
Funciones sturmianas generalizadas en coordenadas … · 2019-07-05 · extrema, llegando al...
80
Dirección: Dirección: Biblioteca Central Dr. Luis F. Leloir, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires. Intendente Güiraldes 2160 - C1428EGA - Tel. (++54 +11) 4789-9293 Contacto: Contacto: bibliotecadigital.exactas.uba.ar Tesis de Grado Funciones sturmianas generalizadas Funciones sturmianas generalizadas en coordenadas esferoidales en coordenadas esferoidales prolatas prolatas López, Faustino Andrés 2017 Este documento forma parte de las colecciones digitales de la Biblioteca Central Dr. Luis Federico Leloir, disponible en bibliotecadigital.exactas.uba.ar. Su utilización debe ser acompañada por la cita bibliográfica con reconocimiento de la fuente. This document is part of the digital collection of the Central Library Dr. Luis Federico Leloir, available in bibliotecadigital.exactas.uba.ar. It should be used accompanied by the corresponding citation acknowledging the source. Cita tipo APA: López, Faustino Andrés. (2017). Funciones sturmianas generalizadas en coordenadas esferoidales prolatas. Facultad de Ciencias Exactas y Naturales. Universidad de Buenos Aires. https://hdl.handle.net/20.500.12110/seminario_nFIS000076_Lopez Cita tipo Chicago: López, Faustino Andrés. "Funciones sturmianas generalizadas en coordenadas esferoidales prolatas". Facultad de Ciencias Exactas y Naturales. Universidad de Buenos Aires. 2017. https://hdl.handle.net/20.500.12110/seminario_nFIS000076_Lopez
Transcript of Funciones sturmianas generalizadas en coordenadas … · 2019-07-05 · extrema, llegando al...

Di r ecci ó n:Di r ecci ó n: Biblioteca Central Dr. Luis F. Leloir, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires. Intendente Güiraldes 2160 - C1428EGA - Tel. (++54 +11) 4789-9293
Co nta cto :Co nta cto : bibliotecadigital.exactas.uba.ar
Tesis de Grado
Funciones sturmianas generalizadasFunciones sturmianas generalizadasen coordenadas esferoidalesen coordenadas esferoidales
prolatasprolatas
López, Faustino Andrés
2017
Este documento forma parte de las colecciones digitales de la Biblioteca Central Dr. Luis FedericoLeloir, disponible en bibliotecadigital.exactas.uba.ar. Su utilización debe ser acompañada por lacita bibliográfica con reconocimiento de la fuente.
This document is part of the digital collection of the Central Library Dr. Luis Federico Leloir,available in bibliotecadigital.exactas.uba.ar. It should be used accompanied by thecorresponding citation acknowledging the source.
Cita tipo APA:
López, Faustino Andrés. (2017). Funciones sturmianas generalizadas en coordenadasesferoidales prolatas. Facultad de Ciencias Exactas y Naturales. Universidad de Buenos Aires.https://hdl.handle.net/20.500.12110/seminario_nFIS000076_Lopez
Cita tipo Chicago:
López, Faustino Andrés. "Funciones sturmianas generalizadas en coordenadas esferoidalesprolatas". Facultad de Ciencias Exactas y Naturales. Universidad de Buenos Aires. 2017.https://hdl.handle.net/20.500.12110/seminario_nFIS000076_Lopez

DEPARTAMENTO DE F ISICA, FACULTAD DECIENCIAS EXACTAS Y NATURALES, UNIVERSIDAD
DE BUENOS AIRES
TESIS DE LICENCIATURA
Funciones Sturmianas Generalizadas enCoordenadas Esferoidales Prolatas
Autor:Faustino Andres Lopez
Director:Darıo Mitnik
Diciembre de 2017

I
Funciones Sturmianas Generalizadas (G.S.F.) enCoordenadas Esferoidales Prolatas
Autor: Faustino Andres Lopez
L.U.: 249/13
Director: Darıo Mitnik
Lugar de Trabajo:Instituto de Astronomıa y Fısica del Espacio IAFE(CONICET-UBA)
Fecha de inicio: Septiembre de 2016
Fecha de finalizacion: Diciembre de 2017
Fecha de examen: 19 de Diciembre de 2017
Informe final aprobado por:
Director: Mitnik Darıo
Tesista: Lopez Faustino A.
Profesores de tesis de licenciatura:V. Bekeris y C. Moreno
Jurado: Arbo Diego
Jurado: Ferraro Marta
Jurado: Llois Ana Marıa

Resumen
A principios de este siglo han surgido diferentes procedimientos teoricos no–perturbativosque permiten resolver en forma completamente cuantica problemas colisionales en atomos sim-ples. La implementacion concreta de estos metodos requiere de una potencia computacionalextrema, llegando al lımite de su aplicacion practica. Aun contando con supercomputadoras ycomputacion de alta performance, los recursos computacionales que demandan son tan altos,que actualmente es practicamente imposible investigar en este marco, a sistemas de mas de trespartıculas.
Cuando nos referimos a problemas fısicos que insumen un costo computacional tan extremo,cualquier metodologıa que reduzca el tamano de los calculos, aun en un porcentaje pequeno, setraduce en un ahorro muy significativo. En algunos casos, esto dictamina la posibilidad concre-ta de que los calculos puedan realizarse o no. Es por eso, que durante los ultimos anos nuestrogrupo de investigacion se ha dedicado al desarrollo e implementacion del metodo de FuncionesSturmianas Generalizadas (GSF). Este metodo presenta una metodologıa elegante y notable-mente eficiente para la solucion de problemas atomicos, tanto de estructura, como colisionales.
Como todo metodo espectral, su fundamento reside en la eleccion de una base, y la repre-sentacion matricial de la ecuacion de Schrodinger en ella. En este marco, la ecuacion diferencialcorrespondiente puede ser resuelta por medio de metodos algebraicos. La principal caracterısti-ca del metodo GSF reside en que la base escogida para la representacion de las funciones deonda, conforma un subespacio reducido, que tiene las propiedades fısicas del problema a con-siderar. De este modo logramos reducir en forma notable las dimensiones de los calculos, dis-minuyendo los recursos computacionales necesarios para la resolucion de estos problemas. Endiversos trabajos, se ha demostrado la factibilidad y la alta eficiencia del metodo, estudiando laestructura y la dinamica de colisiones atomicas en varios sistemas de pocas partıculas.
El objetivo de esta Tesis consiste en extender el rango de aplicacion del metodo GSF a sis-temas moleculares. Para ello, se debe disenar un plan de trabajo que cumpla con las mismasetapas con las que se elaboro el metodo en sistemas atomicos. Estas consisten en el desarrolloteorico y el posterior diseno de los programas computacionales correspondientes. Esto ultimono se limita a la escritura de los codigos, sino al estudio detallado de los diferentes parametrosque se incluyen en los calculos, al analisis de convergencia, y a la evaluacion de los errores ycapacidades del metodo. Para ello, se trabaja con sistemas ligados, de manera de poder contras-tar los resultados con datos conocidos. Solo despues de haber establecido el metodo y su formade implementacion, sera posible pasar a la etapa siguiente, en la cual se estudien problemascolisionales (que ofrecen mayores dificultades en el contraste directo con los experimentos).
II

III
Nuestro trabajo se enmarca en la primer etapa del plan, comenzando con el estudio de lossistemas moleculares mas simples: las moleculas diatomicas. Para ello, es conveniente modifi-car completamente el esquema de calculo, y trabajar en coordenadas esferoidales prolatas, querepresentan naturalmente la geometrıa del sistema. Esto implica un completo desarrollo teori-co, en el cual se debe reformular la teorıa. Y por supuesto, esto implica el desarrollo de nuevosprogramas computacionales que la implementen.
En este trabajo se estudia con particular atencion el ion molecular H+2 , tanto a su estado
fundamental como algunos estados excitados. Ademas de producir resultados muy precisos delas energıas correspondientes, y de estudiar las funciones de onda resultantes, se estudian lasposiciones de los nucleos en equilibrio, y los cambios que se producen al variar los mismos.Tambien se estudian otros iones moleculares diatomicos y monoelectronicos, tales como elHHe+2 y HLi+3.
Desarrollamos dos metodos diferentes dentro del marco GSF. En el primero, se aprovechaque los sistemas estudiados son separables en coordenadas prolatas, por lo que se implementaun metodo iterativo. Las ecuaciones resultantes en cada coordenada son separadas, pero estanacopladas por ciertas constantes de separacion, que asumen inicialmente un valor arbitrario.Se resuelven las ecuaciones, se obtienen nuevos valores de las constantes de separacion, y serepite el proceso hasta obtener convergencia. Explicaremos en detalle como nuestro metodosimplifica notablemente al proceso de iteracion, ya que las funciones GSF traen implıcitamenteresuelta en ellas la mayor parte de las ecuaciones diferenciales. Tambien demostraremos comoal elegir las condiciones asintoticas adecuadas, nuestro metodo tambien acelera notablementela convergencia de estas iteraciones.
El segundo metodo que desarrollamos implica la diagonalizacion directa del Hamiltoniano.Veremos que al escoger las bases Sturmianas generalizadas, la representacion matricial del Ha-miltoniano tiene un rango muy bajo. Esto permite la resolucion del problema en forma numeri-ca, sin necesidad de la utilizacion de grandes recursos computacionales. Con esta implemen-tacion se comprueba claramente como al construir una base en la cual sus funciones absorbenuna gran parte de la fısica del problema a estudiar, los calculos se simplifican notoriamente.

Indice general
1 La molecula de Hidrogeno ionizada 11.1 Antecedentes historicos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11.2 La ecuacion de onda electronica . . . . . . . . . . . . . . . . . . . . . . . . . 31.3 H+
2 en coordenadas prolatas . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51.4 Estado del Arte . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
2 Funciones Sturmianas Generalizadas 82.1 Teorıa . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
2.1.1 Metodos numericos para ecuaciones diferenciales . . . . . . . . . . . . 82.1.2 Funciones Sturmianas . . . . . . . . . . . . . . . . . . . . . . . . . . . 92.1.3 Funciones Sturmianas Coulombianas . . . . . . . . . . . . . . . . . . . 102.1.4 Funciones Sturmianas Generalizadas . . . . . . . . . . . . . . . . . . . 132.1.5 Implementacion: Estados ligados . . . . . . . . . . . . . . . . . . . . . 142.1.6 Implementacion: Estados Continuos . . . . . . . . . . . . . . . . . . . 172.1.7 Problemas Colisionales . . . . . . . . . . . . . . . . . . . . . . . . . . 18
2.2 Aplicaciones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
3 GSF en Esferoidales Prolatas 213.1 Metodo Iterativo (1d) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
3.1.1 Ecuacion angular . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 223.1.2 Ecuacion radial . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
3.2 Metodo Directo (2d) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
4 Resultados 304.1 Metodo Iterativo (1d) para H+
2 . . . . . . . . . . . . . . . . . . . . . . . . . . 304.1.1 Ecuacion angular . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 304.1.2 Ecuacion radial . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 314.1.3 Funciones de Onda . . . . . . . . . . . . . . . . . . . . . . . . . . . . 344.1.4 Distancia internuclear de equilibrio . . . . . . . . . . . . . . . . . . . . 364.1.5 Lımite atomico . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
4.2 Estados excitados H+2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 40
4.3 Metodo Directo (2d): estados excitados del H+2 . . . . . . . . . . . . . . . . . 46
4.4 Otros iones: HHe+2 y HLi+3 . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
IV

INDICE GENERAL V
5 Conclusiones 51
A Combinaciones Lineales de Orbitales Atomicos 53
B Nomenclatura 58
C Coordenadas esferoidales prolatas 62C.1 Laplaciano . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63
D Construccion Numerica de las GSF 65
E Polinomios de Legendre 68

Capıtulo 1
La molecula de Hidrogeno ionizada
1.1. Antecedentes historicosEl problema de la molecula de hidrogeno ionizado tiene una gran importancia ya que cons-
tituye el “atomo de hidrogeno” de las moleculas. Esto ocurre no solo por su simplicidad, sinoporque en las coordenadas adecuadas es separable. Dicha caracterıstica permite que la ecuacionde Schrodinger pueda ser reducida a ecuaciones diferenciales ordinarias, con posibles solucio-nes.
Evitaremos entrar en el debate acerca de si se ha alcanzado o no, una solucion exacta paraesta molecula. Existen quienes dicen que este problema sı admite solucion analıtica, y quienesdicen que cualquier ecuacion diferencial define un polinomio, al que se puede llamar “solucionanalıtica”. En las coordenadas adecuadas tambien se logran relaciones de recursion definidasentre los coeficientes de estos polinomios, lo que podrıa constituir, en principio una “solucionexacta”.
En 1907 J.J. Thomson [1] descubrio que el ion H+2 es una molecula estable. En experimentos
con hidrogeno, observo iones positivos, cuya relacion entre masa y carga era igual a 2.Brasefield [2] observo 45 lıneas de emision en 1927, pero luego se retracto. El ion H+
2 plan-tea una dificultad esencial, ya que no permite la comparacion entre las predicciones teoricas delas energıas de los estados excitados con los resultados experimentales. A diferencia de los sis-temas atomicos, los niveles energeticos de esta molecula no se puede determinar espectroscopi-camente. Esto ocurre porque muchas de las transiciones que deberıan detectarse, relacionan aestados antiligantes. Otro gran grupo de transiciones que involucran estados ligados, son opti-camente prohibidas. Finalmente, las unicas transiciones que son permitidas, y ocurren entreestados ligantes, relacionan estados con distancias internucleares de equilibrio muy diferentes.
El problema teorico fue abordado desde los inicios de la fısica atomica por Pauli [3] yNiessen [4], utilizando la vieja teorıa cuantica de Sommerfield–Wilson. Entre los anos 1923 y1926, se sucedieron una serie de trabajos teoricos y experimentales, estableciendo la energıa deionizacion de la molecula con diferentes valores, entre 11 eV y 23 eV.
Si bien se adjudica a Burrau el primer calculo cuantico del H+2 , la primer publicacion corres-
ponde a Alexandrow [5], en 1926, quien demostro la separabilidad de la solucion. Sin embargo,sus resultados teoricos no coincidıan con los experimentos, Burrau [6], obtiene en 1927 una
1

CAPITULO 1. LA MOLECULA DE HIDROGENO IONIZADA 2
energıa de ligadura de -0.6024 a.u., y una separacion internuclear de 2.0 a.u., en concordanciacon los experimentos.
Con el transcurrir de los anos, este problema fue abordado repetidamente empleando losprincipales metodos que utiliza la cuantica, es decir la solucion directa de la ecuacion deSchrodinger, la teorıa de perturbaciones, la aproximacion WKB, y los metodos variacionales.
Las primeras soluciones directas del estado fundamental, fueron realizadas por Burrau [6](1927), Wilson [7] (1928), Hylleraas [8] (1931), Jaffe [9] (1934) y Bates [10] (1953), quienademas fue el primero en publicar las funciones de onda electronicas. En lo que respecta a losestados excitados, los primeros resultados fueron obtenidos por Teller [11] (1930), Steensholt[12] (1936), Chakravarty [13] (1939) y Bates et al. [14] (1968).

