Leucemia Linfoblástica Aguda (LLA) - cure4kids.org Linfoblástica Aguda (LLA) Módulo 13 -...
19
Leucemia Linfoblástica Aguda (LLA) Módulo 13 - Documento 2 Página 1 de 19 Autoras: Ayda G. Nambayan, DSN, RN, St. Jude Children’s Research Hospital Erin Gafford, Estudiante de Educación de Oncología Pediátrica, St. Jude Children’s Research Hospital; Estudiante de Enfermería, Escuela de Enfermería, Universidad Union Contenido Revisado por: Scott Howard, MD, St. Jude Children’s Research Hospital Gaston K. Rivera, MD, St Jude Children’s Research Hospital Fecha de Publicación en Cure4Kids: 25 de Julio 2008 La Leucemia Linfoblástica Aguda (LLA) es una enfermedad maligna caracterizada por una sobreproducción de linfocitos inmaduros llamados linfoblastos (“blastos”). Como cualquier célula, los leucocitos crecen y se dividen normalmente de un modo ordenado y controlado. En los casos de leucemia, una célula progenitora hematopoyética que tiene un daño genético sufre una transformación maligna que lleva a una división celular no controlada. Esto crea una proliferación de células inmaduras que causan una (A – 1) sobrepoblación en la médula ósea . La sobrepoblación o infiltración medular impide que se produzcan glóbulos blancos, glóbulos rojos y plaquetas normales. La LLA ocurre tanto en adultos como en niños, generalmente de menos de 15 años de edad. El pico de incidencia ocurre a la edad de 4 años, y representa el 75-80% de todas las leucemias infantiles. La incidencia es levemente mayor en el sexo masculino, y en los paises occidentales e industrializados. En los niños, la LLA representa el 25% de todas las enfermedades malignas. La transformación leucémica y la expansión clonal de la LLA puede ocurrir en distintos estadíos de maduración del proceso de diferenciación linfoide (Pizzo, 2002; Pui, 2002). Signos y Síntomas Clínicos: La pérdida de la función de la médula ósea resulta en una disminución del número de glóbulos rojos y plaquetas y en un incremento del número de glóbulos blancos inmaduros. Las manifestaciones comunes de la LLA incluyen: Hemorragias inusuales, generalmente manifestadas por hematomas de fácil aparición sin traumatismo aparente, sangramiento de las encías, sangramiento nasal frecuente, y menstruaciones abundantes en mujeres adolescentes. La hemorragia suele deberse a la disminución de la producción de plaquetas, lo que lleva a bajos recuentos plaquetarios (trombocitopenia) Leucemia Linfoblástica Aguda ( LLA)
Transcript of Leucemia Linfoblástica Aguda (LLA) - cure4kids.org Linfoblástica Aguda (LLA) Módulo 13 -...

Leucemia Linfoblástica Aguda (LLA)
Módulo 13 - Docum
Autoras: Ayda G. NErin Gafford,Children’s RUniversidad U
Contenido Revisado
Fecha de Publicació
La Leucemia Linfobsobreproducción de lcélula, los leucocitoslos casos de leucemiuna transformación mproliferación de célusobrepoblación o infy plaquetas normales
La LLA ocurre tantopico de incidencia ocinfantiles. La incidenindustrializados. Entransformación leucémaduración del proc
Signos y Síntomas C
La pérdida de la funcrojos y plaquetas y emanifestaciones com
Hemorragiastraumatismomenstruaciondisminución(trombocitop
ento 2 Página 1 de 19
ambayan, DSN, RN, St. Jude Children’s Research HospitalEstudiante de Educación de Oncología Pediátrica, St. Judeesearch Hospital; Estudiante de Enfermería, Escuela de Enfermería,nionpor: Scott Howard, MD, St. Jude Children’s Research Hospital
Gaston K. Rivera, MD, St Jude Children’s Research Hospitaln en Cure4Kids: 25 de Julio 2008
lástica Aguda (LLA) es una enfermedad maligna caracterizada por unainfocitos inmaduros llamados linfoblastos (“blastos”). Como cualquiercrecen y se dividen normalmente de un modo ordenado y controlado. En
a, una célula progenitora hematopoyética que tiene un daño genético sufrealigna que lleva a una división celular no controlada. Esto crea una
las inmaduras que causan una (A – 1) sobrepoblación en la médula ósea. Lailtración medular impide que se produzcan glóbulos blancos, glóbulos rojos.
en adultos como en niños, generalmente de menos de 15 años de edad. Elurre a la edad de 4 años, y representa el 75-80% de todas las leucemiascia es levemente mayor en el sexo masculino, y en los paises occidentales e
los niños, la LLA representa el 25% de todas las enfermedades malignas. Lamica y la expansión clonal de la LLA puede ocurrir en distintos estadíos deeso de diferenciación linfoide (Pizzo, 2002; Pui, 2002).
línicos:
ión de la médula ósea resulta en una disminución del número de glóbulosn un incremento del número de glóbulos blancos inmaduros. Lasunes de la LLA incluyen:
inusuales, generalmente manifestadas por hematomas de fácil aparición sinaparente, sangramiento de las encías, sangramiento nasal frecuente, yes abundantes en mujeres adolescentes. La hemorragia suele deberse a lade la producción de plaquetas, lo que lleva a bajos recuentos plaquetariosenia)
Leucemia Linfoblástica Aguda (LLA)

Leucemia Linfoblástica Aguda (LLA)
Módulo 13 - Documento 2 Página 2 de 19
Fatiga persistente, palidez asociada y falta de aire incluso después de mínimos esfuerzos.Esto se debe a la disminución de la producción de glóbulos rojos que causa anemia
Dolor en articulaciones y huesos (especialmente en los huesos largos que contienenmédula) debido a infiltración leucémica en cavidades oseas.
Malestar general y decaimiento, asociado a fiebre, dolor de garganta o bucal; infeccionesa repetición, las cuales son causadas por la falta de leucocitos maduros para combatirprocesos infecciosos.
Pérdida de apetito debido a la fatiga, malestar general, y los efectos sistémicos de laenfermedad maligna.
Linfadenopatías (aumento de volumen de ganglios linfáticos) y visceromegalias (hepatoy esplenomegalia)
En los niños, los síntomas de leucemia a menudo están presentes entre una a seis semanas antesdel diagnóstico.
