
PERFIL DE LOS ÁCIDOS GRASOS DE LA MEMBRANA DE...
56
PERFIL DE LOS ÁCIDOS GRASOS DE LA MEMBRANA DE ERITROCITOS COMO REFLEJO DEL PERFIL DE LOS ÁCIDOS GRASOS DE LOS FOSFOLÍPIDOS HEPÁTICOS EN HÍGADO GRASO NO ALCOHÓLICO Prof. Dra. Julia Araya A. Director de Tesis Prof. Dr. Ramón Rodrigo S. Director de Tesis 2005 ALEJANDRA NOELIA ELIZONDO MUÑOZ TESIS PARA OPTAR AL GRADO DE MAGISTER EN CIENCIAS MÉDICAS MENCIÓN NUTRICIÓN Universidad de Chile Facultad de Medicina Escuela de Postgrado
Transcript of PERFIL DE LOS ÁCIDOS GRASOS DE LA MEMBRANA DE...

PERFIL DE LOS ÁCIDOS GRASOS DE LA MEMBRANA
DE ERITROCITOS COMO REFLEJO DEL PERFIL DE LOS
ÁCIDOS GRASOS DE LOS FOSFOLÍPIDOS HEPÁTICOS
EN HÍGADO GRASO NO ALCOHÓLICO
Prof. Dra. Julia Araya A.Director de Tesis
Prof. Dr. Ramón Rodrigo S.Director de Tesis
2005
ALEJANDRA NOELIA ELIZONDO MUÑOZ
TESIS PARA OPTAR AL GRADO DE MAGISTER EN CIENCIAS MÉDICAS
MENCIÓN NUTRICIÓN
Universidad de ChileFacultad de MedicinaEscuela de Postgrado

1
UNIVERSIDAD DE CHILE FACULTAD DE MEDICINA ESCUELA DE POSTGRADO
INFORME DE APROBACION TESIS DE MAGISTER
Se informa a la Comisión de Grados Académicos de la Facultad de Medicina, que la Tesis de Magister presentada por la candidata
ALEJANDRA NOELIA ELIZONDO MUÑOZ
ha sido aprobada por la Comisión Informante de Tesis como requisito para optar al Grado de Magister en Ciencias Médicas con mención en Nutrición en el Examen de Defensa de Tesis rendido el día 29 de noviembre de 2005
Prof. Dra. Julia Araya A. Prof. Dr. Ramón Rodrigo S. Director de Tesis Director de Tesis Departamento de Nutrición Programa de Farmacología, ICBM Facultad de Medicina, Universidad de Chile Facultad de Medicina, Universidad de Chile
COMISION INFORMANTE DE TESIS
Prof. Dra. Lilia Masson S. Prof. Dra. M. Angélica Carrasco F.
Prof. Dr. Victor Charlin G. Presidente Comisión

2
DEDICATORIA
“Tener siempre horas diarias para trabajar y para jugar, hacer que cada día sea tan
útil como placentero, y apreciar el valor del tiempo aprovechándolo bien. Entonces la
juventud tendrá su encanto, la vejez traerá pocos arrepentimientos y la vida será un
hermoso triunfo.”
Louisa May Alcote
Para mi familia, amigos y a todos quienes estuvieron a mi lado apoyándome y
haciendo de este trabajo una gran experiencia, gracias.

3
AGRADECIMIENTOS
Agradezco todo el apoyo, conocimientos y dedicación que me entregaron mis
directores de tesis: Dra. Julia Araya y Dr. Ramón Rodrigo. Además, agradezco la
colaboración y buena disposición de Diego Soto.

4
ÍNDICE DE CONTENIDOS ÍNDICE DE TABLAS ............................................................................................................ 5 ÍNDICE DE FIGURAS .......................................................................................................... 5 RESUMEN ............................................................................................................................. 6 I. INTRODUCCIÓN: ......................................................................................................... 8
1. ETIOPATOGENIA: ................................................................................................... 9 1.1 Papel del metabolismo hepático de los lípidos: ................................................. 9 1.2 Papel del estrés oxidativo hepático:................................................................ 12 1.3 Perfil de los ácidos grasos de las membranas celulares: ................................. 14
II. HIPÓTESIS DE TRABAJO: ........................................................................................ 16 III. OBJETIVOS: ............................................................................................................ 16
1. OBJETIVOS GENERALES: ................................................................................... 16 2. OBJETIVOS ESPECÍFICOS: .................................................................................. 17
2.1 En Hígado: ....................................................................................................... 17 2.2 En Plasma: ....................................................................................................... 17 2.3 En Eritrocitos: .................................................................................................. 17
IV. METODOLOGÍA: ................................................................................................... 18 1. PACIENTES: ........................................................................................................... 18 2. DISEÑO: .................................................................................................................. 18 3. PROCEDIMIENTOS: .............................................................................................. 19
3.1 Extracción de los lípidos hepáticos totales: ..................................................... 19 3.2 Separación de los fosfolípidos de los lípidos totales del hígado: .................... 19 3.3 Separación de las membranas de los eritrocitos y extracción de sus lípidos: . 20 3.4 Preparación de los ésteres metílicos de los ácidos grasos de: ......................... 21 3.5 Análisis de los esteres metílicos de los ácidos grasos: .................................... 21 3.6 Análisis de lipoproteínas plasmáticas: ............................................................ 22 3.7 Determinación de estrés oxidativo: ................................................................. 22
4. ANÁLISIS ESTADÍSTICO: .................................................................................... 24 V. RESULTADOS: ........................................................................................................... 25
1. Características clínicas y bioquímicas de las pacientes con HGNA y controles: ..... 25 2. Perfil de los ácidos grasos de los lípidos hepáticos totales: ..................................... 25 3. Composición de los ácidos grasos de los fosfolípidos hepáticos: ............................ 25 4. Composición de los ácidos grasos de la membrana de eritrocitos: .......................... 26 5. Perfil de AGPI hepáticos y relación producto/precursor de la serie n-3 y n-6: ........ 27 6. Parámetros relacionados con estrés oxidativo hepático: .......................................... 27
VI. DISCUSIÓN: ............................................................................................................ 40 VII. CONCLUSIONES: ................................................................................................... 44 VIII. REFERENCIAS: ...................................................................................................... 47 IX. ANEXOS: ................................................................................................................. 53 1. CONSENTIMIENTO INFORMADO……………………………………………….53 2. Aprobación Comité de Ética………………………………………………………..56

5
ÍNDICE DE TABLAS
Tabla Nº1: Características bioquímicas de los pacientes con HGNA y controles…………….29 Tabla Nº2: Perfil de los ácidos grasos de los lípidos totales hepáticos………………………..30 Tabla Nº3: Perfil de los ácidos grasos de los fosfolípidos hepáticos………………………….31 Tabla Nº4: Perfil de los ácidos grasos de los fosfolípidos de membranas de eritrocitos……...32
ÍNDICE DE FIGURAS
Figura 1: Cromatograma de los ésteres metílicos de membranas de eritrocitos (A) y de los fosfolípidos de hígado de mujeres controles (B)……………………………..…..........33 Figura 2: Cromatograma de los ésteres metílicos de membranas de eritrocitos (C) y de los fosfolípidos de hígado (D) de pacientes obesas con hígado graso no alcohólico (HGNA)……………………………………………………………………………….34 Figura 3: Cromatograma de los ésteres metílicos de membranas de eritrocitos de pacientes obesas con HGNA, tres meses post-cirugía bariátrica……………………...35 Figura 4: Actividad de enzimas antioxidantes y contenido de glutatión reducido (GSH) en hígado de controles y de pacientes obesas con HGNA……………………………….36 Figura 5: Capacidad antioxidante total del plasma (A), contenido de malondialdehido (B) y contenido de F2-Isoprostanos (C) en controles y pacientes obesas con HGNA….........37 Figura 6: Patrón de ácidos grasos poli-insaturados (AGPI)…………………………….……..38 . Figura 7: Patrón de ácidos grasos poli-insaturados de cadena larga (AGPICL)……………....39 Figura 8: Biosíntesis de los ácidos grasos poli-insaturados de cadena larga en
mamíferos………………………………………………………………………...........45
Figura 9: Factores que conducen a esteatosis no alcohólica y su progresión a esteatohepatitis .………………………………………………………………….........46

6
RESUMEN
Objetivos: Comparar el perfil de los ácidos grasos de los fosfolípidos hepáticos con los de la membrana del eritrocito. Evaluar el efecto del estrés oxidativo sobre la homeostasis de los ácidos grasos poli-insaturados de cadena larga (AGPICL) en eritrocitos y fosfolípidos hepáticos. Implementar un procedimiento no invasivo para estimar en forma indirecta los efectos del estrés oxidativo sobre el perfil de los ácidos grasos de los fosfolípidos hepáticos y evaluar los cambios en el perfil de estos ácidos grasos en la baja de peso post cirugía bariátrica en pacientes obesas con hígado graso no alcohólico (HGNA). Material y Métodos: Se estudiaron 12 pacientes obesas con hígado graso no alcohólico y 8 controles no obesas, sin antecedentes de consumo de alcohol ni de tabaco. Se determinaron parámetros relacionados con resistencia a la insulina (HOMA), daño hepático (transaminasas), estatus antioxidante (capacidad antioxidante del plasma, contenido de glutatión reducido en el hígado, enzimas antioxidantes (catalasa, superóxido dismutasa y glutatión peroxidasa)), lipoperoxidación (F2-isoprostanos en plasma y malondialdehido en hígado) y composición de los ácidos grasos de los fosfolípidos hepáticos y de la membrana del eritrocito. A los 3 meses post-cirugía bariátrica se midieron nuevamente F2isoprostanos y composición de ácidos grasos de la membrana de eritrocitos en 7 de las pacientes con HGNA. Resultados: Las pacientes con HGNA fueron significativamente insulino-resistentes comparadas con el grupo control, evidenciado por una elevación del parámetro HOMA (9,6±3 vs 2.0±0.2). El perfil lipídico y las transaminasas fueron similares entre estos dos grupos. Las pacientes obesas con HGNA presentaron en los fosfolípidos hepáticos, en comparación con el grupo control, un menor contenido (p<0.05) de ácidos grasos poli-insaturados totales (total AGPI), ácidos grasos poli-insaturados n-3 totales (total AGPI n-3), ácidos grasos poli-insaturados de cadena larga n-3 totales (total AGPICL n-3) (27%, 61% y 63% menos, respectivamente) y de los ácidos grasos 20:4 n-6 (ARA), 20:5 n-3 (EPA) y 22:6 n-3 (DHA) (48%, 57% y 70% menos, respectivamente). Acompañándose también de una mayor relación n-6/n-3 AGPICL (211% más) y de un mayor contenido de ácido graso 22:5 n-6 (Osbond). En las membranas de eritrocitos, las pacientes obesas con HGNA, también presentaron un menor contenido (p<0.05) del total AGPI, total AGPI n-3, total AGPICL n-3 (19%, 46% y 50% menos, respectivamente) y de los ácidos grasos 20:4 n-6 y 22:6 n-3 (33% y 52% menos, respectivamente), en comparación al grupo control. Además, también presentaron una mayor

7
relación n-6/n-3 AGPICL (173% más) y mayor contenido del 22:5 n-6. Estos hallazgos fueron similares a los encontrados en los fosfolípidos hepáticos. El grupo HGNA post-cirugía bariátrica presentó cambios en la composición de los ácidos grasos de los fosfolípidos de membrana de eritrocitos asemejándose al grupo control. Estos cambios fueron: aumento de los ácidos grasos 20:4 n-6 (en 51%), 22:6 n-3 (en 142%), total AGPI n-3 (en 39%) y total AGPICL n-3 (en 46%), disminución del 22:5 n-6 y del 18:2 n-6 (en 100% y 58%, respectivamente). La relación n-6/n-3 AGPICL presentó una disminución del 52%, pero sin alcanzar significancia estadística. En los parámetros de estrés oxidativo, las pacientes obesas con HGNA presentaron menor contenido de glutatión (41% menos), menor actividad de enzimas antioxidantes, tales como superóxido dismutasa y catalasa (58% y 30% menos, respectivamente) y menor FRAP (31% menos) comparado con el grupo control (p<0.05%). Además, las pacientes con HGNA presentaron una mayor lipoperoxidación, comparado con el grupo control, evidenciado por el mayor contenido de MDA y de F2I de 185% y 85%, respectivamente. La actividad de la glutatión peroxidasa no fue significativamente diferente del grupo control. El grupo de pacientes HGNA post-cirugía bariátrica presentó una reducción significativa (p<0.05) del estrés oxidativo evidenciado por reducción del 40% en los niveles de F2I, comparado con el grupo inicial de obesas con HGNA (precirugía), alcanzando incluso valores semejantes al grupo control. Conclusiones: Debido a la presencia significativa de resistencia a la insulina, depleción de AGPICL n-3 y de estrés oxidativo en las pacientes obesas con HGNA, se plantea un mecanismo multifactorial en la patogenia del HGNA. Además, la depleción de los AGPICL sugiere que estos resultan ser los marcadores más sensibles a los efectos del estrés oxidativo sobre los fosfolípidos de membranas de hígado y eritrocito. Los datos sugieren que la composición de los ácidos grasos de la membrana del eritrocito podría ser utilizada para estimar los cambios homólogos en el hígado en pacientes con HGNA, lo que constituye un método no invasivo para el estudio de la homeostasis de los ácidos grasos en esta patología.