CAPITULO 1. LA MOLECULA DE HIDROGENO IONIZADA 3
1.2. La ecuacion de onda electronicaLa molecula del ion hidrogenoH+
2 consiste en dos nucleos identicos (protones) y un electron.
Figura 1.1: Geometrıa de la molecula ionizada H+2 con sus dos protones separados una distancia
R y el electron a una distancia r1 del proton 1 y r2 del proton 2.
El potencial de interaccion Epot entre las tres partıculas es
Epot(r1, r2, R) = − 1
r1− 1
r2+
1
R. (1.1)
donde ri son las distancias entre el electron y los nucleos i, y R la distancia internuclear. Se-leccionamos el punto medio entre los dos protones como origen de coordenadas, por lo tantoR1 = −R2. Se obtienen las siguientes relaciones:
r1 = r−R1 ; r2 = r−R2 ; R = |R2 −R1|. (1.2)
Y (1.1) resulta:
Epot(r,R1,R2) = − 1
|r−R1|− 1
|r−R2|+
1
|R2 −R1|. (1.3)
La ecuacion de Schrodinger de este problema de tres partıculas, con dependencia explıcitade los vectores posicion de cada una de ellas, resulta:
H(r,R1,R2)ψ(r,R1,R2) = E ψ(r,R1,R2) , (1.4)
donde
H(r,R1,R2) = − 1
2M∇2(R1)−
1
2M∇2(R2)−
1
2∇2(r) + Epot(r,R1,R2) . (1.5)
Los primeros dos terminos del Hamiltoniano, denotan la energıa cinetica de los nucleos, y eltercer termino, la energıa cinetica del electron.
CAPITULO 1. LA MOLECULA DE HIDROGENO IONIZADA 4
La ecuacion (1.4) no posee solucion analıtica, y al tener nueve variables independientes suestudio resulta complicado. Por tal motivo, en estos problemas se utiliza la aproximacion deBohr–Oppenheimer, que consiste en separar los movimientos de los nucleos del movimientodel electron. Esto es razonable, ya que al ser la diferencia de masas tan grandes, podemosdespreciar en principio la energıa cinetica de los nucleos, y fijar la distancia internuclear Rcomo un parametro libre. Bajo esta aproximacion, y fijando los protones segun R1 = −R
2k y
R2 = R2k, la ecuacion de Schrodinger (1.4) resulta:[
−1
2∇2(r) + Epot(r, R)
]ψ(r, R) = E(R)ψ(r, R) . (1.6)
conEpot(r, R) = − 1
|r + R2k|− 1
|r− R2k|
+1
R. (1.7)
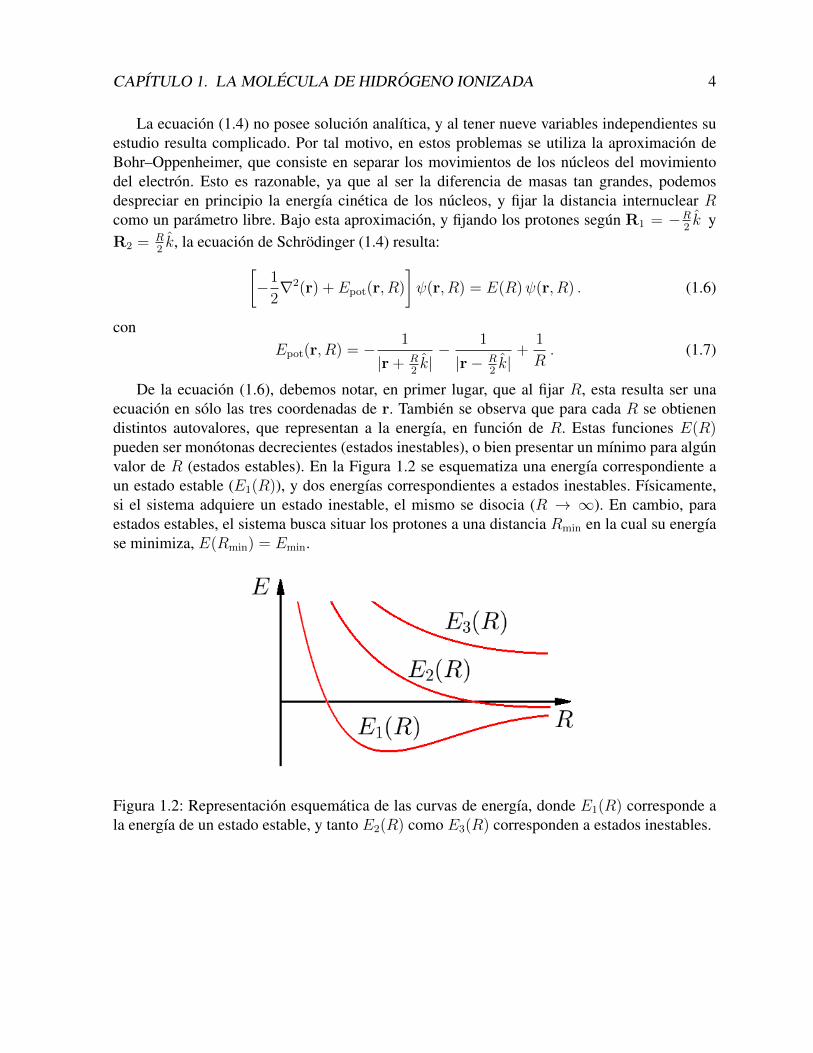
De la ecuacion (1.6), debemos notar, en primer lugar, que al fijar R, esta resulta ser unaecuacion en solo las tres coordenadas de r. Tambien se observa que para cada R se obtienendistintos autovalores, que representan a la energıa, en funcion de R. Estas funciones E(R)pueden ser monotonas decrecientes (estados inestables), o bien presentar un mınimo para algunvalor de R (estados estables). En la Figura 1.2 se esquematiza una energıa correspondiente aun estado estable (E1(R)), y dos energıas correspondientes a estados inestables. Fısicamente,si el sistema adquiere un estado inestable, el mismo se disocia (R → ∞). En cambio, paraestados estables, el sistema busca situar los protones a una distancia Rmin en la cual su energıase minimiza, E(Rmin) = Emin.
Figura 1.2: Representacion esquematica de las curvas de energıa, donde E1(R) corresponde ala energıa de un estado estable, y tanto E2(R) como E3(R) corresponden a estados inestables.

CAPITULO 1. LA MOLECULA DE HIDROGENO IONIZADA 5
1.3. H+2 en coordenadas prolatas
Una de las propiedades fundamentales que tiene la molecula de hidrogeno ionizada, es quesu ecuacion de onda electronica es separable en coordenadas prolatas:
ξ =r1 + r2R
; η =r1 − r2R
; φ = arctan(yx
). (1.8)
donde 1 ≤ ξ < ∞, −1 ≤ η < 1 y 0 ≤ φ < 2π. En el Apendice C se demuestra como sederiva el Laplaciano para cualquier sistema de coordenadas ortogonal, y en particular para estesistema de coordenadas:
∇2 =4
R2(ξ2 − η2)
{∂
∂ξ
[(ξ2 − 1)
∂
∂ξ
]+
∂
∂η
[(1− η2) ∂
∂η
]+
+ξ2 − η2
(ξ2 − 1)(1− η2)∂2
∂φ2
}. (1.9)
En coordenadas prolatas, la ecuacion de Schrodinger independiente del tiempo para loselectrones queda dada por{
− 2
R2(ξ2 − η2)
[∂
∂ξ(ξ2 − 1)
∂
∂ξ+
∂
∂η(1− η2) ∂
∂η+
+ξ2 − η2
(ξ2 − 1)(1− η2)∂2
∂φ2
]+ V
}ψ(ξ, η, φ) = Eψ (ξ, η, φ) , (1.10)
donde el potencial
V (r1, r2) = −Z1
r1− Z2
r2. (1.11)
se escribe en coordenadas prolatas como
V (ξ, η) = − 2
R
(Z1 + Z2)ξ − (Z1 − Z2)η
(ξ2 − η2). (1.12)
Para el caso particular del H+2 se cumple que (Z1 = Z2 = 1), por lo que
V (ξ, η) = − 4
R
ξ
(ξ2 − η2). (1.13)
Esta forma del potencial es la caracterıstica principal que favorece el tratamiento del ion H+2
en coordenadas prolatas. En este caso, la funcion de onda se puede expresar como un productode tres funciones separables:
ψ(ξ, η, φ) = U(ξ)Λ(η)Φ(φ) . (1.14)
La funcion azimutal Φ es la mas simple de resolver, y su tratamiento es completamenteanalogo al que se hace en el atomo de hidrogeno, donde se cumple que
d2Φ
dφ2+m2Φ = 0 , (1.15)

CAPITULO 1. LA MOLECULA DE HIDROGENO IONIZADA 6
y la solucion de esta ecuacion es
Φ(φ) =1√2π
eimφ , (1.16)
donde m = 0,±1,±2,±3, · · · .Una vez separada la parte azimutal, queda el producto de las funciones prolatas, cuya ecua-
cion cumple con{∂
∂ξ
[(ξ2 − 1)
∂
∂ξ
]+R(Z1 + Z2)ξ +
R2E
2ξ2 − m2
ξ2 − 1+ (1.17)
+∂
∂η
[(1− η2) ∂
∂η
]−R(Z1 − Z2)η −
R2E
2η2 − m2
1− η2
}U(ξ)Λ(η) = 0 .
Escrita de esta manera, es obvio que encontramos una parte que solo depende de la coordenadaξ, mientras otra solo depende de η. Esto implica que ambas partes deben ser igual a una cons-tante de separacion, que denominamosA. De esta manera quedan determinadas dos ecuaciones,la que corresponde a U(ξ) se denomina ecuacion radial, mientras que la que corresponde a Λ(η)se denomina ecuacion angular. Las ecuaciones a resolver son[
∂
∂ξ
[(ξ2 − 1
) ∂∂ξ
]+ a2ξ − p2ξ2 −
m2
(ξ2 − 1)+ A
]U(ξ) = 0 , (1.18)[
∂
∂η
[(1− η2
) ∂∂η
]− a1η + p2η2 − m2
(1− η2)− A
]Λ(η) = 0 , (1.19)
donde definimos p2 = −R2E2
, a1 = R(Z1 − Z2) y a2 = R(Z1 + Z2).Las ecuaciones no estan completamente desacopladas sino que estan vinculadas tanto por
el parametro p (que representa a la energıa escaleada) como por la constante de separacion A.Ninguna de estas ecuaciones representa un problema simple de autovectores, ya que tanto pcomo A estan, en principio, indeterminados. Existen varios metodos propuestos para resolvereste problema, destacamos en particular la forma empleada por Hylleraas [8] para encontrar lassoluciones angulares, y el metodo propuesto por Jafee [9], para resolver las radiales.

CAPITULO 1. LA MOLECULA DE HIDROGENO IONIZADA 7
1.4. Estado del ArteAntes de presentar nuestros resultados, es interesante hacer un resumen de los mejores datos
disponibles. Madsen y Peek [15] realizaron en 1970, uno de los calculos mas precisos de lasenergıas del H+
2 , empleado hasta el dıa de hoy como referencia. Esto fue logrado utilizando unaCDC 6600 (la primer supercomputadora de la historia) que implemento por primera vez calcu-los de doble precision. Solucionaron la ecuacion de Schrodinger en coordenadas esferoidalesprolatas, haciendo separacion de variables tal como explicamos en la seccion anterior. Para re-solver la ecuacion radial, realizaron una expansion en serie de potencias hasta orden 47. Pararesolver la ecuacion angular, utilizaron una base de 16 Polinomios asociados de Legendre.
En 2006, Scott et al. [16], implementan el metodo de matematica experimental para hallarun patron analıtico de la solucion de este problema. La idea principal de este metodo es reex-pandir las series resultantes de las soluciones, de manera tal que los coeficientes de esta nuevaexpansion tengan un comportamiento decreciente. Con este metodo, se obtiene una expresionanalıtica (aproximada) de la constante de separacion A en funcion de la energıa p, obteniendoexcelentes resultados en las energıas.
En 2014, Xue–Bin Bian [17] implementa el metodo de Crank–Nicolson con propagacion entiempo imaginario, para obtener tanto las energıas como las funciones de onda del ion molecularH+
2 . Dado que este metodo permite calcular individualmente un estado especıfico (escogiendoun valor de energıa cercano), permite el calculo de estados ligados y tambien del continuo. Enla implementacion numerica del metodo, realiza una expansion de la solucion en 80 B–splinesde orden 7 para la ecuacion radial, y 20 B–splines de orden 7 para la angular. Sus resultadosson extremadamente precisos.
En 2015, Kereselidze et al. [18, 19], generan una base de Funciones Sturmianas Coulombia-nas (CSF) para cada nucleo, resolviendo las ecuaciones Sturmianas correspondientes en coorde-nadas prolatas. Con dichas bases resuelven el problema directo. La eleccion de CSF representauna ventaja respecto a los trabajos anteriores, ya que se consiguen resultados precisos con unabase de 10 funciones por nucleo.
Los resultados obtenidos por todos estos metodos, para la energıa del estado fundamentalde H+
2 se presentan en la Tabla 1.1.
Ref Nro Elementos de base Energıa (a.u.)[15] 47 X 16 -1.1026342144949[16] – -1.102634211[17] 80 X 20 -1.1026342144949[18] 10 CSF por nucleo -1.10261
Este trabajo 6 X 3 -1.1026340
Tabla 1.1: Principales resultados de la energıa del estado fundamental 1σg de H+2 , con R = 2
a.u..

Capıtulo 2
Funciones Sturmianas Generalizadas
En este capıtulo se expone el metodo numerico de Funciones Sturmianas Generalizadaso, por sus siglas en ingles, GSF. En primer lugar, presentaremos los lineamientos basicos dela teorıa. Luego, explicaremos como se implementa esto en los metodos numericos y codigoscomputacionales. Finalmente, nombraremos brevemente algunas de las aplicaciones realizadaspor nuestro grupo de investigacion.
2.1. Teorıa
2.1.1. Metodos numericos para ecuaciones diferencialesPodemos agrupar a los metodos numericos que se utilizan para resolver ecuaciones diferen-
ciales e integrales, en tres grandes grupos:
1. Diferencias Finitas:Estos algoritmos se basan en la discretizacion de las coordenadas, y la representacionde las derivadas por medio de matrices que solo conectan a los puntos proximos (segunel orden de aproximacion de las derivadas). Tienen como ventaja fundamental que sonfacilmente controlables (a traves del tamano de la discretizacion), y son muy utiles paraobtener una estimacion inicial de los resultados. Dado que las discretizaciones solo co-nectan elementos vecinos, las matrices resultantes son ralas. Su gran desventaja radica enlos tamanos de las matrices, que torna imposible su manipulacion, y solo resulta practicoen problemas con muy bajas dimensiones. Una forma de sobreponerse a estas dificultadesconsiste en utilizar metodos especiales para matrices dispersas.
2. Metodos Espectrales:Se basan en representar la solucion incognita por una combinacion lineal de funcionesconocidas. Cuando se reemplaza esta expansion en la ecuacion a resolver, se obtienenecuaciones algebraicas que en general se pueden representar con matrices llenas. La ven-taja principal de estos metodos es su alta precision. En algunos casos, las funciones debase son de una forma determinada, de manera que las integrales resultantes, al calcu-lar los elementos de matriz, tienen una expresion analıtica. Esta caracterıstica se emplea
8