Factores de riesgo:
Los factores de riesgo que se han identificado para la LLA incluyen la radiación ionizante yvarios (A – 2) síndromes genéticos. Otros factores generalmente reconocidos incluyen
Sexo masculino (masculino más que femenino) Edad entre 2 y 5 años Clase socio-económica alta Exposición a rayos X in utero Radioterapia terapeutica postnatal (para el agrandamiento tímico y el tratamiento de la
tinea capitis).
Los factores de riesgo adicionales incuyen
Mayor edad materna al nacer Historia materna de pérdidas fetales Casos múltiples de leucemia reportados dentro de la familia. El riesgo es 2-4 veces mayor
si existe un hermano con LLA (en gemelos monocigóticos el riesgo es mayor durante lainfancia; y existe una posibilidad del 25% de desarrollar leucemia antes de los 7 años devida); grupos dentro de la misma generación y varias generaciones.
Otros factores de riesgo potenciales que no han sido demostrados en forma concluyente oque han sido refutados incluyen las infecciones postnatales, exposiciones ocupacionalesde los padres, padres fumadores, campos eléctricos y magnéticos, y la presencia de radónen el ambiente.
Clasificación de la LLA
Clasificación morfológica
FAB (Clasificación Morfológica Franco-Americana-Británica)

Leucemia Linfoblástica Aguda (LLA)
Módulo 13 - Documento 2 Página 3 de 19
La FAB es un sistema de clasificación para leucemias, basado en morfología de los blastos,estructura y citoquímica (tinciones químicas y actividad de la célula) y el número de células. (A– 3) L1, L2, y L3 comprenden el sistema de clasificación FAB. Una variante de estaclasificación es la (A – 4) variante de células espejo de mano la cual es una variante morfológicainusual de LLA. Esta variante representa menos del 5% de los casos de LLA infantil.
Clasificación Inmunobiológica e Inmunofenotípica
LLA de Linaje B: expresa los antígenos llamados CD19, HLA-DR, y/o CD10 (cALLa –antígeno de LLA común, a menudo correlacionado con pronóstico favorable). La LLA delinaje B tiene una tasa de sobrevida de 75% a 85% con tratamiento moderno. Hay tres tiposprincipales de células de linaje B reflejando el estadío de maduración de la célula B leucémica.Los antígenos e inmunoglobulinas (SIg para inmunoglobulina de superficie, CyIg parainmunoglobulina citoplasmática) estan expresados en la superficie celular dependiendo delestadío de maduración/diferenciación celular.Los tres tipos principales de LLA de linaje B son:
- early pre-B, sin expresión de SIg o CyIg, pero suele expresar el antigeno CD10- pre-B, expresan CyIg, y suele tener CD10- B-cell, expresan SIg, y puede tener CD10
LLA de Células T: Expresan antígenos de superficie de células T; más frecuente en varones; lossíntomas de presentación suelen incluir masa mediastinal, mayor incidencia de leucemia de SNCy, en general, tienen un mayor recuento leucocitario. También se caracterizan por una duraciónsignificativamente más corta de la remisión y de la sobrevida global.
LLA de células nulas (LLA de células no T, no-B): asociada con < 5% de los casos de LLA,los blastos leucemicos en estos pacientes no tienen marcadores celulares para T ni B.
Leucemias Bifenotípicas y de Linaje Mixto: Son leucemias donde las células malignasexpresan características de más de un linaje hematopoyético, o se presentan característicasmieloides y linfoides en las mismas células leucémicas.
Clasificación Genética:
El perfil de expresión genética provee una visión de las células leucémicas que no se limita soloa la patogénesis, sino que también aporta marcadores diagnósticos y objetivos terapéuticos. LaLLA puede ser clasificada genéticamente de acuerdo a:
Número modal de cromosomas
Hiperdiploidía - > 50 cromosomas por célula, index de ADN (ID) 1.16 omayorAsociado con menor edad (1 -10 años),Recuento medio leucocitario bajo,

Leucemia Linfoblástica Aguda (LLA)
Módulo 13 - Documento 2 Página 4 de 19
Mayor sensibilidad a los antimetabolitosSuele mostrar sobre-expresión o mutaciones del FLT3En general, mejor pronostico con mayores tasas desobrevida
Hipodiploidía - < 46 cromosomas por célulaGeneralmente asociada con pobres resultados
Alteraciones genéticas específicas de las líneas progenitoras leucémicas:Gen de fusión TEL-AML1; TEL-AML + LLA creada por la t(12;21)
relativamente buen pronóstico, especialmente conquimioterapia intensiva que incluya asparaginasa
t(1;19) - vista principalmente en pacientes con LLA pre-B,pronóstico moderado
t(8;14), t(2;8), t(8;22) LLA de células B con sobre-expresión de MYC asociado apronóstico favorable
t(9;22) Cromosoma Filadelfia BCR-ABL, t(4;11) - asociados a muy pobrespronósticos a pesar de la terapia intensiva
Expresión deHOX1112 alteración frecuente en LLA de células T infantilNOTCH1 identificado en el 50% de las LLA T, provee un fuerte
fundamento para terapias dirigidas a interferir con elmecanismo de señales NOTCH.
Proceso Diagnóstico
Historia completa de la enfermedad incluyendo una revisión de la incidencia y duraciónde síntomas como dolor, fatiga, infección, fiebre, sangrado y cefaleas; y una revisión delos potenciales factores predisponentes.
Exámen físico que debe enfocarse en las manifestaciones clínicas de la disfunción de lamédula ósea (anemia, trombocitopenia, neutropenia) tales como palidez, petequias, rash,linfadenopatías, cojera, hepatoesplenomegalia, agrandamiento testicular, y cambiosneurológicos debido a compromisodel SNC.
Hemograma con recuento diferencial leucocitario que puede estar bajo, normal o alto;recuento de GR bajo; plaquetas bajas; puede o no haber blastos en sangre periférica, perodeben estar presentes en la médula ósea para diagnosticar leucemia.
Aspirado/biopsia de médula ósea que revela la presencia de blastos (>25% de las célulasnucleadas de la médula)
El inmunofenotipo se realiza para determinar el linaje celular leucémico, y las tincionesespeciales se utilizan para diferenciar entre varios tipos de leucemia.
La inmunohistoquímica es un método no morfológico dirigido a detectar el contenido deantígenos de las células leucémicas que reaccionen con un anticuerpo que se presumeespecífico para dicha sustancia.
