HEMOSTASIA -...
78
HEMOSTASIA 1 1 HEMOSTASIA
Transcript of HEMOSTASIA -...

HEMOSTASIA
11HEMOSTASIA

HEMOSTASIA
E l sistema hemostático constituye, junto al sistema inflamatorio y el inmune, un relevante mecanismo de defensa del organismo. Es bien conocido que la hemorragia ha sido un
elemento muy destacado desde los primeros vestigios de la civilización. Ya en las pinturas del paleolítico que adornan las cuevas de Altamira, el sangrado en los animales es una imagen habitual. Por otra parte, a lo largo de la historia ha existido un interés constante por aclarar los mecanismos responsables de la hemorragia. Este hecho contrasta con el escaso interés despertado por la trombosis durante siglos. Hoy en día, sin embargo, el estudio de la trombo-sis concita grandes esfuerzos al ser la mayor causa de mortalidad1. En definitiva, la forma de expresión clínica de una anomalía del sistema hemostático se manifiesta por situaciones muy distintas, como es la diátesis hemorrágica, o el evento de oclusión vascular tras la generación del trombo. Esas dos manifestaciones clínicas expresan el desequilibrio de uno de los brazos de la balanza hemostática.
Como muchos otros aspectos de la medicina moderna, el verdadero conocimiento de los mecanismos reguladores del sistema hemostático comenzó a ser científicamente entendi-do a lo largo del siglo pasado. Durante los últimos veinticinco años, se ha puesto en eviden-cia la tremenda complejidad del sistema, en el que participan de forma concomitante un amplio número de proteínas plasmáticas y elementos celulares. Precisamente esa comple-jidad ha sido incluso utilizada como argumento de discusión entre las corrientes filosóficas de los ¨creacionistas¨ frente a los ¨evolucionistas¨2.
1.11.1 HEMOSTASIA:FUNDAMENTOS Y BASES FISIOLÓGICAS
Dr. Vicente Vicente
El estudio de la trombosis concita grandes esfuerzos al ser la mayor causa de mortalidad1

HEMOSTASIA
ASPECTOS EVOLUTIVOS
El sistema hemostático más sencillo del que tenemos constancia es el del limulus (Figura 1), un ¨fósil vivo¨ de 500 millones de años de antigüedad3. Su sistema hemostático se circunscribe a sus células circulantes (hemocitos), cuya función es la de formar un tapón reforzado por una proteína gelatinosa conocida como coagulina, ante agresiones externas o en respuesta a una invasión de endotoxina. Un sistema tan simple, presenta ya algunas de las características pro-pias del sistema totalmente desarrollado de los vertebrados superiores.
A la izquierda, limulus o cangrejo de herradura. En la imagen de la derecha se observa su sencillo sistema circulatorio, que va a integrar a dos mecanismos de defensa del organismo, el sistema in-mune y el de coagulación.
FIGURA 1. SISTEMA HEMOSTÁTICO DEL LIMULUS O “CANGREJO DE HERRADURA”

HEMOSTASIA
Por una parte, anuncia la cascada enzimática de la coagulación sanguínea (Figura 2), donde una proteína precursora de la coagulina, el coagulógeno, es sensible a una serín proteasa cono-cida como coagulasa, para dar lugar de forma inmediata a la formación del coágulo y prevenir la exanguinación. Por otra parte, la participación celular, el hemocito, que podemos imaginarlo como un antecesor de la plaqueta.
Representación de un hemocito de limulus. Ante la presencia de un determinado patógeno o agente externo, se produce una cascada enzimática a través de los factores B y C que activan la actividad de la coagulasa para generar coagulina (molécula gelatinosa) a partir de coagulógeno.
Factor C
Factor B
Coagulasa (serinproteasa)
Coagulógeno Coagulina
Amebocito (Hemocito) Agresión de germen Gram (-)
FIGURA 2. LA COAGULACIÓN Y LA INMUNIDAD INNATA EN EL LIMULUS

HEMOSTASIA
Se ha construido una posible ruta de evolución del sistema hemostático (Figura 3), cuyo me-canismo de ensamblaje, mediante la duplicación de genes y la combinación aleatoria de exones, tuvo lugar durante un corto periodo de unos 50 millones de años. Durante los siguientes 450 mi-llones de años el sistema hemostático solamente ha experimentado pequeñas modificaciones3. Estos datos explican el alto grado de homología entre las proteínas que forman parte del propio sistema de coagulación y las pequeñas diferencias observadas entre especies.
FIGURA 3.
EVOLUCIÓN DEL SISTEMA DE COAGULACIÓN SANGUÍNEA
Posible relación evolutiva del siste-ma hemostático en vertebrados a partir de un antecesor putativo co-mún con invertebrados. Datos mos-trados en años.
INVERTEBRADOS
450x106
600 X 106
50 X 106
Peces
AnfibiosReptiles Pájaros Mamíferos
1800 X 106
3500 X 106
4600 X 106

HEMOSTASIA
El sistema hemostático constituye un activo y dinámico mecanismo de defensa del or-ganismo con la función de mantener constantemente permeable la luz vascular, restable-cerla en caso de obstrucción por un fenómeno trombótico y de reparar la lesión en la pared del vaso para impedir una excesiva pérdida sanguínea. Las manifestaciones clínicas de su desequilibrio, como ya hemos indicado, serán la tendencia hemorrágica o la trombótica. El conocimiento de este sistema es todavía limitado, y si bien se van definiendo las bases para su entendimiento, estamos lejos de conseguir asimilar las íntimas y múltiples interacciones entre sus proteínas, agrupadas clásicamente como proteínas procoagulantes, anticoagulan-tes y fibrinolíticas, así como su relación con otros sistemas igualmente complejos como el inmune, inflamatorio, tumoral, etc. En definitiva, el equilibrio hemostático es sin duda alguna, un proceso complejo y dinámico4.
Mantener la hemostasia equilibrada o generar la formación de un trombo requiere una serie de reacciones perfectamente coordinadas donde los protagonistas están localizados en diferentes instancias, como son los receptores de membrana, agonistas o ligandos plas-máticos circulantes o bien liberados de las propias plaquetas o de otros elementos celulares (endotelio, monocitos, etc.), y una articulada transmisión de señales bidireccionales intrace-lulares, etc.5,6
A continuación abordamos los fundamentos y bases fisiológicas del papel que juegan to-dos estos protagonistas con sus consecuentes interacciones. Iniciaremos la descripción co-mentando los principales integrantes que participan en el llamado “componente celular” del sistema hemostático (plaquetas y endotelio vascular), y continuaremos por el componente plasmático de la coagulación (proteínas procoagulantes, anticoagulantes y del sistema fibri-nolítico), pero en ningún momento debemos olvidar que ambos componentes, plasmático y celular, actúan e interaccionan simultáneamente. Sin esta intensa contribución constante el sistema hemostático no podría ejercer su función.
El sistema hemostático constituye un activo y dinámico mecanismo de defensa del organismo con la función de mantener constantemente permeable la luz vascular, restablecerla en caso de obstrucción por un fenómeno trombótico y de reparar la lesión en la pared del vaso para impedir una excesiva pérdida sanguínea

HEMOSTASIA
INTERACCIÓN PLAQUETA/PARED VASCULAR
La integridad vascular es un elemento crucial e indispensable para que se mantenga el equilibrio y no se active el sistema hemostático. La pérdida de la continuidad endotelial pro-picia la exposición de una superficie claramente trombogénica, como es el subendotelio vas-cular, a elementos celulares de la sangre, especialmente plaquetas, iniciando la interacción plaqueta-subendotelio que es un mecanismo desencadenante de la formación del trombo. Es bien conocido que superficies extrañas, sirva de ejemplo los stents, situaciones de desendo-telización o roturas de placas de ateroma, dan lugar a un proceso de generación de trombo donde la activación plaquetaria juega un papel muy relevante. En otras situaciones trombogé-nicas, como es un cuadro séptico, se genera trombina, enzima que tiene capacidad de activar el endotelio vascular y ocasionar la pérdida de su integridad. Diferentes datos experimentales nos muestran que altas concentraciones de trombina son capaces de generar un potente es-tado protrombótico por alteración de la pared vascular, lo que viene a sumarse al efecto de la activación del sistema de coagulación que también tiene lugar.
Diálogo plaqueta-endotelio
En un adulto el número de plaquetas circulantes es de aproximadamente 1x1012 y man-tienen contacto con una superficie revestida de endotelio vascular de unos 1000 m2. Pese a este contacto con el endotelio, pues las plaquetas tienden a circular por las zonas periféricas vasculares, en condiciones normales no hay fenómenos de adhesión, activación y agregación plaquetaria. Las plaquetas en reposo mantienen una forma discoide, y solamente tras su activación cambian drásticamente de forma. El hecho de que no exista activación se debe en buena medida al efecto inhibidor de activación plaquetaria que genera el endotelio, pues sintetiza de forma continua prostaglandina I2 y óxido nítrico y metaboliza los estimuladores fisiológicos de la activación plaquetaria, como ADP y trombina. La prostaglandina I2 y el óxido nítrico son inhibidores fisiológicos de la agregación plaquetaria.
La integridad vascular es un elemento crucial e indispensable para que se mantenga el equilibrio y no se active el sistema hemostático

HEMOSTASIA
Cuando hay una desendotelización o alteración de la pared vascular las plaquetas se ac-tivarán. En primer lugar tendrán un cambio de forma, adquirirán la capacidad de ser una partícula “pegajosa”, con alta capacidad de adherirse a diferentes superficies y dar lugar a un espacio prohemostático-protrombótico. Estos cambios morfológicos y funcionales van a estar regidos en un primer momento por la activación e interacción de diferentes glicopro-teínas de superficie plaquetaria, que ejercen su función como receptores de superficie pla-quetaria (Figura 4). En el inicio del fenómeno de la adhesión plaquetaria participan diferentes mecanismos: reclutamiento y sobreexpresión de receptores, así como formación de nuevos epítopes plaquetarios debido a los cambios generados en esos receptores. Las reacciones moleculares indicadas ocasionan cambio de la forma discoide de la plaqueta en reposo a la aparición de múltiples pseudópodos (plaqueta activada) que precede al fenómeno de secre-ción plaquetaria, que a su vez cierra el círculo de la activación aportando más sustancias que ejercen su función agonista sobre los receptores plaquetarios (Figura 5).
Interacción de los diferentes agonistas plasmáticos (ADP, epinefrina, fibrinógeno, trombina, factor von Willebrand, etc.) y los presentes en la matriz subendotelial (colágeno, factor von Willebrand, etc.) con sus receptores plaquetarios específicos.
PAR-1
Trombina
ColágenoTrombina
ADPEpinefrina
T xA2PAR-4
P 2Y1P 2Y
12
TP
SeñalFuera-Dentro
SeñalDentro-Fuera
FibrinógenoFvW
FvW
GPVI
GPIb/IX/V
α 2A
Gα q
Gα 12
Gα iGα q
Gα 12 Gα q Gα iGα z
α IIb β
3
α 2 β
1ß
Gα q
+
-
Gα 12
FIGURA 4. PRINCIPALES INTERACCIONES LIGANDO RECEPTOR PLAQUETARIOS

HEMOSTASIA
Principales receptores plaquetarios
Los principales receptores plaquetarios con función definida en la formación del trombo son: las glicoproteínas de membrana, algunas de ellas caracterizadas por repeticiones fre-cuentes de leucina en su secuencia de aminoácidos, la familia de las integrinas, los recep-tores de inmunoglobulinas y, finalmente, los receptores para agonistas solubles, conocidos como “ligandos autocrinos”.
FIGURA 5.
ACTIVACIÓN PLAQUETARIA
A) Las plaquetas circulan en sangre con una dis-posición “discoide”.
B) Cuando experimentan activación, sufren una drástica modificación generando pseudópodos y elongaciones que facilitan la interacción con otras plaquetas y células circulantes.
C) Como última fase, se extiende la plaqueta (spreading) en una amplia superficie donde se ha adherido y activado.
A
B
C

HEMOSTASIA
• Receptor rico en leucina: Complejo glicoproteíco Ib-IX-V (GPIb-IX-V)
Cuando la plaqueta en reposo entra en contacto con el subendotelio, se va a poner en marcha de forma inmediata el fenómeno de adhesión plaquetaria al factor von Willebrand (FvW) presente en el subendotelio, y de manera similar al colágeno presente en la matriz subendotelial. La interacción con el FvW tiene lugar a través de un complejo glicoproteico rico en leucina, como es el receptor GPIb-IX-V, que corresponde al primer fenómeno que da lugar a la adhesión y depósito plaquetario sobre la superficie subendotelial. La glicoproteína GPIb-IX-V, presente con unas 25 000 copias en la membrana plaquetaria, se liga al FvW depositado en el subendotelio, y no al circulante en el plasma, pues el presente en la matriz subendo-telial al estar unido al colágeno ha experimentado un cambio conformacional que propicia la interacción FvW-GPIb-IX-V. El FvW, además del que está presente en el subendotelio, se al-macena en los cuerpos de Weibel-Palade, y en los gránulos alfa de las células endoteliales y plaquetas, y también se encuentra circulante en el plasma. El FvW liberado del endotelio es el que se deposita en el subendotelio vascular por la unión con el colágeno tipo I y II allí presen-te. La repercusión clínica del defecto de la interacción GPIb-IX-V y FvW es muy manifiesta y bien conocida. Pacientes con trastornos congénitos del complejo rico en leucina (síndrome de Bernard-Soulier, SBS) o del FvW presente en el subendotelio (enfermedad de von Willebrand) muestran importantes problemas hemorrágicos por un defecto en la adhesión plaquetaria.
Por otra parte, el complejo GPIb-IX-V mantiene una estrecha conexión con el citoesqueleto plaquetario, lo que justifica que los pacientes con SBS muestren plaquetas de gran tamaño (ma-crotrombocitopenia). La interacción FvW con GPIb-IX-V además de ser crucial para el fenómeno de adhesión plaquetaria también es capaz de trasmitir una señal intracelular que ocasiona flujo de calcio y activación de integrinas, aspecto clave en la propagación del fenómeno de adhesión y al inicio de la activación plaquetaria.
Estudios en los últimos años han aportado un papel más extenso a la función del complejo GPIb-IX-V, puesto que se ha comprobado su interacción con varias proteínas plasmáticas y con receptores de otras células. Se ha demostrado el papel que tiene el complejo en el des-plazamiento (rolling) de las plaquetas en el endotelio mediado por selectina P, así como en el control de la interacción plaqueta-neutrófilo.
La interacción FvW con GPIb-IX-V, además de ser crucial para el fenómeno de adhesión plaquetaria, también es capaz de trasmitir una señal intracelular que ocasiona flujo de calcio y activación de integrinas, aspecto clave en la propagación del fenómeno de adhesión y al inicio de la activación plaquetaria

HEMOSTASIA
Además de lo indicado, el receptor GPIb-IX-V tiene alta afinidad por la trombina, esto puede facilitar la activación de la familia de los receptores activados por proteasa (PARs), especial-mente el PAR-1 y PAR-4, y que pueden tener una importante repercusión fisiológica en la ac-tivación del sistema hemostático. Finalmente, otra función relacionada al complejo GPIb-IX-V ha sido su capacidad de ligar proteínas de coagulación como los factores XI, XII, cininógeno de alto peso molecular, y también de actuar como correceptor del FVIIa. En definitiva, el com-plejo rico en leucina queda involucrado en la generación de actividad procoagulante en la superficie plaquetaria, con generación de trombina y fibrina.
Como acabamos de indicar, el complejo GPIb-IX-V además de jugar un papel fundamental en el mecanismo de adhesión plaquetaria (Figura 6), está involucrado en los fenómenos de activación plaquetaria, migración celular (neutrófilos) sobre el endotelio y en la activación del sistema de la coagulación sanguínea.
ADP
GpIIb/IIIa
GpIbTxA2
TROMBINA
Agregaciónplaquetaria
ENDOTELIOColágeno expuesto
ADHESIÓNFvW
Cuando se produce una lesión endotelial, los componentes de la matriz subendotelial quedan en contacto con los elementos formes de la sangre, en concreto con las plaquetas que circulan en la zona periférica de los vasos. De esta forma el factor von Willebrand subendotelial que se encuentra ligado al colágeno se unirá al complejo GPIb-IX-V. Esa interacción será suficiente para trasmitir una señal intraplaquetaria que facilite activación celular que conllevará la modificación conformacional de algunas glicoproteínas plaquetarias, como la GPIIb/IIIa, que será reconocida por sus ligandos plasmáticos (factor von Willebrand y fibrinógeno, especialmente) dando lugar a puentes interplaquetarios, secreción celular y en definitiva a la agregación plaquetaria. El primer estímulo también será un estímulo suficiente para exponer nuevos receptores de membrana que también facilitarán la potenciación de la agregación plaquetaria y la generación de trombina en la superficie celular, dando lugar a la formación del trombo.
FIGURA 6. RUTA DE ACTIVACIÓN PLAQUETARIA

