
PROCEDIMIENTOS DE CALIBRACIÓN
46
- 26 - 4. PROCEDIMIENTOS DE CALIBRACIÓN 4.1 Calibración mediante espectrofotometría UV-visible Procedimiento general Por lo general, para el análisis mediante espectrofotometría UV-visible es posible preparar curvas de calibración "permanentes." Esto se refiere a que una curva de calibración puede usarse con varios lotes de análisis, durante varios meses. Se prepara una nueva curva de calibración, cuando de patrones muestran un cambio de la pendiente o cuando se comienza a usar un nuevo lote de reactivos. La curva estándar inicial debe incluir un blanco reactivo y, por lo menos, ocho estándares que. cubran toda la escala de concentraciones que se usarán en los análisis rutinarios del laboratorio y que permite medir el método analítico. El instrumento debe ponerse en cero con agua destilada. A este punto se denomina "Línea base del blanco." El blanco "de reactivos" se refiere a la respuesta analítica (por ejemplo absorbancia) causada por los reactivos cuando se analiza agua pura que no contiene el parámetro. Cuando se traza la curva de calibración, no se resta el blanco reactivo de las otras lecturas, sino que se trata como un punto correspondiente a la concentración cero (por ejemplo, C = O). Las curvas de calibración pueden ser lineales o no lineales. En la mayoría de casos, cuando la curva es lineal se aplica la Ley de Beer. Para definir la curva de calibración que mejor representa la relación entre la absorbancia (A) y la concentración (C) se pueden usar cálculos de regresión lineal de la siguiente manera: Dadoque A=mC+b donde: A = absorbancia C = concentración
-
Upload
cerecerita -
Category
Documents
-
view
32 -
download
2
description
espectrofotometria
Transcript of PROCEDIMIENTOS DE CALIBRACIÓN

- 26 -
4. PROCEDIMIENTOS DE CALIBRACIÓN
4.1 Calibración mediante espectrofotometría UV-visible
Procedimiento general
Por lo general, para el análisis mediante espectrofotometría UV-visible es posible preparar curvas de calibración "permanentes." Esto se refiere a que una curva de calibración puede usarse con varios lotes de análisis, durante varios meses. Se prepara una nueva curva de calibración, cuando de patrones muestran un cambio de la pendiente o cuando se comienza a usar un nuevo lote de reactivos.
La curva estándar inicial debe incluir un blanco reactivo y, por lo menos, ocho estándares que. cubran toda la escala de concentraciones que se usarán en los análisis rutinarios del laboratorio y que permite medir el método analítico.
El instrumento debe ponerse en cero con agua destilada. A este punto se denomina "Línea base del blanco."
El blanco "de reactivos" se refiere a la respuesta analítica (por ejemplo absorbancia) causada por los reactivos cuando se analiza agua pura que no contiene el parámetro. Cuando se traza la curva de calibración, no se resta el blanco reactivo de las otras lecturas, sino que se trata como un punto correspondiente a la concentración cero (por ejemplo, C = O).
Las curvas de calibración pueden ser lineales o no lineales. En la mayoría de casos, cuando la curva es lineal se aplica la Ley de Beer. Para definir la curva de calibración que mejor representa la relación entre la absorbancia (A) y la concentración (C) se pueden usar cálculos de regresión lineal de la siguiente manera:
Dadoque A=mC+b
donde:
A = absorbancia C = concentración

Se puede demostrar que:
m=
- 27 -
nEAc - LAEc nl:c2 - (LC) 2
b=EA-mEc n n
donde:
y
m = la pendiente de Ja curva de calibración (también denominada "factor de calibración").
b = Ja intersección del valor de absorbancia o del eje "y". n = el número de observaciones (lotes de valores de C y de A).
Aunque estos cálculos pueden realizarse manualmente, resulta mucho más conveniente utili7.ar una computadora o una calculadora manual para elaborar el análisis de regresión.
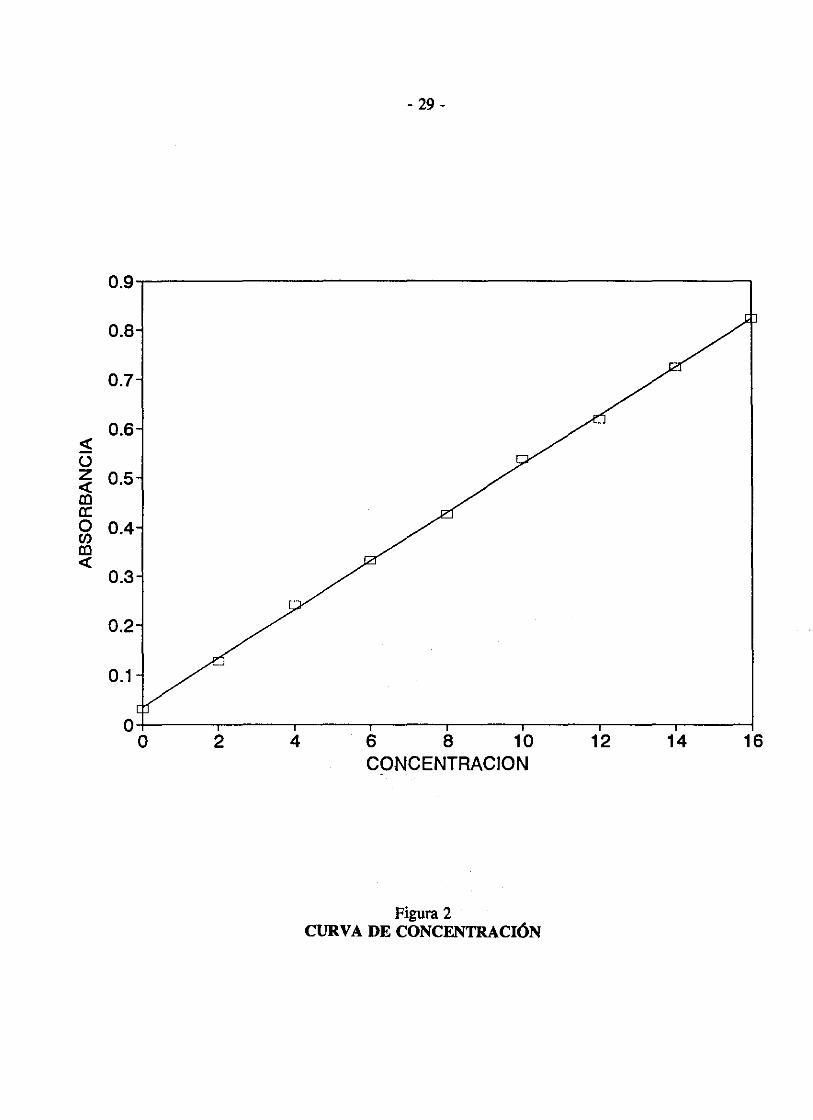
Por ejemplo, dados Jos siguientes resultados de concentración (C) con relación a la absorbancia (A),
e A
o.o 0.032
2.0 0.128
4.0 0.240
6.0 0.331
8.0 0.426
10.0 0.537
12.0 0.618
14.0 0.724
16.0 0.824

se puede calcular,
m b
= (pendiente) = (intersección de y)
- 28 -
= 0.0493 0.035
La ecuación de la mejor línea se da mediante la siguiente ecuación:
A = 0.0493C + 0.035
Con el fin de trazar estos valores en un papel cuadriculado, se sustituye simplemente dos valores para C, se calculan los valores correspondientes a A, se ubican los dos puntos en un gráfico y luego se les une con una línea recta. Por ejemplo, se puede calcular C = O, A = 0.035; y C = 10.0, A = 0.528. Estos valores pueden ser marcados y puede trazarse una línea recta tal como lo indica la figura 2.
Ahora, la curva de calibración está lista para ser usada en el análisis de muestras. Recuerde que la curva fue elaborada sin hacer correcciones en el blanco. Se entiende que las muestras deben analizarse y los valores de la concentración deben calcularse, usando la curva sin hacer, correcciones por el blanco en forma directa. Por regla general, el blanco, los estándares y las muestras deben analizarse exactamente de la misma manera.
En algunos casos, se puede corregir el blanco; entonces, de cierto modo se cambia el procedimiento analítico. En lugar de usar la curva de calibración de la figura 2, que incluye un valor positivo de absorbancia a la concentración cero, sólo se usa el factor de calibración. En otras palabras, en un lote de análisis se procesan tanto las muestras como el blanco, luego se substrae el valor de absorbancia del blanco del mismo valor de la muestra. La concentración de la muestra se calcula por medio de la siguiente ecuación.
A e = factor de calibración
Si se usa el ejemplo anterior, el factor de calibración es 0.0493, por lo tanto, la concentración se puede calcular de la siguiente manera:
A C=---0.0493
Esta ecuación es equivalente y paralela a la curva de la figura 2, pero con la intersección en el origen (A=O, C=O).
- 29 -
0.9
o.a
0.7
0.6 <(
(.) z 0.5 <( m a: o 0.4 en m <(
0.3
0.2
0.1
o o 2 4 6 8 10 12 14 16
CONCENTRACION
Figura 2 CURVA DE CONCENTRACIÓN

- 30 -
Cuando se usa una curva de calibración "permanente" o factor se requiere verificar su exactitud con cada lote de análisis, mediante el análisis de un patrón o estándar de verificación1,
antes de analiz.ar las muestras . Asimismo, se recomienda analiz.ar un blanco antes de la muestra, no para verificar la curva de calibración, sino para observar la pureza y estabilidad de los reactivos. Estos valores del blanco también pueden ser trazados en una carta de control que tiene O como valor central y sirve para controlar los cambios en la pureza de los reactivos. En conclusión, una vez que se ha establecido la curva de calibración, un lote típico de análisis conlleva lo siguiente:
1) Se coloca la absorbancia en O (100% de transmisión) con agua destilada. 2) Se determina la absorbancia del blanco de reactivos. 3) Se analiza el estándar o patrón de verificación para validar la exactitud de la
curva de calibración. 4) Se analizan las muestras (hasta 10). 5) Se repiten los análisis del blanco o testigo de reactivos.
Si se van a analiz.ar más de 10 muestras en un lote, se repiten los pasos del 3 al 5 tantas veces como sea necesario, hasta que se completen los análisis.
En el paso 3 si se estima conveniente, puede analiz.arse más de un patrón o estándar de verificación. Tal como se señaló antes, también éstos pueden usarse en el control de su precisión.
Finalmente, por lote se analizan por los menos 2 blancos para poder calcular el límite de detección. Para mayores detalles, véase el anexo 4.
La concentración del patrón o estándar de verificación, por lo general, es de aproximadamente 0.9 Cm, donde el Cm representa el límite superior de concentración que puede ser medido con el mt!:todo analftico. Esta alta concentración es sensible para detectar los cambios que se producen en la curva de calibración. A menudo, tamb~ se analiza el patrón o estándar de verificación de una concentración menor (0.2 Cm) para controlar niveles mcnora. Ea:tos patrones o estándares de verificación pueden ser usados como muestras de control cuando se miden conjuntamente para verificación de la curva de calibración y para aplicar los datos de las cartas de control de la prcciai6n.

- 31 -
ANEXO 4
ESTIMACIÓN EXPERIMENTAL DE WS LÍMITES DE DETECCIÓN
Por lo general, en métodos que usan espectrofotometría UV-visible, se determina experimentalmente sus límites de deteccióh, por medio de la evaluación de la variabilidad de los análisis del blanco. La teoría del límite de detección adoptada está tratada con detalle en el informe técnico Nº 66 del Water Research Center, pp. 44-49. Como ejemplo se discute la aplicación de esta teoría.
La ecuación básica para calcular el límite de detección es la siguiente:
Límite de detección (LO) = 2.83 lo.1 S.m
en donde
to.1 = el punto del 10% del t de student para pruebas dobles cola y S.m = la desviación estándar del blanco dentro de un grupo.
Con el fin de calcular el valor de S.m, analizar los blancos por duplicado en una serie de lotes. Mientras mayor sea el número de determinaciones (por duplicado) medidas, mejor será la estimación del límite de detección. La ecuación usada para calcular la desviación estándar dentro de un grupo es la siguiente:
en donde
di - la diferencia entre las determinaciones del blanco, dentro del lote. m = el número de determinaciones del blanco por duplicado.
A continuación, se señala un ejemplo de la determinación del límite de detección de los análisis de mercurio con datos generados en el CEPIS/LAB.

- 32 -
Blanco 1 Blanco 2 diferencia di Lote No. (Jlg/l) (Jlg/l) di1
1 .521 .508 .013 1.69 X 10"4
2 .459 .445 .014 1.96 X 10"4
3 .608 .490 .118 139.24 X 10"4
4 .480 .386 .094 88.36 X 10"4
231,25 X 10"4
donde sustituyendo
LD = 2.83 ~. 1 SdB = 2.83 (2.13) (0.0537) • 0.324 µg/l
Nótese que el valor del t de student corresponde al valor que tiene cuatro grados de libertad en el punto del 10% del t de student para las pruebas doble cola. A continuación, se presentan los valores del t de student de uso común en el cálculo del límite de detección.
grados de libertad
1 2 3 4 5 6 7 8
6.31 2.92 2.35 2.13 2.02 1.94 1.89 1.86
grados de libertad
.2 10 12 .u 2Q
1.83 1.81 1.78 1.75 1.72
Para una buena estimación del límite de detección, se debe analizar blancos duplicados en un número mayor de lotes, y no sólo los cuatro utilizados en el ejemplo. Para el cálculo inicial, se sugiere un mínimo de diez lotes de datos, luego se vuelve a calcular, a medida en que se encuentren disponibles más mediciones y, por ende, más grados de libertad.