8
I. INTRODUCCIÓN:
El hígado graso no alcohólico (HGNA) es una enfermedad ampliamente reconocida, debido a
su importancia como causa de daño hepático crónico (DHC), siendo la tercera causa más
común de DHC en EEUU, luego del alcohol y la hepatitis C (1). Se define histológicamente
como la acumulación de grasa en el hígado que excede 5-10% (p/p) estimado por microscopia
óptica (2). El HGNA ha sido considerado un síndrome frecuentemente asociado a obesidad,
diabetes mellitus tipo 2 (DM2) e hipertrigliceridemia (3,4), las cuales forman parte del Síndrome
Metabólico (5), teniendo en común la presencia de resistencia a la insulina. Clínicamente es
semejante al hígado graso alcohólico, pero en individuos que no consumen alcohol
significativamente (<40g/semana) (6,7). La mayoría de estos pacientes son asintomáticos y
presentan un aumento de transaminasas de leve a moderado, hepatomegalia clínica y
características de hígado graso en exámenes de imágenes. El HGNA incluye un amplio
espectro de formas histológicas que van desde esteatosis simple (grasa macrovesicular) a
esteatohepatitis (hepatocitos balonados y cuerpos de Mallory), fibrosis avanzada y cirrosis (8),
pero los mecanismos involucrados en la progresión de este daño no han sido claramente
definidos, por lo que la biopsia hepática sería esencial para la confirmación diagnóstica y
pronóstico de la enfermedad (1), pero no para su manejo (9). La prevalencia del HGNA es difícil
de determinar, ya que los datos varían según el grupo poblacional estudiado y según el método
utilizado. Se estima una prevalencia de HGNA de 16-23% y de esteatohepatitis no alcohólica
(EHNA) de 2-3%, ambos en la población general (10). En grupos de riesgo la prevalencia de
HGNA se incrementa, hasta 75% en DM2 (10), 74% en obesos (11) y hasta 86% en obesos
severos sometidos a cirugía bariátrica, aumentando también en este último grupo la
prevalencia de EHNA a 14% (10). En obesos la prevalencia de cirrosis es 2-3%. La asociación

9
de DM2 y obesidad incrementa considerablemente el riesgo, encontrándose en pacientes
diabéticos severamente obesos que el 100% tenía algún grado de esteatosis, el 50% tenía
EHNA y el 19% tenía cirrosis (11,12). La historia natural de la enfermedad varía de acuerdo al
tipo histológico, por lo que pacientes con esteatosis simple tienen un curso clínico benigno,
generalmente sin progresión de la enfermedad en un periodo mayor a 10 años, sin embargo,
aproximadamente el 25% de los pacientes con EHNA desarrollan fibrosis y 15% desarrollan
cirrosis en el mismo periodo de tiempo, pudiendo desarrollar complicaciones tardías como
carcinoma hepatocelular (8,9,10). La sobrevida de pacientes con EHNA ha sido estimada de 67%
a 5 años y de 59% a 10 años (8). Numerosos estudios han descrito predictores clínicos de
fibrosis avanzada y cirrosis incluyendo: obesidad, DM2, edad>45 años, AST/ALT >1 e
hipertrigliceridemia (8,9,10)
1. ETIOPATOGENIA: 1.1 Papel del metabolismo hepático de los lípidos:
Aunque las alteraciones metabólicas preexistentes como causas HGNA pueden ser
multifactoriales y/o polimorfo-genéticas (8), se ha comprobado que el HGNA se asocia
frecuentemente a un trastorno del metabolismo de los ácidos grasos en personas obesas (13,14).
Alteraciones en el metabolismo lipídico pueden producir un aumento del contenido de lípidos
hepáticos debido a un incremento de la lipogénesis, a un defecto en la β−oxidación
peroxisomal y mitocondrial de los ácidos grasos y/o a una reducida capacidad del hígado para
exportar los lípidos (15,16). En este último caso, la acumulación de lípidos en el hígado
incrementa la tasa de apoptosis celular (15, 17,18) y perjudica el recambio de Apo B100, principal

10
proteína estructural de las VLDL (19). La formación y secreción de las VLDL a nivel hepático
está influenciada por diversos factores incluyendo a la proteína de unión a ácidos grasos
hepáticos (20), proteína citoplasmática que se une a los ácidos grasos poli-insaturados de
cadena larga (AGPICL) con gran afinidad y que es regulada positivamente por éstos (21). La
regulación del metabolismo lipídico celular por las grasas de la dieta puede ocurrir a dos
niveles: a) modulación de la transducción de señales a través de cambios en la composición de
los ácidos grasos de membrana y b) modificación de la transcripción génica (22,23). Estudios
recientes sugieren que los AGPICL y sus productos eicosanoides juegan un papel central
como distribuidores de combustibles metabólicos, favoreciendo la oxidación de ácidos grasos
en el hígado, aumentando el flujo de glucosa a glucógeno y disminuyendo la síntesis de lípidos
y el depósito de triglicéridos hepáticos (17,18,24). Esta acción coordinada de los AGPICL
consistente en suprimir la síntesis de lípidos y estimular la oxidación de ácidos grasos en el
hígado, es ejercida con mayor potencia por los ácidos grasos n-3 que los n-6 (17,18,25,26), pero
aún se desconoce la mejor relación n-6/n-3 AGPI requerida para un óptimo beneficio
metabólico. En concordancia con esto, se ha visto en humanos que incrementos del tamaño de
las gotas de grasa en hepatocitos está asociado con una reducción en el porcentaje del ácido
eicosapentaenoico (20:5 n-3) presente en los triglicéridos (TG) (27). Además se ha observado
una reducción del contenido de TG hepáticos con dietas enriquecidas con aceite de pescado
(28), el cual es rico en AGPICL n-3 que estimulan la oxidación de los ácidos grasos (17). La
deficiencia de ácidos grasos n-3 ha sido sugerida como un síndrome de la sociedad moderna
(29). Diversas fuentes de información sugieren que actualmente los seres humanos consumen
dietas deficientes en ácidos grasos n-3 y con excesivas cantidades de n-6, lo que aumenta
significativamente la relación n-6/n-3 AGPI (30) comparado con la dieta bajo la cual el modelo

11
de control génico humano fue propuesto (31), lo cual jugaría un papel importante en la génesis
del HGNA.
El efecto metabólico de los AGPI requiere un adecuado suministro alimentario de ácidos
grasos esenciales como el ácido linoleico (18:2 n-6) y el ácido α linolénico (18:3 n-3)
(precursores de los AGPICL) (Figura 8), una optima actividad de las desaturasas para que los
ácidos grasos esenciales experimenten delta desaturación (24,18) y un bajo grado de estrés
oxidativo (32) (Figura 9). La pérdida de la actividad de la delta 5 y delta 6 desaturasa ha sido
asociada a obesidad (33) y al desarrollo de hígado graso en humanos (34,35).
El HGNA ha sido consistentemente asociado con resistencia a la insulina y Síndrome
Metabólico(5). La resistencia a la insulina conduce a lipólisis e hiperinsulinemia
compensatoria. La lipólisis incrementa los ácidos grasos libres circulantes y su captación por
los hepatocitos, produciendo una sobrecarga en la β−oxidación mitocondrial y por ende,
acumulación de grasa en el hígado y esteatosis (11). Esto es reconocido como el primer evento
en la génesis del HGNA (1,36). Por otro lado, la hiperinsulinemia incrementa la glicólisis y la
síntesis de ácidos grasos en el hígado y reduce su exportación, por aumento de la degradación
de ApoB100, favoreciendo finalmente la acumulación hepática de TG (11). El segundo evento
condicionante estaría dado por el estrés oxidativo, que actuando sobre un hígado ya
vulnerable, conduciría a EHNA y fibrosis (Figura 9). La severidad de la resistencia a la
insulina, ha demostrado ser paralela a la severidad del daño hepático en el HGNA,
encontrándose mayor prevalencia de DM2 (hasta 55%) en pacientes con EHNA y cirrosis. Al
respecto, la presencia de diabetes en pacientes con EHNA de larga data ha sido sugerida como
un indicador útil para identificar a quienes podrían tener fibrosis hepática severa (37). Estudios
recientes indican que la resistencia a la insulina es un factor de riesgo independiente de

12
fibrosis avanzada, actuando tanto a nivel del primer y segundo evento molecular conducentes
al HGNA. (38)
1.2 Papel del estrés oxidativo hepático:
Inicialmente, la masiva llegada de ácidos grasos libres al hígado debido a la resistencia a la
insulina podría aumentar la β−oxidación mitocondrial y peroxisomal hasta valores de Vmax,
con el consecuente incremento en la producción de especies reactivas de oxígeno (EROs). El
aumento de EROs en un medio rico en lípidos induce peroxidación lipídica, los que pueden
potenciar el estrés oxidativo (8,37). Las principales fuentes de estrés oxidativo identificadas en
HGNA incluyen: citocromo P450, β−oxidación peroxisomal, fuga de electrones de la
mitocondria, desacoplamiento de la fosforilación oxidativa y reclutamiento de células
inflamatorias (8). La mitocondria es la principal fuente de EROs, las cuales podrían gatillar
esteatohepatitis y fibrosis por tres mecanismos: 1) lipoperoxidación, 2) inducción de
citoquinas e 3) inducción de ligandos Fas, que llevan a apoptosis (1).
El desbalance pro-oxidante/antioxidante a favor del primero constituye el fenómeno que
produce el estrés oxidativo, trastorno que genera varios procesos fisiopatológicos en el hígado
(32,39,40) conduciendo a daño en membranas plasmáticas, organelos intracelulares, DNA
mitocondrial y proteínas asociadas a la cadena respiratoria (37), con pérdida de sus funciones
biológicas y de la viabilidad celular. Además, la activación de los factores de transcripción
sensibles a los mecanismos redox, como NF-kB y AP-1 pueden inducir hepatotoxicidad a
través del aumento de la regulación de los mediadores citotóxicos pro-inflamatorios o
fibrogénicos (32,39,40). Estos mediadores junto a los productos de lipoperoxidación son los
principales efectores del segundo evento sobre un hígado vulnerable llevándolo a

13
esteatohepatitis (37) (Figura 9). Recientes estudios en pacientes con HGNA correlacionan
cambios en la homeostasis de los ácidos grasos hepáticos con estrés oxidativo (32,39,40) y con
desbalance dietético (30), En el estudio de Videla et al. 2004a, se observó un deterioro en los
mecanismos de defensa antioxidante evidenciado por menor actividad de enzimas
antioxidantes (superóxido dismutasa y catalasa), de la capacidad antioxidante del plasma
(FRAP: ferric reducing ability of plasma) y del contenido de glutatión reducido (GSH) en
pacientes con EHNA. Esto se ve exacerbado por el aumento de la actividad de la isoenzima
CYP2E1(41,32) de alta acción pro-oxidativa, por tanto se favorecen los procesos mediados por
los radicales libres, lo que conduce a un aumento significativo de la oxidación de lípidos y de
proteínas hepáticas. El aumento del estado pro-oxidante en el hígado de estos pacientes
favorece la lipoperoxidación de los AGPI, siendo más vulnerables los AGPICL, ubicados
preferentemente en las membranas celulares, debido a su mayor grado de insaturación. Esto
constituye un mecanismo alternativo que contribuye al agotamiento de los AGPICL y a la
alteración de la relación n-6/n-3 AGPICL (30,32,39,40). Estos datos evidencian un doble
mecanismo del estrés oxidativo en HGNA: 1) disminuyendo AGPICL n-3, lo que constituye
un mecanismo de señales que contribuye a la esteatosis y 2) contribuyendo a la progresión a
EHNA. (40) (Figura 9). En estudio de Araya J et al 2004, los pacientes con HGNA presentan
una depleción de los AGPI y AGPICL de las series n-6 y n-3 en TG hepáticos y una
aumentada relación n-6/n-3, mientras que en fosfolípidos hepáticos contienen más AGPICL n-
6 y menos n-3 y por ende se produce un marcado aumento en la relación n-6/n-3 AGPICL.
Esto último también se observó en tejido adiposo, junto a aumentados niveles de 18:1 n-9
trans, lo cual es un reflejo de la ingesta. Además, estos resultados se acompañaron de una
mayor oxidación de lípidos y proteínas hepáticas , evidenciados por un mayor contenido de
MDA y carbonilos, respectivamente. Estos hallazgos permiten concluir que la depleción de los

14
AGPICL, principalmente n-3, puede ser resultado tanto de un defecto en la desaturación de
AGPI, debido a una reducida ingesta de precursores n-3 (18:3 n-3) y una mayor ingesta de
18:1 n-9 trans, que conduce a inhibición de las desaturasas, como por un mayor consumo de
los AGPICL debido a estrés oxidativo.
1.3 Perfil de los ácidos grasos de las membranas celulares:
Dado que los ácidos grasos de la membranas celulares derivan del pool plasmático de ácidos
grasos (los cuales a su vez derivan principalmente de las grasas de la dieta), puede ser
hipotetizado que todas las membranas celulares tienen una composición similar de ácidos
grasos y que los cambios generados por el estrés oxidativo afectan de la misma forma al
patrón de ácidos grasos de todas las membranas celulares. Debido a la dificultad para obtener
biopsias hepáticas o de otros tejidos humanos, se ha planteado utilizar glóbulos rojos como
una alternativa representativa y más accesible para el estudio de la composición de ácidos
grasos en membranas celulares (42). De esta manera, recientes estudios in vivo han demostrado
una significativa correlación entre la composición de ácidos grasos de membranas
eritrocitarias y de células musculares, variando según el tipo de acido graso (43,44). Al respecto
el estudio de Felton C.V. et al 2004, realizado en hombres adultos sanos, demostró una
asociación positiva entre membranas eritrocitarias y de músculo esquelético en el porcentaje
de ácido palmítico (16:00), araquidónico (ARA) (20:4n-6), eicosapentanoico (EPA) (20:5 n-3)
y docosapentanoico (22:5 n-3). Además, en el subgrupo de sujetos con sensibilidad a la
insulina normal / baja, encontraron una correlación positiva entre la concentración del ácido
araquidónico y en AGPI totales. En estudio de Baur L.A. et al 2000, realizado en infantes < 2
años, también se demostró una correlación entre composición de ácidos grasos de membranas

15
eritrocitarias y de músculo, pero de menor magnitud, evidenciado en el total de AGPI n-3 y en
la relación n6/n3 AGPI y en DHA. Sin embargo, se requieren más estudios que avalen esta
relación entre membranas celulares, ya que en otro reciente estudio los hallazgos fueron
contrarios a lo esperado, demostrando que la composición de ácidos grasos de membranas
musculares y de eritrocitos son significativamente diferentes (42), aunque este estudio no
evaluó las correlaciones en estos dos tejidos. Connor et al 1990 demostraron la similitud entre
el perfil de los ácidos grasos del cerebro con los de la membrana de los eritrocitos de monos,
en condiciones de deficiencia de n-3, lo que fue revertido con una dieta adecuada en estos
ácidos grasos. Por otro lado, aún no existen datos que comparen la composición de ácidos
grasos de la membrana de eritrocitos con la de hepatocitos.
Sobre la base de este paradigma, en la presente tesis se postula la hipótesis que el perfil de
ácidos grasos de los fosfolípidos de la membrana del eritrocito refleja el perfil de los ácidos
grasos de los fosfolípidos hepáticos, lo que puede ser utilizado como un método no invasivo
para estimar los cambios en la homeostasis de los ácidos grasos de los fosfolípidos hepáticos
en el HGNA y como seguimiento de los cambios en los lípidos hepáticos en la baja de peso
post cirugía bariátrica en las pacientes obesas.