CAPITULO 2. FUNCIONES STURMIANAS GENERALIZADAS 9
fundamentalmente en el tratamiento de problemas moleculares, mediante el uso de basesGaussianas.
3. Elementos Finitos:Estos metodos son similares en su filosofıa a los metodos espectrales, pero, al igual queen los metodos de diferencias finitas, se emplean sub–intervalos. Las funciones de baserepresentan a la solucion localmente dentro de estos intervalos, generalmente a traves depolinomios. Fuera de estos intervalos, las funciones de base se anulan. Estos metodospresentan como ventaja que, dado que las funciones de base son diferentes de cero enuna region pequena, las matrices resultantes son ralas, se pueden resolver en un tiempomucho menor al que requieren las matrices densas, y requieren mucho menos memoria.Si bien estos metodos tienen menos precision que los metodos espectrales, son muy utilesen casos de geometrıas irregulares, donde el dominio se puede descomponer en muchossubespacios.
Esta clasificacion no debe ser tomada muy rigurosamente, ya que existen numerosos al-goritmos que combinan componentes de los distintos metodos. En general, cuando se tienenproblemas dependientes del tiempo y del espacio, se resuelve la parte espacial del problemamediante metodos espectrales, y se propaga en el tiempo con diferencias finitas. Nuestro meto-do de GSF es un metodo espectral, porque utilizamos bases Sturmianas. Pero por otra parte,generamos estas bases mediante diferencias finitas.
2.1.2. Funciones SturmianasPara resolver un problema atomico con metodos espectrales, es logico proponer como fun-
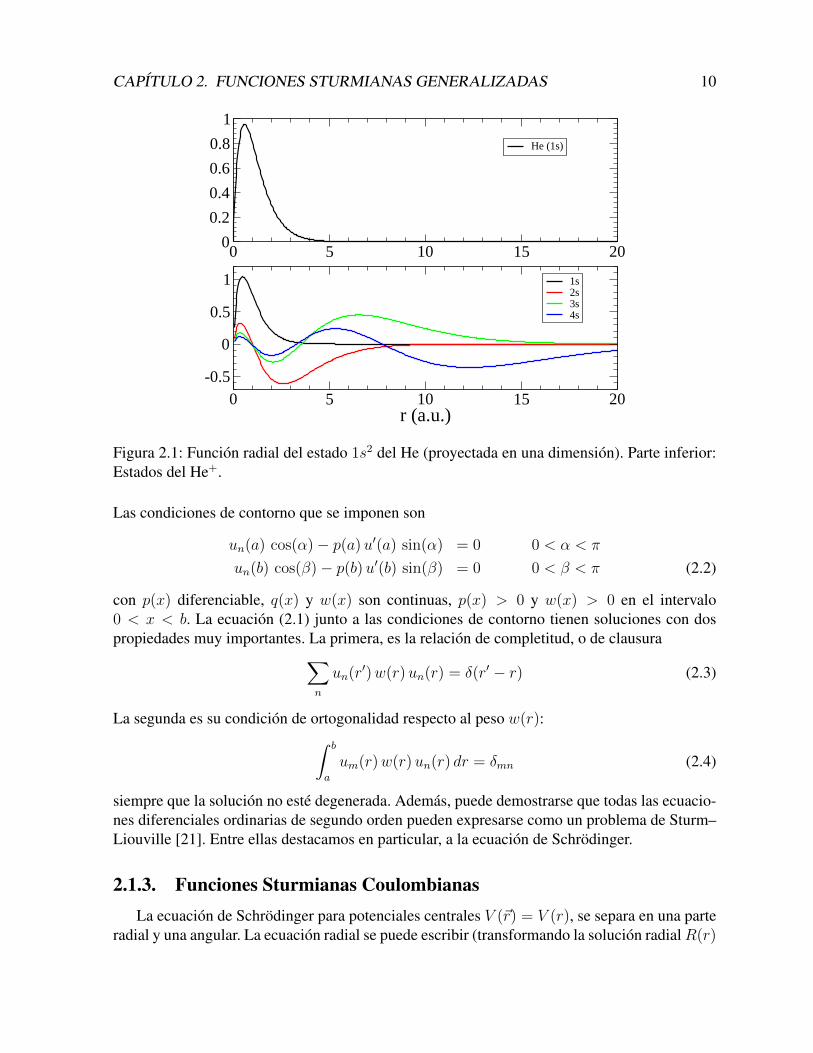
ciones bases a las funciones hidrogenicas, que incluyen, en forma natural, las caracterısticasmas importantes del problema a tratar. Sin embargo, muy rapidamente se revelo que estas fun-ciones no son adecuadas. En primer lugar, los estados ligados del hidrogeno no forman una basecompleta. La completitud ocurre cuando se agrega el continuo, lo que dificulta enormementelos calculos. En segundo lugar, los orbitales hidrogenicos se extienden espacialmente con uncomportamiento proporcional a n2. Al aumentar este numero cuantico, las funciones hidrogeni-cas se alejan del nucleo, y no pueden servir como base. Para ilustrar esto, mostramos, en la partesuperior de la Figura 2.1, la solucion radial del estado fundamental del Helio (proyectada en unadimension). En la parte inferior de la misma figura, se muestran los primeros estados ligadosdel He hidrogenico (He+). Claramente, se ve que a medida que aumenta el numero cuanticoprincipal, las funciones hidrogenicas son muy similares en el origen, y presentan sus maximoscada vez a mayores distancias. Ciertamente, no pueden expandir en forma eficiente un estadoligado como el 1s2 del He, que se encuentra contenido en solo 5 a.u..
Para rectificar esta deficiencia, Høloien, Shull and Lowdin [20] propusieron en 1959, loque hoy se conoce como funciones Sturmianas. Estas son funciones que conforman una basecompleta, que obedecen ciertas condiciones de contorno. Las funciones Sturmianas adquierensu nombre por ser las soluciones de la ecuacion de Sturm–Liouville, que se escribe como[
d
dr
(p(r)
d
dr
)− q(r) + λnw(r)
]un(r) = 0. (2.1)
CAPITULO 2. FUNCIONES STURMIANAS GENERALIZADAS 10
0 5 10 15 200
0.2
0.4
0.6
0.8
1
He (1s)
0 5 10 15 20r (a.u.)
-0.5
0
0.5
1 1s2s3s4s
Figura 2.1: Funcion radial del estado 1s2 del He (proyectada en una dimension). Parte inferior:Estados del He+.
Las condiciones de contorno que se imponen son
un(a) cos(α)− p(a)u′(a) sin(α) = 0 0 < α < π
un(b) cos(β)− p(b)u′(b) sin(β) = 0 0 < β < π (2.2)
con p(x) diferenciable, q(x) y w(x) son continuas, p(x) > 0 y w(x) > 0 en el intervalo0 < x < b. La ecuacion (2.1) junto a las condiciones de contorno tienen soluciones con dospropiedades muy importantes. La primera, es la relacion de completitud, o de clausura∑
n
un(r′)w(r)un(r) = δ(r′ − r) (2.3)
La segunda es su condicion de ortogonalidad respecto al peso w(r):∫ b
a
um(r)w(r)un(r) dr = δmn (2.4)
siempre que la solucion no este degenerada. Ademas, puede demostrarse que todas las ecuacio-nes diferenciales ordinarias de segundo orden pueden expresarse como un problema de Sturm–Liouville [21]. Entre ellas destacamos en particular, a la ecuacion de Schrodinger.
2.1.3. Funciones Sturmianas CoulombianasLa ecuacion de Schrodinger para potenciales centrales V (~r) = V (r), se separa en una parte
radial y una angular. La ecuacion radial se puede escribir (transformando la solucion radialR(r)

CAPITULO 2. FUNCIONES STURMIANAS GENERALIZADAS 11
en una solucion reducida P (r) ≡ rR(r) ) como:[−1
2
∂2
∂r2+l(l + 1)
2r2+ V (r)
]Pnl(r) = En Pnl(r) . (2.5)
Para el problema Coulombiano, esta ecuacion se escribe[−1
2
∂2
∂r2+l(l + 1)
2r2− Z 1
r
]Pnl(r) = En Pnl(r) .
Esta ecuacion es un problema de autovalores, donde lo que debemos hallar son los autovaloresEn, y las autofunciones Pnl(r). Los terminos marcados en rojo son la carga Z (que podemosconsiderar un parametro fijo), y la energıa En. Las soluciones de este problema son
Pnl(r) = rNnl (2Z
nr)l e−
Znr1F1(l + 1− n|2l + 2|2Z
nr) , (2.6)
donde
Nnl =2(Z
n)3/2
(2l + 1)!
√(l + n)!
n(n− l − 1)!, (2.7)
es un factor de normalizacion, y 1F1(a|b|x) es la funcion hipergeometrica confluente. Algunasde estas soluciones son las que hemos presentado en la Figura 2.1.
Las funciones Sturmianas Coulombianas se generan haciendo un simple intercambio entrelos terminos marcados en rojo. Es decir, se intercambian los roles de la carga y la energıa. Laenergıa deja de ser un autovalor, y pasa a ser un parametro fijo Es. En su lugar, la carga pasa aser el autovalor del problema:[
−1
2
∂2
∂r2+l(l + 1)
2r2− Es
]Scnl(r) = Znl
1
rScnl(r) . (2.8)
Las autofunciones de este problema Scnl(r) se denominan Funciones Sturmianas Coulom-bianas (CSF). Esta sustitucion fue estudiada por primera vez por M. Rotenberg [22, 23], para elcalculo de problemas colisionales del tipo (e−−H) y (e+−H). La ecuacion Sturmiana Coulom-biana (2.8) tiene solucion analıtica, y las autofunciones son iguales a las (2.6), exceptuando elcambio de Z
n⇔ k, donde k2
2= Es:
Scnl(r) = rNnl (2kr)l e−kr 1F1(l + 1− n|2l + 2|2kr) , (2.9)
y
Nnl =2(k)3/2
(2l + 1)!
√(l + n)!
n(n− l − 1)!. (2.10)
Como puede verse, la energıa tiene un rol particular, que permite contraer las solucionesa traves del factor k en el exponente de (2.9). Para ilustrar esta particularidad, mostraremoscomo se contraen estas soluciones en los primeros estados ligados del hidrogeno. En la Figura
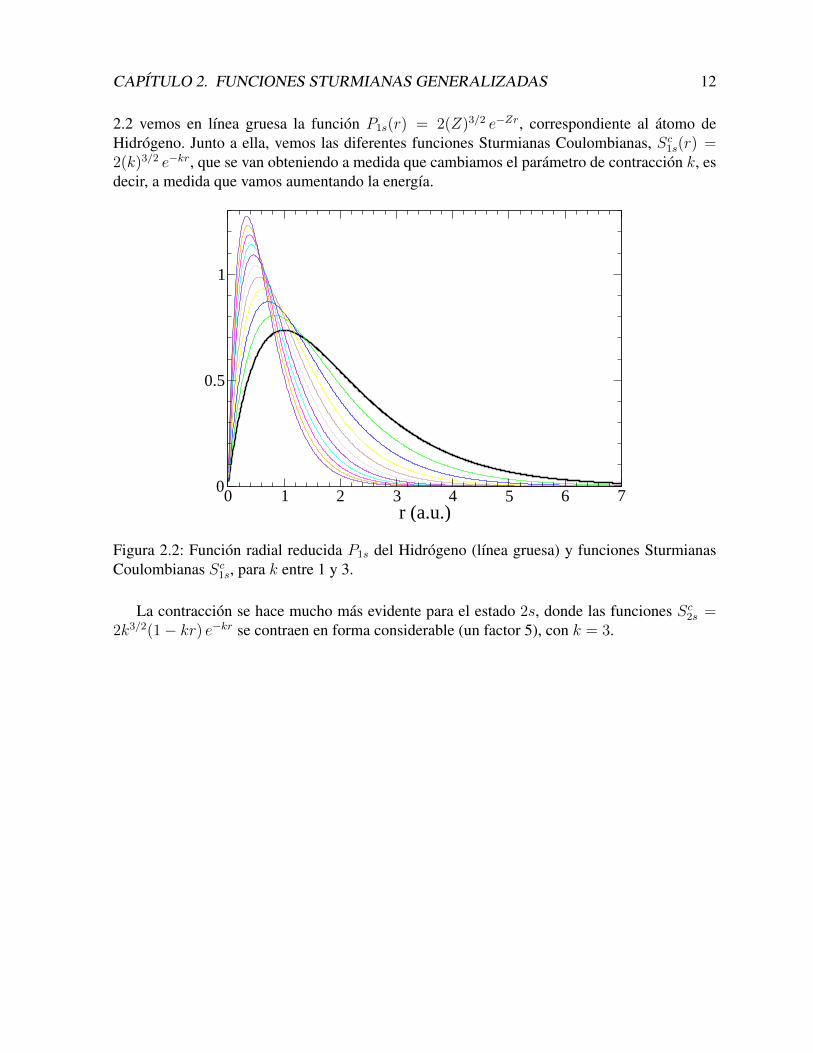
CAPITULO 2. FUNCIONES STURMIANAS GENERALIZADAS 12
2.2 vemos en lınea gruesa la funcion P1s(r) = 2(Z)3/2 e−Zr, correspondiente al atomo deHidrogeno. Junto a ella, vemos las diferentes funciones Sturmianas Coulombianas, Sc1s(r) =2(k)3/2 e−kr, que se van obteniendo a medida que cambiamos el parametro de contraccion k, esdecir, a medida que vamos aumentando la energıa.
0 1 2 3 4 5 6 7r (a.u.)
0
0.5
1
Figura 2.2: Funcion radial reducida P1s del Hidrogeno (lınea gruesa) y funciones SturmianasCoulombianas Sc1s, para k entre 1 y 3.
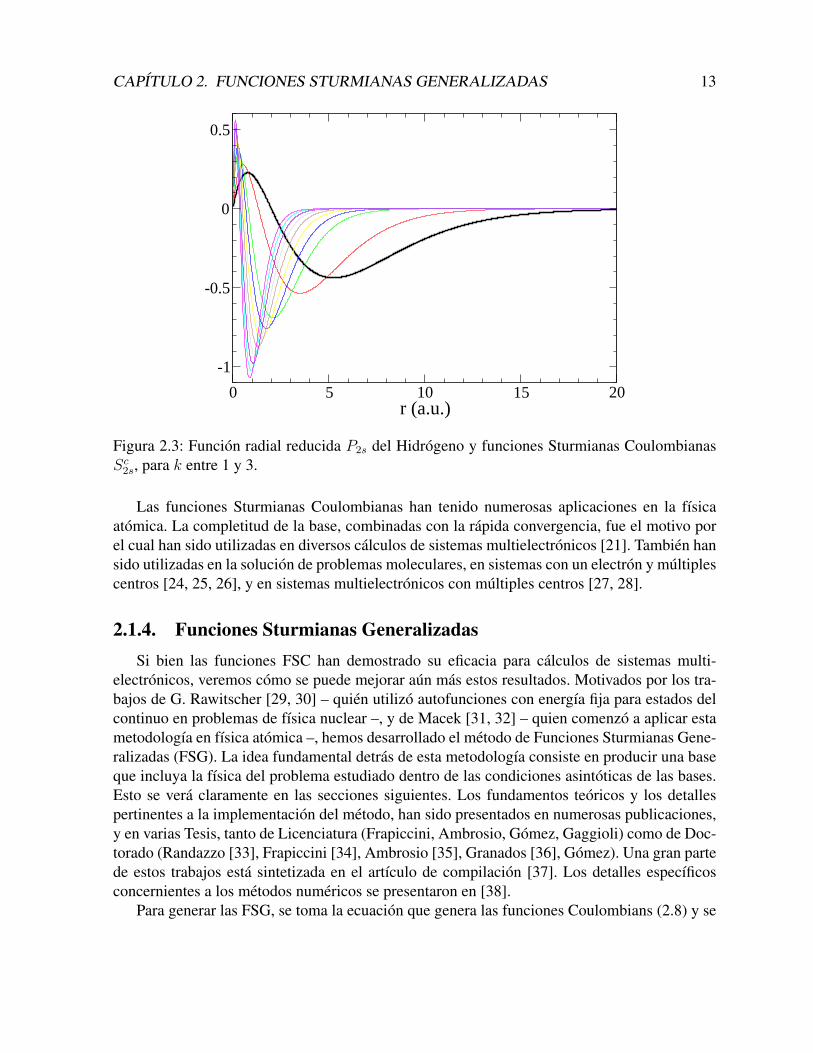
La contraccion se hace mucho mas evidente para el estado 2s, donde las funciones Sc2s =2k3/2(1− kr) e−kr se contraen en forma considerable (un factor 5), con k = 3.
CAPITULO 2. FUNCIONES STURMIANAS GENERALIZADAS 13
0 5 10 15 20r (a.u.)
-1
-0.5
0
0.5
Figura 2.3: Funcion radial reducida P2s del Hidrogeno y funciones Sturmianas CoulombianasSc2s, para k entre 1 y 3.
Las funciones Sturmianas Coulombianas han tenido numerosas aplicaciones en la fısicaatomica. La completitud de la base, combinadas con la rapida convergencia, fue el motivo porel cual han sido utilizadas en diversos calculos de sistemas multielectronicos [21]. Tambien hansido utilizadas en la solucion de problemas moleculares, en sistemas con un electron y multiplescentros [24, 25, 26], y en sistemas multielectronicos con multiples centros [27, 28].
2.1.4. Funciones Sturmianas GeneralizadasSi bien las funciones FSC han demostrado su eficacia para calculos de sistemas multi-
electronicos, veremos como se puede mejorar aun mas estos resultados. Motivados por los tra-bajos de G. Rawitscher [29, 30] – quien utilizo autofunciones con energıa fija para estados delcontinuo en problemas de fısica nuclear –, y de Macek [31, 32] – quien comenzo a aplicar estametodologıa en fısica atomica –, hemos desarrollado el metodo de Funciones Sturmianas Gene-ralizadas (FSG). La idea fundamental detras de esta metodologıa consiste en producir una baseque incluya la fısica del problema estudiado dentro de las condiciones asintoticas de las bases.Esto se vera claramente en las secciones siguientes. Los fundamentos teoricos y los detallespertinentes a la implementacion del metodo, han sido presentados en numerosas publicaciones,y en varias Tesis, tanto de Licenciatura (Frapiccini, Ambrosio, Gomez, Gaggioli) como de Doc-torado (Randazzo [33], Frapiccini [34], Ambrosio [35], Granados [36], Gomez). Una gran partede estos trabajos esta sintetizada en el artıculo de compilacion [37]. Los detalles especıficosconcernientes a los metodos numericos se presentaron en [38].
Para generar las FSG, se toma la ecuacion que genera las funciones Coulombians (2.8) y se

