
Problemas de Enzimología
30
Problemas de Enzimología Hoja 1 1." Vol = 0,1 ml Pm (NR) = 500.000 Da Vmax = 20 mol/5ml [E] = 0,5 mg/ml (de ella sola al estar en homogeneidad) Vfinal de reacción = 1 ml a) Para definir la actividad de la reacción, en este caso para una enzima purificada a homogeneidad, se parte siempre de un dato de velocidad, Vmax. Este parámetro es la condición en la que normalmente se define la cantidad de enzima que hay en una preparación, midiendo la velocidad en condiciones de saturación por sustrato. Por esto se sobrentiende que el dato que nos da el problema de velocidad es el de Vmax. Por lo tanto, para definir la actividad (Act) en U/ml, partimos de la equivalencia de los apuntes: Lo que nos piden con la actividad es la cantidad de enzima que hay en la preparación de NR. Lo que nosotros hacemos es ensayar en un volumen total de reacción de 1 ml un volumen de enzima de 0,1 ml, con lo que 1/10 del volumen será enzima. Lo que nos piden es la cantidad de enzima en 1ml, que es la misma que hay en los 0,1 de preparación de enzima homogénea. Para calcular el dato comenzamos con la Vmax expresada en min"1: Esta velocidad, es la que presentan 0,1 ml de la muestra de enzima. Como nos piden las Unidades por mililitro de la reacción, no hay más que realizar una regla de tres: b) En este caso nos piden la actividad específica (Ae), que se trata de la actividad referida a la cantidad total de proteína. La forma habitual de expresar la Ae es en U/mg de proteína. Como ya sabemos la actividad, y nos dan la concentración de proteína. En este caso, al encontrarse la enzima purificada a homogeneidad, los mg de proteína que nos dan están referidos únicamente a la NR. Ya que la Ae nos define un valor de concentración de enzima, y toda la proteína es ella misma, el dato de Ae que nos da, es el máximo alcanzable. El valor máximo que obtenemos es: c) Los Katales son una medida de actividad, definida por los moles transformados por segundo. Po lo tanto, no se trata más que de transformar las unidades:
Transcript of Problemas de Enzimología

Problemas de EnzimologíaHoja 11."Vol = 0,1 mlPm (NR) = 500.000 Da Vmax = 20 mol/5ml[E] = 0,5 mg/ml (de ella sola al estar en homogeneidad)Vfinal de reacción = 1 mla) Para definir la actividad de la reacción, en este caso para una enzima purificada a homogeneidad, se parte siempre de un dato de velocidad, Vmax. Este parámetro es la condición en la que normalmente se define la cantidad de enzima que hay en una preparación, midiendo la velocidad en condiciones de saturación por sustrato. Por esto se sobrentiende que el dato que nos da el problema de velocidad es el de Vmax.Por lo tanto, para definir la actividad (Act) en U/ml, partimos de la equivalencia de los apuntes:
Lo que nos piden con la actividad es la cantidad de enzima que hay en la preparación de NR. Lo que nosotros hacemos es ensayar en un volumen total de reacción de 1 ml un volumen de enzima de 0,1 ml, con lo que 1/10 del volumen será enzima. Lo que nos piden es la cantidad de enzima en 1ml, que es la misma que hay en los 0,1 de preparación de enzima homogénea. Para calcular el dato comenzamos con la Vmax expresada en min"1:
Esta velocidad, es la que presentan 0,1 ml de la muestra de enzima. Como nos piden las Unidades por mililitro de la reacción, no hay más que realizar una regla de tres:
b)En este caso nos piden la actividad específica (Ae), que se trata de la actividad referida a la cantidad total de proteína. La forma habitual de expresar la Ae es en U/mg de proteína. Como ya sabemos la actividad, y nos dan la concentración de proteína. En este caso, al encontrarse la enzima purificada a homogeneidad, los mg de proteína que nos dan están referidos únicamente a la NR. Ya que la Ae nos define un valor de concentración de enzima, y toda la proteína es ella misma, el dato de Ae que nos da, es el máximo alcanzable.El valor máximo que obtenemos es:
c)Los Katales son una medida de actividad, definida por los moles transformados por segundo. Po lo tanto, no se trata más que de transformar las unidades:
d)En este apartado se nos pide la actividad molecular de la enzima, o lo que es lo mismo, Kcat. Como ya vimos en teoría, la constante catalítica sale de la expresión de Vmax, siendo la constante de proporcionalidad que multiplica a [E]T para darnos Vmax. Ya que tenemos el dato de Ae máxima, podemos calcular directamente Kcat, sin más que multiplicar por el peso molecular.

Esto se debe al hecho de estar purificada a homogeneidad, de modo, que la Ae que hemos hallado es velocidad de transformación por mg de proteína, y como toda la proteína es enzima queda en moles al multiplicarlo por el peso molecular. Hay que tener en cuenta que este dato solo es aplicable en el caso de tener un dato de Ae de la enzima pura. Esto se demuestra fácilmente, mediante las unidades.
Esto puede ser útil en muchas ocasiones, pero siempre hay que tener en cuenta que necesitamos un dato de Vmax obtenido de la enzima purificada. Una vez sabido esto, calculamos Kcat.
e)Si nos confirman que la enzima presenta dos centros activos, sabríamos que la Kcat hallada es Kcat molecular, al haber utilizado en la expresión del apartado anterior el peso del oligómero. De modo que, como vimos en teoría, lo único que hay que hacer para calcular Kcat C.A. es dividir por el nº de C.A.. En este caso Kcat C.A.= 666,6 s"1/2 = 333,3 s"1.f)El tiempo del ciclo catalítico (tcc) no es más que la inversa de Kcat. Ya que tenemos una Kcat C.A. es preferible utilizar este valor, al garantizarnos un tiempo de catálisis por C.A. tcc = 1/333,3 s"1 = 3·10"3 s = 3 milisegundos.En esta segunda parte del ejercicio se nos dan unos datos de purificación, a través de los cuales debemos hallar el grado de purificación alcanzado. En esta tabla se nos dan los valores de Volumen, Actividad y Cantidad de proteína, suficientes para calcular el resto de la siguiente forma: Actividad Total: Se define como las Unidades totales del extracto crudo. Para calcularla se multiplica la actividad por el volumen (U/ml · ml totales = unidades totales). Ejemplo: 1,72 U/ml · 14,080 ml = 24,217 U totales Actividad específica: Actividad del extracto crudo. Como ya hemos mencionado muchas veces, no implica más que dividir la actividad entre la cantidad de proteína (U/ml ÷ mg/ml = U/mg). Ejemplo: 1,72 U/ml ÷ 31,4 mg/ml = 0,0547 U/mg Rendimiento: Se trata de comparar el paso con el extracto crudo. El parámetro a comparar es la actividad total, de modo que efectuamos un cálculo de tanto por ciento haciendo que la actividad total del extracto crudo sea el 100%. Ejemplo:

Grado de purificación: a diferencia del anterior, esta comparación se realiza con la actividad específica y no se da en tanto por ciento, sino en veces, resultado de dividir Aedel paso entre Aecrudo. Ejemplo: Ae (paso II)/Ae (crudo) = 0,3615 U·mg"1/0,0547 U·mg"1= 6,6 veces
El resultado de calcular esto para todos los pasos se presenta en la siguiente tabla.
NºCromatografía DEAE celulosa
Paso de purificación
I II III IV V Filtración en gel de agarosa
Cromatografía DEAE c
Nº Extracto crudo(NH4)2SO4 (30"45 %)
Calentamiento a 62 ºC
1 Volumen (ml) 14080 1600 163 188 94
2 Actividad (U/ml) 1,72 11,10 52,58 29,60 41,37
3 Proteína (mg/ml) 31,40 30,70 7,31 1,43 1,58
4 Actividad total 1·2 24217 17760 8570 5564
5 Rendimiento (%)UT Vs UTcrudo
100 73,3 35,3 22,9
6Actividad específica (U/mg)
2÷3 0,0547 0,3615 7,19 20,7
7Grado de purificación (veces)
Ae Vs Aecrudo
" 6,6 131,5 378 478
[penicilina]
Velocidad [S]/V
0,1 0,11 0,91
0,3 0,25 1,2
0,5 0,34 1,47
1 0,45 2,22
3 0,58 5,17
5 0,61 8,19
Evidentemente, a simple vista parece asemejarse bastante a una hipérbola cuadrangular, con una Vmax que debe rondar 0,6 nmol · min"1. Para asegurarse, lo que se hace es representrar la linearización de los datos, y si estos se ajustan a una recta, es que efectivamente estamos ante una cinética M&M.
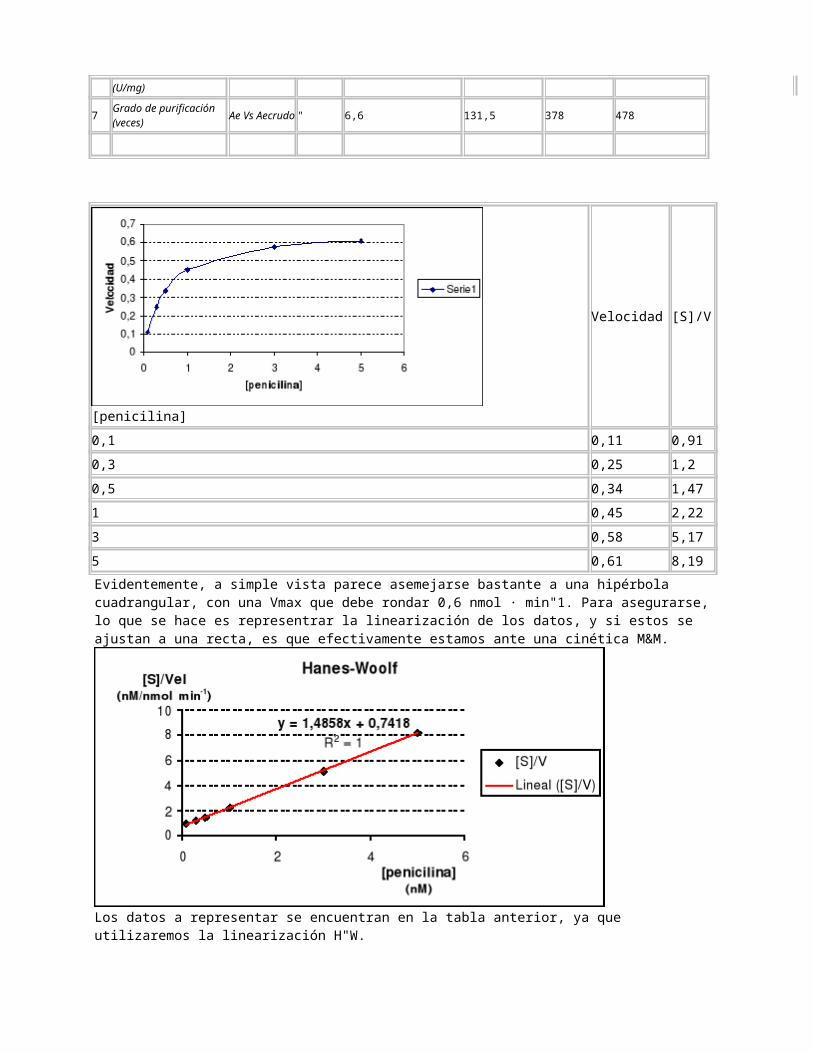
Los datos a representar se encuentran en la tabla anterior, ya que utilizaremos la linearización H"W.El Excel no nos representa el corte en el eje X, pero nos da la ecuación de la recta, con la que podemos calcular todos los parámetros:
Km: Cuando [S]/Vel = 0. . Km = 0,5 nM; Km = 0,5 · 10"9 M . Si nos fijamos, este dato encaja con lo que se obtendría en la gráfica anterior. Vmax: Dato que se puede averiguar por el corte en el eje Y, además de por la pendiente. Lo haremos por los dos métodos.Pendiente: Ya nos la dan en la ecuación de la recta: tg = 1,4858. Su inversa será Vmax. Vmax = 0,673 nmol · min"1. Ordenada en el origen: Se trata del valor de y cuando x = 0.
Con estos dos datos, hacemos la media para obtener que Vmax = 0,6735 · 10"9 mol · min"1. Kcat: Definimos esta constante como la constante de proporcional para [E]T de la ecuación de la veloc
idad máxima. Por esto, podemos definirla como . Podemos definir Kcat porque tenemos nuestra proteína purificada y podemos hallar la concentración total, de otra forma sería imposible. Pero, ya definimos otra forma de calcular Kcat, a través de la actividad específica. El cálculo de Ae lo efectuamos dividiendo el valor de Vmax (mol · min"1) por los mg de proteína. Esta actividad ya es la actividad de la enzima pura, por haberla hallado con el dato de Vmax del extracto puro.
3."
Respuesta
Explicación

