
Técnicas Electroquímicas Para El Control y Estudio de La Corrosión - Joan Genesca
273
, , EC ICA LECT OQ IMICAS RA EL CoNTROL Y EsTUDIO DE LA CoRROSióN Y. MEAS F. J. RoDRíGuEz J. GENESCÁ J. MENDOZA R. DURÁN AuTORES: -. J. URUCHURTU J. M. MALO ·HACÓN AONA F. M. AtMERAYA J. G. GoNZÁLEZ
-
Upload
cesar-a-rodriguez -
Category
Documents
-
view
548 -
download
77
description
electroquimica
Transcript of Técnicas Electroquímicas Para El Control y Estudio de La Corrosión - Joan Genesca

, , EC ICA LECT OQ IMICAS /\RA EL CoNTROL Y EsTUDIO
DE LA CoRROSióN
Y. MEAS
F. J. RoDRíGuEz
J. GENESCÁ
J. MENDOZA
R. DURÁN
AuTORES:
-.............. J. URUCHURTU
J. M. MALO ·HACÓN
AONA
F. M. AtMERAYA
J. G. GoNZÁLEZ

TÉCNICAS ELECTROQUÍMICAS PARA EL
CoNTROL Y EsTuDio DE LA CoRRosióN
EDITOR JuAN GENESCÁ LLONGUERAS
CoMITE EDITORIAL ]OSÉ M. MALO TAMAYO
YUNNY MEAsVoNG
jORGE URUCHURTU CHAVARfN
]UAN GENESCÁ LLONGUERAS
PRIMERA jORNADA SOBRE TÉCNICAS ELECTROQUÍMICAS
PARA EL CoNTROL Y EsTUDIO DE LA CoRRosióN
XVI CoNGREso SociEDAD MEXICANA ELECTROQUíMICA
QUERETARO, 23 MAYO 2001

UNIVERSIDAD NACIONAL AUTÓNOMA DE MÉXICO
TÉCNICAS ELECTROQUÍMICAS PARA EL CoNTROL Y EsTUDIO
DE LA CoRROSióN
EDITOR
JuAN GENESCÁ LLONGUERAS
Ciudad Universitaria, 2002

Responsable de edición: ]UAN GENESCÁ LLONGUERAS Primera edición: diciembre del 2002
TfcNICAs ELECTROQUíMICAS PARA EL CoNTROL Y EsTUDIO DE LA CoRROSióN
D.R. ©Facultad de Química, UNAM
Derechos exclusivos de edición reservados para todos los países de habla española. Prohibida la reproducción total o parcial por cualquier medio sin autorización escrita del editor.
Facultad de Química, UNAM Ciudad Universitaria, C.P. 04510 México, D.F. www.fquim. unam.mx
ISBN: UNAM 970-32-0540-2
HECHO EN MÉXICO
Este libro se publicó con apoyo financiero del Programa Universitario de Ciencia e Ingeniería de Materiales. Por estos apoyos, la Facultad de Química expresa su agradecimiento.


ÍNDICE Página
1. PROLOGO .......................................................................... 7
2. TÉCNICAS ELECTROQUÍMICAS DE CORRIENTE
DIRECTA PARA EL ESTUDIO DE LA CORROSION. 9
2.1 TEORIA ELECTROQUÍMICA DE LA CORROSION. Y. Meas ................................................................................. 9
2.2 RESISTENCIA DE POLARIZACION. F.J. Rodríguez . 28
2.3 EXTRAPOLACION DE TAFEL (INTERSECCIÓN).
J. Genescá . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 40
3. ESPECTROSCOPIA DE IMPEDANCIA ELECTROQUÍMICA EN CORROSION.
J. Mendoza, R. Durán y J. Genescá ................................... 53
4. LA TÉCNICA DE RUIDO ELECTROQUÍMICO PARA EL ESTUDIO DE LA CORROSION.
J.M. Malo y J. Uruchurtu ................................................... 93
5. TÉCNICAS ELECTROQUÍMICAS APLICADAS AL ESTUDIO DE LA CORROSION EN LA INDUSTRIA NUCLEAR . ....................................................................... 119
5.1 SENSIBILIZACION. E.A. Martínez .............................. 119
5.2 EVALUACION DE LA TEMPERATURA CRITICA DE PICADO.
E.A. Martínez . ................................................................... 129
5.3 CORROSION POR PICADURAS Y POR HENDIDURAS.
C. Arganis . .......................................................................... 137
5

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
6. APLICACIÓN DE LA TÉCNICA DE ESPECTROSCOPÍA DE IMPEDANCIA ELECTROQUÍMICA (EIS) EN EL ESTUDIO DE LA CORROSIÓN DEL ACERO DE REFUERZO EMBEBIDO EN CONCRETO.
T. Pérez .............................................................................. 143
7 . OXIDACION EN ALTA TEMPERATURA. A.Martínez, J.G. Chacón, C. Gaona, F. M. Almeraya y J.G. González . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 179
8. APÉNDICE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 245
8.1 CONVENTIONAL DC ELECTROCHEMICAL TECHNIQUES IN CORROSION TESTING.
J. Genescá, J. Mendoza, R. Durán and E. García . ........... 245
6

PRÓLOGO
Durante e l p asado Congreso de la Sociedad Mexicana de Electroquímica celebrado en Querétaro durante 2001 , un grupo de colegas me pidió que organizara una Jornada dedicada a las técnicas electroquímicas para el estudio y control de los procesos de corrosión. Gracias al apoyo entusiasta de los organizadores del congreso y de la Sociedad Mexicana de Electroquímica, esta jornada se pudo celebrar sin contratiempos y con una buena asistencia de público que contribuyó a dar realce al evento.
Ya desde el inicio se creyó oportuno darle la mayor difusión posible a esta jornada mediante la edición de las notas con que cada uno de los participantes contribuyó. Este CD es el primer esfuerzo, al cual le seguirá la edición en forma de libro a cargo del Programa Universitario de Materiales de la UNAM, gracias al generoso ofrecimiento de su actual coordinador, el Dr. Julio A. Juárez Islas.
Siempre es dificil cuando se organiza una jornada de este tipo decidir quienes serán los ponentes. Me atribuyo toda la responsabilidad en cuanto a la selección de los mismos y es a mi persona, en cuanto a editor, que deben dirigirse todas las quejas que tengan los futuros lectores de estas notas. No son, sin embargo, objeto de una decisión unipersonal. Me asesoré en un comité o consejo editorial, formado por personas con una larga trayectoria académica y científica en el área, todos ellos, además, excelentes amigos. Así, Yunny Meas, Jorge Uruchurtu, José María Malo y Juan Genescá nos dimos a la tarea de llevar adelante la selección primero y después la edición de estos apuntes que esperamos sirva como material de apoyo para los cursos de licenciatura, pero especialmente de posgrado relacionados con las aplicaciones de las técnicas electroquímicas en el estudio de los procesos de corrosión.
Quiero agradecer a todos los participantes el esfuerzo realizado y su desinteresada participación, ya que tanto el CD como el libro se van
7

TÉCNICAS ELECTROQUíMICAs PARA EL CoNTROL Y Esrumo DE LA CoRROSIÓN
a distribuir gratuitamente a todas las instituciones y universidades participantes, así como a todas aquellas que lo requieran o piensen utilizarlo.
El contenido temático del CD es el que creo podía esperarse de una jornada de este tipo. Una revisión de la teoría electroquímica de la corrosión a cargo de Yunny Meas, seguido de dos aplicaciones de las técnicas de corriente directa, la de resistencia de polarización, escrito por Francisco Javier Rodríguez y el método de Tafel a cargo de Juan Genescá. Seguidamente se presenta la técnica de espectroscopía de impedancia electroquímica, por Juan Mendoza, Rubén Durán y Juan Genescá, para posteriormente continuar con la técnica de ruido electroquímico, a cargo de José María Malo y Jorge Uruchurtu. Las aplicaciones de las técnicas electroquímicas en la industria nuclear están a cargo de Enrique Martinez y Carlos Arganís, mientras que en el caso del concreto, fueron escritas por Tezozomoc Pérez. Por último y aunque no se presentaron durante la mencionada jornada, el Comité Editorial decidió incluir, por su relevancia e interés, un capítulo dedicado a la oxidación a alta temperatura, redactado por el grupo que dirige Alberto Martínez en el C IMAV de Chihuahua.
En esta primera presentación publica de los apuntes de esta jornada a través del CD que, querido lector, tienes entre tus manos, no se ha realizado una homogeneización de la nomenclatura utilizada por los diferentes autores y se ha respetado la propia de cada quién. Algunos temas incluso pueden repetirse y presentarse con diferentes enfoques. Espero que esto sirva para enriquecer la discusión y el aprendizaje de estas técnicas electroquímicas.
Por último, cabe resaltar que estas notas sólo pretender ser un material de apoyo para los profesores que enseñan corrosión a nivel licenciatura, maestría y doctorado y que su lectura requiere de unos buenos conocimientos previos de los fundamentos electroquímicos de la corrosión. Hay excelentes libros en el mercado y estas notas deben ser un complemento a los mismos. Cualquier comentario u opinión será muy bien recibido, así como las necesarias correcciones que agradecería enviarán al editor.
Ciudad Universitaria D.F. Junio 2002
8
Juan Genescá Llongueras Editor

TÉCNICAS ELECTROQUÍMICAS DE CORRIENTE DIRECTA
PARA EL EsTuDio DE LA CoRROSióN
2.1 TEORÍA ELECTROQUÍMICA DE LA CORROSIÓN
Yunny Meas. Centro de Investigación y Desarrollo en Electroquímica (CIDETEQ), Parque Tecnológico, Sanfandila,
Pedro Escobedo, C.P. 76700, Estado de Querétaro.
La aplicación de técnicas de polarización, tales como la polarización potenciostática y la potenciodinámica ha sido muy exitosa en la evaluación de la velocidad de corrosión y también en el estudio de los fenómenos involucrados en la reacción de corrosión. Este éxito parece lógico si se considera que los procesos involucrados son de naturaleza electroquímica.
El uso de estas técnicas se ha vuelto rutinario, sobre todo con la ayuda de las computadoras y de los programas que permiten llevar a cabo de manera automática los experimentos y también el análisis de los resultados obtenidos. El uso de estos programas simplica enormemente el trabajo de obtención de las curvas experimentales, con el peligro potencial, sobre todo para los principiantes, de considerar que estas pruebas "estándares" pueden aplicarse a todos los sistemas con éxito.
El propósito de esta presentación es hacer un recordatorio de los conceptos teóricos sobre los cuales se fundamentan estas técnicas, así como definir sus alcances para su uso apropiado.
l.-Teoría
1.- Ley de Tafel La observación y el establecimiento de la relación existente
entre la corriente y el potencial del electrodo datan de ya casi un siglo, desde que Tafel, en 1905, dedujo de manera teórica, para la
9

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
reacción de reducción del protón, la ecuación de polarización de un electrodo reversiblemente polarizable, en el cual se desarrolla un sistema casi reversible. Esta ley se expresa por las relaciones siguientes :
- para una reacción anódica (i>O) : 11. = a + b lag i (b>O) (1)
- para una reducción catódica (i<O): 11c = a' - b' lag 1 i 1 (b'>O) (2)
En coordenadas semi-logarítmicas, estas relaciones se representan por las rectas de la Figura 1:
Curva real
log 11
Figura 1. Rectas de Tafel
Estas fórmulas son válidas solamente para una corriente i suficientemente grande, es decir para un sistema bastante alejado del equilibrio. Lo que es evidente, puesto que se debe obtener a i=O un sobrepotencial 11 = O y no 11 = ±oo como lo indicaría la ley de Tafel.
Si las reacciones anódicas y catódicas consideradas son las dos reacciones inversas de un mismo sistema casi reversible, las dos rectas de Tafel se cruzan sobre el eje de las abscisas para una densidad de corriente 10. Se tiene entonces:
-a 1 b = - a' 1 b' = log Í0 (3)
10

Con las mismas coordenadas semi-logarítmicas, la curva de polarización real está constituida de dos ramas, una anódica y otra catódica, siendo cada una asintótica de la recta de Tafel correspondiente.
En coordenadas ordinarias, las curvas de Tafel anódicas y catódicas para un mismo sistema tiene la forma representada en la Figura 2:
rr, 1 1 1 1 '1-'
e 1
1 /
' 1 ,
Figura 2. Curvas de Tafel.
Esta ley de Tafel tiene un papel importante en electroquímica y es en la búsqueda de su justificación teórica que se han podido encontrar las hipótesis correctas para el mecanismo de una reacción electroquímica.
(N ota: Es interesante sab er, desde punto de vista históri co; que la lry de Taj e! fue deducida teóricamente a partir de una hip ótesis errónea:
El razonamiento que sugirió Taj e! para la interpretaá ón de la reacción de reducá ón del protón j ué el siguiente:
La formación del hidrógeno implica 2 etapas: -una etapa electrqquí mica:
H+ + e � H (1) -una etapa química:
2 H ---+ H2 (2)
11

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL Y EsTUDIO DE LA CORROSIÓN
C ada una de estas etapas tiene su propia velocidad y en el caso general, las dos velocidades intervienen para fij ar la velocidad del proceso glob al. Es lo que se llama un rég imen cinético mix to. En este caso, se muestra en cinética general que el proceso glob al tiende hacia un régimen estacionario donde las velocidades de las dos etapas son ig uales, es decir, en donde la concentración de los átomos de H en la supetj icie y a no varia. Este régimen se alcanza teóricamente a un tiempo infinito.
S in emb argo, si una de las etapas es mucha más rápida que la otra, el proceso correspondiente alcanza inmediatamente su estado de equilib rio y es el otro proceso el que impone su veloc idad a la reacci ón glob al (p roceso limitan te). S e dice que el régimen á nético es puro y el estado estacionario está alc anzado desde el inicio de la reacción electroquímica.
Sin emb argo, en el caso de l a reacción de hidrógeno, el primer proceso es una reacción entre iones y electrones, el segundo es una reacción entre átomos neutros y desde Arrhenius hasta 19 20 y un poco mas, se pensab a que las reacciones del primer tip o eran mucho más rápidas que las reacciones del segundo tipo. Entonces, esta primera hipótesis ha llevado de manera natural a suponer que es la etapa de recomb inación de los átomos la que determina la velocidad glob al. En estas condiciones y considerando que el estado estacionario está alc anzado desde el inicio, se tien e:
d i H I/ d t = (d i H I 1 d t)¡ + ( d I H 1; d t)2 = o (4)
La velocidad de formación de los átomos de H segú n la primera etapa es proporcional a la densi dad de corriente que atraviesa el electrodo:
(5)
La velocidad de desaparición de estos mismos átomos en la segunda etapa, considerando ésta como de orden 2, es:
S e tiene, entonces:
(7)
12

Por otro lado, como el primer proceso está en equilibrio, el potencial tomado por el elect rodo es el da do por la ecu ació n de Nernst para la concentración en átomos de H
y la presión de gas H2 ex istente sob re el electrodo:
e=RT/F I n IH+ 1 / IH I (8)
es decir:
o sea: e= RT/2F I n (kF) - 0 .06 pH - RT/2F I n 1 i l (10)
el sob rep otencial vale entonces:
11 = RT/2F Ln (kF) - RT/2F I n 1 i l (11)
Este sobrep otencial tiene la form a dada por la lry de T ifel, 11 = a - b log 1 i 1 , p ero el coefi ciente b correspondiente vale O. 06/2 en lugar de 2 x O. 06 t' omo lo ob tenido en el experimento.
La hipótesis no ha sido satisf actoria. Sin embargo, la teorí a tuvo el mé rito d e ab rir el camino hacia una teorí a corre..ta, admitiendo que la hip ótesis inicial fue errónea y aceptar que, en ciertos casos, es la etapa electrónica la que impone su velocidad a la reacción glob al. La teorí a moderna, fundada sob re esta hip ótesis, es la que veremos ahora, con la fórmul a de B ut ler - V olmer.
2. - Ecuación de Butler -Volmer
Esta teoría que se encontraba en germen bajo una forma incompleta e incorrecta en los trabajos de Butler (1924), fue propuesta en su forma casi definitiva en 1930 por Erdey-Cruz y Volmer. Pero es solamente en Rusia, bajo el impulso de Frumkin, que ha sido aplicada y desarrollada desde sus orígenes. En los otros países, y aún con los esfuerzos de Audubert en Francia y de Bowden en Inglaterra, esta teoría había quedado prácticamente ignorada y empezó a atraer la atención solamente hasta 1949, fecha de la creación del "Comité Internacional de Thermodynamique et Cinétique Electrochimique" (C.I.T.C.E.), actualmente conocido como la "International Society of Electrochemistry" (ISE) .
13

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
Esta teoría permite obtener la velocidad de un proceso electroquímico, es decir la relación i= f(e) en el caso donde la transferencia de los electrones (etapa electroquímica) sea la única que controla la velocidad del proceso global. Un tal régimen cinético se llama régimen puro de transferencia electrónica o régimen de activación.
Considerando la reacción:
<=> Red (3)
Esta reacción se lleva a cabo en el electrodo, en un sentido o el otro. Supondremos para empezar que esta reacción es de primer orden con respecto a Ox y Red. La corriente total que atraviesa el electrodo es la suma algebraica de dos corrientes, una de reducción (negativa), correspondiente a la de izquierda a derecha y una de oxidación (positiva), correspondiente a la de derecha a izquierda:
con: 1 = nF k 1 Red 1 i =
o: nF k Tox 1 red red (14)
(12)
(13)
Las constantes de velocidades k0, y kred pueden ser expresadas a partir de la teoría del complejo activado de Eyring (1935) :
k = (kT /h) . 1 Red* l / 1 Red 1 k:e: = (kT/h) .1 Ox' l / 1 Ox 1
(15)
(16)
donde k es la constante de Boltzmann (k= R/N) h es la constante de Planck ( 6.62 x 10-34 joule .segundo) 1 Red* l y 1 Ox* l son las concentraciones·de los complejos
activados para la oxidación y reducción respectivamente.
Los cocientes 1 Red* 1 / 1 Red 1 y 1 Ox* 1 / 1 Ox 1 son iguales a las constantes de acción de masa de los equilibrios:
1 Red 1 <=> 1 Red* 1 (6)
1 Ox 1 <=> 1 Ox* 1 Cf)
14

Considerando �G* ox y �G*,ed como las entalpías libres correspondientes ( de izquierda a derecha) ( son también las entalpías libres de activación de oxidación y de reducción)
ln ( 1 Red* 1 / 1 Red 1) = - �G* /RT (17) ln ( 1 Ox* 1 1 1 Ox 1 ) = -�G*,e)RT (18)
obtenemos:
i = ox
1 = red nF (kT/h) 1 Red 1 exp(-�G*jRT) nF (kT /h) 1 Ox 1 exp(-�G*,jRT)
(19) (20)
La transferencia de la carga nF a través de la barrera �� (potencial absoluto del electrodo) corresponde a una energía ± nF �� dada al sistema según el sentido de la transferencia.
La hipótesis fundamental de la teoría consiste en admitir que una fracción a de esta energía contribuye a disminuir la entalpía libre de la oxidación y que una fracción l3 de la misma energía incrementa la entalpía libre de activación de la reducción. Se puede entonces escribir:
(21) (22)
Los coeficientes a y 13 son llamados coeficientes de transferencia de carga de las reacciones de oxidación o reducción respectivamente.
(�G*0,\ y (�G*,ed)o representan las entalpías libres de activación de las reacciones para un potencial absoluto nulo.
Como el potencial absoluto no es medible, se introduce el potencial relativo e del electrodo que difiere de �� , solamente por una constante. Entonces las entalpías libres de activación toman la forma:
1 5
(23) (24)

TÉCNICAS ELECTROQUÍMICAS PARA EL CoNTROL Y EsTUDIO DE LA CoRROSIÓN
y las expresiones de i0, y ircd son:
i = ox i = red
nF (kT/h) 1 Red 1 exp(- NjRT) exp (anF e/RT) nF (kT /h) 1 Ox 1 exp(-N,ed /RT) exp (-�nF e/RT)
agrupando las constantes, se obtiene:
i = ox 1 = red
nF kox 1 Red 1 exp (anF e/RT ) nF kred 1 Ox 1 exp (-l3nF e/RT)
por lo tanto, obtenemos:
(25) (26)
(27) (28)
i = nF [ kox 1 Red 1 exp (anF e/RT ) - k red 1 Ox 1 exp (-�nF e/RT) ] (29)
Cuando el electrodo está en equilibrio, es decir que su potencial es igual al potencial termodinámico, ( e=e, ) , la densidad de corriente total que atraviesa el electrodo es nula y las densidades de corriente parcial, i0, y i,ed son iguales en valor absoluto a una densidad de corriente i0, llamada densidad de corriente de intercambio.
i0 = nF kox 1 Red 1 exp ( nF a e.) = nF k.,d 1 Ox 1 exp ( - nF � e, ) (30) RT RT
e = RT (In kred + In 1 Ox 1 ) (31) ' a+ 13 nF k 1 1 ox Red
suponiendo que a y l3 son independientes del potencial.
Considerando que el sobrepotencial: 11 = e - e,
La ecuación (29) se simplifica en:
i = i. [ exp ( a n F11) RT
exp (-13 n F11)] RT
(32)
Esta fórmula es conocida como la ecuación de Butler- Volmer y expresa la ley fundamental del sobre-potencial en régimen puro de transferencia electrónica.
1 6

2.1 -Caso de altos sobre-potenciales. Aproximación de Tafel.
Si el sobre-potencialr¡ es alto, uno de los términos de la fórmula de Butler -Volmer se vuelve despreciable con respecto al otro.
Por ejemplo, para un valor de 11 positivo y grande, se puede escribir:
ó
i = i = i exp ( a n Fr¡ ) ox o RT
RT RT , - -- lni + -- ln i • 1 - - a nF o a nF
(33)
(34)
esta ecuación tiene la forma prevista por la ley experimental de Tafel.
De la misma manera, para sobre-potenciales r¡ negativos y suficientemente grandes en valor absoluto, obtenemos:
r¡ = - � ln i + RT
ln 1 i l (3nF
o (3nF (35)
En coordenadas semi-logarítmicas, estas dos corrientes están representadas por las dos rectas de Tafel (ver figura 1)
La aproximación de Tafel corresponde a despreciar a uno de los términos de la ecuación de Butler-Volmer, o sea a despreciar a una de las corrientes parciales. Esta aproximación es válida a 1% , por eJ· emplo del lado anódico, si i > 102 1 i d 1 o sea si
ox re
012 r¡ > -- volts .
n
2.2 -Caso de sobre-potenciales bajos. Aproximación al origen.
Si el sobrepotencial es pequeño, se puede desarrollar en serie los exponenciales, limitándose al primer orden. Si consideramos que a +¡3 = 1 para e= e, , obtenemos:
i nF . o 1= -- r¡
RT (36)
que corresponde a la ecuación de la tangente al origen de la curva de polarización.
1 7

TÉCNICAS ELECTROQUÍMICAS PARA EL CoNTROL Y EsTumo DE LA CoRROSióN
Si consideramos la corriente total que atraviesa el electrodo de superficie S:
I = i S
De la ecuación (36) se obtiene la relación
11 = RT 1 - -I nF i S o
(37)
Se puede escribir esta relación en forma de ley de OHM de la manera siguiente:
RT 1 R*= --
nF i S o
(38)
R* es análogo a una resistencia, la cual se conoce como "resistencia de polarización" del electrodo.
Hay que tener cuidado que esta aproximac10n al origen es válida solamente en un intervalo estrecho de potencial y depende del número de electrones n. Por ejemplo, para n=1, el error es menor a 1% si 1111 es inferior a 0.01 V
Podemos observar que existe un intervalo de potencial entre los dominios de validez de las dos aproximaciones anteriores.
3.- Procesos competitivos y no-competitivos
En los párrafos anteriores se ha considerado el caso de una sola reacción que se lleva a cabo en el electrodo. Ahora, consideraremos el caso de varias reacciones que se llevan a cabo simultáneamente. Vamos a introducir el concepto de procesos competitivos y procesos no-competitivos.
Dos procesos son llamados no-competitivos si se llevan a cabo simultáneamente en un mismo sitio activo del electrodo. El resultado es el mismo si los sitios, en lugar de ser idénticos para los dos procesos, son diferentes, pero repartidos en numero suficientes de manera regular y constante sobre la superficie del electrodo.
18

Dos procesos son competitivos, en el caso contrario, si cada uno de ellos se lleva a cabo en una fracción de la superficie. Esta fracción puede variar con el tiempo.
En el primer caso, es decir de procesos no-competitivos, las densidades de corriente correspondientes a cada uno de los dos procesos, para un potencial dado, se suman sencillamente, como Wagner y Traud lo han indicado por primera vez en 1938.
i = i + i t 2. (39)
Si cada uno de los dos procesos está en estado estacionario, el elec'trodo también lo estará y podremos trazar sin dificultad la curva de polarización resultante.
En el otro caso, correspondiente a procesos competitivos, podremos deducir la densidad de corriente total a partir de las corrientes parciales de cada uno de los procesos si conocemos las superficies correspondientes a cado uno. Tendremos:
s, . S2 i = i, - + 1z - ( 40)
S S Pero como S1 y S2 varían normalmente con el potencial y el tiempo, el electrodo está generalmente en un estado evolutivo.
Los ejemplos mas conocidos de procesos no competitivos son los que corresponden a dos procesos con polaridades diferentes. Es decir, uno es anódico y el otro es catódico. Es el , por ejemplo, de la corrosión del hierro en un medio ácido.
Un caso típico de procesos competitivos corresponde al de disolución anódica de un metal con dos grados de oxidación diferente, como puede ser la disolución anódica de una aleación binaria. También, como en el caso de un solo metal para el cual el grado de oxidación superior aparece en forma de óxido o hidróxido y no en forma iónica, es decir en presencia de un fenómeno de pasivación. El electrodo está en un estado evolutivo y es el caso, en general, para dos procesos competitivos.
En este capítulo, veremos solamente la teoría para los procesos nocompetitivos.
19
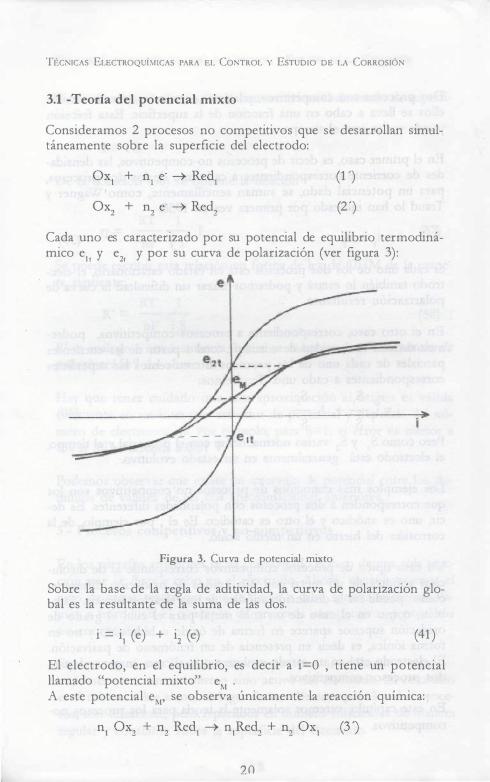
TÉCNICAs ELECTROQUÍMICAS PARA EL CoNTROL v EsTuDio DE LA CoRROSIÓN
3.1 -Teoría del potencial mixto
Consideramos 2 procesos no competitivos que se desarrollan simultáneamente sobre la superficie del electrodo:
(1 ')
(2')
Cada uno es caracterizado por su potencial de equilibrio termodinámico eit y e2, y por su curva de polarización (ver figura 3):
Figura 3. Curva de potencial mixto
Sobre la base de la regla de aditividad, la curva de polarización global es la resultante de la suma de las dos.
i = i1 (e) + i2 (e) (41)
El electrodo, en el equilibrio, es decir a i=O , tiene un potencial llamado "potencial mixto" eM . A este potencial eM, se observa únicamente la reacción química:
20

Las densidades de corriente correspondientes a los dos procesos son respectivamente:
i1 = (i)1 { exp [ a,n, F ( e - e1 ,)] - exp [ -�,n, F (e - elt)] } (42) RT RT
i2 = (i)2 { exp [ a,n, F ( e - e) ] - exp [ - �,n, F (e - e2.) ] } ( 43) RT RT
El potencial mixto al equilibrio está definido por:
y tomando : e
F 0.06 X = exp (_e_) = 10
RT obtenemos una ecuación del tipo:
(44)
Afortunadamente, una resolución general de esta ecuación no es necesaria si se considera que los potenciales de equilibrio de los 2 sistemas e1 , y e2, son suficientemente diferentes para que la aproximación de Tafel sea válida en el potencial mixto eM . La densidad de corriente i1 es entonces igual a la densidad de corriente de oxidación de la reacción (1 ') y la densidad de corriente i2 es igual a la densidad de corriente de reducción de la reacción (2').
El potencial mixto está entonces definido por la ecuación:
(i \ exp [� ( e -e ) ] - (i \_, exp [-�'n' F (eM-e,_,)] (46) ,,1¡ RT M lt
ol RT del cual, se deduce:
RT (i)z + ln ( a,n, + A,n, )F (1. \ 1-' oll (47)
Se puede observar que, si las densidades de corriente de intercambio (iJ1 y (i0)2 son iguales, el potencial corresponde al baricentro de los potenciales de equilibrio de los procesos (1 ') y (2 ') afectados de los coeficientes a1n1 + f3p2•
21 ------------------

TÉCNICAS ELECTROQUÍMICAS PARA EL CoNTROL v EsTuDio DE LA CoRROSIÓN
Corriente de corrosión
Si las reacciones (1 ') y (2 ') corresponden a las reacciones que se llevan a cabo durante la corrosión de un metal, por ejemplo, el Fe en una solución ácida:
(4')
(S')
El potencial mixto de abandono (i=O), se llama potencial de corrosión y el valor absoluto común de las dos corrientes correspondientes a cada proceso (4') y (5') a este potencial mixto es la corriente de corrosión.
La densidad de corriente de corrosión puede ser calculada a partir de las corrientes de cada proceso. En el caso general, de la fórmula (42), que expresa la corriente anódica, basta remplazar el potencial e por el potencial mixto eM
Se obtiene:
In (1·. \ + _E_ o.t n! ¡3,n, ( ) o/2 A ez, - e¡, RT a,n,+ ..,,n,
(48)
Esta densidad tle corriente de corrosión aumenta con las densidades de corriente de intercambio y con la diferencia ( e2, - e1,) .
4.- Determinación grafica de eM e i corr
Los cálculos realizados anteriormente, con la aproximación de Tafel, pueden ser resueltos gráficamente de manera sencilla. En esta aproximación, las curvas de polarización de cada proceso, en coordenadas semi-logarítmicas, son rectas cuyas intersecciones dan de inmediato los valores de eM y log icorr'
22

V e
(]
e • • Q,.J - - - - - - !-
.a,.e. L-------::-------;1:;--:1 _ ,
1 ! lO@ 1 )>
logl0 Figura 4. Determinación grafica del potencial mixto y de i,o"
5.- Validez de la aproximación de Tafel
En los cálculos anteriores, la aproximación de Tafel suponía que, para cada proceso, una de las corrientes es efectivamente despreciable con respecto a la otra. Esta aproximación es valida a 1%, si la corriente que se desprecia es inferior a la centésima parte de la otra, o sea:
��n� exp [ - -- F ( eM e11 ) ] RT
a,n, exp [ - RT F (eM e11) ]
o sea:
<
<
-2 a,n, 1 O exp [ -- F (e M - e1r ) ] RT -2 (Jznz 1 O exp [ -- F (e M - e2, ) ] RT
0.12 0.12 n, n,
(49)
(50)
(51)
ecuación que muestra que eM debe ser suficientemente alejado de e1 y e2.
Podemos anotar que el error sobre el valor de eM es, en general, menor que los errores cometidos sobre i1 e i2, cuando que aplica
23

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
la aproximación de Tafel. Debido a que la concavidad opuesta de las 2 curvas, los errores pueden compensarse. Sin embargo, el error sobre i queda del mismo orden que sobre i e i .
corr 1 2
7. -Calculo de eM e icorr con la aproximación en el origen.
Cuando los potenciales de equilibrio termodinámico e y e están 1t 2t bastante cercanos, la desigualdad (51) ya no está respetada y la aproxi-mación de Tafel ya no es valida. Sin embargo, podemos pensar en utilizar la aproximación al origen. Reconsiderando la ecuación (36) y tomando r1 y r2 como las resistencias de polarización por unidad de superficie de cada una de las reacciones, tenemos:
RT 1 e= e11 + r1i1 con r¡ ---- (52)
n1F (i)l RT 1
con = ---- 1 e= e2, + r2i2 rz (53) n2F (i)z
De estas relaciones, y considerando que i1=i?=i a e=eM, pode- 1 mos deducir: - corr
r1e2, + r2e" (54) eM = r¡ + r?
icorr = e2, _ e1,
(55) r¡ + rz
Como lo hemos visto, esta aproximación al origen es valida a 1% en un dominio estrecho de potencial ( T] < 10 m V para n = 1) .
8. -Factores que influyen sobre la medición de la corriente de corrosión
8. 1 Efecto de la ca(da óhmica En el desarrollo teórico que hicimos, no hemos considerado
la resistencia del electrolito que puede existir entre el electrodo de referencia y el electrodo de trabajo sobre el cual se lleva a cabo la reacción de corrosión estudiada. En la práctica, esta resistencia existe.
24
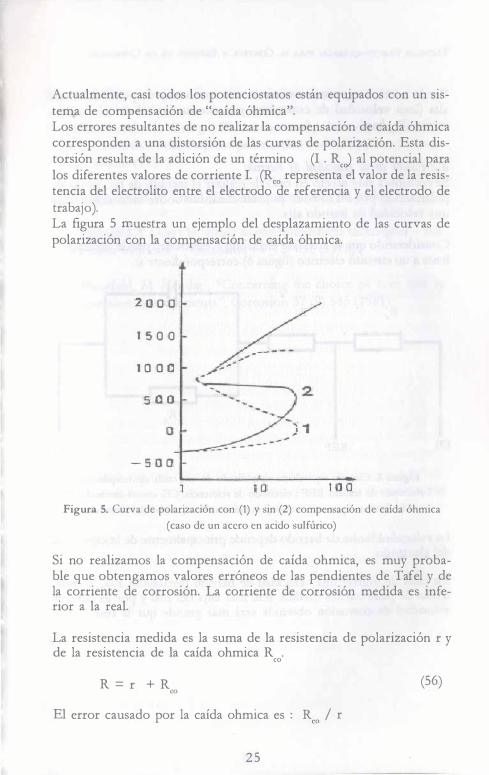
Actualmente, casi todos los potenciostatos están equipados con un sistema de compensación de "caída óhmica". Los errores resultantes de no realizar la compensación de caída óhmica corresponden a una distorsión de las curvas de polarización. Esta distorsión resulta de la adición de un término (I . RcJ al potencial para los diferentes valores de corriente I. (Reo representa el valor de la resistencia del electrolito entre el electrodo de referencia y el electrodo de trabajo). La figura 5 muestra un ejemplo del desplazamiento de las curvas de polarización con la compensación de caída óhmica.
lOO O
1500
1 O O G
500
-50(]
100
Figura 5. Curva de polarización con (1) y sin (2) compensación de caída óhmica (caso de un acero en acido sulfúrico)
Si no realizamos la compensación de caída ohmica, es muy probable que obtengamos valores erróneos de las pendientes de Tafel y de la corriente de corrosión. La corriente de corrosión medida es inferior a la real.
La resistencia medida es la suma de la resistencia de polarización r y de la resistencia de la caída ohmica R . ca
R = r + R (56) ca El error causado por la caída ohmica es : Reo / r
2 5

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
Por lo tanto, este error es mínimo si la resistencia de polarización es alta (baja velocidad de corrosión) y la conductividad del electrolito es alta (Reo baja).
8.2-Ejecto de la velocidad de barrido Se considera este efecto en caso de realizar el trazado de las curvas de polarización por un método potenciodinámico sobre todo si se utiliza una velocidad de barrido alta.
Considerando que la interfase electrodo - electrolito puede ser equivalente a un circuito eléctrico (figura 6) correspondiente a:
11 11
R,, R <O
1-
R p
CE REF ET
Figura 6. Circuito equivalente simplificado de una celda electroquímica
(ET:electrodo de trabajo. REF : electrodo de referencia, CE: contra-electrodo, R'": caída óhmica, R
r: resistencia de polarización, C: capacidad del electrodo)
La velocidad limite de barrido depende principalmente de la capacidad del electrodo.
En caso de utilizar una velocidad de barrido demasiado alta, la resistencia de polarización medida será mas baja del real y por lo tanto la velocidad de corrosión obtenida será más grande que la real.
26

Bibliografía:
1 .- M. Stern and A.L. Geary, " Electrochemical polarization. I. A theoretical analysis of the shape of polarization curves". J. Electrochem. Soc. 104.56 (1957)
2.- C. Wagner and W Traud, Z. Elektrochem., 44, 391 (1938).
3.- F. Mansfeld, " Don't be afraid to electrochemical techniques- but use them with care ", Corrosion 44 (12) 856 (1988)
4.- F. Mansfeld, M. K.endig , "Concerning the choice of sean rate in polarization measurements", Corrosion 37 (9) 545 (1981 ) .
27

TÉCNICAS ELECTROQUÍMICAS DE CORRIENTE DIRECTA
PARA EL EsTuDio DE LA CoRROSióN
2.2 RESISTENCIA A LA POLARIZACIÓN
Francisco Javier Rodríguez Gómez*
Depto. Ing. Metalúrgica, Facultad Química, UNAM Ciudad Universitaria. 04510 - México D.F. MÉXICO
Tel. + 52 55 5622 5225 Fax + 52 55 5622 5228
Introducción
La Resistencia a la Polarización (Rp) o Polarización lineal es una de las técnicas electroquímicas que ha sido más utilizada en los últimos 50 años. Con el paso del tiempo se han desarrollado herramientas experimentales que son más complejas que la Rp, aportan información mecanística e implican el uso de instrumentación cara y sin embargo, no han conseguido desplazar a esta técnica de un lugar importante en el ámbito de la Ingeniería de Corrosión. Ciertamente tiene limitaciones, pero ¿cuál de los métodos electroquímicos no las tiene? El conocimiento de las limitaciones de la Rp y de sus ventajas y bondades, resultará en un uso correcto y una clara interpretación de los resultados que se obtengan al aplicar esta técnica en sistemas simples y complejos. Es importante conocer los pormenores de un método electroquímico que se ha empleado para determinar la velocidad de corrosión en sistemas tan diversos como el hormigón, pinturas y hasta en alimentos, lo que es demostración de la versatilidad de la Rp.
Generalidades
En un artículo publicado en Joumal of Electrochemical Society en 1957, Stem y Geary escribieron que " . . . se deriva una ecuación relacionando la pendiente de esta región con la velocidad de corrosión y las pendientes de Tafel . . . Oo cual) es una nueva aproximación experimental al estudio de la electroquímica de los metales corroyéndose . . . ".
28

La ecuación desarrollada por Stern y Geary toma el nombre de los investigadores y se presenta a continuación:
i = B 1 R (1) rorr p
B = ( b ab c) 1 [ 2 .303 ( ba + be) ] (2)
donde ba y be son las pendientes de Tafel anódica y catódica, respectivamente.
La ecuación de Stern y Geary (ec. 1) establece la relación entre la densidad de corriente de corrosión (icorr), es decir, la velocidad de corrosión, con la resistencia a la polarización. Esta ecuación recuerda la conocida Ley de Ohm, donde se establece que la diferencia de potencial entre dos puntos resulta en un flujo de corriente limitado por la presencia de una resistencia.
La constante B en la ecuación (2) sirve para pasar de la proporcionalidad a la igualdad y presenta una relación existente entre las pendientes de Tafel. Queda claro a partir de esta relación, que es indispensable que exista el comportamiento tafeliano en el sistema electroquímico, para que la ecuación de Stern y Geary pueda ser aplicable.
¿Se debe realizar entonces de manera obligatoria primero la Extrapolación de Tafel para determinar las pendientes y a continuación determinar la Rp y calcular la velocidad de corrosión?
La respuesta a esta pregunta depende del experimentador. Por supuesto, lo recomendable sería realizar Extrapolación de Tafel para determinar los valores de las pendientes anódica y catódica; en ocasiones basta saber que la velocidad de corrosión está aumentando en función del tiempo, por lo que sería suficiente conocer el comportamiento de la Rp contra el tiempo de manera que, al ser inversamente proporcionales, será muy fácil determinar la variación de icorr.
Por otra parte, se ha dicho que la mayor parte de las pendientes de Tafel oscilan entre valores de 60 y 120 m V 1 década, de donde se ha optado por hacer una aproximación suponiendo que ambas pendientes tuvieran el segundo valor. Así, se obtiene una B = 26 m V
29

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL y ESTUDIO DE LA CORROSIÓN
que puede emplearse con reserva, ya que las pendientes del sistema bajo estudio no tienen por qué tener esos valores. En cualquier caso, si se aplica este valor de constante B a un estudio, los valores de velocidad de corrosión serán comparables y éstos serán al menos del mismo orden de magnitud que si se emplearan las pendientes de Tafel reales.
Lo que muchas veces se ignora en referencia al artículo original de Stern y Geary Qunto con los otros dos artículos publicados en continuación a su propuesta de Polarización Lineal), es que ellos aclaran que esta aproximación es válida solo bajo ciertas circunstancias claramente descritas. El desconocimiento de estas limitaciones ha llevado a algunas personas a declarar que se trata de una metodología que no aporta resultados coherentes y concordantes con las otras técnicas experimentales, cuando la realidad es que se está trabajando en condiciones muy alejadas de las ideales para que se cumpla el comportamiento predicho por Stern y Geary.
Las condiciones bajo las cuales la ecuación de Stern y Geary es válida y, en consecuencia, puede ser aplicada sin restricción, son muy simples y pueden resumirse de la siguiente manera: son todas las condiciones que limitan a la ecuación de Butler- Volmer.
La técnica electroquímica llamada Resistencia a la Polarización es el resultado de la aproximación de bajo campo a la ecuación de ButlerVolmer y por este motivo, se debe aplicar solamente cuando exista control activacional o por transferencia de carga. Posteriormente en este capítulo se discutirá una aproximación que a nivel ingenieril puede aplicarse si el sistema se encuentra controlado por transporte de masa o difusión. Sin embargo, debe quedar claro, que se trata solamente de una aproximación y debe ser tomada con reserva.
Los materiales que son susceptibles de ser evaluados mediante la aplicación de la Polarización Lineal deben estar inmersos en electrolitos conductores, es decir, que la resistividad de las soluciones no sea alta, y deben encontrarse libres de películas resistivas, pues la Rp es una técnica inclusiva que globaliza todas las contribuciones resistivas y no alcanza a discriminarlas. Si se trabaja con sistemas altamente resistivos, se recomienda mejor que la Rp, el uso de la Espectroscopia de Impedancia Electroquímica.
30

La aproximac10n de bajo campo implica que, aunque el sobrepotencial y la corriente se relacionan a través de una ecuación que implica la diferencia entre dos exponenciales (ec. Butler-Volmer), en la inmediación del potencial de corrosión se puede asumir que el comportamiento es lineal. Por supuesto, esta supuesta linealidad no existe en esa zona de la curva E-I, por lo que es bueno recordar que la Rp también se ha definido como:
Rp =(BE 1 BIL .. o (3) o bien
(11 Rp) = (BII BE)Ecorr (4)
O dicho en otras palabras, la tangente a la curva de polarización evaluada en el potencial de corrosión o cuando la corriente tiende a cero.
Difusión
En los sistemas electroquímicos controlados por difusión, se presenta un fenómeno especial en el que la corriente catódica se ve limitada por la concentración de los reactivos catódicos, es decir, por el transporte de masa a través del electrolito (difusión). En esas condiciones, lo mejor sería no emplear esta técnica, pero la tentación de utilizar un método tan rápido y sencillo es muy grande y se ha realizado una aproximación que se muestra a continuación. No es ocioso recordar al lector que se trata de una aproximación y que los resultados obtenidos con ella, solo pueden ser un indicativo de los valores de velocidad de corrosión que se tendrían determinados por otros métodos más adecuados en las condiciones planteadas.
Cuando hay control difusional, se dice que se tiene una corriente límite, con una pendiente de Tafel catódica que tiende a infinito. La ecuación de Stern-Geary se ve reducida a la siguiente expresión:
icorr = ba /(2.303Rp) (5)
Procedimiento experimental
Para realizar un estudio experimental de Resistencia a la polarización, lo mejor es acudir a la norma ASTM G 59-91 "Práctica estándar para realizar medidas de resistencia a la polarización potenciodinámica"
3 1

TÉCNICAS ELECTROQUÍMICAS PARA EL CoNTROL v EsTUDIO DE LA CoRROSIÓN
Los pasos a seguir de acuerdo con la norma son los siguientes:
- Usar un puente salino que acerque el electrodo de referencia a 2 o 3 mm de la superficie del electrodo de trabajo.
- Registrar el potenciad de corrosión después de S minutos. - Aplicar un potencial 30 m V más negativo que el potencial regis-
trado. - Un minuto después de que se aplique el potencial, se inicia un
barrido en dirección anódica a 0.6 V /h (10 m V /min), registrando el potencial y la corriente continuamente.
- Terminar el barrido cuando se alcanza un valor de 30 m V más positivo que el potencial de corrosión.
- Graficar la curva de polarización - Determinar gráficamente la resistencia a la polarización, Rp,
como la tangente a la curva en el origen de la gráfica (flE=O) .
Consideraciones experimentales
Como se indicó en el inciso anterior, la perturbación puede realizarse en potencial registrando la respuesta en corriente. La curva de polarización lineal puede obtenerse de manera potenciostática aplicando escalones equipotenciales y esperando a que se alcance el estado estacionario antes de registrar la respuesta en corriente. La función de transferencia entre estímulo y respuesta es la Rp. Si por el contrario se realiza la curva de manera potenciodinámica, se puede aplicar la metodología descrita en el inciso anterior. Varios detalles de esta metodología se discutirán en apartados posteriores.
No se acostumbra realizar la perturbación en corriente y registrando el potencial (galvanostática o galvanodinámica), sin embargo este tipo de perturbación es muy útil cuando se tiene un sistema en el cual el potencial no es estable. En las curvas potenciostáticas y potenciodinámicas, es indispensable determinar el potencial de corrosión y que éste sea estable. Cuando el sistema presenta una deriva importante, se recomienda realizar una perturbación en corriente. Sin embargo, aunque en la literatura se menciona esta posibilidad, no se habla acerca del intervalo en el cual se puede considerar la validez del bajo campo.
32

Lo mejor sería correr una prueba preliminar de Rp de manera potenciostática para determinar el orden de magnitud de la corriente y con base en este dato, determinar la perturbación que llevará a mantener la linealidad indispensable en esta técnica. Como una ventaja adicional,Jones asegura que se reduce enormemente la duración de la prueba.
Ventajas de la Rp
- Se trata de una técnica no destructiva pues emplea bajas perturbaciones.
- Proporciona velocidad de corrosión instantánea. - No hace falta instrumentación muy sofisticada pues solamente
se necesita un potenciostato y, en consecuencia, es una técnica econórruca.
- Para aplicar la metodología no hace falta personal altamente especializado.
Limitaciones de la Rp
- Es necesario que el potencial sea estable. - Caída óhmica en sistemas altamente resistivos, lo que la hace
poco recomendable. - Es necesario seleccionar una velocidad de barrido adecuado.
Velocidad de barrido
Mientras que la norma ASTM sugiere 0.6 V /h como ya fue mencionado, investigadores reconocidos como Mansfeld y Kendig sugieren que la máxima velocidad de barrido tiene que ser seleccionada en función de una frecuencia característica correspondiente con el "break point" en baja frecuencia en un diagrama de Bode, lo que implicaría que para correr la prueba de Rp no bastaría con hacer el ensayo, sino que a las pruebas paralelas de extrapolación de Tafel para determinar las pendientes, habrá que correr una prueba de Espectroscopia de Impedancia Electroquímica. En la siguiente ecuación se menciona que en función de la constante de tiempo depende del valor de la resistencias del electrolito y a la polarización y. de la capacitancia de la doble capa electroquímica.
33