- 33 -
4.2 Calibración de autoanalludores
4.2.1 Introducción
El uso de autoanali7.adores para el análisis del agua es cada vez mayor; además, existen procedimientos disponibles para calibrar y obtener buenos resultados analíticos a partir de las lecturas instrumentales. Esta sección describe los procedimientos recomendados por el Water Research Centre (WRC) de .Inglaterra, mediante su Informe Técnico Nº 32.
Aquí sólo se incluyen aquellos proredimientos que se usan para la calibración y evaluación del sistema Autoanalizadot 11 (AAII). Para mayores detalles puede consultarse el mencionado informe.
El uso de métodos de calibración y evaluación apropiados depende de la exactitud requerida para los resultados analíticos de un programa en particular (algunos parámetros pueden requerir procedimientos diferentes), y del tipo de instrumento que se use (por ejemplo el AAI o el AAII). Por ello, no es posible usar un mismo procedimiento para todas las situaciones; en su lugar, se pone énfasis en principios generales y sugerencias sobre los procedimientos que pueden usarse en diversas aplicaciones.
4.2.2 Calibración
la respuesta de los sistemas AAI se obtiene en unidades % T (porcentaje de transmisión), mientras que la respuesta para los sistemas AAII se obtiene en absorbancia. Esta diferencia puede determinar el uso de diferentes métodos en algunos aspectos de su calibración. El sistema AAII es más simple y su uso se está difundiendo; éste se describe a continuación.
Sistema AAII
El procedimiento que se recomienda para cada lote de muestras en las que se va a medir un parámetro particular es el convencional, el que consiste en calibrar el equipo mediante el análisis de soluciones patrón, y luego el de muestras. El análisis de patrones se repite periódicamente. Generalmente se incluye uno de concentración cero y otro de concentración parecida a la de las muestras que se van a medir. Los distintos aspectos de este método se discuten en las secciones 2.1.1 a la 2.1.6, las cuales pueden estudiane conjuntamente con la figura 3, como una manera de verificar los pasos seguidos para lograr una calibración óptima.

- 34 -
4.2.2.1 Estabilización preliminar del sistema
Al inicio, debe medirse la solución de lavado en forma continua hasta que la respuesta del instrumento sea estable. La experiencia indicará el tiempo requerido; sin embargo, éste puede variar de parámetro a parámetro.
4.2.2.2 Selección de las soluciones patrón o estándares de calibración
En la calibración inicial, por lo general deben emplearse seis soluciones patrón o estándares de calibración (incluido el blanco*). Estos estándares deben tener una estrecha relación con las concentraciones de O.O Cm, 0.2 Cm, 0.4 Cm, 0.6 Cm, 0.8 Cm y 1.0 Cm, en donde Cm representa la mayor concentración que debe cubrir la curva de calibración.
En un sistema particular es posible reducir el número de soluciones patrón o estándar. Por ejemplo, si los análisis muestran una curva de calibración recta, los estándares correspondientes a O.O Cm, 0.5 Cm y 1.0 Cm son suficientes1
.
Las soluciones patrón o estándares tienen que ser preparadas siguiendo instrucciones claras, tantas veces como sea necesario, de acuerdo a la estabilidad de las soluciones.
4.2.2.3 Respuesta inicial para los patrones o estándares O.O Cm y 1.0 Cm
Si los cambios en las respuestas de los patrones o estándares O.O Cm y 1.0 Cm (durante el análisis del conjunto de muestras) no tienen importancia, se puede colocar el instrumento en cero en el caso del blanco y en un nivel de respuesta máxima para el caso del estándar 1.0 Cm. Luego, las concentraciones de las muestras se pueden leer directamente.
Si hay cambios apreciables en las respuestas de los estándares O.O Cm y 1.0 Cm, es mejor registrar la respuesta. En el caso del blanco, en una escala ligeramente superior a cero y, en el caso de la solución patrón o estándar 1.0 Cm, un valor ligeramente menor al máximo de la escala. Con estas mediciones y experiencia, se puede determinar los valores correctos para realizar los ajustes.
El término "blanco" se refiere a una solución de composición idéntica a las otras soluciones patrón o estándares, sin el analito. Se analiza exactamente como los otros patrones o estándares, es decir, se coloca en el recipiente para muestras (sobre la rejilla para las rnuestras).

- 35 -
4.2.2.4 Orden de los análisis de los patrones o estándares de calibración
El orden adecuado para el análisis de los patronr,s o estándares de calibración depende de b naturaleza y magnitud de la cor.laminación cru•.ada 1 entre una solución y otra. Es ideal que los sistemas se seleccionen y operen empleando una relación muestra/agua tal que la contaminación cruzada carez.ca de importancia. Sin embargo, es necesario conocer: (i) la relación muestra/agua de dilución, (ii) el número y estabilidad de las muestras a ser analizadas, y (iii) la exactitud requeridll (error aceptado). Con frecuencia aparece alguna de las siguientes contaminaciones cruzadas:
a) Contaminación cruzada de poca importancia
En este caso, por lo general, el orden de los análisis carece de importancia; sin embargo, si lo tomamos en cuenta el proceso se hace más simple. Para ello, podríamos seguir el siguiente orden: O.O Cm, 0.2 Cm, 0.4 Cm, 0.6 Cm, 0.8 Cm, 1.0 Cm y O.O Cm. Estos siete análisis deben llevarse a cabo después del control inicial del agua de dilución y de la solución patrón o estándar 1.0 Cm.
b) Contaminación cruzada de porcentaje constante
Cuando una solución se mezcla parcialmente con la solución que la precede y el grado de mezcla es constante, el orden del análisis (a) de la figura 2 producirá una curva de calibración con una pendiente menor que la real. Este efecto, a menudo, puede carecer de importancia. Sin embargo, se puede reducir mediante el siguiente orden en los análisis de los patrones o estándares O.O Cm, 0.4 Cm, 0.2 Cm, 0.8 Cm, 0.6 Cm, 1.0 Cm, O.O Cm. Este orden asegura la anulación de los errores positivos y negativos producidos por contaminación cruzada.
c) Contaminación cruzada debido a la absorción
En algunos autoanalizadores, los compuestos de color suelen ser absorbidos en la superficie del tubo, celda de flujo continuo, etc. Por ello, resulta común que al pasar de una solución de alta concentración a una de baja concentración, aumente la contaminación cruzada. Esto se puede apreciar observando los residuos que quedan en la parte superior. En estas condiciones, es mejor analizar los estándares en orden ascendente, es decir O.O Cm, O.O Cm, 0.2 Cm, 0.4 Cm, 0.6 Cm, 0.8 Cm, 1.0 Cm, O.O Cm y O.O Cm. Si la contaminación fuese de grandes proporciones, podría resultar necesario analizar tres blancos sucesivos cada vez que se
La mayor contaminación cruzada puede ocurrir cuando las muestras contienen componentes absorbidos en las superficies internas de los sistemas de los autoanalizadores. Por ello, debe prestarse especial atención a los posibles efectos debidos a la contaminación cruzada. Por ejemplo, cuando se analizan afluentes que contienen aceites y grasas, la magnitud de la contaminación cruzada puede evaluarse mediante el análisis de tres porciones de un estándar 0.9 Cm, tres porciones de un estándar O. 1 Cm y tres porciones adicionales del estándar de 0.9 Cm.

- 36 -
analice una solución de concentración alta; la respuesta del primer blanco se usa para evaluar los resultados del tercer blanco de cada grupo de tres.
d) Otros tipos de contaminación cruzada
Si la contaminación cruzada es de importancia, pero su naturaleza es diferente a la de (b) y (c), se requiere de otro orden de análisis, seleccionado de acuerdo con la experiencia local.
4.2.2.5 Controles regulares de calibración durante cada período
Lo ideal sería que la curva de calibración no sufra cambios de consideración durante un período completo de trabajo, por ello, vale la pena tratar de mejorar el rendimiento analítico para lograr estabilidad2 en las mediciones. Sin embargo, esto no siempre puede lograrse, aún así, la estabilidad siempre debe confirmarse en forma experimental mediante el control periódico de la calibración.
a) Contaminación cruzada de poca importancia
En este caso, por lo menos se necesitan, dos soluciones patrón o estándar para el control de la calibración; un blanco para verificar el cero y una solución patrón o estándar para verificar la pendiente de la curva de calibración. En forma regular y periódica se debe analizar un patrón o estándar de 1.0 Cm y otro de O.O Cm en este orden3
• El número de muestras que se debe analizar entre cada control4 se trata en la sección 3.
b) Contaminación cruzada actual
Se debe tratar en lo posible que las muestras que se analizan en cada lote tengan una concentración bastante similar; aunque, obviamente, dicha concentración puede variar de un lote a otro. Cuando se adopta este método, por lo general resulta conveniente que se realicen algunos cambios en los procedimientos de (a).
i. La concentración del patrón o estándar de control debe ser similar a la de las muestras de su lote. Por ello, puede necesitarse soluciones patrón de control con diferente concentración, de acuerdo al lote de muestras a analizar; o sea, las concentraciones de las soluciones patrón de control pueden variar de un grupo a otro.
3
•
Los fabricantes de Autoanalizadores Technicon exigen que la desviación de lo1 1i1temu AAll no sea mayor al 2~ por hora; en caso de que la desviación fuera mayor, la compañía Technicon debe recomendar medidas al respecto.
Por lo general, se espera que el estándar de 1 .O Cm, que es el de mayor sensibilidad, permita detectar los cambios en la pendiente de la curva de calibración .
De aquí en adelante, usaremos el término grupo o lote para referirnos a los análisis que se realicen entre los controles de calibración sucesivos.

- 37 -
1. ESTA8ILiliCI<li PRELIMINAR
(5occ1dn 2.1.1)
An41 lsls d9 la aolucldn da lavado
axitlr1.m huta ...- •t• 111: rmd==nta
atlble.
2. AJUSTlt INICIAL DE LAS RESIUSTAS PARA LDS ESTAhOAOES
t.IC..yl.IC.
(5ec:cl6n 2.1.3)
"°'' uyor ccnc91"1tracldn dal r-v:> • ser cal lbrado
1 SI la re~ta a •t.ble a travH dial .-11sls de 1• .,..tras
., ....... 1"9Spuata _,.....,. --- ---1.1 c. con> ••• Co
... Co escala ... Co total
3. ANALISIS DE LDS ESTAllJARES DE
CALillRACION
(S.Ccl&a 2.1.4)
FIGURA 2
1
r.-punta
ligera > e""'
~· do la ucala
PROCEDIMIENTOS PARA LA CALIBRACION DE LOS SISTEMAS AA11

(a)
- 38 -
'
C.OOt• lnac ldn (b) 1 r.onstante (c) Contamlnacl6n Cd) otros tipos de ln8dvertlbl• ccnt•inecidn cruzada por cont•lnacl6n
cruz- .tsorcl6n cruz-
(1) ••• c. (i) ••• c. Ci) 1.1 C• SI hay c:ontMlna-¡m 0.2 c. Oll e.e c. (TI~ 1.11 C. cl(n cruz.ta dlfe-
( li) t.4 Ca Clil} 9.4 C• 011 e.2 c. ,..,t. de <•>1 (b) e lv> 1.6 c. ( lv~ 0,2 C. Ctv~ e.4 c. o Ce) =r (V) 9.& ÚI (y 0.8 c. Cv 1.6 C• otras de
CvT> 1.9Ca Cvl l e.e c. Cvl> G,8 C. an6ll1l1 da acuerdo (vil) 9.1 C. {vil) 1.8 C. (vlil) e.• c. a I• experi.-.cia
(vi i i) 1.11 C• Cix> e.e C. local. Clx) 1.8 C.
1 1
•• CtEQ..W REGULAR DE LA CALIBRACION
1 Si hay cont•lnKidn cruzada inadvertible
~ (f} 1.8 ÚI Cii) '·"Ca
DURANTE CADA CDliIDA
CS.ix:ión 2.1.5)
1
Ccm:> ~ra general, I• pn.Jllb•
de cal ibr-.cidn dupuá dti lotas
de 15 .WI Isla Caiestru y pru.r
ba de control de sal ida) se
cW:ien ru I 1 zar •
Cl.llndo • poslbl•, con cada lot•
las -..estrn deben ••r de c:on
centrac i 6n sl•i1ar y dos porcio
nes de la prlHra -...tra deben
ser .anal Izadas en al lote,
(1) 1.1 Cb• (ii) ••• c.
(111) •.•c.
-Cb: ooncentraclón de las -.
tras .,,, el lote,
5. CtEWED DE LA CALIBRACICll FINAL
CS.c:cl6n 2.1.6)
Anal izar nuw...,,t• •I Jusgo completo de estlndares de callbr..:ldn cc.:i ....