HEMOSTASIA
• Integrinas plaquetarias
Los receptores plaquetarios que forman parte de la familia de las integrinas son requeridos para una adhesión estable de las plaquetas a la pared del trombo, así como para el propio crecimiento del mismo. La estructura de las integrinas se refleja por complejos heterodimé-ricos, cadenas alfa y beta, unidas de forma no covalente con localización transmembrana, con un corto tallo intracelular y uno largo extracelular. Un hecho común a las integrinas es que para que adquieran su capacidad funcional necesitan un cambio conformacional.
- La integrina plaquetaria más relevante es GPαIIbβ3, (conocida también como GPIIb/IIIa). Su presencia en la membrana plaquetaria sobrepasa las 80 000 copias. Hay un amplio número de proteínas que son los ligandos fisiológicos del complejo glicoproteico, como el fibri-nógeno, FvW plasmático, fibronectina, vitronectina y CD40L. La activación de la GPIIb/IIIa también se consigue con un buen número de agonistas solubles, como el ADP, epinefrina, tromboxano A2 (TxA2), trombina y colágeno. La unión con sus ligandos, en especial con el FvW plasmático y fibrinógeno, es un elemento indispensable para se lleve adelante el fenómeno de la agregación plaquetaria. La ausencia del complejo GPIIb/IIIa clínicamen-te se expresa por un cuadro hemorrágico grave, que es conocido como tromboastenia de Glazmann. Precisamente, el papel crucial que tiene el complejo GPIIb/IIIa para que haya un adecuado funcionalismo plaquetario, explica que esa integrina se use como diana para terapia antiagregante, ejemplos son los fármacos como abciximab, eptifibatide y tirofibán.
- La integrina GPα2β1, (también conocida como GPIa/IIa), es un receptor que está presente en la membrana plaquetaria entre 1500 y 4000 copias. Esta glicoproteína cuando se activa liga a diferentes tipos de colágeno, y en la formación del trombo se ha visto que potencia la acción de adhesión plaquetaria ejercida por la GPIb-IX-VI y la GPVI.
En la superficie plaquetaria también están presentes otras integrinas, como la GPα2β1 y la GPα6β1, capaces de ligar vitronectina y fibrinógeno la primera, y laminina la segunda. El papel de ambas integrinas en el mecanismo de adhesión plaquetaria no parece ser muy relevante.
La ausencia del complejo GPIIb/IIIa clínicamente se expresa por un cuadro hemorrágico grave, que es conocido como tromboastenia de Glazmann

HEMOSTASIA
• Familia de receptores de inmunoglobulinas y otros receptores adhesivos: GPIV y PECAM-1
La glicoproteína VI (GPVI) pertenece a la superfamilia de las inmunoglobulinas y es el prin-cipal receptor del colágeno presente en la superficie plaquetaria. La deficiencia de GPVI conlleva la existencia de un cuadro hemorrágico, aunque no muy intenso. Estudios in vivo como in vitro han mostrado que la GPVI es un receptor crucial para la formación del trom-bo. El hecho de que la deficiencia de GPVI sea extremadamente rara justifica el poco avance conseguido en el conocimiento detallado del papel que juega en la hemostasia humana.
La GPVI CD36 se expresa en la superficie plaquetaria 10 000 a 25 000 copias, y también está presente en células mononucleares de sangre periférica, en macrófagos tisulares y células endoteliales. Entre otras funciones se le conoce como receptor de la trombospondina-1, y también interacciona con lipoproteínas de baja densidad y otros lípidos. Su papel funcional se circunscribe en la adhesión plaquetaria y también juega un papel en la generación del trombo, especialmente en situaciones donde existe dislipemia y estrés oxidativo.
Las moléculas de citoadhesión endotelial plaquetaria o receptores PECAM-1 participan ac-tivamente en la interacción en la adhesión celular plaqueta-endotelio. Adicionalmente hay datos experimentales que confirman que también PECAM-1 puede actuar como un regulador negativo de la agregación plaquetaria.
La deficiencia de GPVI conlleva la existencia de un cuadro hemorrágico, aunque no muy intenso

HEMOSTASIA
• Receptores plaquetarios para agonistas solubles (ligandos solubles): ligando autocrinos
La activación plaquetaria puede inducirse por mediadores plasmáticos que se generan de diferentes matrices proteicas extracelulares. Esos mediadores, con propiedad activadora, tienen una actividad transitoria, y la mayor parte se degradan o inactivan rápidamente. Ejemplos de estas sustancias son los nucleótidos ADP y ATP (que se liberan al plasma de los gránulos densos plaquetarios), la serotonina, tromboxano A2 y trombina, que se genera en la superficie plaquetaria. El TxA2 es producido por la vía de la ciclooxigenasa plaquetaria (trom-boxano-sintetasa) tras el inicio de la activación plaquetaria.
Todos los agonistas solubles activan las plaquetas a través de la vía de proteínas G (GPCR).
• P2Y12: receptor del ADP que constituye una diana de primer orden en la terapia antiagregan-te en patología cardiovascular. Tiene un mecanismo de inhibición del receptor; un ejemplo son los fármacos: clopidogrel, prasugrel y ticagrelor que tienen una clara función inhibito-ria en la formación del trombo.
• P2Y1: otro receptor del ADP que se acopla a la vía de proteína Gq, y juega un papel predomi-nante en la respuesta inicial plaquetaria, a diferencia de P2Y12 que tiene su mayor actividad en fases más tardías.
Por otra parte, las plaquetas humanas tienen dos receptores para la trombina, el PAR-1 y el PAR-2. Su estimulación, especialmente de PAR-1, da lugar a una inducción muy potente de activación plaquetaria, en gran medida vehiculizada por la vía de la proteína G4. Reciente-mente se ha abierto un nuevo campo de búsqueda de agentes de inhibición de la activación plaquetaria utilizando antagonistas de PAR-1.
La plaqueta cuenta con un receptor para el tromboxano A2, que es inhibido por la ASS, y otro también para la serotonina, denominado receptor 5-HT2A, que también se acopla a la vía de proteína Gq.

HEMOSTASIA
• Proteínas adhesivas
En numerosas ocasiones la formación del trombo se genera por la interacción plaquetaria con un vaso dañado. En esa interacción se desencadenan múltiples reacciones de forma concomi-tante, y a veces mediadas por un mismo estímulo que sigue distintas direcciones. Por una parte la plaqueta inicia su reacción con el fenómeno de adhesión que conlleva su activación inmedia-ta, que a su vez se potencia por la presencia de agonistas solubles. Las sustancias activadoras provienen en buena medida de las propias proteínas secretadas por la plaqueta, así como por enzimas activadas generadas en la activación del sistema de la coagulación, especialmente la trombina y otros factores de coagulación activados. Todo ello ocasiona el ambiente adecuado para generar la formación del trombo que, en definitiva, es el producto de adhesión/agrega-ción plaquetaria a la pared vascular, generación de actividad procoagulante, y como paso final la generación de fibrina (Figura 7) que constituirá la “cimentación” del trombo.
La agregación plaquetaria es un elemento importante que contribuye de forma decisiva a la gene-ración de trombina in situ lo que da lugar a la formación del trombo. Los agregados plaquetarios se encuentran atrapados por redes de fibrina.
El trombo es el producto de adhesión/agregación plaquetaria a la pared vascular, la generación de fibrina que constituirá la “cimentación” del mismo
FIGURA 7. COÁGULO DE PLAQUETAS-FIBRINA

HEMOSTASIA
Como ya indicamos previamente el FvW es una proteína con un especial protagonismo en el mecanismo de la formación del trombo. El FvW es una proteína constituida por múltiples subunidades, y cada subunidad contiene 2050 aminoácidos. Las subunidades están enlazadas por puentes disulfuro y dan lugar a grandes multímeros con una elevada masa molecular. El FvW que circula en una forma globular, y en situaciones de alta turbulencia adopta una disposición filamentosa o lineal adquiriendo una gran afinidad para ligarse al complejo GPIb-IX-V. Las grandes formas filamentosas tienen una actividad hemostáticamente mucho más activa que las globulares, puesto que tienen posibilidad, a través de diferentes dominios de su molécula, de interaccionar con plaquetas, endotelio y colágeno subendotelial. Deficiencias cuantitativas o cualitativas del FvW dan lugar a los diferentes tipos del trastorno hemorrágico conocido como enfermedad de von Willebrand.
Deficiencias cuantitativas o cualitativas del FvW dan lugar a los diferentes tipos del trastorno hemorrágico conocido como enfermedad de von Willebrand
En el plasma contamos con un mecanismo de seguridad para que no exista una hiperacti-vidad de las formas de alto peso molecular del FvW, se trata de la proteasa ADAMTS-13. Esa proteasa, de origen hepático, rompe los grandes multímeros del FvW en formas más pe-queñas con reducida actividad hemostática. Modificaciones de los niveles circulantes plas-máticos de ADAMTS-13 tienen su expresión clínica, pues un incremento de la proteasa puede interferir la adhesión plaqueta-FvW, y conlleva un mayor riesgo hemorrágico, mientras que una clara disminución de ADAMTS-13, que acontece en diferentes microangiopatías, incre-menta la interacción con plaquetas y por tanto un mayor riesgo oclusivo.
El colágeno fibrilar tipo I y IV que forman parte de la matriz subendotelial son potentes activadores plaquetarios interaccionando con los receptores plaquetarios GPVI y GPα2β1, fa-voreciendo la generación y crecimiento del trombo.
Una de las proteínas con mayor presencia cuantitativa en el plasma es el fibrinógeno, y también está presente en los gránulos alfa plaquetarios y en megacariocitos. El fibrinógeno participa de forma importante en la agregación plaquetaria estableciendo puentes inter-plaquetarios al ligarse a la integrina GPIIb/IIIa de plaquetas cercanas. La fibronectina y la vitronectina también son ligandos de las integrinas plaquetarias y juegan un papel en la acti-vación plaquetaria. Clínicamente el defecto más relevante de estas proteínas adhesivas es la deficiencia de fibrinógeno, debido especialmente a la incapacidad de poderse generar fibrina, aspecto crucial para la formación de un trombo estable.
Otras dos proteínas con propiedades adhesivas y que tienen un papel bien definido en el diálogo plaqueta-pared vascular son la trombospondina-1 y la laminina.

HEMOSTASIA
• La trombospondina-1 se libera de los gránulos alfa y se liga a la GPIV, contribuyendo a la acti-vación plaquetaria. Adicionalmente, la trombospondina-1 participa en el control del tamaño de los multímeros de FvW, y tiene otras funciones vinculadas a la cicatrización de heridas y en el proceso de angiogénesis.
• La laminina, que tiene lugar su síntesis en la célula endotelial y se deposita en la matriz extrace-lular y membrana basal, queda expuesta a plaquetas tras la lesión vascular. Cuando esto sucede, la laminina interacciona con las plaquetas a través de la α6β1 y facilita la relación con la GPVI.
En este sentido, debemos considerar que la aplicación de reciente metodología de proteó-mica, metabolómica o genómica están ayudando a conocer mejor la interacción plaque-ta-plaqueta, plaqueta-endotelio y en definitiva la formación del trombo. De igual manera, se está produciendo un notable avance en el conocimiento de detalles de la relación plaquetas y endotelio con monocitos, y con proteínas del sistema de la coagulación. Puede servir de ejemplo el importante papel que se le va confiriendo en estas relaciones al factor tisular (FT), liberado por los monocitos y circulante en plasma con las micropartículas, que son captura-das por el trombo en formación interaccionando con la selectina P, que se expresa en super-ficie de plaquetas activadas, y con el PSGL-1 presente en las propias micropartículas.
En los últimos años va tomando protagonismo el papel del material extracelular nuclear expulsado de neutrófilos, y presumiblemente de otras células, (NETs, Neutrophils Extracellu-lar Traps) como mecanismo de iniciación y progresión del crecimiento del trombo7. El material liberado, ADN y otras proteínas como histonas y serín-proteasas, han mostrado su capacidad de activar el sistema de coagulación y las plaquetas. Estos hallazgos no solamente abren nue-vas vías de estudio de los mecanismos de generación del trombo, sino que también se puede plantear una nueva vía sugerente de una potencial profilaxis y tratamiento de la trombosis.

HEMOSTASIA
En este apartado nos hemos centrado específicamente en los aspectos que, a nuestro en-tender, son los más relevantes en los fundamentos y bases fisiológicas de la conocida, durante años, como “hemostasia primaria”, si bien sería mejor definirla como el “compartimento celu-lar” del sistema hemostático, ya que el fenómeno de la hemostasia y generación de trombo no conoce de apartados independientes, sino concomitantes y sinérgicos.
Las reacciones “fisiológicas” están a su vez moduladas y aceleradas por circunstancias pa-tológicas muy relevantes, como puede ser la lesión aterosclerótica y la ruptura de placa.
La lesión aterosclerótica conlleva una combinación de daño endotelial, depósito extenso de material lipídico en la íntima, activación inmune/inflamatoria mantenida, proliferación de célu-las de músculo liso vascular, remodelamiento de la matriz extravascular y todo ello asociado con las posibles alteraciones reológicas en el flujo vascular que puede condicionar la limitación de la permeabilidad vascular. Obviamente, aunque no es objetivo de este capítulo abordar este importante asunto, debemos tener presente todos estos hechos pues tienen una alta influencia en el funcionamiento del sistema hemostático8.
El hallazgo de todas las múltiples y complejas interacciones que modulan el diálogo de pla-quetas con el endotelio, entre las mismas plaquetas y con otros elementos celulares circulan-tes, están ayudando de forma notable a entender la patogénesis de los trastornos trombóticos y, de igual forma, a plantear una terapia antitrombótica más específica. Hemos comentado un buen listado de moléculas que participan activamente en la formación del trombo, pero no cabe duda que todavía quedan actores por identificar.

HEMOSTASIA
PRUEBAS FUNCIONALES PLAQUETARIAS
Podemos avanzar que las pruebas funcionales plaquetarias durante muchos años han ido más orientadas como herramienta de diagnóstico para trombopatías hereditarias o adquiri-das y para la identificación del riesgo de sangrado, que para discriminar el riesgo trombótico9. En este apartado veremos los ensayos tradicionalmente utilizados (Figura 8 A y B), separados en tres apartados: métodos globales, otros más específicos y finalmente los aplicados recien-temente como la tromboelastografía y pruebas similares.
Pruebas globales
Una prueba clásica utilizada para evaluar globalmente el buen funcionamiento plaquetario ha sido el tiempo de sangrado o tiempo de hemorragia (TH), pero es bien conocido que tiene importantes limitaciones. De hecho se considera una prueba pobremente reproducible, y no predice el riesgo de sangrado quirúrgico. Es una prueba que ya se encuentra en claro retro-ceso de utilización.
El sistema PFA-100 (DadeBehring Inc., Deerfield, IL, EE.UU.) intenta simular lo que sucede en hemostasia primaria a alto flujo (Figura 8C). Pequeñas cantidades de sangre citratada se aspiran a un flujo de 5000-6000 s-1 a través de un capilar. La sangre llega a una membrana con una apertura central de 150 μm recubierta con colágeno-epinefrina o con colágeno-ADP. La adhesión y agregación plaquetaria causan la oclusión de la apertura. Los resultados se informan como tiempos de oclusión en segundos. Actualmente, se considera que este ensayo carece de la sensibilidad y especificidad requeridas para su uso rutinario como test de criba-do. Tampoco tiene un papel definitivo ni establecido en la evaluación del riesgo de sangrado en relación con la terapia antiplaquetaria, ya que la aspirina puede prolongar el tiempo de oclusión con colágeno-epinefrina, pero el ensayo es insensible a las tienopiridinas.

HEMOSTASIA
Pruebas específicas
El estudio de la agregación plaquetaria (Figura 8D) es el método de referencia y el más em-pleado en la identificación y diagnóstico de la disfunción plaquetaria. El tipo de anticoagulante utilizado en la extracción de la muestra, los agonistas y las concentraciones utilizadas, la con-centración de plaquetas y el tipo de agregómetro son variables que tienen influencia sobre la prueba. Por otra parte, también puede cambiar su interpretación el parámetro resultado elegi-do en la prueba: porcentaje de agregación máxima, tiempo de inicio de agregación o lag-phase, y la pendiente o velocidad de agregación. En definitiva, pese al tiempo transcurrido desde el inicio de la aplicación de la agregación plaquetaria, siguen existiendo bastante cuestiones rela-cionadas con su estandarización. Si bien se consiguen parámetros reproducibles y útiles para el diagnóstico de trombopatías hereditarias, su utilidad es más que cuestionable para la detec-ción de hiperactivación plaquetaria, y en definitiva para detectar riesgo trombótico.
En un intento de solventar esos problemas, se han desarrollado métodos de “point of care”, más estandarizados y sencillos (Figura 8E). Veamos algunos de ellos:
• El ensayo rápido de funcionalidad plaquetaria (RPFA; VerifyNowAccumetrics, San Diego, CA) es un test que valora la agregación en sangre total de pequeñas esferas de poliestireno recubiertas de fibrinógeno en respuesta a un agonista, TRAP o ADP. Se comercializa como método de detección de resistencia a la aspirina y al clopidogrel, bien correlacionado con los ensayos de agregación y es sensible en la determinación de los efectos del bloqueo de GPIIb/IIIa, pero no puede emplearse para valorar otros aspectos de la hemostasia primaria. En definitiva, aunque es un test ampliamente probado no existe un consenso generalizado para su uso en las situaciones previamente comentadas.
• Platelet Works (Helena Laboratories, Beaumont, TX) también es un ensayo que analiza la agregación plaquetaria en sangre total, comparando los recuentos plaquetarios antes y tras la agregación con ADP o colágeno. También parece existir una buena correlación entre Platelet Works y la agregación convencional. Al igual que otras pruebas ya comentadas, se ha empleado para la detección de resistencia a la terapia antiplaquetaria, pero no ha podido ser validado convenientemente.
• El ensayo ImpactCone and Plate(let) Analyzer (CPA) (DiaMed, Israel) emplea sangre total que se expone a un flujo uniforme en una cubeta sobre la que gira un cono a gran velocidad, analizándose la adhesión plaquetaria a la superficie de la cubeta y la altura de los trombos. Este ensayo es sencillo de usar, y emplea condiciones de flujo fisiológicamente relevantes, siendo capaz de monitorizar el antagonismo de GPIIb/IIIa, al igual que aspirina y clopi-dogrel. Sin embargo, son necesarios estudios adicionales que determinen su papel en el seguimiento de la terapia antiplaquetaria.