16
II. HIPÓTESIS DE TRABAJO:
Se conoce que la presencia de radicales libres puede modificar la composición de los ácidos
grasos de las membranas biológicas en general. En la presente tesis se tratará de demostrar la
hipótesis que:
1. Los AGPICL resultan ser los marcadores más sensibles de los efectos del estrés oxidativo
sobre los fosfolípidos de las membranas de hígado y eritrocito.
2. El estrés oxidativo produce en el hígado una alteración en el perfil de los ácidos grasos de
los fosfolípidos que también queda reflejada en el eritrocito, permitiendo que este último
pueda ser utilizado como biomarcador de los cambios en el perfil de los ácidos grasos
hepáticos a través de un procedimiento no invasivo.
III. OBJETIVOS:
1. OBJETIVOS GENERALES: En pacientes obesas sometidas a cirugía bariátrica:
1.1 Comparar el perfil de la composición de los ácidos grasos de los fosfolípidos del hígado
con el de la membrana de eritrocitos.
1.2 Evaluar el efecto del estrés oxidativo sobre la homeostasis de los AGPICL.
1.3 Implementar un procedimiento no invasivo para estimar en forma indirecta los efectos
del estrés oxidativo sobre el perfil de los ácidos grasos hepáticos.

17
2. OBJETIVOS ESPECÍFICOS: 2.1 En Hígado: • Determinar el perfil de los ácidos grasos en los fosfolípidos de membranas celulares.
• Identificar el papel del estrés oxidativo en el contenido de los ácidos grasos de los
fosfolípidos y lípidos totales.
• Determinar la actividad de enzimas antioxidantes.
• Determinar el contenido de glutatión reducido (GSH).
• Determinar la lipoperoxidación expresada en la producción de MDA.
2.2 En Plasma: • Determinar el perfil lipídico.
• Determinar la capacidad antioxidante total (FRAP).
• Determinar los niveles de F2-isoprostanos libres.
2.3 En Eritrocitos: • Determinar el perfil de los ácidos grasos de los fosfolípidos de la membrana eritrocitaria.

18
IV. METODOLOGÍA:
1. PACIENTES: Se seleccionaron 12 pacientes obesas con un IMC promedio de 43,8 kg/m2 (35-57,4) y edad
promedio de 41 años (27 a 56), quienes fueron sometidas voluntariamente a cirugía bariátrica.
Para el grupo control se seleccionaron 8 pacientes con IMC promedio 22,7 kg/m2 (20 a 24,7) y
edad promedio 44 años (23 a 63) quienes fueron sometidas a cirugías electivas abiertas
(colecistectomía, cirugía anti-reflujo, por cáncer gástrico, de esófago y cólon). El protocolo
fue explicado en detalle a las pacientes, quienes firmaron un consentimiento informado para
participar en este estudio y los procedimientos que implicó. Todas las pacientes seleccionadas
fueron no fumadoras y con ingesta alcohólica < 40 gr semanales.
Se consideraron criterios de exclusión: valores plasmáticos anormales de aminotransferasas y
gamaglutamil transpeptidasa (5 veces sobre el valor normal), serología positiva para virus
hepatitis B y C, glicemia de ayuno > 126 mg/dl, presencia de enfermedades crónicas,
tabaquismo y consumo de 3 ó más bebidas alcohólicas a la semana (> 40 g/semana). La
resistencia a la insulina fue calculada por el análisis de HOMA en base a los valores de
insulina y glicemia de ayuno (45).
2. DISEÑO: Las pacientes seleccionadas, 2 días previos a la cirugía , recibieron una dieta de 25 kcal/kg
con 30% de las calorías como lípidos y 15% de las calorías como proteínas. Durante la cirugía
se tomaron muestras de tejido hepático de aproximadamente 2 cm3, para diagnóstico
histológico, inmunohistoquímica, determinación de la composición de los ácidos grasos de los
fosfolípidos de membranas, parámetros de estrés oxidativo y de enzimas a nivel biomolecular.

19
Las muestras hepáticas fueron fijadas en formaldehído al 10%, incluidas en parafina y teñidas
con hematoxilina-eosina y Van-Gieson para estudio histológico, siendo agrupadas de acuerdo
a la presencia de esteatosis macrovesicular sola (Esteatosis) o esteatosis más inflamación
lobular con hepatocitos balonados (Esteatohepatitis) (46).
El protocolo del estudio fue aprobado por el Comité de Ética de la Investigación en Seres
Humanos de la Facultad de Medicina de la Universidad de Chile
3. PROCEDIMIENTOS:
3.1 Extracción de los lípidos hepáticos totales: Los lípidos totales del hígado se extrajeron según el método de Bligh y Dyer (47). En presencia
de BHT (hidroxitolueno butilado) como antioxidante. Un gramo de tejido se homogenizó en
una mezcla fría de cloroformo/metanol (2:1,v/v) que contenía 0.01 % (g/v) de BHT,
utilizando un homogenizador Ultraturrax (Janke & Kunkel, Stufen ,Germany).
3.2 Separación de los fosfolípidos de los lípidos totales del hígado: Para separar los diferentes lípidos extraídos del hígado y particularmente los fosfolípidos
hepáticos se aplicó cromatografía en capa fina recomendada por Skipski VP (48). Se utilizaron
láminas de aluminio cubiertas con sílica gel 20 x 20 60 F254, las que fueron activadas
previamente en un horno eléctrico a 105ºC durante una hora antes de aplicar las muestras
(50uL) de lípidos hepáticos totales solubilizados en una mezcla de metanol/cloroformo
(50uL). Como sistema de solventes para cromatografía en capa fina, su usó una mezcla de
hexano/dietiléter/ácido acético (80:20:1). Se aplicó simultáneamente a la placa cromatográfica
un estándar de fosfatidilcolina para identificar y comparar con la altura alcanzada por las

20
manchas de fosfolípidos de las muestras sembradas en la lámina. Después del desarrollo de las
láminas, los solventes impregnados en éstas se evaporaron bajo corriente de nitrógeno y las
bandas de los fosfolípidos fueron visualizadas exponiendo las láminas a una lámpara
ultravioleta CAMAG UV (250nm) diseñada especialmente para su uso en cromatografía en
placa fina. El sistema de solventes empleado separa fosfolípidos del colesterol, de los
triglicéridos y de los ésteres de colesterol basado en el orden de su relativa movilidad
ascendente en la mezcla de solventes empleados (49). La remoción de las manchas
correspondiente sólo a los fosfolípidos se raspó desde la lámina y se transfirió a un tubo de
centrífuga con una espátula pequeña. Cada muestra de polvo de sílica gel fue eluída con una
mezcla de cloroformo/metanol/agua (10:10:1 por vol). Después de centrifugar por 10 min. a
2500 rpm, el solvente fue removido cuidadosamente con una pipeta capilar. El solvente se
transfirió de inmediato directamente a los matraces aforados de 25 ml para derivatizar los
ácidos grasos a sus ésteres metílicos. Los ésteres metílicos fueron extraídos dos veces en
hexano (grado HPLC). Alícuotas del hexano se inyectaron con una pipeta Hamilton a un
cromatógrafo gaseoso (HP6890).
3.3 Separación de las membranas de los eritrocitos y extracción de sus lípidos: La sangre recién extraída con EDTA al 5% como anticoagulante , se centrifugó a 2500 rpm,
durante 15 minutos para obtener los glóbulos rojos. Las membranas de los glóbulos rojos se
separaron según Huertas et al (50), y los lípidos de las membranas se extrajeron según Bligh y
Dyer (47)

21
3.4 Preparación de los ésteres metílicos de los ácidos grasos de: Los ácidos grasos de los lípidos totales de hígado, de los fosfolípidos de hígado y de los
lípidos de membrana de glóbulos rojos, fueron metilados con Na/metanol 0.2 M por 30
minutos a 40ºC y luego con H2SO4/metanol 0.2 M como ha sido descrito para la metilación
alcalina.
3.5 Análisis de los esteres metílicos de los ácidos grasos:
• de los lípidos totales de hígado
• de los fosfolípidos del hígado
• de los lípidos de membranas aisladas de glóbulos rojos.
Los ésteres metílicos de los ácidos grasos de todas las muestras se analizaron por
Cromatografía Gaseosa, empleando un Cromatógrafo Hewlett-Packard modelo 6890,
equipado con una columna capilar apolar (50 m, x 0.22 mm; BPX 70) para separar los ésteres
metílicos. La temperatura fue programada desde los 65ºC hasta 230ºC como temperatura final,
a una velocidad de 2ºC/min con el propósito de separar los ésteres metílicos del C 10:0 hasta
el C 22:6n-3. Las temperaturas del detector e inyector fueron de 240ºC. Se usó Hidrogeno
como gas carrier a un flujo de 1.5 ml/min y un radio de split de 1:80. Los ésteres metílicos de
los respectivos ácidos grasos fueron identificados comparando sus tiempos de retención con
los de auténticos estándares y se cuantificaron usando un integrador Hewlett-Packard (HP
3396 Series III) (51). La cuantificación se hizo por la integración del área del pico de cada éster
metílico y los resultados fueron expresados como porcentajes del área total. El límite de
detección fue de 0.05% del área total.

22
El orden de aparición de los ácidos grasos fue el siguiente:
(1) 10:0; (2)12:0; (3) 14:0; (4) 14:1n-5; (5) 15:0; (6) 16:0; (7) 16:1n-7; (8) 17:0; (9) 18:0; (10) 18:1 n-9; (11) 18:2 n-6; (12) 18:3 n-6; (13) 18:3 n-3; (14) 18:4 n-3; (15) 20:0; (16) 20:1 n-11; (17) 20:1 n-9; (18) 20:1 n-7; (19) 20:2 n-6; (20) 20:3 n-6; (21) 20:4 n-6; (22) 20:3 n-3; (23) 20:4 n-3; (24) 20:5 n-3; (25) 22:0; (26) 22:1 n-9; (27) 22:2 n-6; (28) 22:4 n-6; (29) 22:5 n-6; (30) 22:5 n-3; (31) 24:0; (32) 22:6 n-3; (33) 24:1 n-9.
3.6 Análisis de lipoproteínas plasmáticas: Se determinaron en plasma: colesterol total, triglicéridos y colesterol-HDL por método
enzimático (Boehringer-Mannheim, Roche Diagnostic GMBH. D-68298, Mannheim,
Germany). El colesterol-LDL fue calculado por la formula:
Colesterol LDL(mg/dl)= colesterol total – HDL – TG/5
3.7 Determinación de estrés oxidativo:
3.7.1 Capacidad antioxidante total del plasma (FRAP):
Se midió la habilidad del plasma para reducir el hierro férrico a ferroso siguiendo la
cinética a 593 nm (52).
3.7.2 Lipoperoxidación:
Se midió el efecto del estrés oxidativo sobre la producción de peróxidos de lípidos de
membranas, expresado como nmol de malondialdehido (MDA)/mg proteína de
acuerdo a Ohkawa H. (53)

23
3.7.3 F2 isosprotanos:
Se determinaron en plasma, por espectrometría de masas y cromatografía de gas, los
productos formados in vivo de la peroxidación no enzimática del ácido araquidónico
catalizada por radicales libres (54).
3.7.4 Contenido GSH en hígado:
Medido por el método de Tietze F. (55) y los resultados fueron expresados como
nmol/mg proteína. Las proteínas fueron medidas de acuerdo al método de Lowry O.H.
et al (56).
3.7.5 Actividad de enzimas antioxidantes en hígado:
Se estudio estas enzimas con el propósito de relacionar su actividad con las
alteraciones encontradas en el perfil de los ácidos grasos de los fosfolípidos de
membranas del hígado y del glóbulo rojo:
a) Catalasa: Se determino por método espectrofotométrico siguiendo la cinética de
descomposición del peróxido de hidrogeno a 240 nm (57) y se calculo la constante
cinética para la reacción de primer orden (k).
b) Superóxido dismutasa (SOD): Se midió la cinética de activación de la auto-oxidación
de catecoles tetracíclicos, por espectrofotometría a 525 nm (58).
c) Glutatión peroxidasa: Se midió por técnica espectrofotométrica la cinética de
oxidación de NADPH acoplada a la reducción de GSSG a GSH, a 340 nm (59).

24
4. ANÁLISIS ESTADÍSTICO:
Los resultados fueron expresados como promedios ± SEM para el número de pacientes
indicados. El análisis estadístico de los datos fue realizado usando el sistema de análisis
estadístico SPSS versión 12.0. Para comparación entre pacientes controles y pacientes HGNA
pre-cirugía en composición de ácidos grasos de los lípidos totales, fosfolípidos hepáticos,
fosfolípidos de eritrocitos y parámetros de estrés oxidativo se uso la prueba de Mann-Whitned.
Para comparación entre pacientes con HGNA pre y post-cirugía bariátrica en composición de
fosfolípidos de eritrocitos y parámetros de estrés oxidativo se usó la prueba de Wilcoxon. Las
diferencias fueron consideradas estadísticamente significativas con p<0,05.

