
Me O H O O OHC H Me OH Abisomicina C Me … · estado de transición 15 mediante el cual opera,...
164
SINTESIS SINTESIS TOTALES TOTALES-1 Miguel Carda y Eva Falomir Miguel Carda y Eva Falomir Universidad Jaume I Universidad Jaume I OH Me O O O O O Me Me H H Abisomicina C Alociatina B 2 OHC OH O O Me O O O H ent -Clavilactona B
Transcript of Me O H O O OHC H Me OH Abisomicina C Me … · estado de transición 15 mediante el cual opera,...

SINTESIS SINTESIS TOTALESTOTALES --11
Miguel Carda y Eva FalomirMiguel Carda y Eva FalomirUniversidad Jaume IUniversidad Jaume I
OH
Me
O O
O
O
O
Me
Me
H
H
Abisomicina C
Alociatina B2
OHC
OH
O
O
Me
O
O
OH
ent-Clavilactona B


Tabla de contenidos
1. Síntesis de abisomicina C (Nicolaou, 2007) 1
2. Síntesis de adociacetileno B (Trost, 2006) 25
3. Síntesis de alociatina B2 (Trost, 2005) 33
4. Síntesis de aureotina (Baldwin, 2005) 51
5. Síntesis de broussonetina G (Trost, 2003) 65
6. Síntesis de ciantivigina U (Phillips, 2005) 81
7. Síntesis de cilindrociclofano F (Smith, 2001) 91
8. Síntesis de citostatina (Boger, 2006) 105
9. Síntesis de ent-clavilactona (Barret, 2006) 127
10. Síntesis de clavosólido A (Smith, 2006) 139


Síntesis de abisomicina C 1
SÍNTESIS DE ABISOMICINA C
Aislamiento: La abisomicina C se ha aislado del filtrado AB 18-032 de la bacteria Verrocosispora cultivada en un sedimento marino recogido en los fondos oceánicos del mar de Japón. Actividad biológica: La abisomicina C ha demostrado ser un potente antibiótico puesto que es activa contra Staphylococcus aureus resistente a meticillina (MRSA, 4 µg/ML) y contra Staphylococcus aureus resistente a vancomicina (MVSA, 13 µg/ML).
Retrosíntesis En el esquema 1 se indica el análisis retrosintético de la abisomicina C que se
inicia con la desconexión del doble enlace C8-C9, que se instalará mediante una reacción de metátesis intramolecular sobre el intermedio 1.1 La desconexión de la agrupación vinílica genera el aldehído 2, que se desconecta en el enlace C2-C3 para dar los intermedios 3 y 4. La conexión de estos dos fragmentos se llevará a cabo por reacción del aldehído quiral 3 con el agente organometálico 4, que se preparará por metalación del compuesto 5. La desconexión del puente éter (enlace C12-O) conduce al epóxido 6, cuya unidad de ácido tetrónico se desconecta en el enlace C2-C16 para desvelar la agrupación acetato (C2) y la agrupación éster (C16) que presenta el intermedio 7. Este compuesto derivará del compuesto hidroxiciclohexénico 8, que a su vez se obtendrá por hidroxilación del ciclohexenenocarboxilato 9. El anillo ciclohexénico que presenta este compuesto permite su desconexión, basada en una reacción Diels-Alder, al dieno 10 y al acrilato 11.
1 K. C. Nicolaou, S. T. Harrison, J. Am. Chem. Soc. 2007, 129, 429.

Síntesis de abisomicina C
2
Esquema 1
Síntesis
1. Preparación del sistema ciclohexenónico mediante reacción Diels-Alder: síntesis del intermedio 18
Para la construcción del anillo ciclohexánico que contiene la abisomicina C
se recurrió a un proceso de cicloadición Diels-Alder, para lo cual se empleó el

Síntesis de abisomicina C 3
alcohol quiral 13 (véase esquema 2) como equivalente sintético del dieno 10 que aparece en el análisis retrosintético. El empleo del alcohol quiral 13 como dieno en la proyectada reacción Diels-Alder debería permitir la obtención no racémica del correspondiente cicloaducto. Por otro lado, la conjunción de la agrupación feniltio éter y el grupo hidroxilo se corresponde con un doble enlace latente, que se desvelará en el momento adecuado de la ruta sintética.
De acuerdo con este planteamiento se inició la síntesis del dieno 13 por adición de derivado lítico del tioanisol a la amida de Weinreb 11 (esquema 2). Esta reacción proporcionó la feniltiometilcetona 12, que se transformó enantioselectivamente en el alcohol 13 mediante aplicación del método de Corey-Bakshi-Shibata.
N Me
O
MeO
Me
PhSCH3, DABCO
n-BuLi, THF, 0ºC(80%)
Me
O
PhS-78ºC
(95%, 90%ee)
catecolborano(R)-CBS, CH2Cl2
Me
OH
PhSMeMgBr, CH2=CHCOOMe
OH
NO
R2N
Mg
MgOPhS
Me
O
O
Me++
++
COOMe
Me
BrMgO
SPhMe
O
PhS
O
H
HLiHMDS
luego O2 y P(OEt)3Me
O
PhS
O
H
OH
16
15
13
109
13
10
13
10
14
14
15
10
139
9
9
13
10
9
10
13
15 14
910
13
15
16
16
1112
13
14
15
16 17 18(74%)
16
Esquema 2
La reacción Diels-Alder entre el dienol 13 y el acrilato de metilo se intentó
en primer lugar con el método de Ward, que emplea bromuro de metilmagnesio como plantilla para conseguir una reacción de cicloadición intramolecular (véanse

Síntesis de abisomicina C
4
comentarios).2 Sin embargo, este método proporcionó el cicloaducto 17 con tan solo un 25% de rendimiento, basado en dieno recuperado. Después de una considerable experimentación se encontró que la adición de 10 equivalentes de acrilato de metilo a una mezcla constituida por 1 equivalente del dieno 13, 3 equivalentes del ligando 14 y 4 equivalentes de MeMgBr en tolueno a 0ºC, seguida de calentamiento a 55ºC durante 24 horas proporcionaba, el producto de cicloadición 17, con un 80% de rendimiento. En el esquema 2 se representa el estado de transición 15 mediante el cual opera, probablemente, la reacción de cicloadición anterior. El proceso forma el cicloaducto intermedio 16, que por lactonización in situ se convierte en la lactona bicíclica 17. La α-hidroxilación de la parte lactónica se consiguió mediante enolización de 17 con LiHMDS en THF seguida de burbujeo de oxígeno y adición de fosfito de trietilo a la mezcla de reacción (véanse comentarios).
2. Preparación del sistema tricíclico: síntesis del intermedio 5
OH
Me
O O
O
O
O
Me
Me
H
H
H
abisomicina C
Me
O
PhS
O
H
OH
16
15
13
109
9
13
15
16
OH
Me
O O
O
HH
16
9
13
15
1211
1
2
12
1211
5 18
La síntesis del compuesto tricíclico 5 se inició con el tratamiento de la feniltiometil γ−lactona 18 con el anión radical derivado de 4,4´-di-ter-butilbifenilo y litio metal (esquema 3). La reacción con 4,4´-di-ter-butilbifeniluro de litio provocó la apertura reductiva de la lactona con formación concomitante del doble enlace (véanse comentarios). El tratamiento de la mezcla de reacción con yoduro de metilo condujo al α−hidroxiéster metílico 19, que se epoxidó regio y estereoselectivamente con VO(OEt)3 y t-BuOOH para dar el oxirano 20 con un 90% de rendimiento. El empleo del método tradicional, que hace uso de VO(acac)2/t-BuOOH, proporcionó el epóxido 20 con rendimientos del orden del 55-60%. La acetilación de 20 llevó al acetato 7, que a continuación se trató con la
2 D. E. Ward, M. S. Abaee, Org. Lett. 2000, 2, 3937.

Síntesis de abisomicina C 5
base LiHMDS, en THF a -78ºC. Después de calentar a temperatura ambiente se adicionó cloruro amónico acuoso y se calentó a reflujo durante dos horas. En esta secuencia de reacciones se provocó, en primer lugar, el proceso de ciclación mediante la participación del enolato lítico 21. Esta reacción forma la β−cetolactona 22 que, en el proceso de calentamiento a reflujo con cloruro amónico acuoso, experimenta la apertura intramolecular del anillo oxiránico para conducir al compuesto 5.
Me
O
PhS
O
H
OH1. Li, (4-t-Bu-C6H4)2
THF, -40ºC
2. K2CO3, MeI, 60ºC(97% dos pasos) Me
MeO O
H
OHVO(OEt)3,t-BuOOH
Me
O
H
OH
O
MeO
Ac2O, Et3N,DMAP (93%)
Me
O
H
O
O
MeO
O
CH3
LiHMDS
-78ºC 25ºC
O
Me
O
O CH2O
OMe
Li
O
Me
O
O
H
H
O
HH
O
Me
O
O
O
HH
H
OH
Me
O O
O
HH
NH4Cl
H2O
OH
Me
O O
O
HH
18 19 20
72122
23 24 5H
H
- LiOMe
25ºC, (90%)
Esquema 3

Síntesis de abisomicina C
6
3. Síntesis del fragmento C3-C7 de la cadena de abisomicina C
El fragmento quiral C3-C7 de la abisomicina C se preparó a partir del meso-
diol 25 mediante esterificación enzimática con acetato de vinilo en presencia del enzima lipasa AK en THF, seguida de oxidación a aldehído (esquema 4).
Esquema 4
4. Etapas finales
Después de la obtención del compuesto tricíclico 5 y del fragmento acíclico
quiral 27, la síntesis se continuó con el acoplamiento de estos dos fragmentos. Para ello, el compuesto 5 se protegió en el hidroxilo y el derivado sililado se metaló en C2 por reacción con LiHMDS (esquema 5). Este proceso generó la especie organo-lítica 4 a la cual se le adicionó el acetoxialdehído 27, lo que condujo a la obtención de la mezcla de alcoholes diastereoisoméricos 29. La oxidaciónde esta mezcla con

Síntesis de abisomicina C 7
ácido o-yodoxibenzoico en DMSO proporcionó la cetona 30 (para la oxidación con IBX, véanse comentarios).
Esquema 5

Síntesis de abisomicina C
8
La reacción con 1,2-etanoditiol del carbonilo cetónico y la eliminación concomitante del grupo TES, lo que condujo al el compuesto 31. La saponificación de la agrupación acetato, seguida de oxidación con IBX, llevó al aldehído 33, que reaccionó con cloruro de vinilmagnesio para dar lugar a la mezcla de alcoholes epiméricos 34. Sobre este sustrato se llevó a cabo el proceso de metátesis ciclante mediante reacción con el catalizador de Grubbs de 2ª generación, en diclorometano a reflujo durante 1 hora. En estas condiciones se obtuvo el compuesto de ciclación con un 85% de rendimiento.
La oxidación con IBX en DMSO proporcionó la cetona 35, que por destiocetalización con trifluoroacetato de fenilyodonio (PIFA, PhI(CF3COO)2), véanse comentarios) se transformó en el compuesto 36. Sin embargo, los datos espectroscópicos de este compuesto no coincidían en su totalidad con los descritos para la abisomicina C natural. La solución al enigma vino de la mano de los espectros de Rayos X de la abisomicina C natural y del compuesto 36, que revelaron que este compuesto era un atropoisómero del producto natural. 5. Interconversión atropo-abisomicina C/abisomicina C
El atropoisomerismo surge como consecuencia de la rotación impedida
alrededor de un enlace simple. Muchas veces tiene su origen en bloqueos estéricos provocados en la libre rotación de enlaces, como ocurre en muchos sistemas biarílicos.
Los primeros intentos de conversión de la atropo-abisomicina C (36) en la abisomicina C, mediante calentamiento en metanol o tolueno a reflujo, no produjeron ningún cambio. Cuando el calentamiento se llevó a cabo a 180ºC, en xilenos o mediante irradiación con microondas se provocó la descomposición del producto de partida. Sin embargo, la medición de los espectros de RMN de la atropo-abisomicina C (36), disuelta en CDCl3 no estabilizado, provocó la lenta

Síntesis de abisomicina C 9
conversión de ésta en abisomicina C, obteniéndose una mezcla de abisomicina C/atropo-abisomicina C en relación 2:1, después de 24 horas de la disolución en CDCl3. Este resultado permitió concluir que la interconversión estaba provocada por trazas de HCl presentes en el CDCl3.
En el esquema 6 se indican tres posibles vías para la conversión de la atropo-abisomicina C en la abisomicina C.
Esquema 6
En la primera de ellas (ruta a) se produce la protonación en la parte de
tetronato, lo que convierte a este grupo en un grupo saliente. El subsiguiente ataque del grupo hidroxilo en C11 forma el intermedio 38, que ha sido postulado como

Síntesis de abisomicina C
10
precursor biogenético de la abisomicina C. La flexibilidad conformacional de este intermedio permite la rotación de los enlaces C6-C7 y C7-C8 antes de que se produzca la reprotonación a 39.
Una segunda posibilidad es la que se indica en la ruta b, que se inicia con la protonación de C2, lo que lleva a la formación del catión oxocarbenio 40. La protonación convierte al carbono sp2 de C2 en un carbono tetraédrico, lo que disminuye la tensión en el anillo macrocíclico y facilita la rotación de los enlaces σ.
Una última posibilidad es la que se indica en la ruta c, en la cual la protonación del átomo de oxígeno de la parte de tetronato convierte al carbono C16 en electrónicamente deficiente, lo que provoca la subsiguiente atracción de la densidad electrónica del doble enlace C8-C9. La formación del puente C8-C16 aproxima C8 a la parte de tetronato, lo que disminuye la tensión y favorece la rotación.
Comentarios
1. Reducción enantioselectiva de Corey-Bakshi-Shibata
Este método permite la reducción enantioselectiva de determinado tipo de
cetonas por reacción con BH3, en presencia de una oxazaborolidina quiral derivada de prolina.3 En el esquema 7 se indica la ruta sintética para la preparación de la oxazaborolidina de configuración S.
Esquema 7
3 a) E. J. Corey, R. K. Bakshi , S. Shibata, J. Am. Chem. Soc. 1987, 109, 5551. b) E. J. Corey, R. K. Bakshi, S. Shibata, C.-P. Chen, V. K. Singh, J. Am. Chem. Soc. 1987, 109, 7925. c) E. J. Corey, S. Shibata, R. K. Bakshi, J. Org. Chem. 1988, 53, 2861.

Síntesis de abisomicina C 11
La oxazaborolidina enantiomérica (R)-CBS se obtiene mediante un proceso similar que implica una etapa de resolución (esquema 8).
Esquema 8
El ciclo catalítico que explica la reducción CBS se inicia con la formación
del complejo I (véase esquema 9), que se genera por coordinación del catecolborano con la oxazaborolidina. A continuación, la cetona se coordina con el átomo de boro de la oxazaborolidina, colocando su grupo más voluminoso (agrupación dieno) en una posición de mínima interferencia estérica (intermedio III ).
N BO
H PhPh
MeL2HB
Estado de transición favorecido
ataque a la cara Re del grupo carbonilo
I
II
IV
NB
N BO
H PhPh
Me
PhS
O
R
O
BL2
O
H
MePhS
R III
NB
O
L2B
O
H
MeSPh
R
PhS
OBL2
R
OB
OH
12
Esquema 9

Síntesis de abisomicina C
12
La transferencia intramolecular de hidruro en el intermedio III se produce mediante la intervención un estado de transición cíclico de seis eslabones. El resultado de esta reacción es la formación del intermedio IV , el cual forma el producto de reducción, en forma de alcoxiborano, y regenera el catalizador. 2. Síntesis del compuesto bicíclico 17 mediante reacción Diels-Alder
La síntesis del compuesto 17 estuvo precedida de una experimentación a fondo a fin de determinar las mejores condiciones de reacción. El primer ensayo de cicloadición se llevó a cabo entre el dienol 43 y el acrilato de metilo, en tolueno, en presencia de 1 equivalente de MeMgBr y de 1 equivalente de n-BuOH (véase esquema 10). La cicloadición transcurre a través del estado de transición 45, en el cual el átomo de magnesio del alcoxilato actúa como ácido de Lewis. El proceso genera el intermedio 46, que cicla espontáneamente a la lactona 47.
Esquema 10
Sin embargo, cuando estas condiciones se aplicaron sobre el dienol 13 la reacción no funcionó, debido seguramente al aumento del impedimento estérico provocado por la agrupación tiofenilmetilo. Al aumentar la temperatura a 55ºC, y después de 24 horas de reacción, se obtuvo el producto de cicloadición 17 con un 30% de rendimiento (véase entrada 1 de la tabla 1).

Síntesis de abisomicina C 13
La reacción en presencia de otros ácidos de Lewis, como TiCl4, AlCl3, Zn(OTf)2, MgBr2·EtO/DIPEA y MeAlCl2, tampoco mejoró los resultados anteriores.
Tabla 1
Entr. auxiliar base tiempo Rto
1 ninguno MeMgBr (1 eq.) 24 h 30% 2 BINOL Me2Zn (1 eq.), MeMgBr (1. eq.) 24 h < 5% 3
OH
OH
Me2Zn (1 eq.), MeMgBr(1. eq.)
36 h 35%
4
OH
NH2
MeMgBr (2 equiv.)
24 h 49%
5 14 MeMgBr (2 equiv.) 48 h 55% 6 14 (2 equiv) MeMgBr (3 equiv.) 48 h 70% 7 14 (3 equiv.) MeMgBr (4 equiv.) 12 h 80%
El método de cicloadición de Ward también conoce su variante
enantioselectiva, en la que se emplea BINOL como fuente de quiralidad.4 Sin embargo, con esta metodología sólo se obtuvieron trazas del producto de cicloadición 17 (véase entrada 2 de la tabla 1).
Cuando el BINOL se reemplazó por catecol el rendimiento aumentó al 35% (entrada 3 de la tabla 1). El grupo de Nicolaou pensó que la reacción se debería facilitar en presencia de aminofenoles, puesto que la mayor basicidad de la agrupación amino debería favorecer la coordinación de ésta con la sal de magnesio (II), mientras que el grupo hidroxilo fenólico debería coordinarse con el dienófilo, orientándolo adecuadamente en la reacción de cicloadición (véase 5 en el esquema 2). Así, cuando la reacción se llevó a cabo en presencia de 2-aminofenol el rendimiento aumentó hasta el 49% (entrada 4 de la tabla 1). Cuando la reacción se efectuó en presencia del aminofenol 14 el rendimiento subió al 55% en presencia de 2 equivalentes de MeMgBr. El mejor rendimiento, que llegó al 80%, se consiguió cuando se emplearon 3 equivalentes de 14 y 4 equivalentes de MeMgBr (entrada 7 de la tabla 1).
4 D. E. Ward, M. S. Souweha Org. Lett. 2005, 7, 3533.

Síntesis de abisomicina C
14
3. α. α. α. α-Hidroxilación de la agrupación lactónica: síntesis del compuesto 18
La reacción de α−hidroxilación se llevó a cabo mediante enolización de una disolución tetrahidrofuránica de la lactona 17, con LiHMDS a -78ºC, seguida de adición de (EtO)3P y burbujeo subsiguiente de oxígeno a la mezcla de reacción. La adición de HCl acuoso y extracción con acetato de etilo proporcionaron el compuesto 18 con un 74% de rendimiento. En el esquema 11 se indica el mecanismo de α-hidroxilación del compuesto 17.
Me
O
PhS
O
H
H NLi
SiMe3
Me
O
PhS
OLi
H Me3Si NH SiMe3+
Me
O
PhS
O
H
O O
Li
1)
2)
Me
O
PhS
O
H
O OLi
SiMe3
Me
O
PhS
O
H
O OLi
3)P(OEt)3
Me
O
PhS
O
H
OLiP
O
EtO
OEt
OEt+
Me
O
PhS
O
H
OLi H3O+
4)
Me
O
PhS
O
H
OH
17
18
48
48
49 50
50
49

Síntesis de abisomicina C 15
Esquema 11 La reacción de α-hidroxilación se inicia con la enolización de la lactona 18,
lo que lleva a la formación del enolato lítico 48. El ataque nucleofílico del enolato al oxígeno molecular genera el peróxido lítico 49, que es reducido por el fosfito de trietilo al alcóxido lítico 50, el cual se convierte en el compuesto neutro 18 por adición de HCl acuoso a la mezcla de reacción 4. Eliminación reductiva en el compuesto 18
La síntesis de alquenos mediante eliminación reductiva de sulfonas recibe el nombre de eliminación de Julia. Para este proceso se emplean metales activos amalgamados, como la amalgama de Na(Hg). La reacción ajustada para este proceso se indica a continuación:
El mecanismo consta de dos procesos redox (véase esquema 12).

Síntesis de abisomicina C
16
Esquema 12
En el primero de ellos la sulfona acepta un electrón del metal, produciéndose, a continuación, la ruptura reductiva del enlace C-S. Esta reacción forma el anión fenilsulfinato y un radical centrado en el carbono. En el segundo proceso redox el radical acepta un electrón del metal, lo que genera un carbanión que se transforma en el alqueno por expulsión del anión X-.
La ruptura reductiva del compuesto 18 se intentó mediante su conversión en sulfona seguida de reacción con Na(Hg), o con la combinación Mg/HgCl, pero los resultados fueron nulos. Sin embargo, la ruptura reductiva se consiguió directamente sobre el feniltioéter 18 por reacción con el anión radicalario 4,4´-di-ter-butilbifeniluro de litio (esquema 13).

Síntesis de abisomicina C 17
Me
O
PhS
O
H
OH
Me
O O
H
OH
18
Li + 1e + Li
+ 1e
Me
O
O
H
OH+
1er proceso redox-eliminación
1)
2)
2º proceso redox-apertura de lactona
Li + 1e + Li3)
Me
OO
H
OH4)
+ 1e
Me
OO
H
OH
PhS
LiDBB
51
51 52 53
LiDBB
Esquema 13 En el esquema anterior se indica el mecanismo de la reducción con LiDBB,
que actúa como dador monoelectrónico. El sustrato 18 capta el electrón procedente del LiDBB y experimenta la ruptura reductiva del enlace C-S, lo que lleva a la formación del anión feniltiolato y del radical 51. Una segundo proceso redox convierte al radical 51 en el anión 52, que se transforma en el carboxilato diolefínico 53 por apertura intramolecular del anillo lactónico. La esterificación de este compuesto proporciona el hidroxiéster metílico 19. 5. Oxidación con IBX (ácido o-yodoxibenzoico)
El ácido o-yodoxibenzoico se emplea en la oxidación de alcoholes a compuestos carbonílicos. Este reactivo se prepara por oxidación del ácido o-yodobenzoico con bromato potásico y ácido sulfúrico, o con oxono. El IBX es el precursor del peryodinano de Dess-Martin, que se obtiene por reacción del IBX con anhídrido acético en acido acético (esquema 14).