a) Verdadero
Porque aunque la expresión de Km incluye las concentraciones (como Ks aparente),
se trata de una razón, con lo que si aumenta [E] libre, disminuye [ES] y viceversa, manteniendo a Km constante.. Km sólo depende de las constantes cinéticas de los pasos de unión, disociación y transformación de sustrato, que serán función de la energía libre que se libere. Vmax si depende de la cantidad de enzima.
b) Falso
Esto se comprueba matemáticamente llegando a la misma expresión de dos formas diferentes. Decíamos que la constante de especificidad es la razón ente Km y Kcat, y que se trata la constante de la unión entre la enzima y el sustrato. Ahora, tratamos de llegar a la misma expresión partiendo de la ecuación de Km y de la velocidad genérica (independiente de [E]). Sustituyendo [ES] en la ecuación de velocidad obtenemos la misma ecuación, ahora con la certeza de su independencia de [E].
c) Verdadero
Hablamos de condiciones saturantes, con lo que implica, pero lo demostramos calculando al porcentaje de Vmax que llegamos con una [S] de 100 Km.
Si V/Vmax = 0,99 esta relación es igual a Kcat·[ES]/Kcat · [E]T (recordemos las ecuaciones). Esto nos dice que [ES]/[E]T = 0,99, con lo que la enzima libre tiende a 0.
d) FalsoYa que definimos Kcat como la relación de las constantes implicadas en transformación. En el caso de una reacción monosustrato Kcat sería k2 , sin incluir k1 y k"1, que formarían parte de Km y Ks.
Prácticas de Metodología Y Bioquímica estructural Cromatografía de cambio ionicoSe basa en el proceso de intercambio de iones, en disolución, de la misma naturaleza, sobre una matriz o resina cargada con iones de signo contrario. El intercambio puede ser entre aniones o entre cationes, dando nombre a las resinas que promueven uno y otro proceso. Las fuerzas que actúan son de tipo electrostático y, por tanto, la preferencia de la resina por el contraión (aquel ión de la misma naturaleza, signo, que el inicial que rodeaba las cargas de la matriz), será la base para la separación diferencial.Los dos tipos de resinas pueden ser débiles o fuertes dependiendo del equilibrio de disociación de los grupos unidos covalentemente a las mismas, como se muestra en la tabla.
FUERTES DÉBILES
CATIÓNICOS SO3* H+ COO* H+
ANIÓNICOS +NH3*(CH3) X* NH3 X*
Iones a intercambiar. Fijado covalentemente
La gran utilidad de esta técnica es el poder separar compuestos cargados selectivamente, dependiendo de la naturaleza de los mismos en distintas condiciones (pH, fuerza ionica, tamaño, posición de los grupos cargados, temperatura).La cromatografía de intercambio ionico se realiza en dos partes: Retención estable de los componentes a separar por parte de la resina. Hay que añadir la muestra a la columna, de modo que la muestra presente más o menos las mismas probabilidades de quedar retenida en la columna.

Cambio selectivo de las condiciones del medio. Con lo que eluirán los componentes de forma diferencial.1.1.Separación de la muestraUna de las mayores utilizaciones que se le dan e este tipo de cromatografía es la separación de aminoácidos. Esta separación se basa en las propiedades de éstas moléculas, sobre todo en que son anfipáticos, lo que implica que presentan diferente carga a valores de pH diferentes.Los tres tipos de aminoácidos son los reflejados arriba, de los cuales exponemos sus equilibrios de disociación, haciendo hincapié en la carga que flanquea a cada uno de ellos. En este tipo de cromatografía utilizaremos el equilibrio entre la forma cargada +1 y la neutra o zwitterion 0, de modo que en el paso que definimos antes como 1 todos estarían cargados positivamente, y por lo tanto a un pH inferior al menor de los puntos isoeléctricos (pI) de los aminoácidos de la muestra. Recordemos que el pI es aquel pH para el cual la molécula (aminoácido o proteína) se encuentra en toda su concentración con carga neta 0. Al estar en un principio cargados positivamente, todos los componentes de la muestra quedarán retenidos por la resina (utilizaremos una resina intercambiadora de cationes). En el paso 2 iremos variando el pH, de modo, que se valla acercando a los pI de los aminoácidos, y como éstos son distintos irán eluyendo a diferentes pH, lo que implica que podremos recogerlos en distintas fracciones.En éste experimento contamos con una muestra a separar compuesta por un azúcar y dos aminoácidos con pI distintos. Las variaciones del medio, las realizaremos con tampones citrato a distinto pH y finalmente con hidróxido sódico (NaOH). Se irán recogiendo fracciones y analizando para localizar, en primer lugar la salida del azúcar, con carga 0 y, por tanto, no retenido por la resina en el paso 1. Para ello utilizaremos el ensayo con licor de Fehling. Para la detección de los aminoácidos a distintos pH utilizaremos la ninhidrina.MaterialResina Dowex 50Tampón citrato 50 mM, pH = 3Tampón citrato 50 mM. pH = 5Solución de ninhidrina (0,2 % p/v de ninhidrina en 95 % de etanol)Papel indicador de pHNaOH 2MHCl 2MMezcla problemaMezclar 0,5 ml de glucosa 4 %, 1ml de lisina 2% y 1 ml de aspártico 2%. Todas estas sustancias se disuelven inicialmente en HCl 0,1 M. Añadir 2,5 ml de tampón citrato 50 mM (pH = 3), con lo que finalmente tendremos un volumen de 5 ml. Utilizar 0,25 ml de la muestra por columna.Preparación de la columna Pesar 7 g de resina Dowex 50 y disolverla en 25 ml de ClH 2M. agitar y dejar decantar. Retirar la fase acuosa superior y lavar la resina con 25 ml de H2O(destilada), tantas veces como sea necesario para que el pH sea igual al del agua (* 6). Obtuvimos unas medidas de:Lavados 1er 2º 3er 4º
pH medido * 1 * 2 4 - 5 * 6
Una vez con el pH del agua añadir 25 ml de NaOH 2M, agitar y dejar decantar. Retirar la fase acuosa superior y añadir agua destilada igual que antes, hasta que el pH vuelva a ser el del agua. Las medidas obtenidas fueron:Lavados 1er 2º 3er 4º 5º
pH medido * 10 * 9 * 8 * 7 - 8 * 6
Finalmente resuspender la resina en 25 ml de tampón citrato 50 mM (pH = 3).Una vez preparada la resina (se trataba de limpiarla de posibles contaminantes de anteriores experimentos y de calibrarla), se añade en una columna de vidrio, en la cual habíamos añadido un tapón de lana de vidrio en el fondo, para evitar la elución de la resina. El vertido de la resina en la columna implica que esta se encuentra con la salida cerrada por la presión de la pinza sobre la goma, y éste debe de hacerse con cuidado. En el caso de necesitar eliminar tampón para terminar de echar la resina, se abre la pinza, dejando que eluya el tampón, teniendo en cuenta que la resina nunca debe estar seca. Cerrar la salida de la columna cuando quede aproximadamente 1 cm de tampón sobre el frente de empaquetado de la resina.Separación de los componentes de la muestra.