La citogenética se usa para diagnosticar (A – 5) alteraciones cromosómicas en muchasleucemias más del 90% de los niños con LLA tienen alteraciones citogenéticas,

Leucemia Linfoblástica Aguda (LLA)
Módulo 13 - Documento 2 Página 5 de 19
específicamente alteración del número de cromosomas (ploidía) y translocacionescromosómicas. Estas alteraciones se pueden detectar por el análisis cromosómicotradicional (cariotipo) así como por técnicas más sensitivas tales como la reacción depolimerasa en cadena por transcriptasa reversa (RT-PCR) e hibridización confluorescencia in situ (FISH).
El perfil de expresión génica (análisis molecular) se utiliza para identificar subtipos deleucemia específicos con mayor precisión, y para seleccionar tratamientos dirigidos a laslesiones moleculares subyacentes o a sus alteraciones consecuentes.
Punción lumbar es necesaria para descartar compromiso del sistema nervioso central(SNC); la presencia de blastos en el recuento diferencial indica enfermedad del SNC.
Se realiza una RX de tórax para evaluar la presencia de masa mediastinal y para evaluarel estado de la vía aérea para los requerimientos de sedación para los procedimientos.
Los niveles de inmunoglobulinas séricos están bajos en el 30% de los pacientes con LLAal diagnóstico.
Los análisis químicos séricos proveen información basal para la evaluación decomplicaciones tales como el síndrome de lisis tumoral y la insuficiencia renal. La LDHpuede proveer información acerca de la carga tumoral (mayores niveles de LDHsignifican mayor carga tumoral).
El panel hepático (enzimas hepáticas y bilirrubina) provee datos para evaluar lacapacidad del hígado de metabolizar las drogas quimioterápicas. Esta información seutiliza para decisiones terapéuticas.
Tratamiento Adaptado al Riesgo para Leucemia Linfoblástica Aguda Infantil
El tratamiento adaptado al riesgo utiliza las características del paciente y la enfermedad(variables) que los estudios clínicos de investigación han relacionado a mejores o peoresresultados del tratamiento. Los criterios para estratificación de riesgo del Instituto Nacional delCáncer (NCI)/Roma usan la edad y el recuento leucocitario al diagnóstico como variablespredictivas del resultado de la enfermedad.
Riesgo estándar = edad 1 a 9.99 años y GB <50,000/µLRiesgo Alto = edad 10 años y/o GB 50,000/µL
Los resultados del tratamiento de la LLA dependen no solo de la terapia utilizada sino también,en forma más importante, de la biología subyacente del tumor y del huésped. Aunque los dosfactores predictivos pronósticos más importantes son la edad y el recuento leucocitario aldiagnóstico, estudios recientes sugieren que otras variables como el sexo, inmunofenotipo,perfiles genéticos como el número modal de cromosomas (hiper o hipodiploidía), la presencia deenfermedad del SNC al diagnóstico, y la evaluación de EMR pueden todas influenciar y/opredecir el resultado de la LLA.
El Children’s Oncology Group (COG) propuso un nuevo sistema de clasificación basado en lareevaluación de muchas variables. Basado en la edad del paciente, el recuento leucocitario inicialy el inmunofenotipo, el paciente será asignado a uno de los cuatro grupos iniciales detratamiento:
LLA de células T

Leucemia Linfoblástica Aguda (LLA)
Módulo 13 - Documento 2 Página 6 de 19
LLA del lactanteLLA precursor B de Alto RiesgoLLA precursor B de Riesgo Estándar
Al final de la inducción, los pacientes con LLA precursor B serán reclasificados en bajo riesgo,riesgo estándar, alto riesgo y muy alto riesgo, basado en los hallazgos moleculares del blasto, larespuesta al tratamiento de inducción (morfología de la médula ósea el día 8, 15 y 29, y laenfermedad mínima residual (EMR) el día 29).
Tratamiento
El tratamiento de la LLA ha sido profundamente influenciado por la naturaleza heterogénea de laenfermedad y la capacidad de estratificar a los pacientes de acuerdo a sus riesgos. En la LLA, lossiguientes principios guían el plan de tratamiento:
Determinación del tratamiento basado en los factores pronósticos individuales(terapia riesgo-dirigida)
Uso de la profilaxis temprana del SNC en el curso del tratamiento Uso de quimioterapia combinada para mantener la remisión Prevención y manejo de las complicaciones del tratamiento
Existen tres fases en el tratamiento quimioterápico de la LLA: inducción (inducción de laremisión), consolidación/intensificación tardía (reinducción) y mantenimiento o terapia decontinuación. Muchos pacientes también reciben tratamiento de quimioterapia intratecal paraprevenir la diseminación de la leucemia al sistema nervioso central.
Terapia de Inducción:
El objetivo de la inducción de la remisión es lograr una remisión completa erradicando el 99% delas células leucémicas dentro de aproximadamente 6 semanas, reestablecer la hematopoyesisnormal (recuento absoluto de granulocitos >5x109/L y recuento plaquetario >100x109/L) tanrápido como sea posible, y un estado clínico normal. La respuesta medular temprana secorrelaciona con un pronóstico favorable entre todos los grupos de riesgo. Aunque más del 95%de los niños entran en remisión morfologica dentro de 6 semanas, aun persisten célulasleucemicas no detectables al microscopio de hasta 1 × 1010 (10,000,000,000) células leucémicas(enfermedad residual), necesitando terapia continuada para erradicar completamente las célulasleucémicas y lograr una cura permanente. La terapia de inducción incluye 2 fases:
La fase 1 incluye las primeras 4 semanas del tratamiento. Las drogas más comunmente utilizadasdurante esta fase incluyen vincristina, dexametasona o prednisona, y (A – 6) asparaginasa. Losantraciclínicos (daunorrubicina o doxorrubicina) se agregan al regimen de tratamiento para lospacientes de mayor riesgo. Para los niños entre 1-9 años, la dexametasona puede ser sustituidapor prednisona para reducir el riesgo de recaída en el SNC, ya que la dexametasona tiene mayorpenetración en el LCR y vida media más larga.

Leucemia Linfoblástica Aguda (LLA)
Módulo 13 - Documento 2 Página 7 de 19
La fase 2 de la inducción (conocida también como intensificación) ocurre en las últimas 2semanas; las drogas comunmente utilizadas durante esta fase son ciclofosfamida (Cytoxan), 6MP (Purinetol), y Ara-C (Citarabina; Cytosar).
Terapia de consolidación:
La terapia de consolidación se da para reforzar la remisión y proveer tratamiento directo al SNCy otros santuarios antes de comenzar la terapia de mantenimiento. El propósito del tratamiento deconsolidación es destruir cualquier célula leucémica remanente que puede no estar activa peroque podria comenzar a crecer nuevamente y provocar una recaída.
