 9001:2008 que se refiere a los
requisitos del sistema de gestión de la calidad y la ISO 15189:2012 que se
refiere al sistema de gestión de calidad en los Laboratorios Clínicos.

2
1.1.1. Formulación del problema
El Laboratorio de Biomedicina siendo un centro de diagnóstico biomolecular de
enfermedades infecciosas y servicio a la comunidad, necesita implementar un
sistema de calidad que normalice todos sus procesos, los cuales llevaran a una
mejora continua.
¿Será posible que el Laboratorio del Instituto De Biomedicina De La
Universidad Católica de Santiago de Guayaquil alcance la certificación y
acreditación en base a las normas ISO 9001:2008 e ISO 15189:2012?

3
1.2. Objetivos 1.2.1 objetivo general
Evaluar en base a la norma ISO 9001:2008 e ISO 15189:2012 el estado inicial
del Laboratorio de Biomedicina de la Universidad Católica Santiago de Guayaquil,
2014 en base a listas de verificación.
1.2.2. Objetivos específicos
1. Analizar el estado actual de los documentos que se encuentran en el
Instituto de Biomedicina de la UCSG en relación a la norma ISO 9001:2008 e ISO
15189:2012
2. Realizar una lista de verificación para el diagnóstico de la norma ISO
9001:2008 e ISO 15189:2012.
3. Determinar el nivel de conocimiento del personal del laboratorio sobre la
acreditación y desarrollo de un sistema de gestión de calidad en el que
desenvuelven habitualmente su labor profesional, a través de una encuesta.

4
1.3. Justificación e importancia
Debido a las exigencias actuales de los organismos certificadores y
acreditadores nacionales “Servicio de Acreditación Ecuatoriano e Instituto
Ecuatoriano de Normalización” es necesario la implementación de la calidad y
competencia en los distintos laboratorio de diagnóstico.
La importancia de evaluar un sistema de gestión de calidad en base a la a
norma ISO 9001:2008 e ISO 15189:2012 permitirá al Laboratorio determinar
cuáles son los puntos de mejora con el objetivo de que la alta dirección emplee
las acciones correctivas pertinentes para brindar un mejor servicio de
diagnóstico.
Revisar y documentar los procedimientos que son aplicables al Laboratorio
de diagnóstico es fundamental para poder emitir un resultado confiable y de
calidad (Terrez- Speziale, 2006).
Variable dependiente.-: Instituto de Biomedicina de la Universidad Catolica de
Santiago de Guayaquil
Variable independiente.- Norma ISO 9001:2008 e ISO 15189:2012

5
2. MARCO TEÓRICO
2.1. Introducción
La salud es un derecho universal y permite mejorar la calidad de vida de una
población. En la actualidad la calidad y el cambio hacia la mejora continua es una
prioridad en las empresas, en Ecuador falta mucho por hacer, debido a que no
existe organización dentro de los laboratorios que regule y organice este ámbito.
Los Laboratorios encargados de emitir resultados clínicos, tienen cada día más
relevancia como herramienta de diagnóstico de enfermedades y, a través de sus
resultados debería haber confiabilidad hacia el usuario otorgando un servicio de
calidad. Por esta razón resulta de vital importancia la documentación de los
procedimientos y el registro de cada uno de los análisis.
La norma internacional ISO 15189 provee los requisitos generales para el
sistema de gestión de la calidad (SGC), incluyendo confidencialidad, formación y
competencia técnica, en lo que se refiere a los laboratorios (Estupiñan, 2006).
Garantizar la calidad en el proceso de análisis de muestras biológicas ha
constituido una de las funciones más importantes, siendo cada vez más evidente
que un Laboratorio Clínico además de poder realizar ensayos con calidad, deberá
demostrarlo, es así que un modelo de gestión se muestra como una vía para
poder alcanzar este fin.
"Los Laboratorios juegan un papel fundamental, de la calidad de los servicios
de Laboratorio dependerá la calidad de la información para la toma oportuna de
decisiones" (OPS, 2006).

6
El concepto de calidad no es un término nuevo, los principios y expectativas
con respecto a la calidad han sido siempre establecidos, sin embargo no todas las
organizaciones disponen de esta implementación.
La Norma ISO 15189 tiene como objetivo satisfacer las necesidades de los
clientes, haciendo que los profesionales de los Laboratorios conozcan la manera
de proceder y puedan efectuar el seguimiento de sus procesos, revelar problemas
y ampliar las medidas oportunas para solucionarlos y prevenirlos.
La propuesta de un sistema de gestión de calidad es importante ya que nos
permite el desarrollo de nuevas estrategias las cuales pueden desarrollar, nuevos
procedimientos que permitan la identificación de problemas analíticos llegando
incluso a prevenir errores en beneficio del Laboratorio y de la comunidad que
solicita el servicio.
2.2. Antecedentes
La ISO “Organización Internacional para la Estandarización” con sede en
Ginebra-Suiza es una entidad encargada de favorecer la normalización en el
mundo. Es una organización no gubernamental fue fundada el 23 de febrero de
1947. Su misión es promover el desarrollo de la estandarización.
En el pasado, el control de calidad se limitaba solamente a los productos
terminados, con base en la calificación cuantitativa y/o cualitativa de las
características del producto y su comparación con los requerimientos del
cliente.

7
Posteriormente, la aplicación del Control de la Calidad se extendió hacia la
ejecución de los procesos, con el objeto de asegurar que la calidad esté
presente en cada una de sus etapas, promoviendo la mejora continua del
sistema de producción.
En las últimas décadas el Laboratorio Clínico ha experimentado notables
cambios. Comenzando desde la década de los 70 donde fueron las técnicas
manuales, en los 80 la de la automatización y la de los 90 es la de la
informatización. De igual modo las fuentes de error se han minimizado pero no
quiere decir que no existan, es por ello una buena política de calidad debe
abarcar todas las actividades y procesos de Laboratorio.
En México la EMA ha acreditado a los Laboratorios Clínicos desde 1999
bajo las Normas aplicables a Laboratorios de ensayo anteriores como son: la
NMX-CC-13-1992, NMX-EC-025-IMNC-2000 y NMX-EC-17025-IMNC-2000; y
debido al resultado del trabajo del GTLC, en septiembre 2005 se realizó la
primera evaluación bajo la norma ISO 15189:2003. Como apoyo a esta
iniciativa, el gobierno federal aprobó el proyecto que bajo la administración de
la Beneficencia Pública dio financiamiento para la conformación de esta
actividad, y donde la pequeña industria conformada por los Laboratorios
Clínicos pudo solicitar apoyo para la visita de pre-evaluación. Así mismo,
después de realizar el proceso de evaluación y acreditación correspondiente,
en noviembre del 2005 y en febrero del 2006, dos Laboratorios Clínicos del
país recibieron la primera acreditación bajo la norma ISO 15189:2003 por la
EMA (Sierra -Amor Rosa Isabel, 2008).
En Canadá, la acreditación de los Laboratorios Clínicos está bajo la
responsabilidad de las diferentes provincias y hasta ahora la norma ISO 15189
es obligatoria.

8
2.3. Definiciones
2.3.1. Calidad
El concepto de Calidad varia según cada autor es por ello que revisaremos
sus diferentes conceptos para poder comprender la definicion de la misma.
Kaoru Ishikawa (Japón 1915 - 1989): este autor pone claro que la prueba para
una calidad alta es la satisfaccion de cualquier cambio en las perspectivas del
cliente su teoría se relaciona simplemente en lo siguiente en "desarrollar, diseñar,
manufacturar y mantener un producto de calidad.Para Ishikawa la calidad debía
estar definida compresivamente , no bastaba con decir que el producto era de alta
calidad. Se debe enfocar en la calidad de cada area de la organización.
Phillip B. Crosby: Él implementa la palabra de la Prevención como una palabra
clave en la definición de la calidad total. El paradigma que Crosby quiere eliminar
es el de que la calidad se da por medio de inspección, de pruebas, y de revisiones,
esto originaría pérdidas tanto de tiempo como de materiales, ya que con la
mentalidad de inspección esto está preparando al personal a fallar, así que “hay
que prevenir y no corregir”. Crosby propone 4 pilares que debe incluir un programa
corporativo de la calidad, los cuales son: Participación y actitud de la
administración, Administración profesional de la calidad, Programas originales y
Reconocimiento (Garcia, 2012).
William Edwards Deming, exponia que la calidad no implica lograr la perfección,
implica la producción eficiente de la calidad que el mercado demanda,
mencionando como argumento principal que la calidad tiene que estar definida en
términos de satisfacción al cliente (Garcia, 2012).

9
Otras definiciones de organizaciones reconocidas y expertos del mundo de
la calidad son:
• Real Academia de la Lengua Española: “Propiedad o conjunto de
propiedades inherentes a una cosa que permiten apreciarla como igual, mejor
o peor que las restantes de su especie”.
• Definición de la norma ISO 9001: “Calidad: grado en el que un conjunto
de caracteristicas inherentes cumplen con los requisitos.
La calidad implica la capacidad de satisfacer los deseos de las personas
dentro de su estilo de vida, esto involucra un equilibrio entre lo objetivo/tangible
y lo subjetivo/intangible, ofrecer características beneficiosas y saludables para
las personas y su entorno. Para efectos de este trabajo consideraremos una
definición de calidad que combine el enfoque de cumplimiento de requisitos y el
enfoque de satisfacción del cliente. Calidad será entender los necesidades del
cliente y proveer los procesos que satisfagan dichas necesidades de manera
coherente y sostenida, a través del cumplimiento de los requisitos de la Norma
INTE-ISO/IEC 15189:2008.
2.3.2. Enfoque mediante procesos.
Los sistemas de gestion de la calidad trabajan con modelos enfocados en
procesos. Para que la organización funcione de manera eficaz, se deben
determinar numerosas actividades que van a estar relacionadas entre si. Se
puede considerar como un porceso, una actividad o el conjunto de ellas, que
utilizan recursos y que se gestionan con la finalidad que los elementos de
entrada se transformen en resultados.

10
La ventaja de un enfoque basado en procesos es el control continuo que
porporiciona sobre los vínculos entre los procesos individuales dentro del
sistema de procesos. El Ciclo de mejora continua PHVA (planificar- hacer-
verificar – actuar) puede emplearse a todos los procesos,. el PHVA es una
metodología dinámica que puede ser extendida de cada uno de los procesos
de la organización.
a) Planificar crear los objetivos y procesos necesarios para lograr
resultados de acuerdo con los exigencias del cliente y las politicas de la
organización.
b) Hacer realizar e implementar lo planificado
c) Verificar efectuar el seguimiento y la medicion de los procesos y los
productos referente a las politicas, los objetivos y los requisitos para el
producto y comunicar sobre los resultados.
d) Actuar llevar la toma de las acciones para mejorar continuamente el
desempeño de los procesos y volver al paso 1 planificar.
2.3.3. Los 14 Puntos de Deming
Los catorce prinicipios de Deming son fundamentales para la gestión y la
tranformación en la eficacia de una empresa, los principios para cambiar la
gestión de las empresas son ser competitivo y productivo para mantener una
calidad.

11
1. Crear y dar a conocer: se debe dar a conocer a todos los empleados
una descripción clara de los objetivos y propósitos de la empresa.
2. Aprender la nueva filosofía, desde los altos ejecutivos hasta las bases
de la empresa.
3. Comprender el propósito de la inspección, para una mejora de los
procesos y reducción de los costos.
4. Competir con la calidad y no con el precio de venta.
5. Mejorar continuamente el sistema de producción y servicio.
6. Ofrecer capacitación a los trabajadores.
7. Aprender el liderazgo y fomentar el trabajo en equipo.
8. Eliminar el miedo en la organizacion. Crear confianza. Crear el ambiente
adecuado para la innovación.
9. Eliminar barreras entre departamentos. Optimizar los procesos en busca
del logro de los objetivos y propósitos mediante el esfuerzo de equipos.
10. Eliminar slogans
11. Eliminar estandares para la producción. Eliminar la administración por
objetivos y aprender las capacidades de los procesos y como
mejorarlos.
12. Motivar al personal para que se sienta orgulloso de su trabajo.
13. Fomentar la educación y la automejora en los trabajadores.
14. Poner a todos a cooperar para llevar a cabo la transformación.

12
2.3.4. Control de Calidad
Años atrás el Control de Calidad solo se limitaba a los productos terminados,
con base en la calificación cuantitativa y/o cualitativa de las características del
producto y su comparación con los requerimientos del cliente. Posteriormente, la
aplicación se extendió hacia la ejecución de los procesos, con el objeto de
asegurar que la calidad esté presente en cada una de sus etapas, promoviendo la
mejora continua del sistema de producción. A principios de los años 70, un gran
número de problemas industriales evidenció la necesidad de adoptar un
planteamiento normalizador para la administración de la calidad.
Control de calidad puede definirse como: “Un conjunto de tecnicas y
actividades de carácter operativo, utilizadas para verificar los requisots a la
calidad de las pruebas de un laboratorio”
(http://dspace.espoch.edu.ec/bitstream/123456789/3018/1/85T00281.pdf).
2.3.5. Aseguramiento de la Calidad
El aseguramientos de Calidad es el conjunto de acciones planificadas y
sistemáticas, implementadas en el Sistema de Calidad, que son necesarias
para proporcionar la confianza adecuada de que un producto satisfará los
requisitos dados sobre la calidad (rey, 2006).
2.3.6. Gestion de la Calidad
Se entiende por Gestion de la Calidad el conjunto de caminos mediante los
cuales se consigue la calidad, incorporándolo por tanto al proceso de gestion.
De este modo una posible definicion seria le modo en que la direccion planifica

13
el futuro, implanta los programas y controla los resultados de la funcion calidad
con vistas a su mejora permanente (Durán, 1992).
2.3.7. Sistema de Gestion de la Calidad
Un sistema de Gestion de Calidad es la estructura organizativa, las
responsabilidades, los procedimientos, los procesos y los recursos necesarios
para llevar a cabo la Gestion de la Calidad (Gonzalez, 2007).
Las normas que corresponden a cumplir para el diseño e implementación
del SGC son sujetas a evidencia a través de una auditoria para alcanzar una
certificación. Según la norma ISO 9001:2008 tienen por objeto establecer
procedimientos para la Gestión de Calidad y los fundamentos en los que se
basa el SGC.
La certificación para un Laboratorio Clínico en ISO 9001:2008; se refiere al
reconocimiento formal de la aptitud de la organización para gerenciar la
calidad, puede ser para todos los servicios o solo para una parte de ellos, y
garantiza que el laboratorio tiene implementado y activo un SGC y que cumple
los requisitos de la norma (Gonzalez, 2007).
En cambio la Acreditación ISO 17025 o 15189 reconoce la organización y
competencia técnico-profesional del Laboratorio Clínico para los servicios
concretos acreditados pero también garantizar el funcionamiento adecuado del
SGC ISO 9001 implementado y activo (Gonzalez, 2007).

14
2.3.8. Políticas de Calidad
Se entiende por Políticas de Calidad a los diferentes planteamientos
globales, así como las directrices de una organización, referentes a la calidad,
tal como expresan formalmente por la alta dirección de la empresa. En otras
palabras podemos decir que la política de calidad establece el marco sobre el
cual una organización desea moverse, y se puede definir teniendo en cuenta
las metas de la organización y las expectativa, necesidades de los clientes.
2.3.9. Planificación de la Calidad
Se puede definir como el establecimiento y el desarrollo de los objetivos y
requisitos para la calidad: requisitos del producto/servicio y requisitos para la
aplicación de los sistemas de calidad. Cubre tanto la planificación del producto
o servicio como la planificación empresarial y operacional.
2.3.10. Requisito
Un requisito es una necesidad o expectativa establecida, generalmente
implícita u obligatoria. "Generalmente implícita" significa que es habitual o una
práctica común para la organización, sus clientes y otras partes interesadas.
Pueden utilizarse calificativos para identificar un tipo específico de requisito,
por ejemplo, requisito de un producto, requisito de la gestión de la calidad,
requisito del cliente. De este modo, un requisito especificado es aquél que se
declara, por ejemplo, en un documento. En una gran mayoría de los casos los
requisitos pueden ser generados por las diferentes partes interesadas
(http://www.expero2.eu/expero1/hypertext/documenti/govaq/GLOSARIO_DE_T
ERMINOLOGIA_SOBRE_CALIDAD.pdf).

15
2.4. Normas ISO
2.4.1. Evolución de las Normas ISO 9000. Primera Edición de la Norma ISO 9000.
Aparecieron 3 modelos en el año de 1987 para diseñar sistemas de
aseguramiento de calidad:
1. ISO 9001 Modelo para el Aseguramiento de la Calidad en diseño,
desarrollo, producción, instalación y servicio.
2. ISO 9002 Modelo para el Aseguramiento de la Calidad en producción,
instalación y servicio.
3. ISO 9003 Modelo para el Aseguramiento de la Calidad en inspecciones
y pruebas.
La Segunda Edición de las Normas Iso 9000.
En el año de 1994 se revisan las tres normas anteriores y se publica la 2ª
edición de las normas ISO 9001, ISO 9002 e ISO 9003 y estuvieron
vigentes hasta el 14 de diciembre del 2003 (sayce, 2004).
La Tercera Edición de las Normas Iso 9001.
Durante el año 2000 se revisaron las normas ISO 9001:1994, ISO
9002:1994 e ISO 9003:1994 y el 15 de diciembre de ese año se publicó la

16
norma ISO 9001:2000 Sistemas de gestión de la calidad. Esta norma
contempló un único modelo que remplaza a los tres modelos anteriores (sayce,
2004).
La norma ISO 9001:2000 no solamente incorporó un cambio en su nombre
sino un cambio radical haciendo mucho énfasis en la efectividad del sistema de
gestión de la calidad y el mejoramiento del desempeño de las organizaciones.
Algunos expertos consideran que la norma ISO 9001:2000 evolucionó del
concepto de “conformance” a “performance”, es decir, se evolucionó de
demostrar el cumplimiento de requisitos al mejoramiento del desempeño de las
organizaciones (sayce, 2004).
La Cuarta Edición de la Norma ISO 9001.
La norma ISO 9001:2000 fue revisada en el año 2008 y el 15 de noviembre
de este año se publicó la cuarta edición. El 14 de noviembre de 2008 a través
del boletín 1180 informa que la edición ISO 9001:2008 no contiene nuevos
requisitos comparada con la 3ª edición de ISO 9001 y solamente proporciona
aclaraciones de los requisitos existentes de ISO 9001:2000 basadas en los 8
años de experiencia de la implementación de esta norma a nivel mundial e
introduce cambios con la intención de mejorar la consistencia con la norma de
gestión ambiental ISO 14001:2004 para facilitar la integración de sistemas de
gestión de calidad y sistemas de gestión ambiental. (sayce, 2004)
2.4.2. Norma ISO 9001:2008
La Norma ISO 9001 elaborada por la Organización Internacional para la
Estandarización, especifica los requisitos para un Sistema de Gestión de

17
Calidad que toman utilizarse para su aplicación interna por las organizaciones,
para certificación o con fines contractuales.
De acuerdo a (EQA, 2007), la actual versión de ISO 9001 data
de noviembre de 2008, por ello se expresa como ISO 9001:2008, y está
estructurada en 8 capítulos:
a. Cap.1 al 3: Guías y descripciones generales, no se enuncia ningún
requisito.
b. Cap.4 Sistema de gestión: contiene los requisitos generales y los
requisitos para gestionar la documentación.
c. Cap.5 Responsabilidades de la Dirección: contiene los requisitos que
debe cumplir la dirección de la organización, tales como definir la política,
asegurar que las responsabilidades y autoridades están definidas, aprobar
objetivos,el compromiso de la dirección con la calidad, etc.
d. Cap.6 Gestión de los recursos : la Norma distingue 3 tipos de recursos
sobre los cuales se debe actuar: RRHH, infraestructura, y ambiente de
trabajo. Aquí se contienen los requisitos exigidos en su gestión.
e. Cap.7 Realización del producto: aquí están contenidos los requisitos
puramente productivos, desde la atención al cliente, hasta la entrega del
producto o el servicio.
f. Cap.8 Medición, análisis y mejora : aquí se sitúan los requisitos para los
procesos que recopilan información, la analizan, y que actúan en
consecuencia. El objetivo es mejorar continuamente la capacidad de la

18
organización para suministrar productos que cumplan los requisitos. El
objetivo declarado en la Norma, es que la organización busque sin
descanso la satisfacción del cliente a través del cumplimiento de los
requisitos.
2.4.3. Norma ISO 15189:2012
La Norma ISO 15189: 2012 fue desarrollada con la meta de establecer
requisitos para acreditar el Sistema de Gestión de Calidad y la Competencia
Técnica de los Laboratorios Clínicos, abarcando desde la etapa pre hasta la
postexamen. Desde el punto de vista médico, lo más sobresaliente de la nueva
norma es la necesidad de que los laboratorios generen resultados que sean
médicamente relevantes, por lo que es recomendable que los profesionales del
laboratorio, además de vigilar la confiabilidad de los estudios, nos involucremos
más en la adecuada utilización e indicación de las pruebas y en la correcta
interpretación y utilización de los resultados (Speziale-Terrés, 2007).
2.4.3.1. Componentes del Sistema de Gestion de Calidad. (ISO 15189)
Los componentes de un Sistema de Gestion de la Calidad ISO 15189 se basan
en los siguen puntos:
1. Objeto y campo de aplicación
2. Normas de referencia
3. Terminos y definiciones
4. Requisitos de gestion
4.1. Organización y gestion
4.2. Sistema de gestion de la calidad

19
4.3. Control de los documentos
4.4. Revision de solicitudes, ofertas y contratos
4.5. Analisis efectuados por laboratorios de referencia
4.6. Servicios externos y suministradores
4.7. Servicios de asesoramiento
4.8. Resolución de conflictos y quejas
4.9. Identificacion y control de las no conformidades
4.10. Acción correctiva
4.11. Acción preventiva
4.12. Mejora continua
4.13. Registros tecnicos y de la calidad
4.14. Auditorías internas
4.15. Revisión por la direccion
5. Requisitos técnicos
5.1. Personal
5.2. Ubicación y condiciones ambientales
5.3. Equipos de Laboratorio
5.4. Procedimientos preanalíticos
5.5. Procedimientos analiticos
5.6. Aseguramiento de la calidad de los procedimientos de analisis
5.7. Procedimientos postanaliticos
5.8. Informe de los resultados
2.4.4. Evolución de la Acreditación de los Laboratorios
En Suecia en 1992 se acreditó el primer Laboratorio Clínico con la finalidad de
establecer y obtener un Sistema de Gestión de la Calidad. La acreditación se dio
acorde con la Guía ISO/IEC 25:1990, Requisitos generales de competencia para
Laboratorios de ensayo y calibración, y la EN 45001, Criterios generales para el
funcionamiento de los laboratorios.

