
SINTESIS TOTALES -2 - pmcarda.files.wordpress.com · Para la síntesis del compuesto 1 se eligió...
144
SINTESIS SINTESIS TOTALES TOTALES-2 Miguel Carda y Eva Falomir Miguel Carda y Eva Falomir Universidad Jaume I Universidad Jaume I Me H H Me Me Me O O OH Me Elisabetina A
Transcript of SINTESIS TOTALES -2 - pmcarda.files.wordpress.com · Para la síntesis del compuesto 1 se eligió...

SINTESIS SINTESIS TOTALESTOTALES --22
Miguel Carda y Eva FalomirMiguel Carda y Eva FalomirUniversidad Jaume IUniversidad Jaume I
Me
HHMe
Me
Me
O
O OH
Me
Elisabetina A

Síntesis de (+)-dicroanona 2
Tabla de contenidos
1. Síntesis de (+)-dicroanona (Stolz, 2006) 1
2. Síntesis de elisabetina A (Mulzer, 2003) 19
3. Síntesis de β-eritroidina (Fukumoto, 2006) 35
4. Síntesis de estemospironina (Williams, 2001) 47
5. Síntesis de FR901464 (Koide, 2006) 57
6. Síntesis de guanacastepeno (Overman, 2006) 71
7. Síntesis de harzifilona (Sorensen, 2004) 87
8. Síntesis de ipomoeasina E (Fürstner, 2007) 93
9. Síntesis de manassantina A (Hanessian, 2006) 115
10. Síntesis de (±)-merrilactona A (Frontier, 2007) 131

Síntesis de (+)-dicroanona 1
SÍNTESIS DE (+)-DICROANONA
Aislamiento: La dicroanona se ha aislado de la especie vegetal Salvia dichroantha procedente de Turquía. Actividad biológica: La dicroanona pertenece a una clase de productos naturales, descubiertos recientemente, que poseen un núcleo tricíclico [6-5-6]. Su actividad biológica todavía se está estudiando, pero ya se ha demostrado que son capaces de inhibir el enzima aromatasa y, por tanto, podrían utilizarse como agentes terapéuticos para carcinomas cuyo desarrollo depende del nivel de estrógenos.
Retrosíntesis
La primera operación del análisis retrosintético de la dicroanona convierte el anillo quinónico de ésta en un anillo aromático (véase el intermedio 1 del esquema 1).1
Esquema 1
1 M. Fadden, B. M. Stolz, J. Am. Chem. Soc. 2006, 128, 7738.

Síntesis de (+)-dicroanona 2
El areno 1 se preparará, vía benzoanelación, a partir de la enona 2, que se obtendrá de la alilcetona 3 mediante oxidación de Wacker seguida de condensación aldólica. El fragmento alílico que contiene la cetona 3 se instalará mediante un proceso de alilación enantioselectiva en el carbonato mixto 4, que se sintetizará a partir de la 2,2,6-trimetilciclohexanona, compuesto comercialmente accesible.
Síntesis
1. Síntesis del areno 1
Para la síntesis del compuesto 1 se eligió como material de partida la 2,2,6-trimetilciclohexanona, la cual, por enolización y O-acilación con cloroformiato de alilo, se convirtió en el carbonato de enol 4 (véase esquema 1). Este compuesto se sometió a la reacción de alilación asimétrica Tsuji-Trost con el catalizador de paladio(0) Pd2(dba)3 en presencia del ligando quiral (S)-t-Bu-PHOX. Esta reacción proporcionó la α-alilcetona 3 con un 91% de exceso enantiomérico (véanse comentarios). La oxidación de Wacker de 3 (véanse comentarios) proporcionó la dicetona 5, cuyo tratamiento básico dio lugar a la enona 2. A continuación, se llevó a cabo una adición Michael del enolato de litio derivado de la enona 2 a la metil vinil cetona, lo que condujo a la dicetona 6, la cual, por reacción con potasa dio lugar a la enona 7. El tratamiento de la enona 7 con LDA generó el correspondiente enolato cinético, que se capturó con N-feniltriflimida para dar el triflato de enol 8. El triflato 8 se convirtió en el compuesto 9 mediante reacción de acoplamiento de Kumada con bromuro de isopropenilmagnesio (véanse comentarios). La aromatización de 9 inducida por HCl proporcionó el compuesto 1.

Síntesis de (+)-dicroanona 3
Esquema 1
2. Pasos finales En el esquema 2 se indican los pasos finales en la síntesis de la dicroanona.
La oxidación del anillo aromático se consiguió de forma indirecta por formilación. Así, el areno 1 se trató con diclorometil metil éter en presencia de TiCl4, lo que generó una mezcla 10:1 de benzaldehídos, que fueron separados por cromatografía (véanse comentarios). El isómero mayoritario 10 se convirtió en el fenol 11 por oxidación de Baeyer-Villiger (reacción de Dakin, véanse comentarios). El tratamiento de 11 con IBX 12, que se transformó en el fenol 14 por reacción con pentafluorotiofenol. La oxidación completa de 14 con oxígeno molecular en medio básico (véanse comentarios) condujo a la formación de 15 el cual experimentó, in

Síntesis de (+)-dicroanona 4
situ, la saponificación de la unidad de tioéster vinílogo, para dar lugar a la dicroanona sintética.
Esquema 2

Síntesis de (+)-dicroanona 5
Comentarios 1. Obtención de αααα-alilcetonas mediante alilación asimétrica de Tsuji-Trost
R1R2
O1.base
2. O
ClOR1
R2
O O
O
"Pd(0)"
(S)-L*R1
R2
O(S)
La α-alilación de cetonas catalizada por paladio se basa en la metodología, desarrollada por Trost denominada alilación asimétrica cinético-dinámica (DYKAT, véase síntesis de broussonetina G).
Cuando se utilizan enol-carbonatos de alilo como sustratos en la alilación DYKAT, el contratión que se genera al formarse el complejo catiónico π-alílico entre el Pd(0) y el alilo es el anión enolato, que se coordina con el Pd formando un complejo neutro. Éste experimenta un proceso de eliminación reductora que conduce a la formación de la α-alilcetona y a la regeneración del catalizador (esquema 3).
La diferencia entre el mecanismo general de las reacciones DYKAT y el mecanismo de alilación de enolatos reside en el hecho de que en el primero se produce un ataque directo del nucleófilo al catión π-alílico, mientras que en el segundo la alilación tiene lugar dentro de la esfera de coordinación del metal mediante un proceso de eliminación reductora
Esquema 3

Síntesis de (+)-dicroanona 6
En este punto hay que señalar que la eliminación reductora forma el compuesto resultante de la C-alquilación del enolato, en detrimento del producto de O-alquilación.
Algunos de los ligandos quirales que se emplean en las reacciones DYKAT, diseñados por Trost y Scholtz, se indican en la figura 1.
Ph2P N
O
t-Bu
(S)-t-Bu-PHOX
O
NH
PPh2
Ph Ph
HN
O
Ph2P
PPh2
PPh2
(R)-BINAP(S,S)-ligando de Trost
OMe
PPh2
(R)-MOP
N
PPh2
(R)-QUINAP
P P
(R,R)-Me-DUPHOS
O
O PPh2
PPh2
(R,R)-DIOP
Figura 1 2. Oxidación de Wacker
R
PdCl 2 (cat)
DMF/H2OR
OCuCl (1 eq), O 2
La oxidación de Wacker se emplea en la conversión de alquenos terminales en metilcetonas. La reacción se lleva a cabo mediante tratamiento del alqueno con agua y oxígeno en presencia de cantidades catalíticas de PdCl2 y de CuCl. Esta reacción fue descubierta por Smidt y col. en la empresa Elecktrochemie Industrie G.M.BH, en Munich (Alemania) empresa subsidiaria de Wacker Chemie. Hoy en día se utiliza en la producción industrial de acetaldehído (15.000 toneladas/año), que se obtiene por oxidación Wacker del etileno.
Cuando el proceso Wacker se aplica en la escala de laboratorio, con una mezcla de H2O/DMF como disolvente, se denomina de Wacker-Tsuji.

Síntesis de (+)-dicroanona 7
El mecanismo de esta reacción ha sido ampliamente estudiado y se ha podido demostrar que:
a) Cuando la reación se lleva a cabo con C2D4 en agua se obtiene CD3CDO, y con C2H4 en D2O se obtiene CH3CHO. Por tanto, en la reacción no se produce intercambio H/D por lo que hay que descartar que se produzca un proceso de tautomerización ceto-enólica
b) No se ha observado un efecto isotópico cinético apreciable con reactivos totalmente deuterados (kH/kD=1.07), de lo que se deduce que la etapa de transferencia de hidruro no es la determinante de la velocidad global del proceso.
c) Si que se ha observado un efecto isotópico competitivo con C2H2D2 ((kH/kD=~1.9), de lo que cabe deducir que la etapa determinante de la velocidad global del proceso tiene que tener lugar antes de la formación del producto de oxidación.
El catalizador de paladio que opera en el proceso es el tetracloropaladiato I (véase el esquema 5).
R
Cl
2 CuCl
2 CuCl2
2 HCl + 1/2 O2
H2OPd
Cl
Cl Cl
Cl
PdCl
Cl ClII
H2O
2-
Cl
PdCl
Cl
II
H
PdCl
Cl
OHR
II
R
H3C R
O
+
adiciónsin
I
II
III
V
VII
II
OH2
PdCl
Cl
R
-H
IV
II
OH
PdCl
Cl
R
H2O
R
HOH2O-H2O
PdCl
ClVI
OHH
-
R
PdCl
ClVIII
CH2
H
R
OH
-
- -
- -
Esquema 5
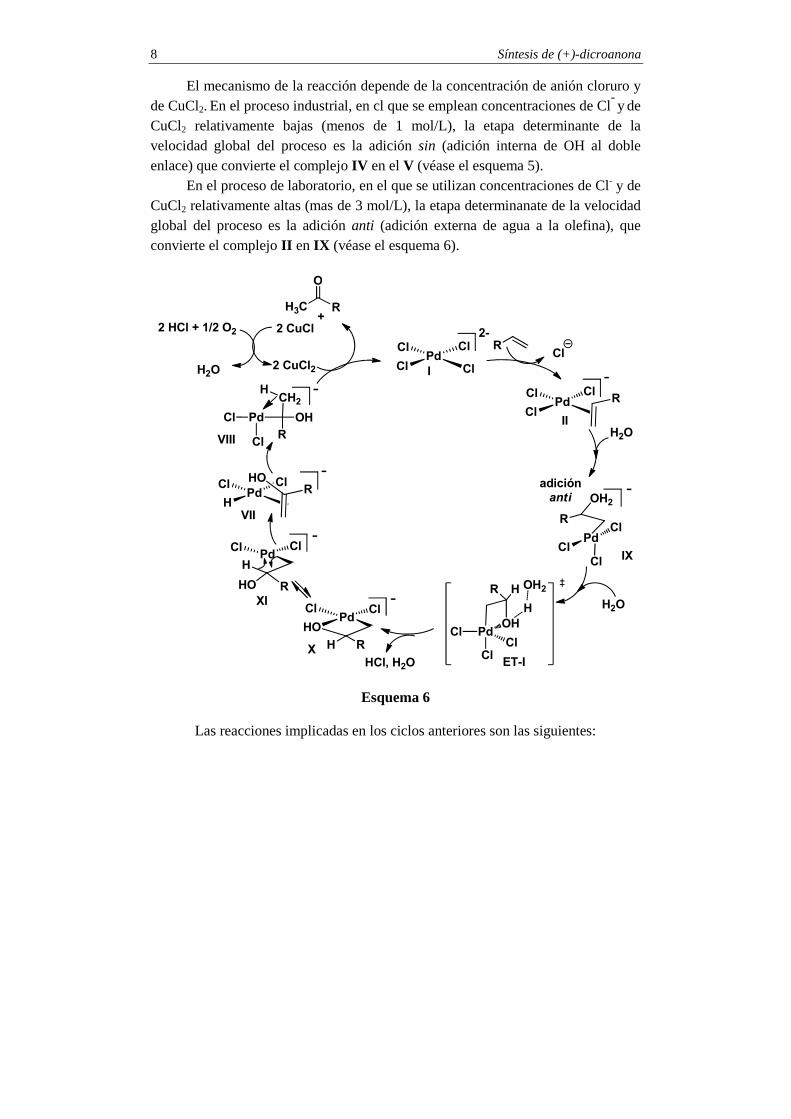
Síntesis de (+)-dicroanona 8
El mecanismo de la reacción depende de la concentración de anión cloruro y de CuCl2. En el proceso industrial, en cl que se emplean concentraciones de Cl
- y de CuCl2 relativamente bajas (menos de 1 mol/L), la etapa determinante de la velocidad global del proceso es la adición sin (adición interna de OH al doble enlace) que convierte el complejo IV en el V (véase el esquema 5).
En el proceso de laboratorio, en el que se utilizan concentraciones de Cl- y de CuCl2 relativamente altas (mas de 3 mol/L), la etapa determinanate de la velocidad global del proceso es la adición anti (adición externa de agua a la olefina), que convierte el complejo II en IX (véase el esquema 6).
R
2 CuCl
2 CuCl2
2 HCl + 1/2 O2
H2OPd
Cl
Cl Cl
Cl
PdCl
Cl Cl
H2O
2-
Cl
PdCl
R
H3C R
O
+
I
II
ET-I
IX
PdCl
ClCl
OH
H
OH2
Cl
Cl
OH2
PdCl Cl
HO
PdCl Cl
HO
H
H
R
R
R
R H
PdH
Cl ClR
HO
VII
PdCl
ClVIII
CH2
H
R
OH
H2O
HCl, H2O
adiciónanti
-
-
-
-
-
-
X
XI
Esquema 6
Las reacciones implicadas en los ciclos anteriores son las siguientes:

Síntesis de (+)-dicroanona 9
La reacción ajustada es muy simple: el alqueno se transforma en la metilcetona por reacción con el oxígeno molecular.
El Pd(0) que se genera en la etapa de acoplamiento reductivo (véase el esquema 7) es oxidado por el CuCl2, que se convierte en CuCl. Éste, a su vez, es oxidado por el oxígeno molecular y reoxidado a CuCl2.
En el esquema 7 se indica el mecanismo del proceso de formación del doble enlace carbonílico. Se ha propuesto que la migración de hidruro desde la agrupación OH tiene lugar mediante la intervención de un estado de transición de 4 eslabones (véase ET-II del esquema 7).
Esquema 7
Sin embargo, estudios computacionales recientes han demostrado que la interveción de un estado de transición de 4 eslabones es energéticamente desfavorable, por lo que se ha propuesto un mecanismo alternativo para el proceso de eliminación reductora, en el cual el atomo de hidrógeno se coordina con el átomo de cloro, que a su vez se encuentra asistido por una molécula de agua (véase ET-III del esquema 8).2
2 J. A. Keith, J. Oxgaard, W. A. Goddard, J. Am. Chem. Soc. 2006, 128, 3132.

Síntesis de (+)-dicroanona 10
Esquema 8
3. Acoplamiento de Kumada
R2Pd(0) cat.
XR1 MgX MgX2++ó
Ni(II) cat.
R1 R2
El acoplamiento de reactivos de Grignard con haluros de vinilo o arilo se describió en 1972 por los grupos de Kumada y Corriu.3 A pesar de la gran economía atómica de este proceso, la reacción está limitada a haluros organicos que no reaccionen con los reactivos organomagnesianos, como los haluros de alquenilo (véase el esquema 9).
Esquema 9
Los complejos de paladio(0) se introdujeron como catalizadores en los acoplamientos de Kumada en el año 1975.4
El ciclo catalítico de la reacción de Kumada es el típico de las reacciones de acoplamiento catalizadas por paladio. Se inicia con la inserción oxidante del haluro de alquilo al complejo de paladio(0) (véase esquema 10). A continuación, se produce la etapa de transmetalación con el reactivo de Grignard, que va seguida de
3 a) K. Tamao, K. Sumitani, M. Kumada, J. Am. Chem. Soc. 1972, 94, 4374. b) R. J. P. Corriu, J. P. Masse, J. Chem. Soc. Chem. Commun. 1972, 144. 4 M. Yamamura, I. Moritani, S-I. Murahashi, J. Organomet. Chem. 1975, 91, 39.

Síntesis de (+)-dicroanona 11
la etapa de eliminación reductora que origina el producto de acoplamiento y regenera el catalizador.
Esquema 10 En la síntesis de la dicroanona el ciclo catalítico que se establece es el que
aparece en el esquema 11.
Esquema 11 El tratamiento del producto de acoplamiento con una disolución acuosa de
ácido clohídrico permitió la obtención del compuesto 1 a través de reacciones

Síntesis de (+)-dicroanona 12
ácido-base, como las que se muestran en el esquema 12, y que conducen a la aromatización del anillo de seis miembros.
Esquema 12
4. Formilación del areno 1 La reacción del compuesto 1 con diclorometil metil éter y TiCl4 genera,
mayoritariamente, el derivado formilado 10.
La reacción que tiene lugar es una sustitución electrofílica aromática, en la cual se genera el electrófilo en el medio de reacción a partir del diclorometil metil éter y TiCl4 (véase el esquema 13).
La reacción SEAr entre 1 y II puede formar tres intermedios regioisoméricos, de los cuales, III y IV son los más favorecidos electrónicamente, ya que presentan un mayor número de estructuras resonantes. Desde el punto de vista estérico la formación del intermedio III está mucho más favorecida que la formación de IV , hecho que se comprueba experimentalmente, ya que la reacción genera una mezcla de regioisómeros en relación 10:1.
Finalmente, la hidrólisis del producto de la reacción SEAr conduce al compuesto formilado.

Síntesis de (+)-dicroanona 13
TiCl4
1
OMe
Cl
Cl OMe
Cl
ClCl4Ti OMe
ClTiCl5 +
III
1. Generación de la especie electrofílica
2. Sustitución electrofílica aromática
OMe
Cl II
HCl
OMe
10 : 1
Cl
MeO
HIII IV
ClOMe
Cl
MeO
H2O
HCl
CHO
9
ClOMe
+
10 : 13. Hidrólisis
MeOH
OMeCl
+
Esquema 13
El método de formilación empleado en la síntesis de la dicroanona es la aplicación concreta de un método diseñado originalmente para la o-formilación de fenoles electrónicamente ricos (esquema 14).5
OHR Cl2CHOCH3
1. TiCl4
OHR
CHO+ + CH3OH + 2 HCl
2. H2O
Esquema 14
5 O. García, O. E. Nicolás, F. Albericio Tetrahedron Lett. 2003, 44, 4961.

Síntesis de (+)-dicroanona 14
La elevada regioselectividad de este método se debe a la coordinación del titanio con los átomos de oxígeno del fenol y del diclorometil metil éter, tal y como se muestra en el esquema 15. Esta coordinación no solo favorece la regioselectividad sino que, además, incrementa la electrofilia del diclorometil metil éter, que resulta atacado por el anillo aromático y da lugar al intermedio II . El tratamiento acuoso de la reacción transforma el intermedio II en el o-formilderivado.
OH
RCl2CHOCH3
TiCl4 O
R
H
Ti
O
Cl Cl
ClCl
Cl Cl
CH3
R
O Ti
O
H Cl ClCl
Cl
CH3
Cl
SEAr
-HCl
H2O
R
O
O
H
H
Cl
+ CH3OH
OH
R
CHO-HCl
TiCl4
II
III
I
Esquema 15 5. Reacción de Dakin
RH2O2
R
OH
NaOHR'
O
R'
O
HO+
La reacción de Dakin se emplea en la conversión de ariladehídos o arilcetonas en fenoles, mediante la reacción de aquéllos con peróxido de hidrógeno en presencia de una base. El mecanismo de la reacción se muestra en el esquema 15 y es similar al de la oxidación de Baeyer-Villiger. La adición del anión peróxido al carbonilo del sustrato genera, después de un proceso de transposición, un intermedio de tipo arilformiato el cual, por saponificación se convierte en el correspondiente fenol.

Síntesis de (+)-dicroanona 15
Esquema 15 6. Oxidación de fenoles con IBX
ROOH
+OI
O
O OH
OR OH
I
O
+
Las o-quinonas se pueden obtener por oxidación de fenoles con IBX. En el esquema 16 se indica el mecanismo de esta reacción.
Esquema 16 7. Conversión de catecoles en quinonas por reacción con oxígeno molecular

Síntesis de (+)-dicroanona 16
R
OHO O
R
OHO2
NaOH/MeOH
La reacción de catecoles con oxígeno molecular en presencia de una base permite la oxidación de éstos en quinonas
En el esquema 17 se indican las reacciones redox que tiene lugar en este proceso.
R
OHO O
R
OH
+ 2 OH + 2 H2O + 2e
O2 + 2 H2O + 4e 4 OH
[oxidación]
[reducción]
2 x
R
HO OH
2 + O2
O O
R
2 + 2 H2O
Esquema 17
En el esquema 18 se indica el mecanismo de la reacción de oxidación.
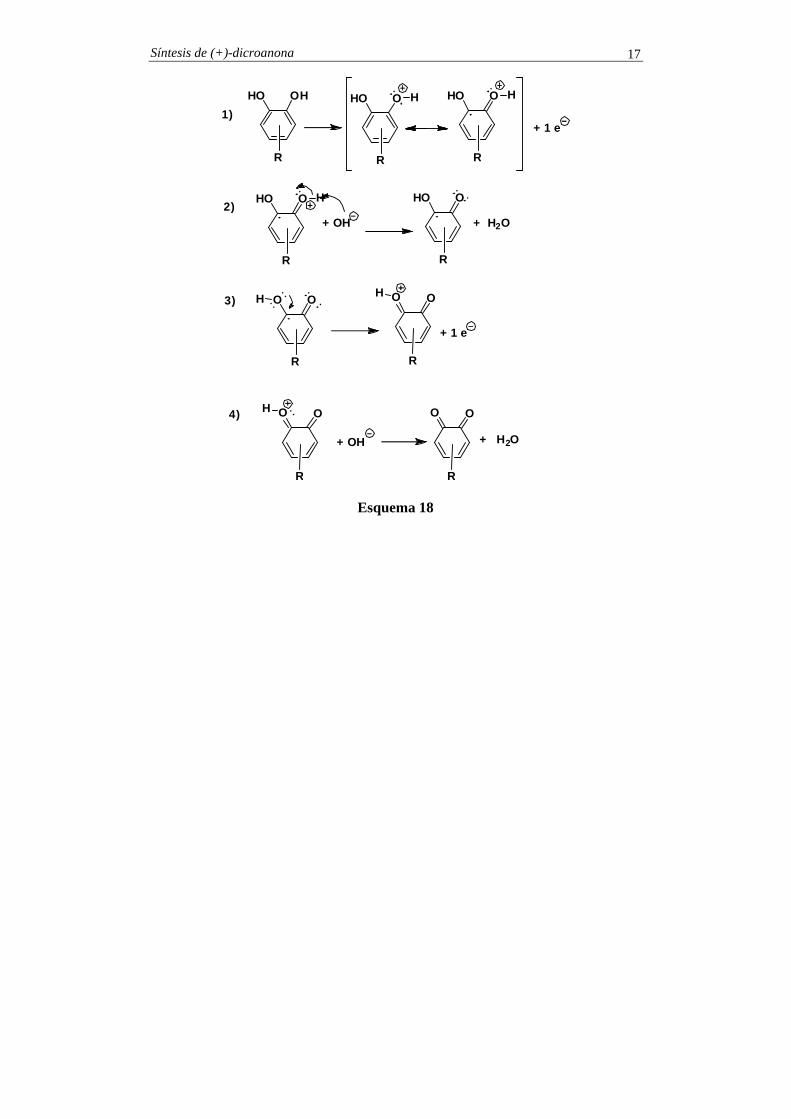
Síntesis de (+)-dicroanona 17
R
O
HO
O
R
OH
+ H2O
R
HO O H
+ 1 e
+ OH
R
HO O
R
HO O H
R
HO O H
+ H2O
R
O O O O
R
H
+ 1 e
O O
R
H
+ OH
1)
2)
3)
4)
H
Esquema 18


Síntesis de elisabetina A 19
SÍNTESIS DE ELISABETINA A
Aislamiento: El diterpeno marino elisabetina A se ha aislado de una gorgonia del Caribe denominada Pseudopterogorgia elisabethae (Octoralia). De este alga se ha aislado también la pseudopterosina, un producto natural que ha demostrado su capacidad para cicatrizar las heridas y reducir la inflamación. Otro de sus derivados, la metopterosina, está siendo analizado por el Centro de Biotecnología y Biomedicina Marina, de la Universidad de California, como un posible tratamiento contra la artritis, la psoriasis y el asma. Actividad biológica: No se han publicado detalles de la actividad biológica de la elisabetina A, pero se sabe que algunos miembros de la clase de las elisabetinas son activos contra Mycobacterium tuberculosis, y también contra algunas líneas de células tumorales.
Retrosíntesis El análisis retreosintético de la elisabetina se indica en el esquema 1, y se
inicia con la adición de un doble enlace entre los carbonos C4-C5.6 Esta es una operación retrosintética denominada adición de grupo funcional (AGF), que aumenta la complejidad estructural del compuesto, en contraposición a lo que es habitual en la retrosíntesis, en la que cada operación va acompañada de una disminución de la complejidad funcional y estructural del sustrato analizado. Sin embargo, la operación AGF efectuada en la elisabetina A, que conduce al intermedio 1, permite el análisis del sistema anular mediante una operación retro-Diels-Alder. Esta operación conduce al intermedio 2, el cual, mediante desconexión de la agrupación isopropilideno y aromatización del anillo de quinona, se transforma en el derivado 3. A continuación, la desconexión del enlace C8-C9
6 T. J. Heckrodt, J. Mulzer, J. Am. Chem. Soc. 2003, 125, 4680.

