Tesis Doctorado en Bioquimica Diego Rojas Rivera
131
UNIVERSIDAD DE CHILE FACULTAD DE CIENCIAS QUIMICAS Y FARMACEUTICAS “TMBIM3/GRINA: UN GEN REGULADO POR LA RESPUESTA A PROTEÍNAS MAL PLEGADAS (UPR) QUE INHIBE LA APOPTOSIS MEDIANTE LA MODULACIÓN DE LA HOMEOSTASIS DEL CALCIO” Tesis presentada a la Universidad de Chile para optar al grado de Doctor en Bioquímica por: DIEGO ANDRES ROJAS RIVERA Director de Tesis Dr. CLAUDIO HETZ SANTIAGO- CHILE 2012
Transcript of Tesis Doctorado en Bioquimica Diego Rojas Rivera

UNIVERSIDAD DE CHILE
FACULTAD DE CIENCIAS QUIMICAS Y FARMACEUTICAS
“TMBIM3/GRINA: UN GEN REGULADO POR LA RESPUESTA A PROTEÍNAS MAL PLEGADAS (UPR) QUE INHIBE LA APOPTOSIS MEDIANTE LA MODULACIÓN DE
LA HOMEOSTASIS DEL CALCIO”
Tesis presentada a la Universidad de Chile para optar al grado de Doctor en Bioquímica por:
DIEGO ANDRES ROJAS RIVERA
Director de Tesis
Dr. CLAUDIO HETZ
SANTIAGO- CHILE 2012

2
UNIVERSIDAD DE CHILE FACULTAD DE CIENCIAS QUÍMICAS Y FARMACÉUTICAS
INFORME DE APROBACIÓN
TESIS DE DOCTORADO
Se informa a la Dirección de Postgrado de la Facultad de Ciencias Químicas y Farmacéuticas que la Tesis de Doctorado presentada por el candidato:
DIEGO ANDRES ROJAS RIVERA
Ha sido aprobada por la Comisión Informante de Tesis como requisito para optar al grado de Doctor en bioquímica, en el examen de defensa de Tesis rendido el día _____________________ Directores de Tesis: Dr. Claudio Hetz. ___________________________ Comisión Informante de Tesis: Dr. Andrew Quest (Presidente). ___________________________ Dra. Julieta González. ___________________________ Dr. Juan Olate. ___________________________ Dr. Ariel Orellana ___________________________

3
Agradecimientos.
1. Quisiera agradecer en primer lugar a mi familia: Mis papás y mis hermanos.
2. A mi tutor de tesis, el Dr. Claudio Hetz. Por su guía, entusiasmo y compromiso en
la búsqueda de nuevos resultados.
3. A los científicos que pusieron su conocimiento en disposición para abordar desde
distintos tópicos, las preguntas que surgieron en el desarrollo de esta tesis.
Especialmente a:
Dr. Ricardo Armisen (Universidad de Chile).
Ana Luisa Eguiguren (Universidad de Chile).
Dr. Andrés Stutzin (Universidad de Chile).
Dra. Jimena Sierralta (Universidad de Chile).
Dra. Alicia Colombo (Universidad de Chile).
Dr. Rosario Rizzuto (Universita degli studi di Padova).
Dr. Geert Bultynck y Santeri Kiviluoto (Katholieke Universiteit Leuven).
4. A todo el laboratorio de “Estrés Celular y Biomedicina”. Especialmente a Fernanda
Lisbona, Sebastián Zamorano (los “UPR”) y Gabriela Martínez. Además de Pamela
Valdés, Karen Castillo, Vicente Valenzuela y Melissa Calegaro. Todas estas personas
contribuyeron enormemente al trabajo, tanto en el diseño de los experimentos, así como
en la discusión e interpretación de los resultados. Además de su apoyo, compañía,
motivación y amistad.
5. A mis amigos que fueron un apoyo en todo momento. Especialmente a: Dra. Paula
Rodas, Dr. Ariel Contreras, Barbra Toro, Dr. Fabian Jaña, Alexis Ahumada, Carlos Basulto
y Carlos Olivares.

4
6. A Cecilia Zúñiga por su ayuda técnica en el citómetro de flujo, a Fabian y Pablo por
su ayuda técnica en los experimentos con Drosophila melanogaster y Alvaro Díaz en los
experimentos con pez zebra.

5
Financiamiento.
Esta tesis de doctorado fue realizada en el laboratorio de “Estrés Celular y Biomedicina”,
perteneciente a:
1) Instituto de Ciencias Biomédicas. Facultad de Medicina. Universidad de Chile.
2) Centro FONDAP de Estudios Moleculares de la Célula. Facultad de Medicina.
Universidad de Chile.
3) Instituto Milenio de Morfogénesis Neural (NEMO). Facultad de Medicina. Universidad
de Chile.
Además, conto con el siguiente financiamiento:
1) Proyecto FONDAP 15010006 (a CH, AS y RA).
2) Beca para la realización de doctorado en Chile CONICYT 2007-2011 (D.R.R.).
3) Beca MECESUP para la realización de pasantía en el extranjero 2010 (D.R.R.).
4) Beca de apoyo para la realización de tesis doctoral CONICYT 2009 y 2010 (D.R.R.).
5) Beca CONICYT de asistencia a congreso internacional 2010 (D.R.R.)
6) Beca “Keystone Symposium”, por trabajo destacado en presentación paneles 2010
(D.R.R).
7) Proyecto FONDECYT 11090324 (A.C)

6
Actividades y material de difusión:
1) Publicaciones:
• Rojas-Rivera D, Armisén R, Colombo A, Eguiguren AL, Martínez G, Díaz A,
Kiviluoto S, Rodríguez D, Patron M, Rizzuto R, Bultynck G, Concha ML, Sierralta J,
Stutzin A, Hetz C. TMBIM3/GRINA is a novel Unfolded Protein Response (UPR)
target gene that controls apoptosis through the modulation of ER calcium homeostasis. Cell death and differentiation. En prensa.
• Rodriguez D, Zamorano S, Lisbona F, Rojas-Rivera D, Arminsen R, Cubillos-Ruiz
JR, Cheng E, Letek M, Vaisar T, Henriquez DR, Irrazabal T, Heinecke JW,
Gonzalez-Billault C, Gupta S, Letai A, Pimentel-Muiños FX, Samali A, Kroemer G,
Hetz C. BH3-only proteins control the sustained signaling of the Unfolded
Protein Response sensor IRE1α . EMBO Journal. En prensa.
• Castillo K, Rojas-Rivera D, Lisbona F, Caballero B, Nassif M, Court F, Schuck S,
IbarC, Walter P, Sierralta J, Glavic A, Hetz C. BAX inhibitor-1 regulates
autophagy by controlling the IRE1α : branch of the unfolded protein response
EMBO Journal, 30 (21): 4465-4478, 2010.
• Rodríguez D, Rojas-Rivera D, Hetz C. Integrating stress signals at the
endoplasmic reticulum: The BCL-2 protein family rheostat. Biochim Biophys
Acta, 1813 (4): 564-574, 2010.
• Rojas-Rivera D, Benjamin Caballero, Sebastian Zamorano, Fernanda Lisbona and
Claudio Hetz; Alternative functions of the BCL-2 protein family at the
endoplasmic reticulum; The BCL-2 protein Family: From mechanisms to biomedical. Adv Exp Med Biol, 687: 33-47, 2010.
• Lisbona F, Rojas-Rivera D, Thielen P, Zamorano S, Todd D, Martinon F, Glavic A,
Kress C, Lin JH, Walter P, Reed JC, Glimcher LH, Hetz C. BAX Inhibitor-1 Is a Negative Regulator of the ER Stress Sensor IRE1. Mol Cell, 33: 679-691, 2009.

7
2) Presentaciones a congresos internacionales:
• Rojas-Rivera D, Armisén R, Colombo A, Eguiguren AL, Martínez G, Díaz A,
Kiviluoto S, Rodríguez D, Patron M, Rizzuto R, Bultynck G, Concha ML, Sierralta J,
Stutzin A, Hetz C. TMBIM3/GRINA is a conserved Unfolded Protein Response
(UPR) target gene that controls apoptosis through the modulation of ER
calcium homeostasis. Metabolism and Cancer. American Association for Cancer
Research. Sheraton Inner Harbor Hotel, Baltimore, Estados Unidos. 2011
• Rojas-Rivera D, Peter Thielen, Fernanda Lisbona, Alexis Martínez, Sebastián
Zamorano, Melissa Calegaro, Jimena Sierralta, Sergio Lavandero and Claudio
Hetz. TMBIM3: A new and conserved member of the Bax-inhibitor-1 family
that regulates ER stress-mediated apoptosis. Cell Death Pathways: Apoptosis,
Autophagy and Necrosis (X3). Keystone Symposium. Vancouver, British Columbia,
Canadá. 2010
• Lisbona F, Rojas-Rivera D, Zamorano S, Thielen P, Walter P, Reed JC, Glimcher
LH y Hetz C. BAX Inhibitor-1 is a negative regulator of the ER stress sensor
IRE1 and is a component of the UPRosome: A new function beyond apoptosis. Protein in Growth, Development & Disease. Estados Unidos. 2009
• Lisbona F, Rojas-Rivera D, Zamorano S, Thielen P, Walter P, Reed JC, Glimcher
LH y Hetz C. BI-1 is a negative regulador of IRE1. Cell Death Pathways, Whistler
Conference Centre, Whistler, British Columbia. Estados Unidos. 2009.
• Lisbona F, Rojas-Rivera D, Thielen P, Kress C, Reed J, Walter P, Glimcher L,
Hetz C. Bax-inhibitor 1 is a negative regulator of the ER stress sensor IRE1: a
new functions beyond apoptosis. Gordon conference: Molecules that specify the
variety of cell death mechanisms. Italia. 2007.
3) Presentación en congresos nacionales:
• Rojas-Rivera D, Armisén R, Colombo A, Eguiguren AL, Martínez G, Díaz A,
Kiviluoto S, Rodríguez D, Patron M, Rizzuto R, Bultynck G, Concha ML, Sierralta J,

8
Stutzin A, Hetz C. TMBIM3/GRINA is conserved unfolded protein response (UPR)
target gene that controls apoptosis through the modulation of ER Calcium
homeostasis. XXV Reunión Anual de la Sociedad de Biología Celular de Chile.
Gran Hotel Pucón, Chile, Noviembre.
• G Martínez, D Rojas-Rivera, A Colombo, F Lisbona, M Concha and C Hetz. A new
animal model of ER stress: Zebrafish UPR. Gabriela Martínez, Institute of
Biomedical Sciences, FONDAP Center for Molecular Studies of the Cell,
Millennium Nucleus for Neural Morphogenesis, U. of Chile, Santiago, Chile. XXIV
Reunión Anual de la Sociedad de Biología Celular de Chile. Gran Hotel Pucón,
Chile, Noviembre.
• Rojas-Rivera D, Armisen R, Eguiguren AL, Martínez G, Colombo A, Stutzin A,
Patron M, Rizzuto R, Sierralta J, Concha M, and Hetz C. TMBIM3: An ancestral
regulator of apoptosis that controls ER calcium. homeostasis. Fondap Center for
Molecular Studies of the Cell, Millennium Nucleus for Neural Morphogenesis,
ICBM, U. de Chile, and U. of Padova. XXIV Reunión Anual de la Sociedad de
Biología Celular de Chile. Gran Hotel Pucón, Chile, Noviembre.
• Diego Rojas-Rivera. "Reguladores ancestrales de la apoptosis: El mundo pre-
familia BCL-2". Reunión anual del Centro FONDAP de Estudios Moleculares de la
Célula. San Bernardo. Santiago.
• K Castillo, D. Rojas-Rivera, B. Caballero, F. Lisbona, A. Glávic, and C. Hetz.The
ER located protein Bax-inhibitor 1 (Bi-1) regulates autophagy (La proteína
inhibidora de Bax (BI-1) del RE regula la autofagia). XXII Reunión Anual de la
Sociedad de Biología Celular de Chile. Gran Hotel Pucón, Chile, Noviembre.
• Diego Rojas-Rivera, Peter Thielen, Alexis Martínez, Ricardo Armisen, Jimena
Sierralta, Sergio Lavandero and Claudio Hetz. GRINA: A new and conserved
member of the Bax-inhibitor-1 family that regulates ER stress mediated apoptosis.

9
XXII Reunión Anual de la Sociedad de Biología Celular de Chile. Gran Hotel
Pucón, Chile, Noviembre.
• Fernanda Lisbona, Diego Rojas-Rivera, Sebastián Zamorano, Alvaro Glavic, and
Claudio Hetz. BAX Inhibitor-1 regulates the inactivation phase of the Unfolded
Protein Response (Bax Inhibitor-I regula la fase de inactivación de la respuesta a
proteínas mal plegadas) XXII Reunión Anual de la Sociedad de Biología Celular de
Chile. Gran Hotel Pucón, Chile.
• Rojas-Rivera D, Lisbona F, Zamorano S, Hetz C. GRINA: a new and conserved
anti-apoptotic protein located at the endoplasmatic reticulum. XXI Reunión Anual
de la Sociedad de Biología Celular de Chile. Gran Hotel Pucón, Chile.
• Lisbona F, Rojas-Rivera D, Glavic A, Walter P, Reed J, Thielen P, Glimcher L,
Hetz C. A new component of the UPRsome: Bax Inhibitor-1 is a negative regulator
of the ER stress sensor IRE1alpha. XXI Reunión Anual de la Sociedad de Biología
Celular de Chile. Gran Hotel Pucón, Chile.
• Rojas-Rivera D, Díaz-Elizondo J, Salas D, Chiong M, Lavandero S. “ERK1/2 on
the protection of cardiac myocyte to hyposmotic stress”. Centro FONDAP Estudios
Moleculares de la Célula, Facultad de Ciencias Químicas y Farmacéuticas y
Medicina. Universidad de Chile 8389100. XX Reunión Anual de la Sociedad de
Biología Celular de Chile. Gran Hotel Pucón, Chile.
• Lisbona F, Rojas-Rivera D, Matus S, Stanley Korsmeyer, Glimcher L, Hetz C.
Regulation of the unfolded protein response and organelle physiology by
components of the apoptosis machinery. XX Reunión Anual de la Sociedad de
Biología Celular de Chile. Gran Hotel Pucón, Chile.
4) Pasantías Internacionales:

10
Estudios del efecto de la expresión de TMBIM3 en el contenido de Calcio en el Retículo
Endoplasmático. Laboratorio de Rosario Rizzuto, Universidad de Padova, Veneto, Italia.
Beca MECESUP, Doctorado en Bioquímica, Universidad de Chile. Junio-Julio 2010.

11
1. Índice General.
2) Índice de Figuras. ...................................................................................................... 13 3) Abreviaciones. ........................................................................................................... 16 4) Resumen. ................................................................................................................... 22 5) Summary .................................................................................................................... 16 6) Introducción. .............................................................................................................. 24 6.1) El Retículo Endoplasmático. .................................................................................. 24 6.2) El estrés de RE. ....................................................................................................... 24 6.3) La UPR. ..................................................................................................................... 25 6.4) La apoptosis ............................................................................................................ 29 6.5) La Familia BCL-2 ..................................................................................................... 30 6.6) La Apoptosis y la UPR ............................................................................................ 31 6.7) TMBIM6/BI-1 ............................................................................................................. 34 6.8) TMBIM6/BI-1 y el estrés de RE. .............................................................................. 35 6.9) Las proteínas de la familia TMBIM: nuevos reguladores de la muerte celular? 37 7) Hipótesis .................................................................................................................... 42 8) Objetivo General ........................................................................................................ 42 9) Objetivos específicos: .............................................................................................. 42 10) Materiales y Métodos ............................................................................................ 43 11) Resultados ............................................................................................................. 54 11.1) TMBIM3 y el control de la apoptosis ................................................................ 54
11.1.1) Sobreexpresión de TMBIM3 y muerte celular inducida por estrés de RE. ..... 54 11.1.2) Efecto de la deficiencia de TMBIM3 y TMBIM6 en la viabilidad celular. ......... 59 11.1.3) Efecto de la deficiencia de TMBIM3 en la muerte celular inducida por distintos estímulos de estrés. .................................................................................................... 61
11.2) TMBIM3 y su regulación transcripcional ............................................................ 65 11.2.1) Regulación de la expresión de TMBIM3 bajo condiciones de estrés de RE. .. 65 11.2.2) Regulación de la expresión de tmbim3 inducida por estrés de RE in vivo. .... 70
11.3) TMBIM3 y el control de la homeostasis del Ca2+ ................................................ 73 11.3.1) La expresión de TMBIM3 controla la homeostasis del calcio del RE. ............ 73 11.3.2) La sobreexpresión de TMBIM3 protege frente a la muerte celular inducida por sobrecarga de Ca2+. .................................................................................................... 76 11.3.3) La sobreexpresión de TMBIM3 no altera los niveles de Ca2+ RE ni la expresión de diferentes chaperonas en el RE. ........................................................... 77 11.3.4) TMBIM3 interactúa con IP3-R y modula su actividad. .................................... 80 11.3.5) Efecto de la liberación de Ca2+ desde el RE en la muerte inducida por estrés de RE .......................................................................................................................... 82 11.3.6) Efecto del Ca2+ en la inhibición de la apoptosis mediada por TMBIM3 .......... 83
11.4) Rol de TMBIM3 in vivo .......................................................................................... 85 11.4.1) TMBIM3 y TMBIM6 tienen actividades antiapoptoticas sinérgicas in vivo en un modelo de ER estrés en Drosophila melanogaster. .................................................... 85 11.4.2) Expresión de tmbim3 en pez zebra y tejidos de ratón. ................................... 90

12
11.4.3) Deficiencia de tmbim3 incrementa la apoptosis durante el desarrollo del pez zebra. .......................................................................................................................... 92 11.4.4) Generación de un modelo de estrés de RE en pez zebra in vivo. .................. 98 11.4.5) TMBIM3 protege del estrés de RE en pez zebra in vivo. ................................ 98
12) Discusión. ............................................................................................................. 101 12.1) TMBIM3 es parte de una familia conservada de proteínas con actividad anti-apoptotica: el mundo pre-bcl2. .................................................................................. 101 12.2) Regulación del tmbim3, uno nuevo blanco de la UPR ................................... 106 12.3) Regulación del Ca2+ mediada por tmbim3, el vínculo con el control de la apoptosis inducida por estrés de RE. ....................................................................... 107 12.4) TMBIM3 y TMBIM6: compañeros en el control de la muerte celular. ........... 109 12.5) TMBIM3 y su función en el sistema nervioso. ................................................ 110 12.6) TMBIM3: un nuevo blanco conservado de la UPR que inhibe la apoptosis inducida por estrés de RE mediante el control de la homeostasis del Ca2+. ........ 112
13) Conclusiones. ...................................................................................................... 115 14) Bibliografía. .......................................................................................................... 115

13
2. Índice de Figuras.
Figura1: La Respuesta a Proteínas mal Plegadas. ...................................................... 28
Figura 2: Apoptosis inducida por estrés de RE. ........................................................... 34
Figura 3: Proteínas de la familia TMBIM. ..................................................................... 38
Figura 4: TMBIM3/GRINA es una proteína conservada. .............................................. 40
Figura 5: Sistema de cruzas para obtención de moscas iRNA. ................................... 50
Figura 6: Localización sub-celular de TMBIM3 en el RE y aparato de Golgi. .............. 55
Figura 7: Expresión estable de TMBIM3 en células MEFs. .......................................... 55
Figura 8: Efecto de la sobreexpresión de TMBIM3 en la muerte celular inducida por diferentes tipos de estrés celular. ................................................................................. 56
Figura 9: Efecto de la sobreexpresión de TMBIM3 en la apoptosis inducida por estrés de RE.. .......................................................................................................................... 57
Figura 10. Figura 10. Efecto de la sobreexpresión de TMBIM3 en la sobrevida bajo condiciones de estrés de RE. ....................................................................................... 58
Figura 11. Efecto de la sobreexpresión de TMBIM3-MYC en la proliferación celular. . 58
Figura 12: Generación de células “knock down” para tmbim3. .................................... 59
Figura 13: La disminución de los niveles de tmbim3 y su efecto en la muerte celular. 60
Figura 14: Efecto de la doble deficiencia de TMBIM3 y TMBIM6 en la apoptosis espontánea en células MEFs. ...................................................................................... 61
Figura 15: Efecto de la deficiencia de TMBIM3 y/o TMBIM6 en la muerte celular inducida por estrés de RE.. .......................................................................................... 62
Figura 16: Efecto de la disminución de tmbim3 en los niveles de muerte de células MEFs expuestas a estímulos que inducen apoptosis.. ................................................. 63
Figura 17: Efecto de la sobreexpresión de TMBIM3-MYC en células TMBIM6 KO y muerte celular inducida por estrés de RE. .................................................................... 63
Figura 18. Estudio de la posible formación de un complejo proteico entre TMBIM3-MYC y TMBIM6-HA forman un complejo. ..................................................................... 64
Figura 19: Cambios en los niveles de el ARNm de tmbim3 en condiciones de estrés de RE en células MEFs. .................................................................................................... 65
Figura 20: Efecto de la deficiencia del sensor de estrés de RE PERK en el incremento del ARNm de tmbim3 inducido por estrés de RE.. ....................................................... 66
Figura 21: Niveles de los blancos transcripcionales de la UPR en las células PERK WT y KO expuestas a estrés de RE.. .................................................................................. 67
Figura 22: Re-expresión de IRE1α en células deficientes para IRE1α.. ...................... 68

14
Figura 23: Efecto de la expresión del sensor de estrés IRE1α en la regulación del ARNm de tmbim3 bajo condiciones de estrés de RE.. ................................................. 68
Figura 24: Efecto de la deficiencia de PERK en la viabilidad de las células expuestas a estrés de RE. ................................................................................................................ 69
Figura 25: Efecto de la sobreexpresión de TMBIM3 en marcadores de la UPR.. ........ 70
Figura 26: Estudio de los niveles de ARNm de tmbim3 en riñones de ratones expuestos a estrés de RE in vivo.. ............................................................................... 71
Figura 27: ATF4 y el control de la expresión de tmbim3 en respuesta a estrés de RE in vivo. .............................................................................................................................. 72
Figura 28: TMBIM3-MYC y la regulación de la homeostasis del Ca2+. ......................... 74
Figura 29: Efecto TMBIM3-MYC en la salida de Ca2+ desde el RE inducida por la inhibición de la bomba SERCA.. ................................................................................... 75
Figura 30: Efecto de la deficiencia de TMBIM3 en la salida de Ca2+ desde el RE. ...... 75
Figura 31: La sobreexpresión TMBIM3 disminuye la muerte por sobre-carga de Ca2+. ...................................................................................................................................... 76
Figura 32: Determinación del contenido de Ca2+ del RE. ............................................. 77
Figura 33: Efecto de la sobreexpresión de TMBIM3 en el contenido de Ca2+ en el RE. ...................................................................................................................................... 78
Figura 34: Efecto de la sobreexpresión de TMBIM3 en los niveles de expresión de diferentes proteínas residentes del RE.. ....................................................................... 79
Figura 35: Efecto de la sobreexpresión de TMBIM-MYC en la liberación de Ca2+ desdedesde el RE inducida por IP3 ……….………………………………………………81
Figura 36: Co-inmunoprecipitación de TMBIM3-MYC con IP3-R y BCL-2. .................. 81
Figura 37: La expresión de TMBIM3 no genera un cambio en la expresión de algunas proteínas involucradas en el control del Ca2+ RE.. ....................................................... 82
Figura 38: El estrés de RE y su efecto en los niveles de Ca2+ en el RE. ..................... 83
Figura 39: Efecto del Ca+2 en la muerte inducida por estrés de RE. ............................ 83
Figura 40: Papel del Ca2+ en el control de la apoptosis ejercida por TMBIM3.. ........... 84
Figura 41: Generación de un modelo deficiente en tmbim3 y tmbim6 en Drosophila melanogaster. ............................................................................................................... 85
Figura 42: Efecto de la deficiencia de dTmbim3 y dTmbim6 en la generación de moscas adultas.. ........................................................................................................... 86
Figura 43: La deficiencia de tmbim3 y tmbim3 y su efecto en la apoptosis espontánea y en respuesta a Tm en Drosophila melanogaster.. ..................................................... 87
Figura 44: Efecto de la deficiencia de dTmbim3 y dTmbim6 en la generación de moscas adultas a partir de larvas crecidas en presencia o ausencia de Tm.. ............. 89
Figura 45: Expresión de z-Tmbim3 en distintos estados de desarrollo del pez zebra. 90

15
Figura 46: Perfil de expresión de z-tmbim3 en el desarrollo de pez zebra.. ................ 90
Figura 47: Patrón de expresión de zTmbim3 en pez zebra.. ........................................ 91
Figura 48: Expresión de tmbim3 en tejidos de ratón. . ................................................ 92
Figura 49: Efecto de la deficiencia de z-tmbim3 en la muerte celular durante el desarrollo del pez zebra. .............................................................................................. 93
Figura 50: Cuantificación de la tinción con naranjo de acridina en la médula espinal de los peces zebra inyectados con morfolino destinado contra zTmbim3. ....................... 94
Figura 51: Cuantificación de los niveles de caspasa 3 activada en la medula espinal de embriones de pez zebra inyectados z-Tmbim3-MO.. ................................................... 95
Figura 52: Efecto de la deficiencia de tmbim3 en pez zebra y la generación de individuos normales. ..................................................................................................... 95
Figura 53: Deficiencia de Tmbim3 y la formación de estructuras cerebrales en pez zebra.. ........................................................................................................................... 96
Figura 54. Sobreexpresión de m-tmbim3 y la letalidad inducida por z-Tmbim3-MO en pez zebra. ..................................................................................................................... 97
Figura 55: UPR en peza zebra. .................................................................................... 98
Figura 56: Analisis de la expresión de m-Tmbim3 en pez zebra. ................................. 99
Figura 57: tmbim3 y el control de la muerte celular inducida por Tg en pez zebra in vivo.. ............................................................................................................................. 99
Figura 58: tmbim3 y el control de la muerte celular inducida por Tg en pez zebra in vivo. ............................................................................................................................ 100
Figura 59: Modelo propuesto: TMBIM3 es un blanco de la UPR que controla la apoptosis mediante la inhibición del receptor de IP3. ................................................ 113

16
3) Abreviaciones.
2-APB: Inhibidor farmacológico del receptor de IP3
ADN: Ácido desoxiribonucleico.
AIF: Apoptotic Inducing Factor.
APAF1: Apoptotic Protease Activating Factor 1.
ARNm: Ácido Ribonucleico mensajero.
ASK1: Apoptosis signal-regulating kinase 1
ATF4: Activating Transcription Factor 4.
ATF6: Activating Transcription Factor 6.
ATF6f: fragmento de ATF6.
ATP: Adenosín trifosfato.
BCL-2: B-cell lymphoma 2 protein.
BH3: BCL-2 homology domain 3.
BH4: BCL-2 homology domain 4.
BIP: Immunoglobulin-Binding Protein.
BI-1: Bax Inhibitor 1 protein.
CaMKII: Ca2+/Calmodulin-Dependent Protein Kinase II.
CHOP: C/EBP-homologous protein.
DIABLO: Direct IAP-binding protein with low PI.
DIG: digoxigenin-UTP
dIRE1α: IRE1α expresado en Drosophila melanogaster.

17
D.mel: Drosophila melanogaster (mosca).
D.rerio: Danio rerio (pez zebra).
dTMBIM6: TMBIM6 expresado en Drosophila melanogaster.
dTMBIM3: TMBIM3 expresado en Drosophila melanogaster.
eIf2α: eukaryotic translation initiation factor α 2.
ERAD: Endoplasmic Reticulum associated degradation.
ERO1: ER oxidase 1.
Est: Estaurosporina
Eto: Etopósido.
GAAP: Golgi Anti-Apoptotic Proteín
GADD34: growth arrest and DNA damage-inducible protein-34.
GBP: Glutamate binding protein.
GFP: Proteína Fluorescente verde, del inglés Green fluorescent protein.
GHITM: Growth-Hormone Inducible Transmembrane Protein.
GRINA: Glutamate Receptor, Ionotropic, N-methyl D-asparate-Associated protein 1.
HA: Péptido señal Human influenza hemagglutinin.
hpf: horas pos-fertilización.
hTMBIM6: Proteína TMBIM6 expresada en humano.
hTMBIM3: Proteína TMBIM3 expresada en humano.
IP3: inositol 1,4,5-trifosfato.
IP3-R1: Isoforma 1 del receptor de IP3.

18
IP3-R3: Isoforma 3 del receptor de IP3.
IRE1: Inositol requering protein-1.
JNK: Stress-activated protein kinases SAPK/Jun N-terminal kinases.
KO: knock out.
LFG: Lifeguard.
MEFs: Células embrionarias de fibroblasto, del inglés Mice embrionary fibroblast.
Mis-MO: Morfolino en sentido inverso.
MYC: Péptido señal derivado del producto génico del gen c-myc.
M. mus: Mus musculus.
mTmbim3: gen de ratón que codifica para TMBIM3.
PBS: Solución tampón de fosfato.
PCR: Reacción en cadena de la polimerasa.
PDI: Protein disulfide isomerase.
PERK: Protein kinase RNA (PKR)-like ER kinase.
PFA: Paraformaldehido.
PI: Ioduro de propidio.
qPCR: PCR cuantitativo.
RE: Retículo endoplasmático.
RECS1: Responsive to Centrifugal force and Shear tress gene 1.
r.p.m.: Revoluciones por minuto.
ROI: Región de interés.

19
SERCA: bomba Ca2+/ATPasa.
shARN: ARN pequeño tipo horquilla, del inglés short hairping.
SMAC: Second Mitochondria-derived Activator of Caspases.
SP1: Site 1 protease.
SP2: Site 2 protease.
TEGT: Testis enhanced gene transcript.
Tg: Tapsigarguina.
Tm: Tunicamicina.
TMBIM: TransMembrane Bax-Inhibitor 1 Motive.
TNF: Tumour necrosis factor.
TRAF2: TNF receptor-associated factor 2.
UPR: Respuesta a proteínas mal plegadas por sus siglas en inglés Unfolded protein
response.
WT: Wild Type.
xbp-1: x-box binding protein-1.
XBP-1s: Proteína XBP-s, traducida a partir del ARNm procesado de xbp-1, s del inglés
splicing.
z-Tmbim3: ARNm de TMBIM3 expresado en pez zebra.
zTmbim3-MO: Morfolino destinado contra z-Tmbim3.

20
4) Resumen.
La familia de proteínas TMBIM (del inglés TransMembrane Bax-Inhibitor 1 Motive)
es altamente conservada y ha sido pobremente caracterizada. Los genes de la familia
TMBIM están presente en mamíferos, insectos, plantas y virus y no presentan
homología aparente con los miembros de la familia BCL-2, los cuales clásicamente
han sido reconocidos como los reguladores maestros de la muerte celular
programada. Al igual que para los miembros de la familia BCL-2, para algunos
miembros de la familia TMBIM ha sido descrito que presentan actividad antiapoptótica
y la capacidad de modificar la homeostasis del Ca2+. El miembro más estudiado de la
familia TMBIM es TMBIM6/BI-1. Esta proteína puede inhibir la muerte celular bajo
condiciones de estrés de RE, el cual es una condición inducida por la acumulación de
proteínas mal plegadas en el interior del RE. El miembro de la familia TMBIM menos
estudiado es TMBIM3/GRINA y existen antecedentes de que se expresa
principalmente en el sistema nervioso central. Sin embargo, se desconoce si comparte
alguna de las actividades descritas para los miembros de la familia TMBIM.
En esta tesis, se propuso estudiar el papel de TMBIM3/GRINA en la muerte
inducida por estrés de RE. La hipótesis propuesta fue determinar si TMBIM3/GRINA
puede inhibir la muerte celular inducida por estrés de RE mediante un mecanismo
dependiente del control de la homeostasis del Ca2+.
Los resultados obtenidos muestran que TMBIM3 inhibe la muerte celular inducida
por estrés de RE y la apoptosis mediada por sobrecarga de Ca2+, sin afectar la muerte
celular inducida por estímulos intrínsecos de apoptosis. La disminución de la
expresión de TMBIM3 en células deficientes en TMBIM6 indujo apoptosis espontánea,
lo cual podría sugerir actividades antiapoptóticas complementarias. Los niveles del

21
ARNm de tmbim3 son altamente incrementados bajo condiciones de estrés de RE,
tanto en modelos celulares, como en modelos animales. Esta regulación de los niveles
del ARNm de tmbim3 son controlados por la vía de PERK/ATF4, la cual es activada
bajo condiciones de estrés de RE.
Además, TMBIM3 controla la homeostasis del Ca2 e interactúa físicamente con el
receptor de IP3, lo cual podría explicar su actividad antiapoptótica bajo condiciones de
estrés de RE.
Estudios de pérdida de función en D. melanogaster revelaron que TMBIM3 y
TMBIM6 tienen actividades sinérgicas respecto a la protección frente a estrés de RE.
De manera similar, la deficiencia de TMBIM3 en embriones de pez zebra generó
apoptosis espontánea durante el desarrollo y drásticas alteraciones en la morfología
del cerebro y la viabilidad neuronal. Además, la sobreexpresión de TMBIM3 protegió a
los embriones de pez zebra en condiciones experimentales de estrés de RE.
Todos estos resultados permiten establecer un rol crítico de TMBIM3 en el control
de la apoptosis inducida por estrés de RE, lo cual sugiere la existencia de un grupo
conservado de reguladores funcionales de la muerte celular, más allá de la familia de
proteínas BCL-2.

22
5) Summary.
The TMBIM proteins family is a highly conserved group of proteins that are poorly
characterized. The TMBIM genes are present in mammals, insects, plants and viruses
and have no apparent homology with members of the BCL-2 family, which have
classically been recognized as the master regulators of programmed cell death.
Similarly to members of BCL-2 family, some TMBIM family members have anti-
apoptotic activity and the ability to alter Ca2+ homeostasis. The most studied member
of the TMBIM family is TMBIM6. This protein can inhibit cell death under conditions of
ER stress, which is induced by the accumulation of misfolded proteins within the ER.
The TMBIM family member that is less studied is TMBIM3/GRINA and it is expressed
mainly in the central nervous system. However, it is unknown if it shares some of the
activities described for other TMBIM family members.
In this thesis, we studied the role of TMBIM3/GRINA in cell death induced by ER
stress. The hypothesis proposed was that TMBIM3/GRINA inhibits cell death induced
by ER stress through a mechanism dependent of homeostasis-Ca2+ controls.
The results obtained showed that TMBIM3 inhibits cell death induced by ER stress
and apoptosis mediated by overload of Ca2+, without affecting the cell death induced
by other intrinsic apoptosis stimuli. Knock down of TMBIM3 in TMBIM6 deficient cells
induced spontaneous apoptosis, which may suggest complementary anti-apoptotic
activities. Tmbim3 mRNA levels are highly increased under ER stress conditions, both
in cellular models and in animal models. This regulation of mRNA levels of tmbim3 is
controlled by the PERK/ATF4 signaling branch, which is activated under conditions of
ER stress. In addition, TMBIM3 controls Ca2+ homeostasis and physically interacts

23
with the IP3 receptor, which could explain its anti-apoptotic activity under conditions of
ER stress.
Loss of function studies in D. melanogaster revealed that TMBIM6 and TMBIM3
have synergistic activities on the protection against ER stress. Similarly, deficiency
TMBIM3 zebrafish embryos resulted in spontaneous apoptosis during development
and drastic alterations in the morphology of the brain and neuronal viability. In addition,
TMBIM3 over-expression protected zebrafish embryos in experimental conditions of
ER stress.
All these results indicate a critical role of TMBIM3 in apoptosis controlling induced
by ER stress, suggesting the existence of a group of conserved functional regulators of
cell death, beyond the family of BCL-2 protein.