CAPITULO 2. FUNCIONES STURMIANAS GENERALIZADAS 14
realiza la siguiente modificacion[−1
2
∂2
∂r2+l(l + 1)
2r2− Es + Uaux(r)
]Snl(r) = −βnl Vgen(r)Snl(r) , (2.11)
en la cual el potencial Coulombiano ha sido reemplazado por un potencial Vgen, de corto al-cance, llamado potencial generador, y se ha agregado, del lado izquierdo un potencial Uaux
llamado potencial auxiliar. Las soluciones Snl que se obtienen, conforman la base de funcionesSturmianas generalizadas, que cumplen con la condicion de completitud y ortogonalidad (2.4)con el potencial generador como funcion de peso. Los algoritmos numericos que utilizamospara resolver la ecuacion (2.11), se encuentran brevemente detallados en el Apendice D.
La inclusion de estos potenciales producen dos efectos fundamentales:
1. El potencial generador permite controlar la contraccion de las bases.
2. El potencial auxiliar, permite determinar las condiciones asintoticas de las bases.
Para ilustrar el primer efecto, tomemos como ejemplo un potencial Coulombiano. A partirde ahora, facilitaremos la notacion eliminando los ındices correspondientes al numero cuanti-co angular l (que suponemos fijo, por lo cual su posterior inclusion es trivial). Sin perder lageneralidad, tomaremos el caso l = 0. La Figura 2.4 muestra en primer lugar las primeras 10funciones ligadas Pn del hidrogeno. Como se aprecia en la figura, estas se extienden hasta 250a.u. En la parte media de la figura, se muestran las 10 primeras funciones de la base CSF, dondehemos utilizado la ecuacion (2.8), con el parametro de energıa E = −2 a.u.. Estas funcionesestan localizadas en un rango aproximadamente igual a n (en este caso, hasta r = 10 a.u.), y sonmuy diferentes a cortas distancias. En la parte inferior de la figura, se muestran las funcionesGSF, correspondientes a la misma energıa E = −2 a.u., con un potencial auxiliar
Uaux(r) = −Zr
(2.12)
con Z = 0, y un potencial generador de Yukawa
Vgen(r) = −e−αr
r(2.13)
con α = 0,5. En este caso la base se contrae hasta 4 a.u., y podrıa hacerse aun mas compacta,aumentando el valor de α.
El segundo efecto, que trata acerca de la determinacion de las condiciones asintotitas, secomprendera mejor en el marco de las soluciones del continuo, que veremos mas adelante.
2.1.5. Implementacion: Estados ligadosEl metodo GSF es espectral. Esto significa, que la solucion buscada se expande en la base
elegida. Para simplificar la explicacion, trataremos un problema de dos cuerpos, aunque su gene-ralizacion es (solo en el aspecto teorico!) trivial. En este caso, la ecuacion radial de Schrodingeres
H Pn(r) =
[−1
2
∂2
∂r2+l(l + 1)
2r2+ V (r)
]Pn(r) = En Pn(r) (2.14)
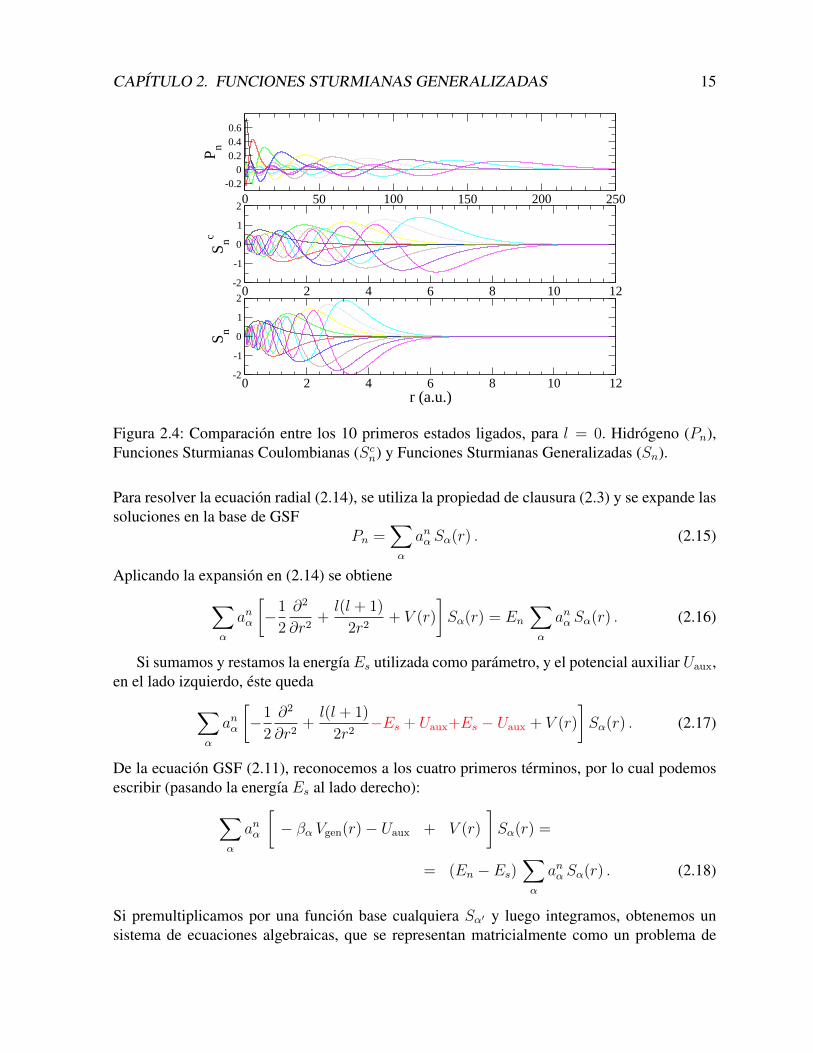
CAPITULO 2. FUNCIONES STURMIANAS GENERALIZADAS 15
0 50 100 150 200 250
-0.2
0
0.2
0.4
0.6
Pn
0 2 4 6 8 10 12-2
-1
0
1
2
Sn
c
0 2 4 6 8 10 12
r (a.u.)
-2
-1
0
1
2
Sn
Figura 2.4: Comparacion entre los 10 primeros estados ligados, para l = 0. Hidrogeno (Pn),Funciones Sturmianas Coulombianas (Scn) y Funciones Sturmianas Generalizadas (Sn).
Para resolver la ecuacion radial (2.14), se utiliza la propiedad de clausura (2.3) y se expande lassoluciones en la base de GSF
Pn =∑α
anα Sα(r) . (2.15)
Aplicando la expansion en (2.14) se obtiene∑α
anα
[−1
2
∂2
∂r2+l(l + 1)
2r2+ V (r)
]Sα(r) = En
∑α
anα Sα(r) . (2.16)
Si sumamos y restamos la energıa Es utilizada como parametro, y el potencial auxiliar Uaux,en el lado izquierdo, este queda∑
α
anα
[−1
2
∂2
∂r2+l(l + 1)
2r2−Es + Uaux+Es − Uaux + V (r)
]Sα(r) . (2.17)
De la ecuacion GSF (2.11), reconocemos a los cuatro primeros terminos, por lo cual podemosescribir (pasando la energıa Es al lado derecho):∑
α
anα
[− βα Vgen(r)− Uaux + V (r)
]Sα(r) =
= (En − Es)∑α
anα Sα(r) . (2.18)
Si premultiplicamos por una funcion base cualquiera Sα′ y luego integramos, obtenemos unsistema de ecuaciones algebraicas, que se representan matricialmente como un problema de

CAPITULO 2. FUNCIONES STURMIANAS GENERALIZADAS 16
autovalores generalizados
Ban = λn San (2.19)
donde B es la matriz
[B]α′α =
∫Sα′(r) (−βα Vgen(r)− Uaux + V (r) )Sα(r) dr , (2.20)
S es la matriz de solapamiento
[S]α′α =
∫Sα′(r)Sα(r) dr , (2.21)
an son las autofunciones con las que se obtienen finalmente los coeficientes de la expansion
an = (an1 , an2 , a
n3 , . . .)
T =
an1an2an3. . .
(2.22)
y λn son los autovalores correspondientes con los cuales se hallan las energıas
λn = En − Es . (2.23)
Los calculos se pueden simplificar en forma considerable, eligiendo al potencial auxiliarigual al potencial Uaux(r) = V (r), con lo cual la matriz se simplifica:
[B]α′α =
∫Sα′(r) (−βα)Vgen(r)Sα(r) dr =
= −βα∫Sα′(r)Vgen(r)Sα(r) dr =
= −βα δα′α = −β , (2.24)
donde hemos utilizado las condiciones de ortogonalidad de las funciones Sturmianas (2.4). Lamatriz β es diagonal,y sus elementos son las cargas βα.
En las aplicaciones al metodo GSF, se ha demostrado que la contraccion de las funcionespermite una mejor representacion de los estados ligados, proporcionando resultados mas pre-cisos con bases de menor tamano. Esta cualidad otorga una gran efectividad al metodo, sobretodo para el calculo de sistemas complejos, que demandan altısimos recursos computaciona-les, donde es crucial la economıa de memoria y tiempo de calculo. Sin embargo, la aplicacionmas importante de este metodo se da en los problemas colisionales, donde se tratan los estadoscontinuos, tal como veremos a continuacion.

CAPITULO 2. FUNCIONES STURMIANAS GENERALIZADAS 17
2.1.6. Implementacion: Estados ContinuosEl potencial generador de corto alcance no solo se utiliza para compactar la base. Al anu-
larse a distancias superiores a su rango, todo el comportamiento asintotico de las bases quedadeterminado unicamente por el potencial auxiliar. Eso significa, que es posible elegir un poten-cial auxiliar que determine las mismas condiciones asintoticas del problema fısico a resolver. Esdecir, las condiciones asintoticas no se forman con una combinacion de las funciones base (loque requiere de una enorme cantidad de funciones, de manera que esta forma perdure a grandesdistancias), sino que se obtienen naturalmente ya que todos los elementos de la base tienen estacondicion.
Para ilustrarlo con un ejemplo, veamos las diferencias entre las bases Sturmianas Coulom-bianas (Scn) y nuestras funciones Sturmianas Generalizadas (Sn). En el primer caso, si quere-mos encontrar las soluciones del continuo, hacemos Es > 0 en la ecuacion (2.8). La repetimosaquı con βnl en lugar de Znl, para enfatizar que estas cargas son los autovalores de la ecuacion,y no un parametro fijo.[
−1
2
∂2
∂r2+l(l + 1)
2r2− Es
]Scn(r) = βn
1
rScn(r) . (2.25)
No vamos a extendernos en la descripcion de las funciones del continuo Coulombiano, perodestacamos que se pueden encontrar dos soluciones linealmente independientes que tienen elsiguiente comportamiento
lımr→∞
Scn(r) ∝ e±ikr−βnk
ln (2kr) , (2.26)
donde los ındices ± estan asociados al flujo, determinando si la funcion es saliente o entrante.El punto importante a destacar es que a pesar que estas soluciones tienen todas la misma energıaEs, requieren de una cantidad muy grande de elementos de base para poder expandir un estadode continuo, aun uno con esta misma energıa Es. El motivo de esta dificultad radica en que cadauna de estas funciones base, se encuentran defasadas por un factor que depende del autovalorβn.
Veamos ahora que ocurre con las funciones Sturmianas generalizadas Sn. Para resolver elmismo problema, utilizamos como potencial auxiliar al potencial Coulombiano (Uaux = −Z
r)
en la ecuacion (2.11):[−1
2
∂2
∂r2+l(l + 1)
2r2− Es −
Z
r
]Sn(r) = −βn Vgen(r)Sn(r) . (2.27)
La eleccion del potencial generador no es muy importante, solo es necesario que tenga un rangoadecuado (que corresponda a la region donde todavıa no se obtienen las condiciones asintoti-cas), y que se anule a distancias mayores que este. En este caso, las soluciones tienen el mismocomportamiento asintotico senalado anteriormente, solo que ahora esta dictado completamentepor el potencial auxiliar
lımr→∞
Sn(r) ∝ e±ikr−Zkln (2kr) , (2.28)
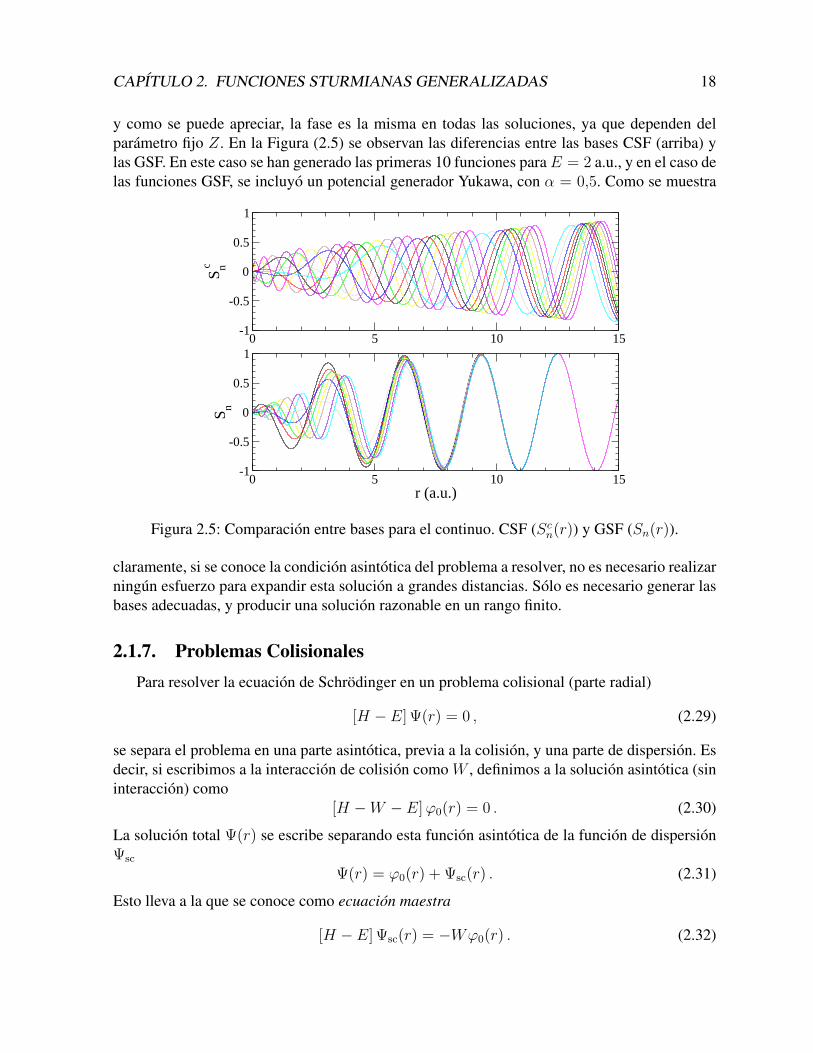
CAPITULO 2. FUNCIONES STURMIANAS GENERALIZADAS 18
y como se puede apreciar, la fase es la misma en todas las soluciones, ya que dependen delparametro fijo Z. En la Figura (2.5) se observan las diferencias entre las bases CSF (arriba) ylas GSF. En este caso se han generado las primeras 10 funciones paraE = 2 a.u., y en el caso delas funciones GSF, se incluyo un potencial generador Yukawa, con α = 0,5. Como se muestra
0 5 10 15-1
-0.5
0
0.5
1S
nc
0 5 10 15
r (a.u.)
-1
-0.5
0
0.5
1
Sn
Figura 2.5: Comparacion entre bases para el continuo. CSF (Scn(r)) y GSF (Sn(r)).
claramente, si se conoce la condicion asintotica del problema a resolver, no es necesario realizarningun esfuerzo para expandir esta solucion a grandes distancias. Solo es necesario generar lasbases adecuadas, y producir una solucion razonable en un rango finito.
2.1.7. Problemas ColisionalesPara resolver la ecuacion de Schrodinger en un problema colisional (parte radial)
[H − E] Ψ(r) = 0 , (2.29)
se separa el problema en una parte asintotica, previa a la colision, y una parte de dispersion. Esdecir, si escribimos a la interaccion de colision como W , definimos a la solucion asintotica (sininteraccion) como
[H −W − E]ϕ0(r) = 0 . (2.30)
La solucion total Ψ(r) se escribe separando esta funcion asintotica de la funcion de dispersionΨsc
Ψ(r) = ϕ0(r) + Ψsc(r) . (2.31)
Esto lleva a la que se conoce como ecuacion maestra
[H − E] Ψsc(r) = −Wϕ0(r) . (2.32)