HEMOSTASIA
A) Recuento y morfología plaquetaria; B) Estudio del tiempo de hemorragia (hoy ya en desuso por su falta de reproducibilidad y aportación clínica); C) Sistema PFA-100 intenta simular lo que sucede en hemostasia primaria a alto flujo; D) Agregación plaquetaria utilizando diferentes agonistas (ADP, epinefrina, colágeno, trombina, tromboxano, etc.); E) Citometría de flujo que aporta información de la presencia de receptores específicos en la membrana plaquetaria.
A
C D
E
B
FIGURA 8.
LAS HERRAMIENTAS CLÁSICAS PARA EVALUAR LA NORMALIDAD DE LA “HEMOSTASIA PRIMARIA”

HEMOSTASIA
Tromboelastografía y similares
La tromboelastografía ya fue utilizada hace varias décadas y cuantifica en sangre total, la fuerza y estabilidad de un trombo formado por la interacción de plaquetas, factores de coa-gulación, inhibidores y proteínas fibrinolíticas. Analiza tanto propiedades bioquímicas (for-mación del trombo y disolución), como mecánicas. En determinados lugares, especialmente en el ambiente de anestesia y quirúrgico, se aplica la tromboelastografía para predicción del sangrado perioperatorio durante el bypass cardiopulmonar, monitorización de la hiper- o hipo-coagulabilidad tras cirugía general y la monitorización de agentes farmacológicos que afecten a la coagulación (heparina, HBPM, FVIIa), a las plaquetas (inhibidores de GPIIb/IIIa, aspirina) o a la fibrinólisis (aprotinina, estreptoquinasa). Hay varias modalidades de aparatos en tromboe-lastografía.
El tromboelastógrafo TEG (Haemoscope) mide la fuerza elástica (rigidez) de un trombo que se forma en el interior de un contenedor oscilante, mediante cambios en la fuerza de torsión que se trasmite a un sensor.
El ROTEG® (Pentapharm) es similar al anterior, excepto que un sistema óptico mide las os-cilaciones de un sensor en un contenedor estático, y la formación del coágulo se acelera por el uso de ácido elágico o factor tisular.
El Sonoclot® (Sienco) mide las propiedades viscoelásticas de la formación del trombo y su retracción mediante impedancia a un sensor en vibración.
El PAS (Platelet Analysis System, Hemodyne) mide la fuerza de contracción plaquetaria (di-nas) de un trombo formado entre dos superficies, estando la superior conectada a un trans-ductor.
Habiendo revisado previamente los diferentes procedimientos de estudio del funcionalismo plaquetario y su papel en la generación del trombo, en términos generales, podemos indicar que aún no disponemos de buenas herramientas que nos ayuden a establecer el riesgo trombótico o controlar con precisión el efecto de los fármacos antiagregantes.
A continuación abordaremos el estudio del componente plasmático de la coagulación, que acompaña a la activación del componente celular y da lugar a que se lleven adelante las fun-ciones que tiene encomendadas el sistema hemostático.

HEMOSTASIA
COMPONENTE PLASMÁTICO DE LA COAGULACIÓN
Tal como hemos indicado, el sistema hemostático tiene tres funciones fundamentales: ce-rrar un vaso dañado, mantener la sangre en un estado fluido y retirar los coágulos una vez restaurada la integridad vascular. En los años sesenta se describe un modelo enzimático para explicar el proceso de la coagulación sanguínea, que recibió el nombre de cascada de la coagulación sanguínea, el cual estaba compuesto por una vía denominada intrínseca, otra extrínseca y una vía final común para ambos caminos de activación. Una amplia mayoría de los factores de la coagulación son proenzimas que se convierten en enzimas activas gracias a su interacción con otros factores previamente activados, dando lugar a una reacción en cade-na. El objeto final de esta cascada enzimática es la activación de la protrombina (factor II) y su paso a trombina, que ocasionará el paso de fibrinógeno (proteína soluble) a fibrina10.
El estudio de la coagulación in vitro definió dos vías de activación de la coagulación, la vía extrínseca y la intrínseca.
• La vía extrínseca se activa al añadir al plasma un extracto de cerebro animal (tromboplas-tina tisular).
• Por otra parte, se observó que la sangre recogida en un tubo de cristal se coagulaba por sim-ple contacto con su pared, y a ese proceso enzimático se denominó vía intrínseca. La fase de activación inicial, en este caso con el vidrio o con otras superficies, se conoce como fase de contacto de la coagulación sanguínea. Sin embargo, hoy sabemos que las llamadas vías intrínseca y extrínseca no funcionan de forma independiente in vivo, sino que sus meca-nismos están íntimamente imbricados y podemos indicar que durante años ha sido utilizado este esquema como una forma académica para describir un complejo sistema como es el de la coagulación sanguínea. Hay que indicar, que pese a que estos esquemas se consideran lejanas de la realidad fisiológica, se siguen manteniendo por su utilidad para la exploración in vitro del sistema de la coagulación sanguínea.
Como hemos visto antes, una vez que se daña el vaso, las plaquetas sufren el proceso de adhe-sión, activación y agregación. A su vez, el factor tisular subendotelial expuesto al fluido sanguí-neo genera trazas de trombina, la cual actuará activando diferentes factores de la coagulación. La activación del sistema de la coagulación estará facilitado también por las propias plaquetas, y se irá haciendo de forma exponencial, hasta conseguir un trombo estable de fibrina10.
El objeto final de esta cascada enzimática es la activación de la protrombina y su paso a trombina, que ocasionará el paso de fibrinógeno (proteína soluble) a fibrina10

HEMOSTASIA
Factores de la coagulación
El sistema de la coagulación está formado por una serie de proteínas plasmáticas deno-minadas factores de la coagulación.
La mayoría son proteínas que circulan en la sangre como zimógenos inactivos, los cuales se activan y transforman en enzimas con actividad serín-proteasa, es decir, con potencial pro-teolítico. Otros factores no tienen actividad proteolítica y actúan como cofactores, facilitando la eficacia de la reacción enzimática.
Nomenclatura de los factores de coagulación
• La designación de los factores de coagulación por un número romano indica el orden de descubrimiento de la proteína, no debiéndolo confundir con su cronología de participa-ción en la secuencia de la reacción enzimática. Por ejemplo, el factor III corresponde al FT y el factor IV se identifica con iones de calcio, esenciales para la progresión de la cas-cada del sistema de coagulación. Aunque, el factor VI entra en juego antes, esta proteína fue confirmada tiempo después.
• Cuando queremos representar la forma activada de un factor, a la derecha del número romano se coloca una “a”, por ejemplo el factor X activo lo identificamos como FXa.
• La mayor parte de los factores de coagulación han sido descubiertos al caracterizar su responsabilidad funcional de una diátesis hemorrágica congénita concreta.
A continuación enumeramos los factores con actividad proteolítica y aquellos con función favorecedora (cofactores) de la reacción enzimática del sistema de la coagulación sanguínea.
• Enzimas proteolíticas (serín-proteasas): son las proteínas que circulan como precursores enzimáticos (zimógenos) y se transforman en enzimas activas durante la coagulación. A este grupo pertenecen la calicreína y los factores II, VII, IX, X, XI y XII, así como la proteína C, si bien esta última es un inhibidor o anticoagulante natural como veremos más adelante.
• Cofactores: son necesarios para formar complejos con las enzimas y así fijarlas en la su-perficie celular, así como acelerar la reacción enzimática. En este grupo se incluyen: el cininógeno de alto peso molecular, el FT, los fosfolípidos, el factor VIII y el V y la proteína S, también esta última con función inhibidora.
• Otras proteínas. Hay otros dos factores que intervienen directamente en las formación y es-tabilización del depósito de fibrina, como son el fibrinógeno y el factor XIII.

HEMOSTASIA
La mayor parte de los factores se sintetizan en el hígado, y algunos necesitan la presencia de la vitamina K para su normal constitución. Éstos son llamados factores vitamina K depen-dientes, y corresponden a los factores II, VII, IX y el X, y también lo son las proteínas inhibidoras C y S. La misión de la vitamina K es intervenir en la adición de grupos carboxilo a los extre-mos aminoterminal del ácido glutámico presente en estas proteínas, para originar residuos gamma-carboxiglutámicos (Gla). Los residuos Gla son fundamentales para que esas proteínas se unan a los iones calcio y a los fosfolípidos,y de esta forma permitir que se lleve adelante el proceso coagulativo13. Ese proceso enzimático es la base de la terapia con antivitaminas K, pues las cumarinas (warfarina o el acenocumarol) inhiben la función de la vitamina K epóxido reduc-tasa (Figura 9).
La vitamina K participa en el proceso de gamma-carboxilación de los factores de coagulación. Los fár-macos antivitamina K o cumarínicos actúan a nivel de la recuperación de la vitamina K activa, desde la forma vitamina K epóxido, para el siguiente proceso de carboxilación.
Residuo Glaa. glutámico
CUMARINICOS
FACTORES II, VII, IX Y X, PROTEÍNAS C Y S
Gamma-glutamil-carboxilasa
Epóxido-reductasasQuinona-reductasas
VITAMINA K ACTIVA
QUINONA
EPÓXIDO
Residuo Glaa. carboxiglutámico
FIGURA 9.
METABOLISMO DE LA VITAMINA K

HEMOSTASIA
Sistema de la coagulación
Como veremos a continuación, el inicio de la cascada de la coagulación por el FT unido al factor VII es esencial para el mantenimiento de la hemostasia fisiológica, sin embargo la ex-presión inadecuada de FT (por ejemplo en una placa aterosclerótica) puede provocar trombosis. Por el contrario, la fase de contacto de la vía intrínseca, en concreto el papel de los factores XI y XII parecen tener un papel menor en mantener una hemostasia normal, pero cada vez hay más datos que apuntan que pueden modular la génesis del mecanismo trombótico10-12.
Una visión más reciente del sistema de la coagulación sanguínea lo divide en tres fases o periodos: fase de iniciación, periodo de amplificación y fase de propagación, con una participa-ción mixta de elementos celulares, especialmente de las plaquetas y el endotelio, y proteínas plasmáticas. Dado que participan conjuntamente la activación del sistema enzimático y la inte-racción con elementos celulares, especialmente las plaquetas, se ha reconocido a este sistema como modelo celular de la coagulación sanguínea10.
Dado que participan conjuntamente la activación del sistema enzimático y la interacción con elementos celulares, especialmente las plaquetas, se ha reconocido a este sistema como modelo celular de la coagulación sanguínea10

HEMOSTASIA
• Fase de iniciación
Sería en buena medida equivalente a la vía extrínseca clásica. Se inicia cuando el vaso se rompe y las células subendoteliales, como las células musculares lisas o los fibroblastos, se exponen al torrente sanguíneo. Estas células expresan FT, el cual es clave para el inicio de la coagulación12. En condiciones normales el endotelio y los monocitos no expresan FT, pero pueden hacerlo en respuesta al daño endotelial o a la presencia de estímulos inflamatorios como toxinas, citoquinas, etc. El FT se une al factor VII provocando su proteólisis y activación, transformándose en factor VII activado (FVIIa). El complejo FT/FVIIa ejerce una acción proteo-lítica sobre los factores IX y X transformándolos en proteínas activas (FIXa y FXa). En la super-ficie celular el FXa se une al cofactor FVa y forma el complejo protrombinasa para catalizar el paso de protrombina (factor II) en trombina (Figura 10)12.
Fase de iniciación del proceso de coagulación. Ante un estímulo, FT se une al factor VII y lo activa, FVIIa provoca la activación secuencial de FXa y FIXa. FXa unido a FVa forman el complejo protombinasa que cataliza la síntesis de trombina.
ESTÍMULO
PROTROMBINA
FT
Xa
IX
VIIaFT
TROMBINA
Cel. con FT
VIIa
IXa
X
FIGURA 10. COMIENZO Y PRODUCCIÓN INICIAL DE TROMBINA

HEMOSTASIA
• Fase de amplificación
Las trazas de trombina formadas en la fase anterior, van a ser capaces de activar las pla-quetas adheridas y convertir el factor V en FVa así como el factor VIII en FVIIIa (Figura 11). El primero va a favorecer la activación del complejo protrombinasa, mientras que el FVIIIa actúa como cofactor para que el FIXa induzca la generación de más cantidad de FXa. Además, la trombina es capaz de convertir el factor XI en FXIa.
Fase de amplificación. La trombina producida en la fase anterior, es capaz de activar el resto de factores de la coagulación e incluso contribuir a una mayor activación plaquetaria, por activación del factor XI.
PLAQUETA
XIa
IXaVaVIIIa
IXV
FvW-VIII
ESTÍMULO
PROTROMBINA
FT
Xa
IX
VIIaFT
TROMBINA
Cel. con FT
VIIa
IXa
X
FIGURA 11. INICIO Y AMPLIFICACIÓN

HEMOSTASIA
• Fase de propagación
Esta fase ocurre en superficies con fosfolípidos procoagulantes expuestos, como por ejemplo las membranas de plaquetas activadas (Figura 12). El FXIa convierte el factor IX en FIXa el cual se une al FVIIIa, ambos junto a los fosfolípidos de la superficie celular forman el complejo tenasa, que ca-taliza el paso de factor X en FXa, el cual unido al FVa producen suficiente cantidad de trombina para formar grandes cantidades de fibrina. El último paso es la activación del factor XIII por la trombina. El FXIIIa cataliza la formación de enlaces covalentes entre las cadenas de fibrina dando lugar a un trombo de fibrina polimerizado y estable.
El fibrinógeno es una proteína dimérica formada por tres pares de cadenas polipéptídicas: alfa, beta y gamma, que están unidas entre sí por puentes disulfuro. La trombina actúa sobre el ex-tremo amino terminal de las cadenas alfa y beta rompiéndolas y liberando dos péptidos de bajo peso molecular, denominados fibrinopéptido A y B. Así la molécula de fibrinógeno se trasforma en monómeros de fibrina que polimerizan de forma instantánea. Sobre el polímero de fibrina actúa el FXIIIa, catalizando la formación de enlaces amida entre residuos de glutamina y lisina de las moléculas de fibrina para darles estabilidad.
Por tanto la fibrino-formación tiene tres etapas:
1) acción de la trombina sobre el fibrinógeno, generando los fibrinopéptidos A y B y monó-meros de fibrina
2) polimerización espontánea de los monómeros de fibrina
3) estabilización del coágulo de fibrina gracias a la formación de enlaces covalentes entre los monómeros de fibrina por acción de una transglutaminasa, el factor XIIIa

HEMOSTASIA
Fase de propagación. Esta ocurre en superficies que contienen fosfolípidos procoagulantes, como por ejemplo las plaquetas activadas. El FXIa convierte el factor IX en FIXa el cual se une al FVIIIa, ambos junto a los fosfolípidos de la superficie celular forman el complejo tenasa, catalizando el paso de factor X en FXa, el cual unido al FVa producen suficiente cantidad de trombina para formar grandes cantida-des de fibrina.
PLAQUETAACTIVADAPROTROMBINA
FIBRINÓGENO
TROMBINA
FIBRINA
FIBRINAESTABLE
Va IXa VIIIaXa
XIIIa
X
XIII
FIGURA 12.
PROPAGACIÓN

HEMOSTASIA
• Fase de contacto
Según el modelo de coagulación vigente (Figura 13), la activación del factor XII y XI (fase de contacto) tan solo actuaría en la fase de amplificación11.
Estudios con ratones han demostrado que a la vez que se activa el factor VII también lo hace la fase de contacto. Estos modelos animales han demostrado que mientras la fase de contac-to no juega ningún papel en la hemostasia, los pacientes o animales con deficiencia de FXII no sangran, y sin embargo si desarrollan trombosis.
Tres son los activadores que se han descrito capaces activar la fase de contacto: el coláge-no, los polifosfatos presentes en la superficie plaquetar y el ADN plasmático y NETS7. El FXIIa activaría a la precalicreína en calicreína, la cual unida al cininógeno de alto peso molecular, sería capaz de activar al factor XI11.
La activación del FXII y FXI por todos esos elementos parece justificar el papel que juega la vía de contacto promoviendo trombosis, y como su papel en la hemostasia parece ser mínimo, se convierten en una atractiva diana terapéutica. Recientemente se ha publicado un ensayo donde un oligonucleótido anti-factor XI es efectivo reduciendo el riesgo de tromboem-bolismo venoso tras cirugía ortopédica sin incrementar el riesgo hemorrágico14.
Por otro lado,la calicreína es capaz de inducir la liberación de bradiquinina que induce a la célula endotelial a liberar activador tisular del plasminógeno (que como veremos a continua-ción participa en el proceso fibrinolítico), formar superóxido y generar óxido nítrico con efecto vasodilatador.