24
6) Introducción. 6.1) El Retículo Endoplasmático
Cerca del 70% de las proteínas sintetizadas en una célula están destinadas a los
distintos sistemas membranosos intracelulares o a su exportación al medio extracelular. El
proceso de síntesis y maduración de estas proteínas se caracteriza por su paso a través
de una serie de organelos que comienza en el retículo endoplasmático (RE), desde que
se inicia el proceso de traducción de las proteínas en los complejos ribosomales hasta
que se encuentran completamente plegadas y funcionales en su sitio final de destinación.
Por lo tanto, el RE es un organelo fundamental en la ruta secretora de las proteínas. ¿Qué
características tiene el RE que permite el correcto plegamiento de las proteínas? En el RE
endoplasmático se localizan múltiples proteínas con actividad chaperona y foldasas,
además de un ambiente reductor que facilita la formación de puentes di-sulfuros, así
como complejos proteicos que permiten la glicosilación de proteínas, con la consecuente
reducción de la polaridad de la cadena aminoacídica, favoreciendo el plegamiento
(Gorlach et al, 2006). Por otra parte, el RE presenta una compleja y tradicional arquitectura que varía
según los requerimientos celulares, estando formado por una serie de estructuras
membranosas tubulares continuas y dinámicas que se organizan en subdominios. Estos
subdominios incluyen el RE rugoso, RE liso, RE transicional y la envoltura nuclear
(Borgese et al, 2006).
Facilitar la síntesis de proteínas no es la única función del RE, puesto que además
es clave en la síntesis de los fosfolípidos (Fagone & Jackowski, 2009; Jesch et al, 2006),
es el reservorio intracelular mas importe de Calcio (Ca2+) (Berridge, 2002; Borgese et al,
2006; Gorlach et al, 2006) y bajo determinadas circunstancias, se generan señales
celulares desde el RE que pueden activar o inhibir la muerte celular (Berridge, 2002;
Tabas & Ron, 2011).
6.2) El estrés de RE
Distintas condiciones que perturban la función del RE, y que tengan como
consecuencia un aumento en la síntesis de proteínas o la generación de proteínas mal
plegadas al interior del RE, pueden generar una condición denominada estrés de RE (Ron

25
& Walter, 2007; Schroder & Kaufman, 2005a). El estrés de RE puede ser, por ejemplo,
desencadenado por alteraciones en la homeostasis del Ca2+, estrés oxidativo y/o
alteraciones en el balance redox en el lumen del RE (Ron & Walter, 2007; Woehlbier &
Hetz, 2011). En eucariontes multicelulares, el estrés de RE es sensado por al menos tres
proteínas que controlan la activación de tres vías de traducción de señales complejas, las
cuales promueven una serie de eventos que pueden contribuir a la recuperación de la
homeostasis pérdida (Hetz & Glimcher, 2009; Schroder & Kaufman, 2005b). La activación
coordinada de estas tres vías constituye una respuesta especifica al estrés de RE, la cual
ha sido denominada Respuesta a Proteínas Mal plegadas o UPR, por sus siglas en inglés:
Unfolded Protein Response. 6.3) La UPR
La acumulación de proteínas mal plegadas en el RE es presumiblemente
detectada, directa o indirectamente, por al menos tres proteínas residentes del RE con un
dominio transmembrana, los cuales han sido denominados “sensores de estrés de RE”
(Cox et al, 1993; Ron & Walter, 2007). Cada uno de esos tres sensores comunica el
estado de plegamiento de las proteínas desde el RE hasta el citoplasma y el núcleo,
controlando la expresión de factores de transcripción específicos en conjunto con
múltiples efectos no transcripcionales (Hetz & Glimcher, 2009; Ron & Walter, 2007). La vía de la UPR mas conservada en la evolución involucra la activación
combinada de una proteína con actividades nucleasa y quinasa, denominada IRE1 (del
inglés Inositol requering protein-1) (Back et al, 2005; Calfon et al, 2002; Tirasophon et al,
1998). En mamíferos, existen dos isoformas: IRE1α, cuya expresión es ubicua, e IRE1β,
cuya expresión se restringe al tejido gastro-intestinal y a las vías respiratorias (Ron &
Hubbard, 2008). La activación de IRE1α es iniciada en el lumen del RE por la disociación
de la chaperona BIP (del inglés immunoglobulin-binding protein o Grp78) la cual se une
preferentemente a las proteínas mal plegadas en el lumen del RE (Bertolotti et al, 2000;
Kimata et al, 2003; Oikawa et al, 2009). Otro fenómeno que ha sido asociado
indirectamente a la activación de IRE1α, es la interacción directa de IRE1α con proteínas
mal plegadas, lo cual favorecería su homo-oligomerización espontánea (Hetz & Glimcher,
2009; Ron & Hubbard, 2008; Zhou et al, 2006). Estos procesos en el lumen del RE
controlan la activación del dominio citosólico de IRE1α, favoreciendo su auto-

26
transfosforilación y unión de un nucleótido (revisado en Hetz & Glimcher, 2009). Estos
eventos en su conjunto activan el dominio RNAsa y promueven una de sus funciones
efectoras en el citoplasma: el procesamiento no convencional del ARN mensajero (ARNm)
de un factor transcripcional denominado x-box binding protein-1 (xbp-1) (Calfon et al,
2002). El procesamiento del ARNm de xbp-1 induce la escisión de 26 nucleótidos,
generando, consecuentemente, un corrimiento en el marco de lectura y la generación de
un codón de stop río arriba del original, lo que induce la traducción de una proteína de
mayor tamaño que contiene un dominio de transactivación transcripcional, el cual actúa
como un potente factor transcripcional denominado XBP-1s (s del inglés splicing). XBP-1s
controla la expresión de un gran número de genes involucrados, en su mayoría, en el
restablecimiento de la homeostasis pérdida en el RE asociadas al plegamiento,
degradación de proteínas, traslocación al RE y síntesis de lípidos (Lee et al, 2003). Bajo
ciertas condiciones, la actividad nucleasa de IRE1α también puede degradar y bloquear la
traducción de una gran variedad de ARNm que codifican proteínas difíciles de plegar (Han
et al, 2009; Hollien et al, 2009; Hollien & Weissman, 2006), aunque el posible rol
fisiológico o patológico de esta actividad es aún desconocida.
Una segunda vía de la UPR es iniciada por la activación de la quinasa PERK (del
inglés Protein kinase RNA (PKR)-like ER kinase), la cual es activada de manera similar a
IRE1α, favoreciéndose bajo condiciones de estrés de RE su auto-transfosforilación y su
homo-oligomerización (revisado en Ron & Walter, 2007 y Bertolotti et al, 2000). La
proteína PERK fosforila a la sub-unidad α del factor de iniciación de la traducción, eIf2α
(del inglés eukaryotic translation initiation factor α 2), con la consecuente inhibición de la
traducción (Shi et al, 1998). Sin embargo, la traducción de otro ARNm que codifica para el
factor de expresión ATF4 es inducida, favoreciéndose la expresión de múltiples genes
blancos de ATF4 (Harding et al, 2000a), los que regulan el metabolismo de aminoácidos
(Harding et al, 2000a; Harding et al, 2003), homeostasis redox (Harding et al, 2003),
autofagia (Rzymski et al, 2009) y apoptosis (Kim et al, 2006) (revisado en Tabas & Ron,
2011). Un importante blanco de ATF4, el cual ha sido ampliamente descrito en la literatura
es chop (C/EBP-homologous protein; también conocido como GADD153) (Harding et al,
2000a). Usualmente, CHOP puede interactuar con otros factores de transcripción
regulando genes blancos de la UPR que favorecen la restauración de la homeostasis

27
pérdida en el lumen del RE (Tabas & Ron, 2011). Por otra parte, CHOP y ATF4 favorecen
la expresión de GADD34 (del inglés growth arrest and DNA damage-inducible protein-34),
una fosfatasa que desfosforila a eIF2α, previamente fosforilado por PERK, restaurándose
la traducción global de proteínas y suprimiéndose la expresión de ATF4 hasta niveles
basales (Novoa et al, 2001).
Una tercera vía de la UPR involucra a la proteína transmembrana ATF6. Bajo
condiciones de estrés de RE, ATF6 es localizada en el RE asociado a BIP (Shen et al,
2005; Sommer & Jarosch, 2002). Sin embargo, bajo condiciones de estrés de RE ATF6 es
translocada al aparato de Golgi (Chen et al, 2002; Shen et al, 2002), en donde es activado
mediante una proteólisis efectuada por las proteasas SP1 y SP2 (del inglés site 1
protease y site 2 protease) (Haze et al, 1999). Este procesamiento tiene como
consecuencia la liberación al citoplasma del fragmento citosólico correspondiente al N-
terminal de ATF6 (ATF6f) y su posterior translocación al núcleo, donde favorece la
expresión de chaperonas residentes del RE (Haze et al, 1999) y genes relacionados con
el sistema de degradación de proteínas asociada al RE o ERAD (del inglés Endoplasmic
Reticulum associated degradation) (Okada et al, 2002), así como la expresión de xbp-1
(Lee et al, 2002; Yoshida et al, 2001) y chop (Okada et al, 2002).
Estas tres vías son activadas bajo diversas condiciones fisiológicas (por ejemplo:
diferenciación de linfocitos B (Iwakoshi et al, 2003) o patológicas (por ejemplo: diabetes,
cáncer o enfermedades neurodegenerativas (revisado en Tabas & Ron, 2011) lo que ha
sido asociado con la transición entre la adaptación y apoptosis. Sin embargo, las cinéticas
de sus respectivas activaciones e inhibiciones difieren drásticamente en condiciones de
estrés de RE crónico. Aunque ha sido descrito que el proceso de atenuación es
secuencial para IRE1α, ATF6 y PERK, el contexto celular parece ser fundamental en las
respectivas cinéticas de activación y atenuación (Lin et al, 2007). Esto puede tener
consecuencias en la restauración de la homeostasis pérdida en el RE, favoreciéndose o
no, la atenuación de la condición de estrés mediante un proceso de adaptación celular.
En resumen, como consecuencia de la activación de los sensores de estrés
XBP1s y ATF6f, se regulan genes relacionados con chaperonas residentes del RE,
componentes del ERAD, síntesis de lípidos, tamaño del RE y aparato de Golgi, con el
consecuente aumento de la capacidad de plegamiento y degradación de proteínas (Figura

28
1). Por otra parte, ATF4 controla positivamente la expresión de genes que modulan el
estado redox y el metabolismo, así como foldasas, generando un medio-ambiente
favorable para el plegamiento oxidativo de proteínas (Figura 1). Colectivamente, estos
tres procesos reducen la cantidad de proteínas mal plegadas al interior del RE
favoreciendo la restauración de la homeostasis celular perdida, es decir forman parte de
una respuesta adaptativa (Woehlbier & Hetz, 2011). Sin embargo, bajo determinados
contextos fisiológicos y patológicos, por ejemplo cuando el estrés de RE es fuerte y
crónico, la UPR puede ser insuficiente para restablecer la homeostasis pérdida,
desencadenando eventos que favorecen la eliminación de las células que han sufrido un
daño irreversible, mediante la inducción de muerte celular por apoptosis (ver a
continuación la descripción de apoptosis) (Tabas & Ron, 2011).
Figura1: La Respuesta a Proteínas mal Plegadas. La acumulación de proteínas Induce la activación de los sensores de estrés de RE. Las quinasas PERK e IRE1α dimerizan y se autotransfosforilan, mientras que ATF6 es translocado al aparato de Golgi. Posterior a su fosforilación y dimerización, IRE1α procesa al ARNm del factor transcripcional xbp-1, mediante su actividad endonucleasa y degrada el ARNm de proteínas difíciles de plegar. PERK fosforila al factor iniciador de la traducción eIF2α bloqueando la traducción general de proteínas y favoreciendo la traducción preferencial del ARNm del factor transcripcional
ATF6 procesado
Aparato de Golgi

29
ATF4. Por su parte, el dominio citoplasmático de ATF6 es liberado, mediante una proteólisis efectuada por las proteasas S1P y S2P en el aparato de Golgi. El fragmento liberado es traslocado al núcleo, donde actúa como factor transcripcional. Los factores transcripcionales en conjunto, activan la expresión de un grupo de genes relacionados. Esta respuesta a proteínas mal plegadas o UPR contrarresta el estrés de RE producido, aumentando la expresión de genes relacionados con metabolismo y balance redox, chaperonas, foldadas, la degradación de proteínas asociada al RE, el volumen del RE, trafico vesicular e induciendo la autofagia y la apoptosis (La fiigura fue modificada a partir de una publicada en The Unfolded Protein Response: Integrating stress signals through the stress sensor IRE1a, de los autores Hetz C, Martino F, Rodriguez, and Glimcher LH. Physiological Reviews. 2011).
6.4) La apoptosis
La apoptosis es un tipo de muerte celular programa conservada a través de todos
los metazoos. Es un proceso esencial en la mantención de la homeostasis celular en los
tejidos en desarrollo y a lo largo de la adultez de los organismos multicelulares. A través
de ensayos de ganancia y pérdida de función de genes involucrados en la regulación de
la apoptosis en ratones y estudios genéticos en pacientes, se ha visto que la
desregulación de este proceso puede desencadenar diferentes patologías. Por ejemplo,
una disminución de la apoptosis puede contribuir a la formación de tumores debido a la
inmortalización celular y la replicación descontrolada de la célula, llevando al cáncer o
enfermedades autoinmunes. Por otra parte, el aceleramiento de la apoptosis podría
conducir al desarrollo de enfermedades neurodegenerativas, la inmunodeficiencia y
también a la infertilidad (Youle & Strasser, 2008).
Quizás, la característica más importante, y la cual algunos autores utilizan para
definir la apoptosis, es la activación de unas proteínas con actividad cisteín-proteasas
denominadas caspasas, las cuales se encuentran en las células animales como pro-
enzimas (Youle & Strasser, 2008). Por otra parte, la apoptosis también puede ser
clasificada de acuerdo a características morfológicas, como el redondeo de las células,
retracción de los pseudópodos, una reducción del volumen celular, la condensación de la
cromatina, la fragmentación nuclear, aparición de cambios en la ultra-estructura de los
organélos, generación de estructuras lobulares en la membrana citoplasmática (en inglés
“blebs”). Además, existen características no morfológicas tales como la caída del
potencial mitocondrial, la exposición de fosfatidil serina, una masiva activación de
caspasas y la fragmentación del ADN (Kroemer et al, 2009).

30
Respecto a los mecanismos que pueden promover la apoptosis, esta puede ser
inducida por señales desde la superficie celular mediante receptores activados por
ligando, tales como el receptor de Fas o el receptor del Factor de Necrosis Tumoral (TNF,
del inglés tumour necrosis factor) (vía extrínseca) o por varios agentes genotóxicos, daños
metabólicos o señales transcripcionales (vía intrínseca). En la vía extrínseca, los
receptores de muerte contienen un dominio de muerte intracelular que recluta a diferentes
moleculas adaptadoras, lo que induce la formación de un complejo denominado DISC (de
sus siglas en inglés Death-Inducing Signalling Complex). El DISC favorece la unión y
activación conformacional de caspasas iniciadoras, como la caspasa-8. Esto inicia una
cascada amplificadora de la señal mediada por la activación por proteólisis de caspasas
efectoras, como caspasa-3, -6 o -7, las que ejecutan la muerte celular. En el caso de la
vía intrínseca, la apoptosis puede ocurrir por estímulos intracelulares como infección viral,
daño del ADN, privación de factores de crecimiento, estrés de RE (ver más adelante) o
activación de programas genéticos, entre otros. Como se verá más adelante, estos
estímulos llevan finalmente a la activación de la caspasa iniciadora, caspasa-9, con la
posterior activación proteolítica de caspasas ejecutoras (Green, 2000).
La vía de apoptosis intrínseca también es conocida como la vía mitocondrial, ya
que los estímulos que causan este tipo de apoptosis llevan a la permeabilización de la
membrana externa de la mitocondria, lo que a su vez produce la liberación de proteínas
como SMAC/DIABLO (del inglés Second Mitochondria-derived Activator of
Caspases/Direct IAP-binding protein with low pI), AIF (del inglés Apoptotic Inducing
Factor) y citocromo c desde el espacio intermembrana de la mitocondria hacia el
citoplasma. En el citoplasma, el citocromo c une a una proteína adaptadora llamada
APAF1 (de las siglas en inglés Apoptotic Protease Activating Factor 1) formando un anillo
heptamérico que recluta a pro-caspasas-9. Este complejo es conocido como apoptosoma
y es esencial para la activación de la apoptosis (Bao & Shi, 2007). La unión de la pro-
caspasa-9 al anillo heptamérico provoca un cambio conformacional en la pro-caspasa-9,
permitiendo su dimerización, generando caspasa-9 activa. Esta caspasa iniciadora, luego
activa directamente a las caspasas efectoras caspasa-3 y caspasa-7 mediante corte
proteolítico (Wang, 2001).
6.5) La Familia BCL-2.

31
Diversas proteínas han sido identificadas como protagonistas en la regulación de
la apoptosis, entre las cuales destacan las proteínas de la familia BCL-2. Los integrantes
de esta familia modulan la activación de las caspasas, proteínas iniciadoras y/o ejecutoras
de la apoptosis. Dentro de la familia BCL-2 se encuentran proteínas antiapoptóticas,
definidas por la presencia de 4 dominios conservados denominados BH1, BH2, BH3 y
BH4 (del inglés BCL-2 homology) y proteínas pro-apoptóticas, las cuales pueden ser
subdivididas en aquellas que presentan multidominios homólogos a BH1, BH2 y BH3 (por
ejemplo BAX y BAX) o miembros que solo poseen el dominio BH3, denominados “BH3-
only” (como BIK, BIM, PUMA y NOXA). La activación de las proteínas BAX y BAK es
esencial para inducir la apoptosis, ya que en ellas convergen múltiples estímulos pro-
apoptóticos intrínsecos. Por otra parte, las proteínas BH3-only actúan río arriba de BAX y
BAK promoviendo su activación mediante un cambio conformacional, proceso que es
inhibido por las proteínas antiapoptóticas como BCL-2 y BCL-XL (Wei et al, 2001). La
activación de BAX y BAK se asocia a sus homo-oligomerizaciones, proceso que precede
a la permeabilización de la membrana mitocondrial y la posterior liberación de proteínas
mitocondriales hacia el citoplasma, como citocromo c, la cual gatilla la activación de
caspasas provocando la muerte celular (Danial & Korsmeyer, 2004).
Proteínas de los tres subgrupos de la familia BCL-2, también se localizan en la
membrana del RE (Oakes et al, 2006). Se ha propuesto que la familia BCL-2 regula en
este organelo distintas respuestas a condiciones de estrés (Hetz & Glimcher, 2008).
Algunas proteínas pro-apoptóticas BH3-only son fuertemente inducidas bajo condiciones
de estrés de RE, lo que esclarece en parte, el mecanismo por el cual se gatilla la
apoptosis en condiciones de daño al RE (Li et al, 2006; Puthalakath et al, 2007). Por otra
parte, proteínas de la familia BCL-2 también participan en el control de la homeostasis del
Ca2+ (Rong & Distelhorst, 2008) y en la regulación directa de la vía de IRE1α (Hetz et al,
2006).
6.6) La Apoptosis y la UPR
Previamente se comentó que un estrés de RE prolongado puede desencadenar la
apoptosis. Este proceso ha sido asociado directamente a una activación sostenida de
IRE1α y CHOP en determinados tipos celulares bajo ciertas condiciones fisiológicas o
patológicas. En un contexto fisiológico, la apoptosis inducida por la UPR podría tener

32
como consecuencia la eliminación de aquellas células expuestas al estrés de RE de
manera de seleccionar aquellas células adaptadas a la condición de estrés. La
eliminación de las células dañadas mediante apoptosis, tiene como consecuencia la
evasión de procesos inflamatorios asociados a muerte no-apoptótica, dado que la
apoptosis se caracteriza por una eficiente fagocitosis, lo cual previene la necrosis tardía
secundaria a la apoptosis, con la consecuente respuesta antiinflamatoria (Woehlbier &
Hetz, 2011).
En contraste con el escenario fisiológico donde la apoptosis es limitada y selectiva,
el estrés de RE crónico puede inducir una apoptosis patológica generalizada. La
apoptosis inducida por un estrés de RE no resuelto comienza a ser reconocido en la
literatura como un importante factor patogénico en un gran número de enfermedades,
entre las que se incluyen las enfermedades neurodegenerativas, diabetes, aterosclerosis
y enfermedades renales (Tabas & Ron, 2011).
Existen efectos no transcripcionales regulados por la vía de IRE1α, los cuales han
sido reconocidos como parte de la respuesta adaptativa a una condición de estrés de RE.
Sin embargo, una activación prolongada de IRE1α podría promover la apoptosis. Algunos
estudios en cultivos celulares expuestos a estrés de RE han identificado que IRE1α puede
asociarse a la proteína TRAF2. TRAF2 interactúa con la pro-caspasa12 y promueve
reclutamiento y su activación en respuesta al estrés de RE, fenómeno que podría ligar la
vía de IRE1α con inducción de apoptosis. Sin embargo, la participación de caspasa 12 en
la inducción de apoptosis es contradictoria. Por otra parte, la formación del complejo
IRE1α/TRAF2/ASK1 regula la activación de JNK (Urano et al, 2000a; Urano et al, 2000b),
lo cual también podría inducir apoptosis (Li et al, 2011a; Xia et al, 1995), de una manera
análoga a la que ha sido descrita para el receptor de muerte de TNFα. La fosforilación de
JNK podría tener como consecuencia la activación de BAX y BAK, mecanismos por el
cual podría ser amplificada la señal pro-apoptótica de IRE1α (Woehlbier & Hetz, 2011).
Sin embargo el mecanismo que relacione esos dos fenómenos no ha sido esclarecido.
Por otra parte, la inducción de CHOP mediada por un estrés de RE prolongado,
suprime la expresión de BCL2, lo cual ha sido asociado al efecto pro-apoptótico de
CHOP, presumiblemente mediante la interacción con uno o más factores transcripcionales

33
que reprimen la transcripción de bcl-2. Además, CHOP regula el aumento transcripcional
de bim, así como otras proteínas BH3-only pueden ser reguladas tanto
transcripcionalmente como no transcripcionalmente bajo condiciones de estrés de RE,
favoreciendo en última instancia, la activación de BAX y BAK, con la consecuente
inducción de apoptosis (revisado en Tabas & Ron, 2011).
Por otra parte, el estrés de RE también puede inducir muerte celular mediante un
mecanismo que involucra cambios en la dinámica del Ca2+, puesto que se ha observado
que en condiciones de estrés de RE existe una desregulación del Ca2+ contenido en el
lumen del RE, observándose un aumento en la concentración de Ca2+ citoplasmático.
Esto último da cuenta de la activación de caspasas y otras proteasas, como las calpaínas,
que se activan en respuesta a diferentes concentraciones de Ca2+ citoplasmático y que se
han reportado como moduladores de la caspasa-12 y de la cascada ASK-1/JNK (Tan et
al, 2006). Se ha observado, además, que la inducción de estrés del RE con agentes
farmacológicos estimulan la proteólisis y activación de caspasa-12 por reclutamiento a la
membrana del RE e interacción con la caspasa-7 (Rao et al, 2001).
Otro mecanismo que liga las señales de Ca2+ provenientes del RE con la muerte
inducida por estrés de RE, fue recientemente descrita en un estudio que demostró que la
inducción de la muerte inducida por CHOP es dependiente de Ca2+, mediante un
mecanismo que involucra la activación de CaMKII (Timmins et al, 2009). Además, también
se describió que CHOP podría regular la apoptosis mediante el control transcripcional de
ERO1 (de sus siglas en ingles ER oxidase 1), regulando la liberación de Ca2+ desde el RE
a través del receptor de IP3 (IP3-R), el cual es crucial en los efectos del Ca2+ en los
procesos de muerte celular (Li et al, 2009). Por otra parte, un reciente estudio demostró
que mediante un mecanismo dependiente de BIP, IP3-R1 puede modular la muerte
celular inducida en neuronas expuestas a estrés de RE y en un modelo de enfermedad de
Hungtinton (Higo et al, 2010). Sin embargo el papel de IP3-R en la muerte inducida por
estrés de RE aun no ha sido completamente descifrado.

34
Figura 2: Apoptosis inducida por estrés de RE. El estrés de RE induce la activación de distintas señales que pueden desencadenar apoptosis. PERK induce la expresión de ATF4 que en conjunto con ATF6 controla la expresión de chop. CHOP puede inhibir la expresión de bcl-2, así como inducir la expresión de las proteínas pro-apoptóticas BH3-only: bim, puma y noxa. A su vez, la activación de caspasa-2 puede activar, mediante un procesamiento proteolítico a la proteína BH3-only BID, la cual, en conjunto con NOXA, PUMA y BIM pueden inducir la oligomerización de BAX y BAK en la mitocondria, lo que favorece la salida de citocromo c, con la consecuente activación de caspasas e inducción de apoptosis. Paralelamente, TRAF2 se asocia a IRE1α, lo que favorece el reclutamiento de ASK1 y la activación, mediante fosforilación, de JNK. JNK induce la activación de BAX y BAK. Por último, bajo condiciones de estrés de RE, IP3R favorece la salida de Ca2+, proceso el cual es regulado por BCL-2 y que podría ser regulado por proteínas de la familia TMBIM (La fiigura fue modificada a partir de una publicada en The Unfolded Protein Response: Integrating stress signals through the stress sensor IRE1a, de los autores Hetz C, Martino F, Rodriguez, and Glimcher LH. Physiological Reviews. 2011).
6.7) TMBIM6/BI-1
De manera similar a las proteínas de la familia BCL-2, Bax Inhibitor-1 (BI-1) o
también llamado TMBIM6 (del inglés Tansmembrane BAX inhibitor motif containing 6)
ha sido relacionado funcionalmente con BCL-2, sin poseer una homología de secuencia
primaria aparente con los dominios conservados de la familia BCL-2. TMBIM6/BI-1

35
interactúa funcional y estructuralmente con BCL-XL y BCL-2 (Xu & Reed, 1998). A
continuación, se describen los antecedentes más relevantes sobre TMBIM6/BI-1 y de
proteínas que han sido identificadas como homólogos de secuencia de TMBIM6/BI-1.
TMBIM6/BI-1 fue el primer integrante descrito de una familia de proteínas con
destinación a membranas y con aparente actividad antiapoptótica. TMBIM6/BI-1 es una
proteína con 6 a 7 dominios hipotéticos para segmentos transmembranas, localizada en el
RE (Xu & Reed, 1998). TMBIM6/BI-1 se identificó inicialmente como “TEGT” (testis
enhanced gene transcript) (Walter et al, 1995) y posteriormente, en un estudio que
buscaba identificar posibles genes inhibitorios de la apoptosis inducida por la
sobrexpresión de Bax humano en levadura. En ese estudio, la co-expresión de
TMBIM6/BI-1 rescató del efecto letal de BAX, en forma similar a lo observado por la
sobreexpresión de BCL-2 (Xu & Reed, 1998). Luego en diversos estudios en células
humanas se demostró que la sobreexpresión de TMBIM6/BI-1, al igual que BCL-2,
protege frente a la muerte inducida por privación de nutrientes, daño al DNA, estrés de RE
y estrés oxidativo (vías intrínsecas) (Chae et al, 2004; Xu & Reed, 1998). Sin embargo,
TMBIM6/BI-1 no afectaba la apoptosis activada por receptores de muerte como Fas y
TNF-α (Xu & Reed, 1998). El análisis filogenético de la secuencia de TMBIM6/BI-1
estableció que esta proteína es altamente conservada en diferentes especies; incluyendo
levaduras, plantas e insectos (Chae et al, 2004; Reimers et al, 2008), sugiriendo que su
efecto antiapoptótico podría estar conservado en la evolución, desde organismos
unicelulares a complejos organismos multicelulares.
6.8) TMBIM6/BI-1 y el estrés de RE.
Estudios in vivo utilizando ratones deficientes para TMBIM6/BI-1 mostraron que
esta proteína es un importante regulador de la apoptosis iniciada bajo condiciones de
estrés en el RE (Bailly-Maitre et al, 2006; Chae et al, 2004). Asimismo, líneas celulares de
fibroblastos embrionarios de ratón (MEFs) carentes de TMBIM6/BI-1 presentaron una
mayor susceptibilidad a la apoptosis inducida por drogas como tunicamicina (Tm), la cual
gatilla estrés por inhibición de la glicosilación de proteínas (Chae et al, 2004).
Posteriormente, se describió que ratones deficientes en TMBIM6/BI-1 presentan hiper-
activación de la vía de IRE1α en respuesta a estrés de RE, tanto en el hígado como en
riñones de ratones sometidos a isquemia y reperfusión (Bailly-Maitre et al, 2006),

36
sugiriéndose que TMBIM6/BI-1 podría poseer una actividad inhibitoria de IRE1α. En esta
línea, nuestro laboratorio describió que proteínas pro-apoptóticas como BAX y BAK
también regulan a IRE1α mediante interacción directa (Hetz et al, 2006). Nuestro
laboratorio también ha caracterizado la respuesta frente a estrés de RE en células MEFs
carentes en TMBIM6/BI-1, observándose una mayor sensibilidad tanto en la activación de
la UPR en respuesta a estrés de RE como a la apoptosis (Lisbona et al, 2009).
Posteriormente, esto fue corroborado por otro grupo in vivo, en donde se asocio la
inhibición de IRE1α con la disminución de obesidad y inducida por resistencia a la
insulina y tolerancia a la glucosa (Bailly-Maitre et al, 2010). Paradójicamente, ambas
actividades parecen oponerse parcialmente, dado la correlación propuesta entre la
activación de IRE1α y la sobrevida celular (Lin et al, 2007). En concordancia con estos
hallazgos, resultados de nuestro laboratorio, sugieren un papel dual para TMBIM6/BI-1
respecto a su actividad antiapoptótica y capacidad para modular la UPR mediante
inactivación de IRE1α, siendo actividades independientes entre sí (Bailly-Maitre et al,
2010; Bailly-Maitre et al, 2006; Lisbona et al, 2009). Esta aparente capacidad de
TMBIM6/BI-1 de modular la activación de la UPR y sobrevida celular en respuesta al
estrés de RE, también es compartida con múltiples proteínas de la familia Bcl-2 (Hetz &
Glimcher, 2008). Sin embargo, se desconoce si otras proteínas de la familia de
TMBIM6/BI-1 presentan estas características
Por otra parte, la sobreexpresión de TMBIM6/BI-1, al igual que BCL-2 y BCL-XL,
disminuye los niveles de Ca2+ luminal del RE (Xu et al, 2008) así como la cantidad de
Ca2+ liberado desde el RE en respuesta a estímulos (Chae et al, 2004; Westphalen et al,
2005; Xu et al, 2008). Un estudio mostró que TMBIM6/BI-1 controla la homeostasis de
Ca2+ mediante la misma vía regulada por BCL-2 y BCL-XL, donde TMBIM6/BI-1 se ubica
como un modulador río abajo de BCL-2 (Xu et al, 2008). Se ha postulado que la
regulación del Ca2+ mediada por la familia BCL-2 y TMBIM6/BI-1 impactan
específicamente en la muerte celular inducida por la liberación de Ca2+ debido a su efecto
directo sobre la mitocondria (revisado en (Henke et al, 2011), ya que la sobrecarga de
Ca2+ en la mitocondria estimula la liberación del citocromo c mediada por el poro de
transición permeable y la posterior activación de las caspasas (Pinton et al, 2008).

37
Los estudios in vivo aportan una aproximación más fisiológica de la función de
genes, en particular en el contexto de un organismo complejo. Bajo esta premisa se
evalúo el efecto de la sobreexpresión de TMBIM6/BI-1 (dTMBIM6/BI-1) en Drosophila
melanogaster (D.mel). Larvas de moscas que poseen una mutación puntual que afecta la
expresión de dBI-1, resultó en la sobreexpresión de dTMBIM6/BI-1. En este sistema, se
evalúo la actividad de la vía IRE1α en larvas crecidas en presencia de Tm. En este
ensayo se observó una clara disminución del splicing del ARNm dxbp-1 en el tejido
larvario frente a condiciones de estrés en el RE (Lisbona et al, 2009). La correlación entre
el aumento de TMBIM6/BI-1 y disminución de la vía dIRE1α/XBP-1, permite establecer
otro vínculo entre TMBIM6/BI-1 y esta vía.
6.9) Las proteínas de la familia TMBIM: nuevos reguladores de la muerte celular?
Diversos análisis bio-informáticos han identificado posibles homólogos para
TMBIM6/BI-1 en mamíferos (Hu et al, 2009; Reimers et al, 2007; Reimers et al, 2008;
Zhou et al, 2008) y plantas (Huckelhoven, 2004), los cuales constituyen una familia
putativa de proteínas conservadas que incluyen: TMBIM6/BI-1, Lifeguard (LFG/TMBIM2),
Growth-Hormone Inducible Transmembrane Protein (GHITM/TMBIM5), Responsive to
Centrifugal force and Shear tress gene 1 (RECS1/TMBIM1), Golgi Anti-Apoptotic Proteín
(GAAP/TMBIM4) y Glutamate Receptor, Ionotropic, N-methyl D-asparate-Associated
protein 1 (GRINA/TMBIM3). La familia de proteínas TMBIM, comparte entre sí un dominio
denominado APF0005, altamente hidrofóbico que posee secuencias de destinación a
membranas. Sin embargo, la localización subcelular de los integrantes de la familia
TMBIM parece no ser compartida entre estos. Por ejemplo, TMBIM6/BI-1 (Xu & Reed,
1998) y TMBIM4/GAAP (Gubser et al, 2007) se identificaron como proteínas de la red RE-
Golgi; TMBIM2/LFG se expresa principalmente en la capa citoplasmática, en el RE y en la
membrana perinuclear (Gubser et al, 2007); TMBIM1/RECS1 se encuentra asociada a
lisosomas y membrana citoplasmática (Zhao et al, 2006b) y TMBIM5/GHITM se describió
como una proteína asociada a la membrana interna mitocondrial (Oka et al, 2008).
La capacidad de formar complejos con proteínas de la familia BCL-2, ha sido
estudiada sólo para TMBIM6/BI-1 y TMBIM2/LFG. Mediante co-inmunoprecipitación y
cross-linking se determinó que TMBIM6/BI-1 interactúa físicamente con BCLXL y BCL-2,
pero no con BAX o BAK (Xu & Reed, 1998). Contrariamente a lo establecido para

38
TMBIM/6BI-1, TMBIM2/LFG parece interaccionar con BAX y BAK, pero no con BCL-XL
(Hu et al, 2009). La probable interacción del resto de la familia TMBIM con proteínas de la
familia BCL-2 queda aún por ser determinada. Aunque las proteínas de la familia TMBIM
se definieron como homólogas de secuencia primaria, poco se sabe de su homología
funcional en términos de posibles propiedades antiapoptóticas. Por ejemplo, la
sobreexpresión de TMBIM2/LFG protege de la muerte inducida por el ligando FasL en
diferentes modelos celulares (Fernandez et al, 2007; Somia et al, 1999). La
sobreexpresión de TMBIM4/GAAP promueve la sobrevida celular frente a la
sobreexpresión de BAX o la exposición de células a ceramidas, estaurosporina o
cisplatino (Gubser et al, 2007). En el caso de TMBIM6/BI-1, su actividad antiapoptótica
protege a las células frente a un amplio espectro de estímulos deletéreos como Tm,
estaurosporina (Est), etopósido (Eto), doxorrubicina, privación de nutrientes, hipoxia y la
sobrexpresión de BAX (Chae et al, 2004; Xu & Reed, 1998). Por otra parte, aún se
desconoce si TMBIM1/RECS1, TMBIM5/GHITM o TMBIM3/GRINA presentan homología
funcional con TMBIM6/BI-1 en términos de la promoción de la sobrevida celular,
modulación de la UPR o actividades sobre la homeostasis del Ca2+.
Figura 3: Proteínas de la familia TMBIM: El árbol filogenético muestra los miembros descritos para la familia TMBIM en humano. TMBIM1 también ha sido nombrado como RECS1; TMBIM2, como LFG; TMBIM3, como GRINA; TMBIM4 como GHTIM; TMBIM5 como GAAP y TMBIM6 como BI-1. Además, se muestra la proteína putativa del gen Yn1305c de levadura, la cual ha sido sindicada como un putativo ancestro en común para toda la familia TMBIM.
Inicialmente un trabajo identificó a TMBIM3/GRINA como una proteína asociada al
receptor de NMDA (Kumar et al, 1991), lo que hace presumir que se expresa en el
TMBIM6TMBIM5
TMBIM4
TMBIM1TMBIM3
TMBIM2
Yn1305c

39
sistema nervioso. Recientemente, esto fue corroborado en un trabajo donde se describió
que TMBIM3/GRINA se expresa principalmente en el sistema nervioso central, con un alto
grado de expresión en el hipocampo. Además, mediante estudios de inmunofluorescencia
se demostró que TMBIM3 colocaliza con GM130 (marcador de aparato de Golgi) y
parcialmente con BIP (RE) (Nielsen et al, 2011). En este trabajo, se describió que la
generación del ratón deficiente en tmbim3 no se manifiesta en un fenotipo evidente
respecto a la supervivencia o letalidad del animal. En otro trabajo, recientemente se
describió que la sobreexpresión de un fragmento de TMBIM3 inhibe la muerte inducida
por la expresión de una toxina, mediante la inhibición del tránsito desde el RE al aparato
de Golgi de su receptor Globotriaosylceramida (Gb3) (Yamaji et al, 2010). Sin embargo, la
expresión de la proteínas completa no presentó la misma actividad del fragmento más
pequeño, pero sí otros miembros de la familia TMBIM.
Un resultado indirecto podría vincular a TMBIM3/GRINA y el estrés de RE. En un
modelo de retinitis pigmentosa, el cual involucra respuestas a estrés de RE (Lin et al,
2007), se determinó un incremento en los niveles de microRNAs (miR-96, miR-183, miR-
1, miR.133), entre los cuales TMBIM3/GRINA figura como potencial blanco (Loscher et al,
2007). TMBIM3/GRINA es la proteína de la familia TMBIM menos estudiada y posee un
grado de similitud e identidad semejante a los que presentan todos los miembros de la
familia TMBIM. TMBIM3/GRINA tiene un perfil de hidrofobicidad similar al de TMBIM3,
con 6 a 7 dominios putativos para transmembrana cercanos al extremo C-terminal,
basado en criterios de análisis Kyte-Doolittle para hidrofobicidad (Kyte & Doolittle, 1982)
con un dominio N-terminal más largo que TMBIM6/BI-1.
En mamíferos, TMBIM3 es una proteína conservada, con un altísimo grado de
identidad en mamíferos. Nuestros análisis filogenéticos muestran la presencia de
proteínas hipotéticas en gusano (Bombix mori), el pez zebra (Danio rerio), xenopus
(Xenopus leavis y Xenopus tropicales) y la mosca (D. melanogaster), con niveles de
conservación de secuencias similares al grado de identidad que presenta TMBIM6 en
diferentes especies.
Cabe destacar que el alto grado de identidad y similitud que conservan
hTMBIM6/BI-1 y hTMBIM3/GRINA en múltiples especies. Por ejemplo, en D.
melanogaster corresponden a 19 y 40% de identidad, y 34 y 55% de similitud,

40
respectivamente. Por otra parte todos los miembros de la familia TMBIM están presentes
en D. melanogaster o en pez zebra, lo que hace presumir su relevancia en la biología
celular. Esto último contrasta fuertemente con el hecho de que solo existen 2 miembros
de la familia BCL2 descritos en D.melanogaster. Esto sumado a la presencia funcional,
recientemente descrita, de la vía IRE1α/XBP-1 en D. melanogaster (Plongthongkum et al,
2007; Souid et al, 2007) y al papel descrito para TMBIM6/BI-1 como inhibidor de la vía
IRE1α/XBP-1 (Lisbona et al, 2009), hace posible proponer el uso D. melanogaster como
un buen modelo para estudiar la función de esta familia de proteínas Otro modelo in vivo
que posee grande ventajas, dado la fácil manipulación genética y la rapidez en el
desarrollo son los estudios en pez zebra, vertebrado que presenta los 5 miembros de la
familia TMBIM, sin embargo, ni uno ha sido estudiado.
Figura 4: TMBIM3/GRINA es una proteína conservada. Las proteínas correspondiente a los homólogos putativos de TMBIM3/GRINA en pez zebra (D.rerio), Drosophila melanogaster (D.mel), ratón (M.mus) y humano (H.sapiens) fueron alineados con el programa ClustalW. Junto con el nombre, entre paréntesis, se indica el número de aminoácido con el que se inicia cada fila, al igual que arriba de cada fila grupal, el cual es indicado en azul. La figura muestra en letra blanca y fondo negro los aminoácidos

41
idénticos conservados, letra negra y fondo gris los aminoácidos similares y en letra gris y fondo blanco los aminoácidos no conservados entre las especies analizadas.
A la fecha, no se ha estudiado en profundidad si TMBIM3/GRINA posee actividad
antiapoptótica frente a estímulos de apoptosis intrínseca o extrínseca, así como la
regulación de su expresión o su relevancia en el control de la apoptosis in vivo. Además,
aún se desconoce si TMBIM3/GRINA puede regular la homeostasis del Ca2+, tal como ha
sido descrito para otros miembros de la familia TMBIM. El probable control sobre la
homeostasis del Ca2+ podría dar cuenta de un probable mecanismo que explicará el
efecto antiapoptótico de los miembros de la familia TMBIM y específicamente de TMBIM3.
Mediante estrategias genéticas, en esta tesis se desarrollaron distintos modelos celulares
y animales, para evaluar la posible actividad antiapoptótica de TMBIM3 y su interacción
con TMBIM6, así como su regulación transcripcional y efecto en la homeostasis del Ca2+,
de manera de responder la siguiente hipótesis.

42
7. Hipótesis
La hipótesis propuesta para este trabajo fue: “TMBIM3/GRINA es regulado por la
Respuesta a Proteínas Mal plegadas que inhibe la apoptosis inducida por estrés de
retículo endoplasmático mediante el control de la homeostasis del Calcio”.
8. Objetivo General
El objetivo general de este trabajo es estudiar si TMBIM3/GRINA es un blanco de la
Respuesta a Proteínas Mal plegadas que presenta actividad antiapoptótica frente a estrés
de RE, así como su posible papel en el control de la homeostasis del Ca2+ in vitro e in
vivo.
9. Objetivos específicos:
1) Determinar si TMBIM3 tiene actividad antiapoptótica frente a condiciones de estrés de
RE.
2) Estudiar la posible regulación de la expresión de TMBIM3 frente a estrés de RE.
3) Evaluar si TMBIM3 puede regular las señales de Ca2+ desde el RE, así como el rol del
Ca2+ en la muerte inducida por estrés de RE.
4) Estudiar la probable función de TMBIM3 en modelos de estrés de RE in vivo usando
D.melanogaster y pez zebra.