CAPITULO 2. FUNCIONES STURMIANAS GENERALIZADAS 19
que es la ecuacion que vamos a resolver en todos los problemas colisionales. La unica diferenciaentre ellos, es la forma del Hamiltoniano, y la parte W del mismo, que se toma para resolver elestado inicial ϕ0.
La implementacion del metodo GSF en este tipo de problemas se realiza mediante la expan-sion de la funcion de dispersion en una base GSF
Ψsc(r) =∑n
an Snl(r) , (2.33)
donde las GSF cumplen[H − E]Sn(r) = −βn V(r)Sn(r) , (2.34)
siendo V(r) un potencial de corto alcance.Reemplazando en (2.32), se obtiene∑
n
an [H − E] Sn(r) =∑n
an(−Wϕ0(r))Sn(r) o,∑n
anβn V(r)Sn(r) =∑n
anWϕ0(r)Sn(r) . (2.35)
multiplicando a izquierda por una funcion Sturmiana arbitraria e integrando se obtiene∑n
an
∫Sm(r) βn V(r)Sn(r) dr =
∑n
an
∫Sm(r)Wϕ0(r)Sn(r) dr ,
que define un sistema de ecuaciones lineales, que se escribe matricialmente como
(B−W0) a = 0 (2.36)
donde donde B es la matriz
[B]mn =
∫Sm(r) βn V(r)Sn(r) dr , (2.37)
W0 es la matriz
[W0]mn =
∫Sm(r)Wϕ0(r)Sn(r) dr , (2.38)
y an son las autofunciones con las que se obtienen finalmente los coeficientes de la expansionde la solucion (2.33)
a = (a1, a2, a3, . . .)T =
a1a2a3. . .
. (2.39)

CAPITULO 2. FUNCIONES STURMIANAS GENERALIZADAS 20
2.2. AplicacionesDurante los ultimos anos se han publicado numerosos trabajos utilizando el metodo GSF.
En esta seccion haremos una mencion muy breve de las aplicaciones mas importantes. Para unareferencia mas completa, referimos al trabajo [39], donde se podran encontrar los detalles yreferencias de cada una de estas aplicaciones.
1. Estados Ligados
a) Estados fundamentales del He y H−
b) Estados excitados del He
c) Ion Positronio (e−,e+,e−), y sistemas exoticos de tres partıculas (µ−,µ−,3He+2),(e−,µ−,AHe+2)
d) Estado fundamental del Ion Hidrogeno molecular H+2
e) He confinado en Fullereno
f ) He confinado en cavidad impenetrable
2. Problemas Colisionales
a) Ionizacion en problemas modelos con solucion analıtica
b) Modelo esferico de Ionizacion (e, 2e) de Hidrogeno (Temkin–Poet)
c) Ionizacion del Hidrogeno
d) Ionizacion doble del He (modelo Temkin–Poet)
e) Ionizacion doble (e, 3e) del He por electrones muy energeticos.
f ) (e, 3e) en coordenadas hiperesfericas
g) Doble fotoionizacion del He
h) Doble ionizacion del He por impacto de protones

Capıtulo 3
GSF en Esferoidales Prolatas
En este capıtulo se presenta el metodo de Funciones Sturmianas Generalizadas en coorde-nadas esferoidales prolatas. Para comprobar los resultados, dominar los parametros a utilizar,y evaluar el metodo, estudiamos iones moleculares diatomicos, con un electron. Para estos sis-temas, hemos desarrollado dos metodos de calculo. El primer metodo resuelve las ecuaciones(1.18) (radial), y (1.19) (angular). En una de ellas se fija la energıa p2 y con ella se obtiene laconstante de separacion A, y en la otra se fija ese A obtenido, para calcular con ella p2. Dadoque repetimos este proceso hasta obtener convergencia, llamamos a este metodo Iterativo (1d).En el segundo metodo, producimos dos bases GSF (una con cada una de estas ecuaciones),y con la combinacion de estas bases resolvemos la ecuacion de Schrodinger completa (1.10).Denominamos a este metodo Directo (2d).
3.1. Metodo Iterativo (1d)El problema de iones moleculares diatomicos monoelectronicos puede tratarse como un pro-
blema iterativo, solucionando en forma separada las ecuaciones (1.18) y (1.19). Si bien estasecuaciones estan separadas, las mismas no estan completamente desacopladas, ya que compar-ten la constante de separacion A y la Energıa E, que junto con la funcion de onda, forman lasincognitas del problema.
El metodo propuesto para solucionar estas ecuaciones, consiste en asignar un valor inicialpara la energıa E (para ser mas precisos p2), en la ecuacion (1.19) correspondiente a la variableangular η. Se resuelve esta ecuacion como un problema de autovalores, donde los autovaloresa encontrar son las constantes de separacion An. Cada constante corresponde a diferentes au-tovectores, y los vamos a identificar de acuerdo a la cantidad de nodos. En el caso del estadofundamental, este no tiene nodos, por lo que llamamos al autovalor simplemente A.
El autovalor hallado se introduce en la ecuacion radial (1.18), que ası se convierte en unaecuacion de autovalores, de la cual se deben obtener los autovalores En. Nuevamente, se selec-ciona el valor que corresponde al numero de nodos buscado, con lo que se obtiene el valor dep2. Este ultimo valor, ingresa nuevamente como parametro en (1.19), y se continua la iteracionhasta que las diferencias en las energıas entre un paso y el anterior sean menores a un cierto
21

CAPITULO 3. GSF EN ESFEROIDALES PROLATAS 22
criterio de aceptacion.Si bien resolvemos las ecuaciones Sturmianas utilizando diferencias finitas, este procedi-
miento no serıa muy eficiente para resolver el problema que nos concierne. En primer lugar,ambas ecuaciones son muy dependientes de las condiciones iniciales. Con pequenas diferen-cias en estas condiciones, se obtienen divergencias a muy cortas distancias del origen de lapropagacion, por lo cual el calculo se hace inestable, a menos que se utilice una grilla numericamuy densa. Por otro lado, una grilla con muchos puntos implica un calculo costoso, que noserıa una dificultad mayor si solo se resuelven las ecuaciones una unica vez, pero resulta en unadificultad muy seria si esto debe hacerse en forma iterativa, en numerosos pasos. Veremos enlas secciones siguientes como se resuelven estas dificultades utilizando el metodo de FuncionesSturmianas Generalizadas.
3.1.1. Ecuacion angularPara simplificar la exposicion, asumiremos que el numero cuantico azimutal m = 0. En este
caso, la ecuacion angular se escribe[∂
∂η
[(1− η2
) ∂∂η
]− a1η + p2η2 − A
]Λ(η) = 0 . (3.1)
Como hemos desarrollado en el capıtulo anterior, la solucion se expande en una base
Λ(η) =∑j
cj Sj(η) . (3.2)
La ecuacion Sturmiana que se propone para generar esta base es
∂
∂η
[(1− η2
) ∂∂η
]Sj(η) = −βj Sj(η) . (3.3)
Escogemos esta ecuacion porque sus soluciones son los polinomios de Legendre Sj(η) =Pj(η), y los autovalores son βj = j(j+ 1). En el Apendice E, se sintetizan algunas propiedadesimportantes de estos polinomios. En la Figura (3.1) se muestran los primeros elementos de estabase.
Realizando el reemplazo mencionado, la ecuacion (3.3) queda∑j
cj
[∂
∂η
[(1− η2
) ∂∂η
]− a1η + p2η2
]Sj(η) =
=∑j
cj[−βj − a1η + p2η2
]Sj(η) = A
∑j
cj Sj(η) (3.4)
Si multiplicamos a la izquierda por Si(η) e integramos, obtenemos una ecuacion de autova-lores generalizados
Mc = ABc (3.5)
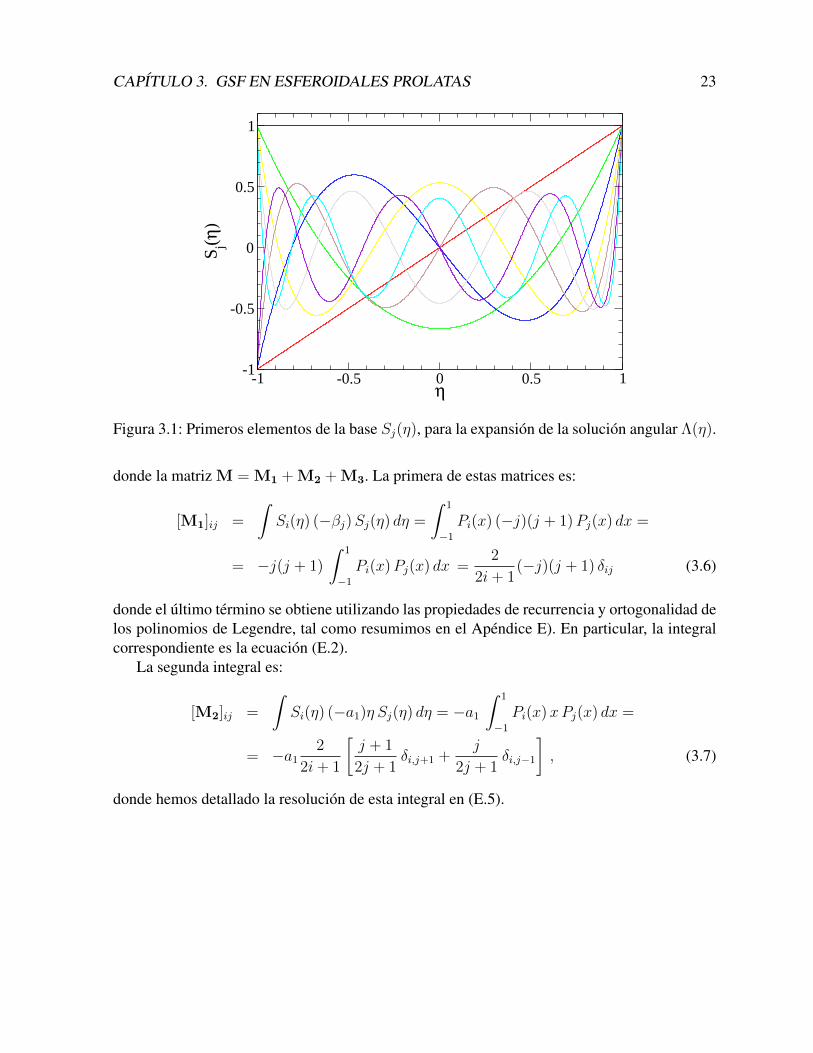
CAPITULO 3. GSF EN ESFEROIDALES PROLATAS 23
-1 -0.5 0 0.5 1η
-1
-0.5
0
0.5
1
Sj(
η)
Figura 3.1: Primeros elementos de la base Sj(η), para la expansion de la solucion angular Λ(η).
donde la matriz M = M1 + M2 + M3. La primera de estas matrices es:
[M1]ij =
∫Si(η) (−βj)Sj(η) dη =
∫ 1
−1Pi(x) (−j)(j + 1)Pj(x) dx =
= −j(j + 1)
∫ 1
−1Pi(x)Pj(x) dx =
2
2i+ 1(−j)(j + 1) δij (3.6)
donde el ultimo termino se obtiene utilizando las propiedades de recurrencia y ortogonalidad delos polinomios de Legendre, tal como resumimos en el Apendice E). En particular, la integralcorrespondiente es la ecuacion (E.2).
La segunda integral es:
[M2]ij =
∫Si(η) (−a1)η Sj(η) dη = −a1
∫ 1
−1Pi(x)xPj(x) dx =
= −a12
2i+ 1
[j + 1
2j + 1δi,j+1 +
j
2j + 1δi,j−1
], (3.7)
donde hemos detallado la resolucion de esta integral en (E.5).

CAPITULO 3. GSF EN ESFEROIDALES PROLATAS 24
La tercer integral es:
[M3]ij =
∫Si(η) p2η2 Sj(η) dη = p2
∫ 1
−1Pi(x)x2 Pj(x) dx =
= p22
2i+ 1
[j + 1
2j + 1
j + 2
2j + 3δi,j+2 +
+
(j + 1
2j + 1
j + 1
2j + 3+
j
2j + 1
j
2j − 1
)δi,j +
+j
2j + 1
j − 1
2j − 1δi,j−2
], (3.8)
y esta integral esta detallada en (E.9).El lado derecho, a su vez, es
[B]ij =
∫Si(η)Sj(η) dη =
∫ 1
−1Pi(x)Pj(x) dx =
=2
2i+ 1δij . (3.9)
Como se puede apreciar, la construccion de estas matrices es bastante sencilla y rapida (yaque no requiere la realizacion de ninguna integracion numerica). Recordemos que en este pro-blema se asume un valor de p2, por lo que el problema de autovalores generalizados consiste enobtener, para las matrices M y B dadas, el autovector de coeficientes c y el autovalor corres-pondiente A. La matriz M3 es la unica que se debe calcular en cada iteracion, ya que dependedel parametro p. Todas las otras matrices se calculan una unica vez. Hemos encontrado que conunos pocos elementos de base, se obtienen soluciones muy precisas.
3.1.2. Ecuacion radialUna vez obtenido el valor de A de la ecuacion angular, se procede a obtener la energıa p2 de
la ecuacion radial, [∂
∂ξ
[(ξ2 − 1
) ∂∂ξ
]+ a2ξ − p2ξ2 + A
]U(ξ) = 0 . (3.10)
Para resolver este problema, planteamos expandir las soluciones
U(ξ) =∑j
djSj(ξ) , (3.11)
donde las bases Sj(ξ) se obtienen como soluciones de la ecuacion de Funciones SturmianasGeneralizadas [
∂
∂ξ
[(ξ2 − 1
) ∂∂ξ
]+ a2 ξ − p2s ξ2
]Sj(ξ) = αj Vs(ξ)Sj(ξ) , (3.12)
CAPITULO 3. GSF EN ESFEROIDALES PROLATAS 25
donde ps es calculado con la misma expresion que p pero con una energıa prefijada Es, y Vs esun potencial de corto alcance.
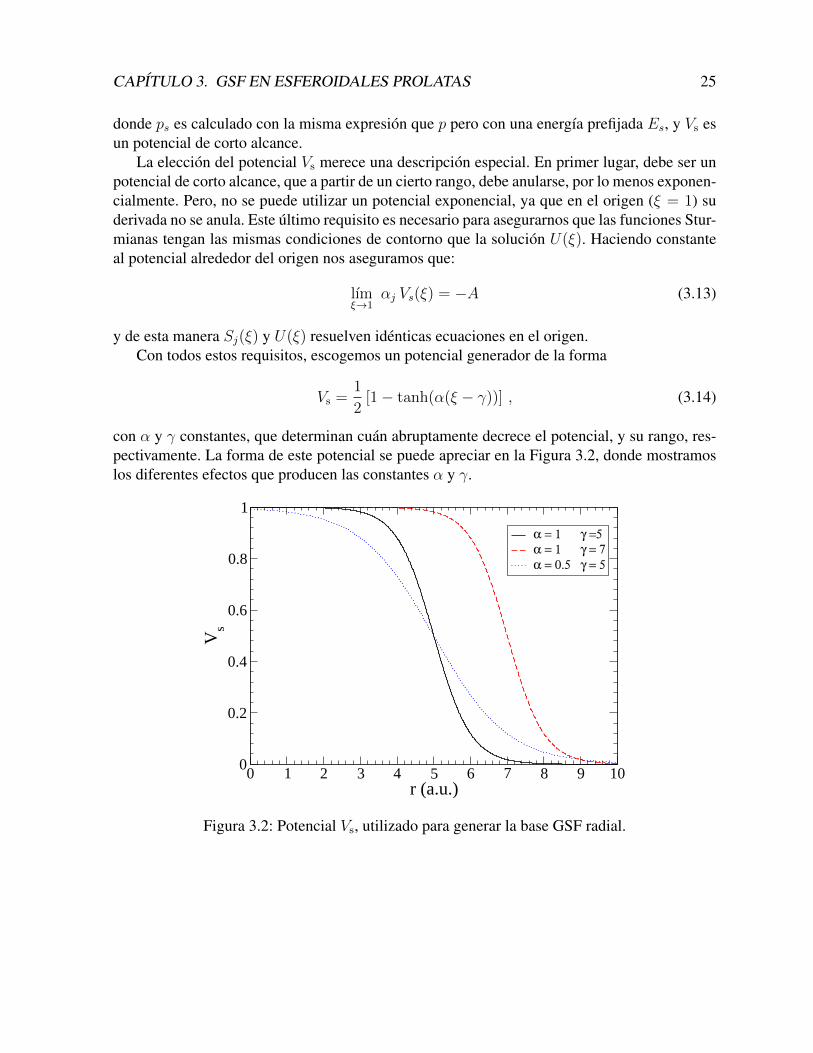
La eleccion del potencial Vs merece una descripcion especial. En primer lugar, debe ser unpotencial de corto alcance, que a partir de un cierto rango, debe anularse, por lo menos exponen-cialmente. Pero, no se puede utilizar un potencial exponencial, ya que en el origen (ξ = 1) suderivada no se anula. Este ultimo requisito es necesario para asegurarnos que las funciones Stur-mianas tengan las mismas condiciones de contorno que la solucion U(ξ). Haciendo constanteal potencial alrededor del origen nos aseguramos que:
lımξ→1
αj Vs(ξ) = −A (3.13)
y de esta manera Sj(ξ) y U(ξ) resuelven identicas ecuaciones en el origen.Con todos estos requisitos, escogemos un potencial generador de la forma
Vs =1
2[1− tanh(α(ξ − γ))] , (3.14)
con α y γ constantes, que determinan cuan abruptamente decrece el potencial, y su rango, res-pectivamente. La forma de este potencial se puede apreciar en la Figura 3.2, donde mostramoslos diferentes efectos que producen las constantes α y γ.
0 1 2 3 4 5 6 7 8 9 10r (a.u.)
0
0.2
0.4
0.6
0.8
1
Vs
α = 1 γ =5
α = 1 γ = 7
α = 0.5 γ = 5
Figura 3.2: Potencial Vs, utilizado para generar la base GSF radial.
CAPITULO 3. GSF EN ESFEROIDALES PROLATAS 26
En el origen, donde se elimina el termino (ξ2 − 1), la ecuacion radial (3.10) se escribe
2ξd
dξU(ξ) =
(p2ξ2 − a2ξ − A
)U(ξ)
dU(ξ)
dξ=
(p2
2ξ − a2
2− A
2ξ
)U(ξ)
U ′(ξ)
U(ξ)=
p2
2ξ − a2
2,A
2ξ(3.15)
cuya solucion determina las condiciones de contorno de las funciones de base:
lımξ→1
U(ξ) = ξ−A2 e
p2
4ξ2−a2
2ξ . (3.16)
y
lımξ→∞
U(ξ) =e−pξ
ξ, (3.17)
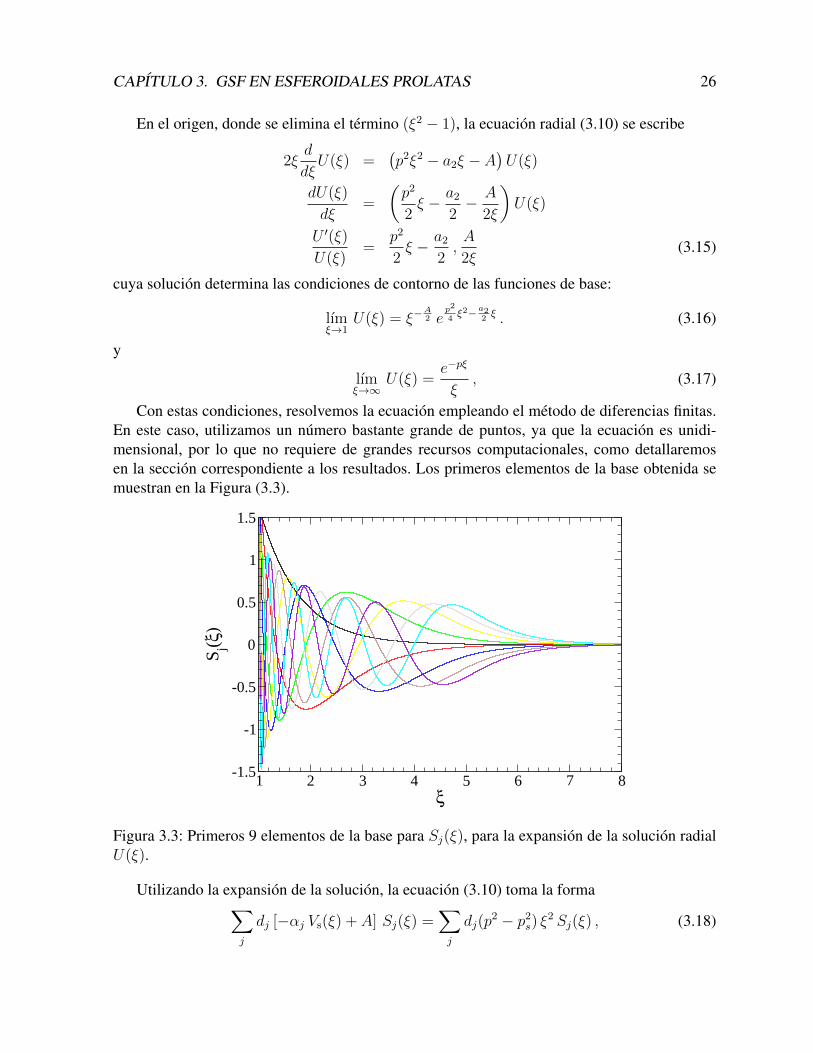
Con estas condiciones, resolvemos la ecuacion empleando el metodo de diferencias finitas.En este caso, utilizamos un numero bastante grande de puntos, ya que la ecuacion es unidi-mensional, por lo que no requiere de grandes recursos computacionales, como detallaremosen la seccion correspondiente a los resultados. Los primeros elementos de la base obtenida semuestran en la Figura (3.3).
1 2 3 4 5 6 7 8
ξ
-1.5
-1
-0.5
0
0.5
1
1.5
Sj(
ξ)
Figura 3.3: Primeros 9 elementos de la base para Sj(ξ), para la expansion de la solucion radialU(ξ).
Utilizando la expansion de la solucion, la ecuacion (3.10) toma la forma∑j
dj [−αj Vs(ξ) + A] Sj(ξ) =∑j
dj(p2 − p2s) ξ2 Sj(ξ) , (3.18)