Del mililitro de muestra que tenemos (compuesta de glucosa, aspártico y lisina), cogemos 250 l y los añadimos con cuidado en la columna, donde previamente hemos dejado con 1 mm de tampón sobre la resina, dejamos que entre y con mucho cuidado vamos añadiendo tampón citrato pH 3 para que no se seque la resina.Una vez añadida la muestra debemos recoger las fracciones, cinco exactamente, que serán de unos 2ml. Cuando tengamos las cinco fracciones, procederemos al análisis de las mismas. Ya que el único componente que no tiene carga es el azúcar (Glucosa), no quedará retenido al no entrar en el intercambio de cationes, donde sí estarán los aminoácidos.Detección de la glucosaLa detección se realiza mediante el licor de Fehling. Añadir a los 2 ml de fracción 2 ml de solución A. Agitar en el vortex. Añadir 2 ml de solución B. Agitar en el vortex. Poner en un baño a 80 ºC y esperar 10 minutos.Se espera ver un precipitado rojo ladrillo en aquella fracción que contenga la glucosa. Si no aparece en las 5 fracciones habrá que seguir eluyendo hasta que aparezca. Hay que realizar un control, y para ello cogeremos 250 l y les añadiremos los 2ml de solución A y de la B, al igual que las fracciones dejaremos que reaccione en el baño a 80 ºC.Principio del licor de Fehling para la detección de azúcares reductoresMuchos reactivos oxidan el grupo aldehído (reductor) de los azúcares. En esto se basa la reacción o prueba de Fehling para distinguir los azúcares reductores.
El tartrato, al unirse al cobre formando un complejo soluble, impide la forma de hidróxido cúprico insoluble que tendría lugar si existiese cobre libre en la solución. La oxidación de la glucosa por este método es más intensa que en el estado de ácido glucónico debido al aporte de cantidades apreciables de Cu2O a partir de los grupos aldehído disponibles. Como criterio de posibilidad de la reacción se utiliza la formación de óxido cuproso rojo insoluble. La prueba de Fehling no es específica. Otras sustancias dan reacción positiva son: fenoles, aminofenoles, benzoína, ácido úrico, catecol, ácido fórmico, hidrazobenceno, fenilhidracina, pirogalol y resorcinol.ComposiciónSolución A: sulfato de cobre Solución B: tartrato sódico-potásico, hidróxido sódico.

Resultado
Control F1 F2 F3 F4 F5
+ +
Detección de los aminoácidosUna vez eluída la glucosa, hay que entrar en la fase 2, separar los aminoácidos selectivamente de la resina. Para ello añadimos ahora tampón citrato a pH 5, abriendo la pinza y recogiendo fracciones. Al añadir el tampón se forma en la columna un gradiente de pH que hará que eluya aquel de los dos aminoácidos que nos quedan retenidos que tenga el pI más cercano a 3 y menor que 5.aá Abr. -R pK1 pK2 pKR pI
Lisina. Lys, K -CH2-CH2-CH2-CH2-+NH3 2,18 8,95 10,53 2,77
Aspártico. Asp, D -CH2-COO- 1,88 9,60 3,65 9,74
En esta tabla se reflejan los puntos isoeléctricos de los dos aminoácidos, y en ellos resalta el del aspártico, con 2,77, ¿no podría ser que hubiera eluído a pH 3?. Teóricamente tendría que haberlo hecho, y posiblemente algo halla salido, pero hay que tener en cuenta que se trata de un equilibrio y, por tanto, para que el aspártico tenga carga neta 0 en gran concentración, el pH de la solución debe ser algo mayor de 3, algo que demostramos haciéndolo eluir cuando añadimos citrato a pH 5, eso si en las primeras fracciones.Base de la reacción de detección de aminoácidos con ninhidrinaEl método se basa en la reacción entre la ninhidrina y el grupo -amino de aminoácidos, péptidos y proteínas, dando un compuesto coloreado azul o azul - violeta.Calentar en presencia de ninhidrina produce la desaminación oxidatíva de los grupos amino en , lo que implica la reducción asociada de la molécula de ninhidrina.
La ninhidrina reducida reacciona con el amonio y con otra ninhidrina oxidada para dar ligar a un compuesto coloreado llamado azul - violeta de Ruhermann.La prolina y la oxiprolina reaccionan con la ninhidrina, pero dan un positivo de color amarillo. La reacción de la ninhidrina puede dar positivo con ciertas aminas, aminas ácidas y algunos otros compuestos.Una vez eluídas tres fracciones se añaden a 250 l de cada una otros 250 de citrato 5, se agita en el vortex y se añaden 500 l de ninhidrina. Se dejan los tubos en un baño a 80 ºC durante 10 minutos.IncidenciasSegún las indicaciones del guión, al comenzar a añadir el citrato a pH 5, no recogimos fracciones, sino que medimos el pH de lo que eluía sin recogerlo, esperando a que fuera de 5 para comenzar a recogerlo. Evidentemente, y como ya hemos dicho el aspártico tiene un pI de 2,77 y, por tanto, coge carga 0 en cuanto el pH sube algo por encima de 3, lo que ocurre nada más añadir el citrato a pH 5 porque se forma un gradiente de pH en la columna.El resultado fue que no pudimos detectar de forma fiable la salida del aspártico en las fracciones, pero si en el líquido que se recogió en un vaso de precipitados y sobre el que efectuamos la detección. El resultado fue un intenso azul-púrpura, que indicaba la presencia

de “algo” en gran cantidad, pero que la escasa limpieza del recipiente no permite afirmar que fuera aspártico.Según los resultados de otras parejas y de acuerdo con la teoría expuesta, el aspártico eluyó en la primera fracción recogida, y por supuesto el control dio positivo.Una vez sabido que ya había eluído al aspártico hay que continuar con la fase 2, lo que implica variar de nuevo el pH de la columna, esta vez lo haremos con sosa, ya que el pI de la lisina es de 9,74. Al igual que con los demás tampones, vamos añadiendo el NaOH a la vez que recogemos las fracciones y con cuidado no se seque la resina.Recogidas 4 fracciones se efectúa la detección de aminoácidos mediante el método de la ninhidrina, con las mismas cantidades de componentes que con el aspártico, salvo que ahora hay que medir el pH con un papel medidor para asegurarnos que no supera las 8 unidades, ya que la ninhidrina se desactiva por encima de ese pH. En nuestro caso ninguna salió con un pH superior.Resultados
Control F1 F2 F3 F4
+
Algo verdoso
Marrón muy clarito
Marrón muy oscuro
Marrón claro
Como se ve en la gráfica, la lisina no eluye en una sola fracción, pero si mayoritariamente en la F3. La razón es similar a la explicada para el aspártico, consistente en la formación del gradiente de pH en la columna, que implica el paso de pH 5 a pH 10, por eso tarda en eluir.Limpiado de la columnaPara ello hacemos eluir secuencialmente por la misma 5 ml de HCl, de H2O, de NaOH y de nuevo de H2O, con el fin de eliminar posibles restos y dejar un pH lo más cercano a 7.2. Cromatografía de filtración molecular.En este tipo de cromatografía los componentes de una muestra se separan de acuerdo a su tamaño y forma molecular. La base de esta cromatografía es el “reparto” que sufren las moléculas entre un solvente y una fase estacionaria contenida en una matriz de porosidad definida (esto es lo que dice el guión, pero en teoría dimos que era de exclusión). La columna se prepara con partículas de una sustancia inerte que contienen poros pequeños. Al pasar una disolución con moléculas de distintas dimensiones a través de la columna, las moléculas con un tamaño mayor que el de los poros se moverán sólo en el espacio que queda entre las partículas, y, por tanto, no sufrirán retraso en su recorrido a través del material de la columna. Sin embargo, las moléculas con un tamaño menor que el de los poros difunden hacia el interior y el exterior de las partículas con una probabilidad que aumenta a medida que disminuye el tamaño molecular. Por lo tanto, las moléculas son eluídas de la columna por orden de tamaños decrecientes.Para un determinado tipo de columna o matriz se puede calcular un parámetro Kav de elución de una determinada molécula. Este parámetro es característico de cada molécula y es diferente para cada columna. La representación gráfica de Kav con respecto al logaritmo del peso molecular da una línea recta. Este parámetro se define como:Kav = (Ve-Vo)/(Vt-Vo) donde,Ve = volumen de disolvente necesario para eluir las moléculas de interés.Vo= volumen vacío o volumen necesario para eluir una molécula que nunca entra en la fase estacionaria.Vt = volumen total de columna.Soportes y partículas Las partículas empleadas requieren:Estabilidad química: Para que no varíen las condiciones durante la cromatografía.Bajo contenido en grupos iónicos: Para que no entren en juego procesos de intercambio.Gran uniformidad de poro y tamaño: No debe haber heterogeneidad para mantener el principio de separación.Que podamos elegir una amplia galería de tamaños de esferas y poros.Los geles o soportes más utilizados son:Columnas que derivan del dextrano SEPHADEX: Se trata de moléculas de glucosa unidas por epicloridrina. Presentan un rango de separación que oscila entre los 700 y los 800.000, aunque este depende del tipo. Cada tipo de resina se nombra como G-X, siendo X los mililitros de agua que es capaz de absorber 1 gr de resina seca. Las resinas Sephadex son de gran resistencia y estabilidad, presentando flujos lentos.Derivados de agarosa Shepharosas: En principio no se diferencia mucho de las derivadas del dextrano, salvo en el rango de tamaños a separar, ahora superior a los 800.000, (mayor que los virus pequeños) y, por tanto, completa a las anteriores. El único problema es que son menos resistentes, ya que no soportan temperaturas superiores a los 30 ºC ni pH fuera del intervalo 4 - 10.Poliacrilamidas Biogel P: Este soporte presenta como gran ventaja el ampliar muchísimo el rango de pH, debido a la gran estabilidad de la acrilamida que ya comentamos en la electroforesis, pudiendo soportar pH del orden de 1 o 10 unidades.Vidrio: se trata de esferas de vidrio con poros. Su mayor ventaja es la mayor resistencia mecánica, seguido de su evidente inercia química, la resistencia a altas temperaturas (100 ºC), que no presentan hinchamiento, y por último que soporta tratamientos ácidos.Factores que condicionan la resolución Tamaño de poro. Tamaño de partícula.