Síntesis de β-eritroidina 20
origina el compuesto carbonílico 4 y al agente alquilante 5. La unión de estos dos fragmentos creará el estereocentro en C9, de ahí que esta reacción se deberá efectuar de forma estereocontrolada. Aunque el fragmento 5 es quiral, la reacción se efectuará mediante la utilización de un auxiliar quiral en la parte nucleofílica (fragmento 4, Xq
*= auxiliar quiral), a fin de asegurar el máximo estereocontrol del proceso.
El intermedio 4 es un arilacetato, que se desconecta al estannano 8 y el bromuro de arilo 9, que se preparará a partir del aldehído aromático 10.
El agente alquilante 5 se convierte, mediante dos desconexiones Wittig consecutivas, en el éster de Roche 7, que será el material quiral de partida para la preparación del fragmento C8-C18.
Esquema 1

Síntesis de elisabetina A 21
Síntesis 1. Síntesis del ácido arilacético 18 a partir del aldehído 10
Me
HHMe
Me
Me
O
O OH
Me
1
45
14
10
17
elisabetina A
18 10
93
10
14
17
21 OTBS
TBSO
Me
OMe
O
HO
14
17
21 CHO
MeO
Me
OMe
Para la síntesis del ácido arilacético 18 se eligió como material de partida el
aldehído aromático 10, comercialmente accesible. Este compuesto se sometió a la reacción de transposición de Baeyer-Villiger mediante tratamiento con ácido m-cloroperoxibenzoico (MCPBA, esquema 2). El producto de la reacción de transposición, el formiato 11, se convirtió en el fenol 12 por saponificación con hidróxido sódico. La desmetilación selectiva del grupo metilo en para respecto del hidroxilo fenólico, se consiguió mediante la aplicación de una secuencia de dos pasos, que se inició con la oxidación del fenol con nitrato de cerio y amonio (CAN, véanse comentarios). En el segundo paso, la quinona 13, resultante del proceso de oxidación, se redujo con ditionito sódico a la hidroquinona 14, que se convirtió en el bis-sililéter 15 por reacción con TBSCl. El tratamiento de 15 con N-bromosuccinimida provocó la reacción de halogenación SEAr, lo que proporcionó regioselectivamente el bromuro de arilo 16, compuesto sobre el que se instaló la cadena de acetato mediante reacción de acoplamiento de Negishi-Reformatsky. Para ello, el bromuro 16 se trató con el 2-(tributilestannil)acetato de etilo (Bu3SnCH2COOEt), en presencia de ZnBr2 y del catalizador de paladio(II) PdCl2(o-tol3P)2, lo que condujo al acetato de arilo 17 con un 71% de rendimiento (véanse comentarios). El arilacetato de etilo 17 se convirtió en el ácido arilacético 18 mediante reducción a alcohol con DIBAL, oxidación de Swern a aldehído y oxidación de éste a ácido carboxílico con clorito sódico en presencia de dihidrógeno fosfato potásico y 2-metil-2-buteno. La conversión del éster 17 en el ácido 18 hubo que efectuarla mediante la secuencia anterior de tres pasos, porque el éster 17 se mostró inerte a la saponificación, incluso bajo calentamiento a reflujo durante 14 horas en una disolución concentrada de hidróxido sódico etanólico.

Síntesis de β-eritroidina 22
Esquema 2 2. Síntesis del yoduro de alquilo 5
Para la síntesis del fragmento 5 se eligió el éster de Roche 7 como material quiral de partida. Este compuesto se convirtió en el aldehído 19 mediante tritilación y reducción (esquema 3). La olefinación del aldehído con el fosfonato 20 condujo a la (E)-amida de Weinreb α,β-insaturada 21, cuya reducción con DIBAL proporcionó el aldehído conjugado 22. La configuración Z del doble enlace restante se consiguió mediante reacción Wittig (véanse comentarios), del aldehído 22 con el fosforano generado por reacción de bromuro de etil trifenil fosfonio con NaHMDS en tetrahidrofurano. Este proceso permitió la obtención del (E,Z)-dieno conjugado

Síntesis de elisabetina A 23
23, el cual, por destritilación con tricloroborano seguida de yodación, proporcionó el yoduro 5.
Esquema 3
3. Acoplamiento de los fragmentos 5 y 18 mediante alquilación estereoselectiva
El acoplamiento de los fragmentos 5 y 18 genera el estereocentro en C9. Para
conseguir la instalación de éste en condiciones estereocontroladas se recurrió a la metodología de Evans. Así, el anhídrido mixto 23, obtenido por reacción del ácido 18 con cloruro de pivaloilo, se hizo reaccionar con la sal lítica 24, generada por ionización de la oxazolidinona derivada de (R)-fenilalaninol, para proporcionar la N-aciloxazolidinona 25. La ionización de ésta con NaHMDS formó el (Z)-enolato sódico 26, que atacó al yoduro 5 desde la cara Si del enolato, estéricamente más accesible que la cara Re (véase el estado de transición 27 del esquema 4). Este proceso condujo al compuesto 28 con un 86% de exceso diastereoselectivo. Hay que señalar que la reacción entre el enolato sódico derivado del éster aquiral 17

Síntesis de β-eritroidina 24
(esquema 2) y el yoduro de alquilo 5 proporcionó el correspondiente producto de C-alquilación con tan solo 42% de exceso diastereoselectivo.
Esquema 4
4. Pasos finales. Reacción Diels-Alder intramolecular

Síntesis de elisabetina A 25
El intermedio 28 posee todos los carbonos del esqueleto de elisabetina a excepción de los de la agrupación isopropilideno. Para la instalación de los carbonos restantes se eliminó reductivamente el auxiliar quiral con LiBH4 (esquema 5). En este punto se obtuvo una mezcla de alcoholes diastereoisoméricos separable por cromatografía de columna (recuérdese que la alquilación transcurre con un 86% de exceso diastereoselectivo). La oxidación de Swern del alcohol mayoritario proporcionó el aldehído 29, que se sometió a olefinación de Wittig con el isopropilidentrifenilfosforano, lo que condujo al trieno 30. La desililación de los hidroxilos fenólicos con TBAF generó la hidroquinona 31, que se oxidó a la quinona 2 con tricloruro de hierro. La quinona se transformó en el compuesto tricíclico 34 al experimentar in situ la reacción de cicloadición Diels-Alder intramolecular. Finalmente, la elisabetina sintética se consiguió mediante O-desmetilación de 25 con tribromuro de boro.
La elección de los grupos protectores de los hidroxilos fenólicos fue clave para el éxito de la síntesis. De hecho, la desmetilación oxidante no se pudo conseguir, con Ag2O/HNO3 o CAN, cuando los hidroxilos fenólicos estaban protegidos en forma de metil éteres. En estas condiciones se provocó la descomposición del material de partida, debido seguramente a la inestabilidad del sistema diénico al medio fuertemente ácido. La protección en forma de TBS éter permitió efectuar la desprotección con TBAF y la subsiguiente oxidación del fenol a p-quinona FeCl3 (véanse comentarios).
El proceso Diels-Alder intramolecular tiene lugar mediante la participación del E. T. endo, indicado como 33 en el esquema 5. En esta reacción se generan los estereocentros C1, C2, C3 y C6. La configuración de C2 es opuesta a la que presenta la molécula de elisabetina y la inversión configuracional se consiguió, después de la hidrogenación del doble enlace C4-C5, mediante enolización-protonación de C2 con NaOH acuosa (control termodinámico).
En el esquema 6 se indican los esqueletos hidrocarbonados de 34 y de 35, después de la modelización molecular. Se aprecia la fusión trans entre el anillo ciclohexénico y el anillo de quinona en el compuesto 34. En el compuesto 35 la fusión del anillo ciclohexánico con el anillo quinónico es cis, por tanto con menor tensión.

Síntesis de β-eritroidina 26
Esquema 5

Síntesis de elisabetina A 27
Comentarios 1. O-Desmetilación selectiva del compuesto 12
La desmetilación selectiva del grupo metoxilo de 12, en para respecto del grupo hidroxilo, se consiguió mediante una secuencia de dos pasos que se inició con la conversión del fenol 12 en la quinona 13. Esta transformación se consiguió mediante oxidación de 12 con nitrato de cerio y amonio en una mezcla de acetonitrilo/agua. Esta reacción fue seguida de reducción de la quinona a la hidroquinona 14 con ditionito sódico. El mecanismo de la oxidación con CAN se indica en el esquema 6.
Esquema 6

Síntesis de β-eritroidina 28
El mecanismo se inicia con la conversión del fenol 12 en el radical I , por oxidación de aquél con nitrato de cerio y amonio. Un segundo proceso redox convierte el radical I en el catión oxonio II , que es hidrolizado en el medio acuoso de la reacción para proporcionar la quinona 13. El grupo metoxilo en orto respecto del hidroxilo fenólico también podría ser hidrolizado puesto que el catión oxonio II se puede describir mediante la contribución de las estructuras resonantes que se indican a continuación:
El ataque nucleofílico de la molécula de agua a la estructura resonante V
conduciría a la formación de la orto-quinona (esquema 7):
Esquema 7
Una explicación para la ausencia de la orto-quinona en la reacción con CAN estaría en la contribución de las estructuras resonantes IV y V al híbrido de resonancia. Estas dos estructuras resonantes colocan la carga positiva al lado del grupo carbonilo, que es electrón-atrayente, y por tanto su contribución al híbrido de resonancia sería mucho menor que la de las estructuras II y III , que serían, en consecuencia, las que resultarían atacadas por el agua.

Síntesis de elisabetina A 29
2. Reacción de acoplamiento de Negishi-Reformatsky
El acoplamiento entre el bromuro de arilo 16 y el 2-(tributilestannil)acetato de etilo se llevó a cabo bajo las condiciones de Negishi, empleando una cantidad catalítica de bromuro de zinc y el catalizador de paladio(II) PdCl2(o-tol)3P, en N,N-dimetilformamida como disolvente. La reacción requiere de paladio(0) coordinativamente insaturado para que el ciclo catalítico pueda funcionar. En las condiciones anteriormente comentadas el paladio(0) se genera a partir de la especie de paladio(II) según el mecanismo que se indica en el esquema 8.
Esquema 8
El ciclo catalítico se inicia con la etapa de inserción oxidante del bromuro de arilo al catalizador de Pd(0). En este proceso se forma el intermedio I (esquema 9). Por otro lado, el bromuro de zinc experimenta una reacción de transmetalación con el 2-(tributilestannil)acetato de etilo, lo que conduce a la formación de bromuro de tributilestaño y BrZnCH2COOEt. Esta especie organometálica es la que participa en el proceso de transmetalación con el intermedio I . En esta reacción se forma la especie de paladio(II) II y bromuro de zinc, que no se consume por tanto en el proceso global de acoplamiento. La última etapa de ciclo catalítico es la eliminación reductora, que proporciona el compuesto 17 y regenera la especie de paladio(0).

Síntesis de β-eritroidina 30
Esquema 9
3. Reacción de Wittig Z selectiva
El mecanismo que se ha propuesto para explicar la reacción de olefinación de Wittig se inicia con la adición nucleofílica del carbanión al carbono carbonílico para formar una betaína (sal interna de alcóxido de fosfonio, véase esquema 10). La existencia de complejos relativamente estables de betainas con sales de litio es uno de los argumentos que se han aducido para proponer su formación como intermedios en el proceso de olefinación. Sin embargo, no hay evidencia experimental de la formación de betaínas en forma no complejada, por lo que, de formarse, la betaína colapsa con gran rapidez para dar un oxafosfetano. La descomposición de éste, cuya existencia real ha sido demostrada mediante experimentos de 31P-RMN, conduce a la olefina final y a óxido de trifenilfosfina.

Síntesis de elisabetina A 31
La elevada energía del enlace P=O (130-140 kcal/mol) constituye la fuerza impulsora del proceso.
Los cálculos computacionales han puesto también de manifiesto que la barrera de activación para el paso final de descomposición del oxafosfetano (25-30 kcal/mol) es marcadamente menor que la de la reversión a iluro y compuesto carbonílico (k3 >> k-1, k 3́ >> k -́1).
R3R1
R2 R4
R1
R2P
+
R4
R3
O
OPk1 (k´1)
k-1 (k´ -1)
OPh 3P
R2 R3R1 R4
OPh3P
R2 R4R1 R3
+k2 (k´2)
k -2 (k´-2)
OPh 3P
R2 R3R1 R4
+
OPh 3P
R2 R4R1 R3
k3 (k´3)
R4R1
R2 R3
+
Esquema 10 Las olefinaciones Wiittig efectuadas con iluros reactivos (no estabilizados)
proporcionan mayoritariamente el isómero Z, sobre todo cuando se eliminan las sales de litio formadas durante la generación del iluro (salt-free conditions). Un método que permite aumentar la proporción de isómero Z consiste en la generación del iluro con bases que no contengan litio, tales como, por ejemplo, NaNH2 o NaHMDS, como en el caso de la conversión de 22 en el dieno conjugado 23.
La formación Z de olefinas con iluros no estabilizados ha sido explicada mediante una aproximación sesgada (gauche) de los dos carbonos, nucleofílico y electrofílico, de ambos reactivos, como se indica en el esquema 11.
olefina Z
δδδδ-δδδδ+ O
H R
R´P
H
O
P
HR
R´
H R R´
H H
Esquema 11
En el estado de transición, los dos átomos cargados (P+ y O-) se sitúan lo más
cerca posible a causa de la atracción colombiana. Por otro lado, el voluminoso fragmento de fósforo, que aún conserva una disposición cuasi-tetraédrica de los

Síntesis de β-eritroidina 32
sustituyentes, tiende a alejarse lo más posible, por razones estéricas, del grupo R del aldehído. A medida que se va formando el nuevo enlace C−C, la progresiva atracción culombiana entre los átomos cargados de P y O hace que se acerquen cada vez más hasta colapsar en un nuevo enlace P−O, sin formación detectable de un intermedio de tipo betaínico. Finalmente se forma un oxafosfetano, en el que los dos grupos R y R´ están del mismo lado del anillo, lo que explica la formación de la olefina Z.
Cuando hay presentes sales de litio, un catión con mucha mayor fuerza ácida de Lewis que el sodio, el proceso se hace mucho más complejo mecanísticamente puesto que el catión litio estabiliza las formas betaínicas a través de algún tipo de agregados o complejos. Es posible entonces que el primer paso del proceso adquiera cierto grado de reversibilidad (k-1 ≈ k2), lo que confiere a su vez a dicho paso un mayor grado de control termodinámico. La consecuencia final es un aumento en la proporción de isómero E, debido a que aumenta la proporción del oxafosfetano trans, termodinámicamente más estable. 4. Oxidación de fenoles con FeCl3
La oxidación de fenoles con FeCl3 acuoso es mecanísticamente similar a la
oxidación con nitrato de cerio y amonio. El catión Fe3+ oxida a uno de los grupos hidroxilo del compuesto 31, formando la especie radicalaria I , que es oxidada de nuevo por el catión Fe3+ para dar lugar a la quinona 2 (esquema 12). La quinona no se aísla, puesto que in situ experimenta la reacción Diels-Alder intramolecular.

Síntesis de elisabetina A 33
Esquema 12

Síntesis de β-eritroidina 35
SÍNTESIS DE ββββ-ERITROIDINA
Aislamiento: La β-eritroidina se ha aislado de las semillas de la planta Erythria sp. Actividad biológica: La β-eritroidina es un relajante muscular con propiedades curaremiméticas, ya que provoca la muerte en mamíferos por parálisis de los músculos del diafragma.
Retrosíntesis El análisis retrosintético de la β-eritroidina se inicia con la desconexión,
basada en una reacción de metátesis de eninos de los anillos A y B (esquema 1).7
Esquema 1
7 H. Fukumoto, K. Takahasi, J. Ishihara, S. Hatakeyama, Angew. Chem. Int. Ed. 2006, 45, 2731.

Síntesis de β-eritroidina 36
Esta operación conduce al azadienino 1, que deriva del β-cetoéster 2. La desconexión del sistema 1,3-dicarbonílico conduce al azodiéster 3, que se obtendrá del amino éster 4 mediante reacciones de N-alquilación. El estereocentro cuaternario que contiene el intermedio 4 se construirá a partir del epoxialcohol 5, que se obtendrá del alcohol alílico 6 mediante reacción de epoxidación. La desconexión de la unidad acetilénica en 6 conduce al yoduro de alquenilo 7, que se obtendrá del alcohol propargílico 8. La fuente de quiralidad de la síntesis será el ácido (S,S)-tartárico 9, a partir del cual se obtendrá el intermedio 8.
Síntesis
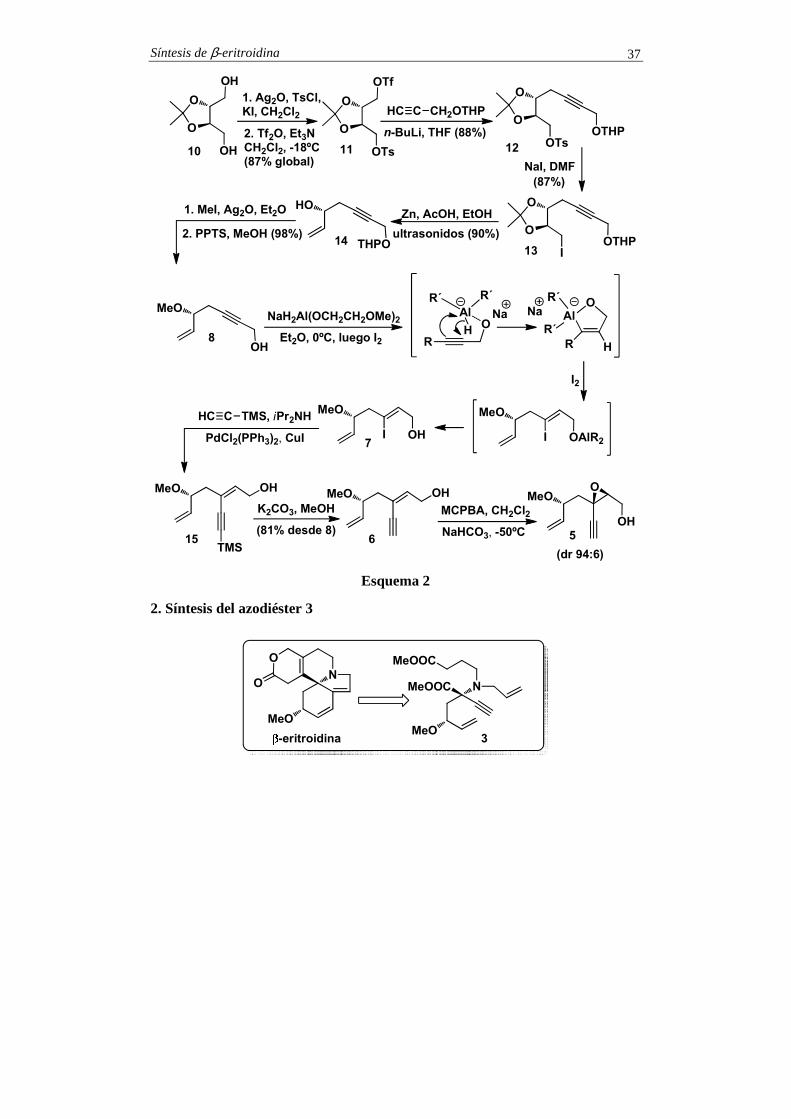
1. Síntesis del epoxialcohol 5
O
NO
MeO
-eritroidina
MeO OH
5
O
La síntesis del compuesto 5 se inició con la tosilación del diol 10, que se obtuvo del (S,S)-tartrato de dietilo por cetalización y reducción (esquema 2). La esterificación del hidroxilo remanente con anhídrido tríflico proporcionó el triflato 11, que reaccionó selectivamente con el alquinuro lítico derivado del alcohol propargílico THP protegido. Esta operación condujo al alquino 12, que se convirtió en el yoduro 13 por desplazamiento nucleofílico del tosilato con yoduro sódico. La reacción de 13 con zinc en ácido acético, en presencia de ultrasonidos proporcionó al alcohol alílico 14 (véanse comentarios), el cual, por O-metilación seguida de metanolisis ácida, condujo al alcohol propargílico 8. La hidroaluminación del triple enlace de 8 con Red-Al (NaH2Al(OCH2CH2OMe)2) seguida de yodonolisis dio el yoduro de alquenilo 7. Este compuesto se sometió a la reacción de acoplamiento de Sonogashira en presencia de PdCl2(PPh3)2 y CuI. Esta operación condujo al dienino 15, que se desililó por reacción con carbonato de potasio en metanol. La epoxidación estereoselectiva de 6 se ensayó con el método de Sharpless (t-BuOOH, T(iPrO)4, tartrato) pero la diastereoselectividad del proceso fue muy baja. Sin embargo, la reacción con ácido m-cloroperoxibenzoico proporcionó, con una relación diastereoisomérica 94:6, el epoxialcohol 5 (véanse comentarios).
Síntesis de β-eritroidina 37
R
OAl
R´
H
R´
Na
I2
R
OAl
R´
R´
Na
H
OH
MeO
I OH
OH
O
O
OTf
OTs
O
O
1. Ag2O, TsCl,
KI, CH2Cl2
2. Tf2O, Et3N
CH2Cl2, -18ºC
(87% global)
HC C CH2OTHP
n-BuLi, THF (88%)OTs
O
OOTHP
NaI, DMF
I
O
OOTHP
Zn, AcOH, EtOH
ultrasonidos (90%)
HO
THPO
MeO
OH
1. MeI, Ag2O, Et2O
2. PPTS, MeOH (98%)
NaH2Al(OCH2CH2OMe)2
HC C TMS, iPr2NH
PdCl2(PPh3)2, CuI
(81% desde 8)
MeO OHMeO
OH
O
(87%)
Et2O, 0ºC, luego I2
TMS
MeO OH
K2CO3, MeOH MCPBA, CH2Cl2
NaHCO3, -50ºC
10 11 12
14
8
7
15 6 5
13
(dr 94:6)
MeO
I OAlR2
Esquema 2
2. Síntesis del azodiéster 3
O
NO
MeO
-eritroidina
MeOOC
MeOOC
N
MeO3
Síntesis de β-eritroidina 38
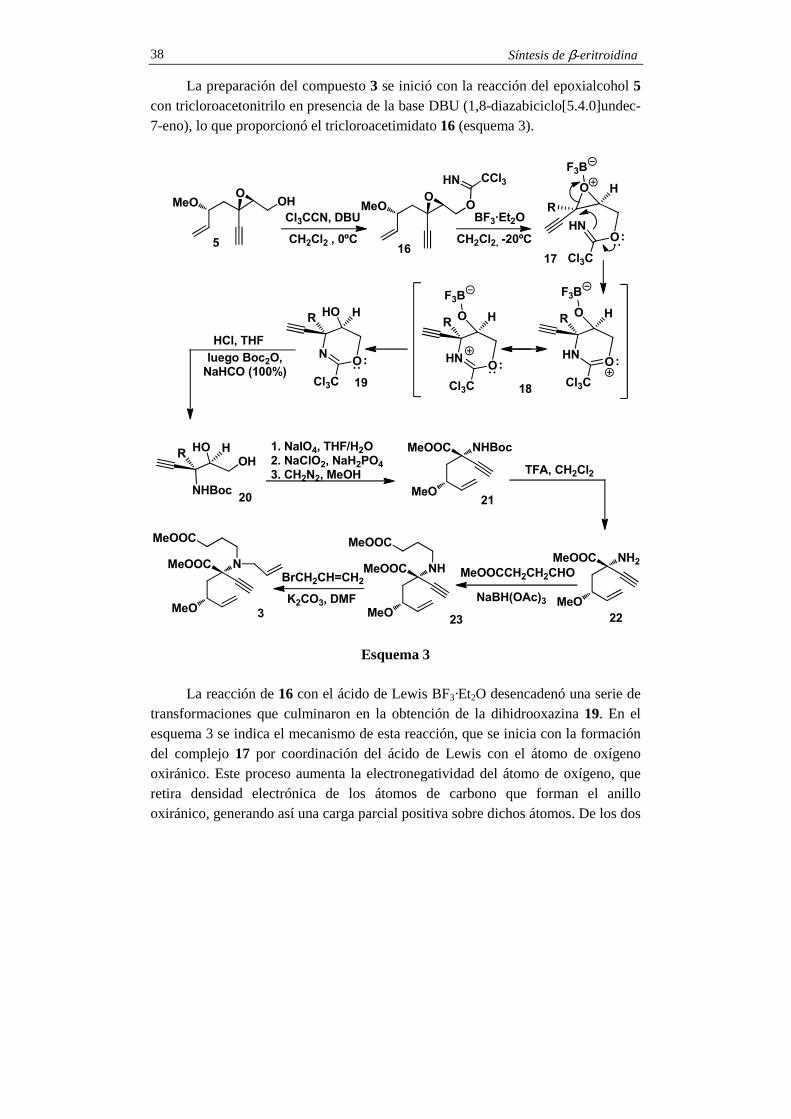
La preparación del compuesto 3 se inició con la reacción del epoxialcohol 5 con tricloroacetonitrilo en presencia de la base DBU (1,8-diazabiciclo[5.4.0]undec-7-eno), lo que proporcionó el tricloroacetimidato 16 (esquema 3).
MeOOC
MeOOC
N
MeO
MeO OH
3
5
OMeO O
O
HN CCl3O
R
H
F3B
OHN
Cl3C
OR H
F3B
OHN
Cl3C
OR H
F3B
OHN
Cl3C
HOR H
ON
Cl3C
HOR H
NHBoc
OH
MeOOC NH2
MeO
MeOOC
MeOOC
NH
MeO
HCl, THF
luego Boc2O,NaHCO (100%)
1. NaIO4, THF/H2O2. NaClO2, NaH2PO4
3. CH2N2, MeOH TFA, CH2Cl2
MeOOCCH2CH2CHOBrCH2CH=CH2
K2CO3, DMF
Cl3CCN, DBU
CH2Cl2 , 0ºC
BF3·Et2O
CH2Cl2, -20ºC
MeOOC NHBoc
MeO
1617
1819
20 21
2223
NaBH(OAc)3
Esquema 3
La reacción de 16 con el ácido de Lewis BF3·Et2O desencadenó una serie de
transformaciones que culminaron en la obtención de la dihidrooxazina 19. En el esquema 3 se indica el mecanismo de esta reacción, que se inicia con la formación del complejo 17 por coordinación del ácido de Lewis con el átomo de oxígeno oxiránico. Este proceso aumenta la electronegatividad del átomo de oxígeno, que retira densidad electrónica de los átomos de carbono que forman el anillo oxiránico, generando así una carga parcial positiva sobre dichos átomos. De los dos