La intensidad de la consolidación varía dependiendo del grupo de riesgo del niño, pero siempreincluye tratamiento del SNC. El agente quimioterápico más importante utilizado durante laconsolidación es el metotrexato a altas dosis (HDMTX). Otros agentes quimioterápicos usadosdurante esta fase incluyen ciclofosfamida, citarabina, asparaginasa, mercaptopurina, tioguanina,epipodofilotoxinas, y quimioterapia intratecal (metotrexato, hidrocortisona, citarabina).
Terapia de re-inducción o de intensificación tardía:
La re-inducción es básicamente una repetición del tratamiento inicial de inducción 3meses luego de la remisión. Este tipo de tratamiento es más beneficioso para lospacientes de riesgo estándar. Estudios recientes también sugieren que la dobleintensificación tardía (a la semana 32 del tratamiento) mejora los resultados de lospacientes con LLA de riesgo intermedio y alto.
Terapia de mantenimiento o continuación:
El objetivo de la terapia de mantenimiento es eliminar las células leucémicas residuales. Es unperíodo prolongado de tratamiento continuo de anti-metabolitos que incluye dosis diarias demercaptopurina o tioguanina oral y dosis semanales de metotrexato oral. Los mejores resultadosclínicos parecen ocurrir si la medicación se administra a los mayores niveles tolerables por elpaciente. La combinación de dosis intermitentes de vincristina y dexametasona o prednisona máslos anti-metabolitos también disminuyen la incidencia de recaídas. Es más, las dosis vespertinasde mercaptopurina oral parecen estar relacionadas a una sobrevida libre de eventos más larga enpacientes con LLA cuando se compara con las dosis matinales.
Terapia del SNC:
Debido a que el SNC es un sitio santuario para las células leucémicas que no fueron detectadasdurante el diagnóstico y fueron protegidas de la quimioterapia sistémica por la barrera hemato-encefálica, el tratamiento de la leucemia subclínica del SNC es esencial. Estudios handemostrado que a menos de que se instituya un tratamiento dirigido al SNC, más del 50% de lospacientes con LLA desarrollarán leucemia del SNC. Por lo tanto, el objetivo de la terapiadirigida al SNC es incrementar la chance de cura previniendo el desarrollo de leucemia meníngea(recaída en SNC) y la necesidad del consecuente tratamiento intensivo.

Leucemia Linfoblástica Aguda (LLA)
Módulo 13 - Documento 2 Página 8 de 19
La terapia del SNC incluye tratamiento triple intratecal con metotrexato, hidrocortisona ycitarabina, y la irradiación intracraneal. Debido a que la irradiación intracraneal puede producirneoplasias secundarias tardías y ha sido asociada a mayores tasas de desempleo y de mortalidad,la mayoría de los protocolos de leucemia limitan el uso de irradiación craneal a grupos de altoriesgo y en dosis bajas. La recomendacion actual es en lo posible reservar la irradiación cranealpara pacientes que a pesar de recibir tratamiento inicial adecuado desarrollan una recaida.
Transplante Alogenéico de Células Progenitoras Hematopoyéticas/Transplante de Médula Ósea(TCPH/TMO):
Aunque el TMO ha sido exitoso en leucemias en estadíos finales, existen controversias en cuantoa sus indicaciones para segundas remisiones; específicamente en temas relacionados al impactodel TMO en las tasas de recaídas, riesgos de los transplantes alogenéicos, SLE globalcomparativa, y las tasas de morbilidad/mortalidad relacionadas al transplante. La controversia esaún más confusa por la falta de estudios randomizados que comparen los resultados del TMO yla quimioterapia en las recaídas de LLA. En la mayoría de los casos, las indicaciones de TMOvarían enormemente entre un centro y otro, dependiendo principalmente de la disponibilidad derecursos.
El transplante alogenéico es aún más limitado por la escasez de hermanos HLA-compatibles ylos riesgos asociados al procedimiento. Es más, la experiencia del equipo de TMO y el tipo detransplante (hermano/donante compatible versus transplantes haploidénticos) a menudoinfluencia los resultados del procedimiento. Actualmente, se están llevando a cabo estudios paradefinir mejor el rol y el tiempo óptimo del TCPH y las medidas profilácticas para controlar laspotenciales complicaciones para los pacientes de alto riesgo y recaídos.
Duración de la terapia:
La LLA de células B maduras se trata con un curso de terapia intensiva de 2 a 8 meses, lograndotasas de curación aceptables.
La LLA de linaje B (pre-B, early pre-B) se trata generalmente entre 2 a 3 años.
La LLA de células T requiere aproximadamente 2-2.5 años de terapia de continuación. Losintentos de reducir la duracion de la terapia han producido mayores tasas de recaídas luego de lasuspensión del tratamiento.
Pronóstico
Ya que el acercamiento terapéutico a la LLA depende del riesgo de recaída del paciente, losresultados para los niños con LLA pueden ser predichos por varios hallazgos clínicos y delaboratorio, tales como el recuento leucocitario inicial, la edad, la presencia de alteracionescitogenéticas, y la respuesta al tratamiento.

Leucemia Linfoblástica Aguda (LLA)
Módulo 13 - Documento 2 Página 9 de 19
Remisión Completa:
Se considera que los pacientes están en remisión si no tienen evidencia de leucemia al examenfísico y en la evaluación hematológica de sangre periférica y médula ósea. Los valores de sangreperiférica deben encontrarse dentro del rango definido de normalidad, y la médula ósea debetener una celularidad normal, con menos de 5% de linfoblastos.
Recaída:
La recaída de LLA o la recurrencia de la enfermedad es a menudo devastadora para el niño y lafamilia. La recaída contribuye en gran medida a la morbilidad y la mortalidad del cáncer infantil.Cerca del 20% de los pacientes recaerán, y aunque el manejo incluye el uso de altas dosis dequimio/radioterapia y transplante de médula ósea, los resultados siguen siendo insatisfactorios.
Una pobre respuesta a las drogas en la fase temprana del curso clínico, y la detección deenfermedad mínima residual luego de la inducción y consolidación están asociadas con un riesgoincrementado de recaída. En general, los niños que tienen múltiples recaídas tienen mayordificultad para lograr la remisión durante la reinducción; y el tiempo entre cada remisión tambiéndisminuye.
Recaída en Médula Ósea (recaída medular):
La médula ósea es el sitio más común de recaída de la LLA y es considerada la principal formade falla de tratamiento en estos pacientes.