HEMOSTASIA
Se muestra dónde actúan cada una de las proteínas anticoagulantes en relación con las distintas fases del proceso coagulatitvo.
IX
X
II
Xa
IIa
IXa
FTVIIa
INICIACIÓN
XIIX
VIII
Va
XIa
IIa
IXa
VIIIa
Va
Activaciónplaquetaria
AMPLIFICACIÓN
X
II
Xa
IIa
IXaVIIIa
PROPAGACIÓN
XaVa
TFPI
PCa-PS
AT
Bloquea la iniciación y el principio de amplificación
Bloquea la amplificación
Bloquea la propagación
INHIBICIÓN
FIGURA 13. TRES FASES Y TRES SISTEMAS PRINCIPALES DE INHIBICIÓN

HEMOSTASIA
REGULACIÓN DE LA COAGULACIÓN
Mecanismos reguladores de la coagulación
Existen dos mecanismos que regulan el proceso de la coagulación10:
• Los inhibidores de las serín-proteasas: inhiben los factores activados. Estos son: la anti-trombina, el cofactor II de la heparina, el inhibidor de la vía del factor tisular (TFPI), el inhi-bidor de la proteína C activada, el inhibidor de C1-esterasa o α1-antitripsina.
- TFPI: proteasa inhibidora tipo Kunitz que juega un papel fundamental al inicio del proce-so de la coagulación12. Circula en plasma unido a lipoproteínas y un 20% de forma libre. Ante la agresión a un vaso se libera tanto de las plaquetas como del endotelio. La hepari-na también incrementa su concentración en plasma. Actúa de dos maneras, inhibiendo directamente el FXa o actuando sobre el complejo FT/FVIIa/FXa. Recientemente se ha descrito que la proteína S actúa como cofactor del TFPI en su inhibición del FXa.
- Antitrombina: principal inhibidor de la trombina y del FXa, aunque también es capaz de inhibir los factores IXa, XIa, XIIa, calicreína y plasmina, así como al FVIIa unido al FT. Su ac-tividad se incrementa mil veces en presencia de heparina. In vivo, esta función la cumplen los proteoglicanos tipo heparina (dermatán sulfato, heparán sulfato y condroitín sulfato) presentes en el endotelio vascular y necesarios para el reconocimiento de la antitrom-bina. La antitrombina presenta una estructura tridimensional donde el centro reactivo se encuentra parcialmente integrado en la molécula circulante. Además, se identifica una zona de unión a un pentasacárido presente en la heparina y a glucosaminoglicanos vasculares. Precisamente, la unión de la heparina a la antitrombina provoca un cam-bio conformacional en la serpina, que libera completamente el centro reactivo. Este cambio tiene importantes consecuencias funcionales, dado que la capacidad inhibitoria de esta conformación es 1000 veces superior a la de la molécula sin heparina. Pero no es este el único cambio conformacional que experimenta la antitrombina. La unión a las proteasas que inhibe, conduce a la ruptura del centro reactivo de la antitrombina. Ese es un proceso clave en el funcionamiento inhibitorio de la serpina que inactiva a la pro-teasa, y forma el casi indisociable complejo proteasa-serpina.

HEMOSTASIA
• Los reguladores de los cofactores, como es el sistema de la proteína C (proteína C, S y trombomodulina) que regula al FVa y FVIIIa.
- Sistema de la proteína C: desempeña un papel esencial en la regulación del proceso de la coagulación sanguínea. La activación de la proteína C conduce a la inactivación de los cofactores de la coagulación activados Va y VIIIa, que son esenciales para mantener la formación de trombina.
El sistema de la proteína C se activa cuando la trombina, se une a la trombomodulina sobre la superficie endotelial. La activación de la proteína C por el complejo trombi-na-trombomodulina es potenciada mediante la unión a su receptor específico, el re-ceptor endotelial de la proteína C (EPCR). La proteína C activada (PCa) unida a la forma soluble del EPCR no tiene propiedades anticoagulantes, lo que sugiere que la PCa debe disociarse del EPCR para poder exhibir su función anticoagulante. Una vez tiene lugar esta disociación, la PCa se une a la proteína S sobre la superficie de la célula endote-lial o de la membrana plaquetaria, e inactiva proteolíticamente a los cofactores de la coagulación Va y VIIIa.

HEMOSTASIA
Sistema fibrinolítico
El sistema hemostático cuenta con un mecanismo de defensa final (Figura 14), como es la eliminación del trombo una vez constituido, después de la reparación del vaso. El principal agente encargado de eliminar el trombo es una enzima proteolítica llamada plasmina. Su precursor, el plasminógeno, se une a la fibrina y al activador tisular del plasminógeno (t-PA). Este complejo lleva a la transformación de la proenzima en su forma activa. La plasmina, en unión a la fibrina, no solo es capaz de romper la red polimérica de esta, sino que también ac-túa sobre el fibrinógeno y otros factores de la coagulación. Al actuar sobre la fibrina libera los productos de degradación de la fibrina, entre los cuales se encuentra el dímero D. El dímero D consiste en dos dominios D procedentes de dos monómeros de fibrina que han sido unidos covalentemente por el FXIIIa.
La plasmina está regulada por las células endoteliales que secretan dos serin-proteasas activadoras del plasminógeno, t-PA y el activador del plasminógeno tipo urokinasa, junto con dos inhibidores (PAI-1 y PAI-2). Además, la plasmina se inhibe por la α2-antiplasmina, la cual actúa rápidamente una vez la plasmina se activa en plasma. La α2-antiplasmina se produce en el hígado y es también secretada por las plaquetas.
Finalmente, existe una proenzima de una carboxipeptidasa-Β llamada inhibidor de la fibrinó-lisis activable por trombina o TAFI, que actúa sobre el complejo trombina-trombomodulina y actúa inhibiendo la fibrinólisis, ya que el trombo parcialmente digerido puede ser estímulo para la activación de más moléculas de plasminógeno. El FXIIIa facilita la unión de TAFI a la fi-brina, evitando que el recién formado coágulo de fibrina sea degradado prematuramente.

HEMOSTASIA
FX Protrombina
Trombina
Fibrinógeno
Agregación plaquetaria
Plasminogeno t-Pa
Plasmina
Productos de degradación de la fibrina
Factor tisular FVIIa
++
+
Complejo protrombínico
PAI-1
α -2-APTAFI
+
+ -
-
TROMBO
Fibrina
FIGURA 14. REPRESENTACIÓN ESQUEMÁTICA DEL PROCESO FIBRINOLÍTICO
FXaFVa
La cascada de reacciones del proceso fibrinolítico se encuentra regulada de forma positiva y negativa por otras moléculas.

HEMOSTASIA
EXPLORACIÓN DE LA FASE PLASMÁTICA DE LA COAGULACIÓN
Para la realización de la mayoría de las pruebas de coagulación se necesita la obtención de plas-ma citratado (citrato trisódico a una concentración 0,129M) pobre en plaquetas. La obtención de la muestra se hace en tubos precargados con citrato sódico que se deben centrifugar a una velocidad de 3000 g durante 15 minutos en frío. La muestra debe ser procesada antes de las 4 horas, si se mantiene a 4-6 ºC o antes de las 2 horas si se mantiene a temperatura ambiente.
La visión académica de la coagulación en una fase intrínseca, extrínseca y común, es útil para la exploración de los tiempos de coagulación y de los factores que de ellos dependen15.
Tiempo de protrombina
Se utiliza para evaluar la vía extrínseca de la coagulación y la vía común, lo cual implica la nor-malidad de los factores II, V, VII, X y fibrinógeno. Se obtiene tras añadir tromboplastina tisular al plas-ma, que aporta factor tisular y fosfolípidos necesarios para iniciar el proceso coagulativo junto con el calcio. Se expresa en segundos, razón normalizada o ratio (tiempo de protrombina del paciente/tiempo de protrombina control) y como actividad (ratio x 100). Es normal una ratio de 0,8-1,2.
Una de las aplicaciones más importantes del tiempo de protrombina es la monitorización de los fármacos anti-vitamina K. El control biológico de los fármacos anti-vitamina debe realizarse con el tiempo de protrombina, expresando su resultado mediante la razón internacional normalizada, más conocida como INR. Para estandarizar el resultado y que el grado de anticoagulación de un paciente sea homogéneo en todos los laboratorios independientemente del reactivo que se use, en 1981 la OMS relacionó todas las tromboplastinas del mercado con tres tromboplastinas estándares prepa-radas por la propia OMS para ello utilizó un índice de sensibilidad internacional (ISI).

HEMOSTASIA
• El ISI corresponde a la pendiente de la recta de regresión que relaciona el resultado obtenido con cualquier tromboplastina frente a la de referencia, que se considera
• La OMS a partir de ese momento recomendaba usar tromboplastinas con ISI<1,5.
• Actualmente se utilizan tromboplastinas recombinantes cuyo ISI es aproximadamente 1.
INR= (TP paciente/TP normal)ISI
La mayoría de los pacientes anticoagulados deben mantener el INR entre 2,0-3,0, salvo los pacien-tes de muy alto riesgo cuyo INR debe estar entre 2,5-3,5 o incluso entre 3,0 y 4,0.
El control de INR puede realizarse mediante:
- Venopunción. Obtención de una muestra de plasma citratado y realización de la prueba en un coagulómetro automatizado.
- Punción capilar. Permite la determinación inmediata del INR tras obtención de sangre total por punción o microcorte en el pulpejo del dedo en un coagulómetro portátil. Utiliza una trom-boplastina con un ISI en torno a 1.
La cuantificación de cada uno de los factores, se hace mezclando la muestra del paciente con un plasma carente del factor a estudiar. La actividad obtenida será por tanto la actividad del fac-tor presente en el plasma a estudio.

HEMOSTASIA
Tiempo de tromboplastina parcial activada
Explora la fase de contacto o vía intrínseca, y su normalidad implica valores normales de los factores XII, XI, IX, X, VIII, V, II y fibrinógeno. Se obtiene tras activar la vía de contacto con caolín, sílice micronizado o ácido elágico y añadiendo fosfolípidos y calcio (Figura 15). Se expre-sa mediante segundos y una razón normalizada. Es normal una ratio de 0,8-1,2.
El TTPA se prolonga ante fármacos con actividad anti-IIa, como es la heparina no frac-cionada. De hecho se utiliza para su monitorización, debiendo estar la ratio entre 1,5-2,5. También por tanto se alarga ante otros fármacos anti-IIa como son las hirudinas o el dabiga-trán. La determinación de los factores de la vía intrínseca, también se realiza con un plasma carente del factor a estudiar, para que así la actividad obtenida corresponda al factor presente en la muestra a estudio.
Tromboplastina (FT+Ca2+)
+Plasma paciente
ALARGAMIENTO DE TP
Déficit de factores II, V, VII ó X
• Congénitos (hace falta tan solo un 5-10% para la hemostasia)
• Alargamiento aislado: déficit de FVII
• Adquirido: déficit de vitamina K, hepatopatía.
• Paciente anticoagulado con dicumarínicos
SISTEMA EXTRÍNSECO TP
FII - FV - FVII - FX
VIIa / factor tisular
Ca2+ PL
Ca2+ PLVaX Xa X
Protrombina TROMBINA
Se obtiene tras añadir tromboplastina tisular al plasma, que aporta factor tisular y fosfolípidos nece-sarios para iniciar el proceso coagulativo junto con el calcio. Se expresa en segundos, razón norma-lizada o ratio (tiempo de protrombina del paciente/tiempo de protrombina control) y como actividad (ratio x 100). Es normal una ratio de 0,8-1,2. Se utiliza para evaluar la vía extrínseca de la coagulación y la vía común, lo cual implica la normalidad de los factores II, V, VII y X. Se resume las principales causas de su alargamiento.
FIGURA 15. REPRESENTACIÓN ESQUEMÁTICA DEL TIEMPO DE PROTROMBINA

HEMOSTASIA
Tiempo de trombina
Se obtiene al añadir trombina al plasma. Es extraordinariamente sensible a la presencia de fármacos anti-IIa. Existe una variedad del mismo, que es el tiempo de reptilase, donde la trombina es sustituida por un veneno de serpiente que es insensible a heparina, por tanto ante la presencia de esta no se alarga el tiempo (Figura 16).
Para la determinación de la actividad anticoagulante del dabigatrán, se ha creado un tiempo de trombina diluido, donde mediante una curva de calibración pueden extrapolarse los tiempos obtenidos a la concentración del fármaco.
Tiempo de ecarina
Otra prueba derivada del tiempo de trombina, es el tiempo de ecarina. Esta prueba está basada en añadir un activador obtenido de serpiente que transforma la protrombina en meizotrombi-na, un precursor lábil de trombina. Ambos compuestos son inhibidos por dabigatrán, prolon-gando el tiempo de coagulación. Existe una correlación lineal entre la prolongación del tiempo de ecarina y la concentración de dabigatrán de la muestra.
Explora la fase de contacto o vía intrínseca, y su normalidad implica valores normales de los factores XII, XI, IX y VIII. Se obtiene tras activar la vía de contacto con caolín, sílice micronizado o ácido elágico y añadiendo fosfolípidos y calcio. Se expresa mediante segundos y una razón normalizada. Es normal una ratio de 0,8-1,2.
Ca+++
Activador & fosfolipidos
+Plasma
paciente
ALARGAMIENTO DE TTPA• La causa más frecuente: MALA EXTRACCIÓN DE LA MUESTRA POR CONTAMINACIÓN
CON HEPARINA• Déficit factorial• Presencia de inhibidor. (A lúpico)• Otros:
- Leucocitosis extremas- Sindrome hiperviscosidad (Waldeström)- Antibióticos (cefalosporinas)- Poliglobulias
SISTEMA INTRÍNSECO TTPa
FVIII - FIX - FXI - FXII
Ca2 + PLVa
VIIIaIX
XI
XII
Ca2+
PK, HMWK
IXa
XIa
XIIa
Protrombina TROMBINA
X Xa X
FIGURA 16. TIEMPO DE TROMBOPLASTINA PARCIAL ACTIVADA

HEMOSTASIA
Determinación de fibrinógeno
Se realiza por el método von Clauss, que se basa en añadir un exceso de trombina a un plasma diluido, de tal forma que el tiempo de coagulación obtenido está en relación lineal con la concentración de fibrinógeno.
Determinación del dímero D
La prueba de referencia y más exacta es la determinación del dímero D mediante ELISA o enzimo-inmuno ensayo. Sin embargo, esta es una técnica larga que no permite una res-puesta rápida del laboratorio. Por tanto, se han buscado técnicas automatizadas. Desde hace años venimos utilizando técnicas basadas en aglutinación con látex. Consiste en enfrentar el plasma del paciente a micropartículas de látex unidas a un anticuerpo frente al dímero D. La unión del dímero D del plasma a su anticuerpo provoca una aglutinación de las partículas de látex y un cambio en la turbidez del plasma.
Actividad anti-Xa
Su determinación se realiza por cuantificación de actividad enzimática mediante métodos cromogénicos. En ella se aporta el factor Xa y se mide la actividad Xa residual no inhibida por el complejo heparina/antitrombina de la muestra. Es útil para el control de tratamiento con heparina de bajo peso molecular (HBPM). Esta misma técnica se emplea para el control de nuevos fármacos anti-Xa usando eso sí, calibradores específicos para cada fármaco.

HEMOSTASIA
PRUEBAS DE LABORATORIO ÚTILES PARA EL CONTROL/MONITORIZACIÓN DE FÁRMACOS ANTICOAGULANTES13,16
• Tiempo de protrombina. Explora la vía extrínseca. Ya que en esta intervienen la mayoría de factores vitamina K dependientes, es ideal para la monitorización de los fármacos anti-vi-tamina K. El resultado se expresa en forma de ratio normalizado o INR.
• TTPA. Explora la vía intrínseca. Es muy sensible a la presencia de fármacos con actividad anti-IIa. Se utiliza para monitorizar la heparina no fraccionada. Se expresa en ratio. Es útil como medida cualitativa de la presencia de dabigatrán, pero no sirve para su cuantificación. Existe una prueba equivalente al TTPA pero “a pie de cama” para evaluar de una forma muy rápida el grado de heparinización del paciente, es un tiempo de coagulación global y con-siste en añadir a sangre total un activador tipo caolín y medir el tiempo de coagulación en segundos. Es útil para quirófanos de cirugía cardiaca.
• Tiempo de trombina diluido. Es una modificación al tiempo de trombina. Útil para la medi-da del efecto anticoagulante de dabigatrán. Permite su cuantificación.
• Tiempo de ecarina. También se puede medir el efecto anticoagulante de dabigatrán me-diante el tiempo de ecarina.
• Determinación de la actividad anti-Xa. Como su nombre indica permite la cuantificación de la actividad anti-Xa y puede ser utilizado para la medida del efecto anticoagulante de HBPM, rivaroxabán, apixabán y edoxabán, en los tres últimos casos es necesario utilizar calibradores adecuados.

