
Herencia Autosomica Dominante
42
HERENCIA AUTOSOMICA HERENCIA AUTOSOMICA DOMINANTE DOMINANTE
-
Upload
marymar-estephania-h-cortes -
Category
Documents
-
view
49 -
download
1
description
descripción acerca de la herencia autosomica dominante
Transcript of Herencia Autosomica Dominante

HERENCIA AUTOSOMICA HERENCIA AUTOSOMICA DOMINANTE DOMINANTE

Un gen presenta una mutación que se expresa en el individuo heterocigoto
EFECTO DOMINANTE el gen mutante se hereda de uno de los progenitores y del otro recibe el normal.


El gen autosómico no se relaciona con la segregación del cromosoma X o Y en los espermatozoides, ambos sexos están afectados por igual.
Mutación de novo ocurrida en uno de los gametos que se unieron para formar el cigoto.

CARACTERISTICAS DE LA HERENCIA CARACTERISTICAS DE LA HERENCIA AUTOSOMICA DOMINANTE AUTOSOMICA DOMINANTE

En el estudio de los caracteres autosomicos dominantes algunos factores pueden modificar la expresión génica:
Los trastornos autosomicos dominantes varían entre los afectados por las diferencias genéticas o del ambiente en el que se desarrolla c/individuo.
*EL RIESGO DE RECIBIR EL GEN MUTANTE Y LA PROBABILIDAD DE PRESENTAR UNA MANIFESTACION CLÍNICA GRAVE DEL PADECIMIENTO.
CAPACIDAD DE UN GEN DE MANIFESTARSE O NO.
CONCEPTO DEL TODO O NADA.

Diferentes genes mutantes puede producir un cuadro clínico similar pero mostrar diferentes
tipos de herencia.

Desde el nacimiento (cromosomopatías o malformaciones congénitas)
ACONDROPLASIA SEUDOACONDROPLASIA
Fenómeno “anticipación” en los padecimientos autosomicos dominantes.
El inicio de la afección en la 1generacion es a una edad mayor que en la generación siguiente y así sucesivamente.

ACONDROPLASIA

HERENCIA DOMINANTE INCOMPLETA
Ningún alelo es dominante sobre el otroLos homocigotos son fenotípicamente diferentes a los heterocigotos.

ACONDROPLASIA
Trastorno esquelético que causa un enanismo con miembros cortos y con cabeza grande.1 en 26 000 recién nacidos90% pacientes: mutaciones de novo y edad paterna avanzada.Mutación Gen FGFR398% cambio de Glicina por Arginina en el nucleótido 1138 2% cambio de Citosina por Adenina

DIAGNOSTICO
Talla baja desproporcionada MICROMELICOMacrocefaliaFrente prominentePuente nasal deprimidoHipoplasia facial mediaPero más largo que la tibia
Tronco largoHiperlordosis lumbarMano en forma de tridente
Desarrollo motor lentoInteligencia normal

Signo de Scott

COMPLICACIONES
HidrocefaliaCompresiones
medularesGenu varum
Infecciones de oído medio

SINDROME DE MARFANSINDROME DE MARFAN

Es un trastorno autosómico dominante del tejido conectivo Existen en todas la etnias que se produce por mutaciones en el gen de la fibrina 1
Incidencia
Mutación novo
Las mutaciones que causan el síndrome de Marfan están diseminadas por todo el gen y cada mutación suele ser característica de esa familia

PATOGENESISPATOGENESIS
Codifica la fibrina 1Micro túbulos tan en tejido elásticos como no elásticos como la adventicia de la aorta, las zonulas ciliares y la piel
Sintesis procesamiento, secrecion, polimerizacion y la estabilidad de la fibrina
Los estudios sobre los depositos de fibrina 1 y su expresion en celulas cultivadas han sugerido una patogenesis dominante negativa
Evidencias mas recientes en modelos de ratones con sindrome de marfan sugieren que la mitad de la cantidad normal de fibrina 1normal es insuficiente para iniciar una reunion microfibrilar efectiva

Sindrome de Marfan neonatalrasgos esqueleticos aisladosEctopia lenticular autosomica
dominanteFenotipo MASS prolapso de la
valvula mitral,miopia,dilatacion aortica no progresiva en el limite de la normalidad y hallazgos dermicos y esqueleticos.
Hasta la fecha no se ha encontrado una correlacion clara entre el fenotipo y genotipo
La variabilidad intra e interfamiliar sugiere que el ambiente y los factores epigeneticos desempeñan un papel un importante papel en la determinacion de fenotipo