Recaída en SNC:
La leucemia meníngea es un impedimento principal al tratamiento y suele ser predictiva derecaída medular. Se observa en <5% de los pacientes, la recaída en SNC es definida como lapresencia de lifoblastos identificados morfológicamente en el extendido de LCR con un recuentode células mononucleares >5/microlitro; o la presencia de infiltración tumoral en el SNC luegode la primera remisión. Aunque antes se correlacionaba con pobres resultados, la sobrevida librede eventos de los pacientes con recaída aislada en SNC es actualmente de cerca de 70% contratamiento intensivo.
Recaída testicular/ovárica:
La recaída testicular/ovárica se define como la evidencia histológica de infiltración linfoblásticaen uno o ambos testículos u ovarios. Puede ser clínicamente detectable u oculta (detectada en labiopsia). Los resultados para los pacientes con recaída testicular (ovárica) parecencorrelacionarse con el tiempo de presentación. Una recaída que ocurre mientras el paciente seencuentra en tratamiento se asocia a pobre pronóstico; sin embargo, con un re-tratamientosistémico intensivo, se puede lograr una sobrevida prolongada. En contraste, una recaídatesticular u ovárica aislada, tardía, que ocurre luego de finalizado el tratamiento, tiene un mejorpronóstico.

Leucemia Linfoblástica Aguda (LLA)
Módulo 13 - Documento 2 Página 10 de 19
Recaída combinada de LLA:
La recaída combinada de LLA se define como uno o más sitios de infiltración linfoblásticaextramedular y > 5% blastos en un aspirado de médula ósea luego de la primera remisión. Lasrecaídas combinadas tienden a ocurrir más tardíamente y tener mejor respuesta al tratamiento.Debido a su naturaleza parcial, las recaídas combinadas (medular y extramedular) tienen mejoresresultados comparadas con la recaída medular aislada.
Direcciones Futuras
Aunque se han conseguido avances dramáticos en el tratamiento y manejo de la LLA, se debensuperar varios desafíos para asegurar tasas de sobrevida más altas para todos los niños con LLA.Los desafíos incluyen:
1. aumento de la comprensión de la leucemogénesis y la susceptibilidad genética2. utilización de la información de la susceptibilidad genética para la detección
temprana3. refinamiento de la terapia dirigida según riesgo para los subgrupos de LLA4. comprensión de las variaciones individuales de la farmacocinética y la
farmacodinámica de las drogas antileucémicas5. desarrollo de estrategias para evitar la resistencia de las drogas6. manejo y superación de los efectos adversos, agudos y a largo plazo, del
tratamiento7. uso de la inmunoterapia para reforzar los efectos antileucémicos durante el
transplante de células progenitoras8. uso de anticuerpos y técnicas de purga basadas en lo molecular para fortalecer el
rol del transplante autólogo de progenitores hematopoyéticos9. control de la adherencia a la quimioterapia oral10. mejoría de los procedimientos para detectar la enfermedad mínima resdual11. adquisición de información confiable con respecto a la quimiosensibilidad in vitro
de las células leucémicas12. diseñar mejores terapias dirigidas en la LLA

Leucemia Linfoblástica Aguda (LLA)
Módulo 13 - Documento 2 Página 11 de 19
Enlaces útiles en la WebNota: Los enlaces en los siguientes sitios en la Web son en inglés, al menos que se indique lo contrario.
Enlaces de Leucemia Linfoblástica Aguda:Sociedad Americana de Cáncer: Leucemia Infantilhttp://www.cancer.org/docroot/CRI/content/CRI_2_2_1X_What_is_childhood_leukemia_24.asp?sitearea=
Sociedad Americana de Hematologíahttp://www.asheducationbook.org/cgi/content/full/2003/1/102
National Cancer Institute - Treatment Statement for Health ProfessionalsLeucemia linfoblástica aguda infantil: Tratamiento (PDQ®)http://www.meb.uni-bonn.de/cancer.gov/CDR0000256671.html
E Medicine.comhttp://www.emedicine.com/ped/topic2587.htm#top