Flujo de elución.Material de la práctica.Tampón PBS (0,1 M de tampón fosfato; 0,15 M de NaCl pH 7.2).Sephadex G-100.Azul dextrano (10 mg/ml). 2.106 DaRojo fenol (10 mg/ml). 345 DaLana de vidrio.Columnas de vidrio.2.1.Preparación de la columna de Sephadex G-100.Nombre comercial de una matriz comercial constituida por polímeros de dextrano entrecruzados por epiclorihidrina. El grado de entrecruzamiento va a determinar el tamaño poro y éste va a determinar el rango de fraccionamiento de tamaños moleculares. Esta resina permite separar proteínas globulares cuyo peso molecular esté entre 4.000 y 150.000 Da. Esto último quiere decir que moléculas mayores de 150.000 Da serán incapaces de entrar en el poro de la matriz.Preparación de la columna en la práctica. Previamente, a 1,5 gramos de resina sephadex G-100 se le añaden 50ml de tampón PBS y se deja esponjar 72 horas. Paso realizado por los profesores de prácticas.El esquema de la columna es:Los pasos que se siguen para preparar la columna para la filtración molecular son: Se añaden a la columna, con la pinza cerrada, 25 ml de PBS y se hace una señal de la altura a la que llegan en la columna esos 25 ml. Esta marca será la altura hasta la que debe llegar la resina en la columna. Poner lana de vidrio en la parte de debajo de la columna, para que, al añadir la resina, esta última quede retenida por la lana. El sephadex suspendido en PBS se echa en la columna, al principio en grandes cantidades y después poco a poco con pipeta Pasteur, hasta que la resina llegue a la marca anteriormente mencionada. Hay que tener extremo cuidado de que la columna no pierda demasiado PBS y se seque, de modo que siempre tiene que haber algo de PBS por encima del frente de compactación de la resina. También hay que tener cuidado al echar la suspensión de la resina para que no se formen burbujas en la columna, ya que estas burbujas dificultarían la elución de la muestra a través de la columna.Lo que sucede al echar la resina en suspensión y abrir la pinza es que el PBS pasa a través de la lana de vidrio y se pierde, mientras que la resina queda retenida por la lana y se va empaquetando poco a poco.Terminado el empaquetamiento, siempre poniendo más PBS por encima del frente del empaquetamiento para que la resina no se seque, se añaden unos ml de PBS por encima de la marca que hicimos. Después se pone parafilm y se deja hasta el día siguiente.Incidencias.Rotura de una pipeta Pasteur. Fragmento de unos 4 ó 5 cm que quedó incluido en la resina, por debajo de la mitad de la columna.A la altura de ese fragmento de la pipeta hay unas burbujas no demasiado grandes y no en demasiada cantidad.En la parte superior de la columna se observan algunas burbujas pero muy separadas y pequeñas. Creemos que no estaban en un número suficiente ni tenían un tamaño suficiente para que la separación de proteínas se viera muy perturbada.Suciedades y partículas, (pelillos o fibras), podrían haber entrado en la columna por falta de limpieza en el material del que disponíamos.2.2. Equilibrado de la columna.Después del empaquetamiento tenemos que equilibrar la columna. Calibrar la columna es definir los valores de Vo y Vt.Procedimiento. Abrimos la llave para que salga el PBS sobrante hasta que queden 1 ó 2 mm por encima del frente de la resina. Se procede a la colocación en la superficie de la resina de 200 l de una mezcla con 40 gr de azul dextrano, cuyo peso molecular es 2 millones g/mol, y 20 gr de rojo fenol, cuyo peso molecular es de 345 g/mol. Se abre la pinza para que entre la muestra en la resina pero teniendo cuidado de que no se seque la resina, puesto que al eluir la muestra estamos eluyendo también el PBS. Tenemos que añadir PBS para que la resina no se seque, pero esperando a que hallan entrado los colorantes. Esta adición de PBS se debe realizar poco a poco, pues si echamos mucho PBS o lo echamos a destiempo podemos diluir la muestra de colorantes, de modo que no entre todo a la vez en la resina y, como consecuencia, la elución no sería correcta. Una vez que se abre la pinza y la muestra ha entrado en la resina comenzamos a recoger fracciones de 1,5 ml en tubos de ensayo hasta que salga la fracción con el rojo fenol. En el caso de que el rojo salga en varias fracciones se coje la que tenga más cantidad de rojo. Esto último fue lo que nos pasó. Después se cuenta el número de fracciones hasta que salga el azul, que debería salir en una sola fracción (que fue lo que nos pasó), y se multiplica ese número de fracción por 1,5, obteniéndose así el volumen de elución del azul dextrano, que tomaremos como el volumen inicial, Vo, el que nos marcará el volumen a partir del cual podremos obtener proteínas al eluir la muestra problema. La misma operación se hace con el rojo fenol, con la única diferencia de que el rojo se recoge en un volumen de elución mucho mayor que el del azul, puesto que al ser el rojo una molécula mucho menor pasa por la columna más despacio. Al multiplicar por 1,5 el número de la fracción con más cantidad de rojo fenol tenemos el volumen de elución del rojo, que nos dice el volumen total, Vt, que nos marcará el volumen hasta el que las proteínas problema pueden eluir, por encima del cual no deberían eluir proteínas.

Los colorantes nos sirven para delimitar el intervalo de volúmenes entre los que deben eluir nuestras proteínas, puesto que sus pesos moleculares se encuentran entre los del azul y los del rojo, y la columna separa las proteínas en función de su peso molecular.Aunque cogimos 2 ml por encima y por debajo de esos Vo y Vt para estar seguros de que recogemos las proteínas, esto no debería ser necesario si la elución es correcta. Resultados del equilibrado de la columna.VeA= Vo= 6 x 1,5 ml = 9 ml, pues recogimos el azul en la fracción 6.VbR= Vt= 16 x 1,5 ml = 24 ml, pues recogimos el rojo en la fracción 16.Según estos datos tendremos que recoger 15+2+2 ml de nuestra muestra para recoger las proteínas.2.3.Separación de proteínas.Procedimiento. Después de recoger el rojo, hay que lavar con PBS la columna para eliminar el resto de rojo y limpiar la columna. La teoría decía que teníamos que lavar con 20 ml de PBS, pero no lo hicimos así, simplemente lavamos con lo que íbamos echando para eluir el rojo y luego añadimos sólo algunos ml de PBS (por indicación del profesor). La metodología para la separación de proteínas es la misma que para el equilibrado. Dejamos eluir el PBS que quedaba después del equilibrado hasta que queden 1 ó 2 mm por encima del frente de la resina. Después aplicamos 200 l de la muestra problema con proteínas. Abrimos la pinza y dejamos que entre la muestra en la resina sin que esta última se seque y sin echar demasiado PBS que diluya mucho la muestra. De esta muestra hay que recoger fracciones de 1 ml en tubos ependorf, estos últimos debidamente marcados con su número de fracción y pareja de prácticas. La columna separará las proteínas en función de su tamaño, de modo que las de mayor tamaño eluirán antes y estarán en las primeras fracciones, y las menores estarán en las últimas. Esto último siempre dentro del intervalo Vo-Vt.Según los volúmenes calculados en el equilibrado nosotros teníamos que recoger la primera fracción 7 ml después de aplicar la muestra en la columna, con lo que recogimos 19 fracciones de 1 ml en el intervalo de volúmenes 7-26 ml.Una vez que recogimos estas 19 fracciones, los ependorf se colocaron en una gradilla y se llevaron las fracciones al frigorífico. El dejar las proteínas a 4ºC se realiza para evitar que las proteínas se desnaturalicen y pierdan su actividad, ya que la filtración molecular separa las proteínas en su estado nativo. El que las proteínas estén en su estado nativo y que conserven su funcionalidad nos sirve para poder identificar en las fracciones determinadas actividades enzimáticas.Con los volúmenes de elución de las fracciones más características, y con los valores de Vo y Vt podemos hallar los valores Kav de las principales fracciones. Con los valores log Pm del azul y el rojo y sus Kav, 0 y 1 respectivamente, hacemos una recta en la que interpolar los valores de log Pm de las proteínas de nuestras fracciones. Las fracciones características fueron escogidas después de realizar todas las pruebas. Las gráficas están en papel milimetrado al final de los apartados 2 y 3.
Valores de Kav
A 0 F6 0,2 F12 0,6
F3 0 F7 0,27 F13 0,67
F4 0,066 F10 0,48 F14 0,733
F5 0,133 F11 0,533 R 1
Incidencias.Quedo algo de rojo fenol entre la lana de vidrio y eluyó junto con las primeras fracciones de la muestra. Como consecuencia de esto en las dos primeras fracciones recogidas había algo de rojo, pero este colorante al no tener naturaleza proteica no interfiere o falsea los datos de la absorbancia obtenida en la prueba de Bradford.La elución del rojo y del azul se hizo bien, pero el rojo al llegar a la altura del trozo de Pasteur rota difundió un poco y no iba tan junto o compacto que al principio, en cambio el azul no sufrió ninguna difusión. No sabemos si la punta pudo influir negativamente en la separación de las proteínas por la columna de resina. 2.4.Regeneración de columna.Paso realizado por los profesores, consistente en vaciar la columna con ayuda de PBS, para que la resina no se seque, para ser reutilizada. Después se quita la lana de vidrio y se lava la columna. 3. Determinación de proteínas y actividades enzimáticas.Método de BradfordEste es uno de los métodos con mayor sensibilidad para la determinación de proteínas.Una de las cuestiones que se nos pedían para este punto es la de encontrar el fundamento de este método, pero a pesar de disponer del artículo original de Bradford y del libro de instrucciones del reactivo de Bradford comercializado por Bio-Rad, nos ha sido imposible saber cuál es el tipo de interacción que le permite al azul brillante de Coomasie unirse a las proteínas, lo cual sí se sabe en los otros métodos de este tipo conocidos (Lowry, Biuret). Está claro que al unirse a la proteína, este reactivo, sufre un cambio en su máximo de absorbancia que es el que medimos con el espectrofotómetro, y que es proporcional, por tanto, a la cantidad de proteína. Así, cuánto más proteína halla en la disolución más cambio en la absorbancia habrá, es decir, más absorción habrá en el nuevo máximo que es 595 nm. El cambio es de 465 a 595 nm.