- 39 -
ii. Se debe analizar sucesivamente dos porciones del blanco en forma conjunta con el patrón de verificación, ya que la primera puede proporcionar una lectura más elevada de lo normal.
iii. Así mismo, es necesario analizar dos porciones sucesivas de la primera muestra del lote, ya que la primera puede dar resultados erróneos.
En este método debe tomarse en cuenta más factores que permitan observar e indicar que la contaminación cruz.ada no tiene mayor importancia.
4.2.2.6 Control final de la calibración
Si no existe la seguridad de que la curva de calibración siga siendo lineal durante todo el período, entonces debe volverse a analizar todo el conjunto de soluciones estándares iniciales de calibración. Sin embargo, siempre que la curva de calibración se prepare al inicio y se verifique durante un período, será suficiente realizar el análisis de las dos soluciones patrón que se especifican en la sección 2.1.5, para conCluir este período.
4.2.3 Análisis de las muestras
4.2.3. l Número de análisis que deben realizarse entre cada control de calibración
El número de análisis que se realice entre cada control de calibración depende tanto de la estabilidad de la calibración, como de la precisión que se requiera. En general, es conveniente realizar 15 análisis entre cada control de calibración, número que puede ser variado de acuerdo a la experiencia. Dentro de estos 15 análisis se anali7.arán conjuntamente muestras y soluciones patrones o estándares de control (véase la sección 5).
4.2.3.2 Orden del análisis de las muestras
Cuando la contaminación cruz.ada en muestras sucesivas carece de importancia, el orden en que se las analiza tampoco tendrá importancia. Por lo tanto, el orden lo determinará la estabilidad de las muestras.
Si la contaminación cruzada es importante, los errores que surjan de esta fuente deben controlarse mediante el análisis de muestras, de acuerdo a un orden predeterminado que permita minimizar los errores de concentración de las muestras sucesivas y los patrones o estándares de control. (Véase también la sección 2.1.5 (b)).

- 40 -
4.2.4 Evaluación
Sistemas AAII
4.2.4.1 Preparación de la curva de calibración
Si la calibración es estable durante una corrida, los valores de la concentración podrán leerse directamente de los registros o de la impresora. En este caso no se requerirá de una curva de calibración. Algunas veces es conveniente usar este método; sin embargo, se prefiere el que se describe en el siguiente párrafo.
Si la curva de calibración vaáa durante un período, se sugiere emplear el siguiente procedimiento. Se trax.a una línea para unir las respuestas de:
(i) el primer blanco, exactamente antes del primer patrón de calibración y (ii) el último blanco de la serie inicial de patrones de calibración (sección 2.1.4).
Las alturas máximas de todos esos patrones o estándares, se miden a partir de la línea base y se grafican contra las concentraciones conocidas. La mejor línea recta que se obtenga de la unión de los puntos, constituye la curva de calibración. Si se sigue el procedimiento que se delineó en la sección 2.1.4 (b), el penúltimo blanco debe registrarse como un punto y la curva de calibración se trax.a a partir del origen.
Cuando los resultados se obtienen con una impresora, las lecturas de los dos blancos, (i) e (ii), de la serie inicial de calibración, deben examinarse con el fin de determinar si es necesario hacer alguna corrección a la desviación de la línea base. Si fuera necesario, las lecturas de la impresora o del cuadro de registros se usan como respuestas analíticas y se c0rrigen sobre la base que se estableció en el párrafo anterior.
4.2.4.2 Verificación de la curva de calibración
Con el fin de definir las respuestas de las determinaciones sucesivas a partir del blanco5
a través de cada corrida, se trax.a una línea apropiada en el registro y a partir de ésta se miden las alturas máximas de las muestras y patrones o estándares de verificación.
Las concentraciones que corresponden a las alturas máximas de los patrones o estándares de verificación deben obtenerse a partir de la curva de calibración. En caso de que las concentraciones se encuentren cerca del valor real, podrá emplearse la calibración inicial para todas las muestras durante la corrida. Si las concentraciones que se obtienen para los patrones o estándares de concentración difieren sistemáticamente del valor real por un gran margen, las
Si se usa el procedimiento delineado en la sección 2.1.5 (b), esta lfnea se debe trazar a partir de las respuestas del segundo blanco de cada par.

- 41 -
respuestas que se obtuvieron para los patrones de verificación deben emplearse en la elaboración de una serie de curvas de calibración.
Cuando los resultados de cada lote de muestras controladas con un patrón de verificación no varían en forma apreciable, se puede usar otra curva de calibración. Cada una de estas curvas de calibración, se obtiene por medio de la altura máxima del patrón de verificación corregido por el blanco graficado contra la concentración de los patrones y dibujando una línea entre el punto del patrón y el origen. Otra vez si se usa impresora y se ve la necesidad de crear una serie de curvas de calibración, éstas deben efectuarse, teniendo en cuenta el proceso anterior.
4.2.4.3 Obtención de resultados analíticos de las muestras
Cada medida de la muestra, corregida por la altura máxima del blanco, se convierte en concentración al usar la curva de calibración apropiada (véase la sección 4.2.2.5 (b), punto (iii)).
4.2.5 Control de calidad analítica
Para estimar la precisión de los resultados analíticos se realizan pruebas experimentales. Estas se discuten en detalle en (1) y (2). Las pruebas requeridas se eligen teniendo en cuenta su aplicación particular; sin embargo, la siguiente información puede resultar de utilidad.
Las soluciones patrón o estándar pueden emplearse para verificar la precisión y algunas fuentes de errores sistemáticos. Si la calibración inicial para analizar muestras se usa durante una corrida, la solución patrón o estándar de verificación (sección 2.1.5) actúa como un estándar de control1
• Por el contrario, si ese patrón o estándar de verificación se usa para definir curvas de calibración (sección 4.1.2) es necesario emplear otro patrón o estándar para el control de calidad. La concentración que constituye el estándar de control, puede ser igual o diferente del estándar de verificación, según se requiera. En documentos como el Technical Memorándum TM 56 (2), se discute sobre la selección apropiada de la concentración del patrón. Si además del patrón o estándar de verificación, se analiza un patrón o estándar de control, éste debe ser el último que se realice antes que se midan los estándares de verificación con cada lote de análisis (sección 2.1.5). Los resultados requeridos para el control estándar, deben evaluarse a partir de la curva de calibración que se emplea para las muestras del mismo lote.
Asimismo, una muestra por lote se analiza por duplicado. Un esquema simple puede ser el analizar una segunda porción de la primera muestra de cada lote. Este análisis se realiza antes del de los patrones o estándares de control y si éstas no se analizan, se efectúa antes de los patrones o estándares de verificación.
El término •estandar de control" se refiere a una solución estandar que se analiza y evalúa como muestra. El resultado de este an.illisis, se emplea en el control de calidad analltica.

- 42 -
Mientras mayor sea la experiencia con respecto a la precisión de los resultados, la confiabilidad de los sistemas analíticos aumenta, pudiéndose reducir pasos dentro del control de calidad analítica. Por ejemplo, tanto las mediciones del estándar de control, como el de la muestra duplicada, pueden aplicarse en lotes alternos y no en cada lote.
La calibración del autoanalizador se efectúa con cada grupo de análisis. Al prepararse (como usualmente se hace) patrones o estándares de control a partir de la misma solución estándar que se usa para preparar los patrones o estándares de calibración, los resultados de los patrones de control no revelarán errores sistemáticos causados, por ejemplo, por el empleo de soluciones patrones o estándar incorrectas o soluciones (reactivos) deterioradas. Sin embargo, se recomienda mantener el control de la calibración y operación de los instrumentos más sensibles. Cambios repentinos o pequeñas desviaciones ameritan una revisión de los patrones o estándares y de los reactivos.

- 43 -
F.JEMPLOS DE AUTOANALIZAOOR CON UN SOPORTE DE MUESTRAS
1. Sistemas AAII
Con el fin de ilustrar los diferentes aspectos vertidos en las secciones 2, 3 y 5, se muestra algunos ejemplos sobre el orden en que deben analizarse las soluciones
2. Contaminación cruz.ada de poca importancia
CALIBRACIÓN ESTABLE CALIBRACIÓN INESTABLE
Prueba No. Solución Prueba No. Solución
l Blanco 1 Blanco 2-6 Patrones de calibración 2-6 Patrones de calibración 7 Blanco 7 Blanco 8-20 Muestras 1-13 8-20 Muestras 1-13 21 Muestra 1 21 Muestra 1 22 Patrones de control 22 Patrones de control 23 Blanco 23 Patrones de calibración 24-37 Muestras 14-26 24 Blanco 38 Muestra 14 25-37 Muestras 14-26 39 Patrones de control 38 Muestra 14 40 Blanco 39 Patrones de control 41 Ciclos repetidos 8-23 40 Patrones de calibración
41 Ciclos repetidos 7-23

- 44 -
3. Contaminación cruzada presente
CALIBRACIÓN ESTABLE CALIBRACIÓN INESTABLE
Prueba No. Solución Prueba No. Solución
1 2 3-8 9 10 11-23 24 25 26 27 28-40 41 42 43
*
4.3
Blanco 1 Blanco Blanco 2 Blanco Patrones de calibración 3-8 Patrones de calibración Blanco 9 Blanco Blanco 10 Blanco Muestras 1-13 11-23 Muestras 1-13 Muestra 1 24 Muestra 1 Patrones de control 25 Patrones de control Blanco 26 Patrones de calibración Blanco 27 Blanco Muestras 14-26 28 Blanco Muestra 14 29-41 Muestras 14-26 Patrones de control 42 Muestra 14 Ciclos repetidos 9-25 43 Patrones de control
44 Patrones de calibración 45 Ciclos repetidos 9-26
Véase la sección 4.2.2.4 donde se establece el orden de los análisis de los patrones o estándares de calibración.
Procedimientos de calibración y de control de calidad mediante la espectrofotometría de absorción atómica
En general, la espectrofotometría de absorción atómica se encuentra libre de muchos de los errores comunes a otros análisis químicos. En lo que respecta al análisis de metales, la técnica es bastante específica y sus mediciones pueden ser muy precisas y estar relativamente libre de errores sistemáticos. Sin embargo, existen ciertas fuentes de error que si se presta atención al proceso de calibración y los procedimientos de control de calidad pueden ser identificados, corregidos y controlados.
Antes de establecer los procedimientos de calibración y de control de calidad, es necesario discutir brevemente sobre algunas definiciones y principios básicos.
En primer lugar, en este documento se define al método analítico como "El grupo de instrucciones escritas que definen completamente al procedimiento que debe adoptar el analista, con el fin de obtener el resultado analítico que requiere". Bajo esta definición, el método

- 45 -
analítico debe ser específico y, si se quiere obtener buenos resultados, el analista debe ceñirse a las instrucciones para desarrollar la medición.
Dado que el método está formado por un "grupo de instrucciones escritas•, no es posible decir, que •el método es preciso•. Mas bien debe decirse que los resultados analíticos tuvieron una precisión de". Diversos laboratorios usan el mismo método, sin embargo, el que obtengan los mismos resultados, tanto para la precisión como para el error sistemático depende de cómo se hayan realizado los análisis. Esta afirmación no excluye que el método pueda tener tendencia inherente a obtener resultados imprecisos o errores sistemáticos. Sin embargo, el grado de error sistemático, así como los valores específicos depende del desempeño de cada laboratorio.
El requerimiento de que todos los blancos, patrones o estándares y muestras se analicen exactamente por el mismo procedimiento, tiene estrecha relación con la definición antes mencionada, puesto que permite reducir el error sistemático asociado a la calibración o blanco. Si los procedimientos que se usan para analizar los blancos, patrones o estándares y muestras difieren en algún punto, el analista deberá determinar experimentalmente si esta diferencia origina errores sistemáticos aceptables.
En el anexo se trata el tema de estimación del "límite de detección• de forma independiente. En los análisis de trazas, generalmente se prefiere "corregir el blanco" de los resultados analíticos de las muestras (p.e. resultado = resultado de la muestra menos el resultado del blanco). En el procedimiento de calibración que se sigue en el CEPIS/LAB primero se coloca la aguja del instrumento en cero con el blanco (p.e. blanco del reactivo), de esta manera, la corrección por el blanco se convierte en una parte inherente del método analítico. Por otro lado, para que el procedimiento de corrección del blanco sea adecuado, es necesario que se corrija el blanco de cada una de las muestras. Si se usa el procedimiento de calibración que se propone, el instrumento debe colocarse en cero con el blanco cada vez que se requiera medir niveles bajos de trazas de metales. Si el resultado de la muestra es mayor que el blanco, no se necesitará realizar ninguna corrección ni repetir el análisis.
En las secciones siguientes sólo se discutirá los procedimientos de calibración que deben seguirse al analizar un grupo de muestras (4 ó más). Cuando se analicen pocas muestras, podrá usarse cualquiera de los métodos que se describen en el capítulo sobre "Recolección y Manejo de Datos".
Una vez que los principios han sido mencionados y comprendidos se procederá a discutir los procedimientos de calibración y de control de calidad específicos.