CAPITULO 3. GSF EN ESFEROIDALES PROLATAS 27
que al multiplicar por el lado izquierdo por un elemento de la base Si, e integrar, se convierteen una ecuacion de autovalores generalizados
Md = λBd (3.19)
donde M = M1 + M2 y
[M1]ij =
∫ ∞1
Si(ξ) (−αj Vs(ξ))Sj(ξ) dξ , (3.20)
[M2]ij =
∫ ∞1
Si(ξ)ASj(ξ) dξ , (3.21)
y
[B]ij =
∫ ∞1
Si(ξ) ξ2 Sj(ξ) dξ , (3.22)
Volvemos a reiterar que en esta ecuacion A es un parametro fijo (obtenido en la solucionangular), y al resolverla se obtienen los coeficientes dj de la expansion de la solucion (3.11)como autovectores, y las energıas p2 = −R2 E
2(a traves del autovalor λ). Hicimos la separacion
de las matrices M, ya que la unica que varıa en las diferentes iteraciones es M2. En rigor,el calculo de esta matriz tambien se realiza una unica vez, ya que consiste en la matriz desolapamiento. En cada iteracion, se la multiplica por un valor diferente de A.
3.2. Metodo Directo (2d)El metodo que se describe en la seccion anterior tiene la ventaja de que al separar el Hamil-
toniano en dos ecuaciones acopladas, ambas son unidimensionales. De este modo, se reduce enforma considerable el costo computacional. Como hemos destacado previamente, al incluir lasbases GSF evitamos tener que resolver las ecuaciones en cada iteracion. Las bases Sturmianasincluyen una gran parte de la solucion (es decir, no se debe resolver la ecuacion diferencial encada paso). Sin embargo, existe un problema con esta metodologıa, que consiste en que paracada estado molecular se deben encontrar las bases correspondientes. Esto significa que se de-ben escoger los autovalores que produzcan soluciones con el numero de nodos buscado, y paracada uno de estos estados moleculares se deben realizar las iteraciones correspondientes.
Proponemos un metodo alternativo, en el cual se resuelve directamente la ecuacion (1.10),que es bidimensional. En este caso, la solucion ψ(ξ, η) se expande en
ψ(ξ, η) =∑ij
aij Si(ξ)Sj(η) (3.23)
donde las bases Sturmianas se obtienen del mismo modo que lo hicimos en las solucionesseparadas (3.12) y (3.3):[
∂
∂ξ
[(ξ2 − 1
) ∂∂ξ
]+ a2 ξ − p2s ξ2
]Si(ξ) = αi Vs(ξ)Si(ξ) , (3.24)

CAPITULO 3. GSF EN ESFEROIDALES PROLATAS 28
∂
∂η
[(1− η2
) ∂∂η
]Sj(η) = −βj Sj(η) . (3.25)
De esta manera, la ecuacion de Schrodinger (recordemos que m = 0) para el H+2{
∂
∂ξ
[(ξ2 − 1)
∂
∂ξ
]+ a2ξ − p2 ξ2 + (3.26)
+∂
∂η
[(1− η2) ∂
∂η
]+ p2 η2
}U(ξ)Λ(η) = 0 .
se escribe∑ij
aij
{αi Vs(ξ) + p2s ξ
2 − βj}Si(ξ)Sj(η) =
∑ij
aij p2 (ξ2 − η2)Si(ξ)Sj(η) . (3.27)
Volvemos a construir el sistema matricial multiplicando por la izquierda por las bases Si′(ξ)Sj′(η),obteniendose un problema de autovalores generalizados
Ma = λBa , (3.28)
donde M = M1 + M2 + M3, siendo
[M1]i′j′,ij =
∫ ∞1
dξ
∫ 1
−1dη Si′(ξ)Sj′(η)αiVs(ξ)Si(ξ)Sj(η) =
=
∫ ∞1
Si′(ξ)αiVs(ξ)Si(ξ) dξ
∫ 1
−1Sj′(η)Sj(η) dη (3.29)
[M2]i′j′,ij =
∫ ∞1
dξ
∫ 1
−1dη Si′(ξ)Sj′(η) p2sξ
2 Si(ξ)Sj(η) =
=
∫ ∞1
Si′(ξ) p2sξ
2 Si(ξ) dξ
∫ 1
−1Sj′(η)Sj(η) dη (3.30)
[M3]i′j′,ij =
∫ ∞1
dξ
∫ 1
−1dη Si′(ξ)Sj′(η) (−βj)Si(ξ)Sj(η) =
= (−βj)∫ ∞1
Si′(ξ)Si(ξ) dξ
∫ 1
−1Sj′(η)Sj(η) dη (3.31)
y B = B1 −B2, siendo
[B1]i′j′,ij =
∫ ∞1
dξ
∫ 1
−1dη Si′(ξ)Sj′(η) ξ2 Si(ξ)Sj(η) =
=
∫ ∞1
Si′(ξ) ξ2 Si(ξ) dξ
∫ 1
−1Sj′(η)Sj(η) dη (3.32)

CAPITULO 3. GSF EN ESFEROIDALES PROLATAS 29
[B1]i′j′,ij =
∫ ∞1
dξ
∫ 1
−1dη Si′(ξ)Sj′(η) η2 Si(ξ)Sj(η) =
=
∫ ∞1
Si′(ξ) Si(ξ) dξ
∫ 1
−1Sj′(η) η2 Sj(η) dη . (3.33)
Se resuelve este problema de autovalores y se obtiene la matriz a, en la cual cada columnaes el vector de coeficientes ~an, que expanden a la solucion de un estado molecular especıfico,cuya energıa corresponde al componente λn = p2n. Para ser mas precisos
a =
a111 a211 a311 . . . aN11a121 a221 a321 . . . aN21. . . . . . . . . . . . . . .a112 a212 a312 . . . aN12a122 a222 a322 . . . aN22. . . . . . . . . . . . . . .a1ij a2ij a3ij . . . aNij. . . . . . . . . . . . . . .
λ =
p21p22. . .p2N
(3.34)
Como vemos, esta metodologıa no solo evita las iteraciones, sino que tambien tiene la ventajade resolver las soluciones para varios estados moleculares simultaneamente. A simple vista, pa-recerıa que los calculos son mucho mas costosos por tratarse de matrices bidimensionales. Sinembargo, todas las integrales son separables, y consisten en productos de integrales unidimen-sionales. Como veremos en el proximo capıtulo, con unos pocos elementos de base se obtienenexcelentes resultados.

Capıtulo 4
Resultados
4.1. Metodo Iterativo (1d) para H+2
El primer paso a realizar para resolver la estructura molecular, consiste en examinar losparametros en los calculos, estudiar sus comportamientos, y analizar las convergencias.
Dado que las ecuaciones separadas (3.1) y (3.10) han sido resueltas por diversos metodos,tomamos los mejores valores de la energıa E del estado fundamental 1σg, y de la constante deseparabilidad correspondiente A (obtenidos por Scott et al., en 2006 [16]). Con estos valorescomo dato, analizaremos los parametros y recursos necesarios para reproducirlos con nuestrosmetodos.
4.1.1. Ecuacion angularComenzamos con la ecuacion angular (3.1). Tal como senalamos, para esta ecuacion se fija
la energıaE (mas precisamente, p2), y con ese valor como parametro, se construyen las matricescorrespondientes (3.5). Destacamos que en el H+
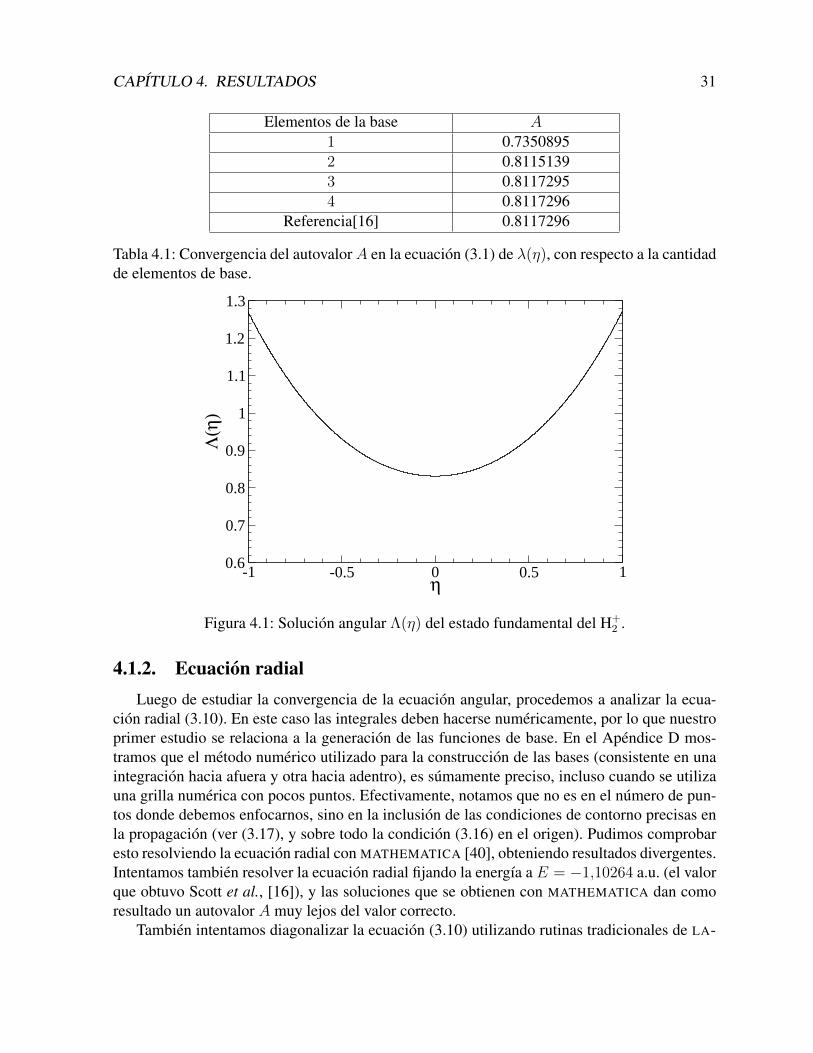
2 , las cargas nucleares son iguales, Z1 = Z2,por lo tanto, el parametro a1 = 0, lo que implica que no se toma en cuenta a la matriz M2.Dado que escogimos como ecuacion Sturmiana a (3.3), y que esta produce naturalmente a lospolinomios de Legendre, no necesitamos calcular analıticamente ninguna integral, ya que todasson analıticas. El unico aspecto a analizar en este caso, es la convergencia de los resultadoscon respecto al tamano de la base utilizada. En la Tabla 4.1, se muestra precisamente estaconvergencia. En estos calculos, asumimos que la distancia internuclear es R = 2 a.u..
El estado fundamental es par en la coordenada ξ. Por lo tanto, solo se incluyen en la expan-sion las funciones pares. Como se observa, con tan solo 4 funciones en la base Sj(η), podemosreproducir los mejores resultados que existen en la actualidad.
Una vez resuelta la ecuacion matricial, con los autovectores obtenemos los coeficientes cjcon los cuales construımos la solucion angular Λ(η) =
∑j cjSj(η), funcion que se muestra en
la Figura 4.1
30
CAPITULO 4. RESULTADOS 31
Elementos de la base A
1 0.73508952 0.81151393 0.81172954 0.8117296
Referencia[16] 0.8117296
Tabla 4.1: Convergencia del autovalor A en la ecuacion (3.1) de λ(η), con respecto a la cantidadde elementos de base.
-1 -0.5 0 0.5 1η
0.6
0.7
0.8
0.9
1
1.1
1.2
1.3
Λ(η
)
Figura 4.1: Solucion angular Λ(η) del estado fundamental del H+2 .
4.1.2. Ecuacion radialLuego de estudiar la convergencia de la ecuacion angular, procedemos a analizar la ecua-
cion radial (3.10). En este caso las integrales deben hacerse numericamente, por lo que nuestroprimer estudio se relaciona a la generacion de las funciones de base. En el Apendice D mos-tramos que el metodo numerico utilizado para la construccion de las bases (consistente en unaintegracion hacia afuera y otra hacia adentro), es sumamente preciso, incluso cuando se utilizauna grilla numerica con pocos puntos. Efectivamente, notamos que no es en el numero de pun-tos donde debemos enfocarnos, sino en la inclusion de las condiciones de contorno precisas enla propagacion (ver (3.17), y sobre todo la condicion (3.16) en el origen). Pudimos comprobaresto resolviendo la ecuacion radial con MATHEMATICA [40], obteniendo resultados divergentes.Intentamos tambien resolver la ecuacion radial fijando la energıa a E = −1,10264 a.u. (el valorque obtuvo Scott et al., [16]), y las soluciones que se obtienen con MATHEMATICA dan comoresultado un autovalor A muy lejos del valor correcto.
Tambien intentamos diagonalizar la ecuacion (3.10) utilizando rutinas tradicionales de LA-