43
10. Materiales y Métodos.
Materiales
Tunicamicina, tapsigargina, estaurosporina, etopósido, actinomicina D y TNFα
fueron adquiridos en Calbiochem EMB Bioscience Inc. El medio de cultivo, sueros fetales
bovinos y antibióticos se obtuvieron en Life Technologies (Maryland, USA). Las sondas
fluorescentes Hoechst, Brefeldina A-BODIPY, y anticuerpos secundarios acoplados a
fluoróforos ALEXA se adquirieron en Molecular Probes. Anticuerpos fluorescentes anti-IgD
y anti-IgM se adquirieron en BD Biosciences.
Cultivo celular y construcciones de ADN
Se generaron células MEFs SV40 y se mantuvieron en cultivo en medio DMEM
suplementado con L-glutamina 1X, aminoácidos no esenciales, penicilina y 5% de suero
fetal bovino, tal como se describió previamente (Cheng et al., 2001; Xu and Reed, 1998).
La proteína BI-1 de humano se clonó en el vector pcDNA3.1 y se fusionó al péptido HA
como se describió con anterioridad (Xu and Reed, 1998; Chae et al., 2003). La proteína
TMBIM3 de ratón se obtuvo desde una biblioteca de ADNc y se clonó en el vector
pCR3.1, y se marcó con el péptido MYC como se describió previamente (Hetz et al.,
2006). Luego, los ADNc fueron sub-clonados en el vector retroviral pMSCV puro
(Clontech).
Las células MEFs deficientes en PERK e IRE1α (Harding et al, 2000b) y sus
respectivos controles fueron obtenidas del laboratorio de David Ron.
Adicionalmente, generamos células MEFs con niveles reducidos, estables y
transitorias, de ARNm de tmbim3 mediante la herramienta de silenciamiento de la
expresión de ARNm, shARN (sh del inglés: short hairping), utilizando el sistema de
expresión lentiviral en el vector pLKO.1, como se describió previamente (Lisbona et al,
2009). Como control del silenciamiento del gen de tmbim3, se utilizó un shARN destinado
contra la secuencia de la luciferasa (Lisbona et al, 2009). Las construcciones para
silenciamiento de ARN, mediante shRNA, fueron generados por The Broad Institute
(Boston, USA) basados en distintos criterios de diseño de shRNA (ver

44
http://www.broad.mit.edu/genome_bio/trc/rnai.html). Se escogieron 5 construcciones de
las generadas y se seleccionaron los dos más eficientes mediante análisis por PCR en
tiempo real (shARN#1y shARN#2). Éstos, correspondieron a las secuencias blanco contra
Tmbim3: 5´-GCTGCATTTCTGTGCCACCTT-3’ y ´5’-CTGGACCATCATTGTCTCCTA-3’.
Inmunofluorescencia indirecta
Las células MEF silvestres fueron sembradas en cubreobjetos de vidrio a una
concentracion final de 2,0 x 105 en placas de 6 pocillos y fueron cultivados en condiciones
normales. 24 h despúes, las células fueron transfectadas con vectores de expresión, ya
sea para TMBIM3-MYC o para TMBIM6-MYC. Luego de 48 h las células fueron incubadas
con Brefeldina-A conjugada al fluoróforo BODIPY 558/568 (3,5 µM) por 45 min a 37ºC y
5% de CO2. A continuación, las células se fijaron con paraformaldehído (4% p/v en PBS,
20 min), se permeabilizaron (PBS 0,3% v/v Tritón X-100, 30 min) y se bloquearon (3% p/v
BSA en PBS, 1 h). Este procedimiento se realizó sobre hielo a 4°C con lavados entre
cada paso, utilizando PBS frio. Posteriormente, las células se incubaron con el primer
anticuerpo, anti-MYC (1:300), diluido en PBS-2% BSA por 1h a 37º C. Luego, las células
se lavaron y se incubaron con el segundo anticuerpo IgG de ratón conjugado a Alexa 488
(1:400), diluidos en PBS 2% BSA y protegidos de la luz. Los núcleos fueron teñidos con
Hoechst por 10 min a Tº ambiente. Finalmente, a los cubreobjetos se les agregó 10 µL de
medio de montaje anti apagamiento (DAKO) para extender el tiempo de decaimiento de
los fluoróforos. Las muestras así tratadas se observaron en microscopio confocal.
Ensayos de viabilidad celular
Se sembraron 2,0 x 104 células en placas de 24 pocillos y se mantuvieron por 24 h
en medio de cultivo DMEM suplementado con suero fetal bovino al 5% y aminoácidos no
esenciales. Se indujo estrés de RE agregando los agentes estresores del RE (Tm y Tg) a
las células en diferentes concentraciones y se mantuvieron por 24 h.
Para inducir privación de nutrientes, el medio de cultivo se reemplazó por EBSS
(Earle's Balanced Salt Solution) o RPMI, sin glucosa y sin suero, y se incubaron por 36,
24, 18, 12 ó 6 h según se indica. A continuación, se monitoreó la viabilidad celular usando
tinción con yoduro de propidio (PI) y análisis por citometría de flujo (FACS Canto A, BD),
como se describió con anterioridad (Hetz et al, 2002a). Para el análisis de las células

45
“hipo diploides” o “Sub-G1”, se incubaron las células por 24 h con los agentes inductores
de estrés de RE. Posteriormente, las células se tripsinizaron, y se fijaron y
permeabilizaron con metanol. Luego, las células se tiñeron con PI y se cuantificó la
población “Sub-G1” mediante análisis de citometría de flujo, como se describió
previamente (Hetz et al, 2002b). En paralelo, la apoptosis fue estimada mediante la
morfología nuclear, la cual se analizó por tinción con Hoechst 333342 (Molecular Probes)
y microscopía de epifluorescencia.
Para los experimentos de re-plaqueo de células, se sembraron 3,0 x 105 células en
placas de 3,5 cm y se trataron por 4 h con Tm 1 µg/mL. A continuación las células se
lavaron con PBS y fueron tripsinizadas. Luego, se sembraron 2 x 105 células MEFs en
placas de 10 cm y se cultivaron por 5 días en medio DMEM normal, suplementado con
suero al 5% y aminoácidos no esenciales. Cinco planos diferentes de cada condición
fueron fotografiados a los 2, 4 y 5 días mediante microscopía de luz y contraste de fase.
Luego, de los cinco días de cultivo en ausencia de Tm, las células cultivadas en las placas
se lavaron 3 veces con PBS, se fijaron con PFA 4% por 30 min a temperatura ambiente y
se tiñeron con cristal violeta por 30 min. Luego se lavaron 3 veces con agua destilada. Las
placas se dejaron secar y luego se escanearon.
Análisis por Western blot
Las células se colectaron en la solución tampón RIPA (Tris 20 mM [pH 8,0], NaCl
150 mM, SDS 0,1%, DOC 0,5%, y Tritón X-100 0,5%) suplementada con una mezcla de
inhibidores de proteasas (Roche, Basel, Switzerland). Luego, las muestras se
homogenizaron por sonicación. La concentración total de proteínas se determinó
mediante el ensayo micro-BCA (Pierce, Rockford, IL) (Hetz et al, 2006). Se cargaron entre
15 a 50 µg de proteínas totales en minigeles denaturantes al 4%, 6%, 8% y 12%,
dependiendo del peso molecular estimado de la proteína a analizar. Los anticuerpos
primarios y sus diluciones respectivas utilizadas, se detallan a continuación: anti-HSP90
1:4000, anti-ATF4 1:2000, anti-CHOP 1:2000, anti-BAX 1:2000, anti-MYC 1:1000, anti-
BCL-2 1:5000 (Santa Cruz), anti-BAK 1:2000 (Upstate Technology), anti-PERK 1:2000,
anti-caspasa-3 activada (Cell Signaling Technology), anti-PDI, anti-ERp57, anti-BiP, anti-
ERp72 1:2000 (StressGene), anti-EDEM 1:2000 (SIGMA), anti-HA 1:5000 (Roche), anti-
SERCA (Affinity Bioreagents), y anti-IP3-R1 y anti- IP3-R3 1:5000 (BD Biosciences). Los

46
anticuerpos secundarios anti-IgG de conejo o ratón (según corresponda) acoplado a la
ezima peroxidasa se utilizaron diluidos 1:3000 (Invitrogen).
Extracción de ARN y análisis mediante RT-PCR
Se extrajo ARN total de células y tejidos utilizando Trizol (Invitrogen, Carlsbad,
CA). El ADNc se sintetizó con SuperScript III (Invitrogen, Carlsbad, CA) utilizando
partidores al azar p(dN)6 (Roche, Basel, Switzerland). Las reacciones de PCR en tiempo
real cuantitativo se realizaron utilizando el agente fluorescente SYBRgreen y el sistema
ABI PRISM7700 (Applied Biosystems, Foster City, CA). Las cantidades relativas de
ARNm se calcularon de los valores de ciclo umbral comparativo utilizando b-actina como
control de amplificación y normalizando los valores del ARNm de interés por el de actina.
Las secuencias de los partidores se diseñaron con el programa Primer Express software
(Applied Biosystems, Foster City, CA) o se obtuvieron de Primer Data bank
(http://pga.mgh.harvard.edu/primerbank/index.html). La mayoría de las reacciones de
PCR en tiempo real fueron descritas previamente (Lee et al., 2005). Adicionalmente,
utilizamos los siguientes partidores: TMBIM3, 5’-CCCTACCCTCAAGGAGGCTAC-3’ y 5’-
CTGGCGAATGTTCTTGTCCC-3’, de ratón; TMBIM3, 5’-GCTTAAGTTGAGGCGCAAAC-
3’ y 5’TTGAAAATCGGGATTCCTTG-3’, de Drosophila melanogaster; TMBIM3 5’-
ATTGTTGGAGTGGATTATGA-3’ y 5’-CAGGTTGTTAATCTGAACAT-3’, de pez cebra;
CHOP, 5’-ACCCTGAATCAGAAGCAGCCG-3’ y 5’-TACGACACGCTCCCACTCCT-3’, de
pez cebra; actina, 5’-ACACGACCCAGAGCATCAGGGAG y 5´-
CCTCTCTTGCTCTGAGCCTCA-3’, de pez zebra.
Para los ensayos de procesamiento del ARNm de xbp-1 en pez cebra se utilizaron
los siguientes partidores: 5´-GTTCAGGTACTGGAGTCCGA-3’ y 5’-
CTCAGAGTCTGCAGGGCCAG-3’.
Clonamiento del gen Tmbim3 de pez zebra (Danio rerio)
El 5’UTR y la parte de la región codificante de tmbim3 del pez cebra (zTmbim3) se
amplificaron por PCR utilizando ADNc de embriones 24 hpf (horas post fertilización). Se
diseñaron los siguientes partidores desde una secuencia conocida de zTmbim3 de pez
zebra (NM_201208):

47
- Sentido: 5´-GGGATGTTTACAGGAAGACGAG-3´.
- Reverso: 5´-ACAAACGCAGTGGTGATGG-3´.
Se clonó el producto de PCR en el vector pCRII-TOPO (Invitrogen, USA) (pCRII-
TOPO-z-tmbim3) y luego se secuenció para comprobar la fidelidad del producto obtenido.
Hibridización in situ e inmunofluorescencia de embriones de pez zebra
Se sintetizó una sonda ARN antisentido contra zTtmbim3, marcada con el
reportero DIG, utilizando la RNA polimerasa T7 y UTPs marcados con DIG (Roche)
usando como templado el pCRII-TOPO-z-tmbim3. La hibridización in situ del embrión
entero se realizó de acuerdo a protocolos estándares para pez zebra (Westerfield 1996).
Después de la tinción, los embriones marcados se montaron en glycerol al 100% y se
analizaron en un microscopio Nikon DIC (Eclipse 80i). Se hicieron ensayos de Caspasa 3
en embriones previamente fijados en paraformaldehído (PAF) al 4% toda lo noche a 4ºC,
y luego se transfirieron a metanol 100% y se dejaron nuevamente toda la noche a 4ºC. Se
utilizó el anticuerpo anti-caspasa 3 activada, concentración 1:200, (Cell signaling) y el
anticuerpo secundario contra conejo conjugado al fluoróforo Alexa-488 1:200 (Molecular
Probes). Para teñir núcleos se utilizó co-tinción nuclear, incubando por 1 h con la tinción
yodada To-Pro-31:1000 (642/661) (Molecular Probes). Los embriones se montaron en
glicerol y se analizaron mediante microscopía confocal en un microscopio Zeiss LSM-140
Axiovert 10.0. Las imágenes se procesaron utilizando el programa Image J
(http://rsb.info.nih.gov/ij/, NIH, USA).
Morfolino contra Tmbim3 de pez zebra
Los morfolinos oligonucleótidos antisentido (MO) fueron diseñados y sintetizados
por Gene Tools (Philomath, OR). Las secuencias son como se describe a continuación: 5’
GCCTGTATCCCTTATTCTGAGACAT-3’ (zTmbim3-MO) y el control 5’-
GCgTcTATCgCTTATTgTcAGACAT-3’ (mal apareamiento de bases, Mis-MO). Las letras
en minúscula indican los cambios de núcleotidos introducidos en la secuencia para
generar el mal apareamiento del morfolino control. La inyección del morfilino antisentido
se hizo a presión en los embriones Tübingen silvestre Tg (HuC::GFP) en estado de una

48
célula (1,5 a 4,5 ng por embrión) utilizando protocolos estándar (Park et al, 2000; Zhao et
al, 2006a).
Síntesis de ARN y ensayo de rescate en pez zebra
Se generaron secuencias de ARN sintético con el sistema T7 mMESSAGE
mMACHINE (Ambion) utilizando como templado el vector TMBIM3-pCT3.1 de ratón, el
cual fue linearizado, previamente con la enzima de restricción XhoI d. El ARNm sentido
(50 pg por embrión) se micro-inyectó en embriones de pez zebra en estado de una célula.
Para experimentos de rescate, los embriones de pez zebra fueron co-inyectados en
estado de una célula con una combinación de 50 pg de ARNm de mTmbim3 + 4,5 ng del
morfolino zTmbim3-MO, ó 50 pg del ARNm de gap43-Cherry-pCS2 + 4,5 ng del morfolino
zTmbim3-MO (o control). Los fenotipos se analizaron a 60-70% de epibolia y 24 hpf y
fueron fotografiados mediante microscopía de luz.
Ensayo de muerte celular in vivo en pez zebra
Para la detección de células apoptóticas in vivo, se inyectaron embriones silvestres
Tübingen en estado de 1 célula, con 1,5 ng del morfolino z-tmbim3-MO o del morfolino
control (mismatch-MO), y se mantuvieron por 24 h en medio E3 a 28,5 ºC. Después de
decorionarse, los embriones se incubaron con medio E3 suplementado con 2 mg/mL de
acridina orange por 30 min a 28,5 ºC, en placas de 24 pocillos (6 embriones por pocillo).
Luego de lavar 8 veces 5 min cada vez con medio E3 (Furutani-Selki M et al, 1996), los
embriones se anestesiaron con tricaína (Sigma-Aldrich) y los resultados se documentaron
mediante microscopía de fluorescencia. Todas las imágenes fueron tomadas exactamente
al mismo tiempo de exposición, ganancia y magnificación. El análisis de la región de
interés (ROI) se realizó en el área de la medula espinal y la cuantificación de la
fluorescencia se llevó a cabo con el programa Image J.
Para demostrar la pérdida de neuronas en el cerebro y la médula espinal, se
inyectaron embriones transgénicos Tg(ngn1::GFP) (Colombo et al, 2006) con 4,5 ng del
morfolino z-tmbim3-MO o morfolino control control-MO. Los embriones se incubaron por
24 h a 28,5 ºC. Para la adquisición de imágenes in vivo los embriones se montaron en
agarosa y se analizaron mediante microscopía confocal (Zeiss LSM-140 Axiovert 10.0).

49
Las imágenes fueron procesadas utilizando los programas Image J y Volocity
(Improvision).
Para evaluar el posible efecto protector de tmbim3 en condiciones de estrés de
retículo endoplásmico, se inyectaron los embriones de pez zebra con 50-100 pg del
ARNm de mTmbim3 o ARNm control (gap43-Cherry-pCS2). A continuación los
embriones se cultivaron en placas de 24 pocillos (6 embriones por placa) a 28,5º C, por 8
y 24 h en medio E3 suplementado con tapsigargina 1-5 µM. Finalmente, las células
apoptóticas se evaluaron en embriones vivos por tinción con acridina orange (8 hpf) o
mediante la determinación de cambios morfológicos en los embriones, asociados a
apoptosis (24 hpf).
Evaluación de toxicidad de Tm en Drosophila melanogaster
Las moscas se mantuvieron a 25º C en medio estándar con ciclos de luz-oscuridad
de 12x12 h. La línea Tub-Gal4/TM3-GFP se obtuvo del Bloomington Drosophila Stock
Center (Bloomington, Indiana). Las líneas UAS-ARNi se obtuvieron del Vienna Drosophila
RNAi Center (VDRC): UAS-TMBIM3RNAi (#28365) y UAS-BI-1RNAi (Dietzl et al, 2007). Para
los experimentos con ARNi, moscas hembra UAS-ARNi se cruzaron con moscas macho
Tub-Gal4/TM3-GFP. Larvas GFP positiva de segundo estadío (que no expresan el ARNi)
se separaron de las larvas GFP negativas (que expresan el ARNi) (Figura 5) y se
alimentaron por separado con Tm o DMSO (vehículo). En los experimentos de
sobrevivencia, un número indicado de larvas de segundo estadío del genotipo indicado
fueron alimentadas con 25 µg/mL de Tm o DMSO en alimento instantáneo de Drosophila
(Carolina Biological Supply 2700 York Burlington, NC). El número total de pupas, moscas
eclosionadas adultas y moscas sobrevivientes después de 5, 10 y 15 días se contaron y
se determinaron los porcentajes respecto del total. Para los experimentos de
inmunohistoquímica, se disectaron larvas en tercer estadío y se marcaron utilizando
protocolos convencionales (Walther & Pichaud, 2006; Wu & Luo, 2006). Brevemente, el
tejido se fijó con paraformaldehído al 4%, preparado en PBS, con Tritón al 0,3% por 30
minutos. A continuación, el tejido se bloqueó en Tritón al 0,3% en PBS y suero de oveja al
0,5% durante 1 hora. El tejido se incubó toda la noche con un anticuerpo anti-caspasa 3
activada 1:3000 (Cell signaling, rabbit) a 4°C, con rotación lenta. Después de lavar, se
agregó el anticuerpo secundario anti-conejo 1:200 conjugado a fluoresceína (Jackson

50
ImmunoResearch, West Grove, PA, USA) y se incubó por 1 a 2 h a temperatura ambiente.
El tejido se montó con DAPI en una cámara y se analizó en un microscopio de spinning
disk.
Figura 5: Sistema de cruzas para obtención de moscas iRNA. Moscas hembra UAS-ARNi, heterocigotas (RNAi/RNAi, a la izquierda) se cruzaron con moscas macho Tub-Gal4/TM3-GFP (Izquierda). Luego, las larvas GFP positiva fueron separadas de las GFP negativas (iRNA) y mantenidas por separado con el fin de estudiar el efecto de la deficiencia del gen de estudio inducido por la expresión del RNAi regulado por la expresión del factor transcripcional Gal4, el cual se expresa bajo control del promotor de tubulina (Tub), de manera de obtener la expresión ubicua del RNAi.
Mediciones de Ca2+ citoplasmático
Las células se crecieron en cubreobjetos de vidrio y luego se cargaron con 1 µM
de la sonda de calcio Fluo-4 AM y se incubaron por 30 min a temperatura ambiente.
Luego se montaron los cubreobjetos en cámaras de 1 mL y se lavaron tres veces con
tampón libre de calcio (NaCl 150 mM, KCl 5 mM, MgCl2 1 mM, HEPES 10 mM, glucosa 10
mM y EGTA 5 mM, pH 7,4). Las señales de fluorescencia se registraron utilizando un
microscopio de fluorescencia invertido IX-81 (DSU, Olympus), equipado con una lámpara
de Xenón de 150-W (Olympus MT-20). La sonda Fluo-4 se excitó y se detectó con un
filtro FITC, utilizando un objetivo de inmersión en aceite a una magnificación de 40x y
apertura numérica 1,4 NA. Los cambios en el calcio citosólico se midieron en el campo
visual completo que contenía entre 15 a 30 células en promedio. Se adquirieron imágenes
cada 5 s y se analizaron utilizando los software Cell R (Olympus) e Image J. Las

51
intensidades promedio de las áreas celulares de interés se colectaron como F(t) y se
sustrajo la intensidad basal, utilizando una misma área de interés en una zona sin células
obteniendo la fluorescencia basal F(t)s. La señal de fluorescencia final se normalizó a la
fluorescencia basal F(0), como [F(t)s − F(0)]/F(0) (Gleason et al, 2004; Varela et al, 2007).
Mediciones de calcio en el retículo endoplásmico
Para mediciones de luminiscencia de Ca+2 se sembraron 5 × 104 células MEFs
silvestres (WT) en cubre-objetos de vidrio de 13 mm. Las células se co-transfectaron con
0,2 µg de la proteína sensora de Ca+2 reticular aequorina-ER (Pinton et al, 1998) y 0,8 µg
de TMBIM3-MYC o control MOCK, utilizando el agente para transfección Lipofectamina
LTX (Invitrogen). En el caso del estudio en las líneas celulares que expresan
establemente TMBIM3-MYC o su control MOCK, se transdujeron con Ad-aequorina-ER.
Luego de 24 h, las células fueron lavadas 3 veces con solución fosfato sin Ca2+, Glucosa
1g/L y 600 µg de EGTA. Luego, se adicionó al medio de lavado Ionomicina 1 µM y las
células se incubaron a 4°C por 5 min. Posteriormente, se agregó al medio celentazina N y
las células fueron incubadas 4°C. Una vez pasados 45 min, las células fueron lavadas 3
veces con solución de fosfato, sin Ca2+, EGTA 100 µM y BSA al 2%. Luego, las células
fueron montadas en el luminómetro y los fotones emitidos antes las distintas soluciones
perfundidas fueron determinados como se describió previamente (Visch et al, 2004). La
secuencia de perfusión fue la siguiente:
1) Solución sin Ca2+ con EGTA 100 µM por 20 s.
2) Solución sin Ca2+ con BSA al 2% por 180 s, EGTA 100 µM 40 s.
3) Solución sin Ca2+ con EGTA 100 µM 40 s.
4) Solución con Ca2+ 10 mM por 120 s.
5) Histamina 100 µM por 40 s.
6) Digitonina + Ca2+ 10 mM 100 s.
Ensayos de flujo de 45Ca2+ unidireccional
Los experimentos se hicieron en células MEFs WT y en aquellas que expresan
establemente TMBIM3-MYC crecidas en monocapas confluentes de células, como se

52
describió previamente (Rong et al, Mol Cell, 2008; Decuypere et al, Cell Calcium 2010). 2
x 105 células se sembraron en las palcas de 12 pocillos y se conservaron por 4 días en
condiciones normales de cultivo. Luego, las células se mantuvieron a 25°C en una placa
con termostato y agitación contante. Posteriormente, el medio de cultivo fue reemplazado
por 10 minutos con una solución compuesta por: KCl 120 mM, imidazole-HCl 30 mM (pH
6,8), MgCl2 2 mM, ATP 1 mM, EGTA 1 mM y saponina 20 µg/ mL. Luego, los reservorios
de Ca2+ no-mitocondriales se cargaron con Calcio radiactivo (45Ca2+) al incubarse las
células por 45 minutos con una solución compuesta por KCl 120 mM, imidazole-HCl 30
mM (pH 6,8), MgCl2 5 mM, ATP 5 mM, EGTA 0,44 mM, NaN3 10 mM y 150 nM de 45Ca2+
libre (28 µCi/mL). A continuación, las células se lavaron dos veces con 1 mL de la
solución de “flujo de salida”, la cual estaba compuesta por: KCl 120 mM, imidazole-HCl 30
mM (pH 6,8) y EGTA 1 mM, más thapsigarguina 4 µM (inhibidor de la bomba de Ca2+
SERCA). Luego, se agregó 500 mL de medio de “flujo de salida” y se reemplazó cada 2
min. Después de 10 min de pasar medio de flujo de salida, se aplicó diferentes
concentraciones de IP3 (pocillos 1 a 10) y 10 mM del ionóforo de Ca+2 A23187 (pocillos 11
y 12) por 2 min. Ocho minutos después, todo el 45Ca+2 remanente en los reservorios, se
liberó incubando con 1 mL de una solución de dodecil sulfato de sodio al 2% (w/v) por
30 min. Las diferencias entre las curvas de respuesta versus concentración se analizaron
mediante ANOVA de dos vías. De manera similar a la experiencia anterior, se realizaron
experimentos para determinar el contenido total de 45Ca2+-reticular entre ambos tipos
celulares. Sin embargo, la solución de “flujo de salida” se perfundio en ausencia de IP3.
Por otra parte, las células se cargaron en presencia de 10 µM del ionóforo de calcio
A23187, para determinar la unión no específica de 45Ca+2. La fuga específica de 45Ca+2
reticular se determinó restando el contenido de 45Ca+2 en presencia de A23187 10 mM al
contenido total de 45Ca+2.
Inmunoprecipitación
Todas las inmunoprecipitaciones se hicieron en tampón CHAPS 1 % (NaCl 150 mM,
KCl 100mM, Tris 50mM) como se describió previamente (Lisbona et al, 2009). En el caso
de las transfecciones transitorias en células HEK 293T, 3 x 105 fueron sembradas en
placas de 35 mm y cultivadas en medio DMEN suplementado con 5% de suero fetal
bovino. Luego de 24 h, las células fueron transfectadas con 2 µg de DNA total (1 µg

53
TMBIM3-MYC y 1 µg de MOCK o 1 µg TMBIM3-MYC y 1 µg de BI-1-HA) utilizando
efectine (Quiagen). Pasadas 48 h, las células fueron lavadas con PBS 1X y
posteriormente, fueron lisadas por 30 min en una solución tampón CHAPS 1% a 4° C.
Después, el lisado celular fue centrifugado por 5 min a 10.000 rpm y el sobrenadante fue
incubado con las esferas de sefarosa conjugadas covalentemente a IgG anti-MYC por 24
h a 4°C. Posteriormente, las esferas de sefarosa fueron centrifugadas, el sobrenadante
descartado, y se procedió a lavar las esferas con solución tampón CHAP 1% por tres
veces. Luego, se eluyeron los complejos proteicos inmunoprecipitados en 30 µL de
solución tampón CHAPS 1%, más péptido MYC 0,66 mg/mL por 30 min a T° ambiente,
seguidos de 5 min a 37°C y por último, 5 min a 4°C. Una vez finalizado esto, se procedió a
centrifugar las esferas de sefarosa por 2 min a 2000 r.p.m. El sobrenadante fue retirado,
descartando las esferas de sefarosa. Luego cada muestra fue analizada mediante
western blot.
Expresión de resultados y análisis estadístico
Los resultados presentados corresponden al promedio ± desviación estandar del
número de experimentos independientes indicados. En algunos casos las figuras o
imágenes corresponden a resultados representativos de cada grupo de experimentos
repetidos al menos 3 veces en forma independiente. La comparación estadistica entre
grupos se realizó usando el test de student; en algunos casos se realizó prueba ANOVA
de una vía con postest de Tukey o ANOVA de dos vías y postest de Bonferroni. Se
consideró como límite de significancia estadística, valores de p<0,05.

54
11) Resultados
11.1) TMBIM3 y el control de la apoptosis
11.1.1) Sobreexpresión de TMBIM3 y muerte celular inducida por estrés de RE.
Para estudiar el posible papel de TMBIM3 en la muerte celular, se procedió a
subclonar desde una biblioteca de cDNA el gen de tmbim3 de ratón en un vector pCR3.1,
el cual permite la expresión transitoria de la proteína TMBIM3 fusionada a un tag MYC.
Posteriormente, se evalúo la expresión y localización sub-celular de TMBIM3-MYC y se
comparó con la de TMBIM6-MYC. Con este fin, se procedió a transfectar fibroblastos
embrionarios de ratón (células MEFs; del inglés Mice embrionary fibroblast). Luego de 24
h pos-transfección, se evaluó su localización sub-celular mediante inmuno-fluorescencia y
microscopia confocal. Además, se realizó una co-tinción con Brefeldina A-bodipy, una
sonda fluorescente que permite teñir de rojo el RE y el aparato de Golgi. Los núcleos
fueron visualizados mediante la tinción con Hoescht (azul). Las imágenes de la figura 6
muestran que TMBIM3 presenta un patrón de localización sub-celular similar al de
TMBIM6, lo cual coincide con los análisis bio-informáticos que predicen la presencia de
una señal de retención en el RE en el c-terminal de TMBIM3 (Zhou et al, 2008).
Una vez determinada la localización sub-celular de TMBIM3 en las células MEFs,
el siguiente paso fue evaluar el efecto de TMBIM3 en la muerte celular. Con este fin, se
sobre-expresó TMBIM3-MYC mediante el uso de un vector retroviral, el cual permite la
expresión estable de la proteína en un grupo de células seleccionadas mediante la adición
al medio de cultivo del antibiótico puromicina. Una vez seleccionadas las células con el
antibiótico, se procedió a evaluar la expresión de TMBIM3 mediante western blot (Figura
7), en donde se inmunodetectó la proteína TMBIM3-MYC solo en las células transducidas
con los retrovirus TMBIM3-MYC, no así en las células transducidas con el vector viral
vacío.

55
Figura 6: Localización sub-celular de TMBIM3 en RE y aparato de Golgi. Para estudiar la localización sub-celular de TMBIM3-MYC y TMBIM6-MYC, se transfectaron las células MEFs con un vector de expresión que permite la expresión transitoria de TMBIM3-MYC o TMBIM6-MYC. Luego de 48 h, se evaluó la localización sub-celular de ambas proteínas mediante inmunofluorescencia indirecta y microscopía confocal. El RE fue teñido con Brefeldina A-Bodipy en rojo y los núcleos fueron teñidos con Hoescht (azul).
Figura 7: Expresión estable de TMBIM3-MYC en células MEFs. Las células MEFs se transducieron con retrovirus control (Mock) o con aquel que permite la expresión estable de TMBIM3-MYC. Luego de 48 h, las células transducidas se seleccionaron mediante la incubación con puromicina (0,3 µg/mL). Una vez seleccionadas las células, se procedió a estudiar la expresión de TMBIM3-MYC mediante western blot en extractos totales de proteínas. 30 µg de proteínas de cada línea celular fueron analizados en geles denaturantes de acrilamida al 12% (ver materiales y métodos). La expresión TMBIM3-MYC fue evaluada usando el anticuerpo 1º de ratón anti MYC (1:1000) y el anticuerpo secundario de conejo anti IgG de ratón (1:3000). Como control de carga, se evaluó la expresión de Hsp90 usando el anticuerpo 1º de conejo anti Hsp90 (1:3000) y el anticuerpo secundario de ratón anti IgG de conejo (1:3000).
Myc ER/Golgi MERGE Hoechst
TMBIM3
-MYC
TMBIM6
-MYC
95
43
TMBI
M3
Hsp90
MYC
MOCK
kDa
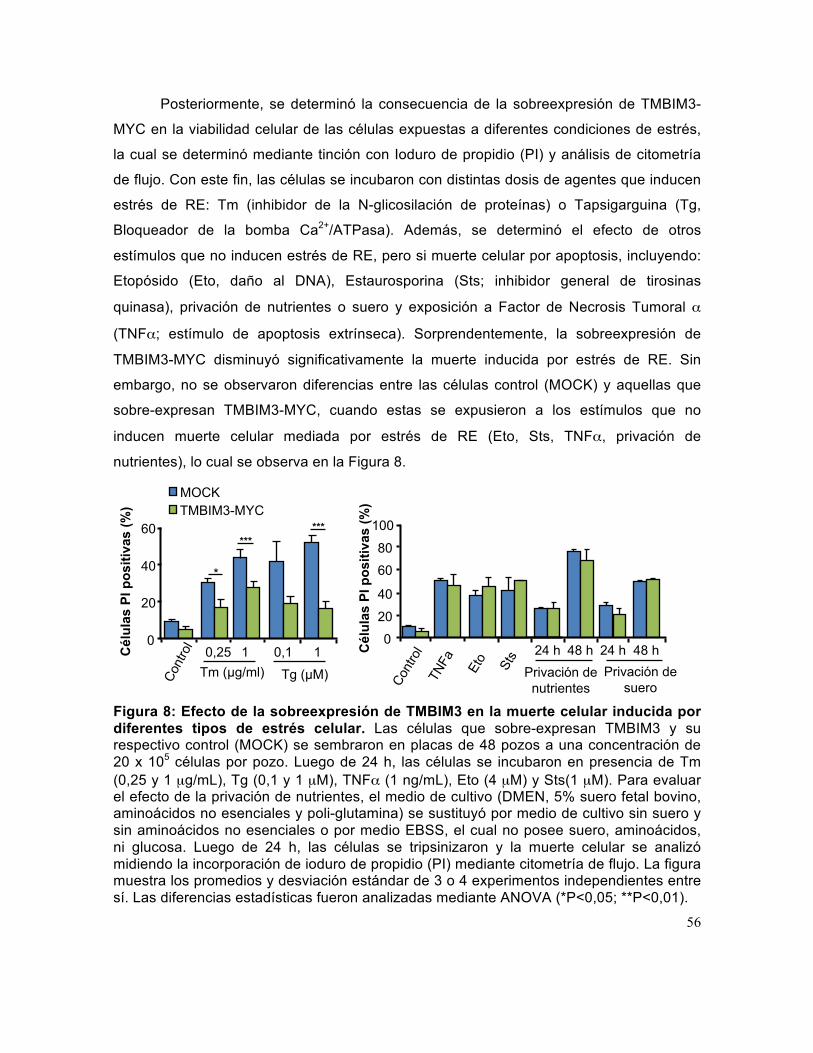
56
Posteriormente, se determinó la consecuencia de la sobreexpresión de TMBIM3-
MYC en la viabilidad celular de las células expuestas a diferentes condiciones de estrés,
la cual se determinó mediante tinción con Ioduro de propidio (PI) y análisis de citometría
de flujo. Con este fin, las células se incubaron con distintas dosis de agentes que inducen
estrés de RE: Tm (inhibidor de la N-glicosilación de proteínas) o Tapsigarguina (Tg,
Bloqueador de la bomba Ca2+/ATPasa). Además, se determinó el efecto de otros
estímulos que no inducen estrés de RE, pero si muerte celular por apoptosis, incluyendo:
Etopósido (Eto, daño al DNA), Estaurosporina (Sts; inhibidor general de tirosinas
quinasa), privación de nutrientes o suero y exposición a Factor de Necrosis Tumoral α
(TNFα; estímulo de apoptosis extrínseca). Sorprendentemente, la sobreexpresión de
TMBIM3-MYC disminuyó significativamente la muerte inducida por estrés de RE. Sin
embargo, no se observaron diferencias entre las células control (MOCK) y aquellas que
sobre-expresan TMBIM3-MYC, cuando estas se expusieron a los estímulos que no
inducen muerte celular mediada por estrés de RE (Eto, Sts, TNFα, privación de
nutrientes), lo cual se observa en la Figura 8.
Figura 8: Efecto de la sobreexpresión de TMBIM3 en la muerte celular inducida por diferentes tipos de estrés celular. Las células que sobre-expresan TMBIM3 y su respectivo control (MOCK) se sembraron en placas de 48 pozos a una concentración de 20 x 105 células por pozo. Luego de 24 h, las células se incubaron en presencia de Tm (0,25 y 1 µg/mL), Tg (0,1 y 1 µM), TNFα (1 ng/mL), Eto (4 µM) y Sts(1 µM). Para evaluar el efecto de la privación de nutrientes, el medio de cultivo (DMEN, 5% suero fetal bovino, aminoácidos no esenciales y poli-glutamina) se sustituyó por medio de cultivo sin suero y sin aminoácidos no esenciales o por medio EBSS, el cual no posee suero, aminoácidos, ni glucosa. Luego de 24 h, las células se tripsinizaron y la muerte celular se analizó midiendo la incorporación de ioduro de propidio (PI) mediante citometría de flujo. La figura muestra los promedios y desviación estándar de 3 o 4 experimentos independientes entre sí. Las diferencias estadísticas fueron analizadas mediante ANOVA (*P<0,05; **P<0,01).
24 h 48 h 24 h 48 h Privación de nutrientes
0
20
40
60
80
100
Cél
ulas
PI p
ositi
vas
(%)
Privación de suero Co
ntro
l TN
Fa
Eto
St
s
***
Tm (µg/ml) Tg (µM) Cont
rol 0,1 1
0
20
40
60
Cél
ulas
PI p
ositi
vas
(%)
*
0,25 1
MOCK TMBIM3-MYC
***

57
El efecto en la muerte celular de la sobreexpresión de TMBIM3 bajo condiciones
de estrés de RE, se validó mediante la cuantificación de la población apoptótica hipo
diploide “sub-G1” a través de la tinción con PI y análisis de citometría de flujo. En esta
experimento, se determinó que las células que sobreexpresan TMBIM3-MYC
experimentan menos apoptosis que las células controles al ser expuestas a estrés de RE.
Del mismo modo a lo observado en la cuantificación de la muerte celular, no se
observaron diferencias significativas entre ambas líneas celulares al ser expuesta a los
estímulos no relacionados con estrés de RE (Figura 9).
Figura 9: Efecto de la sobreexpresión de TMBIM3 en la apoptosis inducida por estrés de RE. Las células TMBIM3 y MOCK se estimularon de la misma forma descrita en la figura 8. La población Sub-G1 se analizó en células fijadas y permeabilizadas con Metanol y teñidas posteriormente con PI. El contenido de DNA se evalúo mediante citometría de flujo. Las figuras representan el promedio y la desviación estándar de 3 o 4 experimentos independientes entre sí. Las diferencias estadísticas fueron analizadas mediante ANOVA y postest de Tukey (*P<0,05).
Por otra parte, se investigó el efecto de la sobreexpresión de TMBIM3-MYC en
células expuestas a una condición aguda de estrés de RE y posterior pos-cultivo en
ausencia de agentes estresores. De esta manera, las células se expusieron a 1 y 2,5
µg/mL de Tm por un periodo de 4 h. Posteriormente, las células se lavaron con PBS, se
tripsinizaron y luego se sembraron (2 x 105 células por pozo) para ser cultivadas en
ausencia de la Tm. Así, se evaluó la proliferación y capacidad de adaptarse y sobrevivir a
una exposición transitoria a un agente inductor de estrés de RE. La figura 10 muestra que
la sobreexpresión de TMBIM3 protegió significativamente de la toxicidad de Tm,
Sub-
G1
(%)
0
20
40
60
MOCKTMBIM3
TgControl Tm
*
Control Eto Sts0
20
40
60
80
100
Sub-
G1
(%)