- 46 -
En un laboratorio se utilizan diversos métodos de análisis, sin embargo, para los propósitos de este estudio, los métodos pueden clasificarse en tres grupos:
(1)
(2)
(3)
Metales disueltos en agua
Cantidad total de metales en el agua
Cantidad total de metales en suelos, sedimentos o lodos
- Filtración a través de un filtro membrana de 0.45 u. e inyección acuosa directa.
- Digestión, seguida de inyección acuosa.
- Digestión vigorosa, seguida de inyección acuosa.
F.n ciertos casos se usa otros métodos de análisis (p.e. extracción por solvente}; al respecto, debe consultarse al Coordinador de Garantía de Calidad en qué condiciones es útil aplicar estos procedimientos, así como todo lo relacionado con los procedimientos de calibración y control de calidad.
F.n el primer caso, se suele preservar las muestras de agua con HNO, (3 mi por litro) 1:1 redestilado o de calidad ultrapuro para análisis de trazas. En consecuencia, todos los blancos y los patrones o estándares deben prepararse con una cantidad similar de HN03; y seguir las instrucciones para la calibración. Esto se hace con el fin de no confundir conceptos sobre la calibración y el control de calidad que se está tratando. Esta elección es arbitraria puesto que, si se desea, los datos de la determinación de los límites de detección en diversos grupos pueden acumularse, conjuntamente con los de la calibración.
El procedimiento de calibración es como sigue:
(1) Se coloca la aguja del instrumento en "cero" con el blanco adecuado al análisis (p.e. blanco con HN03 agua bidestilada o solvente).
(2) Se calibra con un mínimo de cuatro patrones o estándares. (3) Se analiu muestras (hasta nueve). ( 4) Se analiu el patrón o estándar de verificación de concentración equivalente a O. 9
Cm, donde Cm es el límite superior de la concentración que mide el método analítico.
(S} Si es posible, se analiu un segundo estándar de verificación que equivale a 0.2 Cm.
(6) Se repite los pasos (3 al S} hasta terminar de analiur las muestras.
F.n este primer caso, que podría usarse para el análisis de aguas de consumo, u otro tipo de aguas limpias, no habría diferencia entre el análisis de patrones o estándares de calibración y los de verificación.

- 47 -
Los resultados de los análisis de patrones o estándares de verificación se registran en los formularios que tiene el laboratorio para tal fin. Una vez anali:nido un buen número de patrones o estándares de verificación (p.e. 20 o más), se puede preparar las cartas de control, y los subsiguientes patrones o estándares de verificación pueden usarse como estándares de control.
Para el segundo tipo de análisis (p.e. presencia total de metales en el agua) se requiere de digestión. Si, tanto los blancos como los patrones o estándares y las muestras se analizan siguiendo exactamente el mismo procedimiento, la calibración será idéntica a la que se empleó en el caso l. Sin embargo, con frecuencia, se preparan patrones o estándares de calibración directamente con HN03 diluido, mientras que las muestras se digieren antes del análisis. Al seguir este procedimiento, también se debe analiz.ar un patrón o estándar de verificación que haya pasado por el proceso de digestión (p.e. analiz.ar un patrón o estándar de verificación por medio del mismo procedimiento que se usó para la muestra). El patrón o estándar de verificación provee de información sobre la precisión, pero, además, indica si en la calibración hay algún error sistemático. Del mismo modo que el patrón o estándar de verificación, el blanco debe pasar por todo el procedimiento, incluyendo la digestión, con el fin de proveer una base para la corrección del blanco. Se debe enfati7.111' que esto sólo es necesario cuando la calibración de patrones o estándares se realiz:a de manera distinta a las muestras. En suma, se tiene dos posibles procedimientos de calibración para el segundo tipo de análisis:
(a) Cuando los blancos, estándares y muestras se procesan en forma idéntica, tanto la calibración como el control de calidad se efectúan de la misma forma que en el caso l.
(b) Cuando los estándares de calibración se analiz:an en forma diferente que las muestras:
(1) Se coloca la aguja del instrumento en "cero" con el blanco con HN03•
(2) Se calibra directamente con los estándares preparados con HN03•
(3) Se analiz:a las muestras (hasta 9).
( 4) Se analiz:a el patrón o estándar de calibración de la concentración que equivale a 0.9 Cm, utilizando un patrón o estándar de verificación durante todo el procedimiento (p.e. incluye la digestión).
(5) Si es posible se analiz:a un segundo patrón o estándar de verificación de una concentración que equivale a 0.2 Cm, usando el procedimiento descrito en (4).
(6) Se analiz:a el blanco (p.e. usar el blanco durante todo el procedimiento), requerido para hacer la "corrección por el blanco" de los resultados de las muestras. Asimismo, se acostumbra determinar el límite de detección a partir de la variabilidad del blanco; por ello en cada grupo se analiz:a un segundo blanco.

- 48 -
Finalmente, el principio básico que consiste en anali7.ar de la misma forma blancos, patrones o estándares y muestras no es posible en el análisis de suelos, sedimentos y lodos. No hay forma de preparar un blanco o un patrón o estándar de suelo, ya que es imposible tener la seguridad de que el metal de interés esté ausente de la muestra del suelo que se usará para preparar el blanco y el patrón o estándar. Incluso, la mayoría de los suelos, contienen trazas de casi todos los metales de interés.
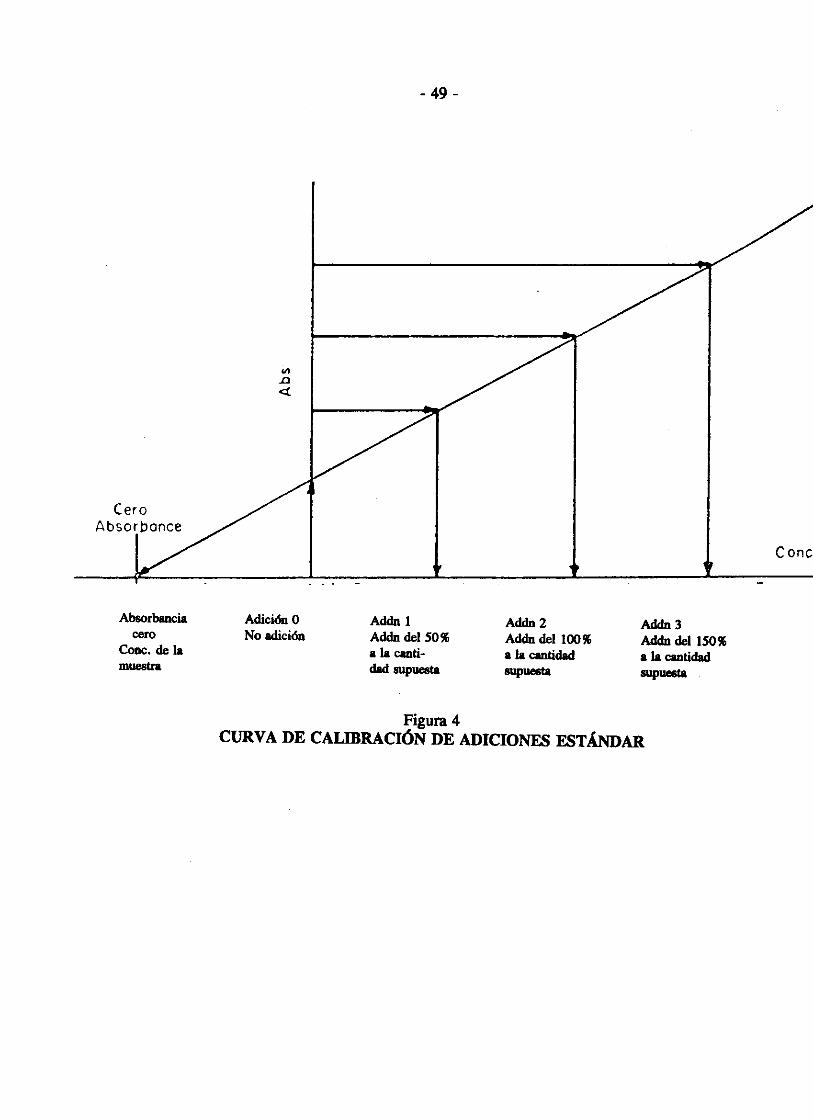
Debido a la complejidad de las matrices de los suelos, es muy frecuente encontrar interferencias químicas, además de la presencia de metales. Por ello, usamos el método de calibración de adición de un patrón o estándar, con el fin de reducir el error por las interferencias. Fn este método se realiza una calibración preliminar con patrones o estándares diluidos con HN03• Luego, se anali7.a la muestra y se registra la absorbancia resultante. Se separa alícuotas de la muestra y se efecttlan adiciones del analito de interés. Estas adiciones corresponden al 50, 100 o 150% de la cantidad de analito que se supone contiene la muestra, las soluciones se anali7.an y se registran las correspondientes absorbancias. Posteriormente, se traza una línea entre las absorbancias y las concentraciones, como se indica en la Figura 1, en la cual puede leerse la concentración de la muestra.
Este procedimiento puede corregir algunos errores sistemáticos causados por las interferencias; sin embargo, no puede corregir todas las fuentes de errores sistemáticos. Para corregir el error por el blanco, se hace por medio del análisis de blancos, de acuerdo al método establecido (p.e. uno debe pasar por el procedimiento de digestión). Fn conclusión, se usa el siguiente procedimiento para el análisis de suelos, sedimentos y lodos.
Cero Absorbonce
Absorbancia cero
Conc. de la muestra
Adición O No adición
- 49 -
Addn 1 Addn del SO 9lí a la canti-dad supuesta
Addn2 Addn del 100 91í a la cantidad supuesta
Addn3 Addn del 1S091í a la cantidad supuesta
Figura 4 CURVA DE CALIBRACIÓN DE ADICIONES ESTÁNDAR
Conc

- 50 -
(1) Se coloca "cero" con el blanco de HN03 diluido. (2) Se calibra directamente con los estándares preparados con HNO, (es suficiente
con una calibración aproximada que usa un patrón o estándar simple). (3) Se anali:z.a la muestra (determinar la absorbancia de la muestra). (4) Se hace tres adiciones estándar sobre la base de la estimación preliminar de la
curva de calibración y se anali:z.an como sigue: (a) adición del 50% a la cantidad supuesta. (b) adición del 100 % a la cantidad supuesta. (c) adición del 150% a la cantidad supuesta.
(5) Se elabora un diagrama que contiene cuatro valores de absorbancia obtenidos experimentalmente, como se indica en la f"igura 3 y se hace una inferencia con el fin de determinar la concentración de la muestra 1.
(6) Se repite los pasos del (3 al 5) con todas las muestras que se van a anali:z.ar.
A pesar de que el procedimiento mencionado se usa para anali:z.ar muestras del suelo1,
sedimento o lodo, también se usa en el análisis de muestras acuosas con matrices complejas. Para determinar la recuperación se usa un estándar de adición simple. Tomando como base esta recuperación se decide si es necesario reali:z.ar el procedimiento completo de adición estándar. Para mayores detalles, vea la sección correspondiente al tema en el Manual de Métodos de la EPA.
Cuando se sigue el procedimiento de adiciones patrón o estándar, no siempre se necesita llevar a cabo el análisis de los patrones o estándares de verificación, puesto que los estándares están incorporados dentro del procedimiento mismo. Sin embargo, es esencial que la preparación de las soluciones patrón o estándar sea exacta, con el fin de evitar errores sistemáticos dentro del procedimiento analítico. Por ello, se pone extremo cuidado en la preparación de las soluciones patrones o estándares y cuando se hacen las adiciones (exactas) a las muestras.
En discusiones precedentes, se ha puesto énfasis en el uso de soluciones patrones o estándares para verificación y control de la precisión de los análisis. El uso de soluciones estándar también puede indicar la presencia de errores sistemáticos en el procedimiento de calibración. Asimismo, los análisis de los blancos constituyen un medio para evaluar, detectar y corregir una posible contaminación de los reactivos, y para estimar el límite de detección.
De otro lado, se puede usar otros procedimientos de control de calidad para ciertos tipos de muestras, como las pruebas de control de precisión y de recuperación, de acuerdo a los objetivos del programa de medición.
La precisión de los análisis de las muestras se evalúan mediante el análisis regular de dos porciones de una muestra real y la gráfica de la diferencia de resultados: de la primera y de la
1 La palabra "suelo" se emplea aqul y en el resto de la discusión para denominar al sedimento, lodo y suelo.

- 51 -
segunda porción, tornando como valor medio cero. El desvío patrón que se usa para fijar, tanto los límites de advertencia como los de rechaw en la carta de control es dos veces mayor a la desviación estándar dentro de cada lote de resultados individuales sujetos a la misma desviación patrón que está siendo graficada. Este tipo de control está limitado a muestras cuyas concentraciones tienen desviaciones patrón similares. Si se dan muchos rangos de concentración o rangos de desviación estándar, se elaboran cartas de control en cada caso.
Las pruebas de control de recuperación de las muestras se realizan con el fin de evaluar errores sistemáticos que tienen su origen en las impurez.as de las muestras. Las recuperaciones que se observan están graficadas en una carta de control, que tiene como valor central la recuperación teórica. Si los resultados de la muestra y la muestra enriquecida constituyen respectivamente R1 y R2 el resultado de la recuperación A es:
SA = sRJ.2 + sRi2
Si Sa depende de la concentración del analito y las concentraciones de las muestras varían apreciablemente, ningún valor de SA se aplicará a los resultados de las pruebas de recuperación. Por ello, este tipo de control se aplica más a muestras cuyas concentraciones no tienen mucha variación. La cantidad del analito que se agrega a la porción "enriquecida" de la muestra es la misma.
:En el capítulo sobre errores analíticos del Manual de Análisis de la Contaminación del agua se presenta una discusión detallada sobre las cartas de control con ejemplos experimentales. Si se requiere de ayuda adicional en la preparación de las cartas de control, se debe consultar al Coordinador de Garantía de Calidad . •
• En vez de preparar un diagrama de los datos como se indica en la figura 3, se puede elaborar una inferencia matemática. Para ello se usa una calculadora manual con capacidad de determinar una ecuación de regresión lineal. En este caso, se ingresa los datos junto con la concentración en el eje de las •y• y la absorbancia en el eje de las •x•. Posteriormente, se calcula la intersección de la •y• de la ecuación de regresión lineal y su valor absoluto se toma como co"espondiente a la concentración de la muestra.