Síntesis de abisomicina C
18
COOH
IKBrO3
H2SO4
ácido o-yodobenzoico ácido o-yodoxibenzoico(IBX)
Ac2O, AcOH
reactivo deDess-Martin
I
O
OAc
OAc
O
O
Ac
I
O
OH
O
O
Esquema 14
El principal inconveniente del IBX es su baja solubilidad en disolventes orgánicos, lo que obliga a su utilización en disolventes polares como el dimetilsulfóxido. Otro de los inconvenientes del empleo del IBX es su supuesta detonación al ser sometido a choques o a calentamientos por encima de los 200ºC. Sin embargo, se ha demostrado que la posibilidad de explosiones con el IBX se debe a la presencia de restos del bromato potásico empleado en su preparación. El IBX comercial se estabiliza mediante la adición de ácidos carboxílicos como el ácido benzoico o el ácido isoftálico.
El IBX existe como mezcla de tautómeros uno de los cuales es la forma de ácido carboxílico.
La acidez del IBX (pKa en agua 2.4; pKa en DMSO 6.65) puede afectar a
sustratos sensibles en el proceso de oxidación. Recientemente se ha propuesto un mecanismo para la oxidación de alcoholes
con IBX. En este mecanismo, basado en cálculos mecano-cuánticos, se demuestra que la etapa lenta, por tanto limitante de la velocidad global del proceso, es la denominada rotación hipervalente,5 que es el giro coordinado de ligandos alrededor del yodo. Este proceso está impulsado por la necesidad de generar la forma estable plana del IBA (ácido yodosobenzoico), que es el producto resultante de la reducción del IBX (véase esquema 15).
5 J. T. Su, W. A. Goddard III, J. Am. Chem. Soc. 2005, 127, 14146.

Síntesis de abisomicina C 19
Esquema 15
El mecanismo de la oxidación comienza con el proceso de intercambio de ligandos, en el que se reemplaza la agrupación hidroxilo del IBX por el alcohol (esquema 15). Esta reacción forma el intermedio 54, que experimenta a continuación la etapa de rotación hipervalente con formación de 55. Sobre este compuesto se produce el paso de eliminación, que tiene lugar mediante un proceso concertado cíclico de cinco eslabones, que conduce al compuesto carbonílico y al ácido yodosobenzoico (IBA).
Para la oxidación del metanol se han determinado las siguientes barreras energéticas que demuestran que la etapa lenta es la de rotación hipervalente (12.1 kcal/mol) (véase esquema 16).

Síntesis de abisomicina C
20
IOO
OOH
IOO
OO
H RH
+
IOO
O
O
H
HR
IOO
OH O
H
RIBX
55
54
intercambio deligandos
9.1 kcal/mol
twisting12.1 Kcal/mol
eliminación4.7 Kcal/mol
IBA
RCH2OH
Esquema 16 La rotación del doble enlace I-O está favorecida por el impedimento estérico
que existe entre el átomo de hidrógeno alílico, que ocupa la posición orto, y los protones de la agrupación alcoxi. En el esquema 17 se indica el proceso de rotación hipervalente para el intermedio resultante del intercambio de ligandos entre el IBX y el metanol. En este esquema se aprecia cómo la interacción estérica entre el átomo de hidrógeno orto (R=H) y el grupo metilo desaparece en el producto que resulta del proceso de rotación.
disminución de lainteracción estérica
H
H
H
OI
O
O
HH
H
RO
posición orto
H
H
H
OI
O
RO
O
H
HH
Esquema 17
De acuerdo con el esquema anterior, los cálculos computacionales han
demostrado que la velocidad de la oxidación aumenta en un factor de 100 cuando se emplea un derivado del IBX que contiene un grupo metilo en la posición orto (R=Me), porque en este derivado se favorece el proceso de rotación hipervalente. Este proceso también explica que el IBX oxide más rápidamente a los alcoholes más voluminosos. En estos alcoholes la mayor interacción estérica con el hidrógeno orto aumenta la velocidad de la etapa de rotación hipervalente.

Síntesis de abisomicina C 21
La aplicación más generalizada del IBX en síntesis orgánica es como oxidante de alcoholes, pero el IBX también es capaz de oxidar metilenos bencílicos, de deshidrogenar compuestos carbonílicos y convertirlos en compuestos carbonílicos α,β-insaturados, y de romper oxidativamente dioles vecinales:
En el esquema 18 se indica el mecanismo de ruptura oxidante del 1,2-diol. El proceso se inicia con la adición del dimetilsulfóxido a la molécula de IBX, lo que genera el intermedio 56 (esquema 18). En el segundo paso se produce el intercambio de ligandos y la generación del intermedio 57.
Esquema 18

Síntesis de abisomicina C
22
3)
I
OOH
O
O
OH
R
R
OH
I
OOH
O
O
RR
O
+ H2O
I
OOH
O
O
RR
O
4)
I
OH
O
O
O
R R
O+ +
58
5857
IBA
Esquema 18 (cont.) El intermedio 57 se transforma en 58 mediante un proceso de deshidratación
intramolecular. Por último, el proceso concertado cíclico sobre 58 lleva a la formación de IBA y los dialdehídos resultantes del proceso de ruptura oxidante. 9. Destiocetalización con bis-(trifluoroacetato) de fenilyodonio, PIFA
La reacción ajustada para el proceso de destiocetalización empleando PIFA
en medio acuoso es la siguiente:

Síntesis de abisomicina C 23
El mecanismo que explica el proceso de destiocetalización se inicia con el ataque nucleofílico de uno de los átomos de azufre del anillo de propilidenditioacetal sobre el átomo de yodo hipervalente del PIFA. Este ataque está favorecido por la elevada nucleofília del átomo de azufre y por la expulsión de un excelente grupo saliente (el anión trifluoroacetato, esquema 19). El catión sulfonio, que se forma en el proceso anterior, experimenta un ataque nucleofílico intramolecular con formación de yodobenceno y un dicatión sulfonio, el cual es atacado a continuación por una molécula de agua. El proceso de hidrólisis conduce finalmente al compuesto carbonílico y a la formación de 1,2-ditiolano.
S S
R R´I
Ph
OCOCF3
OCOCF3
S S
R R´
I
Ph
OCOCF3
S S
R R´
I
Ph
OCOCF3
1)
2)S S
R R´
+ PhI + 2 CF3COO
S S
R R´
3)
OH H
S S
R R´O
H
+ 2 CF3COO + CF3COOH
S S
RR´
OH
4)
R
O
R´+ CF3COOH + S S
CF3COO
CF3COO
CF3COO
CF3COO
Esquema 19


Síntesis de adociacetileno B 25
SÍNTESIS DE ADOCIACETILENO B
Aislamiento: El adociacetileno se ha aislado de la esponja marina Adocia sp., recolectada en Okinawa Actividad biológica: Los compuestos poliacetilénicos naturales, entre los que se encuadra el adociacetileno B, presentan una amplia variedad de actividades biológicas, entre las que destacan la actividad antibiótica y anti-SIDA.
Retrosíntesis La simetría C2 que posee el adociacetileno B simplifica su retrosíntesis
puesto que se puede efectuar la misma desconexión de enlaces a ambos lados de la molécula (esquema 1).6
Esquema 1
6 B. M. Trost, A. H. Weiss, Org. Lett. 2006, 8, 4461.
Síntesis de adociacetileno B
26
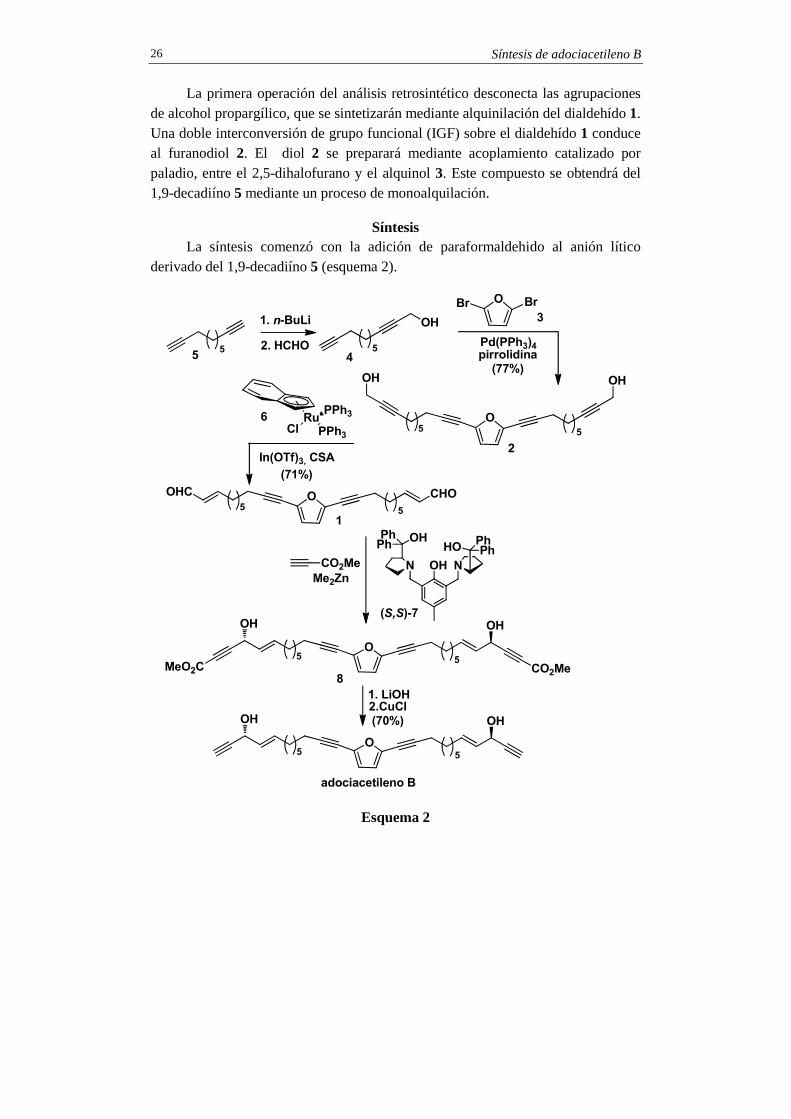
La primera operación del análisis retrosintético desconecta las agrupaciones de alcohol propargílico, que se sintetizarán mediante alquinilación del dialdehído 1. Una doble interconversión de grupo funcional (IGF) sobre el dialdehído 1 conduce al furanodiol 2. El diol 2 se preparará mediante acoplamiento catalizado por paladio, entre el 2,5-dihalofurano y el alquinol 3. Este compuesto se obtendrá del 1,9-decadiíno 5 mediante un proceso de monoalquilación.
Síntesis
La síntesis comenzó con la adición de paraformaldehido al anión lítico derivado del 1,9-decadiíno 5 (esquema 2).
adociacetileno B
5O
OHOH
5
5O
5
2
OH OH
OBr Br
OH
54
5
1. n-BuLi
2. HCHO Pd(PPh3)4pirrolidina
(77%)
5O CHOOHC
51
In(OTf)3, CSA
(71%)
RuPPh3
Cl PPh3
CO2Me
Me2ZnOH N
HOPhPh
N
OHPhPh
6
(S,S)-7
5O
OHOH
5MeO2C CO2Me
1. LiOH2.CuCl(70%)
8
5
3
Esquema 2

Síntesis de adociacetileno B 27
La adición de formaldehído proporcionó el diínol 4, que se sometió al acoplamiento de Sonogashira con el 2,4-dibromofurano, comercialmente accesible. Esta reacción, que se llevó a cabo en ausencia de sales de Cu(I) (véanse comentarios), permitió la obtención del alcohol bispropargílico 2. Este compuesto se convirtió en el dialdehído 1 mediante isomerización redox provocada por el catalizador de Ru(II) 6 en presencia de In(OTf)3 (véanse comentarios). La alquinilación del dialdehído 1, con propargilato de metilo y dimetilzinc, en presencia del ligando quiral (S,S)-7 (véanse comentarios), proporcionó el bispropargilato de dimetilo 8 con un exceso enantiomérico mayor del 99% y una diastereoselectividad de (17:1) (dl:meso). Finalmente, la saponificación de 8 con LiOH, seguida de descarboxilaron con CuCl (véanse comentarios), condujo al (-)-adociacetileno B sintético.
Comentarios
1. Acoplamiento de Sonogashira en ausencia de Cu(I)
+ R2 XR1 R1Pd(0)H R2
amina
La reacción de Sonogashira permite el acoplamiento entre acetilenos terminales y compuestos electrofílicos, en presencia de Pd(0) y Cu(I) a temperatura ambiente. También se ha observado que los triflatos, o haluros de alquenilo, o arilo, reaccionan de forma rápida con alquinos terminales, en ausencia de sales de cobre. En este caso la reacción se tiene que llevar a cabo en disolventes como la pirrolidina o la piperidina.7
En la síntesis de adociacetileno B se efectuó la siguiente reacción de Sonogashira:
7 M. Alami, F. Ferri, G. Linstrumelle Tetrahedron Lett. 1993, 34, 6403.

Síntesis de adociacetileno B
28
El mecanismo de esta reacción se indica en el esquema 3. El ciclo catalítico se inicia con la etapa de adición oxidante, que forma el intermedio I . Por otro lado, la reacción ácido-base entre la pirrolidina y el alquino terminal genera el correspondiente alquinuro. Esta especie interviene en el proceso de intercambio de ligando con el complejo I para generar el intermedio II . Finalmente, la eliminación reductora en II proporciona el producto de acoplamiento y regenera la especie catalítica.
Esquema 3
2. Isomerización redox de alcoholes propargílicos a enales o enonas
R1R2
OH
In(OTf)3, CSA
RuPPh3
Cl PPh36
R1 R2
O
La isomerización de alcoholes propargílicos catalizada por Ru(II) representa un método de síntesis de aldehídos y cetonas α,β-insaturados muy simple, práctico y eficaz. En el esquema 4 se muestra el ciclo catalítico de este proceso de

Síntesis de adociacetileno B 29
isomerización redox.8 La especie que cataliza el proceso es el complejo catiónico I , que resulta de la pérdida de anión cloruro por parte de 6 (IND = indanoílo). El catión I interviene en un proceso de intercambio de ligandos con el alcohol propargílico, lo que genera el intermedio II . Éste experimenta una transposición 1,2 de H y da lugar al intermedio III . Finalmente, una hidrólisis ácida del intermedio III , seguida de coordinación con trifenilfosfina, libera el producto de reacción y regenera la especie catiónica I .
RuPPh3
Cl PPh36
R1 R2
O
IND
Ru
PPh3Ph3P
Cl
R1
R2
OH
R1
R2
O
H
RuH
IND
Ph3P
PPh3
In(OTf) 3
R1 R2
ORuIND
Ph3P
H
PPh3 + H
H
In(OTf) 3
I
II
III
H
H
Esquema 4
El papel de la sal de In(III) todavía no se conoce. Se han propueto dos alternativas: una es la que se muestra en el esquema 3, en la cual el In(III) actúa como “scavenger” del anión cloruro, promoviendo la formación de la especie catalítica I . La otra posiblidad es que el In(III) se coordine con el alcohol propargílico para generar el intermedio IV , que presenta menor tensión estérica que el intermedio II (véase el esquema 5).
8 8 B. M. Trost, R. C. Livingston, J. Am. Chem. Soc. 1995, 117, 9586.

Síntesis de adociacetileno B
30
RuPPh3
ClPPh36
R1 R2
O
IND
Ru
PPh3Ph3P
Cl
R1
R2
OH
PPh3
In(OTf)3
R1 R2
ORu
IND
Ph3P
H
PPh3 + H
H
R1
R2
O
H
RuH
IND
Ph 3PO In
Tf OTf
OTf
R1
R2
O H
TfO InOTf
OTf
I
IVIII
H
H
In(OTf)3 +
Esquema 5
3. Alquinilación asimétrica con el complejo ZnMe2-(S,S)-7
R2
ZnMe2 (3 eq.)
R1CHO +
OH N
HOPhPh
N
OHPhPh
(S,S)-7 OH
R1
R2
En el esquema 6 se muestra el ciclo catalítico para la alquinilación asimétrica
de aldehídos con el complejo Me2Zn-(S,S)-7.9 El ciclo comienza con la coordinación de 2 equivalentes de alquinuro de zinc al complejo. Esta interacción
9 B. M. Trost, A. H. Weiss, A. J. von Wangelin, J. Am. Chem. Soc. 2006, 128, 8.

Síntesis de adociacetileno B 31
genera el intermedio I , que se convierte en el complejo II por coordinación con el aldehído (la coordinación se produce en el átomo de Zn estéricamente más accesible). A continuación tiene lugar la transferencia de una unidad de alquino al carbonilo del aldehído, estableciéndose así la estereoquímica del producto. Finalmente, la transmetalación con una molécula de alquinuro de zinc libera el producto de la reacción, en forma de alcóxido, y regenera el intermedio I .
Esquema 6 4. Descarboxilación de ácidos propargílicos promovida por Cu(I)
El mecanismo de descarboxilación de ácidos propargílicos con sales de Cu(I)
comienza con la formación del carboxilato de cobre I (véase el esquema 7). Sobre el carboxilato de cobre se produce el proceso de descarboxilación, que da lugar al alquinuro de cobre II y a CO2. La protonación del alquinuro, por reacción con una molécula de ácido propargílico, proporciona el alquino terminal, y regenera el carboxilato de cobre I , que inicia un nuevo ciclo catalítico.

Síntesis de adociacetileno B
32
Esquema 7

Síntesis de alociatina B2 33
SÍNTESIS DE ALOCIATINA B 2
Aislamiento: La alociatina B2 se aisló, en 1979, del fruto de Cynanthus earlei. Actividad biológica: La alociatina B2 pertenece a la familia de diterpenos denominados ciatinas. Las ciatinas presentan actividad antifúngica y antibacteriana, especialmente contra las bacterias Gram-positivas y Gram-negativas.
Retrosíntesis
La alociatina B2 presenta un esqueleto tricíclico que contiene fusionados tres anillos de 5, 6 y 7 miembros, con un sistema diénico conjugado y dos grupos metilo en posición 1,4-anti. En el esquema 1 se muestra el análisis retrosintético de la alociatina B2, que se inicia con la desconexión del enlace C11-C12.10 Esta operación, basada en una reacción aldólica, conduce al dialdehído 1. El análisis retrosintético se continúa sobre la forma hemiacetálida del dialdehído 1. Así, el hemiacetal 2 se obtendrá del dialdehído 3 mediante una reacción de Knoevenagel hidroxilante. La desconexión clave del análisis retrosintético es la que convierte el dialdehído bicíclico 3 en el hidroxiéster monocílico 4. En el sentido sintético el compuesto 3 se obtendrá a partir de 4 mediante una reacción de cicloisomerización en el sistema de 1,7-enino que contiene éste. Las desconexiones de los enlaces C4-C5 y C6-C7, basadas en acoplamientos catalizados por paladio, conducen a la ciclopentanona 5. Este compuesto se preparará a partir de 6 mediante adición Michael de dos restos metílicos y alilación enantioselectiva catalizada por paladio. Finalmente, la cetona 6 se obtendrá a partir de la 2-metilciclopentanona.
10 B. M. Trost, G. M. Schroeder, J. Am. Chem. Soc. 2005, 127, 10259.

Síntesis de alociatina B2 34
adición
aldólica
reacción de
Knoevenagel
alociatina B2
OHC
OH
12
3
45
6
9
10
11
12
14
15
16
19
20
OHC
OHC
OHOHO
OHC
1 2
6
11
12
14
15
6
11
12
14
15
cicloisom.
OHC CHO
3
6
11
5
5
10
10
8
8
9
9
Sonogashira
OO
9
17
8
5
alilación
asimétrica
t-BuO
O
6
ad. Michael
6
11
145
10
9
OH
4
CO2R
Negishi
14
ad. Michael
74
7
9
Esquema 1
Síntesis
1. Síntesis del intermedio 4
La síntesis del intermedio 4 comenzó con la reacción de C-formilación de la
2-metilciclopentanona por tratamiento de ésta con formiato de etilo y t-butóxido sódico (esquema 2). El subsiguiente tratamiento ácido proporcionó el ceto-enoléter

Síntesis de alociatina B2 35
6. Este compuesto, por reacción con LDA y cloruro de trimetilestaño, se convirtió en un enolato de estaño, el cual se sometió a un proceso de alilación enantioselectiva mediante tratamiento con acetato de alilo en presencia de (alil)PdCl2 y del ligando quiral (S,S)-L. El resultado fue la obtención de la alilciclopentanona 7 con un 85% de rendimiento químico y un 95% de exceso enantiomérico (véanse comentarios).
Esquema 2

Síntesis de alociatina B2 36
La doble adición Michael de dimetilcuprato de litio a la cetona 7 proporcionó el isopropilderivado 5, que se transformó en el triflato de enol 8. El acoplamiento de Sonogashira entre 8 y el trimetilsililacetileno dio lugar al intermedio 9 (véanse comentarios). La ruptura oxidante del doble enlace condujo al alcohol 10, que se transformó en el yoduro 11 por reacción con yodo molecular y trifenifosfina. La instalación del fragmento que comprende los carbonos C6-C14-C13 se consiguió mediante acoplamiento de Negishi. Para ello, el yoduro de alquilo 11 se convirtió en el reactivo organozíncico 12 mediante intercambio metal-halógeno con t-BuLi seguido de transmetalación con ZnCl2. La reacción de 12 con el yoduro de vinilo 13, en presencia de tetrakistrifenilfosfina paladio(0), proporcionó el producto de acoplamiento 14 (véanse comentarios). La eliminación de la agrupación trimetilsililo fue seguida de carboxilación. lo que condujo, después de un proceso de desililación, al intermedio 4, 2. Construcción del sistema tricíclico y pasos finales
alociatina B2
OHC
OH
5
610
11
12
14
15
OO
17
6
11
12
14
15
10
CN
6
11
145
10OH
4
CO2R
13
13
5
Tal y como se ha indicado en el análisis retrosintético, la construcción del
ciclo de seis miembros se llevó a cabo mediante una reacción de cicloisomerización del compuesto 4 (Esquema 3). Así, el tratamiento del enino 4 con cantidades catalíticas del complejo de Ru(II) CpRu(CH3CN)3PF6 condujo a la obtención del aldehído-éster bicíclico 16 (véanse comentarios). Este compuesto se sometió a la reacción de Knoevenagel hidroxilante, lo que se consiguió con PhS(O)CH2CN y piperidina (véanse comentarios). El nitrilo α,β-insaturado 17, obtenido en la reacción anterior, se convirtió en el nitrilo 18 mediante hidrogenación selectiva. A continuación, la reacción de 18 con DIBAL provocó la reducción simultánea de las agrupaciones nitrilo y éster, y condujo al dialdehído 1, el cual, mediante condensación aldólica intramolecularse trasnformó en la alociatina B2.