HEMOSTASIA
BIBLIOGRAFÍA
1. GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015; 385(9963):117-71.
2. Aird WC. Hemostasis and irreducible complexity. J ThrombHaemost. 2003;1(2):227-30.
3. Wang L, Ho B, Ding JL. Transcriptional regulation of Limulus factor C: repression of an NFB lipopolysac-charide. J BiolChem. 2003; 278(49):49428-37.
4. Vicente V. El equilibrio hemostático, un proceso complejo y dinámico. Haematologica. 2004;89; 87-98.
5. Rivera J, Lozano ML, Navarro-Núñez L, Vicente V. Platelet receptors and signaling in the dynamics of thrombus formation. Haematologica. 2009;94(5):700-11.
6. Versteeg HH, Heemskerk JWM, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev. 2013;93(1):327-58.
7. Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. 2014;123(18):2768-76.
8. Badimon L, Vilahur G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J Intern Med. 2014;276(6):618-32.
9. González-Conejero R, Rivera J, Corral J, Acuna A, Guerrero JA, Vicente V. Biological assessment of aspirin efficacy on healthy individuals: heterogeneous response or aspirin failure?. Stroke. 2005; 36(2):276-80.
10. De Caterina R, Husted S, Wallentin L, Andreotti F, Arnesen H, Bachmann F, et al. Position paper of the ESC working group on thrombosis-task force on anticoagulants in heart disease. General mechanisms of coagulation and targets of anticoagulants. Thromb Haemost. 2013;109(4):569-79.
11. Wu Y. Contact pathway of coagulation and inflammation. Thromb J. 2015;13:17.
12. Owens AP, Mackman N. Tissue factor and thrombosis. The clot stars here. Thromb Haemost. 2010;104(3):432-9.
13. De Caterina R, Husted S, Wallentin L, Andreotti F, Arnesen H, Bachmann F, et al. Position paper of the ESC working group on thrombosis-task force on anticoagulants in heart disease. Vitamin K antagonists in heart disease: Current status and perspectives. Thromb Haemost. 2013;110(6):1087-107.
14. Büller HR, Bethune C, Bhanot S, Gailani D, Monia BP, Raskob GE, et al. Factor XI antisense oligonucleoti-de for prevention of venous thrombosis. N Engl J Med. 2015;372(3):232-40.

HEMOSTASIA
15. Lippi G, Plebani M, Favaloro E. Technological Advances in the Hemostasis Laboratory. Semin Thromb Hemost. 2014;40(2): 178-85.
16. De Caterina R, Husted S, Wallentin L, Andreotti F, Arnesen H, Bachmann F, et al. Position paper of the ESC working group on thrombosis-task force on anticoagulants in heart disease. Parenteral anticoagulants in heart disease: Current status and perspectives. Thromb Haemost. 2013;109(5):769-86.

HEMOSTASIA
L a enfermedad tromboembólica venosa (ETEV) es una enfermedad compleja, multifac-torial, resultado de la interacción de diferentes factores congénitos (se trataría de una
enfermedad poligénica) y ambientales1. Se estima que hasta un 60% de la variación de la sus-ceptibilidad (heredabilidad) a la trombosis venosa es atribuible a factores genéticos2.
El término trombofilia se utiliza para describir un desorden en la hemostasia ligado a la presencia de factores genéticos o adquiridos que condiciona una predisposición para des-encadenar un episodio tromboembólico.
En el presente capítulo primero describiremos en detalle las principales alteraciones trombo-fílicas hereditarias y adquiridas y posteriormente abordaremos el papel actual e indicaciones de los estudios de trombofilia.
TRASTORNOS TROMBOFÍLICOS
1.21.2
Dr. Ramón Lecumberri
Se estima que hasta un 60% de la variación de la susceptibilidad (heredabilidad) a la trombosis venosa es atribuible a factores genéticos

HEMOSTASIA
TROMBOFILIA CONGÉNITA O HEREDITARIA
Introducción
Alrededor del 30-40% de los casos con ETEV que acuden a una puerta de urgencias pre-senta alguna alteración genética que incrementa el riesgo trombótico, como se aprecia en la Tabla 1. Esta tabla también muestra datos de frecuencia y asociación con un primer epi-sodio trombótico3. Las manifestaciones clínicas suelen ser similares en todos los casos de trombofilia congénita. No existen rasgos clínicos claramente diferenciadores (Tabla 2). Clásicamente, se ha establecido que la trombofilia hereditaria se caracteriza por trom-bosis de localización venosa, con historia familiar, habitualmente siguiendo un patrón de herencia autosómica dominante. El primer episodio trombótico puede aparecer en sujetos jóvenes y, en ocasiones, la primera manifestación tiene lugar en localizaciones anatómicas poco frecuentes (trombosis mesentérica, cerebral, etc.).
Antes de describir en detalle los principales estados de trombofilia congénita, es importante destacar:
• Aunque el 30-40% de los casos con trombosis venosa tengan alguna alteración genética protrombótica no se excluye que aquellos pacientes negativos para estas determinacio-nes no tengan algún factor de riesgo genético aún no identificado.
• La identificación de una alteración genética, aunque esté ligada a un importante riesgo trombótico, no siempre explica por sí sola el desarrollo de la oclusión vascular. Un mis-mo defecto trombofílico puede tener diferente penetrancia clínica. La identificación de una alteración trombofílica en un paciente no descarta que pueda tener otra. De hecho, la com-binación de defectos incrementa aditiva o sinérgicamente el riesgo trombótico.
• Diversas situaciones adquiridas ocasionan disminuciones transitorias de proteínas relacio-nadas con riesgo trombótico (por ejemplo, un déficit de antitrombina puede estar ocasio-nado por el tratamiento con L-asparaginasa). Por ello, para considerar un defecto como congénito debe ser permanente y suele afectar a otros miembros de la familia, ya que los casos debidos a mutación espontánea son raros, aunque no inexistentes.
El primer episodio trombótico puede aparecer en sujetos jóvenes y, en ocasiones, la primera manifestación tiene lugar en localizaciones anatómicas poco frecuentes (trombosis mesentérica, cerebral, etc.)

HEMOSTASIA
Población general Pacientes primer episodio ETEV
Riesgo anual de ETEV en sujetos asintomáticos
Sin defecto conocido 85% 65% 0,01%Deficiencia AT 0,01% 0,5-2% 1,7%Deficiencia PC 0,2%-0,5% 2-3% 0,7%Deficiencia PS 0,2%-0,5% 2-3% 0,8%FV Leiden heterocigoto 4% 10-20% 0,1-0,2%PT heterocigoto 2-3% 6-10% 0,1%FV Leiden homocigoto 0,1% 1% 0,8%FV Leiden/PT heterocigoto compuesto 0,1% 2% 0,4%
AT: antitrombina; PC: proteína C; PS: proteína S; FV: factor V; PT: protrombina 20210A; ETEV: enfermedad tromboembólica venosa.
Rasgos clínicos asociados a la trombofilia hereditaria
Trombosis en territorio venoso, en ocasiones en localizaciones atípicas (mesentérica, senos venosos cerebrales, etc.)
Primer episodio en edad joven (<45-50 años)
En mujeres, especial asociación con tratamiento hormonal o con el embarazo
Trombosis de repetición
Historia familiar de trombosis (patrón de herencia autosómico dominante)
Purpura fulminans del recién nacido (deficiencia de PC o PS)
Necrosis cutánea asociada a AVK (deficiencia de PC o PS)
TABLA 2.
CARACTERÍSTICAS CLÍNICAS DE LA TROMBOFILIA HEREDITARIA
TABLA 1. INCIDENCIA Y RIESGO TROMBÓTICO ASOCIADO CON LAS PRINCIPALES TROMBOFILIAS CONGÉNITAS
No se han diferenciado claramente los rasgos clínicos de un episodio de ETEV asociado a una trombo-filia congénita. La tabla refleja la caracterización clásica.

HEMOSTASIA
Deficiencia de anticoagulantes naturales
Englobamos en un mismo apartado las deficiencias de tres anticoagulantes naturales aso-ciadas con trombofilia congénita, antitrombina (AT), proteína C (PC) y proteína S (PS), por-que presentan múltiples similitudes. En primer lugar, fueron las primeras identificadas (AT en 1965, PC y PS en la década de los ochenta). En general, se producen por mutaciones puntua-les que afectan un solo alelo (menos frecuentemente deleciones o inserciones, e inusuales, y siempre con efecto funcional moderado, los casos homocigotos). Dependiendo del efecto final de la alteración genética, se diferencian dos tipos de deficiencia:
• Tipo I, con disminución/ausencia de la proteína mutada en plasma, debido a que el alelo mutado no se traduce o la proteína mutada no se secreta.
• Tipo II, con niveles plasmáticos elevados (no necesariamente iguales a los niveles de la proteína no mutada) de una proteína mutada con menor o nula actividad funcional.
Estas deficiencias se asocian en general con elevado riesgo trombótico y la ausencia com-pleta causa letalidad embrionaria (AT) o púrpura fulminans del recién nacido (PC o PS). Estas características justifican que actualmente sean los defectos trombofílicos cuya identi-ficación, tanto en pacientes sintomáticos como familiares asintomáticos, tiene mayor utilidad clínica en la prevención del primer episodio trombótico mediante profilaxis en situaciones de riesgo4, y reducción de recurrencia por prolongación del tratamiento anticoagulante oral en pacientes con ETEV5 (Tabla 3).

HEMOSTASIA
Efecto de la tromboprofilaxis en situaciones de riesgo a portadores asintomáticos de deficiencia de anticoagulantes naturales sobre el desarrollo de un primer episodio trombótico
TrombofiliaCociente de riesgo (IC 95%)
p Utilidad clínica
ETEV no provocada
Sin deficienciaCualquier deficiencia
Deficiencia ATDeficiencia PCDeficiencia PS
1,0 (referencia)22,3 (2,9-172,7)42,7 (5,2-350,7)
4,4 (0,3-71,7)25,5 (2,9-221,3)
0,003<0,001
0,290,003
La identificación de sujetos con deficiencia de anticoagulantes y
posterior tromboprofilaxis en situaciones de riesgo
reduce la ETEV provocada pero no afecta al riesgo de
ETEV espontáneaETEV provocada
Sin deficienciaCualquier deficiencia
Deficiencia ATDeficiencia PCDeficiencia PS
1,0 (referencia)2,8 (0,9-8,6)
2,5 (0,5-12,9)3,6 (0,9-13,8)2,0 (0,4-10,7)
0,080,280,060,40
Riesgo de primer episodio trombótico y recurrencia
Trombofilia Riesgo anual primer ETEV
Riesgo relativo
Riesgo de recurrencia Utilidad clínica
Deficiencia anticoagulantes (AT, PC o PS)
1,5-1,9% 15-19x 40% a 5 años55% a 10 años
Prolongación del tratamiento anticoagulante para evitar
recurrencia
Polimorfismos protrombóticos (FV Leiden, Protrombina G20210A)
0,3-0,5% 3-5x 11% a 5 años25% a 10 años Nula de forma aislada
AT: antitrombina; PC: proteína C; PS: proteína S; FV: factor V; ETEV: enfermedad tromboembólica venosaAdaptado de Mahmoodi BK, et al. J Thromb Haemost. 2010 y Lijfering WM, et al. Blood. 2009.
TABLA 3. UTILIDAD CLÍNICA DEL DIAGNÓSTICO DE ALTERACIONES TROMBOFÍLICAS4,5

HEMOSTASIA
a) Deficiencia de antitrombina
La AT es una serpina que actúa como excelente y eficaz inhibidor de las principales se-rín-proteasas de la coagulación, especialmente a la trombina y al FXa, pero también al FVIIa, FIXa, FXIa y FXIIa. Este proceso es significativamente potenciado en presencia de glicosami-noglicanos (Figura 1), lo que explica el mecanismo anticoagulante de las heparinas, tanto no fraccionada como de bajo peso molecular.
FIGURA 1. ANTITROMBINA
Antitrombina
SERPINA con efecto anti-IIa, Xa, XIa, XIIa, otros
Síntesis hepática (no dependiente de vitamina K)
Acción biológica potenciada por glicosaminoglicanos (heparinas)
Primera trombofilia descrita: Egeberg 1965
A B C
A: Las moléculas de heparina se unen a la AT a través de una secuencia de 5 azúcares básicos (pen-tasacárido). B: Esta unión induce un cambio conformacional en la molécula de AT, que expone su centro activo. C: La AT activada se une y proteoliza de forma irreversible a la trombina (se precisa la formación de un complejo ternario AT-heparina-trombina) o al factor Xa (en este caso no se precisa la unión simultánea del FXa a la heparina).

HEMOSTASIA
El diagnóstico inicial de deficiencia de AT se lleva a cabo empleando pruebas funcionales, normalmente empleando métodos amidolíticos cromogénicos basados en la cuantificación de la actividad anti-FXa o anti-IIa (Tabla 4 y Figura 2).
En caso de alteración en la prueba funcional, se precisa continuar el estudio determinando los niveles antigénicos y la afinidad por heparina de la AT plasmática (normalmente em-pleando inmunoelectroforesis cruzada en presencia de heparina).
En un tercer paso, el diagnóstico molecular es también interesante (más del 95% de los casos se resuelven secuenciando los 7 exones y regiones flanqueantes del gen codificante, SERPINC1), ya que aporta información relevante sobre el mecanismo responsable de la deficiencia, con potencial valor pronóstico. Hasta la fecha se han descrito más de 250 altera-ciones genéticas diferentes en el gen SERPINC1 asociadas con deficiencia de AT.
TABLA 4. CÓMO REALIZAR ESTUDIO DE TROMBOFILIA: AT
Tipo Deficiencia Cantidad de AT Actividad biológica Observaciones
I Disminuida Disminuida Tipo más frecuente
IIRS Normal Disminuida Anomalía sitio reactivo
IIHBS Normal Normal (en ausencia de heparina)
Anomalía en el sitio de unión heparina-AT
IIPE
Ligeramente Disminuidos Disminuida Disfunción proteica con
baja concentración
1º SCREENING
Test funcional amidolítico (actividad anti-IIa o Xa) con exceso de trombina/FXa en presencia de heparina. La trombina/FXa residual actúan sobre un sustrato cromogénico y es inversamente proporcional a la concentración de AT en la muestra
2º TIEMPO
Determinación antigénica (ELISA...)
Test funcional en ausencia de heparina e incubación prolongada (actividad progresiva)
3º TIEMPO
Estudio genético (gen SERPINC1: cromosoma 1, 7 exones, 13,5 kb)
En el diagnóstico inicial es a través de pruebas funcionales. Si estas pruebas resultan alteradas, se precisa continuar el estudio determinando los niveles antigénicos y la afinidad por heparina de la AT plasmática. En un tercer paso, se realizan pruebas moleculares para el diagnóstico, y tienen un potencial valor pronóstico.

HEMOSTASIA
NORMAL
BAJA
SÍ
SÍ
SÍSÍ NO
NO
NO
NO
Actividad antitrombina
¿En tratamiento actual o reciente
con heparina terapéutica?
¿Presencia de un IDT?
Si se empleó un test basado en trombina, repetir tras la suspensión del IDT
No evidencia de laboratorio de deficiencia de AT
Para confirmar la deficiencia, repetir con nueva muestra y considerar estudiar a
los familiares
Repetir el test cuando el paciente
no reciba dosis anticoagulantes de heparina durante al
menos 1 semana
Repetir el test cuando estas condiciones no estén presentes. Si no es posible,
considerar estudiar a familiares de primer grado
Determinar AT antigénica
¿nivel antigénico bajo?
Deficiencia de AT tipo I
(cuantitativa)
Deficiencia de AT tipo II(cualitativa)
¿Existen datos sugestivos de deficiencia adquirida de AT como trombosis reciente, cirugía, CID,
hepatopatía, embarazo, proteinuria o tratamiento
con L-asparaginasa?
Adaptado de Khor B, et al. Am J Hematol 2010.
Algoritmo de diagnóstico de la deficiencia de AT que destaca la posible interferencia de fármacos en la técnica amidolítica, así como diversas situaciones clínicas que podrían condicionar una deficiencia adquirida de AT.
FIGURA 2.
CÓMO REALIZAR ESTUDIO DE TROMBOFILIA: AT