20
Con el pasar de los años se hizo necesario la acreditacion de un Laboratorio,
sin embargo los Laboratorios creían que la Guía ISO/IEC 25:1990 no tenía la
suficiente aplicación, principalmente en lo que correspondía con la fase
Preanalítica y la Posanalítica.
En 1999 se logro publicar la Norma ISO/IEC 17025 para los Laboratorios de
calibración y ensayos industriales. Sin embargo esta norma debio considerarse de
nuevo, debido a que las relaciones con los pacientes necesitaban ciertas
consideraciones especiales.
Es asi como se planteo de forma específica la Norma EN ISO 15189:2012 como una alternativa a ISO/IEC 17025 e ISO 9001, la cual
porporciona los requisitos relativos a la competencia y calidad que son propios de
los Laboratorios Clinicos (Sans, 1998).
2.4.4.1. Acreditacion en un Laboratorio.
La Acreditación es el proceso mediante el cual un organismo autorizado
realiza la atestación de tercera parte de la competencia de los Organismos de
Evaluación de la Conformidad, (OEC). La autoridad de un organismo de
acreditación generalmente se deriva del gobierno. La acreditación, es la
herramienta establecida a escala internacional para generar confianza sobre la
actuación de un tipo determinado de organizaciones que se denominan de
manera general Organismos de Evaluación de la Conformidad y que abarca a
los Laboratorios de ensayo, Laboratorios de Calibración, Organismos de
certificación, Organismos de Inspección (http://www.acreditacion.gob.ec/que-
es-la-acreditacion/).

21
La Acreditación del Laboratorio Clínico, en su sentido más amplio, cobra una
importancia creciente como instrumento de gestión y como medio para crear
confianza en los resultados. La Norma Internacional ISO/15189 "Laboratorios
Clínicos. Requisitos particulares para la calidad y la competencia" (específica
para los laboratorios clínicos), proporciona los requisitos generales para el
sistema de gestión de la calidad y para la competencia técnica (Manlab, 2007).
2.4.4.2. Preparaciones Y Consideraciones de Acreditacion en base a la Norma ISO 15189
Antes que un Laboratorio quiera buscar una acreditación bajo ISO 15189,
deberá como primer punto integrar un comité de calidad, incorporar un
coordinador de calidad, efectuar un análisis inicial y definir políticas
describiendo procesos y definiendo procedimientos.
El Comité de Calidad deberá realizar las etapas siguientes: hacer un análisis
inicial del laboratorio, posteriormente establecer una política de calidad y
desarrollar un manual de calidad. Se describe cada una de las etapas a
continuación:
Análisis Inicial.-. El Comité de Calidad deberá realizar describir las
políticas, procedimientos o procesos que se tuvieran en marcha. Luego de
realizar esto, habrá que comparar con los requisitos de la Norma Internacional
ISO 15189 y ver las insuficiencias que hay.
Política Global de Calidad.- La política de calidad es una manifestación de
voluntad y forma parte del Manual de Calidad. Deberá describirse lo más
brevemente como sea posible al Laboratorio. Su redacción debe ser general.

22
Una política de calidad es describir que compromisos está deseando realizar el
Laboratorio y como alcanzará esos propósitos.
2.4.4.3. Diferencia entre una Acreditación y Certificación
Es necesario tener bien claro la diferencia entre Certificación y Acreditación. La
Certificación es un procedimiento por el cual una tercera parte da garantia escrita
de que un producto, proceso o servicio es conforme a requisitos especificos. La
Acreditación es el procedieminto mediante el cual un cuerpo autorizado da
reconocimeinto formal de que un organismo o persona es competente para llevar
a cabo tareas especificas (Burnett, 1998).
2.4.4.4. Ventaja De Una Certificación.
a) Permite la aceptación y reconocimiento de resultados de inspecciones,
ensayos y calibraciones.
b) Garantiza la seriedad e idoneidad de un certificado o informe de
resultados.
c) Garantiza que los organismos de certificación que están acreditados
trabajan en forma equivalente.
d) Mejora la calidad de los servicios.
El implementar un Sistema de Gestión de Calidad nos permite asegurar y
mejorar continuamente.

23
2.4.4.5. ¿Que organismos se encargan de la normalización?
En primera instancia en el Ecuador el Instituto Ecuatoriano de Normalización,
INEN, es la entidad adscrita al Ministerio de Comercio Exterior, Industrialización
Pesca y Competitividad, es el organismo encargado de las actividades de
Normalización en el Ecuador.
El INEN representa a la República del Ecuador ante los Organismos
Internacionales de Normalización, Certificación y Metrología, siendo miembro
pleno de la ISO (Organización Internacional de Normalización), de COPANT
(Comisión Panamericana de Normas Técnicas), de SIM (Sistema Interamericano
de Metrología) y miembro corresponsal de la OIML (Organización Internacional de
Metrología Legal) y punto de contacto de la Comisión del Codex Alimentarius
(INEN, 2007).
Los Organismos de Certificación, (OC), para demostrar competencia técnica,
imparcialidad y transparencia de sus operaciones, requieren acreditarse bajo
las siguientes normativas internacionales:
• Organismos de Certificación de Sistema de Gestión Norma NTE
INEN ISO/IEC 17021
• Organismos de Certificación de Productos Norma NTE
INEN ISO/IEC 17065
• Organismos de Certificación de Personas Norma NTE
INEN ISO/IEC 17024
El sistema de acreditación del Servicio de Acreditación Ecuatoriano, SAE,
ofrece a todos los OEC interesados en la acreditación, un procedimiento donde

24
están definidos los requisitos de que deben cumplir los OEC para su
acreditación (http://www.acreditacion.gob.ec/como-acreditarse-2/).
En otros paises tambien se cuenta con ciertos organismos entre ellos tenemos
a la principal ORGANIZACION INTERNACIONAL DE NORMALIZACION (ISO) es
la elaboración de Normas internacionales.
El COMITE EUROPEO DE NORMALIZACION (CEN) es el Organismo
Normalizador de la Comunidad Europea. La Asociación española de
Normalización y Certificación (AENOR) es el Organismo Normalizador Español.
2.4.4.6. Quien se encarga de evaluar la ISO 9001 e ISO 15189
La Iso 15189 es evaluada por organismos de accreditacion los cuales se
encargan de realizar actividades de acreditacion. La Iso 9001 es evaluada por
organismos de certificacion quienes se encargan de realizar actividades
referentes a la certificacion.
El campo de aplicación de las normas ISO es independiente el uno del otro.
La norma ISO 9001 aplica a todas las organizaciones sin importar el tamaño,
tipo o producto. En cambio la ISO 15189 solo especifica los requisitos para la
calidad y la competencia de los Laboratorios Clinicos.
Por ende podemos decir que a diferencia de la certificación ISO 9001, que
es la confirmación de que una empresa ha establecido un sistema de gestión
de la calidad conforme a ciertos requisitos, la acreditación de acuerdo a la
Norma ISO 15189 confirma la competencia técnica del Laboratorio y garantiza

25
la fiabilidad en los resultados de los análisis y ensayos
(http://www.enac.es/web/enac/diferencia_acreditacion_certificacion).
2.5. Antecedente del Instituto de Biomedicina
El Instituto de Biomedicina es una unidad sin fines de lucro adscrita a la
Facultad de Ciencias Médicas de la Universidad Católica de Santiago de
Guayaquil, creada por el Consejo Universitario
(http://www.institutobiomedicina.com/ecuador/el-instituto).
La Universidad Católica Santiago de Guayaquil desde su Misión y Visión es
inherente a la Reforma de la Ley Orgánica de la Educación Superior del
Ecuador y el Plan Nacional del Buen Vivir, por lo que es indispensable
fortalecer el ámbito de la investigación productiva a partir de conocer las
líneas de investigación establecidas, desarrolladas y que los resultados
hayan sido evidencia científica mediante publicaciones nacionales e
internacionales (http://www.institutobiomedicina.com/ecuador/el-instituto).
2.5.1. Misión
Tiene por Misión realizar investigación científica, implementación y
desarrollo de nuevas tecnologías a través de su Laboratorio y personal
especializado dentro de áreas específicas de la Biomedicina y del
conocimiento; prestar servicios profesionales como medio para su
autofinanciamiento y realizar actualización o estudios continuos de postgrado,
dar soporte a la educación de pregrado y colaborar con el Sistema de
Postgrado de la Universidad. Es decir se manejará bajo tres parámetros bien

26
definidos: investigación, docencia y venta de servicios
(http://www.institutobiomedicina.com/ecuador/el-instituto).
2.5.2. Visión
Tiene por visión lograr posicionarse dentro de la investigación científica
nacional e internacional con liderazgo y excelencia académica
(http://www.institutobiomedicina.com/ecuador/el-instituto).
2.5.3. Plan Nacional del Buen Vivir
El Estado y la sociedad hoy demandan que las instituciones de Educación
Superior aporten con nuevos conocimientos para enfrentar los cambios
acelerados que se presentan por la globalización.
Por tanto crear conocimiento es la evidencia científica o de desarrollo e
innovación social que como norma principal determina si una Institución de
Educación Superior ha invertido en investigación productiva, logrando así un
aporte a la solución de los diferentes problemas sociales, económicos, salud y
cultural del país (http://www.institutobiomedicina.com/ecuador/el-instituto).

27
3. Metodología
3.1. Generalidades
El proyecto “Evaluacion de la norma ISO 9001:2008 e ISO 15189:2012
”Laboratorios Clinicos particulares relativos a la calidad y la competencia”
busca aportar al mejoramiento de calidad del Instituto de Biomedicina de la
Universidad Catolica de Santiago de Guayaquil”.
3.2. Diseño de la Investigación
El presente trabajo es de tipo Observacional – descriptiva.
Observacional porque se realizó mediante la observación directa de las
características de la organización, equipo e infraestructura. Descriptivo porque
se realizó una descripción para obtener los datos de las características de
organización de documentos, equipo e infraestructura. Prospectivo porque los
datos se fueron registrando conforme se realizaba la evaluación con el apoyo
de un instrumento de verificación.
Para el desarrollo del proyecto se realizó una evaluación inicial documental
en base a los requisitos de las normas ISO 9001:2008 e ISO 15189:2012 con
el acompañamiento del profesional a cargo de la entidad, con quien se elaboró
una serie de preguntas direccionadas en función a una lista de chequeo,
siendo un instrumento fundamental para el diagnóstico inicia de la
investigación.

28
El criterio establecido para determinar si cumple, no cumple o está en
proceso es el siguiente: Si cumple: cuando el punto o la evidencia se cumple al
100%. No cumple: cuando el punto no lo cumple o la evidencia no existe. En
proceso: cuando el punto o la evidencia se está actualizando, desarrollando o
revisando.
3.3. Procedimiento de la Investigación
1. Evaluación inicial en base a una lista de chequeo.
2. Revisión de la Norma ISO 9001:2008 en el laboratorio de biomedicina de
la universidad católica de Santiago de Guayaquil.
3. Construcción de las interrogantes de la encuesta.
4. Realización de la Encuesta al personal del Laboratorios del instituto de
biomedicina.
5. Revisión de la Norma ISO 15189:2012 en El Laboratorio De Biomedicina
De La Universidad Católica De Santiago De Guayaquil.
6. Análisis de los datos obtenidos en base a la revisión de las normas ISO
9001 E ISO 15189.

29
7. Elaboración de la propuesta.
3.4. Instrumentos de Investigación
Los instrumentos para la recolección de los datos que se utilizaron en la
presente investigación se detallan a continuación:
ü En esta investigación se tomó una Lista de chequeo inicial para ver el
estado general del laboratorio antes de iniciar con la revisión en base a
las normas.
Anexo1 lista de verificación.
ü En esta investigación se tomó como medio de investigación a las
Encuestas, dado que este instrumento permitirá obtener la información
requerida para obtener resultados en torno al tema de la Investigación.
Se llevará a cabo con la correcta formulación de las preguntas, relacionadas
con el tema de investigación, a incluirse en la Encuesta. Cabe indicar que el
tipo de preguntas son en su mayoría, preguntas cerradas.

30
ü Lista de verificación para el cumplimiento de los requisitos establecidos
en la Normas ISO 9001:2008 e ISO 15189:2012
Anexo 2 lista de verificación de la norma ISO 9001:2008
Anexo 3 lista de verificación de la norma ISO 15189:2012
ü Preguntas Para La Encuesta
Anexo 4 lista de preguntas para la encuesta
3.5. Lugar de estudio
Para el desarrollo del presente trabajo se tomó como referencias al Instituto
de Biomedicina de la Universidad Católica de Santiago de Guayaquil, se
encuentra ubicado en la Avenida Carlos Julio Arosemena km 1½ vía Daule,
2º11´1.11´´ latitud sur y 79º54´ 18.11´´longitud occidente.
3.6. Confidencialidad
Para respetar la confidencialidad del Instituto de Biomedicina se firmó un
compromiso de confidencialidad a través de una carta.

31
3.7. Resultados
Dentro de los resultados obtenidos se pudo constatar que el Instituto de
Biomedicina de la Universidad Católica de Santiago de Guayaquil tiene un
Manual de Calidad ISO 9001:2008, consta con un control de documentos, con
un control de registros; de los cuales han sido anexados para su demostración.
Anexo 2 y 3 documentación recopilada.
3.7.1. Diagnóstico respecto a la NORMA ISO 9001:2008
Tabla 1: porcentaje de cumplimiento del instituto de la norma ISO 9001:2008
Requisito de la norma ISO 9001:2008
Porcentaje de cumplimiento
Sistemas de gestión de la calidad 99.29%
Responsabilidad de la dirección 88.94%
Gestión de recursos 98.28%
Prestación del servicio 47.56%
Medición, análisis y mejora 68.18%
Porcentaje de cumplimiento
general
80.45%
32
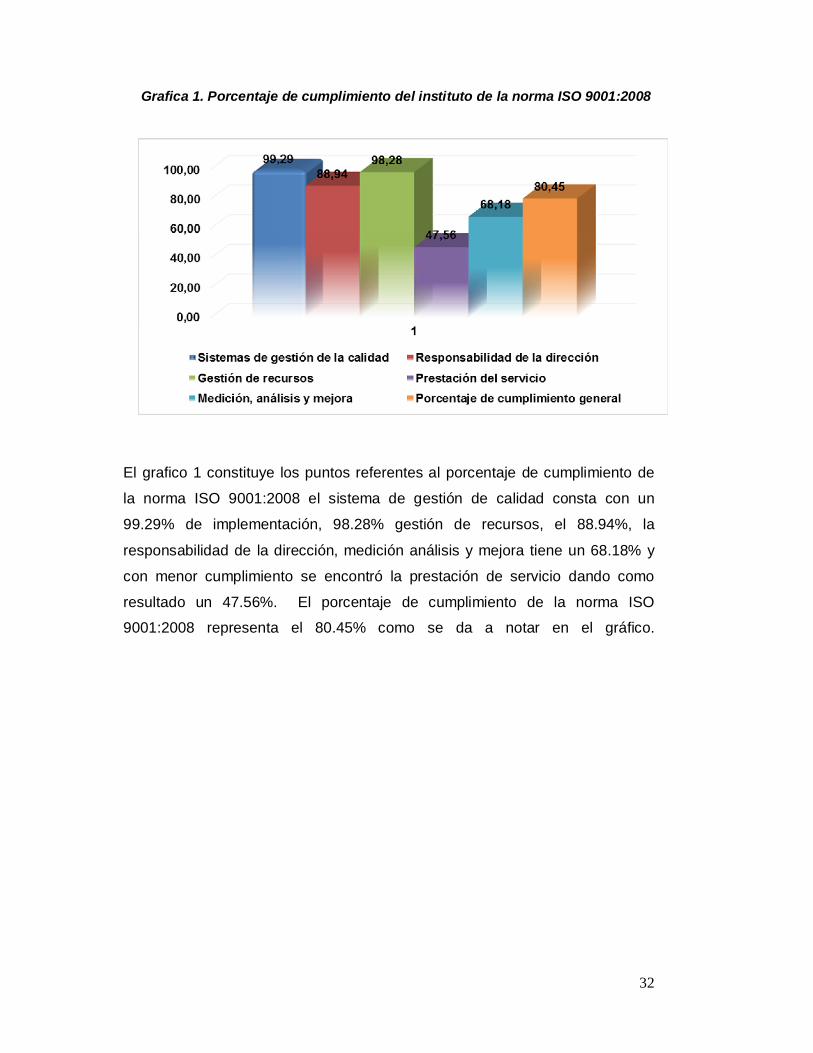
Grafica 1. Porcentaje de cumplimiento del instituto de la norma ISO 9001:2008
3.7.2.
El grafico 1 constituye los puntos referentes al porcentaje de cumplimiento de
la norma ISO 9001:2008 el sistema de gestión de calidad consta con un
99.29% de implementación, 98.28% gestión de recursos, el 88.94%, la
responsabilidad de la dirección, medición análisis y mejora tiene un 68.18% y
con menor cumplimiento se encontró la prestación de servicio dando como
resultado un 47.56%. El porcentaje de cumplimiento de la norma ISO
9001:2008 representa el 80.45% como se da a notar en el gráfico.
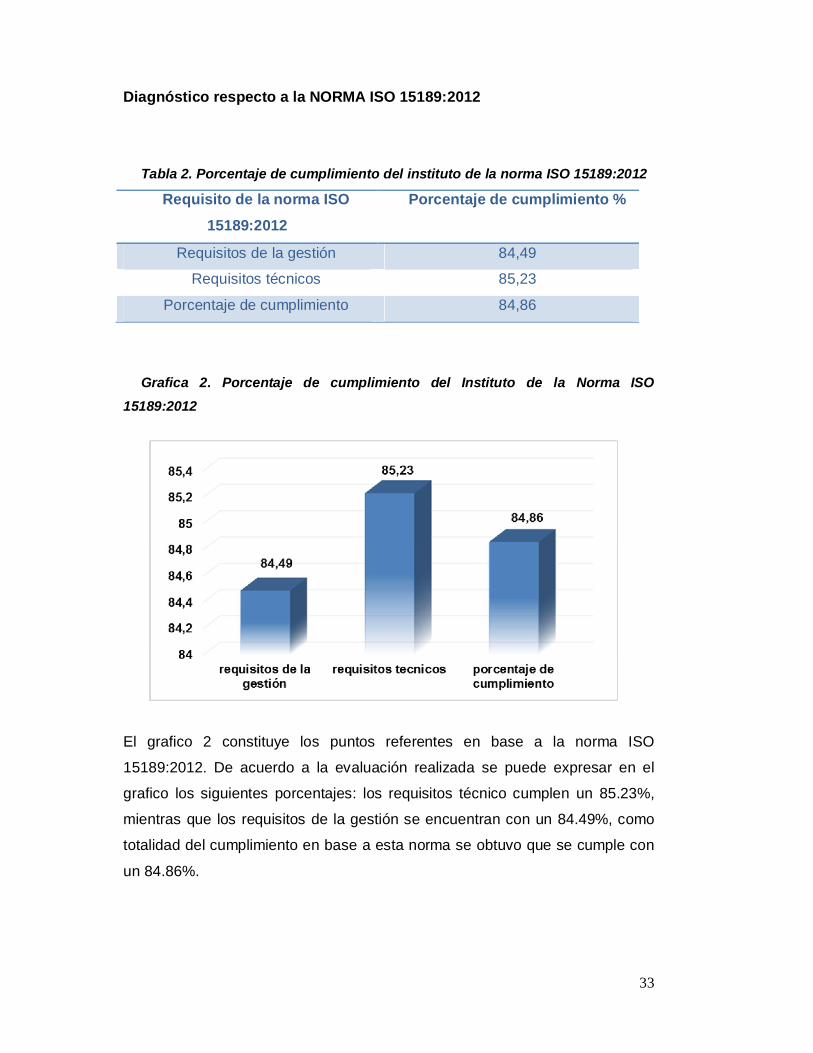
33
Diagnóstico respecto a la NORMA ISO 15189:2012
Tabla 2. Porcentaje de cumplimiento del instituto de la norma ISO 15189:2012
Requisito de la norma ISO
15189:2012
Porcentaje de cumplimiento %
Requisitos de la gestión 84,49
Requisitos técnicos 85,23
Porcentaje de cumplimiento 84,86
Grafica 2. Porcentaje de cumplimiento del Instituto de la Norma ISO
15189:2012
El grafico 2 constituye los puntos referentes en base a la norma ISO
15189:2012. De acuerdo a la evaluación realizada se puede expresar en el
grafico los siguientes porcentajes: los requisitos técnico cumplen un 85.23%,
mientras que los requisitos de la gestión se encuentran con un 84.49%, como
totalidad del cumplimiento en base a esta norma se obtuvo que se cumple con
un 84.86%.
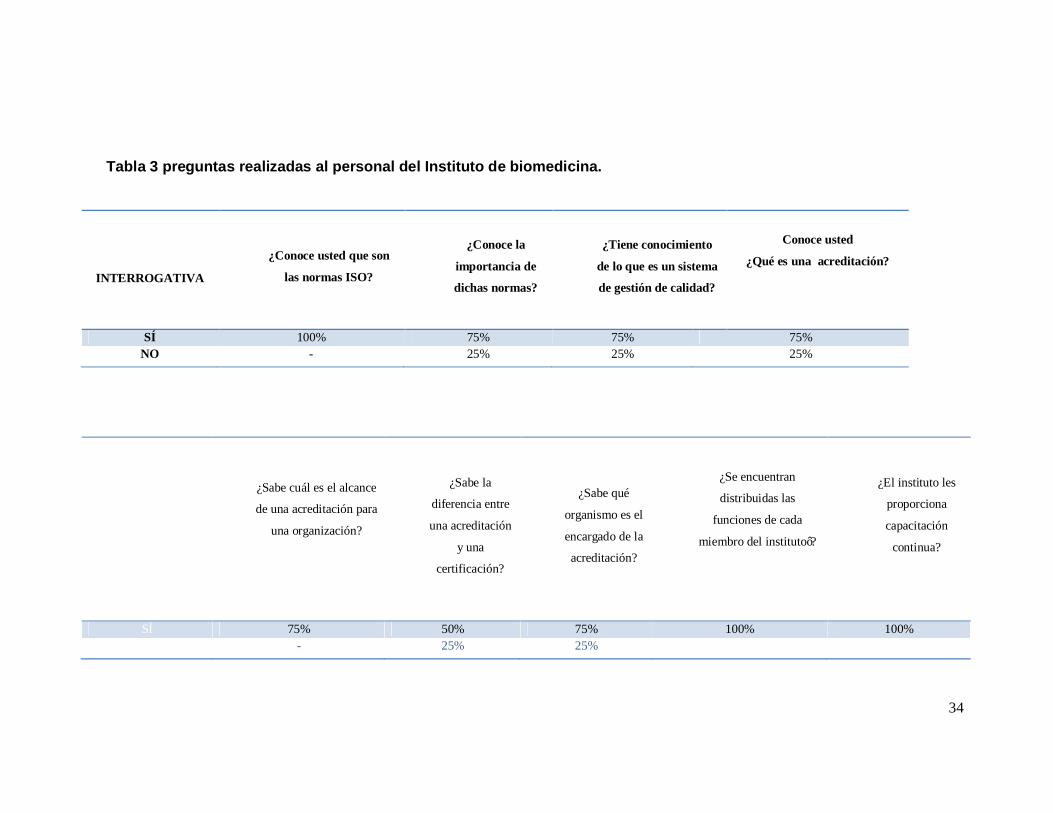
34
Tabla 3 preguntas realizadas al personal del Instituto de biomedicina.
INTERROGATIVA
¿Conoce usted que son
las normas ISO?
¿Conoce la
importancia de
dichas normas?
¿Tiene conocimiento
de lo que es un sistema
de gestión de calidad?
Conoce usted
¿Qué es una acreditación?
SÍ 100% 75% 75% 75% NO - 25% 25% 25%
INTERROGATIVA
RESPUESTAS
¿Sabe cuál es el alcance
de una acreditación para
una organización?
¿Sabe la
diferencia entre
una acreditación
y una
certificación?
¿Sabe qué
organismo es el
encargado de la
acreditación?
¿Se encuentran
distribuidas las
funciones de cada
miembro del instituto’?
¿El instituto les
proporciona
capacitación
continua?
SÍ 75% 50% 75% 100% 100% NO - 25% 25%