TÉCNICAs ELECTROQUÍMICAS PARA EL CoNTROL Y EsTumo DE LA CoRRosióN
Townley asegura que este método subestima la máxima velocidad que se puede emplear. Sugiere emplear la primera frecuencia de "break point" (no la segunda como Mansfeld y Kendig) y medir la Rp no en I=O, sino al final del barrido, sugiriendo que éste sea de 30 m V
Gabriele Rocchini, uno de los teóricos que más han trabajado con la condiciones en las que se puede aplicar esta técnica electroquímica, hace un análisis de series de Fourier, demostrando que cuando las señales s� hacen variar lentamente, una vez que desaparecen los transitorios, la I y el V están en fase. En otras palabras, habría que hacer muy lento el barrido de potencial. Este investigador define el concepto de Rp aparente, es decir, la Rp que depende de la velocidad de barrido en contraposición con la Rp real que sería aquella que no cambia con la velocidad de barrido. Rocchini demuestra también que la histéresis en altas velocidades de barrido de potencial se refleja en un descenso en la Rp aparente.
Macdonald define también la resistencia de polarización aparente (Rpa en algunos artículos) de la misma manera que Rocchini y establece que la Rp no se ve afectada si la velocidad de barrido es menor a 200 m V/ s ; sin embargo Rocchini dice que la Rp no se verá afectada por la velocidad de barrido siempre que ésta sea menor a 1 00 m V/ s.
A pesar de que en algunos lúnites hay divergencias en la literatura, es claro en todos los autores que debe evitarse el uso de altas velocidades de barrido pues el sistema podría no responder espontáneamente y la respuesta ser espúrea, lo que invalidaría los resultados experimentales.
No linealidad
Ya se ha mencionado anteriormente que la supuesta linealidad entre el potencial y la corriente en realidad no existe. Sin embargo, es una aproximación bastante correcta y no implica errores importantes en la técnica. Se puede discutir que la no linealidad invalidaría el empleo de la ecuación de Stern-Geary, pero no ocurre de esta manera de acuerdo con lo mencionado en la literatura . Cabe mencionar
34

además, que en la literatura se manejaron hace años varios programas de cómputo que permitían el cálculo de las pendientes de Tafel a partir de datos pretafelianos (no lineales).
Aplicaciones en Hormigón
Sin duda los trabajos más importantes y con mayor reconocimiento a nivel internacional han sido los desarrollados por Andrade, Feliu y González en España, que han trabajado ampliamente con la Rp, aplicando la técnica y compensando caída óhmica. Sus estudios han dado grandes aportaciones a la ciencia como son el estudio de mecanismos de corrosión, difusión de cloruros, etcétera; por otra parte, a nivel ingenieril sus resultados les permitieron desarrollar un instrumento que es el que se emplea en la mayor parte del mundo para determinar la velocidad de corrosión de acero de refuerzo en estructuras de hormigón. El GECOR es un corrosímetro que emplea la resistencia a la polarización como técnica a aplicar. Cabe mencionar aquí que las estructuras de hormigón son altamente resistivas y por lo tanto, la resistencia a la polarización incluye también esta contribución, lo que resultaría en un valor de Rp mayor al correcto y en consecuencia en una , velocidad de corrosión menor (se subestima la agresividad) . Para evitar esa caída óhmica se procede a realizar una compensación, mediante un método muy simple: se procede a interrumpir la señal y medir el decaimiento del potencial. A partir de esta curva de decaimiento se determina la caída óhmica, que se elimina al valor determinado y así se compensa el valor resistivo del hormigón. Muchos autores han cuestionado este método y otros más que existen en campo para compensar aduciendo que nunca se tiene la seguridad de haber compensado correctamente.
Andrade, Feliu y González opinan que los valores que se obtienen de velocidad de corrosión son adecuados
. cuando se comparan con ensayos gravimétricos. Empleando la Rp han estudiado muchas variables de este sistema tan complejo como interesante, entre otras:
- Distintos arreglos experimentales (geometría de las probetas, ubicación de los electrodos de referencia, etcétera),
- Concentración umbral de cloruros para promover la corrosión, - Uso de anillo guarda para la realización de las medidas, - Distintas dimensiones de muestra.
3 5

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
No son los únicos que han trabajo con la polarización lineal aplicada a este sistema. Macdonald et al han demostrado que en el sistema acero de refuerzo-hormigón, la distribución de corriente se vuelve un problema particular en bajas frecuencias cuando la resistencia a la polarización es alta. Gowers y Millard han realizado "mapeos" de Rp a través de grandes probetas ubicando las zonas en las que hay mayor corrosión, lo que permite trazar lineas de "isocorrosión". Gowers y Millard miden, junto con la Rp, la impedancia a 300 Hz y la resistividad del hormigón p,ara trazar dichos mapas.
G. Song dice, en referencia a la aplicación de la Rp al sistema acero de refuerzo-hormigón: "Aún si la ecuación de Stern-Geary fuera aplicable y los valores de B aceptables (típicamente entre 25-52 m V), se introducirían errores en el cálculo de velocidad de corrosión debido al efecto de sobre o subconfinamiento del anillo guarda" .
Esta polémica no ha terminado aún y creo que eso vuelve más interesante la aplicación de esta metodología en un sistema que necesita ser más estudiado todavía.
Aplicaciones en pinturas
Al respecto existe un artículo sumamente interesante y de lectura obligada para las personas que trabajan con recubrimientos orgánicos. C.W Walter discute el uso de las técnicas de corriente continua en la evaluación de pinturas. Claro que la polarización lineal puede ser aplicada casi sin restricción en el caso de las pinturas ricas en zinc, dado que el contenido de metal permite trabajar casi sin caída óhmica.
Hay que aclarar varias cosas. Un recubrimiento protector intacto puede tener resistencias muy altas y eso podría enmascarar los valores correctos de Rp. Por supuesto, se puede recurrir a la compensación (con todas las reservas mencionadas en el inciso anterior) pero no es la mejor técnica para evaluar pinturas. En cualquier ca.so, algunos autores han reportado que la caída óhmica representa apenas 20% del cambio de potencial.
3 6

Para empezar, el recubrimiento impide o debería impedir idealmen te el contacto del electrolito con el metal, dificultando el establecimiento de un potencial estable, lo que ya es en sí un problema en la medida de Rp.
Walter y otros autores han propuesto que se calcule una "pseudo-Rp", sin compensar la caída óhmica. Walter ha evaluado acero galvanizado pintado inmerso en NaCl 5% wt. mediante el empleo de esta pseudo-Rp, pues evidentemente cuando este valor decrece, se aprecia degradación del sis tema. Hay que considerar que al disminuir la resistencia total del sistema no se puede discernir acerca de cuál de las contribuciones disminuye, pero sea que disminuya la resistencia del recubrimiento o la de transferencia de carga, la protección anticorrosiva está disminuyendo y el sistema degradándose. En 1 990 V. Leal-Mendoza realizó un trabajo muy completo evaluando muchos sistemas de pintura industrial mediante la aplicación de Rp con compensación óhmica en un potenciostato PAR EG&G 173.
Milis y Boden emplean la Rp para evaluar recubrimientos orgánicos desarrollando un ensayo acelerado usando un baño caliente de NaCl 5% wt. Hay que mencionar que estos científicos emplean probetas con incisiones, que suelen utilizarse en ensayos acelerados en Cámara de Niebla Salina. En este caso, las probetas se han rayado a propósito y de manera controlada, para analizar la capacidad de protección de un recubrimiento en presencia de un daño o defecto en la pintura. Los cambios en evaluación visual (de acuerdo a estándares ASTM) son comparables a los resultados de Rp.
Steinsmo y Bardal han realizado mediciones de Rp aplicando velocidades de barrido de 20m V/ min, pero solo cuando el potencial es estable en un epóxico-amida de 50 y 100 mm de espesor de película seca con y sin pigmento de aluminio. Reportan que las mediciones pueden ser potenciodinámicas, pero también galvanodinámicas. En este caso la impedancia total del sistema disminuye porque se está trabajando con probetas con incisión y además se incluye un pigmento metálico como es el aluminio.
37

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
Referencias
l . ASTM G 59-9l ."Standard Practice for Conducting Potentiodynamic Polarization Resistance Measurements".
2. G. Baronio, M. Berra, L. Bertolini, T. Pastare, Cement and Concrete Research 2Q (5), 691-696 (1996).
3. J.-P. Diard, B. Le Gorree, C. Montella, J. Electroanal. Chem. ,432, 27-39 (1997).
4. J.-P. Diard, B. Le Gorree, C. Montella, J. Electroanal. Chem. 432, 41-52 (1997).
5 . J.-P. Diard, B. Le Gorree, C. Montella, J. Electroanal. Chem. 432, 53-62 (1997).
6. S. Feliu, J.A. González, M.C. Andrade, V Feliu, Corrosion Science 29 (1), 105- 1 1 3 (1989).
7. S. Feliu, J.C. Galvan, S. Feliu Jr., J. Simancas, J.M. Bastidas, M. Morcillo, E . Almeida, J. Electroanal. Chem. 381, 1 -4 (1995).
8. M.R. Gennero de Chialvo, A.C. Chialvo, J. Electroanal. Chem. 41 5, 97-106 (1996).
9. J.A. González, A. Coba, M.N. González, S. Feliu, Corrosion Science 43, 61 1 -625 (2001) .
1 O. KR Gowers, G. Millard, Corrosion Science .15. (5-8), 1 593-1600 (1993).
1 1 . P. Gu, B. Arsenault,J.J. Beaudoin,J.G. Legoux, B. Harvey,J. Fournier, Cement and Concrete Research 2.8 (3), 321-327 (1998).
12 D.A. Jones, Corrosion .12, 444 (1983).
1 3. V Leal-Mendoza, Tesis UNAM, Fac. Química, México D.F., 1990.
14. D.D. Macdonald, M. Urquidi-Macdonald, R.C. Rocha-Filho, Y ElTantawy, Corrosion 47 (5), 330-334 (1991).
1 5. D.J. Milis, P.J. Boden, Corrosion Science .3..5. (5-8), 131 1-1318 (1993).
3 8

16. z. Nagy,J.M. Wesson,J. Electrochem. Soc. U2 (5), 1261-1275 (1992).
1 7. z. Nagy, "DC Electrochemical Technique for the Measurement of Corrosion Rate", en Modern Aspects of Electrochemistry, No. 25, edited by John O'M. Bockris et al, Plenum Press, new York, 1 993.
1 8. I.M. Notter, D.R. Gabe, Corrosion Science .11 (5), 851-870 (1993).
19. M.E. Orazem, T. El Moustafid, C. Deslouis, B. Tribollet, J. Electrochem. Soc. 143 (12), 3880- 3890 (1996).
20. G. Rocchini, Corrosion Science .1Q (6), 1063-1076 (1994).
21 . G. Rocchini, Corrosion Science 38 (6), 823-834 (1996).
22. G. Rocchini, Corrosion Science 38 (12), 2095-2109 (1996).
23. G. Rocchini, Corrosion Science 39 (5), 877-891 (1997).
24. G. Rocchini, Corrosion Science :i2 (10-1 1), 1 861 - 1872 (1997).
25. G. Rocchini, Corrosion Science 41, 2353-2367 (1999).
26. A.A. Sagüés, S.C. Kranc, E.I. Moreno, Corrosion 54 (1) , 20-28 (1998).
27. J.R. Scully, Corrosion .5.Q (2), 199 (2000).
28. G. Song, Cement and Concrete Research 22, 407-415 (2000).
29. U. Steinsmo, E. Bardal, Corrosion 48 (1 1), 910-917 (1992).
30. M. Stern, A.L. Geary, J. Electrochem. Soc. 104 (1), 56-63 (1957).
3 1 . J.M. Sykes, Br. Corrosion J. 25 (3), 1 75-183 (1990).
32. D.W Townley, Corrosion 11. (10), 737-741 (1991).
33. EJ. Villalobos-Orozco, Tesis UNAM, México D.F., 1 989.
34. G.W Walter, Corrosion S cien ce 26 (1 ), 39-4 7 (1986).
35. G.W Walter, M.A.D. Madurasinghe, Corrosion Science 29 (8), 1039-1055 (1989).
39

TÉCNICAS ELECTROQUÍMICAS DE CORRIENTE DIRECTA PARA
EL ESTUDIO DE LA CORROSIÓN
2.3 TÉCNICA DE EXTRAPOLACIÓN DE TAFEL O DE
INTERSECCIÓN
J. Genescá. Dpto. Ingeniería Metalúrgica. Facultad Química. UNAM. Ciudad Universitaria. 04510 México D.F.
COEFICIENTES DE TAFEL Si la concentración de los reactivos y de los productos es uniforme en el electrolito, la ecuación de Butler-Volmer toma la forma
1 =lo exp[ �:, ) - lo ex{�: ) Donde 13. y l3c, son los coeficientes de Tafel anódico y catódico respectivamente:
p = R T a anF p = RT e (1 -a )nF
Esta ecuación se aplica a las reacciones electródicas en las cuales la velocidad está controlada por el proceso de transferencia de carga en la interfase electrodo/ electrolito. Esta situación se conoce a menudo como control por activación o control activacional y el sobrepotencial correspondiente como sobrepotencial de activación, Y]A.
El valor de los coeficientes de Tafel , 13. y l3c, depende del mecanismo de las reacciones que tiene lugar en los electrodos, los cuales comprenden muy a menudo varias etapas. Sin embargo, no es necesario conocer este mecanismo para poder utilizar la ecuación de Butler-Volmer, ya que ésta describe la describe la cinética del proceso de transferencia de cargas de una manera global, independientemente del mecanismo, a partir de tres magnitudes fácilmente medibles: j0, 13. y l3c. Las siguientes fórmulas definen los coeficientes de Tafel anódico y catódico.
40

dE P a = d l . ll Ja RECTAS DE TAFEL
Para determinar experimentalmente los parámetros cinéticos j0, 13. y 13c , es recomendable una representación gráfica en la que la densidad de corriente esté en una escala logarítmica, ya que así se pone en evidencia la relación lineal existente entre el logj y el sobrepotencial, r¡, especialmente cuando este último, en valor absoluto, tiene un valor grande.
Se denomina región Ó dominio de Tafel anódico (zona Tafeliana), aquella región en la cual el potencial que corresponde a r¡ / l3 a > > 1 . La ecuación se convierte en
· . . ( ll a ) 1 = 1 a = 1 o exp � Tomando logaritmos se obtiene:
r¡ = -13. lnf, + 13. lnj
Pasando a logaritmos de base diez y definiendo las constantes de Taje/ anódicas, a y b se obtiene la ecuación de Tafel de una reacción anódica, tambi6n u:mada recta de Tafel anódica:
r¡ = a + b log j a a a. = -2.303 13a lnJo
b = -2.303 13 ti ¡¡
De manera análoga, para el dominio catódico de Tafel, r¡/l3c <<1 :
exp (-�) j = j = -j log j ' o A
r¡ = 13 In j - 13 ln 1 J 1 ( o (
4 1
1-',

TÉCNICAS ELECTROQUÍMICAS PARA EL CoNTROL v EsTuDio DE LA CoRROSIÓN
Lo cual permite expresar las constantes de Taje/ catódicas, ae y be de la ecuación de Tafel de una reacción catódica:
r¡ = a + b log j a a
a = -2.303 13 ln}0 a a
b = -2.303 13 a a
Las ecuaciones de T afel describen los limites anódico y catódico de la ecuación de Butler-Volmer. Su descubrimiento a principios del siglo �einte, en el año de 1 904, por Tafel al estudiar el comportamiento del sobrepotencial para la reacción dedesprendimiento de H, en función de la densidad de corriente, precedió durante muchos aftos a la ecuación de Butler-Volmer.
Cuando se obtiene una curva de polarización experimental, la extrapolación de la recta que aparece en el dominio Tafeliano al valor del potencial de equilibrio, permite determinar la densidad de corriente de intercambio, j0, Figura 1 . El inverso del valor de la pendiente de estas rectas proporciona el valor de los coeficientes de Tafel, 13, y 13 e" ECUACION DE BUTLER-VOLMER DE UN ELECTRODO MIXTO
La ecuación de Butler-Volmer se puede aplicar a un electrodo mixto, por ejemplo, al caso concreto de la corrosión del acero en HCl, en ausencia de gradientes de concentración. Tienen lugar simultáneamente dos reacciones:
Fe --+ Fe2+ + 2e reacción anódica
reacción catódica
Si s e toma al acero como elec trodo de trabaj o en una celda electroquímica y se aplica un determinado potencial, resulta en una densidad de corriente medible, cuyo valor corresponde a la suma de las densidades de corriente parciales
. - . + . - . + . + . +" J - )Fe JH - la,Fe Je,Fe la,H le,H
42

Potencial, V
log corriente
pendiente, b = m V /década
Figura 1. Esquema de una reacción anódica en un diagrama de Tafel. E",: potencial de
equilibrio, j0,: densidad corriente intercambio, b: pendiente de Tafel = 2.30313
Los subíndices Fe e H se refieren a las reacciones parciales. Cerca del potencial de corrosión, E , la contribución de j H y j H se puede considerar despreciable. Gc�:rr= ia,H := O) lo que per:rute simplificar la ecuación: . - . + . - . +' J - JFe JH - la,Fe lc,H
En el potencial de corrosión (E = EcoJ , la densidad de corriente) es . cero . . - . +' = o J - la,Fe(Ecorr) lc,H(Ecorr)
Se ha supuesto por una parte que la transferencia de carga en la interfase metal/ solución constituye la etapa limitan te y por otra, que las cinéticas de las dos reacciones parciales son independientes. La densidad de corriente parcial de cada reacción obedece entonces a la ecuación de Butler-Volmer.
· · · · ( TJ Fe J . ( ll Fe J )Fe = Ju,F< + Jc,Fe = Jo,F< eXp -fl- - Jo,Fe exp - -�-
p u ,Fe c ,Fe
· · · · ( 11 H J · exp (- �J lH = la,H + lc,H = J o,H exp 13 a ,H
- J o,H P c,H
43

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
Las mediciones de polarización llevaron al desarrollo, en 1 930, de la teoría del potencial mixto, con el objeto de poder explicar los resultados experimentales obtenidos. La Figura 2a muestra una representación esquemática de una reacción anódica y catódica, típicas de un proceso de corrosión, así como, Figura 2b, la correspondiente curva de polarización experimental que se puede derivar de las mismas.
METODOS ELECTROQUIMICOS DE ESTIMACION DE LA VELOCIDAD DE CORROSION
Cuando toman parte en la reacción especies cargadas, como es el caso de los procesos de corrosión, la barrera de energía que debe vencerse en la transferencia de carga es afectada por el campo eléctrico y puede demostrarse, a partir de la teoría de la cinética electroquímica que las densidades de corriente de los procesos parciales
Se ajustan a las expresiones
. . (anF11 l 1 a = 1 corr exp ---¡¡_;¡:-
. . ( - (1 -a )nF11 ] le = - ¡corr exp
RT
44

1 � .. -
-
-
-
- -
� ¡:: z w f-r
¡ �
i l
Figura 2a. Representación esquemática de la reacciones anódica y catódica de un
proceso de corrosión .
.J � E - - - - - -- -- - --
-- --- - . z '"" "' f-• ..
Figura 2b. Curva de polarización experimental derivada de las reacciones mostradas
en la Figura 2a.
45

TÉCNICAS ELECTROQUÍMICAS PARA EL CoNTROL Y EsTuDio DE LA CoRROSIÓN
Donde T representa la temperatura absoluta, R la constante de los gases perfectos, F la constante de Faraday, n el número de electrones intercambiados en el proceso redox, a y (1 -a) los coeficientes de transferencia de carga, relacionados con la caída de potencial a través de la doble capa que rodea el electrodo (normalmente el valor de ambos es próximo a 1 /2), 11 la polarización (sobrepotencial) aplicada y jcorr la densidad de corriente de corriente de corrosión buscada, que puede transformarse en pérdida de peso o de espesor a partir de la segunda ley de Faraday.
El equilibrio eléctrico existente en el potencial de corrosión libre, expresado como:
I = I + I = O t a e
I = I I I = I a e corr
impide la determinación directa de jcorr" Cuando se rompe dicho equilibrio, imponiendo una polarización al electrodo, se aprecia experimentalmente una corriente externa, resultante de la suma algebraica de las correspondientes a los procesos parciales:
. . . . [ (anFTJ a ) (- (1 -a )nFTJc )] lt = la + le = lcorr exp RT - exp R T
Partiendo de esta ecuación, conocida como de Wagner y Traud (1), se consigue, a través de dos casos límite, la aproximación de alto campo o alto sobrepotencial (extrapolación de Tafel o intersección) y la de bajo campo o bajo sobrepotencial (método de resistencia de polarización), una estimación de jcorr' es decir, de la velocidad de corrosión.
46

METODO DE EXTRAPOLACIÓN DE TAFEL O DE INTERSECCION
Para polarizaciones suficientemente grandes
> RT 11 - a nF
o bien: RT -r¡
�
----
(1 -a )nF la ecuación general de Wagner y Traud se reduce a las ecuaciones de las semireacciones anódica y catódica que se corresponden con las rectas de Tafel respectivas, con pendientes
b = 2 .3RT o bien: b = - 2.3RT
a anF e (I -a)nF Para comprobarlo basta tomar logaritmos en cualquiera de las citadas ecuaciones, por ejemplo en la reacción anódica
de donde:
1 . 1 . anF og ;a = og ;corr +
2.3RT r¡
- 2.3RT . 2.3RT . . r¡ = 10g Jcorr + 1og Ja = a + b0 10g )0 anF anF Entonces, para un sistema metal/ electrolito dado, j tiene un valor determinado y el primer término del segundo ';iembro puede englobarse en la constante a de la ley de Tafel. En las proximidades de Ecorr' para polarizaciones pequeñas, los procesos anódicos y catódicos se influyen mutuamente y las curvas de polarización experimentales se apartan del curso semilogarítmico previsto por la ley de Tafel. Sin embargo, para T]=O (es decir, cuando E = E ), de cualquiera de las ecuaciones resulta:
cocr
i, = JU = jcorr
lo cual permite determinar la J. buscada sin mas que extrapol:tr corr
cualquiera de las rectas de Tafel obtenidas a polarizaciones grandes has-ta el valor E=E tal como se esquematiza en la Figuras 2b y 3a. En la Figura 3a
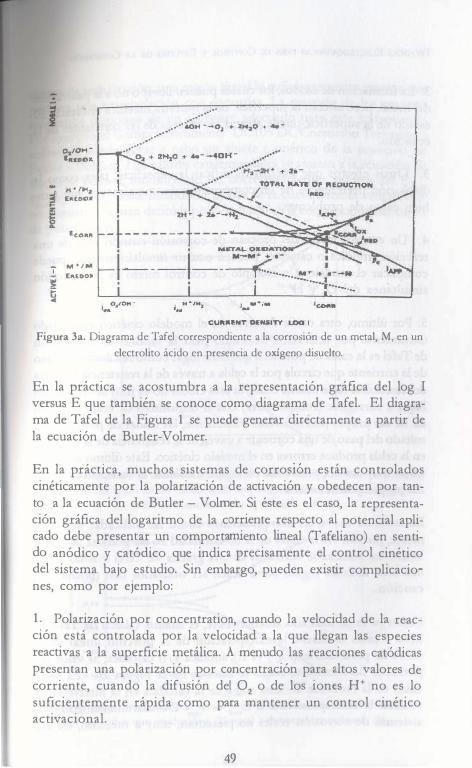
co;� presenta el diagrama de Tafel para el proceso de corrosión de un metal en un medio ácido con oxígeno disuelto,
47

TÉCNICAS ELECTROQUÍMICAS PARA EL CoNTROL Y EsTuDIO DE L\ CoRROSió:-�
mientras que en la Figura 3b se reproduce el mismo diagrama, pero tomando en consideración lo que suele ocurrir frecuentemente en los casos reales, cuando el proceso catódico de reducción del O, está controlado por difusión. La Figura 4 reproduce un diagrama d� Tafel experimental correspondiente al proceso de corrosión de un acero dulce en una salmuera. La principal ventaja y desventaja de este método, derivado en principio por Wagner y Traud (1) y Evans y Hoar (2), y que tiene una amplia aplicabilidad en la práctica, reside en la necesidad del trazado' completo de las curvas de polarización que por una parte posibilita un análisis electroquímico de la cinética del proceso de corrosión y, por otra, puede dar lugar a modificaciones de la superficie del electrodo por efecto de las elevadas polarizaciones aplicadas. Inversamente, si se llega a una concordancia entre los valores de las velocidades de corrosión derivadas de los ensayos gravimétricos de pérdidas de peso y las obtenidas con la utilización de este método, puede concluirse que el proceso de corrosión se realiza según un mecanismo electroquímico característico. El cálculo de la velocidad de corrosión por el método de extrapolación de Tafel, conocido también como método de intersección, se basa en la extrapolación de la zona lineal o de Tafel en un diagrama experimental E vs. log j. Según este método se puede obtener la jcorr sin más que extrapolar cualquiera de las rectas de Tafel hasta el valor del E , tal como se representa en la Figura 1 . P�;� un buen desarrollo experimental de esta técnica s e recomienda consultar las siguientes normas ASTM: • ASTM G 3 - 89. Standard Practice for Conventions Applicable to
Electrochemical Measurements in Corrosion Testing.
• ASTM G 5 - 94. Standard Reference Test Method for making Potentiostatic and Potentiodynamic Anodic polarization Measurements.
• ASTM G 59 - 97. Standard Test Method for Conducting Potentiodynamic Polarization Resistance Measurements.
• ASTM G 102 - 89. Standard Practice for Calculation of Corrosion rates and Related Information from Electrochemical Measurements.
48
..•. ·······��-:·:�, • � ... o • 441 -
.......... ··· OziOJot -��IDDX
� H • tlia Ellll tDC)Jt
¡:: z
� E.coRPII
� · / M 1 Elllt.DO:. � t; ... 1_ ......... 'eooo•
CUitKI!N'r DEMSITY LOa 1
Figura 3a. Diagrama de Tafel correspondiente a la corrosión de un metal, M, en un
electrolito ácido en presencia de oxígeno disuelto.
En la práctica se acostumbra a la representación gráfica del log I versus E que también se conoce como diagrama de Tafel. El diagrama de Tafel de la Figura 1 se puede generar directamente a partir de la ecuación de Butler-Volmer.
En la práctica, muchos sistemas de corros10n están controlados cinéticamente por la polarización de activación y obedecen por tanto a la ecuación de Butler - Volmer. Si éste es el caso, la representación gráfica del logaritmo de la corriente respecto al potencial aplicado debe presentar un comportamiento lineal (fafeliano) en sentido anódico y catódico que indica precisamente el control cinético del sistema bajo estudio. Sin embargo, pueden existir complicaciones, como por ejemplo:
1 . Polarización por concentration, cuando la velocidad de la reacción está controlada por la velocidad a la que llegan las especies reactivas a la superficie metálica. A menudo las reacciones catódicas presentan una polarización por concentración para altos valores de corriente, cuando la difusión del 02 o de los iones H+ no es lo suficientemente rápida como para mantener un control cinético activacional.
49

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL Y EsTUDIO DE LA CORROSIÓN
2. La formación de óxidos, los cuales pueden llevar o no a la pasivación del metal, pero alteran la superficie de la muestra metálica ensayada. El estado de la superficie puede afectar los valores de las constantes de la ecuación.
3. Otros efectos que puedan modificar la superficie, tales como la disolución preferencial de un componente de una aleación son también causa de problemas.
4. Un control mixto del proceso de corrosión cuando más de una reacción, anódica o catódica, pueden ocurrir simultáneamente, puede complicar el modelo. Un ejemplo de control mixto es la reducción simultánea del 02 y H+.
5. Por último, otra causa de error en el modelo cinético controlado únicamente por activación necesario para la validez de la ecuación de Tafel es la caída de potencial que tiene lugar como resultado del paso de la corriente que circula por la celda a través de la resistencia eléctrica del electrolito de la misma celda. Si este efecto no es muy severo puede llegar a corregirse (compensarse) con el dispositivo de compensación de IR del propio potenciostato. Finalmente, las caídas de potencial resultado del paso de una corriente a través de la resistencia de la solución en la celda produce errores en el modelo cinético. Este último efecto, si no es muy severo, puede ser corregido mediante la compensación IR del propio potenciostato.
En muchos casos, las complicaciones como las indicadas anteriormente, pueden ser la causa de la no-linealidad en las gráficas de Tafel. Todos los resultados derivados de una gráfica de Tafel que no tenga una bien definida región lineal deben ser utilizados con mucha precaución . .
Como ya s e ha indicado anteriormente, el análisis clásico de Tafel se lleva a cabo mediante la extrapolación de la porción lineal de la curva de polarización (log j vs E), anódica y/ o catódica, al valor del potencial de corrosión, como se muestra en la Figura 2b. El valor tanto de la corriente anódica como de la catódica en la intersección, es decir la correspondiente al E , es 1 . Desafortunadamente, los sistemas de corrosión reales n�0presed��n, muy a menudo, en sus

curvas de polarización, una región lineal lo suficientemente extensa que permita una extrapolación con garantías. La mayoría de los equipos modernos disponen de un software adecuado para este tratamiento. Por ejemplo, el Gamry Instruments' DC105 DC Corrosion Techniques software, puede llevar a cabo un ajuste numérico de la ecuación de Butler - Volmer. Los valores experimentales se ajustan a la ecuación de Butler - Volmer, modificando los correspondientes valores del E I , b , and b . Este método de ajuste de la curva tiene la ventaja q�� n'�
"requiere u�a bien definida porción lineal en la correspondiente curva de polarización.
Potential (V va SHE)
-5.5 4.5 Log (�A))
-3.5 .J ·2.5 ·2
Figura 3b. Curva de polarización (diagrama de Tafel) correspondiente al proceso de corrosión del acero en un medio ácido con oxígeno disuelto, estando el proce
so catódico controlado por la difusión del 02.
-0.35
-0.40
-0.45 w u " > ·0.50 "' w
-0 .55
-0 .80
1 E -0 8
· • A Z 5 p p m
-- e Z 5 p p m
1 E-05
Aftu 24 h r , O rpm
1 E -04 1 E-03 1 E -02 I (A c m "2 )
Figura 4. Curva de polarización de un acero 1018 en una solución de salmuera con inhibidor tipo imidazolina (25 ppm) a temperatura ambiente (3).
5 1

TÉcNICAS ELECTROQUíMICAs PARA EL CoNTROL Y EsTuDio DE LA CoRROSióN
REFERENCIAS
1 . C. Wagner y W Traud, Z. Elektrochem. 44, 391 (1 938).
2 . U.R. Evans y T.P. Hoar, Proc. Rqy. Soc. A. , 1 37, 34.3 (1 932).
3. J. Mendoza, R. Duran and E. Garcia, Corrosion'2002, Paper # 02491 . Houston (2002).

3. EsPECTROSCOPÍA DE IMPEDANCIA
ELECTROQUÍMICA EN CORROSIÓN
NOTAS
Juan Mendoza Flores1 Rubén Durán Romero1
Juan Genescá Llongueras2
1) Instituto Mexicano del Petróleo.
2) Facultad de Química, UNAM.

TÉCNICAS ELECTROQUÍMICAS PARA EL CoNTROL Y EsTuDio DE LA CoRROSIÓN
CONTENIDO.
Introducción.
Teoría básica.
Impedancia de una reacción simple de transferencia de carga
Presentación de datos.
Gráfico de Nyquist.
Gráficos de Bode.
Análisis de Resultados.
• Circuitos eléctricos equivalentes para sistemas en corrosión.
• Análisis gráfico.
• Análisis mediante circuitos eléctricos equivalentes.
Comparación de la EIS con R . p
Instrumentación
Consideraciones p ara la medición de EIS en celdas electroquímicas.
• Rango de frecuencia.
• Linearidad.
• Señales espurias.
• Número de datos.
• Promedio de la señal.
BIBLIOGRAFÍA RECOMENDADA.
SITIOS DE INTERNET DE INTERÉS.
5 4

ESPECI'ROSCOPÍA DE IMPEDANCIA ELECTROQUÍMICA
Introducción.
La técnica de Espectroscopía de Impedancia Electroquímica (EIS, por sus siglas en inglés), es un método electroquímico utilizado en estudios de corrosión, el cual se basa en el uso de una señal de corriente alterna (CA) que es aplicada a un electrodo (metal en corrosión) y determinando la respuesta correspondiente.
En el procedimiento experimental más comúnmente usado, se aplica una pequeña señal de potencial (E) a un electrodo y se mide su respuesta en corriente (I) a diferentes frecuencias. No obstante, en ciertas circunstancias, es posible aplicar una señal pequeña de corriente y medir la respuesta en potencial del sistema. Así, el equipo electrónico usado procesa las mediciones de potencial - tiempo y corriente - tiempo, dando como resultado una serie de valores de impedancia correspondientes a cada frecuencia estudiada. Esta relación de valores de impedancia y frecuencia se denomina "espectro de impedancias".
En el caso de los estudios de corrosión que utilizan la técnica de EIS, los espectros de impedancia obtenidos suelen ser analizados mediante circuitos eléctricos, compuestos por componentes tales como resistencias (R), capacitancias (C), inductancias (L), etc. Combinados de tal manera que reproduzcan los espectros de impedancia medidos. Estos circuitos eléctricos son denominados "circuitos eléctricos equivalen tes".
La impedancia es un término que describe la resistencia eléctrica (R), utilizado en circuitos de corriente alterna (CA) . En un circuito de corriente directa (CD) la relación entre la corriente (I) y el potencial (E) esta dada por la ley de ohm.
[1 ] E = IR
En donde E es en volts, I en amperes y R en ohms. En el caso de un señal alterna la expresión equivalente es la siguiente.
[2] E = IZ
5 5

TÉCNICAS ELECTROQUÍMICAS PARA EL CoNTROL Y EsTuDio DE LA CoRRosióN
En la ecuación [2] Z representa la impedancia del circuito, con unidades de ohm. Es necesario hacer notar que a diferencia de la resistencia, la impedancia de un circuito de CA depende de la frecuencia de la señal que sea aplicada. La frecuencia (f) de un sistema de CA se expresa en unidades de hertz (Hz) o número de ciclos por segundo (s-1) .
De esta manera, es posible definir la admitancia (Y) de un circuito de CA. La admitancia es el recíproco de la impedancia y es un parámetro de importancia en los cálculos matemáticos que involucra la técnica y por otra parte, los equipos usados en estudios de EIS miden en realidad la admitancia.
[3] Y = � = _!_ Z E
La impedancia de un sistema a cada frecuencia esta definida por, la razón entre la amplitud de la señal de corriente alterna y la amplitud de la señal de potencial alterno y el ángulo de fase. Un listado de estos parámetros a diferentes frecuencias constituye el "espectro de impedancia". El desarrollo matemático de la teoría que fundamenta la técnica de EIS permite describir la impedancia de un sistema en términos de un componente real y un componente imaginario (asociado a la raíz cuadrada de -1) .
Teoría básica.
La presente sección tiene como finalidad aportar al lector, ideas básicas que le permitan comprender la teoría que apoya la técnica de EIS. Debido a que la técnica de EIS se basa en el estudio de redes eléctricas, existe una gran información en la bibliografía referente a circuitos eléctricos.
En la comprensión de la teoría que soporta la técnica de EIS, es conveniente describir a la corriente y al voltaje como vectores giratorios o "fasores", los cuales pueden ser representados en un plano complejo o "Diagrama de Argand".
Un voltaje sinusoidal puede ser representado por la siguiente expresión.
[4] E = L1E sen mt
5 6

En donde E es el valor instantáneo del potencial, DE es la amplitud máxima y w es la frecuencia angular, misma que se relaciona con la frecuencia f de acuerdo a:
[S] ro = 2nf
DE puede entenderse como la proyección, sobre el eje O del fasor E en un diagrama polar. Ver figura l .
7t
2
o
3 - 7t 2
Figura 1. Diagrama del fasor correspondiente al potencial alterno de la ecuación [4] .
En la mayoría de los acasos, la corriente (I) asociada a una señal de potencial sinusoidal, es también sinusoidal, de la misma frecuencia (w) pero de amplitud y fase diferente a la del potencial. Esto puede ser representado de acuerdo a la siguiente expresión.
[6] 1 = �1 sen (rot + �)
Lo anterior significa que, en términos de fasores, los vectores giratorios están separados en el diagrama polar por un ángulo f. Esta situación se ilustra en la Figura 2.
5 7

TÉCNICAS ELECTROQuíwcAs PAR.� EL CoNTROL Y EsTUDIO DE LA CORROSIÓN
7t
2
o
3 - 7t 2
Figura 2. Fasores de corriente (I) y potencial (E) separados por un ángulo de fase �-
La respuesta a un potencial E, de un circuito simple con una resistencia pura R, puede ser descrita por la ley de Ohm [1 ] . Esto, en términos de fasores, corresponde a una situación en donde el ángulo de fase � = O.
Cuando un capacitor se considera en el circuito eléctrico diferentes aspectos deben de tomarse en cuenta. El concepto de "capacitancia" (C), puede definirse a partir de la relación entre el potencial E, aplicado entre las placas del capacitor y la carga (q) en las mismas, de acuerdo a:
[7] q = CE
Considerando de la corriente I que circula por el capacitor puede expresarse como:
[8] 1 = dq dt
En donde t es el tiempo, entonces:
[9] 1 = CdE dt
5 8

y considerando la ecuación [4] , puede obtenerse:
[10] l = w C �E coswt
Si el término 1 /wC es reemplazado por Xc (denominado reactancia capacitiva) se tiene la siguiente expresión:
[1 1 ] �E ( n) 1 = - sen wt + -Xc 2
La ecuación [1 1 ] tiene una forma similar a la ley de Ohm, únicamente reemplazando R por Xc y considerando un ángulo de fase diferente a cero e igual a rt/2. Como el ángulo de fase es positivo se dice que la corriente está adelantada con respecto al potencial.
Con el fin de simplificar la notación matemática, se puede definir el número j = �-1 . Tanto el potencial como la corriente pueden ser representados como fasores (vectores rotatorios) . Así, el fasor E se define como:
[1 2] E = E sen wt
En términos matemáticos los componentes real e imaginario, del fasor E y del fasor I, pueden representarse en un diagrama de Argand, con eje de las abscisas correspondiente alcomponente real y el eje de las ordenadas correspondiente al componente imaginario. Las siguientes figuras muestran la representación de los fasores E y I para un circuito puramente resistivo (Figura 3) y para un circuito con una capacitancia reactiva (Figura 4) .
El lector puede encontrar descripciones más detalladas de los aspectos matemáticos presentados en esta sección en la bibliografía espe
cífica (Kerchner - Corcoran, Circuitos de Corriente Alterna) .
5 9

TÉCNICAS ELECTROQUÍMICAS PARA EL CoNTROL Y EsTuDio DE LA CoRROSIÓN
"'
- , " 1 \ 1 \ 1 \ 1 1 1 1
1
1 1
1 1
\ 1 1 \ 1 ' - "
- , " \ 1 \ 1 \ 1 1 1 1 1
------� - - - - - - - - - � E
\ \ '
Figura 3. Representación en fasores de la corriente (I) y el potencial (E) con el
tiempo (t) , para un sistema resistivo puro con ángulo de fase (<jl) = O.
' 1 \
\ \ 1 1
1 1 1 1 1 1 \ \ \
\ ' " ... ....
1 1
1
1 1
1 1
F-- - - - � - - - - - - - - ·
1 1 1 1 \ 1 \ \ \
' ....
Figura 4. Representación en fasores de la corriente (I) y el potencial (E) con el
tiempo (t), para una relación entre corriente y potencial en un circuito con una
reactancia capacitiva de ángulo de fase (f) = 90 O.
60

En notación de fasores, la caída de potencial total potencial para una resistencia (R) y un capacitor (C) es:
O bien:
[14] E = IZ
En donde el término Z = (R - jXC) recibe el nombre de "impedancia". Así el ángulo de fase (f) puede definirse como:
[1 5] X 1 tan $ = --º- = -R oo RC La relación descrita por las ecuaciones [1 3] a [1 5] puede representarse de manera gráficaen la Figura 5.
E = ZI Figura 5. Diagrama fasorial para una resistencia (R) y un capacitor (C) en serie.
E corresponde al potencial total a través de la combinación de R y C, RI repre
senta el componente resistivo y jXCI representa el componente capacitivo.
Impedancia de una reacción simple de transferencia de carga.
Considerando el sistema electroquímico más sencillo de analizar es, una reacción rápida al equilibrio, con potencial de equilibrio Ee y corriente neta cero. Bajo una excitación alterna de baja amplitud (menor a 5 m V), la corriente neta sigue siendo igual a cero y la relación entre la corriente y el potencial puede considerarse lineal.
6 1

TÉCNICAS ELECTROQUiMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
Considerando la siguiente reacción electroquímica, en la cual tanto los reactivos (O) como los productos (R) son solubles:
Con el fin de evaluar la respuesta a la excitación alterna dos tipos de información son necesarios:
a) Las velocidades de reacción en cada sentido (oxidación y reducción), o bien, la densidad de corriente de intercambio (io) . Es necesario mencionar que este último parámetro es, usualmente, un dato que desea obtenerse a partir de los resultados de los ensayos de EIS.
Las velocidades de difusión de las especies O y R en la vecindad del b) electrodo de trabajo, en función del tiempo durante el ciclo alterno.
Cuando la señal alterna se aplica, en primer instancia, se crea una capa de difusión que depende del tiempo. Debido a que la corriente neta que circula en el sistema es cero, el estado estacionario se alcanza después de unos cuanto ciclos. Esta situación de difusión doble y electroquímica ha sido resuelta de la siguiente manera.
Considerando que la impedancia puede ser expresada como una combinación en serie de una resistencia y un capacitar (ver Figura 6), se puede demostrar que:
[17]
[1 8]
1 es = --1 ,-2 croo
Figura 6. Arreglo en serie de resistencia (Rs) y pseudo capacitancia (Cs).
62

Rct se denomina la "resistencia de transferencia de carga" y Cs es una
pseudo capacitancia. Recordando que en la vecindad del potencial Ee, la relación entre la corriente y el potencial puede considerarse lineal, se puede demostrar que para una difusión hacia una superficie plana:
[19) R = RT
el F. n l o
En donde A es el área del electrodo, D es el coeficiente de difusión para las especies en solución, Cb,O es la concentración en el seno de la solución de la especie O y Cb,R es la concentración en el seno de la solución de la especie R. Por lo tanto Rct es un parámetro que está determinado por la corriente de intercambio (io) y por lo tanto por las velocidades de reacción de oxidación y reducción Por otra parte, s está relacionado con los parámetros difusiones del sistema.
Es posible demostrar que:
(21 ] R _ _ 1_ = R = RT S e el F.
(1) S n 10
Por lo tanto, un gráfico de Rs contra 1 /w1/2, para el sistema en cuestión, debe de producir una linea recta de pendiente s y ordenada al origen igual a Rct, a partir de la cual el valor de io puede ser estimado.
La impedancia total del sistema (Z), estará dada por:
(22] 1 (J (J Z
= Rs + -. --= Rel + ---v2 + -:---1f2 jroCs ro jffi
La ecuación [22) es la suma de dos términos. Un primer término resistivo simple, el cual es pequeño cuando io es grande (ver [1 9]) y un segundo término que puede ser considerado como una resistencia dependiente de la frecuencia. Este último término se denomina
6 3

TECNICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
"Impedancia de Warburg" (Zw). Cuando Rct se aproxima a cero, entonces, la impedancia total del sistema es igual a Zw. La magnitud de Zw estará dada por:
Ya que tanto el componente real como el componente complejo de Zw son iguales, esta impedancia se caracteriza por un ángulo de fase constante e igual a n/4 (45°), independiente de la frecuencia.
La magnitud relativa de Re, y Zw a una frecuencia dada es una medida del balance entre el control por transferencia de carga y el control por difusión del proceso electroquímico. Si el valor de i0 es muy grande entonces Re, � O y será muy difícil de medir, de tal manera que, sólo la impedancia de Warburg podrá ser observada. Por otra parte, una reacción electroquímica lenta tendrá un alto valor de Re, asociado (el cual puede ser difícil de medir), este valor de Rct será el término dominante.
El análisis anterior no ha considerado el hecho de que todos los electrodos muestran una capacitancia, denominada "capacitancia de la doble capa" (CdJ' la cual es independiente de reacciones faradaicas, las cuales contribuyen con una pseudo capacitancia (C.) a la impedancia total de un sistema.
Por otra parte, en una celda electroquímica existe también una resistencia eléctrica, asociada a la resistencia del electrolito, entre el punto en al cual se mide el potencial (usualmente la punta del capilar de Luggin) y el electrodo de trabajo, (R,). Esta resistencia también se hará manifiesta en impedancia total del sistema.
Los efectos de Cdl y Rsol pueden ser considerados en los análisis de impedancia si sus magnitudes son conocidas. También pueden ser determinados mediante mediciones en ausencia del par de especies electroactivas 0/R, ecuación [1 6] . Sin embargo, determinar los valores de Cdl y Rsol de manera separada, incrementa considerablemente
64

la complejidad de la experimentación y el análisis de información. Un
método de análisis que permite evitar la necesidad de hacer mediciones separadas, se deriva de un proceso ampliamente usado en ingeniería eléctrica y que fue adaptado a aplicaciones electroquímicas por Sluyters y colaboradores. Este método se denomina "análisis de impedancia en el plano complejo"
Considerando un circuito simple en sene de una resistencia y una capacitancia, con una impedancia igual a:
[24] 1 Z = R + jroC
Puede verse que la parte real de Z es simplemente R y que la parte imaginaria correspondiente es 1 /wC.
Si el comportamiento descrito por la ecuación [24] se representa en un diagrama de Z = Z'+Z" (diagrama de Argand), en donde Z' =
componente real de la impedancia total y Z" = componente imaginario de la impedancia total se obtendrá el gráfico de la Figura 7. En este caso, la gráfica correspondiente es una serie de puntos a diferentes valores de w, el valor de la componente imaginaria de la impedancia (Z") tiende a cero a medi,da que la frecuencia se hace muy grande (tiende a infinito), situación en la cual la capacitancia se puede considerar como en corto circuito.
Nota: es necesario mencionar que en los estudios electroquímicos la componente imaginaria de la impedancia total (Z') suele presentarse en los gráficos correspondientes, multiplicado por -1 . Lo anterior debido a que, en estricto rigor matemático, en la mayoría de los sistemas electroquímicos Z" tiene valores negativos.
6 5
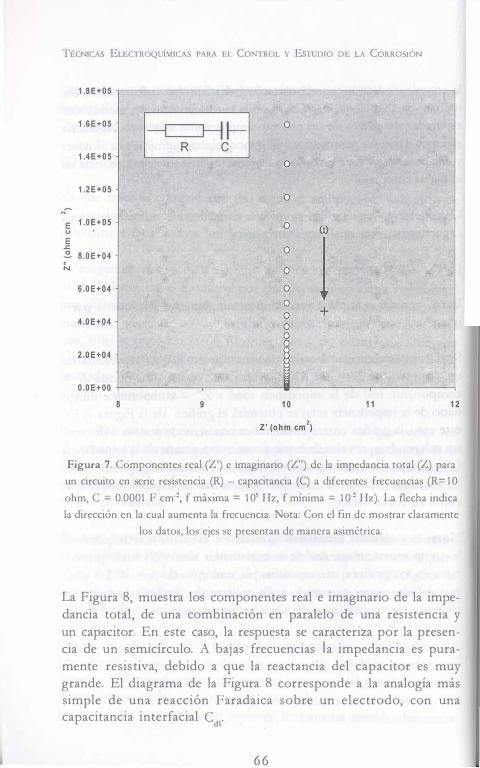
TÉCNICAS ELECTROQUÍMICAS PARA EL CoNTROL v EsTuDio DE LA CoRROSIÓN
Figura 7. Componentes real (Z') e imaginario (Z") de la impedancia total (Z) para
un circuito en serie resistencia (R) - capacitancia (C) a diferentes frecuencias (R= 1 0
ohm, C = 0.0001 F cm·�, f máxima = 1 05 Hz, f mínima = 1 0·� Hz). La flecha indica
la dirección en la cual aumenta la frecuencia. Nota: Con el fin de mostrar claramente
los datos, los ejes se presentan de manera asimétrica.
La Figura 8, muestra los componentes real e imaginario de la impedancia total, de una combinación en paralelo de una resistencia y un capacitar. En este caso, la respuesta se caracteriza por la presencia de un semicírculo. A bajas frecuencias l a impedancia es puramente resistiva, debido a que la reactancia del capacitar es muy grande. El diagrama de la Figura 8 corresponde a la analogía más simple de una reacción Faradaica sobre un electrodo, con una capacitancia interfacial cdl'
6 6

6
<o = --
o RCdl �
o t o e o o
o o 1 o n 1
o /ro o o o 00 0 ! o
o o 1
"'E 4 " E
f uo
l ro � o l � ---------- 1 --...___.¡ o
o 2 4 6 8 1 0
z • ( o h m c m2)
Figura 8. Componentes real (Z') e imaginario (Z") de la impedancia total (Z) para un circuito en paralelo resistencia (R) - capacitancia (C) a diferentes frecuencias (R=10 ohm, C = 0.0001 F cm·2, f máxima = 1 05 Hz, f núnirna = 1 0·2 Hz).
La flecha indica la dirección en la cual aumenta la frecuencia.
El siguiente paso que permite obtener un símil de una reacción electroquímica y que complica el análisis es, agregar una resistencia en serie al circuito paralelo RC, misma que puede representar la resistencia de la solución, R,01 (Figura 9). Esta situación tiene el efecto de transportar el semicírculo, a valores mayores en el eje de la impedancia real (Z') del gráfico.
() 7

6 � ~ e
o o o o o o
o o o o
o o E e
o o o o
o ' � '
..e: � ¡.,¡ 2
o o 2 4 6 8 1 0 r 1 2
R + Rsol Figura 9. Componentes real (Z') e imaginario (Z") de la impedancia total (Z)
para un circuito en paralelo resistencia (R) - capacitancia (C) , que considera la
resistencia de la solución (R,J, a diferentes frecuencias (R,01 :::: 1 ohm, R=10 ohm, C = 0.0001 F cm·2, f máxima :::: 1 05 Hz, f rrúnima :::: 1 0-2 Hz). La flecha indica
la dirección en la cual aumenta la frecuencia.
Los ejemplos anteriores, permiten construir un modelo más realista de un proceso electroquímico simple, si C es considerada como la capacitancia de la doble capa (Cd¡), la cual siempre estará en paralelo con la impedancia de la reacción. Por lo tanto, R puede ser considerada como la "Resistencia de Transferencia de Carga" (RJ.
Un circuito eléctrico equivalente de una reacción electroquímica simple es el denominado "Circuito de Randles" (ver Figura 10) . En este circuito la Re, se encuentra en serie con la impedancia de Warburg.