Síntesis de alociatina B2 37
Esquema 3
Comentarios 1. Alilación asimétrica Tsuji-Trost de enolatos catalizada por Pd(0)
R1R2
O
LDA, Me 3SnCl R1
R2
O
[(alil)PdCl] 2, (S,S)-L*
OAc
La α−alilación de enolatos derivados de cetonas catalizada por paladio se basa en la metodología, desarrollada por Trost, de alilación asimétrica cinético-dinámica (DYKAT).11
La metodología DYKAT aplicada a enolatos de cetonas permite obtener α−alilcetonas quirales mediante tratamiento directo de la cetona con una base (LDA), un ácido de Lewis (Me3SnCl), que estabiliza el enolato, un complejo de paladio(0) y un ligando quiral. Esta metodología es la que se emplea en la trasformación de la cetona 6 en la α−alilcetona 7.
11 a) B. M. Trost, G. M. Schroeder J. Am. Chem. Soc. 1999, 121, 6759: b) Para una revisión sobre la metodología DYKAT y su aplicación en la síntesis de productos naturales véase: B. M. Trost, D. R. Fandrick, Aldrichimica Acta 2007, 40, 59.

Síntesis de alociatina B2 38
Durante el desarrollo de esta metodología se observó que la naturaleza de los
cationes asociados al enolato influye tanto en la reactividad como en la enantioselectividad. Este hecho experimental lleva a pensar que la estructura del nucleófilo que interviene en el proceso no es un éter de enol, sino que, probablemente, se trate de un complejo ato.
La especie catalíticamente activa se genera por intercambio de ligandos entre el complejo de Pd(0), bis[alilcloropaladio(0)], y la fosfina quiral (S,S)-L:
Por otro lado, la cetona 6 es convertida en un enolato de litio, por reacción
con LDA, que, a su vez, es transmetalado por el Me3SnCl al enolato de estaño A.
El ciclo catalítico comienza con la formación del acetato π-alílico quiral II por adición oxidante del acetato de alilo al complejo I (esquema 4). El siguiente paso del ciclo implica el intercambio de acetato por enolato, proceso que se podría denominar transmetalación y que conduce a la formación del enolato π−alílico

Síntesis de alociatina B2 39
quiral III . A continuación se produce el ataque nucleofílico enantioselectivo del enolato al catión π−alílico III , para dar lugar a la α−alilciclopentanona 7 y al catalizador de paladio I , que reinicia el ciclo catalítico.
Esquema 4
2. Acoplamiento de Sonogashira
TMS
9
OTf
8CuI, BuNH 2
(85%)
TMSHPd2(dba) 3, Ph3P
El esquema 5 muestra todos los pasos del doble ciclo catalítico que se
establece en la reacción de Sonogashira entre 8 y trimetilsililacetileno.

Síntesis de alociatina B2 40
Esquema 5
3. Acoplamiento de Negishi
TMS
ZnCl12
Pd(PPh3)4
IOTPS
TMSOTPS
14
13
El esquema 6 muestra todos los pasos del ciclo catalítico que se establece en
la reacción de Negishi entre 12 y 13.

Síntesis de alociatina B2 41
Esquema 6
4. Cicloisomerización de 1,n-eninos catalizada por Pd(0) vs. Ru(II)
n n
[Ru] ó [Pd]
Las cicloisomerizaciones de 1,n-eninos se emplean en la construcción de
anillos debido a su elevada economía atómica. El mecanismo de estos procesos, catalizados por Pd(0) o por Ru(II), depende del metal y, en consecuencia, el producto de la reacción es función, en gran medida, del catalizador utilizado.12
12 B. M. Trost, F. D. Toste, J. Am. Chem. Soc. 2002, 124, 5025.

Síntesis de alociatina B2 42
En el esquema 7 se muestra el ciclo catalítico para la cicloisomerización catalizada por paladio. La especie catalíticamente activa resulta de la protonación del complejo de Pd(0), intermedio catiónico I , que se coordina con el enino para formar el intermedio II . A continuación, la hidropaladación de la unidad acetilénica lleva a la intermedio vinílico III . Este proceso va seguido de una carbopaladación intramolecular, que conduce al complejo IV . Finalmente, una reacción de β-eliminación de hidruro completa el ciclo catalítico.
Tal y como se muestra en el esquema 6 la β-eliminación puede dar lugar a un 1,3-dieno o a un 1,4-dieno. Experimentalmente se ha observado que el proceso transcurre con excelente regioselectividad y la formación del 1,3-dieno o del 1,4-dieno depende de la estructura del sustrato de partida.
Esquema 7
En la cicloisomerización catalizada por rutenio, la especie catalíticamente activa es el intermedio catiónico I , que resulta de la descoordinación del Ru(II) (esquema 8). En el esquema 8 se muestra la estructura orbitálica del complejo catiónico de Ru(II) que se emplea como precatalizador.

Síntesis de alociatina B2 43
Esquema 8
En este complejo el Ru(II) forma tres enlaces σ con tres moléculas de
acetonitrilo, en las cuales el par solitario del N del acetonitrilo solapa frontalmente

Síntesis de alociatina B2 44
con un orbital spd2 vacío del rutenio. En este solapamiento cada molécula de acetonitrilo cede dos electrones al Ru. Por otra parte, el anión ciclopentadienilo (Cp) se coordina al metal compartiendo sus electrones π, que pasan a ocupar 3 orbitales híbridos spd2 del metal. En el complejo catiónico resultante, CpRu(CH3CN)3
+, el átomo de Ru presenta 18 electrones en su capa de valencia (6 propios del metal + 2x3(CH3CN) + 6 del Cp= 18). La descoordinación de las 3 moléculas de acetonitrilo genera un complejo de Ru(II) insaturado, que es la especie que interviene en el ciclo catalítico.
En el esquema 9 se muestra el ciclo catalítico para la cicloisomerización de eninos catalizada por el complejo catiónico insaturado I . Este complejo se coordina con el enino para formar el intermedio II . A continuación, tiene lugar una cicloadición oxidante del enino al Ru(II) que lleva al intermedio III , con el Ru en estado de oxidación IV. Una β−eliminación de hidruro forma el intermedio IV , el cual, por eliminación reductora, regenera el catalizador y da lugar al producto de reacción, que en este caso es un 1,4-dieno. La elevada regioselectividad en la β−eliminación de hidruro, y por tanto, la formación exclusiva del 1,4-dieno se debe a que el proceso de β−eliminación del hidruro del metalaciclo (Hb) está muy desfavorecido debido a motivos puramente geométricos.
Esquema 9

Síntesis de alociatina B2 45
En la síntesis de la alociatina B2 se ensayó la cicloisomerización con ambos metales y se observó que la estereoquímica del producto de reacción dependía del catalizador empleado. Este hecho experimental es el primer ejemplo descrito en la literatura que demuestra el diferente comportamiento del Ru y Pd en este tipo de procesos.
A continuación, en el esquema 10 se muestra la estereoquímica de los compuestos que se obtienen según se empleen el Ru o el Pd como catalizadores de la reacción de cicloisomerización:
Esquema 10
El esquema 11 muestra el ciclo catalítico cuando se emplea Pd(0) como
catalizador. Después de la coordinación del hidruro de paladio(0) al enino se produce la hidropaladación que, en este caso, puede formar los intermedios II y III , en los cuales el Pd se coloca perpendicular al grupo isopropilo para evitar las interacciones estéricas. En el intermedio II , el enlace σ C-Pd y el enlace π del alqueno se colocan en disposición sin-coplanar, tal y como se requiere para que tenga lugar la carbopaladación. Así pues, esta orientación es energéticamente favorable y conduce al producto de reacción, que presenta los dos metilos en posición realtiva 1,4-sin.
Por otro lado, el intermedio III presenta el enlace σ C-Pd y el enlace π del alqueno en disposición ortogonal, lo que desfavorece el proceso de carbometalación e impide que se obtenga el producto de configuración relativa 1,4-anti.

Síntesis de alociatina B2 46
Pd(0)
coordinación
carbopaladacióndel alqueno
hidropaladación
del alquino
H Pd0
H
OHPdo
H
-eliminación
de hidruro
10
614
9
6
14
510
89
OH
6
14
9
OH
6
14
9
5
614
9 OH
5
14
9
PdII
OH
6
H
OH
6
14
510
89
OH
DESFAVORABLE
I
II
III
IV
PdII
PdII
Esquema 11
El esquema 12 muestra el ciclo catalítico cuando se emplea Ru(II) como
catalizador. En este caso, el proceso de coordinación del rutenio al enino lleva a dos confórmeros I y II . El confórmero I presenta una orientación sin-coplanar del doble y del triple enlace, mientras que en el confórmero II dicha orientación es ortogonal. La disposición sin-coplanar de los enlaces múltiples favorece el proceso de cicloadición oxidante y, por tanto, la formación del rutenaciclopenteno III , que es el que conduce a la formación del producto de reacción con los dos grupos metilo en posición relativa 1,4-anti.

Síntesis de alociatina B2 47
Esquema 12
5. Reacción de Knoevenagel hidroxilante
base R CNO
RH
CN
S(O)Ph+
OH
La reacción de aldehídos o cetonas con α-(fenilsulfinil)acetonitrilo, en presencia de una base, permite obtener directamente γ-hidroxialquenonitrilos. Este proceso recibe el nombre de reacción de Knoevenagel hidroxilante
El mecanismo se inica con la ionización del α-(fenilsulfinil)acetonitrilo por reacción con la piperidina (esquema 12). El intermedio aniónico I se adiciona al carbonilo aldehídico lo que conduce, después de la protonación, a la formación del intermedio III . El proceso de eliminación sobre el intermedio III forma el sulfoxinitrilo insaturado IV , que experimenta una migración del doble enlace, por transposición 1,3 de hidruro. Esta reacción forma el compuesto IV , que por transposición sigmatrópica del grupo sulfinilo proporciona el producto de reacción.

Síntesis de alociatina B2 48
Esquema 13
La reacción de Knoevenagel hidroxilante que se efectúa en la síntesis de la alociatina B2, tiene lugar con total estereocontrol, y sólo se observa la formación del diastereoisómero 17.

Síntesis de alociatina B2 49
En el esquema 14 se muestran los dos posibles estados de transición para la
reacción de transposición sigmatrópica. El estado de transición más favorable es el ET-I , debido a su menor compresión estérica, y es el que conduce a la formación de 17.
Esquema 14


Síntesis de aureotina y N-acetil-aureotamina 51
SÍNTESIS DE AUREOTINA y N-ACETIL-AUREOTAMINA
Aislamiento: La aureotina se ha encontrado en el micelio de algunos actinomicetos, mientras que la N-acetil-aureotamina se ha aislado de Streptomyces netropsis. Actividad biológica: La aureotina presenta actividad antitumoral, antifúngica y pesticida, mientras que la N-acetil-aureotamina ha demostrado ser un agente altamente selectivo y potente contra Helicobacter pylori, la bacteria causante de la gastritis crónica.
Retrosíntesis
La retrosíntesis se indica en el esquema 1 y se inicia con la desconexión del grupo metilo del sistema diénico conjugado, que se instalará en la última etapa de la síntesis de aureotina mediante un acoplamiento cruzado, catalizado por paladio, entre el bromuro de alquenilo 1 (electrófilo) y un nucleófilo del tipo metil-metálico.13 La desconexión sp2-sp2 del sistema diénico, basada en una reacción de acoplamiento cruzado de Suzuki, conduce a la gem-dibromoolefina 3 (electrófilo) y al alquenilboronato 2 (nucleófilo). El doble enlace del intermedio 2 se desconecta, mediante metátesis cruzada, al metilentetrahidrofurano 4, y a un boronato de alquenilo, no indicado en el esquema retrosintéico. El anillo tetrahidrofuránico de 4 se construirá mediante una ciclación [3+2], catalizada por paladio, entre el acetato alílico 5 y el aldehído 6, que se sintetizará a partir de la pirona 7.
13 M. F. Jacobsen, J. E. Moses, R. M. Adlington, J. E. Baldwin, Org. Lett. 2005, 7, 641.

Síntesis de aureotina y N-acetil-aureotamina 52
Me O
O
R
O
Me Me
OMe
aureotina (R=NO2)N-acetil-aureotamina (R=NHCOCH3)
O
O
R
O
Me Me
OMe
acoplamientoSuzuki
O
O
R
O
Me Me
OMeBr
Br
(RO)2B+
metátesiscruzada
O
O
O
Me Me
OMe
ciclación [3+2]
O
O
O
Me Me
OMeH
O
O
Me Me
OMe
Me
1
23
4
5
6
Br
AcO SiMe3
7
Esquema 1
El análisis retrosintético de la pirona 7 se indica en el esquema 2, y se simplifica enormemente si se tiene en cuenta que ésta se puede obtener mediante la O-metilación del hidroxilo enólico de la estructura 8, que a su vez no es mas que el producto de ciclación intramolecular del β,δ-dicetoéster 9. La desconexión de la parte de 1,3-dicetona de 9 conduce al sinton catiónico 10, cuyo equivalente sintético es el propionato de etilo, y al sintón aniónico 11, cuyo equivalente sintético es el dianión 12, derivado del 2-propanoilpropanoato de etilo.

Síntesis de clavosólido A
Esquema 2
Síntesis 1. Síntesis de la pirona 7
La preparación de la pirona 7 se indica en el esquema 3 y su síntesis se inició con la autocondensación de Claisen del propanoato de etilo, lo que proporcionó el β-cetoester 13 (esquema 3). La reacción de acilación del β-cetoester 13 tiene que ser regioselectiva, puesto que el carbono que debe experimentar el proceso de acilación es el numerado como C4 en la estructura 13. Sin embargo, los hidrógenos más acidos del β-cetoéster 13 son los de C2, al estar flanqueados por la agrupación éster y por el grupo carbonilo. Para conseguir la ionización de C4 se llevó a cabo una primera ionización del β-cetoester 13 con NaH en THF, lo que formó el enolato de sodio 14. A continuación, se añadió la base fuerte n-BuLi, que convirtió el enolato 14 en el dienolato 12 por captura de uno de los protones en C4. El dienolato 12 no es más que la estructura resonante del dianión 12, indicado en el esquema retrosintético 2.

Síntesis de aureotina y N-acetil-aureotamina 54
O
O
Me Me
OMe
Me
O
O
Me Me
OH
Me
OCOOEt
O
Me Me
Me
78
O
Me
OEtCOOEt
Me
O
Me
NaH, THF
Me
O
Me
17
O Na
OEt
Me
O
Me
O
OEt
NaLi
1
23
4
1
2
3
4
1
2
3
4
1
2
34
EtOH
NaOEt
O
MeOEt
COOEt
Me
O
MeMe
O
O
O
Me Me
MeO
OEt
H
O
O
Me Me
O
Me
SO4Me2
16
13 14
15 12
n-BuLi
Esquema 3
Después de la generación del dianión, o dienolato 12, se añadió a la
disolución que lo contenía el reactivo electrofílico, que en este caso fue el propanoato de etilo. El propanoato de etilo es atacado regioselectivamente por el átomo de carbono C4, que es el más nucleofílico del dienolato, para dar lugar, después de la hidrólisis de la mezcla de reacción, al dicetoéster 15. La estructura 16, que es la forma enólica de 15, contiene un hidroxilo nucleofílico, que ataca intramolecularmente al carbonilo del éster para formar el compuesto cíclico 17. Finalmente, la O-metilacióne 8, la forma enólica de 17, proporcionó la pirona 7.
2. Síntesis del intermedio tetrahidrofuránico 4
Me O
O
O2N
O
Me Me
OMe
aureotina
O
O
O
Me Me
OMe
4

Síntesis de clavosólido A
La síntesis del intermedio 4 se inició con la deshidrogenación de la cadena etílica de la pirona 7 (véase esquema 4). Así, la reacción de la pirona 7 con la base KHMDS generó el enolato potásico 17, que se hizo reaccionar con bromuro de fenilselenilo para dar el selenocompuesto 18. La oxidación del átomo de selenio con NaIO4 generó el selenóxido 19, que experimentó, in situ, la reacción de eliminación sin concertada para dar la olefina 20. La ruptura oxidante selectiva del doble enlace lateral se consiguió mediante dihidroxilación, seguida de reacción del diol 21 con NaIO4, y condujo aldehído 6.
O
O
Me Me
OMe
Me 7
KHMDS, PhSeBr
THF, -78ºC 82%) O
O
Me Me
OMe
Me
K
Ph Se Br
O
O
Me Me
OMe
Me
PhSe
NaIO4, NaHCO3
MeOH, ac.
O
O
Me Me
OMeSe
O
Ph
H
O
O
Me Me
OMe
OsO4, NaIO4
ac. THF
O
O
Me Me
OMe
O
H
Me3Si OAc
Pd(PPh3)4, In(acac)3,
tolueno, reflujo
O
O
Me Me
OMe
O
O
O
Me Me
OMeHO
HO
4
21
17 18
1920
5
6
Esquema 4
Para la construcción del anillo tetrahidrofuránico se recurrió a la metodología de ciclación [3+2] desarrollada por Trost que utiliza el acetato de 2-trimetilsilanilmetil-propen-2-ol 5. Este reactivo aporta 3 carbonos a la formación del ciclo. El segundo componente en esta metodología de ciclación es un aldehído, que en el caso de la síntesis de la aureotina fue el aldehído 6, que incorpora los 2 átomos del grupo carbonilo en la formación del anillo de tetrahidrofrurano. La reacción de ciclación se llevó a cabo en presencia de 5 mol% de Pd(PPh3)4 y de 10

Síntesis de aureotina y N-acetil-aureotamina 56
mol% de In(acac)3 en tolueno a reflujo, y proporcionó el compuesto 4 con un 93% de rendimiento (véanse comentarios). Hay que hacer constar que en este punto de la síntesis se genera el único estereocentro de la aureotina de forma no enantioselectiva y, en consecuencia, el compuesto 4, y la aureotina final, se obtendrán en forma racémica.
3. Pasos finales
En el recuadro anterior se indican las fuentes de carbono necesarias para culminar la síntesis de la aureotina a partir del tetrahidrofurano 4. En el esquema 5 se describen las etapas finales, que se iniciaron con la reacción de metátesis cruzada entre el alquenil boronato 22 (2 equivalentes) y el compuesto 4, en presencia del catalizador de Grubbs de 2ª generación, en diclorometano a reflujo (véanse comentarios). La reacción, que proporcionó con un rendimiento del 89% la olefina cruzada 23, no fue estereoselectiva, puesto que este compuesto se obtuvo como mezcla de alquenos E/Z en relación 1:1.2. Por ello, la siguiente etapa sintética, que implicaba una reacción de acoplamiento cruzado de Suzuki, se llevó a cabo con la mezcla de olefinas. Así, la mezcla de alquenos 23 se hizo reaccionar con el dibromocompuesto 3, en presencia del catalizador Pd(Ph3P)4 y de etóxido de talio en tetrahidrofurano acuoso (véanse comentarios). En estas condiciones se obtuvo regioselectivamente el producto 24, como mezcla E/Z en el doble enlace, que refleja la proporción de estos isómeros en el alquenilboronato 23. La purificación cromatográfica de la mezcla de alquenos 24 condujo a la obtención del bromodieno 25 puro. Este compuesto se sometió a la reacción de acoplamiento de Negishi con dimetilzinc en presencia de Pd(tBu3P)2, lo que proporcionó la aureotina racémica con un rendimiento del 95% (véanse comentarios).
La N-acetil-aureotamina racémica se obtuvo por reducción el grupo nitro de la aureotina seguida de N-acetilación.

Síntesis de clavosólido A
Esquema 5
Comentarios
1. Reacción de cicloadición [3+2]
La síntesis de metilentetrahidrofuranos, mediante cicloadición [3+2] entre aldehídos y silil-acetatos alílicos, ha sido diseñada por Trost y col., quienes han demostrado que las sales de In3+ actúan de cocatalizadores del proceso.14 El mecanismo de la reacción se indica en el esquema 6, y se inicia con la coordinación
14 B. M. Trost, S. Sharma, T. Schmidt, J. Am. Chem. Soc. 1992, 114, 7903.

Síntesis de aureotina y N-acetil-aureotamina 58
del complejo de paladio(0) al doble enlace del sistema de acetato alílico. Esta coordinación provoca la formación de la sal de acetato y complejo π−alílico de paladio (intermedio I ). El ataque nucleofílico del anión acetato sobre el grupo trimetilsililo del intermedio I conduce a la formación de la betaína II , que en el siguiente paso ataca nucleofílicamente al carbonilo aldehídico para formar el intermedio III . Por último, el intermedio III experimenta el ataque nucleofílico intramolecular del alcóxido a la parte de catión alílico, lo que forma el compuesto tetrahidrofuránico y regenera el catalizador.
El efecto de la sal de In3+ en este proceso no está aclarado, pero se apunta a su participación en la etapa de ciclación, o incluso a la formación de una especie bimetálica paladio-indio que sería el verdadero catalizador del proceso.
Esquema 6 2. Reacción de metátesis cruzada
Las reacciones de metátesis ruzadas (CM, cross-metathesis) son reacciones
de metátesis termodinámicamente controladas. Para que el proceso tenga éxito

Síntesis de clavosólido A
(reacción 1 del esquema 9), y no se forme una mezcla estadística del producto de metátesis cruzada y de los productos de homoacoplamiento, se requiere que:
a) La olefina más reactiva que va a experimentar la reacción de cruce (R1CH=CH2 en el esquema 7) genere un homodímero que sea consumible en una subsiguiente reacción de metátesis cruzada con el otro alqueno (véase la secuencia 2 del esquema 7). b) La olefina menos reactiva R2CH=CH2 no experimente homoacoplamiento, o lo haga de forma muy lenta (reacción 3 del esquema 7).15
Esquema 7
La reacción de metátesis cruzada entre el furano 4 el pinacol boronato 22 se llevó a cabo en presencia del catalizador de Grubbs de 2ª generación y proporcionó una mezcla de alquenos E/Z en relación 1:1.2 (esquema 8).
Esquema 8
15 Para modelos empíricos que explican la selectividad en las reacciones de metátesis cruzada véase: A. K. Chatterjee, T.-L. Choi, D. P. Sanders, R. H. Grubbs, J. Am. Chem. Soc. 2003, 37, 11360.