25
V. RESULTADOS:
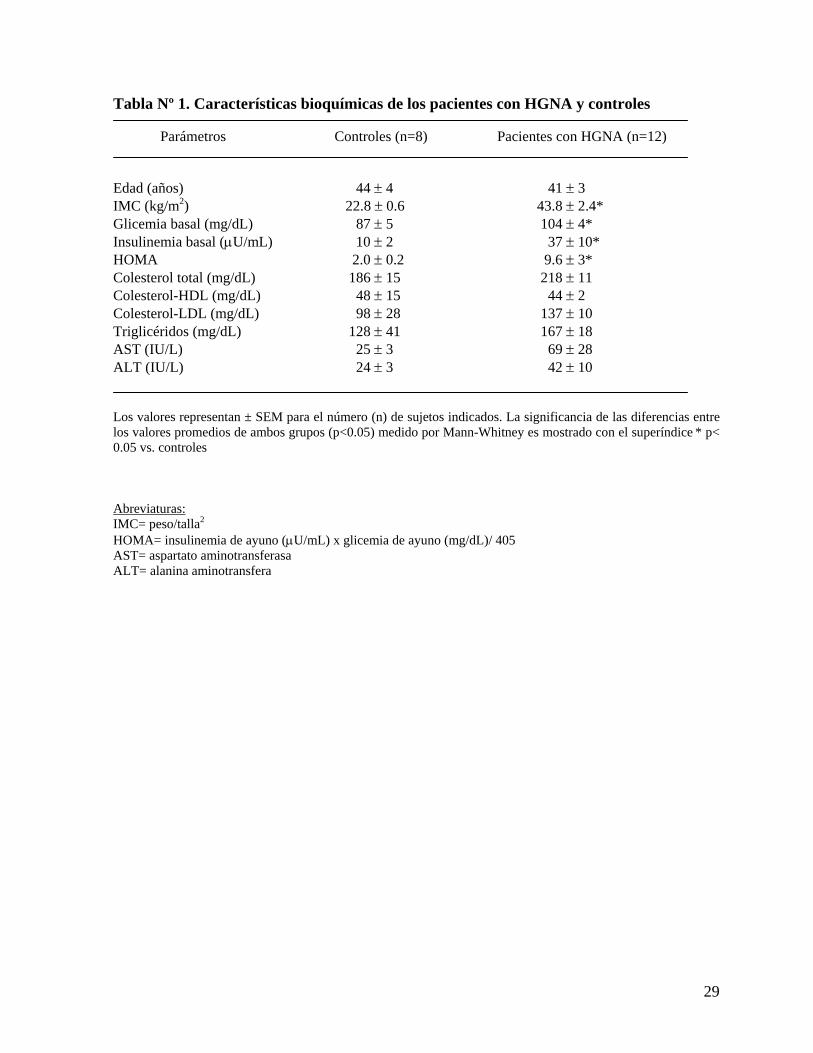
1. Características clínicas y bioquímicas de las pacientes con HGNA y controles: Se distinguieron obesas y controles estimado por IMC (43.8 ± 2.4 vs 22.8 ± 0.6, p<0.05). Las
pacientes obesas con HGNA presentan mayores niveles de insulinemia en ayuna (37 ± 10 vs
10 ± 2) y una marcada resistencia a la insulina calculada por HOMA (9.6 ± 3 vs 2.0 ± 0.2,
p<0.05). La glicemia de ayunas está levemente aumentada en estas pacientes con HGNA, pero
sin alcanzar rangos de diabetes (104 ± 4 vs 87 ± 5, p<0.05). No se encontraron diferencias
significativas en el contenido de lipoproteínas plasmáticas (colesterol total, HDL, LDL,
triglicéridos) y en las transaminasas hepáticas (AST y ALT) entre el grupo HGNA y el grupo
control. Los resultados se muestran en la Tabla 1.
2. Perfil de los ácidos grasos de los lípidos hepáticos totales: La composición de los ácidos grasos de los fosfolípidos hepáticos en las pacientes con HGNA,
en comparación con el grupo control, revela un menor contenido de los AGPI 20:4 n-6 (32%
menos) y 22:6 n-3 (48% menos), mayor contenido del ácido graso 22:5 n-6 y una mayor
relación n-6/n-3 AGPI (124 %más). Los resultados se muestran en la Tabla 2.
3. Composición de los ácidos grasos de los fosfolípidos hepáticos: La composición de los ácidos grasos de los fosfolípidos hepáticos en las pacientes con
HGNA, en comparación con el grupo control, revela que los primeros tienen un mayor
contenido de ácidos grasos saturados totales (32% más) y un menor contenido de ácidos
grasos poli-insaturados con un 45% menos de ácido linoléico (18:2 n-6), 48% menos de ácido

26
araquidónico (20:4 n-6), 57% menos de ácido eicosapentaenoico (20:5 n-3), 70% menos de
ácido docosahexaenoico (22:6 n-3), 27% menos del total de AGPI, 61% menos del total AGPI
n-3 y 63% menos del total AGPICL n-3, acompañado de una relación n6/n3 AGPICL 211%
mayor (p<0.05) y presencia de un ácido graso poli-insaturado de la serie n-6 que no se
evidenció en el grupo control, el ácido graso Osbond (22:5 n-6). Las pacientes con HGNA no
mostraron diferencias significativas con el grupo control en el contenido total de ácidos grasos
monoinsaturados, en el ácido graso α linolénico (18:3 n-3), en el total AGPI n-6 y en el total
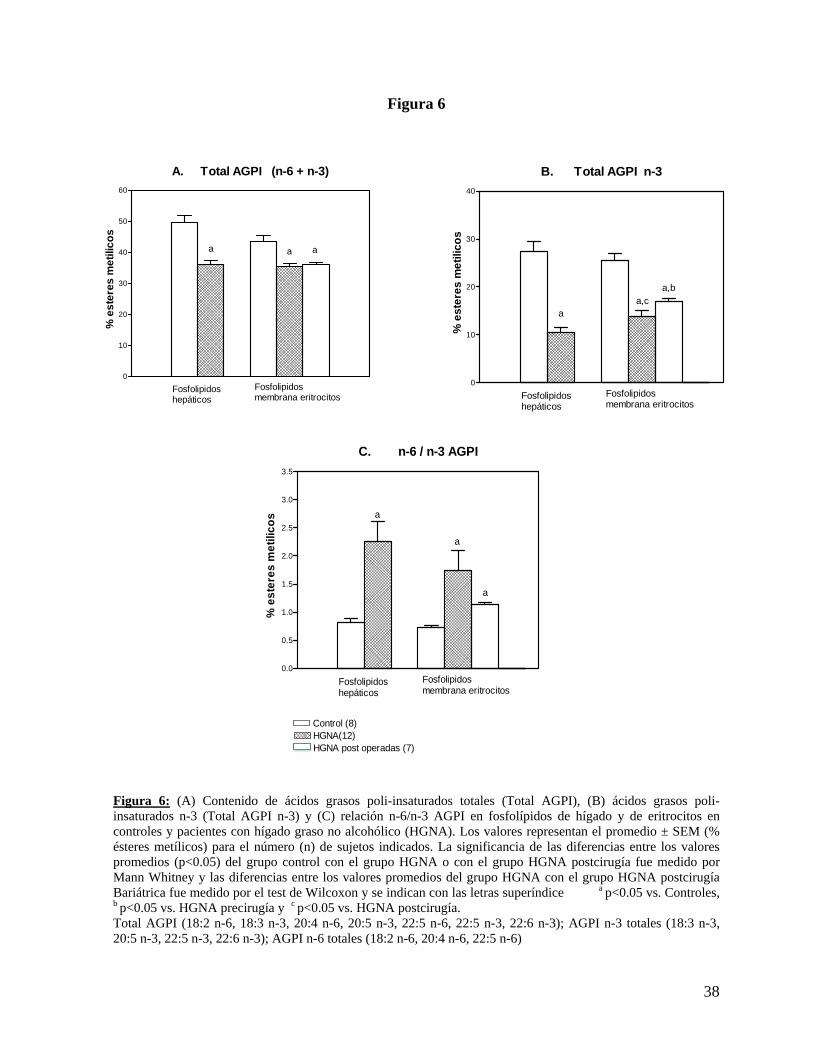
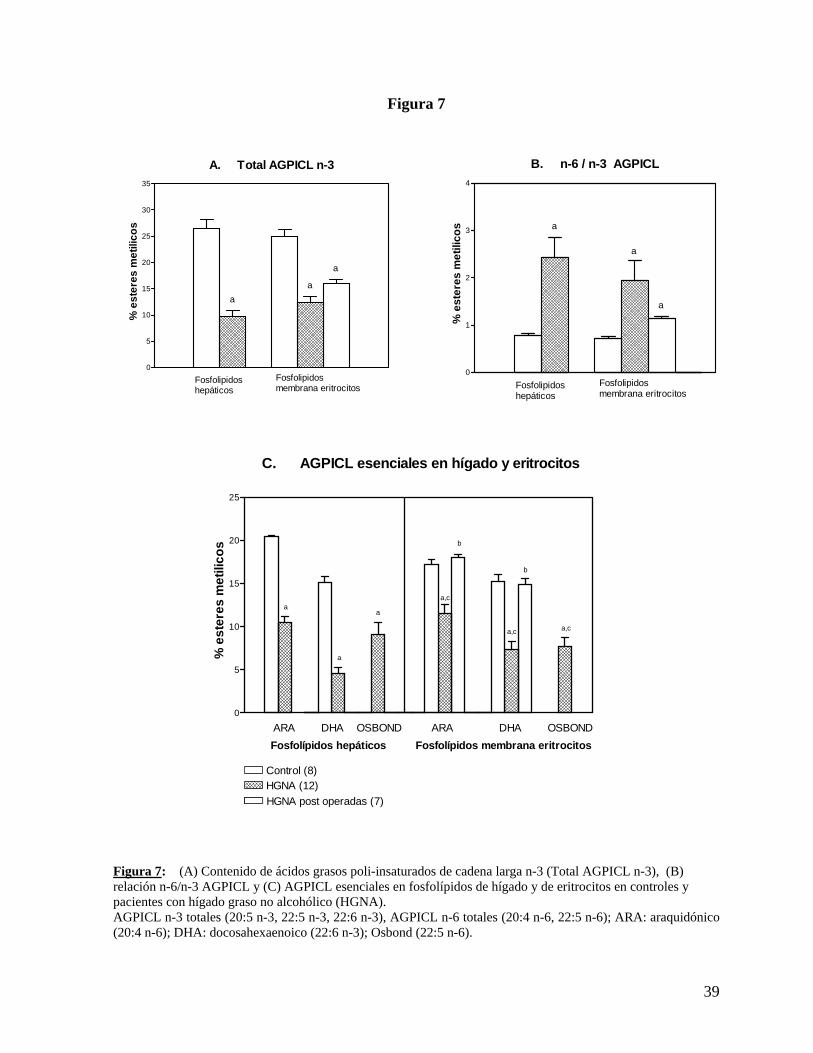
AGPICL n.-6. Los resultados se muestran en la Tabla 3 y Figuras 6 y 7.
4. Composición de los ácidos grasos de la membrana de eritrocitos: Con respecto al grupo control, las pacientes con HGNA presentan un contenido de AGPI
totales, AGPI n-3 totales, AGPICL n-3 totales 19%, 46% y 50% menor, respectivamente y un
contenido de los ácidos grasos 20:4 n-6 y 22:6 n-3 33% y 52% menor, respectivamente.
Además, estos cambios se acompañan de una mayor relación n-6/n-3 AGPICL (del 173%) y
presencia del ácido graso 22:5 n-6 en el grupo HGNA. Estos hallazgos son similares a los
encontrados en los fosfolípidos hepáticos. Otro hallazgo significativo fue el mayor contenido
total de ácidos grasos monoinsaturados (69% mayor) en estos pacientes. En cuanto a los
ácidos grasos esenciales precursores sus niveles son mayores en las pacientes con HGNA, con
una diferencia de 170% en el 18:2 n-6 y de 157% en el 18:3 n-3. Las pacientes con HGNA no
mostraron diferencias significativas, con el grupo control, en el contenido de 20:5 n-3, total de
ácidos grasos saturados, total AGPI n-6 y total AGPICL n-6.
Las pacientes con HGNA post-cirugía bariátrica presentaron cambios en la composición de los
ácidos grasos de membrana de eritrocitos, comparado con las pacientes con HGNA pre-