Orpha.net, París, Franciahttp://www.orpha.net/data/patho/GB/uk-ALL.pdf
Seminarios relacionados en www.cure4kids.orgNota: Los siguientes Seminarios están disponibles en inglés, al menos que se indique lo contrario.
Seminario #770 Manejo general de la LLAChing Hon Pui, MDhttp://www.cure4kids.org/seminar/770
Seminario #51 Terapia dirigida al SNCWren Kennedy, PNP/O, John Sandlund, MD and Ching-hon Pui, MDhttps://www.cure4kids.org/seminar/51
Seminario #50 Recaída en médula ósea en la LLAGaston K. Rivera, MD and Wren Kennedy, PNP/Ohttp://www.cure4kids.org/seminar/50
Seminario #267 Recaída en médula ósea – en portuguéshttp://www.cure4kids.org/seminar/267
Seminario #448 LLA recaídaNobuko Hijiya, MDhttp://www.cure4kids.org/seminar/448
Seminario #255 Tratamiento de la LLA infantil – Intensificación del tratamientoGaston Rivera, MDhttp://www.cure4kids.org/seminar/255
Seminario #270 Tratamiento de la LLA – en españolhttp://www.cure4kids.org/seminar/270
Seminario #914 Leucemia linfoblástica aguda de células TEduardo Delgado, MD and Fred Behm, MDhttp://www.cure4kids.org/seminar/914
Leucemia Linfoblástica Aguda (LLA)
Módulo 13 - Documento 2 Página 12 de 19
Apéndice:
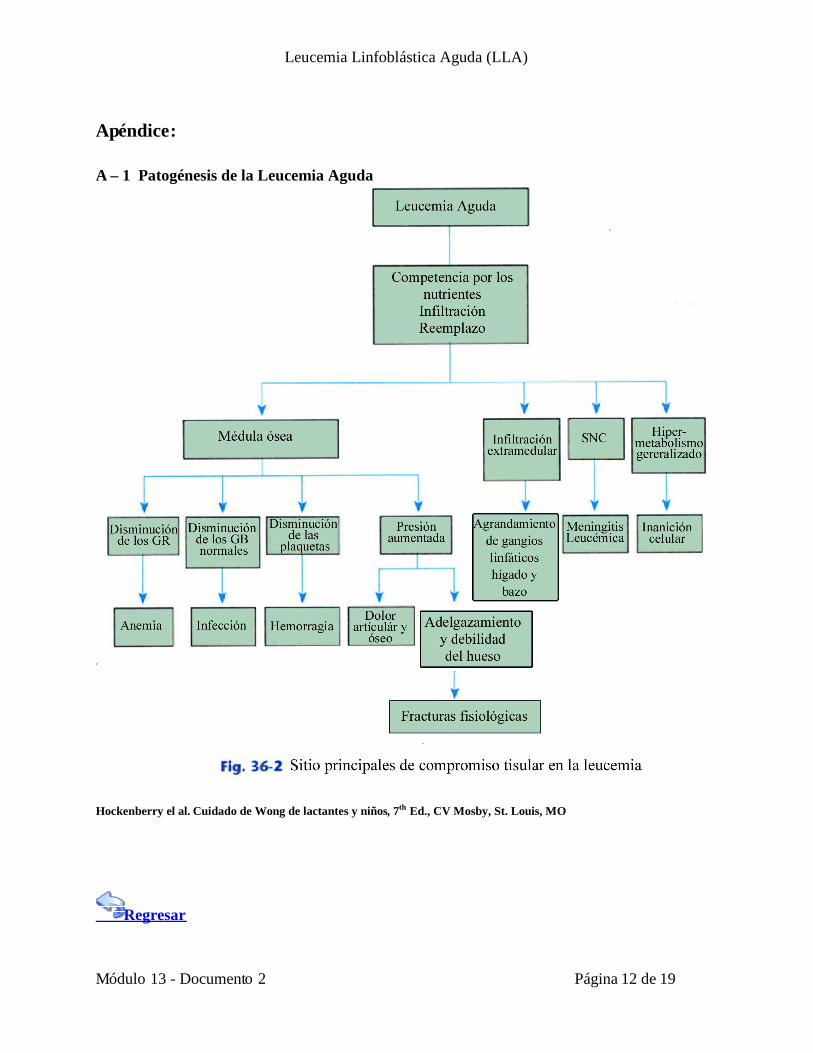
A – 1 Patogénesis de la Leucemia Aguda
Hockenberry el al. Cuidado de Wong de lactantes y niños, 7th Ed., CV Mosby, St. Louis, MO
Regresar

Leucemia Linfoblástica Aguda (LLA)
Módulo 13 - Documento 2 Página 13 de 19
A – 2 Síndromes Genéticos Asociados con LLA:
Trisomía 21 (síndrome de Down): los pacientes con síndrome de Down tienen una posibilidadhasta 15 veces mayor de desarrollar leucemia
Síndrome de Klinefelter: es una condición congénita en la cual los hombres tienen doscromosomas sexuales X y un Y. los lactantes parecen normales al nacer, pero en la pubertadtienen desarrollo tardío de las características sexuales secundarias; suelen ser infértiles. Lossignos y síntomas incluyen un pene pequeño, testículos firmes, disminución del vello púbico,axilar y facial, disfunción sexual, aumento del tejido mamario (ginecomastia), estatura elevadacon proporciones corporales anormales (piernas largas, tronco corto) y dificultades deaprendizaje.
Síndrome de Bloom: una enfermedad genética autosómica recesiva causada por la mutación delgen BLM. Los pacientes con síndrome de Bloom tienen un tamaño corporal pequeño,fotosensibilidad e infertilidad.
Anemia de Fanconi: una enfermedad hereditaria que afecta primariamente la médula ósea,causando disminución en la producción de todas las células de la sangre. Ochenta por ciento delos pacientes con anemia de Fanconi poseen cambios en la pigmentación de la piel tales comoáreas hiperpigmentadas oscuras, vitiligo, manchas café-con-leche. Tienen baja talla conalteraciones esqueléticas como alteraciones de los miembros superiores (dedos faltantes osupernumerarios, huesos ausentes o con escaso desarrollo), escoliosis, alteraciones de la cadera,pierna y hallux, alteraciones faciales como alteraciones oculares, palpebrales y auriculares,sordera, y alteraciones anatómicas tales como malformaciones renales, gastrointestinales ycardiopulmonares. Los lactantes presentan retardo mental y fallo de medro.
Ataxia-Telangiectasia: un desorden autosómico recesivo caracterizado por fragilidadcromosómica, resultado de mutaciones en el gen ATM. El signo más obvio es la presencia demúltiples telangiectasias (lesiones vasculares formadas por la dilatación de los pequeños vasossanguíneos) que son fácilmente visibles en el blanco de los ojos y la piel del rostro, pelo grisáceoy disminución en la coordinación de los movimientos, especialmente en la infancia tardía. Otrossíntomas incluyen deambulación tardía, movimientos torpes, infecciones respiratoriasrecurrentes, fallo de medro y disminución del desarrollo mental.