R
t- w e
Figura 10. Circuito de Randles, equivalente eléctrico de un proceso
electroquímico simple.
El análisis del circuito serie - paralelo de Randles, presenta dos casos límite.
a) A frecuencias bajas, cuando w ® O, los componentes real (Z') e imaginario (Z") de la impedancia total del circuito (Z) son:
[25] Z' R R -1t2 =
sol + ct + 0'0)
De donde se obtiene que:
La ecuación [27] corresponde a la expresión para una linea recta de pendiente unitaria y con intersección en el eje real (Z') de Rsol + Rct - 2s2Cdl.
a)A altas frecuencias, alas cuales la impedancia de Warburg un valor muy pequeño en comparación con el valor de Rct, los componentes real e imaginario de la impedancia son:
[28]
69

TÉCNICAS ELECTROQUÍMICAS PARA EL CoNTROL v EsTUDIO DE LA CoRROSióN
De donde:
[30] (R;, r = ( Z'-Rsol - R;, r + (Z" )2
La cual es la ecuación de un círculo de centro:
[3 1 ] Z'= R + Rct sol 2
y un radio de:
[32] � 2
Un gráfico de Z" contra Z' tendría la forma esquematizada en la Figura 1 L En esta figura, aparecen tanto la región controlada por transferencia de carga (semicírculo) , como la región controlada por difusión (lineal, de pendiente unitaria) . En este caso, Rct puede ser calculada, extrapolando los puntos experimentales sobre el semicírculo, hasta la intersección con el eje Z'.
Z"
� (Ru + Rct - 2cr2Cd1 )
(J) aumenta
/
Z'
Figura 11. Representación en plano complejo de la impedancia para el circuito
de la Figura 1 O.
7 0

Sin embargo, en un sistema real, dentro de un rango realista de frecuencias (por ejemplo 0.01 Hz a 104 Hz), puede mostrar sólo la región controlada por transferencia de carga o la región controlada por difusión. Esto puede ser observado en la Figura 12, que muestra la impedancia obtenida para un electrodo de acero inoxidable (31 6) inmerso en un electrolito de CuS04 - H2S04 a 65 oC. En la Figura 12 puede observarse claramente la parte lineal correspondiente al control por difusión (pendiente unitaria), no así el semicírculo a altas frecuencias.
1400
1200
1 000
0./
/ ;f V
N
E 800 u e
.S:: � Ñ 600
/ 0.317 Hz / / {:{
400
200
o
/ / �
� · ¿t:4 nz
o 200 400 600 800 1 000 1 200 1 400 Z' (ohm-cm2)
Figura 12. Representación en el plano complejo de la impedancia de un electrodo de acero inoxidable 3 1 6 en un electrolito de CuS04 - H2S04, 65 oc.
7 1

TÉCNICAS ELECTROQuí�IICAS PARA EL CoNTROL Y EsTumo DE LA CoRROSIÓN
Presentación de datos.
Los datos obtenidos en los ensayos de espectroscopía de impedancia electroquímica, son reportados por los equipos comerciales en una de dos formas:
a) Módulo de la impedancia ( 1 Z 1 ) y ángulo de fase (f).
b) Componente real de la impedancia total (Z') y componente imaginaria de la impedancia total (Z") .
Estos dos métodos de describir los datos de impedancia son la base de dos maneras comunes de presentar los datos, denominados gráficos de Nyquist y de Bode.
El módulo de la impedancia 1 Z 1 , el ángulo de fase (f) y los componentes real e imaginario de la impedancia total, se relacionan entre sí de acuerdo a las siguientes exprestones.
[33]
[34]
[35]
[36]
Z" tan "' = -'1' Z' Z' = ¡z¡ COS$
Z" = ¡z¡ sen$
Gráfico de Nyquist.
Este tipo de gráfico, también conocido como gráfico en plano complejo, ha sido presentado en el texto anterior (ver Figura 8), y corresponde a graficar -Z" contra Z'.
Gráficos de Bode.
Los gráficos de Bode son representaciones de diferentes parámetros de la impedancia contra frecuencia y existen diferentes variantes.
7 2

Los gráficos de Bode más comunes son:
a) Logaritmo base 1 0 del módulo de la impedancia ( 1 Z l) Contra logaritmo base 10 de la frecuencia (f) .
b) Ángulo de fase (<!>) contra logaritmo base 10 de la frecuencia (0 .
A diferencia de los gráficos de Nyquist, las representaciones de Bode contienen toda la información de un ensayo de EIS, ya que las frecuencias se identifican claramente. Las Figuras 13 y 14, corresponden a los gráficos de Bode correspondientes a los datos mostrados en la Figura 9.
Rsot = 1 ohm, R,, = 10 ohm, Cd1 = 0.0001 F cm·2
0.1 +-����������������r-������ 0.01 0.1 10 100 1000 1 0000 100000
f (Hz)
Figura 13. Gráfico de Bode de IZI vs f, correspondiente a la impedancia de circuito serie - paralelo a diferentes frecuencias, que considera R", C"' y R,01 (Figura 9).
7 3

TECNICAS ELECTROQUÍ�IICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
50
40
20
10
Rsol = 1 ohm, R,, = 10 ohm, Cd1 = 0.0001 F cm·2 o610o o o o o
o o o o
o o
o o
o
o o
o o
o o o
o o o o o o o
o rommmmmmmmm���������=r�� 0.01 0.1 10 1 00 1000 1 0000 1 00000
f (Hz) Figura 14. Gráfico de Bode de ángulo de fase ($) vs f, correspondiente a la impedan
cia de circuito serie - paralelo a diferentes frecuencias, que considera R", C di y R,01 (l'iguras 9 y 1 3) .
Otras representaciones de Bode de utilidad corresponden a las mostradas en las Figuras 1 5 y 1 6, las cuales muestran los componentes real e imaginario de la impedancia total del sistema, Z' y Z" respectivamente, en función de la frecuencia (f).
74

R.o, = 1 ohm, R,, = 10 ohm, c., = 0.0001 F cm"'
0.1 +-�������r-���--���----�mr����--��� 0.01 0.1 10 100 1000 10000 1 00000
f (Hz)
Figura 15. Gráfico de Bode del componente real de la impedancia total (Z') contra la frecuencia (f), correspondiente a un circuito serie - paralelo que conside
ra R", Cd, y R,01 (Figuras 9).
"e 0.1
u E .e .2. � 0.01
0.001
R.a� a: 1 ohm. Re, = 10 ohm, cd, = 0.0001 F cm·2
0.0001
0.01 0.1 1 0 100 1 000 10000 1 00000
f (Hz) Figura 16. Gráfico de Bode del componente imaginario de la impedancia total (Z") contra la frecuencia (f), correspondiente a un circuito serie - paralelo que considera
R", Cd, y R,01 (Figuras 9).
7 5

TECNICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
Análisis de Resultados.
El análisis de la información generada por la técnica de espectroscopía de impedancia electroquímica depende, como en la mayoría de las técnicas electroquímicas, del tipo de información que el usuario requiera. Dos tipos de análisis pueden considerarse:
a) Gráfico. b) Ajuste de los datos obtenidos, a la respuesta de circuitos eléctri
cos equivalentes.
Existen casos en los cuales, la mayoría de la información requerida por el usuario puede ser obtenida mediante la inspección de los gráficos correspondientes conjuntamente con cálculos simples. Lo anterior es particularmente cierto cuando, por ejemplo, la estimación de la velocidad de corrosión ('I.T ) de un sistema dado, es el parámetro \ " con:
de interés para el usuario.
• Circuitos eléctricos equivalentes para sistemas en corrosión.
Un circuito equivalente es una combinación de elementos pasivos (resistencias, capacitancias, inductores y otras formas de impedancias distribuidas) que dan la misma respuesta, a toda frecuencia, de una celda de corrosión.
Cuando el análisis de los datos de EIS se realiza mediante un ajuste de los datos experimentales a un circuito eléctrico equivalente, se obtienen valores de diferentes parámetros eléctricos. Estos valores son utilizados para obtener información, tanto de velocidades de corrosión como de mecanismos de corrosión.
El número de circuitos equivalentes que pueden cumplir el comportamiento de una celda de corrosión es prácticamente infinito. No obstante, existe una condición esencial para la selección de un circuito equivalente: tanto los componentes del circuito, como el circuito eléctrico en sí mismo, deben tener explicación física. Esto es de particular importancia ya que usualmente pueden existir varios circuitos equivalentes que describan con la misma exactitud los datos experimentales.
7 6

Como ejemplo de esto, los circuitos equivalentes de las Figuras 1 7 y 1 8, generan el mismo espectro de impedancia, no obstante, los componentes eléctricos del circuito de la Figura 17 (mismo de la Figura 9) pueden ser asociados a las diferentes partes de un sistema electroquímico. En contraste, para el circuito de la Figura 1 8 es muy difícil, si no imposible, dar una explicación física a cada uno de sus componentes.
Figura 17. Circuito equivalente.
Figura 18. Circuito equivalente.
• Análisis gráfico.
La exposición que describe los componentes real (Z') e imaginarlo (Z") de la impedancia (mostrada en la Figura 9) hace referencia a un circuito simple. En estudios de corrosión, el análisis gráfico de un espectro de impedancia permite obtener parámetros relacionados con la cinética de corrosión de un metal en un medio dado.
A partir de un diagrama de Nyquist es posible estimar el valor de la resistencia de la solución (R'"� ' como el limite a alta frecuencia de Z'.
7 7

Tl�Cl'IIC.�S ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
-La suma de la "resistencia a la polarización" (R.� y R,01, es igual al límite de Z' a bajas frecuencias. La capacitancia del sistema (asociada a la doble capa) cdl' puede ser calculada a partir de la frecuencia en la cima del senucírculo del diagrama de Nyquist y del valor de R . Este tipo de análisis se muestra en la Figura 19.
P
Es necesario indicar que, estrictamente, en el ensayo electroquímico de polarización lineal, no es posible separar el valor de R del valor
p de R sol:
6 .-���-.��--�--------r-------�-------.-------.
e O O O \O O O e 1 E
4 t-------�-oo����------:t=J�==�--��o�t-------l, u o 1 o ] e ro = RpCdl � � o o � 2 +-----rio�r-------,_-------+------_,--------+-�o�--�
o +------',.......__' f----..r--......¡...__,..._.._�-------r------��')--...---1 q 2 4 6 8 1 0 l� Z' (ohm c m2) : R.,, :·--
--------------------------------------+ R,
1 2
Figura 19. Análisis gráfico d e la respuesta de impedancia de u n sistema de corrosión.
R,., = 1 ohm, RP
= 1 0 ohm, Cd1 = 0.0001 F cm·2
Algunas referencias sugieren que el cálculo de Cdl puede hacerse en el diagrama de Bode de 1 Z 1 contra f, ya que a altas frecuencias 1 Z 1 está dada por la ecuación 37, siempre y cuando la resistencia de la solución (R,J sea pequeña.
[37] log iZI = -logro - logCd,
La ecuación (37] indica que, a frecuencias altas, la relación entre 1 Z 1 y f (a partir de w) es una linea recta de pendiente - l . A fln de obtener el valor de Cdl dicha línea recta puede ser extrapolada al valor de frecuencia correspondiente y entonces:
7 8

[38] ¡z¡ = - logCdl
No obstante, este procedimiento solo es válido si la resistencia de la solución es pequeña, de lo contrario el error asociado puede ser considerable. Este problema puede ser afrontado si el diagrama de Bode de Z" contra el logaritmo de f es usado. Debido a que la resistencia de la solución no altera Z", la extrapolación del gráfico de Z" contra el logaritmo de f (a la frecuencia correspondiente) da como resultado:
[39] Z"= -logCd1
Un problema que puede presentarse comúnmente en los estudios de EIS es la estimación de parámetros, a partir de datos que no alcanzan el limite a bajas frecuencias y que por lo tanto muestran un semicírculo parcial. Esta situación se presenta cuando la relación RC es grande. Este comportamiento puede presentarse en metales pasivos o metales en los cuales se forma una película conductora porosa de superficie grande y capacitancia alta (ejemplo: hierro cubierto por una capa de sulfuro) . En casos como el descrito, el uso de gráficos de admitancia puede auxiliar en el análisis de los datos.
• Análisis mediante circuitos eléctricos equivalentes.
En el uso de circuitos eléctricos equivalentes para el análisis de datos de EIS es necesario considerar que, como se mencionó con anterioridad, suele existir un gran número de configuraciones de circuitos que pueden reproducir, con la misma precisión, la respuesta que se obtiene experimentalmente de una celda electroquímica.
Por ejemplo, las figuras 20 a 22 muestran, en diferentes representaciones, los resultados obtenidos en un ensayo de EIS en donde aparecen dos semicírculos en el espectro de Nyquist
7 9

-
60 �
( o ) o o
o o N 40
o o E u o o E
..e o o � o Ñ 2 0
o o V
'O � 1� � o � "
o 2 0 4 0 60 8 0 1 0 0 1 20
Figura
E u E
.r: .2. !:i
Z' (o hm cm 2)
20. Espectro de EIS (representación de Nyquist) que muestra la presencia de
dos semicírculos
1 0 0
1 0
00 o
o o o o o o o o
9¡,'-1 +-������������������������QID 0.0001 0.001 0.01 0.1
f (Hz)
1 0 100 1 0 00 1 0000 1 00000
Figura 21. Espectro de EIS (representación de Bode, 1 Z 1 vs f) correspondiente a
los datos de la figura 20.
80
'. Í

90
80
70
60
;; 50 o "C . � 40
30
20
1 0
0.0001 0.001 0.01 0.1 10
f {Hz)
a o
a o
o a
a o
· a o o
1 00
a o o o o a o o a Q �
1 000 1 0000 1 0 0000
Figura 22. Espectro de EIS (representación de Bode, f vs f) correspondiente a los
datos de la figura 20 y 21 .
Los espectros mostrados en las Figuras 20 a 21 pueden ser descritos, con precisión similar, por los seis circuitos de la Figura 23.
(1)
Figura 23. Algunos circuitos eléctricos equivalentes que permiten describir los
datos de las Figura 20, 21 y 22.
8 1

TÉCNICAS ELECTROQUÍMICAS PARA EL CoNTROL Y EsTuDio DE LA CORROSIÓN
Los circuitos (1) y (2) de la Figura 23 son configuraciones que han sido propuestas para describir los espectros de impedancia que se obtienen en metales con recubrimientos en la superficie, en los cuales un par de elementos RC es asociado al proceso de corrosión y otro al recubrimiento.
Por otra parte los circuitos (3) a (5), no obstante que pueden describir adecuadamente los resultados de las Figuras 20 a 22, no tienen una explicación física clara.
En la actualidad, existen varios programas de cómputo que permiten simular y ajustar datos experimentales de EIS, a la respuesta de impedancia de circuitos eléctricos complejos (diseñados por el usuario) de manera sencilla, mediante diferentes métodos numéricos. Estos programas permiten realizar inferencias sobre el proceso de corrosión, con relativa sencillez, no obstante la matemática compleja que es requerida
En sistemas reales los datos de EIS, representados en un diagrama de Nyquist, suelen mostrar una depresión por debajo del eje real. Este comportamiento no se ha podido explicar totalmente y suele ser asociados a fenómenos tales como: diseño de celda no adecuado, rugosidad superficial, porosidad superficial o reacciones que suceden en varios pasos.
A fin de ajustar espectros de EIS con depresión a un circuito eléctrico equivalente, suelen utilizarse "elementos de fase constante" (CPE, por sus siglas en inglés) .
Un elemento de fase constante es, en realidad, una expresión matemática que representa varios elementos eléctricos. De manera formal, la impedancia de un CPE (ZCPE) está dada por la ecuación [40] .
Cuando n = O, entonces el CPE es una resistencia con R= Z0• Si n = 1 el CPE es un capacitar con C = zo-I. La impedancia de Warburg (a altas frecuencias) es un caso especial y sucede cuando n = 0.5 .
8 2

No obstante, ya que el origen físico de la depresión de los semicírculos de EIS, no es claro, el significado del parámetro "n" tampoco se ha podido definir con certeza. Una consideración práctica es que, si en valor de n es mayor a 0.8, entonces el CPE puede ser considerado como un capacitar y por lo tanto la capacitancia puede ser estimada a partir de Zo.
La Figura 24 muestra espectros de EIS, calculados para un circuito serie paralelo (ver Figura 25), que considera un CPE en lugar de un capacitar con diferentes valores de n.
--- n = 1
100 ,-------·-·-·-----�---�---� -D-n = 0.9
_._ n = O.B
80
NE 60 u E .<: .!!. Ñ 40
20
o o 20 40 60 80 100 120 140 160 180
Z' (ohm cm2)
Figura 24. Impedancias de un circuito serie paralelo que considera un CPE con
diferentes valores de n. R001 = 1 ohm, R = 170 ohm, Rango de frecuencias: 1 o 1 06 Hz
a 1 o 10·3 Hz.
La Figura 25 muestra los datos de un espectro de EIS, en representación de Nyquist, obtenidos experimentalmente para un acero al carbón, inmerso en ácido clorhídrico 0.5M, que muestra un cierto grado de depresión. EN esta misma figura se muestra el ajuste de los datos a un circuito eléctrico equivalente serie paralelo que considera un CPE (en lugar de un capacitar) . En este caso, el procedimiento de ajuste de los valores experimentales, da como resultado un valor de n = 0.9. Por lo tanto, el CPE del circuito puede ser considerado como un capacitar.
8 3

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
� 80
60
E u
E 40 .<:: 2. ;.,
/ .I\. _.....u ..J)
.)Y
CPE
O Datos ..r. -o..... �
-Ajuste
� 1--
� � 20
1 \ o
20 40 60 80 100 1 20 1 40 1 60 1 80
Z' (ohm cm')
Figura 25. Datos experimentales de EIS (puntos), obtenidos para un electrodo de acero al carbón inmerso en una solución aereada de HCl O.SM. Los valores obtenidos mediante ajuste matemático a un circuito serie - paralelo, que considera un
CPE (linea continua) corresponden a un valor de n = 0.9.
Existen algunos tipos de espectros de impedancia que, en representaciones de Nyquist, pueden presentar semicírculos en el cuarto cuadrante o bien pequeños círculos anexos a uno principal, mismos que incluso pueden envolver al principal y continuar hasta el segundo cuadrante. Estos comportamientos han sido asociados a fenómenos de adsorción - desorción de especies en la superficie del electrodo, fenómenos que alteran el potencial del electrodo y la velocidadde corrosión del metal. Los circuitos equivalentes que han sido propuestos para modelar este tipo espectros de impedancia, pueden incluir elementos inductivos o bien combinaciones RC con valores negativos.
Comparación de la EIS con R . p
No obstante que la cantidad de información electroquímica que la técnica de EIS puede proveer es abundante, esta técnica puede ser utilizada para el seguimiento de la velocidad de corrosión de un metal
84
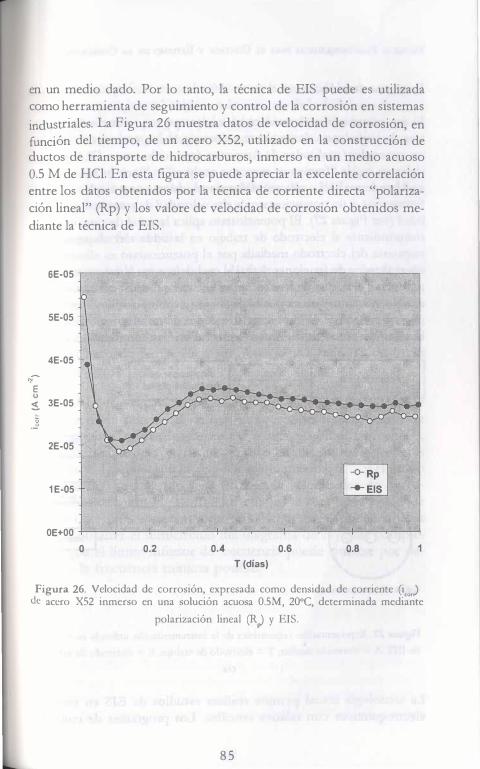
en un medio dado. Por lo tanto, la técnica de EIS puede es utilizada como herramienta de seguimiento y control de la corrosión en sistemas industriales. La Figura 26 muestra datos de velocidad de corrosión, en función del tiempo, de un acero X52, utilizado en la construcción de duetos de transporte de hidrocarburos, inmerso en un medio acuoso 0.5 M de HCL En esta figura se puede apreciar la excelente correlación entre los datos obtenidos por la técnica de corriente directa "polarización lineal" (Rp) y los valore de velocidad de corrosión obtenidos mediante la técnica de EIS.
SE-05
5E-05
4E-05
� E u :!. 3E-05
o u
2E-05
1 E-05
OE+OO
o 0.2 0.4
T (dias)
0.6
� �
0.8
Figura 26. Velocidad de corrosión, expresada como densidad de corriente (i'") de acero X52 inmerso en una solución acuosa O.SM, 20°C, determinada mediante
polarización lineal (R� y EIS.
8 5

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
Instrumentación.
La instrumentación electrónica básica, que permite obtener los espectros de impedancia de un sistema electroquímico, está constituida por un generador/ analizador de funciones (conocido como FRA por sus siglas en inglés "frequency response analyser" o "lock-in amplifier), el cual puede analizar o aplicar señales sinusoidales, en un amplio rango de frecuencias, a un potenciostato de alta velocidad de respuesta y sensibilidad (ver Figura 27) . El potenciostato aplica la señal de sinusoidal correspondiente al electrodo de trabajo en la celda electroquímica. La respuesta del electrodo mediada por el potenciostato es alimentada a un analizador de funciones digital el cual determina la respuesta de impedancia y el ángulo de fase correspondientes a cada frecuencia estudiada. No obstante que es posible utilizar un potenciostato manual, de manera general, la captura o registro de los datos, el almacenamiento y la manipulación de los mismos se efectúa en una computadora dedicada.
Potenciostato t. l(t)
t. E(t)
Celda Electroqu fmica
Analizador de funciones digital
Computadora
Figura 27. Representación esquemática de la instrumentación utilizada en estudios
de EIS. A = electrodo auxiliar, T = electrodo de trabajo, R = electrodo de referen
cia.
La tecnología actual permite realizar estudios de EIS en sistemas electroquímicos con relativa sencillez. Los programas de computo
8 6

disponibles simplifican considerablemente la operación de los equipos electrónicos y, en general, requieren únicamente de la definición de unos cuantos parámetros por parte del usuario. Lo anterior ha hecho que la obtención experimental de espectros de impedancia sea relativamente sencilla.
Cons ideraciones p ara la medición de EIS en celdas
electroquímicas.
• Rango de frecuencia.
Es recomendable que el rango de frecuencia usado sea lo más amplio posible. Idealmente esto implica un rango de 6 a 7 décadas (por ejemplo 10·2 a 105 Hz), si se utilizarán herramientas matemáticas como el análisis de Kramers - Kroning (KK) . N o obstante, muchos sistemas de corrosión no permiten hacer un análisis en un rango extenso de frecuencias, sin obtener una cantidad de ruido considerable.
Un posible criterio para definir el rango de frecuencias, puede provenir de la necesidad de que el c<;>mponente imaginario de la impedancia (Z") se aproxime a O, a altas y a bajas frecuencias, en un diagrama de Nyquist. La Tabla 1 indica, para diferentes sistemas, la frecuencia a la cual sucede la máxima Z" y el rango de frecuencias requerido para diferentes velocidades de corrosión (considerando Cdl = 1 00 ¡..t.F cm-� . De esta tabla se puede Inferir que, para sistemas con una V corr baja, puede no ser posible obtener el semicírculo del diagrama de Nyquist completo, ya que el límite inferior de frecuencia puede situarse por debajo de la frecuencia mínima posible.
87

TÉCNICAS ELECTROQUÍMICAS PARA EL CoNTROL Y EsTuDio DE LA CoRROSIÓN
Tabla 1.
R V f a Z" Rango de p corr (ohm cm2) aproximada máxima (Hz) Frecuencia Ejemplo
(mm año-1) (Hz)
Acero al carbón 20 1 4 80 0.8-8000 en ácido fuerte
so·o 0.032-320 Acero al carbón 0 .6 3 .2 en agua natural Acero al carbón
1 0,000 0.03 0. 1 6 0.001 6-1 6 agua natural inhibida
1'000,000 0.0003 0.0016 0.000016-0. 1 6 Metal pasivo
En algunos casos, es aconsejable utilizar inicialmente un rango de frecuencias amplio, que permita determinar el tiempo en el cual puede obtenerse un espectro representativo y definir si es posible prescindir de datos a bajas frecuencias, en posteriores experimentos.
• Linearidad.
La teoría que fundamenta la técnica de EIS se soporta en teorías lineales. No obstante, los procesos electroquímicos son, estrictamente, no lineales. Lo anterior implica que, para que la teoría que soporta la técnica de EIS pueda ser utilizada en el estudio de procesos electroquímicos, la amplitud de la señal que se use de be de mantenerse lo suficientemente pequeña de manera que la linearidad requerida se cumpla. Para lograr lo anterior, se recomienda generalmente el uso de una amplitud de 10 m V Un análisis más detallado requeriría determinar la respuesta del sistema a diferentes amplitudes y determinar si la linearidad del sistema se conserva.
• Señales espurias.
La técnica de EIS es particularmente sensible a la presencia de señales espurias que pueden alterar las mediciones. Por lo anterior, se debe prestar particular atención a las conexiones eléctricas, a un adecuado sistema de tierras y a un buen diseño de celda experimental (celda electroquímica). Un correcto diseño de la celda electroquímica puede
8 8

ayudar a aliviar problemas con señales espurias. El factor más importante en las mediciones de EIS es, obtener una distribución de corriente uniforme sobre la superficie del electrodo de trabajo. Algunos puntos que permiten obtener un diseño de celda adecuado son:
a) El electrodo de trabajo y el contra electrodo (electrodo auxiliar) deben de estar dispuestos simétricamente, de tal manera que se obtenga una distribución de corriente uniforme.
b) La punta capilar del electrodo de referencia (Luggin) debe colocarse lo suficientemente cerca de la superficie del electrodo de trabajo, para que permita compensar la caida óhmica de la solución y que no altere la distribución de corriente uniforme.
e) Con el fin de disminuir la resistencia asociada al electrodo de referencia, la punta capilar usada debe ser preferentemente recta y corta. Así mismo, debe evitarse una punta capilar de calibre muy fino.
d) Debido a la necesidad de mantener la amplitud de la señal de entrada en un valor muy pequeño, para asegurar la linearidad del sistema, es posible que señales eléctricas externas pueden alterar los resultados del sistema. Lo anterior hace necesario un aislamiento cuidadoso, tanto de los componentes electrónicos usados, como de todas las conexiones eléctricas.
• Número de datos.
Entre mayor es el número de frecuencias en un espectro de impedancia, tanto mayor es la exactitud de cualquier análisis de datos. No obstante el tiempo total requerido para la obtención de los datos se incrementa. En general es recomendable obtener entre 7 y 10 puntos por década de frecuencia, lo cual representa una relación adecuada entre exactitud y tiempo para la mayoría de los casos.
• Promedio de la señal.
Este parámetro comúnmente no es considerado por el usuario, y existen diferentes opciones para promediar la señal del sis tema.
89

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
Este procedimiento suele denominarse "tiempo de integración' debido a que el cálculo de la impedancia involucra la integración de la respuesta de corriente en un cierto número de ciclos. Entre mayor es el número de ciclos alternos que son promediados para la obtención de un punto, menor el ruido que se obtenga. No obstante, a mayor número de ciclos mayor es el tiempo necesario para obtener el espectro del impedancia del sistema.
BIBLIOGRAFÍA RECOMENDADA.
• Cottis R., Turgoose S.; Electrochemical Impedance and Noise; serie Corrosion Testing Made Easy; Syrett B.C editor; NACE Internacional; USA; 1999.
• Greef R., Peat R., Peter L.M., Pletcher D.; Robinson J., (Southampton Electrochemis try Group) ; Instrumental Methods in Electrochemistry; Ellis Horwood Ltd; UK..
• Bard A.J., Faulkner L.; Electrochemical Methods, Fundamentals and Applications; J ohn Wiley and Sons; Singapore; 1980.
• S cully J .R . , S ilverman D. C . , K.endig M.W, (editores); Electrochemical Impedance, Analysis and Interpretation; ASTM, STP 1 188; USA; 1 993.
• ASTM; Standard Practice G106-89; Verification of Algorithm and Equipment for Electrochemical Impedance Measurements; USA; 1999.
• Cogger N.D., Evans N.J.; An Introduction to Electrochemical Impedance Measurement, Technical Report No. 006; Solartron Instruments; USA; 1996.
• Cogger N.D., Webb R.V; Frequency Response Analysis, Technical Report No. 10; Solartron Instruments; USA.
• Jones DA.; Principies and Prevention of Corrosion; 2"d ed.; Prentice - Hall Inc.; USA; 1996.
90

SITIOS DE INTERNET DE INTERÉS.
• h ttp: // www. solartronanalytical.com/ mainpages / corrosion.h tml • http: //www.potentiostat.com
• http: //www.scribner.com
• http :/ /www.cp.umist.ac.uk/CPC/
• http :/ /www.nace.org
9 1


4. LA TÉCNICA DE Rumo ELECTROQUíMico PARA
EL ESTUDIO DE LA CORROSIÓN
J. M. Malo Tamayo y J. Uruchurtu Chavarín Instituto de Investigaciones Eléctricas
Introducción
Ruido es un término común que significa sonido no deseado, en términos científicos es aquello que no puede ser explicado con relación a la variabilidad de las mediciones obtenidas en un experimento. Sin embargo, esto no quiere decir que no contenga información
En el estudio del ruido electroquímico no se trata con señales audibles, sino con oscilaciones en el potencial y corriente electroquímicas. El ruido electroquímico en potencial se define como las oscilaciones estocásticas del potencial electroquímico de un electrodo respecto a un electrodo de referencia, mientras que el ruido electroquímico en corriente es la oscilación estocástica de una corriente electroquímica. La medición del ruido electroquímico es relativamente simple, aunque lo importante es la obtención de la información relevante que puede ser en muchos casos mas problemática.
Medición de Ruido Electroquímico
La medición del ruido electroquímico de potencial y corriente puede hacerse de manera simultáneamente. El ruido en potencial se realiza a través de la medición de las oscilaciones del potencial de corrosión respecto a un electrodo de referencia, o bien de un electyrodo nominalmente "idéntico". El ruido en corriente se obtiene midiendo las oscilaciones de la corriente entre dos electrodos idéntico o de un solo electrodo bajo control potenciostático (Figura 1).
La medición simultánea permite obtener por analogía con la Ley de Ohm la resis tencia de ruido electroquímico y mediante análisis espectral la impedancia de ruido electroquímico (ver Figuras 2 a 4) .
9 3
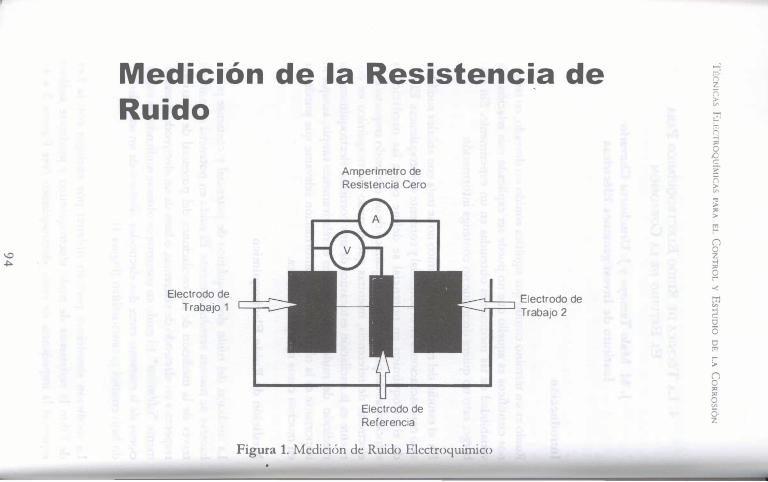
\0 �
Med ición de la Resistencia de Ru ido
Electrodo de Trabajo 1
Amperímetro de
Resistencia Cero
Electrodo de
Referencia
L...---t-L--, Electrodo de
Trabajo 2
Figura 1. Medición de Ruido Electroquímico
>-l rnn z ñ > "'
trJ r-1"'1 n ;1 o D e §:' ñ > "'
;;; " > m r n o z .., " o r-><
t;J ¿ t:J 6 o m
!;: n o " " o "'
O· z

Pot
\0
1 �
(m V) c.----, . _--,-,--,-. · . . . · � ::¡ -260 r -- - 1 ;, _ j 1 ' -1
,,· .. ;.\)\ _ _,� i f' . . :::¡ ¡_ --· . . :. -- - . · ·------· ¡'t-.; ' -:¡ --265 1
� --- . .. . . . .
. 1 � 1 J ·· u -; -270 ¡ --
- - - -- - - ; - �[�-�=-:- _ - J �-· 3
-285 1' ·· ····
r ,1111 / =1 �1 1 ' ,J ' ,; :J --- / : ¡_ .. Jn._) --\/ · · · -··; . ... _ _______ ___ , _ _ -"] / 'kV. ! 3 . 1¡ f � - -: 1�, - - ,J-\r -- · - - - -- · ; - - - 1 l. 1 ,_, �
¡ � !1 '1' ··N¡ : ¡ : '
-290 t -/ �' - - J,J+ - - ' . - -- -···· . l - l 1 ¡ � ···..¡.;,¡
5 1 0 T
1 5 (mins)
20 25
Figura No. 2 Serie de Potencial-Tiempo
30

r
\O 0\
Corr í'"1� �,--,-· ¡ 1 1.,---ry-__,
(JJA) ·. . . " . 1 • � • 1 1 -�� �-���f ,--;-F·,-,-l\ , · �, 11. '\v'y\,..Y�
'
. .. . . · , \ '" . u - 1 1 -- -· ...
-1 5 1 --• !"
- -- -! : �
1MMoOoOO• -·· ., .
-· ·· · · · ·
¡.. 1 ¡f ' : . ! ¡ ll .. ft.11¡ J�w
J ··· · · : · :�H ·v � r� Y 1 ·2 5 ' ��}"*� 1 .. -3 1 --· - ·· Jt{ r'
-3 . 5 [·¡�- -- - -- • · · ···---- ·· ·-·-··-······
l. 11 5 1 0
T 1 5
(mins) 20
Figura No. 3 Serie de Corrientes-Tiempo
25 30
.....¡ tn· () z ñ > "' tTl ' Pi ;l o () e ¡:· ñ > "'
;; "' > tn r-n o z ;l o ' -<
w e o 5 o tn
� n o � o tn
o z

..0 .._¡
Resist (ohms.cm2) 1 . ... , _ , . r r · · --;-"""�:-- . _ _ ,_ J I · ' 1 - , J ¡_ , ¡ J ¡ t. _ ¡ 1 · 1 • o LL¡CI ¡.o • '"T'""ro-T:T=---<��
! : ; ! -- . ·- . , . - 1
3 sooE+os l . ... . . . .. _ : ... -HI ' 1-j !¡¡� ' i -·
. ...
3 .000E+Os [: - - · · - ····- 1 • •• ·----··- - ••• __ ----�- �Jj ¡ \ ______ Jt .. J.i - ·- · · . �1 ¡ � \ ; 1 11 J 1 jl � 2 .500E+05 · · · · ....
. � � 1 lf! l\l 2.000E+05 r - - ·- - . . ..... .. . _ _
i ¡ -. ·.1 i J "l ¡ , , 1 500 i lry,l : 1 ' . E+OS I: - · - · · ··- -----· +.· · · ·--·-·· .. ·-· · ... ;. _ . _____ . 1 �-�- -� __ _ ________ ;_ _______ , . . • 1 • - -·---------· ------· ·· ·-
: 1,'1 i ¡Ji�' ! i : , ¡;¡\1r"'11 1 1/' ; i . 1 .oooE+osl� -- -p:-r�·h --- - -· · · · ·- _j__ : 1 '
"lyl
i r ;
¡ ··-- ···--··-···· j ··--·----- ---+ · ·- ·· - ·-r-;-· .. --···- �
5 : ' ' ' . . . ' ¡ ' ' ' ' 1 _; 1 0 1 5 20 T (mins)
25 30 Figura No. 4 Serie de Resistencia-Tiempo

TECNICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
Esta resistencia es equivalente a la resistencia de polarización, y en ella se incluyen oscilaciones debidas a la transferencia de carga (cinética electroquínúca, o bien por ejemplo oscilaciones de la resistencia de la solución debidas a la nucleación, crecimiento y desprendimiento de burbujas.
Características del Ruido Electroquímico
Las características del ruido electrroquínúco varían frecuentemente a lo largo del tiempo, siendo por esto la señal no estacionaria. La mayor información se obtiene de las oscilaciones en baja frecuencia (menor de 10 Hz). La amplitud de las oscilaciones es pequeña, siendo su desviación estándar del orden de !J. V a m V para el ruido electroquínúco en potencial, y de nA a IJ.A para el ruido electroqumico en corriente. El línúte inferior de sensibilidad está determinado por el ruido del instrumento. Considérese un sistema de dos electrodos de acero inmersos en una solución de cloruro de sodio. Se mide el ruido en potencial entre un electrodo de acero y un electrodo de referencia de calomel. El ruido en corriente se mide entre dos electrodos a través de un amperímetro de resistencia cero, suponiéndolo ideal (sin caída de voltaje), comportándose los dos electrodos de acero utilizados para medir la corriente efectiva, como un solo electrodo del doble del área de uno de ellos.
Información del Ruido Electroquímico
La Figura 5 presenta el registro de la variación de potencial y corriente en el tiempo, del acero inmerso en cloruro de sodio que se conocen como series de potencial/corriente-tiempo o simplemente series de tiempo. Se puede observar que se puede dividir por lo menos en dos partes el comportamiento registrado y presente en las series de tiempo. La primera parte es el comportamiento para período largo que consiste en un decaimiento transitorio o corrimiento de la señal de potencial, a medida que se desarrolla el producto de corrosión o película sobre la superficie la cual suministra una barrera creciente al oxígeno. La segunda parte o característica de la serie
9 8

w u 11) ,; > > ' ] " G o 0..
�.52 0.00005
0.00003 ·0.54
0.00001
-<1.56
�.58
.0.00005 -0.6
-0.00007
-0.62 -!---�---�--�---�--�---l. ·0.00009 �o 500 750
Tlmt / s
A ) Pot�ncial y Corriente
o
·0. 1 .
-0.2
-0.3 -
-0.4
-0.5
·0.6
1000 1250 1 500
0.00005
o
-<l.OOOOS
-0.0001
-0.0001 5
.().0002
.().00025
-0.0003
·0.00035
-0.7 -0.0004 o 1 000 2000 3000 4000 5000 6000 7000 8000 9000
nme / s
B ) Transi torios
< ' 'é .. t 3 u
Figura No. 5 Ruido electroquímico de acero en cloruro de sodio
99

TÉCNICAS ELECTROQUíMICAs PARA EL CoNTROL v EsTuDIO DE LA CoRROSióN
de tiempo presenta oscilaciones a una escala de tiempo menor en su estructura. Ciertamente la primera parte contiene información acerca del proceso de corrosión, pero debe tenerse cuidado al utilizar muchos de los métodos de análisis estadísticos que suponen que las propiedades del sistema son constantes en el tiempo, lo cual no es el caso para un comportamiento transitorio.
Las oscilaciones de potencial y corriente para períodos mas cortos de tiempo, son el verdadero ruido electroquímico, pero aún aquí las propiedades del ruido cambiarán con el paso del tiempo cuando la película de productos de corrosión se desarrolle.
En el caso del acero en solución salina no existe una estructura definida en el ruido, ni que procesos físicos lo están produciendo. Mientras la reacción de corrosión en este caso la controla el transporte de oxígeno a la superficie metálica, las causas probables del ruido observado se pueden asociar con fluctuaciones en el transporte de oxígeno. Se puede observar esto, si se rasga la superficie de uno de los electrodos con un instrumento punzante (Figura Sb). Notar que las escalas en la gráfica son muy diferentes que en la Figura Sa, que es por lo que el ruido electroquímico de fondo parece diferente.
Así como se demuestra el posible papel que juega el transporte de oxígeno en la generación de ruido electroquímico, también ejemplifica que es muy sencillo que influencias externas produzcan efectos en la señal de ruido registrada. Esto hace muy difícil separar estos efectos de la señal de ruido electroquímico después de realizada la medida, siendo necesario tener cuidado para evitarlas en la etapa de medición. Si esto no es posible se requiere los métodos de análisis de la señal que se verán mas adelante.
El ruido electroquímico permite obtener información acerca de la cinética de reacción, o sea la velocidad de corrosión; siendo posible la identificación del tipo de corrosión ya sea: uniforme, generalizada o localizada. Además es posible obtener información acerca de los mecanismos de reacción.
1 00

Técnicas de Registro
La técnica analógica de registro contínuo mas sencilla es simplemente utilizar un graficador, aunque este enfoque es limitado ya que no es posible el análisis de la señal. Otro enfoque es el de la adquisición discreta de datos muestreados, con la utilización de técnicas digitales. El ancho de banda de la señal está limitada en altas frecuencias por el llamado límite de Nyquist que es la mitad de la frecuencia de muestreo (f max = 1 1 2LlT); y en bajas frecuencias por el inverso del tiempo de duración de la serie de tiempo, o sea el inverso del número de muestras multiplicado por el período de muestreo (f mm = 1 1 NilT). Se puede obtener información redundante en altas frecuencias por el muestreo demasiado rápido ("aliasing'), requiriendo la señal un ftltrado antes de realizar el muestreo Se requiere un tratamiento antes de analizar la señal. Este consiste en remover el corrimiento en DC mediante el ajuste de una recta por el método de mínimos cuadrados.
Métodos de Análisis
Series de Tiempo
El método mas simple de análisis y el mas directo, es el examinar las series de tiempo para la identificación de detalles que son característicos de los tipos de corrosión particulares. Por ejemplo, la detección visual de transitorios de rompimiento y repasivación o de oscilaciones asociadas a resquicios o corrosión por picadura.
El ruido electroquímico producido por algunos sistemas puede mostrar una estructura más definida en la serie de tiempo. Por ejemplo, la Figura 6 presenta el ruido electroquímico producido por el acero inmerso en una solución saturada de Ca(OH)2 mas la adición de NaCl. En este caso el acero tiende a formar una capa pasiva debido a la alcalinidad de la solución, pero los cloruros causan rompimiento localizado de la película pasivante, observándose corrosión por picadura. Esto da como resultado, diversos transitorios de potencial y corriente cuando las picaduras se 1U1Clan, crecen y se
1 0 1

....... o N
� � 1 l.
Mild Steel in Ca(OH)2sat.+1 :1 CI:Ca Cut en ( aner 1 tiJUI)
.0.
- ""'
Mild Steel in Ca(OH)2sat.+ 1 :1 CI:Ca Polerfial (after1 hwl)
..- :tal D 1CGa tm l'looCOICI
Mild Steel in Ca(OH)2sat.+ 1 :1 CI:Ca Culrerc (after 11 h:>uts)
··�------------------------¡
JO) ·• .a: - 10CII) \XIO T-t-�
. Mild Steel in Ca(OH)2sat.+ 1 :1 Cl:Ca Corren! ( allet 22 hctn)
lDO CIO a ao u;m tD:> --
Mild Steel in ca(OH)2sa!.+ 1 :1 CI:Ca f'o{eni¡¡j (illtl 22 ho<JIS)
""'
Figura No. 6 Ruido Electroquímico del acero en hidróxido de calcio + cloruro de sodio a diferentes tiempos de inmersión
.....¡ tn· n

repasivan, modificando la estructura de las series de tiempo en función del tiempo de inmersión .. Debido a la estructura definida de la serie de tiempo y la relación establecida entre las características de la serie de tiempo y los procesos físicos subyacentes, es fácil obtener la información que puede extraerse de la serie de tiempo.
Métodos Estadísticos
Los métodos estadísticos simples tratan a la serie de tiempo como una colección de potenciales o corrientes individuales, ignorando la relación entre un valor y el siguiente (muestra de la población). La serie de tiempo está definida completamente por la distribución de sus valores. La media o promedio es el mas común de los parámetros, y el potencial promedio no se considera como parte del ruido electroquímico, aunque se utiliza para calcular otros parámetros de interés como el de corrosión localizada. Las fluctuaciones de la media en largos períodos de tiempo están directamente relacionados a cambios en los procesos de corrosión. El ruido en corriente teóricamente presentaría un valor de cero entre dos electrodos idénticos. En la práctica esto no es así, debido a pequeñas diferencias en su comportamiento frente a la corrosión.
La varianza de una señal depende del rango de frecuencias de ésta, y corresponde a la potencia del ruido. Se espera que la varianza de la corriente aumente a medida que la velocidad de corrosión se incremente; así como también la corrosión se haga más localizada. En contraste la varianza del potencial disminuye a medida que la corrosión aumenta, pero se incrementa a medida que el ataque se. hace mas localizado. Esto se cumple para la mayoría de los tipos de corrosión.
La desviación estándar es simplemente la raíz cuadrada de la varianza, y su cálculo e interpretación son básicamente la misma. Es un parámetro mas comúnmente utilizado y habla del ancho del trazo de la señal o dispersión de los datos de ruido. La raíz cuadrática media (rms) es la raíz cuadrada del valor promedio del cuadrado del
103