La fibrina 1 no es solo una proteina estructural que el sindrome de marfan no tampoco es resultado de una debilidad estructural de los tejidos .
Las microfibrilas d la fibrina 1 se enlazan y reducen la concentracion y la actividad de los factores de crecimiento en la superfamilia TGFB.
La perdida de FIBRINA 1 aumenta la señalizacion de TGFB libre

Es un trastorno multisistémico con anomalías esqueléticas, oculares, cardiovasculares, pulmonares, dérmicas y durales.
Las anomalías esqueléticas incluyen: Estatura desproporcionalmente alta Aracnodactilia Deformidades de esternón Escoliosis Articulaciones laxas Paladar estrecho.
Las anomalías oculares incluyen: Ectopia lenticular Corneas aplanadas Incremento de longitud del globo ocular Iris hipoplásicos.
Fenotipo e historia naturalFenotipo e historia natural

Las anomalías cardiovasculares incluyen: Prolapso de la válvula mitralRegurgitación aorticaDilatación y disección de la aorta ascendente.
Las anomalías pulmonares incluyen: Neumotórax espontaneoBurbujas apicales.
Las anomalías dérmicas incluyen:Estrías atróficasHernias recurrentes
Las anomalías de la dura incluyen: Ectasia dural lumbosacra

Muchas de las características Muchas de las características del síndrome de marfán se del síndrome de marfán se desarrollan con la edad. desarrollan con la edad.

Las principales causas de muerte prematura son el fallo cardiaco debido al a regurgitación aórtica y la disección y rotura de la aorta.
A medida que el tratamiento quirúrgico y médico de la dilatación aórtica ha ido avanzado, también ha mejorado la supervivencia.
Entre 1972 y 1992, la edad a la que se predecía
que el 50% de los pacientes estuviera vivo subió
de los 49 años hasta los 74 años en las mujeres
y de 41 hasta los 70 en los varones

Control y tratamientoControl y tratamiento El diagnóstico del síndrome de marfán es un diagnóstico
clínico.
La confirmación mediante la identificación de mutaciones en FBN1 no resulta práctico debido a la extrema variabilidad alélica que hace que la identificación de la mutación causante en cada familia sea una labor difícil; así como la falta de correlación fiable entre el genotipo y el fenotipo

No existe tratamiento curativo para el síndrome de Marfán. No existe tratamiento curativo para el síndrome de Marfán. Por tanto, la terapia se centra en la prevención y el Por tanto, la terapia se centra en la prevención y el tratamiento sintomáticotratamiento sintomático


Los cambios hemodinámicas asociados a la gestación pueden precipitar la dilatación progresiva de la aorta y su disección.
Se piensa que la disección aortica es secundaria a los cambios hormonales, de volumen sanguíneo y de eyección cardiaca que se asocian al embarazo y al parto.
El riesgo de embarazo no es aceptable cuando el tronco de la aorta mide mas de 4 cm.
Las mujeres pueden elegir realizarse una sustitución aortica que conserve la válvula
antes de quedarse embarazadas.
Los pacientes con síndrome de marfán tienen un 50% de riesgo de tener hijos afectados por esa enfermedad

NEUROFIBROMATOSIS NEUROFIBROMATOSIS 11
PRINCIPIOSExpresividad variablePleiotropia extrema un solo gen es responsable de efectos fenotípicos o caracteres distintos y no relacionadosGen supresor de tumoresMutaciones de perdida de funciónHeterogenicidad genéticaMutaciones de Novo
CARACTERISTICAS FENOTIPICAS PRINCIPALES:
•Edad de inicio prenatal o hasta el final de la infancia•Manchas café con leche •Pecas axilares e inguinales•Neurofibromas cutáneos•Nódulos de lisch•Glioma óptico•Lesiones Oseas especificas

Historia & hallazgos Historia & hallazgos clínicosclínicosNiño 2 años es llevado a la consulta por:
√ 6 manchas café con lecheX pecas axilares ni inguinales X malformación oseaX neurofibromas
• el examen físico de sus padres no revela datos de NF1
• REMITEN al niño por no cumplir los criterios clínicos de NF1