Síntesis de aureotina y N-acetil-aureotamina 60
En el esquema 9 se indica el ciclo catalítico de este proceso, que está sometido a control termodinámico. La formación de la mezcla de alquenos no se debe al empleo de la mezcla de boronatos E/Z, puesto que la configuración de este doble enlace es irrelevante. El empleo del boronato de propenilo 22, en lugar de un boronato de etenilo, tiene que ver con la necesidad de marcar diferencias de reactividad entre las dos olefinas que participan en la metátesis cruzada, y evitar con ello la formación de productos de homoacoplamiento.
Esquema 9
El primer ciclo catalítico de la reacción de metátesis cruzada comienza con la cicloadición del catalizador 24 al doble enlace exocíclico del compuesto 4. Esta reacción genera el rutenaciclobutano I , el cual, mediante eliminación de estireno se convierte en el carbeno de rutenio II . La cicloadición de este intermedio al doble enlace del boronato 22 forma el rutenaciclobutano III , el cual, mediante

Síntesis de clavosólido A
ciclorreversión, proporciona el producto de metátesis cruzada 23 y genera el carbeno de rutenio Ru=CHCH3. Este catalizador es el que en realidad interviene, a continuación, en todos los siguientes ciclos catalíticos. 3. Acoplamiento de Suzuki
El acoplamiento de organoboranos, ácidos organobóricos y ésteres borónicos con haluros o triflatos de vinilo o arilo, bajo catálisis con paladio, recibe el nombre de reacción de Suzuki.
En la síntesis de la aureotina se llevó a cabo el acoplamiento de Suzuki entre
el alquenilborano 23 y el bromuro de alquenilo 3 (esquema 10).
Esquema 10
El ciclo catalítico para la formación de 24 se indica en el esquema 11. En
este ciclo se aprecia el papel que juega la base, etóxido de talio, que provoca la cuaternización del boronato 23 y la formación del borato (intermedio II ), que es la especie que interviene en la etapa de trasmetalación. El proceso de acoplamiento se inicia con la adición oxidante del derivado dibromado 3 al catalizador de paladio(0). Esta reacción forma el complejo I , que es el que experimenta la reacción de transmetalación con el borato II . Finalmente, el complejo III , generado en la reacción de transmetalación, mediante el proceso de eliminación reductora, proporciona el producto de acoplamiento 24, junto con el catalizador de paladio(0), el cual inicia un nuevo ciclo catalítico.

Síntesis de aureotina y N-acetil-aureotamina 62
Esquema 11 En esta reacción en particular se ha demostrado que la base no sólo provoca
la cuaternización del boronato sino que, además, influye en la estereoquímica y regioselectividad del proceso de acoplamiento. Así, cuando se emplea el etóxido de talio se obtiene el producto deseado 24 un 72% de rendimiento. Sin embargo, cuando se utiliza el NaOH no se obtiene el producto de monoalquilación, sino el producto de acoplamiento-eliminación 25, mientras que el empleo de Tl2CO3 conduce a la obtención de una mezcla formada por el producto 24, el producto de alquenilación-eliminación 25 y el producto de dialquenilación 26 (esquema 12).

Síntesis de clavosólido A
Esquema 12
4. Acoplamiento de Negishi
Br O
O
O2N
O
Me Me
OMe
Me2Zn, Pd(tBu3P)2THF, 25ºC (95%)
Me O
O
O2N
O
Me Me
OMe
aureotina
24
El acoplamiento de Negishi es mecanísticamente similar al acoplamiento de Stille y emplea como especie nucleofílica un compuesto orgonozíncico. El ciclo catalítico se inicia con la etapa de inserción oxidante del bromuro de alquenilo al catalizador de paladio(0) (esquema 13). El complejo de paladio(II) experimenta el

Síntesis de aureotina y N-acetil-aureotamina 64
proceso de transmetalación por reacción con Me2Zn, y el intermedio resultante proporciona, mediante el proceso de eliminación reductora, el producto de acoplamiento y regenera el catalizador.
Br O
O
O2N
O
Me Me
OMe
transmetalación
Pd(0)
adiciónoxidante
L
Leliminaciónreductora
Pd O
O
O2N
O
Me Me
OMe
Br
L LII
Me2ZnMeZnBr
Pd O
O
O2N
O
Me Me
OMe
Me
L LII
Me O
O
O2N
O
Me Me
OMe
23aureotina
Esquema 13

Síntesis de broussonetina G 65
SÍNTESIS DE BROUSSONETINA G
Aislamiento: Los alcaloides polihidroxilados denominados broussonetinas se han aislado de las hojas de Broussonetia kazinoki. Se conocen un total de 29 miembros de esta familia de productos naturales, que se caracterizan todos ellos por la presencia en su estructura de un núcleo de pirrolidina polihidroxilada, al que va unida una cadena que contiene un total de trece átomos de carbono, con diferentes grados de funcionalización según la brousonetina de que se trate. Actividad biológica: Las brousonetinas presenten un enorme potencial como agentes antitumorales y anti-SIDA debido a su capacidad como inhibidores de las glicosidasas.
Retrosíntesis
La primera operación del análisis retrosintético convierte la broussonetina G en el intermedio 1 (esquema 1).16 En el sentido sintético el producto natural se obtendrá por epoxidación de 1, seguida de hidrólisis del anillo oxiránico. Esta secuencia epoxidación-hidrólisis deberá permitir la instalación de los grupos hidroxilo en C3 y C4 en posición relativa trans. La desconexión del enlace C1´-C2´ genera el aldehído 2 y el fragmento organometálico 3.
Esquema 1
16 B. M. Trost, D. B. Horne, M. J. Woltering, Angew. Chem. Int. Ed. 2003, 42, 5987.

Síntesis de broussonetina G 66
El esquema 2 muestra los análisis retrosintéticos de los fragmentos 2 y 3. El aldehído 2 se obtendrá del alcohol 4, que se generará a partir del dieno 5 por reacción de metátesis ciclante. La síntesis de 5 efectuará enantioselectivamente mediante la aplicación de dos procesos DYKAT consecutivos sobre el vinilepóxido 6. Por otra parte, el fragmento organometálico 3 se preparará a partir del alcohol 8, que se obtendrá del triol 9 mediante un proceso de espiroacetalización catalizada por paladio. El triol 9 se formará por adición del anión derivado de 10 al óxido de etileno. El compuesto 10 se sintetizará a partir de 11 por reducción enantioselectiva del grupo carbonilo e isomerización del triple enlace y, finalmente, la cetona 11 se obtendrá por reacción de la valerolactona con el anión derivado del 1-pentino.
Esquema 2

Síntesis de broussonetina G 67
Síntesis 1. Síntesis del fragmento 2
O
O
M
NOP OP'
+
brousonetina G 2 3
NOH OHH
O
O
H
OHHO
2
3 4
5
1´6´
13´
10´2
3 4
5
1´6´
10´
13´
2´
H
La síntesis del fragmento 2 comenzó con un proceso DYKAT (alilación
asimétrica dinámico-cinética catalizada por paladio; véase síntesis de alociatina B2).
17 Este proceso se llevó a cabo por reacción de la ftalimida y el monóxido de butadieno racémico y se efectuó en presencia de bis(alilcloropaladio)(II) ([(alil)PdCl]2), la fosfina quiral (R,R)-L y carbonato sódico en diclorometano a temperatura ambiente (véase esquema 3). En estas condiciones se consiguió la apertura regioselectiva del anillo oxiránico y la obtención enantioselectiva del aminoalcohol 12 con un rendimiento casi cuantitativo (véanse comentarios). A continuación, la escisión del sistema de diimida de 12, mediante reacción con etilendiamina en etanol a reflujo durante 4 horas, condujo al vinilglicinol 7. Este compuesto, por reacción con trifosgeno, dio lugar a la oxazolidinona 13. Una nueva alilación asimétrica DYKAT del monóxido de butadieno con la oxazolidinona 13 proporcionó, de forma altamente diastereoselectiva, el compuesto 14. En este caso el proceso se efectuó en presencia de DBU, tris(dibencilidenacetona)dipaladio(0) (Pd2(dba)3) y la fosfina quiral (R,R)-L. La bencilación, seguida de reacción de metátesis ciclante con el catalizadorde Grubbs de segunda generación, condujo al dihidropirrol 16, el cual se convirtió en la dihidropirrolidina 4 por saponificación de la unidad de oxazolidinona, seguida de N-Cbz protección. Este compuesto se convirtió en la amida 17 por oxidación del hidroxilo libre con TEMPO y reacción de amidación con N-metil-O-metilhidroxilamina.
17 Para una revisión sobre la metodología DYKAT y su aplicación en la síntesis de productos naturales véase B. M. Trost, D. R. Fandrick, Aldrichimica Acta 2007, 40, 59.

Síntesis de broussonetina G 68
Esquema 3
2. Síntesis del fragmento 3
O
O
M
NOP OP'
+
Brousonetina G 23
NOH OHH
O
O
H
OHHO
2
3 4
5
1´6´
13´
10´2
3 4
5
1´6´
10´
13´
2´
H
La síntesis del fragmento 3, que contiene doce de los trece átomos de
carbono de la cadena lateral de la broussonetina G, comenzó con la apertura del anillo de la valerolactona por reacción con el anión lítico derivado del 1-pentino

Síntesis de broussonetina G 69
(esquema 4). La hidroxicetona resultante se convirtió en el compuesto 11 por tritilación. La reducción de Noyori de la cetona 11 proporcionó, de forma enantioselectiva, el alcohol 18 (véanse comentarios), que se transformó en el alquino terminal 10 mediante isomerización del triple enlace por reacción con 1,3-diaminopropano en presencia de hidruro potásico (véanse comentarios). La protección temporal del hidroxilo, en forma de tetrahidropiranil éter, permitió la ionización del alquino terminal con la combinación n-BuLi/Me3Al. El reactivo alquinil-metálico resultante se adicionó al óxido de etileno, en presencia de trifluoruro de boro-eterato, para dar lugar a un alcohol homopropargílico que por desprotección ácida se convirtió en el triol 9. Este compuesto se sometió a un proceso de espiroacetalización regioselectiva promovida por paladio (véanse comentarios), que condujo a la obtención de 8 con una diastereoslectividad, debida al efecto anomérico, de 97:3. Finalmente, la sustitución nucleofílica del hidroxilo por bromo proporcionó el derivado 19.
Esquema 4

Síntesis de broussonetina G 70
3. Unión de los fragmentos 2 y 3 y pasos finales
La unión entre los fragmentos 2 y 3 se inició con la metalación del bromuro
19, que se llevó a cabo por reacción con magnesio en THF. El correspondiente reactivo organomagnésico se adicionó a la amida de Weinreb 17 para proporcionar la cetona 20.
Esquema 5
La reducción de la cetona 20 con hidruro de diisobutilaluminio, en éter a 0ºC, condujo a una mezcla de alcoholes diastereoisoméricos en proporción 4,3:1 en favor del diastereoisómero 1 (véanse comentarios). Cuando este compuesto se hizo reaccionar con oxono en trifluoroacetona se obtuvo una mezcla de epóxidos

Síntesis de broussonetina G 71
diastereoisoméricos que se sometió a reacción de hidrólisis con ácido trifluoroacético en tetrahidrofurano acuoso. La apertura ácida del anillo oxiránico condujo al trans-diol 22 como único diastereoisómero (véanse comentarios). La broussonetina G se obtuvo por eliminación hidrogenolítica de las agrupaciones Bn y Cbz en el compuesto 22.
Comentarios
1. Alilación asimétrica cinético-dinámica de aminas con vinil epóxidos
La metodología DYKAT general emplea como catalizador un complejo de paladio quiral y como material de partida un compuesto alílico racémico. El compuesto alílico de partida, por coordinación con el catalizador de paladio, origina un complejo catiónico π−alílico de paladio(II) (esquema 6).
Si el compuesto alílico de partida es un mezcla racémica y el catalizador de paladio es quiral, cada uno de los enantiómeros de la mezcla racémica se ionizará bajo la influencia del catalizador quiral para dar lugar a una mezcla de complejos π−alílicos de paladio(II) (A y B en el esquema 6), que serán diastereoisómeros entre sí. Además, como en cualquier reacción con doble inducción asimétrica, uno de los dos enantiómeros se ionizará a través de una vía consonante, mientras que el otro lo hará de manera disonante, lo que implica que uno de los dos complejos diastereoisoméricos se formará con mayor facilidad que el otro. Si se deja que se alcance el equilibrio entre los dos complejos diastereoisómeros π−alílicos A y B a través de un mecanismo π−σ−π, el complejo menos estable B se irá transformando en A y éste será el que resulte atacado preferentemente por el nucleófilo, para dar lugar a un único enantiómero como producto de reacción.

Síntesis de broussonetina G 72
Esquema 6
La aplicación de esta metodología a la reacción de adición de aminas a vinil epóxidos racémicos genera vinilaminoalcoholes de forma enantioselectiva. En el esquema 7 se muestra el ciclo catalítico de este proceso.
Esquema 7

Síntesis de broussonetina G 73
En primer lugar se produce la complejación de la olefina con el paladio. Esta interacción genera el intermedio I , que interviene en un proceso de adición oxidante para dar lugar a una mezcla de complejos catiónicos π−alílicos diastereoisoméricos II y III , que se equilibran en favor del más estable. A continuación, tiene lugar la adición nucleofílica de la amina al catión π−alílico con formación del intermedio IV el cual, finalmente, se transforma en el aminoalcohol por descomplejación del metal.
2. Reducción Noyori de inonas
La reducción de Noyori de inonas con un complejo quiral de Ru(II) en
presencia de isopropanol se produce mediante un proceso de transferencia de hidrógeno desde el isopropanol a la inona. En el esquema 8 se indica el ciclo catalítico que sigue este proceso.
Esquema 8

Síntesis de broussonetina G 74
3. Isomerización de triple enlace
KHR
H2N NH2R
La reacción de alquinos internos con 1,3-diaminopropano, en presencia de
una base fuerte, permite la isomerización del triple enlace desde su posición interna a la posición terminal. La reacción tiene lugar en pocos segundos y a 0ºC, pero contrasta con el hecho de la mayor estabilidad termodinámica de los alquinos internos frente a los terminales, razón por la cual a esta isomerización se le denomina contratermodinámica. Se supone que la fuerza impelente del proceso es la formación del correspondiente alquinuro metálico que precipita en el seno de la reacción.
La base que se emplea para este proceso es el 3-aminopropilamiduro de potasio (KAPA) una superbase que exhibe una actividad excepcional en reacciones prototrópicas. En el esquema 9 se describe el mecanismo de esta reacción.
Esquema 9

Síntesis de broussonetina G 75
Esquema 9 (cont.) En el primer paso del mecanismo se genera el aleno I por reacción con la
base KAPA. En el paso 2 se produce la desprotonación-protonación del aleno, de nuevo merced a la intervención de la base, que conduce a la formación del alquino II . Los pasos 1 y 2 sobre el alquino II convierten a este compuesto en el alquino III , el cual, mediante la repetición de la misma secuencia de reacciones se transforma en el alquino terminal IV . Este intermedio forma el alquinuro potásico V por reacción con la base. En el proceso de work-up hidrolítico el alquinuro se convierte en el alquino terminal. 4. Espiroacetalización promovida por paladio
En el esquema 10 se muestra el mecanismo de espiroacetalización de
alquinos. El proceso comienza con la activación de la electrofília del triple enlace por coordinación con el complejo de Pd(II). El intermedio I que se forma en la coordinación se transforma en el catión oxonio II por adición 6-exo-dig del hidroxilo al triple enlace. La protonación del enlace C-Pd conduce a la olefina exocíclica III y libera el catalizador. La regioselectividad de la ciclación 6-exo-dig,

Síntesis de broussonetina G 76
frente a la posible ciclación 5-endo-dig, se explica por la adición trans muy preferente, del grupo hidroxilo al triple coordinado con el metal.
Esquema 10
Finalmente, tiene lugar la adición del segundo hidroxilo al catión oxonio IV . La reversibilidad de este proceso forma el compuesto 8, que está estabilizado por el efecto anomérico (esquema 11).
Esquema 11
5. Estereocontrol en la reducción de la cetona 20

Síntesis de broussonetina G 77
La reducción de la cetona 20 se ensayó con dos reductores de tamaños bien distintos. Uno de ellos fue el borohidruro sódico y el otro el voluminoso hidruro de diisobutilaluminio. Con este último, se obtuvo una mezcla de diastereoisómeros 1/23 en proporción 4,3:1, mientras que con el borohidruro de sodio la relación 1/23 fue de 1:2,7 (véase esquema 12).
Esquema 12
La estereoselectividad conseguida con estos dos reductores se puede explicar del siguiente modo. Cuando se emplea borohidruro de sodio la reacción transcurre mediante la intervención de un estado de transición de tipo Felkin-Ahn (véase esquema 13). Sin embargo, cuando se utiliza el voluminoso DIBAL el ataque mayoritario no tiene lugar sobre la cara Re de la cetona, porque el estado de transición Felkin-Ahn asociado a este ataque presenta una importante interacción estérica entre el nucleófilo entrante y la cadena R de la cetona. Para evitar esta interacción desestabilizante el ataque del DIBAL, que tiene lugar mediante la participación de un estado de transición de cuatro eslabones, se produce sobre la conformación que expone la cara Si del carbonilo cetónico, como se indica en el esquema 13. De esta forma se evita la interacción estérica con la cadena lateral R y con el anillo dihidropirrolidínico, al colocarse la agrupación diisobutilalumino en una posición de máxima separación con respecto a este anillo.

Síntesis de broussonetina G 78
NOBn OCbz
O
ODIBAL, Et2O, 0ºC
NOBn OHCbz
O
O
H
20
1
NOBn OHCbz
O
O
H
23
NaBH4, Et2O, 0ºC
N
BnO
O
O
R
H
AlR'2
H
ET de cuatro eslabonesfavorecido con
nucleófilos voluminosos(ataque a la cara Si)
N
BnO
O
RH
Felkin-Ahn favorecido connucleófilos pequeños(ataque a la cara Re)
O
NaBH4
Esquema 13 6. Epoxidación con oxono
La composición del oxono es 2KHSO5.KHSO4
.K2SO4. El agente oxidante del oxono es el monoperoxosulfato de potasio (KHSO5). El proceso de epoxidación con oxono supone la generación in situ de un dioxirano por oxidación de una cetona presente en el medio de reacción, usualmente acetona o trifluoroacetona. El dioxirano transfiere el oxígeno a la olefina para generar el producto de epoxidación y regenerar la cetona inicial, lo cual permite el empleo de cantidades catalíticas de la cetona. En el esquema 14 se muestra el ciclo catalítico que explica la formación del dioxirano y la reacción de epoxidación. El proceso se inicia con la generación del intermedio I por adición nucleofílica del monoperoxosulfato de potasio a la

Síntesis de broussonetina G 79
trifluoroacetona. Este intermedio se transforma, en el medio básico de la reacción, en el trifluorodimetildioxirano IV por eliminación intramolecular de sulfato. La reacción con la olefina forma el epóxido y regenera la trifluoroacetona.
Esquema 14
7. Apertura de epóxido mediante hidrólisis ácida
TFA, THF/H2ON
OBn OHCbz
O
O
H
O
NOBn OHCbz
O
O
H
OHHO
(dr > 99%)21223 4 3 4
La epoxidación proporciona una mezcla de epóxidos diastereoisoméricos, los cuales, por tratamiento con ácido trifluoroacético en tetrahidrofurano acuoso, condujeron a la obtención del compuesto 22. El mecanismo de esta transformación se inicia con la protonación de la mezcla de epóxidos (esquema 15). Los epóxidos protonados experimentan la apertura del anillo oxiránico como consecuencia del ataque trans de la molécula de agua. Este ataque es regioselectivo en cada uno de los intermedios I y II . La regioselectividad se debe a la aproximación de la

Síntesis de broussonetina G 80
molécula de agua desde el lado del anillo oxiránico que presenta menor impedimento estérico.
Esquema 15

Síntesis de ciantivigina U 81
SÍNTESIS DE CIANTIVIGINA U
Aislamiento: La ciantivigina U, un diterpeno que pertenece a la familia de las ciantinas, ha sido aislado de la esponja Myrmekioderma styx. Actividad biológica: Algunos metabolitos de la familia de las ciantinas inhiben el crecimiento de la bacteria Mycobacterium tuberculosis y son capaces de estimular el factor de crecimiento nervioso.
Retrosíntesis El análisis retrosintético de la ciantivigina U se inicia con una operación de
intercambio de grupos funcionales (IGF), lo cual conduce a la hidroxicetona 1 (esquema 1).18 La desconexión de la agrupación isopropilo de 1 genera la dicetona 2, que se desconecta en los enlaces dobles a la dicetona tetraolefínica 3. En el sentido sintético la obtención de 2 se llevará a cabo mediante una reacción de metátesis ciclante sobre el intermedio 3. La reconexión de los carbonos olefínicos C1 y C14 en el intermedio 3 proporciona el compuesto bicíclico 4. En el sentido sintético la formación de 3 se conseguirá con la apertura, mediante metátesis, del anillo ciclohexénico que contiene 4, el cual, por desconexión de los restos vinilo, lleva al dialdehído bicíclico 5. A continuación, una etapa de interconversión de grupos funcionales conduce al compuesto bicíclico 6, cuyo anillo ciclohexénico se desconecta, mediante una operación retro-Diels-Alder, a la enona 7 y al ciclohexadieno 8. La parte Xq que contiene 7 representa al auxiliar quiral que será responsable de la inducción asimétrica en el proceso de cicloadición. La desconexión del doble enlace de 7, basada en una reacción de metátesis cruzada, conduce al alqueno 9 y a la enona 10.
18 M. W. B. Pfeiffer, A. J. Phillips, J. Am. Chem. Soc. 2005, 127, 5334.