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
potencial o corriente (sin substraer la media) . Es la medida de la potencia disponible de la señal, incluyendo el efecto DC o del promedio del potencial o corriente. La rms, varianza y desviación estándar están relacionadas por lo que no se necesitan calcular las tres.
El sesgo (skew) es la medida de la simetría de la distribución. Un valor de cero implica una distribución simétrica alrededor de la media, mientras que un valor positivo indica un sesgo en dirección positiva y un valor negativo un sesgo negativo respecto a la media. Una serie de tiempo que presenta transientes unidireccionales presentará un sesgo cargado en una dirección, y esto es una medida útil para transitorios asociados a picaduras metaestables. La medida del sesgo en corriente indica: si es unidireccional que solo un electrodo está activo, y si es bidireccional ambos lo están.
La kurtosis es una medida de la forma de la distribución comparada con la distribución normal. Una kurtosis de cero implica una forma similar a la distribución normal (pero no necesariamente una distribución normal), mientras que un valor positivo implica una forma con mayor pico y un valor mas negativo una forma mas plana. En términos generales el ruido electroquímico no presenta una forma o distribución normal, o sea presenta sesgo y kurtosis.
El coeficiente de variación o variabilidad es la desviación estándar dividida por la media y es la medida de la cantidad de ruido comparado con la media. Para algunos procesos estocásticos se espera que sea constante. No es de extrañar que pueda ser un indicador del tipo de proceso, utilizándolo con las corrientes de corrosión aunque tratando las reacciones anódicas y catódicas separadamente. En consecuencia la corriente promedio que dividimos, es la suma de las magnitudes de las dos corrientes (2Ico); y no la diferencia entre ellas, que se miden como corrientes galvánicas acopladas.
A partir de las desviaciones estándar de potencial y corriente, por analogía con la ley de Ohm se calcula la mal llamada resistencia de ruido y equivalente a la resistencia de polarización. Se relaciona con
1 04

la velocidad de corrosión total uniforme o generalizada. La resistencia de ruido incorpora fluctuaciones de la resistencia de la solución, a pesar de que no se aplica una corriente a la celda. Por lo tanto, se puede obtener en soluciones de alta resistividad (ver Figura 4) .
Dominio de la Frecuencia
El estimado espectral es el proceso de cálculo de la potencia presente en varias frecuencias en un registro infinito de datos. Cuando se analizan las frecuencias presentes en una señal compleja, se divide la potencias entre las varias frecuencias. La gráfica de potencia en función de la frecuencia se conoce como espectro en potencia (ver Figura 7) .
20 ,----------------------,
" � � -20
-40 '-------,.-,------,-,-:-----:--,.-,-----_J o 400 800 1 200 160C
� - 1 20 '--------------------------'------' 0.0001 0.001 0.01 0.1 1 .C
Time, s Frequency, Hz
Figura 7- Serie de Potencial-tiempo y Densidad Espectral
Existen dos métodos utilizados para la estimación del espectro en potencia en estudios de ruido electroquímico: La transformada de Fourier y el método de máxima entropía. El primero produce un espectro ruidoso mientras que el segundo produce un espectro mas liso, siendo ambos equivalentes.
Para aplicar este tipo de análisis, se requiere que la señal sea estacionaria (con una media que tiende a cero) lo cual no siempre es cierto en sistemas de corrosión, especialmente al inicio de las exposiciones lo que da problemas con la estimación espectral. Por esto, se utiliza un artificio matemático que es la remoción de la tendencia o corrimiento en DC, usualmente en la forma de una substracción de los datos mediante una regresión lineal. Posteriormente se aplica una
105

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
función ventana para escoger secciones de la serie de tiempo (suponiéndola infinita) que quiere analizarse. La ventana reduce la extensión de las frecuencias a lo largo del espectro. Una vez realizado esto se aplica el algoritmo transformada rápida de Fourier (FFf) o de máxima entropía (MEM) para obtener el espectro (equivalente a la varianza). El espectro que se obtiene es el de un comportamiento 1/f en el ancho de banda de bajas frecuencias considerado, presente en muchos procesos de la naturaleza incluyendo el de corrosión.
Si se derivan los espectros de potencia de potencial y corriente, se dividen para cada frecuencia para obtener el espectro de potencia de la impedancia y obteniendo la raíz cuadrada se deriva la densidad espectral o espectro de la impedancia de ruido electroquímico (Z,), comparable a la impedancia electroquímica convencional para bajas frecuencias (menores a 1 Hz). Zn mide el módulo de la impedancia electroquímica del sistema a muy bajas frecuencias, aunque el ángulo de fase no es posible estimarlo.
Las características de los espectros incluyen una meseta a bajas frecuencias seguida de una pendiente y posiblemente una segunda meseta para mas altas frecuencias. Se han utilizado las modificaciones de la pendiente para caracterizar los tipos de corrosión. La meseta en bajas frecuencias parecen estar asociados a fenómenos de transporte mientras que la meseta en altas frecuencias puede confundirse con el nivel de ruido del instrumento y el efecto producida por los datos redundantes (aliasing).
Si una serie de tiempo es producida por la suma de una secuencia de eventos transitorios idénticos aleatorios, el espectro de la serie de tiempo será de la misma forma que el espectro de eventos individuales pero con un aumento proporcional al número de eventos. Si los eventos difieren en amplitud pero no en duración, el espectro completo seguirá manteniendo la misma forma. Sin embargo si los eventos tienen diferente duración, el espectro se distorsiona y la forma ya no será la misma que el del espectro del evento individual. Consecuentemente, la pendiente del espectro se relaciona con la forma de
1 06

Jos transitorios elementales pero estará influenciado también por la distribución de la duración de los tra.nsitorios (Figura 8) . Los cambios en la pendiente y la pendiente del espectro contienen información del tipo de proceso de corrosión.
Es muy importante distinguir entre los espectros de ruido en potencial, corriente y de impedancia, evitando compararlos, y mostrar sus diferencias entre sí (Figuras 9 a 1 1) .
Análúis Discriminante
En muchas mediciones de corrosión, se necesita discriminar entre dos o mas estados o condiciones. Por ejemplo, entre un estado de picado y otro de pasividad. La técnica de ruido permite la posibilidad de derivar muchos parámetros, por lo que en esta situación es dificil decidir el mejor método para discriminar entre los dos estados.
Una aproximación estadística al problema determina la separación de los valores promedio de un parámetro·dado para los dos estados, relativos a la desviación estándar de ese parámetro. Si se utiliza el espectro de potencia a partir de la FFf, se puede derivar una función discriminante única a partir de la relación:
D(f) = [ (PSD/ PSD� - (PSD/PSD¡) -2] 0 5
donde PSD1 y PSD2 son los espectros de los dos estados para cada frecuencia j Por lo tanto, mientras mayor sea el valor de D(f) a esa frecuencia, se puede discriminar entre los dos estados. Se puede derivar el espectro de la función D(f) siendo posible detectar aquellas frecuencias en las cuales es fácil de discriminar entre los dos estados (Figura 1 2) .
1 07

TÉCNICAs ELECTROQUÍMICAS PARA EL CoNTROL v EsTuo1o DE LA CoRROSIÓN
Gl , .E ii e
2 �----------------------------------------,
1.5
< ' 0.5
�.s +-------r-----�------�------�------�----� o 200
100
10
0 .1
600 lime (a )
800 1000 1 200
0.01 +----------.---------....--------1 0.001 0.01
Frequency 1 Hz (b)
0.1
Figura No. 8 Serie de tiempo y espectros

..... o \0
m V. Hz-�
1 .000E+05 ¡ 1
1 .000E+04 ,
1 . 000E+03
1 .000E+02
1 1 1 1
1 .000E+0 1 L,1 1 .OOO E-03
----,------..-·-··-,------: -, -¡· ·
i ·;\, ... :, ! �fi·':';\};,'ü ,, - ' :l i%T�f.,i· , - '
1 .OOOE-02 Frt¡c Hz
1 , }l�l¡\W�� 1
1 .OOOE-01
Figura No. 9 Espectro de Potencial
i · -=¡
�
j 1
·2._

,_. ,_. o
¡JA.Hz-�
1 .000E+04 !
1 .000E+03
1 1 .000E+02 ¡ -
1 .000E+01 ···
1 . 000E+OO _
L
,.. 1
'
�-1 .000E-03
-..,--.,------····-· --,---· r -····· r ·1 ·- ------- T. -·----
. .. 1 ¡ ! · �¡ -
· · :: !'\, ·� ·,¡¡ . . , ¡J •r,.-�w�' il1: ti "� 1
¡ 1 1� ' n 1 . 1 1
1 .000E-02 Free Hz
1 .000E-01
Figura No. 10 Espectro de Corriente
. -
l ---:
1 1
...., m· n z ñ > V> tiJ rm
� o (.) e �-� en
:;; "' >
¡:1 (") o z g r-<
� e o i5 o m
� (") o "' "' o V>
6-z

...... ...... ......
ohms.cm2.Hz-Y2 ...,l
i 1 .000E+06 i
1 1 ,.... ... , ' / � .. ...... ··
1 .000E+05 · ·-
i 1 .000E+04 l
;- 1 1 1 .000E-03
---····- l
: \ 1 ' 1
� : � ' " 1 1 1 • '• , ' •• ¡
� :: r:/� .
·· ¡ :. ��� . ' . , .. . ,\ � 1 i . 11";: 1 \¡ ' !•
! � , 1 rf�l: 1 i 1 q
1 . 000E-02 F ree Hz
Figura No. 11 Espectro de Impedancia
j -¡ -1 ...j
1
1 .000E-0 1

TÉCNICAS ELECTROQUÍMICAS PARA EL CoNTROL Y EsTuDIO DE LA CoRROSIÓN
D % r
11.) ... e:: <:.: e: · a · ¡:¡
o u VJ . .... "O
g e:: -o
· o e:: ::l u ¡¡.. e N
....... � ó z
� '"' ") ::l l:lll ....
¡¡..
( m ) p
1 1 2

Análúú de Pulsos de Descarga
Es común describir al ruido por analogía con los colores de la luz como:
Ruido blanco con un espectro constante en el rango de frecuencias Ruido rosa con un espectro que disminuye al aumentar la frecuencia (1 / f)
O bien como:
movimiento browniano con oscilaciones aleatorias ruido térmico (Johnson) que consiste en el movimiento aleatorio de electrones activado térmicamente que causa la separación de carga a través de una resistencia. Tiene un espectro similar al ruido blanco, con un nivel muy pequeño comparadocon el ruido electroquímico. Ruido de descargas (Shot noise) es debido a movimientos de portadores de carga de corriente en circuitos eléctricos (electrones en metales y iones en soluciones acuosas ) a través de materiales conductores Para una corriente dada, el promedio del número de portadores de carga se define por la corriente promedio dividida por la carga del portador de carga. Sin embargo, el número de portadores que pasa por un punto en un intervalo de tiempo dado, tiene una distribución estadística alrededor del valor promedio, lo que produce el ruido en la corriente observada.
Si el movimiento individual de portadores de carga es independiente de otros portadores de carga, un análisis estadístico sencillo, lleva a la siguiente expresión para el ruido de descarga:
Potencia de ruido en corriente = 2elb
dónde e es la carga del portador, I es la corriente promedio y b es el ancho de banda.
1 1 3

TÉcNICAs ELECTROQUÍMICAS PARA EL CoNTROL v EsTUDIO DE LA CoRROSióN
En la conducción en soluciones iónicas y semiconductores, debe asumirse que obedece a la ley de la fórmula de ruido de descarga. La carga relevante en soluciones iónicas es la carga promedio de iones que portan la corriente y que puede ser mas de un electrón.
Análisis de Ruido de Descarga Aplicado a Ruido Electroquímico
Ruido electroquímico en corrosión, comúnmente aparece como eventos discretos como la generación de transitorios de corriente asociados a crecimiento y muerte de picaduras metaestables. Supóngase que se muestrea lo suficientemente lento de tal manera que se capture la potencia de los transitorios individuales; de tal manera que, en cada periodo de muestreo se captura un número aleatorio de transitorios. Considerando la señal de corriente, cada transitorio contribuye a cierta cantidad de carga q y, dentro del intervalo de la muestra la carga que pasa será q veces el número de transitorios m. Para conocer cuanto ruido se produce, se necesita conocer la desviación estándar de m, la que será la desviación estándar de la serie de tiempo. Si los transitorios son eventos aleatorios independientes, la distribución es de un proceso de Poisson, donde la varianza es igual que el valor medio de m.
Si la corriente promedio es I, entonces el promedio del número de transitorios en un intervalo de muestreo de flf es claramente la carga promedio en el intervalo de muestreo Ifl t dividida por la carga en el transitorio q. Entonces, la varianza de la corriente será la varianza de mq dividida por flt2 para convertir de carga a corriente:
Si se analiza sobre un número rasonablemente grande de muestras, se incluyen frecuencias desde cero hasta la frecuencia de Nyquist 1 / (2flt) en la medición. Se substituye 1 / (2flt) por b , el ancho de banda de la medición, obteniendo la formula de ruido por descarga (shot noise).

En electrónica la carga es simplemente la carga del electrón, pero en el caso de los transitorios en corrosión es mucho mayor la carga en cada transitorio. Es importante saber que la I en la fórmula de ruido por descarga es el promedio que corresponde al paso de los transitorios. Si el interés es en relación con picaduras metaestables anódicas, la corriente es la corriente anódica promedio. En condiciones de corrosión libre será la J,." o corriente de corrosión. Si solo se midió el ruido en corriente, se tienen dos incógnitas ( I,."Y q) y solo un valor medido, por lo que no puede resolverse para J,."Y q. Sin embargo puede medirse el ruido en potencial para obtener un segundo valor.
Se supone que la mitad de la corriente producida por los transitorios en un electrodo pasa al segundo electrodo. Dividir la corriente también divide la desviación estándar, por lo que se divide la varianza por 4 (en este caso, como las dos corrientes se correlacionan la regla normal de que las corrientes se suman no aplica). Esta corriente es la que en realidad se mide como ruido electroquímico en corriente, que polariza al segundo electrodo, produciendo el ruido en potencial:
A primera vista, se introduce una tercera incógnita Rp, pero ésta se puede eliminar suponiendo que la ecuación de Stern-Geary se aplica:
Icorr = B/Rp
donde B es el coeficiente Stern-Geary. Sustituyendo Rp se obtiene:
Si se supone que ambos electrodos de trabajo producen transitorios, esto incrementa la potencia del ruido en potencial y en corriente por un factor de 2(suponiendo que el ruido en corriente asociado a ambos electrodos no está correlacionado) . Por lo tanto se tiene (para toda la celda):
1 1 <;

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
I 2 prom = I qb n corr
E 2 pro m = qbB2 /I n corr
Resolviendo ambas ecuaciones para J"'" y q se tiene:
Con lo que se observa que Rn y Rp son lo mismo. En cierto sentido esto es correcto, aunque en la derivación se supone válido que se utilice Rp para analizar señales de ruido electroquímico sin probar la validez de la suposición. A bajas frecuencias, y utilizando el límite de frecuencia del espectro de potencial y corriente en lugar de las varianzas, se asegura cumplir con este requerimiento y la b en las últimas ecuaciones desaparece.
La bibliografía presentada a continuación incluye una extensa revisión de los trabajos publicados en la materia hasta la fecha; así como detalles teórico-prácticos de los temas tratados aquí.
Conclusiones
Las aplicaciones de ruido electroquímico incluyen:
1 . Corrosión uniforme y generalizada. 2. Corrosión localizada 3. Recubrimientos metálicos 4. Corrosión en películas delgadas S. Soluciones de baja conductividad 6. Estudios mecanisistas.
La resistencia de ruido es equivalente a la Rp, y la impedancia de ruido mide la impedancia de la interfase, sujeto a las siguientes condiciones: linearidad, estacionalidad, para electrodos similares. La técnica detecta el tipo de corrosión, pero no existen reglas generales para su análisis.
1 1 6

Bibliografía.
1. R., A. Cottis, S. Turgoose, Electrochemical Impedance and Noise; Corrosion Testing Made Easy Series; Ed. B. Syrett, NACE, 1 999.
2. J. Uruchurtu-Chavarín, J. M. Malo, ''Eiectrochemical Noúe as a Poweiful Elutrochemical Technique for Corrosión Studies "; Research Trends, Trends in Corrosion Research, Vol. 2, 1997, p.49.
3. R. A. Cottis; "Interpreta/ion of Electrochemical Noise Data "; Corrosion, Vol. 57. 3, 2001 , p.265.
4. D. E den; "Electrochemical N oise-The Firs t Two Octaves" NACE Corrosion 98, San Diego, 1998, paper 386.
5. C. Gabrielli, F. Huet, M. Keddam, '1nvestigations of Metallic Corrosion !¿y Electrochemical Noise Techniques "; Electrochemical and Optical Techniques for the Study and Monitoring of Metalhe Corrosion, Kluger Academic Pub. ,Vianna do Castelo, 1989, p. 1 35 .
6. C. Gabrielli, F. Huet, M. Keddam, "Review of Applications of Impedance and Noise Ana!Jsis to Uniform and Localized Corrosion "; Corrosion Vol. 48, 10, 1 992, p.794.
1 1 7


TÉCNICAS ELECTROQUÍMICAS APLICADAS AL
EsTuDio DE LA CoRROSióN EN LA INDUSTRIA NucLEAR
5.1 SENSIBILIZACIÓN
(Susceptibilidad a la Corrosión Intergranular)
Introducción
Enrique Augusto Martínez Martínez. (IMICORR) MEXICO D.F.
A finales de la década de los 60's y principio de los años 70's, se detectaron una gran cantidad de agrietamientos, en las soldaduras de diferentes componentes de Reactores Nucleares tipo ''Agua en Ebullición" (BWR), fabricados con Aceros InoxidablesC1l, lo cual generó una gran cantidad de trabajos de investigación para determinar las causas y generar métodos de prevención y control.
Las investigaciones efectuadas, permitieron establecer que el mecanismo responsable del problema era el conocido como "Corrosión lnter-granular Bajo Esfuerzo" (Ínter granular Stress Corrosión Cracking = IGSCC), el cual involucraba tres factores fundamentales:
• Presencia de Esfuerzos de Tensión, residuales y/ o aplicados, que podían es tar muy por abajo del Esfuerzo de Cedencia del material.
• La Química del Agua empleada como enfriador-moderad
• Un Material Sensibilizado (Sensitizado) .
La Sensibilización o Sensitización, fue explicada mas tarde sobre la base de la Teoría mixta, en la cual se postulaba que el fenómeno ocurre en el intervalo de temperatura de 400 a 850°C, debido a la precipitación de Carburos de Cromo en los límites de grano y la
1 1 9

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
creación de una pequeña zona empobrecida de Cromo en las regiones adyacentes, con lo cual se generan, por un lado micro celdas de Corrosión y por el otro regiones en las cuales la capa de Oxido pierde sus características protectoras por deficiencia de Cromo<2l.
Técnica de Reactivación Electroquímica Potenciodinámica. (EPR)
(Electrochemical Potentiodynamic Reactivation Method)
La creciente cantidad de fallas por IGSCC en las soldaduras de tuberías en plantas generadoras de energía, crearon la necesidad de desarrollar u.n método para determinar el Grado de Sensibilización (DOS) , que fuera rápido, cuantitativo y no destructivo, ya que en ese momento solamente se disponía de los métodos descritos en la Práctica Recomendada ASTM A-262<3l. de los cuales solamente uno era no destructivo, pero no cuantitativo (práctica A) y los otros cuatro eran cuantitativos pero destructivos y muy lentos.
Clarke, et.al . <4•5l, evaluaron, compararon y seleccionaron la técnica EPR, propuesta por Prazak<6l CihaWl , y Novak<8l, como la mas adecuada para cumplir el ob je tivo propuesto y establecieron los parámetros y el procedimiento óptimo (con base en un gran número de pruebas) , mismo que se resume a continuación:
El espécimen, como electrodo de trabajo, un contra electrodo y un electrodo de Calomel, deben conectarse a un Potenciostato acoplado a un integrador de corriente.
El método se inicia estableciendo el Potencial de Corrosión de la mues tra a evaluar, en una solución de O.SM H2SO 4 + 0.0 1 M de KSCN y a temperatura de 30° ± 1 °C.
La muestra es posteriormente polarizada a un potencial de +200 m V [vs. electrodo de calomel saturado (ECS)] durante 2 minutos
1 20

e inmediatamente después el potencial se revierte (reactivación) hacia el potencial de corrosión, a una velocidad de 6V /hr (1 .67 m V / seg).
La reactivación conlleva el rompimiento (o disolución) preferencial de la capa pasiva en las zonas empobrecidas en Cromo del material sensibilizado y como resultado se genera un incremento de corriente que forma una curva en la gráfica E vs. Log i.
El área bajo la curva, debido a que el potencial decrece a velocidad constante, es proporcional a la cantidad total de carga eléctrica "Q" (amperes x segundo = Coulombios), que pasa a través de la superficie expuesta de la muestra.
En materiales no sensibilizados la capa pasiva permanecerá intacta y la carga Q será pequeña, en función del tamaño de la curva generada.
Debido a que la carga "Q" depende de la superficie expuesta al electrolito y del tamaño de grano del material, Clarke propuso adicionalmente que la carga "Q" obtenida, debería ser normalizada al tamaño de grano, para lo cual se derivo la siguiente expresión:
Pa = Q / GBA En donde:
Pa = Parámetro ajustado al tamaño de grano en Coulombios / cm2. Q = Carga medida. GBA = Area total de limite de grano obtenida de la ecuación
GBA = As [5 .09544 x 10·3 exp (0.34696X)]
En la cual:
As = Area expuesta de la muestra. X = Tamaño de grano según ASTM determinado a 1 00 aumentos.
1 2 1

TÉCNICAS ELECmOQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
Reproducibilidad
Para evaluar la reproducibilidad se efectuaron pruebas comparativas (round robin) . entre 8 de los Laboratorios Nacionales de los Estados Unidos, utilizando muestras preparadas y estandarizadas por ASTM. En el estudio se evaluó la influencia de:
• El equipo utilizado. , • El acabado superficial de la muestra.
• La velocidad de barrido de Potencial. • El Tamaño de grano del material. • Diversas condiciones de Tratamiento Térmico
(Sensibilización) . • La Temperatura. • La Concentración de los constituyentes del electrolito.
De donde se determinó que:
El acabado superficial, la velocidad de barrido y la temperatura influyen fuertemente en la cantidad de carga "Q" que se genera durante la reactivación para la misma condición de Sensibilización.
El efecto del tamaño de grano es minimizado por medio de la normalización propuesta por Clarke.
El equipo empleado en las pruebas, mientras este calibrado adecuadamente, no influye en los resultados.
La concentración inicial del electrolito y sus modificaciones, en función de la temperatura de prueba y el tiempo de uso afectan adversamente los resultados.
1 22

Como resultado, los parámetros óptimos para la prueba, fueron establecidos como se indica a continuación.
Parámetro '
Caracte'ñsiica"i '� _,
Electrolito. O.SM H2S04 + O.OlM KSCN
Temperatura. 30 ± l0C.
Acabado Superficial. 1 ).! con pasta de diamante.
Velocidad de Reactivación. 6 V /hr.
Potencial de pasivación. + 200 mV
Tiempo de permanencia en Dos minutos.
el potencial de pasivación
Deareación. Nitrógeno continuo por S minutos.
Carga catódica. (Solamente si es necesaria para establecer
el potencial de corrosión) -600 mV.
Electrodo auxiliar. Platino.
Electrodo de Referencia. Calomel.
Sistema de polarización. Cualquiera con capacidad coulombimétrica,
Normalización de datos. Ajuste con GBA.
Criterio de evaluación Sensibilizado cuando Q � 0.20 c.
Técnica de Reactivación Electroquímica de Doble Curva
(EPR-DL).
A pesar de que el desarrollo de la técnica EPR representó un gran avance en la determinación cuantitativa, rápida y no destructiva del grado de Sensibilización de Aceros Inoxidables y de que se utilizó por cerca de 20 años, la influencia de factores como el acabado superficial, la temperatura y el tamaño de tamaño de grano, la hacían relativamente complicada para ser efectuada en campo, debido a la dificultad para controlar tales factores.
1 23

TECNICAS ELECTROQUiMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
En 1980 Akashi <9l desarrolla un nuevo procedimiento para la prueba en el cual el barrido de potencial de reactivación es precedido por un barrido anódico (polarización anódica) desde el potencial de corrosión hasta el de pasivación.
Como resultado se obtienen dos curvas, una correspondiente a la polarización anódica y otra resultante de la reactivación y en vez de utilizar la carga integrada bajo la curva, se emplea, como criterio de evaluación, el radio de las corrientes máximas de reactivación (Ir) y anódica (la) obtenidas.
La principal ventaja del nuevo método es que la superficie de prueba no debe, necesariamente, ser terminada con pasta de diamante de 1 f.!, estableciéndose que un acabado con lija grado 100 (140J.!) es suficiente, ya que estudios realizados por Akashi, indicaron que el acabado superficial no tenia, en este caso, una influencia determinante en los resultados obtenidos.
La técnica de Reactivación Electroquímica de Curva Doble, se desarrolla bajo las mismas condiciones básicas de la técnica EPR, esto es, utilizando una solución de O.SM H2SÜ4 + 0.01M de KSCN deareada con Nitrógeno y a temperatura de 30° ± 1 °C; el barrido de potencial se realiza a la misma velocidad (6V /hr).
La prueba se inicia con la determinación del Potencial de Corrosión, pudiendo realizarse un barrido catódico previo, lo cual ayuda a estabilizar el E .
corr
Una vez establecido el Potencial de Corrosión la muestra se polariza anódicamente hasta +300 mV vs. ECS, a la velocidad de 6 V / h, pre-establecida y en cuanto éste es alcanzado la velocidad de barrido se revierte (reactivación), a la misma velocidad, hasta alcanzar nuevamente el Potencial de Corrosión.
Las corrientes anódica (I.) y de reactivación (I) son determinadas de la curva E vs. Log i, (ó E vs. I) y la relación 1/I. obtenida se
1 24

ernplea como criterio de evaluación, habiéndose establecido que la frontera entre un material sensibilizado y uno no sensibilizado esta en el valor de 1/I. = 0.001 .
Reproducibilidad.
La reproducibilidad de la prueba y el efecto de las variables involucradas ha sido evaluado por diferentes autores (10,1 1 ,12), los resultados se resumen a continuación.
La reproducibilidad de la prueba de doble curva es mejor que la de curva sencilla; en la prueba de doble curva se observa que la magnitud de la corriente anódica es independiente del grado de sensibilización mientras que la corriente de reactivación se ve considerablemente afectada pudiendo ser muy pequeña (del orden de 1 Q·6 a 1 Q·4) para materiales no sensibilizados o tan grande como 10·2, para materiales severamente sensibilizados.
Los valores de corriente de la curva de reactivación en esta prueba son muy cercanos a los encontrados en la prueba EPR, lo cual es de esperarse si se considera que todas las condiciones de las pruebas son fundamentalmente iguales, aún el tiempo de permanencia en el potencial de pasivación, ya que los dos minutos a +200 m V de la prueba EPR son equivalentes al tiempo que transcurre de los +200 a los +300 m V ( a velocidad de 6 V / h) que se aplican en la prueba EPR-DL.
Efecto de la Velocidad de Barrido.
Para todos los niveles de sensibilización, se ha encontrado que la relación Ir/la aumenta al disminuirse la velocidad de barrido, el aumento es debido a un incremento en la corriente de reactivación ya que, además de una mayor disolución de material en las regiones de límite de grano, hay también un mayor ataque por Corrosión Uniforme en el resto del material. La velocidad de barrido de 6 V /h, parece ser el mejor compromiso entre una baj a velocidad de Corrosión Uniforme y un elevado grado de ataque en límites de grano.
1 25

TÉCNICAS ELECTROQUÍMICAS PARA EL CoNTROL v EsTuDIO DE LA CoRROsióN
El efecto de la velocidad de barrido en la prueba de doble curva es, s· embargo, menor que en la prueba de curva simple, dando con esto u ventaja adicional.
Efecto del Acabado Superficial.
Pruebas realizadas en muestra con acabado superficial desde 1 f.l hasta lija 100, indican que los resultados son independientes del acabado superficial debido a que durante el barrido anódico toda la superficie de la muestra se activa y disuelve a velocidad elevada, con lo cual las primeras capas superficiales son removidas y entonces el barrido de reactivación se efectua en una superficie "limpia".
Efecto de la Temperatura.
La relación de corrientes obtenidas, indica que en materiales no sensibilizados la temperatura no tiene un efecto considerable; sin embargo en materiales sensibilizados muestra un marcado incremento entre los 25 y 40°C, en una relación aparentemente lineal con una pendiente de 0.004 / °C, para materiales fuertemente sensibilizados y de 0.001 / °C, para materiales medianamente sensibilizados.
Efecto de la Concentración de KSCN.
El Tiocianato de Potasio se adiciona al electrolito de prueba, como un activador o despasivador selectivo del límite de grano, con la finalidad de aumentar la corriente de reactivación.
Se ha encontrado que por arriba de 0.03 mol / 1, de KSCN, no se tiene un aumento adicional en la relación I / I de materiales
r a
sensibilizados.
A la concentración especificada de 0.01M una variación de ± 10% afecta la relación de corrientes en ± 0.003 para materiales severamente sensibiliz ados y en ± 0 .0003 para materiales medianamente sensibilizados. En contraste, en la prueba EPR de curva simple, la misma variación en la concentración de KSCN produce un cambio de Pa de 4 y 0.4 C / cm2 respectivamente.
1 26

Bibliografía
1 . A. Taboada y L. Frank. "Intergranular Corrosion in Nuclear Systems", Intergranular Corrosion in Stainless Alloys, ASTM STP 656. ASTM, 1 978.
2. A.J. Giannuzzi, D.C. Bertossa, J .C. Danko, Materiafs Peiforman.-e 17 (1 1) 32 (1978).
3. ASTM Book of Standards, Vol. lO, A-262, 1 978.
4. WL. Clarke, R.L. Cowan, WL. Walker. "Comparative Methods for Measuring Degree of Sensitization in Stainless Steels". Intergranular Corrosion of Stainless Alloys. ASTM STP 656, 1 978.
5. WL. Clarke, VM. Romero, J.C. Danko. "Detection of Sensitiza tion on Stainless S teel Using Electrochemical Techniques" . NUREG 0251 - 1 , US Nuclear Regulatory Commission. 1 978.
6. M. Prazak. Corrosion 1 9 (3) 75 (1963) .
7. V. Cihal, A. Desestret. Communication au 5ieme Congress Europeanne de la Corrosion, FEC. 1973.
8. P. Novak, et.al. "Testing the Susceptibility of Stainless Steel to Intergranular Corrosion by a Reactivation Method". Department of Chemical metallurgy, The Prague Institute of Chemical Technology and Corrosion, Vol.3 1 , No. 10, 1975.
9 . M. Akashi, T. Kawamoto, F. Umemura, Corrosion Engineering 29 (1 980) .
10 . A.P. Majidi, M.A. Streicher, Corrosion 40 (8) 393 (1984).
1 1 . Ibid. Corrosion 40 (9) 445 (1984).
12 . Ibid. Corrosion 40 (1 1) 584 (1984).
1 27

TÉCNICAS ELECTROQUÍMICAS APLICADAS AL EsTUDIO DE LA CORROSIÓN EN LA INDUSTRIA NUCLEAR
5.2 EVALUACIÓN DE LA TEMPERATURA CRiTICA DE PICADO POR MEDIO DE TÉCNICAS ELECTROQUÍMICAS.
Introducción
Enrique Augusto Marúnez Marúnez. (IMICORR) MEXICO D.F.
La Corrostón por Ptcaduras, definida como "el ataque localizado, que conlleva a la formación de hoyos en la superficie metálica", es una de las formas de Corrosión mas destructivas y dificiles de detectar, ya que el matertal y por lo tanto cualquier componente, puede fallar sin una pérdida de peso o volumen aparente.
;\ través del tiempo se han desarrollado diferentes técnicas para detectar la ocurrencia de picaduras en materiales metálicos, pero no es, sino hasta el desarrollo de las técmcas Electroquímicas, que es posible la determinación de la resistencia al picado de materiales de mterés industria l.
Desde la década de los 50's, todas las evaluaciones y caracterización de metales y aleaciones a nivel laboratorio, se realizan empleando métodos electroquímicos, los cuales establecen el comportamiento de Potencial Vs. Corriente<t .2l, permitiendo determinar parámetros tales como el Potenctal de Picado, el Potencial de re-pasivación y la htstérests de potencial, los cuales, si bien son importantes, son a su vez difíciles de traducir e interpretar en términos ingenieriles.
Temperatura Crítica de Picado.
El concepto de Temperatura Critica de Picado (CPT), fué desarrollado por Brtgham y Tozer en 1 973<3:, con la finalidad de comparar aceros inoxidables de resistencia variable al picado, uttlizando la temperatura como único parámetro de evaluación, mismo que es mas fácil de entender a nivel práctico.
El primer método experimental para la determinación del CPT, esta
1 29

T i,cNJCAS En:cmoQUÍMJCAs J>,\RA m. CosTROL Y Esn:DJo DI·. L.� CoRROSJós
basado en la técnica electroquímica de barndo potenciodinámico, por medio de la cual se determina una serie de curvas de polarización anódica, a illferentes temperaturas, en una solución de Cloruro de Soillo a pH neutro.
La temperatura crítica de picado se establece como la temperatura mas baja a la cual se genera p1cado, (abajo del potencial de transpasivación) y la ocurrencia de picado se cuantifica a partir del Potencial de Ruptura (Eb), el cual esta definido como el potencial en el cual la corriente es mayor a un valor límite, normalmente establecido como 100 �mperes/cm2.
En 1 980, Bernhardsson, et.al<4>., propusieron una modificación al método, la cual no requiere del uso de especímenes múltiples, por medio de la aplicación de un principio Potenciostático. El matenal inmerso en la soluciÓn de prueba se mantiene a un potencial electroquímico constante y la temperatura se va incrementando en
gradientes de unos cuantos grados. Después de cada cambio de temperatura se aplica el potencial pre-establecido y manteniéndose constante por al menos 1 5 rmnutos, se monitorea la corriente; s1 no se detecta la ocurrencia de picado, la temperatura se incrementa en
unos cuantos grados mas y el proceso se repite, hasta la obtenerse el potencial de ruptura.
Prueba Potenciodinámica
En esta prueba, se generan curvas de polarización anódica, a una velocidad de barrido de potencial de 20m V /mio, en el intervalo de 400 a 1 200mV. Vs. Electrodo de Calomel y utilizando como electrolito una solución 1M de NaCl.
Las curvas se obtienen en un intervalo de temperatura determinado, típicamente entre 1 5 y 40°C en gradientes de aproximadamente loC y de ellas se determina el potencial de ruptura con base en el criteriO de 100�/cm2•
Graficando el potencial de ruptura en función de la temperatura de prueba, se obtiene la temperatura crítica de picado, como aquella en la cual el potencial cae unos cuantos milivolts en el mtervalo entre el potencial transpasivo y el potencial de p1cado.


Prueba Potenciostática
Esta prueba implica un cambio constante de temperatura y se emplea una sola muestra. Inicialmente la soluCiÓn, que se llene a alrededor de 0°C, es deareada con Nttrógeno y el sistema se deja estabilizar a 0°C a Potencial de Corrosión.
Una vez que el sistema se estabiliza, a potencial abierto, se aplica un potencial de 700 m V Vs. SCE., de acuerdo a la literatura'51 y después de 1 minuto de la aplicactón del potencial, la temperatura se varia a una velocidad de 1°C/min, manteniéndose el potencial constante.
La corriente es momtoreada continuamente durante el barrido de temperatura y la CPT, defintda como aquella temperatura a la cual la cornente aumenta ráptdamente y rebasa los 100¡lA/ cm2
En ambas pruebas la presencia de picaduras debe ser comprobada uuhzando un microscopio estereoscópico a 20 aumentos.
En la Figura 1 , se muestran resultados típtcos de la prueba potenciodinámica, de la cual se establece un CPT de aproximadamente 30°C.
En la Figura 2, se muestran resultados de la prueba potenciostática, de donde se observa que la corriente obtenida rebasa los 100 )!A/ cm2 a una temperatura (CPT), de 28°C.
Reproducibilidad
La desviación estandard de los valores de CPT, obtenidos en numerosas pruebas, empleando los dos métodos evaluados y diferentes materiales, ha sido reportada, en promedio, como de 1 .7°C Y 3°C, para las pruebas potenciodtnámica y potencwstática respectlvamen te.
También se ha establecido, de los trabajos originales de Brigham y Tozcr y de Bernhardsson, et.al., que la prueba potenciodinámica determina una temperatura crítica de picado independiente del Potencial electroquímico, mientras que la prueba potenctOstática detcrmma una CPT dependiente del potencial electroquímico.

TÉCNICAS ELECTROQUÍMICAS PARA EL CoNTROL v EsTUDIO DE LA CoRROSIÓN
Pistorius y Burstein<6>, a partir de diversos estudios concluyeron que, el proceso de picado está dividido en dos etapas: una asociada a la formación de picaduras metaestables y otra asociada a la propagación de picaduras estables.
Una picadura metaestable es aquella que se desarrolla y crece, solamente por un corto periodo de tiempo antes de re-pasivarse (del orden de segundos o aún menos) y puede formarse a potenciales por abajo del potencial de picado (Ep), [el cual está asociado a la formación de picaduras estables].
1200 ... '"" "-._
"" •
1000 800 � � 600
w 400 200
o 20 22 24 26 28 30 32 34
Temperatura, oC
Figura 1 . Determinación de la TCP por el método potenciodinámico. Acero inoxidable 3 16. CPT 30°C.
250
200 ·e CJ 150 'a. E e( 100 o ..... CJ ·e 50 ·-
o
: 1 J rl _/ 5 1 0 1 5 20 25 30 �5 -50
Temperatura, oC
Figura 2. Determinación de la TCP por el método potenciostático. Acero inoxidable 316. CPT = 28°C.

Los eventos de rompimiento de películas pasivas (nucleaciÓn de picaduras metaestables) y re-pasivación se caracterizan por transientes de potencial, en la dirección actlva a circuito abierto o ba¡o una corriente anódica aplicada o por transientes de corriente anódica bajo un potencial aplicado.
El promedio de la densidad de corriente en estos casos ha sido relacionada de manera directamente proporcional al potencial aplicado, sugtriendo un control ohmico resultante de la de la resistencia asociada a la película pasivanteC7;(control por transporte de masa).
Por otro lado, una picadura estable se comporta como inestable cuando es de tamaño pequeño, sm embargo a medida que crecen, no re-pasivan, alcanzando un estado de crecimiento estable, el cual esta controlado por difusión. El transporte de los cauones que se producen por la dlsolue1Ón del metal es función de su solubilidad y de la formación de un anol1to saturado en la picadura y por lo tanto es también fuertemente dependiente de la temperaturaC8l.
En base al análisis antenor es posible establecer que solamente en casos en los cuales el tamaño de las picaduras sea pequeño, estás se comportaran como metaestables y entonces la temperatura crítica de picado será determinada prácticamente igual por cualesquiera de los dos métodos utilizados en este trabajo; sm embargo las condiciones de picadura estable deberían aparecer de alguna forma cuando el método potenc10stát1co es aplicado.
Para corroborar la condición establecida, se realizaron experimentos adicionales, en los cuales se empleó el método potenciostático pero Variando el potencial aplicado en el intervalo de 300 a 900 m V. Como puede observarse en la Figura 3, existen dos regiones claramente definidas, una por aba¡o de los 550-600 m V en la cual la CPT es dependiente del potencial y otra por arriba de dicho valor en la cual
CPT es mdependiente de potencial, caractenzando de esta forma etapas de picadura metaestable y picadura estable.
1 ? ?

TÉCNICAs ELECTROQuíMICAS PARA EL CoNTROL v EsTUDIO De LA CoRROSióN
0+---�----�----�--�-----r----�--�r---� 300 400 500 600 700 800 900 1 000 1 100
E, mV
Figura 3. Influencia del potencial electroquímico en la CPT evaluada potenciostáticamente. Acero inoxidable 3 1 6 .
Conclusiones
• Existen dos Temperaturas Críticas de Picado (CPT), una que es dependiente del potencial electroquímico y está asociada al proceso de nucleación de picaduras metaestables y otra, independiente del potencial, asociada al proceso de propagación de picaduras estables.
• La Temperatura Crítica de Picado (CPT), para picaduras estables, (independiente del potencial), obtenida por los métodos potenciostático y Potenciodinámico es prácticamente igual.
• Para la determinación de la CPT, independiente del potencial, el método potenciostático, presenta mejores alternativas por ser mas rápido y económico, ya que requiere de una sola muestra para la evaluación.
• El método a utilizar debe ser cuidadosamente seleccionado, en función de las etapas de picado que se deseen evaluar.
134

Bibliografía
1 .- M. A, Streicher.: Journal oJ the Electrochemical S ocie (y, 103, (1953), p.375.
2.- M. Pourbaix, et. al.: Corrosion Science 3, (1 963), p.239.
3.- R. J, Brigham y E. W, Tozier.: Corrosion 29, (1973), p.33.
4.- S, Bcrnhardsson, R, Mcllstrom y B. Brox.: Proc. Corrosion'80. N1\CE, Chicago, (1980). Paper 85.
5.- R. Qvarfort.: Corroszotz Scunce 28, 2, (1988), p. 1 35.
6,- P. C. Pistorius y G. T, Burstein.: Corrosion Science 33, 12, (1992), p. 1 885.
7.- G. S, Frenkel, et.al.: Corrosion 43, (1987), p.429.
8.- T. J. IIakkarainen, O. Var¡onen y A. M. Mahiout.: Proc. 12'h Scandmav1an Corrosion Congress & Eurocorr'92. (NKM12), Espoo, (1992), p.7 1 .
1 35


TÉCNICAS ELETROQUÍMICAS APLICADAS AL ESTUDIO DE LA CoRROSióN EN LA INDUSTRIA NucLEAR
5.3 CORROSIÓN POR PICADURAS Y POR HENDIDURAS
Carlos Arganis.
Instituto Nacional Investigaciones Nucleares
Siendo ambas, tipos de corrosión localizada, requieren del empleo de técnicas específicas. Una forma muy simple de evaluar la suscep tibilidad a las picaduras, es mediante una curva simple de Polanzación anódica, en donde se puede determinar el potencial de picaduras E , potencial en el cual la densidad de corriente se incrementa despJ'és de una zona pasiva, como se muestra en la Figura 1) . Este E , marca el potencial por arriba del cual las picaduras nuclean y s� propagan.
Ep
E Ecorr
Log i Figura 1. Curva de Polarización Anód1ca
La dtferencia entre el potencial de corrosión E y el E , determina� , . corr p ra la facthdad con que el matenal en el medJ.o acuoso dado, tenderán a presentar este fenómeno.
1 37

Ti'.cSIC.\S Eu.r.TROQt;i\1IC.\S 1'.\R.\ EL Co:-:TROL Y EsTUDIO DE 1 A CoRROSIÓN
Sin embargo, la corros1on por hendidura se propaga con el mismo mecanismo que el de una picaduras ya nucleada, por lo que se puede considerar como tal. De este modo, se requiere saber cuando una p1cadura nucleada previamente o una hendidura, tenderá a activarse y mantendrá su propagación, por lo que se requiere un nuevo parámetro y una nueva técnica.
Polarización Cíclica. En esta técmca se aplica un barndo de potencial anódico, desde el
potencial de corrosión, Ecorr' hasta el potencial al cual se alcanza el valor de corriente de 5 mA/ cm2, llamada corriente de reversa i . A
rcv
este potencial se le llama potenctal de Reversa. Ere,· En este punto el potencial se invierte, barriéndose en reversa (dirección catódica), hasta cruzar la curva de anódica o hasta que la corriente tienda a cero. A este potencial se le llama Potencial de protección E como se mues·
prot. tra en la Figura 2.
E
E o
Log i
Propagación ....¡ .. -···-�·
1 Epp 1 p · · . . _.;... roteccton 1
Activación
l n�lllnidad '
rrev
Figura 2. Curva de PolanzaCIÓn Cíclica.
Ahora tenemos un nuevo mtervalo de potenciales relacionados con la propagaciÓn. A partlr del potencial de picaduras EP, hasta el E" .
tenemos la zona donde las picaduras nuclean y se propagan. Sin embargo entre el potencial E y el E , tenemos una zona, donde
p prot
138

nuevas picaduras no nuclean, pero las que esten presentes pueden propagarse. Por debajo del Error' las picaduras no nuclean ni crecen.
Al aphcar el barndo en reversa pueden presentarse tres casos. El Primero cuando el E cruza la curva de ida en esta zona pasiva y pro1 por lo tanto la picadura iniciada se deuene por la pasivación de la misma, o sea que se reconstruye la capa pasiva (En este caso se podría hablar de un potencial de pasivación). El segundo caso, cuando la picadura no se deuene, si no hasta un valor cercano al Ecorr, o menor, en tal caso, se puede decir que la picadura sólo se detiene al regresar al equilibrio o cuando el material es proteg¡do catódicamente .
. En este caso estamos hablando de una susceptibilidad muy grande a la corrosión localizada, como es el caso de la corrosión en Hendiduras, Picaduras o Agrietamiento por corrosión ba¡o esfuerzo.
Cuando la curva regresa por el mismo cammo o por un valor de corriente menor, el material no presenta tendencia a la corrosión localizada, es decir que el incremento de corriente no se debe a la corrosión localizada, si no a alguna otra reacción anódica, ya que el área permanece constante.
Esta técnica, si se aplica a diversos valores de concentrac10nes de agentes agresivos, por ejemplo cloruros, nos permite obtener diagramas predictlvos de comportam1ento de corrosión localizada, en donde tendremos zonas de nucleación y propagación, zonas de propagación y zonas de protección e inmunidad.
Choi y \\fas 1 han dado tratamientos matemáucos para obtener un parámetro de velocidad de crecimiento de picaduras PVCP y la velocidad de creclffiÍento promedio VCP basados en las cargas Ql desde E hasta E y Q2, desde E hasta E como se muestra en la p rcv re\· prot Figura 3. Así
1 39

Tt':cstC:.\S Eu·:r.mOQL'i\ttC.\S P.\R.\ El. CosTROI. Y EsTUDIO DE LA CORROSIÓ-.:
E
Ecorr
Log i Figura 3. Dtagrama de Cargas normalizadas propuesto por Choi y Was .<'l
e B (E E )·2/3 ·2/3 1/3 V P= o pe- p k d
Donde Bo es una constante, k es la relación ancho w profundidad h (h/w) de las picaduras y d la densidad de picaduras. De tal forma que:
PVCP=VCP(Q2-Q1 /Q1) . . . (5)
O sustituyendo 4 en 5.
PGR = s[ Q2; Qi ]rEpr - Ep j"'
Otro parámetro que se puede obtener es el pH crítico de despas1Vación o dpH, el cual es el valor del pH, por abajo del cual la curva de polarización del sistema en estudio, presenta la zona acth·o pasiva. Este valor de pfl, define el valor al cual la película pasiva vuelve inestable. Entre más alto es este valor, mayor suscept1 a las picaduras tendrá el material.
1 40

Técnicas electroquímicas aplicadas en la industria nuclear.
La mdustria nuclear, al igual que la industria convencional, sufre de problemas de corrosión. La Figura 4, muestra los principales mecanismos de daño presentes en los reactores de agua en ebullición.
Corrosión Localizada
0%
Corrosión Golpe de Ariete Generalizada 3%
Erosión 34%
scc 22%
Figura 4. Principales mecamsmos de daño en reactores nucleares tipo B\X'R, de
1993 a 1999 Fuente O lEA
En el caso de los reactores de agua en ebullición, con enfriamiento de agua de mar, como el caso de la Central Laguna Verde se puede hablar de dos medios diferentes, Figura S.
En el llamado circuito primario y en la vasija del reactor, el medio es agua desrruneralizada con una conductividad cercana a 0.1 J.!S/ cm a 288°C y 8 Mpa de presión. En este sistema de alta reststencia óhmica, pocas técnicas electroquímicas pueden funcionar y se requit;ren diseños especiales de electrodos de referencta y el uso de aislantes eléc
del tipo cerámico. Una de las técnicas electroquímicas más es el uso de Ruido Electroquímico para detectar el
nto por corrosión bajo esfuerzo.
los sistemas secundarios, como el sistema de mtercambto de cay el condensador, que maneJan agua de Mar, el t1po de corrostón
que se presenta en medios marinos y las técnicas electroquímicas · pueden aplicarse. De hecho por encontrarse en la
del golfo de México, los problemas de corrosión en las estruccivil y soporteria son iguales a las que sufre la industria conven
en esta región.
141
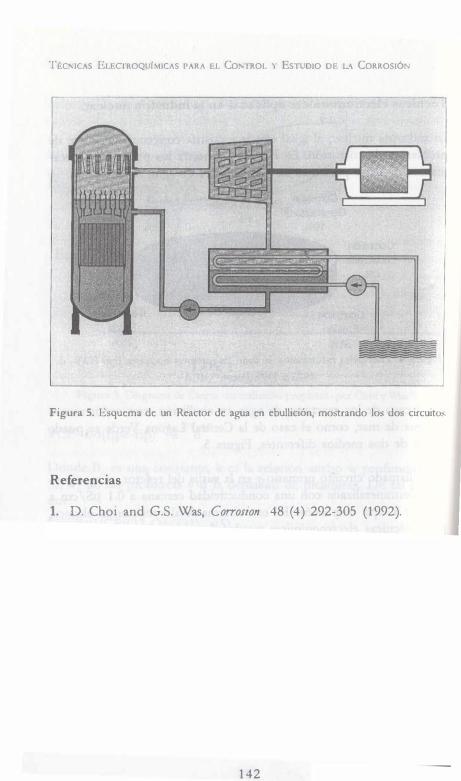
Tt':c:-.:IC:As ELECTROQUI\IICAS PARA I'.L Co:-.:TROL Y Esn.:DIO DE t_\ CoRROSió:-.:
Figura 5. Esquema de un RL':tctor de agua en ebullición, mostrando los dos circuito;
Referencias
1. D. Choi and G.S. Was, Corro.rion 48 ( 4) 292-305 (1992).
1 42