Niño regresa a los 5 años de edad presentando:
Nódulos de lisch 2 ojos12 manchas color caféPecas axilares bilaterales
• DX: NF1 mutación de Novo• Riesgo de recurrencia bajo

PatogénesisPatogénesisNF1 gen que codificaproteina
neurofibrominaNeurofibromina GTPasa Ras supresor
de tumores
Manifestaciones clínicas provienen de una perdida de la función de producto génico
80% mutaciones causan un truncamiento de la proteína

Criterios
• 6> manchas café con leche de más de 5 mm en su diámetro mayor en individuos prepúberes y más de 15 mm en su diámetro mayor en individuos luego de la pubertad.
• 2> neurofibromas, de cualquier tipo, o un neurofibroma plexiforme.
• Pecas en la región axilar o inguinal.• Glioma óptico.• Dos o más nódulos de Lisch.• Una lesión ósea como la displasia esfenoidea o un
estrechamiento del cortex de los huesos largos con o sin pseudoartrosis.
• Un pariente directo con NF1 con los criterios enunciados anteriormente.

Las Manchas Café con Leche (90%)
Son parches planos color café con leche en la piel. Ser redondos y con los bordes lisos u ovalados con bordes irregulares. Son causados por un aumento en lacantidad melanina.Las manchas no tienen tratamiento y no predisponen a cánceres de piel.
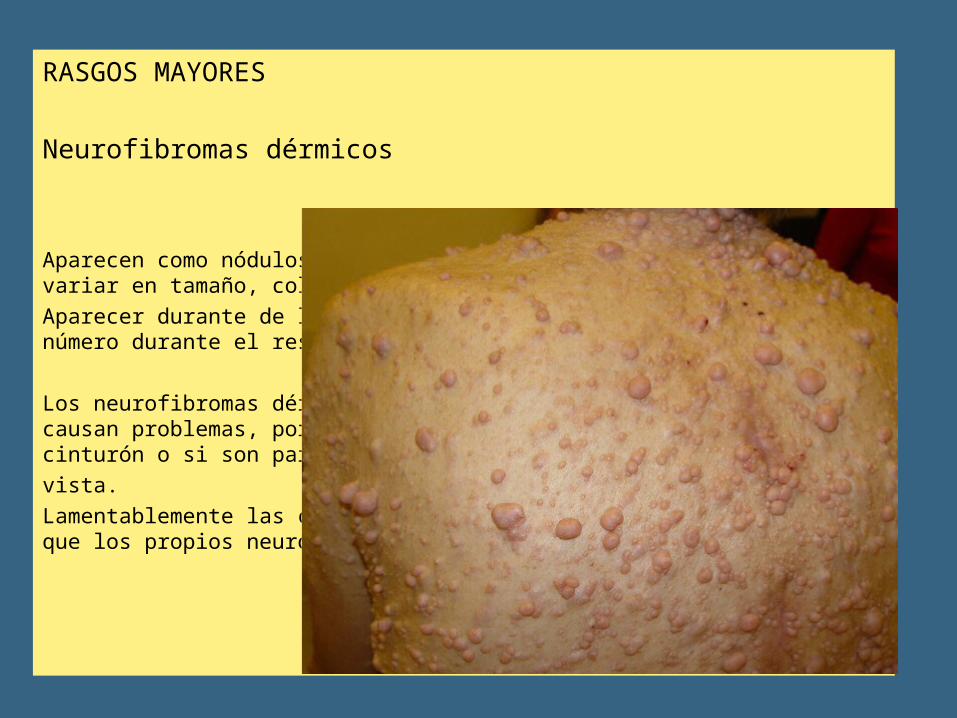
RASGOS MAYORES
Neurofibromas dérmicos
Aparecen como nódulos pequeños en la piel (dermis), pero pueden variar en tamaño, color y forma.Aparecer durante de la pubertad y tienden a incrementarse en número durante el resto de la vida.
Los neurofibromas dérmicos pueden ser fácilmente extirpados cuando causan problemas, por ejemplo, cuando quedan en la línea del cinturón o si son particularmente desagradables para lavista.Lamentablemente las cicatrices pueden generar más desfiguración que los propios neurofibromas.