27
cirugía, asemejándose al perfil del grupo control. Estos cambios fueron: aumento del ácido
graso 20:4 n-6 (en 51%), 22:6 n-3 (en 142%), total AGPI n-3 (en 39%) y total AGPICL n-3
(en 46%) y la disminución de los ácidos 22:5 n-6 y 18:2 n-6 (en 100% y 58%,
respectivamente). La relación n-6/n-3 AGPICL presentó una disminución del 52%, pero sin
alcanzar significancia estadística. No mostraron cambios en el contenido de ácidos grasos
saturados. Los resultados se muestran en la Tabla 4 y Figuras 6 y 7.
5. Perfil de AGPI hepáticos y relación producto/precursor de la serie n-3 y n-6: Los pacientes con HGNA presentan una relación producto/precursor (20:5 n-3 + 22:6 n-3)
/18:3n-3 57% menor en los fosfolípidos hepáticos. Esto se acompañó también de una menor
relación producto/precursor 20:4n-6/18:2n-6 y (20:5n-3 + 22:6n-3) /18:3 n-3 en la membrana
de eritrocitos del 90% y 78%, respectivamente. El grupo de pacientes con HGNA postcirugía
bariátrica presentó un aumento significativo (p<0.05) de esta relación producto/precursor tanto
para los AGPI n-3 como para los n-6 en la membrana de eritrocitos, al compararse con el
grupo HGNA precirugía.
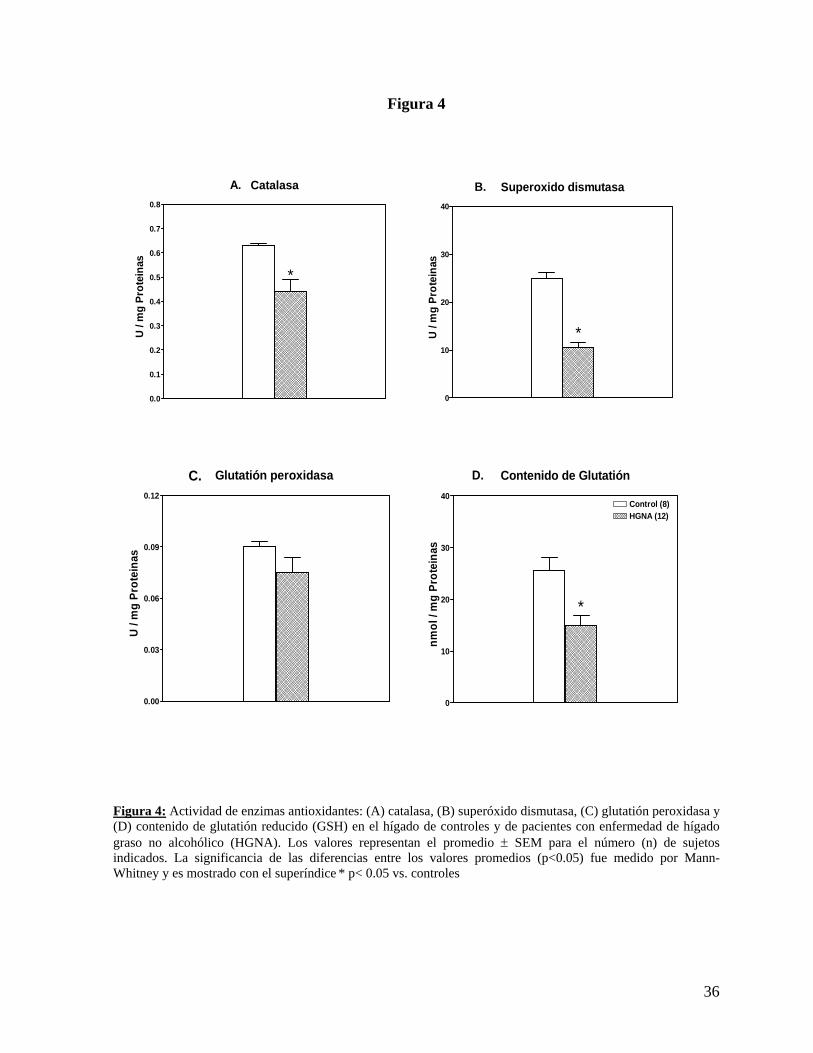
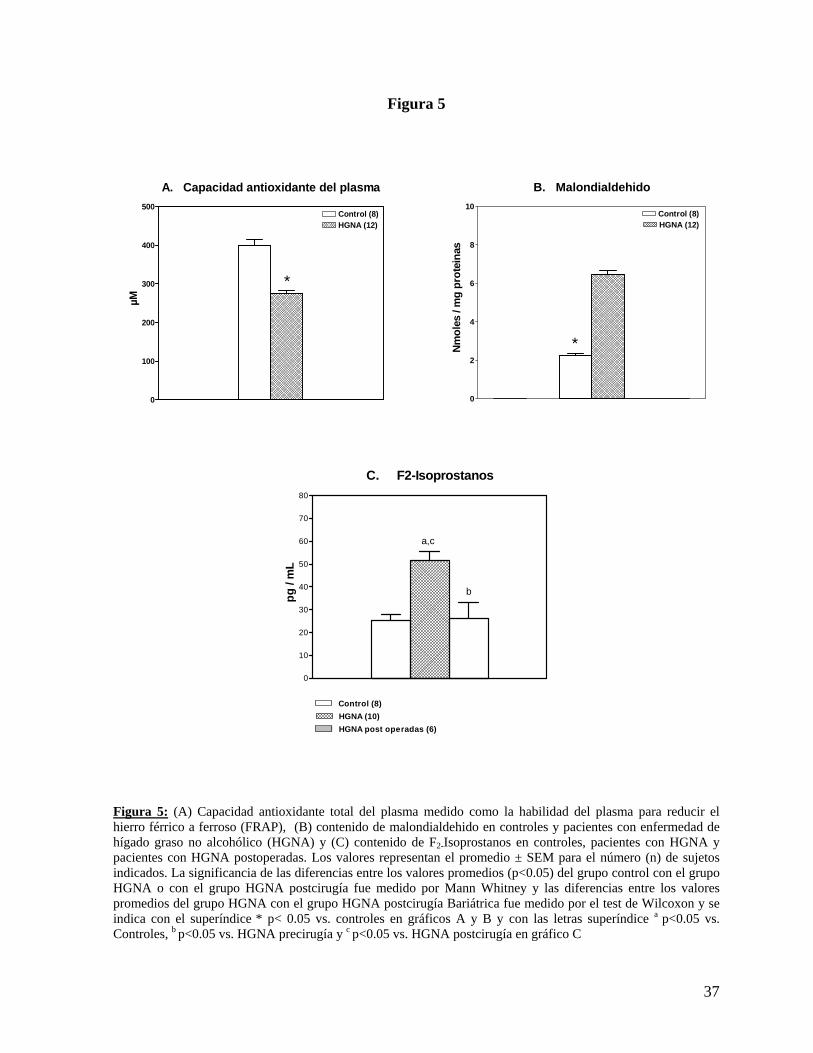
6. Parámetros relacionados con estrés oxidativo hepático: Las pacientes obesas con HGNA presentan un significativo mayor nivel de estrés oxidativo,
evidenciado por menor contenido de glutatión (41% menos), menor actividad de enzimas
antioxidantes, tales como superóxido dismutasa y catalasa (58% y 30% menos,
respectivamente) (Figura 4) y menor FRAP (31% menos) comparado con el grupo control
(p<0.05%) (Figura 5 A). Además, presentan un mayor índice de lipoperoxidación, evidenciado
por un mayor contenido de MDA y de F2I (185% y 85% más elevados, respectivamente),

28
comparado con el grupo control (Figura 5 B y C). El grupo de pacientes HGNA post-cirugía
bariátrica presento una reducción significativa (p<0.05) del estrés oxidativo evidenciado por
reducción del 40% en los niveles de F2I, comparado con el grupo HGNA pre-cirugía,
alcanzando incluso valores semejantes al grupo control.
29
Tabla Nº 1. Características bioquímicas de los pacientes con HGNA y controles
Parámetros Controles (n=8) Pacientes con HGNA (n=12) Edad (años) 44 ± 4 41 ± 3 IMC (kg/m2) 22.8 ± 0.6 43.8 ± 2.4* Glicemia basal (mg/dL) 87 ± 5 104 ± 4* Insulinemia basal (μU/mL) 10 ± 2 37 ± 10* HOMA 2.0 ± 0.2 9.6 ± 3* Colesterol total (mg/dL) 186 ± 15 218 ± 11 Colesterol-HDL (mg/dL) 48 ± 15 44 ± 2
Colesterol-LDL (mg/dL) 98 ± 28 137 ± 10 Triglicéridos (mg/dL) 128 ± 41 167 ± 18 AST (IU/L) 25 ± 3 69 ± 28 ALT (IU/L) 24 ± 3 42 ± 10 Los valores representan ± SEM para el número (n) de sujetos indicados. La significancia de las diferencias entre los valores promedios de ambos grupos (p<0.05) medido por Mann-Whitney es mostrado con el superíndice * p< 0.05 vs. controles Abreviaturas: IMC= peso/talla2 HOMA= insulinemia de ayuno (μU/mL) x glicemia de ayuno (mg/dL)/ 405
AST= aspartato aminotransferasa ALT= alanina aminotransfera

30
Tabla Nº 2. Perfil de los ácidos grasos de los lípidos totales hepáticos Ácidos grasos Controles (8) Pacientes con HGNA(12) (% ésteres metílicos) 18:1n-9 16.47 ± 0.53 18.36 ± 1.1 18:2n-6 0.47 ± 0.15 0.67 ± 0.08 18:3n-3 0.3 ± 0.1 0.48 ± 0.09 20:4n-6 (ARA) 7.99 ± 0.71 5.4 ± 0.69 * 20:5n-3 (EPA) 0.83 ± 0.42 0.7 ± 0.19 22:5n-6 (Osbond) 0 3.07 ± 1.08 * 22:6n-3 (DHA) 6.87 ± 1.26 3.55 ± 0.67 * Total Saturados 60.01 ± 2.76 62.03 ± 2.45 Total Monoinsaturados 23.54 ± 4.52 23.09 ± 1.37
Total AGPI 16.45 ± 1.88 14.88 ± 2.07 AGPI n-6 totales 8.46 ± 0.73 9.13 ± 1.58 AGPI n-3 totales 8.0 ± 1.48 4.73 ± 0.87
AGPI n-6/n-3 1.21 ± 0.27 2.72 ± 0.92 * Los valores representan el promedio ± SEM (% ésteres metílicos) para el número (n) de sujetos indicados. La significancia de las diferencias entre los valores promedios (p<0.05) medido por Mann-Whitney es mostrado con el superíndice * p< 0.05 vs. controles Abreviaturas: AGPI=ácidos grasos poli-insaturados Total Saturados (14:00, 16:00, 18:0, 20:00, 22:00, 24:00) Total Monoinsaturados (14:1 n-7, 16:1 n-7, 18:1 n-9, 22:1 n-9, 24:1 n-9) Total AGPI (18:2 n-6, 18:3 n-3, 20:4 n-6, 20:5 n-3, 22:5 n-6, 22:6 n-3, 22:2 n-9) AGPI n-6 totales (18:2 n-6, 20:4 n-6, 22:5 n-6) AGPI n-3 totales (18:3 n-3, 20:5 n-3, 22:6 n-3) AGPI n-6/n-3 (18:2 n-6+20:4 n-6+22:5 n-6 / 18:3 n-3+20:5 n-3+22:6 n-3)

31
Tabla Nº 3. Perfil de los ácidos grasos de los fosfolípidos hepáticos Ácidos grasos Controles (n=8) Pacientes con HGNA (n=12) (% ésteres metílicos) 14:0 0.68 ± 0.15 1.31 ± 0.28 14:1n-7 0.32 ± 0.16 0.88 ± 0.31 16:0 13.1 ± 1.71 16.35 ± 0.77 16:1n-7 3.72 ± 0.08 4.68 ± 0.96 18:0 9.0 ± 1.49 15.46 ± 0.82 * 18:1n-9 8.26 ± 0.73 10.91 ± 0.84 18:2n-6 1.7 ± 0.42 0.92 ± 0.14 * 18:3n-3 1.05 ± 0.32 0.81 ± 0.11 20:0 1.96 ± 0.55 2.45 ± 0.41 20:4n-6 (ARA) 20.43 ± 0.2 10.55 ± 0.67 * 20:5n-3 (EPA) 4.8 ± 0.89 2.04 ± 0.23 * 22:0 2.94 ± 0.75 1.8 ± 0.29 22:5n-6 (Osbond) 0 9.07 ± 1.47 * 22:5n-3 6.53 ± 1.33 3.14 ± 0.64 * 22:6n-3 (DHA) 15.14 ± 0.73 4.58 ± 0.64 * Total Saturados 31.03 ± 1.89 41.06 ± 0.93 * Total Monoinsaturados 19.33 ± 1.09 22.8 ± 1.5
Total AGPI 49.64 ± 2.15 36.14 ± 1.4 * AGPI n-6 totales 22.13 ± 0.39 20.54 ± 1.37 AGPI n-3 totales 27.51 ± 1.99 10.57 ± 1.04 *
AGPI n-6/n-3 0.82 ± 0.06 2.26 ± 0.35 * Total AGPICL 46.89 ± 1.62 29.38 ± 1.16 * AGPICL n-6 totales 20.43 ± 0.20 19.62 ± 1.38 AGPICL n-3 totales 26.46 ± 1.72 9.76 ± 1.06 *
AGPICL n-6/n-3 0.78 ± 0.05 2.43 ± 0.42 * Los valores representan el promedio ± SEM (% ésteres metílicos) para el número (n) de sujetos indicados. La significancia de las diferencias entre los valores promedios (p<0.05) medido por Mann-Whitney es mostrado con el superíndice * p< 0.05 vs. controles Abreviaturas: AGPI=ácidos grasos poli-insaturados AGPICL=ácidos grasos poli-insaturados de cadena larga. Total Saturados (14:00, 16:00, 18:0, 20:00, 22:00, 24:00) Total Monoinsaturados (14:1 n-7, 16:1 n-7, 18:1 n-9, 22:1 n-9, 24:1 n-9) Total AGPI (18:2 n-6, 18:3 n-3, 20:4 n-6, 20:5 n-3, 22:5 n-6, 22:5 n-3, 22:6 n-3) Total AGPICL (20:4 n-6, 20:5 n-3, 22:5 n-6, 22:5 n-3, 22:6 n-3) AGPI n-6 totales (18:2 n-6, 20:4 n-6, 22:5 n-6) AGPI n-3 totales (18:3 n-3, 20:5 n-3, 22:5 n-3, 22:6 n-3) AGPICL n-6 totales (20:4 n-6, 22:5 n-6) AGPICL n-3 totales (20:5 n-3, 22:5 n-3, 22:6 n-3)

32
Tabla Nº 4. Perfil de los ácidos grasos de los fosfolípidos de membranas de eritrocitos Ácidos grasos Controles HGNA precirugía HGNA postcirugía (% ésteres metílicos) (n=8) (n=12) (n=7) 14:0 0.88 ± 0.14 1.15 ± 0.37 0.7 ± 0.24 14:1n-7 1.08 ± 0.1 2.0 ± 0.44 1.39 ± 0.27
16:0 16.81 ± 0.86 13.86 ± 1.02 16.87 ± 1.15 16:1n-7 2.27 ± 0.19 2.94 ± 0.3 c 6.16 ± 1.66 ab 18:0 19.51 ± 1.37 15.42 ± 1.13 21.66 ± 1.63 18:1n-9 7.81 ± 0.44 10.1 ± 0.67 a 7.92 ± 0.77 18:2n-6 0.85 ± 0.12 2.29 ± 0.41 ac 0.97 ± 0.23 b 18:3n-3 0.58 ± 0.1 1.49 ± 0.26 a 0.89 ± 0.28 20:0 1.24 ± 0.37 2.86 ± 0.48 a 0.62 ± 0.47 20:4n-6 (ARA) 17.21 ± 0.54 11.56 ± 0.98 ac 18.02 ± 0.38 b 20:5n-3 (EPA) 2.61 ± 0.55 2.87 ± 0.45 1.1 ± 0.26 a 22:0 1.32 ± 0.26 2.71 ± 0.49 a 0.95 ± 0.2 22:5n-6 (Osbond) 0 7.75 ± 1.02 ac 0 b 22:6n-3 (DHA) 15.25 ± 0.86 7.37 ± 0.92 ac 14.95 ± 0.6 b Total Saturados 42.12 ± 1.75 40.45 ± 1.38 45.72 ± 2.06 Total Monoinsaturados 14.26 ± 0.68 24.07 ± 1.66 a 18.36 ± 1.49 a
Total AGPI 43.62 ± 1.89 35 49 ± 1.07 a 35.93 ± 0.91 a
AGPI n-6 totales 18.06 ± 0.57 21.6 ± 1.97 18.99 ± 0.36 AGPI n-3 totales 25.56 ± 1.4 13.89 ± 1.4 ac 16.94 ± 0.76 ab
AGPI n-6/n-3 0.73 ± 0.04 1.84 ± 0.35 a 1.13 ± 0.05 a Total AGPICL 42.18 ± 1.78 31.12 ± 1.68 a 34.07 ± 0.76 a AGPICL n-6 totales 17.21 ± 0.54 19.31 ± 1.64 18.02 ± 0.37 AGPICL n-3 totales 24.97 ± 1.4 12.4 ± 1.15 a 16.05 ± 0.63 a
AGPICL n-6/n-3 0.71 ± 0.04 1.94 ± 0.43 a 1.13 ± 0.05 a Los valores representan el promedio ± SEM (% ésteres metílicos) para el número (n) de sujetos indicados. La significancia de las diferencias entre los valores promedios (p<0.05) del grupo control con el grupo HGNA o con el grupo HGNA postcirugía fue medido por Mann Whitney y las diferencias entre los valores promedios del grupo HGNA con el grupo HGNA postcirugía Bariátrica fue medido por el test de Wilcoxon y se indican con las letras superíndice a p<0.05 vs. Controles, b p<0.05 vs. HGNA precirugía y c p<0.05 vs. HGNA postcirugía Abreviaturas: AGPI=ácidos grasos poli-insaturados AGPICL=ácidos grasos poli-insaturados de cadena larga. Total Saturados (14:00, 16:00, 18:0, 20:00, 22:00, 24:00) Total Monoinsaturados (14:1 n-7, 16:1 n-7, 18:1 n-9, 22:1 n-9, 24:1 n-9) Total AGPI (18:2 n-6, 18:3 n-3, 20:4 n-6, 20:5 n-3, 22:5 n-6, 22:5 n-3, 22:6 n-3); Total AGPICL (20:4 n-6, 20:5 n-3, 22:5 n-6, 22:5 n-3, 22:6 n-3) AGPI n-6 totales (18:2 n-6, 20:4 n-6, 22:5 n-6) AGPI n-3 totales (18:3 n-3, 20:5 n-3, 22:5 n-3, 22:6 n-3) AGPICL n-6 totales (20:4 n-6, 22:5 n-6) AGPICL n-3 totales (20:5 n-3, 22:5 n-3, 22:6 n-3)
33
Figura 1
Figura 1: Resolución de los ésteres metílicos de membranas de eritrocitos (A) y de los fosfolípidos de hígado aislados de los lípidos de mujeres controles (B) en una columna capilar. (S) solvente, BHT (hidroxitolueno butilato), EI (estandar interno). Los picos incluyen a los ácidos grasos desde el C10 al C24:1 n-9. La detección de los ácidos grasos poli-insaturados de cadena larga n-6 y n-3 corresponden a los números en paréntesis: 20:4 n-6 (21), 20:5 n-3 (24), 22:5 n-6 (29), 22:5 n-3 (30), 22:6 n-3 (32) Los tiempos de retención fueron evaluados basados en el análisis de estándares comerciales auténticos. La cuantificación de los ésteres metílicos se efectuó integrando el área del pico de cada metil éster en un integrador HP 3396, Hewlett-Packard Series III USA.
A
B