Síntesis de β-eritroidina 39
átomos de carbono del anillo oxiránico es el cuaternario el que soporta una mayor carga parcial positiva, y es por tanto éste el que resulta atacado intramolecularmente por el átomo de nitrógeno. Además, este ataque invierte la configuración del carbono epoxídico porque se produce desde el lado opuesto al del puente oxiránico (véase estructura 17 del esquema 3). El resultado es la formación del compuesto heterocíclico 18, que por descomplejación se transforma en la dihidrooxazina 19. La hidrólisis ácida, seguida de N-Boc protección, condujo al aminodiol 20, el cual, por ruptura oxidante con peryodato sódico, oxidación a ácido carboxílico y esterificación se convirtió en el aminoéster N-Boc protegido 21. La preparación del compuesto 3 se completó con las reacciones de N-alquilación, que se iniciaron con la obtención del aminoéster 22 por N-Boc desprotección con ácido trifluoroacético. La aminación reductiva con 4-oxobutirato de metilo y triacetoxiborohidruro sódico proporcionó el aminodiéster 23, que por N-alilación con bromuro de alilo y carbonato potásico, se convirtió en el compuesto 3.
3. Pasos finales: construcción del sistema tetracíclico de la ββββ-eritroidina
O
NO
MeO
-eritroidina
MeOOC
MeOOC
N
MeO3
A
B
CD
El asalto final a la β-eritroidina comenzó con la construcción del anillo C,
que se consiguió mediante reacción de condensación de Dieckmann del aminodiéster 3, por calentamiento en tolueno a reflujo en presencia de metóxido sódico (Esquema 4). El producto de condensación se obtuvo bajo su forma enólica 24 (para la forma de β-cetoéster véase la estructura 2 del análisis retrosintético). La reducción selectiva de la función éster se consiguió mediante conversión de 24 en el correspondiente enolato sódico, seguida de reducción in situ con alano lo que llevó a la hidroxicetona 25 (véanse comentarios).
Para la construcción del anillo D se recurrió en primer lugar a la reacción Horner-Wadsworth-Emmons intramolecular, lo que implicaba la preparación del fosfonato 26. Sin embargo, este compuesto no se pudo obtener porque la acilación del cetol 25 con (EtO)2P(O)CH2COOH, o con (EtO)2P(O)CH2COCl, provocó la deshidratación de 25. Para solucionar este problema se cambió la estrategia sintética y se recurrió a la reacción de Reformatsky intramolecular. Así, el cetol 26,

Síntesis de β-eritroidina 40
por reacción con cloruro de bromoacetilo en presencia de piridina se transformó en 27, el cual, por reacción con SmI2 en THF a temperatura ambiente, proporcionó la hidroxilactona 28 como mezcla de epímeros (6:4), junto con una pequeña cantidad de producto de deshidratación. El tratamiento de la mezcla con cloruro de tionilo provocó la reacción de deshidratación y la formación de una mezcla inseparable formada por la lactona α,β-insaturada 29 y la lactona β,γ-insaturada 1, en relación 74:26. La proporción de la lactona 1 se aumentó mediante saponificación de la mezcla, seguida de acidificación. En estas condiciones se obtuvo una mezcla de lactonas 29/1 en relación 6:94.
Esquema 4

Síntesis de β-eritroidina 41
Finalmente, para la construcción de los anillos A y B se recurrió a la proyectada reacción de metátesis ciclante. El proceso se llevó a cabo por tratamiento de la mezcla de lactonas con 0.1 equivalentes del catalizador de Grubbs de 1ª generación, en diclorometano a temperatura ambiente durante 6.5 horas. En estas condiciones se obtuvo un 42% de β-eritroidina, junto con un 4% del compuesto 30 y 31% del producto de partida (véanse comentarios). Hay que señalar que la reacción de metátesis con el catalizador de Grubbs de 2ª generación fue mucho menos eficiente, puesto que proporcionó la β-eritroidina con tan sólo el 30% de rendimiento.
Comentarios
1. Apertura reductiva en el yodoacetal 13
El mecanismo que explica la conversión del yodoacetal 13 en el alcohol alílico 14 se indica en el esquema 5. El proceso se inicia con el ataque del metal, que provoca un proceso de ruptura de enlaces que conduce a la formación del alcoxilato de zinc 31. La protonación de éste lleva al hemiacetal 32, que se transforma en el alcohol alílico 14 y acetona.
Esquema 5
Síntesis de β-eritroidina 42
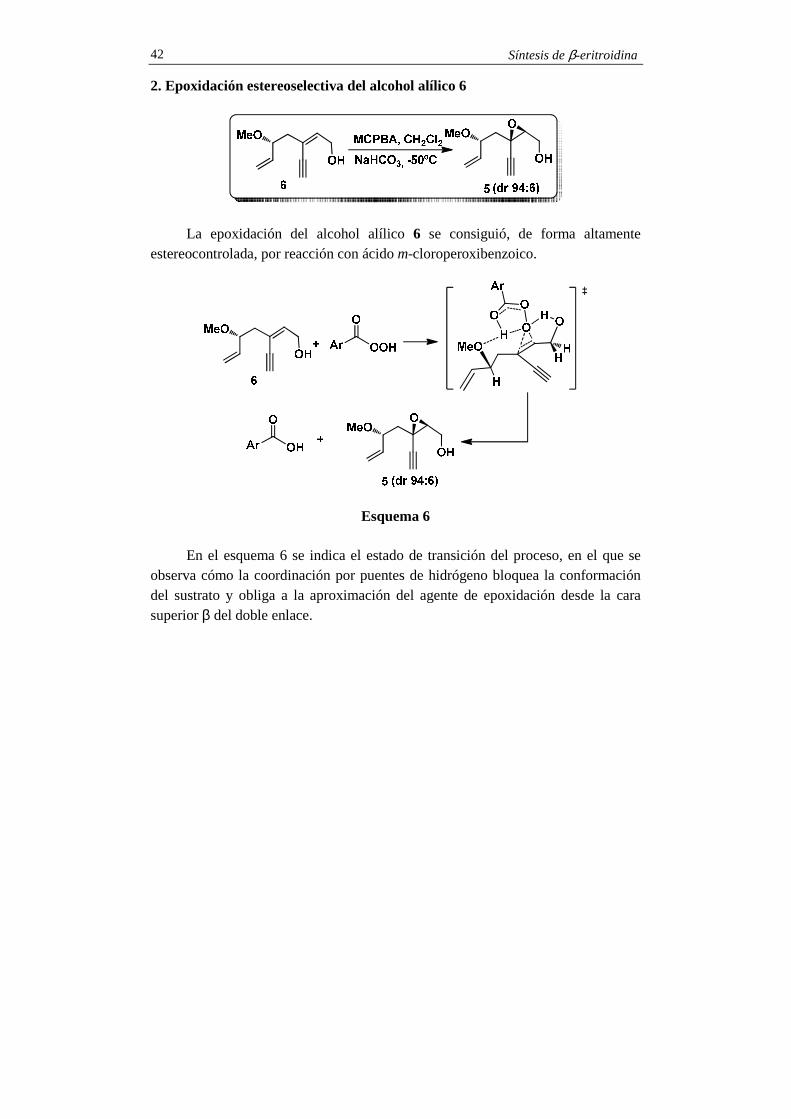
2. Epoxidación estereoselectiva del alcohol alílico 6
La epoxidación del alcohol alílico 6 se consiguió, de forma altamente estereocontrolada, por reacción con ácido m-cloroperoxibenzoico.
Esquema 6 En el esquema 6 se indica el estado de transición del proceso, en el que se
observa cómo la coordinación por puentes de hidrógeno bloquea la conformación del sustrato y obliga a la aproximación del agente de epoxidación desde la cara superior β del doble enlace.

Síntesis de β-eritroidina 43
3. Reducción quimioselectiva del enoléster 24
HO
MeOOC
N
MeO
ON
MeO
HO
NaH, THF
luego AlH3
24 25
La transformación del enoléster 24 en el cetol 25 se consiguió por conversión
en el enolato sódico 33, seguida de adición de alano a la mezcla de reacción (esquema 7). Este procedimiento experimental protege, de forma provisional, el grupo carbonílico cetónico del tautómero 2 en su forma de enolato sódico, evita las etapas de protección-desprotección de la función de cetona, y permite la reducción quimioselectiva de la agrupación éster.
Esquema 7
4. Reacción de Reformatsky inducida por SmI2

Síntesis de β-eritroidina 44
El estado de oxidación más estable de los lantánidos es +3 y por tanto las especies divalentes de estos elementos funcionan como agentes reductores suaves. El SmI2 fue introducido en síntesis orgánica por Kagan, en 1980, como agente reductor mono-electrónico. Este compuesto es capaz de generar radicales alquilo, arilo y alquenilo a partir de los correspondientes haluros, así como radicales cetilo a partir de aldehídos y cetonas. Su débil carácter reductor le permite promover reacciones de creación de enlaces C-C, tales como acoplamientos pinacolínicos, ciclaciones radicalarias, acoplamientos cetilo-olefina, reacciones de Barbier y Reformatsky, adiciones conjugadas, etc.
En el esquema 8 se indica el mecanismo para la conversión del bromoéster 27 en la hidroxilactona 28. El SmI2 rompe reductivamente el enlace C-Br y genera un enolato de samario(III) que, a continuación, ataca intramolecularmente al carbonilo cetónico para formar el producto de ciclación 28.
Esquema 8
5. Metátesis de eninos
La β-eritroidina se obtuvo por reacción de metátesis ciclante en el azadienino
1. En esta reacción se construyeron, en un solo paso operativo, los anillos A y B del alcaloide. En el esquema 9 se indica el ciclo catalítico de este proceso que se inicia con la cicloadición intermolecular del catalizador, simbolizado en el esquema como Ru=CH2, al triple enlace del enino. Esta reacción genera el intermedio I , que experimenta ciclorreversión para formar el carbeno de rutenio II . La adición intramolecular al doble enlace forma el rutenaciclobutano III , que por

Síntesis de β-eritroidina 45
ciclorreversión conduce al intermedio IV y al catalizador Ru=CH2. Un segundo proceso de cicloadición intermolecular forma el rutenaciclobutano V, que por ciclorreversión genera el carbeno de rutenio VI y etileno. La cicloadición intramolecular lleva a la formación de VII , que experimenta ciclorreversión para dar lugar a la β-eritrodidina y al catalizador, que inicia un nuevo ciclo de metátesis.
Esquema 9 El subproducto 30 que se obtiene en la reacción del dienino 1 es el resultado
de la metátesis ciclante entre las dos agrupaciones olefínicas.

Síntesis de β-eritroidina 46

Síntesis de estemospironina 47
SÍNTESIS DE ESTEMOSPIRONINA
Aislamiento: La estemospironina se ha asilado de las raíces y rizomas de plantas de la familia Stemonacae. Actividad biológica: Los extractos de las plantas Stemonacae se han utilizado en la medicina tradicional china y japonesa en el tratamiento de enfermedades respiratorias. Los extractos de estas plantas también tienen actividad insecticida y antihelmíntica.
Retrosíntesis
El análisis retrosintético de la estemospironina se muestra en el esquema 1 y comienza con la desconexión de los enlaces C3-N, C5-N y C14-O.8 Estas desconexiones se basan en reacciones de alquilación de amina y adición a doble enlace, respectivamente, y conducen a la estructura 1, la cual, por escisión del doble enlace C3-C14 lleva a los intermedios 2 y 3. El intermedio 2 se preparará a partir del epóxido 4 por apertura regioselectiva del anillo oxiránico con anión azida y reducción del doble enlace. La desconexión del doble enlace de 4 conduce al alcohol alílico 5, que se obtendrá mediante adición conjugada del reactivo de Grignard 7 al triple enlace del compuesto 6. Finalmente, 6 se sintetizará mediante adición del anión derivado de propiolato al 4-hidroxibutanal protegido.
8 D. R. Williams, M. G. Fromhold, J. D. Earley, Org. Lett. 2001, 3, 2721.

Síntesis de estemospironina 48
Esquema 1
Síntesis Síntesis del compuesto 19
La síntesis de la estemospironina comenzó con la obtención de la inona 9,
que se preparó por adición del bromuro de etinilmagnesio al 4-benciloxibutanal seguida de oxidación (esquema 2). La reducción Midland de la inona 9 con (R)-Alpino-borano dio lugar al alcohol 10 con un 88% de exceso enantiomérico (véanse comentarios). La protección de la agrupación hidroxilo en forma de triisopropil silil éter (TPS) éter, seguida de metalación con n-BuLi y carboxilación

Síntesis de estemospironina 49
con cloroformiato de isopropilo condujo al éster 11 que, por adición conjugada del reactivo de Grignard 79 al éster 11 proporcionó el (E)-enoato de isopropilo 12 (véanse comentarios).
OBn
O
OBn
H
O1.
2.Swern(68%, dos pasos)
MgBr
OBn
OH(+)-pineno, 9-BBN
(95%, 88% ee)
1. TPSCl
OBn
OTPS
(72%)
CO2i-Pr
2. n-BuLi,ClCO2i-Pr
MEMO MgBr
CuBr·SMe2 (70%)
MEMO
CO2i-Pr
H
OBn
TPSO1. TBAF
2. NaH, MeI3. DIBALH(71%, 3 pasos)
MEMO
H
BnO
MeO
OH
MEMO
H
BnO
MeO
OH
Ti(iPrO)4, CaH2, SiO2
(-)DIPT, t-BuOOH(90%, 4:1)
O
1. Dess-Martin
2. Ph3P=CHCO2Me(60%, dos pasos)
MEMO
H
OBn
MeO
O
CO2Me
1. H2,Rh/Al2O3
2. LiBH4
3. PivCl
MEMO
H
BnO
MeO
O
PivO
LiN3,NH4Cl
DMPU(83%)
MEMO
N3
BnO
MeO
OH
PivO
MEMO
OBn
MeO
O
N3
OH
Ph3P
OBzI
tBuOK (77%)
MEMO
N3
OBn
MeO
OH
BzO
(70%)
1. LiOH
2. Swern(95%)
8 9 10
1112
7
13 14
1516
17
1819
3
Esquema 2
9 Preparado en forma ópticamente pura a partir de (S)-3-hidroxi-2-metilpropionato de metilo

Síntesis de estemospironina 50
La eliminación del grupo t-butildifenilsililo (TPS), O-metilación y reducción, condujo al alcohol 13, que se convirtió en el epoxialcohol 14 mediante la modificación de Zhou10 del método de Sharpless. Esta modificación experimental, que consiste en llevar a cabo la epoxidación en presencia de CaH2 y SiO2, aumenta la velocidad del proceso sin merma del rendimiento estereoquímico del mismo. La oxidación de 14 con el reactivo de Dess-Martin, seguida de olefinación Horner-Wadsworth-Emmons, llevó al enoato 15, cuya hidrogenación en presencia del catalizador Rh/Al2O3 permitió la reducción quimioselectiva del doble enlace. La reducción subsiguiente de la función éster con borohidruro de litio, seguida de pivaloilación, proporcionó el epóxido 16. La apertura regioselectiva del anillo oxiránico se consiguió por reacción con azida de litio y cloruro amónico en presencia de DMPU (1,3-dimetil-3,4,5,6-tetrahidro-2(1H)-pirimidinona) y llevó al azidoalcohol 17. La saponificación del éster, seguida de oxidación Swer dio una mezcla de hemiacetales diastereoisoméricos 18, la cual, por reacción con el trifenilfosforano derivado de 3,11 proporcionó el (Z)-alqueno 19. 2. Pasos finales: síntesis del intermedio 1 y su conversión en estemospironina
La conversión del azidoalcohol 19 en la estemospironina comenzó con la
eliminación del grupo metoximetilo (MEM), que fue seguida de eliminación del grupo benzoilo mediante hidrólisis básica (esquema 3). El diol 20, resultante de la desprotección, se oxidó con CrO3 disuelto en acetona acuosa en presencia de H2SO4 (reactivo de Jones), lo que provocó la oxidación de los grupos hidroxilo primarios en C12 y C17 hasta ácido carboxílico, y la subsiguiente lactonización por ataque intramolecular del grupo hidroxilo libre en C9. La esterificación de la mezcla de oxidación proporcionó el compuesto 1 que, por desbencilación quimioselectiva con tricloruro de boro y oxidación Dess-Martin, se convirtió en el aldehído 21.
10 Z.-M. Wang, W.-S. Zhou, G.-Q. Lin, Tetrahedron 1987, 43, 2935. 11 Preparado a partir de (2S)-2-metil-4-penten-1-ol mediante formación del benzoato, ozonólisis, reducción con NaBH4 y reacción con I2 y PPh3.
Síntesis de estemospironina 51
MEMO
N3
OBn
MeO
OH
BzO
1. HCl
2. LiOH(75%,dos pasos)
HO
N3
OBn
MeO
OH
HO
1. Jones
2. CH2N2
(80%)
ON3
OBn
MeO
OCO2Me
H
1. BCl3 (60%)
2. Dess-Martin(80%)
ON3
CHO
MeO
OCO2Me
H
I2 (30%)
O
MeO
O
H
NH
OMeOI
H
estemospironina
N O
MeOH H
O
OO
HH
PPh3
NaBH4
(60%)
19 20
121
22 24
O
MeO
O
MeO2CH
NH
I
O
MeO
O
CO2MeH
NH I
H
23
O
MeO
O
CO2MeH
NO
N
CHO
MeO
O
CO2MeH
2526
27
PPh3
129a9
8
5
17 9a9
8
1217
1712
9a
9
5
5
88
9
9a
17
5
5
9a
9 9
5
9a
9
9
H
H
HRNH
II
NH
H-I
OO
H
OO
H
24
3 14
3 14
3
14
H H
R
I
Esquema 3

Síntesis de estemospironina 52
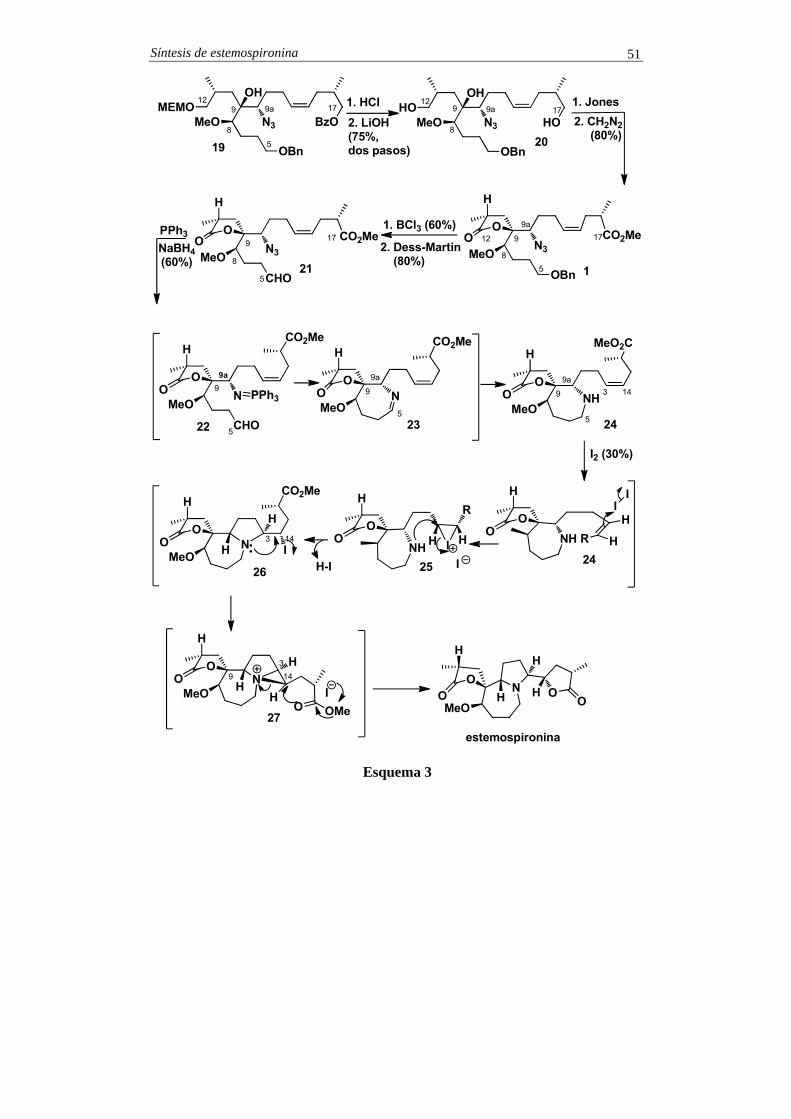
El anillo de azepina se construyó del siguiente modo. La reacción de 21 con trifenilfosfina, en THF a temperatura ambiente y durante 16 h, generó el azailuro 22 (véanse comentarios) que se convirtió en la imina 23 por reacción aza-Wittig intramolecular. A continuación, a la mezcla de reacción se le añadió borohidruro de sodio en metanol, lo que provocó la reducción in situ de la función imina de 23, con la subsiguiente formación de la azepina 24. Finalmente, el tratamiento de 24 con yodo molecular provocó una doble ciclación que llevó a la obtención de la estemospironina de forma regio y estereoselectiva. Este proceso engloba los siguientes pasos:
a) En primer lugar, se produce una adición electrofílica del yodo al doble enlace para formar la sal de yodonio 25 que sufre un ataque intramolecular y regioselectivo de la amina para formar la 2,5-trans-yodopirrolidina 26. Esta reacción puede considerarse estereoespecífica, ya que se ha observado que a partir de alquenos de configuración Z se obtiene la 2,5-trans-yodopirrolidina, mientras que con alquenos de configuración E se genera la 2,5-cis-yodopirrolidina. En el caso de la pirrolidina 26, el estereocentro en C9a es el responsable de la configuración de los estereocentros C3 y C14 que se generan en la reacción de aminoyodación.
b) En segundo lugar, se produce una sustitución nucleofílica intramolecular de la amina terciaria sobre el átomo de yodo en C14. Esta reacción conduce a la formación estereoespecífica de la sal de aziridinio 27. Finalmente, el ataque del anión yoduro al éster metílico provoca la apertura estereoespecífica del anillo de aziridinio para formar el anillo de lactona y dar lugar a la estemospironina. Este proceso de lactonización a partir de la yodopirrolidina 26 tiene lugar con retención de la configuración del estereocentro C14, puesto que se producen dos inversiones de configuración sucesivas.
Comentarios
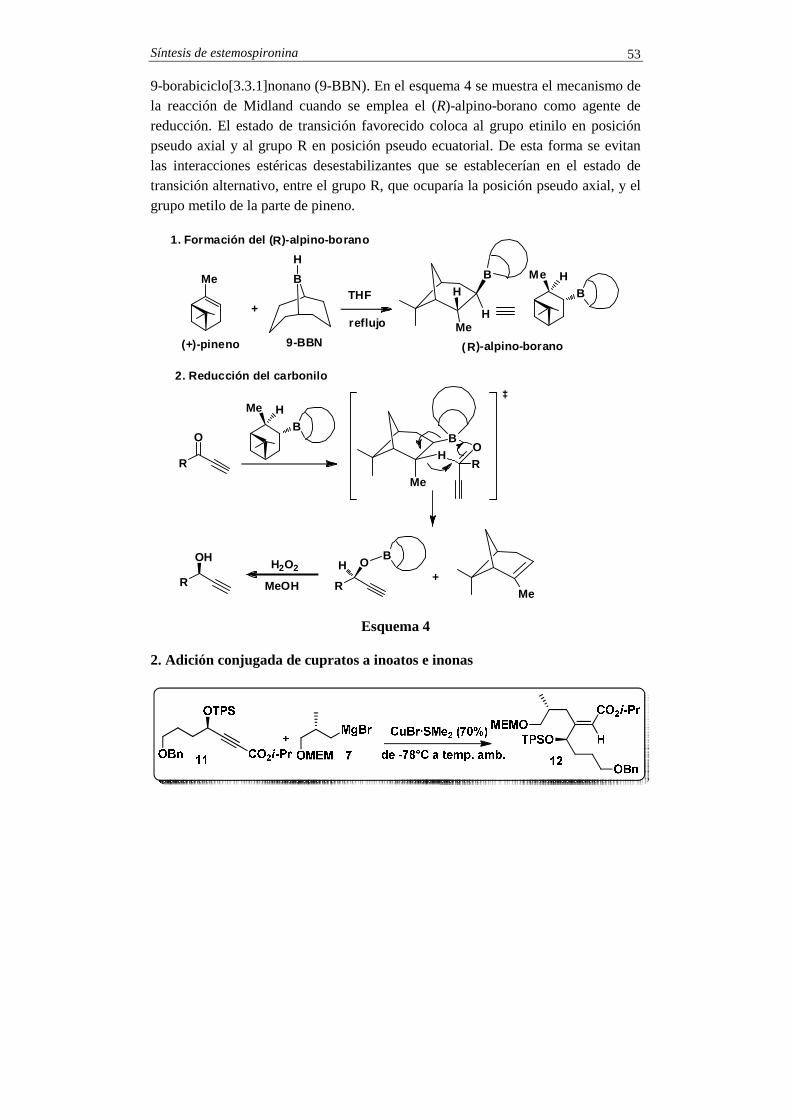
1. Reacción de Midland (reducción enantioselectiva de inonas conjugadas)
La reacción de Midland permite la obtención enantioselectiva de alcoholes propargílicos por reducción de inonas con B-3-pinanil-9-borabiciclo[3.3.1]nonano (alpino-borano), que se prepara por hidroboración del correspondiente pineno con
Síntesis de estemospironina 53
9-borabiciclo[3.3.1]nonano (9-BBN). En el esquema 4 se muestra el mecanismo de la reacción de Midland cuando se emplea el (R)-alpino-borano como agente de reducción. El estado de transición favorecido coloca al grupo etinilo en posición pseudo axial y al grupo R en posición pseudo ecuatorial. De esta forma se evitan las interacciones estéricas desestabilizantes que se establecerían en el estado de transición alternativo, entre el grupo R, que ocuparía la posición pseudo axial, y el grupo metilo de la parte de pineno.
R
O
R
OH
B
H
THF
reflujo+
(+)-pineno 9-BBN
Me HB
(R)-alpino-borano
1. Formación del ( R)-alpino-borano
2. Reducción del carbonilo
Me BH
Me
R
OHH2O2
MeOH+
H
Me HB
BH
Me
OR
Me
B
Esquema 4
2. Adición conjugada de cupratos a inoatos e inonas