Complicaciones de la NF1
Los neurofibromas plexiformes son generalmente mucho más grandes y su forma es más indefinida, tendiendo a mezclarse con el tejido que lo rodea, lo que hace que sea muy difícil extraerlos completamente. Pueden estar asociados con pigmentación e hipercrecimiento de la piel que los rodea o, si están en una extremidad, con los huesos debajo del mismo.
Los neurofibromas plexiformes pueden estar presentes en el nacimiento o aparecer durante la infancia temprana.
Pueden encontrarse en cualquier lugar del cuerpo, pero en un pequeño grupo de pacientes se encuentran en la cara, generando un serio problema estético.

Complicaciones de la NF1
Cáncer
El riesgo de desarrollar cáncer es alrededor del 5%.
Los 2 tipos de cáncer particularmente asociados con la NF son:
“Tumores embrionarios" en la infancia que aparecen en tejidos embrionarios primitivos
Neurosarcomas tumores malignos de los nervios, generados por cambios en el comportamiento de un neurofibroma preexistente.

Complicaciones de la NF1
Dificultades de Aprendizaje
Aproximadamente un tercio de las personas con NF1 tiene dificultades de aprendizaje específicas, experimentan problema al aprender a leer o escribir o la aritmética.

Complicaciones de la NF1
Problemas Ortopédicos
Existen dos problemas importantes que pueden aparecer con la NF1 y que están relacionados con defectos congénitos en el hueso.
• EscoliosisEs una curvatura hacia el costado de la columna que, generalmente, se desarrolla en la adolescencia temprana. Algunas personas con NF1 tienen una escoliosis leve que no cambia, pero en otras la curva se vuelve mucho más pronunciada y requiere tratamiento con un corsé o una operación.
• Curvatura congénita de la Tibia o Peroné (Pseudoartrosis)Esto ocurre cuando los huesos debajo de la rodilla (tibia y peroné) se curvan, tienden a ser más finos que lo normal y, por lo tanto, se quiebran fácilmente. Cuando esto ocurre, la unión de los huesos puede ser demasiado lenta o incompleta (pseudoartrosis). Este problema también puede ocurrir en el brazo con el radio o el cúbito.

Complicaciones de la NF1
Glioma OpticoEs un tumor del nervio óptico. El glioma óptico aparece generalmente en la niñez, y se nota porque se deteriora la visión del niño o porque un ojo se vuelve más saliente.
Se debe consultar a un especialista si un niño o adulto con NF1 desarrolla estrabismo, doble visión o visión borrosa. Muchos gliomas ópticos crecen muy despacio, o crecen inicialmente y luego no cambian, y no necesitan tratamiento. Aquellos que crecen rápido son tratados con una combinación de cirugía, radioterapia o quimioterapia.

Complicaciones de la NF1
HipertensiónTanto adultos como niños con NF1 deben tener su presión controlada regularmente. Hay 2 complicaciones poco comunes de la NF1 que pueden generar hipertensión. -Estenosis de la arteria renal -Feocromocitoma, un tumor raro de la glándula suprarrenal. Ambos problemas se tratan con éxito mediante operación.
EpilepsiaExiste la probabilidad de un pequeño incremento en pacientes con NF1 de tener epilepsia y suocurrencia no indica necesariamente la presencia de un tumor cerebral.

Control y tratamiento
Diagnóstico clínicoNo hay tratamiento curativo
El seguimiento de un paciente con NF1 debe incluir:Examen físico anual realizado por alguien familiarizado con la patologíaEvaluación oftalmológica anual durante la infanciaEvaluación periódica del desarrollo en la infanciaMedición periódica de la T/A
Neurofibromas plexiformes, intervención quirúrgica, pero esta se puede complicar, puesto que a menudo se encuentran ligados a nervios y tienen tendencia a recurrir en el sitio donde ha sido removido

Riesgo de Herencia
Los individuos con NF1 tienen un 50% de riesgo de tener un hijo infectado por NF1.
El dx prenatal es posible para las familias cuya mutación en el gen NF1 ha sido identificada.
El dx prenatal es preciso, no proporciona mucha información pronóstica, debido a la gran variabilidad fenotípica de la enfermedad.
Los padres de un hijo afectado que no presenta signos de la enf. Tiene aun así una ligera elevación del riesgo de recurrencia en la siguiente gestación debido a la posibilidad de que posean un mosaicismo de la línea germinal.


