Según lo encontrado en la bibliografía utilizada, podemos decir de este método que:Es un ensayo de unión proteína colorante basado en el diferencial cambio de color de un colorante en respuesta a varias concentraciones de proteína. Este es un método de determinación de proteínas que implica la unión de Azul Brillante de Coomasie G-250 con las proteínas. Esta unión del colorante con las proteínas provoca un cambio en el máximo de absorción del colorante desde 465 a 595 nm. Por lo tanto, se basa en la observación de que el Azul Brillante de Coomasie G-250 existe en dos formas con colores diferentes, rojo y azul. La forma roja se convierte en azul cuando se une el colorante a la proteína. El complejo colorante-proteína tiene un alto coeficiente de extinción lo cual es imprescindible aumentar la sensibilidad en la medición de proteínas. La unión colorante-proteína es un proceso muy rápido (2min) y el complejo permanece estable durante un tiempo relativamente largo, una hora. Haciendo de este un proceso muy rápido, y que no requiere un tiempo crítico para el ensayo. Se pueden utilizar un gran número de muestras (muy reproducible) y es adaptable a la automatización.Es un ensayo que elimina casi todos los problemas que había anteriormente con es tipo de ensayos.Las interferencias o no existen o son mínimas por cationes tanto sodio como potasio y carbohidratos como la sacarosa. Otras sustancias que interfieren en el ensayo son los agentes fuertemente alcalinos (cuestión fácilmente solventable con la utilización de tampones). Los únicos componentes encontrados que dan una excesiva interferencia en el color del ensayo, son grandes cantidades de detergentes como el SDS, Triton X-100, etc.Preparación del colorante. Azul Brillante de Coomasie G-250 (100mg) es disuelto en 50 ml de etanol 95%. A esta disolución se le añade 100ml de ácido fosfórico 85% p/v. La solución resultante se diluye llevándola hasta un litro de volumen final. La concentración en la disolución final es 0,01% p/v de azul, 4,7% p/v de etanol y 8,5 % p/v de ácido fosfórico.Una dificultad observada en la realización del ensayo es la tendencia del complejo colorante-proteína a unirse a las cubetas. Esto resulta en una cubeta coloreada de azul: la cantidad de unión es poco importante en lo que se refiere al resultado del ensayo. Se suele lavar el tubo o cubeta con agua destilada entre medidas de diferentes muestras. Este azul se puede quitar y las cubetas se reutilizan sin problemas. La sensibilidad del ensayo depende de la naturaleza de la proteína, y en muestras complejas (aunque no es nuestro caso), en las cuales las proteínas se diferencian ampliamente en su respuesta al ensayo, puede ser difícilmente cuantificable. Sin embargo, recientes modificaciones en el ensayo han mejorado su eficacia en estas aplicaciones. Se aplica una cierta cantidad de SDS, el cual por si mismo causa una falta de desarrollo de color, es añadido a la muestra o al colorante. El SDS iguala la reactividad de las distintas proteínas, así que el ensayo puede ser utilizado para mezclas de proteínas. Esta igualdad produce tanto una disminución de la reactividad de proteínas con alta respuesta, como la albúmina, como un incremento en la reactividad en las proteínas con baja respuesta.Aplicaciones: Cromatografía, electroforesis, soluciones de proteínas complejas, química clínica, adaptable a la automatización.Material de la práctica.
Reactivo de BradfordPBSAlbúmina bovina.Fracciones de proteína.Espectrofotómetro.Tubos de espectrofotómetro.
Procedimiento. Preparar las muestras que tenemos que medir para la determinación de proteínas. Tenemos que tomar alícuotas de 500 l de cada fracción.(5). Añadir a esas alícuotas 3,5 ml de PBS. Añadir 1ml de reactivo de Bradford. (6). Después de esto tenemos en cada tubo 5 ml. Esto último es importante, ya que todos los tubos deben tener la misma concentración para que la medida de la concentración de proteína sea correcta. Después ésta disolución se mezcla bien en el vortex. Preparar una curva patrón en la que conocemos las cantidades de proteína que ponemos y medimos su absorbancia. Esta nos servirá para interpolar, a partir de la curva, los valores de concentración de proteína en las fracciones, sabiendo sus valores de absorbancia. La curva se prepara a partir de una disolución stock que tiene 100 gr/ml, de modo que preparamos seis tubos con cantidades crecientes de albúmina(uno sin nada que utilizaremos como nuestro cero), y los llevamos a 4 ml con PBS y después les añadimos 1 ml de reactivo Bradford. Es muy importante añadir el Bradford a la vez en todos los tubos, tanto en los tubos de la curva patrón como en los de las fracciones. Así todos los tubos se disparan a la vez y la medición de la absorbancia es fiable y correcta. Este hecho es una consecuencia de la cinética característica de esta reacción, ya que cuanto más tiempo transcurre más intenso es el color de la disolución. Por eso, se preparan todos los tubos y después se añade a todos los tubos el reactivo. Medir los valores de absorbancia de los distintos tubos. Entre medida y medida al cambiar de fracción o muestra hay que lavar el tubo de espectrofotómetro con agua destilada. Esto no hay que hacerlo al medir la recta patrón puesto que vamos en orden creciente

de concentración de proteínas. Representar con los datos de D.O.: 1. D.O. frente nº fracción; 2. D.O. frente [prot]; 3. [prot] frente nº fracción. Resultados del patrón obtenidos a partir de la mesa 3D.Tubo Concentración Albúmina ABS595
1 0 0
2 20 g/ml, 200l de stock 0,336
3 40 g/ml, 400l de stock 0,542
4 60g/ml, 600l de stock 0,662
5 80 g/ml, 800 l de stock 0,701
6 100g/ml, 1000l de stock 1,02
Resultados de absorbancia de las muestras.
Tubo Absorbancia Tubo Absorbancia
1 -0,123 11 0,370
2 -0,212 12 0,313
3 0.783 13 0,272
4 -0,192 14 0,178
5 -0,182 15 -0,014
6 -0,197 16 -0,062
7 0,82 17 -0,125
8 -0,011 18 -0,158
9 0,074 19 -0,183
10 0,280
Incidencias.Problemas con los espectrofotómetros, pues algunos no funcionaban (incomprensiblemente) y otros daban medidas extrañas e ilógicas. Además de la falta de aparatos disponibles. Al final medimos en uno que medía perfectamente, aparentemente.Fallo al preparar las muestras de las fracciones 4,5, y 6. Este fallo nos impidió ver si tenían proteína esas fracciones y complicó mucho la realización del cuaderno. Deducimos que debían tener proteínas a partir de la gráfica D.O. frente al número de fracción, pues estas fracciones quedaban dentro del primer pico que aparecía en esta gráfica. Después comprobamos que si tenían proteínas al correr el gel de acrilamida.Entendimos mal la aclaración del profesor, en la que decía que había que diluir 1/10 las muestras de la curva patrón si la segunda (la primera después del blanco) daba un valor de absorbancia de 1,3 o 1,4, aproximadamente. Pero nosotros al entender mal, diluimos las muestras patrón, 1/10, al obtener en la tercera muestra 1,2. Después de esta dilución, las medidas resultaron totalmente incoherentes. Por la falta de tiempo y por los nervios, no fuimos capaces de resolver este problema y decidimos tomar, como nuestra, la recta patrón de la mesa 3D, y medir con su mismo aparato y con el mismo tubo.3.2. Determinación de la actividad catalasa.Un método rápido para la determinación de la proteína catalasa es la observación de su actividad por la formación de burbujas en una solución que contiene H2O2. Procedimiento. Preparar un tubo con 1 ml de una solución constituida por 1 ml de PBS + 10% de H2O2. Añadir 100l de las fracciones que, después de la realización del método de Bradford, sepamos que contienen un alto contenido en proteínas. Aquellas fracciones que contengan catalasa producirán un burbujeo instantáneo en la solución.Resultados de la prueba de la catalasa.Tubo Actividad Catalasa Tubo Actividad catalasa
Muestra + + + + + + + + + + 10 -
3 + + + + + + + + 11 -
4 + + + + + + + + 12 -
5 + + + + 13 -