CAPITULO 4. RESULTADOS 32
PACK [41], de manera de obtener directamente los autovalores y autovectores en forma numeri-ca, y tampoco hemos obtenido resultados satisfactorios. Esto se debe a las dificultades de intro-ducir las condiciones de contorno explıcitamente en las matrices. Sin embargo, con el metodode resolucion presentado en el Apendice, obtenemos excelentes funciones de base, aun utilizan-do un numero pequeno de puntos (500) y un primer orden en las diferencias finitas. En nuestroscalculos aprovechamos que las integrales son unidimensionales, y a pesar de no ser estrictamen-te necesario, utilizamos grillas numericas del orden de 104 puntos, sin mayores dificultades.
Despues de obtener las bases, pasamos a analizar la convergencia de la expansion (3.11) dela funcion U(ξ). En la Tabla 4.2 se muestran los diferentes valores de energıas E que se obtie-nen, fijando la constante de separacion A = 0,8117296, para distintos numeros de funciones debase Si(ξ).
Elementos de la base E (a.u.) E* (a.u.)1 -1.0 -1.13 -1.1 -1.10266 -1.1024 -1.10263409 -1.1026 -1.1026346
Referencia[17] -1.1026342
Tabla 4.2: Convergencia de la energıa E en la ecuacion (3.10) de U(ξ), con respecto a la canti-dad de elementos de base. ∗: Base recalculada.
No hemos mencionado todavıa el valor de la energıa Es que se toma como parametro ex-terno para generar las bases (3.12). Si bien esa energıa es arbitraria, es conveniente escogerlalo mas cercanamente posible a la energıa del estado. Por lo tanto, asignamos el valor arbitra-rio E = −1 a.u. en primera instancia, obteniendo los resultados que se detallan en la segundacolumna de la tabla. Con ese parametro externo, obtuvimos una energıa E = −1,1026 a.u.. Esposible mejorar estos resultados, generando nuevamente las bases Si(ξ), pero ahora, cambiandoel valor de la energıa a Es = −1,1026 a.u.. La nueva convergencia esta detallada en la tercercolumna de la tabla, senalandola como “base recalculada”.
Una vez resuelta la ecuacion matricial, con los autovectores obtenemos los coeficientes dicon los cuales construımos la solucion angular U(ξ) =
∑i diSi(η), funcion que se muestra en
la Figura 4.2
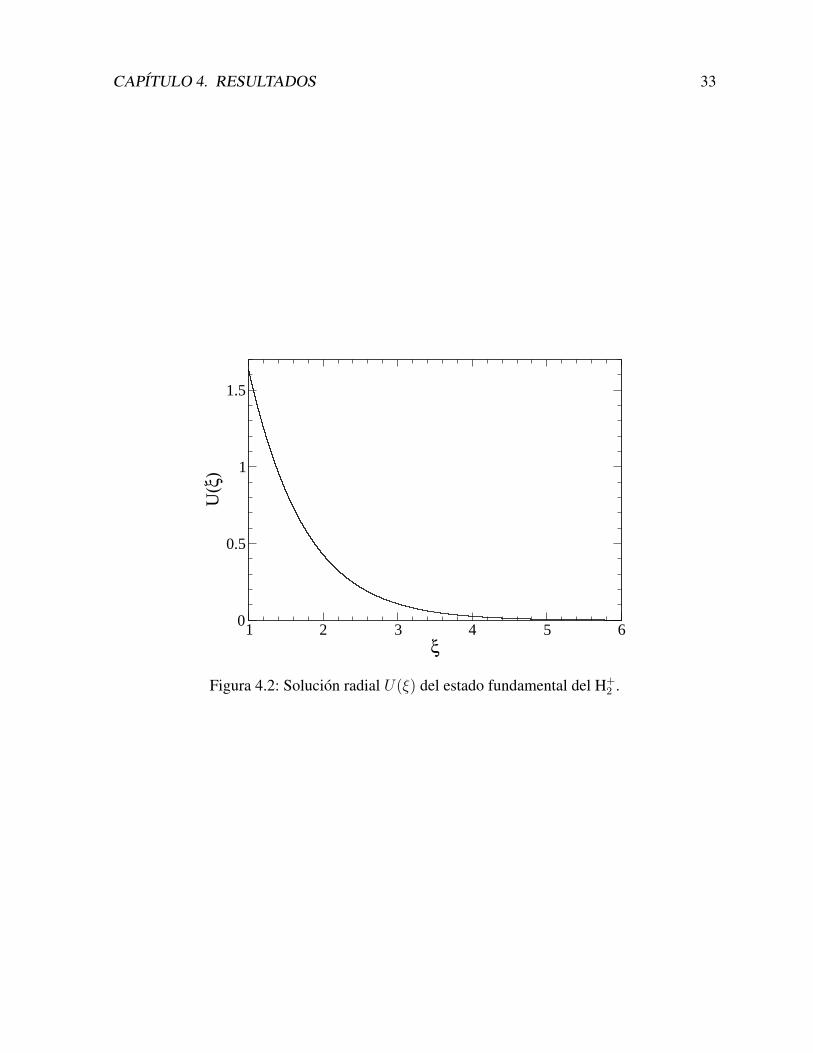
CAPITULO 4. RESULTADOS 33
1 2 3 4 5 6
ξ
0
0.5
1
1.5
U(ξ
)
Figura 4.2: Solucion radial U(ξ) del estado fundamental del H+2 .

CAPITULO 4. RESULTADOS 34
4.1.3. Funciones de OndaCon las soluciones separadas, procedemos a calcular la funcion de onda total que se obtiene
al combinar la solucion radial y la angular
ψ(ξ, η, φ) = U(ξ)Λ(η)Φ(φ). (4.1)
Para visualizar los resultados, conviene convertir la funcion total en coordenadas prolatas acoordenadas cartesianas, tal como explicamos en el Apendice C. Recordemos que las distanciasdel electron a los nucleos esta dada por
r1(x, y, z) =
√x2 + y2 +
(z +
R
2
)2
r2(x, y, z) =
√x2 + y2 +
(z − R
2
)2
. (4.2)
De aquı podemos entonces hacer la conversion de coordenadas,
ξ =r1(x, y, z) + r2(x, y, z)
R
η =r1(x, y, z)− r2(x, y, z)
R
φ = arctan(yx
). (4.3)
En la Figura 4.3 mostramos la solucion total ψ1σg = Λ(η)U(ξ), en coordenadas cartesianas,y para un angulo φ fijo (para los estados con m = 0, existe simetrıa rotacional respecto al ejez, por cual los resultados no dependen del valor del angulo φ). Incluımos en otros colores, eldetalle de las proyecciones en los diferentes ejes.
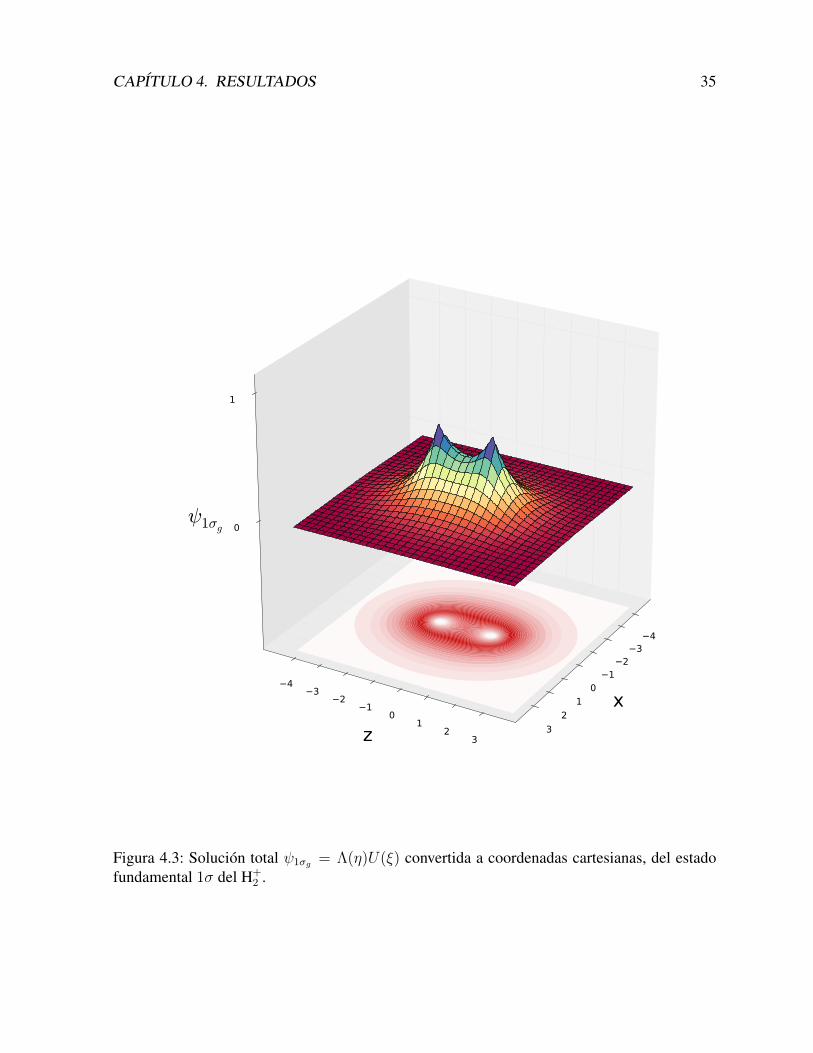
CAPITULO 4. RESULTADOS 35
x
−4−3
−2−1
01
23z
−4−3
−2−1
01
23
ψ1σg 0
1
Figura 4.3: Solucion total ψ1σg = Λ(η)U(ξ) convertida a coordenadas cartesianas, del estadofundamental 1σ del H+
2 .

CAPITULO 4. RESULTADOS 36
4.1.4. Distancia internuclear de equilibrioHemos calculado la distancia internuclear de equilibrio, en la aproximacion de Born–Oppenheimer.
Para ello, calculamos las energıas E(R) del estado fundamental a diferentes distancias R, y encada caso calculamos la energıa total
Etot(R) = E(R) +1
R. (4.4)
Utilizamos las bases encontradas en el punto anterior, pero ahora debemos tener en cuentaque cuando resolvemos la ecuacion Sturmiana radial (3.12),[
∂
∂ξ
[(ξ2 − 1
) ∂∂ξ
]+ a2s ξ − p2s ξ2
]Sj(ξ) = βj Vs(ξ)Sj(ξ) , (4.5)
consideramos aa2s = Rs(Z1 + Z2) , (4.6)
y agregamos el subındice s para explicitar que ese calculo se hace a una distancia fija y prede-terminada. Como la ecuacion de Schrodinger radial (3.10) se calcula ahora para una distanciaa2 variable, esta se modifica haciendo:[
∂
∂ξ
[(ξ2 − 1
) ∂∂ξ
]+ a2ξ + a2sξ − a2sξ − p2ξ2 + A
]U(ξ) = 0 , (4.7)
con lo cual, en la ecuacion matricial (3.19), a la matriz M se le agrega un termino
[M3]ij =
∫ ∞1
Si(ξ) (a2 − a2s) ξ Sj(ξ) dξ , (4.8)
En general, hemos utilizado una unica base, calculada para Rs = 2 a.u., pero para valoresde R muy altos, generamos nuevas bases, con otros valores de a2s. La Figura (4.4) presenta losresultados obtenidos, que a simple vista no parecen tener un mınimo definido. En el recuadrose nota claramente que el mınimo de energıa para la molecula ionizada H+
2 se obtiene en R =1,99704 a.u., y es Etot = −0,602635 a.u., que se corresponde con el valor aceptado [42].
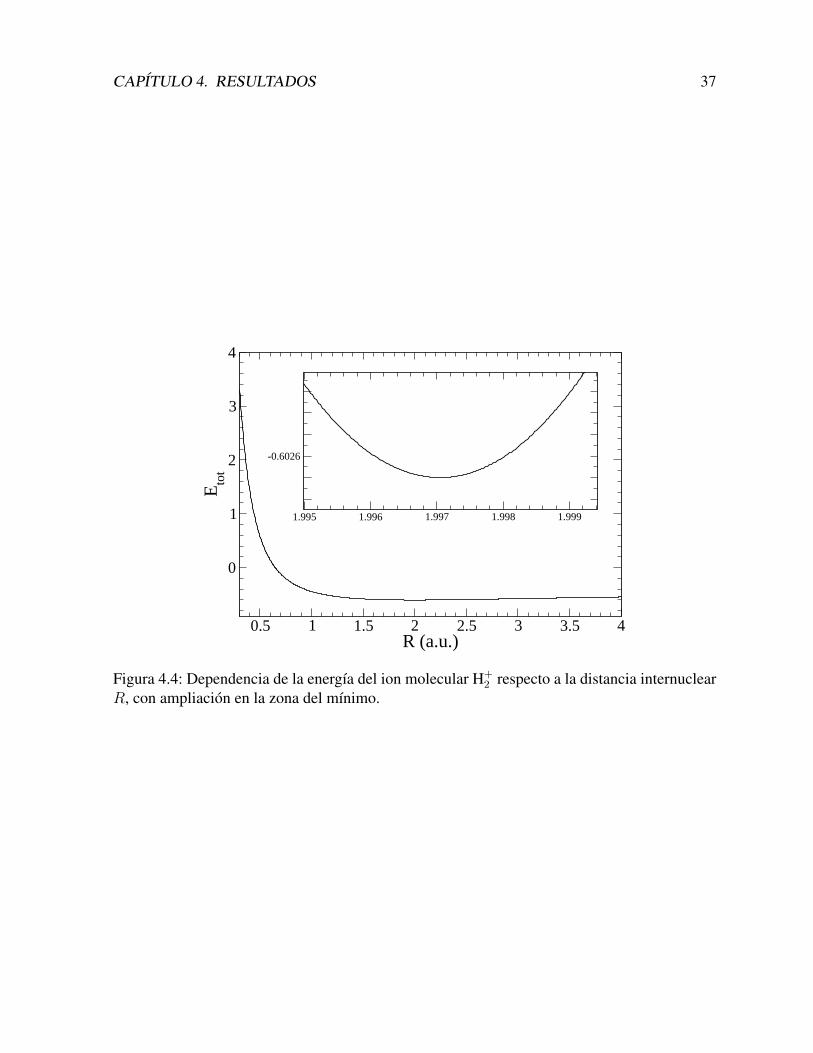
CAPITULO 4. RESULTADOS 37
0.5 1 1.5 2 2.5 3 3.5 4R (a.u.)
0
1
2
3
4
Eto
t
1.995 1.996 1.997 1.998 1.999
-0.6026
Figura 4.4: Dependencia de la energıa del ion molecular H+2 respecto a la distancia internuclear
R, con ampliacion en la zona del mınimo.