- 52 -
ANEXOS
ÚMITE DE DETECCIÓN
La teoría sobre los límites de detección detalla, en el Informe Técnio Nº 66 del Water Research Centre, pp. 44-49. Aquí sólo se discute la aplicación de esta teoría en la espectrofotometría de absorción atómica y se pone énfasis en las mediciones y en el método de cálculo.
El fundamento para establecer el límite de detección es la variabilidad de la medición del blanco. Con el fin de realizar este cálculo, la variabilidad se mide como la desviación estándar dentro de un lote. Generalmente, una mejor estimación se obtiene por medio de la combinación de las desviaciones estándar que se obtuvieron a partir de las mediciones del blanco por duplicado de varios lotes. Para obtener una estimación preliminar del límite de detección, se mide la variabilidad del blanco dentro de un mismo lote. Ambos procedimientos se discuten en este documento. El analista debe elegir cuál es el procedimiento más apropiado y conveniente para el laboratorio.
Antes de proceder a medir el blanco, el analista debe calibrar el instrumento. Esto es necesario si se necesita convertir los valores de respuesta del blanco a unidades de concentración (claro está que para el registro directo de la concentración, la respuesta luego de la calibración se da en unidades de concentración).
El analista decide el procedimiento de calibración que usará. El criterio primario que debe considerar es la capacidad para hacer el análisis de suficientes soluciones patrones o estándares, de tal forma que la variabilidad de la respuesta de los patrones o estándares no afecte la exactitud del cálculo del límite de detección.
Si el límite de detección se estima a partir de mediciones del blanco medidos dentro de un mismo lote, se prefiere calibrar por medio de mediciones repetitivas (cuatro o más) de una misma solución patrón o estándar, cuya concentración es aproximadamente 20 veces el valor del límite de detección probable. Luego, el resultado promedio de estas mediciones repetitivas se emplea para calibrar la respuesta del blanco. El que se utilice para calibrar un estándar de una concentración 20 veces el valor del límite de detección probable, se debe a que a menores concentraciones el error aleatorio de las mediciones podáa generar errores inaceptables en la calibración.
Si el límite de detección se estima a partir de determinaciones del blanco (tomados por duplicados) en varios lotes de análisis, la calibración se debe realii:ar de manera normal (p.e. medición de por lo menos cuatro soluciones patrones o estándares, con el fin de determinar el factor de calibración).
Para calibrar, se coloca el instrumento en cero, de acuerdo con el procedimiento descrito en la parte principal de este capítulo. Para ello se emplea un blanco (de reactivos). En algunos

- 53 -
casos es más conveniente usar agua destilada en vez del blanco. Esta elección no debe afectar el resultado final, porque lo que interesa es la variabilidad del blanco y no los valores absolutos.
La estimación del límite de detección, a partir de la variabilidad del blanco dentro de un mismo lote, se debe realizar con once mediciones por lo menos (diez grados de libertad). El cálculo de la desviación estándar dentro del lote es como sigue:
donde:
L,x~ - <L,xifn s da = ='----==-..;.._n-1
X; = valores para las respuestas del blanco individuales n = número de los resultados
Para estimar el límite de detección de varios lotes a partir de determinaciones del blanco por duplicado, se calcula la desviación estándar dentro del lote de la siguiente manera:
donde:
-~di2 s,. -2m
di - diferencia entre las determinaciones del blanco dentro de un mismo lote m - número de determinaciones del blanco medido por duplicado
Para mayores detalles sobre el uso de esta fórmula para el cálculo de desviación estándar, véase el anexo 4 del capítulo "Procedimientos de Calibración•. -Visible".
Finalmente, el límite de detección también puede calcularse por medio de la combinación de estimaciones de la desviación estándar procedentes de lotes que contienen distinto número de determinaciones del blanco.
En este caso, las desviaciones estándar se calculan para los lotes individuales y se aplica la ecuación (1) ya mostrada. Luego, estos valores individuales se combinan por medio de la siguiente ecuación:
(3)

- 54 -
donde:
v; = Número de los grados de libertad de las soluciones individuales usadas para estimar S;. La estimación de S, tiene V; grados de libertad. Por ejemplo, si se dan las siguientes mediciones del blanco en dos lotes:
Lote 1: 0.29, 0.04, 0.13, 0.22, µgil Lote 2: 0.35, 0.14, 0.03, 0.26, 0.08, 0.02, 0.17, 0.31, 0.07 µgil
La estimación de las desviaciones estándar para cada grupo es como sigue:
S1 = 0.1086 y S2 = 0.1225
Las desviaciones del estándar del blanco combinadas dentro del mismo lote, se calculan de la siguiente forma:
3.(0.1086)2 + 8(0.1225)2 = 0.1189
11
Después de calcular la desviación estándar del blanco, el límite de detección se calcula por medio de la siguiente fórmula:
donde:
LD to.1 sdB
= = =
L = 2.83 t0.1 S46
límite de detección coeficiente de la "t" de student al 10% para pruebas bilaterales con 11 g.l. desviación estándar del blanco dentro de un lote.
De los datos del ejemplo, se puede calcular el límite de detección:
LD = 2.83 (1.795) (0.1189) 6 LD • 0.6 µg/l
Nótese que los grados de libertad, en todos los casos, son iguales a la suma de los grados de libertad de cada lote individual y que los grados de libertad para cada lote es un grado menor que el número de mediciones en el lote.

- 55 -
ANÁLISIS DE MERCURIO POR ABSORCIÓN ATÓMICA
Proce<limiento cuando no se reali?.a corrección del blanco
1) Se corre los patrones o estándares (el blanco y cuatro patrones o estándares en el rango analítico). Se registra los valores de porcentaje de absorción.
2) Se corre las muestras desconocidas. Se registra los valores de porcentaje de absorción. 3) Se convierte los valores de porcentaje de absorción en absorbancia (A) usando la tabla. 4) Se tra7.a la línea de A con relación a la de concentración (C) de los estándares. (Se
aplica la regresión lineal para establecer la línea de mejor ajuste. Se puede reali7.ar esta operación con una minicomputadora y así se obtiene una copia del gráfico).
5) Se lee los valores de la concentración de las muestras desconocidas, usando los valores de absorbancia (A).
Procedimiento cuando se realiza corrección del blanco
(Para concentraciones bajas, es necesario reali?.ar correcciones del blanco, con el fin de mejorar la exactitud de la medida).
1) Se corre los patrones o estándares (el blanco reactivo y cuatro patrones o estándares en el rango analítico). Se registra los valores de porcentaje de absorción.
2) Se corre muestras desconocidas y el blanco para la corrección de la absorción base. Se registrar los valores del porcentaje de absorción.
3) Se convierte los valores del porcentaje de absorción en absorbancia (A) usando la tabla. 4) Se corrige los valores de absorbancia (A) de las muestras desconocidas, substrayendo a
las absorbancias de las muestras la absorbancia del blanco. 5) Se tra7.a la línea de A con relación a la de concentración (C) para los patrones o estánda
res (se aplica la regresión lineal para establecer la línea de mejor ajuste). Se puede reali7.ar esta operación en una minicomputadora, para obtener una copia del gráfico.
- 56 -
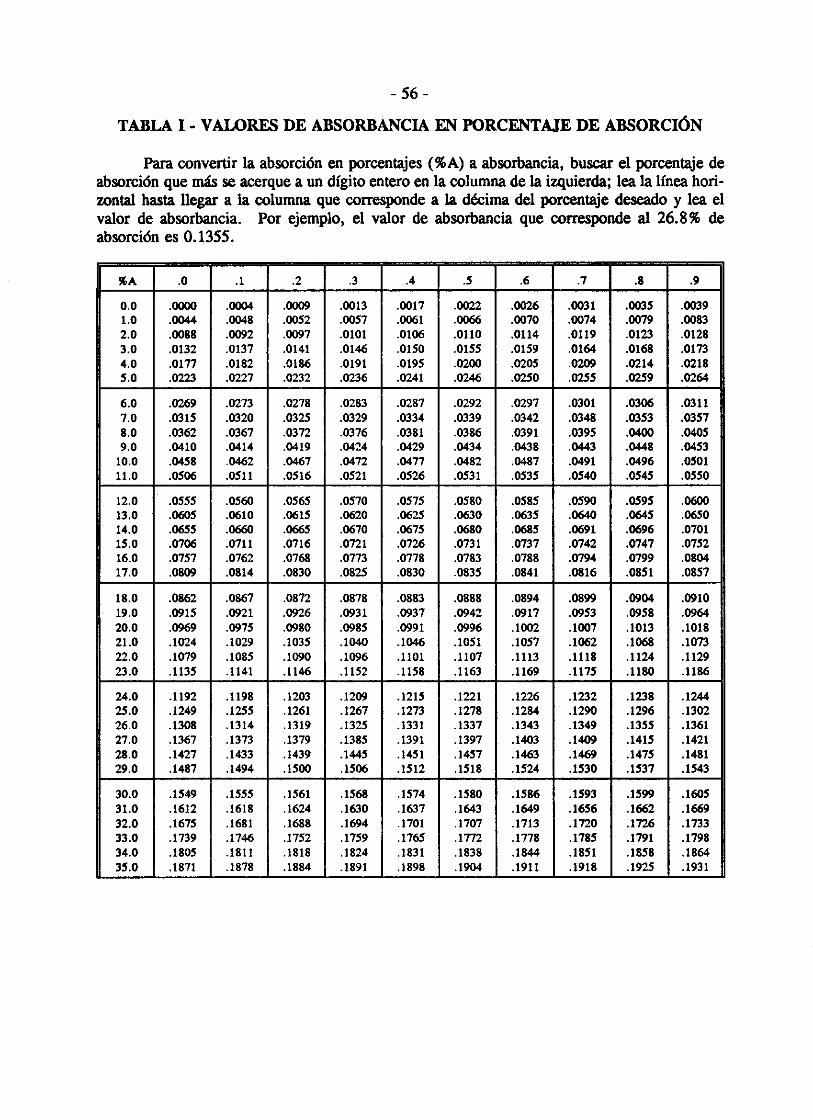
TABLA 1 - VALORES DE ABSORBANCIA EN PORCENTAJE DE ABSORCIÓN
Para convertir la absorción en porcentajes (%A) a absorbancia, buscar el porcentaje de absorción que más se acerque a un dígito entero en la columna de la izquierda; lea la línea horirontal hasta llegar a la columna que corresponde a la décima del porcentaje deseado y lea el valor de absorbancia. Por ejemplo, el valor de absorbancia que corresponde al 26.8% de absorción es 0.1355.
%A .o .1 .2 .3 .4 .s .6 .7 .8 .9
o.o .0000 .0004 .0009 .0013 .0017 .0022 .0026 .0031 .0035 .0039 1.0 .0044 .0048 .0052 .0051 .0061 .0066 .0070 .0074 .0079 .0083 2.0 .0088 .0092 .0097 .0101 .0106 .0110 .0114 .0119 .0123 .0128 3.0 .0132 .0137 .0141 .0146 .0150 .OISS .0159 .0164 .0168 .0173 4.0 .0177 .0182 .0186 .0191 .0195 .0200 .0205 .0209 .0214 .0218 s.o .0223 .0227 .0232 .0236 .0241 .0246 .0250 .0255 .0259 .0264
6.0 .0269 .0273 .0278 .0283 .0287 .0292 .0297 .0301 .0306 .o311 7.0 .0315 .0320 .0325 .0329 .0334 .0339 .0342 .0348 .0353 .0357 8.0 .0362 .0367 .0372 .0376 .0381 .0386 .0391 .0395 .0400 .0405 9.0 .0410 .0414 .0419 .0424 .0429 .0434 .0438 .0443 .0448 .0453
10.0 .0458 .0462 .0467 .0472 .0477 .0482 .0487 .0491 .0496 .OSO! 11.0 .0506 .0511 .0516 .0521 .0526 .0531 .0535 .0540 .OS4S .osso
12.0 1
.0555 .0560 .0565 .0570 .0575 .0580 .0585 .0590 .0595 .0600 13.0 .0605 .0610 .0615 .0620 .0625 .0630 .0635 .0640 .0645 .0650 14.0 .0655 .0660 .0665 .0670 .0615 .0680 .0685 .0691 .0696 .0701 15.0 .0706 .0711 .0716 .0721 .0726 .0731 .0737 .0742 .0747 .0752 16.0 .0151 .0762 .0768 .0773 .0778 .0783 .0788 .0794 .0799 .0804 17.0 .0809 .0814 .0830 .0825 .0830 .0835 .0841 .0816 .0851 .0857
18.0 .0862 .0867 .0872 .0878 .0883 .0888 .0894 .0899 .0904 .0910 19.0 .0915 .0921 .0926 .0931 .0937 .0942 .0917 .0953 .0958 .0964 20.0 .0969 .0975 .0980 .0985 .0991 .0996 .1002 .1007 .1013 .1018 21.0 .1024 .1029 .1035 .1040 .1046 .1051 .1057 .1062 .1068 .1073 22.0 .1079 .1085 .1090 .1096 .1101 .1107 .1113 .1118 .1124 .1129 23.0 .1135 .1141 .1146 .1152 .1158 .1163 .1169 .1175 .1180 .1186
24.0 .1192 .1198 .1203 .1209 .1215 .1221 .1226 .1232 .1238 .1244 25.0 .1249 .1255 .1261 .1267 .1273 .1278 .1284 .1290 .1296 .1302 26.0 .1308 .1314 .1319 .1325 .1331 .1337 .1343 .1349 .1355 .1361 27.0 .1367 .1373 .1379 .1385 .1391 .1397 .1403 .1409 .1415 .1421 28.0 .1427 .1433 .1439 .1445 .1451 .1457 .1463 .1469 .1475 .1481 29.0 .1487 .1494 .1500 .1506 .1512 .1518 .1524 .1530 .1537 .1543
30.0 .1549 .1555 .1561 .1568 .1574 .1580 .1586 .1593 .1599 .1605 31.0 .1612 .1618 .1624 .1630 .1637 .1643 .1649 .1656 .1662 .1669 32.0 .1675 .1681 .1688 .1694 .1701 .1707 .1713 .1720 .1726 .1733 33.0 .1739 .1746 .1752 .1759 .1765 .1m .1778 .1785 .1791 .1798 34.0 .!SOS .1811 .1818 .1824 .1831 .1838 .1844 .1851 .1858 .1864 35.0 .1871 .1878 .1884 .1891 .1898 .1904 .1911 .1918 .1925 .1931