HEMOSTASIA
La deficiencia de AT tipo I, cuantitativa, se asocia con elevado riesgo trombótico, mientras que la deficiencia tipo II, cualitativa, presenta mayor heterogeneidad clínica.
En las deficiencias tipo II se pueden diferenciar tres subgrupos:
- Defectos del sitio reactivo. Causadas por mutaciones que afectan directamente al centro reactivo de la molécula e interfieren o anulan la capacidad anticoagulante de la AT. Pre-sentan gran heterogeneidad clínica, desde muy moderados como la AT Cambridge II (Ala-384Ser) a muy severos como la AT London (del Arg393).
- Defectos del sitio de unión a heparina. Reducen la afinidad por heparina y, por tanto, la activación de la AT por glicosaminoglicanos. Generalmente son las que menor riesgo trom-bótico tienen.
- Defectos pleiotrópicos. La mutación genera variantes con afinidad por heparina y mecanis-mo inhibitorio afectados. Suelen ser graves desde un punto de vista clínico.
Aunque el aumento del riesgo trombótico asociado a la deficiencia de AT sea alto (x 10-50 ve-ces), la muy baja incidencia del déficit de este anticoagulante en la población general (0,02%) explica que sean pocos los pacientes con ETEV que presentan esta trombofilia congénita (0,5-2%) (Tablas 1 y 3). Sin embargo, la elevada tasa de recurrencia en portadores y las ventajas clínicas de su diagnóstico justifica su estudio tanto en pacientes con trombosis venosa como en sus familiares (Tabla 3). En la Figura 3 se muestra la probabilidad de evento trombótico en sujetos portadores de alteraciones trombofílicas congénitas a diferentes edades, y en ella se puede constatar que la deficiencia de antitrombina es la que se asocia con mayor riesgo trombótico a cualquier edad6.
Aunque el aumento del riesgo trombótico asociado a la deficiencia de AT sea alto, la muy baja incidencia del déficit de este anticoagulante en la población general (0,02%) explica que sean pocos los pacientes con ETEV que presentan esta trombofilia congénita (0,5-2%)
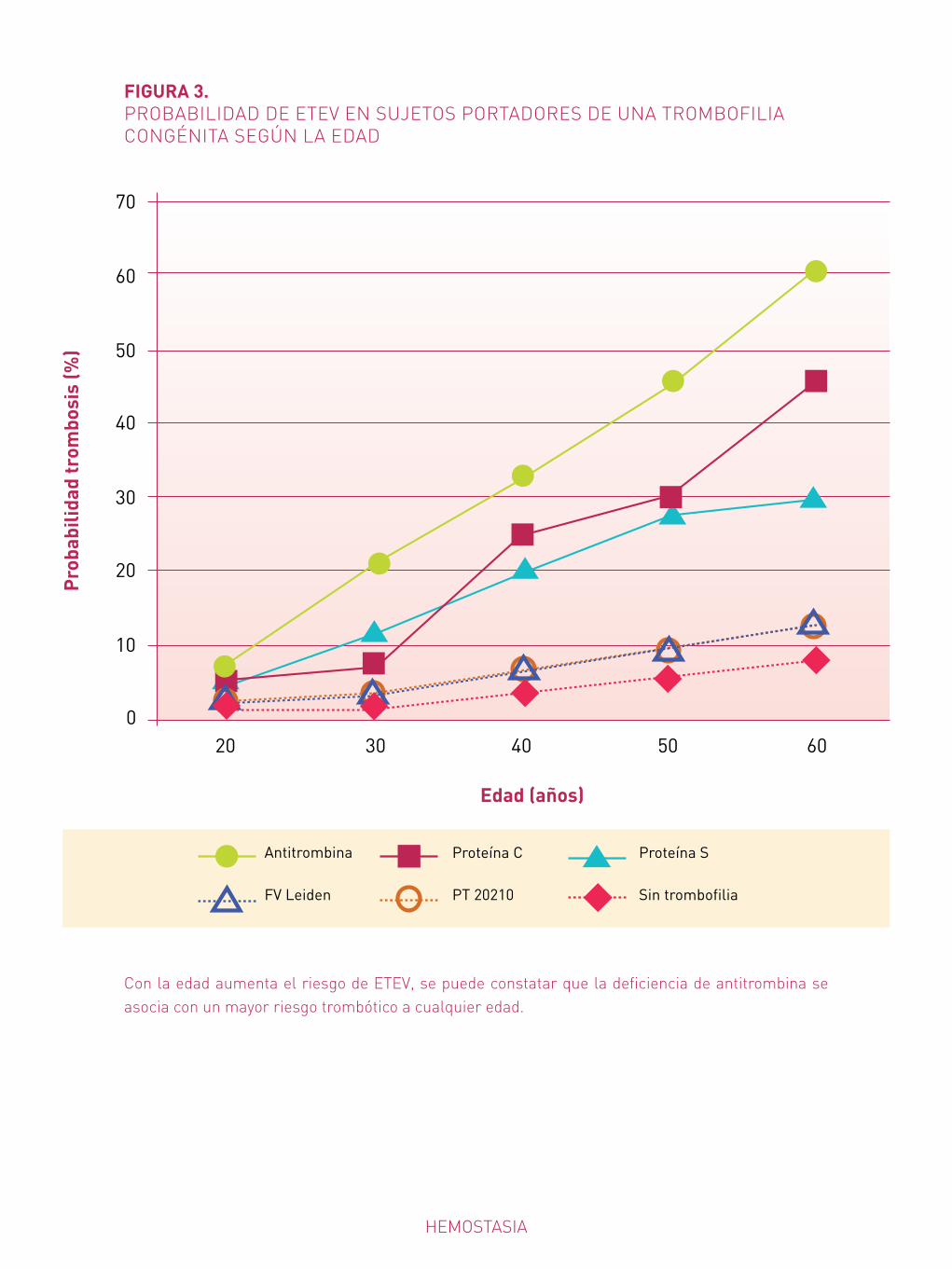
HEMOSTASIA
FIGURA 3. PROBABILIDAD DE ETEV EN SUJETOS PORTADORES DE UNA TROMBOFILIA CONGÉNITA SEGÚN LA EDAD
70
Pro
babi
lidad
trom
bosi
s (%
)
Edad (años)
60
50
40
30
20
20 30 40 50 60
10
0
Antitrombina
FV Leiden
Proteína C
PT 20210
Proteína S
Sin trombofilia
Con la edad aumenta el riesgo de ETEV, se puede constatar que la deficiencia de antitrombina se asocia con un mayor riesgo trombótico a cualquier edad.

HEMOSTASIA
b) Deficiencia de proteína C
El segundo sistema anticoagulante más importante del organismo es el de la PC. Una vez activada por la trombina en presencia de trombomodulina, la PC activada (PCa) ejerce su papel anticoagulante inactivando los factores VIIIa y Va (Figura 4). Por ello, la deficiencia heterocigota de PC implica un importante riesgo trombótico. La deficiencia homocigota (ex-traordinariamente infrecuente) causa un cuadro de púrpura fulminans en neonatos.
APC
PC
EPCRIIa
EPCR
PC
PCa Vai
V
PCa
PSPS
Xa IXaX
VIIIai
II
Sistema de la proteína C
La PC y PS son proteínas de síntesis hepática (dependiente de vitamina K)
La PC es activada por complejo Trombina-Trombomodulina (facilitada por el EPCR)
La PCa inactiva a los factores Va y VIIIa
La PS actúa como cofactor (favorece su unión a fosfolípidos de carga) y circula en forma libre o unida a
C4bBP (60%, no actividad cofactor-PCa)
FIGURA 4. SISTEMA DE LA PROTEÍNA C
A.
TM
B.
La deficiencia homocigota (extraordinariamente infrecuente) causa un cuadro de púrpura fulminans en neonatos
A. La trombina (IIa), además de inducir la transformación del fibrinógeno en fibrina, puede unirse a una proteí-na de membrana del endotelio, la trombomodulina (TM). La proteína C circulante se une al complejo trom-bina-trombomodulina (esta unión se ve muy favorecida en presencia del receptor endotelial de la proteína C: EPCR) y es activada.
B. La proteína C activada (PCa) en presencia de PS degrada a los factores Va y VIIIa.

HEMOSTASIA
El diagnóstico de deficiencia de PC se realiza inicialmente mediante determinaciones fun-cionales (tanto coagulantes como cromogénicas) (Tabla 5), y antigénicas, que permiten iden-tificar y diagnosticar los dos tipos de deficiencia de PC:
• Tipo I (cuantitativa), que es el más común.
• Tipo II (cualitativa).
El estudio genético también es relativamente sencillo de realizar (el gen contiene solo 8 exones codificantes), pero hay menos datos sobre la utilidad clínica de dicha información. Se han descrito más de 270 mutaciones diferentes en el gen que codifica la PC en pacientes con deficiencia de este anticoagulante.
Como sucede con la deficiencia de AT, la prevalencia de la deficiencia de PC en la po-blación general es baja (0,2-0,4%), mientras que en pacientes con ETEV está entre el 2,5-6% (Tablas 1 y 3). De nuevo, el riesgo de trombosis tanto relativo como absoluto es alto (10-15 veces y 7 veces, respectivamente) y aumenta con la edad (Figura 4). Finalmente, la tasa de recurrencia, es también elevada. Todos estos datos y las ventajas clínicas de su diagnóstico justificarían su estudio, tanto en pacientes con trombosis venosa como en familiares asintomáticos7.
Deficiencia tipo I: Cuantitativa Deficiencia tipo II: Cualitativa
( Funcional y antigénica) ( Funcional)
TABLA 5. CÓMO REALIZAR ESTUDIO DE TROMBOFILIA: PC
SCREENING
Test funcional empleando plasma deficiente en PC + activador específico de PC (Protac)
La actividad de la PC es proporcional a la prolongación del TTPa (test coagulativo) o se determina mediante sustrato cromogénico. - Valor normal: 70-120% - Mayoría heterocigotos: 30-60% - Homocigotos o dobles heterocigotos: <25%
2º TIEMPO
Determinación cuantitativa (ELISA)
3er TIEMPO
Estudio genético (Gen PROS 1: cromosoma 2; 8 exones)

HEMOSTASIA
c) Deficiencia de proteína S
La proteína S participa como cofactor de la PCa (Figura 4). Por tanto, su déficit afecta prin-cipalmente al mecanismo anticoagulante de la PC. Aproximadamente, el 60-80% de la PS en plasma forma un complejo con una proteína reguladora del complemento (C4bBP). Solo la fracción de PS libre tiene actividad anticoagulante. Empleando sistemas de cuantificación de niveles antigénicos totales y libres de proteína S, así como pruebas funcionales que evalúan la actividad de la proteína S como cofactor de la PCa se pueden definir tres tipos de deficiencia de PS (Tabla 6):
• Tipo I. Niveles antigénicos de PS total y libre reducidos. Por supuesto, asocian baja actividad funcional. Son la mayoría, dos tercios de los casos de déficit de PS.
• Tipo II. Niveles antigénicos (totales y libres) normales, pero baja actividad funcional como cofactor de la PCa. Es el tipo más raro.
• Tipo III. Niveles de proteína total normales, con niveles de proteína libre bajos, debido al au-mento de PS unida a C4bBP. Se identifican en un tercio de los casos con déficit de PS.
TABLA 6. CÓMO REALIZAR ESTUDIO DE TROMBOFILIA: PS
Deficiencia de PS I II III
PS funcional
PS antigénica total Normal NormalPS antigénica libre Normal
SCREENING
Test funcional coagulativo con plasma deficiente en PS + PCa + FVa. Posibilidad de artefacto en caso de resistencia a la PCa/FV Leiden
2º TIEMPO
Determinación cuantitativa (Inmunoturbidimétrico/ELISA) de PStotal y PSlibre
3er TIEMPO
Estudio genético (gen PROS 1: cromosoma 3; 15 exones + pseudogen PROSP2)

HEMOSTASIA
A diferencia de los otros dos anticoagulantes naturales, para los que las deficiencias adquiri-das son relativamente poco frecuentes (con la excepción de hepatopatías), en el caso de la PS, además, los anticonceptivos orales y el tratamiento hormonal sustitutivo reducen sus nive-les. El embarazo también causa una progresiva reducción de los niveles de PS. En pacientes con anticuerpos antifosfolípidos se ha observado un estado adquirido de deficiencia de PS, así como en casos de coagulación intravascular diseminada y enfermedad hepática. Por este moti-vo, con frecuencia se llega a diagnósticos incorrectos de deficiencia congénita de PS (Tabla 7).
Problema PC PS
Estudios funcionales coagulativos
Anticoagulante lúpico Sobreestimación
Altas concentraciones de heparina Sobreestimación
Inhibidores directos de IIa/Xa Sobreestimación
Niveles elevados de FVIII (>250%) Infraestimación
FV Leiden Infraestimación
Estudio funcional amidolítico
Presencia de enzimas que actúan sobre el sustrato cromogénico
Sobreestimación N.A.
Mutaciones en -carboxilación o alteración de la unión a fosfolípidos
Resultado normal a pesar de mutación N.A.
Diagnóstico genético Pseudogen PROSP
Situaciones que afectan a los niveles de PC/PS
Embarazo Elevación en las primeras semanas
Disminución
Anticonceptivos/terapia hormonal sustitutiva Disminución
Edad infantil Disminución Disminución
Deficiencia adquirida
Tratamiento con AVK Disminución
Hepatopatía Disminución
Consumo (ETEV, CID) Disminución
Autoanticuerpos (lupus, sepsis, VIH, cáncer, etc.) Disminución
TABLA 7. DIFICULTADES EN EL DIAGNÓSTICO DE DEFICIENCIAS DE PC Y PS
Adaptado de Bereczky S, et al. Clin Chem Lab Med. 2010

HEMOSTASIA
La prevalencia del déficit de PS en la población general no se conoce con exactitud, se estima entre el 0,03 y el 0,13%, mientras que asciende hasta el 1-3% en pacientes con ETEV. La defi-ciencia normalmente es heterocigota, siendo muy infrecuente la deficiencia en homocigosis. Al igual que con la deficiencia de PC, el déficit homocigoto causa púrpura fulminans en neonatos. El riesgo relativo de trombosis venosa en individuos con deficiencia hereditaria de PS es de 11,5, con alta incidencia de recurrencias tras finalizar el tratamiento anticoagulante (Tablas 1 y 3).
El análisis genético de la PS es más complejo, pues existen dos genes homólogos para la proteí-na: PROS1 (responsable de la mayoría de las deficiencias de tipos I y II) y el pseudogén PROSP. Ade-más, solo un 50% de los pacientes con deficiencia de PS tienen alguna mutación en el gen PROS1. Se han descrito más de 234 mutaciones puntuales diferentes8.
Tanto la PC como la PS son dos proteínas vitamina K dependientes. En pacientes con deficien-cia de alguna de ellas, al iniciar tratamiento anticoagulante con acenocumarol o warfarina puede desencadenarse un cuadro de necrosis cutánea, ya que el inicial descenso acusado de estas proteínas, inducido por el antagonista de la vitamina K (AVK), favorece la formación de coágulos de fibrina en los pequeños vasos de la piel. Para evitar el desarrollo de esta complicación, en los pacientes con deficiencia conocida de PC o PS es imprescindible asegurar una correcta anticoa-gulación con heparina (no fraccionada o de bajo peso molecular) antes del inicio del tratamiento con AVK. Además, el solapamiento de ambos tratamientos debe ser prolongado, comenzando con dosis de AVK inferiores a las habituales.
La prevalencia del déficit de PS en la población general no se conoce con exactitud, se estima entre el 0,03 y el 0,13%, mientras que asciende hasta el 1-3% en pacientes con ETEV
En los pacientes con deficiencia conocida de PC o PS es imprescindible asegurar una correcta anticoagulación con heparina (no fraccionada o de bajo peso molecular) antes del inicio del tratamiento con AVK

HEMOSTASIA
Polimorfismos protrombóticos
En la década de los noventa se produjo una revolución en la visión de la trombofilia con la identificación por parte del grupo de la Universidad de Leiden (Holanda) de los polimorfismos del factor V Leiden y la protrombina G20210A. Ambos son cambios frecuentes en la pobla-ción general (>1%), con efecto funcional e incrementan de forma moderada (en heterocigosis) el riesgo trombótico. Con estos descubrimientos, la trombosis venosa pasó a ser considera-da una enfermedad poligénica.
Pese a la elevada prevalencia de estos polimorfismos en pacientes con ETEV, aspecto que junto a la sencillez diagnóstica han propiciado una elevada tasa de solicitudes de detección de estos defectos en los laboratorios, su trascendencia clínica es mucho más cuestionada, y se restringe, incluso con poca evidencia bibliográfica, a los defectos en homocigosis o las heterocigosis compuestas en pacientes con trombosis venosa y familiares asintomáticos. El estudio de estos polimorfismos en otras circunstancias como complicaciones obstétricas, trombosis arterial o exposición a situaciones de alto riesgo trombótico, se restringe a los ca-sos con historia personal o familiar de trombosis venosa significativa9.
El estudio de estos polimorfismos en otras circunstancias como complicaciones obstétricas, trombosis arterial o exposición a situaciones de alto riesgo trombótico, se restringe a los casos con historia personal o familiar de trombosis venosa significativa9

HEMOSTASIA
a) Factor V Leiden y resistencia a la proteína C activada
El factor V Leiden constituye la trombofilia hereditaria más frecuente en nuestro medio. Es el resultado de una mutación puntual que cambia la arginina por glutámico en el primer sitio de ruptura proteolítica del FV activo por la PCa (Arg506Gln). La consecuencia funcional de este simple cambio de aminoácido es un FVa más resistente a la inactivación por la PCa (Figura 5). De esta forma, el tiempo de tromboplastina parcial activado (TTPa) de un portador de la mutación apenas se prolonga tras la adición de PCa, lo que se denomina resistencia a la PCa. Aunque todos los casos con FV Leiden presentan resistencia a la PCa, existen casos de resistencia a la proteína C activada (RPCa) por otras causas diferentes (Tabla 8).
GEN FV LEIDEN
Factor V Leiden
FLVFXa y/o Trombina ??
FLVa
Arg 306
NH2 A11 303 317 656 709 1546 2196
A2 A3, C1, C2 COOH
G1n 506 Arg 679
FVi
A
PCa
E1
Exón 10
1487 1691 1701
5’ 3’
E25
PROTEÍNA FV LEIDEN
FIGURA 5. FACTOR V LEIDEN
El factor V Leiden es el resultado de una mutación puntual (adenina por guanina) en el nucleótido 1691, en el exón 10. Este simple cambio de nucleótido implica el cambio del aminoácido arginina por glutámico en el primer sitio de ruptura proteolítica del FV activo por la PCa. La consecuencia funcio-nal de este simple cambio de aminoácido es un FVa más resistente a la inactivación por la PCa.