6. APLICACIÓN DE LA TÉNICA DE ESPECTROSCOPiA DE IMPEDANCIA ELECTROQUÍMICA (EIS) EN EL ESTUDIO DE LA CORRO
SIÓN DEL ACERO DE REFUERZO EMBEBIDO EN CONCRETO
T. Pérez. Programa de Corrosión del Golfo de México, Universidad Autó
noma de Campeche. Av. Agustín Melgar S/N, Ciudad Universitaria, Campeche, Cam., C. P. 24030. Tel. 01 (9) 8 1 1 1 7 60 ; fax: 01
(9) 8 1 1 22 53. Correo electrónico: [email protected]:un.mx
RESUMEN
Se presentan la técnica de EIS como una herramienta para el estudio del proceso de corrosión en concreto, con una revisión breve de los fundamentos de la técnica y diversos estudios con propuestas de modelos de circuitos eléctricos e interpretación de resultados. Son mostradas las ventajas del uso de EIS para la estimación del grado de corrosiÓn así como para proponer modelos y mecanismos del proceso de corrosión del acero de refuerzo embebido en concreto.
INTRODUCCIÓN
La preservación de la infraestructura civil como puentes, carreteras, muelles, etc. es de vital 1mportanc1a económica y social, puesto que brinda servicio y segundad para el desarrollo de las sociedades. En las últimas décadas, el material de construcción más utilizado en obras civiles es el concreto reforzado, cuyas propiedades mecántcas Y químtcas le confieren una durabilidad prolongada. Un concreto
hbre de contaminación, tiene un medio alcalino que produuna capa superficial sobre el acero y lo mant1ene pas1vo, con una
-�•-•u••u de disolución metálica muy pequeña. Sm embargo, se han IDOrt<>d ..... casos de detenoro de concreto debido a la corrosiÓn de la
embebida, lo que ha propiciado el estudio de las causas que y favorecen el avance del ataque al refuerzo de acero. Se ha
que la penetración de cloruro a través del concreto por ocación de la estructura en zona marina o el uso de sales
ntes, así como la reducción del pii natural de la disolución
1 41

'J'!�C:,IIC:AS ELECTROQUh11CAS PARA FL CO�OL Y Esn.:DIO DE LA CORROSIÓN
poro del concreto por su interacción con el dióxido de carbono son las causas de mayor nnportancia para la despasivación de la varilla y su consecuente corrosión. Una vez que se ha iniciado, los productos de reacción son más volummosos que el acero, generando esfuerzos de tensión desde el interior y postenores gnetas y desprendimientos, que comprometen la integridad de la estructura.
Para la inspecciÓn y evaluación del grado de deterioro de estructuras de concreto, es fundamental utilizar técnicas no destructivas, a fin de evitar daños. Esto ha dado la pauta para proponer el uso de técnicas electroquímicas en la supervisión del estado de corrosión del acero de refuerzo. La norma ASTM -C-876-91 1 establece la medición del potencial de media celda como un parámetro de estimación de la probabilidad de ocurrencia de corrosión. Aunque se ha cuestionado su validez, sigue s1endo empleada para mediciones en condiciones reales.
En investigaciones de laboratorio se han usado técmcas de corriente directa como polarización lineal, voltametría cíclica, pulsos coulostático y galvanostático. Una técnica que utiliza señales de corriente alterna es la conocida como espectroscopia de impedanCia electroquínúca (EIS, por sus sigla en inglés), que ha mostrado excelentes resultados en el análisis de los componentes del concreto armado, permitiendo aproximar mecanismos de reacciÓn sin alterar prácticamente las condiciones del sistema. En este documento se descnben de manera breve las características de la técnica de EIS, mostrando resultados que permiten exhibir su potencialidad en el estudio la corrosión del acero de refuerzo embebido en concreto.
FUNDAMENTOS DE ESPECTROSCOPiA DE IMPEDANCIA ELECTROQUÍMICA.
El uso de la 1mpedanc1a en cornente alterna ya se conocía y apl.tcaba en el área de lrlgeruería eléctnca. Se comenzó a utilizar para el estuCI.io y caracte· rizac1ón de materiales, hasta la década de 1970, y alcanzó gran aceptación por su característica de señales en función de la frecuencta, lo que permite
analizar un sistema y sus componentes.
La causa del prácticamente nulo empleo de la técruca, era lo tedioso de
1 44

recolecCIÓn y tratarruenro de la 111formación, ya que era de manera manual y se
trazaban las gráficas en papel polar. Este problema fue superado con el avance de
los sistemas computarizados que facilitan la captura y orgaruzaoón de los datos
obterudos.
La corriente alterna tiene como una ventaJa ser funciÓn del uempo y de la
frecuencia; cuando se apl..tcan frecuencias de orden de kl [z, la onda de tipo
scn01dal es tan ráp1da que Circula a través de un med10 como SI estuvtese en
corto circulto. Ilustrando el modelo de la Interfase electrificada, se llenen
d1ferentes componentes que se asemejan a elementos eléctricos como
n.:s1stores, capac1tores e mductores (figura 1).
Metal x1 x2
� Rct
Figura 1.- S1mtliwd de la interfase electrificada con un c1rcu1to eléctrico et(uivalcntc
este caso, el más sunple, la zona compacta ttene similitud con un ..:T .. ... ,·,vl (Cdl); el electrohto (zona difusa) una resistencia al paso de
y la resistencia característica de un metal a reacciones se con la resistencia a la transferencia de carga(Rct) .
�ecesario para el anáhs1s de un diagrama generado con datos de te alterna, conocer las respuestas de un elemento, sabiendo
una unpedanc1a (Z) es una oposiciÓn al paso de la señal eléctrt-
145

Tl'.cNIC.:\s Eu:c rROQuiMICAs PARA '''- Co:--;TRoL Y EsTUDIO DE 1-' CoRROSJós
ca. La analogía con la corriente directa se da como un límite en que la frecuencia es cero.
Esta es la ley de Ohm:
R = V/1
Z = V(w)/I(w)
Cuando se aplica la señal de voltaje en cornente alterna, se obtiene una respuesta en corriente de acuerdo a las ecuactones:
V = Vmax sen wt
I = Imax sen (wt+8)
donde 8 es el ángulo de la fase y es muy útil para aproximar las características de los elementos eléctricos a diferentes frecuencias.
De esta manera, la señal circula a través de la resistencia del electroluo (Re) y del capacitar Cdl, y se registrará la Rct (figura 2).
""Cdl 1 �'----
R..ct Figura 2.- Circuito de una interfase ckctnficada al p<ISO de
una señal senoidal de alta frecuencia.
El ángulo de fase toma valores cercanos a cero, e mdtca que Z (u11-pedancia) medtda corresponde a una resistencia; al dtsminuir la frecuencia, el capacttor tarda más en permitir el flujo de corriente Y tendrá una caída de potencial. Si esa caída de potertcial es menor qu: la Rct (resistencia a la transferencia de carga), la corriente circulara por el capacitar iniciándose un decremento del ángulo de fase. Al continuar disminuyendo la frecuencia, la pérdida de potencial sera
mayor en el capacitar y llegará el momento en que compita con ]A Rct; a estos valores de frecuencia, el ángulo de fase alcanza un máXÍ· mo y toma valores menos negativos.
146

Para otras frecuencias, la resistencia es sustituida por la expresión apropiada de la reactanc1a, X, del elemento eléctriCO se tiene:
vplcO = i pico X
La reactancia de un capacitar o de un inductor puede ser expresado de varias formas, por ejemplo:
Xc = 1 /ju-C para un capacitar C XL = jwL para un inductor L
donde w es la frecuencia angular y j = "-1 . Esta notación permite la representación de una reactancia o la unpedancia de una combinación de éstas como un vector en el plano real-imaginario en la forma de un diagrama de Argand. De este modo una impedancia puede ser defimda Indicando SU magnitud 1 Z 1 y el ángulo e especificando las magnitudes de los componentes real e imaginario, como se muestra en la figura 3. Se pueden escribir las siguientes equivalencias:
z' = z cos e y z " = z sen e
Z = Z' + ¡Z»
Las expres10nes contienen 111 y por lo tanto la respuesta del circUlto depende de la frecuencia de la señal. Esta dependencia con la frecuencia está representada en los diagramas de Nyquist. De este modo, la respuesta a cada frecuencia aparece como un componente real e imagmario. Un ejemplo es el de un circuito de una resistencia Y un capacitar en paralelo. Para el caso de sistemas electroquímicos se ha encontrado un circuito que parece representar a sistemas cuya corrosión está controlada por activaciÓn, esto se muestra en la figura 4. En este caso la resistencia R tu , corresponde a la resistencia que presenta una disolución de io'h�s. La resistencia Rct y Cd1 ltpresentan a la interfase bajo corrosión, donde el pnmer térrruno
la resistencia de transferencia de carga y el otro término, el pa�iuvo corresponde a la doble capa formada en la interfase. El
tno Rct está directamente relacionado con la velocidad de y es mdicativo de la velocidad de transferencia de carga .
. un término que tiene su análogo con la Rp de la técnica de teneta de polarizaciÓn.
147

Tf.:csiC.Is ELECrROQL·huc.ls P. IR\ H. Cos-1 ROl \' E�Tt.:mo DE 1 .\ CoRROSIÓ!'<
z·
Figura 3.- Diagrama de s\rgand
R c t
Figura 4.- CircuitO para un Sistema
controlado por acuvaCJÓn.
Lo antenor tiene validez solo si se considera R" y Cu1 como elemcn tos lineales. La respuesta del circutto de Randles en el diagrama tle Nyquist puede verse en la figura 5. En altas frecuencias, el capacttor aparece en cortocircuito, es decir, conduce libremente obteméndosc solo la contribución de la R h de la soluciÓn. Es así como se mide la
o m
resistividad de un electrolito. A frecuencias bajas el capacttor actúa como si estuviera abierto y se obtiene R 1 y R . La capacidad de O\ m te la doble capa eléctrica corresponde al térnuno C y puede ser calcu-lada considerando el valor de la frecuencia por el punto más alto del semicírculo. Todo lo expuesto antenormente tiene sentido para
sistemas controlados por activación; para sistemas controlados por difusiÓn se ha propuesto un elemento llamado Warburg, \'</, cuya impedancia descnbe la resistencia que ofrece el sistema por la dlfu· sión de especies a la mterfase. Este es un térnuno que se mantficsta a bajas frecuencias.
El circutto equivalente que mcluye W se muestra en la figura 6 Y el dtagrama de Nyquist de este elemento se representa como una línea recta a 45°, como se muestra en la figura 7.
La técntca de impedancia es una herramienta poderosa que ha trado ser útil en sistemas donde otras técnicas han tenido
1 4R

-jZ
1 Rohm
= --
1 z Rtc
Fag·ur.t S.· Di:-�grama Uc Nv�ut\t un '''tema
conlfolado por act1\·ac1• '"
-jZ"
e
Rtc
Figura 6.· (¡rcultn �qUivalentc cuanOn c�tá prcc;cntc un proceso de c.lafus1ún.
Z'
Figura 7.- Dtagrama de Nyquisr para un sistema controlado por difustón.
Con estos prmctpios y análisis de respuesta a una señal de corncnte alterna, tnvestigadores de diversas partes del mundo, han elaborado modelos para expl.tcar el fenómeno de corrosión en diferentes sistemas, como en recubrtmtentos orgánicos (ptnturasY 3 y el que se trata en este documento: corrosión en concreto�· 5•6·
John y colaboradores7 realizaron estudtos en muestras de concreto lrmado en agua de mar y agua destilada. Con esta expertmentaClÓn de cspectroscopía de impedancia electroquírruca mostraron las ven
y potencialidades de la técnica con respecto al uso de corriente Proponen un ctrcuito equivalente que asigna elementos eléc
como reststencias y capacitares a elementos físicos del concre-como lo son las interfases óxido-concreto y acero-concreto. Indi
ta diferencta en los dtagramas para casos en que se tenga la activa y pastva medtante la tnterpretactón de los dtagramas
U 1St.
1 49

TI�C:-;Ir_\S EtECIROQl:i\IIC.\S r.\R.\ El. Co:-;TROL Y Esn:Dio DE 1 A CoRROSió:-;
Hope et aJ.S en sus estudios encuentran resultados similares y mencionan la aproxtmactón a un mecanismo de difusión en ba¡as frecuencias, en que aparece una región lineal en el diagrama de Nyquist, que forma un ángulo de 45°. Wenger et al.9, Sagoe-Crentsil et al.10, Avila-Mendoza et al. 1 1 , Andrade et al.12, Matsuoka et al.U han usado muestras de mortero para sus estudios, como aproximación a piezas de concreto, probando acle más geometrías cilíndricas y prismáticas.
Los intervalos de frecuencias utilizados van desde 0.1 mHz hasta 10 kHz9, encontrándose que de 100 Hz a 10 mHz son las frecuenctas en las cuales es posible caracterizar a la interfase acero-concreto14• 4
El circuito propuesto por John et ai.7 (figura 8) ha sido modificado por diferentes investigadores que proponen un mayor ajuste a la tn formación experimental, aunque se continúa considerando los elementos básicos lineales:
' resistencias y capacitorcs acomodados de
distinta forma.
�� Rf Rct Difusión deWalburg
Figura 8.- :'\1odclo propuesto por John ct aJ.1
Wenger et al.9 proponen otra constante de ttempo (RoCa), como respuesta de una reacción intermedia o absorción superficial de alguna especie, esto no es observado en sistemas pasivos (figura 9).
Cdl
Figura 9.- Modelo de Wcngcr ct al 9
Newton y Sykes1s proponen un circuito (figura 10)_, pero con relajación en alta frecuencia en el concreto (ReCe), y men que no es necesarto el término de difusión durante la pastvidad.
1 50

::J-Rct Difusión
de Wa.lburg Figura 10.- Modelo de Ncwton y Sykcs 11•
Sagoe-Crentsil y colaboradores10 hacen patente que a elevadas frecuencias la matriz de concreto presenta una constante de tiempo y la incorporan a su modelo; lo nombran: resistencia y capacitancia de la matriz de concreto (figura 1 1).
CHF RHF
Rct Figura 11.- Modelo de Sagoc-Crcntsil ct al 10
Feliú et al.11' ocupan una combinación en paralelo diferente, incorporando efectos dieléctricos en alta frecuencia en la masa de concreto, como lo muestra al figura 12.
R.ct Figura 12.- 1\loddo de l·cliú ct al 16.
eléctncos propuestos, se ajustan mediante cálculos s, buscando aproximar los elementos eléctricos asigna
por el modelo a los diferentes elementos del concreto. En este se han desarrollado metodologías para el análisis de circui
entes. Un paquete comercial que procesa información tal y ajusta valores para la Rtc, fue elaborado por Boukamp
1 51

'l'Er.:-;IC:IS EI.I.CIROQU\11(:\s P.IR.I El. Co:-;rROL Y Es1cmo m: 1.1 CoRROSió:-;
(EQIVCRT)17, que ajusta circuitos propuestos y es ampliamente utilizado. Combinó con el método Monte Carlo y evaluó los valores de los componentes de un circmto equivalente de resultados experimentales. Otro paquete mas reciente es el Zview1S, que realiza ajustes y simulac1o nes de circuitos eléctricos y es muy sencillo de uttlizar.
También han surgido controversias con respecto al uso de paquetes de computación opcionales. Vilche19 menciOnÓ que cada centro debe desarrollar su prop1o método de cálculo como requisito de for mación académica e investigación. Hace notar la posibilidad de graves errores en la interpretaciÓn de circuitos equivalentes.
McDonald20 resalta la importancia de tocar aspectos electroquímicos y mecanísticos, y lo contraproducente que puede llegar a ser simu lar sm estudtar previamente un proceso de corrosiÓn.
Como ventajas de EIS, son de mencionar la capacidad para eliminar errores de medición deb1dos a la resistencia del electrolito e información para aproximar mecanismos de reacción, además de ser no destructiva y factible de usar para realizar seguimientos en el uempo. Las desventajas principales, son el alto costo del equipo ) el tiempo relativamente largo de experimentaciÓn.
ANÁLISIS DE ENSAYOS REALIZADOS EN MUESTRAS DE CONCRETO.
Análtsis de los dtagramas de EIS.
Para la interpretación de los diagramas de EIS, se han propuesto a
los componentes del concreto armado como un circuito eléctriCO equivalente, en el cual se representan los diferentes integrantes del
sistema como elementos eléctricos. \Venger et al.9, Sagoe-Crentsil el al 10, Hachini et al.21 y Dhouib1- l lach1nt et al.22 , plantean una re ¡.,resentación como la esquematizada en la figura 13. Se definen rres regiones: la matriz de concreto, la capa porosa (que es una películs
de Ca(OH)2 depositada sobre la superficie metálica) y la mter
acero-capa porosa.
1 52

Película interfacial {Ccp}
Doble capa � capacitiva Matriz de concreto
{ Cdl,� ¡
! Rcp Ro Rtc
Figura 13.- Componentes del concreto armado.
En el caso del sistema mostrado en la figura 13, la facubtlidad de suministrar señales de tipo seno1dal dentro de un tntervalo de frecuencias de vanos órdenes de magn1tud, hace posible 1denuficar las diferentes zonas en regtones de frecuencia característicos. En altas frecuencias, mayores de 1 O ki Iz, la respuesta del sistema se atribuye a propiedades dicléctncas del concreto111•12• La lectura de la resistencia del concreto, RO, se determma en el cambio de pendiente que se presenta cercano a 10 kiiz de los diagramas de Nyquist, Bode-módulo de Impedancia y Bode-ángulo de fase111•1'. Para frecuencias intermedias, el análisis del sistema corresponde a la película porosa. Wengcr et al.9, señalan como frecuencias intermedias de 10 kiiz a
Hz, mientras que Gu et al.23 anotan de 10 ki Iz a 1 00 Hz y Andrade 2l no precisan valores, solo menciona de kJ Iz a unas decenas de La %ona de bajas frecuencias atañe a los procesos de transferen-
de carga y de difusión la interfase acero-productos de corrosiÓn; se llevan a cabo las reacctones de corrosiÓn del acero embe
De acuerdo a los autores menctonados9.2'. el intervalo de bajas ·
va del orden de 10 I Iz hasta 1 mHz. 1\ndradc ct al.'' barndos hasta 50 f.!F proponiendo el análisis como la rcac
dl' reducciÓn de hcmatita a magnetita.
1 5 3

'J'U;:-;1c.�s ELEC.TROQUÍ\m��s P.\RA H. CosTROL Y ESTUDIO DE u CoRROSIÓ:-J
El circuito eléctrico equivalente para las zonas del concreto se muestra en la figura 14.
Cdl Ccp
Ro w
Rtc Rcp Figura 14.- Circuito eléctrico equivalente de los componentes
del concreto armado.
Con el modelo de circuito eléctrico asociado a los componentes dd concreto armado, se hacen los análtsis de los diagramas de EIS expt.:nmentales al aplicar señales de potencial senoidal en un intervalo de frecuencias de varios órdenes de magnitud.
En el diagrama de Nyquist, se describen semicírculos que corrt.:sponden a un arreglo de resistencia y capacitar en paralelo (tambtén llamado ctrcuito RC o constante de t1empo).
En el máximo del semicírculo se obttene la mayor reactancta capacitiva de la constante de tiempo. La extrapolaciÓn de los extremos del semicírculo haCia el eje real corresponden a valores de resiStencias. El ángulo de fase en el diagrama Bode-ángulo de fase descrl· be curvas variantes con la frecuencia, cuyos valores mínimos corres· ponden a la reactancia capacitiva y los mayores a las resistencias de las constantes de tiempo del sistema. Para el diagrama de Bode-mÓ· dulo de impedancta, se observa una recta de pendiente negatt\'a en
los intervalos de frecuencia de predominancia de la reactancta
capacitiva y lineas que tienden a la horizontal para las frecuenctaS
en que la respuesta es resistiva.
1 54

Las figuras 1 5 a 29 exhiben el seguun.iento del estado de corrosiÓn de la varilla medJante los ensayos de EIS, representados por dJagramas de Nyquist (raíz real vs raíz imaginaria) y de Bode (log de la frecuencia vs módulo de impedancia y log de la frecuencia vs ángulo de fase). Son resultados de ensayos realizados a muestras cilindricas de concreto coladas con 3% en peso de cloruro de sodJo en el agua de amasado25 durante el periodo de curado y exposición en tres diferentes condic10nes: ciclos de inmerstón en agua de mar-tntemperte, inmersión permanente en agua de mar e tntempertsmo.
Curado. El segutm1ento durante esta etapa expertmental se presenta en las figuras 1 5 a 20. Se notan cambios en la forma y magmtud de los parámetros de los diagramas de Nyquist y de Bode durante al avance del proceso de curado.
El análisis de las reacciones de hidrataciÓn del cemento que se anotan a continuación son un apoyo para la explicación de) comportamiento de los resultados electroquímicos.
Componentes
2(3Cao�S10)+61 rp Silicato tndlcico+agua
2(2Ca'Si0)+41I,0 S1licato d1cálico+agua
4Cao• ,\1203* Fc203+ 101 120+
Productos de h1dratae�ón
---+ 3Cao�Si0,*3I rp+3Ca(OII)2 +-- Gel de tobcrmortta+h1dróx1do de
Calc1o
---+ 3Ca'2Si02'311,0+ 3Ca(Oll)2 +-- Gel de tobermonta+hidróxido de
Calc1o
2Ca(OI I)2 ---+ +-uminato tctracálcico+agua+
6Ca0 · Al20/ Fe2C\ 121 120 Fcrnalumtnato hcxacálc1co
h1dratado hidróxido de calcio
3CaO• Al201 + 1 OII20 + Aluminato tridlcico+agua +
h1dróxido de Calcio
3CaO• ;\l2C\+ 1 0 1 120+ CaS0(21120
inato tncálcico+agua+ycso
1 5 5
3CaO· Al ,O,· Ca(OI 1)2* 1 21 120 ;\luminato trtcálcico hidratado
3Cao• AI201• Caso; 12 1 120 l\!onosulfoalummato cálc1co

TEQ>IC:.\S EL L:c;rRoc�ehHc,,s 1'.\R\ u. Co:-;IROl. Y Es1uDIO t>l' 1 ' CoRROSió:-:
Dado que las varillas fueron decapadas, se encontraban activas, es decir, sin productos de corrosión superficiales al momento de colocarlas en los moldes y vaciar el concreto. En el diagrama de la figura 1 5, que corresponde al ensayo llevado a cabo inmediatamente después de retirar al cilindro del molde y marca el princip10 del segUImiento, se observan en el diagrama de Nyquist dos semicírculos, con tendencia a cerrarse a frecuencias menores de 100 mi Iz; el diagrama de log frecuencia vs 1 Z 1 señala una tendencia a formar una honzontal en ba¡as frecuencias y el diagrama de log frecuenCia n
ángulo de fase (O) adquiere valores cercanos a 10 grados en el mtervalo de frecuencias menores de 100 mHz. Lo antenor es mdicto de un comportamiento resistivo en la región de la interfase por lo cual se afirma <.¡ue las reacciones de hidrataciÓn del cemento durante el tiempo de fraguado no generan las condiciones suficientes para productr la pasivactón de la vanlla.
La sccuencta de dtagramas 15 a 19 exhibe el comportamiento de la vanlla embebida en los cilindros de concreto en el transcurso del curado. Se hace notar en el diagrama de Nyquist cómo se inicia con la figura de un semiCÍrculo que tiende a cerrarse para tr abriendo con el avance del tiempo, lo que es un indicio de que la varilla se va pastvando.
El dtagrama de raíz real (Z ') contra raíz imaginaria (Z' ') o dtagra
ma de Nyquist es el más utilizado para reportar los resultados de los ensayos de EIS. Sm embargo, los dtagramas de Bode son muy uules para complementar la información de los experimentos, ya que se cuentan con mayores elementos para aproximar lo que sucede en el sistema acero-concreto.
En cuanto al diagrama Bode fase al intcio se ttene un ángulo de fase núntmo de cercano a 25° en la regtón de baja frecuencia (de 100mi!z a 1 miiz), que es reportada como la región de la mterfase metal·
concreto \.'\ mcrementándose hasta llegar a 10°. Esta curvatura Y el ángulo de fase que tiende a cero mdican un comportamiento
resistivo en la interfase metal-concreto, permitiendo proponer una varilla en estado acttvo. Con el avance del curado el ángulo de fase dismmuye en bajas frecuencias, hasta llegar a valores cercanos
a -70°. Se advierte la aparición de la curvatura descendente en 5
1 56

ángulo de fase en bajas frecuencias que señala un cambio en la condición de la mterfase, en la que se minimiza el comportamiento eststivo y predomina el capacitivo. Este cambio corresponde a la ansición acttvo-pastva de la varilla que forma la película pastva so-re la superficte, reduciendo el intercambio de carga y por lo tanto cinética del proceso de corrosiÓn. En la figura 20, se presenta la áfica de EIS resumida en la que se sobreponen los resultados de O 10 días de curado. Se aprecia claramente el mcremento de la res-
uesta capacitiva con el lapso de prueba en el diagrama de Nyqu1st n el avance del tier.1po. El ángulo de fase disminuye, indicando la
preponderancia de la reactancia capacitiva, y, por lo tanto, la pasivación de la varilla.
Jafar et al.�¡, realizaron un segmmtento similar, con mediciones de EIS durante la etapa de curado a probetas preparadas con y sin NaCI efl el agua de amasado. Los resultados que presentan son semeJantes a los obtenidos en el presente trabajo con diagramas que muestran '*tiaciones en la forma y magnitud de los diagramas con el avance cltl periodo experimental. Determinaron en su análisis un tiempo • curado necesario para pasivar la varilla de 6 días, apuntando que » nr•••:�•·ncta de cloruros retarda la formación de la película pasivante
explican en el hecho de la competencia de reacciones de forma-IH1 .. r,,ntura de película por presencia de cloruros.
seguimiento resalta la valiosa informaciÓn que se obtiene con la de EIS, dado que permite aproximar el estado de corrosión
guarda un elemento metálico embebido en concreto. Se advierposibilidad de conocer el tiempo de curado en el cual se alcanza
d del refuerzo de acero, lo cual es importante desde el .de vista práctico, porque asegura que el refuerzo cuenta con
química que le confiere el concreto, lo que favorecerá ades contra la corrosión.
Los diagramas 21 a 27, muestran los cambios (¡ue se duran te la exposición a ciclos de inmersión en agua
intempene de las mismas muestras de concreto armado. cómo en los diagramas de Nyqlllst el arco capacttivo que
a 259 días de expostctón modifica su forma al llegar a 280 este lapso el ángulo de fase a frecuencias menores de 1 00
1 57

Ti'C.SI<. \S ELECil\OQl!L\IICAS 1'.\R.\ 11. Co"'rROL Y EsTUDIO Dll t.\ CoRROSió:-:
mHz tiende a ser muy negativo, cercano a -80°, lo que señala un comportamiento capacitivo en la interfase metal-concreto y que la varilla está pastvada. El diagrama del dia 294 muestra en la zona de frecuencias de la interfase ángulos de fase menores de -20°, que indica la predominancia de un comportamiento resis tivo sobre uno capacitivo. En el día 308 el diagrama de Bode-fase muestra un decremento en el ángulo de fase a bajas frecuencias, llegando hasta . 30°, esbozándose una curvatura en esa zona. Para los días 280 al 308, en el diagrama de Nyquist el valor de la raíz real es mayor <-¡ue la imaginana en las bajas frecuencias, correspondiendo en la propuesta de una transición de comportamiento capacitivo a resistt,·o en la interfase.
En los diagramas de Nyquist a 322 dias, se advierte que el diagrama adquiere una forma que semeja un semicírculo, indicio de actl\·actón en el sistema. El dtagrama de Bode-módulo de frecuencia muestra una curvatura en las bajas frecuencias, con tendencia a disminuir. En ambos diagramas es posible observar la disminución en los valores de la Rtc en un orden de magmtud. Para 336 días, los diagramas tienen una definición más clara hacia una varilla acm·ada. El diagrama 27 exhibe la secuencia de los diagramas con el annce del tiempo, en el cual se advierten los cambios de la forma de los gráficos y los valores de tmpedancia obtemdos.
Este seguimiento tiene analogía con el que se reporta para el curado, solo que se tienen dos procesos inversos, uno descnbe la formactón de una barrera protectora del acero y el segundo trata de la ruptura de esa película pasiva.
De acuerdo a los diagramas de EIS, se mtció la despastvactón de la
vanlla entre los días 259 y 280 de exposición. Los diagramas que abarcan desde 280 a 308 dias, se proponen como la etapa de transt· ción en la que se ataca la película pasiva del acero y se establece una
competencia de reacctones de estabilidad de los compue$tos en la
interfase. El potencial de corrostón toma fluctuaciones de -250 a -450 m V en este periodo, lo que es indicio un camb10 en las condictones estado termodinármco superfictal de la varilla. El lapso de 259 a
1 58

días señala que el proceso para la despasivaciÓn no se presenta de manera súbtta una vez que se alcanzan condicwnes propicias, sino que se lleva a cabo en forma paulatina hasta completarse. En este
periodo, se establece una competencia de reacciones entre la
pasivación y despasivación del acero, en la que finalmente prevalece la corrosión acelerada por la inestabilidad de la película pasiva inducida por la presencia de cloruros, tanto los adicionados en el agua de amasado como los que ingresaron en el lapso de exposición.
En la determinación de la penetración de cloruro para los cilindros colados en paralelo con las mismas condiciones, se hace notar que a 90 días la concentración a nivel de la varilla ya superaba la señalada como crítica para el inicio de corrosión del refuerzo. De acuerdo a los resultados de EIS, el proceso comienza más de 100 días después, lo que hace notar que la despasivación del acero embebido se induce por la presencia de cloruros, pero la concentraciÓn crítica que toman varias normativas mternacwnales, que van de 0.1 5°1o a 1%2., 0.4%2\ de 0.35 a 0.9529, 0.6311 no es necesariamente la condiciÓn suficiente para dar inicio a la corrosión acelerada de la varilla. Esta es una información relevante que se obtiene mediante EIS, puesto que aporta datos acerca de la condición de la varilla embebida.
lomersión. La figura 28 muestra los resultados de EIS para la sene expuesta a inmersión en el periodo de 259 a 336 días. En el diagrama
Nyquist, se observa la tendencia a cerrar un semicírculo en la de bajas frecuencias, lo cual es indicio del estado activo de la El diagrama de Bode-ángulo de fase exhibe a 259 días una
con tendencia a incrementar su valor lo que apunta hacia comportanuento resistivo de la varilla, aún con un valor cercano
. Gu et al.23 mencionan en su investigaciÓn, realizada en disode Ca(OJ I), con 10% de NaCI en peso, que la vanación
ángulo de fase-es la respuesta más sensible a los cambios en
.� ....... �.•v••o::� de la interfase acero-concreto. En sus resultados, el de fase pasa de 58.22° a 35.6° en 120 horas de expostción,
como evidencia de la activación del acero. Jafar et aJ.26•
relacwnan la dismmución en el ángulo de fase a bajas con la activaciÓn de la vanlla.
1 59

Tt·:C:NICAS ELE<:rROQUÍ\II<:As PARA r:1. Co:-.TROl. Y EsTUDIO DI( t.\ CoRROSIÓS
Por su permanencia en agua de mar, los especimenes expuestos en In· mersión permanente alcanzan las mayores concentraciones de cloruro al nivel de la varilla. En esta conclición, la estabilidad de la película pas1va se ve comprometida, abriendo la posibilidad de inicio del proceso de corrosión. Entonces, el control anóclico del sistema se p1erde, dando paso al dominio de la cinética del proceso a las reacc10nes de reducción de oxígeno y de hematita11 .24
Dado que la concentración de oxígeno clisuelto en el agua de mar no rebasa de Smg/100 g de agua, la reacción de reducción de la hemattta es la causante de que la icorr sea mayor que en ciclos de inmersiónatmósfera.
Intemperie. En la figura 29 se muestra la información obtenida por EIS de las probetas expuestas a la atmósfera en el lapso de 259 a 336 días. En el cliagrama de Nyquist no se advierte la tendencia a cerrar un semicírculo que inclique activación del proceso de corrosión. Se observa la predominancia de la vanable imaginaria sobre la real, señal de un comportamiento capacitivo en la interfase del acero, y, por lo tanto, de la pasividad del acero. El cliagrama de Bode-ángulo de fase exhibe valores cercanos a -70°, confirmando el estado de pasividad superficial de la varilla. No obstante que las probetas fue· ron coladas con cloruro en el agua de mezclado y el acceso de oxíg�· no a través de los poros del concreto es factible, no son una condición suficiente para promover la corrosión acelerada del refuerzo de acero. La estabilidad de la película pas1va establece como factor controlante la condición de proceso anódico, con velocidades de corrosiÓn muy pequeñas, que no representan riesgo para el desarrollo acelerado de la estructura de concreto.
Las secuencias de los seguimientos de los procesos de curado y s1ción presentados, exhiben la potencialidad de la técnica espectroscopia de impedancia electroquímica en el análisis de propiedades de los cliferentes componentes del concreto en especial del estado de corrosión que guarda el acero de Así mismo, es posible aproximar los mecanismos que nr.�cH
.)n'""'
para el inicio y desarrollo del proceso de corrosión de la varilla bebida en concreto.
1 60

- 1 0000 ,--------------------,
-7500
Ñ -5000
-2500
O días
o '-·--�---L--�----�--�---L--�--�
íil
o 2500 5000 Z'
7500 1 0000
1 04 �----------------------------------� o dlas
-60 r-----------------. -50
g -40 � -30 ICII ...... -20 ._.
- 1 0 o ������u=���������� 1 0'3 1 0"2 1 0"1 1 0° 1 01 1 02
Frecuencia (Hz) Figura 15.- Dtagramas de Nyqutst, Bode y Bode fase.
Tiempo de curado O días. Ecorr . -410 m V vs l�cs.
1 6 1

Tí;c�JCAS ELECTROQUÍ\tJCAs rARA EL CoNTROL v EsTUDIO DE LA CoRROSJó:-:
-7500 ,.-------------------, 3 días
-5000
-2500
Z'
3 días
. . . . . . . . . . . . . .
102
1 0-3 1 0-2 1 0-1 1 0°
1 01 1 02 1 03 104
Frecuencia (Hz) -75
íii' o -50 � - o o o o
"O 111 .... s _2s Cb
o 1 0-3 1 0-2 10-1 10
° 1 01 1 02 1 0
3 104
Frecuencia (Hz)
Figura 16.- Diagramas de Nyquist, Bode y Bode-fase.
Tiempo de curado : 3 días.
Ecorr : -422 m V vs ECS.
162

-7500 .-------------------, 5 días
-5000
-2500
<e>J": O L--��--�--�-----L----�--� o 2500 Z' 5000 7500
1 04.-----------------------------------� � 5 dfas
-50 ¡;¡ .......
-40 o
111 .g -30 la s -20
� - 1 0
o o o o o o o o o o o o o o o o • •
o ����wW���LU�wu�����aw•' 10"3 10"2 1 0"1 1 0° 1 01 1 02 1 03 1 04
Frecuencia (Hz) Figura 17.- Dtagramas de NyqUlst, Bode y Bode-fase.
Tiempo de curado : S días.
Ecorr . -432 m V vs ECS.
163

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA (ORROSIÓ:-.J
-30000 ,.--------------------., 7 dfas
-20000
- 1 0000
o J ..
o 1 0000 Z' 20000 30000
1 05
7 dfas [¡]
1 04
N 1 03
• • • • • • • o • • • o • • o • •
1 02
1 0-3 1 0-2 1 0"1 1 0° 1 01
1 02 Frecuencia (Hz)
-75
íi) [¡] . . . o -50 "O I'CI .... s .2s q,
o ���������LU��·�·�·��UU���m,
10"3 1 0"2 10'1 1 0° 1 01 1 02 1 03 1 04
Frecuencia (Hz)
Figura 18.- Diagramas de Nyquist, Bode y Bode-fase.
Tiempo de curado · 7 días.
Ecorr : 423 m V vs ECS.
1 64

-75000 .--------------------. 1 0 días
-50000
-25000
.i 0 �-�---L--�--�--�-� o 25000 75000
Z' 50000
1 05�----------------------------------· � - 1 0 días
. . . . . . . . . . . . . . · • ·
1 02 ��WL��L-�wm-L���ww��wL��
10"3 1 0"2 1 0"1 1 0°
1 01
1 02 1 03 1 04
Frecuencia (Hz) -75
.-------------------,
íñ Fl · . . . . o -50
-o � S -25 �
o �����_w���WL�����-�·��· 1 0"
3 10"2 10"1 10
° 1 0
1 1 02 1 0
3 1 04
Frecuencia (Hz) Figura 19.- Diagramas de Nyqutst, Bode y Bode-fase.
Tiempo de curado · 1 O días.
Ecorr -446 m V vs ECS.
1 65 -
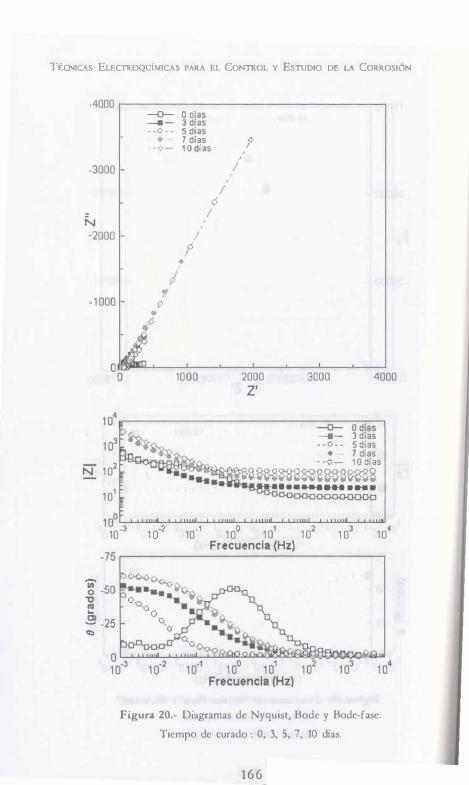
TÉCNICAS ELECTROQUfMICAS I'ARA EL CONT!lOL y EsTUDIO Df. u CORROSIÓN
-4000 .--------------------.,
-3000
N -2000
-o- O dlas - - 3 dlas - -o - - 5 dlas • 7 días - -<>- 1 0 días
ó 1 1
f ,, . 1 ,
1000
1 1
?
2000 Z'
3000 4000
1 04 r-------------------------------�� -o- O dias _._ 3 días - - o - - 5 días
• 7 días F.""''0(]1J13t}j�íio¡L<,.,..,.o. - -o- 1 o dlas N 1 02
-�-um�esaeecsm� 1 01 ....... .... . _ .
1 �������������w_����
10'3
10'1 1 0° 1 01 1 02 1 03 10
4
Frecuencia (Hz) -75 .----------------�
íii' o -50
"tl IV ... S -25 �
Figura 20.- Dmgramas de Nyquist, Bode y Bode-fase.
Tiempo de curado : O, 3, 5, 7, 10 días.
166
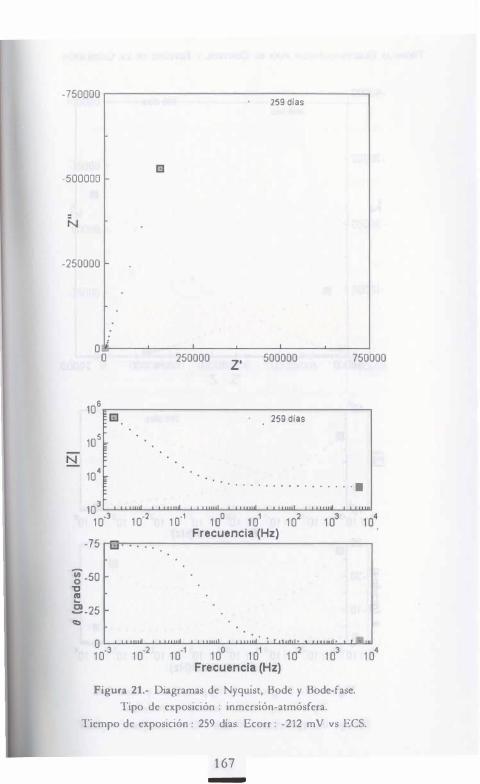
-750000 r-----------------, 259 dlas
-500000
-250000
oi o
106 1::1
105 N
104
103 1 0-3
-75 �
� -50 "O (1;1 .9-25 �
250000 Z'
500000 750000
259 dlas
. . . . . . . . . . . . . . . . · •
1 0-2 1 0� 1 � 1� 1 � Frecuencia (Hz)
Figura 21.- Diagramas de Nyqutst, Bode y Bode-fase. Tipo de exposición : inmerstón-atmósfera
Tiempo de exposición : 259 días. Ecorr : -212 m V vs ECS.
1 67 -

Tfc.:-;IC\S 1-.i.I.CrRO<,¡ll \IIC.\S PARA EL CONTROL Y ESTUDIO DE LA (ORROSIÓ!'I
-40000 .-------------------, 280 días
-30000
Ñ l -200 r.
l
-1 0000 � �---· ·· · · o - 1 -- __._ _ _..J... _ ___._ _ ___l. _ __.. _ __,
10000 2C001 300CO z·
40000 50000
1�.------------------------------� 280 días
. . .
� -20 -e "" .... S -10
. . . .
Figura 22.- Diagramas de :-...yqUist, Bode y Bode-fase.
Tipo de exposición mmer>IÓn-atmósfcra.
Tiempo de exposiCIÓn : 280 días. l;corr : -167 m V vs ECS.
1 68
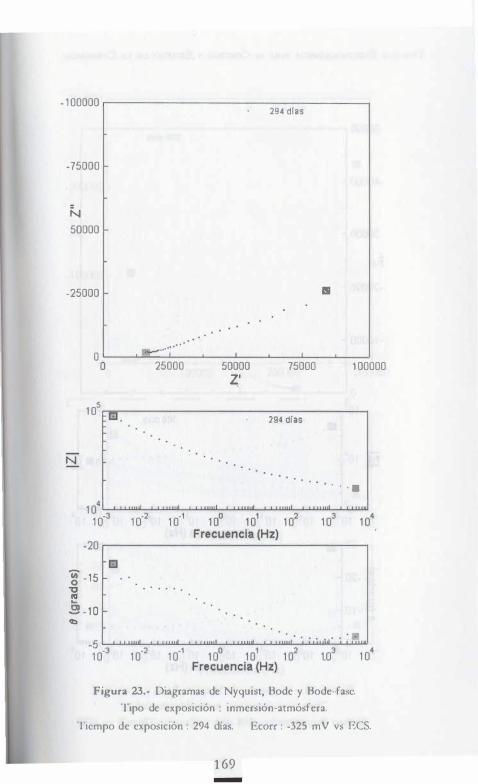
- 1 00000 .------------------, 29• dlas
-75000
N 50000
-25000
o o
1 05
: I:J.
,..,.., ... 25000
11
50000 75000 1 00000 Z'
294 dlas
. . · •
-20 .----------------�
� - 15 "' � s -1o q,
Figura 23.- Dmgramas de Nyqutst, Bode y Bode fase.
Tipo de exposición · inmersión-atmósfera
Tiempo de exposición : 294 dias. Ecorr : -325 m V vs ECS.
1 69 -

Tf-:c-.:IC\S EI.El.TROQt.:I\IIOS r.\RA EL CosTROL Y EsTt.:DIO DE LA CoRROSIÓN
-50000 .--------------------, 308 días
-40000
-30000
-20000
- 1 0000
o ��--L�-·-·_···�··-�-��--L-�-��
1 05 Q . 308 días
. . . . . . . . . . . . . . . · •
-30 n��.----------------------------.
íii o -20
, 111 .... S - 1 0 �
. . . . . . . . . . . . .
Figura 24.- Diagramas de NyqUist, Bode y Bode-fase. Tipo de exposición : Inmersión-atmósfera
T!Cmpo de exposiCIÓn : 308 días. Ecorr : -220 m V vs ECS
1 70
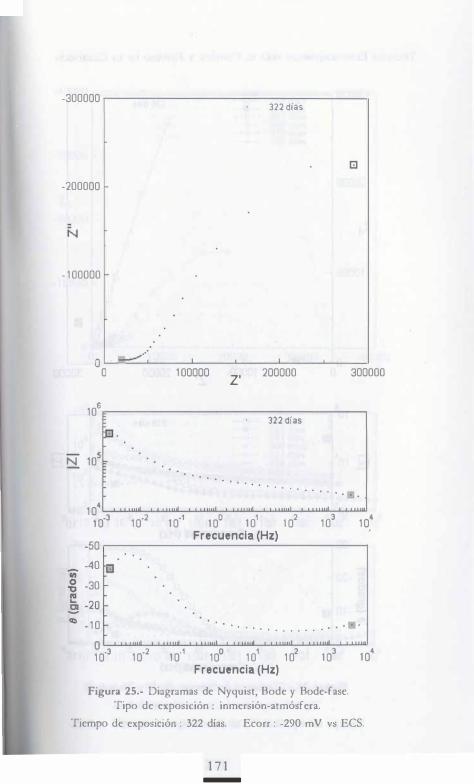
-300000 .------------------.,
-200000
- 1 00000
o ..__.,-·· o
106
:O . N 10
5
1 04
1 0·3
-50
íii' -40 l8
o -30 "O IU s -2o <t> - 1 0
o 1 0.3
1 0·2
1 0-2
322 días
1 00000 Z'
200000
1 0·1
1 0-1
322 días
. . . . . . . . • • • • o . . . . .
1 0° 1 01 1 02 1 03
Frecuencia (Hz)
. . . . . . . . • o • • • • •
10° 1 01 1 02 1 03
Frecuencia (Hz)
Figura 25.- Diagramas de Nyquist, Bode y Bode-fase. Tipo de exposiCIÓn Inmersión-atmósfera.
o
300000
1 04
1 04
Tiempo de exposición : 322 días. Ecorr : -290 m V vs ECS.
1 7 1 -

TÉCNICAS EJ.ECTROQUi\tiCAS PARA EJ. CoNTROL v EsTUDIO DE LA CoRROSIÓN
-30000 ,...------------------, 336 dfas
-20000
-1 0000
N
o 'U/.. o 1 0000 Z' 20000 30000
105 336 dlas
�- . 1 04
• • • o • • • • • . . . . . . � . . .
-30 r-----------------�
. . . . . . . . o �.WU-�WL����U-�WL�UW��·�ud��,UU� 1� 1� 1� 1� 1� 1� 1� 1� 1� Frecuencia (Hz) Figura 26.- Diagramas de Nyquist, Bode y Bode-fase.
Tipo de expostciÓn : tnmersión-atmósfera.
Tiempo de exposición . 336 días. Ecorr : -433 m V vs ECS.
172

-40000 r-----------------------------------�
-30000
N -20000
- 10000
1 0000
-o-- -- -0 - -
.. - -<)-•
20000 Z'
259 dfas 280 dfas 294 dfas 308 dfas 322 dfas 336 dfas
30000 40000
1 02 ��ü_���uw�ww�_u���wL�uw��w 1 � 1 � 1 � 1 � 1 � 1 � 1 � 1 �
Frecuencia (Hz) -75 ����-----------------------¡
1 0"2 10"1 1 0° 101 102 Frecuencia (Hz)
Figura 27.- Diagramas de Nyquist, Bode y Bode-fase.
Tipo de exposición : inmerstón-atmósfera.
Tiempo de exposictón . de 259 a 336 días.
173