58
habiéndose evaluado la proliferación celular a los 2, 4 y 5 días post-exposición al agente
estresor y finalmente, se realizo la tinción de la placa de cultivo con cristal violeta. Dado la
diferencia observada entre ambos tipos celulares, se evaluó sí la sobreexpresión de
TMBIM3-MYC incrementa por sí sola la proliferación de las células MEFs en condiciones
basales. No se observaron diferencias (Figura 11) en la proliferación de ambos tipos
celulares. Considerando estos resultados en conjunto, se sugiere un papel especifico de
TMBIM3 en el control de la muerte celular inducida por estrés de RE.
Figura 10. Efecto de la sobreexpresión de TMBIM3 en la sobrevida bajo condiciones de estrés de RE. Se sembraron 3.5 x 105 células en placas de 6 pozos. Después de 24 h, las células se incubaron por 4 h con las dosis indicadas de Tm. Posteriormente, las células se lavaron 3 veces con PBS, se tripsinizaron y 2.0 x 105 células se sembraron y cultivaron en ausencia de Tm. Luego, las placas se fotografiaron y el número de células por mm2 fue cuantificado a los 2, 4 y 5 días. Al finalizar el experimento, se procedió a fijar las células en la placa con PAF y posteriormente, se tiñeron las células con cristal violeta. El grafico de la derecha muestra el promedio y desviación estándar de 3 experimentos independientes entre sí. Las diferencias estadísticas fueron analizadas mediante ANOVA y postest de Tukey (*P<0,05).
Figura 11. Efecto de la sobreexpresión de TMBIM3-MYC en la proliferación celular. Para medir la proliferación celular de la línea celular control y la línea celular MOCK y aquella que sobreexpresa TMBIM3-MYC, se sembraron 5 x 104 células en placas de 12 pozos. Luego, las células se tripsinizaron y se contaron cada 24 h en una cámara de Nuebaur, previa tinción con azul de tripán. La figura representa el promedio y la desviación estandar de tres experimentos independientes entre sí.
MOCK
TMBIM3-MYC
Control 1 2,5
Tm (µg/ml)
0
100
200
300
2 4 5Tiempo (Días) Nú
mer
o de
cél
ulas
/mm
2
*MOCKTMBIM3
0
50
100
150
200
1 2 3 4
TMBIM3MOCK
Tiempo (Días)
Núm
ero
de c
élul
as
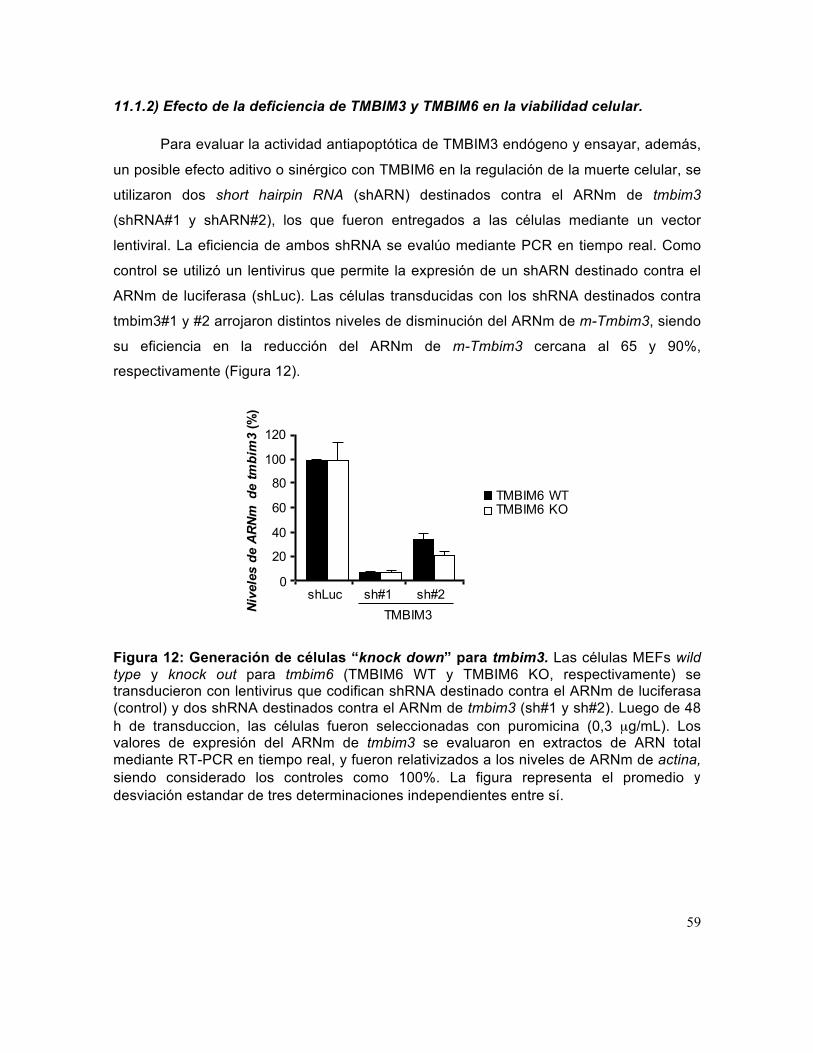
59
11.1.2) Efecto de la deficiencia de TMBIM3 y TMBIM6 en la viabilidad celular.
Para evaluar la actividad antiapoptótica de TMBIM3 endógeno y ensayar, además,
un posible efecto aditivo o sinérgico con TMBIM6 en la regulación de la muerte celular, se
utilizaron dos short hairpin RNA (shARN) destinados contra el ARNm de tmbim3
(shRNA#1 y shARN#2), los que fueron entregados a las células mediante un vector
lentiviral. La eficiencia de ambos shRNA se evalúo mediante PCR en tiempo real. Como
control se utilizó un lentivirus que permite la expresión de un shARN destinado contra el
ARNm de luciferasa (shLuc). Las células transducidas con los shRNA destinados contra
tmbim3#1 y #2 arrojaron distintos niveles de disminución del ARNm de m-Tmbim3, siendo
su eficiencia en la reducción del ARNm de m-Tmbim3 cercana al 65 y 90%,
respectivamente (Figura 12).
Figura 12: Generación de células “knock down” para tmbim3. Las células MEFs wild type y knock out para tmbim6 (TMBIM6 WT y TMBIM6 KO, respectivamente) se transducieron con lentivirus que codifican shRNA destinado contra el ARNm de luciferasa (control) y dos shRNA destinados contra el ARNm de tmbim3 (sh#1 y sh#2). Luego de 48 h de transduccion, las células fueron seleccionadas con puromicina (0,3 µg/mL). Los valores de expresión del ARNm de tmbim3 se evaluaron en extractos de ARN total mediante RT-PCR en tiempo real, y fueron relativizados a los niveles de ARNm de actina, siendo considerado los controles como 100%. La figura representa el promedio y desviación estandar de tres determinaciones independientes entre sí.
shLuc sh#1 sh#2
Nive
les
de A
RNm
de tm
bim
3(%)
0
20
40
60
80
100
120
TMBIM3
TMBIM6 WTTMBIM6 KO
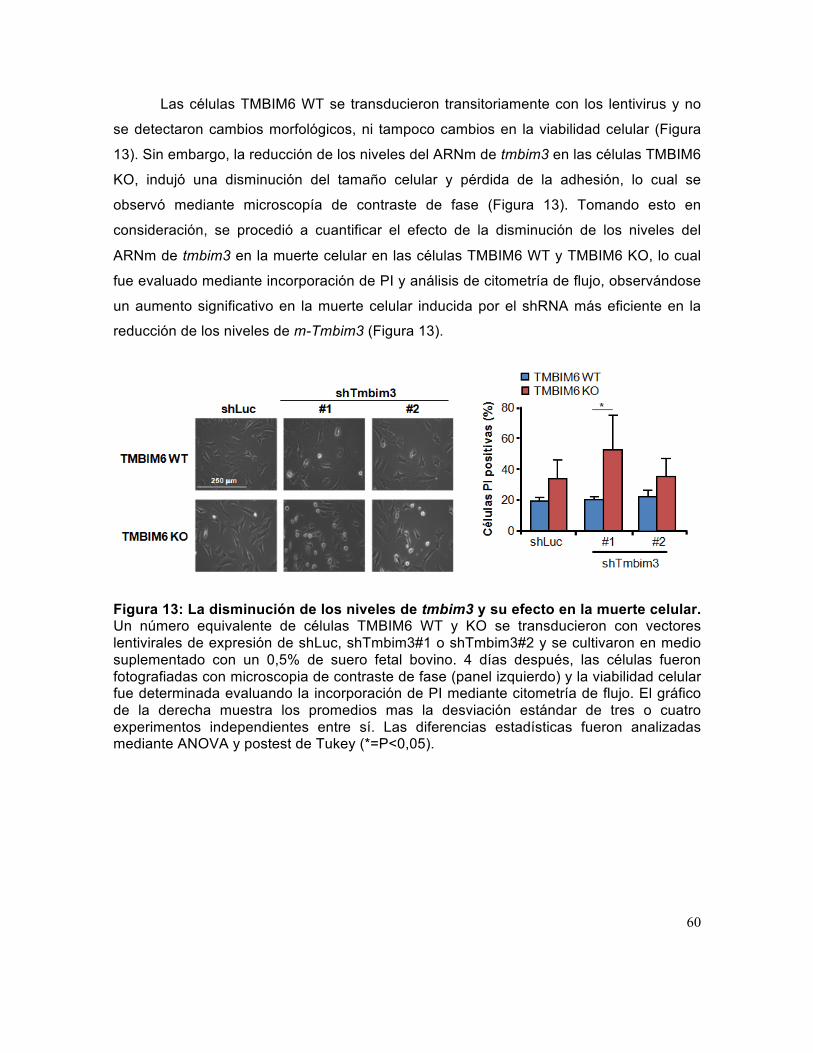
60
Las células TMBIM6 WT se transducieron transitoriamente con los lentivirus y no
se detectaron cambios morfológicos, ni tampoco cambios en la viabilidad celular (Figura
13). Sin embargo, la reducción de los niveles del ARNm de tmbim3 en las células TMBIM6
KO, indujó una disminución del tamaño celular y pérdida de la adhesión, lo cual se
observó mediante microscopía de contraste de fase (Figura 13). Tomando esto en
consideración, se procedió a cuantificar el efecto de la disminución de los niveles del
ARNm de tmbim3 en la muerte celular en las células TMBIM6 WT y TMBIM6 KO, lo cual
fue evaluado mediante incorporación de PI y análisis de citometría de flujo, observándose
un aumento significativo en la muerte celular inducida por el shRNA más eficiente en la
reducción de los niveles de m-Tmbim3 (Figura 13).
Figura 13: La disminución de los niveles de tmbim3 y su efecto en la muerte celular. Un número equivalente de células TMBIM6 WT y KO se transducieron con vectores lentivirales de expresión de shLuc, shTmbim3#1 o shTmbim3#2 y se cultivaron en medio suplementado con un 0,5% de suero fetal bovino. 4 días después, las células fueron fotografiadas con microscopia de contraste de fase (panel izquierdo) y la viabilidad celular fue determinada evaluando la incorporación de PI mediante citometría de flujo. El gráfico de la derecha muestra los promedios mas la desviación estándar de tres o cuatro experimentos independientes entre sí. Las diferencias estadísticas fueron analizadas mediante ANOVA y postest de Tukey (*=P<0,05).
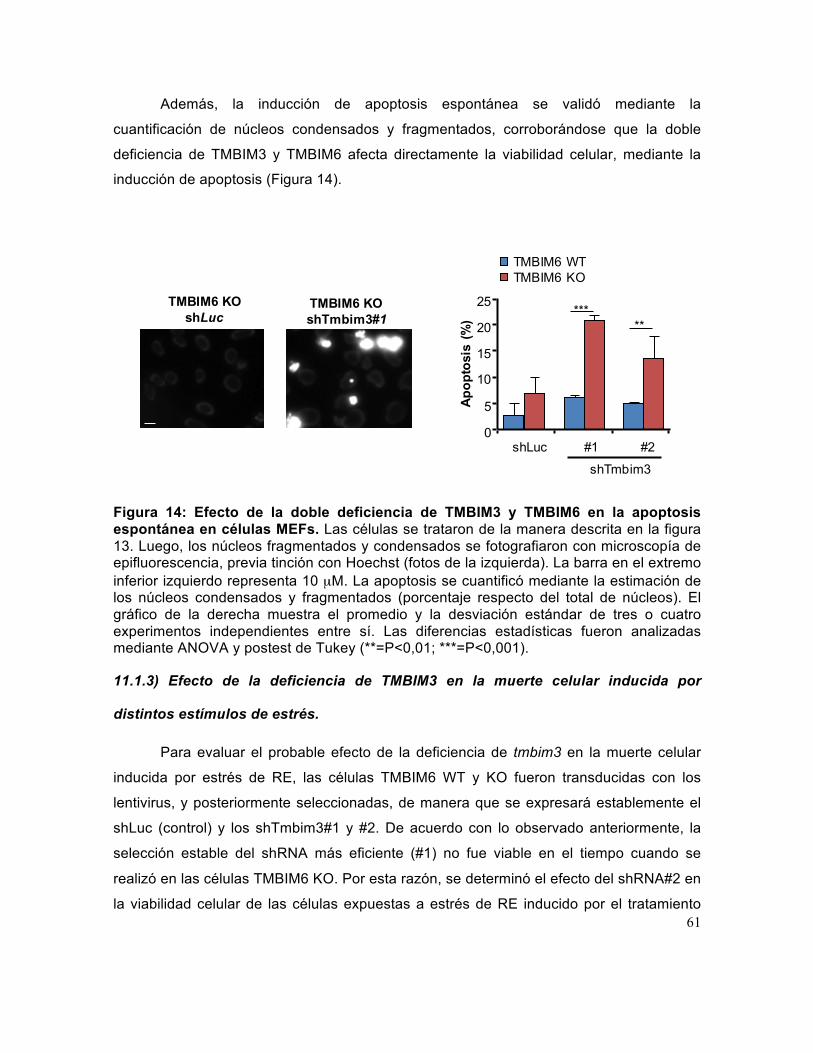
61
Además, la inducción de apoptosis espontánea se validó mediante la
cuantificación de núcleos condensados y fragmentados, corroborándose que la doble
deficiencia de TMBIM3 y TMBIM6 afecta directamente la viabilidad celular, mediante la
inducción de apoptosis (Figura 14).
Figura 14: Efecto de la doble deficiencia de TMBIM3 y TMBIM6 en la apoptosis espontánea en células MEFs. Las células se trataron de la manera descrita en la figura 13. Luego, los núcleos fragmentados y condensados se fotografiaron con microscopía de epifluorescencia, previa tinción con Hoechst (fotos de la izquierda). La barra en el extremo inferior izquierdo representa 10 µM. La apoptosis se cuantificó mediante la estimación de los núcleos condensados y fragmentados (porcentaje respecto del total de núcleos). El gráfico de la derecha muestra el promedio y la desviación estándar de tres o cuatro experimentos independientes entre sí. Las diferencias estadísticas fueron analizadas mediante ANOVA y postest de Tukey (**=P<0,01; ***=P<0,001).
11.1.3) Efecto de la deficiencia de TMBIM3 en la muerte celular inducida por
distintos estímulos de estrés.
Para evaluar el probable efecto de la deficiencia de tmbim3 en la muerte celular
inducida por estrés de RE, las células TMBIM6 WT y KO fueron transducidas con los
lentivirus, y posteriormente seleccionadas, de manera que se expresará establemente el
shLuc (control) y los shTmbim3#1 y #2. De acuerdo con lo observado anteriormente, la
selección estable del shRNA más eficiente (#1) no fue viable en el tiempo cuando se
realizó en las células TMBIM6 KO. Por esta razón, se determinó el efecto del shRNA#2 en
la viabilidad celular de las células expuestas a estrés de RE inducido por el tratamiento
TMBIM6 KO shLuc
TMBIM6 KO shTmbim3#1
Apop
tosi
s (%
)
0
5
15
20
25
shLuc #1 #2
*****
10
shTmbim3
TMBIM6 WTTMBIM6 KO
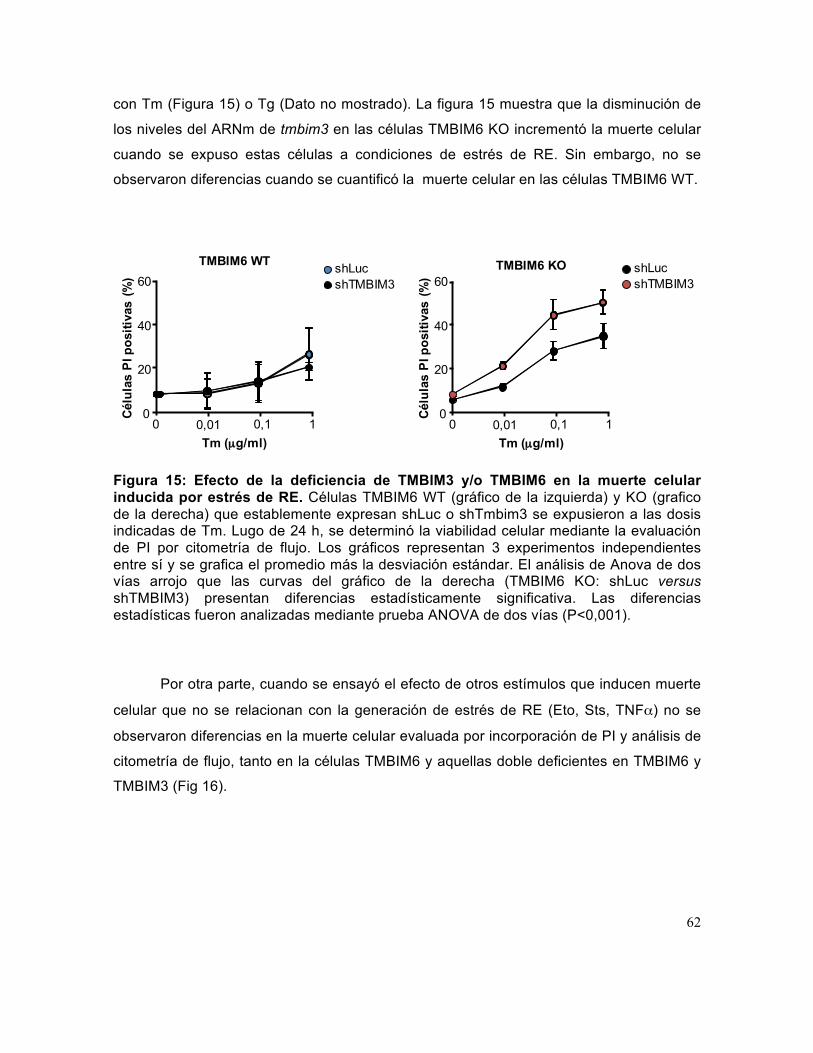
62
con Tm (Figura 15) o Tg (Dato no mostrado). La figura 15 muestra que la disminución de
los niveles del ARNm de tmbim3 en las células TMBIM6 KO incrementó la muerte celular
cuando se expuso estas células a condiciones de estrés de RE. Sin embargo, no se
observaron diferencias cuando se cuantificó la muerte celular en las células TMBIM6 WT.
Figura 15: Efecto de la deficiencia de TMBIM3 y/o TMBIM6 en la muerte celular inducida por estrés de RE. Células TMBIM6 WT (gráfico de la izquierda) y KO (grafico de la derecha) que establemente expresan shLuc o shTmbim3 se expusieron a las dosis indicadas de Tm. Lugo de 24 h, se determinó la viabilidad celular mediante la evaluación de PI por citometría de flujo. Los gráficos representan 3 experimentos independientes entre sí y se grafica el promedio más la desviación estándar. El análisis de Anova de dos vías arrojo que las curvas del gráfico de la derecha (TMBIM6 KO: shLuc versus shTMBIM3) presentan diferencias estadísticamente significativa. Las diferencias estadísticas fueron analizadas mediante prueba ANOVA de dos vías (P<0,001).
Por otra parte, cuando se ensayó el efecto de otros estímulos que inducen muerte
celular que no se relacionan con la generación de estrés de RE (Eto, Sts, TNFα) no se
observaron diferencias en la muerte celular evaluada por incorporación de PI y análisis de
citometría de flujo, tanto en la células TMBIM6 y aquellas doble deficientes en TMBIM6 y
TMBIM3 (Fig 16).
0
20
40
60
0 0,1 1Tm (µg/ml)
Célu
las
PI p
ositi
vas
(%)
0,01
shLucshTMBIM3
TMBIM6 WT TMBIM6 KO
0
20
40
60
0 0,1 1Tm (µg/ml)
Célu
las
PI p
ositi
vas
(%)
0,01
shLucshTMBIM3
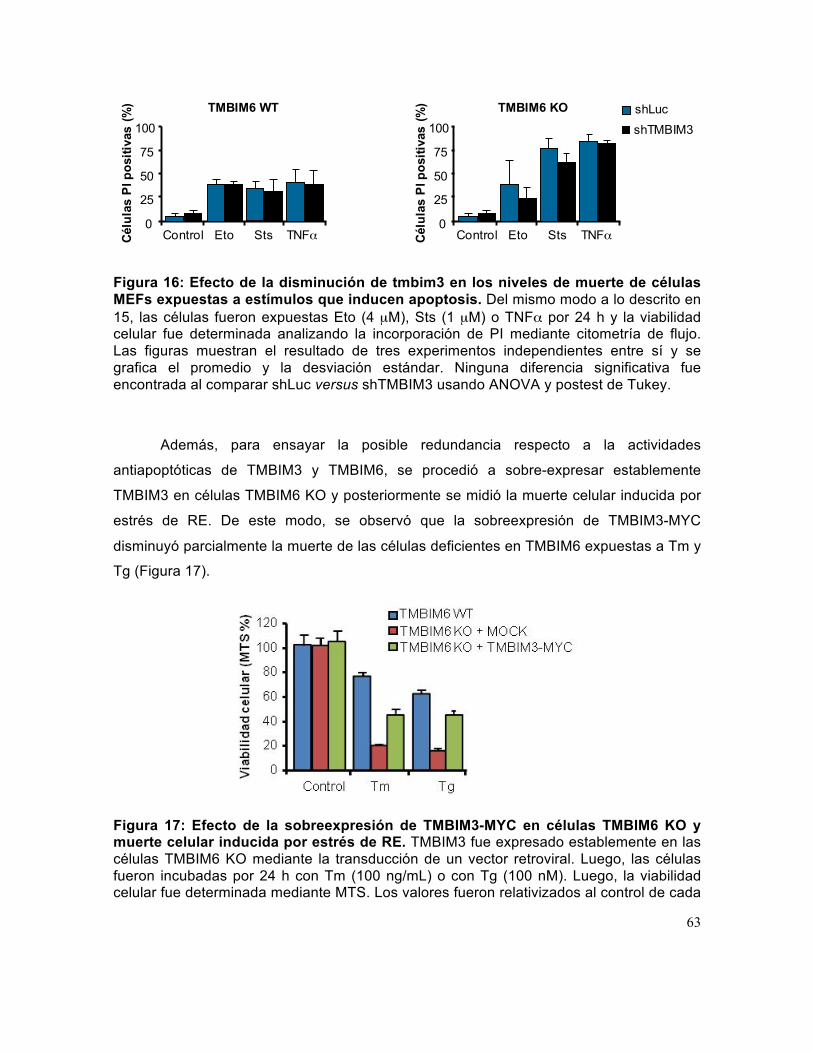
63
Figura 16: Efecto de la disminución de tmbim3 en los niveles de muerte de células MEFs expuestas a estímulos que inducen apoptosis. Del mismo modo a lo descrito en 15, las células fueron expuestas Eto (4 µM), Sts (1 µM) o TNFα por 24 h y la viabilidad celular fue determinada analizando la incorporación de PI mediante citometría de flujo. Las figuras muestran el resultado de tres experimentos independientes entre sí y se grafica el promedio y la desviación estándar. Ninguna diferencia significativa fue encontrada al comparar shLuc versus shTMBIM3 usando ANOVA y postest de Tukey.
Además, para ensayar la posible redundancia respecto a la actividades
antiapoptóticas de TMBIM3 y TMBIM6, se procedió a sobre-expresar establemente
TMBIM3 en células TMBIM6 KO y posteriormente se midió la muerte celular inducida por
estrés de RE. De este modo, se observó que la sobreexpresión de TMBIM3-MYC
disminuyó parcialmente la muerte de las células deficientes en TMBIM6 expuestas a Tm y
Tg (Figura 17).
Figura 17: Efecto de la sobreexpresión de TMBIM3-MYC en células TMBIM6 KO y muerte celular inducida por estrés de RE. TMBIM3 fue expresado establemente en las células TMBIM6 KO mediante la transducción de un vector retroviral. Luego, las células fueron incubadas por 24 h con Tm (100 ng/mL) o con Tg (100 nM). Luego, la viabilidad celular fue determinada mediante MTS. Los valores fueron relativizados al control de cada
0
25
50
75
100
Control Eto Sts TNFαCélu
las
PI p
ositi
vas
(%)TMBIM6 WT TMBIM6 KO shLuc
shTMBIM3
0
25
50
75
100
Control Eto Sts TNFαCélu
las
PI p
ositi
vas
(%)

64
línea celular (100%). El grafico, muestra el promedio ± desviación estándar de un experimento representativo hecho en triplicado (n=2).
Debido al efecto sinérgico en la regulación de la muerte celular observado entre
TMBIM3 y TMBIM6, se procedió a evaluar la posible interacción entre ambas proteínas
mediante un ensayo de co-inmunoprecipitación. Con este fin, se expresó transitoriamente
TMBIM3-MYC y TMBIM6-HA en células HEK293T y se procedió a inmunoprecipitar
TMBIM3-MYC desde un extracto total de proteínas. Posteriormente, se evaluó la co-
inmunoprecipitación de TMBIM6 mediante Western blot, como lo muestra la figura 18, lo
cual sugiere que ambas proteínas forman parte de un complejo proteico.
Figura 18. Estudio de la posible formación de un complejo proteico entre TMBIM3-MYC y TMBIM6-HA forman un complejo. Las células HEK 293T fueron transfectadas con vectores de expresión para TMBIM3-MYC y TMBIM6-HA. 48 h despues, las células fueron lisadas en una solución de lisado (CHAPS 1%, NaCl 150 mM, Kcl 40 mM, ) por 30 min a 4ºC con agitación constante. Luego, este lisado fue centrifugado por 5 min a 10000 r.p.m. y el sobrenadante fue incubado por 2 h con esferas de sefarosa acopladas covalentemente a un anticuerpo IgG anti-MYC, de manera de inmunoprecipitar TMBIM3-MYC. Posteriormente, las esferas de sefarosa fueron lavadas 3 veces con la solucion CHAPS (1%) y se procedio a eluir los complejos proteicos acoplados a los anticuerpo IgG anti-MYC por adición del péptido MYC 10 μM (30 min a temperatura ambiente con agitación constante). Luego, el sobrenadante fue removido de las esferas de sefarosa y las proteínas inmunoprecipitadas fiueron analizadas por western blot en geles de acrilamida al 12%. La co-inmunoprecipitación de TMBIM6-HA fue determinada usando el anticuerpo de raton 1º anti-HA (1:2000, Roche) y la inmunoprecipitación de TMBIM3-MYC fue determinada usando el anticuerpo 1º de ratón anti-MYC (1:2000, santa cruz). El asterisco (*) muestra la señal dada por la cadena liviana de la IgG utilizada en la inmunoprecipitación.
20
20
43 IP: MYC WB:Myc
IP: MYC WB:HA
*
*
WB:HA Extracto total
pCDNA3 TMBIM3-MYC
TMBIM6-HA
+ - - + + +
kDa
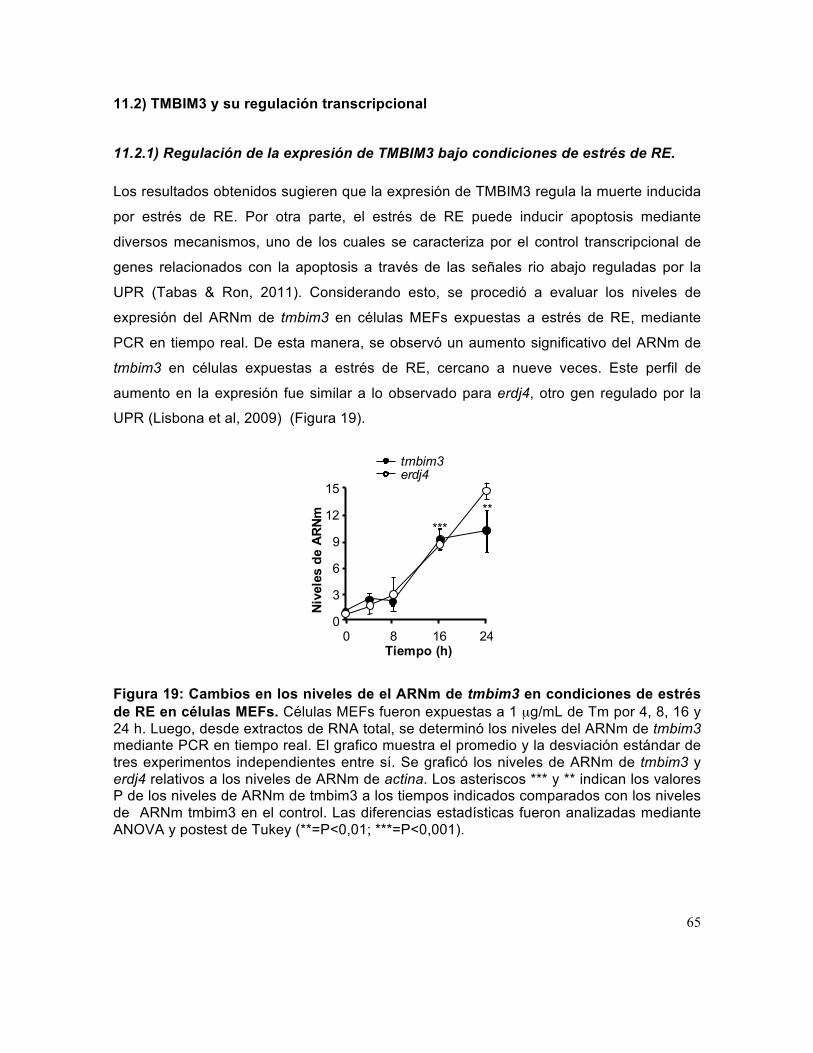
65
11.2) TMBIM3 y su regulación transcripcional
11.2.1) Regulación de la expresión de TMBIM3 bajo condiciones de estrés de RE.
Los resultados obtenidos sugieren que la expresión de TMBIM3 regula la muerte inducida
por estrés de RE. Por otra parte, el estrés de RE puede inducir apoptosis mediante
diversos mecanismos, uno de los cuales se caracteriza por el control transcripcional de
genes relacionados con la apoptosis a través de las señales rio abajo reguladas por la
UPR (Tabas & Ron, 2011). Considerando esto, se procedió a evaluar los niveles de
expresión del ARNm de tmbim3 en células MEFs expuestas a estrés de RE, mediante
PCR en tiempo real. De esta manera, se observó un aumento significativo del ARNm de
tmbim3 en células expuestas a estrés de RE, cercano a nueve veces. Este perfil de
aumento en la expresión fue similar a lo observado para erdj4, otro gen regulado por la
UPR (Lisbona et al, 2009) (Figura 19).
Figura 19: Cambios en los niveles de el ARNm de tmbim3 en condiciones de estrés de RE en células MEFs. Células MEFs fueron expuestas a 1 µg/mL de Tm por 4, 8, 16 y 24 h. Luego, desde extractos de RNA total, se determinó los niveles del ARNm de tmbim3 mediante PCR en tiempo real. El grafico muestra el promedio y la desviación estándar de tres experimentos independientes entre sí. Se graficó los niveles de ARNm de tmbim3 y erdj4 relativos a los niveles de ARNm de actina. Los asteriscos *** y ** indican los valores P de los niveles de ARNm de tmbim3 a los tiempos indicados comparados con los niveles de ARNm tmbim3 en el control. Las diferencias estadísticas fueron analizadas mediante ANOVA y postest de Tukey (**=P<0,01; ***=P<0,001).
0
3
6
9
12
15
0 8 16 24Tiempo (h)
*****
Nive
les
de A
RNm
erdj4tmbim3
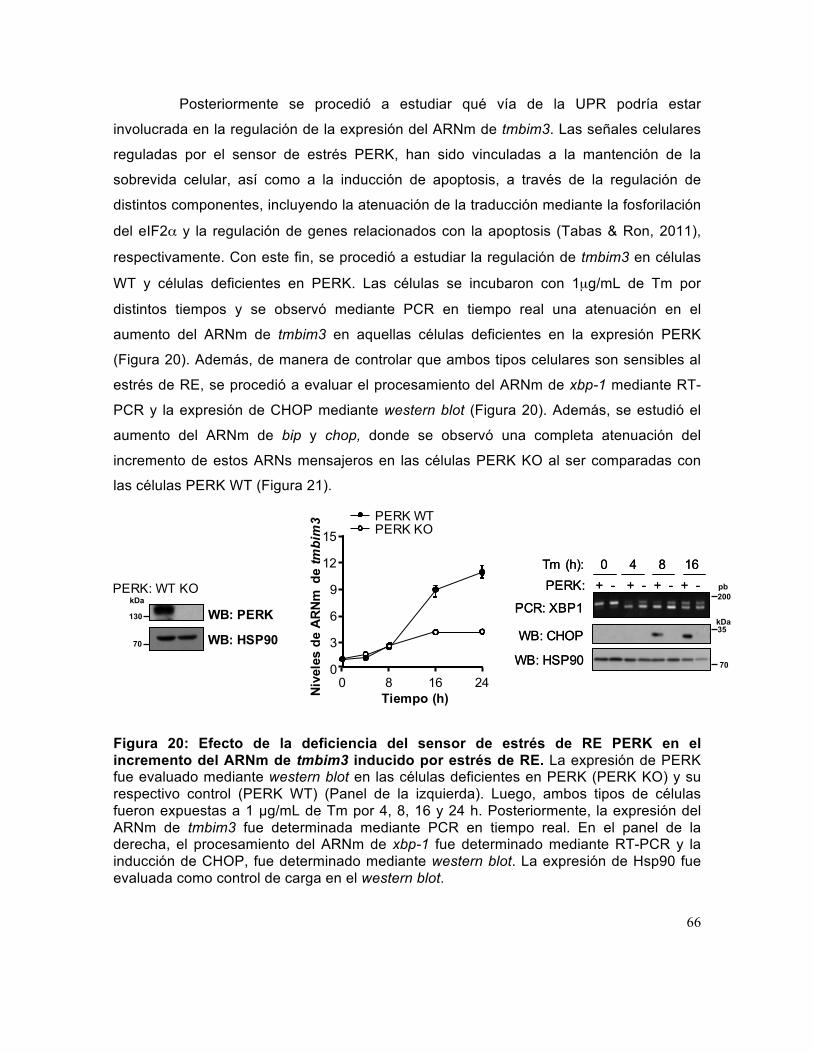
66
0
3
6
9
12
15
0 8 16 24Tiempo (h)N
ivel
es d
e A
RN
mde
tmbim3
PERK KOPERK WT
WB: HSP90
WB: PERK
PERK: WT KO
!
kDa
70
130 kDa
70
35
PERK: + - + - + - + -Tm (h): 0 4 8 16
WB: CHOP
WB: HSP90
PCR: XBP1
!
PERK: + - + - + - + -Tm (h): 0 4 8 16
WB: CHOP
WB: HSP90
PCR: XBP1
!
pb 200
Posteriormente se procedió a estudiar qué vía de la UPR podría estar
involucrada en la regulación de la expresión del ARNm de tmbim3. Las señales celulares
reguladas por el sensor de estrés PERK, han sido vinculadas a la mantención de la
sobrevida celular, así como a la inducción de apoptosis, a través de la regulación de
distintos componentes, incluyendo la atenuación de la traducción mediante la fosforilación
del eIF2α y la regulación de genes relacionados con la apoptosis (Tabas & Ron, 2011),
respectivamente. Con este fin, se procedió a estudiar la regulación de tmbim3 en células
WT y células deficientes en PERK. Las células se incubaron con 1µg/mL de Tm por
distintos tiempos y se observó mediante PCR en tiempo real una atenuación en el
aumento del ARNm de tmbim3 en aquellas células deficientes en la expresión PERK
(Figura 20). Además, de manera de controlar que ambos tipos celulares son sensibles al
estrés de RE, se procedió a evaluar el procesamiento del ARNm de xbp-1 mediante RT-
PCR y la expresión de CHOP mediante western blot (Figura 20). Además, se estudió el
aumento del ARNm de bip y chop, donde se observó una completa atenuación del
incremento de estos ARNs mensajeros en las células PERK KO al ser comparadas con
las células PERK WT (Figura 21).
Figura 20: Efecto de la deficiencia del sensor de estrés de RE PERK en el incremento del ARNm de tmbim3 inducido por estrés de RE. La expresión de PERK fue evaluado mediante western blot en las células deficientes en PERK (PERK KO) y su respectivo control (PERK WT) (Panel de la izquierda). Luego, ambos tipos de células fueron expuestas a 1 µg/mL de Tm por 4, 8, 16 y 24 h. Posteriormente, la expresión del ARNm de tmbim3 fue determinada mediante PCR en tiempo real. En el panel de la derecha, el procesamiento del ARNm de xbp-1 fue determinado mediante RT-PCR y la inducción de CHOP, fue determinado mediante western blot. La expresión de Hsp90 fue evaluada como control de carga en el western blot.
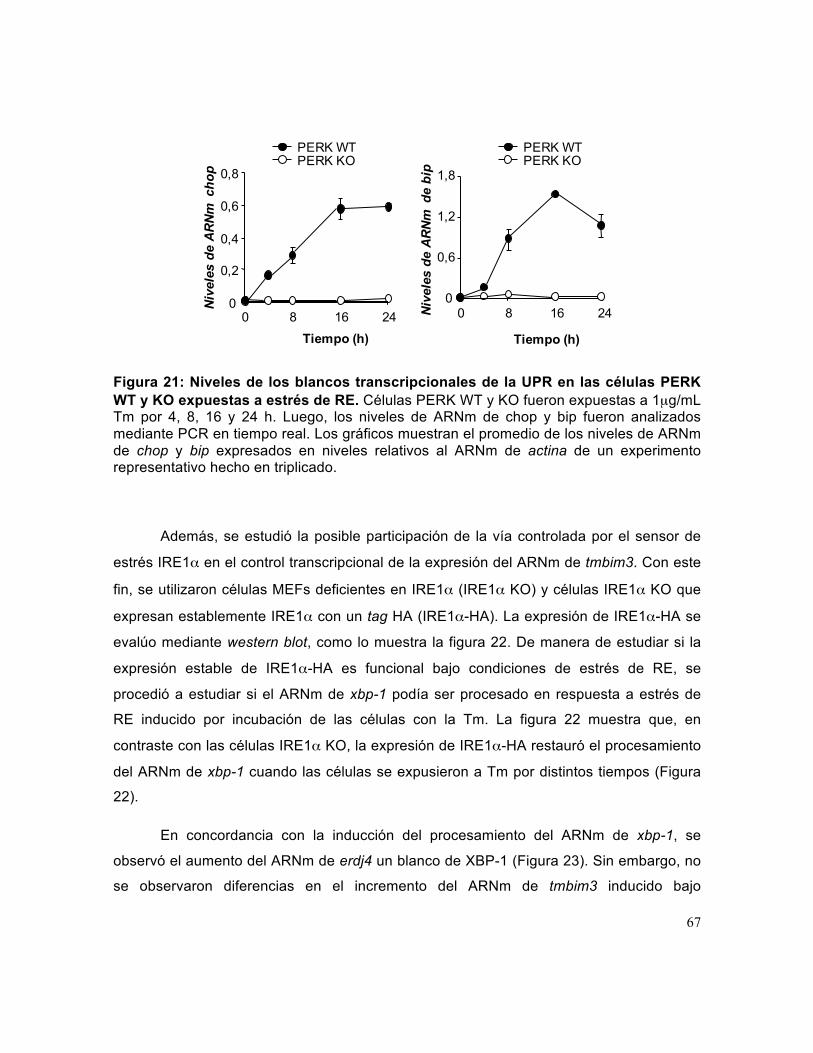
67
Figura 21: Niveles de los blancos transcripcionales de la UPR en las células PERK WT y KO expuestas a estrés de RE. Células PERK WT y KO fueron expuestas a 1µg/mL Tm por 4, 8, 16 y 24 h. Luego, los niveles de ARNm de chop y bip fueron analizados mediante PCR en tiempo real. Los gráficos muestran el promedio de los niveles de ARNm de chop y bip expresados en niveles relativos al ARNm de actina de un experimento representativo hecho en triplicado.
Además, se estudió la posible participación de la vía controlada por el sensor de
estrés IRE1α en el control transcripcional de la expresión del ARNm de tmbim3. Con este
fin, se utilizaron células MEFs deficientes en IRE1α (IRE1α KO) y células IRE1α KO que
expresan establemente IRE1α con un tag HA (IRE1α-HA). La expresión de IRE1α-HA se
evalúo mediante western blot, como lo muestra la figura 22. De manera de estudiar si la
expresión estable de IRE1α-HA es funcional bajo condiciones de estrés de RE, se
procedió a estudiar si el ARNm de xbp-1 podía ser procesado en respuesta a estrés de
RE inducido por incubación de las células con la Tm. La figura 22 muestra que, en
contraste con las células IRE1α KO, la expresión de IRE1α-HA restauró el procesamiento
del ARNm de xbp-1 cuando las células se expusieron a Tm por distintos tiempos (Figura
22).
En concordancia con la inducción del procesamiento del ARNm de xbp-1, se
observó el aumento del ARNm de erdj4 un blanco de XBP-1 (Figura 23). Sin embargo, no
se observaron diferencias en el incremento del ARNm de tmbim3 inducido bajo
Nive
les
de A
RNm
chop
Tiempo (h)
0
0,2
0,4
0,6
0,8
0 8 16 240
0,6
1,2
1,8
Nive
les
de A
RNm
de b
ip
0 8 16 24
Tiempo (h)
PERK KOPERK WT
PERK KOPERK WT
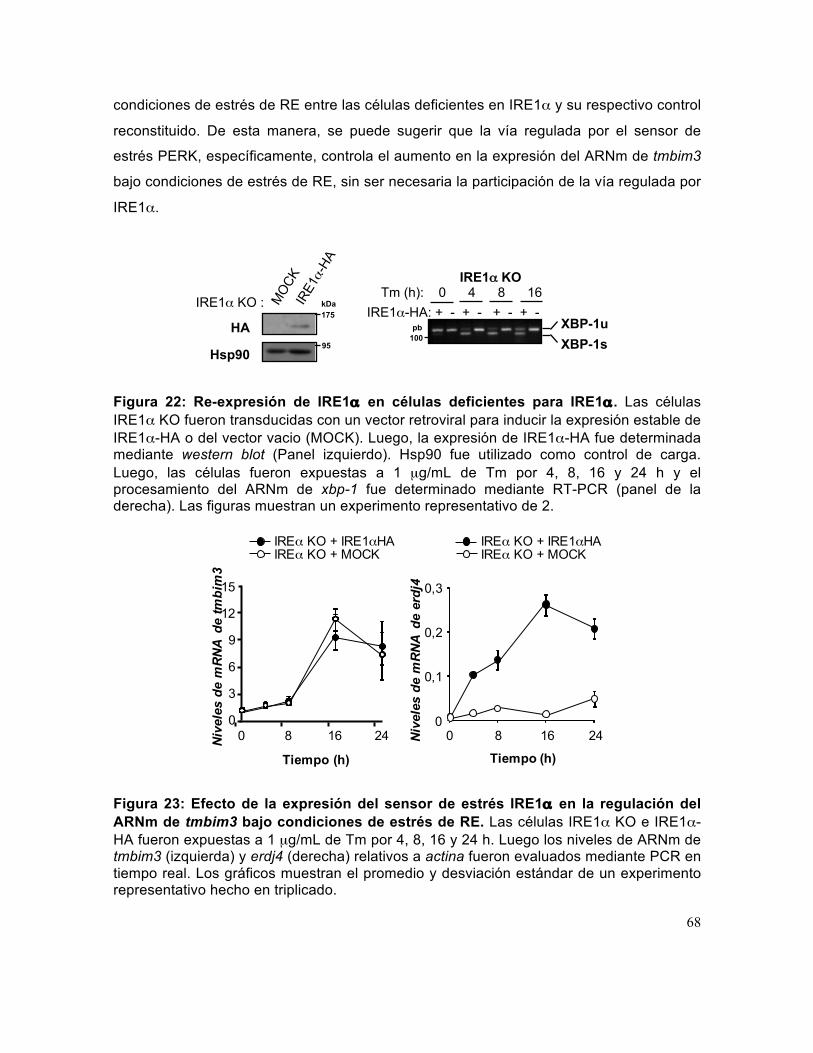
68
condiciones de estrés de RE entre las células deficientes en IRE1α y su respectivo control
reconstituido. De esta manera, se puede sugerir que la vía regulada por el sensor de
estrés PERK, específicamente, controla el aumento en la expresión del ARNm de tmbim3
bajo condiciones de estrés de RE, sin ser necesaria la participación de la vía regulada por
IRE1α.
Figura 22: Re-expresión de IRE1α en células deficientes para IRE1α . Las células IRE1α KO fueron transducidas con un vector retroviral para inducir la expresión estable de IRE1α-HA o del vector vacio (MOCK). Luego, la expresión de IRE1α-HA fue determinada mediante western blot (Panel izquierdo). Hsp90 fue utilizado como control de carga. Luego, las células fueron expuestas a 1 µg/mL de Tm por 4, 8, 16 y 24 h y el procesamiento del ARNm de xbp-1 fue determinado mediante RT-PCR (panel de la derecha). Las figuras muestran un experimento representativo de 2.
Figura 23: Efecto de la expresión del sensor de estrés IRE1α en la regulación del ARNm de tmbim3 bajo condiciones de estrés de RE. Las células IRE1α KO e IRE1α-HA fueron expuestas a 1 µg/mL de Tm por 4, 8, 16 y 24 h. Luego los niveles de ARNm de tmbim3 (izquierda) y erdj4 (derecha) relativos a actina fueron evaluados mediante PCR en tiempo real. Los gráficos muestran el promedio y desviación estándar de un experimento representativo hecho en triplicado.
Nive
les
de m
RNA
de tm
bim
3
Tiempo (h)
0
3
6
9
12
15
0 8 16 24 Nive
les
de m
RNA
de e
rdj4
0
0,1
0,2
0,3
IREα KO + IRE1αHAIREα KO + MOCK
0 8 16 24
Tiempo (h)
IREα KO + IRE1αHAIREα KO + MOCK
95
175
Hsp90
HA
IRE1α KO : IRE1α-HA: + - + - + - + - Tm (h): 0 4 8 16
MOCK
IR
E1α-
HA
XBP-1u XBP-1s
IRE1α KO
100 pb
kDa
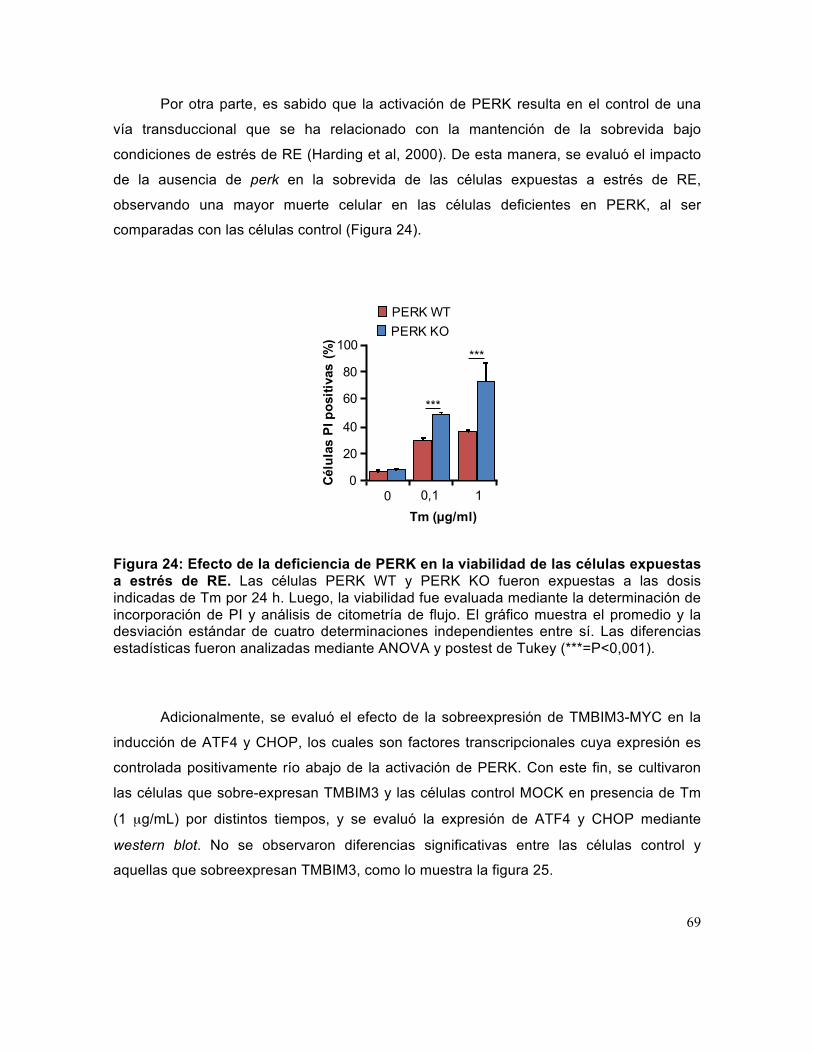
69
Por otra parte, es sabido que la activación de PERK resulta en el control de una
vía transduccional que se ha relacionado con la mantención de la sobrevida bajo
condiciones de estrés de RE (Harding et al, 2000). De esta manera, se evaluó el impacto
de la ausencia de perk en la sobrevida de las células expuestas a estrés de RE,
observando una mayor muerte celular en las células deficientes en PERK, al ser
comparadas con las células control (Figura 24).
Figura 24: Efecto de la deficiencia de PERK en la viabilidad de las células expuestas a estrés de RE. Las células PERK WT y PERK KO fueron expuestas a las dosis indicadas de Tm por 24 h. Luego, la viabilidad fue evaluada mediante la determinación de incorporación de PI y análisis de citometría de flujo. El gráfico muestra el promedio y la desviación estándar de cuatro determinaciones independientes entre sí. Las diferencias estadísticas fueron analizadas mediante ANOVA y postest de Tukey (***=P<0,001).
Adicionalmente, se evaluó el efecto de la sobreexpresión de TMBIM3-MYC en la
inducción de ATF4 y CHOP, los cuales son factores transcripcionales cuya expresión es
controlada positivamente río abajo de la activación de PERK. Con este fin, se cultivaron
las células que sobre-expresan TMBIM3 y las células control MOCK en presencia de Tm
(1 µg/mL) por distintos tiempos, y se evaluó la expresión de ATF4 y CHOP mediante
western blot. No se observaron diferencias significativas entre las células control y
aquellas que sobreexpresan TMBIM3, como lo muestra la figura 25.
Tm (µg/ml)
0
20
40
60
80
100Cé
lula
s PI
pos
itiva
s (%
)PERK WTPERK KO
10,1
***
*
0
***
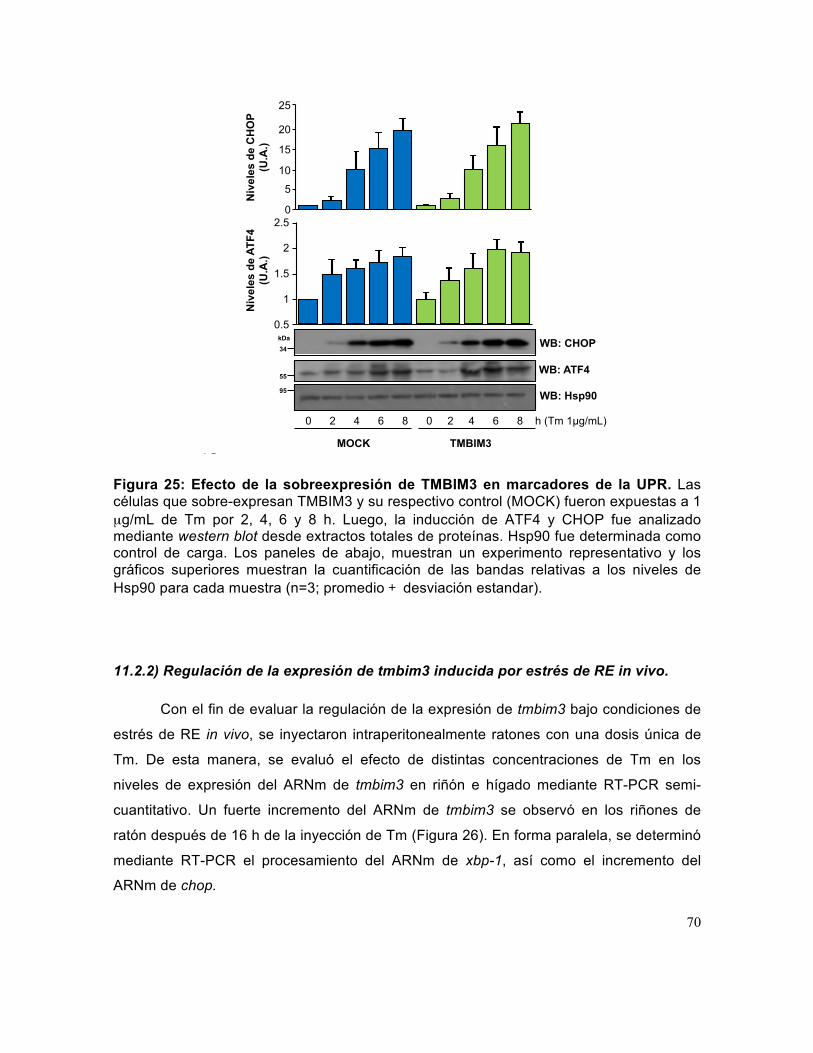
70
Figura 25: Efecto de la sobreexpresión de TMBIM3 en marcadores de la UPR. Las células que sobre-expresan TMBIM3 y su respectivo control (MOCK) fueron expuestas a 1 µg/mL de Tm por 2, 4, 6 y 8 h. Luego, la inducción de ATF4 y CHOP fue analizado mediante western blot desde extractos totales de proteínas. Hsp90 fue determinada como control de carga. Los paneles de abajo, muestran un experimento representativo y los gráficos superiores muestran la cuantificación de las bandas relativas a los niveles de Hsp90 para cada muestra (n=3; promedio + desviación estandar).
11.2.2) Regulación de la expresión de tmbim3 inducida por estrés de RE in vivo.
Con el fin de evaluar la regulación de la expresión de tmbim3 bajo condiciones de
estrés de RE in vivo, se inyectaron intraperitonealmente ratones con una dosis única de
Tm. De esta manera, se evaluó el efecto de distintas concentraciones de Tm en los
niveles de expresión del ARNm de tmbim3 en riñón e hígado mediante RT-PCR semi-
cuantitativo. Un fuerte incremento del ARNm de tmbim3 se observó en los riñones de
ratón después de 16 h de la inyección de Tm (Figura 26). En forma paralela, se determinó
mediante RT-PCR el procesamiento del ARNm de xbp-1, así como el incremento del
ARNm de chop.
1.5 1.5
0 2 4 6 8 0 2 4 6 8 h (Tm 1µg/mL)
MOCK TMBIM3
0.5 1
1.5 2
2.5 0 5
10 15 20
Niv
eles
de
CH
OP
(U.A
.)
Niv
eles
de
ATF4
(U
.A.)
25
55
95
34 kDa
WB: ATF4
WB: CHOP
WB: Hsp90