Síntesis de ciantivigina U 82
Esquema 1
Síntesis 1. Síntesis del dialdehído 5
La secuencia sintética que condujo al dialdehído 5 contiene, como paso clave, una reacción de cicloadición Diels-Alder asimétrica. Este proceso se llevó a cabo mediante la metodología desarrollada por Palomo y colaboradores, que emplea enonas quirales derivadas del alcanfor como dienófilos en el proceso de

Síntesis de ciantivigina U 83
cicloadición.19 Así, el (1R)-(+)-alcanfor 11, por reacción con 1-litio-1-metoxialeno, generado mediante metalación del metoxialeno, se transformó en el compuesto 12. La hidrólisis ácida de 12 proporcionó la α−hidroxienona 10.
Esquema 2
La reacción de metátesis cruzada entre la enona 10 y el alqueno 9 se llevó a
cabo en presencia del catalizador de Grubbs de 2ª generación, y condujo a la α-hidroxienona quiral 7, con un 93% de rendimiento y una relación E/Z de >99:1. La cicloadición Diels-Alder se llevó a cabo por reacción de 7 con el 1,4-dimetilciclohexadieno 8, a -78ºC en presencia de ácido tríflico, y proporcionó, con un 70% de rendimiento, el cicloaducto 6 como único diastereoisómero (véanse comentarios). La eliminación oxidante del auxiliar quiral, por reacción de 6 con nitrato de cerio y amonio (CAN) llevó al ácido 13 el cual, por tratamiento reductivo con aluminio hidruro de litio, dio el diol 14. Finalmente, el dialdehído 5 se obtuvo mediante oxidación Swern del diol 14.
19 C. Palomo, M. Oiarbide, J. M. García, A. González, A. Lecumberri, A. Linden, J. Am. Chem. Soc. 2002, 124, 10288.

Síntesis de ciantivigina U 84
3. Pasos finales: síntesis de la dicetona tricíclica 2 y su transformación en ciantivigina U
La transformación del dialdehído 5 en la ciantivigina U se inició con la reacción de aquél con bromuro de vinilmagnesio en presencia de tricloruro de cerio (esquema 3). Este proceso proporcionó el bis-alcohol alílico 15 que, por oxidación Swern, condujo a la bis-enona 4.
OHH
HOHH
O
O
HH
O
2 (43% desde 5)
3
12
12
3
3 2
1
12
1314
2
Me
Me
H
H
CHO
OHC
1
143
12
6
9
5
CeCl3
CH2=CHMgBr
Me
Me
H
H
1
143
12
6
9
HO
HO13
Dess-Martin
2
Me
Me
H
H
1
143
12
6
9
4
O
O13
15
etileno,tolueno
LiAlH4
H
HOH
H
H 3
12
OH
OHO
HH
ciantivigina U
3
12
(CH3)2CHLi
CeCl3
PCC
O MeLi
(92%)
(90% dos etapas)
(100%)
1617
1
69
cat. Grubbs 2ª gen.
Esquema 3

Síntesis de ciantivigina U 85
El paso clave en las etapas finales de la síntesis fue la obtención de la dicetona 2. Para ello, la bis-enona 4 se trató, bajo atmósfera de etileno, con un 20 mol% del catalizador de Grubbs de 2ª generación. En estas condiciones el compuesto 4 se transformó directamente en la cetona bicíclica 2, con un 43% global desde el dialdehído 5 (véanse comentarios). La reducción regioselectiva de la dicetona 2 con aluminio hidruro de litio condujo al alcohol alílico 16, que por reacción con isopropil-litio, en presencia de tricloruro de cerio, dio el diol 17. La reacción de este compuesto con clorocromato de piridinio provocó la oxidación del grupo hidroxilo de C12 y la reacción de transposición alílica oxidante del sistema de alcohol alílico condujo a la obtención de la bis-enona 1 (véanse comentarios). Finalmente, la adición estereoselectiva de metil-litio al carbonilo cetónico en C12 proporcionó la ciantivigina U sintética (véanse comentarios).
Comentarios
1. Síntesis de 6 mediante reacción de cicloadición Diels-Alder
El compuesto 6 se obtuvo mediante la reacción de cicloadición Diels-Alder entre el dienófilo 7 el dieno 8 en presencia de ácido tríflico. La inducción asimétrica de este proceso corre a cargo de la parte de alcanfor que transporta el dienófilo. Por otro lado, es muy conveniente comentar que la reacción de cicloadición se llevó a cabo sin la intervención de catalizadores metálicos. En su lugar se emplea el ácido tríflico, que juega un doble papel: activa al dienófilo y bloquea el movimiento conformacional de éste mediante la coordinación vía puentes de hidrógeno, tal y como se representa en el complejo 18 del esquema 4. La conformación que adquiere el sistema de enona en el complejo 18 impide el acceso del dienófilo a la cara superior, que está ocupada por la parte de alcanfor.
El ET-1 del esquema 4 representa la aproximación endo del dienófilo desde la cara inferior del doble enlace, y explica la formación del cicloaducto 6.

Síntesis de ciantivigina U 86
ET-1
Me Me
Me
OH
O
OPiv
MeMe
Me
Me
H
H
PivO
Me
Me
Me
OH
O
6 97 8
14 1
6
9
1
14
8
7
3
4
5
5
4
3
7
8
6
TfOH
18
R
H COXq
H
Me
Me
87
9
61
14
34
5
Me Me
Me
O
O
H
H O
S
O O
CF3
R H
H
Me Me
Me
O
O
H
H O
S
O O
CF3
R H
H
Esquema 4
2. Síntesis de la dicetona 2 mediante apertura de anillo/metátesis ciclante en la dienona 4
O
HH
O
2
3 2
1
12
1314
2
Me
Me
H
H
1
143
12
6
9
4
O
O13
N N
RuCl
Cl
PCy3
Ph
Mes Mes
tolueno, etileno
La formación de la dicetona 2, por reacción de 4 con el catalizador de Grubbs de 2ª generación, se explica en el ciclo catalítico del esquema 5. El catalizador, simbolizado como Ru=CH2, se cicloadiciona al doble enlace C1-C14 de la molécula para formar el intermedio rutenaciclobutánico I , cuya ciclorreversión origina el carbeno de rutenio II . A continuación, un proceso de cicloadición intramolecular sobre el sistema de enona, seguido de ciclorreversión, regenera el carbeno de rutenio y forma el compuesto bicíclico III . La cicloadición-ciclorreversión del catalizador al doble enlace más nucleofílico de III genera el

Síntesis de ciantivigina U 87
carbeno de rutenio IV , que se transforma en la dicetona 2 mediante una nueva secuencia de cicloadición intramolecular-ciclorreversión.
Esquema 5
Un mecanismo alternativo se indica en el esquema 6. La principal diferencia con el mecanismo dibujado en el esquema 5 radica en la primera etapa, en la cual la orientación alternativa del catalizador, respecto del doble enlace C1-C14, lleva a la formación del rutenaciclobutano V. La etapa de ciclorreversión forma el intermedio VI , que se convierte en el intermedio VII mediante una secuencia de cicloadición intramolecular-ciclorreversión. La subsiguiente generación del intermedio VIII desemboca finalmente en la formación de la dicetona 2.

Síntesis de ciantivigina U 88
Esquema 6
3. Síntesis de la dicetona 1 por oxidación alílica con clorocromato de piridinio
El tratamiento del diol 17 con clorocromato de piridinio (PCC) proporcionó directamente la dienona 1. El compuesto 17 contiene dos grupos hidroxilo. El situado en C12 es secundario y su oxidación produce la correspondiente cetona (compuesto 19 del esquema 7).

Síntesis de ciantivigina U 89
Esquema 7
Por otro lado, el hidroxilo en C3 es terciario. Este tipo de agrupaciones son
resistentes a la oxidación, pero el grupo hidroxilo en C3 está situado en posición alílica respecto del doble enlace C1-C2. Cuando estos alcoholes alílicos se tratan con determinados agentes oxidantes, como los derivados de Cr(VI), experimentan un proceso de transposición oxidante que conduce a la formación de enonas conjugadas. Así, la reacción de 19 con clorocromato de piridinio forma el cromato 20, que experimenta un proceso de transposición [3,3] sigmatrópica que conduce a la formación del cromato 21. La subsiguiente oxidación de este intermedio proporciona la dienona 1. 4. Adición regio y estereoselectiva de metil-litio a la dienona 1

Síntesis de ciantivigina U 90
La última etapa en la síntesis de la ciantivigina U fue la adición regio y estereoselectiva de metil-litio a la dienona 1. En el esquema 8 se indica la conformación de mínima energía que adquiere la dienona 1. En el estado de transición ET-2 se aprecia cómo la conformación doblada de 1 bloquea el acceso del reactivo a la cara superior endo, produciéndose el ataque desde la cara inferior exo. Esta conformación también explica el bloqueo estérico del grupo carbonilo en C1, puesto que, en este caso, la cara superior queda inaccesible al ataque endo del reactivo nucleofílico, mientras que el ataque alternativo exo está impedido por la presencia del grupo metilo angular en C9.
Esquema 8

Síntesis de cilindrociclofano F 91
SÍNTESIS DE CILINDROCICLOFANO F
Aislamiento: El cilindrociclofano F se ha aislado de la cianobacteria Cylindrospermum licheniforme. Actividad biológica: El cilindrociclofano F presenta citotoxicidad frente al cáncer nasofaríngeo y de colon.
Retrosíntesis
La retrosíntesis del cilindrociclofano F se inicia con una etapa de adición de grupo funcional (AGF), que conduce, por instalación de dos enlaces dobles entre los carbonos C4-C5 y C17-C18, al intermedio 1 (esquema 1).20 Los enlaces dobles, junto con la simetría C2 que presenta el compuesto 1, permiten su desconexión a la estructura fenólica 2. El análisis retrosintético de 2 no es evidente. Baste comentar en este punto que este compuesto se obtendrá mediante un proceso de anelación entre el alquino 3 y la ciclobutenona 4. Por un lado, el compuesto de partida para la preparación de 3 será el hexanoato de etilo, mientras que la ciclobutenona 2 se obtendrá por adición de un compuesto organometálico derivado del yoduro 8 a la 3-etoxi-2-ciclobutenona 7.
20 A. B. Smith III, S. A. Kozmin, C. M. Adams, D. V. Paone, J. Am. Chem. Soc. 2001, 123, 5925.

Síntesis de cilindrociclofano F 92
cilindrociclofano F
OHHO
HO OH
OPPO
PO OP
AGF
1
2
PO OP
O
OP'
+
OEt
O
+
I
CO2Et
CO2Et
4
3
5
6 7 8
metátesis cruzada
metátesis ciclante
anelación de
Danheiser
homologación
adición
alilación
1
4
2 5 7
1415
17
18
4
5
17
18
Esquema 1
Síntesis 1. Síntesis del intermedio 3
La síntesis del fragmento 3 comenzó con la obtención de la N-hexanoil oxazolidinona 10, que se consiguió mediante N-metalación de la oxazolidinona de Evans 9 y reacción subsiguiente con hexanoato de etilo (esquema 2).

Síntesis de cilindrociclofano F 93
Esquema 2
La enolización de 10 con hexametildisililamiduro de sodio (NaHMDS) generó un Z-enolato sódico, que atacó nucleofílicamente al bromuro de alilo desde la cara opuesta a la ocupada por los grupos metilo y fenilo (véase estructura 11 en el esquema 2). Este proceso proporcionó, con un exceso diastereoselectivo de más del 97%, el compuesto 12. La saponificación de 12 con hidroperóxido de litio condujo al ácido 13, que se convirtió en el éster etílico 5 por reacción con yoduro de etilo y carbonato potásico. La reacción del éster con dibromometano, en presencia de 2,2,6,6-tetrametilpiperiduro de litio llevó a la dibromocetona 13, la cual se trató secuencialmente con hexametildisililamiduro de litio, n-butil-litio y triflato de triisopropilo. Esta secuencia de reacciones condujo a la obtención del alquinolato 3 (véanse comentarios).

Síntesis de cilindrociclofano F 94
2. Síntesis de la ciclobutenona 4
cilindrociclofano F
OHHO
HO OH
O
OEt
O
+
I47 8
La síntesis de la ciclobutenona 4 se inició con la C-alilación del enolato sódico derivado de la N-propanoiloxazolidinona 14 (esquema 3). Para ello, 14 se trató con hexametildisililamiduro de sodio, y el (Z)-enolato sódico resultante se hizo reaccionar con bromuro de alilo.
Esquema 3 En el esquema 3 se indica el estado de transición de la reacción de C-
alilación, en el que se aprecia cómo el bromuro de alilo se aproxima al enolato desde la cara opuesta a los grupos metilo y fenilo de la oxazolidinona. El producto

Síntesis de cilindrociclofano F 95
de alilación 16, que se obtuvo con mas del 98% de exceso diastereoselectivo, se convirtió en el alcohol 17 por eliminación reductiva del auxiliar quiral. La reacción de la agrupación hidroxilo con yodo y trifenilfosfina proporcionó el yoduro de alquilo 18, que se convirtió en el alquil-litico 19 por intercambio metal–halógeno con t-BuLi. El compuesto organometálico 19 se adicionó a la etoxiciclobutenona 7, que se obtuvo mediante cicloadición [2+2] entre la cetena y el etoxiacetileno. La reacción de adición nucleofílica condujo a la 3-alquil-2-ciclobutenona quiral 4. 3. Unión de los fragmentos 3 y 4: anelación de Danheiser
Para la síntesis del compuesto fenólico 2 se recurrió a la metodología de
anelación de Danheiser (esquema 4).21 Para ello, el alquino 3 y la ciclobutenona 4 se disolvieron en tolueno y la mezcla se calentó durante dos horas a 80°C. En estas condiciones se obtuvo el fenol 25, el cual, por desililación y O-metilación, se transformó en el compuesto 2.
En el esquema 4 se indican los intermedios implicados en la formación del fenol 25. La ciclación se inicia con un proceso de electrorreversión térmica de la ciclobutenona 4, que conduce a la cetena 21. A continuación, tiene lugar una cicloadición [2+2] entre la cetena 16 y el alquino 3, que da lugar a la ciclobutenona trisustituida 22. Un segundo proceso de electrorreversión convierte la ciclobutenona 22 en la cetena 23, que forma la ciclohexadienona 24 mediante un proceso de electro-ciclación 6π concertado. Finalmente, la tautomerización de la ciclohexadienona proporciona el compuesto aromático 25, el cual, por tratamiento con TBAF, conduce al fenol 26. Es notable el rendimiento global del proceso de anelación-desililación, que fue del 70%. Hay que tener en cuenta que la formación
21 a) R. L. Danheiser, S. K. Gee, J. Org. Chem. 1984, 49, 1670. b) R. L. Danheiser, A. Nishida, S. Savariar, M. P. Trova, Tetrahedron Lett. 1988, 29, 4917.

Síntesis de cilindrociclofano F 96
de 26 requiere un total de seis transformaciones químicas, lo que implica casi un rendimiento medio del 95% en cada una de las reacciones que intervienen en la síntesis de 26 a partir de la ciclobutenona 4 y el alquino 3.
O
4
25
HO OTPS
OTPS
3+
tolueno
80°CO
4
OTPS
3
electrociclo
reversión
4
21
(S)
OTPS
3
·
O
cicloadición[2+2]
O
OTPS
22
electrociclo
reversión
4
23
·O
OTPS
electro-
ciclación
6
24
·O OTPS
tautom.
(70% global)
26
HO OH
MeI, K2CO3
(91%)
2
MeO OMe
TBAF
Esquema 4

Síntesis de cilindrociclofano F 97
4. Dimerización del intermedio 2: reacciones de metátesis en cascada
cilindrociclofano F
OHHO
HO OH
2
MeO OMe
1
OHHO
HO OH
La dimerización del compuesto 2 se consiguió de forma directa mediante reacción de metátesis. Cuando se empleó el catalizador de Grubbs de 1ª generación (20 mol %) en diclorometano, durante 75 horas a 20ºC, se obtuvo el compuesto 28 con un 61% de rendimiento. La reacción con el catalizador de Grubbs de 2ª generación (15 mol %) en diclorometano, a 40ºC durante 27 horas, llevó al compuesto 28 con un 58% de rendimiento (véase el esquema 5). Sin embargo, los mejores resultados se obtuvieron cuando la dimerización se llevó a cabo con el catalizador de Schrock (30 mol %), en benceno a 20ºC durante 2 h. Esta reacción dio lugar, con un 72% de rendimiento, al (E,E)-dieno 28. Hay que hacer constar que la dimerización de 2 puede formar un total de siete isómeros. Por ello, y con el fin de averiguar que isómero, de los siete posibles, era el más estable termodinámicamente, se realizaron cálculos de modelización molecular que demostraron que el más estable era el isómero 28. Estos resultados apoyan un mecanismo mediante una cascada de metátesis reversibles de olefinas, que llevan finalmente a la formación del producto termodinámicamente más estable. Esta hipótesis se confirmó al efectuar la metátesis del isómero (Z)-27, que llevó también a la formación del compuesto 28.
Finalmente, el cilindrociclofano F sintético se consiguió mediante hidrogenación de los enlaces dobles de 28, seguida de O-desmetilación por tratamiento con tribromuro de boro.

Síntesis de cilindrociclofano F 98
Esquema 5
En relación con los catalizadores de Grubbs de 1ª y 2ª generación conviene indicar que el catalizador de 2ª generación es más estable témicamente, y también más reactivo que el catalizador de 1ª generación. Los estudios de tipo cinético sobre la metátesis RCM con el catalizador 2 han puesto de manifiesto que el paso inicial es de tipo disociativo, produciéndose en primer lugar la expulsión reversible de una de las fosfinas para dar la especie de 14 electrones I (véase el esquema 6), que es la verdadera especie catalítica. El ciclo catalítico se inicia, a continuación, con la coordinación de la olefina en el puesto vacante. El valor más importante para medir la efectividad del catalizador es la relación entre k-1 (velocidad de regresión de la especie I al catalizador de partida) y k2 (velocidad de coordinación

Síntesis de cilindrociclofano F 99
de la olefina). Cuanto más elevada sea esta relación, menos eficaz es el catalizador de partida, pues significa que éste tiende a permanecer menos tiempo en el ciclo catalítico.
1ª generación L = PCy 3
Ru
LPh
ClCl
PCy3
NN MesMes
k1
k-1
- PCy3
16 e-
Ru
LPh
ClCl
I (14 e )
olefinak2
Ciclo catalítico
-olefinak -2
2ª generación L =
PCy3
Esquema 6
El catalizador de 1ª generación tiene una mucha mayor velocidad de iniciación que el de 2ª generación (k1 unas 100 veces mayor), pero por contra exhibe una relación k-1/k2 que es unas 104 veces mayor que el catalizador de 2ª generación. Este último es más eficiente, no porque facilite la salida del ligando fosfina, sino porque impide el regreso del mismo.
Comentarios
1. Reacción de homologación de Kowalski La reacción de ésteres con dibromometano y tetrametilpiperiduro de litio
forma dibromometilcetonas, las cuales, por tratamiento básico y quench con triflato de silil, proporcionan O-sililalquinoatos. Por otra parte, si el quench de la reacción se efectúa con etanol en medio ácido el producto de la reacción de homologación es un éster etílico. Este método sintético recibe el nombre de homologación de Kowalski (esquema 6).
R
OTPS
quench, EtOH, H
RO
OEt
1. CH2Br 2, LiTMP
2. LiHMDS, n-BuLi
quench TPSOTfO
OEtR
O
CHBr 2R
2. LiHMDS, n-BuLi
Esquema 6

Síntesis de cilindrociclofano F 100
La homologación de Kowalski se empleó en la conversión del éster 5 en el alquinoato 3 (esquema 7).22
Esquema 7
En el esquema 8 se indica el mecanismo que explica la conversión del éster en la dibromocetona. El proceso comienza con la generación del anión dibromometilo, que tiene lugar por desprotonación del dibromometano con el 2,2,6,6-tetrametilpiperiduro de litio. El anión dibrometilo, mediante adición-eliminación sobre el grupo carbonilo del éster, conduce a la dibromocetona (intermedio I del esquema 8).
O
OEtRO
CHBr2R
N
O
Li
CH2Br2 + CHBr2 +LiNH
O
1) Reacción ácido-base
2) Adición al éster
Li CHBr2
O
OEtR
CHBr2
Li
I
+ LiOEt
Esquema 8
La reacción de la dibromocetona con LiHMDS genera un dibromoenolato de litio, que se convierte en el dianión II del esquema 9 por ataque básico del n-BuLi. El dianión experimenta una reacción de transposición de Arndt-Eistert, lo que lleva a la formación del alquinoato de litio III .
22 C. J. Kowalski, M. S. Haque, K. W. Fiekds, J. Am. Chem. Soc. 1985, 107, 1429.