35
Encuesta realizada al personal del laboratorio del Instituto de Biomedicina.
75%
25%
SINO
Gráfico 1
El 75% del personal que labora en las instalaciones tiene el conocimiento claro del
proceso que se está llevando a cabo, el 25 % desconoce de ciertas interrogantes de la
encuesta.

36
4. Propuesta
Para el presente proyecto” EVALUACION DE LA NORMA ISO 9001 E ISO
15189 EN EL INSTITUO DE BIOMEDICINA DE LA UNIVERSIDAD CATOLICA
DE SANTIAGO DE GUAYAQUIL, 2014”; dada la finalidad y las funciones que
desempeña la organización se ha considerado que el siguiente paso que le
corresponde sería optar por la implementación de un Sistema de Gestión de la
Calidad formulado con base en la norma INTE/ISO 15189:2012. Por lo que
tomando como referencia los capítulos anteriores sobre el grado de
cumplimiento y la totalidad de requisitos que la empresa no cumple de la
norma, se propone utilizar una herramienta Seis Sigma para desarrollar una
propuesta organizacional con el fin de subsanar aquellos requisitos cuya
implementación se encontraba parcial o sin implementar, queda a criterio y
competencia del departamento de la Calidad, la Gerencia Técnica y la
Gerencia General, evaluar la aplicación del diagrama de Gantt.
Para poder evaluar el avance se realizara una nueva evaluación documental
en seis meses para ver el progreso de la organización en cuanto a la
implementación de los puntos restantes de la Norma ISO 9001:2008 e ISO
15189:2012, la cual le permitirá al Laboratorio tener una mayor confianza en
cuanto a los resultados de sus análisis.
Para el desarrollo de una metodología de mejora continua es fundamental el
claro compromiso y apoyo de la institución para con el proceso, proporcionando
los recursos necesarios para mantener la motivación en el mismo.
Se plantea la posibilidad de que el personal del laboratorio sea capacitado en
nuevos estilos de trabajo y que se implemente Sistema de Gestión de Calidad
que garantice un trabajo organizado, sustentable, sistémico y reproducible a
través de la elaboración de Manuales, Guía de Elaboración de Documentos,
Procedimientos de Análisis y Formatos de trabajo que conllevan a lograr un
Plan de mejora continua.

37
Conclusión
1. De acuerdo al análisis realizado en base a la lista de verificación de la
norma ISO 9001:2008 se identificó que ciertos puntos de la Norma
están en proceso para su posterior implementación actualmente hay
un 80.45% de cumplimiento de los requisitos de la norma.
2. Respecto al análisis en base a los requisitos de la Norma ISO 15189,
actualmente hay un 84.86% de cumplimiento de los requisitos de la
Norma.
3. El resultado de las encuestas índico que un gran porcentaje del
personal si cuenta con la información y el conocimiento apropiado
para el proceso de la certificación, sin embargo un 25% no tuvo el
conocimiento de todas las interrogantes realizadas en la encuesta.
4. De acuerdo al análisis realizado en base a las normas ISO el
Laboratorio de Biomedicina de la Universidad Católica Santiago de
Guayaquil, se puede decir que faltan ciertos puntos de la norma de ser
implementados para lograr la acreditación, así mismo se observó la falta
del trabajo en equipo dando como un posible atraso la implementación
de la norma.

38
Recomendaciones
1. Elaborar planes de capacitación continua para el personal administrativo
y técnico del instituto de biomedicina en el conocimiento de la norma
ISO 9001:2008 e ISO 15189:2012.
2. Brindar más soporte y liderazgo para fortalecer el trabajo en equipo.

39
5. BIBLIOGRAFÍA
1. Brizuela O, C. (2011). La acreditacion en el laboratorio clinico. revista IBEROLAB .
2. Burnett, D. (1998). acrediatcion del laboratorio clinico. En D. Burnett. reverte.
3. criterios de evaluacion del ruido. Legislacion . (s.f.). Obtenido de http://rabfis15.uco.es/lvct/tutorial/1/paginas%20proyecto%20def/(5)%20Criterios%20de%20evaluaci%C3%B3n/organismos%20de%20normalizacion.htm
4. Durán, M. U. (1992). Gestion de Calidad. Madrid, España: ediciones Diaz de Santos.
5. EQA. (2007). ISO 9001. Obtenido de http://www.eqa.org/productos/9001.htm
6. Estupiñan, E. B. (2006). Etica en el laboratorio clinico. publi lab .
7. Garcia, F. J. (2012). diagnostico del cumplimiento de la norma ISO 15189 en la empresa Laboratorios Guerrero. veracruz.
8. Gonzalez, C. (2007). gestion de la calidad, concepto, enfoque, modelos y sistemas. españa: pearson prentice hall.
9. http://www.enac.es/web/enac/diferencia_acreditacion_certificacion. (s.f.). ENTIDAD NACIONAL DE ACREDITACION. Obtenido de http://www.enac.es/web/enac/diferencia_acreditacion_certificacion
10. http://www.expero2.eu/expero1/hypertext/documenti/govaq/GLOSARIO_DE_TERMINOLOGIA_SOBRE_CALIDAD.pdf. (s.f.). Obtenido de http://www.expero2.eu/expero1/hypertext/documenti/govaq/GLOSARIO_DE_TERMINOLOGIA_SOBRE_CALIDAD.pdf
11. http://www.institutobiomedicina.com/ecuador/el-instituto. (s.f.). Instituto de biomedicina de la universidad catolica de santiago de guayaquil. Obtenido de http://www.institutobiomedicina.com/ecuador/el-instituto
12. INEN. (2007). BOLETÍN OFICIAL DEL INSTITUTO ECUATORIANO DE NORMALIZACION. (43).
13. lindsay, J. R. (2005). Administración y control de la calidad. Recuperado el 17 de agosto de 2014, de http://jorriveraunah.files.wordpress.com/2011/06/capitulo-3-filosofias-y-marcos-de-referncia-de-la-calidad.pdf
14. Manlab, D. B. (OCTUBRE de 2007). CALIDAD EN EL LABORATORIO CLINICO MAS ALLA DE LAS NORMAS. REVISTA DE BIOANALISIS , 20-21-22.
15. Marambo, C. (14 de ENERO de 2013). REVISTA CERTIFICACION. Recuperado el 06 de JULIO de 2014, de http://www.revistacertificacion.cl/category/articulo-tecnico/

40
16. MEDICA, I. (s.f.). REQUISITOS GENERALES PARA LA COMPETENCIA DE LOS LABORATORIOS DE ENSAYO Y CALIBRACION. Obtenido de http://www.icsa.es/consultora-ISO-15189
17. OPS, ". h. (3 de enero de 2006). ORGANIZACION PANAMERICANA DE LA SALUD. Recuperado el 26 de JUNIO de 2014, de http://www.paho.org/bol/index.php?option=com_content&view=article&id=583:ops-hora-darle-un-fuerte-impulso-laboratorios&Itemid=1
18. rey, s. l. (2006). implantacion de un sistema de calidad. españa: ideaspropias.
19. Sans, C. (1998). las normas ISO. REVISTA BIBLIOGRAFICA DE GEOGRAFIA Y CIENCIAS SOCIALES (129).
20. sayce. (2004). evolucion de las normas iso 9000. Recuperado el 28 de septiembre de 2014, de http://www.sayce.com.mx/index.php?id=57
21. Sierra -Amor Rosa Isabel, M. D. (2008). acreditacion de laboratorios clinicos ISO 15189-2003. revista Bioquimia , 33 (3).
22. Terrez- Speziale, A. M. (2006). Estimacion de la incertidumbre de la variabilidad total en el laboratorio clinico. rev Mex Patrol Clin , 53 (4), 185-196.
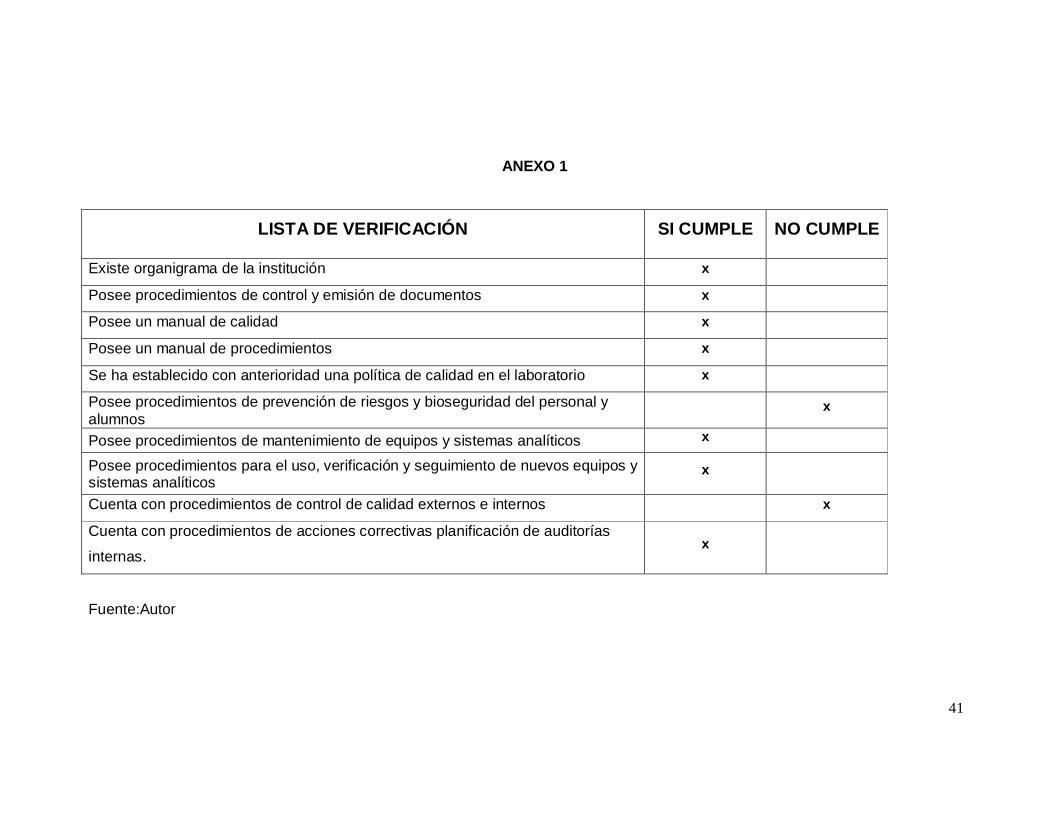
41
ANEXO 1
LISTA DE VERIFICACIÓN SI CUMPLE NO CUMPLE
Existe organigrama de la institución x
Posee procedimientos de control y emisión de documentos x
Posee un manual de calidad x
Posee un manual de procedimientos x
Se ha establecido con anterioridad una política de calidad en el laboratorio x
Posee procedimientos de prevención de riesgos y bioseguridad del personal y alumnos
x
Posee procedimientos de mantenimiento de equipos y sistemas analíticos x
Posee procedimientos para el uso, verificación y seguimiento de nuevos equipos y sistemas analíticos
x
Cuenta con procedimientos de control de calidad externos e internos x
Cuenta con procedimientos de acciones correctivas planificación de auditorías
internas. x
Fuente:Autor
42
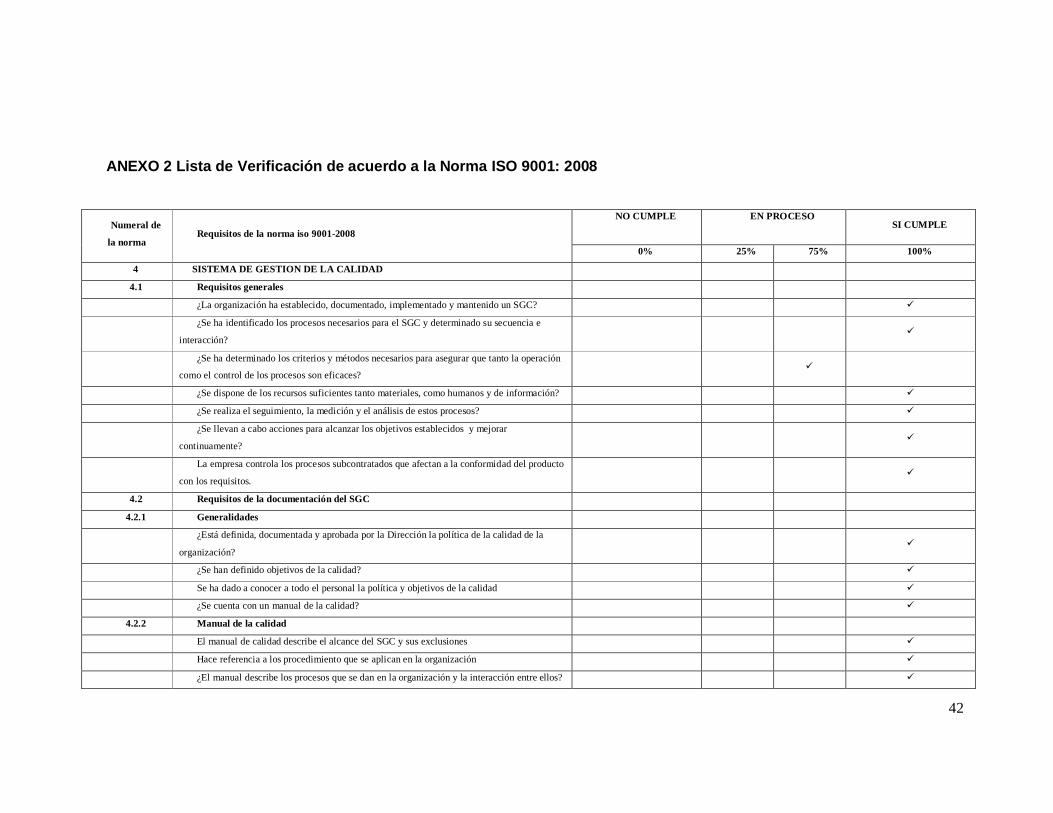
ANEXO 2 Lista de Verificación de acuerdo a la Norma ISO 9001: 2008
Numeral de
la norma Requisitos de la norma iso 9001-2008
NO CUMPLE
EN PROCESO
SI CUMPLE
0% 25% 75% 100%
4 SISTEMA DE GESTION DE LA CALIDAD
4.1 Requisitos generales
¿La organización ha establecido, documentado, implementado y mantenido un SGC? ü
¿Se ha identificado los procesos necesarios para el SGC y determinado su secuencia e
interacción? ü
¿Se ha determinado los criterios y métodos necesarios para asegurar que tanto la operación
como el control de los procesos son eficaces? ü
¿Se dispone de los recursos suficientes tanto materiales, como humanos y de información? ü
¿Se realiza el seguimiento, la medición y el análisis de estos procesos? ü
¿Se llevan a cabo acciones para alcanzar los objetivos establecidos y mejorar
continuamente? ü
La empresa controla los procesos subcontratados que afectan a la conformidad del producto
con los requisitos. ü
4.2 Requisitos de la documentación del SGC
4.2.1 Generalidades
¿Está definida, documentada y aprobada por la Dirección la política de la calidad de la
organización? ü
¿Se han definido objetivos de la calidad? ü
Se ha dado a conocer a todo el personal la política y objetivos de la calidad ü
¿Se cuenta con un manual de la calidad? ü
4.2.2 Manual de la calidad
El manual de calidad describe el alcance del SGC y sus exclusiones ü
Hace referencia a los procedimiento que se aplican en la organización ü
¿El manual describe los procesos que se dan en la organización y la interacción entre ellos? ü
43
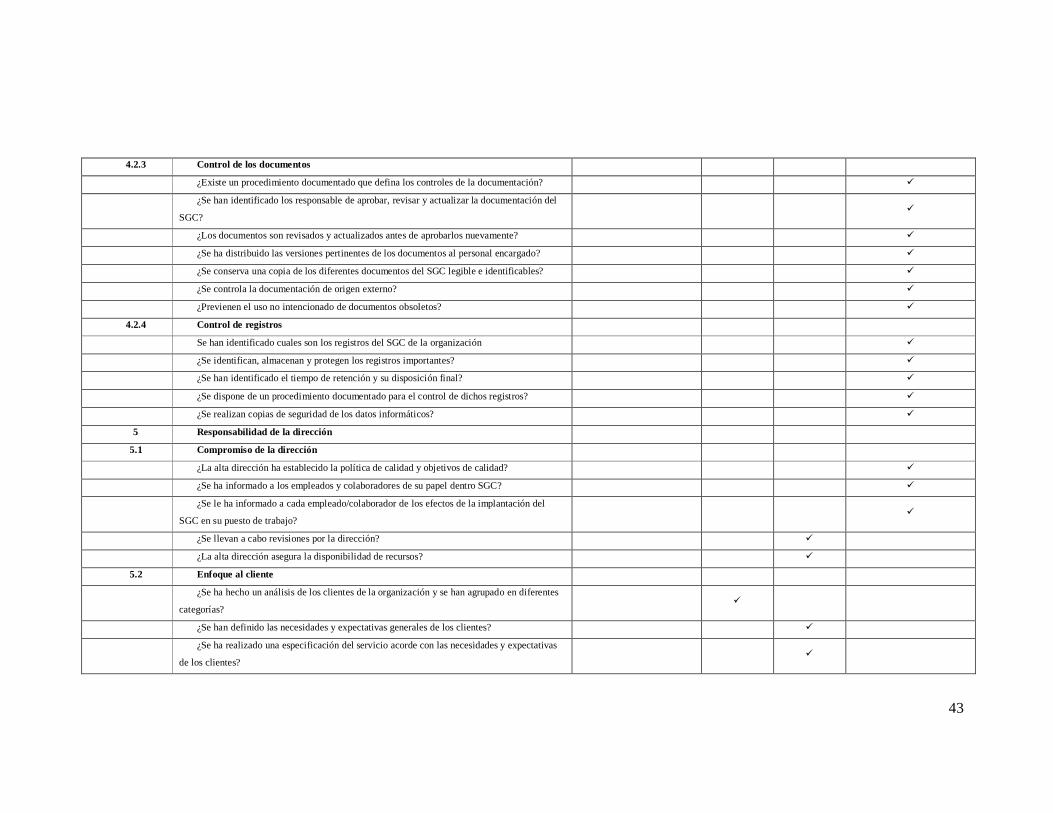
4.2.3 Control de los documentos
¿Existe un procedimiento documentado que defina los controles de la documentación? ü
¿Se han identificado los responsable de aprobar, revisar y actualizar la documentación del
SGC? ü
¿Los documentos son revisados y actualizados antes de aprobarlos nuevamente? ü
¿Se ha distribuido las versiones pertinentes de los documentos al personal encargado? ü
¿Se conserva una copia de los diferentes documentos del SGC legible e identificables? ü
¿Se controla la documentación de origen externo? ü
¿Previenen el uso no intencionado de documentos obsoletos? ü
4.2.4 Control de registros
Se han identificado cuales son los registros del SGC de la organización ü
¿Se identifican, almacenan y protegen los registros importantes? ü
¿Se han identificado el tiempo de retención y su disposición final? ü
¿Se dispone de un procedimiento documentado para el control de dichos registros? ü
¿Se realizan copias de seguridad de los datos informáticos? ü
5 Responsabilidad de la dirección
5.1 Compromiso de la dirección
¿La alta dirección ha establecido la política de calidad y objetivos de calidad? ü
¿Se ha informado a los empleados y colaboradores de su papel dentro SGC? ü
¿Se le ha informado a cada empleado/colaborador de los efectos de la implantación del
SGC en su puesto de trabajo? ü
¿Se llevan a cabo revisiones por la dirección? ü
¿La alta dirección asegura la disponibilidad de recursos? ü
5.2 Enfoque al cliente
¿Se ha hecho un análisis de los clientes de la organización y se han agrupado en diferentes
categorías? ü
¿Se han definido las necesidades y expectativas generales de los clientes? ü
¿Se ha realizado una especificación del servicio acorde con las necesidades y expectativas
de los clientes? ü