34
Figura 2
Figura 2: Resolución de los ésteres metílicos de membranas de eritrocitos (C) y de los fosfolípidos de hígado (D) aislados de los lípidos de pacientes obesas con hígado graso no alcohólico, en una columna capilar. (S) solvente, BHT (hidroxitolueno butilato), EI (estandar interno). Los picos incluyen a los ácidos grasos desde el C10 al C24:1 n-9. La detección de los ácidos grasos poli-insaturados de cadena larga n-6 y n-3 corresponden a los números en paréntesis: 20:4 n-6 (21), 20:5 n-3 (24), 22:5 n-6 (29), 22:5 n-3 (30), 22:6 n-3 (32) Los tiempos de retención fueron evaluados basados en el análisis de estándares comerciales auténticos. La cuantificación de los ésteres metílicos se efectuó integrando el área del pico de cada metil éster en un integrador HP 3396, Hewlett-Packard Series III USA
C
D

35
Figura 3
Figura 3: Resolución de los ésteres metílicos de membranas de eritrocitos de pacientes obesas con hígado graso no alcohólico, tres meses post cirugía bariátrica (E), en una columna capilar. (S) solvente, BHT (hidroxitolueno butilato), EI (estandar interno). Los picos incluyen a los ácidos grasos desde el C10 al C24:1 n-9. La detección de los ácidos grasos poli-insaturados de cadena larga n-6 y n-3 corresponden a los números en paréntesis: 20:4 n-6 (21), 20:5 n-3 (24), 22:5 n-6 (29), 22:5 n-3 (30), 22:6 n-3 (32) Los tiempos de retención fueron evaluados basados en el análisis de estándares comerciales auténticos. La cuantificación de los ésteres metílicos se efectuó integrando el área del pico de cada metil éster en un integrador HP 3396, Hewlett-Packard Series III USA.
E
36
Figura 4
Figura 4: Actividad de enzimas antioxidantes: (A) catalasa, (B) superóxido dismutasa, (C) glutatión peroxidasa y (D) contenido de glutatión reducido (GSH) en el hígado de controles y de pacientes con enfermedad de hígado graso no alcohólico (HGNA). Los valores representan el promedio ± SEM para el número (n) de sujetos indicados. La significancia de las diferencias entre los valores promedios (p<0.05) fue medido por Mann-Whitney y es mostrado con el superíndice * p< 0.05 vs. controles
Superoxido dismutasa
0
10
20
30
40
*U /
mg
Prot
eina
s
B.
Glutatión peroxidasa
0.00
0.03
0.06
0.09
0.12
U /
mg
Prot
eina
s
C. Contenido de Glutatión
0
10
20
30
40Control (8)HGNA (12)
*
nmol
/ m
g Pr
otei
nas
D.
Catalasa
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
*
U /
mg
Prot
eina
s
A.
37
Figura 5
Figura 5: (A) Capacidad antioxidante total del plasma medido como la habilidad del plasma para reducir el hierro férrico a ferroso (FRAP), (B) contenido de malondialdehido en controles y pacientes con enfermedad de hígado graso no alcohólico (HGNA) y (C) contenido de F2-Isoprostanos en controles, pacientes con HGNA y pacientes con HGNA postoperadas. Los valores representan el promedio ± SEM para el número (n) de sujetos indicados. La significancia de las diferencias entre los valores promedios (p<0.05) del grupo control con el grupo HGNA o con el grupo HGNA postcirugía fue medido por Mann Whitney y las diferencias entre los valores promedios del grupo HGNA con el grupo HGNA postcirugía Bariátrica fue medido por el test de Wilcoxon y se indica con el superíndice * p< 0.05 vs. controles en gráficos A y B y con las letras superíndice a p<0.05 vs. Controles, b p<0.05 vs. HGNA precirugía y c p<0.05 vs. HGNA postcirugía en gráfico C
A. Capacidad antioxidante del plasma
0
100
200
300
400
500Control (8)HGNA (12)
*
µM
B. Malondialdehido
0
2
4
6
8
10Control (8)HGNA (12)
*Nmol
es /
mg
prot
eina
s
C. F2-Isoprostanos
0
10
20
30
40
50
60
70
80
a,c
HGNA post operadas (6)
Control (8)HGNA (10)
bpg /
mL
38
Figura 6
Figura 6: (A) Contenido de ácidos grasos poli-insaturados totales (Total AGPI), (B) ácidos grasos poli-insaturados n-3 (Total AGPI n-3) y (C) relación n-6/n-3 AGPI en fosfolípidos de hígado y de eritrocitos en controles y pacientes con hígado graso no alcohólico (HGNA). Los valores representan el promedio ± SEM (% ésteres metílicos) para el número (n) de sujetos indicados. La significancia de las diferencias entre los valores promedios (p<0.05) del grupo control con el grupo HGNA o con el grupo HGNA postcirugía fue medido por Mann Whitney y las diferencias entre los valores promedios del grupo HGNA con el grupo HGNA postcirugía Bariátrica fue medido por el test de Wilcoxon y se indican con las letras superíndice a p<0.05 vs. Controles, b p<0.05 vs. HGNA precirugía y c p<0.05 vs. HGNA postcirugía. Total AGPI (18:2 n-6, 18:3 n-3, 20:4 n-6, 20:5 n-3, 22:5 n-6, 22:5 n-3, 22:6 n-3); AGPI n-3 totales (18:3 n-3, 20:5 n-3, 22:5 n-3, 22:6 n-3); AGPI n-6 totales (18:2 n-6, 20:4 n-6, 22:5 n-6)
C. n-6 / n-3 AGPI
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
Control (8)HGNA(12)HGNA post operadas (7)
Fosfolipidoshepáticos
Fosfolipidosmembrana eritrocitos
a
a
a
% e
ster
es m
etili
cos
A. Total AGPI (n-6 + n-3)
0
10
20
30
40
50
60
Fosfolipidoshepáticos
Fosfolipidosmembrana eritrocitos
a a a
% e
ster
es m
etili
cos
B. Total AGPI n-3
0
10
20
30
40
Fosfolipidoshepáticos
Fosfolipidosmembrana eritrocitos
aa,c
a,b
% e
ster
es m
etili
cos
39
Figura 7
Figura 7: (A) Contenido de ácidos grasos poli-insaturados de cadena larga n-3 (Total AGPICL n-3), (B) relación n-6/n-3 AGPICL y (C) AGPICL esenciales en fosfolípidos de hígado y de eritrocitos en controles y pacientes con hígado graso no alcohólico (HGNA). AGPICL n-3 totales (20:5 n-3, 22:5 n-3, 22:6 n-3), AGPICL n-6 totales (20:4 n-6, 22:5 n-6); ARA: araquidónico (20:4 n-6); DHA: docosahexaenoico (22:6 n-3); Osbond (22:5 n-6).
A. Total AGPICL n-3
0
5
10
15
20
25
30
35
Fosfolipidoshepáticos
Fosfolipidosmembrana eritrocitos
aa
a
% e
ster
es m
etili
cos
B. n-6 / n-3 AGPICL
0
1
2
3
4
Fosfolipidoshepáticos
Fosfolipidosmembrana eritrocitos
a
a
a
% e
ster
es m
etili
cos
0
5
10
15
20
25
Control (8)HGNA (12)
ARA DHA OSBOND ARA DHA OSBOND
a
a
a
a,c
b
a,c
b
a,c
HGNA post operadas (7)
Fosfolípidos hepáticos Fosfolípidos membrana eritrocitos
C. AGPICL esenciales en hígado y eritrocitos
% e
ster
es m
etili
cos

40
VI. DISCUSIÓN:
Los datos obtenidos en este estudio revelan que los pacientes con HGNA tienen una
importante depleción de AGPI, particularmente los AGPICL de la serie n-3 en los fosfolípidos
hepáticos, con una elevada relación n-6/n-3 de AGPICL. La depleción hepática de los
AGPICL presente en estos pacientes puede ser un importante factor patogénico en el
desarrollo de esta enfermedad, debido a que condiciona un trastorno en la capacidad de
regulación del metabolismo lipídico (18,24), ya que en condiciones normales los AGPICL son
responsables de la repartición de los combustibles metabólicos. Esto último es logrado a través
de 2 mecanismos: el primero dado por inhibición de la transcripción de genes lipogénicos y
glicolíticos (60,61), ya sea por regulación negativa de la expresión de la proteína 1 de unión al
elemento regulador del esterol (SREBP-1) y/o por daño en el procesamiento de SREBP-1 y
un segundo mecanismo dado por la inducción de la trascripción de genes lipolíticos, es decir,
que codifican para enzimas de oxidación de los ácidos grasos, los que actúan como ligandos
activadores del receptor α activador de la proliferación de peroxisomas (PPAR α) (62,63). La
activación del PPARα por los AGPICL también resulta en un aumento de la secreción de
ApoB-100, apoproteína fundamental para la formación de las lipoproteínas hepáticas,
permitiendo la exportación de los TG hepáticos a la circulación. Por ende, en condiciones que
implican una depleción de AGPICL tales como el HGNA, es esperable encontrar un aumento
de los depósitos de TG hepáticos por favorecerse la síntesis de ácidos grasos y deposito de los
TG por sobre la oxidación de ácidos grasos y la exportación de los TG en las lipoproteínas,
hallazgos observados en estudio previo de Araya et al. 2004 (30). Por otro lado, los hallazgos de
una mayor relación n-6/n-3 de AGPI y de AGPICL en los fosfolípidos hepáticos en los
pacientes con HGNA versus los sujetos controles, es concordante con la visión de una

41
deprimida oxidación y secreción de lípidos hepáticos, ya que los AGPICL n-3 son más
potentes que los n-6 como activadores de PPAR α (17,64). Se postulan diversos mecanismos
implicados en la depleción de los AGPICL en el hígado de pacientes con HGNA incluyendo
daño en las vías de desaturación y elongación de los ácidos grasos esenciales 18:2 n-6 y 18:3
n-3 (requeridos para la síntesis de los AGPICL), un desbalance dietético y aumento del estrés
oxidativo. La baja relación producto/precursor de los AGPI encontrada en el hígado de los
pacientes con HGNA podría sugerir una regulación negativa en la delta 5 y delta 6
desaturasas, enzimas claves en la síntesis de los AGPICL n-3 y n-6, lo que ocurre
fundamentalmente en el hígado (65). Estudios previos han demostrado una asociación negativa
entre obesidad y actividad de delta 5 desaturasa con una menor relación producto/precursor de
AGPI (33). Es importante considerar que una dieta desbalanceada por un mayor aporte de AGPI
n-6 puede constituir un factor limitante en la producción de los AGPICL n-3 (65) y un mayor
aporte dietético de ácidos grasos trans puede producir un defecto en la actividad de las
desaturasas hepáticas (65,66) resultando en una menor producción de AGPICL y una menor
relación producto/precursor de los AGPI (30). Sin embargo, para establecer la contribución de
una desaturación defectuosa en la patogenia del HGNA es necesario determinar la actividad de
las desaturasas hepáticas.
Por otra parte, el estrés oxidativo, un desbalance redox entre la tasa de producción de especies
reactivas de oxigeno y nitrógeno y su consumo por mecanismos antioxidantes, favoreciendo la
generación de pro-oxidantes (67), en el HGNA está relacionado con la inducción del citocromo
P450 2E1 (68) lo que conlleva una mayor actividad pro-oxidante (69). Los datos obtenidos en
este estudio revelan un aumento del estado de estrés oxidativo en el hígado de los pacientes
con HGNA, evidenciado por el marcado aumento de la lipoperoxidación medido como MDA