71
Posteriormente, se determinó la contribución de la vía de PERK y ATF4 en el
control transcripcional del ARNm de tmbin3 in vivo. Con este fin, se expusieron ratones
atf4-/- y ratones atf4+/+ a Tm inyectada intraperitonealmente y se analizó la expresión del
ARNm de tmbim3 mediante RT-PCR semi-cuantitativo y PCR en tiempo real. La figura 27
muestra la inducción del mensajero de tmbim3 en los ratones atf4+/+ expuestos a Tm. Sin
embargo, este incremento se ve disminuido en los ratones atf4-/-. Como control se
determinaron los niveles de actina como control de carga. El mismo efecto en la inducción
del ARNm de tmbim3 se observó en la cuantificación hecha mediante PCR en tiempo real,
como lo muestra la figura 27, en donde se observa una diminución significativa de los
niveles de ARNm de tmbim3 en los ratones atf4-/- comparados con los ratones atf4+/+ al
ser expuestos a Tm mediante inyección intraperitoneal.
De esta manera, de logró determinar que el mRNA de tmbim3 es incrementado
bajo condiciones de estrés de RE y que esta regulación depende de la vía regulada por
PERK y del factor transcripcional ATF4. Este fenómeno ocurre tanto en cultivos celulares
de células MEFs expuestas a estrés de RE inducido por Tm, así como en riñones de
ratones expuestos a inyecciones intraperitoneales de Tm.
Figura 26: Estudio de los niveles de ARNm de tmbim3 en riñones de ratones expuestos a estrés de RE in vivo. Ratones fueron inyectados intraperitonealmente con Tm a las dosis indicadas. 16 h después, los riñones fueron extraídos y el RNA total fue aislado. Posteriormente, se procedió a determinar los niveles de ARNm de tmbim3 y chop mediante RT-PCR. El procesamiento del ARNm de xbp-1 fue analizado para controlar la inducción de estrés de RE mediante RT-PCR, así como los niveles de ARNm de chop Y los niveles de ARNm de actina fueron determinados como control de carga. La figura muestra el resultado de un experimento realizado.
Riñón (Tm µg/g)
Actina
pb
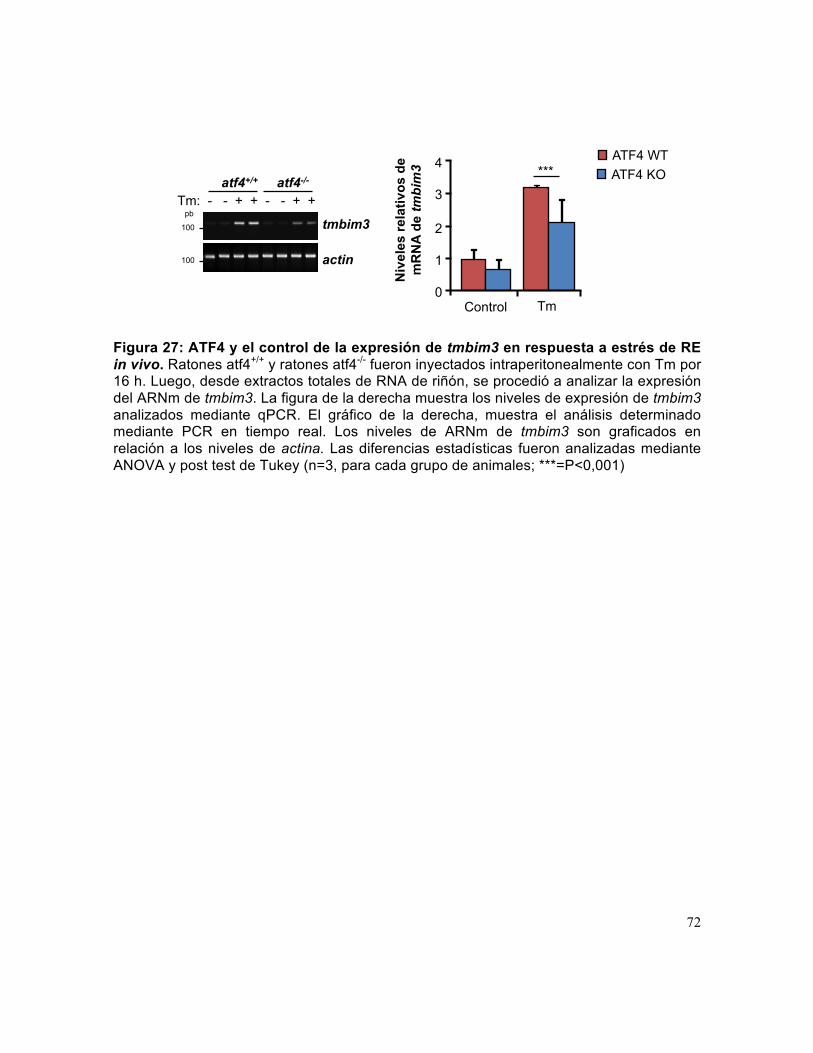
72
Figura 27: ATF4 y el control de la expresión de tmbim3 en respuesta a estrés de RE in vivo. Ratones atf4+/+ y ratones atf4-/- fueron inyectados intraperitonealmente con Tm por 16 h. Luego, desde extractos totales de RNA de riñón, se procedió a analizar la expresión del ARNm de tmbim3. La figura de la derecha muestra los niveles de expresión de tmbim3 analizados mediante qPCR. El gráfico de la derecha, muestra el análisis determinado mediante PCR en tiempo real. Los niveles de ARNm de tmbim3 son graficados en relación a los niveles de actina. Las diferencias estadísticas fueron analizadas mediante ANOVA y post test de Tukey (n=3, para cada grupo de animales; ***=P<0,001)
Niv
eles
rel
ativ
os d
e m
RN
A d
e tm
bim
3
pb
Tm Control 0
1
2
3
4 *** ATF4 WT ATF4 KO
actin 100
tmbim3 100
atf4+/+ atf4-/- Tm: - - + + - - + +

73
11.3) TMBIM3 y el control de la homeostasis del Ca2+
11.3.1) La expresión de TMBIM3 controla la homeostasis del calcio del RE.
Previamente, se demostró que TMBIM6 y TMBIM4 pueden modular negativamente la
liberación de Ca2+ desde el RE, fenómeno que se relaciona directamente con sus
respectivas actividades antiapoptóticas (Chae et al, 2004; Gubser et al, 2007). Por esta
razón, se monitorizó el Ca2+ citosólico en células que sobre-expresan TMBIM3-MYC
expuestas a estímulos que inducen la salida de Ca2+ desde el RE. Con este fin, las células
se incubaron con el sensor de Ca2+ Fluo-4 y posteriormente y en ausencia de Ca2+
extracelular, se determinaron por microscopia de spinning disc (ver metodología) los
cambios en el Ca2+ citosólico inducido por ATP, el cual favorece la producción de IP3
(Rizzuto et al, 2009), con la consecuente liberación de Ca2+ mediada por el receptor de
IP3R. La figura 28 muestra una cinética temporal en donde se observó una significativa
disminución del aumento de Ca2+ citosólico en las células que sobre-expresan TMBIM3-
MYC. Del mismo modo, se observó una disminución del máximo de la señal en aquellas
células que sobre-expresan TMBIM3-MYC. Por otra parte, cuando las células fueron
estimuladas con H2O2 (Figura 28), el cual induce un aumento del Ca2+ citosólico por la
generación de IP3 (Rizzuto et al, 2009), se observó que la sobreexpresión de TMBIM3-
MYC también se traduce en una disminución de la señal de Ca2+ inducida por H2O2.
Por otra parte, se analizó el efecto del tratamiento con Tg, el cual induce un
aumento del calcio citosólico, por inhibición de la bomba SERCA. La figura 29 muestra
que la adición de Tg induce un menor aumento del Ca2+ citosólico en las células que
sobre-expresan TMBIM3-MYC, respecto a las células control, fenómeno que puede
observarse tanto en la cinética, como en la cuantificación del máximo de fluorescencia
inducido por la adición de Tg (Figura 29).
Para complementar estos experimentos, se estudió el efecto en las señales de
Ca2+ de Tg y ATP en células deficientes en TMBIM6 (TMBIM6 KO) transducidas
establemente con el shRNA destinado contra el ARNm de tmbim3. De manera opuesta a
lo observado en las células que sobre-expresan TMBIM3-MYC, la reducción de los niveles
de tmbim3 se reflejó en un aumento en el máximo de Ca2+ liberado desde el RE inducido
con ATP o con Tg. La figura 30 muestra un experimento representativo del efecto inducido
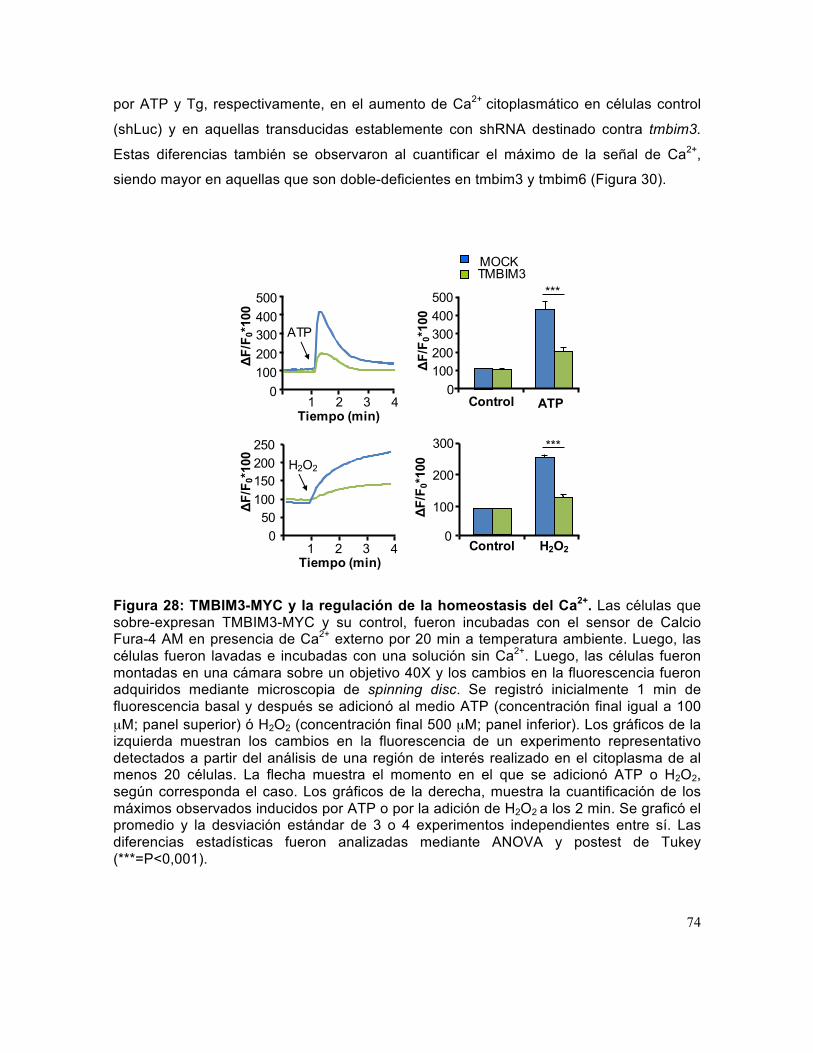
74
por ATP y Tg, respectivamente, en el aumento de Ca2+ citoplasmático en células control
(shLuc) y en aquellas transducidas establemente con shRNA destinado contra tmbim3.
Estas diferencias también se observaron al cuantificar el máximo de la señal de Ca2+,
siendo mayor en aquellas que son doble-deficientes en tmbim3 y tmbim6 (Figura 30).
Figura 28: TMBIM3-MYC y la regulación de la homeostasis del Ca2+. Las células que sobre-expresan TMBIM3-MYC y su control, fueron incubadas con el sensor de Calcio Fura-4 AM en presencia de Ca2+ externo por 20 min a temperatura ambiente. Luego, las células fueron lavadas e incubadas con una solución sin Ca2+. Luego, las células fueron montadas en una cámara sobre un objetivo 40X y los cambios en la fluorescencia fueron adquiridos mediante microscopia de spinning disc. Se registró inicialmente 1 min de fluorescencia basal y después se adicionó al medio ATP (concentración final igual a 100 µM; panel superior) ó H2O2 (concentración final 500 µM; panel inferior). Los gráficos de la izquierda muestran los cambios en la fluorescencia de un experimento representativo detectados a partir del análisis de una región de interés realizado en el citoplasma de al menos 20 células. La flecha muestra el momento en el que se adicionó ATP o H2O2, según corresponda el caso. Los gráficos de la derecha, muestra la cuantificación de los máximos observados inducidos por ATP o por la adición de H2O2 a los 2 min. Se graficó el promedio y la desviación estándar de 3 o 4 experimentos independientes entre sí. Las diferencias estadísticas fueron analizadas mediante ANOVA y postest de Tukey (***=P<0,001).
***
ΔF/F
0*10
0
0100200300400500
1 2 3 4Tiempo (min)
ATP
0100200300400500
ΔF/F
0*10
0
TMBIM3MOCK
Control ATP
050100150200250
1 2 3 4Tiempo (min)
ΔF/F
0*10
0
0
100
200
300
ΔF/F
0*10
0
Control H2O2
H2O2
***
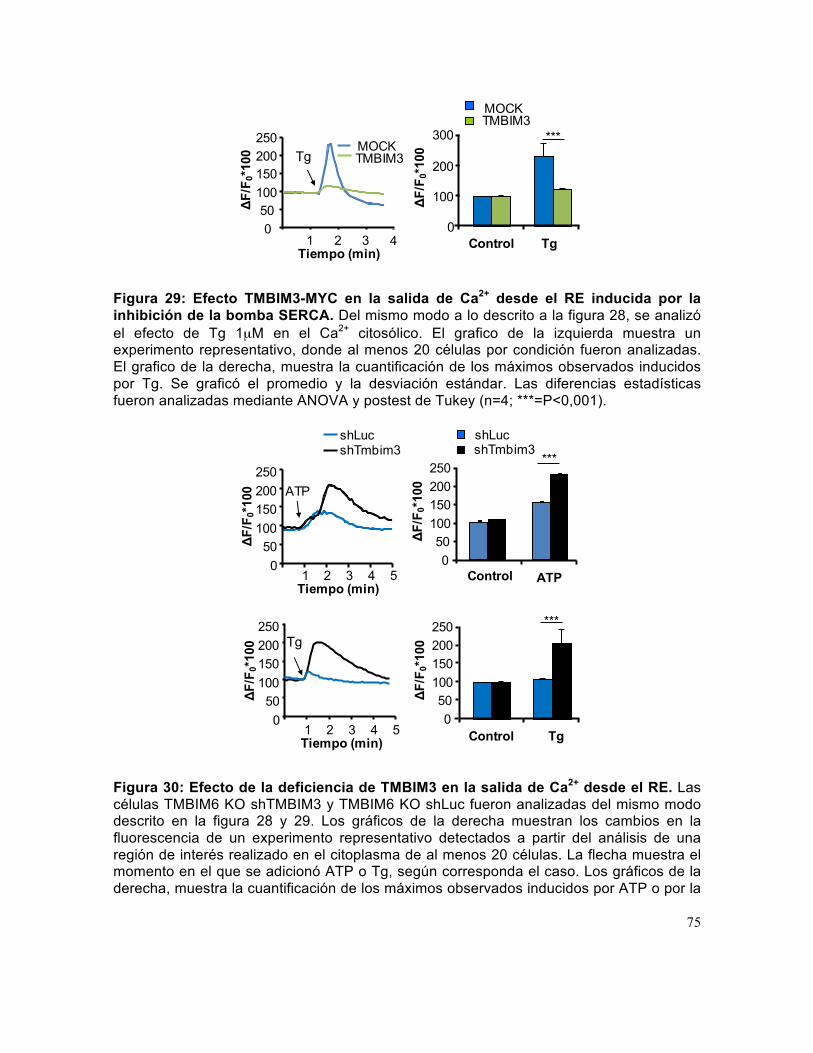
75
Figura 29: Efecto TMBIM3-MYC en la salida de Ca2+ desde el RE inducida por la inhibición de la bomba SERCA. Del mismo modo a lo descrito a la figura 28, se analizó el efecto de Tg 1µM en el Ca2+ citosólico. El grafico de la izquierda muestra un experimento representativo, donde al menos 20 células por condición fueron analizadas. El grafico de la derecha, muestra la cuantificación de los máximos observados inducidos por Tg. Se graficó el promedio y la desviación estándar. Las diferencias estadísticas fueron analizadas mediante ANOVA y postest de Tukey (n=4; ***=P<0,001).
Figura 30: Efecto de la deficiencia de TMBIM3 en la salida de Ca2+ desde el RE. Las células TMBIM6 KO shTMBIM3 y TMBIM6 KO shLuc fueron analizadas del mismo modo descrito en la figura 28 y 29. Los gráficos de la derecha muestran los cambios en la fluorescencia de un experimento representativo detectados a partir del análisis de una región de interés realizado en el citoplasma de al menos 20 células. La flecha muestra el momento en el que se adicionó ATP o Tg, según corresponda el caso. Los gráficos de la derecha, muestra la cuantificación de los máximos observados inducidos por ATP o por la
Tiempo (min)
050100150200250
1 2 3 4
TMBIM3MOCK
ΔF/F
0*10
00
100
200
300
ΔF/F
0*10
0
Control Tg
Thg
***TMBIM3MOCK
050100150200250
1 2 3 4 5
ΔF/F
0*10
0
Tiempo (min)
050100150200250
ΔF/F
0*10
0
Control Tg
Thg
***
shTmbim3shLuc
050100150200250
1 2 3 4
ΔF/F
0*10
0
Tiempo (min)5
050100150200250
ΔF/F
0*10
0ATP
***
Control ATP
shTmbim3shLuc
Tg
Tg
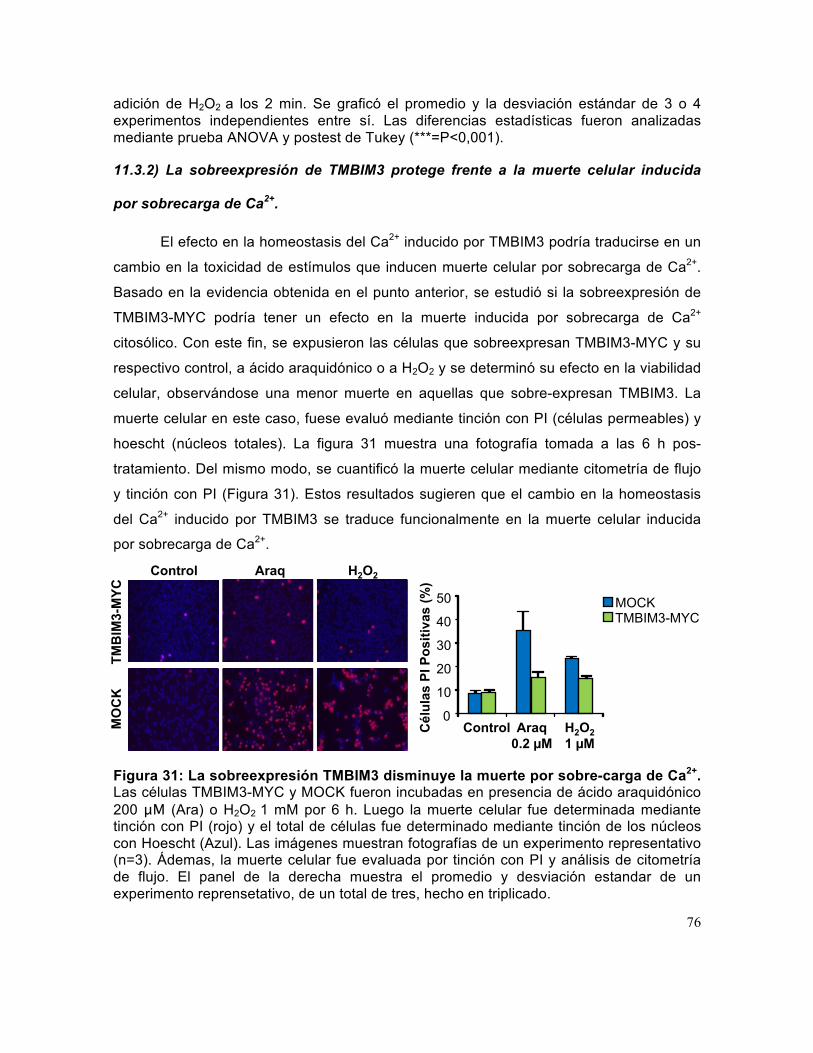
76
adición de H2O2 a los 2 min. Se graficó el promedio y la desviación estándar de 3 o 4 experimentos independientes entre sí. Las diferencias estadísticas fueron analizadas mediante prueba ANOVA y postest de Tukey (***=P<0,001).
11.3.2) La sobreexpresión de TMBIM3 protege frente a la muerte celular inducida
por sobrecarga de Ca2+.
El efecto en la homeostasis del Ca2+ inducido por TMBIM3 podría traducirse en un
cambio en la toxicidad de estímulos que inducen muerte celular por sobrecarga de Ca2+.
Basado en la evidencia obtenida en el punto anterior, se estudió si la sobreexpresión de
TMBIM3-MYC podría tener un efecto en la muerte inducida por sobrecarga de Ca2+
citosólico. Con este fin, se expusieron las células que sobreexpresan TMBIM3-MYC y su
respectivo control, a ácido araquidónico o a H2O2 y se determinó su efecto en la viabilidad
celular, observándose una menor muerte en aquellas que sobre-expresan TMBIM3. La
muerte celular en este caso, fuese evaluó mediante tinción con PI (células permeables) y
hoescht (núcleos totales). La figura 31 muestra una fotografía tomada a las 6 h pos-
tratamiento. Del mismo modo, se cuantificó la muerte celular mediante citometría de flujo
y tinción con PI (Figura 31). Estos resultados sugieren que el cambio en la homeostasis
del Ca2+ inducido por TMBIM3 se traduce funcionalmente en la muerte celular inducida
por sobrecarga de Ca2+.
Figura 31: La sobreexpresión TMBIM3 disminuye la muerte por sobre-carga de Ca2+. Las células TMBIM3-MYC y MOCK fueron incubadas en presencia de ácido araquidónico 200 μM (Ara) o H2O2 1 mM por 6 h. Luego la muerte celular fue determinada mediante tinción con PI (rojo) y el total de células fue determinado mediante tinción de los núcleos con Hoescht (Azul). Las imágenes muestran fotografías de un experimento representativo (n=3). Ádemas, la muerte celular fue evaluada por tinción con PI y análisis de citometría de flujo. El panel de la derecha muestra el promedio y desviación estandar de un experimento reprensetativo, de un total de tres, hecho en triplicado.
TMB
IM3-
MYC
M
OC
K
Control Araq H2O2
MOCK TMBIM3-MYC
0
10
20
30
40
50
Control Araq 0.2 µM
H2O2 1 µM
Cél
ulas
PI P
ositi
vas
(%)

77
11.3.3) La sobreexpresión de TMBIM3 no altera los niveles de Ca2+ RE ni la
expresión de diferentes chaperonas en el RE.
Existen reportes que indican que TMBIM6 y TMBIM4 pueden modular la
homeostasis del Ca2+ RE alterando el contenido neto de Ca2+ en el RE (Chae et al, 2004;
Gubser et al, 2007). Debido a esto, se estudió si la sobreexpresión de TMBIM3 podría
afectar los niveles de Ca2+ presentes en el RE. Con este fin, se transdujeron células
control (MOCK) y aquellas que sobre-expresan TMBIM3-MYC con un adenovirus que
permite la expresión de Aequorina-ER, una proteína destinada al RE y la cual emite luz en
presencia de Ca2+. De esta manera, se determinó que ni la sobreexpresión constitutiva, ni
la sobreexpresión transitoria de TMBIM3-MYC se traduce en un cambio en los niveles de
Ca2+ en el RE (Figura 32). La sobreexpresión de TMBIM3-MYC tampoco se tradujó en un
cambio en la liberación pasiva de Ca2+ desde el RE. Esto último fue demostrado con un
método cuantitativo usando Ca2+ radiactivo (45Ca2+). A partir de monocapaz de membrana
plasmática de ambos tipos celulares, los reservorios de Ca2+ no mitocondriales fueron
cargados con 45Ca2+ y se determinó la salida pasiva de 45Ca2+ (Figura 32). De esta
manera, se demuestró que la expresión de TMBIM3-MYC no alterá ni los niveles de Ca2+
contenidos en el RE, ni la salida pasiva de Ca2+ desde el RE hacia el citoplasma.
Figura 32: Determinación del contenido de Ca2+ del RE. Las células MOCK y TMBIM3 (expresión estable) fueron transducidas por 24 h con un vector adenoviral que permite la expresión de la proteína Aequerina fusionada a una secuencia de destinación al RE (Aequorina-ER). Para estudiar el efecto de la expresión transitoria de TMBIM3, las células MEFs WT fueron transfectadas por 24 h con un vector de expresión plasmidial que permite la expresión de Aequorina-ER. El contenido de Ca2+ en RE fue determinado en un luminómetro de acuerdo a lo descrito en materiales y métodos. El grafico muestra el promedio y desviación estándar de 4 experimentos independientes entre sí. Las diferencias estadísticas fueron analizadas mediante prueba t.
0
100
200
300
[Ca2
+ ]R
E µ
M
Expresión transitoria
Expresión estable
TMBIM3 MOCK MOCK
TMBIM3
0 20 40 60 80
100 120
0 5 10 15 20 Tiempo (min)
Con
teni
do d
e 45
Ca2
+
liber
ado
(% to
tal)
n.s.
n.s.
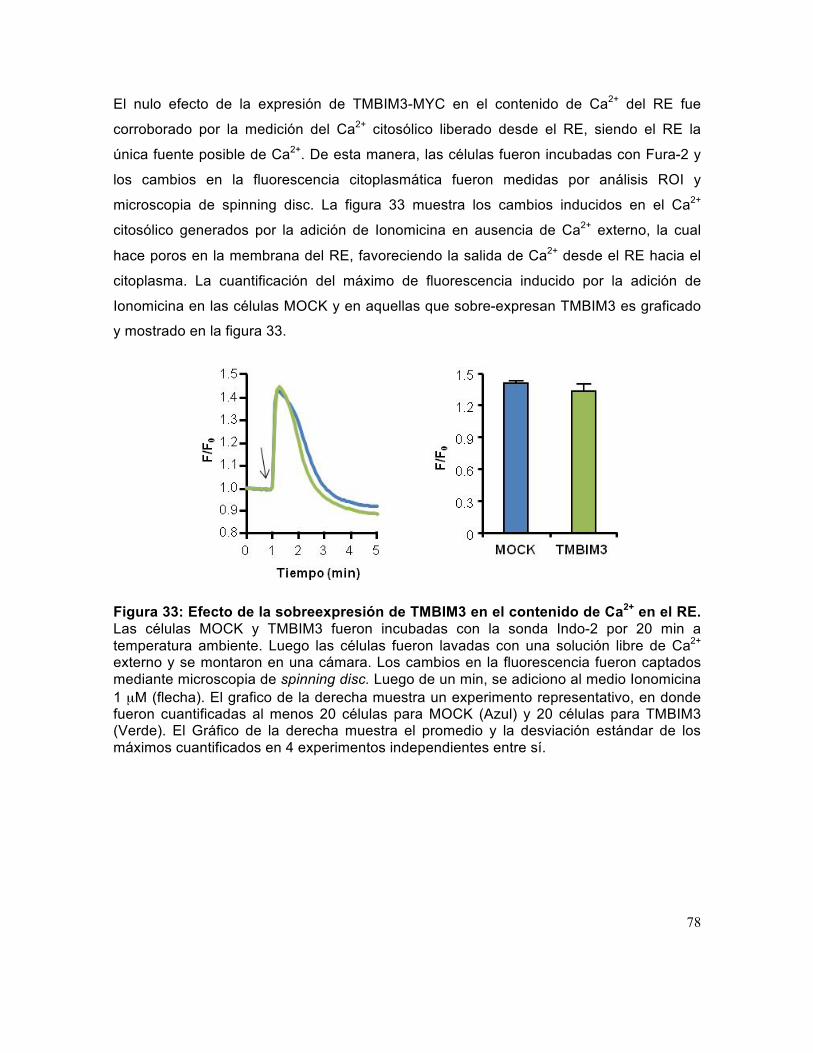
78
El nulo efecto de la expresión de TMBIM3-MYC en el contenido de Ca2+ del RE fue
corroborado por la medición del Ca2+ citosólico liberado desde el RE, siendo el RE la
única fuente posible de Ca2+. De esta manera, las células fueron incubadas con Fura-2 y
los cambios en la fluorescencia citoplasmática fueron medidas por análisis ROI y
microscopia de spinning disc. La figura 33 muestra los cambios inducidos en el Ca2+
citosólico generados por la adición de Ionomicina en ausencia de Ca2+ externo, la cual
hace poros en la membrana del RE, favoreciendo la salida de Ca2+ desde el RE hacia el
citoplasma. La cuantificación del máximo de fluorescencia inducido por la adición de
Ionomicina en las células MOCK y en aquellas que sobre-expresan TMBIM3 es graficado
y mostrado en la figura 33.
Figura 33: Efecto de la sobreexpresión de TMBIM3 en el contenido de Ca2+ en el RE. Las células MOCK y TMBIM3 fueron incubadas con la sonda Indo-2 por 20 min a temperatura ambiente. Luego las células fueron lavadas con una solución libre de Ca2+ externo y se montaron en una cámara. Los cambios en la fluorescencia fueron captados mediante microscopia de spinning disc. Luego de un min, se adiciono al medio Ionomicina 1 µM (flecha). El grafico de la derecha muestra un experimento representativo, en donde fueron cuantificadas al menos 20 células para MOCK (Azul) y 20 células para TMBIM3 (Verde). El Gráfico de la derecha muestra el promedio y la desviación estándar de los máximos cuantificados en 4 experimentos independientes entre sí.