Síntesis de estemospironina 54
La adición de especies organocúpricas a enlaces triples activados permite el acceso a olefinas trisustituídas con excelentes rendimientos químicos y elevadas regio y estereoselectividades. La estereoquímica del doble enlace depende de las condiciones de reacción. Así, cuando la adición se efectúa a baja temperatura se forma exclusivamente la olefina cis, mientras que a temperatura ambiente se forma mayoritariamente la olefina trans.
En el esquema 5 se muestra el mecanismo de estas adiciones. A baja temperatura, se forma un intermedio de tipo cis-alquenilcobre (intermedio cis-I ), que en el quench de la reacción da lugar a la olefina cis.
Si se eleva la temperatura el intermedio cis-I se isomeriza, vía alenolato de litio II , al trans-alquenilcobre trans-III , que por hidrólisis se transforma en la olefina trans.
R1Z
O R2CuLi
R1
R2 H
OZ
-20°C
Cu
temp.amb.
cis trans
R1Z
OR2 R2
R1
R2 Cu(R2)Li
OZ
cis-I
H
·
R1
R2
O
Z
LiR1
R2
Cu(R2)LiII
OZ
trans-III
H
R1
R2
H
OZ
Li
Esquema 5

Síntesis de estemospironina 55
3. Transformación de la azida 22 en el iminofosforano 23
O N3
CHO
MeO
O
O
H
OMePPh3
225
8
9
17
5
9a
9
23
O N
CHO
MeO
O
CO2MeH
PPh 3
La reacción de 22 con trifenilfosfina forma el azailuro 23 mediante el
mecanismo que se muestra en el esquema 6. La trifenilfosfina reacciona con la azida para generar una fosfazida I , que pierde nitrógeno y da lugar al azailuro de fosforo II . La forma resonante del azailuro de fosforo II es precisamente el iminofosforano 23.
R'
NR
R'N
R
PPhPh
Ph
NN P Ph
Ph
Ph R'N
R
N N
P PhPh
Ph
R'N
R
PPhPh
Ph
N2
I
II
1. Formación de una fosfazida
2. Eliminación de N 2: formación del azailuro
22
23
R'
NR
N N
P PhPh
Ph
I
R'N
R
N N
P Ph
PhPh
−−−−δδδδ ++++δδδδ
Esquema 6


Síntesis de FR901464 57
SÍNTESIS DE FR901464
Aislamiento: El FR901464 se ha aislado de cultivos de una bacteria de Pseudomonoas sp. Actividad biológica: El FR901464 es un agente antitumoral que regula la transcripción de los oncogenes y de los genes supresores de tumores.
Retrosíntesis El análisis retrosintético del FR901464se inicia con la desconexión del doble
enlace (E)-disustituido, según se indica en el esquema 1. Esta desconexión, basada en una reacción de metátesis cruzada, conduce a los fragmentos 1 y 2.12
Esquema 1
12 B. J. Albert, A. Sivaramakrishnan, T. Naka, K. Koide, J. Am. Chem. Soc. 2006, 128, 2792.
Síntesis de FR901464 58
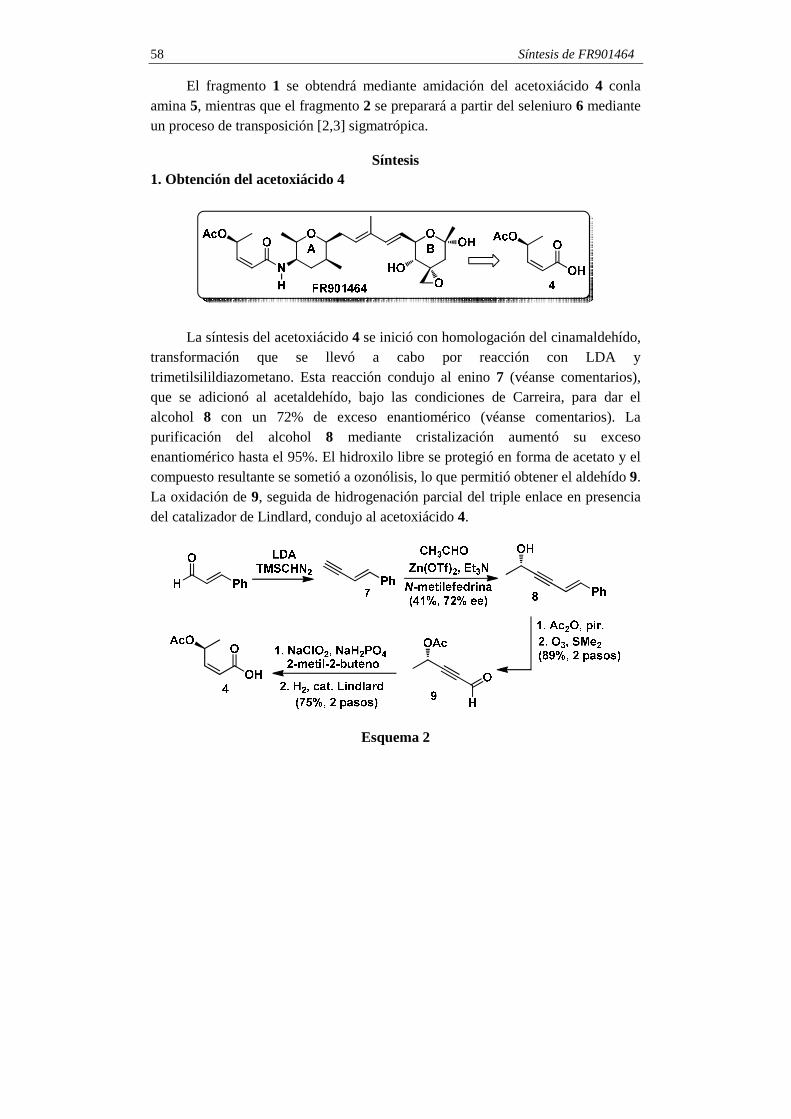
El fragmento 1 se obtendrá mediante amidación del acetoxiácido 4 conla amina 5, mientras que el fragmento 2 se preparará a partir del seleniuro 6 mediante un proceso de transposición [2,3] sigmatrópica.
Síntesis 1. Obtención del acetoxiácido 4
La síntesis del acetoxiácido 4 se inició con homologación del cinamaldehído,
transformación que se llevó a cabo por reacción con LDA y trimetilsilildiazometano. Esta reacción condujo al enino 7 (véanse comentarios), que se adicionó al acetaldehído, bajo las condiciones de Carreira, para dar el alcohol 8 con un 72% de exceso enantiomérico (véanse comentarios). La purificación del alcohol 8 mediante cristalización aumentó su exceso enantiomérico hasta el 95%. El hidroxilo libre se protegió en forma de acetato y el compuesto resultante se sometió a ozonólisis, lo que permitió obtener el aldehído 9. La oxidación de 9, seguida de hidrogenación parcial del triple enlace en presencia del catalizador de Lindlard, condujo al acetoxiácido 4.
Esquema 2

Síntesis de FR901464 59
2. Obtención del fragmento 5
Para la síntesis del fragmento 5 se utilizó como material de partida el N-Boc-
L-treoninato de metilo 10, compuesto comercialmente accesible (esquema 3).
Esquema 3

Síntesis de FR901464 60
La protección de 10 en forma de aminal, seguida de reducción de la función éster y de metilenación Wittig, condujo al compuesto 11. La metanolisis ácida de la función aminal liberó el grupo hidroxilo, que se sometió a O-alquilación por reacción con bromuro de metalilo en presencia de óxido de plata. Esta secuencia de reacciones proporcionó el compuesto diolefínico 12, el cual, por reacción de metátesis ciclante inducida por el catalizador de Grubbs de segunda generación, se transformó en el dihidropirano 13. La oxidación con dicromato de piridinio (PDC), seguida hidrogenación en presencia de PtO2, proporcionó una mezcla de lactonas diastereoisoméricas, en relación 10:1, mayoritaria en la lactona 14. A continuación se llevó a cabo un proceso de alilación anomérica estereocontrolado, que se efectuó del siguiente modo. La lactona 14, por reacción con bromuro de alilmagnesio, se transformó en una mezcla de hemiacetales anoméricos 15, los cuales se convirtieron, estereoselectivamente, en el tetrahidropirano 5 por reacción con trietilsilano en presencia de trifluoruro de boro-eterato. En el esquema 3 se indica el mecanismo de este proceso que se inicia con la formación del catión oxonio 16. Este intermedio es reducido estereoselectivamente por el ataque axial del hidruro proporcionado desde el Et3SiH. 3. Obtención del fragmento 2
La síntesis del fragmento 2 comenzó con el acoplamiento, inducido por zinc,
entre el bromuro de metalilo y el alcohol propargílico (véase esquema 4 y comentarios). Esta reacción generó el alcohol alílico 17 sobre el que se efectuó una reacción de epoxidación asimétrica de Sharpless que condujo al epoxialcohol 18 con un 95% de exceso enantiomérico. La oxidación de 18 dio el epoxialdehído 19, el cual se sometió a un proceso de alquinilación promovido por Ag/Zr (véanse comentarios). Esta reacción proporcionó una mezcla de inoésteres diastereoisoméricos, en relación 7.5:1, mayoritaria en el compuesto 20. La reducción estereoselectiva del triple enlace con Red-Al, seguida de protección del hidroxilo, permitó la obtención de la (E)-éster α,β-insaturado 21. El tratamiento de 21 con DIBAL proporcionó un alcohol alílico que se transformó en el arilseleniuro 6 (véanse comentarios). La oxidación del seleniuro con agua oxigenada generó un

Síntesis de FR901464 61
arilselenóxido, el cual experimentó un proceso de transposición sigmatrópica que condujo al alcohol alílico 22 (véanse comentarios). Este compuesto se convirtió en la epoxicetona 23 por sililación del hidroxilo seguida de ruptura oxidante del doble enlace. Cuando el compuesto 23 se sometió a hidrólis ácida se provocó la eliminación de los grupos TES y la subsiguiente formación del hemiacetal 2.
2
HO
OOH
O
HOBr
Zn (93%)
HO Ti(OiPr)4, TBHP
(+)-DIPT
HO
O
Dess-Martin(81%)
OHC
O
(90%, 95%ee)
Ag CO2Me
Cp2ZrCl2, AgOTf(84%; de 7.5:1)
OMeO2C
OH1. Red-Al
2. TESCl(81%)
O
MeO2C
OTES
1. DIBAL
2. SeCN
Bu3P (95%)
O
OTES
SeAr
H2O2
DMAP(96%
O
OTES
OH
1. TESCl (95%)
2. OsO4,NMOPb(OAc)4 (86%)
OO
OTES
TESO
AcOH
THF/H2O(91%)
17 18
1920
21 6
23 22
NO2
Esquema 4
4. Unión de los fragmentos 4, 5 y 6 y pasos finales
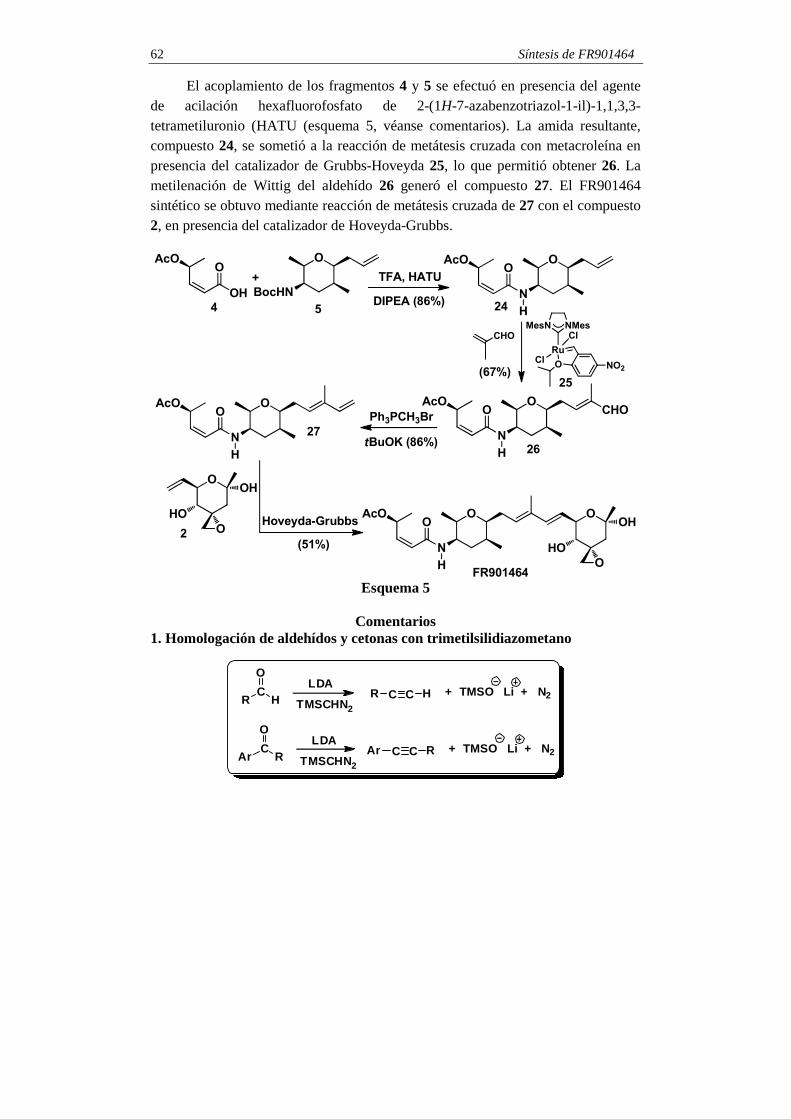
Síntesis de FR901464 62
El acoplamiento de los fragmentos 4 y 5 se efectuó en presencia del agente de acilación hexafluorofosfato de 2-(1H-7-azabenzotriazol-1-il)-1,1,3,3-tetrametiluronio (HATU (esquema 5, véanse comentarios). La amida resultante, compuesto 24, se sometió a la reacción de metátesis cruzada con metacroleína en presencia del catalizador de Grubbs-Hoveyda 25, lo que permitió obtener 26. La metilenación de Wittig del aldehído 26 generó el compuesto 27. El FR901464 sintético se obtuvo mediante reacción de metátesis cruzada de 27 con el compuesto 2, en presencia del catalizador de Hoveyda-Grubbs.
O
N
O
HO
OAcO
H
OH
O
OAcO
OH4
BocHN
O
5
HO
OOH
O2
+ TFA, HATU
DIPEA (86%)
O
N
OAcO
H
CHO
O
N
OAcO
H
CHO
(67%)
Ph3PCH3Br
tBuOK (86%)
O
N
OAcO
H
Hoveyda-Grubbs
24
25
26
(51%)
Ru
O
Cl
ClNO2
NMesMesN
FR901464
27
Esquema 5
Comentarios
1. Homologación de aldehídos y cetonas con trimetilsilidiazometano
O
CHR
LDAR C C H + TMSO Li + N2
O
CRAr
LDAAr C C R + TMSO Li + N2
TMSCHN2
TMSCHN2

Síntesis de FR901464 63
La reacción de arilalquilcetonas y aldehídos con trimetilsildiazometano y LDA permite la obtención de alquinos homólogos.13 El mecanismo de la reacción se indica en el esquema 6. La base (LDA) ioniza al trimetilsildiazometano, que se adiciona al grupo carbonilo para generar un α-diazoalcóxido. A continuación se produce la eliminación de trimetilsilanolato de litio, lo que genera un diazoalqueno, el cual se transforma en el alquino homólogo mediante eliminación de nitrógeno seguida de transposicion.
O
CHR
R C C H + N2
1. Ionización del TMSCHN 2
MeSi
MeMe C
HN N
N
MeSi
MeMe C N N
Li Li
NH
+
2. Ataque nucleofílico al carbonilo
MeSi
MeMe C N N
Li
O
CH
RC
Si
NN
+ Me3SiO Li
O
C
HR
C
Si
NN
3. Eliminación de Me 3SiOLi
CH
RC N N C
H
R
C N N
4. Transposición y eliminación de N 2
αααα-diazoalcóxido
αααα-diazoalqueno
Li
Li
CH
RC N N
Esquema 6
13 K. Miwa, T. Aoyama, T. Shioiri, Synlett 1994, 107.

Síntesis de FR901464 64
2. Alquinilación asimétrica de Carreira
O
R1 HR2 H R1
R2
OH
+
HO NMe2
Ph CH3(22 mol%)
Zn(OTf)2 (20 mol%)Et3N (50 mol%)tolueno, 60ºC
En el método de alquinilación asimétrica de Carreira se consigue la adición asimétrica de reactivos alquinil-zíncicos a aldehídos mediante adición de aquéllos en presencia de N-metilefedrina.14 La enantioselectividad que se puede conseguir con este método es elevada, del orden del 90-99% ee, sobre todo cuando la adición se lleva a cabo sobre aldehídos alifáticos a 60ºC. Sin embargo, el método no funciona con aldehídos aromáticos debido a que un importante porcentaje del aldehído experimenta la reacción de Cannizaro en las condiciones del proceso.
El mecanismo que se ha propuesto para explicar la enantioselectividad del método de alquinilación de Carreira se indica en el esquema 7. El proceso se inicia con la generación de un alquinilzincico que completa su esfera de coordinación con los centros básicos de la molécula de N-metilefedrina y con Et3N. El intercambio de ligandos en la especie de zinc quiral permite la coordinación con el compuesto carbonílico. La coordinación de un segundo equivalente del reactivo alquinilzíncico con el átomo de oxígeno de la parte de efedrina dispone al sistema para la adición al grupo carbonilo, que expone la cara Re al ataque del nucleófilo, tal y como se indica en el esquema 7.15
14 a) N. K. Anand, E. M. Carreira, J. Am. Chem. Soc. 2001, 123, 9687; b) D. Boyall, D. E. Frantz, E. M. Carreira, Org. Lett. 2002, 4, 2605. 15 A. Fettes, M. Carreira, J. Org. Chem. 2003, 68, 9274.

Síntesis de FR901464 65
R H + Zn(OTf) 2 + Et3N R ZnOTf + Et 3NHOTf
R ZnOTf
(R)(S)
HOMe2N
CH3Ph
H
H+
(-)-N-metilefedrina
O
N
CH3Ph
H
HZn
R
Et3N
Et3NMe
Me
R ZnOTf
O
N
CH3Ph
H
HZn
R
O
MeMe
Zn
ROTf
R'CHO
O
N
CH3Ph
H
HZn
R
O
MeMe
H
R'
H
R'
ataque a la cara Re
O
N
CH3Ph
H
HZn
R
O
MeMe
Zn
OTf
H
R'
R
R'
OH
R
Esquema 7
3. Alquinilación de Koide
El método de alquinilación de aldehídos de Koide emplea acetiluros de plata
en presencia de complejos de Zr(IV).16 La ventaja de este método de alquinilación es doble. Por una parte no se requiere el uso de bases fuertes para la ionización del derivado acetilénico, ya que el acetiluro de plata se genera, de forma cuantitativa,
16 S. P. Shahi, K. Koide, K. Angew. Chem., Int. Ed. 2004, 43, 2525.

Síntesis de FR901464 66
utilizando nitrato de plata. Por otra, no se requiere trabajar en condiciones de sequedad.
Se ha observado también que la presencia de sales de plata, como el AgOTf,
acelera el proceso de adición, por lo que la reacción se suele efectuar en presencia de una cantidad catalítica de AgOTf. El mecanismo de la reacción se inicia con la formación del alquinuro de zirconio I , que se genera por transmetalación del alquinuro de plata con Cp2ZrCl2 (esquema 8). El triflato de plata se coordina con el aldehído y el complejo resultante es atacado por el alquinuro de zirconio para dar lugar al producto de la reacción.
Esquema 8 En la síntesis del FR901464 se adiciona el alquinuro de plata al α-
alcoxialdehído quiral 17. El proceso transcurre con una diastereoselectividad 7.5:1 en favor del diastereoisómero 18 (esquema 9). La formación de 18 se explica mediante la participación de un estado de transición, que se muestra en el esquema 9, en el cual el aldehído adopta una conformación Felkin-Ahn.
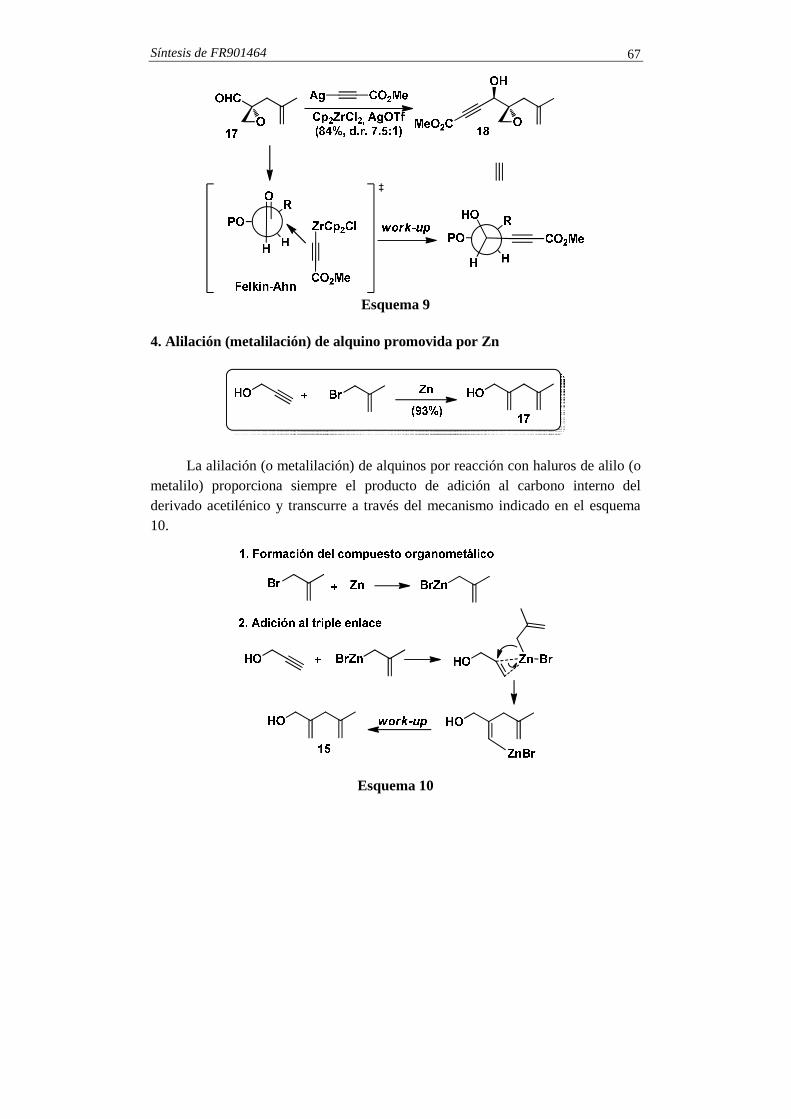
Síntesis de FR901464 67
Esquema 9
4. Alilación (metalilación) de alquino promovida por Zn
La alilación (o metalilación) de alquinos por reacción con haluros de alilo (o metalilo) proporciona siempre el producto de adición al carbono interno del derivado acetilénico y transcurre a través del mecanismo indicado en el esquema 10.
Esquema 10

Síntesis de FR901464 68
El Zn se inserta al enlace C-Br, generando el correspondiente compuesto organozíncico, que se adiciona regioselectivamente al triple enlace para formar un alquenilzinc que es hidrolizado en el proceso de work-up.
5. Reacción de selenilación de alcoholes
La reacción de alcoholes con o-nitrofenilselenocianato en presencia de tributilfosfina proporciona los correspondientes aril alquil seleniuros. El mecanismo del proceso se muestra en el esquema 11.
Esquema 11
6. Transposición sigmatrópica [2,3] de selenóxidos alílicos
La oxidación de seleniuros alílicos con peróxido de hidrógeno genera selenóxidos, que experimentan espontáneamente un proceso de transposición

Síntesis de FR901464 69
sigmatrópica [2,3] para dar lugar a aril selenil éteres, cuya hidrólisis proporciona alcoholes alílicos (esquema 12).
Esquema 12 7. Reacción de amidación promovida por HATU
La reacción de amidación promovida por hexafluorofosfato de O-(1H-7-
azabenzotriazol-1-il)-N,N,N',N'-tetrametiluronio (HATU) se caracteriza por la escasa toxicidad del agente acilante y la eficacia con la que transcurre el proceso, sobre todo, en los casos de elevado impedimento estérico de los sustratos. En el caso de sustratos quirales la acilación transcurre sin epimerización. El mecanismo de la reacción se muestra en el esquema 13.