6 + + 14 -
7 +
Según los resultados obtenidos en la prueba tenemos que la catalasa se encuentra en las primeras fracciones sobre todo en la 3 y en la 4 que se corresponde con el mililitro 9 y 10. Esto nos dice que la catalasa es una proteína de gran peso molecular al eluir pronto en la columna. Con la columna separamos las proteínas en fracciones según su peso y después con experimentos como este y como el siguiente de la lisozima, identificamos la presencia de determinadas proteínas en esas fracciones.3.3. Determinación de la presencia de lisozima.Un método rápido para la determinación de la actividad lisozima es la medida de la disminución de turbidez de una suspensión de pared bacteriana al agregar dicha proteína. Al incubar la suspensión con lisozima la pared celular se hidroliza originando fragmentos pequeños, reduciéndose la dispersión de la luz lo que se manifiesta en una reducción de la absorbancia a la longitud de onda de 450 nm. Así, la actividad enzimática será, por lo tanto, proporcional a la disminución en absorbancia.En nuestro caso concreto se va a analizar la presencia de esta enzima en las mismas fracciones en las que realizamos el ensayo de la catalasa.Procedimiento. Colocar 100l de la fracción sobre un tubo que contenga 5 ml de suspensión de pared bacteriana, mezclar e incubar a temperatura ambiente durante 15 min. Como tubo “blanco” tomamos uno solamente con la suspensión que nos dará el máximo de absorbancia. Para el ajuste a 0 del densiómetro o espectrofotómetro se utilizará el tubo en el que se colocaron 100l de la muestra inicial, que lógicamente tendrá un máximo de actividad lisozima (sí es que esta proteína existiera en la muestra). Medir DO450.La suspensión de pared bacteriana se prepara a partir de la pared del microorganismo Micrococcus lysodeikticus (preparado comercial). 20 mg de este preparado se mezclan con 100 ml de tampón fosfato (100 mM; pH 6.2). (A nosotros se nos dio la suspensión ya preparada).Los resultados de este ensayo.Tubo Absorbancia Actividad Tubo Absorbancia Actividad
Muestra 0,00 100% Blanco 0,38 0%
3 0,38 0% 10 0,285 25%
4 0,378 0,52% 11 0,28 26,3%
5 0,373 1,8% 12 0,242 36,3%
6 0,375 1,3% 13 0,28 26,3%
7 0,377 0,78% 14 0,307 19,21%
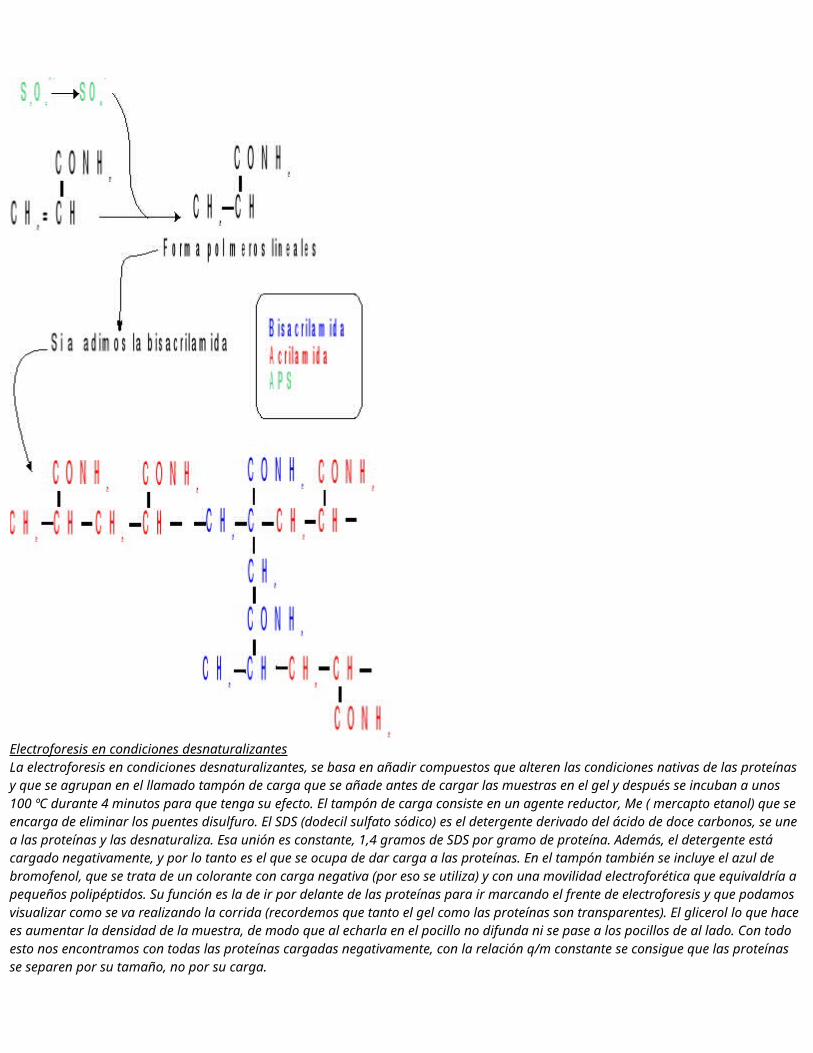
Para calcular la actividad de cada fracción tenemos que utilizar una regla de tres inversa.Se puede ver como en este caso la actividad lisozima se encuentra en las fracciones 10, 11, 12, 13, 14 que se corresponden con los mililitros 16, 17, 18, 19, 20. Esta es, por tanto, una proteína de menor tamaño que la catalasa. El hecho de identificar en estos dos grupos de fracciones dos proteínas, mediante los ensayos de actividad, no quiere decir que no pueda haber más proteínas en esas fracciones, pues al separar, durante la cromatografía, las proteínas en función del peso molecular, puede haber en esas fracciones otras proteínas a parte de las ya mencionadas, con pesos parecidos. Los dos grupos de fracciones se corresponden, como ya vimos, con dos picos en la gráfica de D.O. frente a número de fracción, que son las fracciones donde hay mayor concentración de proteínas y con las que tenemos que trabajar para ver cuántas y cuáles son las proteínas presentes en ellas.Las gráficas donde quedan reflejados los resultados de estos dos apartados se encuentran a continuación, y las conclusiones sobre ellas se pueden ver la parte final del cuaderno, sección 7 de conclusiones.4. Electroforesis en geles de Poliacrilamida-SDSEl principio básico de la electroforesis es el movimiento que experimenta una partícula cargada en un determinado medio cuando se ve sometida a la acción de un campo eléctrico. Dada una partícula de carga neta Q sometida a un campo eléctrico de intensidad E, se moverá por acción de una fuerza igual a:La técnica se utiliza para separar proteínas y ácidos nucleicos, y uno de los medios soporte más utilizados es el gel de poliaclilamida.Geles de acrilamidaPermite gran cantidad de tratamientosPermiten regular el tamaño de poro.Muy estables.Permite separaciones más rápidas.Casi no presenta endósmosis.Son transparentes, con lo que permiten hacer estudios de densidometría.Para realizar estos geles se parte de cuatro componentes líquidos:

- AcrilamidaCH2=CHCONH2- Agente entrecruzante: bisacrilamidaCH2=CHCONH2CH2 CONH2CH=CH2TEMED (Tetrametilen, etilen diamina)(CH3)2CHNHCH(CH3)2APS (Persulfato amónico)S2O8(NH4)2
Electroforesis en condiciones desnaturalizantesLa electroforesis en condiciones desnaturalizantes, se basa en añadir compuestos que alteren las condiciones nativas de las proteínas y que se agrupan en el llamado tampón de carga que se añade antes de cargar las muestras en el gel y después se incuban a unos 100 ºC durante 4 minutos para que tenga su efecto. El tampón de carga consiste en un agente reductor, Me ( mercapto etanol) que se encarga de eliminar los puentes disulfuro. El SDS (dodecil sulfato sódico) es el detergente derivado del ácido de doce carbonos, se une a las proteínas y las desnaturaliza. Esa unión es constante, 1,4 gramos de SDS por gramo de proteína. Además, el detergente está cargado negativamente, y por lo tanto es el que se ocupa de dar carga a las proteínas. En el tampón también se

incluye el azul de bromofenol, que se trata de un colorante con carga negativa (por eso se utiliza) y con una movilidad electroforética que equivaldría a pequeños polipéptidos. Su función es la de ir por delante de las proteínas para ir marcando el frente de electroforesis y que podamos visualizar como se va realizando la corrida (recordemos que tanto el gel como las proteínas son transparentes). El glicerol lo que hace es aumentar la densidad de la muestra, de modo que al echarla en el pocillo no difunda ni se pase a los pocillos de al lado. Con todo esto nos encontramos con todas las proteínas cargadas negativamente, con la relación q/m constante se consigue que las proteínas se separen por su tamaño, no por su carga.
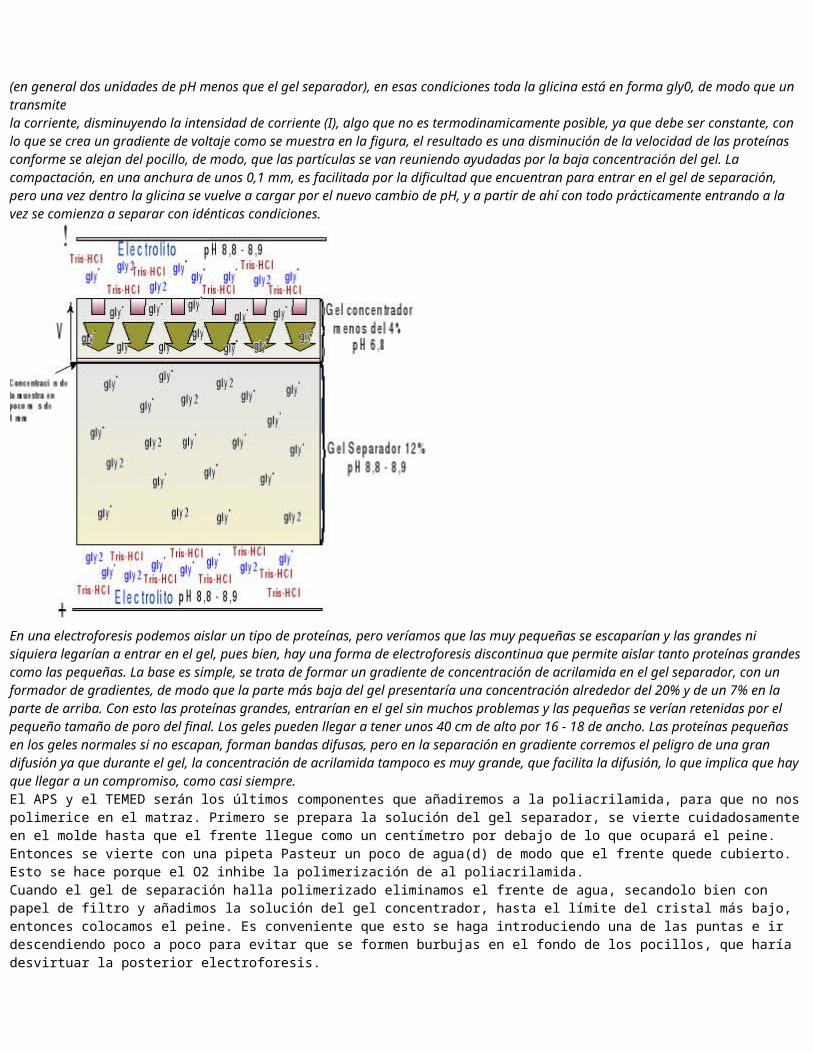
4.1. Determinación del peso molecular de proteínas mediante electroforesis en geles de poliacrilamidaDeterminación del peso molecular mediante electroforesis con SDSEste tipo de electroforesis permite separar entonces las proteínas por tamaño, lo que implica que podemos conocer la relación existente entre y el peso molecular de las moléculas, o lo que es lo mismo calcular el Pm de distintas subunidades de proteínas, ya que recordemos que en condiciones desnaturalizantes se eliminan los conjuntos de monómeros. La relación se consigue poniendo en el gel unos marcadores de Pm, y una vez corrido el gel medimos (como distancia al pocillo) y representamos la gráfica (log Pm / ).Una vez representados los marcadores, medimos de las distintas bandas y las hacemos corresponder un valor del Pm a través del antilogaritmo como se ve en la gráfica.Preparación del gel de poliacrilamidaStock de acrilamida 30 % (29,2 g de acrilamida: 0,8 g de bisacrilamida.Tris-HCl 1M. pH 6,8.Tris-HCl 1M. pH 8,8.SDS 10 %.Persulfato amónico (APS) 10 %TEMEDTampón de electroforesis (Tris 0,025 M; glicina 0,192 M; SDS 0,1%; pH 8,3).Tampón de carga 2x (Tris-HCl 0,125 M (pH 6,8; SDS 4 %; glicerol 20 %; 2--mercaptoetanol 10 %; EDTA 15 nM; azul de bromofenol 0,008 %.Limpiar los cristales con agua y etanol, colocar los separadores con algo de vaselina en la mitad que se orientará hacia fuera, montar los cristales y sellarlos bien con vaselina. Antes de echar la acrilamida comprobar que el molde no tiene escapes vertiendo un poco de agua. Haremos un gel discontinuo, con el gel separador y el de empaquetamiento.Electroforesis Discontinua Por esta causa se diseñó la electroforesis discontinua, con el fin de minimizar las diferencias de distancia en la salida para que la separación nos permita obtener la mayor resolución. El nombre le viene del propio método, que implica la polimerización de dos geles con diferente concentración de acrilamida y distintas condiciones de pH. El electrolito se compone de una mezcla de Tris-HCl y glicina, siendo el aminoácido la clave, ya que a pH de 8,8 - 8,9 se encuentra cargado al 15% en forma de gly", de modo que transmite la corriente, al entrar en el gel concentrador, el pH varía a 6,8 (en general dos unidades de pH menos que el gel separador), en esas condiciones toda la glicina está en forma gly0, de modo que un transmite