'I'f.Q>IC\S ELECI'ROQL'Í\IICAS 1'.\R.\ 1'.1. co�TROI \ EsTUDIO DE LA CoRROSIÓN
-30000 .-------------------<r-----�25�9���$----------,
-20000
-1 0000 {/ // r o
-- 280días --o-- 294 cfas • 306 cias - ·()- 336 cias
l 0 '�--�----�----�----�--�----� o 1 0000 20000 30000
Z'
1 0�
�--------------------��---------. -<r- 259 días - 280 días - -0 - - 294 días - • 306 días - ·(.·- 3J6cfes
102 1 0'
3 1 0-2 10·1
10° 101 1 02 103 -75 Frecuencia (Hz)
00o -¡;; 00� 0.� o -50 Q� 'O
10 � ... s.2s �' <t> �� e
o 1 0-3 1 0-2 1 0-1 1 0
° 1 01 1 02 1 0
3
Frecuencia (Hz)
Figura 28.- Dtagramas de NyqUtst, Bode y Bode-fase.
Tipo de exposición atmosférica.
Tiempo de exposiciÓn : de 259 a 336 días.
174

-5000 .----------�-:;--------, -o- 259 dlas
-4000
-3000
N -2000
- 1 000
o o 1 000
1 04
N
1 02
10-3
1 0-2 1 0-1
-75
i -50 "O !U ... s .25 �
o
1 0-3 1 0-2 1 0-1
- 280 áas / -0- 304 cfas
• 336áas .
/ / •
1 IJII
- .o - - - -o' r.; .. "o
•
. . . •
2000 3000 z·
-o- 259dias -- 280 dóas - -0- - 304 c:tíes
• 336 dóas
4000
�
1 0° 1 01 1 02 1 03
Frecuencia (Hz)
1 0°
1 01 1 02 Frecuencia (Hz)
5000
...
1 04 1 0�
....
1 04 1 05
Figura 29.- Dmgramas de Nyqutst, Bode y Bode-fase.
Tipo de exposiciÓn : inmersión.
Tiempo de cxpos1ción de 259 a 336 días.
175

TÉCSICAS ELF.c:TROQUÍMIC.\S PARA EL co�TROL y EsTuDIO DI! LA CORROSIÓN
CONCLUSIONES.
La técnica espectroscop1a de impedancia electroquímica (EIS) presenta importantes ventajas para el estudio del proceso de corrosión del acero de refuerzo embebido en concreto. La información que aporta es útil para estimar el estado de corrosión que guardan las varillas de acero embebidas en concreto.
Con la interpretación de los diagramas de Nyquist, Bode-ángulo de fase y Bode-módulo de impedancia es posible aproximar mecantsmos de las reacciones que se llevan a cabo en la interfase acero-concreto y con ello establecer el efecto de diferentes parámetros que actúan en la corrosión del acero de refuerzo.
La EIS se muestra como una técnica muy conveniente para realizar seguimientos en estructuras de concreto y contar con información confiable del grado de corrosión de la varilla para tomar medidas pertinentes.
REFERENCIAS BIBLIOGRÁFICAS
1.- ASTM-C-876-91. "Standard Test Method for Half-Cell Potentials of Reinforcing Steel in Concrete". Philadelphia, U.S.A., 1992. 2.- Walter, G. W "A review of impedance plot methods used for corrosion performance analysis of painted metals". Corrosion Science, Vol. 26, no. 9, pp. 681-703, 1986. 3.- Mansfeld, F, Shih, H., Greene, H. and Tsai, C. "Analysis of EIS data for common corrosion processes". ASTM STP 1 188. Pp. 37-53, 1993. 4.- Beavers, J. A., Thompson, N. G. and Silverman, D. C. "Corrosíon engineering applications of electrochemical techniques : Laboratory testing". Corrosion 93, NACE, Paper No. 348. 5.- Sagüés, A. "Corrosion measurement techniques for steel tn con· crete". Corrosion 93, NACE, Paper No. 353.
176

6.- Hladki, K., Callow, L. M. a11d Dawson, J. L. "Corrosion rates from impedance measurements : .. n introductton". Brit1sh Corros10n Journal, Vol. 1 5, No. 1 , pp. 20-25, 1980. 7.- John, D., Searson, P. C. and Dawson, J. L. "Use of AC impedance techmque in studies on steel m concrete in immersed conditions", Bricish Corrosion Jo urna!, Vol. 1 6, N o. 2, pp. 102-106, 1981 . 8.- I lope, B., Page, J . A. and Ip, A. "Corrosion rates o f steel in concrete". Cement and Concrete Research, Vol. 16 , pp. 77 1 -781, !986. 9.- Wcnger, F., Gallar.d, J. a:1.d Lemoine, L. "Application o f electrochemical impedance mca.mrements to the monitor.ing o f corrosion o f retnforced concrete �tructures in marine environmt·nt". ECROCOR '87, Karlsruhe, Ger'Tiany, 1 987. 1 0 . - Sagoe-Crentstl, K. K., G lasser. F. P. and Irvine J T. "Elcctrochemical charactenstics of reinforced concrete corrosH.n as deterrruncd by impedance spectroscopy" . Bntish Corrosion Jút:fc.tl, Vol. 27, No. 2, pp. 1 1 3 1 1 8, 1992. 1 1 .- Avila-Mendoza, J., Cano, C. and Flores, J .M, "Effect • > f superficial oxides on corrosion of steel retnforcement embeddcd in concrete", Corros10n Vol. SO,No. 1 1 , 879-885, 1994. 12.- .\ndrade, C., Soler, L , Alonso, C., Nóvoa, X. and Keddam. ''The tmportancc of geometrical considcrattons in thc measurements of stecl corrosion in concrete by mea1s uf AC impedance". Corrvswn SClence, Vol. 37, no. 12, pp. 2013-2023, 1995. 1 3 . - Matsuoka, K., Kihtra, H. and Murata, T. "Monitoriug o f �orrosion of reinforcing bar in c•>ncrete". Procecdings ó f the Corroston/87. Symposium on corroston of metals tn concrete. NACE UNIT commtttee T-3K. 14.- I Iope, B. B., Pagc, J. A. and Ip, A. K. "Corrosion of sted in c:>ncrete". Cement and Concrete Rescuch, V16, pp 771-781, 1986. 1 5.- Newton C.J. y Sykes, J.M., Cot rosion Science, Vol. 28, p.p. ] )51 , 1988. ll ·- Feliú, S., González, J. A., and Ar drade, C. Corrosion Sctence, Vol. pp. 961-970, 1986. 17.- Boukamp, B. A . . "Equivalent Circntt (EQl.IIVCRT.PAS)" , SecOild Edttton, University of Twente, Enschedc, The Netherlands,
89.
1 77

TI'.CNICAS ELECTROQUiMIC.\S 1'.\RA EL CoNTROL Y EsTUDIO DE L� CoRROSIÓ:-.1
18.- Johnson, D., "ZVIEW", Scriber Associates Inc., 1996. 19.- Comunicación personal. Vilche, J. "Curso de Espectroscopía de Impedancia Electroquímica", U.A.M.I., 29 y 30 de noviembre-1-3 de diciembre de 1992 20.- McDonald, D. D. "Sorne Adventages and Pitfalls o f Electrochemical Impedance Spectroscopy", Corrosion, Vol. 46, No. 3, pp. 229-242, 1990. 2 1 . - Hachtni, L., Fiaud, C. , Triki, E. and Raharinaivo, A. "Characterisatton of steel/ concrete interface by electrochemical impedance spectroscopy", British Corrosion Journal, Vol 29, No. 2, pp. 122-127, 1994. 22.- Dhouibi-I Iachini, L., Trikt, E., Grandet, J. and Raharinatvo, A . "Comparing the steel-concrete interface state and tts electrochemical impedance", Cement and Concrete Research, Vol. 26, No. 2, pp.253-266, 1996. 23.- Gu, P. , Fu, Y, Xie, P, and Beaudoin, J. "Characterization of surface corrosion of reinforcing steel in cement paste by low frequency impedance spectroscopy". Cement and Concrete Research, Vol. 24, No 2, pp. 231-242, 1994. 24.- Andrade, C . , Soler, L. and Nóvoa, X. R . "Advances tn electrochemical impedance measurements in reinforced concrete" Materials Science Forum, Trans. Tech. Publications, Switzerland, Vols. 192-194, pp 843-856, 1995. 25.- T. Pérez, "Estudio de la cinética de corrosión del refuerzo de acero embebido en concreto en diferentes condiciones de exposictón en medio marino" Tesis Doctoral, Facultad de Química, UNAM. México, 2000. 26.- Jafar, M. 1 . , Dawson, J. L., and John, G. D. "Electrochemtcal impedance and harmonic analysis measurements on steel in concrete". Electrochemical impedance : Analysis and Interpretation, ASTM STP 1 1 88. J. R, Scully, D. C. Silverman and M. W Kendin Eds., American Society for Testing and Materials, Philadelphia, P 384-403, 1993. 27.- Norma ACL 318, E.U. 28.- Norma EH91, España. 29.- Norma CP 1 10, Inglaterra. 30.- Norma NS 3474, Noruega.
178

7. ÜXIDACIÓN EN ALTA TEMPERATURA
A. Martfnez Villafañe J. G. Chacón Nava C. Gaona Tiburcio F. M. Almeraya Calderón J. G. González Rodríguez
179

TI'.<:SICAS Et.F-CTROQUi.\IICAs I'AB 1-.1. CoNTROL Y EsTUDIO DE LA CoRROSióN
OXIDACION EN ALTA TEMPERATURA
l.- INTRODl.JCCIÓN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 181
Il.- TERMODINÁMICA Y TERMOQUÍMICA . . . . . . . . . . . . . . . . . . . . . . 183
III.- CINÉTICA DE LA OXIDACIÓN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 198
íV- DIFUSIÓN . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 204
V- OXIDACIÓN INICIAL . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 209
VI.- OXIDACIÓN DE ALEACIONES . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 213
VII.- DISTRIBUCIÓN DE CATIONES EN SOLUCIONES SOLIDASDE OXIDOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 229
VIII.-COMPORTAMIENTO DE RUPTURA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 235
IX.- EFECTO DE TIERRAS RARAS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 236
X.- METODOS MAS COMUNES PARA DETERMINAR EL ESPESOR DE OXIDOS FORMADOS . . . . . . . . . . . . . . . . . . . . . . . . 240
XI.- REFERENCIAS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 242
1 80

OXIDACION EN ALTA TEMPERATURA
1.- INTRODUCCION.
Cuando un metal es expuesto a un gas oxidante a temperaturas elevadas la corrosión puede ocurrir por la reacción directa con el gas sin la presencia de un electrolito liquido. Este tipo de corrosión se refiere como un manchado, oxidación en alta temperatura o costrado. La veloctdad de ataque se mcrementa sustancialmente con la temperatura, la película superficial típicamente aumenta como un resultado de la reacción en las mterfases óxido/gas u óxido/metal debtdo al transporte de cationes o aniones a través del óxido, el cual se comporta como un electrolito sólido. Para un óxido no poroso el transporte i6nico a través de la costra es la veloctdad que controla el proceso. La estabilidad termodinámica, la estructura con defectos iónicos y ciertos rasgos morfológtcos de la costra formada son factores claves que determinan la resistencia de una aleación a un medio ambiente específico.
El crecimiento inicial de la película es comúnmente rápido. Si la costra es un sólido no poroso y cubre totalm
.ente la superficie del metal,
la velocidad de reacción decrecerá cuando el espesor llegue a unos miles de Angstroms tanto como el transporte de espectes reacuvas a través de la película sean los que controlen el proceso. 1 ,a subsecuente velocidad de corrosión dependerá de mecanismos de transporte, los cuales pueden ser debtdos a potenctales eléctricos o gradtcntes de concentración o a emigración a lo largo de tta};ectorias preferenciales, y de este modo pueden seguir una o "arias leyes de velocidad.
Cuando un proceso de difusión es controlantc de la vdoctdad la cinética usualmente stgue una ley de veloctdad parabóltca, y ésta progresivamente decrece con el tiempo, se forma una capa compacta y continua protectora de Cr203 para aquellas aleaciones con contenidos suficientes de cromo. St la costra es porosa (o es formada por especies en fase vapor) o no cubre completamente la superficiesc tiene que una velocidad de reacción lineal es la que se cumple. Esta Ultima circus tancia puede determtnarse de la relactón de
18 1

TÉCNICAS ELECTROQUiMICAS PARA m. Co:-.·rnot Y Esr�_;o1o DJ! LA CoRROsiós
Pilling-Bedworth, la cual es el cociente del volumen del óxido producido al metal consumido por oxidación; valores de 1 ó mayores resultan en un Óxtdo que cubre completamente la superficte y usualmente con un comportamiento protector. A altas temperaturas el crecmúento de los óxidos protectores puede ser tan rápido que los esfuerzos compresivos resultantes de una relactón Pilling-Bedworth mayores a 1 resulten lo suficientemente grandes tal que la costra (o aleactón) se deforme y posiblemente se rompa como un mecanismo de alivio; en algunos casos la protección ofrecida por tales costras puede ser baja.
Las características deseadas para una costra de óxido protector son:
• Alta estabilidad termodinámica (energías libres de formación de Gibbs altamente negativas) de manera que integre perfectamente otro posible producto de reacción.
• Baja presión de vapor de manera que el óxido se forme como un sólido y no evapore dentro la atmósfera.
• Relación Pilling-Bedworth mayor que 1 , de modo que el óxido cubra completamente la superficie metálica.
• Bajo coeficiente de dtfustón de las especies reactantes (cationes metálicos y aniones del corrodente) de manera que la costra tenga una velocidad de crecimiento lent0.
• ¡\lta temperatura de fusión.
• Buena adherencia al metal base; el cual usualmente involucra un coefictente de expanstón térmtca cercano al del metal, y suficiente plasticidad en alta temperatura para resistir fractura con los esfuerzos de expansión térmica diferenciaL
El costrado en alta temperatura usualmente se piensa como óxidos pero también pueden ser sulfuros, carburos o mezclas de esas especies. Oxtdos y sulfuros son componentes no estequiométricos y
1 82

semiconductores, estos semiconductores pueden ser: tipo-p (o portador positivo), el cual puede tener vacancias en su red metálica, 0 un exceso de aniones en posiciones intersticiales. Tipo-n (o portador negauvo), el cual puede tener un exceso de iones metálicos contenidos intersticialmente, o vacancias aniónicas en sitios de red. Para castrado controlado por difusión, la velocidad de crecimiento de la costra puede alterarse por la modificación de la concentración de defectos involucrados. Por ejemplo óxidos tipo-p muestran un mcremento en la velocidad de transporte catióntco (incremento en la velocidad de oxidación) a un incremento de las presiones de oxidactón, mientras que el transporte de óxidos tipo-n es esencialmente tndependiente de la presión de oxígeno. Ambos tipos de óxido pueden ser dopados por la adición de iones específicos para la red del óxido. Por e¡emplo, la adición de cationes de mayor valencia que los cationes nativos resulta en un incremento en el número de vacancias catiónicas y por consiguiente un incremento en la velocidad de oxidación, por lo que la adición de cationes de baja valencia tiene el efecto contrario. Los sulfuros típicamente muestran una mayor velocidad intrinsica de transporte de aniones y cationes que los óxidos del mismo metal y es por eso que son menos protectores que los óxidos.
11.- TERMODINAMICA Y TERMOQUIMICA.
Termodinámica de la oxidación.- Cuando un metal se oxida se produce un cambio en la energía libre, G, del sistema que es igual al trabajo realizado o absorbido durante el proceso, este es máximo cuando el proceso se verifica reverstblemente, el cambio en la energía libre del sistema es la fuerza motora de la reacción y representa la fracción máxuna de energía que puede convertirse en traba¡o, acompañado por una disminución en la energía libre del sistema ( óG) ya que de lo contrario, la reacción no podría tener lugar. El cambio de la energía libre, óG, está representado por:
óG = G(productos) - G(reactantes) .. . .. ... ... . . . . . ... . . . . . . . . . . . . . . . .. (1)
E.! cambto en energía libre estándar para la formación de casi todos los oxidas metálicos es negativo, esto es, los óxidos son termodiná-
1 83

TIL"r-�s ELECTROQCÍMICAS PARA EL CoNrROL Y Esll.!DIO DL LA CoRRO�Iús
micamente estables en atmósferas de oxígeno, mientras que tos metales no lo son, por consiguiente, tenderá a producirse la oxidactón para la reacción:
xMe + 02 � Me.02 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . (2)
de acuerdo a la ley de ace1ón de masas, la constante de equíiibm. k, <'S :
k=---- . . . . . .. . . . . . . . . . . .. . -· . . . . .. . . . . . . . . . . .. . .. . . . . . . . . 0)
clunde aMexoz y aMex representan las actividades del 6xtdo y Jd tll':t:tl �ólido respectivamente. Estas actividades son iguales a la urudad para rases condensadas puras y po; representa la presión parcial de oxigeno en condiciones de equilibno. Sí el oxígeno está presente en la atmósfera, la constante de equilibno se convterte en:
1 k - . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ��
p
La constante de equilibrio de una reacción guarda relactón con el camb10 de ener:,ria libe;· de la 3tguientc forma:
�G=--.ZTlnk + RTLn!npo, . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . (5) p -
donde el térrrino RTinlnpo2 define el estado inicial y final del �istema y en el que n y p representan, respectivamente, d número de moles y la presión de las sustancias participantes en h reacción.
A d!fercncta de la constante Ó"! equilibno, estos términos snn variables
<;í la presión de oxígeno es la atmosférica, RTL:nlnpo� se hace igual
a cero para dar una simple reacctón de oxidación, entonces:
�G0 =-RTlnk . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. (b p
dond(· �G0 define el cambio de energía libre estándar de la re>� · lD
1 84

En el transcurso de una reacción quúnica, las masas de los reactantes y el producto de la reacción disminuyen y se incrementan respectivamente. Puesto que la energía interna de las sustancias disminuirá y la de los productos de la reacción aumentará, el término potencial quúnico f.l., se utiliza para indicar el cambio de energía libre al cambiar el número de moles n de una sustancia en una reacción en la que se mantienen constantes la temperatura, la presión y el número de moles de las demás sustancias. Así:
f.l., = f.l-01 + RTlna 1
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. . . . . . . . . . (7)
donde a1 es la actividad del material y f.l-01
el potencial químico de un mol para la activida unitaria.
El cambio de la energía libre la reacción de oxidación Me + 02 --+ MeO 2, es igual a :
ó LlG = -RTlnkr - RTlnpo2 . . . . • . • . • . . . • • . . . . • . . . . . . . . . . . . . . • . . . . . . . • . (8)
ilG=RTlnp'02 - RTln p"o2 . . . . . . . • . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ... (9)
donde p'o2
es la presión de oxígeno en equilibrio, y p"o2 la presión inicial de oxígeno en el instante en que da comienzo la reacción.
�G = O ocurre cuando la presión inicial de oxígeno coincide con la presiÓn parcial de oxígeno representada en la constante de equilibrio, bajo estas condiciones no existe fuerza motora para la reacción, el óxido y el metal son igualmente estables; si la presión desciende por debajo de aquel valor, el óxido se disocia, a dicho valor crítico de la presión que es función de la temperatura se le llama
de disociación del óxido. En caso de formarse varios óxidos un metal, cada uno tendrá presiones de disociación diferentes
es normal que el óxido más rico en oxígeno se disocie para dar a un óxido de menor contenido en oxígeno y no al metal o directamente.
partir de la ecuación (9), se deduce que una elevación de la presión de oxígeno lo bastante por encima de la atmosférica se
1 85

Tí:C.--IC\S Et.i'.GrROQlJÍMICAS I'ARA EL CONTROL \ ESTUDIO Dh LA CORROSIÓS
traductrá en valores cada vez menos positlvos del cambio de energía libre que podrá llegar a hacerse negativo, en cuyo caso el óxido será estable.
Análisis termoquímico.- Uno de los problemas más 1mportantes de los materiales metálicos y no metálicos es su reactividad química en alta temperatura, en vacío, en oxígeno y en mezclas de gases oxidantes y reductores. Sí la reactividad química puede predecirse sobre bases teóricas, la preparación y limitaciones sobre el uso de materiales pueden definirse. Se encuentra que el análisis termoquímico es la disc1phna más utilizada para hacer tales predicciones. La actividad del metal y el gas, así como también el medio ambiente y propiedades termoquimicas de los materiales pueden considerarse en el análisis.
Los análisis termoquímicos utilizan datos de energía libre ÓG0, y datos de la constante de equilibrio, log kP, para evaluar:
• Los potenciales de oxidación de mezcla de gases reactivos.
• La estabilidad termoquímica de las fases condensadas metal y óxido.
• Las presiones de equilibrio de las espec1es volátiles sobre las fases condensadas como función de la temperatura y de los potencia les de oxígeno en la mezcla de gases.
Las ecuaciones básicas para derivar y usar datos termoquímicos son las sigu1entes:
óG = 6G0 + RT lnK . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . (10) óG = óH - TóS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . (1 1) 6G0= - RT lnK ó 6G0 = -4.575T log K .. . . . . . . . . . . . . . . . . . . . . (12) p p 6GR0 = IóG0(produc.) - IóG0 (react.) . . . . . . . . . . . . . . . . . . . . . . . . . . (13) logKR = LlogKP (produc.) - LlogKP (react.) . . . . . . . . . . . . . . . . . . . . (14)
la ecuación (10) es la expresión general para la energía libre de G1bbs,
1 86

tiG, en ténrunos de la constante K de la ley de acción de masas y la energía libre estándar f1G0 ; la ecuación (1 1) es la expresión para la energía libre de Gibbs, é1G, en términos de la entalpía, f1H, y entropía liS ; la ecuación (12) es la expresión para la energía libre estándar é1G0, en términos de la constante de equilibrio K ; las ecuaciones (13) y (14) son expresiones para energías libres estáridar f1G0 R y log � para una reacción química en términos de los valores de f1G0 y log K de los
p p productos y reactantes.
Interpretación termodinámica.
La tabla I muestra los principales tipos de reacciones que pueden ocurrir en la oxidactón de aleaciones Fe-Cr-tierras raras, y éstas de terminan en parte la construcción de la costra de óxido formada en alta temperatura.
A. Oxidación Directa. Fe(s) + 1 /2 02(g) = FeO(s) 3 FeO(s) + 1/2 02(g) = Fe304(s) 2 Fe304(s) + 1/2 02(g) = 3Fe203(s) 2 Cr(s) + 3/2 02(g) = Cr2Ü3 (s) 2 Pr(s) + 3/2 02(g) = Pr2Ü3 (s) 2 Nd (s) + 3/2 02(g) = Nd203(s)
B. FormaciÓn de Espinelas. 2 Fc(s) + 02(g) + Cr203(s) = 2 FeCr204(s) 4Fco Cr203(s) + 02(g) = 2 Fe203 4Cr203(s) 1/2 Pr703(s) + 3/4 02(g) + Fe (s) = Pr Fe 03(s) 1/2 Nd203(s) + 3/4 02(g) + Fe (s) = Nd Fe03(s) 1 /2 Prp,(s) + 3/4 02(g) + Cr (s) = PrCr03 (s) 1/2 Nd,03(s) + 3/4 02(g) + Cr(s) = Nd Cr03(s)
(s) = sólido (g) = gas
l. Principales reacciones que ocurren en aleaoones Fe-Cr-tierras raras.
1 87

Tu:.,11.1s Eu:cmo<�CI\IIC:.IS PIR.I u. Co:-.JROL \' EHLDIO 01. 1.1 CORROSIÓ'
La tabla 11 , muestra logarítmos de las presiones de disociación de los óxidos desde 25° a 1000°C calculados de las ecuaciones dadas por Kubaschewskt y Evans 1 • Todos los óxtdos son estables en las atmósferas y temperaturas empleadas en los experimentos de oxida-ción.
Temperatura (0C) Fe203 Cr203 Pr203 Nd203 25 85.5 121 .8 201.78 201.78
200 50.8 73.4 123.60 123.60 400 33.0 48.9 84.08 84.08 600 23.4 35.6 62.68 62.68 800 17.4 27.3 49.27 49.27
1000 13.3 21.6 40.06 40.06
TABLA I I . Presiones de dJsociacJÓn de los óxidos -log Po2 (atmósferas,
Dtagramas de Elhngham.
Los diagramas de Ellingham ofrecen un método reconocido para presentar mformac1ón termodinármca, una aplicación de este es que nos perrmte predecir la posibilidad de reducir un óxido por despla· zamiento con otro metal o sin necesidad de calentarlo hasta que el óxido se vuelva inestable. Una gráfica de RT lnp02 (óG0) contra
temperatura para algunos elementos en el sistema metal-óxido se
muestran en la figura 1 .
188

.t
T l"'C j FIGURA 1.- D1agrama de Ellmgham.
DIAGRAMAS DE FASES.
En princtpio, uno puede calcular el diagrama de fases si existen datos termodinám1cos apropiados; estos diagramas son usados .com0 punto de partida para un estudio de la cinética de c.•xidación y ml ca ntsmos de reacción.
Diagrama de fases del ststema binario hierro-oxígeuo (2) •
H sistema contiene tres CJx�dos: wü!>tita (FeO), magnetita (Fe10) }' htmatita (Fe20J. La wustiol\ y magneúta son ambas cúbtcas centrada en las caras basadas sobr-: una red de oxígeno; la wüstita tiene la {Structura del l\:.1Cl figura �; (en la esuuctura NaCl los antones son cúbicos de emp,1quetamie1 r) compa•·to y los pequeños cauoncs ocupan los intersticios oct; .!.:cos, cada catión está rodt>ado por 'ts ion<:s oxígeno).
189

"fl:csiCAS ELECTROQI:iMICAs PARA I'.L Co:-�TROL Y EsTUDIO DE LA CoRROSIÓN
Qarion FIGURA 2.- Estructura de Cloruro de Sodio.
La magnetita tiene la estructura de espinela (en la estructura de esp1· nela los átomos de oxigeno forman una red cúbica de empaque· tamiento compacto y los iones metálicos ocupan ambos saws tetraédricos y octaédricos, y la celda unitaria consiste de 32 sitio; octaédricos y 64 tetraédricos).
La hematita tiene la estructura de corundum (para recordar esta e;·
tructura se considera que los iones oxígeno forman una red hexagonal de empaquetamiento compacto con los átomos trivalentes M ocu· panda 2/3 de los sttlos octaédricos). En la figura 3 se muestra uc diagrama de fases del sistema binario Fe-O.
1 90

F ... - 1. + LIOU!CO
re ( ____ .,.. - 'HUSTIT.\
'C
10''0 - - - - .O"- ti -- - - - 10::¡4--- -· - - .• \ -- - - -:g�F-- - - - - � -
- - - - -· __ tO_�_
re- 0::. • MAGNETITA
'Y. PESO
FIGURA 3.- Diagrama de fases Fe-O
19-� - - !O"� - - 12: ''
10•11 - - - io·to loii.GNETITA + HEMATITA
Diagrama de fases del sistema binario Cromo-Oxígeno (2)
El sesquióxido de cromo Cr201 es el único óxido de cromo el cual es estable a altas temperaturas y a bajas temperaturas (<400-500°C) Varias otras fases ricas en oxígeno existen y la composición de las fases de óxido en la región Cr203 - Cr03 es Cr0265 (bióxido), Crüw (Y-óxido), Cr6 015, Cr5012, Cr02 • El Cr203 posee la estructura de corundum. En la figura 4 se muestra el sistema Cr-0 en el intervalo Cr-Cr203.
1 9 1
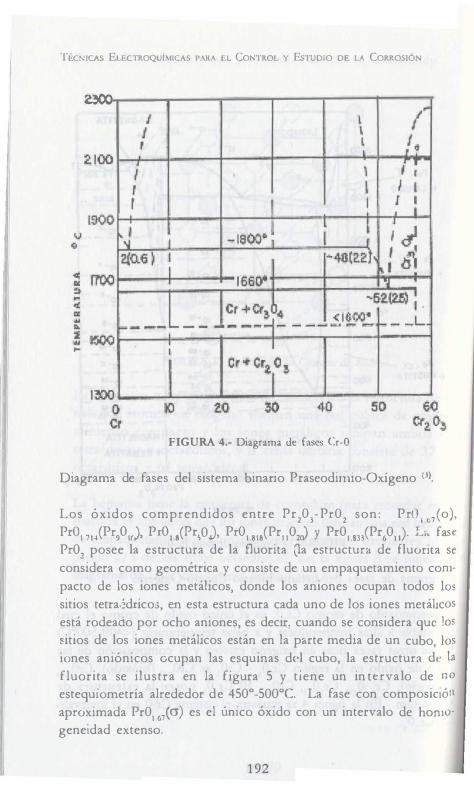
Tfo.(_-..;¡c\S ELEGTROQl'hiiC:.\S 1'.\RA 1'1. CONTROL Y ESTUDIO DE l.\ (ORROSIÓ"
�
2. 100 • 1 r r
1 J ,
FIGURA 4.- Diagrama de fases Cr-0
L \ 1 ..
� }o
Diagrama de fases del sistema binario Praseodirruo-Oxígeno l1>.
Los óxidos comprendidos entre Pr203- Pr02 son: Pr<\ �7(o), PrOI 714(Pr?OI,), PrOLB(Pr,OJ, Pr01 818(Pri i02J Y Pr0Lm(Pr601 1). L fase Pr02 posee la estructura de la fluorita Oa estructura de fluonta se considera como geométrica y consiste de un empaquetamiento compacto de los iones metálicos, donde los aniones ocupan todos los sitios tetra.fdrico;;, en esta estructura cada uno de los iones merálJcos está rodeado por ocho aniones, es decir. cuando se considera que !os sitios de los iones metálicos están en la parte media de un cubo, los wnes anión1cos ocupan las esquinas d<'l cubo, la estructura de la fluorita se ilustra en la figura 5 y tiene un in ter va lo de n o
estequ10metría alrededor de 450°-500°C. La fase con composietón aproximada Pr01 6/cr) es el úruco ÓXIdo con un intervalo de homogeneidad extenso.
192

• (•1' ,.
FIGURA 5.- Estructura cristalina de fluorita (CaFJ.
El Pr201 cristaliza con la estructura tipo A de sequióx.ido de tierra rara, aquí se sugiere que la fase puede considerarse como de estructura sequióx.ida con átomos de oxígeno interticiales. El diagrama de fase Pr-O se presenta en la figura 6.
1 000
o �- eoo oc => 1-< ffi 600 Q. ::1: w 1-
, 00
200 15
1 714
t6 1 7
1 1 1778�.8181 1 800 1833 1 8 19
OXYGEN/PRASEOOYMIUM RAT I O FIGURA 6.- S1stema Binano Pr-O.
1 93
2 0

Tí;c:NICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
Diagrama de fases del sistema binario Neodimio�Oxígeno <4l.
Se presenta un equilibrio para un sistema tentativo Nd-Nd203, incorporando NdO como se muestra en la figura 7, el sistema se estu dió por análisis térmico y metalográfico de aleaciones preparadas de 99% Nd y Nd203, los componentes Nd6011 y Nd02 han side reportados. El punto de fusión de Nd203 es 2272°C, el NdO es cúbico centrado en las caras y tiene una estructura de NaCl; Nd203 existe en
dos modificaciones: tipo A altas temperaturas (estructura hexagonal) y t1po e bajas temperaturas (estructura hexagonal) y tipo e bajas temperaturas (estructura cúbica centrada en el cuerpo); la tempera· tura de transformación está dada en 600°C.
••• •
...
o • 1eo .
o - no .
.. , . 1 1 11 1 1 1 11 f l l l O UICh:iJ • tO tf u •
1 1 --'¡--'- -, ;
. � . ----t - 1
_.. UJI!ll' ' ¡/' "'"� --...... ...... 1 -
...... ...... 1 -- 1 0�� ... -� �!f.� �=--1·=-"'--= -- -1- - -
...
•
-- --
,. ..
- -
•• .. • •
P I I C i l l l 1 l f i . I C I 11 UIUU ,;--Jo
FIGURA 7.- Sistema Bmario Nd-0
Dtagrama de fases Hierro-Cromo (S) •
Una aleación Fe-13Cr cae en la región a del sistema de aleactone! Fe-Cr en el intervalo de temperaturas de estudio (600° - l l00°C. apenas afuera de la región bifásica (a + y). El sistema Fe-Cr forflll una serie continua de soluciones sólidas, ya que el Cr (est. cúbiCI centrada en el curpo) expande el intervalo de estructura cúbica cett trada en el cuerpo y contrae el intervalo de estructura cúbica cenut da en las caras. El diagrama de fases del sistema binario Fe-Cr '1 presenta en la figura 8.
1 94

o . • Z400 ;
... • t. E 2000 �
1500
FIGURA 8.- Diagrama de fases para el sistema binario Fe-Cr
Sistema binario Cromo-Neodimio <4> .
La solubilidad de Nd en Cr a 1260°C se estima metalográficamente mucho menor que 0.30% en peso y decrece con la temperatura.
La solubilidad de Pr en Cr a 1260°C se estima metalográficamente mucho menor que 0.37 % en peso y decrece con la temperatura.
Sistema bínario Neodimio-Praseodimio <•> •
Este sistema cuyos componentes son Nd y Pr forma una solución ideal en los estados líquido y sólido.
Constitución de fases en el sistema Fe-Cr-0 <6>
En este sistema los óxidos FeO, Fe304, Fe203, Cr203 y FeCr2Ü4, pueden presentarse dependiendo de la composición y presión parcial de oxígeno.

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
Para este sistema se encontró que únicamente dos clases de costra pueden formarse en equilibrio con aleaciones binarias Fe-Cr: Hierro, cromita o espinela alrededor de 13% Cr y Cr203 por arriba de esta composición.
Sección isotérmica del sistema Fe-Cr-0 en 1 300°C. La primera linea horizontal superior unida por Fe203 y Cr2 03 representada la serie continua de soluciones sólidas formadas por esos dos óxidos, justamente abajo de ésta unión horizontal entre Fe304 y FeCr204 (S�, esta última composición está denotada en el punto sl en la figura 9, la unión Fe304 tiene una amplitud finita.
El diagrama puede describirse mejor refiriéndose a tres campos de fase, partiendo de la izquierda con yFe, FeO y espinela, continuando con a.Fe, yFe y espinela, finalmente espinela.
FIGURA 9.- Diagrama de fases isotérmico a 1300"C para el sistema Fe-Cr·O
Constitución de fases en el sistema Pr-Fe-0 (7) .
El equilibrio de fases y la termodinámica de fases coexistentes en el sistema Pr-Fe-0 está limitado por el intervalo Prp3-Pr02-Fe203-Fe para la construcción del diagrama de equilibrio, el cual define el carácter
1 96

de cambio de fases en el sistema durante el cambio de presión parcial de ÓXIdos, temperatura y composición de la mezcla de óxidos iniciales y evalúa la estabilidad térmica de los productos intermedios, orto ferritas PrFe03 con respecto a la oxidación.
Para la construcción de este diagrama se utilizan datos reportados en la literatura de los sistemas binarios Pr-O y Fe-O, así como también datos sobre las propiedades termodinámicas de PrFe03.
Una sección transversal isotérmica a1300°K del diagrama de fases para Pr-Fe-0 se muestra en la figura 10.
FIGURA 10.- Diagrama de fases isotérmtco a 1 300"C para el sistema Pr-Fe-0
197

TECNIC\S ELECTROQUiMICAS PARA EL CONTROL Y EsTUDIO DE LA CORROSIÓN
En la tabla III se muestran las fases coexistentes en d sistema Pr-Fe-0. La conclusión que se obtiene de este sistema es que únicamente un componente ternarto de ortoferrita PrFe03 es estable a cualquier valor de po2.
Región
I Pr203 + PrFe03 + Fe II PrFe03 + Fe<1 •lO + Fe m PrFe03 + Fe(l xP IV PrFe03 + Fe3 04 + Fe<1 ,p V PrFe03 + Fe203 + Fe(l-x)O VI Pr203 + "Pr01 16" + PrFe03 VII ''Pr01 16" + PrFe03 VIII "Pr01 16" + PrFe03 + PrO (2 •) IX PrO r- •l + Fe203 + PrFe03 X PrO <2 •) + Pr02 + Fe203
TABLA 111.- Fases coexistentes en el ststema Pr-Fc-0
111.- CINETICA DE LA OXIDACION
Los datos cinéticos son una prueba directa de la medida de la veloc1 dad de oxidación como función de la temperatura y composición de la aleación. Los mecanismos dentro de los cuales un metal puro o aleación se oxida a elevadas temperaturas puede interpretarse como una serie de pasos sucesivos, como sigue:
L Adsorción de un componente gaseoso. 2. Dtsoctaaón de la molécula gaseosa y transferencia de electrones. 3. Nucleación y crecimiento de cristales. 4. Dtfusión y transporte de catiónes, aniónes y electrones a través
de la costra.
En un proceso cinético el paso más lento es el que controla la velocidad de reacción. La experiencia muestra que esencialmente tres
1 98

opos de ecuaciones son observadas en o:r.idación en alta temperatura:
1 . Ecuación de velocidad de reacción logarítmica :
X = K.,.ln(t +t) + A . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . (15) ó
X= �,Jn(Bt + 1) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. (16)
donde A y B son constantes, K., es la constante de reacción, t es el tiempo y x puede representar el espesor del óxido consumido por unidad de superficie ó la ganancia de peso por unidad de área. 2.Ecuación de velocidad de reacción parabólica :
ó
dx KP - = - . . . . . . . . . . . .. . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. . . . . . . . .. (17) dt X
2 X =Kpt+ep . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ... (18)
donde Kp es la constante de reacáón y ep una constante de integraáón.
3. Ecuación de velocidad de reacción lineal :
dx K, . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . (19) ó
- =
dt x = K, t + e, . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . (21J)
donde K1 es la constante de reacción y e, es una constante de Integración.
La ecuación de velocidad logarítrruca usualmente representa los estados iniciales de oxidación a bajas temperaturas; la ecuación de reacción parabólicas princtpalmente se ajusta a los procesos controlados por difusión de especies; la ecuación de reacción lineal (costra no protectora) representa una velocidad constante del crecimiento del óxido aplicable a muy altas temperaturas ó cuando esfuerzos COntinuos rompen la costra, exponiendo una área superficial relatilrnente constante al medio ambiente, resultando en un crecimien
to acelerado de la costra.
1 99

TÉCNICAS ELECTROQUIMICAS PARA EL CONTROL Y ESTUDIO DE LA CORROSIÓN
La figura 1 1 presenta un esquema general del comportamiento cinético de aleaciones Fe-Cr y aceros inoxidables. Estudios realizados por G.C. Wood (S) sobre la cinética de crecimiento de estos materiales postulan que la mayoría de las relaciones comunes de crecimiento tienen que observarse considerando que existe un gran intervalo de aleaciones, de atmósferas y de condiciones que pueden presentarse. No obstante, para intentar alguna generalización, puede usarse la curva esquemática de ganancia de peso por unidad de área contra tiempo mostrada en la figura 12.
FIGURA 11.- Relaciones oxidación - tiempo para las cinéticas de cioxidación más comúnmente observadas.
ganancia en peso
tiempo, h
e
o
FIGURA 12.- Curva de crecimiento típico para la oxidación de aleaciones Fe-Cr y aceros inoxidables.
200

Bajo condiciones poco agresivas, una película protectora se forma con una velocidad inversamente proporcional al tiempo transcurrido de acuerdo con la curva OAD de la figura 1 1 ; cuando la atmósfera es un poco más agresiva y después de un período inicial OA (que algunas veces llamado "período de mducción"), hay un repentino incremento en la velocidad de oxidación, segmento AB (break-through). Este estado es frecuentemente seguido por una reducción en la velocidad de oxidación, segmento BC (auto-curarniento), pero en algunos casos donde el óxido permanece corno no protector, el comportamiento BE puede sustituir a BC. En una atmósfera muy agresiva no se observan períodos de inducción y AB sigue directamente. Las longitudes y formas de las diversas porciones de las curvas de crecimiento dependen críticamente de la aleación y medio ambiente en forma compleja.
Recientes estudios reahzados para la oxidaciÓn de aleaciones Fe-Cr conteniendo 0.2-1 0% Cr y 13 25% Cr en oxígeno y aire en un intervalo de temperatura de 750° - 1025°C, muestran que el cornportanuento cinético sigue una. ley parabólica en todos los casos. El modo de oxidarse de aleaciones Fe-Cr en aire a una temperatura dada, se estudia por métodos terrnogravirnétricos, rnetalográficos y particularmente por microsonda; con este equipo es posible seguir en detalle los procesos que ocurren durante el desarrollo de costras protectoras, crecimiento nodular estratificado y espesor de la costra estratificada.
Rickett y Wood (9) oxidaron aleaciones Fe-12-18% Cr en oxígeno, de 980°-1090° C y encontraron espinelas de FeCr2 04 y Crp3 sobre la parte interior de la superficie del metal y Fe203 sobre la parte exterior de la costra, la naturaleza y número de las capas dependen del contenido de Cr y la temperatura. McCoullough, Fontana y Beck <10) encontraron que para aceros inoxidables calentados en oxígeno a 980°C se formaron espinelas sobre el metal pero a medida que la oxidación proseguía una canudad de Fe203 predominaba en la costra. Caplan y Cohen <1 1l usaron aleaciones hierro-cromo comerciales con Fe-1 1-26% Cr y 0.3% Si en un intervalo de temperatura de 871°-1093°C en aire seco y húmedo.
201 ..

TF.C:->IC..AS ELEC!llOQUÍMICAS PARA fl CO:-J"ll\OL y EsTUDIO DE. LA CORROSIÓN
Se observó Cr203 y espinelas en varias proporciones como componentes principales en la costra. Moreau <1
2> estudió un amplio inter
valo de aleaciones Fe-Cr de 800-1250°C y es uno de los pocos autores que reportan al FeO como óxido estable (mezclado con FeCr20) sobre el metal. Yearian, Randell y Longo (IJ) encontraron prácticamente todos los óxidos dependiendo de los parámetros de composición y temperatura. Principalmente simplificaron el problema a dos tipos de costras:
a) (baJas veloctdades de ataque) consisten principalmente de Cr203 con algún Fe203 disuelto.
b) costras donde el ataque es severo, y en la interfase M/MO las espinelas del tipo FeCr204 predominan.
Wood y Whittle (l•l reportaron dos óxidos inicialmente habilitados para formar:
a) una costra que consiste de Cr203, que es protectora para largos períodos después de que ocurre una falla (rompimiento de la película de óxtdo).
b) un óxido el cual no es protector o únicamente es protector para períodos cortos, el óxido es una espinela del tipo Fe-Fe(2 ,1Cr,04•
Otros autores solo mencionan que Fe203 y Cr203 forman una sene continua de soluciones sólidas. Factores que afectan la cinética de oxidación. Un proceso de oxidación depende de muchos factores. Entre los más importantes podemos citar los siguientes:
1 . Temperatura.- Stendo la oxidación un proceso cinético es de esperarse que se cumpla una ecuación del tipo de Arrhenius:
K= K0.exp(-Q/R1) . . 0 0 . 0 0 . 0 0 . 0 0 • • • • 0 0 . 0 0 . 0 0 . 0 0 . 0 0 . 0 0 . 0 0 . 0 0 . 0 0 . ... (21)
donde K es la constante de reacción, K0 una constante. Q la energía de
202

activación, R es la constante de los gases y T la temperatura absoluta.
2. Presión.- Si el factor que deterrrúna la velocidad de reacción es la adsorción, entonces la reacción mostrará una gran dependencia con la presión de oxígeno; cuando la presión de oxígeno es muy baja la velocidad de reacción es proporcional al número de moléculas de 02 que llegan a la superficie, cuando la presión de 02 es muy muy elevada la velocidad de reacción es proporcional a P 02. Si la presión es controlada por un proceso de difusión en el estado sólido, el sumtnistro de 02 a la superficie no es un factor limitante ya que la reacción en la interfase es más rápida que la difusión de las especies.
3. Gas.- Existen diferencias en la velocidad de reacción st se trata con diferente gas (aire, C02, H20, S02 ,etc.).
4. Pureza del gas.- Los contarrúnantes alteran la velocidad de reacción (el vapor de agua y sales influyen considerablemente.)
5. Velocidad del flujo del gas.- Su importancia aumenta cuando la adsorción es limitante.
6. Pureza del metal.- Porosidad, tamaño de grano, impurezas.
7. Acabado de la superfície.- Una superficie completamente lisa tiene una energía disponible menor que una rugosa.
Tamaño y geometría de la muestra-relación área-volúmen.Romanski <15•16J investtgó la influencia de la geometría en las muestras y concluyó que los factores geométricos más importan-
efecto de la razón área de la muestra a su masa (área específica) sobre la inestabilidad térmica de la reacciÓn y así sobre la velocidad de reacción, espectalmente sobre los estados iniciales.
203

•
T(.c;¡.;rc:As ELECfROQUiMrCAS PARA EL CONTROr. Y Es·r u uro DE LA CoRROSIÓN
b) efecto de la forma geométrica y tamaño del espécimen sobre los procesos de oxidación.
e) efecto de la forma de determinar la ganancia de masa por unidad de área del espécimen oxidado.
IV.- DIFUSION
Teoría de Wagner de la oxidación parabólica (t7) •
Cuando la oXIdación de metales da como resultado la formación de una costra de óxido compacta y el suficiente oxigeno está dispomble en la superficie, la velocidad de reacción está gobernada por eLfustán en el estado sól.Jdo a través de la costra de óxido, el incremento del espesor del óxido incrementa la distancia de difusión, la velocidad de reacción decrece con el tiempo. Cuando la difusión es
homogénea, es decir, cuando las fronteras de grano y difusión de corto alcance pueden despreciarse, la velocidad de crecimiento del espesor del óxido, x, es inversamente proporcional al espesor de óxido:
dx - a dt 1 dx _ K _!_
X � dt - P X
. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ... ( 22)
integrando esta ecuación resulta:
X 2 = 2 K/ + eo = K�t + e: , _ . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . (Z3)
donde eo y e: son constantes de mtegractón. El óx1do crece en espesor parabólicamente con el tiempo y la oxidación es llamada parabólica.
La figura 1 3 ilustra esquemáticamente los procesos de difusión que
204

tienen lugar en la oxidación parabólica de metales en alta temperatura. Los iones metálicos y electrones emigran hacia afuera a través de la costra, desde el metal hasta la superficte del óxido y los iones oxígeno emigran en la dirección opuesta. Cuando la difusión se realiza a través de sttios vacantes, los sitios vacantes del metal y/o oxígeno emigran en direcctón opuesta de los iones metal y oxígeno respectivamente.
ior.e s -M
M i ones-O
e lectrone s
FIGURA 13.- Proceso de dtfustón a través de una costra compacta en oxtdación
parabólica de metales en alta temperatura.
En ambiente de presión de oxígeno constante, la presión parcial de oxigeno en la parte interna y externa de las fronteras de fase está fija, Y es igual, respectivamente, a la presión del ambiente en la frontera de fase M0/02 y la presión de disociación del óxido en equilibrio COn el metal en la frontera de fase M/MO.
Durante el crecimiento del óxido, no hay flujos de corriente a través de la costra, por lo tanto 1, = O pero:
1, = Id + I.oo =0 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .. (24)
205