Síntesis de FR901464 70
Esquema 13

Síntesis de guanacastepeno 71
SÍNTESIS DE GUANACASTEPENO
Aislamiento: El guanacastepeno se ha sido aislado de un hongo endofítico17 que crece sobre el árbol Daphnosis americana en Costa Rica. Actividad biológica: El guanacastepeno presenta actividad antibacteriana contra Staphylococcus aureus resistente a meticilina y contra Enterococcus faecium resistente a vancomicina.
Retrosíntesis
El análisis retrosintético del guanacastepeno se describe en el esquema 1. El proceso se inicia con la desconexión del anillo lactónico D, lo que conduce al intermedio tricíclico 1.18
OOO
AcOOH
guanacastepeno
O
AcO
COOBn
TfO
O COOBn
acoplamiento Heck
O
Met
adiciónconjugada
+
O
HO
OH
+
1 2
3465
AB
C
D1
2
10
11 11
10
1011
Esquema 1 17 Los hongos endofíticos son aquellos que invaden los tejidos internos de las plantas sin provocar síntomas de enfermedad. 18 S. Limura, L. E. Overman, R. Paulini, A. Zakarian, J. Am. Chem. Soc. 2006, 128, 13095.

Síntesis de guanacastepeno 72
La operación clave del esquema retrosintético es la que desconecta el enlace C1-C2 en el intermedio 1. Esta desconexión se basa en una reacción de acoplamiento Heck y conduce al intermedio bicíclico 2, que se desconecta en el enlace C10-C11 para conducir a los fragmentos monocíclicos 3 y 4. Como se explicará a continuación en la parte de síntesis, el intermedio ciclohexenil-metálico 3 se obtendrá del 3-metil-2-ciclohexen-1-ol 6, mientras que la ciclopentenona 4 se sintetizará a partir de la 3-hidroxi-2-ciclopenten-1-ona 5.
Síntesis
1. Síntesis de la ciclopentenona 4
OOO
AcOOH
guanacastepeno
O O
HO4 5
AB C
D
Como se ha indicado en la parte retrosintética, la ciclopentenona 4, que
constituirá el anillo A del guanacastepeno, se obtendrá a partir de la 3-hidroxi-2-ciclopenten-1-ona 5 (esquema 2). Para conseguir una síntesis no racémica de 4 se recurrió a la conversión del compuesto aquiral 5 en un sustrato quiral mediante reacción con mentol 7 en benceno a reflujo, en presencia de ácido p-toluensulfónico. Esta reacción proporcionó el enol éter quiral 8, el cual , mediante enolización cinéticacon LDA en THF a -78ºC, seguida de transmetalación del enolato lítico a enolato de zinc con dietilzinc, y adición de yoduro de isopropilo a la mezcla de enolización, proporcionó el compuesto de C-alquilación 9. Este compuesto se obtuvo como mezcla de diastereoisómeros en proporción 1.5:1. La separación mediante HPLC de la mezcla de diastereoisómeros permitió la obtención del compuesto 10 puro.
La adición de metil-litio al carbonilo cetónico en el sustrato 10 condujo al ciclopentenol 11, el cual, por reacción con ácido sulfúrico acuoso se transformó en la ciclopentenona 4, mediante la participación de los intermedios 12-13-14-15 que se indican en el esquema 2.

Síntesis de guanacastepeno 73
Esquema 2 2. Síntesis del compuesto organometálico 3 (Met=Cu(CN)Li)
La preparación del intermedio organometálico 3 (Met=Cu(CN)Li), que constituirá el anillo C del guanacastepeno, se inició con la esterificación enzimática enantioselectiva de la mezcla racémica de ciclohexenoles 6 (esquema 3).
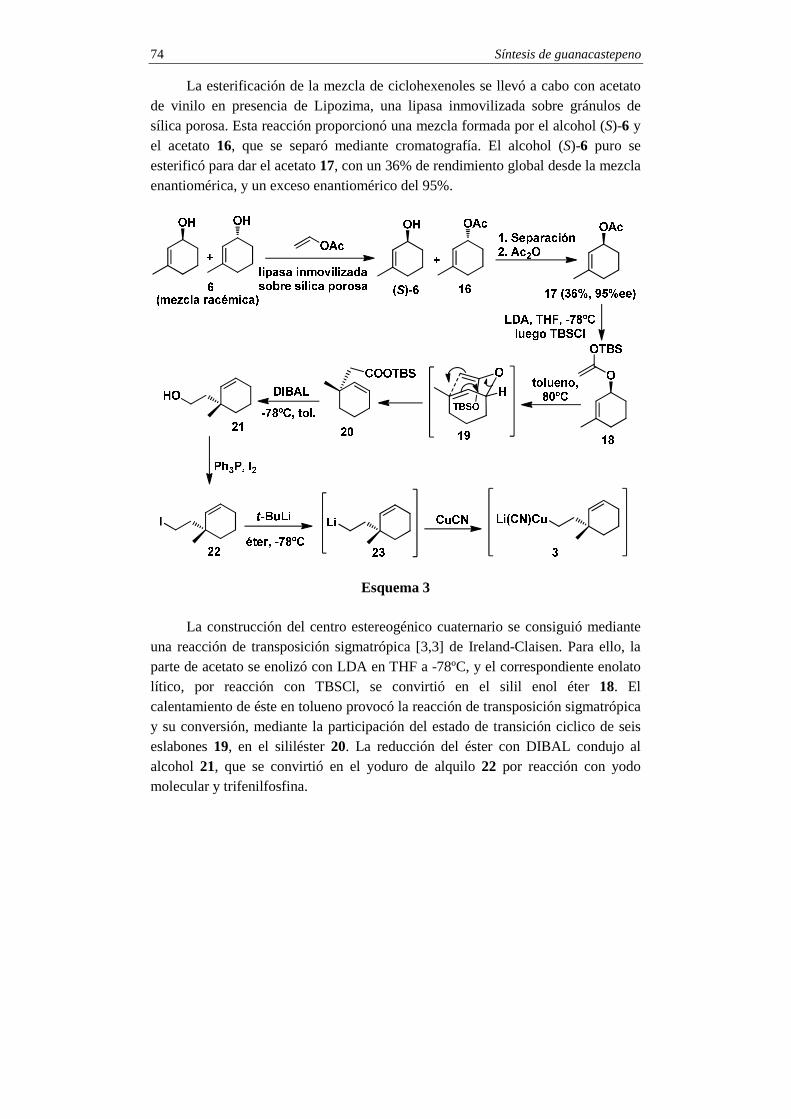
Síntesis de guanacastepeno 74
La esterificación de la mezcla de ciclohexenoles se llevó a cabo con acetato de vinilo en presencia de Lipozima, una lipasa inmovilizada sobre gránulos de sílica porosa. Esta reacción proporcionó una mezcla formada por el alcohol (S)-6 y el acetato 16, que se separó mediante cromatografía. El alcohol (S)-6 puro se esterificó para dar el acetato 17, con un 36% de rendimiento global desde la mezcla enantiomérica, y un exceso enantiomérico del 95%.
Esquema 3 La construcción del centro estereogénico cuaternario se consiguió mediante
una reacción de transposición sigmatrópica [3,3] de Ireland-Claisen. Para ello, la parte de acetato se enolizó con LDA en THF a -78ºC, y el correspondiente enolato lítico, por reacción con TBSCl, se convirtió en el silil enol éter 18. El calentamiento de éste en tolueno provocó la reacción de transposición sigmatrópica y su conversión, mediante la participación del estado de transición ciclico de seis eslabones 19, en el sililéster 20. La reducción del éster con DIBAL condujo al alcohol 21, que se convirtió en el yoduro de alquilo 22 por reacción con yodo molecular y trifenilfosfina.

Síntesis de guanacastepeno 75
La conversión del yoduro 22 en el compuesto organometálico 3 se hizo del siguiente modo. En primer lugar el yoduro 22 se convirtió en el alquil-litico 23 por reacción de intercambio metal-halógeno con t-BuLi. A continuación se procedió a la etapa de transmetalación por reacción del organolítico 23 con 1 equivalente de CuCN, lo que condujo a la formación del organocuprato 3. Por supuesto, este intermedio no se aisló sino que se hizo reaccionar, inmediatamente después de ser generado, con la ciclopentenona 4, tal y como se describe en la siguiente sección.
3. Síntesis del intermedio 1
La unión de los fragmentos 3 y 4 se consiguió mediante la adición conjugada
del organocuprato 3 al sistema enónico de la ciclopentenona 4 (esquema 4). Este proceso, que generó el enlace C10-C11, se produjo de forma altamente estereocontrolada, puesto que el fragmento organometálico se adicionó al doble enlace desde la cara opuesta a la ocupada por el grupo isopropilo. La reacción se llevó a cabo en presencia de TMSBr, lo que provocó la O-sililación del enolato metálico generado en el proceso de adición conjugada. De esta forma se obtuvo el silil enol éter 24, que se hizo reaccionar, in situ, con la sal de Eschenmoser, yoduro de metilen dimetilamonio, para formar la β-aminocetona 26. La N-metilación con yoduro de metilo proporcionó la sal de amonio 27, la cual, mediante eliminación de trietilamina por reacción con carbonato de potasio, condujo a la α-metilenciclopentanona 28. El rendimiento global en la obtención de 28 a partir de la ciclopentenona 4 fue del 53-58%.
La formación del enlace C1-C2 mediante acoplamiento de Heck exigía la manipulación sintética de la parte de anillo ciclohexénico. Este proceso se inició con la epoxidación del doble enlace por reacción con ácido m-cloroperoxibenzoico (MCPBA), lo que proporcionó una mezcla de epoxicetonas diastereoisoméricas 29. La reacción fue totalmente quimioselectiva, puesto que el doble enlace exometilénico, conjugado con el carbonilo cetónico, no reaccionó con MCPBA a causa de su baja nucleofília.

Síntesis de guanacastepeno 76
Esquema 4 Antes de continuar con la manipulación sintética del anillo ciclohexánico se
procedió a la protección del carbonilo cetónico mediante reducción de Luche de 29 con NaBH4/CeCl3 seguida de O-sililación. A continuación, el sililepóxido 30 se trató con trietilborohidruro de litio, lo que provocó la apertura reductiva regioselectiva del anillo oxiránico, obteniéndose una mezcla diastereoisomérica de alcoholes que se oxidó a la cetona 31. La enolización de 31 con LiN(TMS)2, en

Síntesis de guanacastepeno 77
THF a -78ºC, seguida de adición de cianoformiato de bencilo, provocó la C-alcoxicarbonilación del enolato lítico, para dar lugar al β-cetoéster 32 como mezcla tautomérica de isómeros ceto y enol. El triflato de enol 33 se obtuvo mediante enolización de 32 con KN(TMS)2, en THF a -78ºC, seguida de una rápida adición de anhídrido tríflico a la mezcla de reacción. Finalmente, el sustrato para el acoplamiento de Heck (intermedio 1) se obtuvo por desililación de 33 con HF en acetonitrilo acuoso, seguido de oxidación con TPAP y NMO. 4. Pasos finales: construcción de los anillos B y D
El asalto final al guanacastepeno se inició con la construcción del enlace C1-C2 mediante reacción de Heck intramolecular, para lo cual se empleó como catalizador el generado in situ por interacción entre el complejo Pd2(dba)3·CHCl3 y bis(difenilfosfino)butano (dppb) (véase esquema 5 y comentarios).
El producto de la reacción de Heck intramolecular 34, que se obtuvo con un 75% de rendimiento, se convirtió en el silil enol éter 35 por reacción con TESOTf y trietilamina. La epoxidación regioselectiva de 35 con dimetildioxirano generó el epóxido 36, que experimentó la transposición de Rubottom, mediante la participación del intermedio 37, para formar la sililoxicetona 38. La hidrólisis de este compuesto proporcionó una mezcla de hidroxicetonas, epiméricas en C13, en relación 9:1 a favor de 39. La acetilación de la mezcla de epímeros facilitó su separación y la obtención de la acetoxicetona 40 pura con un 67% de rendimiento global desde 34.
La construcción del anillo D requería la conversión de la parte de éster bencílico en ácido carboxílico. Sin embargo, esta transformación no se pudo conseguir bajo ninguna de las reacciones ensayadas. No obstante se descubrió que la reacción de 40 con Pd(OAc)2, Et3SiH y trietilamina proporcionaba, con un 77% de rendimiento, la lactona tetracíclica 41 como mezcla de los cuatro posibles diastereoisómeros (véanse comentarios).
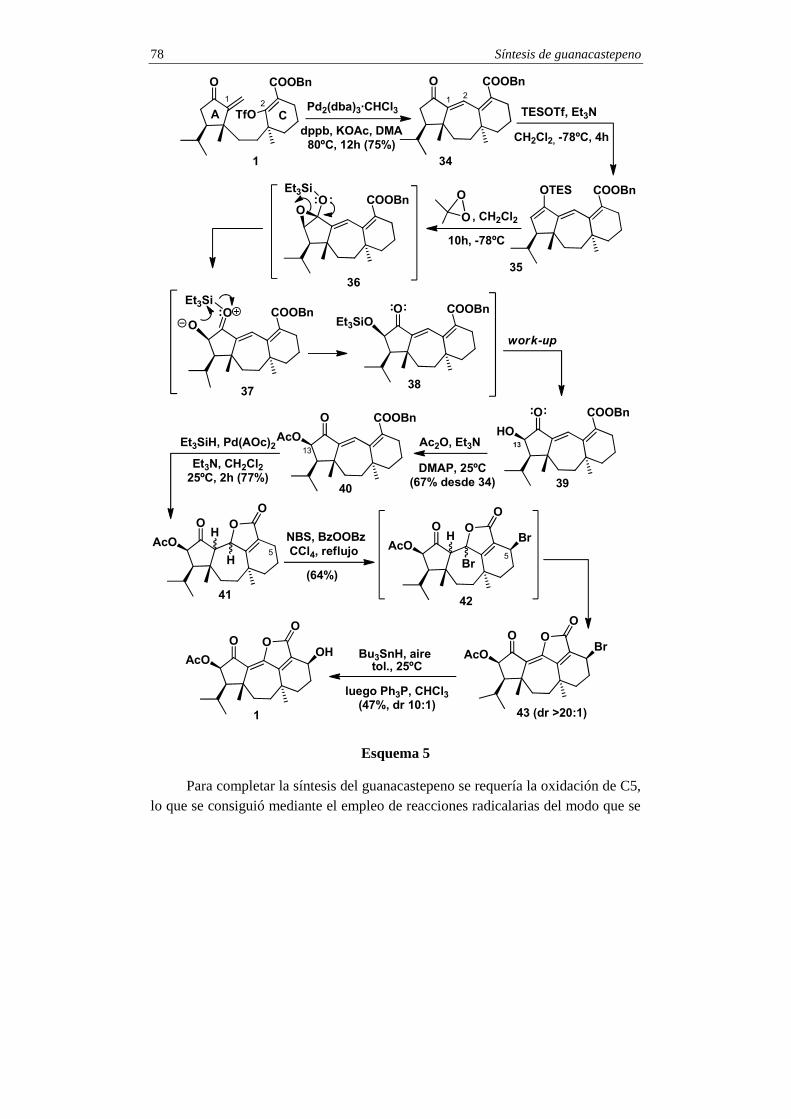
Síntesis de guanacastepeno 78
TfO
O COOBn
1
A C
12 Pd2(dba)3·CHCl3
dppb, KOAc, DMA
80ºC, 12h (75%)
O COOBn
12
TESOTf, Et3N
CH2Cl2, -78ºC, 4h
OTES COOBn
O
O
, CH2Cl2
10h, -78ºC
O COOBnO
Et3Si
O COOBnO
Et3SiO COOBn
Et3SiO
work-up
O COOBn
HOAc2O, Et3N
DMAP, 25ºC
(67% desde 34)
34
35
36
3738
39
O COOBn
AcO
40
NBS, BzOOBz
CCl4, reflujo
O
AcO
Et3SiH, Pd(AOc)2
Et3N, CH2Cl225ºC, 2h (77%)
O
O
H
H
41
O
AcO
O
O
H
Br
Br
O
AcO
O
O
BrBu3SnH, aire
luego Ph3P, CHCl3(47%, dr 10:1)
O
AcO
O
O
OH
42
43 (dr >20:1)1
1313
55
(64%)
tol., 25ºC
Esquema 5 Para completar la síntesis del guanacastepeno se requería la oxidación de C5,
lo que se consiguió mediante el empleo de reacciones radicalarias del modo que se

Síntesis de guanacastepeno 79
explica a continuación. En primer lugar la mezcla de lactonas 41 se calentó a reflujo en CCl4, en presencia del iniciador radicalario peróxido de benzoílo, y N-bromosuccinimida (NBS). Esta reacción provocó dos cambios funcionales en el sustrato de partida: por un lado se instaló un átomo de bromo en C5 y por otro se formó el doble enlace C1-C2, muy probablemente por eliminación de HBr en el intermedio dibromado 42 (véanse comentarios). El bromocompuesto 43 se convirtió en el guanacastepeno sintético por reacción con hidruro de tributilestaño en presencia de aire, y reducción del peróxido alílico intermedio con trifenilfosfina (véanse comentarios).
Comentarios
1. Acoplamiento de Heck intramolecular
El acoplamiento sp2-sp2 de haluros y triflatos de alquenilo con olefinas,
catalizado por paladio, se conoce como reacción de Heck. Como se ha explicado en la parte sintética, el enlace C1-C2 del guanacastepeno se construyó mediante acoplamiento Heck intramolecular en el sustrato 1. Para esta reacción se empleó el catalizador de paladio 44, generado in situ por reacción entre el complejo de paladio(0) Pd2(dba)3·CHCl3 y dppb, tal y como se describe en el esquema 6.
Esquema 6
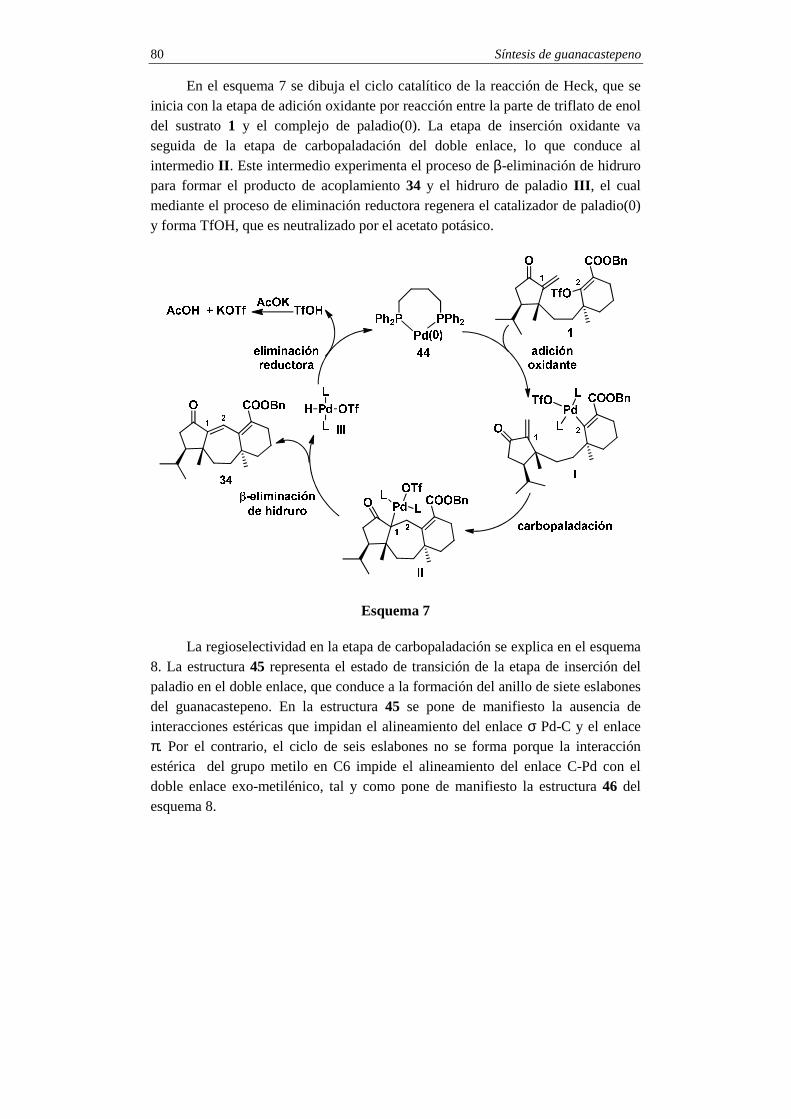
Síntesis de guanacastepeno 80
En el esquema 7 se dibuja el ciclo catalítico de la reacción de Heck, que se inicia con la etapa de adición oxidante por reacción entre la parte de triflato de enol del sustrato 1 y el complejo de paladio(0). La etapa de inserción oxidante va seguida de la etapa de carbopaladación del doble enlace, lo que conduce al intermedio II . Este intermedio experimenta el proceso de β-eliminación de hidruro para formar el producto de acoplamiento 34 y el hidruro de paladio III , el cual mediante el proceso de eliminación reductora regenera el catalizador de paladio(0) y forma TfOH, que es neutralizado por el acetato potásico.
Esquema 7
La regioselectividad en la etapa de carbopaladación se explica en el esquema 8. La estructura 45 representa el estado de transición de la etapa de inserción del paladio en el doble enlace, que conduce a la formación del anillo de siete eslabones del guanacastepeno. En la estructura 45 se pone de manifiesto la ausencia de interacciones estéricas que impidan el alineamiento del enlace σ Pd-C y el enlace π. Por el contrario, el ciclo de seis eslabones no se forma porque la interacción estérica del grupo metilo en C6 impide el alineamiento del enlace C-Pd con el doble enlace exo-metilénico, tal y como pone de manifiesto la estructura 46 del esquema 8.

Síntesis de guanacastepeno 81
Esquema 8
2. Síntesis del anillo D mediante lactonización inducida por paladio
La construcción del anillo D se consiguió mediante reacción de la parte de éster bencílico con diacetato de paladio y trietilamina, en presencia de trietilsilano como dador de hidruro. El mecanismo de esta ciclación se inicia con la reducción de la sal de paladio(II), Pd(OAc)2, por reacción con la trietilamina:
Pd(OAc) 2 + Et3N Pd(0) + Et3NOAc + AcO El ciclo catalítico se inicia con la inserción del paladio(0) en el enlace Bn-O,
que es el enlace más polar del sustrato. Esta reacción genera el intermedio I , que experimenta una reacción de oxigenopaladación del doble enlace. El resultado de este proceso es la formación del intermedio II , que ya contiene el anillo lactónico. A continuación, la transmetalación de II con Et3SiH forma benciltrietilsilano y el intermedio III , el cual, mediante el proceso de eliminación reductora, proporciona el producto de la reacción y regenera el catalizador.

Síntesis de guanacastepeno 82
Esquema 9 3. Bromación radicalaria del intermedio 41
La reacción de la mezcla de diastereoisómeros 41 con N-bromosuccinimida,
en presencia del iniciador radicalario peróxido de benzoilo, conduce a la formación de 43 como único compuesto. En este proceso no sólo se halogena estereoselectivamente C5, sino que además se reintroduce el doble enlace C1-C2 mediante halogenación de C2 seguida de deshidrohalogenación in situ.
Los carbonos C2 y C5 ocupan posiciones alílicas que generan fácilmente, al estar deslocalizados, los correspondientes radicales alilicos. El mecanismo de la

Síntesis de guanacastepeno 83
reacción de halogenación, que se indica en el esquema 10, comprende las etapas propias de las reacciones radicalarias en cadena: iniciación y propagación.
Esquema 10 Así, en la etapa de iniciación se produce la ruptura homolítica del enlace
peróxido del peróxido de benzoilo y se genera el radical benzoiloxi. La primera etapa del proceso de propagación corresponde a la reacción entre
el radical benzoiloxi y uno de los hidrógenos alílicos en C5. Este proceso forma el radical centrado en el carbono A que, a continuación, se halogena por reacción con N-bromosuccinimida para dar lugar al compuesto monobromado B y al radical N-succinimido. En la tercera etapa del proceso de propagación el radical N-succinimido reacciona con 41 para formar succinimida y generar el radical A.
Quede claro que las tres etapas a-b-c del proceso de propagación indicado en el esquema 10 no se repiten en el siguiente ciclo radicalario. Una vez el radical benzoiloxi ha provocado la formación del radical A, ya no se requiere de su

Síntesis de guanacastepeno 84
participación en el proceso de propagación, que solo constará en los siguientes ciclos de las etapas b y c.
El mecanismo de bromación de la posición alílica C2 es similar a la de la posición C5. La bromación en C2 genera el compuesto dibromado 42, que no se llega a aislar, puesto que experimenta un proceso de eliminación formal de HBr, facilitado por la acidez del protón en C1, y se transforma en el compuesto 43 (esquema 11).
Esquema 11
La estereoquímica de la reacción de bromación en C5 viene dictada por la presencia del metilo angular en C8, que obliga a aproximarse a la N-bromosuccinimida, en su ataque al radical A, desde el lado opuesto al ocupado por este grupo metilo. 4. Sustitución radicalaria de bromo por hidroxilo
La sustitución, con retención de la configuración, del átomo de bromo en C5
por el grupo hidroxilo se consiguió en dos etapas. En la primera de ellas el bromocompuesto 43 se transformó en el hidroperóxido 47 por reacción con hidruro

Síntesis de guanacastepeno 85
de tributilestaño y aire. El mecanismo de esta conversión se indica en el esquema 12 y es de tipo radicalario, por tanto similar al que se acaba de explicar en la sección anterior.
Esquema 12
La etapa de iniciación tiene lugar por reacción del hidruro de tributilestaño
con oxígeno, lo que genera los radicales tributilestaño e hidroperóxido. La primera reacción de la etapa de propagación provoca la ruptura
homolítica del enlace C-Br y la formación del radical C, que en la segunda etapa del proceso de propagación reacciona con oxígeno molecular para dar lugar al radical peróxido D. La reducción del radical D, por reacción con hidruro de tributilestaño, conduce al hidroperóxido 47 y regenera el radical tributilestaño, que inicia un nuevo ciclo radicalario.
Al igual que en el proceso de bromación radicalaria, la estereoquímica de la reacción de oxidación en C5 viene dictada por la presencia del metilo angular en C8, que obliga a la aproximación del oxígeno desde el lado opuesto al ocupado por el grupo metilo.