44
5.3 Política de la calidad
¿La política es adecuada al propósito de la organización? ü
¿Existe una política de la calidad, recogida en algún documento y aprobada por la
dirección? ü
¿Incluye un compromiso de cumplir con los requisitos y de mejora de eficacia del sistema
de gestión de la calidad? ü
¿Pueden extraerse o derivarse fácilmente objetivos de calidad? ü
¿Se ha distribuido y explicado al personal la política de la calidad? ü
¿Es revisada para su continua adecuación? ü
5.4 Planificación
5.4.1 Objetivos de la calidad ü
¿Están definidos los objetivos de la calidad? ü
¿Están cuantificados dichos objetivos y por tanto son medibles? ü
¿Son representativos de las características del servicio? ü
Las metas fijadas para dichos objetivos ¿son “a priori” alcanzables? ü
¿Se han comunicado a los empleados y colaboradores dichos objetivos? ü
¿Se les ha explicados como alcanzar dichos objetivos? ü
¿Se realiza una revisión periódica de dichos objetivos? ü
5.4.2 Planificación del SGC
¿La planificación del sistema de gestión de calidad con el fin de cumplir los requisitos
citados en el apartado 4.1 y objetivos de calidad? ü
¿Se mantienen la integridad del sistema de gestión cuando se planifican cambios en este? ü
5.5 Responsabilidad, autoridad y comunicación
5.5.1 Responsabilidad y autoridad
¿Existe un organigrama actualizado de la organización? ü
¿Están definidas por escrito las funciones y responsabilidades del personal? ü
¿Están claras las relaciones entre todo el personal (dependencias, canales de información y
ayuda)? ü
¿Todo el personal conoce sus funciones y responsabilidades? ü
45
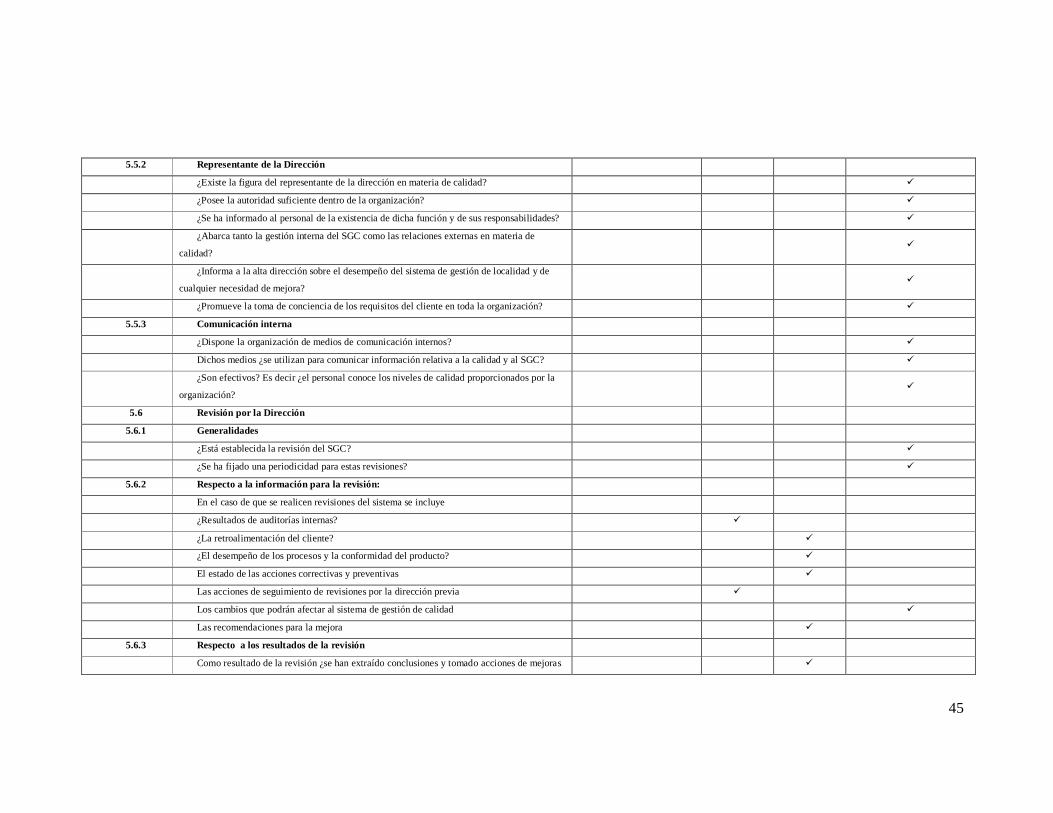
5.5.2 Representante de la Dirección
¿Existe la figura del representante de la dirección en materia de calidad? ü
¿Posee la autoridad suficiente dentro de la organización? ü
¿Se ha informado al personal de la existencia de dicha función y de sus responsabilidades? ü
¿Abarca tanto la gestión interna del SGC como las relaciones externas en materia de
calidad? ü
¿Informa a la alta dirección sobre el desempeño del sistema de gestión de localidad y de
cualquier necesidad de mejora? ü
¿Promueve la toma de conciencia de los requisitos del cliente en toda la organización? ü
5.5.3 Comunicación interna
¿Dispone la organización de medios de comunicación internos? ü
Dichos medios ¿se utilizan para comunicar información relativa a la calidad y al SGC? ü
¿Son efectivos? Es decir ¿el personal conoce los niveles de calidad proporcionados por la
organización? ü
5.6 Revisión por la Dirección
5.6.1 Generalidades
¿Está establecida la revisión del SGC? ü
¿Se ha fijado una periodicidad para estas revisiones? ü
5.6.2 Respecto a la información para la revisión:
En el caso de que se realicen revisiones del sistema se incluye
¿Resultados de auditorías internas? ü
¿La retroalimentación del cliente? ü
¿El desempeño de los procesos y la conformidad del producto? ü
El estado de las acciones correctivas y preventivas ü
Las acciones de seguimiento de revisiones por la dirección previa ü
Los cambios que podrán afectar al sistema de gestión de calidad ü
Las recomendaciones para la mejora ü
5.6.3 Respecto a los resultados de la revisión
Como resultado de la revisión ¿se han extraído conclusiones y tomado acciones de mejoras ü
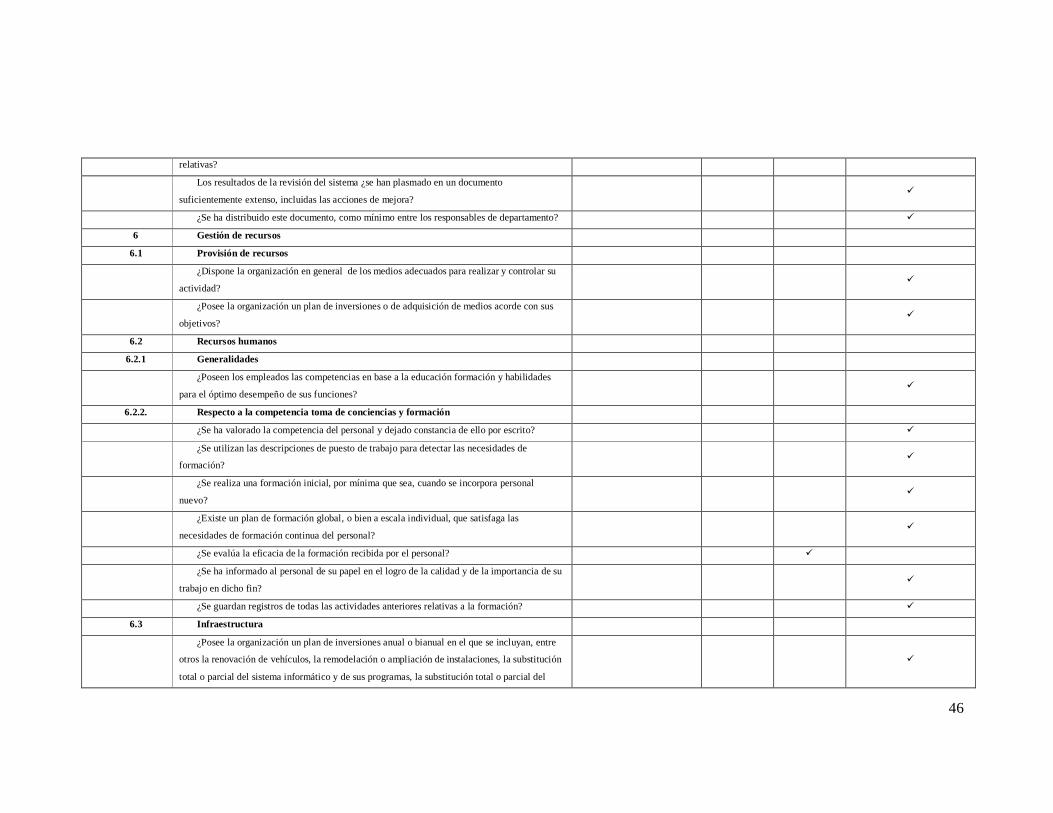
46
relativas?
Los resultados de la revisión del sistema ¿se han plasmado en un documento
suficientemente extenso, incluidas las acciones de mejora? ü
¿Se ha distribuido este documento, como mínimo entre los responsables de departamento? ü
6 Gestión de recursos
6.1 Provisión de recursos
¿Dispone la organización en general de los medios adecuados para realizar y controlar su
actividad? ü
¿Posee la organización un plan de inversiones o de adquisición de medios acorde con sus
objetivos? ü
6.2 Recursos humanos
6.2.1 Generalidades
¿Poseen los empleados las competencias en base a la educación formación y habilidades
para el óptimo desempeño de sus funciones? ü
6.2.2. Respecto a la competencia toma de conciencias y formación
¿Se ha valorado la competencia del personal y dejado constancia de ello por escrito? ü
¿Se utilizan las descripciones de puesto de trabajo para detectar las necesidades de
formación? ü
¿Se realiza una formación inicial, por mínima que sea, cuando se incorpora personal
nuevo? ü
¿Existe un plan de formación global, o bien a escala individual, que satisfaga las
necesidades de formación continua del personal? ü
¿Se evalúa la eficacia de la formación recibida por el personal? ü
¿Se ha informado al personal de su papel en el logro de la calidad y de la importancia de su
trabajo en dicho fin? ü
¿Se guardan registros de todas las actividades anteriores relativas a la formación? ü
6.3 Infraestructura
¿Posee la organización un plan de inversiones anual o bianual en el que se incluyan, entre
otros la renovación de vehículos, la remodelación o ampliación de instalaciones, la substitución
total o parcial del sistema informático y de sus programas, la substitución total o parcial del
ü
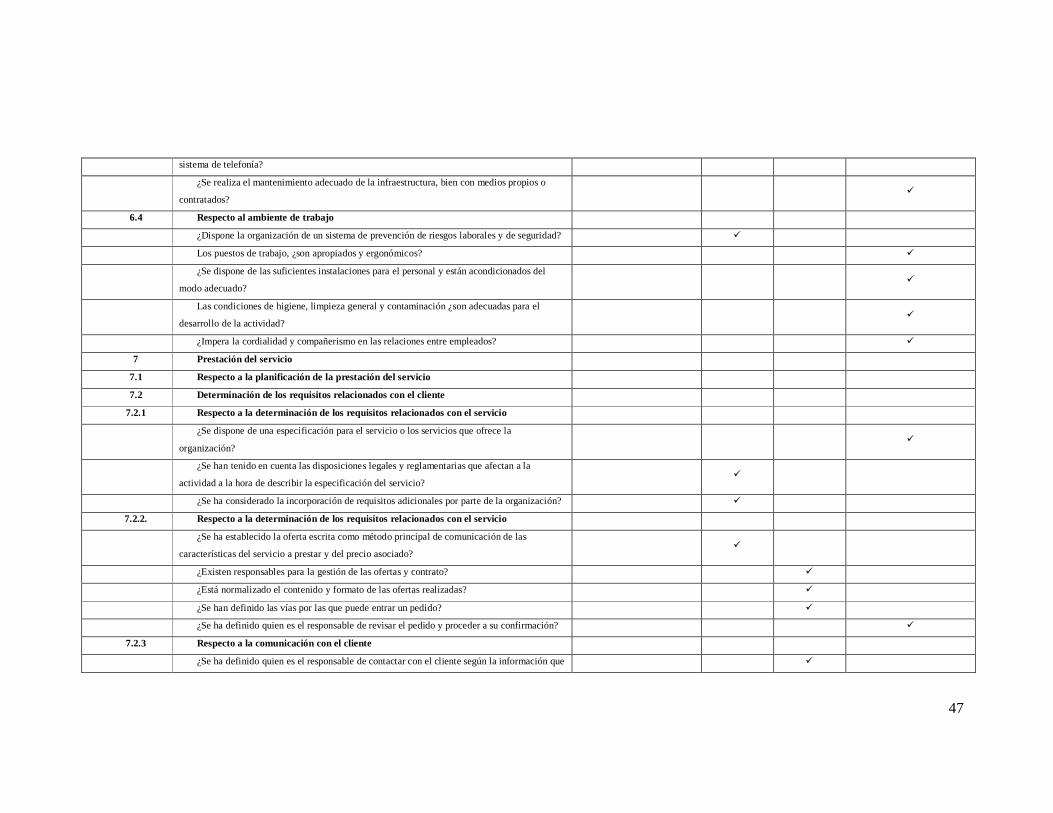
47
sistema de telefonía?
¿Se realiza el mantenimiento adecuado de la infraestructura, bien con medios propios o
contratados? ü
6.4 Respecto al ambiente de trabajo
¿Dispone la organización de un sistema de prevención de riesgos laborales y de seguridad? ü
Los puestos de trabajo, ¿son apropiados y ergonómicos? ü
¿Se dispone de las suficientes instalaciones para el personal y están acondicionados del
modo adecuado? ü
Las condiciones de higiene, limpieza general y contaminación ¿son adecuadas para el
desarrollo de la actividad? ü
¿Impera la cordialidad y compañerismo en las relaciones entre empleados? ü
7 Prestación del servicio
7.1 Respecto a la planificación de la prestación del servicio
7.2 Determinación de los requisitos relacionados con el cliente
7.2.1 Respecto a la determinación de los requisitos relacionados con el servicio
¿Se dispone de una especificación para el servicio o los servicios que ofrece la
organización? ü
¿Se han tenido en cuenta las disposiciones legales y reglamentarias que afectan a la
actividad a la hora de describir la especificación del servicio? ü
¿Se ha considerado la incorporación de requisitos adicionales por parte de la organización? ü
7.2.2. Respecto a la determinación de los requisitos relacionados con el servicio
¿Se ha establecido la oferta escrita como método principal de comunicación de las
características del servicio a prestar y del precio asociado? ü
¿Existen responsables para la gestión de las ofertas y contrato? ü
¿Está normalizado el contenido y formato de las ofertas realizadas? ü
¿Se han definido las vías por las que puede entrar un pedido? ü
¿Se ha definido quien es el responsable de revisar el pedido y proceder a su confirmación? ü
7.2.3 Respecto a la comunicación con el cliente
¿Se ha definido quien es el responsable de contactar con el cliente según la información que ü
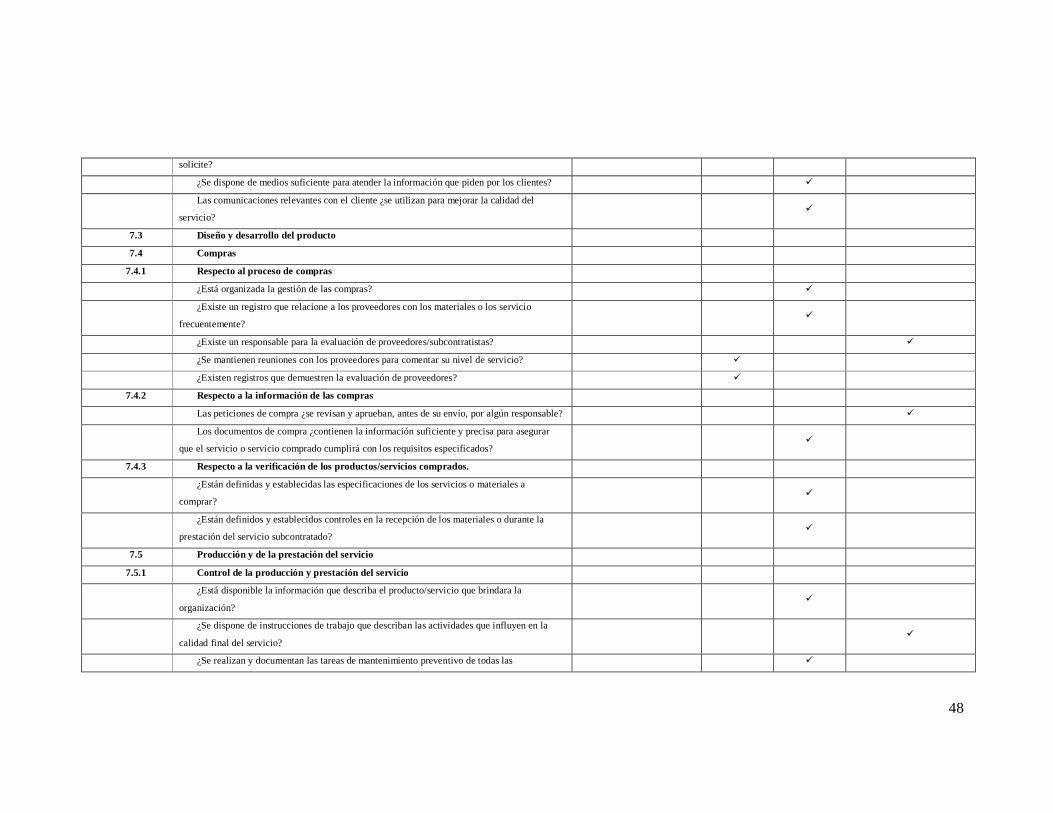
48
solicite?
¿Se dispone de medios suficiente para atender la información que piden por los clientes? ü
Las comunicaciones relevantes con el cliente ¿se utilizan para mejorar la calidad del
servicio? ü
7.3 Diseño y desarrollo del producto
7.4 Compras
7.4.1 Respecto al proceso de compras
¿Está organizada la gestión de las compras? ü
¿Existe un registro que relacione a los proveedores con los materiales o los servicio
frecuentemente? ü
¿Existe un responsable para la evaluación de proveedores/subcontratistas? ü
¿Se mantienen reuniones con los proveedores para comentar su nivel de servicio? ü
¿Existen registros que demuestren la evaluación de proveedores? ü
7.4.2 Respecto a la información de las compras
Las peticiones de compra ¿se revisan y aprueban, antes de su envío, por algún responsable? ü
Los documentos de compra ¿contienen la información suficiente y precisa para asegurar
que el servicio o servicio comprado cumplirá con los requisitos especificados? ü
7.4.3 Respecto a la verificación de los productos/servicios comprados.
¿Están definidas y establecidas las especificaciones de los servicios o materiales a
comprar? ü
¿Están definidos y establecidos controles en la recepción de los materiales o durante la
prestación del servicio subcontratado? ü
7.5 Producción y de la prestación del servicio
7.5.1 Control de la producción y prestación del servicio
¿Está disponible la información que describa el producto/servicio que brindara la
organización? ü
¿Se dispone de instrucciones de trabajo que describan las actividades que influyen en la
calidad final del servicio? ü
¿Se realizan y documentan las tareas de mantenimiento preventivo de todas las ü
49
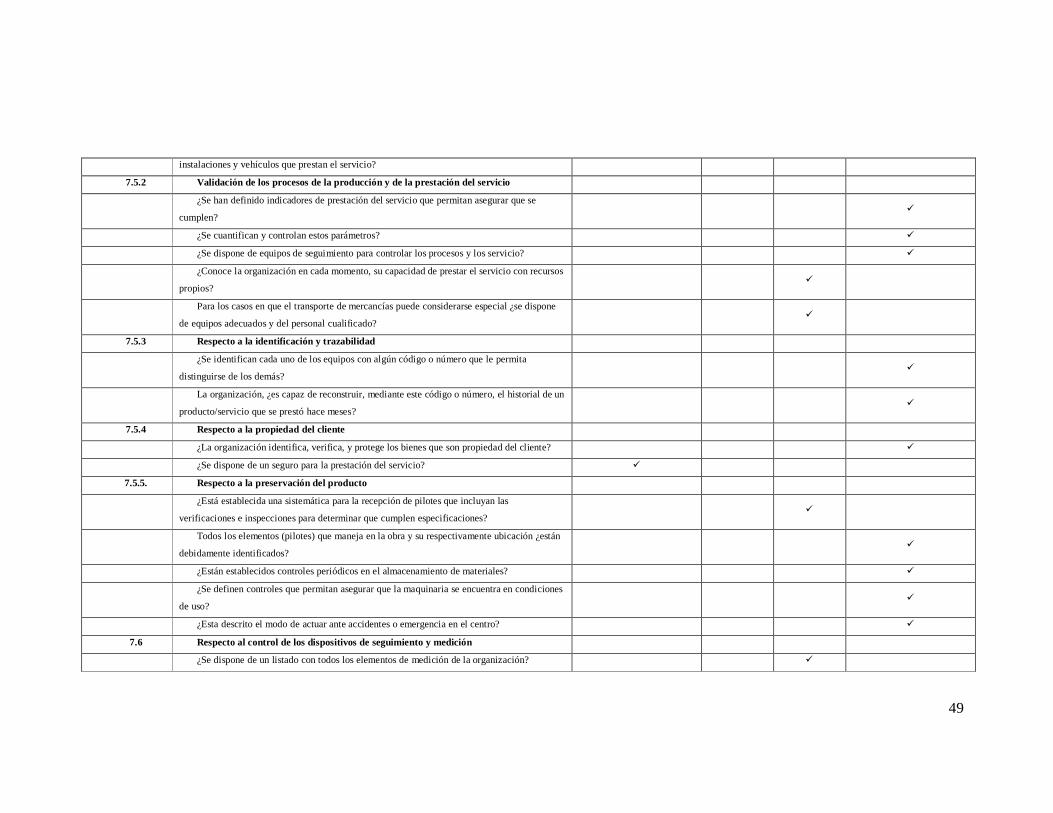
instalaciones y vehículos que prestan el servicio?
7.5.2 Validación de los procesos de la producción y de la prestación del servicio
¿Se han definido indicadores de prestación del servicio que permitan asegurar que se
cumplen? ü
¿Se cuantifican y controlan estos parámetros? ü
¿Se dispone de equipos de seguimiento para controlar los procesos y los servicio? ü
¿Conoce la organización en cada momento, su capacidad de prestar el servicio con recursos
propios? ü
Para los casos en que el transporte de mercancías puede considerarse especial ¿se dispone
de equipos adecuados y del personal cualificado? ü
7.5.3 Respecto a la identificación y trazabilidad
¿Se identifican cada uno de los equipos con algún código o número que le permita
distinguirse de los demás? ü
La organización, ¿es capaz de reconstruir, mediante este código o número, el historial de un
producto/servicio que se prestó hace meses? ü
7.5.4 Respecto a la propiedad del cliente
¿La organización identifica, verifica, y protege los bienes que son propiedad del cliente? ü
¿Se dispone de un seguro para la prestación del servicio? ü
7.5.5. Respecto a la preservación del producto
¿Está establecida una sistemática para la recepción de pilotes que incluyan las
verificaciones e inspecciones para determinar que cumplen especificaciones? ü
Todos los elementos (pilotes) que maneja en la obra y su respectivamente ubicación ¿están
debidamente identificados? ü
¿Están establecidos controles periódicos en el almacenamiento de materiales? ü
¿Se definen controles que permitan asegurar que la maquinaria se encuentra en condiciones
de uso? ü
¿Esta descrito el modo de actuar ante accidentes o emergencia en el centro? ü
7.6 Respecto al control de los dispositivos de seguimiento y medición
¿Se dispone de un listado con todos los elementos de medición de la organización? ü
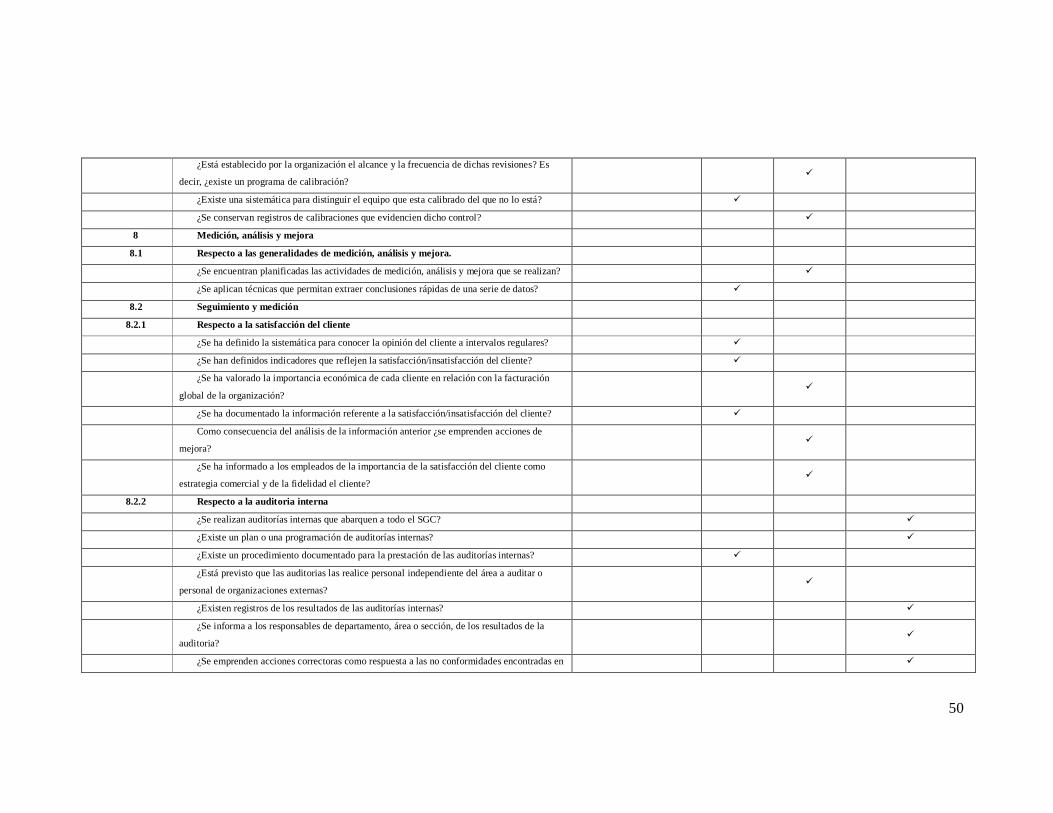
50
¿Está establecido por la organización el alcance y la frecuencia de dichas revisiones? Es
decir, ¿existe un programa de calibración? ü
¿Existe una sistemática para distinguir el equipo que esta calibrado del que no lo está? ü
¿Se conservan registros de calibraciones que evidencien dicho control? ü
8 Medición, análisis y mejora
8.1 Respecto a las generalidades de medición, análisis y mejora.
¿Se encuentran planificadas las actividades de medición, análisis y mejora que se realizan? ü
¿Se aplican técnicas que permitan extraer conclusiones rápidas de una serie de datos? ü
8.2 Seguimiento y medición
8.2.1 Respecto a la satisfacción del cliente
¿Se ha definido la sistemática para conocer la opinión del cliente a intervalos regulares? ü
¿Se han definidos indicadores que reflejen la satisfacción/insatisfacción del cliente? ü
¿Se ha valorado la importancia económica de cada cliente en relación con la facturación
global de la organización? ü
¿Se ha documentado la información referente a la satisfacción/insatisfacción del cliente? ü
Como consecuencia del análisis de la información anterior ¿se emprenden acciones de
mejora? ü
¿Se ha informado a los empleados de la importancia de la satisfacción del cliente como
estrategia comercial y de la fidelidad el cliente? ü
8.2.2 Respecto a la auditoria interna
¿Se realizan auditorías internas que abarquen a todo el SGC? ü
¿Existe un plan o una programación de auditorías internas? ü
¿Existe un procedimiento documentado para la prestación de las auditorías internas? ü
¿Está previsto que las auditorias las realice personal independiente del área a auditar o
personal de organizaciones externas? ü
¿Existen registros de los resultados de las auditorías internas? ü
¿Se informa a los responsables de departamento, área o sección, de los resultados de la
auditoria? ü
¿Se emprenden acciones correctoras como respuesta a las no conformidades encontradas en ü
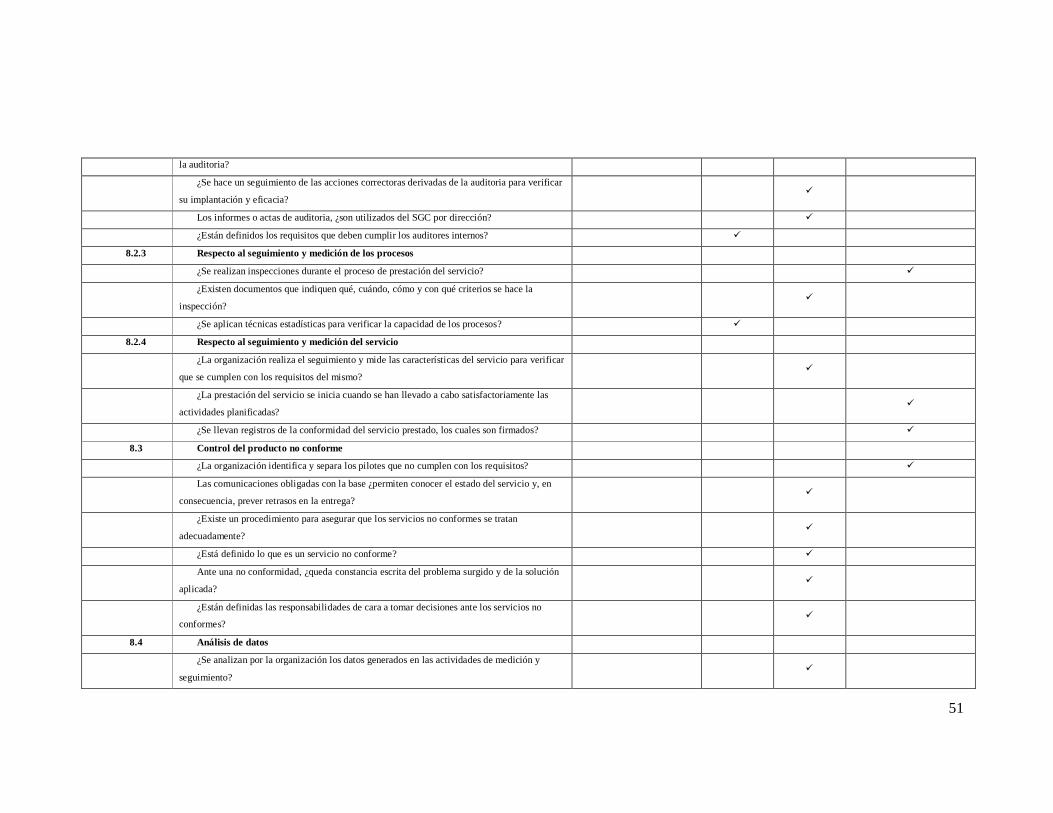
51
la auditoria?
¿Se hace un seguimiento de las acciones correctoras derivadas de la auditoria para verificar
su implantación y eficacia? ü
Los informes o actas de auditoria, ¿son utilizados del SGC por dirección? ü
¿Están definidos los requisitos que deben cumplir los auditores internos? ü
8.2.3 Respecto al seguimiento y medición de los procesos
¿Se realizan inspecciones durante el proceso de prestación del servicio? ü
¿Existen documentos que indiquen qué, cuándo, cómo y con qué criterios se hace la
inspección? ü
¿Se aplican técnicas estadísticas para verificar la capacidad de los procesos? ü
8.2.4 Respecto al seguimiento y medición del servicio
¿La organización realiza el seguimiento y mide las características del servicio para verificar
que se cumplen con los requisitos del mismo? ü
¿La prestación del servicio se inicia cuando se han llevado a cabo satisfactoriamente las
actividades planificadas? ü
¿Se llevan registros de la conformidad del servicio prestado, los cuales son firmados? ü
8.3 Control del producto no conforme
¿La organización identifica y separa los pilotes que no cumplen con los requisitos? ü
Las comunicaciones obligadas con la base ¿permiten conocer el estado del servicio y, en
consecuencia, prever retrasos en la entrega? ü
¿Existe un procedimiento para asegurar que los servicios no conformes se tratan
adecuadamente? ü
¿Está definido lo que es un servicio no conforme? ü
Ante una no conformidad, ¿queda constancia escrita del problema surgido y de la solución
aplicada? ü
¿Están definidas las responsabilidades de cara a tomar decisiones ante los servicios no
conformes? ü
8.4 Análisis de datos
¿Se analizan por la organización los datos generados en las actividades de medición y
seguimiento? ü
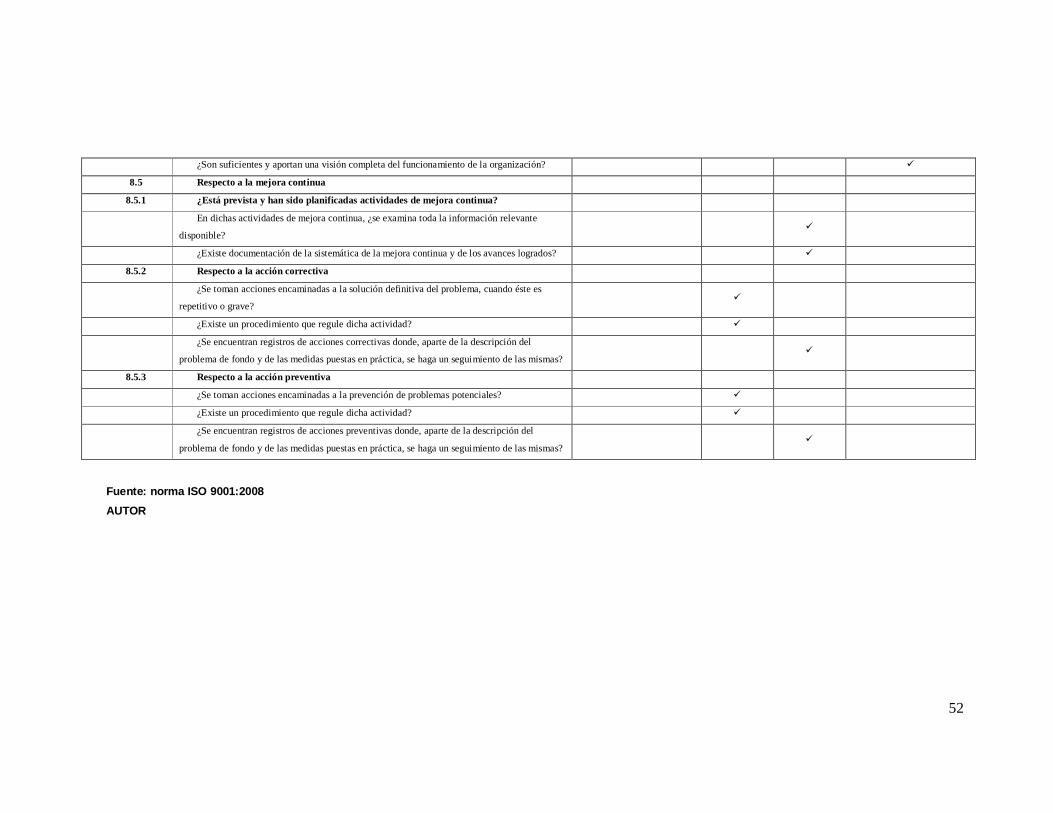
52
¿Son suficientes y aportan una visión completa del funcionamiento de la organización? ü
8.5 Respecto a la mejora continua
8.5.1 ¿Está prevista y han sido planificadas actividades de mejora continua?
En dichas actividades de mejora continua, ¿se examina toda la información relevante
disponible? ü
¿Existe documentación de la sistemática de la mejora continua y de los avances logrados? ü
8.5.2 Respecto a la acción correctiva
¿Se toman acciones encaminadas a la solución definitiva del problema, cuando éste es
repetitivo o grave? ü
¿Existe un procedimiento que regule dicha actividad? ü
¿Se encuentran registros de acciones correctivas donde, aparte de la descripción del
problema de fondo y de las medidas puestas en práctica, se haga un seguimiento de las mismas? ü
8.5.3 Respecto a la acción preventiva
¿Se toman acciones encaminadas a la prevención de problemas potenciales? ü
¿Existe un procedimiento que regule dicha actividad? ü
¿Se encuentran registros de acciones preventivas donde, aparte de la descripción del
problema de fondo y de las medidas puestas en práctica, se haga un seguimiento de las mismas? ü
Fuente: norma ISO 9001:2008 AUTOR