la corriente, disminuyendo la intensidad de corriente (I), algo que no es termodinamicamente posible, ya que debe ser constante, con lo que se crea un gradiente de voltaje como se muestra en la figura, el resultado es una disminución de la velocidad de las proteínas conforme se alejan del pocillo, de modo, que las partículas se van reuniendo ayudadas por la baja concentración del gel. La compactación, en una anchura de unos 0,1 mm, es facilitada por la dificultad que encuentran para entrar en el gel de separación, pero una vez dentro la glicina se vuelve a cargar por el nuevo cambio de pH, y a partir de ahí con todo prácticamente entrando a la vez se comienza a separar con idénticas condiciones.
En una electroforesis podemos aislar un tipo de proteínas, pero veríamos que las muy pequeñas se escaparían y las grandes ni siquiera legarían a entrar en el gel, pues bien, hay una forma de electroforesis discontinua que permite aislar tanto proteínas grandes como las pequeñas. La base es simple, se trata de formar un gradiente de concentración de acrilamida en el gel separador, con un formador de gradientes, de modo que la parte más baja del gel presentaría una concentración alrededor del 20% y de un 7% en la parte de arriba. Con esto las proteínas grandes, entrarían en el gel sin muchos problemas y las pequeñas se verían retenidas por el pequeño tamaño de poro del final. Los geles pueden llegar a tener unos 40 cm de alto por 16 - 18 de ancho. Las proteínas pequeñas en los geles normales si no escapan, forman bandas difusas, pero en la separación en gradiente corremos el peligro de una gran difusión ya que durante el gel, la concentración de acrilamida tampoco es muy grande, que facilita la difusión, lo que implica que hay que llegar a un compromiso, como casi siempre.El APS y el TEMED serán los últimos componentes que añadiremos a la poliacrilamida, para que no nos polimerice en el matraz. Primero se prepara la solución del gel separador, se vierte cuidadosamente en el molde hasta que el frente llegue como un centímetro por debajo de lo que ocupará el peine. Entonces se vierte con una pipeta Pasteur un poco de agua(d) de modo que el frente quede cubierto. Esto se hace porque el O2 inhibe la polimerización de al poliacrilamida.Cuando el gel de separación halla polimerizado eliminamos el frente de agua, secandolo bien con papel de filtro y añadimos la solución del gel concentrador, hasta el límite del cristal más bajo, entonces colocamos el peine. Es conveniente que esto se haga introduciendo una de las puntas e ir descendiendo poco a poco para evitar que se formen burbujas en el fondo de los pocillos, que haría desvirtuar la posterior electroforesis.Una vez polimerizado totalmente el gel, se retira el peine con cuidado de no romper el gel concertador, ya que es muy frágil. El molde se coloca en el aparato de electroforesis, tal y como se muestra en la figura. Una vez fijo, se añade el tampón de electroforesis en las cubetas, teniendo en cuenta que en la de arriba debe entrar en contacto con el gel. Preparación de las muestrasComo ya hemos dicho, las muestras deben incubarse con el tampón de carga para desnaturalizarse, para ello seguimos esta lista: Muestra: 60 l + 30 l de tampón de carga. Fracciones: 60 l + 30l de tampón de carga. Patrón: 25 l + 30 l de tampón de carga.Todo ello en eppendorf, a calentar 5 minutos a 100 ºC para desnaturalizar las proteínas y luego en hielo para mantenerlas

desnaturalizadas. Para que los eppendorf no se abran violentamente durante la incubación se les hace un orificio en la tapa y se cubre todo con parafilm, material elástico, con lo que dará de si y se hinchará, pero no estallará.Cargado de las muestras en el gelUna vez preparadas las muestras, las cargamos en el gel con este orden:
F3 PATRÓN F4 MUESTRA F5 F6 F10 F11 F12 F13 F14 F7
Una vez cargadas las muestras en el gel se desarrolla la electroforesis a una intensidad constante de 15 mA durante toda la noche.Parar la electroforesis al día siguiente, cuando el azul de bromofenol esté a 1cm del final del gel. Desmontarlo y ponerlo a teñir.Tinción del gel de proteínasMaterialSolución fijadora: etanol + acético + agua (en proporción 5/1/5).Solución teñidora: 0,1 5 de azul de Coomassie; 40 % de etanol, y 10 % de ácido acético.Solución desteñidora 7 % de ácido acético.Colocamos el gel de proteínas, con cuidado de no romperlo, en un bandeja donde se añadirá la solución fijadora. Incubamos 30 minutos a 20 ºC con agitación leve. Tras retirar el fijador, añadimos al solución de tinción e incubamos durante 30 minutos a temperatura ambiente. Finalmente, retirar el teñidor y pasar a desteñir. Para acelerar el proceso de destinción añadimos unas bolitas de papel de filtro, que absorberá el azul de Coomassie. Esto se mantendrá así hasta que las bandas de proteínas sean claramente visibles, en este punto, el gel está listo para ser secado sobre el papel W 3MM. Para ello se utilizará un secador de geles.ResultadosUna vez teñido el gel tenía un aspecto similar al de la figura:
F3 PATRÓN F4 MUESTRA F5 F6 F10 F11 F12 F13 F14 F7
Determinación de los pesos moleculares
Banda 1 (cm)
Banda 2 (cm)
Banda 3 (cm)
Banda 4 (cm)
Banda 5 (cm)
Banda 6 (cm)
Patrón 0,7 1,3 1,8 2,4 2,7 3,2
F3 0,7 1
F6 0,8 1