Síntesis de guanacastepeno 86
La reducción del hidroperóxido 47 con trifenilfosfina lleva a la obtención del gunacasterpeno sintético.
El mecanismo de la reducción de hidroperóxidos con Ph3P es el siguiente.
Esquema 13

Síntesis de harzifilona 87
SÍNTESIS DE HARZIFILONA
Aislamiento: La harzifilona se aisló de Trichoderma harzianum, un hongo antagonista de patógenos vegetales, que se encuentra presente en la mayoría de los suelos. Su crecimiento se ve favorecido por la presencia de raíces de plantas, a las cuales coloniza rápidamente. Algunas cepas son capaces de colonizar y crecer en las raíces a medida que éstas se desarrollan. El Trichoderma harzianum presenta excelentes propiedades para el control biológico, siendo especialmente efectivo contra Rhizoctonia, Fusarium y Pythium. Es también un excelente estimulador del crecimiento radicular. Actividad biológica: La harzifilona presenta propiedades antivirales y citotóxicas.
Retrosíntesis El análisis retrosintético de la harzifilona se indica en el esquema 1.
Esquema 1

Síntesis de harzifilona 88
La retrosíntesis de la harzifilona se inicia con la desconexión del anillo oxacíclico.19 Este sistema se sintetizará mediante un proceso de electrociclación 6π en el compuesto 1, que a su vez se obtendrá mediante una reacción de adición Michael intramolecular en el intermedio 2. Éste procederá de la dicetona 3, que se sintetizará mediante una reacción de C-acilación entre el cloruro de ácido 4 y la inona 5. La inona derivará de la dihidroxiciclopentanona 6, que a su vez se obtendrá de la 2-metil-2-ciclopentenona 7, compuesto que es comercialmente accesible.
Síntesis 1. Síntesis del intermedio 3
La síntesis del intermedio 3 se muestra en el esquema 2 y comenzó con la
reducción enantioselectiva de la 2-metil-2-ciclopentenona, para lo cual se empleó el método de Corey-Bakshi-Shibata (véanse comentarios). Este proceso proporcionó el ciclopentenol 8 con un 90% de exceso enantiomérico. La sililación del hidroxilo condujo al siliéter 9, cuya dihidroxilación proporcionó el correspondiente diol, el cual se transformó en el acetónido 10 mediante cetalización con isobutileno en presencia de cantidades catalíticas de ácido canforsulfónico. Este proceso permitió obtener el acetónido 10 con un 98% de exceso diastereoisomérico. La desililación de 10, seguida de oxidación condujo, a la ciclopentanona 11 que se convirtió en el acetato de enol 12 por reacción con anhidrido acético en piridina en presencia de 4-N,N-dimetilaminopiridina (DMAP). La ruptura oxidante del doble enlace, mediante dihidroxilación en presencia de metaperyodato sódico, llevó al aldehído-acido 13, que se transformó en el aldehído-éster 14 por esterificación con trimetilsilildiazometano. Este compuesto se convirtió en el alquino-éster 15 mediante reacción de homologación de Ohira-Bestmann. La ionización de la parte de alquino terminal, por reacción con n-BuLi,
19 L. M. Stara, K. Pekari, E. Sorensen, Proc. Natl. Acad. Sci. USA 2004, 101, 12064.

Síntesis de harzifilona 89
seguida de adición del (E,E)-2,4-hexenal (sorbaldehído), condujo a la obtención del correspondiente alcohol que se sililó para dar el compuesto 16.
O
(S)-CBS
OH
1. OsO4, NMO
2. CH2=C(CH3)2ac. canfor.
(95%, 98%de)
OTBS
O
O
1. TBAF
O
O
O
Ac2O, pir.
DMAP
(84%)
OAc
O
O
K2OsO2(OH)4NaIO4
CHO
CO2HO
O
TMSCHN2 CH3COC(N2)PO(OMe)2
CHO
CO2MeO
OK2CO3, MeOH, (62%)
CO2MeO
O
1. LDA,
O
H
2. TESCl (62%)
CO2MeO
O
OTES
LiHMDS,
O
O
OTES
O
1. TBAF, AcOH
(60%)
O
O
O
O
TFA-H2O
(81%)
HO
HO
O
O
3
8 9
17
101112
13 14
1516
HCl·HN(OMe)Me
TBSCl
imidazol(73%, 2 pasos)
OTBS
7
BH3·SMe2
(90% ee)
2. SO3·pir, Et3N
DMSO, (84%)
(56% 2pasos)
2. Dess-Martin
18
O
O
OTES
O
N
OMe
Me
(55%)
CH2=CHMgBr
19
Esquema 2
Síntesis de harzifilona 90
La introducción de la agrupación vinilo se consiguió por conversión de 16 en la amida de Weinreb 17, seguida de reacción de ésta con bromuro de vinilmagnesio. La desililación de 18, seguida de oxidación condujo a la dicetona 19, que se transformó en el intermedio clave 3 por hidrólisis ácida de la agrupación acetálica. 2. Pasos finales
El tratamiento del intermedio 3 con cantidades catalíticas de 1,4-
diazabiciclo[2.2.2]octano (DABCO) en cloroformo a temperatura ambiente durante 24 horas, proporcionó, directamente, la harzifilona con un 70% de rendimiento (véase esquema 3).
harzifilona
OHO
HO
O
OHO
HO
O
1
N
N
CHCl3, 25°C
(99%)
OHO
HO
O N
N
electrociclación
22
3
HO
HO
O
O
HO
HO
O
O
N
N
C
HO
HO
O
O
N
N
H
6
20
21
Esquema 3

Síntesis de harzifilona 91
La reacción global que tiene lugar en este proceso es una bicicloisomerización que transcurre en varias etapas, tal y como se muestra en el esquema 3. En primer lugar, la base DABCO se adiciona reversiblemente al sistema de enona, lo que genera el intermedio 20. A continuación, un proceso de adición Michael intramolecular sobre el sistema de inona genera la cetena 21, que se transforma en el intermedio 22 mediante una reacción de transferencia protónica intramolecular. La reacción de β−eliminación conduce a la dicetona monocíclica 1, que se convierte en la harzifilona mediante un proceso de electrociclación 6π.
Una alternativa mecanística, que también explica la formación de la harzifilona, se indica en el esquema 4. En este mecanismo el intermedio 22 se transforma directamente en la harzifilona mediante un proceso de eliminación intramolecular concertado de la molécula de DABCO.
Esquema 4
Comentarios 1. Reducción enantioselectiva de Corey-Bakshi-Shibata
Este método permite la reducción enantioselectiva de determinado tipo de cetonas por reacción con BH3, en presencia de una oxazaborolidina quiral derivada de prolina (véase síntesis de abisomicina A).
El mecanismo de la reducción con el método CBS se inicia con la formación de un complejo oxazaborolidina-BH3 en el cual el borano se coordina con el átomo de nitrógeno en la parte convexa, menos impedida, de la oxazaborolidina (intermedio II del esquema 5). A continuación, la cetona se coordina con el átomo de boro de la oxazaborolidina colocando su grupo más voluminoso (RL) en una posición de mínima interferencia estérica (intermedio III) . La transferencia de hidruro se produce a través de un estado de transición cíclico de seis eslabones que conduce a un alcoxiborano IV , cuya eliminación regenera el catalizador.

Síntesis de harzifilona 92
Esquema 5

Síntesis de ipomoeasina E 93
SÍNTESIS DE IPOMOEASINA E
Aislamiento: La glicoresina ipomoeasina E se ha aislado de la planta Ipomoea squamosa, que se encuentra en los bosques tropicales de Surinam. Actividad biológica: Este compuesto inhibe el crecimiento de células de cáncer de ovario humano A2780 en concentraciones de orden namomolar (IC50 = 35 nM).
Retrosíntesis La retrosíntesis de la ipomoeasina E se inicia con una operación de adición
de grupo funcional (AGF), concretamente con la adición de un doble enlace entre los carbonos C8-C9 de la cadena que conecta las dos unidades de carbohidrato (véase esquema 1).20 Esta operación conduce al intermedio 1, que se desconecta en el doble enlace a la diolefina 2. La siguiente operación retrosintética desconecta la cadena de acetoxi-cetoácido para dar lugar al intermedio 3 y al ácido carboxílico 4. Una siguiente desconexión en la parte de glucosa del disacárido lleva al compuesto 5, que se desconecta en el enlace glicosídico a los fragmentos 6 y 7. El fragmento 6 se obtendrá de la D-glucosa mientras que el fragmento 7 se preparará de la fucosa.
20 A. Fürstner, T. Nagano, J. Am. Chem. Soc. 2007, 127, 1906.
Síntesis de ipomoeasina E 94
Esquema 1

Síntesis de ipomoeasina E 95
Síntesis 1. Síntesis de la glucosa funcionalizada 6
La preparación del intermedio 6 se inició con la cetalización de los hidroxilos en C4 y C6 de la D-glucosa, lo que se consiguió por reacción con el dimetilacetal del p-metoxibenzaldehído en presencia de cantidades catalíticas de ácido p-toluensulfónico (esquema 2). La acetilación subsiguiente condujo a la triacetoxiglucosa 8, que se desacetalizó selectivamente en el hidroxilo anomérico por reacción con bencilamina. La activación del hidroxilo anomérico para el proyectado proceso de glicosidación se llevó a cabo por reacción de 9 con tricloroacetonitrilo y carbonato de cesio en diclorometano, lo que condujo a la obtención de un tricloracetimidato, que se corresponde con el intermedio 6 del esquema retrosintético.
Esquema 2

Síntesis de ipomoeasina E 96
2. Síntesis de la fucosa funcionalizada 7
La unidad de mucosa componente del disacárido transporta un fragmento en
el hidroxilo anomérico, que se corresponde con el alcohol (S)-1-hepten-4-ol. Este alcohol se preparó a partir de la (S)-epiclorohidrina 10, que se convirtió en primer lugar en la clorohidrina 11 por reacción con yoduro de etilmagnesio en presencia de cianuro cuproso (esquema 3). El tratamiento básico de 11 dio el epóxido 12, cuya reacción con bromuro de vinilmagnesio y cianuro cuproso condujo al (S)-1-hepten-4-ol 13.
Esquema 3 Por otro lado, el carbohidrato fucopiranosa se convirtió en el tetraacetato 14,
que se transformó en el α-bromofucopiranósido 15 por reacción con bromuro de hidrógeno (esquema 4). La glicosidación de 15 se consiguió por reacción de éste con el alcohol 13 en presencia de triflato de plata y de la base 2,6-di-ter-butilpiridina (véanse comentarios). En estas condiciones se obtuvo el β-glicósido 16, con un 84% de rendimiento, el cual, por saponificación con metóxido de potasio, condujo al intermedio 17. La cetalización selectiva de los hidroxilos en posición relativa cis, en C3 y C4, por reacción con 2,2-dimetoxipropano y ácido p-toluensulfónico proporcionó el acetónido 7.

Síntesis de ipomoeasina E 97
Esquema 4 3. Síntesis del cetoácido 4
La síntesis del cetoácido quiral 4 se inició con la adición del bromuro de butenilmagnesio 19 al furfuraldehído 18 (esquema 5). Esta reacción proporcionó una mezcla racémica de alcoholes furfurílicos 19, que se sometió a un proceso de resolución cinética Sharpless con t-butilhidroperóxido, tetraisopropóxido de titanio y D-(-)-tartrato de diisopropilo. En estas condiciones el enantiómero R se oxida más rápidamente que el S, con lo que después de 24 horas de reacción a -20ºC, se obtuvo el alcohol 21 con un 47% de rendimiento químico y más del 99% de exceso enantiomérico. Hay que señalar que la oxidación del enantiómero S no conduce a ningún producto definido, por lo que la reacción proporciona el alcohol 21 junto

Síntesis de ipomoeasina E 98
con una mezcla de productos no identificados, que se separaron en la cromatografía de columna del crudo de la reacción.
O
O
O
HO
O
CHO
BrMg+ O
HO
O
HO
O
O
HOO
O
O
SiMe3O
O
O
AcO
SiMe3OH
O
O
AcO
THF
(82%)
Ti(iPrO)4, D-(-)-DIPT
t-BuOOH, CH2Cl2, -20ºC
21 (47%, >99%ee)
t-BuOOH, VO(acac)2
CH2Cl2, 25ºC
1. CrO3, H2SO4
2. Zn, AcOH
HOSiMe3
TsOH, CH2Cl2
Ac2O, Et3N
DMAP, CH2Cl2
(93%)
TASF, DMF
(68%)
18 19 20
2223
24 25 4
Esquema 5 El compuesto furánico 21 se sometió a un proceso de transposición oxidante
por reacción con hidroperóxido de t-butilo y acetoacetilacetato de vanadilo (véanse comentarios). Esta reacción proporcionó el hemiacetal 22, que se transformó en la lactona 23 por oxidación con el reactivo de Jones seguida de reducción del doble enlace con zinc en ácido acético. La apertura del anillo lactónico con trimetilsililetanol llevó al hidroxiéster 24, el cual, tras acetilación y reacción con TASF (difluorotrimetilsilicato de tris(dimetilamino)sulfonio, ((CH3)2N)3S
+)(F2Si(CH3)3-), proporcionó el cetoácido 4.
El TASF es una fuente de anión fluoruro anhidro, al contrario que muchos de los agentes empleados en procesos de desililación que suelen contener proporciones variables de agua debido a la extraordinaria basicidad del anión fluoruro. En el TASF el anión fluoruro está enmascarado mediante formación del aducto F2Si(CH3)3
-, mientras que el contraión, el catión sulfonio ((CH3)2N)3S+,
tiene su electrofilia anulada debido al carácter electrón-donante de los tres grupos dimetilamino. El TASF se prepara mediante la siguiente reacción:

Síntesis de ipomoeasina E 99
3 (CH3)2NSi(CH3)3 + SF4 → 2 (CH3)3SiF + [(CH3)2N)3S][F2Si(CH3)3]
En relación con el proceso de apertura lactónica con trimetilsilietanol hay
que señalar que la elección de este reactivo, que condujo al cetoéster 24, permitió, después de la reacción de acetilación, la liberación quimioselectiva del ácido carboxílico con el empleo de TASF, sin reacción concomitante de la función acetato.
4. Glicosidación: unión de los fragmentos de glucosa y fucosa
OO O
MeO
O
PhO
O OH
O
O
O
AcO
O
HOOH
Ipomoeasina E
OO O
MeO
O
PhO
O OH
O
O
O
AcO
O
HOOH
Ipomoeasina E
OO O
MeO
O
PhO
O OH
O
O
O
AcO
O
HOOH
Ipomoeasina E
OO O
MeO
O
PhO
O OH
O
O
O
AcO
O
HOOH
ipomoeasina E
HO
OMe
O
OO
+
O
O
O
AcOAcO
O
Cl3C NH
MeO6
7
La unión entre el derivado de glucosa 6 y de la fucosa 7 se llevó a cabo por reacción con el complejo trifluoruro de boro·eterato, lo que proporcionó el glicósido 5 (véase esquema 6 y comentarios). La saponificación de las funciones acetato con metóxido de potasio condujo al compuesto 26, que se esterificó selectivamente en el hidroxilo C3 de la glucosa por reacción con ácido tíglico en presencia de diciclohexilcarbodiimida (DCC) y N,N-dimetilaminopiridina (DMAP). La protección del hidroxilo libre de 27, por reacción con ter-butildimetilclorosilano y 2,6-lutidina, dio el compuesto 28, cuya reacción con cianoborohidruro de sodio y trimetilclorosilano llevó a la obtención de la mezcla de p-metoxibenciléteres regioisoméricos 29:30 en relación 4:1. Este resultado era opuesto al que se esperaba, puesto que los antecedentes bibliográficos21 de esta clase de reacciones apuntaban a la formación muy preferente del regioisómero 30 (véanse comentarios). El isómero mayoritario 29 se obtuvo, con un 62% de rendimiento, después de la separación cromatográfica de la mezcla de reducción.
21 R. Johansson, B. Samuelsson, J. Chem. Soc. Perkin Trans. I 1984, 2371.

Síntesis de ipomoeasina E 100
Esquema 6

Síntesis de ipomoeasina E 101
5. Síntesis del ácido (Z)-3-dimetil(fenil)silil-2-propenoico 31
El resultado inesperado en la reducción del anillo de p-metoxibencilidenacetal obligó a un cambio en la estrategia sintética, que implicó aprovechar la formación mayoritaria del compuesto 29 para la síntesis del producto natural. En la práctica, la continuación de la síntesis con el regioisómero 29 obligaba a esterificar prematuramente el hidroxilo en C5 con la unidad de cinamoilo. Si el proceso de apertura regioselectiva del p-metoxibencilidenacetal hubiese proporcionado mayoritariamente el compuesto 30, la esterificación con el ácido cinámico se habría llevado a cabo después de la hidrogenación del doble enlace generado en la reacción de metátesis. Esta secuencia de acontecimientos evitaría la hidrogenación concomitante del doble enlace de la parte de cinamoilo, lo que es muy probable que ocurra si ésta función se encuentra presente en el proceso de hidrogenación.
Para solucionar el problema asociado al rediseño de la síntesis se empleo el ácido (Z)-sililpropenoico 31 como sintón del ácido cinámico, puesto que la mayor sustitución del doble enlace hace a éste más resistente al proceso hidrogenante. El ácido 31 se obtuvo mediante la secuencia de reacciones que se indica en el esquema 7. Asi, el alcohol propargílico 32 se sometió a un proceso de hidroaluminación por reacción con Red-Al bis-metoxietoxialuminiohidruro de sodio). En esta reacción se generó el aluminato 34, que reaccionó con yodo molecular para formar el yoduro intermedio 35. La adición de cloruro de dimetilfenilsililo a la mezcla de reacción proporcionó el yodosilil éter 36, el cual, por metalación con n-BuLi generó el alquenil-litico 37, que experimentó una transposición intramolecular del grupo dimetilfenilsililo para dar el alcóxido lítico 38. La protonación de éste proporcionó el alcohol 39, que por oxidación de Swern seguida de oxidación con clorito sódico, dio el sililácido 31.

Síntesis de ipomoeasina E 102
Esquema 7
6. Etapas finales
OO O
MeO
O
PhO
O OH
O
O
O
AcO
O
HOOH
ipomoeasina E
OO O
MeHO
OOTBS
PMBO O
OO
O
29
En el esquema 8 se indican las últimas etapas en la síntesis de la ipomoeasina E. Este proceso se inició con la esterificación del disacárido 29 con el ácido sililpropenoico 31, para lo cual se empleó el método de esterificación de Yamaguchi. El compuesto 41 resultante del proceso anterior se desprotegió en el PMB por reacción con diclorodicianoquinona (DDQ) en diclorometano acuoso para dar el alcohol 42.-
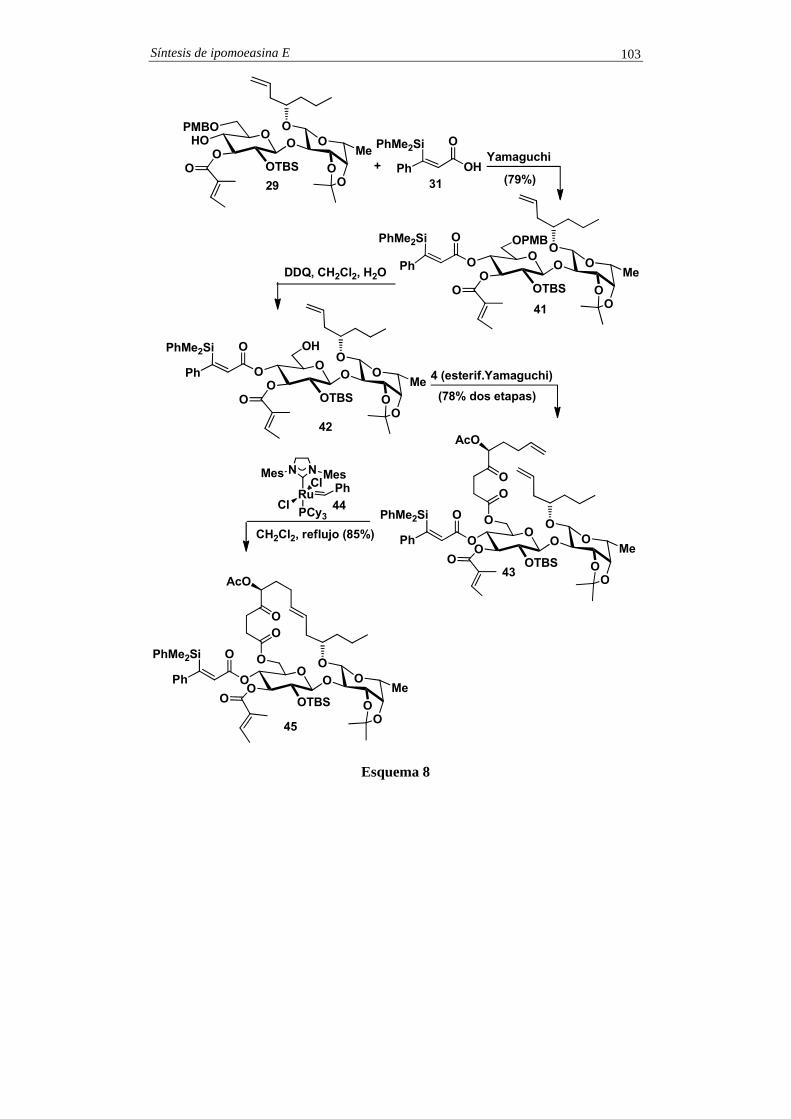
Síntesis de ipomoeasina E 103
OO O
MeHO
OOTBS
PMBO O
OO
O + Ph OH
PhMe2Si O
OO O
MeO
OOTBS
OPMBO
OO
O
PhMe2Si
Ph
O
Yamaguchi
DDQ, CH2Cl2, H2O
OO O
MeO
OOTBS
OHO
OO
O
PhMe2Si
Ph
O
OO O
MeO
O
PhO
O OTBS
O
O
O
AcO
O
OO
4 (esterif.Yamaguchi)
Ru
PCy3
Cl
Cl Ph
NNMes Mes
OO O
MeO
O
PhO
O OTBS
O
O
O
AcO
O
OO
29 31
41
42
43
44
45
(79%)
(78% dos etapas)
CH2Cl2, reflujo (85%)
PhMe2Si
PhMe2Si
Esquema 8
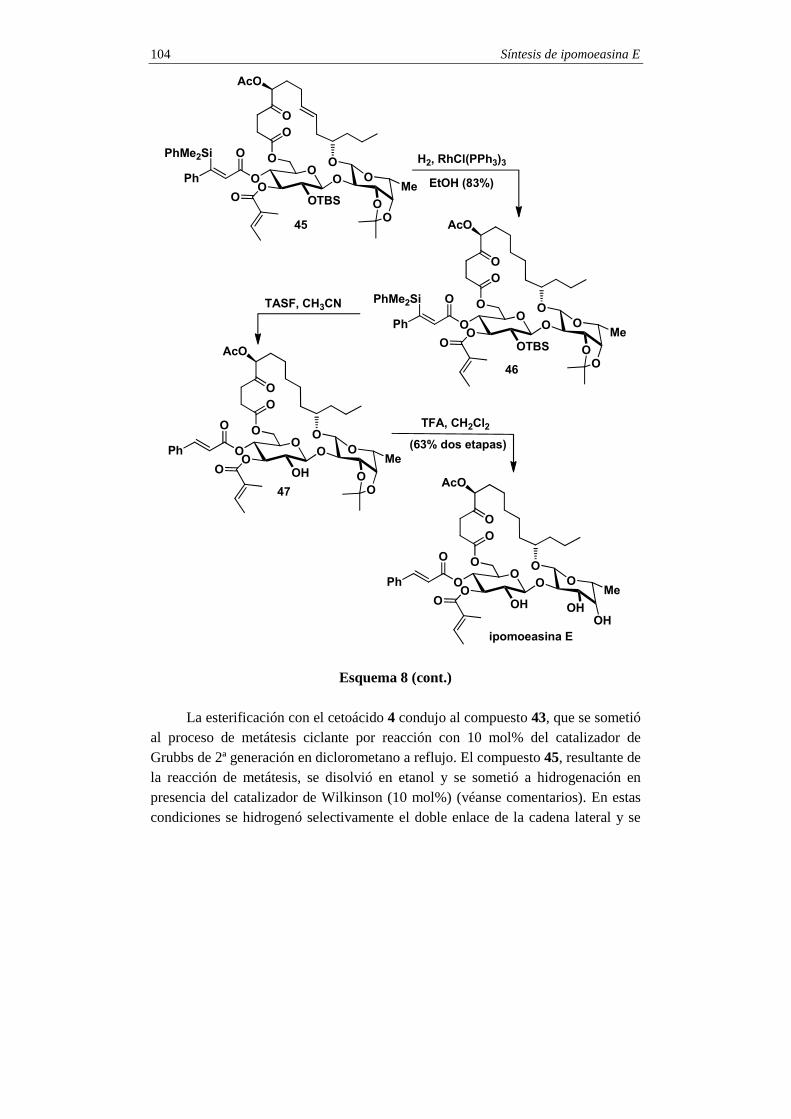
Síntesis de ipomoeasina E 104
OO O
MeO
O
PhO
O OTBS
O
O
O
AcO
O
OO
H2, RhCl(PPh3)3
EtOH (83%)
OO O
MeO
O
PhO
O OTBS
O
O
O
AcO
O
OO
TASF, CH3CN
OO O
MeO
O
PhO
O OH
O
O
O
AcO
O
OO
TFA, CH2Cl2
(63% dos etapas)
OO O
MeO
O
PhO
O OH
O
O
O
AcO
O
OHOH
45
46
ipomoeasina E
47
PhMe2Si
PhMe2Si
Esquema 8 (cont.)
La esterificación con el cetoácido 4 condujo al compuesto 43, que se sometió al proceso de metátesis ciclante por reacción con 10 mol% del catalizador de Grubbs de 2ª generación en diclorometano a reflujo. El compuesto 45, resultante de la reacción de metátesis, se disolvió en etanol y se sometió a hidrogenación en presencia del catalizador de Wilkinson (10 mol%) (véanse comentarios). En estas condiciones se hidrogenó selectivamente el doble enlace de la cadena lateral y se