- 57 -
"A .o .1 .2 .3 .4 .5 .6 .7 .8 .9
36.0 .1938 .1945 .1952 .1959 .1965 .1972 .1979 .1986 .1993 .2000 37.0 .2007 .2013 .2020 .2027 .2034 .2041 .2048 .2055 .2062 .2069 38.0 .2076 .2083 .2090 .2097 .2104 .2111 .2118 .2125 .2132 .2140 39.0 .2147 .2154 .2161 .2168 .2175 .2182 .2190 .2197 .2204 .2211 40.0 .2218 .2226 .2233 .2240 .2248 .2255 .2262 .2269 .2277 .2284 41.0 .2291 .2299 .2306 .2314 .2321 .2338 .2336 .2343 .2351 .2358
2.0 .2366 .2373 .2381 .2388 .2396 .2403 .2411 .2418 .2426 .2434 43.0 .2441 .2449 .2457 .2464 .2472 .2480 .2487 .2495 .2503 .2510 44.0 .2518 .2526 .2534 .2541 .2549 .2557 .2565 .2573 .2581 .2668 4S.O .2596 .2604 .2612 .2620 .2628 .2636 .2644 .2652 .2660 .2749 46.0 .2676 .2684 .2692 .2700 .2708 .2716 .2725 .2733 .2741 .2749 47.0 .2757 .2765 .2774 .2782 .2790 .2798 .2807 .2815 .2823 .2832
48.0 .2840 .2848 .2857 .2865 .2874 .2882 .2890 .2899 .2907 .2916 49.0 .2924 .2933 .2941 .2950 .2958 .2967 .2976 .2984 .2993 .3002 so.o .3010 .3019 .3028 .3036 .3045 .3054 .3063 .3072 .3080 .3089 SI.O .3098 .3107 .3116 .3125 .3134 .3143 .3152 .3161 .3170 .3179 S2.0 .3188 .3197 .3206 .32!S .3224 .3233 .3242 .3251 .3161 .3270 S3.0 .3279 .3288 .3298 .3307 .3316 .3325 .3335 .3344 .33S4 .3363
S4.0 .3372 .3382 .3391 .3401 .3410 .3420 .3429 .3439 .3449 .3458 SS.O .348 .3478 .3487 .3497 .3507 .3516 .3526 .3S36 .3546 .3SS6 56.0 .3565 .351S .3S8S .3595 .3605 .3615 .3625 .363S .364S .3655 S7.0 .3665 .3675 .3686 .3696 .3706 .3716 .3726 .3737 .3747 .3151 SS.O .3768 .3778 .3788 .3799 .3809 .3820 .3830 .3840 .3851 .3862 S9.0 .3872 .3883 .3893 .3904 .3915 .3925 .3936 .3947 .3958 .3969
60.0 .3979 .3990 .4001 .4012 .4023 .4034 .4045 .40S6 .4067 .4078 61.0 .4089 .4101 .4112 .4123 .4134 .4145 .4157 .4168 .4179 .4191 62.0 .4202 .4214 .4225 .4237 .4248 .4260 42.71 .4283 .429S .4306 63.0 .4318 .4330 .4342 .4353 .436S .4377 .4389 .4401 .4413 .4425 64.0 .4437 .4449 .4461 .4473 .448S .4498 .4SIO .4S22 .4S3S .4S47 65.0 .4SS9 .4572 .4584 .4597 .4609 .4622 .4634 .4647 .4660 .4672
66.0 .4685 .4698 .4711 .4724 .4737 .4750 .4763 .4776 .4789 .4802 67.0 .481S .4828 .4841 .4855 .4868 .4881 .4895 .4908 .4921 .4935 68.0 .4948 .4962 .4976 .4989 .5003 .S017 .S031 .5045 .sosa .S072 69.0 .S086 .5100 .5114 .Sl29 .5143 .Sl57 .Sl71 .5186 .5200 .5214 70.0 .S229 .5243 .5258 .S272 .5287 .5302 .5317 .S331 .5346 .5361 71.0 .S376 .S391 .5406 .5421 .5436 .S4S2 .5467 .S482 .5498 .5513
72.0 .5S28 .5S44 .5560 .5515 .S591 .5607 .S622 .5638 .5654 .5670 73.0 .5686 .5702 .5719 .S13S .5751 .S768 .5784 .5800 .S817 .5834 74.0 .58SO .S867 .5884 .S901 .5918 .S93S .S952 .S969 .S986 .6003 7S.O .6021 .6038 .60SS .6073 .6091 .6108 .6126 .6144 .6162 .6130 76.0 .6198 .6216 .6234 .6253 .6271 .6289 .6308 .6326 .634S .6364 77.0 .6383 .6402 .6421 .6440 .6459 .6478 .6498 .6517 .6536 .6556
78.0 .6576 .6596 .6615 .663S .6655 .6676 .6696 .6716 .6737 .6757 79.0 .6778 .6799 .6819 .6840 .6861 .6882 .6904 .9625 .6946 .6968 so.o .6990 .7011 .7033 .7055 .7077 .7100 .7122 .7144 .7167 .7190 81.0 .7212 .7235 .7258 .7282 .7305 .7328 .7352 .7375 .7399 .7423 82.0 .7447 .7471 .7496 .7520 .7545 .7570 .7595 .7620 .7645 .7670 83.0 .7696 .m1 .7747 .7773 .7799 .7825 .7852 .7878 .7905 .7932

- 58 -
ll\A .o .1 .2 .3 .4 .5 .6 .7 .8 .9
84.0 .1959 .7986 .8013 .8041 .8069 .8097 .8125 .8153 .8182 .8210 as.o .8239 .8268 .8297 .8327 .8356 .8386 .8416 .8447 .8177 .8508 86.0 .8539 .8570 .8601 .8633 .8665 .8697 .8729 .8761 .9794 .8827 87.0 .8861 .8894 .8928 .8962 .8996 .9031 .9066 .9101 .9136 .9172 88.0 .9208 .9245 .9281 .9318 .9355 .9393 .9431 .9469 .9508 .9547 89.0 .9586 .9626 .9666 .9706 .9747 .9788 .9830 .9872 .9914 .9951
6) Determinar por medio de los registros de la computadora o por medio de cálculos, la pendiente de la curva de calibración.
G(Y) = 0.01599 + 0.02876 F(X)
donde 0.02876 ~ pendiente = m
7) Una vez que se conoce la pendiente y asumiendo que los valores corregidos del blanco regresen a la concentración cero, cuando la absorbancia es cero, convertir los valores de absorbancia corregidos del blanco en concentración, valiéndose de la siguiente relación:
donde:
e = concentración en µgil
m e= -A
m = pendiente de la curva de calibración A = absorbancia.
Nota: Los valores de concentración que se sitúen por debajo del límite de detección en forma experimental, deben registrarse como < LD (menor que el límite de detección).

- 59 -
4.4 Calibración en cromatografía de gases
Existen dos procedimientos básicos que se usan para la calibración de sistemas de cromatografía de gases (C.O.):
a. Calibración externa b. Estandariz.ación interna
En la calibración externa o directa se inyectan cantidades exactas de la muestra pura. Se grafican los valores de las alturas de los picos (o áreas) contra la cantidad en peso conocida del compuesto inyectado. De esta manera, se obtiene la curva de calibración, la cual debe ser lineal y pasar a través del origen.
Las curvas de calibración de e.o. no son válidas para períodos de tiempo largos. Sin embargo, éstas definen en forma clara el rango lineal de respuesta del detector, que corresponde al rango de concentración sobre el cual el detector mantiene una sensibilidad constante de e.o. Antes de hacer cualquier cuantificación con un detector nuevo o totalmente renovado, se construye las curvas de linearidad para los compuestos de interés, bajo las condiciones de operación prevalentes. Se realiza revisiones frecuentes que aseguran que la operación continua se efectúa dentro de rangos de concentración aceptables.
Cuando no se está trabajando dentro del rango lineal del detector, el procedimiento apropiado para la cuantificación es espaciar las inyecciones de estándar mixto durante todo un día de trabajo, con frecuencia adecuada, para señalar las fluctuaciones de la respuesta. Se hace referencia cuantitativa con un estándar espaciado. Para obtener máxima exactitud, se intercala la inyección de la muestra desconocida con las inyecciones del estándar justo antes y después de la muestra.
La ecuación aplicada en el análisis de e.o. para calcular un pico desconocido a partir de un pico de un estándar de concentración conocida es la siguiente:
donde:
a =
b = c -d = R = e =
R = abe cd
nanogramos del compuesto orgánico desconocido representado por pico estándar altura (o área) del pico de la muestra altura (o área) del pico estándar ml (o gramos) de la muestra original concentración del residuo en partes por millón o billón factor de dilución que se obtiene de la siguiente relación

- 60 -
\/Olumen final del extracto 1
µl inyectados
Esla ecuación básica puede ser simplificada cuando se realizan análisis repetitivos en forma rutinaria.
las unidades que frecuentemente se usan en el CEPIS/LAB en análisis de trazas orgánic& son:
p.g - 1 O..S gramos "g == 10-9 gramos pg - 10-12gramos
ml = lo-3 litros ¡.il = 104.ítros
ppm "" partes por millón = mg/l; µg/g; µg/ml; ng/mg o pg/¡.ig ppb = partes por billón = µgil; ng/g; ng/ml o pg/mg ppt = partes por trillón = pg/g; pg/ml
En el procedimiento de estandarización interna, también conocida como calibración relativa o indirecta, como se conoce la relación de pesos del componente de interés y del estándar (ya sellalado) se prepara la mezcla y se inyecta en el cromatógrafo. La relación de áreas de componente/estándar se grafica contra la relación de peso del componente/estándar. El uso de un estándar interno añadido al iniciar el ensayo puede ayudar a compensar cualquier pérdida que pueda ocurrir. Para producir resultados precisos, el estándar interno debe tener exactamente los mismos cambios de concentración que el analito y la misma sensitividad al detector usado.
Una cantidad exactamente conocida del estándar interno se adiciona a una muestra desconocida y esta mezcla se inyecta al cromatógrafo y se miden las relaciones de área y de la curva de calibración y se determina la relación entre la concentración desconocida de la muestra y el estándar. Como la cantidad de estándar añadido se conoce mediante un cálculo simple, se determina la cantidad del compuesto desconocido presente.
En lugar de graficar las relaciones de área versus las relaciones de peso (o relaciones de concentración) puede hacerse una gráfica de un factor de respuesta (RF) con relación a la concentración del contaminante que ha sido medido. El factor de respuesta se define como:
FR = (As Csi) / (Asi Cs)
Este volumen es en ul para ppb y en mi para ppm

- 61 -
donde:
As - área integrada o la altura de pico del compuesto estándar Asi = pico integrado o la altura del pico para el estándar interno Csi - concentración del estándar interno Cs - concentración del compuesto estándar.
Si se añade una cantidad constante conocida de estándar interno (Csi) a cada muestra o extracto de la muestra, la concentración del compuesto (Co) en la muestra se calcula usando la siguiente ecuación:
Co = (As) (Csi) / (Asi) (RF) (Yo)
donde:
Vo = El volumen (o peso) de la muestra original; los otros términos se definieron anteriormente
Para escoger el patrón o estándar interno se debe tener en cuenta:
a. El pico del patrón o estándar debe encontrarse próximo al pico del compuesto a analizar pero bien separado de ella.
b. Se debe escoger la concentración del estándar interno, de manera que no sea necesario cambiar la atenuación.
c. Se pueden emplear dos o más patrones internos.
d. Se debe cubrir el rango de concentración esperado. No se puede extrapolar los datos de la gráfica.
Si el estándar interno, es elegido de forma adecuada, esta técnica corrige pérdidas durante la extracción y la recolección, y compensa errores debido a diferencias en el volumen de inyección y las variaciones no advertidas por la sensibilidad del instrumento. Una ventaja particular del uso del método de estandarización interna es que la cantidad inyectada no necesita ser medida exactamente. Además, no se necesita conocer la respuesta del detector porque cualquier cambio en la sensibilidad no afectará la relación de las áreas. Si un estándar interno ideal tiene propiedades físicas y químicas similares a aquellos compuestos que nos interesan, se asegura una diferencia despreciable en la extracción, derivatización y factores cromatográficos durante la preparación de la muestra. Sólo en el paso final del análisis debe distinguirse en forma clara el compuesto(s) de interés y el estándar interno.