CIENCIAS BIOLÓGICAS Y DE LA SALUDBIOQUÍMICA IILABORATORIOPRACTICA 1HIDRÓLISIS ENZIMATICA DE UN POLISACARIDO VEGETAL (ALMIDON)PROF. VERONICA SOUZA ARROYOALUMNO:ESPINOSA NEIRA ROBERTOGRUPOBC0518 - JUN - 2002INTRODUCCIÓNAlmidón.Es un hidrato de carbono complejo (C6H10O5), inodoro e insípido, en forma de grano o polvo. El almidón es el principal carbohidrato de reserva en la mayoría de las plantas. En las hojas el almidón se acumula en los cloroplastos, donde es un producto directo de la fotosíntesis. En los órganos de almacenamiento, se acumula en los amiloplastos, en los cuales se forma después de la translocación de sacarosa u otro carbohidrato provenientes de las hojas.En los vegetales, el almidón se encuentra en uno o más granos amiláceos en un plastidio. La cantidad de almidón en diversos tejidos depende de muchos factores genéticos y ambientales. El almidón se acumula a la luz del día cuando la fotosíntesis excede las tasas combinadas de respiración y translocación, después parte de él desaparece por la noche.Se presentan dos tipos de almidón en la mayoría de los granos amiláceos: amilosa y amilopectina, ambos compuestos por unidades de d-glucosa unidas por enlaces ð-1, 4. Las uniones ð-1, 4 hacen que las cadenas de almidón se enrollen en forma de hélices. La amilopectina consta de moléculas muy ramificadas, cuyas ramas se localizan entre el C-6 de una glucosa de la cadena principal y el C-1 de la primera glucosa en la cadena que forma la rama (enlaces ð-1, 6). Las amilosas son más pequeñas y contienen de cientos a miles de unidades de glucosa, numero que depende de la especie y las condiciones ambientales.La formación de almidón ocurre sobre todo por un proceso que implica la donación repetida de unidades de glucosa provenientes de un azúcar nucleotidico similar al UDPG y que se denomina difosfoglucosa de adenosina, ADPG.Hidrólisis del almidón.La hidrólisis implica la ruptura de un enlace mediante la adición en medio del mismo de los elementos del agua. Los polisacáridos de la dieta se metabolizan mediante hidrólisis a monosacáridos.La mayoría de los pasos de la degradación de almidón a glucosa pueden ser catalizados por tres enzimas distintas, si bien hay otras más que se necesitan para completar el proceso. Las tres primeras enzimas son una ð-amilasa, ð-amilasa y almidón fosforilasa. Al parecer solo la ð-amilasa puede atacar gránulos de almidón intactos, por lo que cuando participan la ð-amilasa y la almidón fosforilasa, es probable que actúen sobre los primeros productos liberados por la ð-amilasa. La ð-amilasa ataca de manera aleatoria enlaces 1,4 en las moléculas de amilosa y amilopectina, al principio creando huecos al azar en los granos de almidón y liberando productos que aun son grandes. En cadenas de amilosa no ramificadas, el ataque repetido por la ð-amilasa produce maltosa, un disacárido que contiene dos unidades de glucosa. Sin embargo, la ð-amilasa no puede atacar los enlaces 1,6 localizados en los puntos de ramificación de la amilopectina, por lo que la digestión de amilopectina cesa cuando aun quedan dextrinas ramificadas con cadenas de longitud corta. Muchas ð-amilasas son activadas por Ca+, lo cual es una de las razones por las que el calcio es un elemento esencial.La ð-amilasa hidroliza al almidón en ð-maltosa; la enzima actúa primero solo sobre los extremos no reductores. La ð-maltosa cambia con rapidez, por mutarrotación, para formar las mezclas naturales de isomeros ð y ð. La hidrólisis de amilosa por la ð-amilasa es casi
completa, pero la degradación de amilopectina es incompleta porque no son atacados los enlaces de los puntos de ramificación. La actividad de ambas amilasas implica la incorporación de una molécula de H2O por cada enlace roto, por lo que son enzimas hidrolasas. Las reacciones hidrolíticas no son reversibles, de modo que no se pueden detectar síntesis de almidón por amilasas. Las amilasas están diseminadas en diversos tejidos pero son mas activas en las semillas que están germinando, ricas en almidón. Es probable que la ð-amilasa tenga más importancia que la ð-amilasa para la hidrólisis de almidón. Gran parte de la ð-amilasa se localiza dentro de los cloroplastos, muchas veces unida a los granos de almidón que atacara. Actúa tanto en el día como por la noche aunque, por supuesto, durante la luz de día hay producción neta de almidón por la fotosíntesis.La amilopectina solo es degradada parcialmente por la acción del almidón fosforilasa. La reacción procede de manera consecutiva a partir del extremo no reductor de cada cadena principal o cadena ramificada hasta a unos residuos de glucosa de las uniones ð-1,6 de las ramificaciones, por lo que de nuevo que dan dextrinas. La amilosa, que tiene pocas ramificaciones, se degrada casi por completo, por eliminación repetida de unidades de glucosa a partir del extremo no reductor de la cadena. La almidón fosforilasa esta ampliamente distribuida en la planta y a veces resulta difícil determinar que enzima digiere la mayor parte del almidón en las células de interés.OBJETIVOComprobar que las amilasas producidas por semillas de maíz hidrolizan al almidón, dando como producto final unidades de maltosa siendo este uno de los mecanismos que la semilla utiliza para obtener energía necesaria para germinar.El alumno estudiara las diferencias en la actividad amilolítica, dependiendo de los estadíos de germinación en las semillas y de sus requerimientos energéticos.Se aplicara el método de Nelson-Simogy para la determinación de reductores totales.Determinaremos la actividad enzimática de las semillas de maíz, dependiendo de su tiempo de germinación.MATERIAL1 mortero1 bisturí1 baño de agua hirviendo6 tubos de centrifuga5 pipetas graduadas de 10 ml1 embudo de filtración de 5 cm de diámetro1 caja de Petri de 9 cm1 tripié con tela de asbesto1 mechero25 tubos de ensayo de 15X1501 centrifuga1 espectrofotómetro1 gasa 30 semillas de maízMETODOLOGÍALa preparación de las semillas de maíz, se llevara a cabo cinco días anteriores a la práctica. Se desinfectaran 10 semillas de maíz en cloro (hipoclorito de sodio) durante 20 minutos y las enjuagaremos muchas veces con agua hervida; posteriormente las semillas las pondremos a germinar en un frasco limpio, en el frasco se pondrá una capa de algodón, las semillas, y se humedecerá el algodón. Después el mismo procedimiento de germinación se aplicara a otras cuantas semillas limpias pero tres días antes de la práctica.Para empezar la practica se ponen a remojar en agua 2 semillas de maíz de ningún día de germinación; después se seleccionaron un par de semillas de 5, de 3 y 0 días de germinación, entonces se les quita el embrión y el escueto, para que solo nos quede el endospermo almidonoso. Una vez limpios los maíces prepararemos un extracto enzimático de los respectivos días de germinación; lo anterior se hará macerando el endospermo de 2 semillas de 5 días de germinación, en un mortero de con 5 ml de succinato con pH 5. Se hizo el mismo procedimiento con las semillas de 3 y 0 días de germinación, los productos obtenidos de los macerados se colocaran en tubos de centrífuga, lavando el mortero con el amortiguador en cada uno de los tres casos para centrifugar a 3000 rpm durante 20 minutos. Después de centrifugar se separara el sobrenadante de cada tubo; para ser colocadas en los tubos numerados del 7 al 12, según la relación del formato siguiente.
tubo # dias de germinacion ml extracto diluido
7 0 0.25
8 0 0.25
9 3 0.25
10 3 0.25
11 5 0.25
12 5 0.25

El ultimo paso de la practica será la determinación de los reductores totales, que se llevara a cabo con las diluciones de los extractos enzimáticos de la semilla de diferentes tiempos de germinación; aquí se realizan las reacciones con los diferentes reactivos (I y II) y la determinación de azucares reductores totales que obtuvimos con la hidrólisis enzimática del almidón; también realizaremos la curva patrón. Estas reacciones fueron de acuerdo a la tabla.RESULTADOS Anotaremos las lecturas espectrofotométricas de cada tubo en la tabla siguiente.
TUBODENSIDAD OPTICA (nm)
MUESTRA DUPLICADO MEDIA
1 0 0. 0
2 0.086 0.090 0.088
3 0.377 0.212 0.2945
4 0.297 0.292 0.2945
5 0.339 0.259 0.299
6 0.464 0.505 0.4845
Determinaremos la concentración de maltosa en los tubos 1 al 6 correspondientes a la curva patrón, de acuerdo con la ecuación: (Volumen inicial)(Concentración inicial) = (Volumen final)(Concentración final)(Tubo x)(500 ðg/ml) = (Patrón maltosa + Agua)( X )Se anota la concentración en la columna X de la tabla siguiente y en la columna Y se anotan las lecturas espectrofotométricas obtenidas en los tubos 1 a 6.
Tabla 1. VALORES PARA CURVA PATRON
TUBO #
X Y
[Maltosa ðg/mL]Densidad óptica (520 nm)
1 0 0
2 125 0.088
3 333.33 0.2945
4 750 0.2945
5 2000 0.299
6 0 0.4845
Se trazara la grafica con los datos de las columnas X y Y. Debido a que los datos se presentan como una recta, se ajustan de ser necesario. En el siguiente cuadro anotaremos los resultados obtenidos de las lecturas de densidad óptica de los tubos problema (7-12).
DIASTUBOSPROBLEMA
DENSIDAD OPTICA (nm)
MUESTRA
DUPLICADO
MEDIA
0 7 1.229 0.833 1.031
0 8 0.990 1.218 1.104
3 9 1.252 0.989 1.120
3 10 0.844 1.106 0.975
5 11 1.071 1.200 1.1355
5 12 1.114 1.164 1.139
Tomando en consideración los datos de la curva patrón donde Y ha sido corregida Y´ y recordando nuevamente que y=0.0873x-0.0621; se calcula la concentración de maltosa de los diferentes días de germinación.Los datos se registran en la siguiente tabla.
TUBO # DIAS [MALTOSA ðg]
7 0 0.0279
8 0 0.0342
9 3 0.0356
10 3 0.0230
11 5 0.0370
12 5 0.0373
En la siguiente grafica se encuentra la concentración de maltosa por los días de germinación de las semillas.DISCUSIÓNEn esta práctica se intento tener el mejor rendimiento respecto a los resultados. Creemos que nuestros resultados hubieran estado más precisos si la extracción del almidón no hubiera presentado algunas dificultades para obtener el endospermo. Esto lo pensamos ya que en las semillas de 0 y 3 días el endospermo presentaba partes más duras, que hacían más difícil la extracción total.Otra cuestión a destacar, es la parte en que hacemos la segunda incubación (reactivo 1), pues por un descuido el agua hirvió y se derramo un poco, provocando que a algún tubo le entrase agua y alterara una absorbancia; esto se nota pues el mismo número de tubo pero del duplicado da una lectura más coherente.Por tanto con nuestros resultados y el texto consultado se demuestra que la actividad enzimática en la semilla es mayor si el tiempo de germinación es mayor.CONCLUSIÓNEn esta práctica se esperaba y se observo la actividad enzimática de la amilasa sobre el almidón. Observamos que al tener más tiempo de germinación una semilla, mayor es la actividad enzimática de la amilasa. En los granos de cero días se manifestó una actividad de la enzima muy baja, después existe un incremento en su actividad al usar de semillas de tres días y por supuesto que la mayor actividad se registro en las semillas de 5 días. Todas estas reacciones nos dejan una producción de maltosa, por acción de la hidrólisis enzimática de la amilasa sobre el almidón; esto nos sugiere que si la enzima trabaja mucho, sería porque había mucho almidón para hidrolizar y poder producir maltosa.En resumen y conclusión, la actividad enzimática es directamente proporcional a la producción de maltosa, todo esto con respecto a los días de germinación que tengan las semillas (entre mas días de germinación tenga las semillas, mas almidón habrá, entonces, la actividad enzimática será mucho mayor.)CUESTIONARIO1.- CUAL ES LA ESTRUCTURA DEL ALMIDON?
32.- QUE ES UN AZUCAR REDUCTOR?Cuando en una determinación cuantitativa de los monosacáridos se realiza frecuentemente, basándose en su observación en disoluciones alcalinas mediante Cu2+, Ag+ o ferricianuros, se origina una mezcla de azucares -ácidos. Los azucares son capaces de reducir, a tales oxidantes se les denomina azucares reductores.Se puede definir también a un azúcar reductor como cualquier carbohidrato que tiene libre un grupo carbonio y es susceptible de participar en otra oxidación.3.- QUE TIPO DE ENLACES HIDROLIZAN LAS AMILASAS?ð-Amilasa, ð(14) glucan 4-glucanohidrolasað-Amilasa, ð(14) glucan maltodeshidrogenasa4.- QUE AMILASAS SE ENCUENTRAN EN LOS ORGANISMOS ANIMALES?

ð-D-glucopiranosað-Amilasa, ð(14) glucan 4-glucanohidrolasaBIBLIOGRAFÍASALISBURY, B. FRANK Fisiología Vegetal, Ed. Iberoamericana, 1994.MATHEWS, C. K. Y VAN HOLDE, K. E. Bioquímica, McGraw Hill Interamericana, 2ª. Ed., España, 1998.BRITÁNNICA CD 2000 DELUXE Encyclopædia Britannica, Inc.WHITE, A., HANDLER, P., SMITH, E., HILL, R., LEHMAN, R. Principios de Bioquímica, McGraw Hill, 1989.MICROSOFT® EXCEL 2002 (10.2614.2625)


