Síntesis de ipomoeasina E 105
obtuvo el compuesto 46 con un 83% de rendimiento. El tratamiento del compuesto 46 con TASF en acetonitrilo provocó la eliminación simultánea de los grupos C-sililo y O-sililo y permitió la obtención del compuesto 47. Finalmente, la eliminación de la función acetálica, por reacción de 47 con ácido trifluoroacético en diclorometano, proporcionó la ipomoeasina E sintética.
Comentarios
1. Glicosidación del bromuro de fucopiranosilo 15
La glicosidación del bromuro de fucopiranosilo 15 se llevó a cabo mediante
reacción con el alcohol 13 en presencia de triflato de plata y de la base 2,6-di-ter-butilpiridina.
El mecanismo de este proceso se indica en el esquema 9 y se inicia con la reacción del bromuro de fucopiranosilo con el triflato de plata lo que lleva a la formación de bromuro de plata, del anión trifluroacetato y del catión oxonio 48. El ataque intramolecular del acetato vecinal a la parte de catión oxonio forma el intermedio cíclico 49, que se encuentra estabilizado por la deslocalización de la carga positiva, como se pone de manifiesto con la estructura resonante 50. La formación del intermedio cíclico 49-50 facilita el ataque del alcohol nucleofílico desde la cara exo inferior, lo que conduce a la formación de 51. Finalmente, la base 2,6-di-ter-butilpiridina, altamente impedida desde el punto de vista estérico, elimina el protón del intermedio 51 y lleva a la formación del glicósido 16.
Síntesis de ipomoeasina E 106
Esquema 9
2. Transposición oxidante del alcohol furfurílico 21
O
HO O
O
HO
21
t-BuOOH, VO(acac)2
CH2Cl2, 25ºC
22
Las transposiciones oxidantes de anillos furánicos sustituidos proporcionan sistema 1,4-dicarbonílicos. Estos procesos se suelen llevar a cabo mediante oxidación con ácido m-cloroperoxibenozico (MCPBA) o con fuentes de bromo positivo como la N-bromosuccinimida (NBS). Cuando el compuesto furánico presenta enlaces dobles, que pueden ser atacados por los dos reactivos acabados de comentar, se emplea la combinación hidroperóxido de t-butilo/acetilacetoacetato de vanadilo porque con este método, desarrollado por Sharpless, se consigue la
Síntesis de ipomoeasina E 107
epoxidación selectiva de alcoholes alílicos, mediante el mecanismo que se indica en el esquema 10.
Esquema 10
De acuerdo con el esquema anterior, la reacción del alcohol furfurílico 21
con la combinación t-BuOOH/VO(acac)2 genera quimioselectivamente el oxirano 52 (esquema 11), que experimenta un proceso de transposición oxidante para dar lugar al cetoaldehído 53, que se obtiene en la reacción en la forma hemiacetálica 22.
Esquema 11

Síntesis de ipomoeasina E 108
3. Síntesis del disacárido 5 mediante glicosidación de Schmidt
La unión de los fragmentos de monosacárido se llevó a cabo mediante el
proceso de glicosidación de Schmidt (esquema 12).22
Esquema 12
22 R. R. Schmidt, J. Michel, Angew. Chem. Int. Ed. Engl. 1980, 19, 731.

Síntesis de ipomoeasina E 109
En el proceso de glicosidación de Schmidt se hizo reaccionar la mezcla de tricloroacetimidatos anoméricos 6 con el fucopiranósido 7, en presencia del ácido de Lewis trifluoruro de boro·eterato. El mecanismo de la reacción se inicia con la coordinación de la parte de tricloroacetimato con el ácido de Lewis. Este proceso genera el complejo 54 que conduce al catión oxonio 55 mediante la eliminación del fragmento de tricloroacetamida. El ataque intramolecular del acetato vecinal forma el intermedio catiónico 56, que es atacado por el alcohol desde la cara exterior, más accesible desde el punto de vista estérico. Esta reacción lleva a la formación del glicósido protonado 57, cuya desprotonación permite la obtención del glicósido 5.
4. Apertura reductiva regioselectiva de p-metoxibencilidenacetal 28
NaBH3CN,TMSCl
OO O
MeO
OOTBS
O O
OO
MeOO
OO O MePMBO
OOTBS
HO O
OO
O
OO O
MeHO
OOTBS
PMBO O
OO
O
28
30
29
+
(62%)
Como se ha explicado anteriormente, los antecedentes bibliográficos
apuntaban a una apertura reductiva del anillo de p-metoxibencilidenacetal de 28 a favor del isómero 30. El mecanismo de esta conversión se indica en el esquema 13 y se inicia con la interacción ácido-base de Lewis entre el oxígeno acetálico y el átomo de silicio del Me3SiCl. Este proceso forma el complejo 58, el cual experimenta un proceso de apertura del anillo acetálico activado que conduce a la formación del catión oxonio 59. La reducción de éste con cianoborohidruro sódico forma el p-metoxibenciléter 60, el cual mediante desililación se transforma en el compuesto 30.

Síntesis de ipomoeasina E 110
Esquema 13
Sin embargo, el compuesto 30 fue el producto minoritario de la mezcla de reducción. El compuesto mayoritario fue el regioisómero 29 (relación 4:1), que se forma mediante el mecanismo que se indica en el esquema 14. La diferencia con el mecanismo anterior se debe a la coordinación del ácido de Lewis Me3SiCl con el oxígeno C4 del anillo de glucosa, lo que lleva a la formación del intermedio 61, que se transforma en el catión oxonio 62. La reducción de éste forma el p-metoxibenciliéter 63, cuya desililación proporciona el compuesto 29, mayoritario en la reacción. La regioselectividad de este proceso se achaca a la sililación preferente del hidroxilo C4 del anillo de glucosa, debida al mayor impedimento estérico que presenta el hidroxilo de C6.
La apertura reductiva del anillo de p-metoxibencilidenacetal se intentó también con otros reactivos como la combinación BH3·THF/Cu(OTf)2, BH3·THF/Bu2BOTf, PMSH/AlCl3 y Et3SiH/PhBCl2, pero con estos métodos se
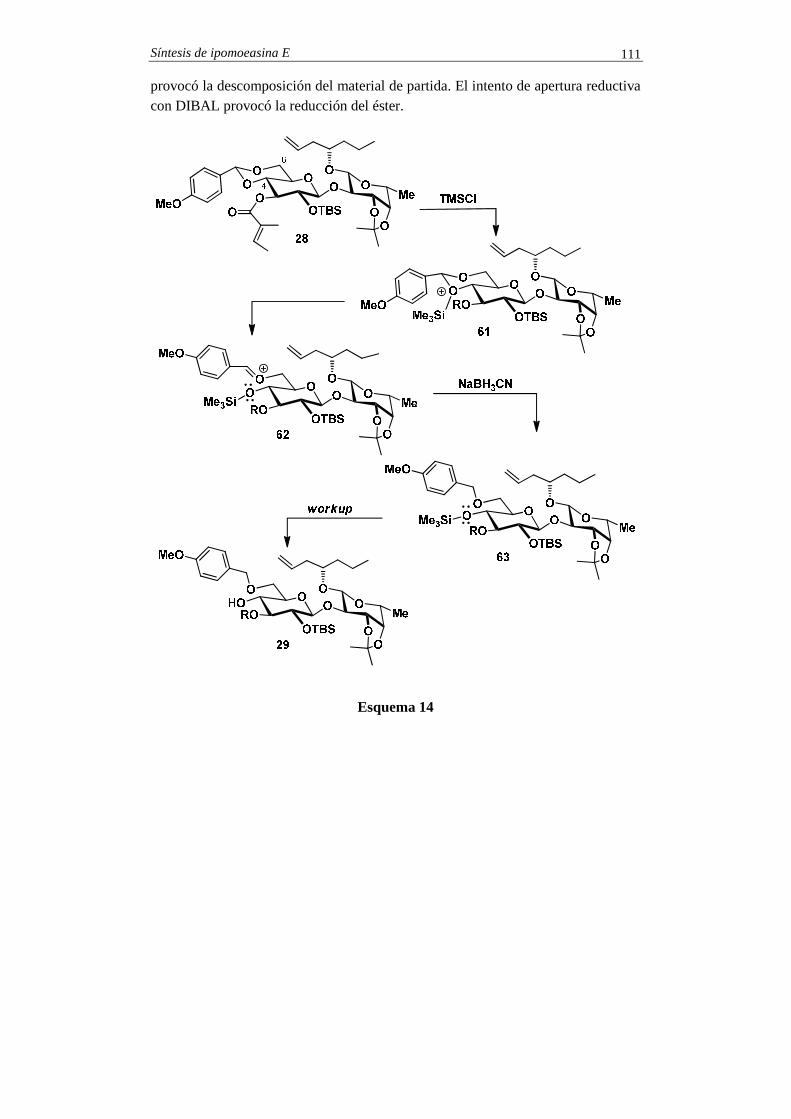
Síntesis de ipomoeasina E 111
provocó la descomposición del material de partida. El intento de apertura reductiva con DIBAL provocó la reducción del éster.
Esquema 14
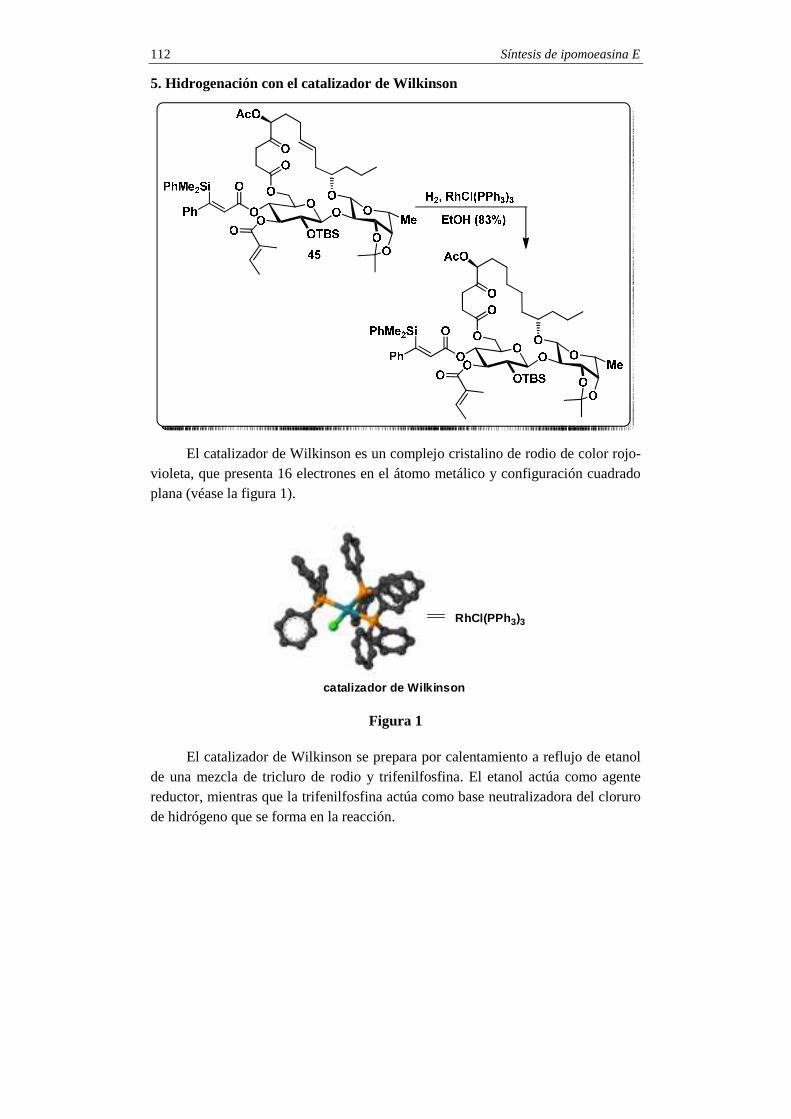
Síntesis de ipomoeasina E 112
5. Hidrogenación con el catalizador de Wilkinson
El catalizador de Wilkinson es un complejo cristalino de rodio de color rojo-violeta, que presenta 16 electrones en el átomo metálico y configuración cuadrado plana (véase la figura 1).
catalizador de Wilkinson
RhCl(PPh 3)3
Figura 1
El catalizador de Wilkinson se prepara por calentamiento a reflujo de etanol de una mezcla de tricluro de rodio y trifenilfosfina. El etanol actúa como agente reductor, mientras que la trifenilfosfina actúa como base neutralizadora del cloruro de hidrógeno que se forma en la reacción.

Síntesis de ipomoeasina E 113
RhCl 3(H2O)3 + CH3CH2OH + 3 PPh 3 RhCl(PPh 3)3 + CH3CHO + 2 HCl + 3 H2O
El catalizador de Wilkinson I se emplea como catalizador en la hidrogenación de alquenos. El mecanismo de este proceso se describe en el esquema 15, y se inicia con la disociación de uno de los ligandos trifenilfosfina para dar lugar al complejo de rodio II (14 electrones), que es la especie de rodio que cataliza el proceso de hidrogenación. A continuación, la inserción oxidante de hidrógeno molecular conduce al complejo III (16 electrones), que se coordina con el alqueno para generar el intermedio IV coordinativamente saturado (18 electrones). La hidropaladiación lleva al intermedio V (16 electrones), que por eliminación reductora forma el alcano y regenera el catalizador.
Rh(I)Ph3P
Ph3P PPh3
ClI (16 e-)
Rh(I)Ph3P
Ph3PCl
II (14 e-)
H2
inserciónoxidante
Rh(III)Ph3P
Ph3PH
H
Cl
R
Rh(III)Ph3P
Ph3PHH
Cl
R
III (16 e-)
Rh(III)Ph3P
Ph3P
H
Cl
R
IV (18 e-)
V (16 e-)
R
eliminaciónreductora
alqueno
alcano
- PPh3
hidrometalación
Esquema 15


Síntesis de manassantina A 115
SÍNTESIS DE MANASSANTINA A
OOMe
OO
OHOH
MeO OMe
OMe
R
R
manassantina A R=OMemanassantina B R=-OCH2O-
Aislamiento: Las manassantinas se han aislado de extractos de las raíces de Saurus cernuus L (Sauraceae), una planta acuática que se encuentra en el Este de Estados Unidos y en países de Extremo Oriente (Saurus chinensis). Actividad biológica: Los extractos de estas plantas se han utilizado en medicina tradicional para combatir una serie de enfermedades. Se sabe que la manassantina B es un potente inhibidor del factor de transcripción HIF-1 (hipoxia-inducible factor-1). La sobreexpresión de HIF-1 en tumores sólidos impide la eliminación del tumor y en consecuencia es la responsable del fracaso de muchos tratamientos anticancerígenos. Las manassantinas A y B inhiben el crecimiento de células PC-3 de cáncer de próstata y de otras líneas celulares con valores de IC50 superiores a los del cisplatino y la doxorubicina.
Retrosíntesis
La manassantina A presenta simetría C2 lo que facilita su análisis retrosintético, que se indica en el esquema 1.23 Éste se inicia con la doble desconexión de los anillos aromáticos, que se instalarán mediante adición del correspondiente reactivo aril-metálico al intermedio 1 (X=H, OR ó NR2), el cual, por desconexión de los enlaces aril-éter, conduce a los fragmentos 2 y 3 (P=tosilato, mesilato o triflato, X=H ó OR). El anillo tetrahidrofuránico de 2 se construirá mediante la reacción SN2 intramolecular en el hidroximesilato 4, que se podría preparar mediante la adición de un reactivo arilmetálico al aldehído 5. El sistema sin 1,2-dimetilo de este compuesto se obtendrá a partir del γ-hidroxiester (E)-insaturado 6, que se desconecta en el doble enlace al α-alcoxialdehído 7, que a
23 S. Hanessian, G. J. Reddy, N. Chalal, Org. Lett. 2006, 8, 5477.

Síntesis de manassantina A 116
su vez proviene de la cianohidrina asimétrica 8. Este compuesto se obtendrá mediante la adición asimétrica de anión cianuro al aldehído 9.
Esquema 1

Síntesis de manassantina A 117
Síntesis 1. Síntesis del intermedio 16
La síntesis de la manassantina A se inició con la cianosililación asimétrica de la O-alilvainillina 9, por adición de cianuro de trimetilsililo en presencia del catalizador 10 (véanse comentarios y esquema 2). La metanolisis la O-sililcianohidrina, en presencia de HCl gaseoso, condujo al correspondiente α-hidroxiéster, que se transformó en el α-sililoxiéster 11 por O-sililación con TBSCl. La reducción de la función éster con DIBAL, seguida de reacción de Wittig con Ph3P=CHCOOMe, proporcionó el (E)-éster α,β-insaturado 6, que se convirtió estereoselectivamente en el β-metiléster 12 por adición conjugada de dimetilcuprato de litio (véanse comentarios). La enolización del éster 12 con KHMDS, seguida de adición de yoduro de metilo a la mezcla de reacción, proporcionó estereoselectivamente el sin-α,β-dimetiléster 13 (véanse comentarios), que se transformó en el aldehído 5 por reducción con DIBAL seguida de oxidación de Dess-Martin. La adición del reactivo de Grignard 14 al aldehído 3 se consiguió, después de numerosos intentos, en presencia de CeCl3. Para ello, el aldehído 5 (1 equiv.) se añadió a una suspensión de CeCl3 anhidro (2.5 equiv.) en THF seco, a 0ºC. Después de agitación a 0ºC durante 30 minutos, enfriamiento a -78ºC, adición del reactivo de Grignard 14 (2.5 equiv.) y agitación durante 2 horas a 0ºC, se obtuvo la mezcla de diastereosiómeos 15 en relación 2.5:1. La fuerte oxofilia del CeCl3 activa la adición del reactivo de Grignard y disminuye las reacciones indeseadas, como la enolización del compuesto carbonílico, provocada muchas veces por la fuerte basicidad del reactivo organometálico.24
24 T. Imamoto, N. Takiyama, K. Nakamura, T. Hatajima, Y. Kamiya, J. Am. Chem. Soc. 1989, 111, 4392.
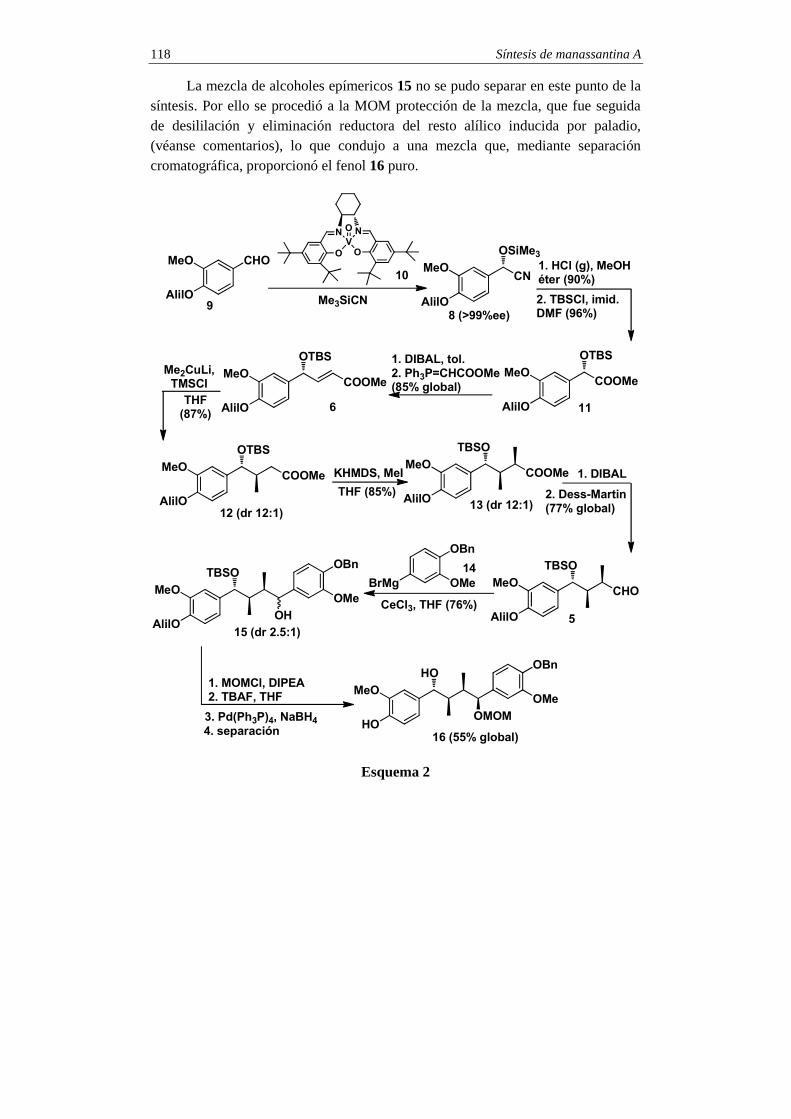
Síntesis de manassantina A 118
La mezcla de alcoholes epímericos 15 no se pudo separar en este punto de la síntesis. Por ello se procedió a la MOM protección de la mezcla, que fue seguida de desililación y eliminación reductora del resto alílico inducida por paladio, (véanse comentarios), lo que condujo a una mezcla que, mediante separación cromatográfica, proporcionó el fenol 16 puro.
CHOMeO
AlilO9 Me3SiCN
1. HCl (g), MeOHéter (90%)
2. TBSCl, imid.DMF (96%)
MeO
AlilO
OSiMe3
CN
MeO
AlilO
OTBS
COOMe
8 (>99%ee)
1. DIBAL, tol.
2. Ph3P=CHCOOMe(85% global)
MeO
AlilO
OTBS
COOMeMe2CuLi,TMSCl
THF(87%)
MeO
AlilO
OTBS
COOMe
12 (dr 12:1)
KHMDS, MeI
THF (85%)
MeO
AlilO
TBSO
COOMe 1. DIBAL
2. Dess-Martin
(77% global)
MeO
AlilO
TBSO
CHOBrMg
OBn
OMe
CeCl3, THF (76%)
MeO
AlilO
TBSOOBn
OMe
3. Pd(Ph3P)4, NaBH4
4. separación
MeO
HO
HOOBn
OMe
OMOM
OH
15 (dr 2.5:1)
16 (55% global)
116
5
13 (dr 12:1)
14
NV
N
O O
O
10
1. MOMCl, DIPEA2. TBAF, THF
Esquema 2
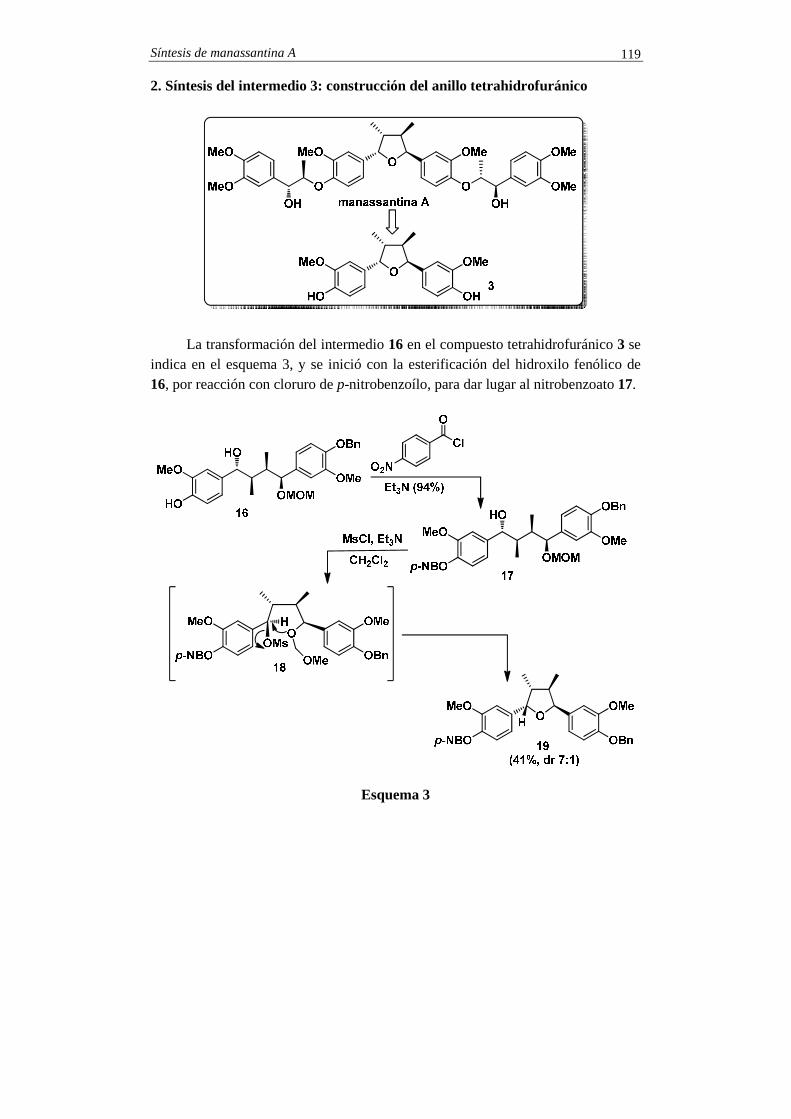
Síntesis de manassantina A 119
2. Síntesis del intermedio 3: construcción del anillo tetrahidrofuránico
La transformación del intermedio 16 en el compuesto tetrahidrofuránico 3 se indica en el esquema 3, y se inició con la esterificación del hidroxilo fenólico de 16, por reacción con cloruro de p-nitrobenzoílo, para dar lugar al nitrobenzoato 17.
Esquema 3

Síntesis de manassantina A 120
Esquema 3 (cont.)
La reacción de 17 con cloruro de metanosulfonilo, en presencia de trietilamina, generó el mesilato 18, que experimentó in situ un desplazamiento intramolecular de tipo SN2, para dar lugar al compuesto tetrahidrofuránico 19, junto con su epímero, en relación 7:1 (véanse comentarios). Por último, la saponificación de 19, seguida de desbencilación hidrogenolítica, condujo al intermedio 2. 3. Pasos finales
La conversión del intermedio bis-fenólico 2 en la manassantina A se inició con la eterificación SN2 por reacción con el triflato 3, derivado de (S)-lactato de metilo. Esta reacción se efectuó en presencia de la base de Schwesinger (2-ter-butilimino-2-dietilamino-1,3-dimetilperhidro-1,3,2-diazafosforina, véanse comentarios) y proporcionó el diéster 21 con un 95% de rendimiento. La conversión de éste en la bis-amida de Weinreb 1, seguida de adición del reactivo de Grignard 22, condujo a la dicetona 23, que se convirtió en la manassantina A por reducción estereoselectiva con el borohidruro soportado 24.