42
y F2I y por la disminución de los mecanismos de defensa antioxidante como el FRAP, la
actividad de enzimas antioxidantes (catalasa, SOD) y el contenido de GSH. Este proceso
podría ser el resultado del aumento de reacciones entre radicales libres y AGPI (67), siendo
concordante con estudios previos en animales experimentales (70) y con pacientes con HGNA
(71,32). Dada la mayor susceptibilidad de los AGPICL a ser oxidados por los radicales, debido a
su mayor insaturación y mayor largo de su cadena (67,72), se podría plantear la lipoperoxidación
dependiente de estrés oxidativo como un mecanismo contribuyente a la depleción de AGPICL
observada en el hígado de pacientes con HGNA. Por lo tanto, el estrés oxidativo constituiría
un mecanismo dual en la patogenia del HGNA, contribuyendo por un lado a la esteatosis
mediante la depleción de los AGPICL n-3 y por otro lado, promoviendo la progresión de la
enfermedad a esteatohepatitis y fibrosis (40).
La marcada resistencia a la insulina encontrada en las pacientes con HGNA jugaría un
importante papel en la patogenia de esta enfermedad, ya que por un lado la resistencia a la
insulina gatilla la masiva llegada de ácidos grasos libres al hígado y su mayor captación por
éste y, por otro lado, la hiperinsulinemia asociada a la resistencia insulínica estimula la
lipogénesis vía SREBP-1 y favorece el depósito de grasa en el hígado, al disminuir los niveles
de Apo B-100.
Resulta de interés destacar que los datos de este estudio también revelan que la membrana del
eritrocito tiene una composición de ácidos grasos comparable a la de los fosfolípidos
hepáticos, reflejando los cambios producidos por el estrés oxidativo en el hígado,
encontrándose en las pacientes con HGNA una significativa disminución del total de AGPI,
AGPICL, AGPICL n-3 y de los ácidos grasos 20:4 n-6 (ARA) y 22:6 n-3 (DHA), asociado a
un aumento de la relación n-6/n-3 AGPICL y el aumento del ácido graso 22:5 n-6 (Osbond).
La aparición del ácido graso Osbond podría usarse como un marcador de la deficiencia de

43
DHA, ya que la delta 4 desaturasa requerida para la síntesis de 22:5 n-6 está normalmente
reprimida por los AGPICLn-3. Este aumento del 22:5 n-6 ha sido sugerido como una
respuesta compensatoria a la deficiencia de 22:6 n-3 (73). Se ha demostrado que la célula
nerviosa y las membranas de sus organelos expuestos a una deficiencia específica de ácidos
grasos n-3 con un suministro adecuado de ácidos grasos n-6 es capaz de conservar la
cantidad de poli-insaturados totales, pero con un alto déficit de 22:6n-3, el que eventualmente
es compensado por un exceso de 22:5n-6 (74). Disturbios en el perfil de los ácidos grasos poli-
insaturados pueden alterar la expresión de enzimas de membranas, y entorpecer la interacción
entre receptor y ligando, alterando el normal funcionamiento de un órgano. (75,76,77). Connor
WE et al 1990 (78), establecieron como marcador bioquímico específico de n-3 la declinación
del contenido de DHA en los fosfolípidos del cerebro y al aumento compensatorio de los n-6
especialmente del 22:5n-6, esto podría constituir un mecanismo para conservar la poli-
insaturación de la membrana de los fosfolípidos tanto como sea posible, a pesar de la
deficiencia de n-3. La deficiencia de ácidos grasos n-3 y el recambio del DHA en el cerebro,
eritrocitos y plasma de monos rhesus fue revertida con una dieta rica en aceite de pescado
(AGPICL n-3).
Finalmente, las pacientes con HGNA post-cirugía bariátrica presentan cambios en el perfil de
ácidos grasos de la membrana del eritrocito y en el nivel de estrés oxidativo, evidenciado por
un significativo aumento de los ácidos grasos 20:4 n-6 (ARA), 22:6 n-3 (DHA), total AGPI n-
3 y total AGPICL n-3, asociado a la disminución del ácido graso Osbond, del ácido linoleico
(18:2 n-6), de la relación n-6/n-3 AGPICL y del contenido de F2I, alcanzando valores
similares al grupo control, lo que sugiere que la membrana del eritrocito refleja los cambios
provocados por el estrés oxidativo en la homeostasis de los ácidos grasos de los fosfolípidos
hepáticos, sugiriendo la evaluación del perfil de los ácidos grasos de las membranas de los

44
eritrocitos como un método no invasivo en la estimación de los cambios hepáticos por la baja
de peso post-cirugía bariátrica en pacientes obesas con HGNA.
VII. CONCLUSIONES:
Debido a la presencia significativa de obesidad, resistencia a la insulina, depleción de
AGPICL n-3 y estrés oxidativo en las pacientes obesas con HGNA se plantea un mecanismo
multifactorial en la patogenia del HGNA. Además, la marcada depleción de los AGPICL
sugiere que estos resultan ser los marcadores más sensibles a los efectos del estrés oxidativo
sobre los fosfolípidos de membranas de hígado y eritrocito. Los datos sugieren que la
composición de los ácidos grasos de la membrana del eritrocito podría ser utilizada para
estimar los cambios homólogos en el hígado en pacientes con HGNA, lo que constituye un
método no invasivo para el estudio de la homeostasis de los ácidos grasos en esta patología.

45
Figura 8
Figura 8: Biosíntesis de los ácidos grasos poli-insaturados de cadena larga en mamíferos

46
Figura 9
Figura 9: Factores que influyen en el agotamiento de los ácidos grasos poli-insaturados de cadena larga n-3 (AGPICL n-3), proceso que conduce a la esteatosis y su progresión a la esteatohepatitis en la enfermedad de hígado graso no alcohólico (HGNA) AG: ácidos grasos, AGL: ácidos grasos libres, TG: triglicéridos, SREBP-1: proteína-1 de unión al elemento regulador del esterol, PPAR-α: receptor activado por proliferadores de peroxisomas α, AP-1: proteína activadora-1, NF-κB: factor nuclear kappa B
inhibición PPAR-α
activación SREBP-1
Activación
NF-κB/AP-1
Desbalance dietético
Depleción AGPICL n-3
Defecto desaturación
Estrés Oxidativo
Esteatohepatitis
Esteatosis Hepática
Mayor síntesis de AG y TG
Menor oxidación AG y menor
exportación TG
Resistencia insulínica
Aumento periférico de lipólisis flujo de AGL y
glicerol al hígado
Estimulación mediadores
proinflamatorios
Hiper-insulinemia

47
VIII. REFERENCIAS:
1. Das K., Kar P. (2005) Non-Alcoholic Steatohepatitis. J.A.P.I. 53, 195-199 2. Cairns Sr., Peters T. (1983) Biochemical analysis of hepatic lipid in alcoholic and diabetic and control subjects. Clin. Sci. 65, 645-6523 3. Neuschwander-Tetri B.A. (2002) Evolving pathophysiologic concepts in nonalcoholic steatohepatitis. Curr. Gastroenterol. Rep. 4, 31-36 4. Haque M., Sanyal A.J. (2002) The metabolic abnormalities associated with non-alcoholic fatty liver disease. Best Pract. Res. Clin. Gastroenterol. 16, 709-731 5. Marchesini G., Brizi M., et al. (2001) Nonalcoholic fatty liver disease. A feature of the metabolic syndrome. Diabetes. 50, 1844-1850 6. de Knegt R.J. (2001) Non-alcoholic steatohepatitis: clinical significance and pathogenesis. Scand. J. Gastroenterol. 234 (Suppl), 88-92 7. Ludwig J., Viggiano R.T., and McGill D.B. (1980) Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. 55, 342-348 8. Neuschwander-Tetri B.A. (2003) Nonalcoholic Steatohepatitis: Summary of an AASLD single topic conference. Hepatology. 37, 1202-1219 9. Yu A.S., Keeffe E.B. (2002) Nonalcoholic fatty liver disease. Rev. Gastroenterol. Disord. 2 (1), 11-19 10. McCullough A.J. (2002) Update on nonalcoholic fatty liver disease. J. Clin. Gastroenterol. 34, 255-262 11. Angulo P. (2002) Nonalcoholic fatty liver disease. New Engl. J. Med. 346, 1221-1231 12. Silverman J.F., Pories W.J., Caro J.F.(1989) Liver pathology in diabetes mellitus and morbid obesity : clinical, pathological and biochemical considerations. Pathol. Annul. 24, 275-302 13. de Almeida I.T., Cortés-Pinto H., Fidalgo G., Rodríguez D., Camilo M.E. (2002) Plasma total and free fatty acid composition in human non-alcoholic steatohepatitis. Clin. Nutr. 21, 219-223 14. Bray G., Lovejoy J., Smith S., DeLany J., Lefevre M., Hwang D., Ryan D., York D. (2002) The influence of different fats and fatty acids on obesity, insulin resistance and inflammation. J. Nutr. 132, 2488-2491

48
15. Clarke S.D., Gasperikova D., Nelson C., Lapillonne A., Heird W.C. (2002) Fatty acid regulation of gene expression. A genomic explanation for the benefits of the mediterranean diet. Ann. NY Acad. Sci. 967, 283-298 16. Eaton S., Zaitoun A.M., Record C.O., Bartlett K. (1996) Beta-Oxidation in human alcoholic and non-alcoholic hepatic steatosis. Clin. Sci. (London) 90, 307-313 17. Clarke S.D. (2001a) Nonalcoholic steatosis and steatohepatitis. I. Molecular mechanism for polyunsaturated fatty acid regulation of gene transcription. Am. J. Physiol. Gastrointest. Liver Physiol. 281, 865-869 18. Clarke S.D. (2001b) Polyunsaturated fatty acid regulation of gene transcription: A molecular mechanism to improve the metabolic syndrome. J. Nutr. 131, 1129-1132 19. Chan D.C., Watts G.F., Barrett P.H., Beilin L.J., Redgrave T.G., Mori T.A. (2002) Regulatory effects of HMG CoA reductase inhibitor and fish oils on apolipoprotein B-100 kinetics in insulin-resistant obese male subjects with dyslipidemia. Diabetes. 51, 2377-2386 20. Lindén D., Lindberg K., Oscarsson J., Claesson C., Asp L., Li L., Gustafsson M., Borén J., Olofsson S.O. (2002) Influence of peroxisome proliferator-activated receptor α agonists on the intracellular turnover and secretion of apolipoprotein (Apo)B-100 and ApoB-48. J. Biol. Chem. 277, 23044-23053 21. Meunier-Durmort C., Poirier H., Niot I., Forest C., Besnard P. (1996) Up-regulation of the expression of the gene for liver fatty acid-binding protein by long-chain fatty acids. Biochem. J. 319, 483-487 22. Sessler A.M, Ntambi J.M. (1998) Polyunsaturated fatty acid regulation of gene expression. J. Nutr. 128, 923-926 23. Ou J., Tu H., Shan B., Luk A., DeBose-Boyd R.A., Bashmakov Y., Goldstein J.L., Brown M.S. (2002) Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR. Proc. Natl. Acad. Sci .USA 22, 6027-6032 24. Nakamura M.T., Cho H.P., Clarke S.D. (2000) Regulation of hepatic delta-6 desaturase expression and its role in the polyunsaturated fatty acid inhibition of fatty acid synthase gene expression in mice. J. Nutr. 130, 1561-1565 25. Clarke S.D. (2000) Polyunsaturated fatty acid regulation of gene transcription: A mechanism to improve energy balance and insulin resistance. Bri. J. Nutr. 83, S59-S66 26. Xu J., Nakamura M.T., Cjo H.P., Clarke S.D. (1999) Sterol regulation element binding protein-1 expression is suppressed by dietary polyunsatured fatty acids. J. Biol. Chem. 274, 23477-23583

49
27. Singer P., Richter-Heinrich E. (1991) Stress and fatty liver—possible indications for dietary long-chain n-3 fatty acids. Med. Hypotheses 36, 90-94 28. Niot I., Gresti J., Boichot J., Sempore G., Durand G., Bezard J., Clouet P. (1994) Effect of dietary n-3 and n-6 polyunsaturated fatty acids on lipid-metabolizing enzymes in obese rat liver. Lipids 29, 481-489 29. Rudin D. (1982) The dominant diseases of modernized societies as omega 3 essential fatty acid deficiency syndrome: substrate beriberi. Med. Hypoth. 8, 17-47 30. Araya J., Rodrigo R., Videla L.A. (2004) Increase in long-chain polyunsaturated fatty acid n-6/n-3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clinical Science 106, 635-643 31. Simopoulus A.P. (2002) The importance of the ratio of omega 6/ omega 3 essential fatty acids. Biomed. Pharmacother. 56, 365-379 32. Videla L.A., Rodrigo R., Orellana M., Fernandez V. et al. (2004a) Oxidative stress-related parameters in the liver of non-alcoholic fatty liver disease patients. Clin. Sci. 106, 261-268 33. Pan D.A., Hulbert A.J., Storlien L.H. (1994) Dietary fats, membrane phospholipids and obesity. J. Nutr. 124, 1555-1565 34. Cho H.P., Nakamura M.T., Clarke S.D. (1999a) Cloning, expression and nutritional regulation of human delta 5 desaturase. J. Biol. Chem. 272, 471-477 35. Cho H.P., Nakamura M.T., Clarke S.D. (1999b) Cloning, expression and fatty acid regulation of human delta 5 desaturase. J. Biol. Chem. 274, 37335-37339 36. Diehl A.M. (1999) Nonalcoholic Steatohepatitis. Sem. Liver Dis. 19, 221-229 37. Medina J., Fernandez-Salazar L.I., García-Buey L., Moreno-Otero R. (2004) Approach to the pathogenesis and treatment of nonalcoholic steatohepatitis. Diabetes Care. 27, 2057-2066 38. Bugianesi E., Manzini P., D’Antico S., Vanni E., Longo F., Leone N., Massarenti P., Piga A., Marchesini G., Rizzetto M. (2004) Relative contribution of iron burden, HFE mutations, and insulin resistance to fibrosis in nonalcoholic fatty liver. Hepatology. 39, 179-187 39. Videla L.A., Rodrigo R., Araya J. (2004b) Oxidative stress and depletion of hepatic long-chain polyunsatured fatty acids may contribute to nonalcoholic fatty liver disease. Free Radical Biology and Medicine. 37, 1499-1507 40. Videla L.A., Rodrigo R., Araya J., Poniachik J. (2004c) Dual mechanistic significance of oxidative stress in human nonalcoholic fatty liver disease (NAFLD). XII Biennial Meeting of