79
Alteraciones directas o indirectas en el contenido de Ca2+ en el RE, podrían
traducirse en cambios compensatorios en la expresión de chaperonas residentes en el
RE. Estos cambios en la fisiología del RE podrían conceder una protección ante estímulos
que inducen estrés de RE. Tomando esto en consideración, se evaluó mediante western
blot el nivel de expresión de foldasas, chaperonas y proteínas involucradas en el control
de calidad de proteínas: PDI, ERp72, BIP/Grp78, ERp57/Grp58 y EDEM. La
sobreexpresión de TMBIM3, no se tradujo en una diferencia significativa en la expresión
de estas proteínas residentes en el RE (Figura 34). Además, se evaluó el efecto de la
sobreexpresión de TMBIM3 en la regulación de la expresión de estas chaperonas
residentes del RE bajo condiciones de estrés RE. Con este fin, se expusieron las células
control y aquellas que sobre-expresan TMBIM3-MYC a Tm por 24 h y luego se evaluó la
expresión de PDI, ERp72, BIP/Grp78, ERp57/Grp58 y de EDEM (Figura 34). De esta
manera, se determinó que la sobreexpresión de TMBIM3-MYC no altera la expresión
basal, ni el aumento que experimentan estas proteínas en respuesta a agentes que
inducen estrés de RE.
Figura 34: Efecto de la sobreexpresión de TMBIM3 en los niveles de expresión de diferentes proteínas residentes del RE. Las células que sobre-expresan TMBIM3 y su control, fueron o no expuestas a 100 ng/mL de Tm por 16 h. Luego, a partir de extractos totales de proteínas, se analizó el contenido de PDI, BIP, ERp72, ERp57 y EDEM mediante western blot. La proteína chaperona residente en el citoplasma, Hsp90, fue determinada como control de carga. Ádemas, se puede observar el cambio en la movilidad electroforética de la proteína EDEM, como consecuencia de la inhibición de la N-glicosilación de proteínas. La figura muestra un experimento representativo de tres hechos de manera independiente.
72
72 BIP
50 PDI
TMBIM3: - + - + Tm
50 ERp57
72
100
ERp72
Hsp90
EDEM
kDa

80
11.3.4) TMBIM3 interactúa con IP3-R y modula su actividad.
Distintas proteínas de la familia BCL-2 pueden regular la homeostasis del Ca2+ en
el RE y su libración desde el RE hacia el citoplasma, proceso el que puede involucrar la
interacción de estas proteínas con IP3-R, lo que podría alterar su actividad (algunos
ejemplos en (Oakes et al, 2005; Rong et al, 2009). Por otra parte, ha sido demostrado que
TMBIM4/GAAP puede interactuar con IP3-R, disminuyendo su actividad (de Mattia et al,
2009). Ademas, dado que la sobreexpresión de TMBIM3-MYC disminuye el incremento de
Ca2+ citosólico inducido por agentes que favorecen la producción de IP3 citoplasmática
(ATP, H2O2), se estudió el efecto de la sobreexpresión de TMBIM3-MYC en la liberación
de Ca2+ inducido directamente por el ligando endógeno del IP3-R, es decir: IP3. A partir
de células cargadas con 45Ca2+, se estudió el efecto de concentraciones crecientes de IP3
en la liberacion de 45Ca2+, observándose que la sobreexpresión de TMBIM-MYC
disminuye significativamente la liberación de 45Ca2+ inducida por IP3 (Figura 35). Esta
experiencia se realizó considerando que el flujo de Ca2+ es unidireccional, debido a la
ausencia funcional de mitocondrias y la bomba SERCA (ver metodología). Tomando esto
en consideración, se estudió la probable asociación entre TMBIM3-MYC y IP3-R mediante
estudios de co-inmunoprecipitación. Con este fin, se procedió a inmunoprecipitar
TMBIM3-MYC desde células MEFs que expresan establemente TMBIM3-MYC,
observándose una débil, pero reproducible co-inmunoprecipitación con el IP3-R3
endógeno (Figura 36). Con el fin de validar este resultado, se procedió a estudiar la
coinmunoprecipitacion entre TMBIM3-MYC y el IP3R endógeno en otros tipos celulares.
De este modo, las células HEK293T y las células Hela, fueron transfectadas con vectores
que permiten la expresión transitoria de TMBIM3-MYC y se determinó que TMBIM3-MYC
puede co-inmunoprecipitar, tanto con IP3-R3, así como con IP3-R1. También se
determinó, mediante inmunoprecipitación en las células MEFs que expresan establemente
TMBIM3-MYC, la asociación entre TMBIM3-MYC y la proteína BCL-2 endógeno, para la
cual ya ha sido demostrado que puede modular la actividad del receptor de IP3
(Vanderheyden et al, 2009).
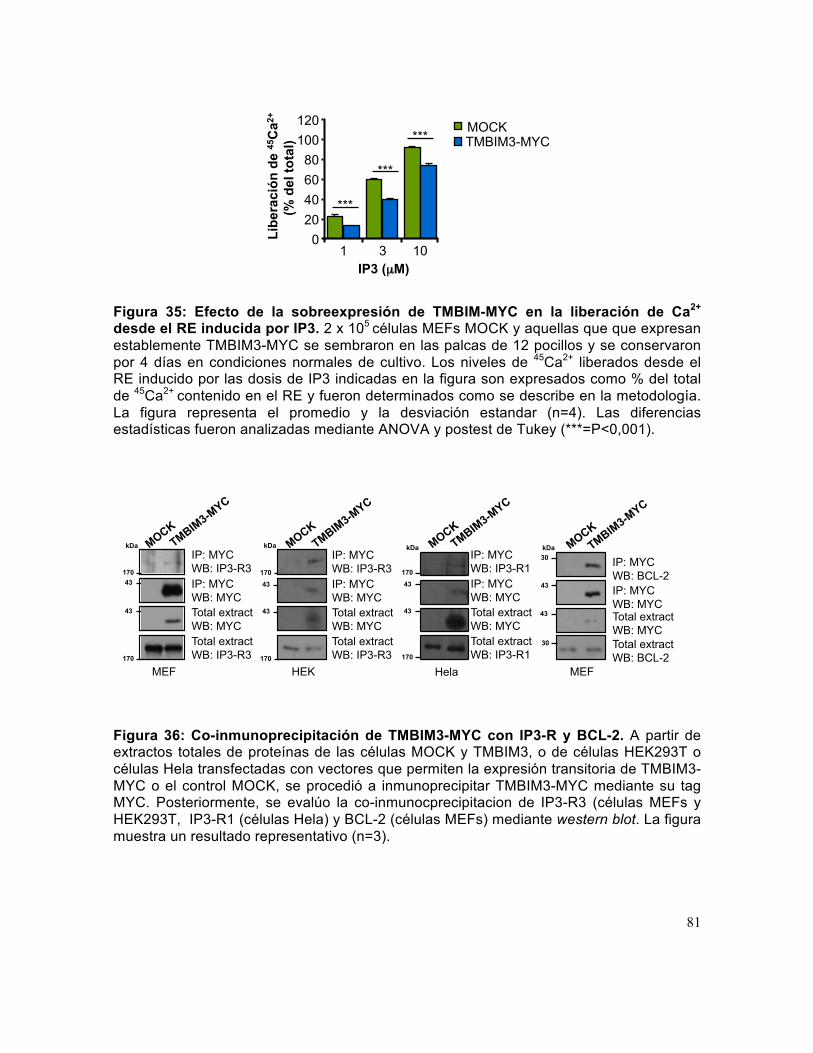
81
Figura 35: Efecto de la sobreexpresión de TMBIM-MYC en la liberación de Ca2+
desde el RE inducida por IP3. 2 x 105 células MEFs MOCK y aquellas que que expresan establemente TMBIM3-MYC se sembraron en las palcas de 12 pocillos y se conservaron por 4 días en condiciones normales de cultivo. Los niveles de 45Ca2+ liberados desde el RE inducido por las dosis de IP3 indicadas en la figura son expresados como % del total de 45Ca2+ contenido en el RE y fueron determinados como se describe en la metodología. La figura representa el promedio y la desviación estandar (n=4). Las diferencias estadísticas fueron analizadas mediante ANOVA y postest de Tukey (***=P<0,001).
Figura 36: Co-inmunoprecipitación de TMBIM3-MYC con IP3-R y BCL-2. A partir de extractos totales de proteínas de las células MOCK y TMBIM3, o de células HEK293T o células Hela transfectadas con vectores que permiten la expresión transitoria de TMBIM3-MYC o el control MOCK, se procedió a inmunoprecipitar TMBIM3-MYC mediante su tag MYC. Posteriormente, se evalúo la co-inmunocprecipitacion de IP3-R3 (células MEFs y HEK293T, IP3-R1 (células Hela) y BCL-2 (células MEFs) mediante western blot. La figura muestra un resultado representativo (n=3).
0 20 40 60 80
100 120
Lib
erac
ión
de 4
5 Ca2
+
(%
del
tota
l) TMBIM3-MYC MOCK
***
1 3 10 IP3 (µM)
***
***
43
43
170
170
MOCK
TMBIM3-M
YC
IP: MYC WB: IP3-R1 IP: MYC WB: MYC Total extract WB: MYC Total extract WB: IP3-R1
43
43
170
170
MOCK
IP: MYC WB: IP3-R3 IP: MYC WB: MYC Total extract WB: MYC Total extract WB: IP3-R3
43
43
170
170
MOCK
TMBIM3-M
YC
IP: MYC WB: IP3-R3 IP: MYC WB: MYC Total extract WB: MYC Total extract WB: IP3-R3
MEF HEK Hela
TMBIM3-M
YC
IP: MYC WB: BCL-2 IP: MYC WB: MYC Total extract WB: MYC Total extract WB: BCL-2
MOCK
TMBIM3-M
YC
43
43
30
30
MEF
kDa kDa kDa kDa

82
Considerando el efecto de la sobreexpresión de TMBIM3 en la homeostasis del
Ca2+, se estudio si este cambio en la expresión de TMBIM3, podría afectar los niveles de
expresión de otras proteínas relacionadas con el control del Ca2+. Con este fin, se
evaluaron los niveles de expresión de BCL-2, SERCA, IP3-R y BAX. No se observaron
cambios significativos entre las células control (MOCK) y las células que sobre-expresan
TMBIM3-MYC (Figura 37).
Figura 37: La expresión de TMBIM3 no genera un cambio en la expresión de algunas proteínas involucradas en el control del Ca2+ RE. A partir de extractos proteicos totales de las células MOCK y de aquellas que sobre-expresan TMBIM3, se procedió a evaluar los niveles de expresión del IP3-R3, SERCA, BCL-2 y TMBIM3 mediante western blot. La expresión de Hsp90 fue determinada como control de carga (n=2).
11.3.5) Efecto de la liberación de Calcio desde el RE en la muerte inducida por
estrés de RE
Hay una asociación directa entre cambios en la homeostasis del Ca2+ relacionados
con el RE y la susceptibilidad al estrés de RE. Aunque inicialmente esta idea parecía ser
especulativa, existen algunos reportes que han podido correlacionar ambos fenómenos.
Con el fin de estudiar si efectivamente el estrés de RE cambia los niveles de Ca2+, se
expusieron las células MEFs WT a estrés de RE por 16 h y se evaluó posteriormente los
cambios en los niveles de Ca2+ citoplasmático inducidos por la adición de Ionomicina en
43
95
26
110 170 IP3-R3
Myc
Hsp90
Bcl2
SERCA
MOCK
TMBIM3-M
YC
kDa
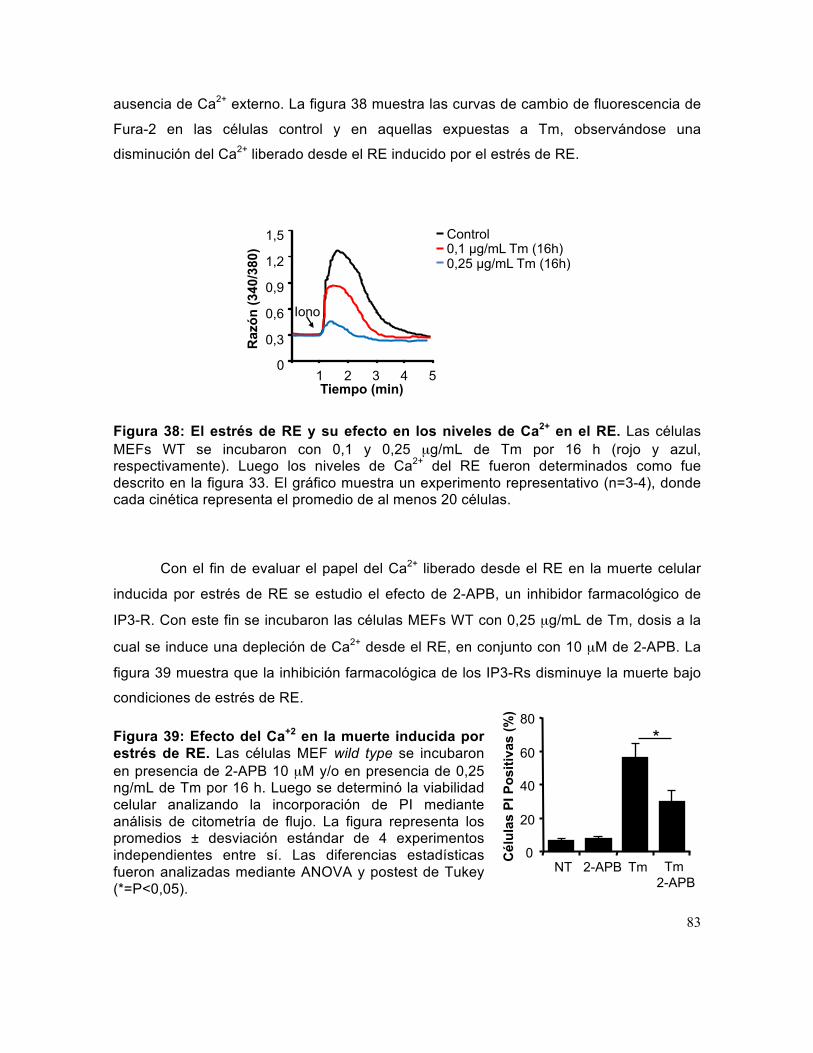
83
ausencia de Ca2+ externo. La figura 38 muestra las curvas de cambio de fluorescencia de
Fura-2 en las células control y en aquellas expuestas a Tm, observándose una
disminución del Ca2+ liberado desde el RE inducido por el estrés de RE.
Figura 38: El estrés de RE y su efecto en los niveles de Ca2+ en el RE. Las células MEFs WT se incubaron con 0,1 y 0,25 µg/mL de Tm por 16 h (rojo y azul, respectivamente). Luego los niveles de Ca2+ del RE fueron determinados como fue descrito en la figura 33. El gráfico muestra un experimento representativo (n=3-4), donde cada cinética representa el promedio de al menos 20 células.
Con el fin de evaluar el papel del Ca2+ liberado desde el RE en la muerte celular
inducida por estrés de RE se estudio el efecto de 2-APB, un inhibidor farmacológico de
IP3-R. Con este fin se incubaron las células MEFs WT con 0,25 µg/mL de Tm, dosis a la
cual se induce una depleción de Ca2+ desde el RE, en conjunto con 10 µM de 2-APB. La
figura 39 muestra que la inhibición farmacológica de los IP3-Rs disminuye la muerte bajo
condiciones de estrés de RE.
Figura 39: Efecto del Ca+2 en la muerte inducida por estrés de RE. Las células MEF wild type se incubaron en presencia de 2-APB 10 µM y/o en presencia de 0,25 ng/mL de Tm por 16 h. Luego se determinó la viabilidad celular analizando la incorporación de PI mediante análisis de citometría de flujo. La figura representa los promedios ± desviación estándar de 4 experimentos independientes entre sí. Las diferencias estadísticas fueron analizadas mediante ANOVA y postest de Tukey (*=P<0,05).
0
0,3
0,6
0,9
1,2
1,5
1 2 3 4 5 Tiempo (min)
Raz
ón (3
40/3
80) 0,1 µg/mL Tm (16h)
0,25 µg/mL Tm (16h)
Control
Iono
0
20
40
60
80
NT 2-APB Tm Tm 2-APB
Cél
ulas
PI P
ositi
vas
(%)
*

84
11.3.6) Efecto del Ca2+ en la inhibición de la apoptosis mediada por TMBIM3
Para evaluar el rol funcional del Ca2+ liberado desde el RE en el control que ejerce
TMBIM3 en la apoptosis inducida por estrés de RE, se estudio en efecto de 2-APB en
conjunto con la disminución de los niveles de tmbim3. La figura 40 muestra que la
disminución de los niveles de tmbim3 aumenta significativamente la apoptosis inducida
por estrés de RE en células TMBIM6 KO. Sin embargo, este aumento significativo se
disminuyó cuando el estrés de RE es inducido en presencia de 2-APB, lo cual demuestra
el efecto protector de TMBIM3 sobre el estrés de RE, a través de la posible modulación de
IP3-R. La figura 39 muestra una tinción vial con Hoechst (azul) y con PI (Rojo), del
experimento previamente descrito. Además, la apoptosis fue evaluada mediante la
cuantificación de los núcleos condensados teñidos con Hoescht, lo que es graficado en la
figura 40.
Figura 40: Papel del Ca2+ en el control de la apoptosis ejercida por TMBIM3. Las células TMBIM6 KO que expresan establemente shLuc o shTmbim3 fueron expuestas a 0,1 µg/mL Tm y/o 10 µM de 2-APB por 16 h. Luego, se evalúo la apoptosis mediante tinción con Hoechst de núcleos fragmentados y condensados y la viabilidad celular, mediante incorporación de PI. Las imágenes de la izquierda muestran un experimento representativo (barra = 60 µm)y el gráfico de la derecha muestra la cuantificación de los núcleos condensados (apoptosis), respecto del total de núcleos por campo. Se graficó el promedio + desviación estándar de 4 experimentos independientes entre sí. Las diferencias estadísticas fueron analizadas mediante ANOVA y postest de Tukey (*=P<0,05; n.s.=no significativo).
0
10
20
30 * n.s.
Apo
ptos
is (%
)
shTmbim3:
+ + +
- + +
- + -
- - +
- - -
Tm: 2-APB:
Hoe
chst
shTmbim3:
+ + +
- + +
- + -
- - +
- - -
Tm: 2-APB:
PI

85
11.4) Rol de TMBIM3 in vivo
11.4.1) TMBIM3 y TMBIM6 tienen actividades antiapoptóticas sinérgicas in vivo en
un modelo de ER estrés en Drosophila melanogaster.
La secuencia primaria de todos los miembros de la familia de proteínas TMBIM es
conservada y presente en distintos organismos, incluyendo Drosophila melanogaster
(Figura 4). La mayoría de los componentes de las vías de la UPR están presentes en
mosca y previamente han sido descritos algunos modelos para estudiar moduladores de
la UPR in vivo (Lisbona et al, 2009; Mori, 2009; Plongthongkum et al, 2007; Ryoo et al,
2007). Para estudiar el efecto de la deficiencia del tmbim3, tmbim6 y la doble deficiencia
de ambos, se usaron moscas transgénicas GAL4/UAS-RNAi que permiten la expresión de
RNA interferente destinados contra el ARNm de tmbim3 o el ARNm de tmbim6. El gen del
factor transcripcional GAL4 se expresa bajo el control del promotor de tubulina, lo cual
permite su expresión ubicua. El factor transcripcional GAL4 induce la expresión de genes
que poseen una secuencia denominada UAS. En este caso, se estableció un sistema de
cruzas que permite la obtención de moscas heterocigotas que poseen GAL4/UAS-RNAi y
un control GFP/UAS-RNAi, el cual no expresa el RNAi interferente. Habiendo establecido
el sistema de cruzas, se procedió a determinar los niveles del ARNm de tmbim3 y tmbim6
en las larvas de moscas de segundo estadio (Figura 41). Además, con el fin de estudiar
posibles roles complementarios entre tmbim3 y tmbim6 en respuesta a estrés de RE in
vivo, se realizó un sistema de cruzas de manera de obtener moscas doble deficientes
para tmbim3 y tmbim6 (Doble RNAi).
Figura 41: Generación de un modelo deficiente en tmbim3 y tmbim6 en Drosophila melanogaster. Extractos de ARN total de larvas de moscas control y de RNAi tmbim3 y
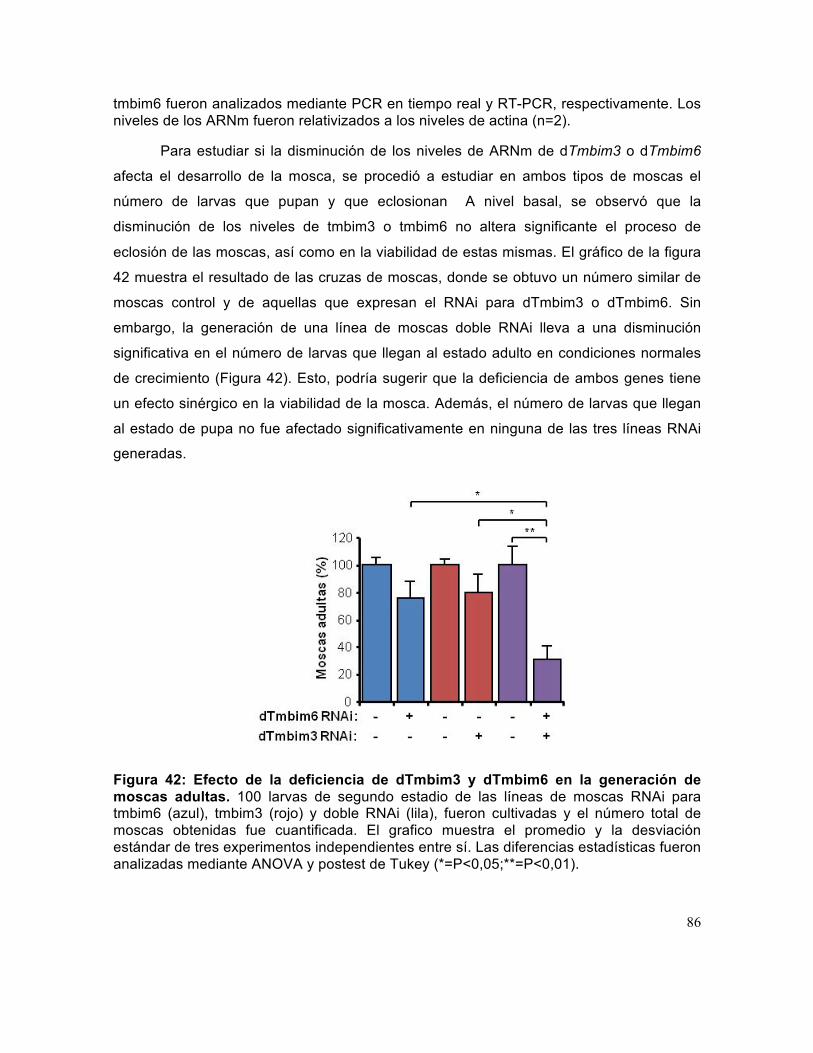
86
tmbim6 fueron analizados mediante PCR en tiempo real y RT-PCR, respectivamente. Los niveles de los ARNm fueron relativizados a los niveles de actina (n=2).
Para estudiar si la disminución de los niveles de ARNm de dTmbim3 o dTmbim6
afecta el desarrollo de la mosca, se procedió a estudiar en ambos tipos de moscas el
número de larvas que pupan y que eclosionan A nivel basal, se observó que la
disminución de los niveles de tmbim3 o tmbim6 no altera significante el proceso de
eclosión de las moscas, así como en la viabilidad de estas mismas. El gráfico de la figura
42 muestra el resultado de las cruzas de moscas, donde se obtuvo un número similar de
moscas control y de aquellas que expresan el RNAi para dTmbim3 o dTmbim6. Sin
embargo, la generación de una línea de moscas doble RNAi lleva a una disminución
significativa en el número de larvas que llegan al estado adulto en condiciones normales
de crecimiento (Figura 42). Esto, podría sugerir que la deficiencia de ambos genes tiene
un efecto sinérgico en la viabilidad de la mosca. Además, el número de larvas que llegan
al estado de pupa no fue afectado significativamente en ninguna de las tres líneas RNAi
generadas.
Figura 42: Efecto de la deficiencia de dTmbim3 y dTmbim6 en la generación de moscas adultas. 100 larvas de segundo estadio de las líneas de moscas RNAi para tmbim6 (azul), tmbim3 (rojo) y doble RNAi (lila), fueron cultivadas y el número total de moscas obtenidas fue cuantificada. El grafico muestra el promedio y la desviación estándar de tres experimentos independientes entre sí. Las diferencias estadísticas fueron analizadas mediante ANOVA y postest de Tukey (*=P<0,05;**=P<0,01).

87
Para estudiar el efecto de tmbim3 o tmbim6 en la muerte inducida por estrés de
RE, se crecieron las larvas de moscas en un medio de crecimiento condicionado con 25
µg/mL de Tm, de manera de inducir estrés de RE en las larvas mediante la incorporación
de la droga a través de la ingesta de comida presente en el medio de crecimiento. Por
esta razón, se evaluó el efecto de la disminución en la expresión de dTmbim3 o dTmbim6
en la inducción de apoptosis en los intestinos de las larvas. Los intestinos de las larvas
fueron disectados y la apoptosis fue evaluada mediante la inmuno-fluorescencia indirecta
de caspasa 3 activada (verde) y el total de las células fue visualizado mediante la tinción
con Hoechst (azul). La figura 43 muestra que en las larvas control no se observó la
detección de caspasa 3 activada, tanto es condiciones basales, como en las larvas
expuestas al tratamiento crónico de una dosis baja Tm. Sin embargo, se detectó
basalmente apoptosis en las líneas RNAi para dTmbim3 y dTmbim6. Este proceso fue
mayor en las larvas expuestas a Tm. Esto sugiere que la disminución de la expresión de
dTmbim3 o dTmbim6 induce la apoptosis, tanto basalmente, como en respuesta a un
estímulo que genera estrés de RE.
Figura 43: La deficiencia de tmbim3 y tmbim3 y su efecto en la apoptosis espontánea y en respuesta a Tm en Drosophila melanogaster. Las larvas de las moscas wild type y de las líneas RNAi y sus respectivos controles fueron crecidas en presencia o ausencia de 50 µg/mL de Tm por 16 h. Luego, en los intestinos de las larvas, se procedió a analizar mediante inmuno-fluorescencia indirecta la presencia de caspasa 3 activa. Los núcleos totales fueron teñidos con DAPI. Las imágenes son parte de un experimento representativo (n=3) y fueron adquiridas mediante microscopía de spinning disc con un objetivo 10x.

88
A demás, se determinó el impacto funcional de la deficiencia de tmbim3 y tmbim6
en la sobrevida de los animales expuestos a estrés de RE inducido por Tm. Con este fin,
se cuantificó el número de larvas que se desarrollan hasta llegar a moscas adultas. La
exposición de las larvas a Tm llevó a una drástica disminución en el número de moscas
que llegan a estado adulto en las líneas que expresan el RNAi destinado contra el ARN
codificante para dTmbim3 y dTmbim6 (38 y 43%, respectivamente). Sin embargo, el
efecto de la Tm en sus respectivos controles, no induce una disminución significativa en
las moscas generadas. Por otra parte, la línea doble RNAi para dTmbim3 y dTmbim6 fue
extremadamente susceptible, tanto en la generación de moscas en condiciones basales,
así como en condiciones de estrés de RE inducido por Tm (Figura 44).
Considerando todos estos resultados, puede sugerirse que la expresión de
dTMBIM3 y dTMBIM6 tienen actividades antiapoptóticas in vivo, y que su expresión es
importante para el desarrollo de la mosca, pues la deficiencia de ambos genes es
parcialmente letal en moscas. Por otra parte, existe un efecto sinérgico en la deficiencia
de ambos genes, respecto a la viabilidad de moscas adultas, así como la resistencia a la
toxicidad en un modelo de estrés de RE in vivo en D. melanogaster.
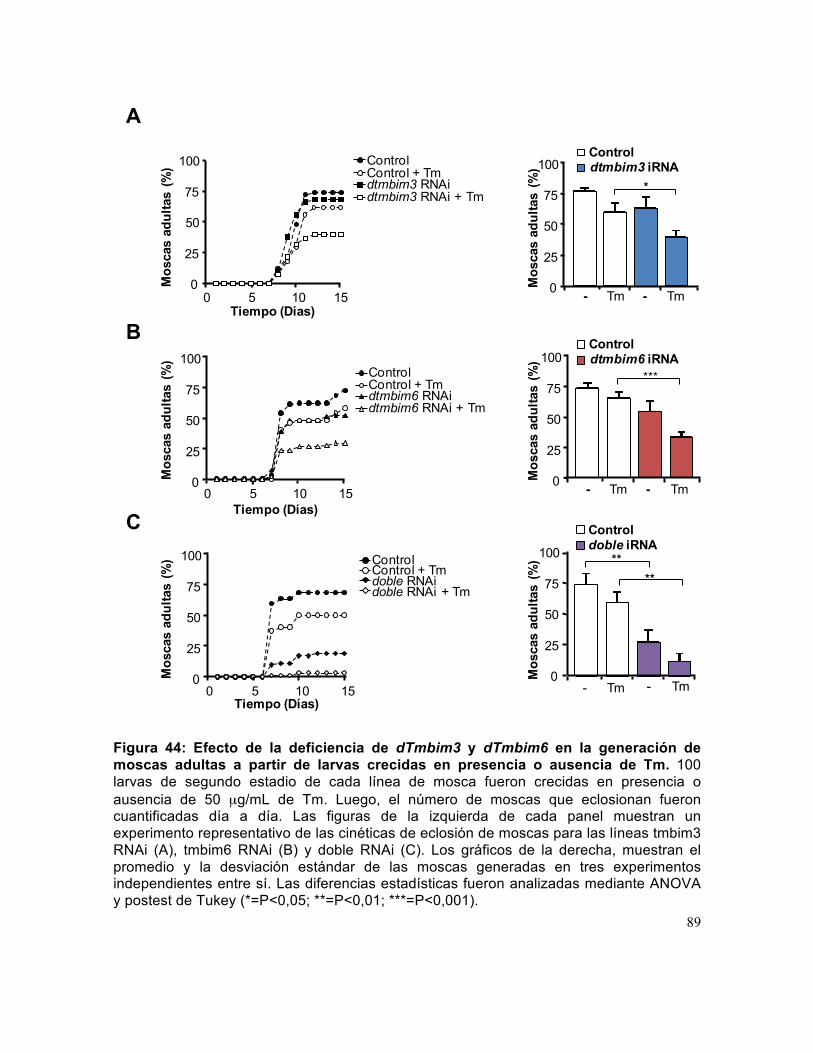
89
Figura 44: Efecto de la deficiencia de dTmbim3 y dTmbim6 en la generación de moscas adultas a partir de larvas crecidas en presencia o ausencia de Tm. 100 larvas de segundo estadio de cada línea de mosca fueron crecidas en presencia o ausencia de 50 µg/mL de Tm. Luego, el número de moscas que eclosionan fueron cuantificadas día a día. Las figuras de la izquierda de cada panel muestran un experimento representativo de las cinéticas de eclosión de moscas para las líneas tmbim3 RNAi (A), tmbim6 RNAi (B) y doble RNAi (C). Los gráficos de la derecha, muestran el promedio y la desviación estándar de las moscas generadas en tres experimentos independientes entre sí. Las diferencias estadísticas fueron analizadas mediante ANOVA y postest de Tukey (*=P<0,05; **=P<0,01; ***=P<0,001).
dtmbim3 iRNA
15Tiempo (Días)
0 5 10
Control Control + Tmdtmbim3 RNAidtmbim3 RNAi + Tm
0
25
50
75
100
- Tm - Tm
*
Control
0
25
50
75
100
0
25
50
75
100
0 5 10 15
Control Control + Tmdtmbim6 RNAidtmbim6 RNAi + Tm
Tiempo (Días)- Tm - Tm
***
0
25
50
75
100
Tiempo (Días)
0
25
50
75
100
0 5 10 15
Control Control + Tmdoble RNAidoble RNAi + Tm
**
- Tm - Tm
**
0
25
50
75
100
A
B
C
Mos
cas
adul
tas
(%)
dtmbim6 iRNAControl
doble iRNAControl
Mos
cas
adul
tas
(%)
Mos
cas
adul
tas
(%)
Mos
cas
adul
tas
(%)
Mos
cas
adul
tas
(%)
Mos
cas
adul
tas
(%)
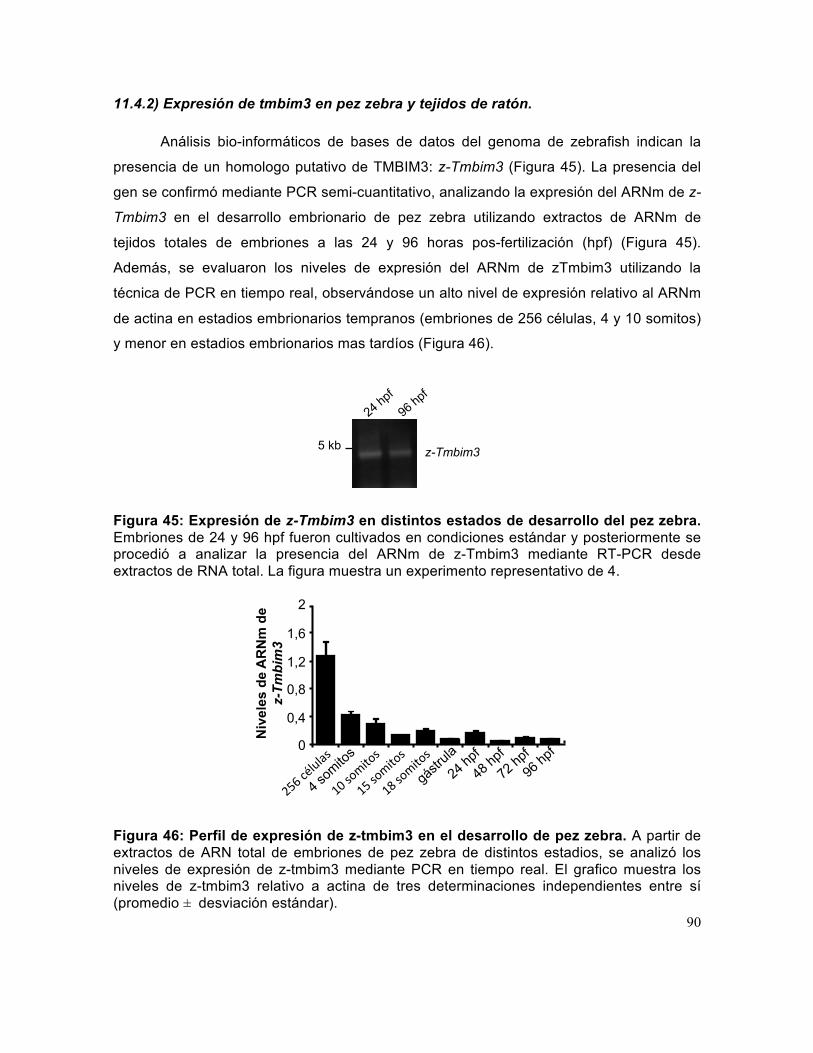
90
11.4.2) Expresión de tmbim3 en pez zebra y tejidos de ratón.
Análisis bio-informáticos de bases de datos del genoma de zebrafish indican la
presencia de un homologo putativo de TMBIM3: z-Tmbim3 (Figura 45). La presencia del
gen se confirmó mediante PCR semi-cuantitativo, analizando la expresión del ARNm de z-
Tmbim3 en el desarrollo embrionario de pez zebra utilizando extractos de ARNm de
tejidos totales de embriones a las 24 y 96 horas pos-fertilización (hpf) (Figura 45).
Además, se evaluaron los niveles de expresión del ARNm de zTmbim3 utilizando la
técnica de PCR en tiempo real, observándose un alto nivel de expresión relativo al ARNm
de actina en estadios embrionarios tempranos (embriones de 256 células, 4 y 10 somitos)
y menor en estadios embrionarios mas tardíos (Figura 46).
Figura 45: Expresión de z-Tmbim3 en distintos estados de desarrollo del pez zebra. Embriones de 24 y 96 hpf fueron cultivados en condiciones estándar y posteriormente se procedió a analizar la presencia del ARNm de z-Tmbim3 mediante RT-PCR desde extractos de RNA total. La figura muestra un experimento representativo de 4.
Figura 46: Perfil de expresión de z-tmbim3 en el desarrollo de pez zebra. A partir de extractos de ARN total de embriones de pez zebra de distintos estadios, se analizó los niveles de expresión de z-tmbim3 mediante PCR en tiempo real. El grafico muestra los niveles de z-tmbim3 relativo a actina de tres determinaciones independientes entre sí (promedio ± desviación estándar).
z-Tmbim3
24 hpf
96 hpf
5 kb
256$células$
4 som
itos
10$somitos$
15$somitos$
18$somitos$
gástr
ula
24 hp
f
48 hp
f
72 hp
f
96 hp
f 0
0,4
0,8
1,2
1,6
2
Niv
eles
de
AR
Nm
de
z-Tm
bim
3

91
Además se estudio el perfil de expresión del ARNm de zTmbim3 en pez zebra.
Con este fin, se realizó una hibridación in situ para analizar el patrón de expresión del
ARNm de z-Tmbim3 en pez zebra, observándose, principalmente, el transcrito en tejido
neuronal, incluyendo cabeza, ojo y medula espinal (Figura 47). La alta expresión del
ARNm de z-Tmbim3 en el sistema nervioso central coincide con lo previamente publicado
para d-Tmbim3 en D. melanogaster.
Figura 47: Patrón de expresión de zTmbim3 en pez zebra. La expresión del ARNm de tmbim3 en pez zebra (z-tmbim3) fue determinada mediante hibridación in situ en embriones de pez zebra en distintos niveles de desarrollo: i: 8 células; ii: 10 somitos y iii, iv y v: 24 hpf. Las imágenes corresponden a visiones laterales (i-iii) y dorsales (iv-v) de los embriones.
En consecuencia con lo anterior, se procedió a analizar los niveles de expresión
del ARNm de m-Tmbim3 en distintos tejidos de ratón mediante PCR en tiempo real
(ovario, útero, bazo, estomago, corazón, hígado, vejiga, cerebro, médula espinal,
cerebelo, riñón, testículos). La figura 48 muestra que tmbim3 en ratón tiene un perfil alto
de expresión relativa en medula espinal, cerebelo, cerebro, riñón y testículos; y bajo en
ovarios, útero, bazo, estómago y riñón.
ISH: z-Tmbim3
i ii
iii iv
v

92
Figura 48: Expresión de tmbim3 en tejidos de ratón. Desde extractos de RNA total de diferentes tejídos de raton, se analizó los niveles de ARNm de tmbim3 de ratón (m-Tmbim3) mediante PCR en tiempo real. El gráfico muestra los niveles de ARNm de m-Tmbim3 relativos al ARNm de actina para dos ratones (promedio ± desviación estándar).
11.4.3) Deficiencia de tmbim3 incrementa la apoptosis durante el desarrollo del pez
zebra.
Con el fin de estudiar el posible efecto de la deficiencia de TMBIM3 en la apoptosis
en pez zebra se diseño un morfolino, el cual actúa como oligonucleótido anti-sentido,
destinado contra la región 5’-UTR y la región ATG del ARNm de zTmbim3. De esta
manera, se procedió a inyectar los ovocitos fertilizados de pez zebra con 1,5 ng de
morfolino destinado contra el ARNm de z-Tmbim3 (z-Tmbim3-MO) o su control
“mismatch”, en el cual se sustituyeron 5 bases de manera de no permitir el correcto
apareamiento entre el oligonucleótido y el ARNm de tmbim3 (Mis-MO). Después de 24 h
pos-inyección, se estudio el patrón y el grado de muerte celular en diferentes tejidos del
animal mediante tinción con la sonda naranjo de acridina, la cual posee un máximo de
excitación/emision de 502/525 nm al estar unida al DNA, proceso que se ve favorecido en
tejido apoptótico. De este modo, la deficiencia en la expresión de z-Tmbim3 se tradujó en
un incrementó del grado de tinción y del número de células teñidas con naranjo de
0
0,05
0,1
0,15
0,2
0,25
Ova
rio
Úter
o Ba
zo
Esto
mag
o Co
razó
n Hí
gado
ve
jiga
Cere
bro
Méd
ula
espi
nal
Cere
belo
Ri
ñón
Test
ícul
o
Niv
eles
de
mR
NA
m-Tmbim3