- 62 -
4.4.1 Control de Calidad en Cromatografía de gases
Además de una calibración correcta en la cromatografía de gases, se requiere de procedimientos de control de calidad para asegurar que se cumple con los requerimientos analíticos de exactitud. Normalmente se hacen tres tipos de pruebas de control:
a. Pruebas de control de la precisión de estándares b. Pruebas de control de la precisión en muestras por duplicado c. Pruebas de control del error sistemático con muestras adicionadas, agregando una
cantidad conocida de estándar a las muestras reales.
Debido a que el laboratorio opera bajo limitaciones de presupuesto y límites de tiempo, es necesario asignar prioridades a los diferentes tipos de pruebas de control. Se asigna prioridad a las pruebas de control de la precisión con soluciones patrones o estándares. Estas soluciones patrones o estándares se preparan independiente de los patrones o estándares de calibración, a fin de proporcionar una evaluación independiente que incluya la exactitud y estabilidad de la solución patrón o estándar de calibración. La siguiente prioridad es para la elaboración de cartas de control de la precisión basadas en el análisis de duplicados de muestras reales. Las pruebas de control de error sistemático son la tercera prioridad y se basan en la recuperación de muestras adicionadas.
4.4.2 Pruebas de control de la precisión
La precisión de un método cromatográfico se determina por el grado de analito recuperado en cada fase del análisis. Para ello, se deben considerar los siguientes puntos: las muestras colectadas deben ser representativas del agua que se ha muestreado. La técnica de colección no debe alterar la concentración de los componentes de la muestra. Debe prevenirse la degradación química o biológica de la muestra antes de su análisis. Las técnicas de limpieza o de preparación de un derivado del analito de la muestra deben proveer una recuperación completa del analito o, por lo menos reproducible.
El adicionar a la muestra cantidades conocidas del analito en varias etapas a través del análisis puede ayudar a localizar en dónde ocurren las pérdidas y ayudar a estimar la precisión.
4.4.2.1 Soluciones estándar
Para evaluar la precisión de los análisis se analiza un patrón o estándar con cada lote de muestras. Generalmente se elige un estándar de concentración de aproximadamente 0.9 Cm, (Cm es el límite máximo de concentración del método, por ejemplo, el límite máximo de la curva de calibración lineal). Mediante el uso de una concentración alta se puede detectar fácilmente cualquier cambio en la precisión. En algunos casos para evaluar la precisión se analiza un segundo estándar que contiene una concentración baja (aproximadamente 0.2 Cm). Si la precisión de los análisis a estas dos concentraciones es aceptable, se asume que la precisión de todos los análisis de concentraciones intermedias también son aceptables.

- 63 -
El patrón o estándar de control se debe analizar con el mismo procedimiento usado para las muestras (incluyendo, extracción, purificación y determinación cromatográfica apropiada). Se pone énfasis en la calibración con un patrón o estándar preparado con un solvente puro y, que cuando contiene al analito a medir por un procedimiento analítico completo, es posible detectar y estimar el error sistemático debido a la calibración. Los errores serios asociados a la calibración indican la necesidad de analizar los patrones o estándares de calibración de manera idéntica a las muestras.
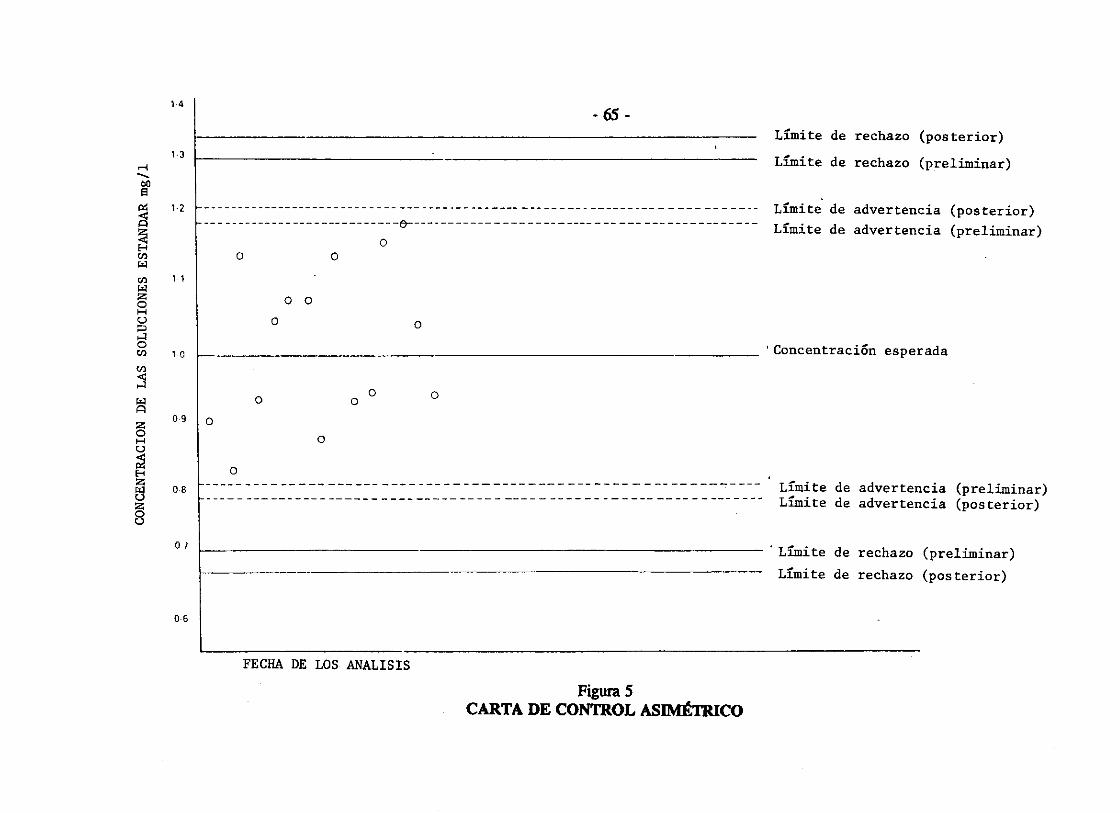
Se registran los resultados de los análisis de los patrones o estándares a medir. Para mejores resultados en el control de calidad se realiza, por lo menos, 20 determinaciones para el cálculo de la desviación estándar S. Estos resultados no necesitan ser obtenidos el mismo día. Con ello se harán las cartas de control que, como ya se ha mencionado, se emplean para controlar y evaluar mediciones repetidas. Con la desviación estándar se construye la carta de control con los límites de "advertencia" dados por X ± 2S; y los límites de "acción" dados por X ± 3S (ver el ejemplo de la figura 5). Como los límites de control se basan en estimados de la desviación estándar, ésta debe actualizarse en forma periódica agrupando estimados preliminares y recientes. Nótese que los límites de advertencia y acción, (en este caso), están graficados en forma simétrica alrededor de la concentración media esperada. Esto porque se conoce la concentración de la solución. Cuando el resultado del estándar está fuera de los límites de "acción•, los análisis están fuera de control, por ello deben repetirse tanto los análisis de todas las muestras como del estándar de control.
4.4.2.2 Duplicados de las muestras naturales
Para evaluar la precisión de los análisis en las muestras naturales, con cada lote de mediciones debe analizarse una muestra por duplicado. Si las concentraciones de la muestra son constantes en todos los lotes, las cartas de control se basan en el control de la desviación estándar dentro del lote. Esta desviación estándar se calcula mediante el uso de la siguiente fórmula:
donde:
s ... di m
---
desviación estándar dentro del lote diferencia entre las mediciones efectuadas por duplicado en cada lote número de lotes (para determinar los límites de control se requiere de un mínimo de 20 mediciones).

- 64 -
En las cartas de control los límites de •advertencia" se establecen por medio del cálculo X ± 2 v'2 s., y los límites de "acción" por X ± 3 v'2 S.,.
Cuando las concentraciones del parámetro a medir varían en forma considerable de muestra a muestra, no es suficiente una sola estimación de la desviación estándar dentro del lote. En este caso, las diferencias de las muestras duplicadas sólo se evalúan en forma semicuantitativa y no son aplicables las cartas de control. Las determinaciones de duplicados sirven para, detectar de manera aproximada los problemas de precisión.
4.4.2.3 Pruebas de control de error sistemático (BIAS)
También se pueden preparar las cartas de control mediante la evaluación de la recuperación del analito en una muestra natural adicionada. Este tipo de control se aplica a muestras cuyas concentraciones no cambian en forma significativa y cuando la cantidad "adicionada" que se recupera es constante.
La recuperación se calcula de la diferencia entre la concentración del compuesto de interés en la muestra adicionada y la muestra original. La desviación estándar de la concentración recuperada puede obtenerse después de haber ejecutado 10 pruebas de recuperación en los análisis de rutina. Con la estimación apropiada de la desviación estándar, se construye la carta de control donde la línea media corresponde a la recuperación teórica µ; el límite de advertencia aµ ± 2S y los límites de acción aµ± 3S. En algunos casos, las cartas de recuperación se expresan en términos de porcentaje de recuperación, donde la recuperación esperada es de 100%.
Cuando no se tiene una carta de control de recuperación en forma preliminar se efectúa un mínimo de muestras adicionadas con matrices típicas para establecer si el método analítico cumple con los objetivos de error sistemático1
•
El procedimiento para evaluar Ja recuperación se presenta en la páginas 107 del Informe N°66 del Water Research Centre, 1978.
, .. - 65 -
'3
1·2 --- - --- - - ---------- -- --- - - - -- --- ---- -- - ------- --- -- - -- - ------ --- - - -- - -- - -- -
- -- - - --- --- -- - --- - ------ -- -- -e----- -- ----- - - -- - - -- - ---- - -- --- - -- - - - - - -- - - - - -o
o o
' ' o o
o o
'o
o o o o 09 o
o
o L _____ - -- - -- --- --- --- --- - - -- - - --- ---- ---- --- ---- - - -- --- ---- ---
O·B .._ _____ ---------- ------ ------------- _ ---- _ ---- __ ------ ---- __ -------
o 1
o 6
FECHA DE LOS ANALISIS
Figura 5 CARTA DE CONTROL ASIMÉTRICO
Límite de rechazo (posterior)
Límite de rechazo (preliminar)
Límite de advertencia (posterior) Límite de advertencia (preliminar)
'Concentración esperada
Límite de advertencia (preliminar) Límite de advertencia (posterior)
' Límite de rechazo (preliminar) Límite de rechazo (posterior)

- 66 -
4.4.2.4 Estimación de la recuperación de muestras adicionadas
En el siguiente ejemplo hay n determinaciones, tanto de muestras adicionadas y no adicionadas, medidas en m lotes. Para mostrar que la recuperación media no difiere significativamente (a un nivel de significancia 0.05) de % de la cantidad añadida (100 ± D), se usa el procedimiento siguiente.
Calcule tanto la recuperación como la diferencia entre los resultados para n pares de muestras adicionadas y no adicionadas en cada lote de mediciones. Calcule la recuperación media R de los mn resultados. También calcule la desviación estándar S de las m recuperaciones medias de los m lotes. La cantidad adicionada será d (en las mismas unidades que R). La recuperación media será aceptable si:
S.t oc R a) R > (1.00 - 0.1 D) d - - 2
- fpara - < (1.00 - 0.01 D)] m d
S.'2.. R b) R < (1.00 + 0.1 D) d + -- fpara - > (1.00 + 0.01 D)]
m d
donde:
~oc = to.05 de student con (m-1) grados de libertad y un nivel de probabilidad de 2oc
4.4.2.5 Determinación de los blancos
Antes de procesar cualquier muestra, se demuestra con el análisis de un blanco (la EPA lo denomina como un método del blanco con agua destilada) que todo el material de vidrio y los reactivos están libres de interferencia. De igual modo, cada vez que se procesa un blanco, o cuando hay cambio de los lotes de reactivos para evaluar la posible contaminación crónica del laboratorio, se analiza un lote de muestras. Los valores de las determinaciones del blanco se usan también para la determinación del límite de detección.
4.4.2.6 Determinaciones de límites de detección
Los límites de detección dependen del tipo de detector, las características y cantidad de los compuestos interferentes presentes, y concentración de la preparación de la muestra. En la ausencia de picos interferentes, el detector de conductividad térmica es capaz de detectar cantidades de material en el rango de microgramo; el detector coulométrico en el rango de nanogramo; el detector de ionización de llama en el rango de 10 pg y los detectores de captura de electrones y termoiónicos en el rango de picogramo.