53
PORCENTAJE DE CUMPLIMIENTO ISO 9001:2008
4 Sistema De Gestión De La Calidad 99.29%
4.1 Requisitos generales 96.43%
4.2 Requisitos de la documentación del SGC 100%
4.2.1 Generalidades 100%
4.2.2 Manual de la calidad 100%
4.2.3 Control de documentos 100%
4.2.4 Control de los registros 100%
5 Responsabilidad de la dirección 88.94
5.1 Compromiso de la dirección 90%
5.2 Enfoque al cliente 58.33%
5.3 Política de la calidad 100%
5.4 Planificación 100%
5.4.1 Objetivos de la calidad 100%
5.4.2 Planificación del SGC 100%
5.5 Responsabilidad autoridad y comunicación 100%
5.5.1 Responsabilidad y autoridad 100%
5.5.2 Representante de la dirección 100%
5.5.3 Comunicación interna 100%
5.6 Revisión por la dirección 85.31%
5.6.1 Generalidades 100%
5.6.2 Respecto a la información para la revisión 64.28
5.6.3 Respecto a los resultados de la revisión 91.66%
6 Gestión de recursos 98.28%
6.1 Provisión de recursos 100%
6.2 Recursos humanos 98.22%
6.2.1 Generalidades 100%
6.2.2 Respecto a la competencia toma de conciencia y formación 96.43%
6.3 Infraestructura 100%
6.4 Respecto al ambiente de trabajo 95%
7 Prestación del servicio 47.56%
7.1 Respecto a la planificación de la prestación del servicio 0%

54
7.2 Determinación de los requisitos relacionados con el cliente 65%
7.2.1 Respecto a la determinación de los requisitos relacionados con el
servicio 50%
7.2.2 Respecto a la determinación de los requisitos con el servicio 70%
7.2.3 Respecto a la comunicación con el cliente 75%
7.3 Diseño y desarrollo del producto 0%
7.4 Compras 74.17
7.4.1 Respecto al proceso de compras 60%
7.4.2 Respecto a la información de las compras 87.5%
7.4.3 Respecto a la verificación de los productos/servicios comprados 75%
7.5 Producción y de la prestación del servicio 83.66
7.5.1 Control de la producción y prestación del servicio 83.33%
7.5.2 Validación de los procesos de la producción y de la prestación del
servicio 90%
7.5.3 Respeto a la identificación y trazabilidad 100%
7.5.4 Respecto a la propiedad del cliente 50%
7.5.5 Respecto a la preservación del producto 95%
7.6 Respecto al control de los dispositivos de seguimiento y medición 62.5%
8 Medición , análisis y mejora 68.18%
8.1 Respecto a las generalidades de medición, análisis y mejora 50%
8.2 Seguimiento y medición 71.46%
8.2.1 Respecto a la satisfacción del cliente 50%
8.2.2 Respecto a la auditoria interna 77.5%
8.2.3 Respecto al seguimiento y medición de los procesos 66.67%
8.2.4 Respecto al seguimiento y medición del servicio 91.67%
8.3 Control de productos no conforme 79.17%
8.4 Análisis de datos 87.5%
8.5 Mejora 52.77%
8.5.1 Respecto a la mejora continua 75%
8.5.2 Respecto a la mejora correctiva 41.66%
8.5.3 Respecto a la mejora preventiva 41.66%
Cumplimiento de la ISO 9001 80.45%
Fuente: Autor
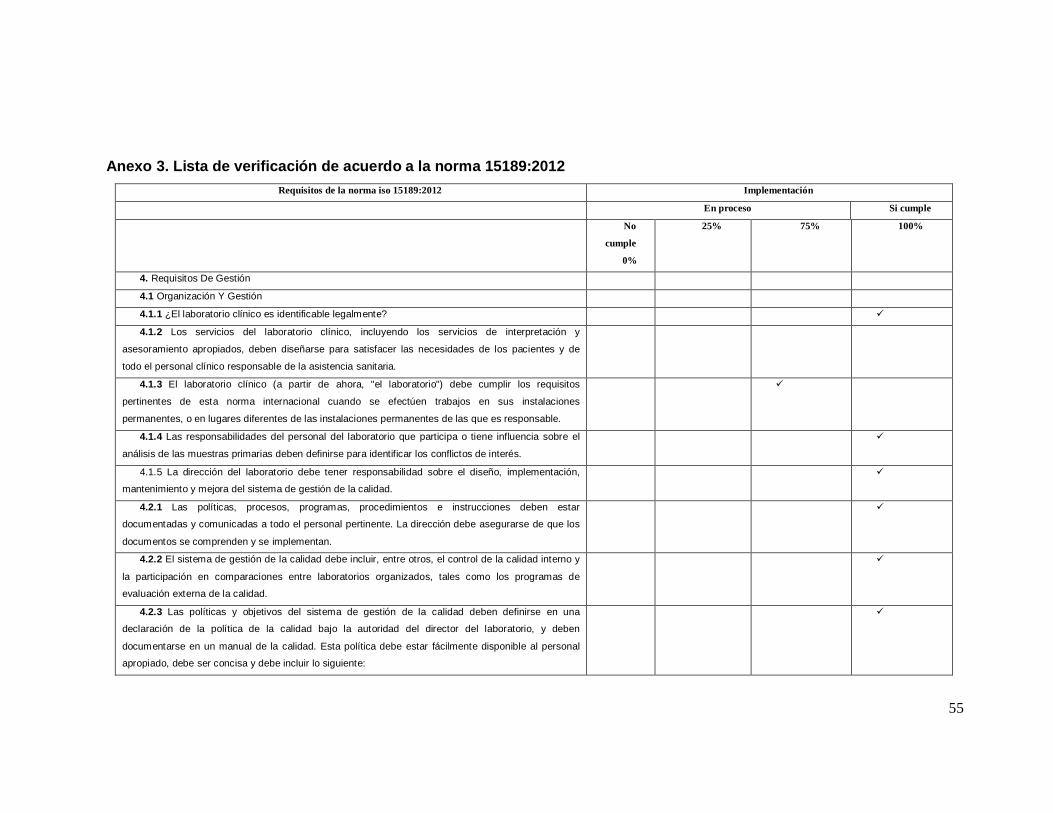
55
Anexo 3. Lista de verificación de acuerdo a la norma 15189:2012 Requisitos de la norma iso 15189:2012 Implementación
En proceso Si cumple
No
cumple
0%
25% 75% 100%
4. Requisitos De Gestión
4.1 Organización Y Gestión
4.1.1 ¿El laboratorio clínico es identificable legalmente? ü
4.1.2 Los servicios del laboratorio clínico, incluyendo los servicios de interpretación y
asesoramiento apropiados, deben diseñarse para satisfacer las necesidades de los pacientes y de
todo el personal clínico responsable de la asistencia sanitaria.
4.1.3 El laboratorio clínico (a partir de ahora, "el laboratorio") debe cumplir los requisitos
pertinentes de esta norma internacional cuando se efectúen trabajos en sus instalaciones
permanentes, o en lugares diferentes de las instalaciones permanentes de las que es responsable.
ü
4.1.4 Las responsabilidades del personal del laboratorio que participa o tiene influencia sobre el
análisis de las muestras primarias deben definirse para identificar los conflictos de interés.
ü
4.1.5 La dirección del laboratorio debe tener responsabilidad sobre el diseño, implementación,
mantenimiento y mejora del sistema de gestión de la calidad.
ü
4.2.1 Las políticas, procesos, programas, procedimientos e instrucciones deben estar
documentadas y comunicadas a todo el personal pertinente. La dirección debe asegurarse de que los
documentos se comprenden y se implementan.
ü
4.2.2 El sistema de gestión de la calidad debe incluir, entre otros, el control de la calidad interno y
la participación en comparaciones entre laboratorios organizados, tales como los programas de
evaluación externa de la calidad.
ü
4.2.3 Las políticas y objetivos del sistema de gestión de la calidad deben definirse en una
declaración de la política de la calidad bajo la autoridad del director del laboratorio, y deben
documentarse en un manual de la calidad. Esta política debe estar fácilmente disponible al personal
apropiado, debe ser concisa y debe incluir lo siguiente:
ü