HEMOSTASIA
La frecuencia de esta alteración en la población general es alta (4% de la población es-pañola, y hasta el 10% en el norte de Europa) y aumenta al 20% en pacientes no seleccio-nados con trombosis venosa, por tanto incrementa de forma moderada el riesgo de trom-bosis venosa (3-7 veces en heterocigotos, aunque en homocigotos el riesgo aumenta hasta 20 veces). El riesgo absoluto de desarrollar trombosis venosa en portadores del FV Leiden es considerablemente inferior al descrito en las deficiencias de proteínas anticoagulantes (2,2 veces en heterocigosis), y el riesgo de recurrencia, aunque estadísticamente significa-tivo (aumenta 1,3 veces), es tan bajo que apenas tiene utilidad clínica10 (Tablas 1 y 3).
b) Protrombina G20210A
Es un cambio de un solo nucleótido (SNP, en inglés) que afecta a la zona 3’ no codifi-cante del gen de la protrombina. Se asocia con ligero aumento de los niveles de esta molécula en plasma. El alelo 20210A está presente en el 3% de la población española sana (<0,1% en homocigosis), pero asciende hasta el 6-9% en los pacientes con ETEV. Por tanto, el riesgo relativo de trombosis venosa en portadores es 3-4 veces mayor que en no portadores, valores que ascienden a 10-20 en homocigotos. El riesgo abso-luto de desarrollar trombosis venosa en portadores del polimorfismo es similar al del FV Leiden y el riesgo de recurrencia bajo (1,2 veces) (Tablas 1 y 3)11.
El diagnóstico de FV Leiden y de la protrombina G20210A es sencillo, realizándose de forma rápida mediante la utilización de kits comerciales para PCR en tiempo real.
TABLA 8. CAUSAS DE RESISTENCIA A LA PCA
Causas de resistencia a la PCa
Congénitas
FV Leiden (la causa más frecuente)
Otras mutaciones puntuales en el gen del FV (Cambridge, Hong Kong, Liverpool)
Haplotipo R2 del FV
Congénitas o adquiridas
Aumento de niveles de protrombina, FVIII, FIX o FX
Descenso de PS, o TFPI (inhibidor de la vía del factor tisular)
Adquiridas
Anticonceptivos hormonales / terapia hormonal sustitutiva
Embarazo
Cáncer
Anticuerpos antifosfolípido y otros autoanticuerpos (anti-PC, anti-PS, anti-FV)
El FV Leiden es la causa mayoritaria de resistencia a la PCa, aunque existen casos de resistencia a la RPCa por otras causas diferentes.

HEMOSTASIA
c) Otras alteraciones genéticas
Existen otras alteraciones genéticas que se han implicado en el incremento de ries-go trombótico, tanto mutaciones poco frecuentes como por ejemplo las responsables de disfibrinogenemia, como polimorfismos que afectan a diferentes elementos del sistema hemostático y muchos otros que se han descrito en estudios de genotipado masivo12 y cuya relevancia clínica y aplicación diagnóstica todavía tienen que contrastarse. En este apartado haremos referencia a dos situaciones antagónicas:
• El grupo sanguíneo ABO, definido por una serie de polimorfismos, clara y consistente-mente se asocia con riesgo trombótico. Los grupos no-O, excepto el A2, asociados con mayores niveles de FVIII y FvW, incrementan casi dos veces el riesgo trombótico, deberían incluirse en estudios de trombofilia13.
• El polimorfismo C677T de la metilen-tetrahidrofolato-reductasa (MTHFR) que, al estar incorporado en sistemas comerciales automatizados de trombofilia congénita, se realiza frecuentemente. Este polimorfismo implica un cambio de alanina por valina en el ami-noácido 223, que condiciona una termolabilidad de la enzima, de forma que a 37°C su actividad es un 50% menor que la de la variante normal. El alelo 677T, sobre todo en homocigosis, se asocia con mayores niveles plasmáticos de homocisteína (Tabla 9). La hiperhomocisteinemia (en la que influyen tanto factores hereditarios como adquiridos) inicialmente se asoció con un mayor riesgo trombótico, sobre todo en territorio arterial, aunque en la actualidad esta asociación es controvertida14. Pero el estudio del polimor-fismo MTHFR-C677T no debería incluirse en los estudios de trombofilia congénita, ya que únicamente aporta preocupación injustificada a los pacientes. En un reciente metaanálisis icluyendo más de 11 000 pacientes con ETEV y 21 000 controles, no se observó una asocia-ción significativa de la homocigosis MTHFR-677TT con el riesgo de trombosis15.

HEMOSTASIA
Factores que influyen en los niveles plasmáticos de homocisteína
Genéticos (mutaciones)
Cistationina β-Sintetasa
MTHFR
Metionina sintetasa
Edad/Sexo
Edad avanzada
Varones
Menopausia
Nutricionales
Deficiencia de folato/vit. B12
Deficiencia de vit. B6
Excesivo consumo de café y alcohol
Enfermedades
Malabsorción intestinal de B12
Fallo hepático
Insuficiencia renal
Psoriasis
Neoplasias
Hipotiroidismo
Diabetes mellitus
Farmacológicos
Metotrexato
Estrógenos
Anticonvulsionantes
Metformina
Antagonistas del folato
Glitazonas (algunas)
Hipolipemiantes (fibratos, colestipol, nicotinato)
Adaptado de Di Minno MN, et al. Thromb Haemost. 2010.
TABLA 9. FACTORES QUE INFLUYEN EN LOS NIVELES PLASMÁTICOS DE HOMOCISTEÍNA
En los niveles plasmáticos de homocisteína influyen múltiples factores, tanto hereditarios como ad-quiridos. La asociación de hiperhomocisteinemia con un mayor riesgo trombótico, sobre todo en territorio arterial, es controvertida. En lo que sí existe unanimidad es en la nula utilidad de la inclu-sión del polimorfismo MTHFR-C677T en los estudios de trombofilia. Por otra parte, hasta la fecha tampoco se ha demostrado que la administración de suplementos de ácido fólico y vitamina B12 a individuos con hiperhomocisteinemia moderada, con el fin de reducir sus niveles, se asocie con una reducción del riesgo de eventos trombóticos.

HEMOSTASIA
El aumento de los niveles plasmáticos de FVIII, FIX y FXI también puede incrementar el riesgo trombótico, si bien hasta la fecha no se ha caracterizado la alteración (o alteraciones) genética responsable. Una pega adicional es que el aumento de riesgo trombótico asociado a la concentración de los factores sería una función continua, por lo que la fijación de puntos de corte no dejaría de tener cierto carácter arbitrario. Además, hay que tener en cuenta el alto número de variables preanalíticas y condiciones clínicas que pueden influir en el resul-tado de una determinación puntual de estos niveles16.
Defectos combinados
Como hemos comentado anteriormente, la trombosis es una enfermedad poligénica y por ello la combinación de defectos trombofílicos, si bien tiene una incidencia baja, aumenta, aditiva o incluso sinérgicamente, el riesgo trombótico. La combinación más relevante, la de los dos polimorfismos más comunes, FV Leiden y protrombina 20210A, incrementa 20 veces el riesgo de trombosis.
En los últimos años estamos asistiendo al desarrollo de algoritmos predictivos que cuan-tifican el riesgo trombótico asociado con perfiles genéticos complejos definidos por múl-tiples polimorfismos y que con seguridad dotarán de mayor aplicación clínica a los estudios de trombofilia (Figura 6)17. Por el contrario, los resultados obtenidos hasta la fecha en ambiciosos estudios tipo Genomic Wide Association Studies (GWAS) han sido francamente desalentadores18.
La combinación más relevante, FV Leiden y protrombina 20210A, incrementa 20 veces el riesgo de trombosis

HEMOSTASIA
Análisis combinado de múltiples SNPs en ETEV de Plataforma Thromboincode
Valoran el posible efecto aditivo de varios SNPs sobre el riesgo vascular
Incluyen algoritmos matemáticos para valorar interacciones gen-gen y gen-ambiente
Parecen mejorar la predicción de riesgo vascular, permitiendo consejo individualizado
SNPs GenPrevalencia en pacientes con
DVTRiesgo relativo
46C>T FXII 6% 5
rs8176719
Grupo ABO(portador A1) nd 2-4
(+ FV Leiden: 4-23)
rs7853989
rs8176743
rs8176750
Arg67Stop Serpin A10 4,4% 3,3
Ala384Ser(Cambridge II) Serpin C1 1,7% 10
Arg506GIn(FV Leiden)
Factor V (FV)
15-25% 5
Arg306Thr(FV Cambridge) nd nd
Arg306GIy(FV Hong Kong) nd nd
Val34Leu Factor XIII (FXIII) 2% Factor de protección
G20210A Factor IIprotrombina (FII) 6-16% 2-3
GENÉTICA
12 VARIANTES ALÉLICAS EN 7 GENES
FIGURA 6.
ANÁLISIS COMBINADO DE MÚLTIPLES SNPS EN ETEV
Ejemplo de plataforma Thromboincode para el análisis combinado de 12 variantes alélicas en 7 ge-nes para la evaluación del riesgo de ETEV.
El desarrollo de algoritmos predictivos que cuantifican el riesgo trombótico asociado con perfiles genéticos complejos definidos por múltiples polimorfismos va a dotar de mayor aplicación clínica a los estudios de trombofilia.

HEMOSTASIA
TROMBOFILIA ADQUIRIDA
Estados de trombofilia adquirida
Los estados de trombofilia adquirida o secundaria deben ser considerados en aquellos pa-cientes con tromboembolismo venoso espontáneo o idiopático, sin un factor desencadenan-te obvio, como la inmovilización, obesidad o cirugía. Hay un número importante de situaciones que se reflejan en la Tabla 10.
Estados de trombofilia adquirida
Síndrome antifosfolípido
Neoplasias
Síndromes mieloproliferativos crónicos
Hemoglobinuria paroxística nocturna
Trombocitopenia inducida por heparina
Síndrome nefrótico
Enfermedad de Behçet
Otras conectivopatías
Cirugías
Infecciones/sepsis
Coagulación intravascular diseminada
Traumatismos
Hiperviscosidad plasmática
Embarazo / tratamiento con estrógenos
Tratamientos antitumorales
Otros
TABLA 10. ESTADOS DE TROMBOFILIA ADQUIRIDA
Existen numerosas situaciones clínicas que condicionan el desarrollo de una situación de hiper-coagulabilidad/trombofilia adquirida, por mecanismos muy variados, asociándose a un incremento significativo del riesgo de ETEV.

HEMOSTASIA
Cáncer oculto
Hasta el 10% de los pacientes que presentan un episodio de ETEV no provocado presentan una neoplasia oculta. La realización sistemática de exploraciones complementarias dirigidas a la detección de cáncer oculto en este contexto es motivo de controversia. Aunque en teoría, cabría pensar que la realización de procedimientos de despistaje de cáncer oculto se asocia-ría con un diagnóstico más precoz, y por tanto una mejoría en el pronóstico de los pacientes, hasta la fecha ningún estudio ha demostrado un impacto positivo en términos de superviven-cia de la implementación de protocolos de este tipo.
Por otra parte, no existe unanimidad acerca de qué exploraciones complementarias se de-berían incluir en el cribado, existiendo discrepancias acerca de su coste/beneficio. Está claro que en todo paciente con un episodio de ETEV no provocada se debería realizar una anamne-sis dirigida y una exploración física completa con el fin de detectar síntomas y signos sugesti-vos de posible neoplasia oculta. En caso de no encontrar alteraciones, algunas guías clínicas como la NICE 2012, recomiendan en pacientes mayores de 40 años completar el estudio me-diante TAC de tórax y abdomen y en las mujeres realizar también una mamografía19.
Recientemente se ha evaluado la potencial utilidad de la tomografía por emisión de posi-trones (PET-CT) como herramienta única para el cribado de cáncer oculto en pacientes con ETEV idiopática. Aunque los resultados iniciales han sido positivos por la elevada sensibilidad de esta técnica diagnóstica, su alto coste supone una importante limitación20.
Algunas guías clínicas como la NICE 2012, recomiendan en pacientes mayores de 40 años completar el estudio mediante TAC de tórax y abdomen y en las mujeres realizar también una mamografía19

HEMOSTASIA
Patología hematológica
Diferentes cuadros hematológicos incrementan el riesgo de aparición de complicaciones tromboembólicas, como los síndromes mieloproliferativos crónicos tipo trombocitemia esencial y policitemia vera. También la hemoglobinuria paroxística nocturna y otros tipos de procesos asociados con hemólisis, como la anemia falciforme. Con frecuencia, en el con-texto de estos procesos las complicaciones trombóticas acontecen en localizaciones inusua-les (territorio esplácnico o senos venosos cerebrales).
La complicación trombótica puede constituir, en ocasiones, la primera manifestación clíni-ca de la enfermedad. Por este motivo, sobre todo en caso de trombosis venosas en territorio esplácnico no justificadas por otras causas, algunos recomiendan la realización del análisis molecular de JAK2. La alteración de este gen (la más frecuente la V617F) es muy caracterís-tica de síndromes mieloproliferativos crónicos y se asocia con un incremento significativo del riesgo trombótico21.

HEMOSTASIA
Síndrome antifosfolipídico
El síndrome antifosfolipídico (SAF) es quizás la principal causa de trombofilia adquirida. Los datos clínicos que sugieren la existencia de SAF incluyen historia de abortos de repeti-ción, historia personal de trombosis arterial o venosa y trombocitopenia (Tabla 11). En estos casos, se recomienda evaluar el anticoagulante lúpico, anticuerpos anticardiolipina y anti-cuerpos anti-β2-glicoproteína I.
Es importante destacar la necesidad de confirmación diagnóstica de la positividad de anti-cuerpo antifosfolipídico (AAF) en una segunda muestra separada al menos 6 semanas de la inicial22. Debido al alto riesgo de recurrencia asociado a los pacientes con diagnóstico de SAF, se recomienda anticoagulación a largo plazo incluso tras el primer episodio de trombosis venosa. Sin embargo, el riesgo de complicaciones trombóticas varía en función del tipo de AAF, siendo claramente superior para los individuos con positividad del anticoagulante lúpico o con triple positividad.
Diagnóstico del síndrome antifosfolipídico
Criterios clínicos
Trombosis en territorio arterial y/o venoso y/o en pequeños vasos
Complicaciones obstétricas (muerte fetal >10 semanas, nacimiento pretérmino <34 semanas, 3 abortos <10 semanas)
Criterios de laboratorio
Anticoagulante lúpico (screening y confirmación)
Anticuerpos anti-cardiolipina (>40 GPL o MPL o percentil>99%)
Anticuerpos anti-β2glicoptroteína I (>40 GPL o MPL o percentil>99%)
Se precisa cumplir al menos 1 criterio clínico y 1 criterio de laboratorio (confirmada la positividad en una nueva muestra separada al menos 12 semanas)
TABLA 11. DIAGNÓSTICO DEL SÍNDROME ANTIFOSFOLIPÍDICO
Criterios de Sidney (2006) para el diagnóstico de síndrome antifosfolipídico (SAF), que son los vigentes en la actualidad. Los datos clínicos que sugieren la existencia de un SAF incluyen historia de abortos de repetición, historia personal de trombosis arterial o venosa y trombocitopenia. En estos casos, se recomienda evaluar el anticoagulante lúpico, anticuerpos anticardiolipina y anticuerpos anti-β2-gli-coproteína I. Es importante destacar la necesidad de confirmación diagnóstica de la positividad de anticuerpo antifosfolipídico en una segunda muestra separada al menos 12 semanas de la inicial.

HEMOSTASIA
PERTINENCIA DE LOS ESTUDIOS DE TROMBOFILIA CONGÉNITA
La perspectiva de los estudios de trombofilia se ha ensombrecido en los últimos años23. De considerarse un cribado prácticamente universal, hemos pasado a cuestionarse por completo la utilidad clínica de estos estudios, si bien existen diferencias en las recomendaciones de dife-rentes guías de práctica clínica (Tabla 12)24. La principal razón es la escasa utilidad clínica de un diagnóstico de trombofilia, especialmente de los polimorfismos protrombóticos, en el manejo de los pacientes (particularmente en lo que a la duración del tratamiento anticoagulante tras un episodio de ETEV respecta). Existen argumentos, tanto a favor como en contra, de realizar estos estudios tanto en pacientes con trombosis venosa como en familiares asintomáticos de pacientes con trombofilia, que se resumen en las Tablas 13 y 1425.
TABLA 12. DIFERENCIAS ENTRE LAS RECOMENDACIONES DE LAS DISTINTAS GUÍAS CLÍNICAS
Valor para determinar la razón de la ETEV
Valor para la predicción de recurrencia tras ETEV no provocada
Valor para la predicción de ETEV y profilaxis en familiares asintomáticos
Valor para la predicción de ETEV y profilaxis en la población general
International Consensus Statement (2005)
Sí, en todos los pacientes (excepto aquellos con primer episodio de ETEV provocada >50 años)
Sí (AT, PC, PS, homocigotos y dobles heterocigotos para FVL y Pt 20210A)
Sí (particularmente en mujeres en edad fértil) (particularmente en mu-jeres en edad fértil)
No
Guía de Consenso Francesa (2009)
Sí, en pacientes con un primer episodio de TVP proximal/EP no provocado >60 años y en pacientes con ETEV recurrente
Sí (AT, PC, PS, homocigotos y dobles heterocigotos para FVL y Pt 20210A)
Sí (posible excepción para familiares de portadores heterocigotos aislados del FVL o de la PT20210A)
No
British Committee for Standars in Hematology (2010)
British Committee for Standars in Hematology (2010)
No (posible excepción en aquellos con fuerte historia familiar de ETEV no provocada recurrente)
No (posible excepción de familiares de probandos con deficiencia de AT, PC o PS)
No
Grupo de Trabajo para la Evaluación de las Aplicaciones Genómicas en la Práctica y Prevención (EGAPP) (2011)
No (análisis limitado a FVL y Pt20210A)
No (análisis limitado a FVL y Pt20210A)
No (análisis limitado a FVL y Pt20210A) No evaluado
NICE (2012)
Sí, en pacientes con ETEV no provocada y con un familiar de primer grado con ETEV<50 años (estudiar deficiencia de AT, PC y PS)
Sí, en pacientes con un familiar de primer grado con ETEV<50 años si se va a suspender el tratamiento anticoagulante (estudiar deficiencia de AT, PC y PS)
No (posible excepción para mujeres en edad fértil que son familiares de primer grado de pacientes con ETEV y trombofilia conocida y están considerando anticoncepción oral o embarazo)
No evaluado
ACCP (2012) No evaluado
No (valor limitado en pacientes seleccionados con parte de una evaluación global del riesgo/beneficio de anticoagulación indefinida)
No evaluado No evaluado
Adaptado de De Stefano V & Rossi, E. Thromb Haemost 2013; 110:97-705.