T ?:cNICAs ELECTROQuíMICAS PARA EL CoNTROL v Esruo1o DE LA CoRROSióN
donde I, es la corriente total, I<� es la corriente electrónica e 1.,0 es la corncnte Jónica. Así, de la anterior ecuación tenemos:
L = -11 = -crt/e t 1V'TJ ' . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . (25) �n e e e
donde crt es la conductividad total, (e) es la carga electrónica, td es el número de transporte de electrones y Y'r¡ � es el gradiente de potencial electroquímico y está dado por:
. . 1 cr 0n V'r¡e = V' f.! e - e'VV = --
t- V'f.l X
. . . . . . . . . . . . . . . . . . . . . . . . . . . ( 26' za O"
introduciendo este valor en la ecuación (25) obtenemos:
1 /ion =27 t.tel '(ion .V'f.lo . . .. . . . . . . . . . . . . . . . . . . . .. . . . . . . . . . . . . . . (Zl
La velocidad de crecimiento de la costra es igual a la corriente total de partículas iónicas. Si la costra formada tiene la fórmula MaXb (aM + bX = MaXb), donde los iones metálicos y no metálicos tienen valencia Zc y Za respectivamente, entonces el número de moléculas de MaXb formadas por unidad de área y por unidad de tiempo está dado por:
1 2 = O" (. (ion .(el' V'f.lx (28) bjZa l e2 . . . . . . . . . . . .. . . . . . . . . . . .
En un óxido M.Ob para el cual jZa l=2, Y flx =f.lo= (1 /2)J.ia: tenemos para la ecuación anterior:
dn _ _ 1_ d(f.l m) 2!!; - 2 O" t · t ion · t el · · · · · · · · · · · · · · · · · · · · · · · · · · · · · . . · .. ( dt 8be dx
integrando la ecuación (29) sobre el espesor instantáneo de la cosW. Dx, se obtiene:
206

dn 1 =
dt 8be2 � 1 1 í'" cr t · t · t · d•• - - K - ( 30 ) �:, ion el r- "2 /1x - t /1x · · · · · · · · · · · ·
donde representan los potenciales qutmtcos de oxígeno en las interfases M0/02 (exterior) y M/MO (interior) respectivamente, Kt es la constante de velocidad parabólica para la reacción. Si el potencial químico de oxígeno está expresado en términos de la presión parcial de oxígeno la ecuación (30) puede expresarse como:
dn RT (óz 1 1 dt = 8be2 JP'O¡ O' t . (ion . te/ . d (In Pol ) /1x = Kt /1x . . . . . . . . . . (31)
donde son las presiones parciales de oxígeno en el exterior e interior de las interfases, respectivamente.
Las ecuaciones (28) a (31) son todas formas de la ecuación de Wagner para la velocidad de crectmiento de costras de óxido compactas y pueden aplicarse a cualquier reacción metal-gas. Podemos notar que aunque la corriente total de partículas es igual a la velocidad de crecimiento de la costra, la determinación de la velocidad de difusión puede ser también la de los iones ó los electrones dependiendo de las propiedades de la costra. Si la costra es un conductor electrónico t,1 �1, el transporte de electrones es el que determina la velocidad. La dependencia de la reacción con la presión de oxígeno dependerá de si el óxido es conductor iónico o electrónico y en aquellos casos que el óxido es conductor tipo p ó n.
La teoría de Wagner está ltmitada para la difusión de la red vía defectos puntuales dentro del sólido, sin embargo, materiales policristalinos también tienen dislocaciones, fronteras de grano y superfictes tnternas y externas; es de esperarse que la difusión a lo largo de esta clase de defectos es considerablemente más rápida que la difusión de la red.
MORFOLOGIA
El primer estado de una reacción metal-gas en alta temperatura tiene una considerable influencia sobre el comportamiento subsecuente.
207

TÉCNICAS ELECTROQUÍMICAS PARA EL CONTROL v EsTUDIO DE LA CoRROSIÓN
La morfología y consecuentemente las propiedades de los óxidos pueden alterarse durante los procesos de nucleación.
I.G. Wright (tS) menciona que cuando una aleación contiene varios componentes y es expuesta en aire u oxígeno a elevadas temperaturas, el primer óxido formado probablemente consta de núcleos de todas las posibles fases de óxido en aproximadamente la proporción de aleantes que tiene. Como la superficie aleada resulta cubierta con óxido, la actividad del oxígeno en la interfase metal óxido decrece a un valor determinado por la presión de disociación de los óxidos presentes, tal que se obtienen las condiciones para la formación selectiva de únicamente los óxidos termodinámicamente más estables. Sin embargo, dado que la oxidación en alta temperatura es un proceso dinámico la secuencia de eventos que ocurren después de los estados de nucleación, están gobernados por la cinética de difusión de los componentes menos nobles desde la mayor parte de la aleación a la interfase metal-óxido. Kubaschewski y Hopkins <19l señalan que sobre la superficie de un metal policristalino con orientaciones no preferenciales, una monocapa saturada también será orientada al azar pero debido a la variación en la velocidad de crecimiento con la orientación, los granos de la costra pueden diferir el espesor.
Una vez que la formación de óxidos se completa, la costra puede crecer en una variedad de configuraciones las que incluyen: cristales columnares, bigotes de gato (whiskers), hojas, plaquetas, ampollas, astillamientos, etc., la forma y crecimiento de esos cristales dependen dela presión, temperatura, tiempo de exposición, flujo del gas, preparación de la superficie, geometría de la muestra, etc .. Otra característica importante en la estructura de las costras es la formación de huecos, que son frecuentemente resultado de la coalescencia de vacancias necesarias para mantener la difusión, cuando el transporte de cationes por un mecanismo de emigración de vacancias es el mecanismo de crecimiento predominante. Otro factor que puede influir en la formación de huecos, es la deformación plástica de la costra para mantenerla íntegra <20l. La figura 14 muestra la morfología de la costra de óxido formada para una aleación Fe-13Cr oxidada en oxígeno a 1000°C.
208

FIGURA 14. Aspecto de los productos de ox1dac1ón en una aleac1ón Fe-Cr oxidada en oxígeno a 1000"C, por 24h.
V.- OXIDACION INICIAL (Transient Oxidation).
Las primeras etapas de una reacción gas-metal en alta temperatura tienen una influencia considerable sobre el comportamiento posterior. La morfología y por lo tanto las propiedades de los óxidos se pueden alterar durante el proceso de nucleación.
Un criterio de la formación inictal del óxido es que se forman partículas aisladas de óxido en lo que parecen ser posiciones al azar en la IUperficic. Aunque todavía no esta claro, esos sitios de nucleación parecen estar asociados a imperfecciones superficiales, impurezas, etc.
Benardl32> ha sugerido que el paso que limita la reacción (el más lento) es la adsorción de oxígeno en la superficie de substrato, el oxíge.Po adsorbido difunde en la superficie hacia los sitios de nucleación
209

T i,CNICAS ELECTROQUíMICAS PARA EL CoNTROL Y EsTUDIO DE u CoRRos1ós
creando así zonas de empobre!=imiento de oxígeno alrededor de cada sitio y por lo tanto evitando que se formen otros sitios de nucleación cercanos. También se ha propuesto que partículas pequeñas que se nuclean cerca de otras partículas grandes se disuelven ya que las primeras tienen un potencial químico más positivo que las partículas grandes.
Kubaschewski y Hopkins<'9l han hecho énfasis en que la superficie de un metal policristalino sin orientación preferencial presenta una monocapa saturada cuya orientación también será al azar pero cuya veloctdad de crecimiento varía con la orientación como se observa en el diagrama esquemático de la figura 15 .
Granos �etálicos
interfase oriqinal
FIGURA 15.- Fe en la cara (100) tiene cien veces más s1tios de nucleación que la
cara (1 1 0).
Examinemos otras dos posibilidades que afectan a la oxidación inicial:
1 . Si la superficie se expone bajando directamente la muestra a la zona caliente que contiene el gas, entonces habrá un período inestable de calentamiento que dependerá de las condiciones del horno. El patrón de crecimiento de la costra se establece durante ese período.
2. Si se eleva la temperatura de la muestra dentro de un sistema de vacío, puede entonces ocurrir una oxidación preferencial al introducirse el gas oxidante y observarse un comportamiento de la costra un tanto alejado de la realidad.
Aleaciones del tipo Ni-Cu (una fase en todo el rango de composición) dan un ejemplo de costreo en estado constante donde Jos
210

óxidos producidos (Cu20, NiO). Son insolubles entre sí [llegado al estado constante el mecanismo de oxidación se mantiene]. La figura 16 da la secuencia de eventos que llevan a un costreo de estado constante para los sistemas Ni-45Cu y Ni-90Cu.
Cu
Ni-45Cu Cu20
---
NiO
-o tl!� :· . .
. ;;; iiil l
Cup f �.J.� ....... tt---------11 NiO
ALEACION
Ni-90Cu Cu,P
NiO Cu.p
NiO
Cu,P
1 -. . .. . . . ' O D O O O O O
FIGURA 16.- Formación de óxtdos en aleaciones N•-45Cu y Ni-90Cu.
Consideremos primero el caso Ni-45Cu:
Los dos componentes están inicialmente en contacto con 02 y se oxidan rápidamente para dar partículas pequeñas de Cu20 y NiO. Aquí, el tiempo es demasiado corto para que se tenga una difusión lpreciable dentro de la aleación o para que haya enriquecimiento de Ni en la costra. el Cu20 crece más rápidamente que el NiO así es fae sobresale a la atmósfera (y tal vez también dentro de la aleación) -ta que cubre completamente al NiO cuando ya se ha formado Completamente la costra, el transporte de iones en la red determina¡ la velocidad de la reacción y el proceso de difusión dentro de la lleación jugará un papel más importante ya que el Ni en la interfase
eación-costra se oxida directamente y también reduce Cu20 que
2 1 1

Tfx:r-.;IcAs ELEClllOQüiMICAs r.�RA EJ. CoNTROL Y 1-.sTt:DIO or. LA CoRROsióN
este en contacto con la aleación por medio de una reacción de desplazamiento. Ftnalmente NiO crece lateralmente hasta que forma una capa completa que se engrosa, y poco Cu se incorporará a la costra ya que la presión de oxigeno efectiva en la interfase aleacióncostra es menor que la presión de disociación de Cu20. El Cu20 crecerá si los iones Cu en el NiO alcanzan una concentraciÓn alta que les permita difundirse rápidamente. El NiO se oxida debido a la difusión hacia el exterior de iones Ni2• a través de vacancias catiómcas. El espesor de la capa de Cu20 permanece constante.
La aleación de Ni-90Cu se comporta inicialmente en forma similar a Ni-45Cu pero no hay suficiente Ni para formar una capa completa de NiO. De esta forma, el Cu20 en la interfase aleación-óxido se puede disociar parcialmente y ceder oxígeno atómico a la aleació�. Este oxígeno difunde hacia adentro y produce partículas mternas de NiO que, al crecer la costra, se incorporan a ésta en sus partes inte· riores haciéndola porosa y permitiendo el acceso fácil de oxígeno. El costreo de estado constante produce un óxido superficial bifásico con una matríz de Cu20 y NiO formandose internamente.
Los parámetros que determinan como se establece el costreo de estado constante son:
1. Las energías libres estándar de formación de los óxidos que son factibles de formarse, permite predecir que óxido será favorecido termodinámicamente.
2. La composición total de aleación determina parcialmente si existe suficiente óxido para formar una capa completa o si se formará un ÓXldo interno sm características protectoras.
3. El coeficiente de interdifusión de la aleación determinará que tan rápido el componente que ha sido oxidado pref, rencialmente puede ser reabastecido a la costra.
4. La solubilidad y difusividad de oxígeno en la aleacJÓn detcrrn.t· nao grandemente las característJcas de la ox1dación mterna.
212

S. La velocidad de crecimiento de los óxidos determina las velocidades de desarrollo relativo y de crecimiento de la nucleación inicial y también la velocidad de avance de la costra en la aleación que absorve el óxido interno antes de que forme una capa completa.
6. Las cond1ciones de oxidación, en particular la temperatura y la presión de oxígeno, así como la forma de elevar la temperatura de la muestra, son importantes y tienen influencia en los factores antes mencionados.
VI.- OXIDACION DE ALEACIONES
Los componentes de una aleación tienen diferentes afmidades hacia el oxígeno y no difunden con la misma velocidad dentro del óxido o en la aleación.
Los óxidos de los componentes pueden ser completamente miscibles entre sí y formar una solución sólida o pueden ser completa o parcialmente inmiscibles produciendo una costra multifás1ca, en este segundo caso se pueden caracterizar de la siguiente manera: en aleaciones A-B donde A es el metal más noble y B el menos noble (se oxtda preferencialmente) se puede encontrar 3 rangos, como se observa esquemáticamente a contmuación:
1 ( 1 ). A
donde
AO
(2). (3) 1 B O B solución solida AO-BO
l . Es un rango pequeño de composición cercana a A pero, donde AO se produce casi exclusivamente por lo menos en el caso
de la costra extenor (se encuentra dopado óxido de B); (dopado = ión extraño que entra al óxido).
2. Es un rango de composición intermedia, hay AO y BO.
3. Es un rango cercano a B pero, donde B se produce exclusivamente (casi no hay dopado de óxido de A).
213

..
TOCNICAS ELECTROQUIMIUS PARA EL CONTROL Y EsnJoto DE LA CORROSIÓN
En (1) y (2) se puede obtener dopado del óxido principal, por ejemplo si AO se produce casi exclusivamente, se pueden disolver concentraciones pequeñas de B dentro de AO aún en el caso de que B tenga diferente valencia de A y BO sea de diferente cristalografía que AO.B cambiaría la velocidad de reacción. En la región (2) AO y BO se pueden cambiar y dar óxidos de composición compleja.
Esquemáticamente, algunos aspectos básicos de la oxidación de aleaciones A-B en la que B es el elemento aleante han sido mencionados y discutidos por Moreau yBenard <21> y Wood (&) se tienen en la Tabla IV a continuación:
TABLA 5- Asoeclos tip,:os de lo cor·ostór ct uno aleac1on A- 6, �n que e5 e .
a t e:n:�. e n prese1cio d t une: atrr�sfera ccn O co,.,J com�onentt o�res1'1 o
t; "'
c�r.o.:.·;u; ;·r::.s O�L ,..� .. ::f.S:l C� :: ... �h�SIO 'i
:1 4:.: �" �u:c· .:. t s - s :1t .... �, -c ... 11:r :o:�tr!t t.:; .. : n�f·: - � ·t ·�.;.�te:: · · J f' �1 C·rxc e r1 ·1w:or .;e �.·e- pq I H I
.. . .. . .. � . _ , ;. e
Tabla IV.- Aspectos típicos de la corrostón de una aleación A -B en la ()UC B e> el elemento alcante, en presencia de una atmósfera de oxígeno como componente agrc>I\'O
214

La clasificación anterior deja ver que no puede haber una sola teoría para la oxidación de metales en altas temperaturas.
Oxidación Interna y Exerna.
Se usa como ejemplo el caso de una aleación A-B donde solo uno de los componentes se puede oxidar en altas temperaturas (B). Si el componente B se puede difundir con suficiente rapidez hasta la superficie se formará una costra completa de BO, pero si la difusión de B es lenta, el oxígeno atómico se difunde dentro de la aleación precipitando BO internamente en los lugares apropiados. De tal forma que la formación interna o externa de BO hacia afuera y el flujo de oxígeno hacia adentro. (A) depende del balance del flujo de B hacia afuera.
Suponiendo la formación de un óxido compacto y libre de poros, la concentración crítl.ca de B por arriba de la cual solo se formará un óxido superficial de BO, esta dada por:
N, � Z:Mo [ ;' ] v' · · ·· · · · · ·· · ·· · ·· · · · · · · · · · · ·· · · · · ·· · ·· · ·� donde
V= volumen molar de la aleación, Z8= valencias de los atamos de B, D= Coeficiente de difusión de B en la aleación, Mo� peso atómico de oxígeno, Kp= cte. de velocidad de oxidación parabólica.
Bajo condiciones úpicas (latm) se requiere aproximadamente 1 0%B .¡tara formar una capa exterior de BO pero este número es muy arbitrario ya que se puede alterar según las condiciones de operación (se �ede reducir el valor si se disminuye Po).
el nivel crítico de B, el volumen del óxido precipitado cerca de la Uperficic es tan grande que las partículas inhiben la difusión de
· eno atómico dentro de la aleación, esas partículas finalmente se utinan e impiden una oxidación interna posterior.
2 15

T!í.ci'ICAs EtEC:TROQUIMICAS PARA EL CoNTROl. Y EsTumo DE LA CoRROSió:--1
Whittle y colaboradores(22) tomaron en cuenta la relación Kp/D y postularon que:
, no hay suficiente B en la aleación para
formar un óxido protector.
( 1 nKp ) y, [ ( 1 nKp) y, ] 2 2. Si: 2o < NB < 1 - 1 - 2o , hay suficiente
B en la aleación para formar BO pero no suficiente para reformarlo si el primer óxido se fractura.
[ ( 1 K ) y, ] 2 [ ( 1 K ) v' l 3 3. Si: 1 - 1 - 2 I1 :; < N B < 1 - 1 - 2 I1 :; J hay suficiente NB para dar una capa de curación si el primer óxido se fractura, pero no hay suficiente para el caso de fracturas sucesivas
[ ( 1 K )y' ] 3 4. Si: N B > 1 - 1 - 2 n :; ,entonces es posible formar tantas
capas de óxido como fracturas ocurran.
En sistemas de aleaciones A-B donde los dos componentes se oxidan, si el nivel de B es alto se forma rápidamente una costra virtualfuente pura de BO, mientras que en concentraciones más bajas B se precipitan internamente partículas de óxido ricas en BO. Al disminuir la velocidad de oxidación y engrosarse la costra, las condictones en la aleación pueden modificarse de tal forma que permitan la formación de una capa (de curación en la base de la costra).
2 16

Un ejemplo de la oxidación interna de aleaciones base níquel.
Las aleaciones con base en níquel son la principal fuente de materiales para uso a elevadas temperaturas en diversas industrias tales como la química, eléctrica, petrolera y nuclear.
El modelo clásico para la oxidación interna es usado para describir el comportamiento en un número de aleaciones diluidas <23-24>. El proceso ocurre típicamente en la oxidación en alta temperatura de aleaciones diluidas, compuestas de un metal noble (solvente), i.e., níquel, cobalto, hierro, cobre o plata y una pequeña cantidad de elemento menos noble (soluto), tales como aluminio, cromo, silicio, vanadio, molibdeno o magnesio. Los principales requisitos, para tal oxidación, son que el soluto forme un óxido más estable que el solvente y que el oxígeno (azufre, halógenos, etc.) tenga una solubilidad y difusividad significativa en la matriz (solvente). Cuando una aleación diluida es expuesta a una atmósfera oxidante en alta temperatura se observa que partículas de óxido del elemento aleante precipitan en la dirección del frente de oxidación dentro de la matriz. Este proceso de precipitación es conocido como oxidación interna.
La situación más simple para analizar este proceso es la oxidación interna de una aleación binaria en ausencia de óxido externo. Esta puede llevarse a cabo por el método conocido como "Rhines Pack" en el cual las muestras son colocadas en una cápsula de cuarzo, evacuada y sellada, que contiene una mezcla de polvos del et
'emento
rnenos noble y su óxido más bajo, de manera que la presión del oxígeno se mantiene en equilibrio a la presión de disociación del óxido<25>. En situaciones prácticas, la oxidación interna se desarrolla debajo de una costra de óxido externo del elemento más noble.
El oxígeno de la cápsula o de la costra de óxido externo, satura la 8\iperficie de la aleación y difunde hacia adentro a través de la zona de oxidación interna y reacciona con el soluto, dando como resultadb la formación de nuevos óxidos internos en la matriz, justamente en la interfase frente de oxidación interna/aleación, el cual avanza internamente. Si se supone que las partículas de óxido interno en la
2 1 7

T�-:c:-;JcAs ELECTROQCí�ucAs PARA llL Co:-�TROL Y EsTUDIO DE LA CoRROSió�
zona de oxidación interna no tienen influencia en el transporte de oxígeno hacia adentro, ecuaciones de transporte pueden utilizarse para relacionar la velocidad de avance del frente de oxidación interna con varios parámetros fundamentales como: solubilidad de oxígeno en la matriz, (No(S>), difusividad de oxígeno a través de la zona de oxidación interna, (Do), (la cual es esencialmente puro solvente, debido a que el óxido del soluto es muy estable), el contenido de soluto de la aleación, (N¡¡), los coeficientes de interdifusión de la aleación y la razón estequiométrica de oxígeno a soluto en los precipitados internos (v).
Los mecanismos de oxidación interna han sido sujetos de numerosos estudios experimentales y teóricos. La primera investigación sistemática sobre este fenómeno fue llevada a cabo por Rhines, Johnson y Anderson<26>, Meijering y Druyvesteyn<27). Años después, la teoría de oxidación interna fue ampliamente desarrollada por Wagner (l7', Maak<28> y Raap(2l) dando como resultado una serie de postulados generales de considerable importancia teórica y práctica. Las mediciones de la penetración interna son utilizadas para determinar la permeabilidad del oxígeno (es decir, No(S)Do) en metales. Sin embargo, trabajos recientes han mostrado que desviaciones significativas del modelo clásico de oxidación interna ocurren y, en particular, valores de No(S>Do calculados de mediciones de penetración de la zona de oxidación interna después de varios tiempos y temperaturas para aleaciones Ni-Al y Ni-Cr, son influenciados por la presencia de los precipitados internos en la zona de penetración interna '�� 29• }O). Esta desviación a sido explicada en términos de un incremento en la difusividad del oxígeno a lo largo de las interfases incoherentes óxido interno/ aleación dentro de la zona de oxidación interna.
Experimentación
Las aleaciones usadas fueron: Ni-0.55%Al, Ni-1 . 15%Al, Ni-2.45%Al, Ni-4. 1 0%Al, Ni-0. 1 2%Cr, Ni-0.56%Cr, Ni- 1 . 1 0%Cr, Ni-3%Cr, Ni-4.66Cr y Ni-7%Cr (todas están expresadas en % en peso). Las aleaciones fueron preparadas en un horno de fusión rápida de vado con materiales de alta pureza, después de vaciados fueron rolados en
2 18

caliente y en frío hasta obtener placas con un espesor de aproximadamente 0.8 mm. Las muestras fueron cortadas y desvastadas en papel de carburo de silicio grado 120 y recocidas en vacío por Sh a 1200°C, después del recocido fueron nuevamente desvastadas en papeles de SiC grados 800 y 1200 hasta obtener las dimensiones finales (10 x 5 x 0.6 mm) y electropulidas en una solución de ácido ortofosfórico/ácido sulfúrico/agua por 20 minutos, finalmente fueron atacadas levemente por 5 seg en una solución de HF al 5% a 20°C. Las muestras fueron oxidadas en una cápsula de cuarzo, sellada y evacuada (Rhines Pack) usando una mezcla de Ni/NiO como agente oxidante.
La actividad de oxígeno dentro de la cápsula es insuficiente para desarrollar una costra de óxido externo (NiO) en la aleación, pero sí es el suficiente para la oxidación interna del elemento más reactivo. La cantidad de polvo de óxido fue el doble de la necesaria para oxidar completamente el elemento reactivo en la muestra. Los polvos y las muestras se colocaron en diferentes regiones de la cápsula, después fue llenada con argón, evacuada y finalmente sellada. Solo muestras de un mismo sistema (es decir, Ni-Al o Ni-Cr) fueron colocadas en una cápsula dada. una vez que se terminó la preparación de las cápsulas, éstas fueron colocadas en la zona de temperatura constante en un horno axial mantenido a la temperatura de experimentación (±2°C) y mantenida por un tiempo determinado.
Seguido a la oxidación, las muestras fueron cortadas transversalmente, montadas en resina y preparadas metalográficamente. La medición de la penetración interna fue realizada en un microscopio óptico. Las morfologías de los óxidos internos fue analizada después de hacer un ataque profundo de la sección transversal usando la técnica de electropulido <31> , esta remueve el metal sin afectar los óxidos internos.
Resultados.
Morfologías de los Oxidas Internos. Los precipitados de los óxidos internos desarrollados en el sistema Ni-Al fueron muy diferentes de los desarrollados en el sistema
219

Tf:c:-..lc:As ELEcrROQUiMI<:As P.\RA m. CoNTROL v EsTUDIO DE LA CoRROSióN
Nt Cr. En las aleaciones Ni-2.45%Al y Ni-4.10%Al los precipitados fueron ctlmdros continuos, desde la superficie de la muestra hasta el frente de oxidación tnterna, para todas las condiciones de temperatura y tiempo de experimentación. Mientras que en la aleación Ni-1 . 15%Al, los precipitados fueron cilindrtcos y continuos después de oxidarlos a 1000°C, en la aleación Ni-0.55%Al la concentración de aluminio fue Insuficiente para completar precipitados continuos, y los óxidos internos fueron alargados pero no continuos, ver figura 17 .
En el sistema Nt Cr los óxidos internos nunca fueron continuos bajo ninguna condición de temperatura y tiempo, en su lugar los prectpitados fueron dtscretos, siendo generalmente pequeños y granulares cerca de la superficie, pero alargados y más irregulares, en forma, cerca del frente de oxidación, ver figura 1 8.
El tamaño específico y forma de los precipttados fue tnfluenciada por la temperatura, naturaleza del soluto y por su concentración.
220

ALEACIONE& Ni-Al
lil%AI
l4 2'iTSikU'"SIU
til%AI FIGURA 17. Dtfercntcs aspectos de la oxidación interna de aleaciones Nt Al en
microscopía óptica y en microscopía de barrido de electrones.
221

T�:.CNICAS ELECfROQuiMICAS PARA EL CoNTR.oL v EsTuDIO DE u CoRROsióN
NI-2%Cr Ni-3%Cr
,�,.. ··A.. • ·� · ·-- �: ... , .. �,.· ., -....•
.
Ni-5%Cr Ni-7%Cr
•
Ni-7%Cr Ní-7°�r
NI-Cr Alloys oxk!ized 1n N"IINiO R�nes Pack at 1 OOOOC for 21ll.
FIGURA 18. Diferentes aspectos de la oxidación interna de aleaciones N1-Cr
222

Penetración de la Zona de Oxidación Interna.
La penetración del frente de oxidación interna uniforme medido desde la superficie externa como una función de la fracción atómica de soluto en la aleación es graficada para ambas aleaciones, Ni-Al, Ni-Cr, y esto se muestra en la figura 19. Las condiciones fueron a 1 1 ÜÜ°C por 1 O h, 1 000°C por 20 h y a 800°C por 1 60 h. En cada caso, excepto para las aleaciones Ni-Al a 800°C, como es de esperar por la teoría, la penetración interna disminuye con el incremento en la concentración de soluto. hay pequeñas pero significativas diferencias en las medidas de penetración interna de las aleaciones NiCr con respecto de las Ni-Al, en las últimas la penetración interna fue mucho mayor que las obtenidas en las aleaciones Ni-Cr. Similar comportamiento fue observado para varios tiempos de exposición a estas tres temperaturas.
Del modelo clásico de oxidación interna(l1l, la razón de penetración del frente de oxidación interna en la aleación es una función parabólica del tiempo, es decir:
� 2 / t = 2 N0<•> D0 /v N t .. ... .. . . .. . . . . . . . . . . . . . . . . . . . . . . . . . . . (33)
donde � es la penetración de la zona de oxidación interna, t es el tiempo de exposición, Do es la difusividad de oxigeno en la red del metal base, N0<•> es la fracción atómica del oxígeno disuelto en la superficie de la aleación; N8 es la fracción atómica del alurt,linio o cromo en la aleación y v es la razón de oxigeno a aluminio o cromo en la estequiometría del óxido interno.
Usando la ecuación (33), valores aparentes de N}'>Do fueron calculados de las medidas de penetración interna para los sistemas Ni-Al y Ni-Cr (figura 20), se supuso que v fue 1 .9 para el sistema Ni-Al (debido a que los óxidos internos fueron NiAl204 hasta ana penetración de 0.9� y Al203 más allá de ésta) y 1 .5 para el sistellla Ni-Cr (ya que sólo fue detectado Cr203 a través de la zona de OXidación interna). Los resultados mostrados en la figura 20 indican que los valores para No<•>Do son dependientes de la naturaleza del IOluto y sobre su concentración en la aleación.
223

Tf.r.siC::,\S Eu'.C. IROQL'i�IIC:.\S P.\RA u. CosTROJ. Y l'.srumo DI. 1., CoRROSIOS
di ii u «
, • .¡¡"..----.----.-----.--.......
.... fl
�lomu.. lr�d on �c,b\t
·� , .. f
V
Nt -�•o Nt - V
• Nt - W
O 01S
FIGURA 19. ( ; ráfica de penetración interna contra fracción atúm1ca dc sol uro
en aleaciones diluidas base níquel
224

urn
�e T �:�r 1601 .. o Nt•At �'r-----.------r-----,--�
,.\ u • X (\ •
�� ·Ct
't, .. wn N! -�o Ni ·'i N i • W
1000• e lor lO "'
100\0 ........ O Ni • AI O t�1 -Ct • t .. ¡ .. Mn )( Ni • M O C. NI- V • NI -W
a. ..-o o ���_o_ • �o
o
--o 6í;----rl�,..---o...lo-,-� __¡ AtOIJIIC. Ir u tior: so.-t tt
!!!)O•� ,., !Qbr o o • X 6 •
o o-----o �Ir� 0-=o--t:J-o 0o�---�o�o�2�s----�o�o�s-----o�o�7�s__J
Atonllc trx.t1on soWtc
Ht· Al Ni ·Cr H I - Mn HI -Mo Ni . v N i -VI
Gráfica de los valores de N.<•>n. aparentes.contra fracc1ón atóm1ca de soluto para las aleaciones diluidas, base níquel.
225

TU;�•c.�s Eu:c..I"ROQL'!�IIc�s 1'.\R.� 1.1 CosTROI. Y Esnm1o DE L� CoRROSIÓS
Discusión
De acuerdo al modelo clásico de ox.1dactón tnterna, la razón de pe netración de los óxidos internos está gobernada por los parámetros indicados en la ecuación (33). No obstante, los resultados obtenidos indican que el modelo clásico no aplica enteramente para este fenó meno, debtdo a que los valores calculados para No<•Do de la penetractón de la zona de oxidactón interna debiera ser tndependiente de la concentraCiÓn de soluto y de la naturaleza de este soluto. De hecho, como se ve en la figura 19, esos valores aumentan con el incremento de la concentraciÓn de soluto para los sistemas estudta dos, hay que hacer notar que este aumento fue mayor para el caso dt las aleaciones Ni-Al que las aleaciones Ni-Cr.
Es posible expltcar estas discrepancias, si se supone que el transpor· te de oxígeno dentro de la zona de ox.1dación interna es influenciado por los prectpttados de óxido tnterno extstentes. En particular, el flujo efectivo de oxígeno a través de la zona de ox.1dactón interna es la suma de los flujos a través de la red, el flu¡o a lo largo de las interfases incoherentes (óxido interno/metal) y el flujo a través del óxido interno. El flujo de oxígeno a través de los óxidos tnternos es muy ba¡o pero a lo largo de la interfase óxido interno/ metal puede aumentar constderablemente. Entonces, el flu¡o efectivo de oxígeno es influenciado por el tamaño, forma y onentación de los óxidos internos. La figura 21 ilustra esquemáticamente este hecho, para un número de posibles morfologías para óxidos tnternos. De esta forma, (a) indtca la situación donde los prectpttados son cilmdros continuos, perpendiculares desde la superficie hasta el frente de oxidactón interna (como ocurre en el caso del sistema Nt-Al) y el flu¡o de oxígeno a tra\·és de la zona es relativamente rápido, debido a la sustanctal contribuctón de las interfases tncoherentes, óxtdo interno/metal. En (b) los prectpttados son continuos pero no per· pendiculares a la superficte, aquí nuevamente el flujo de oxígeno es relativamente alto. De (e) a (f), los precipitados son discretos y no continuos, como ocurrió en el caso del sistema Ni-Cr. Aquí, la> interfases tncoherentes, óxido interno/metal, promueven un mere· mento en flujo de oxígeno a través de la zona de oxidación interna.
226

este incremento es menor que en los casos donde esas interfases son continuas. En (g), donde no hay precipitados, el flujo de oxígeno ocurre totalmente a través de la red del metal. Por último, en ciertos casos, donde la concentración de soluto se aproxima a la requerida para la transición de la formación de óxido interno a externo, los mismos óxidos internos producen un efecto de bloqueo parcial [(h) e (I)] y eventualmente un bloqueo total de fluJo de oxígeno G) en el metal. Este último efecto no fue observado para ninguno de los casos presentados en este trabajo.
SUPERFICIE
'
� :::J 1 (j) '
(e) (f) (g) (i) (e) (d)
(a) (b)
FIGURA 21.- Diagrama c�yuemático mostrando dtstintas postbtlidades de cómo el nu¡o efectivo de oxígeno puede ser innucnctado por el tamaño, forma y
oncnta(iún de los úxidos tntcrnos.
Conclusiones.
l . La oxidación mterna de aleaciOnes Ni Al (0.55 a ..J.. 10%Al) en cápsulas con N10/N1 de 800 a 1 100°C, dw como resultado la formación de óx1dos mternos cilindricos y conunuos. En Slmllares situaciones de oxidación las aleaciones N1-Cr desarrollan precipitados discretos y no continuos.
2. La penetración de la zona de oxidación interna generalmente decrece con el incremento en la concentración del soluto en la aleaciÓn. Para una concentraciÓn de soluto dada, las penetraclom·s son considerablemente mayores en el sistema Ni-1\l que en el Ni Cr.
227 •

Tf..C:"ICAS ELECfROQUiMICAS PARA u. CONTROl. y EsTUDIO DE u. CüRROSIÓ:-.'
3. La relativa penetración interna en los dos sistemas de aleación, puede ser explicada en términos de un aumento en la velocidad de difusión de oxígeno a lo largo de las interfases óxido interno/ metal comparado con la difusión a través de la red de níquel en la zona de oxidación interna. Con base en estas observaciones, se han propuesto modelos <31>, los cuales relacionan el coeficiente de difusión efectivo para oxígeno en la zona de oxidación interna con el coeficiente de difusión de oxígeno en la red de níquel y el aumento del coeficiente de difusión en las interfases. Estos modelos requieren que el coeficiente de difusión efectivo Incremente en forma lineal, aproximadamente, con el incremento en concentración de soluto.
228

VII.- DISTRIBUCION DE CATIONES EN SOLUCIONES SOLIDAS DE OXIDOS
Existe una diferencia en la actividad o pres1ón de 02 a través de la costra que esta fijada por la presión de disociación de óxido en contacto con el metal en la mterfase metal-óxido y por la atmósfera eXlstente en la interface óxido gas. También existe un gradiente en la concentración de vacancias catiónicas.
El trabajo clás-ico de \Vagner demuestra que el crecimiento de la costra puede esperarse que siga un comportamiento parabólico si la chfusión a través de las vacancias catiónicas es el paso que determina la velocidad de la reacción. En esta forma hay dos procesos que mantienen el crecimiento de la costra:
a) Los electrones se mueven rápidamente entre iones y huecos a todo lo largo de la película para ionizar átomos de oxígeno quimiadsorbidos sobre la superficie.
b) Los cauones se mueven más lentamente y determinan la velocidad de la reacción (movimiento a través de vacancias catiónicas). Las vacancias catiónicas se mueven en sentido contrario al de los cationes, i.e. hacia el substrato, por lo que tienen que ser absorbidos por la misma costra o por el metal.
La ley de Wagner no toma en cuenta los defectos en la estructura 01 el efecto causado por pares o límites de grano; sm embargo, da la
se para poder considerar la chstribuc1ón de catiónes a través de la lución sólida de ÓXldos que se forman en aleaciones. El caso más
cilio es el de (Ni, Co )O sobre aleaciones Ni-Co [es similar al so de (Ni-Mn)O sobre Ni-Mn o el de (Cr , Fe)203 sobre Fe-Cr as en Cr].
perfil de concentrac10nes obtenido en la microsonda, figura 22, estra un empobrecimiento de Co y enriquecimiento de Ni dende la aleación justo por debajo de la costra; esto se debe a que el
229
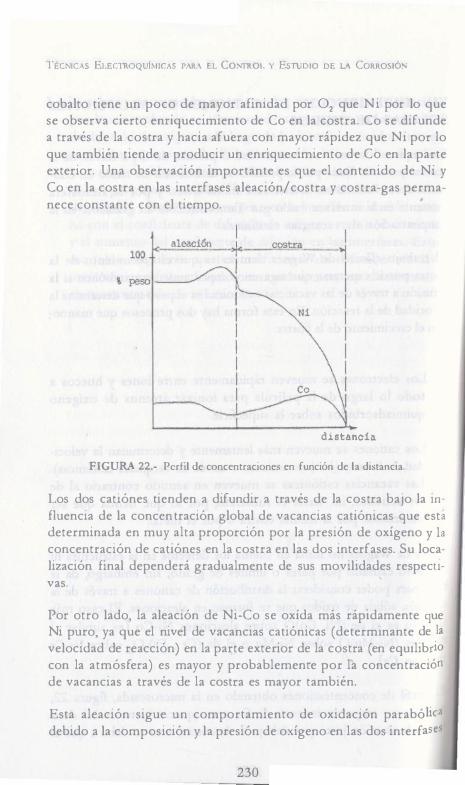
Ti"-:c�IC,\S Eu:r.nta<�Uhll<:.c\s P.\R.\ 11. Co:-;mol. Y EsTCDIO 01. l.\ CoRROSIÓ'
cobalto ttene un poco de mayor afinidad por 02 que Ni por lo que se observa cierto ennquecimicnto de Co en la costra. Co se difunde a través de la costra y hacia afuera con mayor rápidez que N1 por lo que también tiende a producir un ennquecimiento de Co en la parte exterior. Una observaciÓn importante es que el contenido de N1 y Co en la costra en las interfases aleación/ costra y costra-gas perma-nece constante con el tiempo. •
distanch
FIGURA 22.- Perfll de concentraciones en función de la distancia
Los dos catiónes uenden a difundir a través de la costra bajo la 1n
fluencia de la concentración global de vacancias catiónicas que esta determinada en muy alta proporción por la presión de oxígeno y la concentración de catiónes en la costra en las dos interfases. Su loca· lización final dependerá gradualmente de sus movilidades respectt· vas.
Por otro lado, la aleación de Ni-Co se oxída más rápidamente que Ni puro, ya que el nivel de vacancias catiónicas (determmante de la velocidad de reacción) en la parte extenor de la costra (en eqUilibrio con la atmósfera) es mayor y probablemente por la concentración de vacancias a través de la costra es mayor también.
Esta aleación sigue un comportamiento de oxidación parabólica debido a la composición y la presión de oxígeno en las dos interfases
230

de la costra, y por lo tanto la concentración de vacancias catiónicas, permanece constante con el tiempo en esas interfases; esto también quiere decir que la diferencia de concentraciones de vacancias catiónicas permanece constante con el tiempo.
Dopado - (Las reglas de Wagner-Hauffe)
Los óxidos semiconductores normalmente se dividen en dos clases:
1.- Oxidos con defectos catiónicos.
a) Metal-deficiente, con vacancias catiónicas en la subred catiónica(semiconductor tipo-p), por ejemplo NiO, CoO, FeO.
por
y
b)
ejemplo:
FeZ+
c2 -
FeH
2-o
2-0
oz·
Fe 3 1
oz·
o
FeZ +
o2·
FeH
o 2-
2-0
02-
Fe2 + o
z ·
o oz-Fe2 '
O z- Fe >+ o2 -
FeH oz-FeH
Fc2 1 o
z ·
Metal en exceso.- con catiónes intersticiales tipo-n), posiblemente ZnO
Zn2"' 02 -Znzt 2-
Zn2+ o
e z� 2 - z�· 2 - Zn2+ z -
o o o z � e zn21'
0 2-7 2+ 2-
Zn2'�' �n o
a 02- Zn2 t oz·
Znz t o 2 -
8 z�+ 21' z -
ZnH 2-Zn2+
ln o o
231
(semiconductor

TÉC�ICAS ELECTROQUÍ�IICAS PARA EL CONTROL Y Esn:010 DE LA CORROSIÓ�
11.- Oxidos con defectos aniónicos.
a) Oxígeno-deficiente, con vacancias anwntcas en la subred aniónica (semiconductor tipo-n), por ejemplo Nb205, Ta205
�� '
b) Oxígeno en exceso, con 10nes oxígeno intersticiales (semiconductor tipo p), por ejemplo U02, BeO.
En realidad las estructuras son más complejas y los óxidos contienen varios tipos de defectos que algunas veces estan ligados a impurezas.
Cuando un 10n extraño (dopante) se agrega a un óxido no estequiométrico, la electroneutralidad se debe mantener por medio de una redistribución de los defectos iónicos y electrónicos. por ejemplo con un óxido tipo P (metal deficiente) como el NiO
232

el adicionar iones intersticiales come Cr1• aumenta la concentración de vacancias catiónicas pero disminuye la concentración de huecos electrónicos:
3Ni2+ � 2Cr1+ i.e. dos iones Cr pueden sustituir a tres iones Ni, pero al haber más vacancias catiónicas habrá menos conductividad electrónica ya que el transporte de catiónes a través de la costra determina la velocidad de la reacción, en este ejemplo se aumentará la velocidad de oxidación por haber más vacancias catiónicas.
Para óxidos tipo n la adición de catiónes de mayor valencia que óxido inicial dtsminuirán la concentración de vacancias aniónicas o catiónes intersticiales reduciendo así la velocidad de oxidación. Adiciones de valencia menor aumentan la velocidad de reacción.
Sin embargo estas leyes no son una panacea que nos permitan controlar a placer la cinética de los procesos de oxidación; para que estas leyes sean obedecidas es necesario que:
l . La oxidación del metal puro debe seguir el mecanismo de Wagner de oxidación (parabólico). Las leyes no se cumplen para difusión a través de los lirrútes de grano.
2. Se debe entender completamente la estructura (incluyendo defectos): un dopante será agresivo hacia uno de los óxidos y be,néficio hacia el otro s1 se encuentran presentes óxidos del tipo n y p.
3. El dopante debe ser soluble en el óxido inicial; esta es la razón por la cual pequeñas adtcioncs pueden tener éxito.
4. Se debe estar seguro que el dopante tiene valencia esperada cuando se agrega en pequeñas cantidades.
S. En casos ideales se supone que el dopante está distribuido homogéneamente en la costra i.e. los iones iniciales y dopantes se mudan a la misma velocidad, y la velocidad de difusión de los cationes es igual.
233

TÉc:-.:JCAS ELEcnmQubtJC\S r,\RA EL CoNTROL v EsTUDIO DE u CoRROSJó:--�
En realidad, aun en los casos que alguna vez se consideraron como clásicos para apoyar estas leyes (oxidación de Ni ó Co con Cr como dopante) estos se han reconocido como mucho más complejos.
El aumento en la velocidad de reacción es el resultado de un juego muy sutil del dopado u otros efectos en el óxido semiconductor. Otros factores que posiblemente también tengan influencia son efectos de campo eléctrico, efecto de consumo de vacancias por la aleación, oxidación interna y externa, formación de poros en las regiones internas y externas de la costra y, finalmente, la reducción del área seccional dentro de la costra y en la interfase aleación-óxido causada por Cr203 o partículas de NiCr204 ó CoCr204.
Se ha encontrado que aleaciones Ni-Cr que contienen de 1 a 10% Cr a 1000°C y de 2-10% Cr a 1200°C, se oxidan con casi la misma velocidad parabólica pero más rápido que Ni puro. Si el contenido de Cr es mayor, la velocidad de oxidación es menor debido al efecto de bloqueo mecánico de las partículas de NiCr20 4 y Cr203• Un diagrama esquemático de esto se tiene en la figura 23 a continua-ción:
1 'O -
1\ I1S 'O .... (,) o r-l � Q) () �> ·«"i 1 r-i 0,) \Q 'd .Q 1 113 QJ H 1 .¡J (j u o.
2 10 % Cr FIGURA 23.- Constante de velocidad parabólica en función de la concentractón
de Cr (% peso) en el sistema Ni-Cr, en condiciones de oxidación.
234

VIII.- COMPORTAMIENTO DE RUPTURA.
Para explicar este fenómeno tomaremos el ejemplo de aleaciones Fe-Cr (14 a 25%Cr) donde se sabe que inicialmente hay un período de protección donde la costra de Cr203 es dopada con Fe. La clave para explicar el comportamiento de ruptura esta en la consideración de que hay un empobrecimiento de Cr en la aleación durante el crecimiento de Cr203. Un diagrama esquemático que muestra el punto de ruptura de un óxido se tiene en la figura 24.
órn
ruptura del 6xic1o
t FIGURA 24.- Aspecto cualitativo de la ruptura del óxido en una curva cinética.
La evidencia experimental indica que la adhesión entre el 'Cr203 dopado y la aleación se pierde debido a esfuerzos acumulados y aglutinamiento de vacancias produciendo ampollamiento {"ballooning") o deformación de la costra de óxido a menudo sobre los límites de grano, como se muestra en la figura 25.
�---....;>:;.-=::::--Fe-C r- !;p!nel
( b ) Alloy FIGURA 25.- Ampollamiento de la costra de óxido
235

T f:c:-;ICAS ELECTROQUÍMICAS PARA a CoNTROL Y Esn,;o1o DE L/1 CoRROSió�
Si el óxido levantado se rompe o se desprende completamente la aleación empobrecida de Cr será expuesta inmediatamente al medio agresivo. Esta es una situación muy severa ya que probablemente se produzca óxido de Fe en la parte exterior y una capa interior de óxido de Cr y Fe asi como un óx.1do interno de Cr. El óxido de Fe se desarrolla rápidamente y puede o no ser detenido en su desarrollo por el nuevo Cr20\ que crece en la base, o en el aglutinamiento de partículas de Cr20, que servirán como capa de curación, como se observa en la figura 26.
Fe - C r - s p.¡ :.z l e le _ 0
:) o o o c:r--..cr2o3
AHoy � e )
FIGURA 26.- RepresentaciÓn esquemática del rompimiento de la costra en aleaciO
nes Fc-Cr.
IX.- EFECTO DE TIERRAS RARAS.
Por muchos años se ha sabido que el adicionar 1% o menos de alguno de los elementos de los llamados tierras raras a las aleaciones empleadas en alta temperatura mejora notablemente la resistencia a la oxidación de las mismas tanto en situaciones isotérmicas como de ciclo térmico, y en específico mejora la adhesión a la costra. Es necesario sin embargo realizar un esfuerzo mayúsculo dentro de ésta área para comprender el efecto de las tierras raras. La reducción en la velocidad de reacción se ha atribuido al efecto de dopado o a efectos de solución sólida, pero es más probable que sea debido a la formación de capas parciales o completas de compuestos tales como Cr01 que se localizan en la interfase aleación-costra. De cualesquter
236

forma, los beneficios de la adición de tierras raras en aleaciones FeCr y aceros inoxidables son reconocidos <33-36>.
E. J. Felten <37> estudió la cinética de oxtdación de aleaciones Fe-Cr con un contenido de 25 y 37.5% Cr y pequeñas adiciones de Y y tierras raras en un intervalo de temperatura de 700°-l200°C, se observa que para las aleaciones Fe-Cr se cumple la ley parabólica pero pierde su película protectora de óxido con el astillanúento arriba de 1000°C, las aleaciones que contienen Y y tierras raras exhiben buena resistencia a la oxidación y retención de la costra, el aumento de la adhesión de la costra para aleaciones con tierras raras se atribuye a la formación de óxidos internos así como también externos. El óxido externo formado sobre las aleaciones es de Cr203 arriba de 1000°C y la aleación que contiene Y y tierras raras también oxidan internamente, la cantidad y composición del óxido interno depende de la temperatura de oxidación, en 1000°C el principal óxido es del tipo R203 (R. = Y, tierras raras) y en 1200°C es RCr03, éste óxido se encuentra en las fronteras de grano.
Y Nakamura <38> estudió el comportamiento de aleaciones Fe-Cr con adición de Y y tierras raras entre 900° y 1 200°C, trabajó con aleaciones Fe-Cr, Fe-Cr-Y y Fe-Cr-T.R. encontró que la adición de estos elementos aumenta la resistencia a la oxidación y comenta que los óxidos de Y y tierras raras sirven como sitios de rápida nucleación para un óxido protector de cromo y juegan un importante papel en la difusión del cromo.
Otros estudios también han mostrado que adiciones en el rango de 0.01-0.5% en peso de elementos tales como Y, Sm, La, Ce y/o sus óxidos producen un notable incremento en la resistencia a oxidación(39-4
2>. Chevalier y colaboradores<43> así como Bonnet y colabora
dores<4�> han estudiado el efecto de Nd203 en el comportamiento �nte a oxidación en aceros inoxidables. Fernandes y colaboradotts<Hl han estudiado el efecto de la adición de Pr203 en la oxidactón dt una aleación Fe-20Cr. En otro trabajo, Martínez-Villafañe y colaboradores<46> reportaron el efecto de adición de cantidades variables de tierras raras i.e., Nd y Pr en la cinética y morfologías de
237

TÉC:-<IC.\S ELECTROQt:íMICAS PAR.� EL Co:-JTROL Y EsTUDIO DE LA CoRROSIÓN
oxidación de una aleación Fe-13Cr. La figura 27 da un ejemplo de la cinética de oxidación en una aleación Fe-13Cr y de aleaciones Fe-13Cr-X (con X= Nd y Pr en cantidades de 0.01% a 0.03%). Aquí, es claro el efecto de la adición de tierras raras en la reducción de la cinética de oxidación.
• FeCr 1 4 • FeCrNd • • A FeCrPr • 1 2 T FeCrNdPr •
N E 1 0 u e, A E 8 � E 6 <
4
2 l o
o 2 4 6 8 1 o 1 2 1 4 1 6 1 8 20 22 2 4 Tim e, Hours
FIGURA 27.- Comportamiento cinético de alcactones Fc-Cr-X (X=Nd y ¡>r).
La morfología superficial de los productos de oxidación es también afectada. Como ejemplo, la figura 28 presenta diferentes aspectos para una aleación Fe-13Cr a) sin adición de tierras raras, b) con adición de 0.03% Nd y e) con adición de 0.01% (Nd + Pr).
(a)
238