Síntesis de cilindrociclofano F 101
Si a la mezcla de reacción se le adiciona TPSOTf se provoca la O-sililación de III y la formación del alquinoato sililado IV .
Por otro lado, la adición de metanol en medio ácido convierte el alquinoato III en una cetena, que es atacada por el alcohol para dar lugar al éster V.
1. Reacción ácido-base
O
CHBr2R
O
CBr 2R
LiN
SiMe3Me3SiNH
SiMe3Me3Si
2. Reacción ácido-base
O
CBr 2R
Lin-BuLi
O
CBrR
Li
Li
+ n-BuBr
O
CRLi
3. Transposición
Br C CR O
Li
Li + LiBr
EtOH, HR
O
OEt
TPSOTf
4. Quench de la reacción
C CR O Li
H
CR
OH
EtOH
I
II
II III
III
IV
V
Li
C CR OTPS
+
C CR O Li
III
+
Esquema 9

Síntesis de cilindrociclofano F 102
2. Síntesis del ciclofano 28 mediante reacción de metátesis
2
MeO OMe
N
Mo PhO
O
F3C CF3
F3C
F3C
benceno, 20°C(72%)
OMeMeO
MeO OMe
28
La síntesis del compuesto 28 requiere dos reacciones de metátesis: una primera intermolecular y una segunda intramolecular. En el esquema 7 se indica esquemáticamente el ciclo catalítico para la reacción de metátesis cruzada (intermolecular). El catalizador, que es un carbeno de molibdeno, se simboliza en el esquema 10 con la estructura simplificada Mo=CH2.
catalizador
H2C CH2
Mo
Mo
Mo
cicloadición[2+2]
I
II
III
I
IV
V
ciclorreversión [2+2]
cicloadición[2+2]
ciclorreversión[2+2]
Mo=CH2
Esquema 10

Síntesis de cilindrociclofano F 103
En primer lugar el catalizador, mediante una cicloadición [2+2], se adiciona a uno de los dos enlaces dobles del dieno, lo que conduce a la formación del intermedio II . A continuación, se produce un proceso de ciclorreversión que genera etileno y el carbeno de molibdeno III , que se cicloadiciona a otra molécula de dieno para formar el intermedio metalaciclobutánico IV . Finalmente, un proceso de ciclorreversión forma el producto de metátesis cruzada V y regenera el catalizador.
Después del proceso de metátesis cruzada se inicia la reacción de metátesis intramolecular. Este proceso consta básicamente de las mismas etapas que el proceso intermolecular y se indica en el esquema 11.
Esquema 11
En el esquema 12 se dibujan las estructuras de los siete productos de metátesis cruzada-metátesis ciclante que se pueden formar en la reacción del dieno

Síntesis de cilindrociclofano F 104
2. Como se ha indicado anteriormente, el producto deseado, compuesto 28, resultó ser el más estable, por lo cual, el proceso de metátesis, que es reversible cuando se utiliza el catalizador de molibeno, proporcionó mayoritariamente el producto de control termodinámico 28.
28
OMeMeO
MeO OMeOMeMeO
MeO OMe
OMeMeO
MeO OMe
OMeMeOOMeMeO
OMeMeOOMeMeO
OMeMeOOMeMeO
OMeMeOOMeMeO
Esquema 12

Síntesis de citostatina 105
SÍNTESIS DE CITOSTATINA
Aislamiento: La citostatina, producto natural que pertenece a la familia de las fostriecinas, se ha aislado de Streptomyces sp. MJ654-Nf4. Actividad biológica: La citostatina es un potente citotóxico contra la leucemia y el cáncer de melanona (IC50 = 100nM). Provoca apoptosis e inhibe la metástasis del cáncer de pulmón. Es también un potente inhibidor de la proteinfosfatasa 2A (IC50 = 210 nM).
Retrosíntesis El análisis retrosintético de la citosatatina se indica en el esquema 1.
Esquema 1

Síntesis de citostatina 106
El análisis retrosintético se inicia con la desconexión del enlace C11-C12, que se formará mediante la adición del reactivo organometálico 2 al carbonilo aldehídico del compuesto 1.23 Este compuesto contiene la funcionalidad lactónica enmascarada en forma de acetal mixto El proceso de adición del reactivo organometálico 2 deberá ser estereoselectivo, puesto que en el mismo se generará el estereocentro C11, para lo cual será clave la adecuada elección del protector P del compuesto 1 y del metal en el intermedio 2.
El siguiente paso en el análisis retrosintético lo constituye la desconexión del enlace C7-C8. Esta operación conduce al compuesto organometálico 3 y al epóxido 4, que se obtendrá del cis-2-buten-1,4-diol 5.
Por otro lado, 3 derivará de la lactona 6, que se preparará mediante reacción de metátesis ciclante en el acrilato 7. Éste se sintetizará por esterificación del alcohol 8, que se obtendrá mediante un proceso de crotilación del α−metilaldehído quiral 9, cuyo precursor es el éster de Roche 10.
Síntesis 1. Síntesis del yoduro 15, precursor del compuesto organometálico 3
Para la síntesis del yoduro 15 se eligió como producto de partida el éster de
Roche 10, cuya sililación condujo a 11 (esquema 2). La reducción hasta alcohol, con DIBAL, y oxidación con TPAP/NMO proporcionó el aldehído 9. La instalación de los estereocentros en C4 y C5 se consiguió mediante reacción de crotilación de Brown del aldehído 9 con el (Z)-crotilborano quiral, generado por metalación del (Z)-2-buteno con la combinación t-BuOK/n-BuLi, seguida de transmetalación con (+)-Ipc2BOMe. La adición del reactivo así preparado al aldehído 9 proporcionó el alcohol homoalílico 8, con un 63% de rendimiento químico y con una relación de diastereoisómeros de 8:1, a favor del isómero deseado. La esterificación del alcohol 8 con cloruro de acriloílo, seguida de
23 B. G. Lawhorn, S. B. Boga, S. E. Wolkenberg, D. A. Colby, C-M. Gauss, M. R. Swingle, L. Amable, R. E. Honkanen, D. L. Boger, J. Am. Chem. Soc. 2006, 128, 16720.

Síntesis de citostatina 107
reacción de metátesis ciclante condujo a la lactona 6. La función lactónica se protegió, mediante conversión en un acetal mixto, por reacción con DIBAL, fue seguida de la transformación en el acetal 12 por tratamiento con metanol en presencia de p-toluensulfonato de piridinio, a temperatura ambiente durante 10 minutos. Tiempos de reacción más prolongados provocaban la formación de la mezcla de acetales anoméricos en relación 2:1. Desde el punto de vista de la eficiencia sintética la formación de la mezcla de anómeros no supone ningún inconveniente puesto que el estereocentro de C1 se eliminará en pasos mas avanzados de la síntesis. Sin embargo, la afortunada formación de un único isómero evita trabajar con una mezcla de diastereoisómeros, que siempre es problemática cuando se tienen que llevar a cabo los procesos de aislamiento y caracterización de los intermedios sintéticos.
MeO
O OP1. DIBAL
2. TPAP, NMOH
O OTPS(+)-Ipc2B
2. H2O2, NaOH
OTPSOH
O
Cl
OTPSO
O
O
OTPS
(63%, dr 8:1)
(91%)
(89%)O
1. DIBAL
2. MeOH,PPTS
(82%)O
OTPS
MeO
O
OH
MeO O
OTs
MeO O
I
MeONaI
(90%)
TsCl, NaH
(81% desde 12)
TBAF
65
4
546
5
6
6
9
10 P=H11 P=TPS
8
7612
13 14 15
RuPh
Cy3P
Cl
Cl
Cy3P
Esquema 2
La escisión de la función sililéter con TBAF llevó al alcohol 13, que se tenía que convertir en el yoduro 15. Sin embargo, esta, aparentemente, conversión trivial no funcionó con las combinaciones Ph3P/NIS o Ph3P/CBr4 (conversión en bromuro), que dejaron inalterado al alcohol de partida, mientras que la reacción con Ph3P/I2 provocó la descomposición de la mezcla de reacción. Finalmente, la preparación del yoduro 15 se consiguió mediante una secuencia de dos pasos, que se inició con la tosilación de 14, por reacción con cloruro de tosilo y NaH, en

Síntesis de citostatina 108
benceno a temperatura ambiente durante 4 horas, seguida de reacción con NaI en acetona a reflujo durante 12 horas. 2. Síntesis del epóxido 4
OO
H2O3PO HO
citostatina
O OPMB
4
1011
9
8
10
1198
La síntesis del epóxido 4 se inició con la monobencilación del cis-2-buten-1,4-diol 5, que fue seguida de la reacción de epoxidación asimétrica Sharpless, lo que proporcionó el epoxialcohol 17 con un 86% de rendimiento químico y un 89% de exceso enantiomérico (esquema 3).
Esquema 3

Síntesis de citostatina 109
Las epoxidaciones asimétricas Sharpless de alcoholes alílicos de configuración Z son menos enantioselectivas que las que tienen lugar sobre los (E)-alcoholes alílicos homólogos. Por ello, y a fin de aumentar la pureza óptica del epoxialcohol 17, se esterificó la mezcla de epoxialcoholes enantioméricos (89% ee) con cloruro de 3,5-dinitrobenzoilo, y la mezcla de ésteres enantioméricos se purificó por cristalización. La saponificación de los cristales obtenidos en el proceso anterior proporcionó, con un 100% de exceso enantiomérico, el epoxialcohol 17.
La reacción de epoxidación provoca la oxidación de los carbonos C9 y C10
(numeración final de la molécula de citostatina). La configuración del esterocentro C9 es la que se requiere para la síntesis del producto natural, pero la configuración de C10 se tiene que invertir con instalación concomitante de grupo metilo. Para ello se recurrió a un proceso de apertura nucleofílica del anillo oxiránico mediante reacción con yoduro de metilmagnesio, en THF a -40ºC en presencia de cantidades catalíticas de CuI. Este proceso, cuyo regiocontrol se basa en impedimentos estéricos, proporcionó una mezcla de alcoholes regioisoméricos 18/19, en relación 1:3. El tratamiento de la mezcla con NaIO4 provocó la ruptura oxidante del sistema glicólico de 18, lo que permitió la fácil purificación de 19.
La hidrogenolisis de la función benciléter condujo al triol 20, que se convirtió en el acetal 21 por reacción con 3,3-dimetoxipentano en presencia de ácido p-toluensulfónico. La PMB protección del hidroxilo libre de 21, seguida de hidrólisis ácida del acetal llevó al diol 22, que se transformó en el epóxido 4 por reacción con p-tosilimidazol y NaH. 3. Síntesis del bromuro 27, precursor del intermedio organometálico 2
La síntesis de (Z,Z,E)-bromuro vinílico 27 se inició con la adición de metil-litio al tetrafluoroborato de pirilio 23. Esta reacción generó el pirano 24 que experimentó, in situ, un proceso de apertura electrocíclica, que condujo a la formación del (2Z,4E)-2,4-hexadienal 25 (esquema 4). La reacción de éste con tetrabromuro de carbono y trifenilfosfina proporcionó la gem-dibromoolefina 26.

Síntesis de citostatina 110
que se transformó en el bromuro 27 por reacción de Stille con hidruro de tributilestaño en presencia de tetrakis-trifenilfosfinapaladio(0) (véanse comentarios).
O BF4
MeLi, THF
-78ºc (66%) O H
CH3
LiBF4
O HCH3H
CBr4, Ph3P
(97%)Bu3SnH
Pd(PPh3)4
Br
Br
Br
23 24 25
2627
Esquema 4
4. Síntesis del aldehído 1
La síntesis del aldehído 1 comenzó con la metalación del yoduro 15. Este proceso se consiguió por reacción de 15 mediante con t-BuLi, lo que formó el reactivo organolítico 28, seguida de transmetalación de éste con 2-tienil-cianocuprato de litio 29 (esquema 5). Con esta secuencia de reacciones se formó el organocuprato de Mayor Orden 3 (Higher Order cuprate, véanse comentarios), que atacó regioselectivamente al oxirano 4 para proporcionar, con un 84% de rendimiento, el alcohol 30. La protección del hidroxilo como etoxi etil éter condujo al compuesto 31, el cual, por PMB desprotección con DDQ acuosa y oxidación Dess-Martin llevó al aldehído 1.

Síntesis de citostatina 111
Esquema 5
4. Pasos finales: unión de los fragmentos 1 y 2
El asalto final a la molécula de citostatina se inició con la metalación del bromotrieno 27, lo que se consiguió por reacción con t-BuLi, seguida de transmetalación con el complejo CuI·PBu3 (esquema 6). El reactivo organocuprato generado se adicionó al aldehído 1 para dar lugar a una mezcla de diastereoisómeros en relación 7:1 (control mediante β-quelación, véanse comentarios). La separación de la mezcla de diastereoisómeros mediante cromatografía de columna proporcionó el isómero mayoritario 34 puro. La diastereoselectividad en la adición del trieno era función del grupo protector del

Síntesis de citostatina 112
hidroxilo C9. Así, cuando este hidroxilo se protegió con el grupo TES, que no posee capacidad quelante, se obtuvo mayoritariamente el producto de control Felkin-Ahn, tanto en la adición de reactivos organolíticos como organocupratos (véanse comentarios).
La sililación del compuesto 34, seguida de reacción con HCl acuoso (0.02 M, en acetona a 25ºC durante 10 minutos, provocó la escisión quimioselectiva del grupo etoxi etil éter yde la parte de metil acetal, y llevó a la obtención del alcohol hemiacetálico 35. Cuando se emplearon las condiciones usuales de desprotección del grupo etoxi etilo, como AcOH acuoso del 80%, HCl 0.2 M en acetona, o TFA acuoso del 90%, se provocó, junto con la escisión del grupo EE, la eliminación del grupo TBS. La oxidación quimioselectiva de 35 con carbonato de plata sobre celite (oxidación de Fetizon, véanse comentarios) condujo a la lactona 36, que se fosforiló en el hidroxilo por reacción con (i-Pr2NP)(OFm)2 (N-isopropil-bis-(9-fluorenilmetil)fosforoamidito) en presencia de tetrazol (véanse comentarios). Esta reacción proporcionó el compuesto 37, que se transformó en 38 mediante desililación. Por último, la eliminación de los grupos fluorenilmetilo proporcionó la la citostatina (véanse comentarios).
Esquema 6

Síntesis de citostatina 113
citostatina
i-Pr2NP(OFm)2
tetrazol, luego H2O2
(82%)
OO
O OTBSP
OFm
O
FmO
HF-pir. (85%)
OO
O OHP
OFm
O
FmO
OO
O OHP
OH
O
HO
Et3N, CH3CN
(99%)
36
37
38
Esquema 6 (cont.)
Comentarios 1. Reacción de crotilación de Brown
La reacción de crotilación asimétrica de Brown permite la creación simultánea de dos nuevos esterocentros. El reactivo de crotilación se genera in situ mediante la secuencia de reacciones del esquema 7. El proceso se inicia con la metalación del (Z)-2-buteno mediante la combinación t-BuOK/n-BuLi, que va seguida de transmetalación por reacción con el (+)-Ipc2BOMe. Esta secuencia de reacciones conduce a la formación de un borato de potasio, que genera el reactivo de crotilación por transmetoxilación con el ácido de Lewis trifluoruro de boro·eterato.

Síntesis de citostatina 114
Esquema 7
El modelo estereoquímico que explica la esteroselectividad de la reacción de crotilación se basa en el estado de transición de Zimmerman-Traxler (esquema 8)
Esquema 8
La posición relativa sin de los dos nuevos estereocentros depende de la configuración Z del doble enlace del reactivo de crotilación, mientras que la selectividad π−facial se debe a los ligandos quirales sobre el boro, que discriminan

Síntesis de citostatina 115
eficientemente la cara Si del aldehído. En el esquema 8 se dibujan los dos estados de transición alternativos que se generan en la adición del reactivo de crotilación derivado de (+)-Ipc2BOMe a un aldehído RCHO. Como se puede observar, el estado de transición favorable es el que se produce en el ataque a la cara Re, porque este no presenta interacciones estéricas desestabilizantes, mientras que el ataque a la cara Si del aldehído presenta una interacción estérica desestabilizante entre el átomo de hidrógeno ecuatorial del grupo metileno alílico y el metilo de la parte terpénica.
2. Conversión del gem-dibromuro 26 en el (Z)-bromuro de alquenilo 27 mediante reacción de Stille
La preparación del (Z)-bromuro de alquenilo 27 se llevó a cabo mediante
acoplamiento reductivo de Stille del gem-dibromuro 26 con hidruro de tributilestaño en presencia de Pd(Ph3P)4. El ciclo catalítico del proceso se indica en el esquema 9.
Esquema 9 El ciclo catalítico se inicia con la reacción de adición oxidante, lo que genera
el intermedio I , que experimenta el proceso de transmetalación para formar el

Síntesis de citostatina 116
intermedio II . La eliminación reductora en este intermedio proporciona el bromuro de alquenilo 27.
La estereoselectividad del proceso se produce en el paso de acoplamiento oxidante, en el que se forma el intermedio I , que coloca al complejo de paladio trans con respecto a la cadena diénica lateral, por tanto en una situación de poca compresión estérica. La inserción oxidante alternativa, que coloca a la parte de paladio cis con respecto a la cadena diénica, presenta mayor compresión estérica y, por tanto, no compite favorablemente con la colocación trans.
El ciclo catalítico habitual para las reacciones Stille, que se llevan a cabo en presencia de ligandos monodentados, es el que se indica en el esquema 10.
Esquema 10
El ciclo comienza con la etapa de adición oxidante del haluro de alquenilo al complejo de paladio(0), simbolizado en el esquema 10 como PdL2. La reacción de adición oxidante genera el complejo I , cis-cuadrado plano de paladio(II). A continuación se produce una rápida isomerización al complejo trans II , que experimenta el proceso de transmetalación con el alquenilestannano para dar lugar al complejo III , trans-cuadrado plano de paladio(II). Una etapa de isomerización al complejo cis IV , seguida de eliminación reductora forma el producto de acoplamiento y regenera el catalizador. En este ciclo catalítico la etapa determinante de la velocidad de la reacción es la de transmetalación.

Síntesis de citostatina 117
Espinet y Casado en 1998, propusieron una modificación del ciclo catalítico anterior. Así, el complejo trans-cuadrado plano, que resulta de la adición oxidante-isomerización, experimenta la transmetalación mediante un mecanismo de sustitución asociativa de ligando (véase esquema 11).24 Este proceso conduce al complejo VI , tricoordinado de paladio cis, que experimenta directamente el proceso de eliminación reductora irreversible, para dar lugar al producto de acoplamiento.
Esquema 11
En el mecanismo de sustitución asociativa de ligando se produce el ataque nucleofílico del complejo II al alquenilestannano, lo que provoca el desplazamiento de un ligando y la formación del complejo V. Esta reacción, que tiene lugar con la participación de un estado de transición con el átomo de paladio pentacoordinado (véase ET-I del esquema 12), preserva la estereoquímica del paladio, y por tanto R2 se coloca en la posición del ligando saliente. De este modo, el complejo tricoordinado de paladio que se genera en el proceso anterior (complejo VI ), coloca los grupos R1 y R2 en posición cis, y experimenta directamente la etapa de eliminación reductora.
24 A. L. Casado, P. Espinet J. Am. Chem. Soc. 1998, 120, 8978-8985.

Síntesis de citostatina 118
Esquema 12
En una publicación reciente, R. de Lera y colaboradores han investigado
mediante métodos computacionales el mecanismo de la reacción de Stille.25 En presencia de disolventes coordinantes, como la DMF, el ciclo catalítico sería el que se indica en el esquema 13. El disolvente coordinante participa en la isomerización cis-trans del complejo que resulta de la etapa de adición oxidante. La etapa de transmetalación forma el complejo tricoordinado de paladio VIII , el cual experimenta directamente el proceso de eliminación reductora.
Esquema 13
25 R. Álvarez, O. Nieto Faza. A. R. De Lera, D. J. Cárdenas, Adv. Synth. Catal. 2007, 349, 887.

Síntesis de citostatina 119
Según los cálculos computacionales efectuados por el grupo de R. de Lera, la etapa de transmetalación tiene lugar mediante un mecanismo asociativo. La reacción se inicia con la coordinación del complejo VII al doble enlace del alquenilestannano (esquema 14). Esta reacción forma el complejo cuadrado plano IX , el cual, mediante la intervención de un estado de transición de cuatro eslabones se convierte en el intermedio X. Este complejo elimina el haluro de tributilestaño y se transforma en el complejo tricoordinado de paladio VIII .
Esquema 14
3. Síntesis del intermedio 30 mediante metalación del yoduro de alquilo 15 y reacción con el epóxido 4
La reacción de apertura del epóxido 4 se llevó a cabo con el organocuprato de Mayor Orden (Higher Order) generado por metalación del yoduro de alquilo 15 con t-BuLi seguida de transmetalación con 2-tienilcianocuprato de litio (véase esquema 5).
Los compuestos organometálicos de cobre (I) de Menor Orden (Lower Order), también denominados reactivos de Gilman, son sales monoaniónicas

Síntesis de citostatina 120
(R2Cu-Li +) que se preparan a partir de sales de Cu(I) y 2 equivalentes de un reactivo organolítico (RLi) o Grignard (RMgX).
Por el contrario, los compuestos organometálicos de Cu(I) de Mayor Orden (Higher Order cuprates) son sales dianiónicas ([R2Cu(CN)2-]Li 2
2+) que se preparan a partir de organocupratos de bajo orden R2CuLi, mediante la reacción de éstos con CuCN.
Un inconveniente asociado a los reactivos organocupratos, ya sean L.O o H.O., es que solo se aprovecha uno de los dos restos R en el proceso de transferencia, por lo cual la síntesis pierde eficiencia, sobre todo cuando el reactivo RLi es costoso de conseguir (esquema 15).
Síntesis de cupratos L.O.
2 RLi + CuI R 2CuLi + LiI
Síntesis de cupratos H.O.
R2CuLi + CuCN
InconvenientesEn el proceso de transferenciasólo se aprovecha 1 de los 2equivalentes del fragmento R
que transporta el reactivoR2Cu(CN)Li2
Esquema 15
Lipshutz introdujo en 1987 una clase de organocupratos H.O. mixtos, para
los cuales empleó como precursor el 2-tiofenilcianocuprato de litio ((2-Th)Cu(CN)Li).26 Este compuesto se obtiene a partir del tiofeno. Así, la metalación del tiofeno con n-BuLi genera el 2-tiofenil-litio, el cual proporciona el (2-Th)Cu(CN)Li mediante reacción con CuCN (esquema 10). La reacción de (2-Th)Cu(CN)Li con 1 equivalente de RLi (o RMgX) forma el H.O. cuprato [R(2-Th)Cu(CN)2-]Li 2
2+ (o [R(2-Th)Cu(CN)2-](LiMgX 2+). Estos organocupratos presentan máxima eficiencia porque en el proceso de transferencia del resto alquílico se aprovecha en su totalidad el fragmento R que proviene del, muy a menudo, valioso RLi (o RMgX).
26 B. H. Lipshutz, M. Koerner, D. A. Parker Tetrahedron Lett. 1987, 28, 945.