56
4.2.4 El manual de la calidad debe describir el sistema de gestión de la calidad y la estructura de la
documentación utilizada en el sistema de gestión de la calidad.
ü
La lista del contenido de un manual de la calidad de un laboratorio clínico puede ser la siguiente:
a) Introducción.
ü
b) Descripción del laboratorio clínico, su identidad legal, recursos y actividades principales ü
c) Política de la calidad ü
d) Educación y formación del personal ü
e) Aseguramiento de la calidad. ü
f) Control de la documentación ü
g) Registros, mantenimiento y archivo ü
h) Instalaciones y entorno de trabajo ü
i) Gestión de los instrumentos, reactivos u otros materiales fungibles pertinentes ü
j) Validación de los procedimientos analíticos. ü
k) Seguridad ü
l) Aspectos medioambientales ü
m) Investigación y desarrollo. (Si procede). ü
n) Lista de los procedimientos analíticos ü
o) Protocolos de petición, muestra primaria, toma de muestras y manipulación de las muestras de
laboratorio
ü
p) Validación de los resultados ü
q) Control de la calidad ü
r) Sistema de información del laboratorio
s) Informe de los resultados ü
t) Acciones correctivas y gestión de las reclamaciones ü
u) Comunicaciones y otras interacciones con los pacientes, profesionales de la salud, laboratorios
subcontratistas y proveedores.
ü
v) Auditorías internas ü
w) Aspectos éticos ü
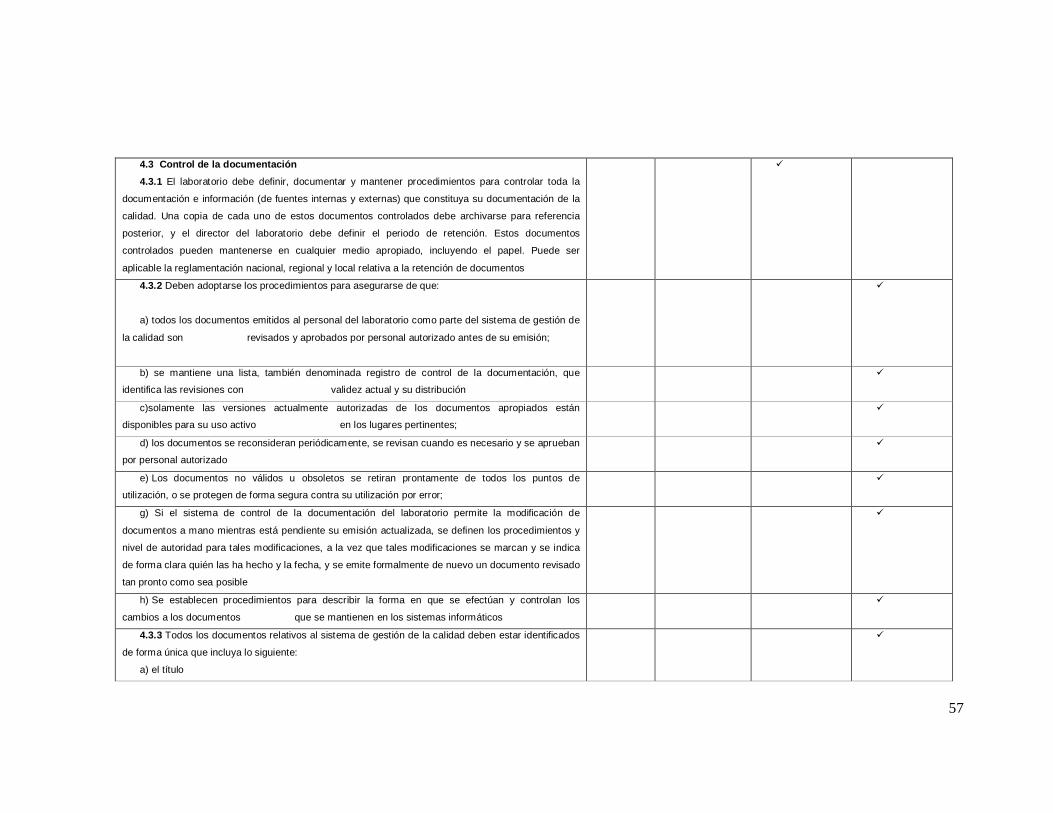
57
4.3 Control de la documentación 4.3.1 El laboratorio debe definir, documentar y mantener procedimientos para controlar toda la
documentación e información (de fuentes internas y externas) que constituya su documentación de la
calidad. Una copia de cada uno de estos documentos controlados debe archivarse para referencia
posterior, y el director del laboratorio debe definir el periodo de retención. Estos documentos
controlados pueden mantenerse en cualquier medio apropiado, incluyendo el papel. Puede ser
aplicable la reglamentación nacional, regional y local relativa a la retención de documentos
ü
4.3.2 Deben adoptarse los procedimientos para asegurarse de que:
a) todos los documentos emitidos al personal del laboratorio como parte del sistema de gestión de
la calidad son revisados y aprobados por personal autorizado antes de su emisión;
ü
b) se mantiene una lista, también denominada registro de control de la documentación, que
identifica las revisiones con validez actual y su distribución
ü
c)solamente las versiones actualmente autorizadas de los documentos apropiados están
disponibles para su uso activo en los lugares pertinentes;
ü
d) los documentos se reconsideran periódicamente, se revisan cuando es necesario y se aprueban
por personal autorizado
ü
e) Los documentos no válidos u obsoletos se retiran prontamente de todos los puntos de
utilización, o se protegen de forma segura contra su utilización por error;
ü
g) Si el sistema de control de la documentación del laboratorio permite la modificación de
documentos a mano mientras está pendiente su emisión actualizada, se definen los procedimientos y
nivel de autoridad para tales modificaciones, a la vez que tales modificaciones se marcan y se indica
de forma clara quién las ha hecho y la fecha, y se emite formalmente de nuevo un documento revisado
tan pronto como sea posible
ü
h) Se establecen procedimientos para describir la forma en que se efectúan y controlan los
cambios a los documentos que se mantienen en los sistemas informáticos
ü
4.3.3 Todos los documentos relativos al sistema de gestión de la calidad deben estar identificados
de forma única que incluya lo siguiente:
a) el título
ü
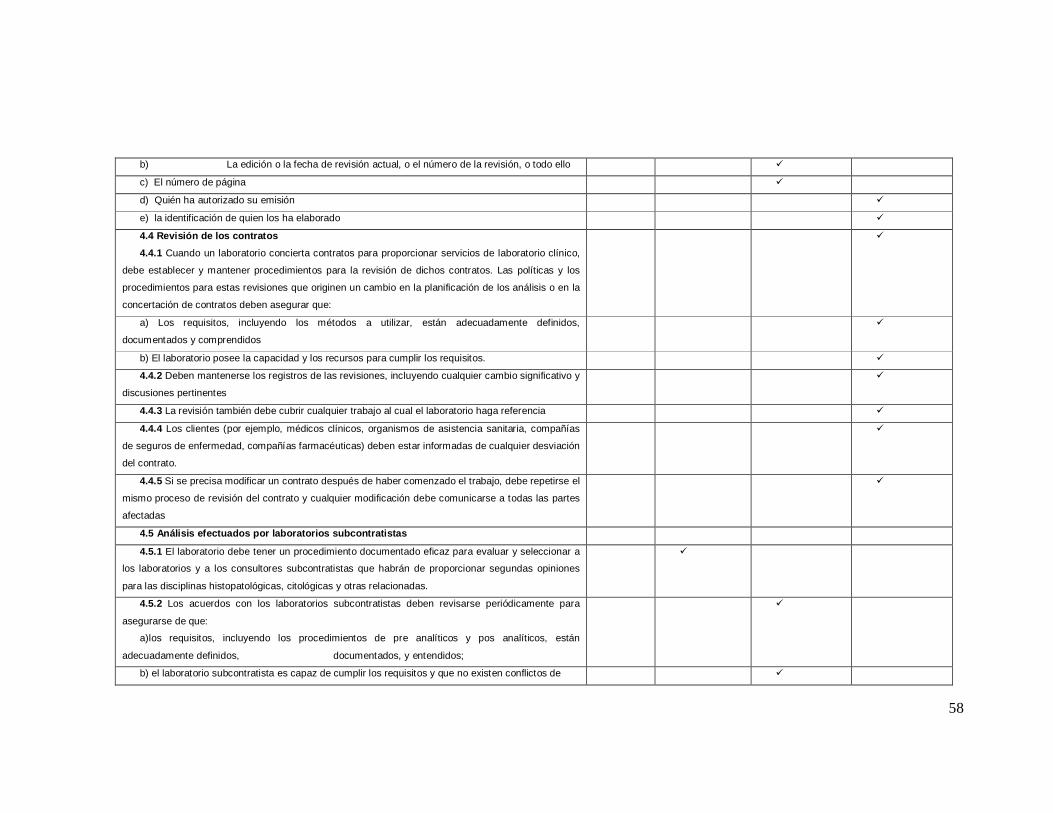
58
b) La edición o la fecha de revisión actual, o el número de la revisión, o todo ello ü
c) El número de página ü
d) Quién ha autorizado su emisión ü
e) la identificación de quien los ha elaborado ü
4.4 Revisión de los contratos 4.4.1 Cuando un laboratorio concierta contratos para proporcionar servicios de laboratorio clínico,
debe establecer y mantener procedimientos para la revisión de dichos contratos. Las políticas y los
procedimientos para estas revisiones que originen un cambio en la planificación de los análisis o en la
concertación de contratos deben asegurar que:
ü
a) Los requisitos, incluyendo los métodos a utilizar, están adecuadamente definidos,
documentados y comprendidos
ü
b) El laboratorio posee la capacidad y los recursos para cumplir los requisitos. ü
4.4.2 Deben mantenerse los registros de las revisiones, incluyendo cualquier cambio significativo y
discusiones pertinentes
ü
4.4.3 La revisión también debe cubrir cualquier trabajo al cual el laboratorio haga referencia ü
4.4.4 Los clientes (por ejemplo, médicos clínicos, organismos de asistencia sanitaria, compañías
de seguros de enfermedad, compañías farmacéuticas) deben estar informadas de cualquier desviación
del contrato.
ü
4.4.5 Si se precisa modificar un contrato después de haber comenzado el trabajo, debe repetirse el
mismo proceso de revisión del contrato y cualquier modificación debe comunicarse a todas las partes
afectadas
ü
4.5 Análisis efectuados por laboratorios subcontratistas
4.5.1 El laboratorio debe tener un procedimiento documentado eficaz para evaluar y seleccionar a
los laboratorios y a los consultores subcontratistas que habrán de proporcionar segundas opiniones
para las disciplinas histopatológicas, citológicas y otras relacionadas.
ü
4.5.2 Los acuerdos con los laboratorios subcontratistas deben revisarse periódicamente para
asegurarse de que:
a)los requisitos, incluyendo los procedimientos de pre analíticos y pos analíticos, están
adecuadamente definidos, documentados, y entendidos;
ü
b) el laboratorio subcontratista es capaz de cumplir los requisitos y que no existen conflictos de ü

59
interés
c) la selección de los procedimientos de análisis es apropiada para su utilización prevista; ü
d) las responsabilidades respectivas para la interpretación de los resultados del análisis están
claramente definidas.
ü
4.5.3 El laboratorio debe mantener un registro de todos los laboratorios subcontratistas que utiliza.
Debe mantenerse un registro de todas las muestras que han sido enviadas a otro laboratorio. El
nombre y dirección del laboratorio responsable del resultado del análisis debe proporcionarse al
usuario de los servicios del laboratorio. Debe retenerse un duplicado del informe de laboratorio en el
historial del paciente y en el archivo permanente del laboratorio
ü
4.5.4 El laboratorio solicitante, y no el laboratorio subcontratista, debe ser responsable de asegurar
que los resultados y hallazgos del análisis del laboratorio subcontratista se suministran a la persona
que solicita el análisis. Si el laboratorio solicitante prepara el informe de laboratorio, debe incluir todos
los elementos esenciales de los resultados comunicados por el laboratorio subcontratista, sin
alteraciones que puedan afectar la interpretación clínica.
ü
4.6 Servicios externos y suministros
4.6.1 La dirección del laboratorio debe definir y documentar sus políticas y procedimientos para la
selección y utilización de los servicios, equipos y materiales fungibles adquiridos externamente que
afecten a la calidad de su servicio.
ü
4.6.2 El equipo y los materiales fungibles comprados que afecten a la calidad del servicio no deben
utilizarse hasta que se haya verificado que cumplen las especificaciones o requisitos normalizados
definidos por los procedimientos correspondientes. Esto puede conseguirse examinando muestras
para control de la calidad y verificando que los resultados son aceptables. La documentación de la
conformidad del proveedor con su sistema de gestión de la calidad también puede utilizarse para la
verificación
ü
4.6.3 Debe existir un sistema de control de inventario para los suministros. Deben establecerse y
mantenerse registros de la calidad apropiados de los servicios externos, suministros y productos
comprados, durante un periodo de tiempo, según se haya definido en el sistema de gestión de la
calidad. Este sistema debería incluir el registro de los números de lote de todos los reactivos,
materiales de control y calibradores relevantes, la fecha de recepción en el laboratorio y la fecha en
ü
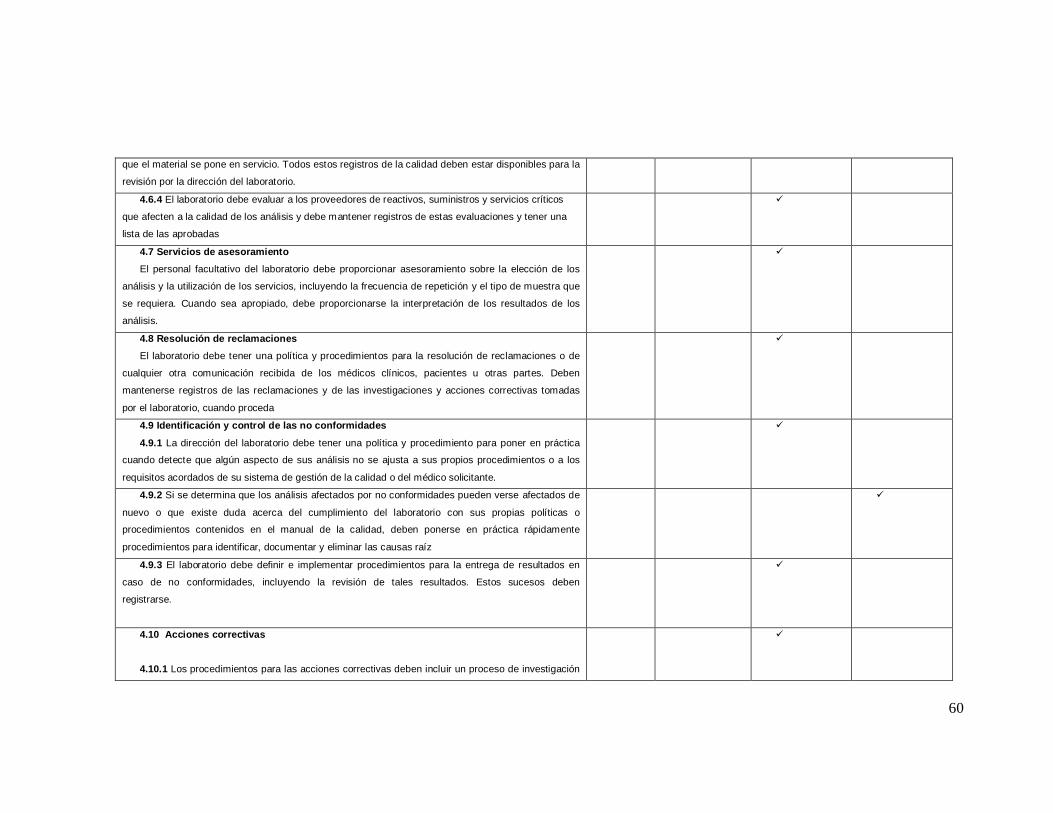
60
que el material se pone en servicio. Todos estos registros de la calidad deben estar disponibles para la
revisión por la dirección del laboratorio.
4.6.4 El laboratorio debe evaluar a los proveedores de reactivos, suministros y servicios críticos
que afecten a la calidad de los análisis y debe mantener registros de estas evaluaciones y tener una
lista de las aprobadas
ü
4.7 Servicios de asesoramiento El personal facultativo del laboratorio debe proporcionar asesoramiento sobre la elección de los
análisis y la utilización de los servicios, incluyendo la frecuencia de repetición y el tipo de muestra que
se requiera. Cuando sea apropiado, debe proporcionarse la interpretación de los resultados de los
análisis.
ü
4.8 Resolución de reclamaciones El laboratorio debe tener una política y procedimientos para la resolución de reclamaciones o de
cualquier otra comunicación recibida de los médicos clínicos, pacientes u otras partes. Deben
mantenerse registros de las reclamaciones y de las investigaciones y acciones correctivas tomadas
por el laboratorio, cuando proceda
ü
4.9 Identificación y control de las no conformidades
4.9.1 La dirección del laboratorio debe tener una política y procedimiento para poner en práctica
cuando detecte que algún aspecto de sus análisis no se ajusta a sus propios procedimientos o a los
requisitos acordados de su sistema de gestión de la calidad o del médico solicitante.
ü
4.9.2 Si se determina que los análisis afectados por no conformidades pueden verse afectados de
nuevo o que existe duda acerca del cumplimiento del laboratorio con sus propias políticas o
procedimientos contenidos en el manual de la calidad, deben ponerse en práctica rápidamente
procedimientos para identificar, documentar y eliminar las causas raíz
ü
4.9.3 El laboratorio debe definir e implementar procedimientos para la entrega de resultados en
caso de no conformidades, incluyendo la revisión de tales resultados. Estos sucesos deben
registrarse.
ü
4.10 Acciones correctivas 4.10.1 Los procedimientos para las acciones correctivas deben incluir un proceso de investigación
ü
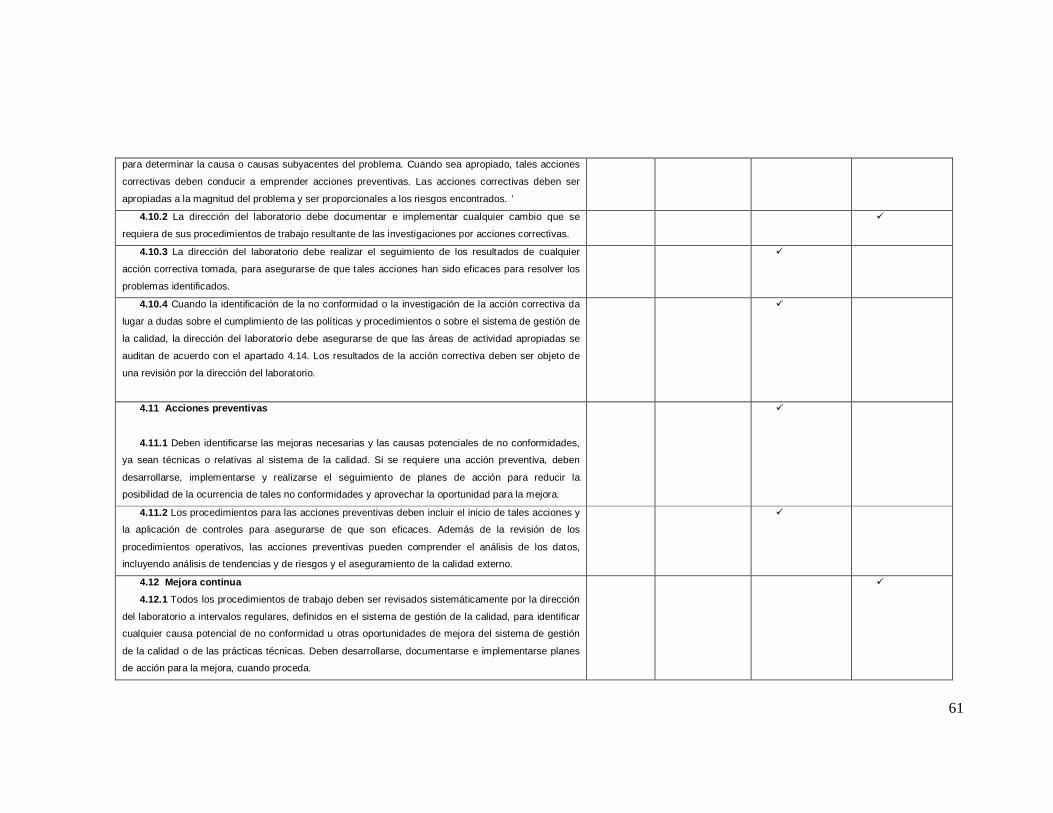
61
para determinar la causa o causas subyacentes del problema. Cuando sea apropiado, tales acciones
correctivas deben conducir a emprender acciones preventivas. Las acciones correctivas deben ser
apropiadas a la magnitud del problema y ser proporcionales a los riesgos encontrados. '
4.10.2 La dirección del laboratorio debe documentar e implementar cualquier cambio que se
requiera de sus procedimientos de trabajo resultante de las investigaciones por acciones correctivas.
ü
4.10.3 La dirección del laboratorio debe realizar el seguimiento de los resultados de cualquier
acción correctiva tomada, para asegurarse de que tales acciones han sido eficaces para resolver los
problemas identificados.
ü
4.10.4 Cuando la identificación de la no conformidad o la investigación de la acción correctiva da
lugar a dudas sobre el cumplimiento de las políticas y procedimientos o sobre el sistema de gestión de
la calidad, la dirección del laboratorio debe asegurarse de que las áreas de actividad apropiadas se
auditan de acuerdo con el apartado 4.14. Los resultados de la acción correctiva deben ser objeto de
una revisión por la dirección del laboratorio.
ü
4.11 Acciones preventivas
4.11.1 Deben identificarse las mejoras necesarias y las causas potenciales de no conformidades,
ya sean técnicas o relativas al sistema de la calidad. Si se requiere una acción preventiva, deben
desarrollarse, implementarse y realizarse el seguimiento de planes de acción para reducir la
posibilidad de la ocurrencia de tales no conformidades y aprovechar la oportunidad para la mejora.
ü
4.11.2 Los procedimientos para las acciones preventivas deben incluir el inicio de tales acciones y
la aplicación de controles para asegurarse de que son eficaces. Además de la revisión de los
procedimientos operativos, las acciones preventivas pueden comprender el análisis de los datos,
incluyendo análisis de tendencias y de riesgos y el aseguramiento de la calidad externo.
ü
4.12 Mejora continua 4.12.1 Todos los procedimientos de trabajo deben ser revisados sistemáticamente por la dirección
del laboratorio a intervalos regulares, definidos en el sistema de gestión de la calidad, para identificar
cualquier causa potencial de no conformidad u otras oportunidades de mejora del sistema de gestión
de la calidad o de las prácticas técnicas. Deben desarrollarse, documentarse e implementarse planes
de acción para la mejora, cuando proceda.
ü