(b)
(e)
FIGURA 28.- Morfologías típicas obtenidas para las aleacidnes a) Fe-13Cr (sin adición de tierras raras), b) Fe-13Cr-0.03Nd, e) Fe-
13Cr-0.01(Nd +Pr),oxidadas a 800°C por 24 h.
En el caso (a), los óxidos formados presentan una morfología en forma cónica, la cual es notoriamente afectada al adicionar tierras raras como se observa en (b) y (e), donde la morfología anterior ya no se observa. En cambio, su apariencia es más uniforme y con un tamaño de grano de óxidos más refinado.
•
239

T f;c:->IC:AS ELECfROQUÍ\tJC.\S PARA EL CosnoL v EsTUDIO DE LA CoRROSIÓN
X.- METODOS MAS COMUNES PARA DETERMINAR EL ESPESOR DE OXIDOS FORMADOS.
1. Colores de interferencia. Cuando un haz de luz blanca se hace caer sobre la película formada sobre un metal o aleación, puede ocurrir interferencia de ciertas longitudes de onda que se encuentran dentro de la región visible del espectro, esa interferencia dependerá del espesor de la película y el ojo la verá como luz coloreada.
Al caer la luz monocromática sobre la película, parte de ella se reflejará a partir de la interfase película/ aire otra será refractada por la película y refleJada por la interfase metal/película. La interferencia ocurre cuando
donde:
2 - Á 3Á Á 5 y
-
2n ' 2n ' 2n · · · · · · · · etc.
y = espesor de la película
Á = longitud de onda de la luz en el aire
n = índice refractivo de la película
El ancho de la banda de interferencia varÍll con el poder de la absor· ción de la película y la reflectividad de las superficies.
Una película transparente perfecta sobre una superficie reflejante perfecta dará una banda de ancho = cero. Mientras mayor sea el poder de absorción y menor la reflectividad más ancha será la banda.
La variación en el ancho de la banda de interferencia afecta el color visto por el ojo.
2. Luz polarizada. Este método depende del hecho de que un haz de luz polarizada reflejada en la superficie de un metal sufre un
240

cambio en su estado de polarizacién que dependerá del ángulo de incidencia, las propiedades ópticas del metal, la presencia de una película en la superficie y del medio ambiente. Si se comparan las propiedades ópticas de una superficie metálica limpia y una cubierta con una película de óxido, es posible obtener el espesor de la película.
3. Análisis químico. Es un método poco confiable que consiste en hacer un análisis químico directo.
4. Trazas radioactivas. Si una proporción del gas se introduce como isótopo radioactiva es posible determinar por medio de un contador, la actividad por unidad de área de la película formada. Si se conoce la relación de los isótopos radioactivos a la de isótopos estables, se podrá calcular el espesor de la película.
S. Método termogravimétrico. Los metales o aleaciones aumentan constantemente su peso durante la oxidación (ignorando factores de volatilización o desprendimiento) por lo que se puede seguir el proceso de oxidación, haciendo mediciones de cambio de peso.
6. Balanza de resorte. La velocidad de la reacción se puede seguir si se miden los cambios en la elongación del resorte.
7. Método manométrico. El consumo de 02 se puede medu: a volumen constante (PV=cte) y se puede relacionar con el espésor de la costra.
241

TÉCNICAS ELECTROQUiMIOS PARA n CoNTROL Y EsTUDIO DE LA CoRROSIÓN
XI.- REFERENCIAS
1 . O. Kubaschewski, E . LL. Evans and C. B. Alcock "Metallurgical Thermochemistry". 4th. ed Pergamon Press (1967).
2. P. Kofstad. ''Nonstoiometry, Diffusion and Electrical Conduccivity m Binary Metal Oxides". Wiley-Interscience (1972).
3. Max Hansen. "Constitution of binary Alloys". McGraw-Hill book Company, Inc. 2nd. Ed. (1958) .
4. R.P. Elliot "First Supplement". McGraw-Hill Book Company (1965).
5 . Lawrence H. Van Vlack., "Materiales para Ingeniería" Compañia Editorial Continental, S. A. (1975)
6. A.U. Seybolt, J. Electrochem. Soc., lQl, 147, (1960).
7 . Y u D. Tretyakov, V. V. Sorokin and A. P. Eras tova, J. of SolidState Chemistry .lit 263-269 (1976).
8. G.C. Wood, Corr. Sci. 2, 1 73, (1961).
9. R. L. Pickett and W D. Wood, Trans. Am. Soc. Met., 22, 347, (1934).
10. H. M. McCullough, M. G. Fontana and F. H. Beck, ibid, 13, 404, (1950).
1 1 . D. Caplan and M. Cohen, Corr. Sct., .Q, 321, (1960).
12. J. Moreau, Comp. Rend., lli, 85, (1953).
13 . H. J. Yearin, E. C. Randall and T. Longo, Corrosion, 12, 51 5, (1 956).
14. G.C. Wood and D.P. Whttde, Corrosion Sctence :1, 263, (196-l).
1 5. J. Romanski, Corr. Sci . .8., 67, (1968).
242

16. J. Romanski, Corr. Sci. �. 89, (1968).
17 . C. Wagner, Z. Elektrochem., 63, 772, (1959).
18 . I.G. Wright, "Oxidation of Iron- Nickel and Cobalt-Base Alloys" MCIC Report 72-07, June (1972)
19 . O. Kubaschewski and B.E. Hopkins "Oxidation of Metals and Alloys" 2nd. ed. Butterworths (1967).
20. Alberto Marúncz Villafañe "Contenido Optimo de Alumimo en la Resistencia a la Oxidación en Alta Temperatura de Aleaciones Hierro-Cromo" Tesis Profesional, Mayo (1978).
2 1 . J. Moreau and J. Benard, J. Chem. Phys., .5.3., 787, (1956).
22. D.P. Whittle, Y. Shida, G.C. \Xfood, F.H. Sttot and B.D. Bastow., Phi!. Mag . .M.Q, 931 , (1982).
23. R.A. Rapp, Corrosion 21, 382, (1965).
24. J.L. Meijering, Advances in Materials Research, Ed. H. Herman, Vol. 5, \Xfiley-Interscience, New York (1971).
25. F.N. Rhines, Trans. AIME ill, 246, (1940).
26. F.N. Rhines, WA. Johnson and WA. Andcrson, Trans. AIME ill, 205, (1942).
27. J.L. Meijcring, and M.J. Druyvesteyn, Philips Res. Rep. 2, 81 , (1 947).
8. F. Maak, Z. Metallkunde .5.2, 545, (1961).
9 . EJI. Stott, G.C. \Xfood, D.P. \Xfhittle, B.D. Bastow, Y. Shida and A. Martinez-Vtllafañe, So lid S tate Ionics 12, 365, (1984).
O. F.H. S.tott, A. Marttnez-Vtllafañe and G.C. \Xfood, Proc, Int. Congrcss on Metallic Corros ion .:2, 317, (1984).
l . A. Martinez-Villafañc, Ph. D. Thesis, University of Manches ter (1 983).
243

Tr.C1'1CAS Eu:CillOQUI�flí�\S PAR.\ 1.1. CosTROI. Y EsTUDIO DE L� CORROSI(>N
32. J. Benard, Lóxydation of Metaux, Gauthier Villars, Paris, (1962).
33. D. P. Whittle and J. Stringer, Phil. Trans. Roy. Soc. London, i\..2.22, 309' (1980).
34. S. KMitra, S. K Roy, and S. K Bose, Oxtd. Met., .14, 101, (1990).
35. M. J. Bennett and D. P. Moon, Mat. Sci. Forum, 5.1, 135, (1999).
36. D. T. Hoelzcr, B. A. Pint and l. G. Wright, J. Nucl. Mat., 2..8..3., 1306, (2000).
37. E. J. Felten, J. Elcctrochcm. Soc., .l.Q.8, No 6, 490, (1961).
38. Y. Nakamura, Mctall. Trans., .5., 903, (1974).
39. N. Hiramatsu and F. H. Stott, Oxid. Mct., 5.1, 479, (1999).
40. H. J. Grabke, Mat. Sci. Forum, 221, 135, (1999).
4 1 . J. Dcakin, V. Prunicr, G. C. Wood and F. H. Stott, Mat. Sci. Forum, 2.5..1, 41, (1997).
42. B.T. Kilbourn, J. of Metals, 22-27, May (1988).
43. S. Chevalicr, P.G. Bonnet, K Przybylski, J.C. Colson and J.P. Larpin, Oxid. Met., .5_1, 527, (2000).
44. G. Bonnet, J. P. Larpin and J. C. Colson, Solid State lomes, 21., 1 1 , (1992).
45. S. M. C. Fernandes and L. V Ramanathan, Surf. Eng., .l.Q, 327, (2000).
46. A Martinez-Villafañe, ]. G. Chacon-Nava, F. Almeraya-Calderon, C. Gaona-Tiburcio, R, Bautista-Margulis and J . G. GonzalezRodriguez, "Oxidation of a Fe-13Cr alloy with small additwns of Nd and Pr", articulo env1ado para publicación en Scripta Materialia., Abril, (2001).
244

8. APENDICE CONVENTIONAL DC ELECIROCHEMICAL TECHNIQUES
IN CoRROSION TESTING
J. Genesca>, J. Mendozab, R. Duranb and E. Garciab
a. Corrosion Laboratory. Department of Mctallurgical Engineenng. Faculty of Chemistry. UNAM. City Univcrsity. 04510 Méxtco D.F. e-mail: [email protected]
b. Instituto Mextcano del Petroleo. Eje Central Lazaro Cardenas, 1 52. 07730 Mexico D.F.
ABSTRACT
Metallic corrosion undcr wct conditions is generally electrochemical, occurring in corrosion cells at the metal surface. Being corroston phenomena in aqueous environments generally of electrochemical nature they are dominated by the corrosion potential, E of
corr,
the metal. Thc mixed-potential theory was developed by C. Wagncr and W Traud and was published in German tn 1 938.
Elcctrochemtcal methods in corrosion testing havc been used ever stncc thc elcctrochemical naturc of corros10n processcs was discovercd. These methods are used both for corros10n momtoring and as laboratory techniques. One of the most important applica�ons has bcen thc esttrnation of corrosion rate instantaneously by mcans of polanzation reststance, bemg of spectal rclevancc the rclationship betwcen mass loss and polarizatton reststance, whtch cannot always be deduced from Tafel parameters, because the constancy of electro�hemical parameters with time cannot a priori be assumed. The polartzation resistance measurements can also be a very useful tech-�que for the dcterrnination of time effect on corrosion. Usually
cctrochcffilcal techniques have been employed to both speed data evelopment and to betfer understand corrosion mechanisms.
� this paper, the use of clectrochemical methods for corrolion testing will be reviewed, with special attention to convcn-
?.4::.

TU�-..:Ir�>\5 ELECTROQUhiiCAS PARA EL CONTROL Y EsTuDIO DE LA CoRROSIÓN
tional de techniques such as linear polarization and Tafel extrapolation. Other techniques, including corrosion potencial measurements and p o larization methods such as p o te n tiodynamic p olarization (for example, cyclic potcntiodynamic polarization, CPP, technique providcs a reasonable, rapid method for qualitatively predicting the propensity of an alloy to suffer localized corrosion in the form of pitting and crevice corrosion), potentiostaircase and cyclic voltammetry will be described, reaching conclusions concerning their use, significance and limitations.
Keywords: DC electrochemical techniques, LPR, Tafel
INTRODUCTION
ASTM defines polarizacion as: "the change from the open-circuit electrode potencial as a result of the passage of current" [1). Polarizacien methods involve changing the potencial of the working electrode and monitoring he current which is produced as a function of time or potential. Applications of electrochemical techniques to corrosion processes have been made by many researchers during the last 75 years. The fundamental equations for this development can be found in many standard reference texts [2-4). Polarization measurements pushed the development in the 1930's of mixed potencial theory, in order for the experimental results to be explained [S] This paper (report) discusses sorne applications of our research group with DC electrochemical techniques for determination of corrosion rate. The example originate from experiments to show the effect of flow condittons on the efficiency of an inhibitor.
The obJectives of corrosion research and testing are to provide information about the reasons for and mechanisms in corrosion processes and the determination of corrosion rates and the efficiency of corrosion protection achieved by the application of appropriate alloys, protective layers, coatings and inhibitors, respeccively. Since corrosion processes are mostly of an electrochemical nature, electrochemical methods play an important role in corrosion research and tes ting.
246

The polarization resistance method, based on electrochemical concepts, enables determination of instantaneous interfacial reaction rates such as corrosion rates and exchange current densities from a single experiment. The estimation of corrosion rate based on polarization resistance (R) measurements has been used more and more extensively. In order to achieve more accurate and reproducible values of the RP severa! authors have applied periodic voltage or current waves to the specimen [6]. Under such conditions, in most cases the response of the metal-clectrolyte system 1s far more complicated than that expected for the single equivalent circuit: polarization resistance-double layer capacitance in parallcl. Nowadays, the study of corrosion mechanisms and the estimation of the corrosion rate by means of electrochemical techniques tend to be increasingly related to each other.
Thts paper studies the extent to which the techniques "Linear Polarization Resistance", LPR, "Tafel Extrapolation Method", can be applied in order to obtain representative values of the corrosion rate in a particular environment. The determination of the c\,rrosion rate at which a metal dissolves in a certain environmen� and conditions is of primary importance in laboratory and field corrosion studies. Through the years severa! techniques used to determine such corrosion rate have been developed: weight loss measurements, Linear Polarization Resistance, LPR, Tafel Extrapolation Method, Electrochemical Impedance Spectroscopy, EIS, and more recently Elec-trochemical Noise. '
The use of different techniques requires different considerations and it is common to obtain results that, at first sight, may not be representative of the system under study. Therefore, it is important to obtain as much information related to the use of different techniques as possible, in order to develop data analysis criteria which lead us to a better understanding of their use, advantages and disadvantages.
247

T(:r:sJr:As 1 �Lr:crnOQu huCAs PARA EL Co�lllOL Y EsTUDIO m: L\ CoRRos1ó:-:
LINEAR POLARIZATION RESISTANCE (LPR)
The "Linear Polarizacion Resistance" techmque used for measuring corrosion rates has been extensively used in the past years either in laboratory or field corrosion studies. This measurement technique is based on the "mixed potenttal" theory proposed by Wagner and Traud [S]. This theory postula tes that it is poss1ble to explain the corrosion reaccions by assuming that cathodic and anodic partía! reaccions are occurring at the metal-electrolyte of a corroding electrode interface at a certain "mixed" or "corrosion" potencial, E . corr
The E'"'' depends on a variety of parameters, like alloy composition, accivity of oxidizable and reducible species in the environment, its flow rate and the overpotential of relevant electrochemical reaccions at the metal/ solution interface. These processes can be influenced strongly by temperature.
The mixed potential theory takes into account two main hypotheses:
a) Any electrochemical reaccion can be divided into two or more parttal cathodic and anodic reaccions,
b) There is no net accumulacion of electric charge during an electrochemical reaccion.
lt has been demonstrated that the slope of the potencial-current curve (polarizacion curve) measured near the mixed or corrosion potenttal Ecorr of a corroding electrode is inversely proporcional to the reaccion or corrosion rate of the electrode.
The net current density, j , which circulates through a metalelectrolyte of a corroding electrode interface is given by the equacion
248

where: j= net current density across the metal-electrolyte interface, icorr = corroswn current density (A/ cm2), 11 = overpotential (E d - E ). Th1s relationship is experimentally observed between mea,urc: corr
applied electrochemical current density and potencial for a corroding elcctrode in the abscncc of competing rcduction-oxidation rcactions [2,3). The applicability of this relationship rclies on the prescncc of a single charge transfer controlled cathodic rcaction and a single chargc transfer controllcd anoche reaction. This rclationship provides the bas1s for the elcctrochcmical polanzauon tcchniquc as applied to a corroding electrode at lts corrosion potenual [7].
P. and Pe are the anodic and cathodic Tafel constants (parametcrs) (8E/8log j) dependent on the reaction mechanism and are given by:
p =
RT a (1 -a )nF p = _ RT
e a nF
(2)
(3)
Equation (1) shows hat the net current, j, is in exponential relationship with the overpotential, 11 and assumes that:
1. The slower stcp in thc electrochemlcal corrosion process is the charge transfcr at thc metal-electrolyte interface (the corrosion process is controlled only by chargc transfer). '
u. Ohmic drops in the electrolyte and surface films can be neglected.
üi. Concentration polarization is absent. 1v. The corros10n potcntial E is far from the reversible ' corr
potentials of the simultaneous anodic and cathodic reactwns occurnng.
v. There are no sccondary electrochemical reactions occurring.
l'he derivation of the polarization resistance
E.quauon (1) can be mathemaucally lmeanzed by taking its senes
249

'lú:.-.;IC.\S Eur.TROQL:huc.�s PARA 1c1. Co:--:rROL Y Esn;01o DE LA CoRROSIÓS
expansion and by neglecting higher terms when l1 /13L 0.1 . The simplified relationship has the following form:
( a¡ ) aE E "'"
= 2.303 · (ba + bJ lcorr b b a e
(4)
whcrc b, and be are the Tafel slopes for anodic and cathodic react1ons respectively, dctermined from the log j vs. E plot and are related to 13 and 13 constants by: b = 2.303 l3. • e
From equation (4) it can be seen that the variation of the current density across the interface with respect to the potencial, ncar the E is a linear function. The slope R of the linear polarization corr, p curve is called the polarization resistance. Dcfining the "Polarization Resistancc", R as:
p,
and the proportionality constant B as:
(6)
the corrosion current density is given by:
B =
which is the well-known Stern-Geary equation [8). Stern and Geary [8] onginally described a region in the vicinity of the corrosion potentíal where a linear dependence of potencial on applied current existed for a corroding electrode. They derived an equation rclating the slope of this linear region to the corrosion rate and Tafel slopes and the method became known as Linear Polarization Resistance,
250

LPR. Equation (7) gives a method of measuring corrosion rate as current denstty, J. provided that the parameters R and B are known corr, p [6-10). The Stern-Geary mathematical treatment [8] requires that polarization affects only the rates of the anodic and cathodic reactions. It can be noted that the proportionality constant, B, is dominated by the smaller of the two anodic and cathodic Tafel slopes, if unequal.
Th fact that the corrosion rate is inversely proportional to the polarization resistance is clearly seen in equation (7). Talcing the logartthm of this equation, it is seen that log Ícorr vs. log RP is linear with a slope of - 1 and has the intercept log B [7]
logRP = logB - log Jcon- (8)
Stern and Wiesert [9] conflrmed such relationship over six orders of magnitude change in corrosion rate for corroding systems.
Stern and Geary were able to elaborate their equation based on Kolotyrlcin's approach [1 1) and because advances in electrochemical lcinetics had by then produced a detailed two-term expression for the whole Tafel line starting from the reversible potentials [12). Stern and Geary simplifies the kinetic expression to provide an approximation to the charge transfer controlled reaction kinetics given by equation (1) for the case of small overpotentials with respect to Ecorr [8, 13- 14) . By applying the full Tafel expression to the case were the corrosion reaction was balanced by the cathodic reaction (reduction of H+ for example) and then malcing sorne simplifying assumptions, they obtained a convenient and convincing expression for the metallic corrosion current, the equation (7). In many cases Ícorr is reasonably insensttive to values of b and b , so that the essence of
• e
the mcthod is the measurement of R from the linear polarization p
regt.on clase to the corrosion potencial, E . In one sense, what Stern corr and Geary accomplished was the quantitative expression of the constant of proportionality between polarization resistance, RP, and corrosion rate, Ícorr
251

Tt.c-.;t<�\S 1 �tt:cmoQUI'IIC:\S P:\R.\ u. CmnRot. Y E .. •iTvoto DE L.\ CoRRostó:-.�
The R value p
In order to obtain the value of Rp it is necessary to measure the relacionship between the potencial and the current denstty which ctrculates across the metal-electrolyte interface, close to the corrosion potencial. Thts rclacionshtp is often approximately linear with potenual withm ± 5-10 m V of Ecorr This is usually done by polarizing the electro de above and bclow ±20 m V [ 16] its corrosion potential Ecorr and measuring the net current, j, circulating between this electrode and an inert or auxiliary electrode. Then the slope of this plot 1s �E/�¡. When determined from a tangent to the E-j curve at E it defines the polarizacion resistance, R . Consequently, this corr, p method is often called the linear polarization method (LPR). The slope is tndependent of the degree of lineanty [7], although the extent of the approximately linear E-j region can vary considerably amongst corroding systems. [7,15]. The constant B can be obtained in several ways:
- Considering a theoretical value of Tafel slopcs; Obtaining the values of Tafel slopes from separare polanzauon expenments in identical condiuons (potentiodynamic polarization scans or by harmonic distortion analysis);
- Obtaining the Tafel slopes by fitting the same curve used to determine the polarization resistance, Rp;
- Taken Tafel values from literature for the same or similar corrosion system or by calibrating the B constant using weight loss measurements.
In the case where the reaction mechanism is known in detall, the Tafel slopes may be estimated from the rate-determinmg step in the mechamsm of reaction [ 16] .
Processes Controlled by Mass Transfer (Diffusion)
Is is tmportant to note that if the anodic or cathodic processes do not satisfy the assumpuons stated before for equation (1), the values of j obtained by LPR technique can be erroneous and some corr
';�tions must be madc. Thc model which leads to equauon (1)
252

can not explatn completely the corrosicn process if phenomena such as active-passive transttions, reduction of oxide films or diffusion of spectes to and away from the metal-electrolyte interface occur.
If the cathodic reaction of the corrosion process is controlled by dtffusion of reactants from the bulk solution to the metal surface (e.g. reduction of oxygen) the cathodic Tafel constant, be, tends to be infimte and equation (6) becomes
B = � 2.303
So equation (7) takes the form:
. ba Jcorr = 2.303Rp
(9)
(10)
Similarly, anodic mass transport control results in [6]
B = � 2.303
(11)
Effect of the electrolyte resistance, Rs, on the LPR measurements
In Linear Polarization Res t s tance measurements it is usually constdered that the value of the solution resistance, R,, surface film reststance and all other elcctrtcal resistances between the tip of the Luggin caplllary and the test metal surface are smaller than thc·valuc of the R measured, but this ts not the general situation. "
Considenng that the soluuon rcststance, R,, ts btgger than the surfacc film reststance and all other electrical resistances between the tip of the Luggin captllary and the test metal surface, the measured polarization resistance, Rmw, ts given by the following equatton:
(12)
253

T�.CNICAS ELocrnOQUi\-!ICAS PARA u.. Co:--TROL v EsTuo1o oF. LA CoRROSióN
where A = area of the electrode. Considering equation (7), the true corrosion current density is given by:
B icorr = R _ AR
(13) p S
From equation (13) it can be seen that an uncompensated R value . p
can lead to smaller values of J corr
If the error, 8, due to an uncompensated IR drop is defined as (17]:
icorr - J;orr = icorr - 1 J;orr J;orr
(14)
where Ícorr is the true corrosion current density without IR drop effects (given by equation 7) and j*corr is the corrosion current density with IR drop effects given by:
The error becomes
. • B lcorr = R + AR p S
(15)
(16)
Equation (16) shows that the error increases with the ratio of ohm!C resistance to polarization resistance, R /R
1 p
lt has been stated that an uncompensated solution resistance can lead to a straight line in the LPR measurements and such linear behavior should not be considered as representative of the system.
ASTM standards D2776 [10] and G59 [ 1 8] describe standard procedures for conducting polarization resistance measurements. Potentiodynamic [19] , potential step, and current-step methods
254

[20,21] have all be en described to determine the linear E-j behavior of an electrode near the E The current step method has been corr cited to be faster than potentiodynamic methods and less suscepti-ble to errors associated with drift tn E [7]. LPR determined by corr potenciodynamic sweep. is likelly the most common method for assessing the corrosion rate.
Electrochemical impedance methods for determination o f p olarization res1stance has been reviewed b y Scully [7] . Determination of R is attainable in media of high resiscivity because
p Rp can be mathemacically separated from R, by taking the difference between Z(ro) obtained at low and high ro.
Errors with polarizacion resistance measurement by LPR method have been collected by Scully in his excellent review about the R
p method for determination of instantaneous corrosion rates [7]. The most common errors involve:
i. invalidation of the results through oxidation of sorne other electroactive species besides the corroding metal in question,
ii. a change in the open-circuit or corrosion potencial during the time taken to perform the measurement,
iii. use of a potential gradient (L'lE) that is too large, invalidacing the assumpcion of a linear relacionship between net current density and potencial required by equation (7),
iv. too fast of a voltage sean rate or insufficient potential hold time,
v. current and potential distributions.
THE TAFEL LINE IN CORROSION SCIENCE
Conversion of polarizacion resistance to corrosion current requires knowledge of the constant, B, of equation (7). Without knowledge of B, corrosion rates can be estimated to within a factor of two (9], assuming no other error exists. If either b. and be are accurately known, and the other esumated, then corrosion current 1s relauvely tnsensitive to errors in the estimated Tafel slope value [9]. Tafel slope values can be esumated from h1gh polanzation data and
255

this reduces the level of uncertainty in calculating corrosion rates via thc polarizatlon resistance method, but the method becomes redundant because corrosion rates can be estimated directly by back cxtrapolation of high polarization data.
An activation controlled systcm or proccss is one where the rates of thc electrochemical processes are controlled by the charge transfer across the metal solution mterface (hence the alternate term charge transfer control).
To experimentally determine kinetic parameters such as ¡· l3 and cote. a l3
c 1t is often convenicnt to produce schematic potential-current
density curves for corroding systems, and because of the exponencial nature of the relationships these are often drawn as E - log lj L curves (Tafel diagrams). Then, plotting E against logújú, activation polarization gives a straight line. For a simple activation controlled metal dissolution (anodic) process, the current density is given by Tafel's Law
· · · (Tla ) 1 = 1 a = 1 corr exp p a
The rate of a corrosion reaction depends on potential according to thc above equation, being the region or anodic Tafel domain (Tafelian zone), thc portian of the polarization curve in which po tential satis fies the condiuon that 11/13. >> l. Taking logarithms:
Working with logarithms to base 1 O and defining the anodzc Taje! constants, a. and b, ,it is possible to get thc Tafel equation for an anodic rcaction, also known as anodic Tafel line:
11a = a a + ba Iog ia where :
a a = -2.303P a In io ba = 2 .303 Pa
256

In the same way, for the cathodic Tafel domain, 11/J3c < < 1 :
. . . ( 'll e J 1 = le = - ¡eorr exp -f3:" 11 e = �e In icorr - �e In j e
that allows to determme the cathodic Taje/ constants, a and b of the e e .
Tafel equation for a cathodic reaction:
11 = a e - be Iog j e where : ac = 2.303 � c In io be = 2.303�c
POTENTIAL-CURRENT DIAGRAM
lt is very useful to know if a metal is immune to corrosion in given c1rcumstances usmg the potential-pH diagram (Pourbaix diagram), but practica! situations most often subject metals to corrosive risks and the rate of corrosion then becomes of great importance. The advent of polarization curves in corrosion science waited until Evans and Hoar [22] had demonstrated that the current in an ordmary corrosion cell was worth cquating to the metal loss. The vo�e then startcd for diagrams with two polarization curves, separate for anodes and cathodes on the corroding metal, each being plotted as a straight line against lmear scales of potencial and current. The anodic and cathodic lines wcre seen to approach each other with increasing currcnt were reached, a situation freely identified with the practica! corrosion cell where the cathodcs and anodes would be virtually short-circuited.
Polanzauon methods involve changing the potential of the workmg clectrode and monitoring the current which is produced as a funct10n of time or poten tia!. The proper method for polarization tcsung of materials is facihtated by the development of standard test
257

TíL"'IC.\S ELECTROQuí�HcAs PARA EJ. Co:-.rrnoL v EsTUDIO DE LA CoRRos1ó:--�
methods such as ASTM G5 "Standard Reference Test Method for Makmg Potentiostatic and Potentiodynamic Anodic Polarization Measurements" [23].
Severa! methods may be used in polarization of specimens for corrosion tes cing [24] . Potentio.rtatic polan·zation 1s a technique where the potential of the electrode 1s changed at a selected rate and involves maintaining a constant potential at the working electrode. Potentiodynamic polarization 1s a technique where the potencial of the electrode is varied at a selected rate by applicacion of a current through the clectrolyte. It is used for determining the corrosion current and to idencify specific corrosion reactions, such as pitcing and crevice corrosion. A variant of potenciodynamic polarization is ryclic potenliodynamic polan·zation, CPP, test technique, described in ASTM Standard Practice G 61 [25] which provides a reasonable, rapid method for qualitatively prediccing the propensity of an alloy to suffer localized corrosion in the form of pitcing and crevice corrosion. This technique was developed for stainless steels and nickelbase alloys but has been increasingly used for other alloys. This ASTM standard provides details on conduccing CPP tests but is very limited with respect to interpretation. Beavers et al [26] have recently published a paper concluding that from the analysis of CPP curves for stainless stecl-aqueous chloride system and other alloyenvironment systems, both a forward and a reverse sean should be performed in order to maximize the informauon on localized corrosion obtainable from the test technique. Thc pitcing potencial, whtch is one parameter obtained from a CPP curve, is a non-conservauve parameter for assessing susceptibility of a metal to pitting corrosion while the proteccion potential is a much more conservative parameter. Silverman [27) outlines the features that have been found useful for interpreting the polarization sean, as well as of sorne of the effects that uncompensated solution resistance, inappropriate sean rate, and improper point of sean reversa! can have on the polarization sean features and how these effects might influence the interpretation. Another variant of potentiodynamic polarization is rydic voltammetry, which mvolves sweeping the potential in a posicive direction until a predetermined value of current or potenttal
258

is reached, then the sean is immediately reversed until the original value of potencial is reached. The potentiostaircase method is another variation of potentiodynamic polarization. This refers to a techmque for polarizing an electrode in a series of potential steps where the time is constant at each potencial, and often the current is allowed to stabilize prior to changing the potencial to the next step. Another m e t h o d u s in g po la r i z a tion me thods i s eleclrochemical
potentzodynamic reactiva/ion (EPR), which measures the degree of sensitization of stain less steels.
EFFECfS OF TURBULENf FLOW ON THE EFFICIENCY OF TRIAZOLE BASED INHIBITORS (28,29)
The effect of movmg environments on the efficiency of corrosion inhtbitors is a research tapie that has been in continuous development over the recent years. The present work explores the effect of turbulent flow on the efficiency of a tnazole-based inhibitor (3-amino-1 ,2,4, tnazole) m a O.SM HCl solution, usmg the otating cylinder electrode, RCE, as hydrodynamic control system. The influence of turbulent flow on the corrosion rate of X52 pipeline steel was investigated using corrosion potencial monitoring (Eco.), polarization curves, Rp and electrochemical impedance spectroscopy, EIS. Rp, EIS and Ecorr monitoring were carried out during a period of 1 day of exposure to the test solution. Anodic and cathodic polarization curves were obtained after the 1 day period. The' experimental technique has been described in sorne detail in recent publications [29,30]. It was found that the inhibiting effect of the triazole is considerably increased by flow.
The relevant experimental parameters for each electrochemical techmque were:
a) Lineal polarization resistance (Rp): potetionstatic, potencial range ± 0.01 V (referred to the Eco.J, potencial sweep rate = 0.017 mV s 1 •
b) Electrochemical impedance spectroscopy (EIS): frequency range: 20,000 to 0.1 Hz, amplitude = 1 x 1 O 2 V.

Ti'.r�"IC.\S ELECJllOQL'ÍMIC:As PARA m.CmrTROL Y Esn:01o DI:'�' CoRROSIÓS
e) Polarization curves: At a rotation rate of O rpm a cathodic and an anodic polarization curves were measured on different steel samples, after 1 da y of exposur.e to the test solution. These Polarization curves were obtained, departing from E , at a cocr sweeping rate of 0.001 V s 1, in cathodic or anodic direction, depending on each case. Therefore, two polarization curves were obtained, one cathodic and one anodic. At a rotation ate of 1000 rpm a single polarizatton curve was obtained after 1 day of exposure time, at each inhibitor concentration. In thts particular case, the polarization curves were started at an overpotential of -0.4 V (with respect to E,0J and stopped at an overpotential of +0.4 V (referred to E ) . corr

RESULTS
Polarization Curves.
In the following section the polarization curves, obtained at different rotation rates an inhibitor concentrations, are shown.
Aft•r 24 hr. o rpm · 0 . 3 5
·O . 4 O
·O .4 5
V ., .. > ·O .5O
� w
-0.55
·0.10
·0.15
1 E ·O 1 1 E-05 1 E · 0 4 1 E -os 1 E ·02
1 (A c m ") Figure 1 - Anodic and cathodic polarization curves, after 1 day (24 hrs) of
cxposurc. Rotatton ratc = O rpm; inhibttor conccntratton = O, 25 and 200 ppm.
-241r,1000rpm 435
. _.,
415+----+-��-��..,._�-...... -�-� 1&47 1&45 -
I(Acm') Figure 2 - Anodic and cathodic polarization curves, after 1 day (24 hrs) of
cxposurc. Rotatton cate = 1000 rpm; tnh1bitor concentration = 25 and 200 ppm.
261

TÉC,tC.\5 Eu:cTROQuhuc\5 PARA EL CoNTROL. v &rumo or: t.A CoRROSIÓN
The following tables summarize the electrochemical parameters: anoclic and cathoclic Tafel slopes (b. and b), corrosion potencial (Eco), corrosion current density Gco) and Stern-Geary constant (B), calcu-lated from the polarization curves shown in F1gures 1 and 2.
Tablc 1 Elcctrochcmical paramctcrs calculatcd from Figure 1 (O rpm, O ppm).
b . b E J((lfT B < con
V dccadc 1 V dccadc -l V VS SCE A cm·2 V
\nodic o 0782 - -0.5003 2.87x10 5 0.0176
Cathodtc o 0843 - -0.5083 7 45x10 1
Table 2. Elcctrochemical parametcrs calculatcd from Figure 1 (O rpm, 25 ppm).
b . b E J((lfT B ' '''"' A cm·2 V dccadc "1 V dccadc "1 V VS SCE V
t\nodic 0.0721 - -0.4905 2.18x 1 0 1 0.0167
Cathodic - 0.0821 -0.5037 4.53x10 1
Tablc 3. Elcctrochcmtcal paramctcrs calculatcd from Figure 1 (O rpm, 200 ppm).
b . b E J,� B < <O"
V dccadc 1 V dccadc -1 V VS SCE A cm·2 V
t\nodtc 0.0729 - -0 4932 1.23x105 o 0163 Cathodic - 0.0776 -0.4997 2.23x1 0 5
Table 4. Elcctrochcrrucal paramctcrs calcuhtcd from Joigurc 1 (1000 rpm, 25 and 200 ppm).
b b E i,.,.,. ' B . ' con
V dccadc "1 V dccadc -l V vs SCE A cm·2 V
25 ppm 0.1 259 0.10 1 1 -0 5057 9.59x 1 0 6 0 0244
200 ppm o 1 1 05 o. 1046 -0.5125 9.48x106 0.0233
262 __.
1

Corrosion rate
Using the data obtamed from the EIS analyses and the data obtained from the l1near polarization resistance (R� experiments, the instantaneous corrosion current density values G ) can be estimated, corr using the values of Stern-Geary constants from tables 1 to 4. The variation of jcorr with time and inhibitor concentration, at O rpm, is sh �
1.2E-04
1.0E-04
8.0E�5 '1 E u � 6.0E�5 J
4.0E�5
2.0E�5
O.OE+OO o 0.2
-----, e O,OR4>
-o-0.0 EIS
0.4 0.6 0.8 1.2 T (days)
Figure 3 - Valucs of j'""' csumatcd by EIS and RP, as a function of time and tnh1b1tor conccntratJon. Rotatlon ratc = O rpm. Lcgcnds: thc fl.rst numbcr indicatcs rotation ratc, thc sccond numbcr indicatcs mhibitor conccntranon, thc lcttcrs md1catc
thc clcctrochcmlcal tcchniquc uscd.
Figure 3 demonstratcs the dependency of the measured jcorr with mhibitor concentrat10n. As the inhibitor concentration incrcases the measured corros10n rate decreases. This is a somehow cxpected rcsult. It is interesting to note the feature that shows that at the bcginning of the experiment jcorr increases with time and then decreases to a steady value. This feature can be observed at O and 25 ppm. However, at 200 ppm of inhtbitor concentratton, the tendcncy of ¡· is to dccrease at the beginning of the experiment the incrcasc curr
263

Tf'.CStC\S Eu.cntOQUiMtCAS PARA EL Co:-.uoL Y Esruoto DE t.A CoRRostó:-:
up to a steady value. This behavior suggests that the surface of the electrode i s changing with time and therefore the inhibitor surface concentration of is also changing with time.
Figure 4 shows the measured dependency of icorr with time and inhibitor concentration, at a rotation rate of 1000 rpm. The axis limits in Figure 3 are maintained in Figure 4, 1n order to facilitate com-panson.
1.2E·04
• 1000,25 Rp
1 .0E·04 --<l- 1000,15 Ell ....,_ 1000,200 Rp ..o0-- 1 000,200 Ell
8.0E·05
.,� u � II.OE-05
.J 4.0E.05
2.0E.05
O.OE+OO +--�-+--�-+--�-+--�-+--�-+--�---1 o 0.2 0.4 o.e 0.8 1.2
T (days) Figure 4 - Values of i,.,,, cstunatcd by EIS and RP, as a function of time and tn·
htbttor conccntration. Rotation ratc = 1000 rpm Lcgcnds: thc first numbcr mdtcatcs rotatton ratc, the sccond numbcr indtcatcs inhibitor conccntration, thc
lcttcrs mdicatc thc clcctrochcmical tcchmquc uscd.
Figure 4 demonstrates that, at 1 000rpm, icorr is independent of the inhibitor concentration. The calculated values of icorr are practically the same, at bulk concentrations of inhibitor of 25 and 200 ppm of inhibitor. These results indicate that, the controlled hydrodynamics favours inhibitor migration from the bulk of the solution towards the surface of the electrode. This increased migration rise inhibttor concentration and coverage at the surface of the electrocle, decreasing lcorr· It is also interesting to note the excellent correlation betwcen thc J. values calculated by EIS and R . corr p
264

Figure 5 shows the calculated instantancous efficiency of thc inhibt tor, referred to the values of icorr at O rpm and O ppm of mhibitor bulk conccntration. This Figure demonstratcs that the calculatcd effictcncy of the inhtbitor reaches a maxtmum value of about 94°'o at a concentration of 200 ppm and O rpm, and then dccreascs with time to values of about 60%.
At a rotation rate of 1000rpm and 25 ppm bulk concentration of inhib1tor, a maximum value of efficiency of about 92°1o is rcachcd and thcn decreascd to a value of 90% approximatcly. This value of efficiency is practically constant during all the tcsting ttme.
At a rotation rate of 1000 rpm and a 200 ppm bulk conccntration of inhibitor, a maximum value of cfficiency of about 95% is reachcd and thcn dccreascd to a value of 92% approximatcly. This value of effictcncy is practically constant during all the tcsting time.
It is clear that the efficicncy of the tested inhibitor ts strongly dependcnt on the flow conditions of the system.
100 90 80 70
,.,
l! 60
.! 50 u 11: w � 40
30 20 1 0
o o
e 0,25 Rp -o-0,25 EIS -e-0,200 Rp -o-0,200 EIS ...... 1000,20 Rp -6- 1 000,20 EIS -...- 1000,200 Rp -1000,200 EIS
0.2 0.4 0.6 T (days)
o.a 1.2
Figure 5 - Estimatcd valucs of cffictcncy of thc tnhtbttor calculatcd by 1 IS and
Rr, as a function of time, inhibttor conccntration and rotauon ratc. Lcgcnus. thc
fir:;t numbcr indicares rotation ratc, thc scconu numbcr mdtcatcs inhtbttor con-
ccntration, thc lcttcrs tndtatc thc clcctrochcmtcal tcchnique uscd.
265

Tl:c:-.itCAs Et.rT.TROQuhuc.\S r,IRA EL Co:--:-rnor. Y EsTuoto 01: •-� CoRRostó:-�
Corrosion potential (E ) . corr
Figure 6 shows the variation of the measured corrosion potential (E ) with time, rotation rate and inhibitor concentration. In gen-con era, at a rotatton ratc of O rpm Ecorr increases as the concentration of 1 : the inhibitor increases. However at a rotation rate of 1000rpm Ecorr decreases dramatically in comparison with the values measured at O rpm. Thts behavior ts conststent with the previous observations.
-0.47
-0.48
-0.49
-0.50
¡¡¡ u -0.51 (/) .. > � -0.52 w
-0.53
-0.54
-0.55
tJ. .t. .t. tJ. • ()
-o.oRp -O,OEIS
e 0.25 Rp O 0.25 EIS .t. 0,200 Rp tJ. 0,200 EJS • 1000.25 Rp O 1000,25 EIS • 1000,200 Rp <> 1000.200 EIS
-0.56 -+--....--t----,--t-�--+--,--+--..,..--+--.....---1 o 0.2 0.4 0.6
T (days) 0.8 1.2
Figure 6 - Mcasurcd valucs of E"'" as a funcuon of umc, mhibttor conccntrauon and rotation ratc. Lcgcnds: thc flrst numbcr indtcatcs rotatton ratc, thc sccond
numbcr indicatcs inhtbitor conccntration, thc lcttcrs mdicatc thc clectrochcmical tcchniguc tn whtch thc E'"" was mcasured
266
'

CONCLUSIONS
The LPR or polanzatlon resistance method enables reliable (consistent) determinations of instantaneous corrosion rates.
The results obtained in this work support the following conclusions, regarding the behav10ur of the 3-amio-1 ,2,4, triazole, as a corrosion inhibitor.
The inhibiting effect of the tested substance 1s strongly dependent on the hydrodynamic conditions of the environment. As the turbulence of the environment increases, the cfficiency of the inhibitor also increases. This increment is associated to an increased migration of the inh1bitor, from the bulk of the solution towards the surface of the corroding electrode. This increased migration can genera te a better covcrage of the surface of the corroding electrode and thercfore a decrement of i .
corr
When the efficiency of this inhibitor is estimated, controlled hydro-dynamic conditions must be taken into account. Otherwise the value of efficiency is calculated can be underestimated.
REFERENCES
[1 ] ASTM G 15 (latest rev1sion), "Standard Termmology Relating to Corrosion and Corrosion Testing", Annual Book of.ASTM Standards, Volume 3.02, Metal Corrosion, Amencan Society for Testing Materials, West Conshohocken.
[2] J.O'M. Bockris and A.K.N. Reddy, Modern Electrochemistry, Vol. 2, Plcnum Press, New York (1 977).
[3] A .J. Bard and L. R. Faulkner, Electrocbemzcal Metbods, J oh n Wiley and Sons, Ncw York (1 980) .
[4] E. Gilcad1, Electrode Kinctics for Chcmists, Chem1cal Englneers and Materials Scicntists, VCI I Publishers (1993).
[S] C. Wagner and \XI. Traud, Z. Elektrochem. 44, 391 (1938). [6] l. Epelboin, C. Gabriclli, M. Keddam and Il. Takenouti, "Al
ternating-Current Impedance Measurements Applied to Cor-
267

T !;c.,:tC.\S EuxrnOQUÍMIC.A.S 1'.\RA EL Co�oL Y Esruo1o DE LA CoRRos•ó�
rosion Studies and Corrosion-Rate Determination" In: Electrochemical Corrosion Testing; ASTM STP 727, (Eds. F. Mansfeld and U. Bcrtocci), American Society for Testing Materials, Philadelphia 1981 , pp. 1 50-166.
[7] J.R. Scully, "The Polarization Resistance Method for Determination of Instantaneous Corrosion Rate: A review", NACE Corrosion/98, Houston (1998). Paper # 304.
[8] M. Stern and A.L. Geary, J. Electrochem. Soc. 104, 56 (1957.) [9] M. Stern and E.D. \Veisert, "Experimental Observations on
the Relation Between Polanzation Resistance and Corrosion Rate", in ASTM Proceedings, Vol. 59, p. 1280, ASTM, (1959).
[10] ASTM D 2776 (latest revision), "Test Methods for Corrosivity of Water m the Absence of Heat Transfer", Annual Book of ASTM Standards, Vol. 03.02, Metal Corrosion, ASTM. Wes t Conshohocken
[ 1 1 ] Y a.M. Kolotyrkin and A.N. Frumkin, Dokl. Akad. N a u k. SSSR 33, 445 (1941) .
[ 12) J.O'M. Bockris, Modern Aspects of Electrochemistry, Chapter IV, page 1 80 et seq, Butterworth, London (1954) cited by E. C. Potter in Electrochemistry p. 236).
[13] M. Stern and R.M. Roth,J. Electrochem. Soc. 104, 390 (1957). [ 14] M. Stcrn and A.L. Geary, J. Electrochem. Soc. 105, 638 (1958). [ 1 5] D.A. Jones, Principies and Preven/ion of Corrosion, Macmillan
Publishing, New York (1 992). [16) ASTM G 102 (latest revision), "Standard Practlce for Calcula
tion of Corrosion rates and Related Information from Electrochemtcal Measurements", Annual Book of ASTM Standards, Vol. 03.02, Metal Corrosion, ASTM. West Conshohocken.
[ 17) M.G. Fontana and R.W Staehle, Advances in Corrosion Scunce and Technology, Vol. 6, Plenum Press, (1 976)
[ 18) ASTM G59 (latest revision), "Standard Practice for Conductmg Potentiodynamic Polarization Resistance Measurements", Annual Book of ASTM Slandards, Volume 3.02, Metal Corrosion,
American Society for Testing Materials. \Vest Conshohocken. [ 1 9] F. Mansfeld, in Electrochemical Techniques for Corrosion, R.
Babotan, Ed. NACE, Hosuton (1 977), p. 18 . [20] D.A. Jones and N. D. Greene, Corrosion 22, 198 (1966).
268

(21] D.A. Jones, Corros10n 39, 444 (1?83). (22] U.R. Evans and T.P. Hoar, Proc. R. Soc. 137, 343 (1932). (23] ASTM GS (latest revision), "Standard Reference Test Method
for Making Potentiostatic and Potcntiodynamic Anodic Polarization Measuremcnts" Annual Book of ASTM Standards, Vol
ume 3.02, Metal Corrosion, American Society for Testing Materials, \X'est Conshohocken.
(24] A.C. Van Orden, "Applications and Problems Solving usmg the Polanzation Technique", NACE Corrosion/98, I Iouston (1998). Paper # 301.
(25] ASTM G 61 (latest revision), "Conduc t1ng Cyclic Potentiodynamic Polarization Measurements for Localized Corrosion Susceptibility of Iron-, Nickel-, and Cobalt Based Alloys", Annual Book of ASTM Standards, Volume 3. 02, Metal
Corrosion, American Society for Testing Materials. West Conshohocken.
[26] J.A. Beavers, C.L. Durr and N.G. Thompson, "Unique Interpretations of Potentiodynamic Polarization Rechnique", NACE Corrosion/98, Houston (1998). Paper # 300.
(27] D.C. Silverman, "Tutoría! on Cyclic Potentiodynamic Polarization Technique", NACE Corrosion/98, Houston (1998). Paper # 299.
[28] J. Mendoza, R. Duran and E. Garcia, "Effects of Turbulent Flow on The Efficiency of Triazole Based Inhibitors", NACE Corrosion/2002, Houston (2002). Paper # 02491 .
[29] J. Mendoza, R. Duran, J. Genesca and E . Garcia, "Efecto del flujo turbulento sobre la eficiencia de un inhibidor de corrostón base trtazol", Expo Protection & Anti-Corroston Mextco City, June 1 1 - 1 2, 2002, NACE In ternational (Mexican Section)(2002).
[30] "Techniques for Monitoring Corrosion and Related Parameters in Field Applications" NACE International Publit'ation #T199, Item No. 24203, NACE Internacional, Houston, ( 1999).
269


ESTA EDICIÓN SE TERMINÓ DE IMPRIMIR EN JULIO DE 2003
EN LOS TALLERES DE LITOGRAFIA MIER Y CONCHA, S.A. DE C.V
MÉXICO, D.F.



