93
acridina, fenómeno observado principalmente en la cola y la medula espinal (Figura 49), lo
cual coincide con el perfil de expresión observado mediante la hibridación in situ.
Figura 49: Efecto de la deficiencia de z-tmbim3 en la muerte celular durante el desarrollo del pez zebra. Los embriones de pez zebra en estado de una célula fueron inyectados con 1,5 ng del morfolino control (Mis-Mo) o del morfolino destinado contra ARNm de z-Tmbim3 (z-tmbim3-Mo). Luego de 24 h, se procedió a teñir con naranjo de acridina los embriones y posteriormente, se procedió a fotografiar a los embriones de 24 hpf. Las imágenes corresponden a visiones laterales de los embriones tanto en campo claro (paneles de la izquierda), así como de fluorescencia (paneles de la derecha). Las imágenes son representativas de tres experimentos independientes entre sí.
El nivel de muerte celular evaluado por tinción con naranjo de acridina fue
cuantificado mediante un análisis de fluorescencia en la región medula espinal. La figura
50 muestra que z-Tmbim3-MO incrementa significativamente, respecto al control Mis-MO,
el nivel de muerte celular en la medula espinal de los embriones después de haber sido
inyectados por 24 h con los respectivos morfolinos.
Para complementar estos resultados, se estudio la posible activación de caspasa 3
inducida por la disminución de los niveles de expresión de TMBIM3. La figura 50 muestra
la presencia de caspasa 3 activa en embriones de 24 hpf inyectados con z-Tmbim3-MO,
pero no con Mis-MO. Esto se evalúo mediante estudios de inmuno-fluorescencia indirecta.
El nivel de de activación fue cuantificado, observándose un aumento significativo de la
activación de caspasa 3 inducida por la inyección de z-Tmbim3-MO, respecto a su control
Mis-MO (Figura 51).
Mis-MO z-Tmbim3-MO
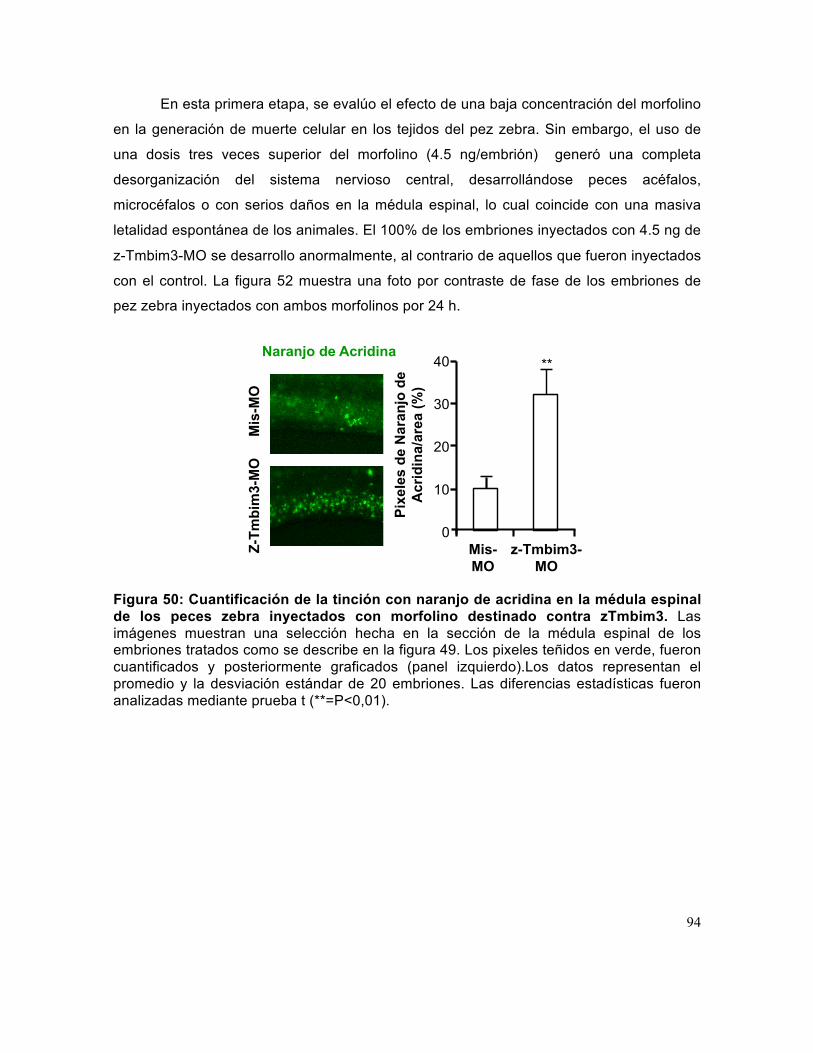
94
En esta primera etapa, se evalúo el efecto de una baja concentración del morfolino
en la generación de muerte celular en los tejidos del pez zebra. Sin embargo, el uso de
una dosis tres veces superior del morfolino (4.5 ng/embrión) generó una completa
desorganización del sistema nervioso central, desarrollándose peces acéfalos,
microcéfalos o con serios daños en la médula espinal, lo cual coincide con una masiva
letalidad espontánea de los animales. El 100% de los embriones inyectados con 4.5 ng de
z-Tmbim3-MO se desarrollo anormalmente, al contrario de aquellos que fueron inyectados
con el control. La figura 52 muestra una foto por contraste de fase de los embriones de
pez zebra inyectados con ambos morfolinos por 24 h.
Figura 50: Cuantificación de la tinción con naranjo de acridina en la médula espinal de los peces zebra inyectados con morfolino destinado contra zTmbim3. Las imágenes muestran una selección hecha en la sección de la médula espinal de los embriones tratados como se describe en la figura 49. Los pixeles teñidos en verde, fueron cuantificados y posteriormente graficados (panel izquierdo).Los datos representan el promedio y la desviación estándar de 20 embriones. Las diferencias estadísticas fueron analizadas mediante prueba t (**=P<0,01).
**
0
10
20
30
40
Pixe
les
de N
aran
jo d
e A
crid
ina/
area
(%)
Mis- MO
z-Tmbim3- MO
Naranjo de Acridina
Z-Tm
bim
3-M
O
Mis
-MO
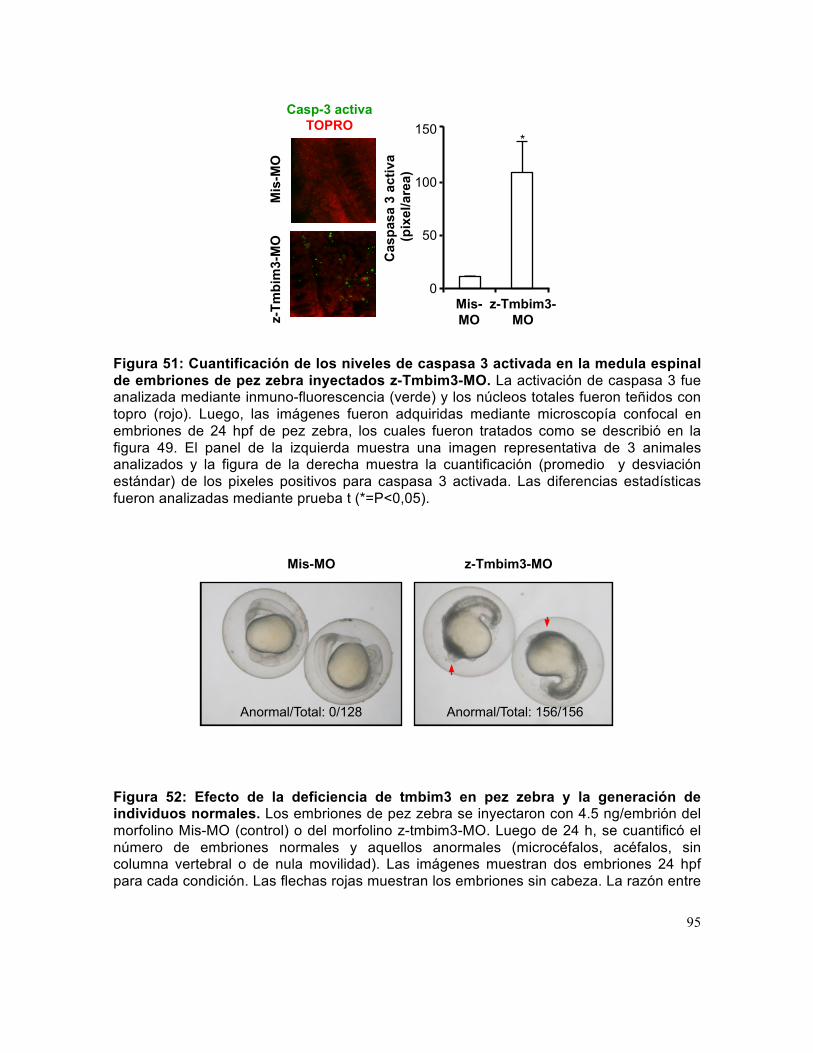
95
Figura 51: Cuantificación de los niveles de caspasa 3 activada en la medula espinal de embriones de pez zebra inyectados z-Tmbim3-MO. La activación de caspasa 3 fue analizada mediante inmuno-fluorescencia (verde) y los núcleos totales fueron teñidos con topro (rojo). Luego, las imágenes fueron adquiridas mediante microscopía confocal en embriones de 24 hpf de pez zebra, los cuales fueron tratados como se describió en la figura 49. El panel de la izquierda muestra una imagen representativa de 3 animales analizados y la figura de la derecha muestra la cuantificación (promedio y desviación estándar) de los pixeles positivos para caspasa 3 activada. Las diferencias estadísticas fueron analizadas mediante prueba t (*=P<0,05).
Figura 52: Efecto de la deficiencia de tmbim3 en pez zebra y la generación de individuos normales. Los embriones de pez zebra se inyectaron con 4.5 ng/embrión del morfolino Mis-MO (control) o del morfolino z-tmbim3-MO. Luego de 24 h, se cuantificó el número de embriones normales y aquellos anormales (microcéfalos, acéfalos, sin columna vertebral o de nula movilidad). Las imágenes muestran dos embriones 24 hpf para cada condición. Las flechas rojas muestran los embriones sin cabeza. La razón entre
0
50
100
150 *
Mis- MO
z-Tmbim3- MO
Cas
pasa
3 a
ctiv
a (p
ixel
/are
a)
Casp-3 activa TOPRO
z-Tm
bim
3-M
O
Mis
-MO
Mis-MO z-Tmbim3-MO
Anormal/Total: 0/128 Anormal/Total: 156/156
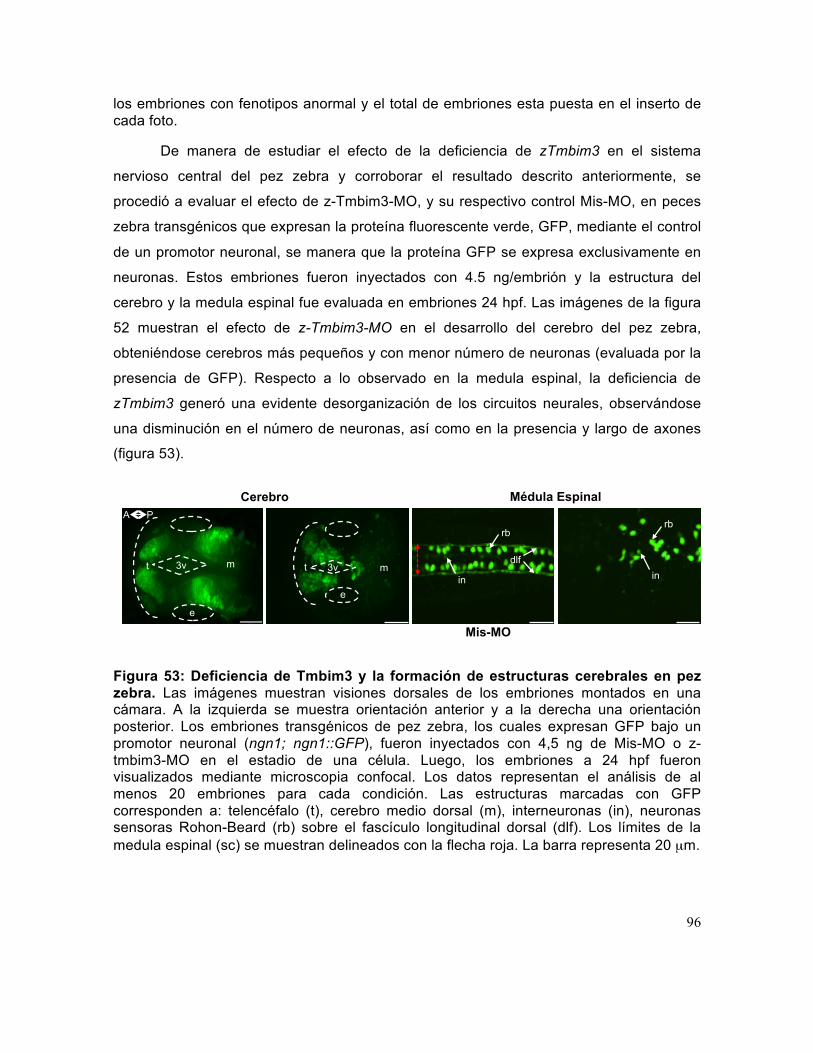
96
los embriones con fenotipos anormal y el total de embriones esta puesta en el inserto de cada foto.
De manera de estudiar el efecto de la deficiencia de zTmbim3 en el sistema
nervioso central del pez zebra y corroborar el resultado descrito anteriormente, se
procedió a evaluar el efecto de z-Tmbim3-MO, y su respectivo control Mis-MO, en peces
zebra transgénicos que expresan la proteína fluorescente verde, GFP, mediante el control
de un promotor neuronal, se manera que la proteína GFP se expresa exclusivamente en
neuronas. Estos embriones fueron inyectados con 4.5 ng/embrión y la estructura del
cerebro y la medula espinal fue evaluada en embriones 24 hpf. Las imágenes de la figura
52 muestran el efecto de z-Tmbim3-MO en el desarrollo del cerebro del pez zebra,
obteniéndose cerebros más pequeños y con menor número de neuronas (evaluada por la
presencia de GFP). Respecto a lo observado en la medula espinal, la deficiencia de
zTmbim3 generó una evidente desorganización de los circuitos neurales, observándose
una disminución en el número de neuronas, así como en la presencia y largo de axones
(figura 53).
Figura 53: Deficiencia de Tmbim3 y la formación de estructuras cerebrales en pez zebra. Las imágenes muestran visiones dorsales de los embriones montados en una cámara. A la izquierda se muestra orientación anterior y a la derecha una orientación posterior. Los embriones transgénicos de pez zebra, los cuales expresan GFP bajo un promotor neuronal (ngn1; ngn1::GFP), fueron inyectados con 4,5 ng de Mis-MO o z-tmbim3-MO en el estadio de una célula. Luego, los embriones a 24 hpf fueron visualizados mediante microscopia confocal. Los datos representan el análisis de al menos 20 embriones para cada condición. Las estructuras marcadas con GFP corresponden a: telencéfalo (t), cerebro medio dorsal (m), interneuronas (in), neuronas sensoras Rohon-Beard (rb) sobre el fascículo longitudinal dorsal (dlf). Los límites de la medula espinal (sc) se muestran delineados con la flecha roja. La barra representa 20 µm.
rb
in
rb
in
dlf
Mis-MO
Cerebro Médula Espinal
t m 3v
A P
e
3v m t
e

97
Por otra parte se estudio si el efecto letal del morfolino destinado contra el ARNm
de zTmbim3 podría generar un daño en estadios más tempranos, previo a las 24 hpf. La
figura 53 muestra que la inyección de 4.5 ng/embrión de z-Tmbim3-MO no afecta el
proceso de gastrulación, el cual fue evaluado en embriones con un 70% de epibolia, lo
cual sugiere que los procesos tempranos de desarrollo no son dependientes de la
expresión de zTmbim3. Dado que cerca a un 80% de los embriones no fueron viables al
ser inyectados con z-Tmbim3-MO, se procedió a evaluar si este fenotipo podría ser
revertido mediante la micro-inyección de ARNm de Tmbim3 de ratón (mTmbim3)
transcrito in vitro. Con este fin, los ovocitos fertilizados de pez zebra fueron inyectados o
con el control Mis-MO, o inyectados simultáneamente con z-Tmbim3-MO más el ARNm
de gap43-cherry (control) o con el ARNm de mTmbim3. La imágenes de la figura 54
muestra que el ARNm de Tmbim3 disminuye la muerte espontánea inducida por la
deficiencia de zTMBIM3, generándose embriones normales a las 24 hpf. Esto se
cuantificó, lo cual es mostrado en el gráfico de la figura 55, donde se observa que el
ARNm de mTmbim3 rescata casi en un 100% el efecto en la sobrevida de los embriones
inyectados con el z-Tmbim3-MO, no así el ARNm de gap43-cherry. Este resultado,
sugiere que las funciones de ambos genes son conservadas
Figura 54. Sobreexpresión de m-tmbim3 y la letalidad inducida por z-Tmbim3-MO en pez zebra. Embriones de pez zebra en estado de una célula fueron inyectados con 4,5 ng de Mis-MO o zTmbim3-MO en presencia o ausencia de 50 µg del ARNm control (Gap43-cherry) o del ARNm de Tmbim3 de ratón (mTmbim3). Las imágenes de la izquierda, muestra los embriones de pez zebra, los cuales fueron fotografiados en campo claro a un 60-70% de epibolia o a 24 hpf. Los datos representan un total de 346 embriones de pez zebra. El gráfico de la derecha, muestra la cuantificación del porcentaje de embriones vivos a las 24 hpf. Los valores representan el promedio (línea) de dos experimentos independientes entre sí (círculos, triángulos y cuadrados; n=127 y 219 embriones fueron analizados en cada experimento, respectivamente).
Mis MO
z-tmbim3-MO + gap43-cherry
z-tmbim3-MO + mTmbim3
20 40 60 80
100
0 Mis MO
gap43 mTmbim3
z-tmbim3-MO
-

98
pb
11.4.4) Generación de un modelo de estrés de RE en pez zebra in vivo.
En este estudio se evaluó la contribución de z-Tmbim3 en la apoptosis generada
por estrés de RE en pez zebra. Con este fin, se generó un modelo de estrés de RE en pez
zebra, mediante la adición de Tg al medio de mantención de los peces de 24 hpf. Usando
este método, se observó una evidente inducción del procesamiento no convencional del
ARNm de xbp-1 a las 4 h de tratamiento con 1 y 5 µM de Tg, lo cual es indicativo de la
generación de estrés de RE. Además, mediante PCR en tiempo real se observó un
aumento de la expresión del ARNm de tmbim3 y chop en los embriones de 24 hpf
expuestos a Tg por 4 h (Figura 55). Estos resultados sugieren que es posible estudiar
UPR en el modelo de estrés de RE en pez zebra in vivo.
Figura 55: UPR en pez zebra: Embriones de pez zebra a las 24 hpf fueron expuestos a Tg 1 y 5 µM por 4 h. Luego, el procesamiento del ARNm de xbp-1 fue determinado mediante RT-PCR. La figura de la izquierda, muestra los fragmentos generados por el PCR correspondientes a la forma procesada (s) y no procesada (u) están indicadas en la figura. Los niveles de actina fueron monitorizados como control de carga. El grafico de la derecha muestra en los embriones descritos anteriormente, los niveles de z-Chop y z-tmbim3 relativos a actina evaluados por PCR en tiempo real. Los datos representan el promedio y la desviación estandar de dos experimentos independientes entre sí realizado en triplicado.
11.4.5) TMBIM3 protege del estrés de RE en pez zebra in vivo.
Para evaluar el efecto de la expresión de TMBIM3 en la muerte celular inducida
por estrés de RE, y considerando el efecto de “rescate” realizado por el ARNm de
mTmbim3 ante la disminución de la expresión de zTmbim3 endógeno, se procedió a
100
zActin
Tg (µM) 0 1 5
Niv
eles
de
mR
NA
0 - Tg - Tg
0.4
0.8
zchop zTmbim3
zXBP-1u zXBP-1s
100

99
sobreexpresar la proteína mediante la micro-inyección del ARNm de mTmbim3 en
ovocitos fertilizados en estadio de una célula. La presencia del ARNm de mTmbim3 fue
corroborada en los embriones de 24 hpf mediante PCR semi-cuantitativo, así como la
presencia del ARNm de zTmbim3 endógeno (Figura 56).
Figura 56: Análisis de la expresión de m-Tmbim3 en pez zebra. Embriones de pez zebra fueron inyectados con 50 pg de ARNm de tmbim3 de ratón sintetizado in vitro (mTmbim3). Luego de 24 h, la presencia del ARNm de mTmbim3 exógeno fue determinado mediante RT-PCR. Los niveles del ARNm de z-tmbim3 endógeno fueron evaluados mediante RT-PCR como control del experimento.
Posteriormente, se evaluó en embriones de 8 hpf, el efecto en la muerte celular
inducido por un tratamiento de Tg por 4 h. Las imágenes de la figura 57 muestran el
efecto en la muerte celular de dos dosis de Tg en los embriones utilizados como control y
aquellos que sobreexpresan mTmbim3. La muerte celular fue evaluada mediante la
tinción de los embriones con naranja de acridina y posteriormente adquisición de las
imágenes con un microscopio de fluorescencia. El inserto en cada imagen, muestra una
foto tomada al mismo plano con la luz transmitida. En la figura 56 se puede observar que
la sobreexpresión de m-Tmbim3 disminuye la muerte ante una exposición aguda a estrés
de RE inducida por Tg in vivo.
100
100 zTmbim3
mTmbim3
Contro
l m
Tmbi
m3
pb

100
Figura 57: tmbim3 y el control de la muerte celular inducida por Tg en pez zebra in vivo. Embriones de pez zebra a las 8 hpf fueron expuestos a Tg por 4 h. Luego la muerte celular fue determinada mediante tinción de los embriones con naranjo de acridina y evaluada mediante fluorescencia. Las imágenes insertas en el extremo superior izquierdo de cada imagen, muestra las fotografías adquiridas en el mismo plano que la fotografía fluorescente, pero con el campo claro. Los datos representan tres experimentos independientes entre sí, donde 3-5 embriones fueron analizados por cada determinación.
Además, se observó que la exposición a Tg por 4 h genera una curvatura en la
cola del pez zebra. Este fenómeno fue revertido completamente en aquellos embriones
que sobre-expresan m-Tmbim3, lo cual sugiere un amplio papel protector de tmbim3 in
vivo (Figura 58). Estos resultados sugieren en conjunto que z-Tmbim3 podría tener un
papel importante en control de la apoptosis durante el desarrollo y en la regulación de la
muerte inducida por estrés de RE in vivo.
Figura 58: c Los cambios en la morfología de las colas de los embriones de pez zebra fueron determinados a las 24 hpf en los embriones tratados como se describió en la figura 54. Los datos son representativos de tres experimentos independientes entre sí, donde un total de 10-15 embriones fueron analizados para cada condición por experimento.

101
12) Discusión.
12.1) TMBIM3 es parte de una familia conservada de proteínas con actividad
antiapoptótica: el mundo pre-bcl2.
La regulación fina de la apoptosis es esencial para el desarrollo de los organismos
multicelulares y la mantención de la homeostasis de los tejidos en el adulto. Respecto a
este punto, gran parte de los estudios de la apoptosis se han enfocado en entender el
papel de las proteínas de la familia BCL-2 en este proceso, y específicamente en definir y
esclarecer los eventos moleculares y las señales bioquímicas que convergen en la
mitocondria. Esto puede ser explicado, en gran parte, por una razón histórica. A finales
de los años 80 y principios de los 90, se identificó el gen bcl-2 (Negrini et al, 1987;
Tsujimoto et al, 1987), cuya sobreexpresión en linfocitos B podría ser la causa de una
forma particular de un linfoma, contribuyendo a la patogénesis en esta forma de leucemia
(Butturini & Gale, 1990; Crescenzi et al, 1988; Ngan & Berinstein, 1990). Posteriormente,
BCL-2 fue identificada como una proteína localizada en la mitocondria (Reed et al, 1991),
hechos que ayudaron a establecer el papel de estas proteínas en la muerte celular
programada y en la carcinogénesis. Posteriormente, el descubrimiento de BCL-2 llevo a la
identificación de distintos homólogos de secuencia primaria, capaces de interactuar entre
ellos lo que llevo a la definición de una familia de proteínas denominada familia BCL-2
(Kozopas et al, 1993; Larsen, 1994; Lin et al, 1996; Meijerink et al, 1995; Reed, 1995;
Takayama et al, 1995; Yang et al, 1995). Aunque las proteínas de la familia BCL-2 son
esenciales en la regulación de la apoptosis, esta actividad ha sido limitada básicamente a
modelos mamíferos, debido a que la mayoría de los miembros de la familia BCL-2 son
pobremente conservados en otras especies, pese a que la muerte celular programada
tiene un papel fundamental en el desarrollo y mantención fisiológica de los organismos
multicelulares (Degterev & Yuan, 2008; Youle & Strasser, 2008). Un ejemplo de esto es la
presencia de solo dos homólogos putativos de componentes de la familia de proteínas
BCL-2 en D.melanogaster, con papeles controversiales en la muerte celular programada
(Galindo et al, 2009; Kornbluth & White, 2005; Sevrioukov et al, 2007; Wu et al, 2010) y
recientemente fueron descritos funcionalmente algunos miembros de esta familia de
proteínas en pez zebra (Jette et al, 2008). Sin embargo, en otros organismos, incluyendo

102
levadura y plantas, ningún homologo funcional de la familia BCL-2 ha sido descrito
(Ameisen, 2002; Hengartner, 1996; Hengartner & Horvitz, 1994; Youle & Strasser, 2008)
pese a ser reconocida diversas formas de muerte celular programada en estos
organismos. Esto, contrasta fuertemente con los estudios genéticos y bio-informáticos que
han descrito a los miembros de la familia TMBIM como genes altamente conservados en
la evolución (Chae et al, 2003; Hu et al, 2009; Reimers et al, 2008), con un posible
ancestro en común en levadura con una controvertida actividad antiapoptótica (Buttner et
al, 2011; Cebulski et al, 2011; Huckelhoven, 2004). Incluso, ha sido descrita la presencia
de homólogos en virus (Gubser et al, 2007) o en bacterias (van Stelten et al, 2009).
La proteína TMBIM6/BI-1 es el miembro de la familia TMBIM mejor caracterizado a
la fecha, y es la única cuya actividad antiapoptótica ha sido validada in vivo. Existen
pocos estudios que se han enfocado en estudiar el posible papel antiapoptótico de otros
miembros de la familia TMBIM en modelos celulares. Sin embargo, ninguno de estos
trabajos ha logrado descifrar el probable mecanismo por el cual ejercen un papel
antiapoptótico, y en ninguno de ellos se ha ensayado la probable interacción entre los
miembros de la familia TMBIM lo cual contrasta fuertemente con toda la literatura
existente para la familia BCL-2.
Actualmente, se ha descrito para TMBIM2/LFG que se localiza principalmente en
membrana citoplasmática y específicamente, en microdominios o balsas lipídicas
denominadas “lipid-raft” y existen varios estudios que muestran que su sobreexpresión
disminuye la apoptosis inducida por el ligando de Fas, con una relevante actividad en
neuronas (Beier et al, 2005; Fernandez et al, 2007; Schweitzer et al, 2002; Somia et al,
1999). Para TMBIM1/RECS1 se describió que se localiza en lisosomas, aparato de Golgi
y membrana plasmática (Zhao et al, 2006b; Zhao et al, 2006c), y solo un estudio ha
demostrado su impacto en la apoptosis inducida por el ligado de Fas (Shukla et al, 2011).
TMBIM4/GAAP se localiza exclusivamente en el aparato de Golgi y su expresión
disminuye la muerte inducida por diversos estímulos intrínsecos (sobreexpresión de BAX
y tratamientos con estaurosporina, cisplatino y ceramida c2) y estímulos que inducen
apoptosis extrínseca (ligando de Fas y TNFα) (de Mattia et al, 2009; Gubser et al, 2007).
TMBIM5/GHITM se localiza en la mitocondria y su expresión previene la fragmentación
mitocondrial y la liberación de citocromo c inducido por tratamientos con actinomicina D

103
(Oka et al, 2008). Respecto a trabajos que vinculen a TMBIM3/GRINA con su probable
papel antiapoptótico, existe un trabajo en donde se analizó una biblioteca de cDNA y se
identificó un fragmento del cDNA de TMBIM3/GRINA como un posible inhibidor de la
muerte inducida por la toxina Shiga, mediante la retención del receptor Gb3 en el aparato
de Golgi. Además, la expresión del TMBIM1,2,4,6, pero no TMBIM5 ni el fragmento
completo de TMBIM3, también presentaron un efecto protector ante la toxina Shiga
(Yamaji et al, 2010). Respecto al papel de TMBIM3 in vivo, recientemente, un trabajo
confirmó la expresión de TMBIM3 en el sistema nervioso central y además publicó la
generación de un ratón deficiente en TMBIM3, el cual no presenta un fenotipo espontáneo
(Nielsen et al, 2011). Sin embargo no se realizaron estudios en donde evaluarán la
susceptibilidad de este ratón ante modelos patológicos, como lo fue descrito para
TMBIM6. Por otra parte, en este estudio se corroboró el tamaño predicho de la proteína
(38 Kda) y su localización tanto en aparato de Golgi, como en RE, mediante estudios de
inmuno-fluorescencia indirecta, lo cual coincide con los resultados presentados en esta
tesis. Respecto al análisis de la actividad antiapoptótica, en este trabajo se presentó un
discreto análisis en donde la expresión transitoria de TMBIM3 disminuyó la muerte
inducida por H2O2 en células COS. Esto coincide con los resultados obtenidos en esta
tesis en donde la sobreexpresión estable de TMBIM3 inhibe la muerte, así como la sobre-
carga de Ca2+ citosólico inducida por H2O2 en las células MEFs.
A la fecha, los estudios existentes que han descrito el papel de algunos miembros
de la familia TMBIM in vivo se han basado en TMBIM6. La expresión de TMBIM6 ha sido
propuesta como un factor importante en el desarrollo del cáncer (Lee et al, 2010; Reimers
et al, 2008), al favorecer la transformación e inmortalización celular, inhibir la apoptosis en
células tumorigénicas y aumentar la metástasis (Lee et al, 2010). Por otra parte, los
ratones deficientes en TMBIM6 son viables y no presentan un fenotipo espontáneo
aparente. Sin embargo, la deficiencia de TMBIM6 en ratones se tradujo en un aumento
del área del tejido renal dañado en respuesta a isquemia y reperfusión (Chae et al, 2004;
Krajewska et al, 2010); así como, paradójicamente, en un aumento en la regeneración del
hígado después de una hepatoctomía parcial (Bailly-Maitre et al, 2007). Por otra parte, la
sobreexpresión de TMBIM6 mediante adenovirus en hígado mejoró el metabolismo
asociado a glucosa en ratones con dieta normal y en ratones con dieta rica en grasa y
revertió la hiperglicemia en ratones espontáneamente obesos (Bailly-Maitre et al, 2010). A

104
demás, mediante el uso de lentivirus codificantes para shRNA destinados contra el RNAm
de TMBIM6 se determinó que la disminución de los niveles de TMBIM6 previene la
proliferación ex vivo de células cancerígenas, así como el crecimiento de los tumores de
carcinoma nasofaringeal (Li et al, 2011b). Por otra parte, los estudios para establecer el
papel de TMBIM6 se han extendido a otros sistemas, por ejemplo, recientemente se
determinó que la expresión de TMBIM6 es necesaria para la inhibición de la apoptosis
regulada por la proteína NIeH durante la infección de Escherichia coli enteropatogenica
(Hemrajani et al, 2010). Ratones deficientes en TMBIM2 también son viables. Sin
embargo la eficiencia de este gen induce un aumento en la susceptibilidad de sufrir
dilatación de la aorta, así como un aumento en la degeneración media cística, lo cual
podría hacer presumir un importante rol de TMBIM2 en la mantención de la homeostasis y
fisiología cardiovascular (Zhao et al, 2006b).
Otro ejemplo de la relevancia de los miembros de la familia TMBIM, así como su
alto grado de conservación es la presencia de un homologo de TMBIM4 en Vaccinia virus,
gen que conserva la actividad antiapoptótica y que además controla positivamente la
virulencia del virus en un modelo de infección murina (Gubser et al, 2007).
Por último, y como otra evidencia de que el alto grado de conservación de TMBIM
no se restringe solamente a su secuencia, recientemente un grupo publicó que el único
gen que ha sido indicado como un homologo putativo para los miembros de la familia
TMBIM en levadura conserva, tanto su localización sub-celular, limitándose al RE, así
como su actividad antiapoptótica frente a distintos estímulos (estrés de temperatura y
metabólico, muerte inducida por etanol y estrés de RE) así como su capacidad de inhibir
la UPR (Cebulski et al, 2011), efecto que previamente había sido descrito por nuestro
laboratorio en un modelo murino (Lisbona et al, 2009) y corroborado posteriormente por el
grupo de Bailly-Maitre (Bailly-Maitre et al, 2010). Todos estos ejemplos, apuntan a un
hecho que con el pasar del tiempo se ha vuelto más evidente, y es que las proteínas de
la familia TMBIM, no solo presentan un alto grado de conservación, si no que presentan
una gran relevancia en diversos procesos biológicos.
En este trabajo, se ha investigado en diversos modelos tanto in vitro como in vivo,
el posible impacto de TMBIM3 en la regulación de la apoptosis. Basado en modelos de
ganancia y pérdida de función, en esta tesis se ha descrito que TMBIM3 presenta una

105
conservada actividad supresora de la apoptosis inducida por estrés de RE, sin afectar la
muerte inducida por otros estímulos que inducen apoptosis, como TNFα, daño al DNA
privación de nutrientes o ausencia de factores de crecimientos. A demás, la reducción de
los niveles de TMBIM3 en ausencia de su ortólogo TMBIM6 induce apoptosis espontánea
en células MEFs e incrementa la susceptibilidad de estas células a la muerte inducida por
estrés de RE, lo cual hace presumir que ambas proteínas sinergizan en sus actividades
antiapoptóticas. De manera similar, en estudios de pérdida de función in vivo, se generó
un modelo en D. melanogaster en donde se evidenció que TMBIM3 y TMBIM6 tienen
potentes y sinérgicas actividades pro-sobrevida bajo condiciones de estrés de RE. Esto,
se suma a la evidencia que muestra que la generación de moscas deficientes en tmbim3 y
tmbim6 induce apoptosis espontanea en los intestinos de las larvas, así como la
disminución de la viabilidad de las moscas, lo cual podría reflejar un papel relevante en la
mantención de la viabilidad celular. Interesantemente, el efecto de TMBIM3 y TMBIM6 en
la mantención de la sobrevida y muerte de los animales contrasta con lo descrito para los
únicos dos ortólogos de BCL-2 y BAX en mosca, en donde la generación de mutantes
para buffy (BCL-2) o debcl (BAX) no se tradujo en un fenotipo espontáneo en D.
melanogaster (Galindo et al, 2009; Sevrioukov et al, 2007). La importancia de la familia
BCL-2 en D. melanogaster en la mantención y control de la muerte celular podría ser
secundaria respecto a la familia TMBIM, dado que todos los miembros descritos de la
familia TMBIM están presentes en mosca y presentan, en algunos casos, hasta el doble
de grado de identidad y similitud respecto a buffy y debcl.
Por otra parte, en esta tesis se estudio la función in vivo de TMBIM3 en un modelo
vertebrado. Con este fin, se corroboró la presencia del z-TMBIM3 en pez zebra, se clonó
el cDNA y se sintetizó un morfolino con el fin de bloquear la traducción del mRNA. La
disminución de la expresión de z-TMBIM3 en pez zebra, generó un aumento de la
apoptosis durante el desarrollo y produjo drásticas alteraciones en la morfología del
cerebro y la sobrevida neuronal. Por otra parte, la expresión de TMBIM3 de ratón en pez
zebra, rescató completamente el fenotipo observado al bloquear la expresión de su
homologo endógeno en pez zebra, lo cual indica que sus funciones son conservadas. En
consistencia con el fenotipo en el sistema nervioso central descrito en esta tesis, destaca
la evidencia que indica que TMBIM3 es uno de los 28 genes que se expresan
preferentemente en la zona marginal en el desarrollo de la corteza cerebral del ratón

106
(Tachikawa et al, 2008), así como el patrón de expresión en el sistema nervioso central
descrito en D. melanogaster (Pellicena-Palle & Salz, 1995) o el estudio de expresión del
ARNm hecho mediante PCR en tiempo real para Tmbim3 en tejidos de ratón durante el
desarrollo de esta tesis.
12.2) Regulación del tmbim3, uno nuevo blanco de la UPR
Nuestros resultados indican que la expresión de tmbim3 en cultivo es
posiblemente regulada bajo condiciones de estrés de RE a nivel transcripcional, y que es
controlada por la vía de la UPR dependiente de PERK/ATF4. Esta regulación
transcripcional podría explicar la actividad antiapoptótica específica de TMBIM3 bajo
condiciones de estrés de RE. Dado que la activación de la vía de PERK ha sido sindicada
como una señal fundamentalmente de adaptación frente a la condición de estrés de RE y
como consecuencia de esto, de sobrevida celular (Harding et al, 2000b), podría
especularse que la regulación de TMBIM3 es un componente protector de esta vía
durante la etapa adaptativa de la UPR. Esta idea está acorde con la observación de una
menor viabilidad celular de células MEFs deficientes en PERK expuestas a una condición
de estrés de RE. Por otra parte, el control sobre la muerte celular ejercido por la vía
PERK/ATF4 ha sido ligado a la regulación de la expresión de miembros de la familia BCL-
2, disminuyendo la expresión de BCL-2 (Puthalakath et al, 2007) e incrementado la
expresión de miembros pro-apoptóticos, como BIM, PUMA y NOXA (revisado en
(Rodriguez et al, 2011).. Si bien, el control que podría ejercer TMBIM3 o cualquiera de los
miembros de la familia TMBIM sobre la apoptosis inducida por los miembros pro-
apoptóticos de la familia BCL-2, aún no ha sido estudiada, la evidencia que relaciona
ambas familias de proteínas se limita, básicamente, a estudios de co-inmunoprecipitación
que establecen que bajo condiciones basales, tanto TMBIM6 (Xu & Reed, 1998), TMBIM2
(Reimers et al, 2006) y TMBIM3 pueden interactuar con BCL-2. Sin embargo, el posible
impacto funcional de estas interacciones no ha sido estudiado y no existen antecedentes
a la fecha que evalúen si la formación de complejos proteicos entre los miembros de la
familia BCL-2 con las proteínas miembros de la familia TMBIM puede ser regulada bajo
condiciones de estrés de RE o si la regulación de ambos grupos de genes podría ser
preponderante en la muerte inducida por los miembros pro-apoptóticos de la familia BCL2.

107
Retomando la relación entre TMBIM3 y la UPR, queda pendiente evaluar si esta
proteína comparte el control que ejerce TMBIM6 sobre la inactivación IRE1α. Tal como se
describió previamente, TMBIM6 inhibe la actividad RNAsa de IRE1α bajo condiciones de
estrés de RE, siendo un factor necesario para el retorno a un estado inactivo de IRE1α
bajo condiciones de estrés de RE crónico (Lisbona et al, 2009). Considerando que la
activación de las vías de la UPR, no desencadena señales unidireccionales que actúan
descoordinadamente y como ejemplo de esto es el control positivo que ejerce ATF6 sobre
la expresión del mRNA de xbp-1, el cual es procesado posteriormente por IRE1α; en este
trabajo, se determinó que la presencia de IRE1α no es necesaria para el incremento del
ARNm de tmbim3 bajo condiciones de estrés de RE, pero no puede descartarse que
TMBIM3 ejerza alguna acción sobre la actividad de IRE1α bajo condiciones de estrés de
RE, al igual como lo que ha sido descrito para TMBIM6
Por otra parte, se evalúo in vivo la regulación de la expresión de tmbim3. Con este
fin, se determinó que el ARNm de tmbim3 fue fuertemente incrementado en los riñones de
ratones expuestos a Tm mediante inyección intra-peritoneal. Sin embargo, no se observó
un incremento en el hígado de los mismos ratones, lo cual contribuye al concepto que las
características de la UPR son fuertemente dependientes del contexto fisiológico y del
tejido en estudio. Además, utilizando ratones deficientes en ATF4 se estableció que la
presencia de este factor transcripcional es necesaria para la regulación de la expresión
del ARNm tmbim3 bajo condiciones de estrés de RE.
12.3) Regulación del Ca2+ mediada por tmbim3, el vínculo con el control de la
apoptosis inducida por estrés de RE.
Dado el alto grado de similitud entre las secuencias primarias los miembros de la
familia TMBIM, se estudio si TMBIM3 presenta un efecto en la regulación de la
homeostasis de Ca2+ que ya ha sido descrito para TMBIM6 (Xu & Reed, 1998) y TMBIM4
(de Mattia et al, 2009). Del mismo modo, se observó que las células que sobreexpresan
TMBIM3 experimentan una drástica disminución del Ca2+ liberado desde el RE. Sin
embargo, los niveles basales de Ca2+ en el RE no fueron afectados. En consecuencia con
esto, se observó que la doble deficiencia de TMBIM3 y TMBIM6 sinergiza respecto al
control del Ca2+ liberado desde el RE. Además del efecto de la sobreexpresión de

108
TMBIM3 en la liberación de Ca2+ desde el RE mediada por IP3, se determinó que TMBIM3
co-inmunoprecipita con el IP3-R1 y con el IP3-R3. Estos resultados permiten especular
que TMBIM3 podría tener un efecto inhibidor sobre el IP3-R al formar un complejo
proteico.
El control en la liberación del Ca2+ ejercido por TMBIM3 podría tener un impacto
significativo en la apoptosis inducida por estrés de RE. Esto fue corroborado en los
experimentos donde se observó una disminución de la muerte inducida por estrés de RE
crónico al usar 2-APB, un inhibidor farmacológico del IP3-R. Sin embargo, el mecanismo
exacto de cómo TMBIM3 regula la homeostasis del Ca2+ aún no ha sido determinada. Por
otra parte, la liberación de Ca2+ desde el RE, en primera instancia podría ser fundamental
para la mantención del potencial mitocondrial, favoreciendo de este modo, la producción
de ATP, lo cual podría ser considerado como un mecanismo que favorece la sobrevida
celular, ante la alta demanda metabólica que podría generarse ante condiciones de estrés
de RE (Bravo et al, 2011). En contraste con esto, debe considerarse que la apoptosis es
un mecanismo de muerte controlado, el cual necesita ATP para la formación del
apoptosoma y posterior activación de las caspasas efectoras (Youle & Strasser, 2008).
Por lo tanto el control de las señales de Ca2+ emanadas desde el RE no pueden ser
consideradas unidireccionales, ya sea señales pro-sobrevida celular o pro-apoptosis. Otro
ejemplo de esto, es el papel que descrito para las proteínas de la familia BCL-2 en el RE,
ejerciendo un control en la homeostasis de Ca2+, en donde BCL-2 puede inhibir al IP3-R,
modulando su fosforilación y a su vez el flujo de Ca2+ desde el RE hacia el citoplasma
(revisado en (Pinton & Rizzuto, 2006) o BCL-XL que regula la frecuencia e intensidad de
estas señales (White et al, 2005). Por otra parte, BAX y BAK sinergizan en el RE, de
modo que la ausencia de ambas proteínas disminuyen el contenido neto de Ca2+ en
lumen del RE (Oakes et al, 2005; Scorrano et al, 2003), similar al fenotipo observado en
células en donde se sobre-expresa BCL-2 (algunos ejemplos en Bassik et al, 2004;
Distelhorst et al, 1996; Lam et al, 1994; Oakes et al, 2003; Pinton et al, 2000). Este efecto
sinérgico parece no ser restringido para la familia BCL-2, dado lo observado para TMBIM3
y TMBIM6, en donde ambas proteínas parecen tener actividades complementarias
respecto del control del Ca2+ y en la apoptosis inducida por estrés de RE.