50
the Society for Free Radical Research International (SFRR), by Medimond Sr. Editores Susana Puntarolu y Alberto Boveris. Bologna, Italia. pp 261-266, 2004. 41. Chalasani N., Gorski J.C., Asghar M.N., et al. (2003) Hepatic cytochrome P450 2E1 activity in nondiabetic patients with nonalcoholic steatohepatitis. Hepatology 37, 544-550 42. Di Marino L., Maffettone A., Cipriano P. (2000) Is the erythrocyte membrane fatty acid composition a valid index of skeletal muscle membrane fatty acid composition ?. Metabolism. 49 (9), 1164-1166 43. Felton C.V., Stevenson J.C., Godsland I.F. (2004) Erythrocyte-derived measures of membrane lipid composition in healthy men: associations with arachidonic at low to moderate not high insulin sensitivity. Metabolism. 53 (5), 571-577 44. Baur L.A., O´Coonor J., Pan D.A. (2000) Relationships between the fatty acid compositon of muscle and erythrocyte phospholipid in young children and the effect type of infant feeding. Lipids. 35 (1), 77-82 45. Matthews D.R., Hosker J.P., Rudenski A.S., Naylor B.A., Treacher D.F., Turner R.C. (1985) Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentration in man. Diabetologia 28, 412-419 46. Bacon R.B., Farahvash M.J., Janney C.G., Neuschwander B.A. (1994) Non-alcoholic steatohepatitis: an expanded clinical entity. Gastroenterology 107,1103-1109 47. Bligh E.G., Dyer W.J. (1959) A rapid method of total lipid extraction. Can. J. Biochem. Physiol. 37, 911-917 48. Skipski VP, Peterson R.F., Barclay. M.(1964) Cuantitative analysis of phospholipids by thin-layer chromatography. Biochem. J. 90 (2), 374-8. 49. Ruiz-Gutiérrez V., Cert A., Rios J.J. (1992) Determination of phospholipid fatty acid and triacylglycerol composition of rat caecal mucosa. J. Chromatogr. 575, 1-6 50. Huertas J.R., Palomino N., Ochoa J.J., Quiles J.L., Ramirez-Tortosa M.C., Battino M., Robles R., Mataix J. (1998) Lipid peroxidation and antioxidants in erythrocyte membranes of full-term and preterm newborns. Biofactors. 8 (1-2), 133-7. 51. Araya J., Rodrigo R., Orellana M., Rivera G. (2001) Red wine raises HDL and preserves long-chain polyunsaturated fatty acids in rat kidney and erythrocytes. Br. J. Nutr. 86, 189-195 52. Benzie I.F.F., Strain J.J. (1996) The ferric reducing ability of plasma (FRAP) as a measure of antioxidant power: the FRAP assay. Anal. Biochem. 239, 70-76 53. Ohkawa H., Ohishi N., Yagi K. (1979) Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal. Biochem. 95, 351-358

51
54. Nourooz-Zadeh J., Gopaul N.K., Barrow S., Mallet A.I., Anggard E.E. (1995) Analysis of F2-isoprostanes as indicators of non-enzymatic lipid peroxidation in vivo by gas chromatography-mass spectrometry: development of a solid-phase extraction procedure. J. Chromatogr. B. Biomed. Appl. 667 (2), 199-208. 55. Tietze F. (1969) Enzymatic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal. Biochem. 27, 502-522 56. Lowry O.H., Rosebrough N.J., Farr A.L., Randall R.J. (1951) Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265-275 57. Aebi H. (1974) Catalase. In: Bergmeyer, H.U., ed. Methods of enzymatic analysis. Vol. New York: Academic Press, Inc., pp. 673-678 58. Nebot C., Moutet M., Huet P., Xu J.Z., and Chaudiere J. (1993) Spectrophotometric assay of superoxide dismutase activity based on the activated autoxidation of a tetracyclic catechol. Anal. Biochem. 214, 442-451 59. Flohé L., and Gunzler W.A. (1984) Assays of glutathione peroxidase. Methods Enzymol. 105, 114-121 60. Osborne T.F. (2000) Sterol regulatory element-binding proteins (SREBPs): key regulators of nutritional homeostasis and insulin action. J. Biol. Chem. 275, 32379-32282 61. Yoshikawa T., Shimano H., Yahagi N., Ide T., Amemiya-Kudo M., Matsuzaka T., Nakakuki M., Tomita S., Okasaki H., Tamura Y. ,et al. (2002) Polyunsaturated fatty acids suppress sterol regulatory element-binding protein 1c promoter activity by inhibition of liver X receptor (LXR) binding to LXR response elements. J. Biol. Chem. 277, 1705-1711 62. Clarke S.D., Thuillier P., Baillie R.A., Cha X. (1999) Peroxisome proliferator-activated receptors: a family of lipid-activated transcription factors. Am. J. Clin. Nutr. 70, 566-571 63. Kersten S. (2002) Effects of fatty acids on gene expression: role of peroxisome proliferator-activated receptor alpha, liver X receptor alpha and sterol regulatory-element-binding protein-1c. Proc. Nutr. Soc. 61, 371-374 64. Kersten S., Seydoux J., Peters J.M., Gonzalez F.J., Desbvergne B., Wahli W. (1999) Peroxisome proliferator-activated receptor alpha mediates the adaptive response to fasting. J. Clin. Invest. 103, 1489-1498 65. Mahfouz M.M., Smith T.L., Kummerow F.A. (1984) Effect of dietary fats on desaturase activities and the biosynthesis of fatty acids in rat-liver microsomes. Lipids 19, 214-222 66. Larqué E., Pérez-Llamas F., Puerta V., Girón M.D., Suárez M.D., Zamora A., Gil A. (2000) Dietary trans fatty acids affect docosahexaenoic acid concentrations in plasma and liver but not brain of pregnant and fetal rats. Pediatr. Res. 47, 278-283

52
67. Sies H. (1986) Biochemistry of oxidative stress. Angew. Chem. Int. Ed. Engl. 25, 1058-1071 68. Weltman M.D., Farrel G.C., Hall P., Ingelman-Sundberg M., Liddle C. (1998) Hepatic cytochrome P4502E1 is increased in patients with nonalcoholic steatohepatitis. Hepatology 27, 128-133 69. Lieber C.S. (1997) Cytochrome P-4502E1: its physiological and pathological role. Physiol. Rev. 77, 517-544 70. Lettéron P., Fromenty B., Terris B., Degott C., Pessayre D. (1996) Acute and chronic hepatic steatosis lead to in vivo lipid peroxidation in mice. J. Hepatol. 24, 200-208 71. Cortez-Pinto H., Filipe P., Camilo M.E., Baptista A., Fernández A., Mopuro M,C. (1999) A peroxidação dos lípidos na esteatohepatite não alcoólica :relação com a esteatose. J. Português Gastroenterol. 6, 234-240 72. Sevanian A., Hochstein P.(1985) Mechanisms and consequences of lipid peroxidation in biological systems. Ann. Rev. Nutr. 5, 365-390 73. Zhou D., Zaiger G., Ghebremeskel K., Crawford M.A., Reifen R. (2004) Vitamin A deficiency reduces liver and colon docosahexaenoic acid levels in rats fed high linoleic and low alpha-linolenic acid diet. Prostaglandins Leukot Essent fatty acids. 71 (6), 383-9 74. Bourre J.M., Durand G., Pascal G., Youyou A. (1989) Brain cell and tissue recovery in rats made deficient in n-3 fatty acids by alteration of dietary fat. J.Nutr. 119, 15-22 75. Clandinin M., Foot M., Robson L. (1983) Plasma membrane: can its structure and function be modulated by dietary fat? Comp. Biochem. Physiol. 76, 335-339 76. Stubbs C.D., Smith A.D. (1971) The modification of mammalian membrane polyunsatured fatty acid composition in relation a membrana fluidity and function. Biochim. Biophys. Acta. 248, 449-454 77. Brenner R.R. (1984) Effect of unsatured acids on membrana structure and enzymes kinetics. Prog. Lipid Res. 23: 69-96 78. Connnor W.E., Neuringer M., Lin D.S. (1990) Dietary effects on brain fatty acid composition: the reversibility of n-3 fatty acid deficiency and turnover of docosahexaenoic acid in the brain, erythrocytes, and plasma of rhesus monkeys. J. Lipid Res. 31, 237-247

53
IX. ANEXOS:
1. CONSENTIMIENTO INFORMADO Grupo Control: La obesidad es una enfermedad crónica que se asocia a Hígado graso, enfermedad hepática, similar a la que ocurre por ingesta excesiva y crónica de alcohol. Para entender mejor esta enfermedad y obtener un marcador no invasivo que refleje lo que ocurre en estos hígados, se llevará a cabo el proyecto de investigación “Perfil de los ácidos grasos de la membrana de eritrocitos como reflejo del perfil de los ácidos grasos de los fosfolípidos hepáticos en hígado graso no alcohólico ”. El estudio incluirá a un número total de 24 pacientes (8 controles y 16 casos) del Hospital Clínico de la Universidad de Chile. Usted pertenecerá al grupo de pacientes control, sin obesidad ni hígado graso, que son sometidas a una cirugía abdominal abierta por patologías como Reflujo Gastroesofágico, Canceres abdominales no hepáticos o Colecistectomía. Si usted acepta participar será sometido a un examen médico para conocer sus antecedentes de enfermedades previas, evaluación nutricional y se le tomará una muestra de sangre de 12 ml. Estos últimos exámenes tomarán aproximadamente 20 minutos y no significarán gran riesgo ni costos adicionales para usted, aparte de los derivados de su enfermedad y de la cirugía a efectuarse. Durante su operación se le realizará una biopsia hepática, la cual consiste en extraer un pequeño trozo de hígado para estudiarlo en el microscopio y ver si tiene alguna alteración. La biopsia hepática tomada durante su operación es de muy bajo riesgo, pudiendo complicarse sólo con hemorragia en menos de 1 por 1000 casos (<0,1%), lo cual se tratará rápidamente sin ningún costo para usted y cualquier gasto adicional será financiado por este proyecto de investigación. Este estudio será beneficioso ya que permitirá en un futuro evaluar la presencia de hígado graso y su disminución por la baja de peso postcirugía bariátrica, a través de solo una muestra de sangre evitando usar nuevamente técnicas invasivas como la biopsia hepática. Usted no recibirá ninguna compensación económica por participar en este estudio. Toda la información derivada de su participación en este estudio será conservada con estricta confidencialidad. Cualquier publicación o comunicación científica de los resultados de la investigación será completamente anónima. Si usted decide no participar en esta investigación o si acepta pero desea retirarse de él en cualquier momento, puede hacerlo, sin que ello signifique modificaciones en el estudio y tratamiento habituales de su enfermedad, los que también pueden tener efectos adversos relacionados a la cirugía. Si usted requiere cualquier otra información sobre su participación en este estudio puede llamar a: Investigador: Dra. Alejandra Elizondo M., Departamento de Nutrición. Facultad de Medicina de la Universidad de Chile. Teléfono: 9786136

54
Directores de la investigación: Dra. Julia Araya. Teléfono: 9786760, Dr. Ramón Rodrigo. Teléfono: 9786126 Después de haber recibido y comprendido la información de este documento y de haber podido aclarar todas mis dudas, otorgo mi consentimiento para participar en este proyecto de investigación ----------------------------------- --------------------------- ---------------- Nombre del paciente firma fecha ----------------------------------- -------------------------- ----------------Nombre del testigo firma fecha ----------------------------------- -------------------------- ----------------Nombre del investigador firma fecha Grupo en Estudio: La obesidad es una enfermedad crónica que se asocia a Hígado graso, enfermedad hepática, similar a la que ocurre por ingesta excesiva y crónica de alcohol. Para entender mejor esta enfermedad y obtener un marcador no invasivo que refleje lo que ocurre en estos hígados, se le invita a participar en el proyecto de investigación “Perfil de los ácidos grasos de la membrana de eritrocitos como reflejo del perfil de los ácidos grasos de los fosfolípidos hepáticos en hígado graso no alcohólico ”. El estudio incluirá a un número total de 24 pacientes (8 controles y 16 casos) del Hospital Clínico de la Universidad de Chile. Si usted acepta participar será sometido a un examen médico para conocer sus antecedentes de enfermedades, evaluación nutricional y se le tomará una muestra de sangre de 12 ml. Estos últimos exámenes tomaran aproximadamente 20 minutos y no significarán gran riesgo ni costos adicionales para usted, aparte de los derivados de su enfermedad y de la cirugía de obesidad (bariátrica) a efectuarse. Durante su operación se le realizará una biopsia hepática, la cual consiste en extraer un pequeño trozo de hígado para estudiarlo en el microscopio y ver si tiene alguna alteración. Además, a los 3 meses postcirugía se le tomara una nueva muestra de sangre de 12 ml para compararla con la inicial. La biopsia hepática tomada durante su operación es de muy bajo riesgo, pudiendo complicarse sólo con hemorragia en menos de 1 por 1000 casos (<0,1%), lo cual se tratará rápidamente sin

55
ningún costo para usted y cualquier gasto adicional será financiado por este proyecto de investigación. Este estudio será beneficioso para usted ya que permitirá evaluar la presencia de hígado graso y su disminución con la baja de peso postcirugía bariátrica, a través de sólo una muestra de sangre evitando usar nuevamente técnicas invasivas como la biopsia hepática. Usted no recibirá ninguna compensación económica por participar en este estudio. Toda la información derivada de su participación en este estudio será conservada con estricta confidencialidad. Cualquier publicación o comunicación científica de los resultados de la investigación será completamente anónima. Si usted decide no participar en esta investigación o si acepta pero desea retirarse de él en cualquier momento, puede hacerlo, sin que ello signifique modificaciones en el estudio y tratamiento habituales de su enfermedad, los que también pueden tener efectos adversos relacionados a la cirugía. Si usted requiere cualquier otra información sobre su participación en este estudio puede llamar a: Investigador: Dra. Alejandra Elizondo M., Departamento de Nutrición. Facultad de Medicina de la Universidad de Chile. Teléfono: 9786136 Directores de la investigación: Dra. Julia Araya. Teléfono: 9786760, Dr. Ramón Rodrigo. Teléfono: 9786126 Después de haber recibido y comprendido la información de este documento y de haber podido aclarar todas mis dudas, otorgo mi consentimiento para participar en este proyecto de investigación ----------------------------------- --------------------------- ---------------- Nombre del paciente firma fecha ----------------------------------- -------------------------- ----------------Nombre del testigo firma fecha ----------------------------------- -------------------------- ----------------Nombre del investigador firma fecha


