Regresar
Leucemia Linfoblástica Aguda (LLA)
Módulo 13 - Documento 2 Página 14 de 19
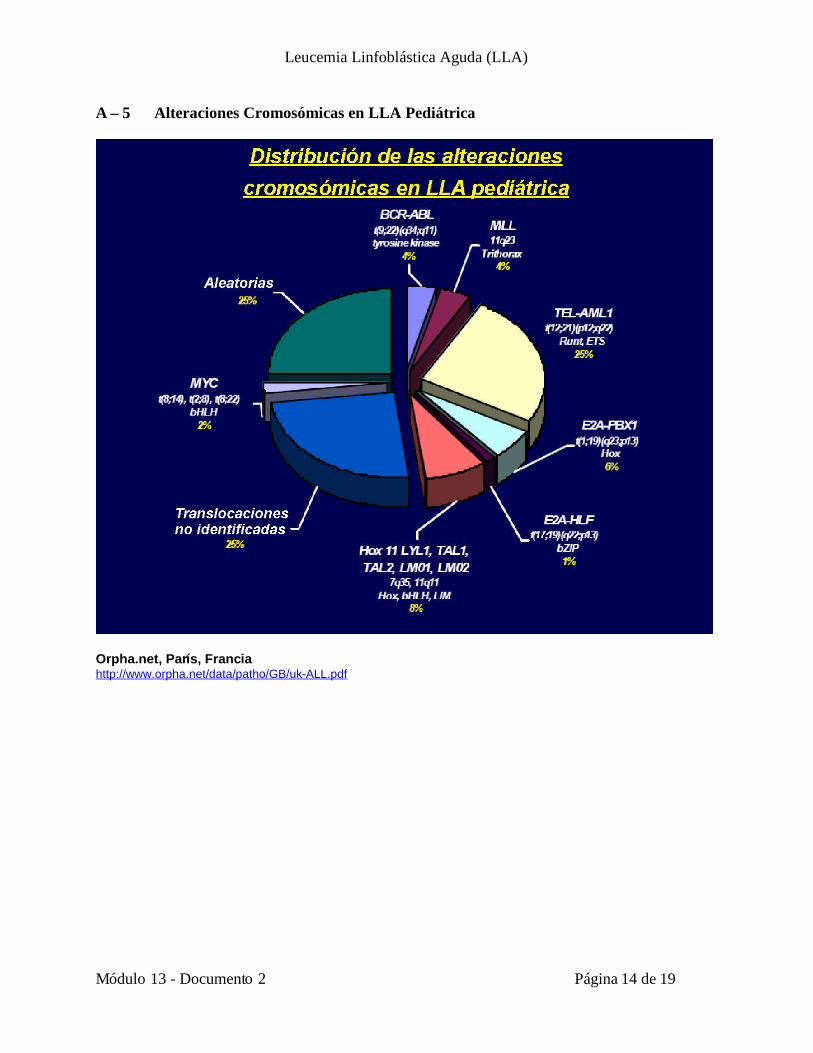
A – 5 Alteraciones Cromosómicas en LLA Pediátrica
Orpha.net, París, Franciahttp://www.orpha.net/data/patho/GB/uk-ALL.pdf
Leucemia Linfoblástica Aguda (LLA)
Módulo 13 - Documento 2 Página 15 de 19
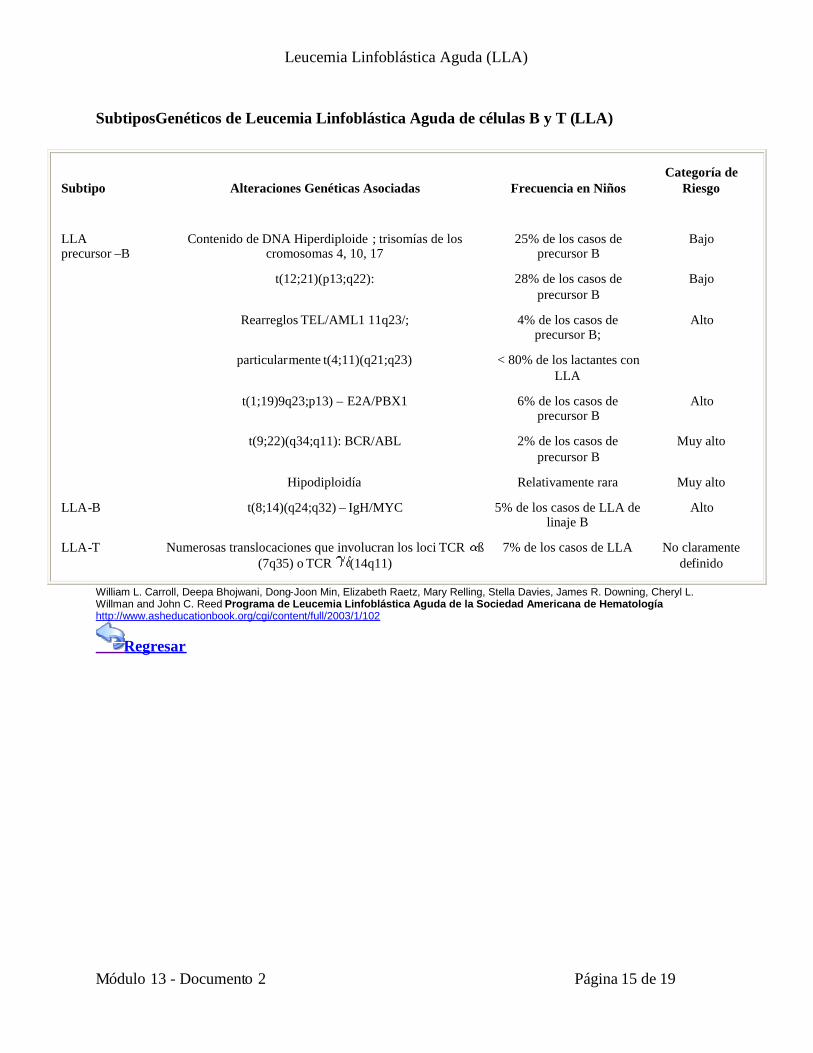
SubtiposGenéticos de Leucemia Linfoblástica Aguda de células B y T (LLA)
Subtipo Alteraciones Genéticas Asociadas Frecuencia en NiñosCategoría de
Riesgo
LLAprecursor –B
Contenido de DNA Hiperdiploide ; trisomías de loscromosomas 4, 10, 17
25% de los casos deprecursor B
Bajo
t(12;21)(p13;q22): 28% de los casos deprecursor B
Bajo
Rearreglos TEL/AML1 11q23/; 4% de los casos deprecursor B;
Alto
particularmente t(4;11)(q21;q23) < 80% de los lactantes conLLA
t(1;19)9q23;p13) – E2A/PBX1 6% de los casos deprecursor B
Alto
t(9;22)(q34;q11): BCR/ABL 2% de los casos deprecursor B
Muy alto
Hipodiploidía Relativamente rara Muy alto
LLA-B t(8;14)(q24;q32) – IgH/MYC 5% de los casos de LLA delinaje B
Alto
LLA-T Numerosas translocaciones que involucran los loci TCR ß(7q35) o TCR (14q11)
7% de los casos de LLA No claramentedefinido
William L. Carroll, Deepa Bhojwani, Dong-Joon Min, Elizabeth Raetz, Mary Relling, Stella Davies, James R. Downing, Cheryl L.Willman and John C. Reed Programa de Leucemia Linfoblástica Aguda de la Sociedad Americana de Hematologíahttp://www.asheducationbook.org/cgi/content/full/2003/1/102
Regresar

Leucemia Linfoblástica Aguda (LLA)
Módulo 13 - Documento 2 Página 16 de 19
A – 3 Sistema de Clasificación FAB
Linfoblastos L1 suelen ser menores que los linfoblastos L2, con escaso citoplasma y nucléolosapenas visibles. Aproximadamente el 85% de los niños con LLA tienen morfología L1.
Linfoblastos L2 son más grandes que los linfoblastos L1 con gran heterogeneicidad en sutamaño, tienen nucléolos prominentes y citoplasma más abundante. La morfología L2 estápresente en el 14% de los casos de LLA.