Síntesis de citostatina 121
Esquema 16 4. Síntesis del alcohol 34 por adición estereoselectiva al aldehído 1
La reacción del aldehído 1 con el organocuprato generado a partir del
bromuro 27 proporcionó mayoritariamente el anti 1,3-diol monoprotegido 34. La formación mayoritaria del isómero anti se explica mediante la participación de un estado de transición que bloquea la libertad conformacional del β-alcoxialdehído mediante la quelación del átomo metálico con el oxígeno carbonílico y con los átomos de oxígeno de la parte de etoxi etil éter. En esta situación, el ataque del reactivo organometálico se produce desde la cara Si del aldehido, que es la que presenta menor impedimento estérico (véase el esquema 17).
Esquema 17

Síntesis de citostatina 122
Si el grupo protector del hidroxilo beta no tiene capacidad quelante, como el grupo TES, la adición del reactivo organometálico proporciona mayoritariamente el producto de control Felkin-Ahn. En este modelo estereoquímico el grupo voluminoso del estereocentro se coloca perpendicular al grupo carbonilo, y anti respecto de la trayectoria del nucleófilo atacante. El estado de transición más favorecido es el que tiene lugar mediante el ataque del nucleófilo a la conformación que coloca al grupo metilo del esterocentro al lado del grupo carbonilo. De esta forma el nucleófilo ataca la cara Re del aldehído, porque en su trayectoria de aproximación la interacción estérica es mínima (esquema 18).
Esquema 18 5. Oxidación de Fetizon (Ag2CO3/celite)

Síntesis de citostatina 123
La reacción de oxidación de alcoholes con carbonato de plata depositado sobre celite recibe el nombre de oxidación de Fetizon. La reacción ajustada, para la oxidación de un lactol a lactona, es la siguiente:
Esquema 19
El mecanismo de este proceso, que se indica en el esquema 20, se inicia con la ruptura homolítica del enlace C-H carbinólico, lo que genera un átomo de hidrógeno, cuya oxidación por el catión plata forma el hidrogenocarbonato de plata y plata metálica. En el segundo paso el radical centrado en el carbono sufre la oxidación por el catión plata para dar lugar a la lactona, a plata metálica y al ácido carbónico
Esquema 20
6. Reacción de fosforilación

Síntesis de citostatina 124
Para la fosforilación del hidroxilo libre del compuesto 36 se empleó una secuencia de dos pasos. En el primero de ellos el alcohol se convirtió en un fosfito por reacción con (i-Pr2N)P(OFm)2 (N,N-diisopropil-bis-(9-fluorenilmetil)-fosforoamidito) en presencia de tetrazol. El mecanismo que explica esta conversión se indica en el esquema 21, y se inicia con la reacción ácido-base entre el (i-Pr2N)P(OFm)2 y el tetrazol. Esta interacción provoca la protonación del átomo de nitrógeno del fosforamidito y la formación del intermedio I (paso 1 del esquema 21). Este intermedio es atacado nucleofílicamente por el alcohol, lo que forma el fosfito protonado II y la diisopropilamina (paso 2 del esquema 21). Por último, el intercambio protónico entre II y la base conjugada del tetrazol proporciona el fosfito III .
Esquema 21
El fosfito formado en el proceso anterior se convierte en fosfato por adición de peróxido de hidrógeno al medio de reacción (Esquema 22).

Síntesis de citostatina 125
P
O
O O
R
+ H2O2 P
O
O O
R
O
+ H2O
fosfito de trialquilo fosfato de trialquilo
Esquema 22 7. Eliminación del grupo fluorenilmetilo: conversión del fosfato de trialquilo en dihidrógeno fosfato de alquilo
La eliminación de los grupos fluorenilmetilo se lleva a cabo, en condiciones
no próticas, por reacción con trietilamina en acetonitrilo. El mecanismo se indica en el esquema 23 y se inicia con la reacción de eliminación E2 por ataque básico de la amina terciaria (paso 1 del esquema 23). Esta reacción conduce a la formación de un fosfato de trietilamonio y 9-metilen-9-H-fluoreno. En un segundo paso se produce la reacción ácido-base entre el anión fosfato y el catión trietilamonio para formar trietilamina y el fosfato de dialquilo.

Síntesis de citostatina 126
Esquema 23
Un nuevo proceso similar al anterior elimina el grupo fluorenilmetilo residual y conduce a la obtención del fosfato de monoalquilo (esquema 24).
Esquema 24

Síntesis de ent-clavilactona B 127
SÍNTESIS DE ent-CLAVILACTONA B
Aislamiento: Las clavilactonas A, B y C se han aislado de cultivos del hongo Clitocybe clavipes. Actividad biológica: Las clavilactonas son potentes inhibidores de tirosina kinasas, los enzimas que modulan la vía de transducción y regulación de los procesos celulares de crecimiento, diferenciación y apoptosis.
Retrosíntesis
La síntesis de la ent-clavilactona B fue llevada a cabo por primera vez por A. G. M. Barret y col.27 Hay que señalar que cuando se inició esta síntesis se desconocía la configuración absoluta de la clavilactona B. De hecho, el compuesto obtenido en el proceso sintético, cuyos datos espectroscópicos eran idénticos a los de la clavilactona B, presentaba una rotación óptica de -105 (c 0.1 en MeOH), en contraposición a la del producto natural, que era de +111 (c 0.1 en MeOH). Por tanto, el compuesto sintético que preparó el grupo de A. G. M. Barret era la ent-clavilactona B, el enantiómero del producto natural.
La retrosíntesis de la ent-clavilactona B se inicia con una operación de intercambio de grupo funcional que transforma el anillo de p-quinona en el anillo p-dimetoxibencénico (vease intermedio 1 el esquema 1) La desconexión del doble enlace trisustituido, basada en una reacción de metátesis ciclante, conduce a la diolefina 2, que por desconexión del anillo lactónico deriva al intermedio 3. Sobre este compuesto tiene lugar la desconexión clave del análisis retrosintético. Esta operación proporciona los fragmentos 4 (un bencino), 5 (un compuesto organometálico) y 6 (un α,β-epoxialdehído). En el sentido sintético el
27 I. Larrosa, M. I. Da Silva, P. M. Gómez, P. Hannen, E. Ko, S. R. Lenger, S. R. Linke, A. J. P. White, D. Wilton, A. G. M. Barrett, J. Am. Chem. Soc. 2006, 128, 14042.

Síntesis de ent-clavilactona B 128
acoplamiento de estos tres fragmentos se efectuará en una sola etapa mediante un proceso que se iniciará con la adición del compuesto organometálico nucleofílico 5 al triple enlace bencínico, lo que formará un compuesto arilmetálico que se adicionará al aldehído 6. Este compuesto se obtendrá, vía alcohol alílico 7, del inoéster 8.
O
O
Me
O
O
O
ent-clavilactona B
H
RCMMeO
MeO O
O
OH
lactonizaciónoxidante
MeO
MeO OH
OH
OH
MeO
MeO
O
OH
OTPS
M
H
OH OTPS
EtOOC
OTPS
1 2
3
4
5
67
+
+
IGF
OMe
OMe
Me
O
O
OH
8 Esquema 1
Síntesis
1. Síntesis del epóxido alílico 6
Para la síntesis del α,β-epoxialdehído 6 se eligió como compuesto de partida
el alcohol propargílico 9, el cual, por O-sililación, ionización del triple enlace

Síntesis de ent-clavilactona B 129
terminal con n-butil-ltio y carboetoxilación con cloroformiato de etilo, se transformó en el inoéster 8 (esquema 2). La adición conjugada del bromuro de butenilmagnesio a 8, en presencia de sales de Cu(I), proporcionó, estereoselectivamente, el compuesto olefínico trisustituido 11 (véanse comentarios). La reducción de éste con DIBAL llevó al alcohol alílico 7, cuya epoxidación asimétrica Sharpless condujo al epoxialcohol 12, con un 93% de rendimiento y con un 97% de exceso enantioselectivo. Finalmente, la oxidación Swern de 12 proporcionó el epoxialdehído 6.
Esquema 2 2. Acoplamiento bencínico: síntesis del intermedio 3
En la parte de análisis retrosintético se ha explicado que el paso clave en la síntesis será la formación del intermedio 3 mediante el acoplamiento bencínico de tres componentes. En la práctica esta reacción se llevó a cabo por conversión del

Síntesis de ent-clavilactona B 130
fluorobenceno 13 en el bencino 4, mediante reacción con la base n-butil-litio, seguida de adición de bromuro de isobutenilmagnesio 5 a la mezcla de reacción (esquema 3). El reactivo nucleofílico se adicionó al triple enlace bencínico para generar el reactivo aril-magnésico 14. La subsiguiente adición del aldehído 6 a la mezcla de reacción provocó el ataque del reactivo de Grignard a aquél, para dar lugar a la mezcla de diastereoisómeros 15/16, cuya separación cromatográfica proporcionó el compuesto 15 puro. Por último, el intermedio avanzado 3 se obtuvo por desililación de 15, con TBAF.
Esquema 3

Síntesis de ent-clavilactona B 131
3. Pasos finales
MeO
MeOHO
OH
OTPSH
O
O
Me
O
O
OH
ent-clavilactona B3
La síntesis de la ent-clavilactona B se completó del siguiente modo (esquema
4). El compuesto 3 se transformó en la lactona 2 mediante oxidación con TEMPO (N-óxido de 2,2,6,6,-tetrametilpiperidilo) en presencia de diacetato de fenilyodonio (véanse comentarios).
OMe
OMe
Me
O
O
OH
TEMPO, PhI(OAc)2
CH2Cl2 (80%)
MeO
MeO O
OH
O
Ru
PCy3
Cl
ClPh
N NMs Ms
O
O
F
F F
F, tol., 80ºC
O
O
Me
O
O
OH
MeO
MeO OH
OH
OH
CAN
(74%)
ent-clavilactona B
CH3CN ac.
32
1
Esquema 4
La reacción de metátesis ciclante sobre 2 se llevó a cabo con el catalizador de Grubbs de 2ª generación, en presencia de tetrafluoroquinona, y proporcionó el compuesto tricíclico 1. Es conocido que la síntesis de olefinas trisustituidas mediante reacción de metátesis es un proceso que no suele proporcionar rendimientos elevados, debido a la descomposición del catalizador como consecuencia de los prolongados tiempos de reacción y de las temperaturas

Síntesis de ent-clavilactona B 132
elevadas que se necesitan en este tipo de metátesis. Parece ser que la descomposición del catalizador está relacionada con la formación de hidruros de rutenio, cuya generación se evita si se añaden quinonas a la mezcla de reacción.28 En la práctica, la síntesis de 1 se consiguió, con un 65% de rendimiento, mediante la adición lenta del catalizador de Grubbs de 2ª generación a una disolución 0.03 M del dieno 2 en tolueno, a 80ºC en presencia de la tetrafluoroquinona, con eliminación concomitante del etileno formado en la reacción. Por último, la ent-clavilactona B se consiguió por oxidación del anillo aromático de 1 con nitrato de cerio y amonio (CAN) (véanse comentarios). 4. Isomerización de 16
La reacción de acoplamiento bencínico proporcionó la mezcla de diastereoisómeros 15/16, mayoritaria en este último, que presenta la configuración del hidroxilo secundario opuesta a la requerida para la síntesis de la ent-clavilactona B. Para aprovechar todo el material obtenido en el acoplamiento bencínico se investigó la epimerización bencílica de la lactona 17 con ácidos de Lewis (esquema 5). Con TiCln(Oi-Pr)4-n (n=0-4), o BX3 (X=Br, F) se provocó la descomposición del material de partida, mientras que con el ácido de Yamamoto MeAl(OAr)2 (Ar=2,6-t-Bu-4-BrC6H2) el compuesto 17 se epimerizó a 2 con un 40% de conversión después de 3 días de reacción. La solución se encontró en el ácido de Lewis 18 (esquema 5), que consiguió la epimerización de la lactona 17 con un 80% de rendimiento.
28 S. H. Hong, D. P. Sanders, C. W. Lee, R. H. Grubbs, J. Am. Chem. Soc. 2005, 127, 17160.

Síntesis de ent-clavilactona B 133
Esquema 5
En la práctica la isomerización se llevó a cabo sobre la mezcla de epímeros 15+16 obtenida en el acoplamiento bencínico. La mezcla se sometía a desililación y oxidación con TEMPO/PhI(OAc)2 y la mezcla de lactonas 2/17, se convertía en 2 por isomerización con el complejo de aluminio 18 (esquema 6).
Esquema 6
El mecanismo que explica la isomerización de la lactona 17 se indica en el esquema 7. El ácido de Lewis se coordina con el oxígeno carbonílico del anillo lactónico, lo que provoca la escisión del enlace C-O bencílico y conduce a un carbocatión bencílico, el cual es atacado intramolecularmente para formar la lactona 2.
La epimerización de 17 en 2 es un proceso en equilibrio que se decanta del lado de la especie termodinámicamente más estable.

Síntesis de ent-clavilactona B 134
Esquema 7
Comentarios
1. Adición conjugada de organocupratos a inoésteres
El mecanismo de adición de organocupratos a inoésteres se inicia con la coordinación de la especie organometálica al triple enlace (esquema 8).
Esquema 8
La etapa de coordinación va seguida de la cis-carbocupración regioselectiva del enlace múltiple, que conduce a la formación del alquenilcuprato 20, el cual se

Síntesis de ent-clavilactona B 135
transforma en la olefina trisustituida.en el proceso de hidrólisis ácida. La regioselectividad en el proceso de carbocupración se explica por la adición del fragmento nucleofílico del organocuprato al carbono β del triple enlace, que es el más electrofílico en el sistema de inoéster. 2. Oxidación selectiva de hidroxilos primarios
TEMPO
PhI(OAc)2R OH
OH
R H
OH O
La oxidación de alcoholes con cantidades catalíticas de TEMPO (N-óxido de
2,2,6,6-tetrametilpiperidilo), en presencia de diacetato de fenilyodonio, permite la oxidación selectiva de hidroxilos primarios en presencia de hidroxilos secundarios.29 El mecanismo de esta oxidación se indica en el esquema 9.
N
O
I
OAc
OAc + 2 RCH2OH
I
OR
OR+ 2 AcOH
1)
N
O
+ + AcOHN
O
N
O
+
H
+ AcO
2)
N
O
N
O
H
O RH
N
O
H
O
RHH
AcO
AcOH + +R
O
H
AcO3)
I
OAc
OAc
N
O
H
+2
4)
N
O
2 + 2 AcOH +
I
Esquema 9
29 A. De Mico, R. Margarita, L. Parlanti, A. Vescovi, G. Piancatelli, J. Org. Chem. 1997, 62, 6974.

Síntesis de ent-clavilactona B 136
El proceso de oxidación comienza con la generación de ácido acético por intercambio de ligandos sobre el átomo de yodo de la molécula de acetato de fenilyodonio (etapa 1 del esquema 9). El ácido cataliza la dismutación de TEMPO a hidroxilamina y sal oxoamónica (etapa 2). La sal oxoamónica es la responsable de la oxidación del alcohol, mediante la intervención de un mecanismo que se inicia con el ataque nucleofílico del alcohol al átomo de nitrógeno (etapa 3). Este proceso va seguido del ataque básico del anión acetato, lo que conduce a la formación del aldehído y de la hidroxilamina. Por último, en la etapa 4 se produce la regeneración del TEMPO por reacción de la hidroxilamina con el diacetato de fenilyodonio.
La selectividad en la oxidación preferente de hidroxilos primarios en presencia de secundarios se explica mediante el mecanismo que se describe en la etapa 3 del esquema anterior, y se debe al fuerte impedimento estérico que presenta la sal oxoamónica al ataque nucleofílico del alcohol, que será mucho más lento si el hidroxilo atacante corresponde al de un alcohol secundario.
En la síntesis de la ent-clavilactona B se somete el diol 3 a la oxidación con el método anterior, lo que lleva a la obtención de la lactona 2. En este caso el hidroxilo primario de 3 se oxida al aldehído 21, que está en equilibrio con la forma hemiacetálica 22, cuya oxidación subsiguiente conduce a la lactona 2 (esquema 10).
Esquema 10

Síntesis de ent-clavilactona B 137
3. Síntesis de quinonas por oxidación de p-dimetoxibencenos con CAN
El nitrato de cerio y amonio (NH4)2[Ce(NO3)]6 (abrevido CAN) es un potente
agente de oxidación (E°~ 0.96 eV). Pocos reactivos estables alcanzan el poder oxidante del CAN, que es incluso más potente que el Cl2 en procesos redox. En estas reacciones el Ce(IV) se convierte en Ce(III) mediante una etapa de transferencia monoelectrónica, que se pone de manifiesto por el cambio de color naranja, de la disolución que contiene el Ce(IV), al amarillo del Ce(III).
El CAN se emplea como oxidante en una gran variedad de procesos como: 1) en la dinitroxilación de alquenos 2) en la oxidación bencílica de metilarenos 3) en la oxidación de alcoholes, fenoles y éteres 4) en la oxidación de hidroquinons y catecoles a quinonas e hidroquiononas El mecanismo que opera en el proceso de conversión de p-dimetoxiariléteres
en quinonas, mediante oxidación con nitrato de cerio y amonio en disolución acuosa, se indica en el esquema 11.
Esquema 11
OMe
OMe
Me
O
O
OH
O
O
Me
O
O
OHCAN
(74%)
ent-clavilactona B
CH3CN ac

Síntesis de ent-clavilactona B 138
O
MeO
Me
O
H
H O
MeO
MeO
H
HO
MeO
Me O H
H
MeO
OH
MeOH
H-
MeO
O
MeO
O
MeO
O
+ 1e (oxidación)
Ce(IV) + 1e Ce(III) (reducción)
3) 2º Proceso Redox
MeO
O
OH
H
MeO
O
O
H
H
MeO
O
OH
H
O
O H
H-
O
O
2) Hidrólisis del cation oxonio
4) Hidrólisis del cation oxonio
MeOH
Esquema 11 (cont.)

Síntesis de clavosólido A
139
SÍNTESIS DE CLAVOSÓLIDO A
Aislamiento: El clavosólido A se ha aislado de la esponja marina Myriastra clavosa procedente de Filipinas.
Retrosíntesis
El clavosólido A presenta una estructura inusual de diólido glicosídico con dos restos de 2,3,4-trimetil−β−D-xilosa y un aglicón con simetría C2. La retrosíntesis del clavosólido A se indica en el esquema 1 y se inicia con la ruptura de los enlaces glicosídicos (véase esquema 1).30 Esta operación origina dos restos de xilosa (intermedio 1) y el aglicón 2, que se obtendrá por dimerización del hidroxiácido tetrahiropiránico 3. La desconexión clave del análisis retrosintético es la que convierte el intermedio 3 en el metilendioxano 4. En el sentido sintético la transformación del dioxano 4 en el compuesto tetrahidropiránico 3 se llevará a cabo mediante una reacción de transposición Ferrier-Petasis. La unidad exometilénica que contiene 4 se introducirá mediante metilenación de la dioxanona 5, que a su vez se sintetizará mediante cetalización del aldehído 6 con el β−hidroxiácido 7.
La desconexión del anillo ciclopropánico en el aldehído 6 conduce al aldol 8. En esta operación retrosintética también se ha invertido la configuración del estereocentro en C9. Esta inversión permitirá instalar, sobre el sistema de alcohol alílico del sustrato 8, el anillo ciclopropánico con la configuración requerida para la síntesis del clavosólido A. El compuesto 8 se preparará mediante reacción aldólica enantioselectiva entre el crotonaldehído 9 y un enolato derivado del compuesto 3 (Xq=auxiliar quiral).
30 A. B. Smith III, V. Simov, Org. Lett. 2006, 8, 3315.