62
4.12.2 Después de haber emprendido las acciones resultantes de la revisión, la dirección del
laboratorio debe evaluar la eficacia de la acción mediante una revisión o auditoría centrada en el área
correspondiente.
ü
4.12.3 Los resultados de la acción tras la revisión deben presentarse a la dirección del laboratorio
para revisión y puesta en práctica de cualquier cambio necesario del sistema de gestión de la calidad.
ü
4.12.4 La dirección del laboratorio debe implementar indicadores de la calidad para realizar el
seguimiento y evaluar sistemáticamente la contribución del laboratorio al cuidado del paciente. Cuando
este programa identifique oportunidades para la mejora, la dirección del laboratorio debe tenerlas en
cuenta sea cual fuere el área en la que aparezcan. La dirección del laboratorio debe asegurarse de
que el laboratorio clínico participa en las actividades de mejora de la calidad relacionadas con las
áreas y resultados pertinentes para el cuidado del paciente.
ü
4.12.5 La dirección del laboratorio debe proporcionar acceso a las oportunidades de educación y
formación adecuadas para todo el personal del laboratorio y para los usuarios pertinentes de los
servicios del mismo
ü
4.13 Registros de la calidad y registros técnicos 4.13.1 El laboratorio debe establecer e implementar procedimientos para la identificación, toma,
indexación, acceso, almacenamiento, mantenimiento y desecho seguro de los registros de la calidad y
de los registros técnicos.
ü
4.13.2 Todos los registros deben ser legibles y almacenarse de forma tal que sean fácilmente
recuperables. Los registros pueden almacenarse en cualquier medio apropiado, sujetos a los
requisitos nacionales, regionales o locales (véase la nota del apartado 4.3.1). Las instalaciones deben
proporcionar un entorno adecuado para impedir el daño, deterioro, pérdida o acceso no autorizado.
ü
4.13.3 El laboratorio debe tener una política que defina el periodo de retención de los diversos
registros relativos al sistema de gestión de la calidad y a los resultados de los análisis. El tiempo de
retención debe definirse según la naturaleza del análisis o específicamente para cada registro.
ü
4.14 Auditorías internas 4.14.1 Para verificar que las operaciones continúan cumpliendo los requisitos del sistema de
gestión de la calidad, deben realizarse auditorías internas de todos los elementos del sistema, tanto de
gestión como técnicos, a intervalos definidos por el propio sistema. La auditoría interna debe tratar
progresivamente estos elementos y poner énfasis en las áreas de importancia crítica para el cuidado
ü

63
del paciente.
4.14.2 Las auditorías deben planificarse, organizarse y llevarse a cabo formalmente por el
responsable de la calidad o el personal cualificado designado. El personal no debe auditar sus propias
actividades. Los procedimientos para las auditorías internas deben definirse y documentarse e incluir
los tipos de auditoría, las frecuencias de las mismas, la metodología y la documentación requerida.
Cuando se detecten deficiencias u oportunidades de mejora, el laboratorio debe emprender las
acciones correctivas o preventivas apropiadas, que deben documentarse y llevarse a cabo dentro de
plazos acordados.
Los principales elementos del sistema de gestión de la calidad deberían normalmente estar sujetos
a auditoría interna una vez cada doce meses.
ü
4.14.3 Los resultados de las auditorías internas deben presentarse a la dirección del laboratorio
para su revisión por la misma.
ü
4.15 Revisión por la dirección
4.15.1 La dirección del laboratorio debe revisar el sistema de gestión de la calidad del laboratorio y
todos sus servicios sanitarios, incluyendo las actividades de realización de análisis y asesoramiento,
para asegurar su continua adecuación y su eficacia en el apoyo al cuidado del paciente y para
introducir cualquier cambio o mejora necesarios. Los resultados de la revisión deben incorporarse a un
plan que incluya los objetivos y planes de acción. Una frecuencia típica para llevar a cabo una revisión
por la dirección es una vez cada doce meses.
ü
4.15.2 La revisión por la dirección debe tener en cuenta, entre otros elementos, los siguientes:
a) las revisiones por la dirección previas;
b) estado de las acciones correctivas tomadas y acciones preventivas requeridas;
c) informes del personal técnico y de gestión;
ü
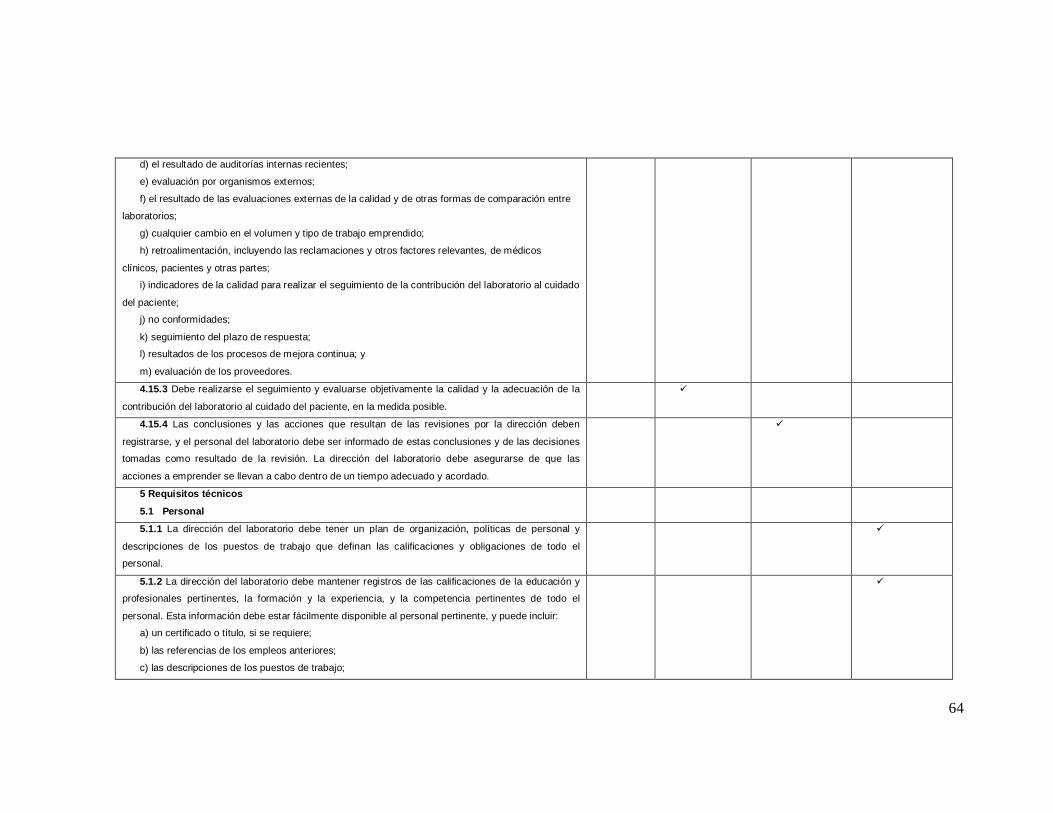
64
d) el resultado de auditorías internas recientes;
e) evaluación por organismos externos;
f) el resultado de las evaluaciones externas de la calidad y de otras formas de comparación entre
laboratorios;
g) cualquier cambio en el volumen y tipo de trabajo emprendido;
h) retroalimentación, incluyendo las reclamaciones y otros factores relevantes, de médicos
clínicos, pacientes y otras partes;
i) indicadores de la calidad para realizar el seguimiento de la contribución del laboratorio al cuidado
del paciente;
j) no conformidades;
k) seguimiento del plazo de respuesta;
l) resultados de los procesos de mejora continua; y
m) evaluación de los proveedores.
4.15.3 Debe realizarse el seguimiento y evaluarse objetivamente la calidad y la adecuación de la
contribución del laboratorio al cuidado del paciente, en la medida posible.
ü
4.15.4 Las conclusiones y las acciones que resultan de las revisiones por la dirección deben
registrarse, y el personal del laboratorio debe ser informado de estas conclusiones y de las decisiones
tomadas como resultado de la revisión. La dirección del laboratorio debe asegurarse de que las
acciones a emprender se llevan a cabo dentro de un tiempo adecuado y acordado.
ü
5 Requisitos técnicos
5.1 Personal
5.1.1 La dirección del laboratorio debe tener un plan de organización, políticas de personal y
descripciones de los puestos de trabajo que definan las calificaciones y obligaciones de todo el
personal.
ü
5.1.2 La dirección del laboratorio debe mantener registros de las calificaciones de la educación y
profesionales pertinentes, la formación y la experiencia, y la competencia pertinentes de todo el
personal. Esta información debe estar fácilmente disponible al personal pertinente, y puede incluir:
a) un certificado o título, si se requiere;
b) las referencias de los empleos anteriores;
c) las descripciones de los puestos de trabajo;
ü
65
d) los registros de la formación continua y el nivel conseguido;
e) las evaluaciones de la competencia; y
f) provisión en caso de incidencias adversas o de informes de accidente.
Otros registros disponibles a las personas autorizadas relacionados con la salud del personal
pueden incluir registros relativos a la de exposición a riesgos laborales y registros del estado de
inmunización
5.1.3 El laboratorio debe estar dirigido por una o varias personas que tengan la responsabilidad
ejecutiva y la competencia para asumir la responsabilidad por los servicios proporcionados,
ü
5.1.4 Las responsabilidades del director del laboratorio o del personal designado deben incluir
cuestiones de tipo profesional, científica, de consulta o asesoramiento de la organización,
administrativas y educativas. Estas responsabilidades deben ser pertinentes para los servicios
ofrecidos por el laboratorio.
ü
5.1.5 Deben existir recursos de personal adecuado para realizar el trabajo requerido y para llevar a
cabo otras funciones del sistema de gestión de la calidad.
ü
5.1.6 El personal debe tener formación específica en el aseguramiento de la calidad y la gestión de
la calidad de los servicios que se ofrecen.
ü
5.1.7 La dirección del laboratorio debe autorizar al personal a realizar tareas particulares tales
como la toma de muestras, el análisis y la utilización de tipos particulares de equipos, incluyendo la
utilización de equipos informáticos en el sistema de información del laboratorio
ü
5.1.8 Deben establecerse políticas que definan quién puede utilizar el sistema informático, quién
puede acceder a los datos de los pacientes y quién está autorizado para entrar y cambiar los
resultados de los pacientes, corregir la facturación o modificar los programas informáticos
ü
5.1.9 Debe existir un programa de formación continua disponible al personal a todos los niveles. ü
5.1.10 Los empleados deben estar formados para prevenir o limitar los efectos de los incidentes
adversos.
ü
5.1.11 La competencia de cada persona para realizar las tareas asignadas debe evaluarse
después de su formación y de forma periódica a partir de entonces. La nueva formación y la re-
evaluación deben realizarse cuando sea necesario.
ü
5.1.12 El personal que emite comentarios profesionales con referencia a los análisis debe tener ü

66
conocimientos previos, prácticas y teorías aplicables, así como experiencia reciente. Los comentarios
profesionales pueden expresarse como opiniones, interpretaciones, pronósticos, simulaciones y
modelos y valores, y deberían estar de acuerdo con la reglamentación nacional, regional y local.
El personal debe tomar parte en programas regulares de desarrollo profesional regular u otros
tipos de relaciones profesionales.
5.1.13 Todo el personal debe respetar la confidencialidad de la información sobre los pacientes. ü
5.2 Instalaciones y condiciones ambientales 5.2.1 El laboratorio debe disponer de un espacio designado de forma que su carga de trabajo
pueda realizarse sin comprometer la calidad del mismo, los procedimientos de control de la calidad, la
seguridad del personal o los servicios de asistencia sanitaria. El director del laboratorio debe
determinar la adecuación de este espacio. Los recursos deben ser suficientes para las actividades del
laboratorio. La funcionalidad y la fiabilidad de los recursos del laboratorio deben mantenerse. Deberían
efectuarse provisiones similares para la toma de muestras primarias y análisis realizados en lugares
diferentes a las instalaciones permanentes del laboratorio.
ü
5.2.2 El laboratorio debe diseñarse para operar de forma eficaz, optimizar la comodidad de sus
ocupantes y reducir al mínimo el riesgo de lesiones y enfermedades profesionales. Los pacientes,
empleados y visitantes deben estar protegidos frente a los peligros reconocidos.
ü
5.2.3 Cuando se proporcionen instalaciones para la toma de muestras primarias, deben tenerse en
cuenta las discapacidades, comodidad y privacidad de los pacientes, además de la optimización de las
condiciones de toma de muestras.
ü
5.2.4 El diseño y el entorno del laboratorio deben ser adecuados para las tareas realizadas. El
entorno en que se efectúa la toma de muestras primarias o en el que se realizan los análisis, o ambos,
no debe invalidar los resultados ni afectar adversamente a la calidad requerida de ninguna medición.
Las instalaciones del laboratorio previstas para realizar los análisis deberían permitir la correcta
realización de los mismos. Tales instalaciones incluyen, entre otras, fuentes de energía, iluminación,
ventilación, agua, residuos y desecho de residuos, y condiciones ambientales. El laboratorio debería
tener procedimientos para comprobar que el entorno no afecta adversamente a la toma de muestras ni
al equipo.
ü
5.2.5 El laboratorio debe realizar el seguimiento, controlar y registrar las condiciones ambientales,
según lo requieran las especificaciones pertinentes o cuando puedan influir en la calidad de los
ü

67
resultados. Debería prestarse atención a la esterilidad, polvo, interferencias electromagnéticas,
radiación, humedad, suministro eléctrico, temperatura y niveles de sonido y vibración, según sea
apropiado a las actividades técnicas correspondientes.
5.2.6 Debe existir una separación eficaz entre las secciones adyacentes del laboratorio en las que
se realizan actividades incompatibles. Deben tomarse medidas para impedir la contaminación cruzada.
ü
5.2.7 Debe controlarse el acceso y el uso de las áreas que afectan a la calidad de los análisis.
Deben tomarse las medidas apropiadas para proteger las muestras y los recursos del acceso no
autorizado.
ü
5.2.8 Los sistemas de comunicación dentro del laboratorio deben ser los apropiados al tamaño y
complejidad de la instalación y que permitan la transferencia eficaz de mensajes ü
5.2.9 Deben proporcionarse espacio y condiciones de almacenamiento pertinentes para asegurar
la continua integridad de muestras, portaobjetos, bloques para histología, microorganismos retenidos,
documentos, archivos, manuales, equipo, reactivos, suministros de laboratorio, registros y resultados.
ü
5.3 Equipo de laboratorio 5.3.1 El laboratorio debe estar dotado de todos los equipos requeridos para proveer los servicios
(incluyendo la toma de muestras primarias, la preparación y procesado de las muestras, el análisis y el
almacenamiento). En aquellos casos en los que el laboratorio necesita utilizar equipos que están fuera
de su control permanente, la dirección del laboratorio debe asegurarse de que se cumplen los
requisitos de esta norma internacional.
Cuando se selecciona equipo, debería tenerse en cuenta el consumo de energía y el futuro
desecho del mismo (respecto al medio ambiente).
ü
5.3.2 El equipo debe haber demostrado (durante su instalación y utilización ordinaria) que es
capaz de alcanzar las prestaciones precisas y debe cumplir las especificaciones pertinentes a los
análisis correspondientes.
ü
5.3.3 Cada unidad del equipo debe estar etiquetada, marcada o identificada de forma única. ü
5.3.4 Deben mantenerse registros para cada unidad del equipo que contribuye a la realización de
los análisis. Estos registros deben incluir al menos lo siguiente:
a) la identificación del equipo;
b) nombre del fabricante, identificación del tipo y número de serie u otra identificación única;
c) persona de contacto del fabricante y número de teléfono, cuando proceda;
ü
68
d) fecha de recepción y fecha de puesta en servicio;
e) lugar en que se encuentra actualmente, cuando sea apropiado;
f) condición cuando se recibe (por ejemplo: nuevo, utilizado o reacondicionado);
g) instrucciones del fabricante, si están disponibles, o referencia al lugar donde se hallan;
h) registros de desempeño del equipo que confirmen la adecuación del mismo para su
utilización;
i) mantenimiento realizado y mantenimiento planificado en fechas futuras;
j) cualquier daño sufrido, defectos de funcionamiento, modificaciones o reparaciones que ha
experimentado el equipo;
k) fecha prevista de sustitución, si es posible.
5.3.5 El equipo debe ser utilizado solamente por personal autorizado. Las instrucciones
actualizadas sobre la utilización y el mantenimiento del equipo (incluyendo cualquier manual de
instrucciones de uso pertinentes proporcionados por el fabricante del equipo) deben estar fácilmente
disponibles al personal del laboratorio.
ü
5.3.6 El equipo debe mantenerse en condiciones de trabajo seguras. Esto debe incluir el examen
de la seguridad eléctrica, los dispositivos de parada de emergencia y la manipulación y desecho
seguros de materiales químicos, radioactivos y biológicos por las personas autorizadas. Deben
utilizarse las especificaciones o las instrucciones del fabricante o ambas, según proceda
ü
5.3.7 Todo equipo defectuoso, debe retirarse del servicio, etiquetarse claramente y almacenarse
de forma apropiada hasta que haya sido reparado y su calibración, verificación o ensayo demuestre
que cumple los criterios de aceptación especificados. El laboratorio debe examinar el efecto de este
defecto en análisis previos e instituir el procedimiento dado en el apartado 4.9. El laboratorio debe
tomar medidas razonables para descontaminar el equipo antes de ponerlo en servicio, repararlo o
retirarlo del servicio.
ü
5.3.8 Debe proporcionarse una lista de las medidas tomadas para reducir la contaminación, a la
persona que trabaja con el equipo. El laboratorio debe proporcionar un espacio adecuado para las
reparaciones y un equipo de protección personal apropiado.
5.3.9 Siempre que sea posible, el equipo bajo el control del laboratorio que requiera calibración o
verificación, debe etiquetarse o codificarse de forma que se indique su estado de calibración o
verificación y la fecha en que habrá de recalibrarse o verificarse de nuevo.
ü
69
5.3.10 Cuando el equipo se retira del control directo del laboratorio o se repara o se pone en
servicio, el laboratorio debe asegurarse de que se comprueba y que demuestra funcionar
satisfactoriamente antes de volverse a utilizar en el laboratorio.
ü
5.3.11 Cuando se utilizan equipos informáticos o equipo de análisis automatizado para la toma,
procesado, registro, informe de laboratorio, almacenamiento o recuperación de los datos de análisis, el
laboratorio debe asegurarse de que:
a) el software informático, incluyendo el que está incorporado en el equipo, se documenta y
valida de forma adecuada para su utilización en la instalación;
b) se establecen e implementan procedimientos para proteger la integridad de los datos en todo
momento;
c) los equipos informáticos y el equipo automatizado se mantienen para asegurar su
funcionamiento apropiado y se proporcionan las condiciones ambientales y de funcionamiento
necesarias para mantener la integridad de los datos;
d) los programas y rutinas informáticos están adecuadamente protegidos para impedir el
acceso, alteración o destrucción por personal ocasional o personas no autorizadas.
ü
5.3.12 El laboratorio debe tener procedimientos para la manipulación, transporte, almacenamiento
y utilización segura del equipo, para impedir su contaminación o deterioro.
ü
5.3.13 Cuando las calibraciones dan lugar a un conjunto de factores de corrección, el laboratorio
debe tener procedimientos para asegurarse de que las copias de factores de corrección anteriores se
actualizan correctamente.
ü
5.3.14 El equipo, incluyendo el hardware, el software, los materiales de referencia, los reactivos y
otros materiales fungibles, y los sistemas analíticos deben protegerse contra desajustes o alteraciones
que puedan invalidar los resultados de los análisis.
ü
5.4 Procedimientos pre analíticos
5.4.1 La hoja de petición debe contener información suficiente para identificar al paciente y al
solicitante autorizado, así como proporcionar los datos clínicos pertinentes. Deben aplicarse los
requisitos nacionales, regionales o locales.
ü
5.4.2 Las instrucciones específicas para la toma y manipulación adecuadas de las muestras
primarias deben documentarse e implementarse por la dirección del laboratorio y deben estar
disponibles a aquéllos responsables de la toma de muestras primarias. Estas instrucciones deben
ü
70
estar contenidas en un manual para la toma de muestras primarias.
5.4.3 El manual para la toma de muestras primarias debe incluir lo siguiente:
a) copias o referencias a:
1) las listas de los análisis de laboratorio disponibles que se ofrecen;
2) los impresos de consentimiento, cuando proceda;
3) la información e instrucciones proporcionadas a los pacientes en relación a su preparación
antes de la toma de la muestra primaria; y
4) la información a los usuarios de los servicios del laboratorio sobre las indicaciones
médicas y la selección apropiada de los procedimientos disponibles;
b) procedimientos para:
1) la preparación del paciente (por ejemplo: instrucciones al personal sanitario y flebotomistas);
2) la identificación de la muestra primaria; y
3) la toma de la muestra primaria (por ejemplo: flebotomía, punción de la piel, sangre, orina y
otros fluidos corporales), con descripciones de los recipientes de la muestra primaria y cualquier
aditivo necesario;
c) instrucciones relativas a:
1) la forma de completar la hoja de petición o el equivalente electrónico,
2) el tipo y cantidad de la muestra primaria a tomar,
3) el momento preciso para realizar la toma, si se requiere,
4) cualquier requisito de manipulación especial entre el momento de la toma y el momento de
recepción en el laboratorio (requisitos de transporte, refrigeración, calentamiento, entrega inmediata,
etc.),
5) el etiquetado de las muestras primarias,
6) la información clínica (por ejemplo, historial de administración de medicamentos),
7) la identificación detallada del paciente del cual se ha tomado una muestra primaria,
8) el registro de la identidad de la persona que toma la muestra primaria, y
9) el desecho seguro de los materiales utilizados en la toma;
d) instrucciones para:
1) el almacenamiento de las muestras analizadas,
ü
71
2) los límites de tiempo para solicitar análisis adicionales,
3) los análisis adicionales, y
4) repetir el análisis debido a un fallo analítico o a análisis posteriores de la misma muestra
primaria.
5.4.4 El manual para la toma de muestras primarias debe ser parte del sistema de control de la
documentación
ü
5.4.5 Las muestras primarias deben poseer trazabilidad, normalmente mediante la hoja de
petición, hasta un individuo identificado. Las muestras primarias que no posean identificación
adecuada no deben ser aceptadas ni procesadas por el laboratorio.
ü
5.4.6 El laboratorio debe asegurarse de que las muestras se transportan al laboratorio:
a) dentro de un intervalo de tiempo apropiado a la naturaleza de los análisis solicitados y a la
disciplina del laboratorio correspondiente;
b) a una temperatura comprendida en un intervalo especificado en el manual para la toma de
muestras primarias y con los conservantes designados para asegurar la integridad de las
muestras; y
c) de manera que se asegure la seguridad del transportista, del público en general y del
laboratorio receptor, de acuerdo con los requisitos reglamentarios nacionales, regionales o
locales.
ü
5.4.7 Todas las muestras primarias recibidas deben registrarse en un libro de entradas, hoja de
cálculo, equipo informático u otro sistema comparable. Debe registrarse la fecha y la hora de recepción
de las muestras, así como la identidad del oficial receptor de las mismas.
ü
5.4.8 Deben desarrollarse y documentarse los criterios para la aceptación o rechazo de las
muestras primarias. Si se aceptan muestras primarias comprometidas, el informe de laboratorio final
debe indicar la naturaleza del problema y, si procede, las precauciones requeridas cuando se
interprete el resultado.
ü
5.4.9 El laboratorio debe revisar periódicamente sus requisitos sobre el volumen de muestras de
flebotomía (y otras muestras, tales como las de líquido cefalorraquídeo) para asegurarse de que no se
toman cantidades insuficientes ni excesivas.
5.4.10 El personal autorizado debe revisar sistemáticamente las hojas de petición y las muestras, y
decidir qué análisis deben realizarse y los métodos a utilizar para ello.
ü