- 67 -
A continuación, se describe el método usado para estimar el límite de detección del método analítico. Este se basa en la variabilidad del blanco, donde el blanco es procesado siguiendo la totalidad del método analítico. El(los) compuesto(s) de interés, como normalmente sucede, no se presentan en el blanco y la respuesta de éste normalmente corresponde al ruido de fondo.
F.n cromatografía de gases, una estimación simple del límite de detección se basa en por lo menos diez análisis de un blanco. Este procedimiento es especificado en varios métodos recomendados por la EPA con la siguiente fórmula:
LD (µo/l) = Á X Átt . X µo// º' BxAtt º'
donde:
LD - límite de detección en µgil u otras unidades de concentración apropiadas A - cinco veces el nivel de ruido en mm a un período exacto de retención
correspondiente al compuesto de interés o el desplazamiento de la línea base en mm para el cero teórico a un período de retención exacto correspondiente al compuesto de interés
B = altura del pico (mm) del patrón o estándar de concentración igual a X µgil Att = factor de atenuación.
Nota: El estándar de concentración X µgil debe ser mínimo 10 veces el valor final estimado del límite de detección.
Muchas veces las interferencias en las muestras incrementan el ruido comparado con el fondo (background) del valor del blanco usado en el cálculo del límite de detección. Si esta interferencia es seria, el límite de detección calculado no es válido. En este caso, si se necesitan hacer mediciones y las concentraciones están cerca del límite de detección, es necesario una extracción más fuerte.
Las constantes interferencias pueden invalidar la utilidad del límite de detección de una muestra específica. Se considera que el valor estimado es de utilidad cuando es característica propia del método analítico e indica que el laboratorio es capaz de cumplirlo bajo circunstancias controladas.
El límite de detección se calcula para un método analítico específico. Por ejemplo, el método puede señalar que se tome un litro de muestra, que se concentre a 10 mi, y que del extracto se inyecte 5 µl. Si se realiza cualquier desviación de las instrucciones del método se debe esperar un cambio en el límite de detección (por un factor correspondiente). Las desviaciones en el método deben ser tomadas en cuenta cuando se reportan los resultados de las muestras naturales.

- 68 -
4.4.3 Guías para la revisión del cromatógrafo de gases
INSTRUMENTO FRECUENCIA COMPUESTO A PROCEDIMIENTO REVISAR
Cromatógrafo de gas con Diaria Resolución y reproducti- El instrumento se acon-detector de captura de bilidad de la relación dicion con un estándar y electrones área/altura del pico luego se calibra. Los están-
dares se repiten cada S muestns para actualizar la calibración.
Mensual Contaminación del DCE' La salida y el fondo del detector se monitorea para detectar posible contamina-ción. Los DCE se limpian térmicamente el fm de semana una vez por mes.
Mensual Resolución Si se conoce el estándar de PCB inyectado y se com-para con la del mes ante-rior.
Cromatógrafo de gas con Anual o cuando sea Contaminación del DIF' Se limpia el detector ma-detector de ioni7.11Ción de necesario nualmente usando un equipo llama deHP
Diaria Resolución y reproducti- El instrumento se evalúa bilidad de la relación con un estándar calibrado. área/altura del pico Los estándares se repiten
cada S muestras para actua-!izar la calibración.
1 DCE: detector de captura de electrones
' DIF: detector de ioni7.11Ción de llama
4.4.4 Revisión de las columnas cromatográficas de gas
La revisión de las columnas cromatográficas se lleva a cabo midiendo el factor de pico asimétrico. Este factor compara la base de la segunda mitad del pico cromatográfico con la primera mitad de éste, tal como se aprecia en la figura 6.
Para cada tipo de columna se deben establecer los valores aceptables del factor de pico asimétrico. Por ejemplo, en la realización de análisis de plaguicidas se acepta si el factor de pico de aldrín es menor que 2.
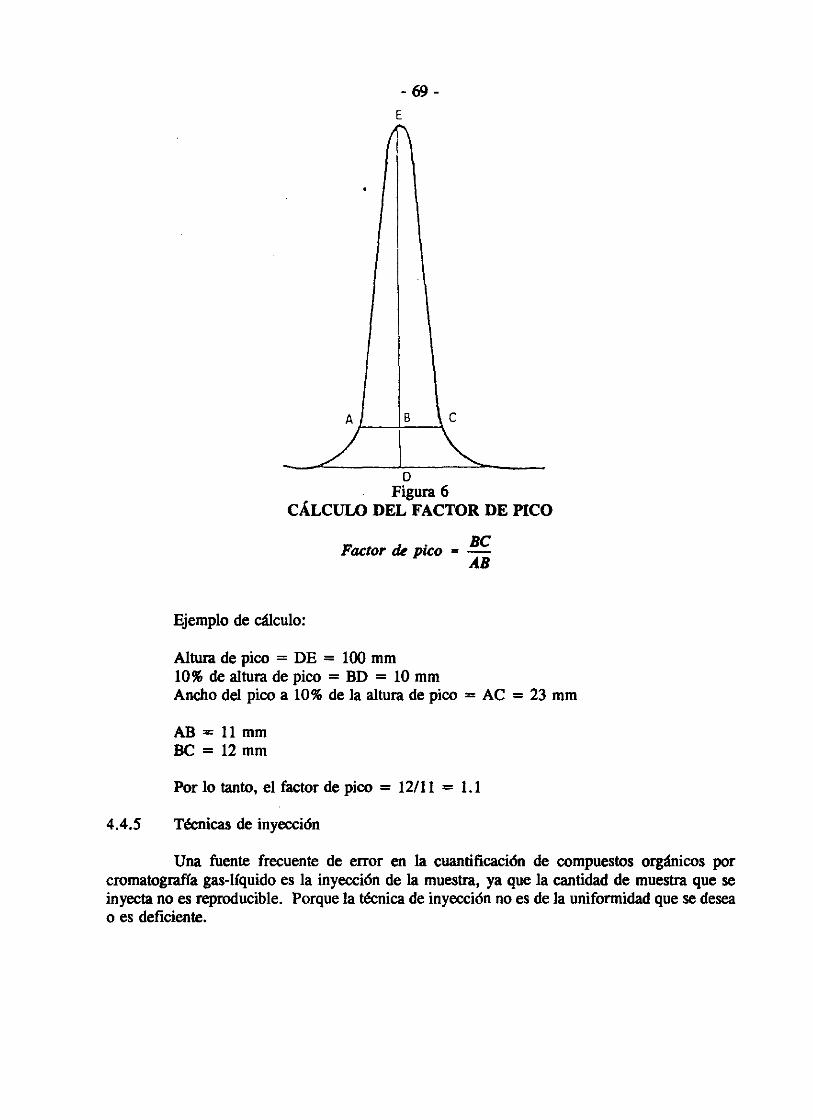
- 69 -E
A B
D Figura 6
CÁLCUW DEL FACTOR DE PICO
Ejemplo de cálculo:
Factor de pico = BC AB
Altura de pico = DE = 100 mm 10% de altura de pico = BD = 10 mm Ancho del pico a 10% de la altura de pico = AC = 23 mm
AB = 11 mm BC = 12 mm
Por lo tanto, el factor de pico = 12/11 = 1.1
4.4.5 Técnicas de inyección
Una fuente frecuente de error en la cuantificación de compuestos orgánicos por cromatografía gas-líquido es la inyección de la muestra, ya que la cantidad de muestra que se inyecta no es reproducible. Porque la técnica de inyección no es de la uniformidad que se desea o es deficiente.

- 70 -
Antes de usar una jeringa se revisa que esté totalmente limpia, libre de residuos, porque Ja falta de limpieza puede estropear completamente el cromatograma.
Se toma un tamaño de muestra moderado en un frasco que Juego es sellado. Si el frasco de la muestra es pequeño y/o un buen número de muestras van a ser colectadas, el frasco es primero presurizado por inyección de un gas inerte (por ejemplo, el gas acarreador) en el frasco. Con una jeringa nunca se debe obtener una muestra cuando hay presión negativa dentro del frasco de la muestra. Se empuja totalmente el émbolo, con cuidado de no tocarlo. Se agita el frasco con la muestra. Se toma la jeringa en forma vertical para atrapar al gas y que éste pueda ascender y escapar. Se inserta la aguja en el frasco, de tal forma que esté en su totalidad inmersa en Ja muestra. En seguida, se mueve el émbolo hacia afuera y a una distancia corta hasta humedecer la superficie interior del émbolo y del cuerpo de la jeringa. Esto neutraliza cualquier movimiento del líquido, debido a la capilaridad. Después, mediante bombeo, se retira el émbolo hasta llenar Ja jeringa entre un 5 y un 10%. Se retira la aguja, se toma Ja jeringa verticalmente, se llena Ja aguja cuidando que no presente burbujas o espuma. Se empuja el émbolo lentamente y sacude el exceso de líquido deteniendo el émbolo en Ja marca previamente seleccionada. El exceso de líquido de Ja aguja es descartado después que ésta se ha sacado del frasco. Si se descarga mientras se encuentra Ja aguja dentro del frasco, puede presentarse una pequeña pérdida de muestra al retirar la aguja. Se debe leer cuidadosamente Ja graduación de la jeringa. La mejor manera es tomar la jeringa frente a los ojos quedando la parte final del émbolo en posición horizontal. Esto elimina cualquier paralelaje que pueda causar lecturas erróneas. También se recomienda que la parte final del émbolo, que contiene un volumen de líquido siempre sea leído en el "tope", es decir, en el borde de la línea de calibración. Esto eliminará cualquier posibilidad de error, debido al espesor de la línea de calibración. Mientras se lee el volumen contenido en la jeringa, se debe revisar de nuevo que no haya en la muestra burbujas o materia extraña. Frote la aguja para limpiarla con un paño, tenga cuidado de no limpiar cualquier muestra fuera o el extremo de la aguja y nos transfiera el calor que puedan generar los dedos a la aguja. En un rápido movimiento, se introduce la aguja a través del septum del cromatógrafo y se inyecta la muestra. Inmediatamente después de inyectar la muestra se retira la aguja para prevenir la vaporización del líquido remanente en la aguja con el consiguiente resto. Después de que la aguja ha sido retirada, la jeringa puede ponerse verticalmente con la aguja hacia arriba y el émbolo empujado hacia afuera a una distancia corta. Esto arrasa con el líquido residual fuera de la aguja al interior de la jeringa, donde este volumen pueda medirse. La cantidad total inyectada puede determinarse restando el volumen residual del volumen original en la jeringa.
Otra técnica que puede usarse es succionar una pequeña cantidad de aire en Ja jeringa, tomar las muestras y finalmente otro poco de aire. Eso nos dará una especie de "sandwich" de líquido entre dos fracciones de aire. El volumen de la muestra líquida puede leerse exactamente por medio de las graduaciones de la jeringa. Después de introducir la aguja a través del septum, el émbolo puede empujarse totalmente. El aire delante de la muestra permite la medición exacta del volumen de líquido contenido y el aire detrás del líquido permite que toda la muestra sea inyectada en el cromatógrafo. No habrá residuos remanentes ni espacios muertos entre el extremo del émbolo y el inicio del contenedor de Ja jeringa. Uno de estos dos métodos

- 71 -
es recomendado particularmente si la jeringa está equipada con una aguja desechable. El método del "sandwich" descrito sólo es usado para muestras que no se contaminen por el aire. Si este último es un contaminante, la muestra puede ser puesta como "sandwich" entre dos capas del gas acarreador.
Para muestras pequeñas de 0.05 a 1.0 microlitros se dispone de jeringas y agujas, con las cuales se toma la muestra líquida. La parte inferior del émbolo y la punta de la aguja forman parte del volumen total contenido en la jeringa. Esto asegura una medición exacta de la cantidad inyectada y esencialmente elimina cualquier efecto final. El volumen de la muestra se lee en el contenedor como una jeringa ordinaria.
Un problema especial es el muestreo de líquidos altamente viscosos. Con frecuencia, la viscosidad puede disminuir por calentamiento de la muestra en una jeringa protegida y mantenida a alta temperatura hasta el momento de la inyección; si ésta no contiene fracciones ligeras, las cuales serán vaporizadas por la alta temperatura. Otra manera de muestrear es por encapsulado en un contenedor de metal o de vidrio.
Otro método de manejo de muestras viscosas es el disolverlas en un solvente neutro de baja viscosidad. La solución puede manejarse fácilmente con una jeringa estándar. El problema es encontrar un solvente que no interfiera. Tal solvente es difícil encontrar para muchas muestras, por lo que este método, que teóricamente es posible, en la práctica tiene poco uso.
Algunas muestras líquidas son manejadas bajo presión para obtener las fracciones de gas en solución. La muestra puede estar contenida en un frasco con un septum fuerte. El problema es que tan pronto como la aguja es retirada del septum del frasco de la muestra, el extremo de la aguja es expuesto a la presión atmosférica. El contenido y los gases presurizados en la muestra líquida se expandirán a través de la aguja y escaparán a la atmósfera. Sin embargo, el diámetro interior de la aguja es pequeño, 0.006 pulgadas o menos. Si se hace rápidamente, puede taparse con un tapón de caucho o con una pieza pesada de material de septum. Para hacer esto se coloca un tapón contra el septum de bote de la muestra antes de insertar la aguja. La aguja perfora el tapón, antes del septum. Cuando la aguja se retira del frasco de la muestra, la punta queda dentro del tapón, entonces ésta se coloca contra el septum del cromatógrafo y la aguja se desliza a través de éste antes de entrar al septum a la columna. Con este procedimiento se mantiene dentro de la jeringa, muchos de los gases aunque algunos se pierden antes y después de los deslizamientos de la aguja.


