HEMOSTASIA
Ventajas Inconvenientes
Identifica factores implicados en el desarrollo de la trombosis Efectos psicológicos negativos en portadores (estigma so-cial, sentimiento de culpabilidad por transmisión a hijo, etc.)
Estimula el cambio de estilo de vida: eliminar o reducir factores de riesgo (anticonceptivos orales, obesidad, sedentarismo)
Las recomendaciones sobre factores de riesgo evitables son las mismas con o sin trombofilia
Facilita recibir una correcta prolifaxis en situaciones de riesgo. Especialmente en déficit de antitrombina
Profilaxis en situaciones de riesgo independiente de la presencia de trombofilia
Puede asistir en el consejo sobre el riesgo de recurrencia y puede influir en el manejo clínico del paciente (terapia anticoagulante prolongada)
El riesgo de recurrencia no es completamente predecible y el riesgo de sangrado puede superar al de recurrencia en tratamientos más prolongados
Facilita la identificación de familiares con potencial riesgo de trombosis
Falsa seguridad en familiares negativos. Posibilidad de detectar no consanguinidad
Facilita una respuesta clínica más rápida ante los primeros síntomas de trombosis
La respuesta clínica debe ser igual independientemente de la trombofilia
No es caro, ni complicado metodológicamente
TABLA 13.
PERTINENCIA DE LA REALIZACIÓN DE ESTUDIOS DE TROMBOFILIA CONGÉNITA EN PACIENTES CON ETEV
TABLA 14. PERTINENCIA DE LA REALIZACIÓN DE ESTUDIOS DE TROMBOFILIA CONGÉNITA EN FAMILIARES DE PORTADORES
Ventajas Inconvenientes
Anima estilo de vida saludablePuede inducir una actitud fatalista y tener efectos psicológicos adversos (ansiedad, culpabilidad, estigmatización
Facilita decisiones sobre factores de riesgo modificables (anticonceptivos orales, terapia hormonal sustitutiva)
Falsa seguridad en sujetos sin alteraciones. Aumento del riesgo de embarazos no deseados
Sensibilización positiva del paciente ante signos iniciales de episodios trombóticos
Aumento de estrés y paranoia
Profilaxis antitrombótica en situaciones de alto riesgo en sujetos con trombofilia severa (deficiencia anticoagulantes o trombofilia combinada)
Los beneficios de la profilaxis todavía no están claramente sustentados y pueden variar atendiendo al defecto trombofílico y la historia familiar de trombosis (excepto deficiencia anticoagulantes)
Facilitan al clínico un diagnóstico y tratamiento precoz del evento trombótico
Es más barato y fácil (búsqueda dirigida)
Argumentos para defender las posturas “a favor” y “en contra” de la realización de estudios de trombofilia hereditaria en pacientes con historia de ETEV.
Argumentos para defender las posturas “a favor” y “en contra” de la realización de estudios de trombofilia hereditaria en familiares asintomáticos de pacientes.

HEMOSTASIA
Sin embargo, la mayoría de las recomendaciones de las guías se basan en evidencia de calidad baja. Existe una relación inversa entre la rareza de una alteración trombofílica y su penetrancia clí-nica. La susceptibilidad a la ETEV se ve modulada por la presencia o ausencia de historia familiar, lo que puede influir en las decisiones terapéuticas. Es poco probable, debido a la relativa infrecuencia de algunos de los defectos trombofílicos, que se realicen estudios aleatorizados controlados que evalúen el impacto de diversas estrategias de tratamiento en portadores de los mismos.
En términos generales, por la relativa sencillez de su estudio y por la información que apor-tan acerca del conocimiento de la enfermedad, los estudios tienen su validez y, en ciertos casos (sobre todo en caso de deficiencias de anticoagulantes y defectos combinados de po-limorfismos protrombóticos), podría tener utilidad clínica: ampliar el periodo de anticoa-gulación en pacientes para evitar recurrencias (siempre dando un peso relevante a otros elementos clínicos como la historia personal y familiar de trombosis, el carácter idiopático, niveles elevados de dímero D) o prevención del primer evento en sujetos asintomáticos en situaciones de riesgo trombótico. En este sentido, recientemente se ha demostrado que el genotipo y forma de presentación clínica en el sujeto probando influye en el riesgo trombótico de los familiares con FV Leiden o protrombina 20210A26. En ningún caso la identificación de un defecto trombofílico modifica la intensidad, esquema o tipo de tratamiento anticoagulante.
En la práctica diaria asistimos a la solicitud de estudios de trombofilia hereditaria en multitud de situaciones en las que no estarían indicados, lo que implica un considerable consumo de recursos27. Recientemente, dentro del programa Choosing Wisely de la Sociedad Americana de Hematología (ASH), dirigido a mejorar la calidad asistencial, se realizó un posicionamiento claro y rotundo en contra de la realización de estudios de trombofilia en pacientes adultos que hayan sufrido un episodio de ETEV en el contexto de un factor de riesgo transitorio “mayor” (cirugía, trauma o inmovilización prolongada)28.
Los estudios de trombofilia deberían ser solicitados por profesionales de la salud que pue-dan asegurar las siguientes condiciones29:
• Selección apropiada del paciente al que se solicita el estudio.
• Proporcionar consejo previo al procedimiento.
• Interpretación adecuada de los resultados.
• Proporcionar adecuada educación al paciente.
Existe una relación inversa entre la rareza de una alteración trombofílica y su penetrancia clínica26

HEMOSTASIA
Merece la pena destacar, que con la excepción de los AAF, no se recomienda la realización de estudios de trombofilia en pacientes con trombosis en territorio arterial (síndrome co-ronario agudo, ictus isquémico o trombosis arterial periférica).
Otra cuestión de interés es establecer el momento óptimo para la realización del estudio de trombofilia. La Tabla 15 resume y justifica los momentos que deben ser elegidos para realizar estos estudios. Igualmente, en la Figura 7 se muestra un algoritmo para orientar el diagnós-tico biológico de los estados de trombofilia.
Estudios funcionales de proteínas C, S y antitrombina
Evitar el estudio en la fase aguda de la enfermedad
El paciente no debe estar bajo tratamiento con antagonistas de la vitamina K (afecta niveles de PC y PS)
El tratamiento con heparina puede modificar los niveles de AT y la resistencia a la proteína C activada
En caso de estar en tratamiento con antagonistas de la vitamina K, pasa transitoriamente a heparina
Confirmar el diagnóstico a los tres meses (o al menos a las tres semanas de finalizar el tratamiento con antagonistas de la vitamina K, o a los dos meses del parto)
Extracción sanguínea para pruebas moleculares (FV Leiden y protrombina 20210 A)
El estudio se puede realizar en cualquier momento
TABLA 15.INDICACIÓN DEL MOMENTO PARA REALIZAR EL ESTUDIO DE TROMBOFILIA HEREDITARIA
Los anticoagulantes orales de acción directa también interfieren con la medición de los niveles funcionales de AT, PC y PS.

HEMOSTASIA
FIGURA 7.
ALGORITMO DIAGNÓSTICO BIOLÓGICO DE TROMBOFILIA
SAF: síndrome antifosfolípido; AL: Anticoagulante lúpico; AA: Anticuerpos antifosfolípido (Anticuerpos an-ticardiolipina y anti b2-GPI); PS: proteína S; PC: Proteína C; AT: Antitrombina; PT: protrombina; RPCa: resistencia a proteína C activada; AO: Anticonceptivos orales; THS: Terapia hormonal sustitutiva; LES: Lupus eritematoso sistémico; CIE: Cross-inmunoelectroforesis.
Algoritmo diagnóstico biológico de trombofilia
Situación clínica
Estudios específicos de trombofilia primaria (subrayado primera elección)
Primaria
Trombosis venosa:<50 años
Recurrente Familiar
Localización inusualSecundaria a AO, embarazo, THS
ATFuncional
(anti-Fxa/IIa)Antígeno/CIE/
secuenc.
PCFuncional
(cromogénico/coagulativo)
Antígeno
PSFuncional
+ Antígeno libre
RPCaFuncional
genético
PT 20210Genético
SAFALAA
Necrosiscutánea
dicumarínicos
Déficit AT, PC, PS
RPCa/FVLPT 20210
SAF
Sin heparina ni DOACs
Sin cumarínicos (al menos 3 semanas) ni DOACs
Déficit PC Déficit PS Déficit AT SAF
¿otros?
No estudio específico
salvo sospecha
Déficit ATDéficit PC Déficit PS
Evaluación rutinaria: TTPA, TP, TT
Trombosiso púrpuraneonatal
Resistenciaa heparina
1) Abortos repetición2) Trombosis arteriales recidivantes o jóvenes
Asociada a factores trombogénicos
Secundaria o mixta
No realizar en el episodio agudo. Si positivo, confirmar en otra muestra y estudios
familiares susceptibles
Estudios familiares ¿solo defecto homocigoto o combinado?

HEMOSTASIA
CONCLUSIONES
• En las últimas décadas se han identificado nuevos factores trombofílicos, muchos de ellos genéticos, y en los próximos años probablemente surgirán más.
• La identificación de una alteración trombofílica supone un mayor riesgo trombótico, pero no necesariamente ha de ser el responsable del desarrollo de la trombosis ni implicar que el portador vaya a tener un episodio trombótico. De hecho, el riesgo trombótico de cada de-fecto es variable, incluso para una misma alteración, como consecuencia tanto de la hete-rogeneidad molecular como, fundamentalmente, del carácter multigénico y multifactorial de la trombosis venosa. Por ello, el análisis de trombofilia no puede entenderse de forma aislada, sino teniendo en cuenta la historia clínica personal y familiar del enfermo.
• Existen diferentes situaciones que generan estados de trombofilia adquirida. La existencia de anticuerpos antifosfolipídicos, en especial el anticoagulante lúpico, es posiblemente el factor de riesgo más relevante y requiere una detenida atención.

HEMOSTASIA
BIBLIOGRAFÍA
1. Dahlbäck B. Advances in understanding pathogenic mechanisms of thrombophilic disorders. Blood. 2008;112(1):19-27.
2. Souto JC, Almasy L, Borrell M, Blanco-Vaca F, Mateo J, Soria JM, et al. Genetic susceptibility to thrombo-sis and its relationship to physiological risk factors: the GAIT study. Genetic Analysis of Idiopathic Throm-bophilia. Am J Hum Genet. 2000;67(6):1452-9.
3. Middeldorp S. Evidence-based approach to thrombophilia testing. J Thromb Thrombolysis. 2011;31(3):275-81.
4. Mahmoodi BK, Brouwer JL, Ten Kate MK, Lijfering WM, Veeger NJ, Mulder AB, et al. A prospective cohort study on the absolute risks of venous thromboembolism and predictive value of screening asymptomatic relatives of patients with hereditary deficiencies of protein S, protein C or antithrombin. J Thromb Hae-most. 2010;8(6):1193-200.
5. Lijfering WM, Brouwer J-LP, Veeger NJ, Bank I, Coppens M, Middeldorp S, et al. Selective testing for throm-bophilia in patients with first venous thrombosis: results from a retrospective family cohort study on absolute thrombotic risk for currently known thrombophilic defects in 2479 relatives. Blood. 2009;113(21):5314-22.
6. Cooper PC, Coath F, Daly ME, Makris M. The phenotypic and genetic assessment of antithrombin deficien-cy. Int J Lab Hematol. 2011;33(3):227-37.
7. Reitsma PH. Protein C deficiency: from gene defects to disease. Thromb Haemost. 1997;78(1):344-50.
8. D’Angelo A, Vigano D’Angelo S. Protein S deficiency. Haematologica. 2008;93(4):498-501.
9. Baglin T, Gray E, Greaves M, Hunt BJ, Keeling D, Machin S, et al. Clinical guidelines for testing for herita-ble thrombophilia. Br J Haematol. 2010;149(2):209-20.
10. Kyrle PA, Rosendaal FR, Eichinger S. Risk assessment for recurrent venous thrombosis. Lancet. 2010;376(9757):2032-9.
11. Vicente V, González-Conejero R, Rivera J, Corral J. The prothrombin gene variant 20210A in venous and arterial thromboembolism. Haematologica. 1999;84(4):356-62.
12. Morange PE, Trégouët DA. Current knowledge on the genetics of incident venous thrombosis. J Thromb Haemost. 2013;11 Suppl 1:111-21.
13. Miñano A, Ordóñez A, España F, González-Porras JR, Lecumberri R, Fontcuberta J, et al. AB0 blood group and risk of venous or arterial thrombosis in carriers of factor V Leiden or prothrombin G20210A polymor-phisms. Haematologica. 2008;93(5):729-34.
14. Di Minno MN, Tremoli E, Coppola A, Lupoli R, Di Minno G. Homocysteine and arterial thrombosis: Challen-ge and opportunity. Thromb Haemost. 2010;103(5):942-61.

HEMOSTASIA
15. Simone B, De Stefano V, Leoncini E, Zacho J, Martinelli I, Emmerich J, et al. Risk of venous thromboem-bolism associated with single and combined effects of Factor V Leiden, Prothrombin 20210A and Methyle-netethraydrofolate reductase C677T: a meta-analysis involving over 11,000 cases and 21,000 controls. Eur J Epidemiol. 2013;28(8):621-47.
16. Nossent AY, Eikenboom JC, Bertina RM. Plasma coagulation factor levels in venous thrombosis. Semin Hematol. 2007;44(2):77-84.
17. Lluis-Ganella C, Subirana I, Lucas G, Tomás M, Muñoz D, Sentí M, Salas E, Sala J, Ramos R, Ordovas JM, Marrugat J, Elosua R. Assessment of the value of a genetic risk score in improving the estimation of coro-nary risk. Atherosclerosis. 2012; 222(2):456-63.
18. Morange PE, Tregouet DA. Lessons from genome-wide association studies in venous thrombosis. J Thromb Haemost. 2011; 9 Suppl 1:258-64.
19. Venous thromboembolic diseases: the management of venous thromboembolic diseases and the role of thrombophilia testing. NICE Clinical Guideline 144. 2012.
20. Alfonso A, Redondo M, Rubio T, Del Olmo B, Rodríguez-Wilhelmi P, García-Velloso MJ, et al. Screening for occult malignancy with FDG-PET/CT in patients with unprovoked venous thromboembolism. Int J Cancer. 2013; 133(9):2157-64.
21. Sekhar M, McVinnie K, Burroughs AK. Splanchnic vein thrombosis in myeloproliferative neoplasms. Br J Haematol. 2013;162(6):730-47.
22. Giannakopoulos B, Krilis SA. The pathogenesis of the antiphospholipid syndrome. N Engl J Med. 2013;368(11):1033-44.
23. Middeldorp S. Is thrombophilia testing useful? Hematology Am Soc Hematol Educ Program. 2011:150-5.
24. De Stefano V, Rossi E. Testing for inherited thrombophilia and consequences for antithrombotic prophylaxis in patients with venous thromboembolism and their relatives. A review of the Guidelines from Scientific Societies and Working Groups. Thromb Haemost. 2013;110(4):697-705.
25. Varga E, Kujovich J. Management of inherited thrombophilia: guide for genetics professionals. Clin Genet. 2012;81(1):7-17.
26. Bucciarelli P, De Stefano V, Passamonti SM, Tormene D, Legnani C, Rossi E, et al. Influence of proband’s characteristics on the risk for venous thromboembolism in relatives with factor V Leiden or prothrombin G20210A polymorphisms. Blood. 2013;122(15):2555-61.
27. Roldan V, Lecumberri R, Muñoz-Torrero JF, Vicente V, Rocha E, Brenner B, et al. Thrombophilia testing in patients with venous thromboembolism. Findings from the RIETE registry. Thromb Res. 2009;124(2):174-7.
28. Hicks LK, Bering H, Carson KR, Kleinerman J, Kukreti V, Ma A, et al. The ASH Choosing Wisely® campaign: five hematologic tests and treatments to question. Blood. 2013;122(24):3879-83.
29. Moll S. Thrombophilia: clinical-practical aspects. J Thromb Thrombolysis. 2015;39(3):367-78.