109
12.4) TMBIM3 y TMBIM6: compañeros en el control de la muerte celular.
La disminución simultánea de la expresión de TMBIM3 y TMBIM6 se traduce en un
fenotipo celular relacionado con la inducción de apoptosis espontanea. Esta observación
sugiere que existe una compleja red de proteínas que podrían ejercer su actividad desde
la membrana del RE, en donde distintos miembros de la familia TMBIM podrían
interactuar funcionalmente y sinergizar desde el RE en el control de la muerte celular.
Esta idea podría ser apoyada por la evidencia que demuestra la existencia de
heterodímeros formados por TMBIM3 y TMBIM6, lo cual fue evaluado mediante co-
inmunoprecipitación de ambas proteínas.
El efecto sinérgico en las proteínas que controlan la apoptosis también ha sido
descrito para los miembros de la familia BCL-2. Cabe destacar que todas las señales
mediadas por los miembros de la familia BCL-2 convergen en la activación de BAX/BAK,
favoreciendo la formación de un poro en la membrana mitocondrial con la consecuente
salida de citocromo c y activación posterior del apoptosoma. Respecto al sinergismo entre
estas proteínas, la evidencia existente en la literatura muestra que la mono-deficiencia de
BAX o BAK no implica un efecto importante en el control de la muerte celular, mientras
que la doble deficiencia de ambas proteínas, tanto en ratones como en células, resulta en
una alta resistencia a estímulos intrínsecos de apoptosis, incluyendo el estrés de RE
(Hetz et al, 2006; Scorrano et al, 2003; Wei et al, 2001). Del mismo modo, las células
deficientes en las proteínas BH3-Only BIM o PUMA, son ligeramente resistente a la
apoptosis (Erlacher et al, 2006; Kim et al, 2009). Sin embargo, esta resistencia se
incrementa enormemente en las células doble deficientes o en aquellas triple deficientes
para BIM/PUMA/BID (Ren et al, 2010). Este efecto podría ser explicado porque las
proteínas BH3-only pueden activar directamente a BAX/BAK o inhibir a los miembros
antiapoptóticos, de manera dependiente de su dominio BH3. Interesantemente, la doble
deficiencia para BAX y BAK ha sido descrita como letal. Sin embargo, el 10% de los
animales es viable (Wei et al, 2001), lo cual sugiere que existen otros reguladores de la
muerte celular programada que pueden compensar o controlar la apoptosis,
independiente de las proteínas de la familia BCL-2. En esta tesis, un fenómeno similar fue
observado en D. melanogaster en donde la doble deficiencia de TMBIM3 y TMBIM6

110
sinergizan en el control de la muerte celular, lo cual se refleja en una disminución
significativa de individuos adultos viables.
Otro punto importante a considerar es la reciente descripción de un dominio BH3
en la proteína Bxi1p/Ybh3p (Buttner et al, 2011), codificada por el gen YNI305C presente
en levadura, el cual es el homologo putativo para los miembros de la familia TMBIM.
Aunque el rol de Bxi1p/Ybh3p en la apoptosis puede ser considerada controversial, dado
que dos trabajos describen actividades contrarias (Buttner et al, 2011; Cebulski et al,
2011), resulta interesante la presencia de un dominio BH3 en las proteínas de la familia
TMBIM, dado su alto grado de conservación y su presencia en virus, plantas, insectos y
mamíferos. En humanos, la presencia de un putativo dominio BH3 ha sido descrito en el
extremo C-terminal de las proteínas TMBIM1, 2 (Buttner et al, 2011). Sin embargo, se
desconoce el posible rol funcional de estos dominios en el control de la apoptosis.
Además, la presencia de Bxi1p/Ybh3p como única proteína en levadura, permite
especular que Bxi1p/Ybh3p pudiera operar como un único miembro de ambas familias en
levadura, mientras que en animales podría haberse desarrollado un sofisticado sistema
regulatorio formado por componentes de la familia BCL-2 y los miembros de la familia
TMBIM. Esta idea, podría ser apoyada por las interacciones descritas entre TMBIM3 y
TMBIM6 con BCL-2, así como con la evidencia que describe que tanto TMBIM6, como
TMBIM5 pueden inhibir la muerte inducida por BAX. Estos dos hechos en conjunto,
apoyan la idea de la existencia de dos familias de proteínas que, coordinadamente,
podrían controlar le muerte celular. Por otra parte, podría ser interesante determinar si, al
igual que los miembros de la familia BCL-2, los miembros de la familia TMBIM poseen
actividades complementarias, sinérgicas o incluso opuestas en consecuencia con la
presencia o ausencia de los dominios BH3 putativos. Como antecedente a esta idea, en
esta tesis se presenta por primera vez el efecto sinérgico en la viabilidad observado ante
la deficiencia de TMBIM3 y TMBIM6, tanto en cultivos celulares mamíferos, como in vivo
en D. melanogaster.
12.5) TMBIM3 y su función en el sistema nervioso.
Basado en el patrón de expresión neuronal de TMBIM3 y la asociación del estrés
de RE y distintas enfermedades neurodegenerativas (Matus et al, 2011), en esta tesis se
evaluó la contribución de TMBIM3 en la fisiología del sistema nervioso además del

111
impacto de TMBIM3 bajo condiciones patológicas. Aproximadamente hace dos décadas
atrás un gen homólogo putativo de TMBIM3/GRINA fue descrito en rata como una
proteína unida al receptor de NMDA, cuyo nombre asignado fue NMDARA1 o GBP, del
inglés Glutamate Binding Protein (Kumar et al, 1991). El empeño de los autores en
identificar un nuevo subtipo de receptores de glutamato fue contrariado ante las dudas
generadas por la falta de evidencias y ante un probable error en el método de clonamiento
(Hollmann & Heinemann, 1994). Esto, habría resultado en la obtención de un gen que
codifica para una proteína con un peso estimado en 56 KDa (Kumar et al, 1991). No
obstante, los estudios bioquímicos arrojaron que el peso molecular aparente de esta
proteína sería cercano a los 70 KDa, lo cual contrasta con el peso predicho para la
proteína codificada por los genes homólogos en ratón o humano. Estas diferencias en el
peso molecular podrían ser explicadas por que la secuencia clonada en 1991 generaría
una proteína con un extremo C terminal adicional.
Previamente se describió que GBP se localiza en cuerpos neuronales y dendríticos
en el cerebro y la medula espinal (Eaton et al, 1990; Mattson et al, 1991; Pal et al, 1999).
Además, se determinó que el ARNm de GBP podía ser incrementado en neuronas
expuestas crónicamente a etanol (Bao et al, 2001; Bhave et al, 1996; Chen et al, 1997), y
que además presenta un posible rol protector ante el daño generado por isquemia y
reperfusión en la retina (Aikawa et al, 2003). En esta tesis, se describió que la expresión
de TMBIM3 es esencial para el desarrollo y generación de estructuras cerebrales en pez
zebra, lo cual podría dar cuenta de un probable papel protector de TMBIM3 en el sistema
nervioso central. Sin embargo, un reciente reporte describe la ausencia de un fenotipo
espontáneo como consecuencia de la generación de un ratón deficiente para TMBIM3
(Nielsen et al, 2011). Por otra parte, el efecto de la deficiencia de TMBIM3 ante
condiciones fisiológicas no fue evaluado en este modelo murino. Por último, pero no
menos relevante, está el antecedente que describe que el gen de TMBIM3 humano está
asociado una duplicación génica en un locus ligada genéticamente a la generación de
epilepsia (Bonaglia et al, 2005; Lewis et al, 1996).
Los antecedentes obtenidos a la fecha acerca de TMBIM3, así como de toda la
familia TMBIM y en conjunto con los resultados obtenidos en esta tesis, hacen necesario
considerar un análisis sistemático para establecer y definir si existe una organización

112
jerárquica entre sus componentes, así como el papel que juega la interacción con los
miembros de la familia BCL-2. Esto podría ser relevante tanto en el desarrollo, como en la
mantención de la homeostasis fisiológica, así como en la patogenia de enfermedades
relacionadas con la alteración en el plegamiento de proteínas.
12.6) TMBIM3: un nuevo y conservado gen regulado por la UPR que inhibe la
apoptosis inducida por estrés de RE mediante el control de la homeostasis del Ca2+.
Los antecedentes reunidos en esta tesis permiten proponer a TMBIM3 como parte
funcional de la familia de proteínas TMBIM, la cual, desde el RE, coordina en conjunto con
TMBIM6 una respuesta antiapoptótica, específicamente, bajo condiciones de estrés de
RE. Estudios en modelos celulares y animales, nos demostraron que los niveles de
expresión del ARNm de tmbim3 son fuertemente incrementados en respuesta a estrés de
RE, como consecuencia de la activación de la vía de la UPR comandada por el sensor de
estrés de RE PERK y el factor de transcripción ATF4. Por otra parte, se describió que
TMBIM3 sinergiza con TMBIM6, respecto al control de la apoptosis, así como en la
sobrevida de animales en estado adulto. Además, se evalúo el efecto de TMBIM3 en el
control de la homeostasis del Ca2+, observándose que TMBIM3 inhibe la salida de Ca2+
desde el RE, efecto que podría estar asociado con la interacción descrita entre TMBIM3 y
el IP3-R evaluado mediante co-inmunoprecipitación (Figura 58).
Los estudios de pérdida de función en D. melanogaster demostraron que TMBIM3
y TMBIM6 tienen actividades pro-sobrevida, las cuales están funcionalmente relacionadas
en respuesta al estrés de RE in vivo. De manera similar, la manipulación de los niveles de
TMBIM3 en pez zebra, reveló un papel esencial de TMBIM3 en el control de la apoptosis
durante el desarrollo neuronal y en modelos de estrés de RE. Todos estos hallazgos en
conjunto, sugieren la existencia de un grupo conservado de reguladores relacionados
funcionalmente con muerte celular.

113
Figura 59. Modelo propuesto: Rol de TMBIM3 en la mantención de la sobrevida celular y como blanco de la UPR que controla la apoptosis mediante la inhibición del receptor de IP3. La expresión de TMBIM3 es escencial para la mantención de la sobrevida celular bajo condiciones normales y durante el desarrollo. Por otra parte, bajo condiciones de estrés de RE se induce la activación del sensor de estrés de RE PERK, favoreciéndose su auto-transfosforilación. Río abajo, se induce la expresión del factor transcripcional ATF4, el cual se transloca al núcleo y regula la expresión de sus genes blancos. Directa o indirectamente regulado por ATF4, se induce la expresión de tmbim3. TMBIM3 dimeriza con TMBIM6 en el RE. A su vez, el flujo de Ca2+ desde el RE hacia el citoplasma mediante el receptor de IP3 (IP3R) es inducido bajo condiciones de estrés de RE favoreciendo la activación de apoptosis, proceso el cual podría ser inhibido por TMBIM3.
Citoplasma

114
13) Conclusiones
Las conclusiones más relevantes del trabajo realizado en esta tesis son:
• TMBIM3 tiene actividad antiapoptótica bajo condiciones basales y de estrés de
RE.
• TMBIM3 y TMBIM6 tienen actividades complementarias.
• TMBIM3 interactúa con TMBIM6.
• Tmbim3 aumenta bajo condiciones de estrés de RE, mediante un mecanismo
dependiente de la vía PERK/ATF4.
• La expresión de TMBIM3 controla la homeostasis del calcio y disminuye la muerte
inducida por estrés de retículo mediada por Calcio.
• TMBIM3 interactúa con BCL-2, IP3R1 e IP3R3.
• TMBIM3 y TMBIM6 tienen actividades antiapoptóticas complementarias (in vitro e
in vivo).
• La deficiencia de Tmbim3 aumenta la apoptosis durante el desarrollo en pez
zebra.
• TMBIM3 tiene actividad antiapoptótica en un modelo de estrés de RE en pez
zebra.

115
14) Bibliografía.
Aikawa H, Tomita H, Ishiguro S, Nishikawa S, Sugano E, Tamai M (2003) Increased expression of glutamate binding protein mRNA in rat retina after ischemia-reperfusion injury. Tohoku J Exp Med 199: 25-33
Ameisen JC (2002) On the origin, evolution, and nature of programmed cell death: a timeline of four billion years. Cell Death Differ 9: 367-393
Back SH, Schroder M, Lee K, Zhang K, Kaufman RJ (2005) ER stress signaling by regulated splicing: IRE1/HAC1/XBP1. Methods 35: 395-416
Bailly-Maitre B, Bard-Chapeau E, Luciano F, Droin N, Bruey JM, Faustin B, Kress C, Zapata JM, Reed JC (2007) Mice lacking bi-1 gene show accelerated liver regeneration. Cancer Res 67: 1442-1450
Bailly-Maitre B, Belgardt BF, Jordan SD, Coornaert B, von Freyend MJ, Kleinridders A, Mauer J, Cuddy M, Kress CL, Willmes D, Essig M, Hampel B, Protzer U, Reed JC, Bruning JC (2010) Hepatic Bax inhibitor-1 inhibits IRE1alpha and protects from obesity-associated insulin resistance and glucose intolerance. J Biol Chem 285: 6198-6207
Bailly-Maitre B, Fondevila C, Kaldas F, Droin N, Luciano F, Ricci JE, Croxton R, Krajewska M, Zapata JM, Kupiec-Weglinski JW, Farmer D, Reed JC (2006) Cytoprotective gene bi-1 is required for intrinsic protection from endoplasmic reticulum stress and ischemia-reperfusion injury. Proc Natl Acad Sci U S A 103: 2809-2814
Bao Q, Shi Y (2007) Apoptosome: a platform for the activation of initiator caspases. Cell death and differentiation 14: 56-65
Bao X, Hui D, Naassila M, Michaelis EK (2001) Chronic ethanol exposure increases gene transcription of subunits of an N-methyl-D-aspartate receptor-like complex in cortical neurons in culture. Neurosci Lett 315: 5-8
Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ (2004) Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J 23: 1207-1216
Beier CP, Wischhusen J, Gleichmann M, Gerhardt E, Pekanovic A, Krueger A, Taylor V, Suter U, Krammer PH, Endres M, Weller M, Schulz JB (2005) FasL (CD95L/APO-1L) resistance of neurons mediated by phosphatidylinositol 3-kinase-Akt/protein kinase B-dependent expression of lifeguard/neuronal membrane protein 35. J Neurosci 25: 6765-6774

116
Berridge MJ (2002) The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32: 235-249
Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D (2000) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2: 326-332
Bhave SV, Snell LD, Tabakoff B, Hoffman PL (1996) Mechanism of ethanol inhibition of NMDA receptor function in primary cultures of cerebral cortical cells. Alcohol Clin Exp Res 20: 934-941
Bonaglia MC, Giorda R, Tenconi R, Pessina M, Pramparo T, Borgatti R, Zuffardi O (2005) A 2.3 Mb duplication of chromosome 8q24.3 associated with severe mental retardation and epilepsy detected by standard karyotype. Eur J Hum Genet 13: 586-591
Borgese N, Francolini M, Snapp E (2006) Endoplasmic reticulum architecture: structures in flux. Curr Opin Cell Biol 18: 358-364
Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M, Quiroga C, Rodriguez AE, Verdejo HE, Ferreira J, Iglewski M, Chiong M, Simmen T, Zorzano A, Hill JA, Rothermel BA, Szabadkai G, Lavandero S (2011) Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci 124: 2143-2152
Buttner S, Ruli D, Vogtle FN, Galluzzi L, Moitzi B, Eisenberg T, Kepp O, Habernig L, Carmona-Gutierrez D, Rockenfeller P, Laun P, Breitenbach M, Khoury C, Frohlich KU, Rechberger G, Meisinger C, Kroemer G, Madeo F (2011) A yeast BH3-only protein mediates the mitochondrial pathway of apoptosis. EMBO J 30: 2779-2792
Butturini A, Gale RP (1990) Oncogenes and leukemia. Leukemia 4: 138-160
Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415: 92-96
Cebulski J, Malouin J, Pinches N, Cascio V, Austriaco N (2011) Yeast Bax Inhibitor, Bxi1p, Is an ER-Localized Protein That Links the Unfolded Protein Response and Programmed Cell Death in Saccharomyces cerevisiae. PLoS One 6: e20882

117
Colombo A, Reig G, Mione M, Concha ML (2006) Zebrafish BarH-like genes define discrete neural domains in the early embryo. Gene Expr Patterns 6: 347-352
Cox JS, Shamu CE, Walter P (1993) Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73: 1197-1206
Crescenzi M, Seto M, Herzig GP, Weiss PD, Griffith RC, Korsmeyer SJ (1988) Thermostable DNA polymerase chain amplification of t(14;18) chromosome breakpoints and detection of minimal residual disease. Proc Natl Acad Sci U S A 85: 4869-4873
Chae HJ, Ke N, Kim HR, Chen S, Godzik A, Dickman M, Reed JC (2003) Evolutionarily conserved cytoprotection provided by Bax Inhibitor-1 homologs from animals, plants, and yeast. Gene 323: 101-113
Chae HJ, Kim HR, Xu C, Bailly-Maitre B, Krajewska M, Krajewski S, Banares S, Cui J, Digicaylioglu M, Ke N, Kitada S, Monosov E, Thomas M, Kress CL, Babendure JR, Tsien RY, Lipton SA, Reed JC (2004) BI-1 regulates an apoptosis pathway linked to endoplasmic reticulum stress. Mol Cell 15: 355-366
Chen X, Michaelis ML, Michaelis EK (1997) Effects of chronic ethanol treatment on the expression of calcium transport carriers and NMDA/glutamate receptor proteins in brain synaptic membranes. J Neurochem 69: 1559-1569
Chen X, Shen J, Prywes R (2002) The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J Biol Chem 277: 13045-13052
Danial NN, Korsmeyer SJ (2004) Cell death: critical control points. Cell 116: 205-219
de Mattia F, Gubser C, van Dommelen MM, Visch HJ, Distelmaier F, Postigo A, Luyten T, Parys JB, de Smedt H, Smith GL, Willems PH, van Kuppeveld FJ (2009) Human Golgi antiapoptotic protein modulates intracellular calcium fluxes. Mol Biol Cell 20: 3638-3645
Degterev A, Yuan J (2008) Expansion and evolution of cell death programmes. Nat Rev Mol Cell Biol 9: 378-390
Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, Couto A, Marra V, Keleman K, Dickson BJ (2007) A genome-wide

118
transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448: 151-156
Distelhorst CW, Lam M, McCormick TS (1996) Bcl-2 inhibits hydrogen peroxide-induced ER Ca2+ pool depletion. Oncogene 12: 2051-2055
Eaton MJ, Chen JW, Kumar KN, Cong Y, Michaelis EK (1990) Immunochemical characterization of brain synaptic membrane glutamate-binding proteins. J Biol Chem 265: 16195-16204
Erlacher M, Labi V, Manzl C, Bock G, Tzankov A, Hacker G, Michalak E, Strasser A, Villunger A (2006) Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J Exp Med 203: 2939-2951
Fagone P, Jackowski S (2009) Membrane phospholipid synthesis and endoplasmic reticulum function. J Lipid Res 50 Suppl: S311-316
Fernandez M, Segura MF, Sole C, Colino A, Comella JX, Cena V (2007) Lifeguard/neuronal membrane protein 35 regulates Fas ligand-mediated apoptosis in neurons via microdomain recruitment. J Neurochem 103: 190-203
Galindo KA, Lu WJ, Park JH, Abrams JM (2009) The Bax/Bak ortholog in Drosophila, Debcl, exerts limited control over programmed cell death. Development 136: 275-283
Gleason MR, Armisen R, Verdecia MA, Sirotkin H, Brehm P, Mandel G (2004) A mutation in serca underlies motility dysfunction in accordion zebrafish. Dev Biol 276: 441-451
Gorlach A, Klappa P, Kietzmann T (2006) The endoplasmic reticulum: folding, calcium homeostasis, signaling, and redox control. Antioxid Redox Signal 8: 1391-1418
Green DR (2000) Apoptotic pathways: paper wraps stone blunts scissors. Cell 102: 1-4
Gubser C, Bergamaschi D, Hollinshead M, Lu X, van Kuppeveld FJ, Smith GL (2007) A new inhibitor of apoptosis from vaccinia virus and eukaryotes. PLoS Pathog 3: e17
Han D, Lerner AG, Vande Walle L, Upton JP, Xu W, Hagen A, Backes BJ, Oakes SA, Papa FR (2009) IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 138: 562-575

119
Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D (2000a) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6: 1099-1108
Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D (2000b) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 5: 897-904
Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619-633
Haze K, Yoshida H, Yanagi H, Yura T, Mori K (1999) Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell 10: 3787-3799
Hemrajani C, Berger CN, Robinson KS, Marches O, Mousnier A, Frankel G (2010) NleH effectors interact with Bax inhibitor-1 to block apoptosis during enteropathogenic Escherichia coli infection. Proc Natl Acad Sci U S A 107: 3129-3134
Hengartner MO (1996) Programmed cell death in invertebrates. Curr Opin Genet Dev 6: 34-38
Hengartner MO, Horvitz HR (1994) Programmed cell death in Caenorhabditis elegans. Curr Opin Genet Dev 4: 581-586
Henke N, Lisak DA, Schneider L, Habicht J, Pergande M, Methner A (2011) The ancient cell death suppressor BAX inhibitor-1. Cell Calcium
Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B, Brandt GS, Iwakoshi NN, Schinzel A, Glimcher LH, Korsmeyer SJ (2006) Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science 312: 572-576
Hetz C, Bono MR, Barros LF, Lagos R (2002a) Microcin E492, a channel-forming bacteriocin from Klebsiella pneumoniae, induces apoptosis in some human cell lines. Proc Natl Acad Sci U S A 99: 2696-2701

120
Hetz C, Glimcher L (2008) The daily job of night killers: alternative roles of the BCL-2 family in organelle physiology. Trends Cell Biol 18: 38-44
Hetz C, Glimcher LH (2009) Fine-tuning of the unfolded protein response: Assembling the IRE1alpha interactome. Mol Cell 35: 551-561
Hetz CA, Hunn M, Rojas P, Torres V, Leyton L, Quest AF (2002b) Caspase-dependent initiation of apoptosis and necrosis by the Fas receptor in lymphoid cells: onset of necrosis is associated with delayed ceramide increase. J Cell Sci 115: 4671-4683
Higo T, Hamada K, Hisatsune C, Nukina N, Hashikawa T, Hattori M, Nakamura T, Mikoshiba K (2010) Mechanism of ER stress-induced brain damage by IP(3) receptor. Neuron 68: 865-878
Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS (2009) Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol 186: 323-331
Hollien J, Weissman JS (2006) Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 313: 104-107
Hollmann M, Heinemann S (1994) Cloned glutamate receptors. Annu Rev Neurosci 17: 31-108
Hu L, Smith TF, Goldberger G (2009) LFG: a candidate apoptosis regulatory gene family. Apoptosis 14: 1255-1265
Huckelhoven R (2004) BAX Inhibitor-1, an ancient cell death suppressor in animals and plants with prokaryotic relatives. Apoptosis 9: 299-307
Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH (2003) Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-1. Nat Immunol 4: 321-329
Jesch SA, Liu P, Zhao X, Wells MT, Henry SA (2006) Multiple endoplasmic reticulum-to-nucleus signaling pathways coordinate phospholipid metabolism with gene expression by distinct mechanisms. J Biol Chem 281: 24070-24083
Jette CA, Flanagan AM, Ryan J, Pyati UJ, Carbonneau S, Stewart RA, Langenau DM, Look AT, Letai A (2008) BIM and other BCL-2 family proteins exhibit cross-species

121
conservation of function between zebrafish and mammals. Cell Death Differ 15: 1063-1072
Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH (2009) Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell 36: 487-499
Kim R, Emi M, Tanabe K, Murakami S (2006) Role of the unfolded protein response in cell death. Apoptosis 11: 5-13
Kimata Y, Kimata YI, Shimizu Y, Abe H, Farcasanu IC, Takeuchi M, Rose MD, Kohno K (2003) Genetic evidence for a role of BiP/Kar2 that regulates Ire1 in response to accumulation of unfolded proteins. Mol Biol Cell 14: 2559-2569
Kornbluth S, White K (2005) Apoptosis in Drosophila: neither fish nor fowl (nor man, nor worm). J Cell Sci 118: 1779-1787
Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW (1993) MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci U S A 90: 3516-3520
Krajewska M, Xu L, Xu W, Krajewski S, Kress CL, Cui J, Yang L, Irie F, Yamaguchi Y, Lipton SA, Reed JC (2010) Endoplasmic Reticulum Protein BI-1 Modulates Unfolded Protein Response Signaling and Protects Against Stroke and Traumatic Brain Injury. Brain Res
Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, Hengartner M, Knight RA, Kumar S, Lipton SA, Malorni W, Nunez G, Peter ME, Tschopp J, Yuan J, Piacentini M, Zhivotovsky B, Melino G (2009) Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 16: 3-11
Kumar KN, Tilakaratne N, Johnson PS, Allen AE, Michaelis EK (1991) Cloning of cDNA for the glutamate-binding subunit of an NMDA receptor complex. Nature 354: 70-73
Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157: 105-132

122
Lam M, Dubyak G, Chen L, Nunez G, Miesfeld RL, Distelhorst CW (1994) Evidence that BCL-2 represses apoptosis by regulating endoplasmic reticulum-associated Ca2+ fluxes. Proc Natl Acad Sci U S A 91: 6569-6573
Larsen CJ (1994) [The BCL2 gene, prototype of a gene family that controls programmed cell death (apoptosis)]. Ann Genet 37: 121-134
Lee AH, Iwakoshi NN, Glimcher LH (2003) XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23: 7448-7459
Lee GH, Yan C, Shin SJ, Hong SC, Ahn T, Moon A, Park SJ, Lee YC, Yoo WH, Kim HT, Kim DS, Chae SW, Kim HR, Chae HJ (2010) BAX inhibitor-1 enhances cancer metastasis by altering glucose metabolism and activating the sodium-hydrogen exchanger: the alteration of mitochondrial function. Oncogene 29: 2130-2141
Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ (2002) IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev 16: 452-466
Lewis TB, Wood S, Michaelis EK, DuPont BR, Leach RJ (1996) Localization of a gene for a glutamate binding subunit of a NMDA receptor (GRINA) to 8q24. Genomics 32: 131-133
Li B, Yi P, Zhang B, Xu C, Liu Q, Pi Z, Xu X, Chevet E, Liu J (2011a) Differences in endoplasmic reticulum stress signalling kinetics determine cell survival outcome through activation of MKP-1. Cell Signal 23: 35-45
Li G, Mongillo M, Chin KT, Harding H, Ron D, Marks AR, Tabas I (2009) Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J Cell Biol 186: 783-792
Li J, Lee B, Lee AS (2006) Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem 281: 7260-7270
Li XY, Lai YK, Zhang JF, Luo HQ, Zhang MH, Zhou KY, Kung HF (2011b) Lentivirus-Mediated RNA Interference Targeting Bax Inhibitor-1 Suppresses Ex Vivo Cell Proliferation and In Vivo Tumor Growth of Nasopharyngeal Carcinoma. Hum Gene Ther

123
Lin EY, Orlofsky A, Wang HG, Reed JC, Prystowsky MB (1996) A1, a Bcl-2 family member, prolongs cell survival and permits myeloid differentiation. Blood 87: 983-992
Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P (2007) IRE1 signaling affects cell fate during the unfolded protein response. Science 318: 944-949
Lisbona F, Rojas-Rivera D, Thielen P, Zamorano S, Todd D, Martinon F, Glavic A, Kress C, Lin JH, Walter P, Reed JC, Glimcher LH, Hetz C (2009) BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1alpha. Mol Cell 33: 679-691
Loscher CJ, Hokamp K, Kenna PF, Ivens AC, Humphries P, Palfi A, Farrar GJ (2007) Altered retinal microRNA expression profile in a mouse model of retinitis pigmentosa. Genome Biol 8: R248
Mattson MP, Wang H, Michaelis EK (1991) Developmental expression, compartmentalization, and possible role in excitotoxicity of a putative NMDA receptor protein in cultured hippocampal neurons. Brain Res 565: 94-108
Matus S, Glimcher LH, Hetz C (2011) Protein folding stress in neurodegenerative diseases: a glimpse into the ER. Curr Opin Cell Biol
Meijerink JP, Smetsers TF, Sloetjes AW, Linders EH, Mensink EJ (1995) Bax mutations in cell lines derived from hematological malignancies. Leukemia 9: 1828-1832
Mori K (2009) Signalling pathways in the unfolded protein response: development from yeast to mammals. J Biochem 146: 743-750
Negrini M, Silini E, Kozak C, Tsujimoto Y, Croce CM (1987) Molecular analysis of mbcl-2: structure and expression of the murine gene homologous to the human gene involved in follicular lymphoma. Cell 49: 455-463
Ngan BY, Berinstein N (1990) Oncogene involvement in lymphoid malignancy. Tumour Biol 11 Suppl 1: 78-93
Nielsen JA, Chambers MA, Romm E, Lee LY, Berndt JA, Hudson LD (2011) Mouse transmembrane BAX inhibitor Motif 3 (Tmbim3) encodes a 38 kDa transmembrane protein expressed in the central nervous system. Mol Cell Biochem

124
Novoa I, Zeng H, Harding HP, Ron D (2001) Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol 153: 1011-1022
Oakes SA, Lin SS, Bassik MC (2006) The control of endoplasmic reticulum-initiated apoptosis by the BCL-2 family of proteins. Curr Mol Med 6: 99-109
Oakes SA, Opferman JT, Pozzan T, Korsmeyer SJ, Scorrano L (2003) Regulation of endoplasmic reticulum Ca2+ dynamics by proapoptotic BCL-2 family members. Biochem Pharmacol 66: 1335-1340
Oakes SA, Scorrano L, Opferman JT, Bassik MC, Nishino M, Pozzan T, Korsmeyer SJ (2005) Proapoptotic BAX and BAK regulate the type 1 inositol trisphosphate receptor and calcium leak from the endoplasmic reticulum. Proc Natl Acad Sci U S A 102: 105-110
Oikawa D, Kimata Y, Kohno K, Iwawaki T (2009) Activation of mammalian IRE1alpha upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp Cell Res 315: 2496-2504
Oka T, Sayano T, Tamai S, Yokota S, Kato H, Fujii G, Mihara K (2008) Identification of a novel protein MICS1 that is involved in maintenance of mitochondrial morphology and apoptotic release of cytochrome c. Mol Biol Cell 19: 2597-2608
Okada T, Yoshida H, Akazawa R, Negishi M, Mori K (2002) Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem J 366: 585-594
Pal R, Eaton MJ, Islam S, Hake-Frendscho M, Kumar KN, Michaelis EK (1999) Immunocytochemical and in situ hybridization studies of the expression and distribution of three subunits of a complex with N-methyl-D-aspartate receptor-like properties. Neuroscience 94: 1291-1311
Park HC, Kim CH, Bae YK, Yeo SY, Kim SH, Hong SK, Shin J, Yoo KW, Hibi M, Hirano T, Miki N, Chitnis AB, Huh TL (2000) Analysis of upstream elements in the HuC promoter leads to the establishment of transgenic zebrafish with fluorescent neurons. Dev Biol 227: 279-293
Pellicena-Palle A, Salz HK (1995) The putative Drosophila NMDARA1 gene is located on the second chromosome and is ubiquitously expressed in embryogenesis. Biochim Biophys Acta 1261: 301-303

125
Pinton P, Brini M, Bastianutto C, Tuft RA, Pozzan T, Rizzuto R (1998) New light on mitochondrial calcium. Biofactors 8: 243-253
Pinton P, Ferrari D, Magalhaes P, Schulze-Osthoff K, Di Virgilio F, Pozzan T, Rizzuto R (2000) Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. J Cell Biol 148: 857-862
Pinton P, Giorgi C, Siviero R, Zecchini E, Rizzuto R (2008) Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 27: 6407-6418
Pinton P, Rizzuto R (2006) Bcl-2 and Ca2+ homeostasis in the endoplasmic reticulum. Cell Death Differ 13: 1409-1418
Plongthongkum N, Kullawong N, Panyim S, Tirasophon W (2007) Ire1 regulated XBP1 mRNA splicing is essential for the unfolded protein response (UPR) in Drosophila melanogaster. Biochem Biophys Res Commun 354: 789-794
Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, Gotoh T, Akira S, Bouillet P, Strasser A (2007) ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129: 1337-1349
Rao RV, Hermel E, Castro-Obregon S, del Rio G, Ellerby LM, Ellerby HM, Bredesen DE (2001) Coupling endoplasmic reticulum stress to the cell death program. Mechanism of caspase activation. J Biol Chem 276: 33869-33874
Reed JC (1995) Regulation of apoptosis by bcl-2 family proteins and its role in cancer and chemoresistance. Curr Opin Oncol 7: 541-546
Reed JC, Talwar HS, Cuddy M, Baffy G, Williamson J, Rapp UR, Fisher GJ (1991) Mitochondrial protein p26 BCL2 reduces growth factor requirements of NIH3T3 fibroblasts. Exp Cell Res 195: 277-283
Reimers K, Choi CY, Bucan V, Vogt PM (2007) The growth-hormone inducible transmembrane protein (Ghitm) belongs to the Bax inhibitory protein-like family. Int J Biol Sci 3: 471-476
Reimers K, Choi CY, Bucan V, Vogt PM (2008) The Bax Inhibitor-1 (BI-1) family in apoptosis and tumorigenesis. Curr Mol Med 8: 148-156

126
Reimers K, Choi CY, Mau-Thek E, Vogt PM (2006) Sequence analysis shows that Lifeguard belongs to a new evolutionarily conserved cytoprotective family. Int J Mol Med 18: 729-734
Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH (2010) BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science 330: 1390-1393
Rizzuto R, Marchi S, Bonora M, Aguiari P, Bononi A, De Stefani D, Giorgi C, Leo S, Rimessi A, Siviero R, Zecchini E, Pinton P (2009) Ca(2+) transfer from the ER to mitochondria: when, how and why. Biochim Biophys Acta 1787: 1342-1351
Rodriguez D, Rojas-Rivera D, Hetz C (2011) Integrating stress signals at the endoplasmic reticulum: The BCL-2 protein family rheostat. Biochim Biophys Acta 1813: 564-574
Ron D, Hubbard SR (2008) How IRE1 reacts to ER stress. Cell 132: 24-26
Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8: 519-529
Rong Y, Distelhorst CW (2008) Bcl-2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu Rev Physiol 70: 73-91
Rong YP, Bultynck G, Aromolaran AS, Zhong F, Parys JB, De Smedt H, Mignery GA, Roderick HL, Bootman MD, Distelhorst CW (2009) The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci U S A 106: 14397-14402
Ryoo HD, Domingos PM, Kang MJ, Steller H (2007) Unfolded protein response in a Drosophila model for retinal degeneration. EMBO J 26: 242-252
Rzymski T, Milani M, Singleton DC, Harris AL (2009) Role of ATF4 in regulation of autophagy and resistance to drugs and hypoxia. Cell Cycle 8: 3838-3847
Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300: 135-139

127
Schroder M, Kaufman RJ (2005a) ER stress and the unfolded protein response. Mutat Res 569: 29-63
Schroder M, Kaufman RJ (2005b) The mammalian unfolded protein response. Annu Rev Biochem 74: 739-789
Schweitzer B, Suter U, Taylor V (2002) Neural membrane protein 35/Lifeguard is localized at postsynaptic sites and in dendrites. Brain Res Mol Brain Res 107: 47-56
Sevrioukov EA, Burr J, Huang EW, Assi HH, Monserrate JP, Purves DC, Wu JN, Song EJ, Brachmann CB (2007) Drosophila Bcl-2 proteins participate in stress-induced apoptosis, but are not required for normal development. Genesis 45: 184-193
Shen J, Chen X, Hendershot L, Prywes R (2002) ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell 3: 99-111
Shen J, Snapp EL, Lippincott-Schwartz J, Prywes R (2005) Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Mol Cell Biol 25: 921-932
Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, Wek RC (1998) Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol 18: 7499-7509
Shukla S, Fujita K, Xiao Q, Liao Z, Garfield S, Srinivasula SM (2011) A shear stress responsive gene product PP1201 protects against Fas-mediated apoptosis by reducing Fas expression on the cell surface. Apoptosis 16: 162-173
Somia NV, Schmitt MJ, Vetter DE, Van Antwerp D, Heinemann SF, Verma IM (1999) LFG: an anti-apoptotic gene that provides protection from Fas-mediated cell death. Proc Natl Acad Sci U S A 96: 12667-12672
Sommer T, Jarosch E (2002) BiP binding keeps ATF6 at bay. Dev Cell 3: 1-2
Souid S, Lepesant JA, Yanicostas C (2007) The xbp-1 gene is essential for development in Drosophila. Dev Genes Evol 217: 159-167
Tabas I, Ron D (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 13: 184-190

128
Tachikawa K, Sasaki S, Maeda T, Nakajima K (2008) Identification of molecules preferentially expressed beneath the marginal zone in the developing cerebral cortex. Neurosci Res 60: 135-146
Takayama S, Sato T, Krajewski S, Kochel K, Irie S, Millan JA, Reed JC (1995) Cloning and functional analysis of BAG-1: a novel Bcl-2-binding protein with anti-cell death activity. Cell 80: 279-284
Tan Y, Dourdin N, Wu C, De Veyra T, Elce JS, Greer PA (2006) Ubiquitous calpains promote caspase-12 and JNK activation during endoplasmic reticulum stress-induced apoptosis. J Biol Chem 281: 16016-16024
Timmins JM, Ozcan L, Seimon TA, Li G, Malagelada C, Backs J, Backs T, Bassel-Duby R, Olson EN, Anderson ME, Tabas I (2009) Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J Clin Invest 119: 2925-2941
Tirasophon W, Welihinda AA, Kaufman RJ (1998) A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev 12: 1812-1824
Tsujimoto Y, Ikegaki N, Croce CM (1987) Characterization of the protein product of bcl-2, the gene involved in human follicular lymphoma. Oncogene 2: 3-7
Urano F, Bertolotti A, Ron D (2000a) IRE1 and efferent signaling from the endoplasmic reticulum. J Cell Sci 113 Pt 21: 3697-3702
Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D (2000b) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287: 664-666
van Stelten J, Silva F, Belin D, Silhavy TJ (2009) Effects of antibiotics and a proto-oncogene homolog on destruction of protein translocator SecY. Science 325: 753-756
Vanderheyden V, Devogelaere B, Missiaen L, De Smedt H, Bultynck G, Parys JB. (2009) Regulation of inositol 1,4,5-trisphosphate-induced Ca2+ release by reversible phosphorylation and dephosphorylation. Biochim Biophys Acta, Vol. 1793, pp. 959-970.

129
Varela D, Simon F, Olivero P, Armisen R, Leiva-Salcedo E, Jorgensen F, Sala F, Stutzin A (2007) Activation of H2O2-induced VSOR Cl- currents in HTC cells require phospholipase Cgamma1 phosphorylation and Ca2+ mobilisation. Cell Physiol Biochem 20: 773-780
Visch HJ, Rutter GA, Koopman WJ, Koenderink JB, Verkaart S, de Groot T, Varadi A, Mitchell KJ, van den Heuvel LP, Smeitink JA, Willems PH (2004) Inhibition of mitochondrial Na+-Ca2+ exchange restores agonist-induced ATP production and Ca2+ handling in human complex I deficiency. J Biol Chem 279: 40328-40336
Walter L, Marynen P, Szpirer J, Levan G, Gunther E (1995) Identification of a novel conserved human gene, TEGT. Genomics 28: 301-304
Walther R, Pichaud F (2006) Immunofluorescent staining and imaging of the pupal and adult Drosophila visual system. Nature Protocol 1: 2635-2642
Wang X (2001) The expanding role of mitochondria in apoptosis. Genes & development 15: 2922-2933
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727-730
Westphalen BC, Wessig J, Leypoldt F, Arnold S, Methner A (2005) BI-1 protects cells from oxygen glucose deprivation by reducing the calcium content of the endoplasmic reticulum. Cell Death Differ 12: 304-306
White C, Li C, Yang J, Petrenko NB, Madesh M, Thompson CB, Foskett JK (2005) The endoplasmic reticulum gateway to apoptosis by Bcl-X(L) modulation of the InsP3R. Nat Cell Biol 7: 1021-1028
Woehlbier U, Hetz C (2011) Modulating stress responses by the UPRosome: A matter of life and death. Trends Biochem Sci 36: 329-337
Wu JN, Nguyen N, Aghazarian M, Tan Y, Sevrioukov EA, Mabuchi M, Tang W, Monserrate JP, White K, Brachmann CB (2010) grim promotes programmed cell death of Drosophila microchaete glial cells. Mech Dev 127: 407-417
Wu JS, Luo L (2006) A protocol for dissecting Drosophila melanogaster brains for live imaging or immunostaining. Nat Protoc 1: 2110-2115

130
Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME (1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270: 1326-1331
Xu C, Xu W, Palmer AE, Reed JC (2008) BI-1 regulates endoplasmic reticulum Ca2+ homeostasis downstream of Bcl-2 family proteins. J Biol Chem 283: 11477-11484
Xu Q, Reed JC (1998) Bax inhibitor-1, a mammalian apoptosis suppressor identified by functional screening in yeast. Mol Cell 1: 337-346
Yamaji T, Nishikawa K, Hanada K (2010) Transmembrane BAX inhibitor motif containing (TMBIM) family proteins perturbs a trans-Golgi network enzyme, Gb3 synthase, and reduces Gb3 biosynthesis. J Biol Chem 285: 35505-35518
Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ (1995) Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 80: 285-291
Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107: 881-891
Youle RJ, Strasser A (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9: 47-59
Zhao C, He X, Tian C, Meng A (2006a) Two GC-rich boxes in huC promoter play distinct roles in controlling its neuronal specific expression in zebrafish embryos. Biochem Biophys Res Commun 342: 214-220
Zhao H, Ito A, Kimura SH, Yabuta N, Sakai N, Ikawa M, Okabe M, Matsuzawa Y, Yamashita S, Nojima H (2006b) RECS1 deficiency in mice induces susceptibility to cystic medial degeneration. Genes Genet Syst 81: 41-50
Zhao H, Ito A, Sakai N, Matsuzawa Y, Yamashita S, Nojima H (2006c) RECS1 is a negative regulator of matrix metalloproteinase-9 production and aged RECS1 knockout mice are prone to aortic dilation. Circ J 70: 615-624
Zhou J, Liu CY, Back SH, Clark RL, Peisach D, Xu Z, Kaufman RJ (2006) The crystal structure of human IRE1 luminal domain reveals a conserved dimerization interface

131
required for activation of the unfolded protein response. Proc Natl Acad Sci U S A 103: 14343-14348
Zhou J, Zhu T, Hu C, Li H, Chen G, Xu G, Wang S, Ma D (2008) Comparative genomics and function analysis on BI1 family. Comput Biol Chem 32: 159-162