Síntesis de manassantina A 121
Esquema 4

Síntesis de manassantina A 122
Comentarios 1. Síntesis asimétrica de cianohidrinas
El ciclo catalítico de la reacción de cianosililación asimétrica implica a tres
complejos diméricos bimetálicos (intermedios III , IV y V del esquema 5).
Esquema 5

Síntesis de manassantina A 123
La formación de los dímeros viene precedida de la conversión del catalizador 10 en el complejo de cianuro I y en el metaloacetal monometálico II . El catalizador activa simultáneamente al cianuro de trimetilsililo y al aldehído, y la etapa clave del ciclo catalítico es la conversión del intermedio III en IV , que implica el ataque intramolecular del cianuro a la cara Re del aldehído, que se encuentra coordinado con el metal.25 2. Adición estereoselectiva de organocupratos a γγγγ-sililoxiésteres (E)-α,βα,βα,βα,β-insaturados
En el esquema 6 se indican tres posibles conformaciones que conducen al aducto anti, que es el que se forma mayoritariamente en la adición de Me2CuLi al éster 6. El estado de transición asociado a la conformación A está claramente desfavorecido por el impedimento estérico que sufre el nucleófilo en su trayectoria de aproximación al doble enlace.
Esquema 6
25 Este mecanismo es traslación del que se representa en el artículo de M. North Tetrahedron:Asymmetry 2003, 12, 147, para la cianosililación asimétrica con un catalizador quiral salen-Titanio(IV).

Síntesis de manassantina A 124
La conformación B es de tipo Felkin-Ahn (grupo OTBS perpendicular al doble enlace y anti respecto de la trayectoria del nucleófilo atacante) y además está favorecida por el efecto electrón-atrayente del grupo γ-sililoxi, debido a la orientación que ocupa en relación con el sistema π del doble enlace. Otro factor favorable de esta conformación es que permite un mejor solapamiento del HOMOdel nucleófilo con el orbital σ* del enlace C-OTBS. Sin embargo, el estado de transición asociado a esta conformación no se beneficia de la estabilización del incipiente orbital σ* C-Cu, puesto que el enlace sigma paralelo implica al grupo sililoxi electronegativo. Por otro lado, la tensión alílica 1,2 entre el grupo R y el H también es un factor desfavorable asociado a esta conformación.
La conformación C permite una interacción favorable del grupo sililoxi con el sistema electrónico π a través de la interacción de dos electrones (p) y de cuatro electrones (π) en el estado fundamental. Además, el orbital σ R-C puede interaccionar con el orbital π*, estabilizando así el sistema α,β-insaturado y convirtiendo al grupo carbonilo del éster en una mejor zona para la quelación. La orientación anti del grupo R en el confórmero C también puede estabilizar el orbital incipiente σ* C-Cu en el complejo d,π*-β-cuprio(III) mediante donación sigma. Por otro lado, la conformación C está libre de tensión alílica 1,2. Así pues el estado de transición asociado a la conformación C es el que explica mejor la formación del aducto anti.
Sin embargo, con grupos R poco voluminosos la diferencia de energía entre la conformación C y las conformaciones D-E-F diminuye, lo que provoca un aumento de la proporción del aducto sin.
3. Alquilación estereoselectiva de ββββ-metilésteres
La formación mayoritaria del aducto sin, en la etapa de alquilación del enolato, se explica mediante la intervención del estado de transición que se dibuja en el esquema 7. La conformación que adopta el enolato coloca al átomo de hidrógeno en la proximidad del grupo OMe, a fin de disminuir al máximo las interacciones estéricas. De esta forma el grupo R voluminoso bloquea la cara Si del

Síntesis de manassantina A 125
enolato y la aproximación del yoduro de metilo se produce desde la cara Re, para dar lugar al aducto sin.
Esquema 7
4. Ruptura reductiva de alil aril éteres con Pd(Ph3P)4/NaBH4
El compuesto 16 se obtuvo a partir de la mezcla de epímeros 15 mediante una secuencia sintética que implicaba, entre otras, una reacción de ruptura de alil aril éter con la combinación Pd(Ph3P)4/NaBH4. El mecanismo de esta reacción, catalítica en Pd(Ph3P)4, se indica en el esquema 8. El proceso se inicia con la adición oxidante del alil aril éter al complejo de Pd(0) coordinativamente insaturado, lo que conduce a la formación del complejo π-alílico de paladio I . La sustitución nucleofílica del grupo arilóxido por hidruro forma el complejo de paladio (II) II , cuya eliminación reductora genera propeno y el catalizador.
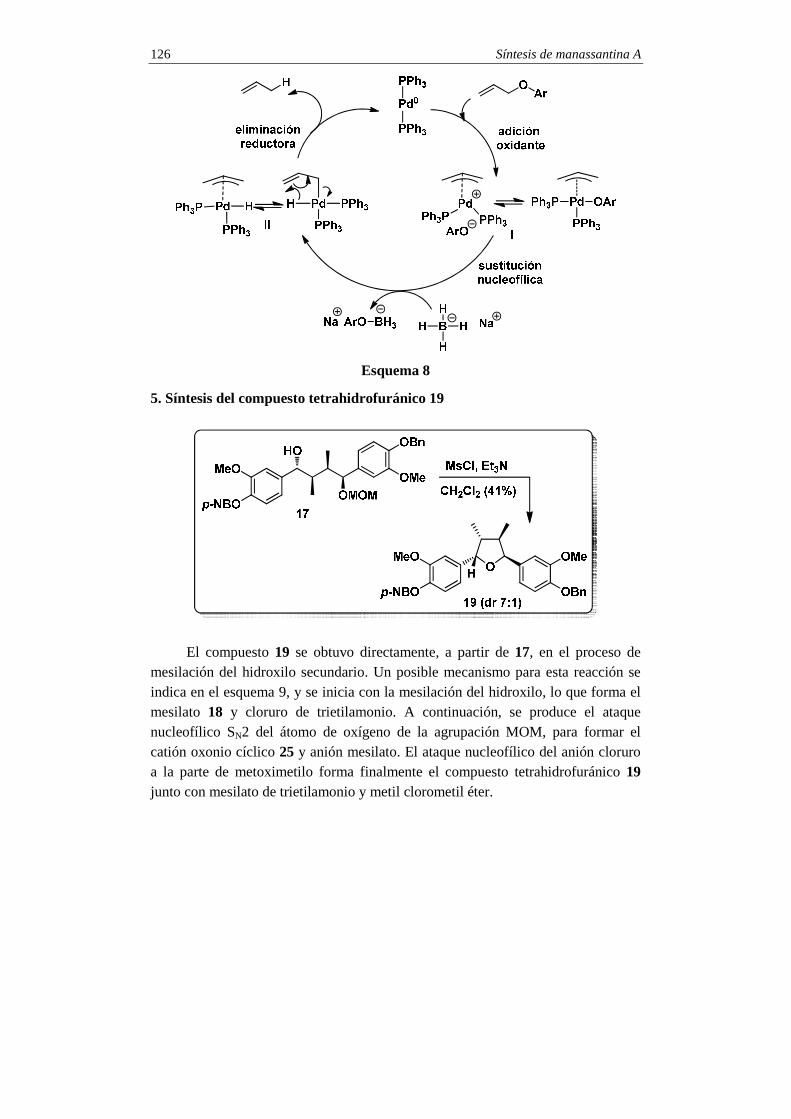
Síntesis de manassantina A 126
Esquema 8
5. Síntesis del compuesto tetrahidrofuránico 19
El compuesto 19 se obtuvo directamente, a partir de 17, en el proceso de mesilación del hidroxilo secundario. Un posible mecanismo para esta reacción se indica en el esquema 9, y se inicia con la mesilación del hidroxilo, lo que forma el mesilato 18 y cloruro de trietilamonio. A continuación, se produce el ataque nucleofílico SN2 del átomo de oxígeno de la agrupación MOM, para formar el catión oxonio cíclico 25 y anión mesilato. El ataque nucleofílico del anión cloruro a la parte de metoximetilo forma finalmente el compuesto tetrahidrofuránico 19 junto con mesilato de trietilamonio y metil clorometil éter.

Síntesis de manassantina A 127
Esquema 9 La reacción de ciclación proporciona alrededor de un 12% del epímero 28,
que no se puede explicar mediante el proceso SN2. La formación de este epímero se puede explicar si se admite que, asociada a la vía mecanística SN2, se produce también el ataque nucleofílico SN1, como consecuencia de la formación del catión bencílico 26, favorecida por la presencia en el anillo aromático del grupo metoxilo electrón-donante (esquema 10).
Esto explica por qué se lleva a cabo el cambio del grupo protector del hidroxilo en posición para de alilo a p-nitrobenzoílo. Este cambio de grupos protectores, que no está justificado en el trabajo original, se lleva a cabo para evitar al máximo la vía mecanística SN1, asociada a la formación del carbocatión bencílico, y en consecuencia a la pérdida de esterecontrol del proceso de ciclación. Al cambiar el grupo alilo (electrón-donante) por el grupo p-nitrobenzoilo (electrón-atrayente) se desfavorece la formación del catión bencílico.

Síntesis de manassantina A 128
Esquema 10 6. Bases de Schwesinger
La necesidad de aumentar la fuerza de las bases, sin aumentar al mismo tiempo su nucleofília, explica el desarrollo de las denominadas superbases, entre las cuales se encuentran las denominadas bases de Schwesinger, algunas de las cuales se dibujan en el esquema 11.
La bases de Schwesinger tienen una fortaleza básica similar a la de las bases organolíticas y son alrededor de 18 órdenes de magnitud más básicas que la DBU o DIPEA. El centro básico de los fosfacenos de Schwesinger es el átomo de nitrógeno, que genera un ácido conjugado estabilizado por deslocalización de la carga positiva.

Síntesis de manassantina A 129
Esquema 11
La base de Schwesinger BEMP se emplea en la síntesis de la manassantina A en la conversión del bis-fenol 2 en el compuesto 21, mediante una reacción tipo SN2 con el triflato 3 (esquema 12).
Esquema 12


Síntesis de (±)-merrilactona
131
SÍNTESIS DE (±)-MERRILACTONA A
Aislamiento: La merrilactona es un sesquiterpeno pentacíclico que se aisló en el año 2000, por Fukuyama y colaboradores, de la planta Illicium merrillianum. Actividad biológica: La merrilactona es un potente factor neurotrófico. Favorece el crecimiento de las neuritas en cultivos de neuronas corticales de fetos de rata, en concentraciones del orden de 0.1µmol/L.
Retrosíntesis
La primera operación retrosintética sobre la merrilactona desconecta el enlace éter del anillo oxaciclobutánico E (esquema 1).
OO
Me
MeO
OO
HO
Me
merrilactona A
OO
Me
MeO
OHO
O
Me
OO
Me
MeO
OHO
Me
OO
Me
MeO
O
O
OP
COOEt
OMe
O
O
OP
OMe
O
O
OP
OMe
O
O
OPO MePO
O
OPO
O
OP
X
X
+ OO
Br
1 2 3
4567
8
9 X=metal)10
A
B CD
E
D
B
A
adición
radicalaria
lactoni.
ciclación de
Nazarov C
SN2
Esquema 1

Síntesis de (±)-merrilactona
132
La desconexión conduce al epoxialcohol 1, que se obtendrá por epoxidación de 2 (esquema 1).26 La isomerización del doble enlace lleva al intermedio 3, que se desconecta en el anillo lactónico D al sistema de γ-alcoxiéster que presenta el intermedio 4. La desconexión de la agrupación etoxicarbonilo origina el intermedio 5, cuya desconexión en el anillo B, que se construirá mediante un proceso de adición radicalaria intramolecular, da lugar al compuesto bicíclico 6. La operación clave del análisis retrosintético es la que desconecta el anillo C, que se instalará mediante un proceso de ciclación Nazarov sobre la furil-enona 7. Este compuesto se obtendrá mediante la reacción del derivado de ácido 8 con el compuesto metal-furánico 9, que a su vez se sintetizará de la bromolactona 10.
Síntesis
1. Síntesis del agente de acilación 8
El agente de acilación 8 se preparó en tres etapas a partir de la hidroxiacetona 11, mediante sililación, seguida de olefinación al éster insaturado 13, y conversión en la amida de Weinreb por reacción con el clorhidrato de N,O-dimetilhidroxilamina y cloruro de isopropilmagnesio (esquema 2).
O
TBSO
EtO
O
OH
TBSCl, imid
DMF
O
OTBS
(EtO)2P(O)CH2COOEt
MeNHOMe·HCl
O
TBSO
NMeO
Me
11 12
138
iPrMgCl, THF
(84%)
t-BuOK
Esquema 2 26 W. He, J. Huang, X. Sun, A. J. Frontier, J. Am. Chem. Soc. 2007, 127, 498.

Síntesis de (±)-merrilactona
133
2. Síntesis del bromofurano 21, precursor del intermedio metal-furánico 9
La síntesis del compuesto furánico 21 se inició con la reacción de metalación del propiolato de etilo 14, lo que generó el intermedio organolítico 15, que se adicionó al aldehído 16 para dar lugar al hidroxiéster diínico 17 (esquema 3). La adición conjugada generó el alquenilestannano 18, que experimentó in situ la reacción de lactonización para proporcionar la estanil-lactona 19. La reacción de ésta con bromo molecular proporcionó la bromolactona 20, que se transformó en el bromofurano funcionalizado 21 por reacción con triflato de triisopropilsililo y trietilamina.
Esquema 3

Síntesis de (±)-merrilactona
134
3. Acilación del anillo furánico: síntesis del intermedio 7
La acilación del intermedio bromofuránico 21 se consiguió por metalación con t-butil-litio, seguida de adición de la amida de Weinreb 8 a la mezcla de reacción. Este proceso proporcionó el furano funcionalizado que se indica en el esquema 4, que se corresponde con el intermedio 7 del análisis retrosintético.
OTIPSO
Br
TMS
t-BuLi, éter OTIPSO
Li
TMS
OTIPSO
O
OTBSTMS(82%)de -50ºC a -20ºC
21 9 7
amida 8
Esquema 4
4. Síntesis del sistema de anillos A-B-C: preparación del intermedio tricíclico 5
El furano funcionalizado 7 contiene todos los carbonos necesarios para la construcción del sistema de anillos A-B-C de la merrilactona. El anillo C se sintetizó mediante reacción de ciclación de Nazarov promovida por el catalizador I [(IrMe(CO)(dppe)(DIB)](BARF)2 (véanse comentarios para la estructura y modo de acción del catalizador). La reacción proporcionó el compuesto bicíclico 22 con un 87% de rendimiento (esquema 5). La eliminación selectiva del grupo trimetilsililo de la parte de acetileno terminal condujo al compuesto 23, que se sometió al proceso de ciclación radicalaria intramolecular por reacción con hidruro de tributilestaño, en benceno a reflujo, en presencia de azo-bis-isobutironitrilo como iniciador radicalario (véanse comentarios). Este proceso permitió la

Síntesis de (±)-merrilactona
135
construcción del anillo B de la merrilactona y generó el alquenil-estannano 24, el cual, por protólisis con ácido p-toluensulfónico, se convirtió en el compuesto tricíclico 25. Finalmente, la desililación inducida por fluoruro de tetra-n-butilamonio condujo a la cetolactona tricíclica 5.27
Esquema 5
5. Pasos finales: construcción de los anillos D y E
En el esquema 6 se indican los pasos finales de la síntesis de la merrilactona A. La secuencia se inició con la construcción del anillo lactónico D. Para ello, el hidroxilo del compuesto 5 se convirtió en el carbonato mixto 26 por reacción con cloroformiato de metilo. El tratamiento con hidruro sódico provocó la reacción de lactonización nucleofílica intramolecular para dar una mezcla 1:1 de la bislactona 28 y el producto abierto 27. La reacción de esta mezcla con ácido p-
27 Todos los compuestos quirales en esta síntesis de merrilactona A se obtuvieron en forma de racematos.

Síntesis de (±)-merrilactona
136
toluensulfónico, en benceno a reflujo, convirtió el hidroxiéster 27 en la bis-lactona 28, lo que proporcionó el compuesto tetracíclico con un rendimiento global del 90% a partir del carbonato 26. La α-metilación de la parte cetónica de 28 se consiguió por ionización con hidruro sódico y reacción con yoduro de metilo en una mezcla de THF y HMPA. Con esta última reacción se completaba el sistema carbonado de la merrilactona quedando por construir el anillo E.
Esquema 6
La síntesis de este anillo se inició con la reducción estereoselectiva del
carbonilo cetónico del compuesto tetracíclico 3. Para este fin se utilizó DIBAL y L-selectride, pero con ambos agentes se provocó la sobre-reducción del sustrato 3. Cuando se empleó borohidruro de sodio se obtuvo, con un 93% de rendimiento, una mezcla constituida por los epímeros 29/30 en relación 1.2:1, cuya cromatografía de columna proporcionó el compuesto 29 puro. Después de la separación, el compuesto 30 puro se oxidó, con el reactivo de Dess-Martin y de forma casi cuantitativa, a la cetona 3. Este procedimiento de reciclado permitió aprovechar todo el compuesto 3 y convertirlo en el alcohol 29. A continuación, el tratamiento de éste con ácido p-toluensulfónico provocó la isomerización del doble

Síntesis de (±)-merrilactona
137
enlace y la obtención del intermedio 2, el cual, se transformó en el oxirano 1 por epoxidación estereoselectiva con ácido m-cloroperoxibenzoico. Finalmente, la merrilactona A sintética se obtuvo mediante tratamiento del epóxido 1 con ácido p-toluensulfónico.
Comentarios
1. Ciclación de Nazarov: construcción del anillo C
La reacción de ciclación de Nazarov, que se conoce desde 1940, permite la conversión de sistemas dienónicos cruzados en ciclopentenonas. La reacción requiere la catálisis mediante ácidos protónicos o ácidos de Lewis. El mecanismo de la reacción de ciclación de Nazarov se inicia con la coordinación del ácido de Lewis, o el protón, con el grupo carbonilo cetónico del sistema dienónico (véase intermedio 33 del esquema 7, LA= ácido de Lewis).
Esquema 7

Síntesis de (±)-merrilactona
138
La coordinación con el ácido de Lewis provoca la reacción de electrociclación 4π conrotatoria, lo cual genera el catión 3-oxiciclopentadienilo 34, que por pérdida de protón se transforma en el dienolato 35. La reprotonación, conduce al compuesto ciclopentenónico 36 con regeneración concomitante del catalizador.
El catalizador que se empleó en la ciclación de Nazarov del compuesto 7 fue el complejo dicatiónico de Ir(III) [(IrMe(CO)(dppe)(DIB)](BARF)2, (DIB=o-diyodobenceno, BARF=B(3,5-((CF3)2)C6H3)4, cuya estructura se indica a continuación:
Catalizador I
La capacidad catalítica del complejo anterior se basa en la débil coordinación
del ligando DIB (o-diyodobenceno), que deja sitios vacantes para la coordinación con el sustrato orgánico. Por ejemplo, en presencia de 2 mol% del catalizador 1 el sustrato 37 se convierte, en menos de 20 minutos, al calentar desde -60ºC a temperatura ambiente, en el compuesto ciclado 38 con un 99% de rendimiento.28
Un mecanismo que explica el modo de acción del catalizador I se indica en el esquema 8. El proceso se inicia mediante el intercambio de ligandos, que está favorecido por la débil coordinación del o-diyodobenceno al catalizador I . Esta reacción conduce al intermedio II que, a continuación, experimenta la reacción de
28 M. Janka, W. He, A. J. Frontier, R. Eisenberg J. Am. Chem. Soc. 2004, 126, 6864.

Síntesis de (±)-merrilactona
139
electrociclación 4π conrotatoria que lleva a la formación del catión oxialilo III . El proceso desprotonación-reprotonación genera IV , cuya reacción de intercambio con el sustrato 37 forma el compuesto 38 y regenera la especie II , que vuelve a intervenir en un nuevo ciclo catalítico.
O
R
O O
OMecat. I
O
R
O O
OMe
LnIr 2+
O
R
O O
OMe
LnIr 2+
electrociclación
4
conrotatoria
H
O
R
O O
OMe
LnIr 2+
O
R
O O
OMe
O
R
O O
OMe
II
IIIIV
37
37
38
- H+
Esquema 8
La presencia de dos centros de coordinación vecinales en el sustrato es vital
para que el catalizador I ejerza su acción en el proceso Nazarov. Esta hipótesis está respaldada por el fallo de la reacción sobre el sustrato 39.
R
O
cat. I
R= 2,4,6-trimetoxifenilo
39
No hay reacción

Síntesis de (±)-merrilactona
140
Los estudios de RMN de 1H han demostrado que el sustrato 39 desplaza al ligando o-diyodobenceno del catalizador I , pero la coordinación subsiguiente forma el intermedio 40, que no experimenta el proceso de ciclación porque los dos grupos vinilo están espacialmente separados, y por tanto en una orientación que imposibilita el proceso de ciclación.
A tenor de los antecedentes acabados de comentar habría que concluir que el
compuesto 7, debido a la ausencia en su estructura de dos centros de coordinación vecinales, no debería ser un sustrato adecuado para el catalizador I . Sin embargo, la reacción de 7 con 2 mol% del catalizador I proporcionó el compuesto ciclado 22 con un 87% de rendimiento. Un mecanismo para esta reacción se indica en el esquema 9. Hay que señalar que el proceso 4π conrotatorio permite la creación estereoespecífica de los estereocentros en C4 y C5 con la configuración relativa requerida para la síntesis de la merrilactona A.
Esquema 9

Síntesis de (±)-merrilactona
141
2. Construcción del anillo B mediante reacción de adición radicalaria intramolecular
OTBS
OMe
O
OTIPS
OTBS
OMe
O
OTIPS
H
Bu3SnH, AIBN
benceno, reflujo
TsOH, 25ºC
SnBu3
(91%)
OTBS
OMe
O
OTIPS
2423 25
AA A
C
BB
CC
La construcción del anillo B se consiguió mediante un proceso de adición radicalaria intramolecular en el sustrato 23 (esquema 10).
Esquema 10

Síntesis de (±)-merrilactona
142
En la reacción se utilizó, como iniciador radicalario, el azo-bis-isobutironitrilo (AIBN), y el hidruro de tributrilestaño como especie dadora de hidrógeno y generadora de radicales tributilestaño. En el esquema 10 se indica el mecanismo radicalario que consta de dos etapas claramente diferenciadas: la de iniciación y la de propagación. La etapa de iniciación comienza con la generación de radicales isobutironitrilo por escisión homolítica del AIBN. La reacción de estos radicales con el hidruro de tributilestaño forma isobutironitrilo y radicales tributilestaño, que son los que intervienen en la etapa de propagación. En el primer paso de esta etapa el radical tributilestaño se adiciona regioselectivamente al triple enlace del sustrato 23 para formar el radical vinílico 42, cuya adición conjugada intramolecular al sistema de enona forma el radical centrado en el oxígeno 43. En el último paso, el radical 43 se reduce, por reacción con hidruro de tributilestaño, para formar producto de ciclación 24 y el radical tributilestaño que inicia un nuevo ciclo de propagación.


