72
5.4.11 Si procede, el laboratorio debe tener un procedimiento documentado para la recepción,
etiquetado, procesado e informe de aquellas muestras primarias recibidas por el laboratorio y
marcadas específicamente como urgentes. El procedimiento debe incluir los detalles de cualquier
etiquetado especial de la hoja de petición y de la muestra primaria, el mecanismo de transferencia de
la muestra primaria al área de análisis del laboratorio, cualquier modo de procesado rápido a utilizar y
cualquier criterio especial a seguir al realizar el informe.
ü
5.4.12 Las porciones de la muestra también deben tener trazabilidad hasta la muestra primaria
original.
ü
5.4.13 El laboratorio debe tener una política escrita sobre las peticiones verbales de análisis de
muestras.
ü
5.4.14 Las muestras deben almacenarse durante un tiempo especificado, en condiciones que
garanticen la estabilidad de las propiedades de la muestra, para permitir la repetición del análisis
después de emitir el informe del resultado o para efectuar análisis adicionales.
ü
5.5 Procedimientos analíticos 5.5.1 El laboratorio debe utilizar procedimientos analíticos, incluyendo aquéllos para
seleccionar/tomar porciones de la muestra, que cumplan las necesidades de los usuarios de los
servicios del laboratorio y sean apropiados para los análisis a efectuar. Los procedimientos preferidos
son aquéllos que han sido publicados en manuales reconocidos, en textos o publicaciones evaluados
por expertos, o en directrices internacionales, nacionales o regionales. Si se utilizan procedimientos
propios, deben estar validados de forma apropiada para su utilización prevista y totalmente
documentados.
ü
5.5.2 El laboratorio debe utilizar solamente procedimientos validados para confirmar que los
procedimientos analíticos son adecuados para su utilización prevista. Las validaciones deben ser todo
lo extensas que sea necesario para cumplir las necesidades de la aplicación o campo de aplicación
dados. El laboratorio debe registrar los resultados obtenidos y el procedimiento utilizado para la
validación.
ü
5.5.3 Todos los procedimientos deben documentarse y estar disponibles en el puesto de trabajo
para el personal pertinente. Los procedimientos documentados y las instrucciones necesarias deben
estar disponibles en un idioma comúnmente entendido por el personal del laboratorio.
ü
5.5.4 Las especificaciones técnicas para cada procedimiento analítico deben corresponder a la ü

73
utilización prevista de tal procedimiento.
5.5.5 Los intervalos de referencia biológicos deben revisarse periódicamente. Si el laboratorio tiene
razones para creer que un intervalo particular ya no es apropiado para la población de referencia,
entonces debe iniciarse una investigación, seguida, si es necesario, de la correspondiente acción
correctiva. También debe efectuarse una revisión de los intervalos de referencia biológicos cuando el
laboratorio cambia un procedimiento analítico o pre analítico, si procede.
ü
5.5.6 El laboratorio debe disponer de su catálogo de prestaciones, incluyendo los requisitos de las
muestras primarias y las especificaciones técnicas y requisitos pertinentes, disponible a los usuarios
de los servicios del laboratorio cuando así lo soliciten.
5.5.7 Si el laboratorio tiene la intención de cambiar un procedimiento analítico de forma que los
resultados o su interpretación puedan ser significativamente diferentes, las implicaciones deben
explicarse por escrito a los usuarios de los servicios del laboratorio antes de la introducción de tal
cambio.
ü
5.6 Aseguramiento de la calidad de los procedimientos analíticos
5.6.1 El laboratorio debe diseñar sistemas de control de la calidad internos que verifiquen que se
consigue la calidad prevista de los resultados. Es importante que el sistema de control proporcione al
personal del laboratorio información clara y fácilmente entendible sobre la que basar las decisiones
técnicas y clínicas. Debería prestarse atención especial a la eliminación de equivocaciones en el
proceso de manipulación de las muestras, peticiones, análisis, redacción de informes de laboratorio,
etc.
ü
5.6.2 El laboratorio debe determinar la incertidumbre de los resultados, siempre que sea pertinente
y posible. Deben tenerse en cuenta los componentes de la incertidumbre que sean importantes. Las
causas que contribuyen a la incertidumbre pueden incluir el muestreo, la preparación de la muestra, la
selección de la porción de la muestra, los calibradores, los materiales de referencia, las magnitudes de
entrada, el equipo utilizado, las condiciones ambientales, la condición de la muestra y los cambios de
operador.
ü
5.6.3 Debe diseñarse y ponerse en práctica un programa de calibración de los sistemas de medida
y verificación de la veracidad para asegurar la trazabilidad de los resultados a las unidades SI o por
referencia a una constante natural u otra referencia declarada. Cuando ninguna de estas referencias
es posible o pertinente, deben aplicarse otros medios para proporcionar confianza en los resultados,
ü

74
incluyendo entre otros los siguientes:
a) la participación en un programa adecuado de comparaciones entre laboratorios.
b) la utilización de materiales de referencia adecuados, certificados para indicar la
caracterización del material;
c) el análisis o la calibración por otro procedimiento;
d) las mediciones por cociente o basadas en relaciones de reciprocidad;
e) Normas o métodos de consentimiento mutuo que estén claramente establecidas,
especificadas, caracterizadas y mutuamente acordadas por todas las partes involucradas;
f) la documentación de las declaraciones sobre los reactivos, los procedimientos o el sistema
analítico cuando la trazabilidad la proporciona el proveedor o el fabricante.
5.6.4 El laboratorio debe participar en comparaciones entre laboratorios tales como las
organizadas en el marco de programas de evaluación externa de la calidad. La dirección del
laboratorio debe realizar el seguimiento de los resultados de la evaluación externa de la calidad y
participar en la implementación de acciones correctivas cuando los criterios de control no se cumplen.
Los programas de comparación entre laboratorios deben cumplir de forma sustancial la Guía ISO/IEC
43-1.
ü
5.6.5 Cuando no esté disponible un programa formal de comparación entre laboratorios, el
laboratorio debe desarrollar un mecanismo para determinar la aceptabilidad de los procedimientos que
no se hayan evaluado. Siempre que sea posible, este mecanismo debe utilizar materiales de control
de origen externo, tales como un intercambio de muestras con otros laboratorios. La dirección del
laboratorio debe realizar el seguimiento de los resultados de este mecanismo de comparación entre
laboratorios y participar en la implementación y registro de acciones correctivas.
ü
5.6.6 Para aquellos análisis realizados utilizando procedimientos o equipo diferentes, o en lugares
diferentes, o ambos, debe existir un mecanismo definido para verificar que los resultados son
comparables en los intervalos clínicamente apropiados. Tal verificación debe efectuarse en periodos
de tiempo definidos apropiados a las características del procedimiento o del instrumento.
ü
5.6.7 El laboratorio debe documentar, registrar y, cuando proceda, actuar rápidamente sobre los
resultados de estas comparaciones. Debe actuarse sobre los problemas o deficiencias identificados y
deben conservarse los registros de tales acciones.
ü
5.7 Procedimientos pos analíticos ü

75
5.7.1 El personal autorizado debe revisar sistemáticamente los resultados de los análisis,
evaluarlos de acuerdo con la información clínica disponible del paciente y autorizar la entrega de los
resultados.
5.7.2 El almacenamiento de la muestra primaria y de las otras muestras de laboratorio debe
hacerse de acuerdo con una política aprobada.
ü
5.7.3 El desecho seguro de las muestras que ya no se requieran para su análisis debe efectuarse
de acuerdo con la reglamentación o las recomendaciones locales sobre gestión de residuos.
ü
5.8 Informe de laboratorio 5.8.1 La dirección del laboratorio debe ser responsable del formato dado a los informes de
laboratorio. El formato del impreso del informe de laboratorio (es decir, en papel o electrónico) y la
forma en que habrá de comunicarse desde el laboratorio deberían determinarse por acuerdo con los
usuarios de los servicios del laboratorio.
ü
5.8.2 La dirección del laboratorio comparte la responsabilidad con el solicitante para asegurarse de
que los individuos apropiados reciben los informes de laboratorio dentro de un intervalo de tiempo
acordado.
ü
5.8.3 Los resultados deben ser legibles, sin errores de transcripción y comunicados a personas
autorizadas para recibirlos y utilizar la información clínica. El informe de laboratorio también debe
incluir, entre otra información, la siguiente:
a) la identificación clara y no ambigua del análisis, incluyendo, cuando sea apropiado, el
procedimiento de medida;
b) la identificación del laboratorio que emitió el informe de laboratorio;
c) la identificación única y la dirección del paciente, cuando sea posible, y destino del informe
de laboratorio;
d) el nombre u otro identificador único del solicitante y la dirección del mismo;
e) la fecha y la hora de toma de la muestra primaria, cuando tal información esté disponible y
sea pertinente para la asistencia al paciente, y la hora de recepción por el laboratorio;
f) la fecha y la hora de la entrega del informe de laboratorio, lo cual, si no aparece en el
informe de laboratorio, debe estar accesible fácilmente cuando se necesite;
g) el origen y el sistema (o el tipo de muestra primaria);
ü
76
h) los resultados del análisis expresados en unidades SI o unidades trazables a unidades SI
(véase la Guía ISO 31), cuando proceda;
i) los intervalos de referencia biológicos, si procede;
j) la interpretación de los resultados, si procede;
k) otros comentarios (por ejemplo: la calidad o idoneidad de la muestra primaria que puede
haber comprometido el resultado, los resultados o interpretaciones de los laboratorios subcontratistas,
la utilización de un procedimiento experimental); el informe de laboratorio debería identificar los
análisis realizados como parte de un programa de desarrollo y para los cuales no hay indicaciones
específicas sobre los resultados de la medición y, cuando proceda, debería proporcionarse la
información sobre el límite de detección y la incertidumbre de medida cuando así se solicite;
l) la identificación de la persona que autoriza la entrega del informe de laboratorio;
m) los resultados originales y los corregidos, si procede;
n) la firma o la autorización de la persona que verifica o entrega el informe de laboratorio,
cuando sea posible.
5.8.4 Cuando proceda, la descripción de los análisis realizados y sus resultados deberían seguir la
nomenclatura y la sintaxis recomendadas por una o más de las organizaciones siguientes:
- Consejo Internacional de Normalización en Hematología (ICSH);
- Sociedad Internacional de Hematología (ISH);
- Federación Internacional de Química Clínica (IFCC);
- Unión Internacional de Química Pura y Aplicada (IUP AC);
- Sociedad Internacional de Trombosis y Hemostasia (ISTH);
- Comité Europeo de Normalización (CEN).
Cuando proceda, la descripción y los resultados deberían seguir la nomenclatura recomendada
por una o más de las organizaciones siguientes:
- Unión Internacional de Bioquímica y Biología Molecular (IUBMB);
- Unión Internacional de Sociedades de Microbiología (IUMS);
- Unión Internacional de Sociedades de Inmunología (IUIS);
- SNOMED International (College of American Pathologists);
ü
77
- Organización Mundial de la Salud (OMS).
5.8.5 El informe de laboratorio debe indicar si la calidad de la muestra primaria recibida fue
inadecuada para el análisis o pudo haber comprometido el resultado.
ü
5.8.6 Las copias o archivos de los resultados indicados en el informe de laboratorio deben ser
retenidos por el laboratorio de forma que sea posible la recuperación rápida de tal información. El
periodo de tiempo durante el que han de retenerse los datos del informe de laboratorio puede variar;
sin embargo, los resultados indicados en el informe de laboratorio deben ser recuperables durante el
tiempo que sean médicamente relevantes o según lo requieran los requisitos nacionales, regionales y
locales.
ü
5.8.7 El laboratorio debe tener procedimientos para avisar inmediatamente a un médico (u otra
persona responsable de la asistencia sanitaria al paciente) cuando los resultados de los análisis
correspondientes a propiedades críticas se encuentran dentro de los intervalos alarmantes
establecidos. Esto incluye a los resultados recibidos sobre muestras enviadas para su análisis a
laboratorios subcontratistas.
ü
5.8.8 Para que puedan satisfacerse las necesidades clínicas locales, el laboratorio debe definir las
propiedades cuyos valores pueden ser alarmantes y los intervalos correspondientes, de acuerdo con
los médicos clínicos que utilizan el laboratorio. Esto es aplicable a todo tipo de análisis, incluyendo los
correspondientes a propiedades nominales y ordinales.
ü
5.8.9 Para los resultados transmitidos como un informe de laboratorio provisional, el informe de
laboratorio final debe siempre ser enviado al solicitante.
ü
5.8.10 Deben mantenerse los registros de las acciones tomadas en respuesta a resultados que
están dentro de los intervalos alarmantes. Tales registros deben incluir la fecha, la hora, el miembro
del personal responsable del laboratorio, la persona a la que se avisa y los resultados del análisis.
Cualquier dificultad encontrada al cumplir este requisito debe registrarse y revisarse durante las
auditorías.
ü
5.8.11 La dirección del laboratorio, en consulta con los solicitantes, debe establecer los plazos de
entrega para cada uno de los análisis. El plazo de entrega debe reflejar las necesidades clínicas.
Debe existir una política para notificar al solicitante el retraso en un análisis. Debe realizarse el
seguimiento, registrarse y revisarse por la dirección del laboratorio los plazos de entrega, así como
cualquier comunicación de los médicos clínicos a tal respecto. Cuando sea necesario, deben tomarse
ü

78
acciones correctivas para afrontar cualquier problema así identificado.
Esto no significa que el personal clínico deba ser notificado de todos los retrasos en los análisis,
sino solamente en aquellas situaciones en las que el retraso pueda comprometer la asistencia
sanitaria. Este procedimiento debería desarrollarse en colaboración entre el personal clínico y el
personal del laboratorio.
5.8.12 Cuando los resultados del análisis realizado por un laboratorio subcontratista necesitan ser
transcritos por el laboratorio que comunicará el resultado al solicitante, deben existir procedimientos
para verificar la exactitud de todas las transcripciones
ü
5.8.13 El laboratorio debe tener claramente documentados los procedimientos para la entrega de
los resultados del análisis, incluidos los detalles de quién puede entregarlos y a quién. Los
procedimientos también deben incluir directrices para la comunicación de los resultados directamente
a los pacientes.
ü
5.8.14 El laboratorio debe establecer políticas y prácticas para asegurarse de que los resultados
comunicados por teléfono u otro medio electrónico lleguen solamente a los receptores autorizados.
Los resultados comunicados verbalmente deben ir seguidos de un informe de laboratorio registrado
apropiadamente.
ü
5.8.15 El laboratorio debe tener políticas y procedimientos escritos respecto a la modificación de
los informes de laboratorio.
Cuando se modifiquen, el registro debe mostrar la hora, la fecha y el nombre de la persona
responsable de tal cambio.
Los datos originales deben permanecer legibles cuando se efectúen modificaciones.
Los registros electrónicos originales deben retenerse y las modificaciones añadirse al registro
utilizando procedimientos de edición apropiados de forma que los informes de laboratorio indiquen
claramente la modificación.
ü
5.8.16 Los resultados que ya se han comunicado para la toma de decisiones clínicas y que han
sido revisados deben retenerse en informes de laboratorio acumulativos subsiguientes, y deben
identificarse claramente como que han sido revisados. Si el sistema de notificación no puede reflejar
las modificaciones, cambios o alteraciones realizados, debe utilizarse un diario de anotaciones de las
mismas.
ü
Fuente: norma ISO 15189:2012

79
Porcentaje de cumplimiento de la norma ISO 15189
4. Requisitos de la gestion 84.49%
4.1. Organización y gestion 95%
4.2. Sistema de gestion de la calidad 94.84%
4.3. Control de los documentos 88.33%
4.4. Revision de solicitudes, ofertas y contratos 100%
4.5. Analisis efectuados por laboratorios de
referencia 75%
4.6. Servicios externos y suministradores 91.67%
4.7. Servicios de asesoramiento 75%
4.8. Resolución de conflictos y quejas 75%
4.9. Identificacion y control de las no conformidades 88.33%
4.10. Accion correctiva 81.25%
4.11. Accion preventiva 75%
4.12. Mejora continua 80%
4.13. Registros tecnicos y de la calidad 100%
4.14. Auditorias internas 91.66%
4.15. Revision por la direccion 56.25
5. Requisitos tecnicos 85.23%
5.1. Personal 89.23%
5.2. Ubicación y condiciones ambientales 97.22%
5.3. Equipos de laboratorio 96.43%
5.4. Procedimientos preanaliticos 87.5%
5.5. Procedimientos analiticos 92.86%
5.6. Aseguramiento de la calidad de los
procedimientos de analisis 42.43%
5.7. Procedimientos postanaliticos 100%
5.8. Informe de los resultados 76.56%
Total de cumplimiento 84.86
Fuente: norma ISO 15189:2012 AUTOR

80
Anexo 4
PREGUNTAS INCLUIDAS EN LA ENCUESTA
§ ¿Conoce usted que son las normas ISO?
§ Conoce la importancia de dichas normas.
§ Tiene conocimiento de lo que es un sistema de gestión de calidad
§ Conoce usted que es una acreditación
§ Sabe cuál es el alcance de una acreditación para una organización.
§ Sabe la diferencia entre una acreditación y una certificación
§ Sabe que organismo es el encargado de la acreditación.
§ ¿Se encuentran distribuidas las funciones de cada miembro del instituto’
§ ¿El instituto les proporciona capacitación continua?
Fuente autor


