Linfoblastos L3 son grandes y se destacan por su profunda basofilia citoplasmática.Frecuentemente poseen vacuolización y son morfológicamente idénticos a las células del linfomade Burkitt. Representan el 1% de la morfología de LLA. Los linfoblastos L3 poseeninmunoglobulinas de superficie y otros marcadores característicos de células B. La LLA L3 tieneel peor pronóstico global.
Orpha.net, París, Franciahttp://www.orpha.net/data/patho/GB/uk-ALL.pdf
Regresar
Leucemia Linfoblástica Aguda (LLA)
Módulo 13 - Documento 2 Página 17 de 19
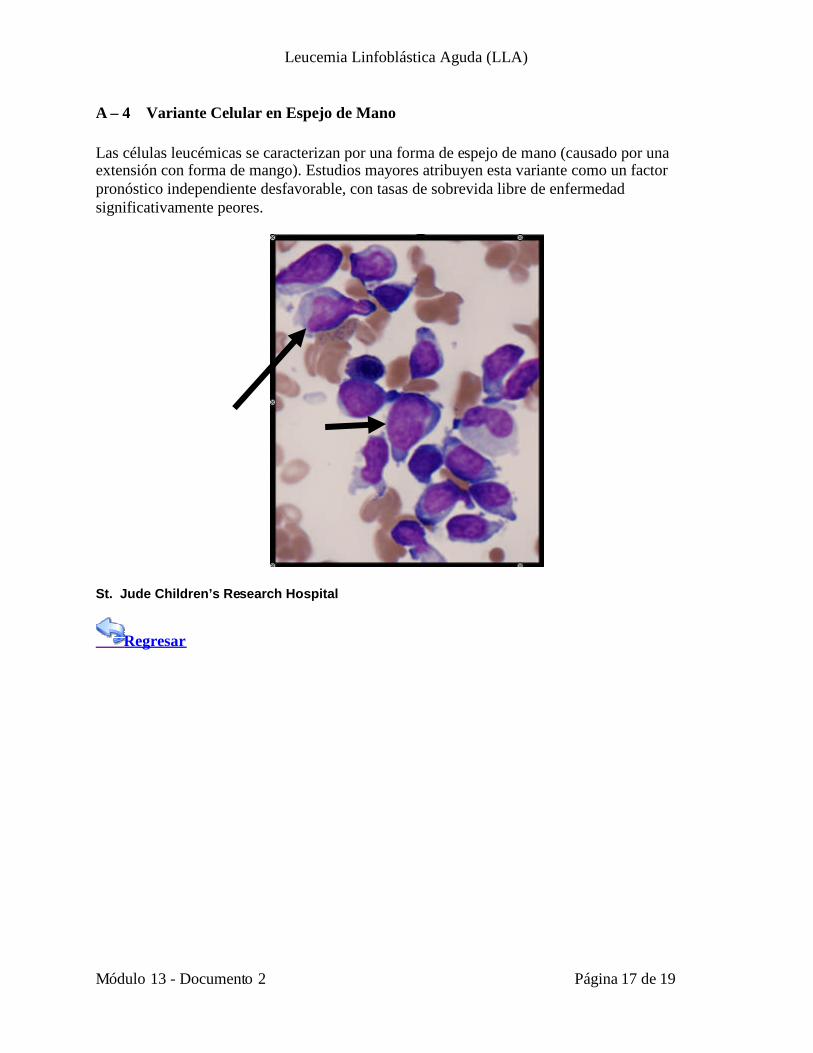
A – 4 Variante Celular en Espejo de Mano
Las células leucémicas se caracterizan por una forma de espejo de mano (causado por unaextensión con forma de mango). Estudios mayores atribuyen esta variante como un factorpronóstico independiente desfavorable, con tasas de sobrevida libre de enfermedadsignificativamente peores.
St. Jude Children’s Research Hospital
Regresar
Leucemia Linfoblástica Aguda (LLA)
Módulo 13 - Documento 2 Página 18 de 19
A – 6 Asparaginasa: No toda asparaginasa es la misma.
Características farmacológicas de las diferentes preparaciones de asparagina
Vida Media Depleción de asparaginaE-Coli* 1.28 ± 0.35 d 14–23 dErwinia 0.65 ± 0.13 d 7–15 dPEG 5.73 ± 3.24 d 26–34 d
Asselin et al J Clin Oncol 11:1786-6, 1993
Equivalent Doses of Commercial Products of Asparaginase
NombreComercial
FuenteBacteriana
Compañía Farmacéutica Dosis Equivalente enUnidades
Erwinase® Erwiniachrysanthemi
Enzon, Ipsen-Speywood 20,000
Elspar® E coli Merck 10,000
Leunase® E coli Medac, Kyowa Hakko 5,000
Oncaspar® E coli pegylated Medac, Rohne-PoulencRorer
500
CH Pui, 2003
Regresar

Leucemia Linfoblástica Aguda (LLA)
Módulo 13 - Documento 2 Página 19 de 19
Agradecimientos:
Autoras: Ayda G. Nambayan, DSN, RN, St. Jude Children’s Research HospitalErin Gafford, Estudiante de Educación de Oncología Pediátrica, St. JudeChildren’s Research Hospital; Estudiante de Enfermería, Escuela de Enfermería,Universidad Union
Contenido Revisado por: Scott Howard, MD, St. Jude Children’s Research HospitalGaston K. Rivera, MD, St Jude Children’s Research Hospital
Traducido por: Damián Nirenberg, MD, Hospital JP Garrahan, Buenos Aires, ArgentinaEditado por: Gaston K. Rivera, MD, St Jude Children’s Research HospitalCure4Kids Release Date: 25 de Julio 2008
Cure4Kids.orgPrograma de Alcance InternacionalSt. Jude Children's Research Hospital332 N. Lauderdale St.Memphis, TN 38105-2794
Usted puede duplicar y redistribuir este contenido en su totalidad con fines educacionales, el contenidoesta hecho libre de cargos. Este contenido no puede ser modificado o vendido. Usted puede ayudarnosen el desarrollo de material adicional de educación gratis enviándonos información acerca de cómo ycuando muestre este contenido y cuantas personas lo vieron. Enviar todos los comentarios y preguntas [email protected]
© St. Jude Children's Research Hospital, 2008
Last printed 18/07/2008 10:06:00 a.m.Last Updated: 18 de Julio 2008: ASX:\HO\IO Edu Grp\Projects\NURSING COURSE\NCSpanish\Module 13\M13 Final Revisions\NSM13D02V05.doc