Síntesis de clavosólido A
140
Por otro lado, el β−hidroxiácido 7 se desconecta al derivado de propionato 11 (Xq=auxiliar quiral) y al β−alcoxialdehído 12. En el sentido sintético la adición aldólica del enolato derivado de 11 al aldehído 12 deberá ser enantio- y diastereoselectiva a favor del isómero anti.
O O
OH
OH
H
PO
H
Me
Me
O O
O
O O
O
Me H
O
H Me
O
Me
H H
Me
HH
O
OMeMeOMeO
OMeO
OMeOMe
clavosólido A
1
2
dimerización
O
OP'
H
H
H
Me
OH OP'
HO
O
Me
6
3
Ferrier-
Petasis
OXq
Me
OHciclopropanación
H
Me
O
adiciónaldólica
Aux
O
Me+
O OP'
H
7
8
10
12
X
O
MeOMeO
MeO
11
Xq
O
Me
9
+
+
134
5
7
13
45
7
9
6
54
31
7
9
12
87
9
12
8
adición
aldólica anti
esterificación
+glicosidación
O OP
OP´
H
H
O
Me
Me 4
13
4
5
6
7O OP
OP´
H
O
H
O
Me
Me5
13
4
5
7 metilenación
cetalización
O O
O
O O
O
Me H
HO
H Me
OH
Me
H H
Me
HH1
Esquema 1

Síntesis de clavosólido A
141
Síntesis 1. Síntesis del aldehído ciclopropánico 6 (fragmento C7-C13)
El aldehído ciclopropánico 6 constituye el fragmento C7-C13 del monómero
del aglicón de clavosólido A. Su síntesis comenzó con la adición aldólica de Nagao entre el enolato de titanio derivado de la tiazolidintiona quiral 10 y el crotonaldehído 9 (esquema 2). Este proceso generó el centro estereogénico en C9 y condujo a la obtención del aldol 8 con un 94% de rendimiento y una relación diastereoisomérica de 18:1 en favor de 8 (véase esquema 2 y comentarios).
Esquema 2
La reacción de aldol 8 con el hidrocloruro de N,O-metilhidroxilamina proporcionó la amida de Weinreb 13, que se convirtió en alcohol ciclopropánico 14 mediante ciclopropanación Simmons-Smith bajo las condiciones de Charette (véanse comentarios). La estereoquímica en C9 era contraria a la del producto natural, por lo que el alcohol ciclopropánico 14 se sometió a un proceso de

Síntesis de clavosólido A
142
inversión de configuración de Mitsunobu, seguido de saponificación, que condujo a la obtención del alcohol 15 (mezcla de epímeros en C9 en relación 12:1). Finalmente, la sililación del hidroxilo en el alcohol 15, seguida de reducción con DIBAL, proporcionó el aldehído 6.
2. Síntesis del ácido 7 (fragmento C1-C5)
O O
O
O O
O
Me H
O
H Me
O
Me
H H
Me
HHO
OMeMeO
MeO
OMeO
OMeOMe
clavosólido A
134
59´
OH OTIPS
HO
O
Me 7
13
45
El ácido 7 se preparó mediante aplicación de la metodología desarrollada por
Masamune que permite la obtención, enantio y diasteroselectiva, de aldoles de configuración relativa anti. Así, el éster quiral derivado de norefedrina 11 se convirtió en el correspondiente enolato de boro, de configuración E, por reacción con diciclohexilboriltriflato y trietilamina. La adición subsiguiente de aldehído 12 a la mezcla de enolización proporcionó el aldol anti 16 como único diastereoisómero (véase esquema 3 y comentarios). El β−hidroxiácido 7 se obtuvo por saponificación del éster 16 con hidróxido de litio.
Esquema 3

Síntesis de clavosólido A
143
3. Síntesis del β−β−β−β−hidroxiácido 3
O O
OH
OH
H
BnO
H
Me
Me
O O
O
O O
O
Me H
O
H Me
O
Me
H H
Me
HH
O
OMeMeO
MeO
OMeO
OMeOMe
clavosólido A 3
134
5
7
9
134
5
7
9
El hidroxiácido 3 es el monómero del aglicón del clavosólido A. Su
preparación incluye la formación del anillo tetrahidropiránico mediante transposición Ferrier-Petasis. Para llevar a cabo esta reacción era necesario la cetalización previa del aldehído 6 con el β−hidroxiácido 7. Con el fin de evitar el consumo del catalizador de cetalización (TMSOTf), por sililación de los hidroxilos libres en el β−hidroxiácido 7, se procedió a la sililación previa de este compuesto mediante reacción con 4 equivalentes de hexametildisililamina, en diclorometano a 35ºC, durante cuatro días (esquema 4). Cuando el bis-sililderivado 17, obtenido en el proceso anterior, se hizo reaccionar con el aldehído 6 en presencia de 0.22 equivalentes de TMSOTf y de 0.05 equivalentes de 2,6-di-t-butil-4-metilpiridina (DtBMP), durante 1.5 h a -78ºC, se obtuvo la dioxanona 16 como una mezcla inseparable de epímeros 7:1 en el carbono C7. La metilenación de la dioxanona 5 con el reactivo de Petasis-Tebbe (dimetiltitanoceno, Cp2TiMe2) condujo al enol éter 4 (véanse comentarios), que se sometió al proceso de transposición Ferrier-Petasis por reacción con 1.3 equivalentes de cloruro de dimetilaluminio, en diclorometano en presencia de tamices moleculares. En estas condiciones se obtuvo la piranona 18 con un 65% de rendimiento (véanse comentarios). La reducción de 18 con borohidruro de sodio condujo al alcohol 19, con una relación diastereoisomérica de 10:1. Finalmente, la protección del hidroxilo en forma de bencil éter, eliminación de los dos grupos silil éter y oxidación selectiva del hidroxilo primario, por reacción con TEMPO e hipoclorito sódico, proporcionó el monómero 3 (véanse comentarios).

Síntesis de clavosólido A
144
Esquema 4
4. Preparación del fragmento de xilosa 1
1O
O
MeOMeO
clavosólido A
CCl3
NHMeOO O
O
O O
O
Me H
O
H Me
O
Me
H H
Me
HHO
OMeMeO
MeO
OMeO
OMeOMe
El derivado de xilosa 1 se preparó a partir de la D-xilosa mediante la
secuencia sintética que se indica en el esquema 5. El proceso se inició con la permetilación de la D-xilosa. Esta reacción proporcionó el correspondiente

Síntesis de clavosólido A
145
metilglicósido, que se convirtió en la 2,3,4-tri-O-metil-D-xilosa 21 mediante hidrólisis con ácido tríflico en ácido acético. La reacción de 21 con tricloracetonitrilo en presencia de DBU proporcionó el tricloroacetimidato 1 como mezcla 6:1 de anómeros α/β.
Esquema 5
5. Síntesis del aglicón 2 y pasos finales
La síntesis del aglicón 2 se inició con la dimerización del hidroxiácido 3
mediante esterificación de Yamaguchi. Así, la reacción de 3 con el cloruro del ácido 2,4,6-triclorobenzoilo y DMAP en tolueno, a 125ºC durante 6 horas en concentración 0.004 M proporcionó el dímero 22 con un rendimiento del 66% (esquema 6). La hidrogenolisis de 22 condujo al diol 2, que se sometió al proceso de bis-glicosidación con el derivado de xilosa 1. Conviene indicar que, en primer lugar, se intentó la glicosidación con triflato de trimetilsililo en acetonitrilo. Sin embargo, en estas condiciones se observó la descomposición del material de partida. Este resultado probablemente se deba a la naturaleza nucleofílica del disolvente, por lo que la reacción se llevó a cabo en diclorometano. En estas nuevas condiciones se obtuvo el clavosólido sintético con un 12% de rendimiento, junto con un 37% del diastereoisómero [α,α] y un 20% del estereoisómero [α,β].

Síntesis de clavosólido A
146
COClCl
Cl
Cl
Et3N, DMAP(66%)
H2, Pd/C
O
OMeO
OMeOMe
NH
Cl3C
TMSOTf, CH2Cl2 (12%)
(64%)
3 22
1
O O
O
O O
O
Me H
O
H Me
O
Me
H H
Me
HH
O
OMeMeO
MeO
OMeO
OMeOMe
clavosólido A
2
O O
O
O O
O
Me H
HO
H Me
OH
Me
H H
Me
HH
O O
O
O O
O
Me H
BnO
H Me
OBn
Me
H H
Me
HH
O O
OH
OH
Me H
BnO
Me
H
Esquema 6
Comentarios 1. Reacciones aldólicas con el auxiliar quiral de Nagao
Las adiciones aldólicas que emplean auxiliares quirales derivados de acetato suelen ser mucho menos estereoselectivas que las correspondientes adiciones aldólicas de propionato. Esta baja selectividad se debe a la ausencia de sustituyentes en alfa en la parte de enolato, hecho que provoca escasa

Síntesis de clavosólido A
147
discriminación energética entre los dos estados de transición competitivos: el estado de transición bote y el de silla (véase esquema 7).
O
R
MLn
O
MLnOR´CHO R´
HR
H
H
R´ R
HO O
R´CHOO
OMLn´R
H
H
H
R
R´ R
HO O
E.T. silla
E.T. bote
+
Esquema 7
Un ejemplo de la falta de estereocontrol en adiciones aldólicas de acetato lo constituyen las N-acetil-oxazolidinonas de Evans, que proporcionan mezclas de aldoles diastereoisoméricos en relaciones comprendidas entre 52:48 y 87:13 (esquema 8).
Esquema 8
La falta de estereoselectividad en las adiciones aldólicas que emplean los
auxiliares quirales de Evans ha sido superada por la metodología de Nagao, que utiliza enolatos de estaño derivados de N-acetil-1,3-tiazolidin-2-tionas para conseguir, con excelentes niveles de estereocontrol, adiciones aldólicas a aldehídos α,β-insaturados. La adición aldólica de Nagao se lleva a cabo mediante el empleo de enolatos de estaño, que se generan por reacción de las tiazolidintionas con

Síntesis de clavosólido A
148
triflato de estaño y una base no nucleofílica.31 La selectividad π-facial de estas adiciones se explica mediante la intervención de un estado de transición cíclico, con quelación intramolecular entre el átomo de azufre de la tiona y el átomo de estaño, que se encuentra en su máximo estado de coordinación, que es de 4 (véase esquema 9). De los dos estados de transición alternativos, el favorecido es el que se genera en el ataque a la cara Re del aldehído porque el grupo isopropilo de la parte de tiazolidintiona apunta hacia fuera del estado de transición, y por tanto se encuentra en una posición de mínima compresión estérica.
Esquema 9
En la síntesis del compuesto 6 se empleó un enolato de titanio, en lugar de un enolato de estaño. Cuando se emplea el enolato de titanio el estado de transición favorecido es también el que se genera en el ataque a la cara Re del aldehído, tal y como se indica en el esquema 10.
31 Y. Nagao, Y. Hagiwara, T. Kumagai, M. Ochiai, T. Inoue, K. Hashimoto, E. Fujita J. Org. Chem. 1986, 51, 2391.

Síntesis de clavosólido A
149
Esquema 10
2. Modificación de Charette de la reacción de ciclopropanación de Simmons-Smith
En 1959 H. E. Simmons y R. D. Smith consiguieron la ciclopropanación de
alquenos mediante la reacción con diyodometano en presencia del par zinc/cobre.32 Debido a los bajos rendimientos y a la falta de reproducibilidad de los que adolece el método anterior, Furukawa desarrolló una variación del mismo que emplea dietilzinc en lugar del par zinc/cobre. Esta modificación permite llevar a cabo la reacción a un mayor rango de temperaturas.33 Más recientemente Friedrich ha demostrado que la ciclopropanación se puede conseguir en presencia de una cantidad catalítica de TiCl4.
34
32 H. E. Simmons, R. D. Smith, J. Am. Chem. Soc. 1959, 81, 4256. 33 J. Furukawa, N. Kawabata, J. Nishimura, Tetrahedron 1968, 53, 24. 34 E. C. Friedrich, S. E. Lunetta, E. J. Lewis, J. Org. Chem. 1989, 54, 2388.

Síntesis de clavosólido A
150
En relación con el mecanismo de estas reacciones se pueden señalar las siguientes características generales:
1) El papel del cobre es el de activador de la superficie del zinc. 2) Todos los dihalometilen-compuestos son capaces de conseguir la
transferencia de metilenos, aunque la transferencia más rápida se consigue con diyodometano.
3) Cuando se emplea la combinación Zn/Cu/CH2I2 la especie reactiva es,
probablemente, ICH2ZnI. Cuando se emplea Et2Zn/CH2I2 la especie que transfiere el metileno es ICH2ZnEt.
4) En disolventes como éter o THF se produce la solvatación del zinc. 5) La presencia de grupos hidroxilo o éter en el sustrato puede dirigir la
estereoquímica de la ciclopropanación del doble enlace carbono-carbono, tal y como se indica en el esquema 11.
Esquema 11
El mecanismo de la reacción se explica mediante un estado de transición que implica la formación y ruptura concertada de enlaces, como se indica en el esquema 12.
Esquema 12

Síntesis de clavosólido A
151
Cuando las condiciones de ciclopropanación de Simmons-Smith se aplican sobre alcoholes alílicos acíclicos, se observa que el nivel de estereoselección es función de la configuración del doble enlace. Así, con enlaces dobles de configuración Z la reacción funciona con elevada diastereoselectividad, pero el estereocontrol disminuye si el doble enlace es de configuración E (véase esquema 13).
Esquema 13 El grupo de Charette ha demostrado que se puede conseguir un elevado nivel
de estereocontrol en la ciclopropanación de alcoholes alílicos de configuración E, si se emplean 5 equivalentes de Et2Zn y 5 equivalentes de CH2I2 a -15ºC.35 En estas condiciones el hidroxilo alílico se convierte en un alcóxico de zinc, que se coordina con el reactivo de ciclopropanación ICH2ZnEt para transferir de forma intramolecular el grupo metileno. El estado de transición que explica el estereocontrol observado en la reacción se indica en el esquema 14.36
H
H3C H
R
HO
Et2Zn, CH2I2
CH2Cl2, -15ºC
H
H3C HO
HR
ZnEt
Zn
CH2
I
Et
H
H3C HOH
H R
CH2
Esquema 14
35 A. Charette, H. Lebel, J. Org. Chem. 1995, 60, 2966. 36 H. Lebel, J-F. Marcoux, C. Molinaro, A. B. Charrette, Chem. Rev. 2003, 103, 977.

Síntesis de clavosólido A
152
El estado de transición dibujado en el esquema anterior es el preponderante en la reacción de ciclopropanación por dos motivos. Por una parte las interacciones de tipo estérico son mínimas. Por otra, el estado de transición se beneficia de un efecto estereoelectrónico estabilizante, puesto que la colocación del oxígeno alcóxidico y del enlace C-R paralelos al orbital π*, promueve la donación electrónica a este orbital vacío, con la consiguiente estabilización adicional del estado de transición (véase el esquema 15).
Esquema 15
3. Reacciones aldólicas anti con el auxiliar quiral de Masamune
El método de Abiko y Masamune para la obtención de aldoles anti emplea
un éster quiral de tipo O-acilnorefedrina 57 el cual, mediante reacción con Chx2BOTf/Et3N, genera estereoselectivamente el enolato de boro de configuración E. La adición de éste a aldehídos alifáticos, aromáticos y α,β-insaturados proporciona los correspondientes aldoles anti con relaciones anti/sin de hasta 99:1.37
37 A. Abiko, J. Liu, S. Masamune, J. Am. Chem. Soc. 1997, 119, 2586.

Síntesis de clavosólido A
153
NO
Ph
Me
Bn
MesSO2
O
(Chx)2BOTf
Et3NXqO
OB(Chx)2
Me
Xq =N
Ph
Me
Bn
MesSO2
RCHO
XqO R
O OH
XqO R
O OH
+
anti / sin (> 98:2)
Esquema 16
Un inconveniente de la metodología anterior es el largo periodo de reacción necesario para la eliminación del auxiliar quiral, que puede llegar a ser de hasta tres días. El otro es la falta de condiciones experimentales que permitan conseguir, a partir del mismo auxiliar quiral, un acceso estereodivergente a ambos aldoles anti.
4. Metilenación Petasis-Tebbe
O OTIPS
OTIPS
H
O
H
O
Me
Me
Cp2TiMe2 O OTIPS
OTIPS
H
H
O
Me
Me
tBuCO2Et
16 17
Determinado tipo de compuestos gem-dimetaloorgánicos M−C−M, o
complejos metal-carbeno M=C, se pueden emplear en la metilenación de compuestos carbonílicos. El reactivo de Tebbe, de fórmula (C5H5)2TiCH2ClAl(CH3)2, se emplea en la metilenación de compuestos carbonílicos.38 Este reactivo es un sólido de color rojo, pirofórico, que se debe manejar bajo atmósfera de N2 o de Ar. El reactivo de Tebbe contiene dos unidades ciclopentadienilo (C5H5
-, Cp) unidas al átomo de titanio, estando los átomos de aluminio y titanio unidos a través de sendos puentes de metileno y cloro. El anillo formado por Ti-CH2-Al-Cl adopta una forma de cuadrado plano (véase la figura 1).
38 F. N. Tebbe, G. W. Parshall, G. S. Reddy, J. Am. Chem. Soc. 1978, 100, 3611.

Síntesis de clavosólido A
154
Reactivo de Tebbe
Figura 1
El reactivo de Tebbe se prepara mediante la reacción del dicloruro de
titanoceno con trimetilaluminio en disolución de tolueno: Cp2TiCl 2 + 2Al(CH3)3 Cp2TiCH2AlCl(CH 3)2 + CH4 + Al(CH3)2Cl
El reactivo de Tebbe, por si mismo, no reacciona con compuestos carbonílicos. Para conseguir la metilenación es necesario el tratamiento previo del reactivo con una base de Lewis, como la piridina, para que aquél se transforme en una especie metalocarbénica, que es capaz de metilenar no sólo aldehídos y cetonas, sino también ésteres y amidas (véase el esquema 17).
Esquema 17 El mecanismo propuesto para explicar la metilenación con la especie
metalocarbénica se indica en el esquema 18. Así, el carbeno se adiciona al grupo carbonilo mediante una reacción de cicloadición [2+2]. A continuación, la ciclorreversión en el intermedio oxa-titanaciclobutánico forma el compuesto metilénico y Cp2TiO.

Síntesis de clavosólido A
155
Ti CH2
1)O
H R+ Ti
OR
2)
TiO
R Ti O H2CR
H+
Esquema 18 Petasis desarrolló, en 1990, una alternativa al reactivo de Tebbe con el
dimetiltitanoceno, Cp2TiMe2 (véase la figura 2), que se prepara fácilmente por interacción de Cp2TiCl2 con dos equivalentes de MeLi.39 Este reactivo no comparte la extrema sensibilidad del reactivo de Tebbe a la humedad, es más estable y es capaz también de metilenar aldehidos, cetonas, ésteres y lactonas.
Reactivo de Petasis
Figura 2
Cuando el reactivo de Petasis se somete a calentamiento experimenta la
eliminación de metano con formación de la especie metalocarbénica Cp2Ti=CH2, que reacciona con el grupo carbonilo, según se indica en el esquema 15, para dar lugar al correspondiente producto de metilenación.
Esquema 19
39 N. A. Petasis, E. I. Bzowej, J. Am. Chem. Soc. 1990, 112, 6392.

Síntesis de clavosólido A
156
5. Reacción de Ferrier-Petasis
En 1979 Ferrier demostró que determinados enol éteres derivados de
carbohidratos se podían convertir en ciclohexanonas por reacción con sales de mercurio. Desde entonces este proceso se conoce como reacción de Ferrier tipo II. El mecanismo de esta transformación se indica en el esquema 20, y se inicia con la formación del hemiacetal lI por hidroximercuriación regioselectiva del doble enlace en el carbohidrato I . La descomposición del hemiacetal forma el intermedio dicetónico III , el cual experimenta una adición aldólica intramolecular que conduce a la formación de la ciclohexanona IV .
Esquema 20 La transposición de Ferrier se enmarca dentro de las denominadas
transposiciones O→C. La expresión general de estas transformaciones, que se definen como de ruptura endocíclica de acetal, se describe en el esquema 21.40
Esquema 21 40 S. J. Meek, J. P. A. Harrity, Tetrahedron 2007, 63, 3081.

Síntesis de clavosólido A
157
La fornulación de la reacción de transposición O→C de Ferrier-Petasis, que se clasifica como de ruptura endocíclica de acetal dioxano/dioxolánico, se indica en el esquema 22.
Esquema 22
La aplicación del mecanismo anterior al acetal dioxolánico 4 explica la formación de la piranona 18 (esquema 23). Así, la complejación del oxígeno acetálico con el ácido de Lewis genera el intermedio V, que experimenta el proceso de apertura endocíclica del acetal para dar lugar a la betaína VI . El ataque nucleofílico intramolecular del enolato sobre la parte de catión oxonio forma el compuesto 18.
La estereoselectividad en la formación del centro estereogénico en C7 se debe a la disposición ecuatorial del sustituyente R`, como se pone de manifiesto en el intermedio VI del esquema 23.
Esquema 23

Síntesis de clavosólido A
158
6. Oxidación selectiva de hidroxilos primarios en presencia de hidroxilos secundarios
En 1987 Anelli y colaboradores demostraron que los alcoholes primarios se
podían oxidar a aldehídos, o a ácidos carboxílicos, por reacción con hipoclorito sódico en una mezcla bifásica CH2Cl2/H2O, en presencia de bicarbonato sódico, bromuro potásico y una cantidad catalítica de 4-metoxi-2,2,6,6-tetrametilpiperidina-1-oxilo (4-MeO-TEMPO).41 La oxidación se podía parar en la etapa de aldehído si se acortaba el tiempo de la reacción. De manera alternativa, la oxidación hasta ácido carboxílico se podía conseguir de forma rápida por adición de un catalizador de transferencia de fase.
El mecanismo de la oxidación de Anelli se indica en el esquema 24 y se
inicia con la formación de la sal oxoamónica I por oxidación del 4-MeO-TEMPO con el hipoclorito. El ataque nucleofílico del alcohol a la sal oxoamónica genera la especie N-oxoamónica II , que origina el aldehído y la hidroxilamina III . La oxidación de la hidroxilamina por reacción con el ácido hipobromoso regenera la sal oxoamónica (el HOBr es mejor oxidante para la regeneración de la sal
41 P. L. Anelli, C. Biffi, F. Montanari, S. Quici, J. Org. Chem. 1987, 52, 2559.

Síntesis de clavosólido A
159
oxoamónica que el HOCl). El ácido hipobromoso se produce en el ciclo catalítico secundario por oxidación del bromuro con hipoclorito.
La reacción es bastante lenta al pH del hipoclorito sódico (pH=12.7) y mucho más rápida a pH=8.6, que es el que se genera cuando se adiciona NaHCO3. A pH muy básico la concentración de HOBr, el oxidante que regenera la sal oxoamónica, es muy baja en relación a la concentración del anión hipobromito.
N
OH
N
O O
H R
H
N
O Br
RCH2O
N
O
HOBr
BrNaOCl
oxidante
estequiométrico oxidante
catalítico
R
O
H
R
OH
H
OHR
O
OH
H2O
OMeOMe OMe
Cl
BrOel mismo mecanismo
que en la oxidación de
alcoholes a aldehídos
OMe
III
III
NaOCl
Esquema 24
Paradójicamente, la velocidad de la oxidación disminuye al incrementar la
temperatura. Este hecho se debe a la descomposición de la sal oxoamónica, que es muy estable a 0ºC, pero que descompone rápidamente en presencia de agua a 25ºC.
Por otro lado, la oxidación del aldehído al ácido carboxílico está mediada también por la sal oxoamónica, mas que por el hipoclorito. De hecho, en ausencia de los radicales TEMPO, la velocidad de oxidación de aldehídos hasta ácidos carboxílicos es muy lenta.
Aunque el método original de Anelli utilizaba 4-MeO-TEMPO, otros derivados como 4-OH-TEMPO, 4-AcNH-TEMPO o el propio TEMPO, son igualmente eficaces en la reacción de oxidación. Usualmente un 1 mol% del radical TEMPO es suficiente para que la oxidación transcurra de forma eficiente.

Síntesis de clavosólido A
160
Una limitación importante del método de oxidación de Anellli es la utilización de una cantidad estequiométrica de NaClO, un reactivo que puede provocar cloraciones en determinados sustratos.

















![(53 6LVWHPD GH 3ODQLILFDFLyQ GH UHFXUVRV ...d o } À Z _ µ o } t' ^d/ME ^ Zs/ /K^ ] y } o ( o À ] ] } ï ð d o } À Z _ µ o } t' ^d/ME 'ZhW : ^ ï ñ d o } À Z _ µ o } t' ^d/ME](https://static.fdocuments.ec/doc/165x107/5ecbbe6505a88e62d61df9ea/53-6lvwhpd-gh-3odqlilfdflyq-gh-uhfxuvrv-d-o-z-o-t-dme-zs.jpg)