CAPITULO 4. RESULTADOS 38
4.1.5. Lımite atomicoUna prueba adicional que se puede realizar a nuestro metodo, consiste en calcular las
energıas del H+2 para distancias R muy pequenas. Este calculo, deberıa dar en el lımite R → 0
la solucion correspondiente al atomo de Helio ionizado He+, cuya energıa es
EHe+ = −1
2Z2 = −2 a.u. . (4.9)
En la Tabla (4.3) se muestran las energıas del estado fundamental obtenidas para distintosvalores de R.
R (a.u.) E (a.u.)2 -1.10263401 -1.45178230.4 -1.8007540.1 -1.97825520.025 -1.99841130.008 -1.9998307He+ -2.0
Tabla 4.3: Energıa del estado fundamental del sistema H + H + e−, en funcion de la distanciainternuclear R.
Por otro lado, la funcion de onda tambien deberıa coincidir con la del ion atomico, es de-cir, la probabilidad deberıa centrarse entre ambos nucleos, a diferencia del caso molecular, en elcual se concentra alrededor de cada uno de ellos. En la Figura 4.5 mostramos como se va produ-ciendo esta transicion, a medida que la distancia disminuye. Para que quede mejor establecidaesta transicion, hemos normalizado las funciones de onda.
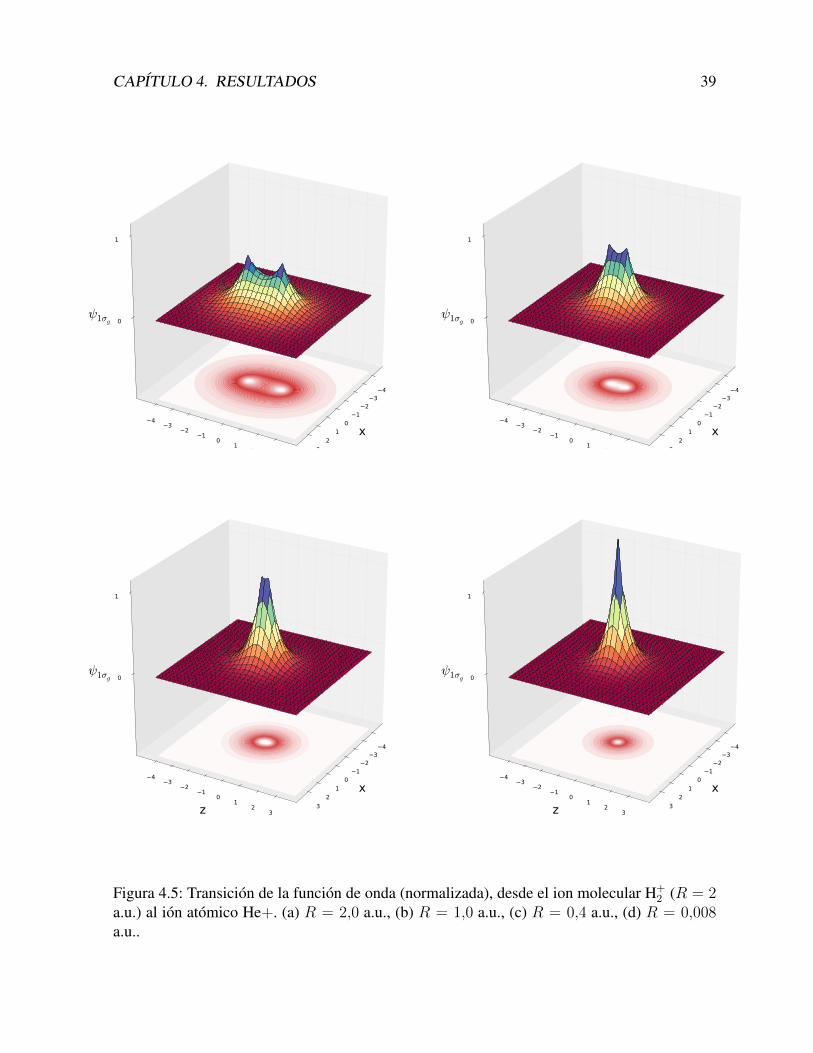
CAPITULO 4. RESULTADOS 39
x
−4−3
−2−1
01
23z
−4−3
−2−1
01
23
ψ1σg 0
1
x
−4−3
−2−1
01
23z
−4−3
−2−1
01
23
ψ1σg 0
1
x
−4−3
−2−1
01
23z
−4−3
−2−1
01
23
ψ1σg 0
1
x
−4−3
−2−1
01
23z
−4−3
−2−1
01
23
ψ1σg 0
1
Figura 4.5: Transicion de la funcion de onda (normalizada), desde el ion molecular H+2 (R = 2
a.u.) al ion atomico He+. (a) R = 2,0 a.u., (b) R = 1,0 a.u., (c) R = 0,4 a.u., (d) R = 0,008a.u..

CAPITULO 4. RESULTADOS 40
4.2. Estados excitados H+2
El metodo de GSF permite no solo obtener el estado fundamental, sino tambien los estadosexcitados y del continuo. Variando la cantidad de nodos admitidos para las autofunciones U(ξ)y Λ(η) segun corresponda para cada caso, se obtienen los estados excitados. En primer lugar,estudiamos como converge el metodo para el calculo del primer estado excitado (1σu, o 2pσu,segun el esquema de notacion – ver Apendice B).
En primer lugar, generamos las bases angulares y radiales. Para el caso de la base radial,recordemos que estas funciones se calculan introduciendo a la energıa como parametro externo,que en principio es un valor arbitrario. Asumiendo que no disponemos de la informacion acercade cual es el valor mas adecuado a asignar, utilizamos como parametro al valor de energıa queobtuvimos para el estado fundamental ( ps = 1,485015 o Es = −1,10263 a.u. ). Generamosentonces 3 bases angulares (solo impares), y 6 radiales.
Con estas bases, realizamos el proceso de iteracion, resolviendo la ecuacion angular, ob-teniendose con ella el autovalor A. Este, a su vez, se ingresa como parametro en la ecuacionradial, y de esta se obtiene la energıa p. En tan solo 8 pasos de iteracion conseguimos un resul-tado con 6 cifras significativas correctas. Los detalles de la convergencia estan presentados enla Tabla 4.4. Sin embargo, tal como vimos en el caso del estado fundamental, podemos utilizarlos ultimos valores hallados en este calculo, (p∗s = 1,154791, es decir E∗s = −0,666771 a.u.),y recalcular la base radial. Utilizando esta base optimizada, hemos obtenido una convergenciamucho mas rapida, y en solo 4 iteraciones llegamos a coincidir con los resultados de Scott [16].
Iteracion p E p∗ E∗
0 1.485015 -1.10263 1.154791 -0.6667712 1.175548 -0.690957 1.155444 -0.6675254 1.155869 -0.668017 1.155451 -0.6675346 1.154793 -0.6667738 1.154791 -0.666771
Referencia[16] 1.155452 -0.667534
Tabla 4.4: Convergencia de los valores de energıa (en a.u.), para el primer estado exciado 1σu(con L=1 y m=0) del H+
2 .
En la Figura 4.6 se muestra la funcion de onda 1σu(x, z). Recordamos que esta funcion esinvariante ante rotaciones en el eje z, por lo tanto resulta suficiente el dibujo en un unico plano.
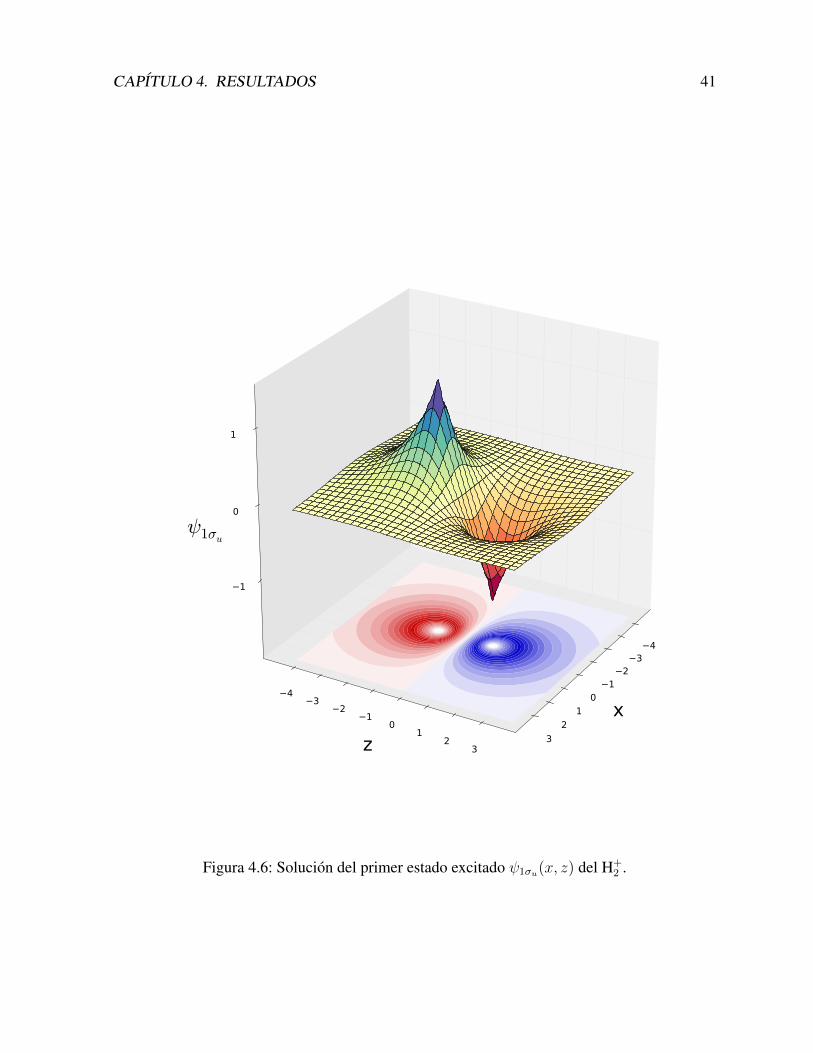
CAPITULO 4. RESULTADOS 41
x
−4−3
−2−1
01
23z
−4−3
−2−1
01
23
ψ1σu
−1
0
1
Figura 4.6: Solucion del primer estado excitado ψ1σu(x, z) del H+2 .
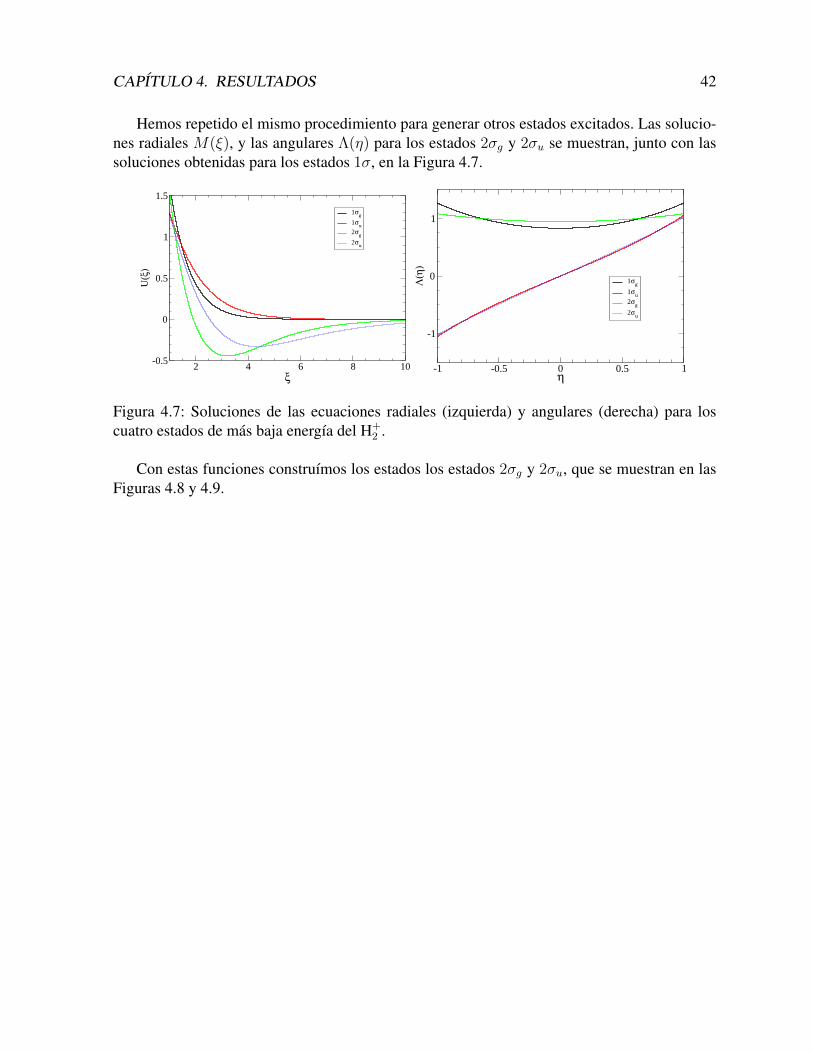
CAPITULO 4. RESULTADOS 42
Hemos repetido el mismo procedimiento para generar otros estados excitados. Las solucio-nes radiales M(ξ), y las angulares Λ(η) para los estados 2σg y 2σu se muestran, junto con lassoluciones obtenidas para los estados 1σ, en la Figura 4.7.
2 4 6 8 10
ξ
-0.5
0
0.5
1
1.5
U(ξ
)
1σg
1σu
2σg
2σu
-1 -0.5 0 0.5 1η
-1
0
1
Λ(η
)
1σg
1σu
2σg
2σu
Figura 4.7: Soluciones de las ecuaciones radiales (izquierda) y angulares (derecha) para loscuatro estados de mas baja energıa del H+
2 .
Con estas funciones construımos los estados los estados 2σg y 2σu, que se muestran en lasFiguras 4.8 y 4.9.
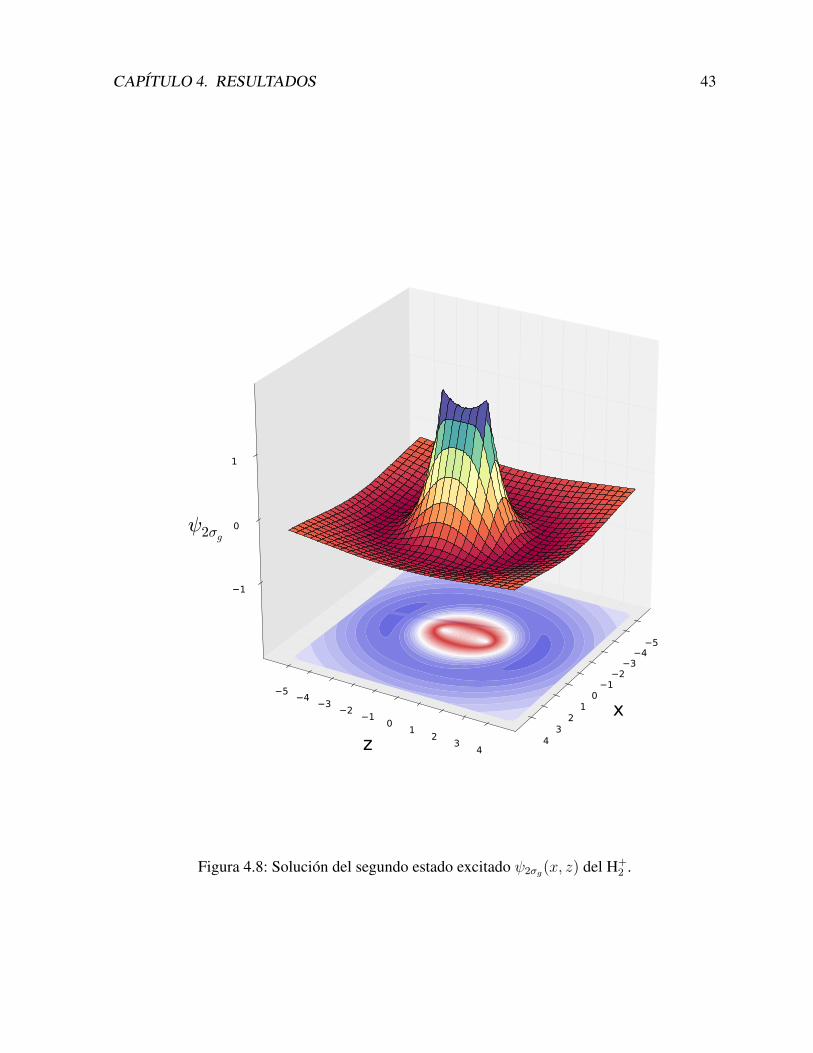
CAPITULO 4. RESULTADOS 43
x
−5−4
−3−2
−10
12
34z
−5 −4 −3 −2 −1 0 1 2 3 4
ψ2σg
−1
0
1
Figura 4.8: Solucion del segundo estado excitado ψ2σg(x, z) del H+2 .
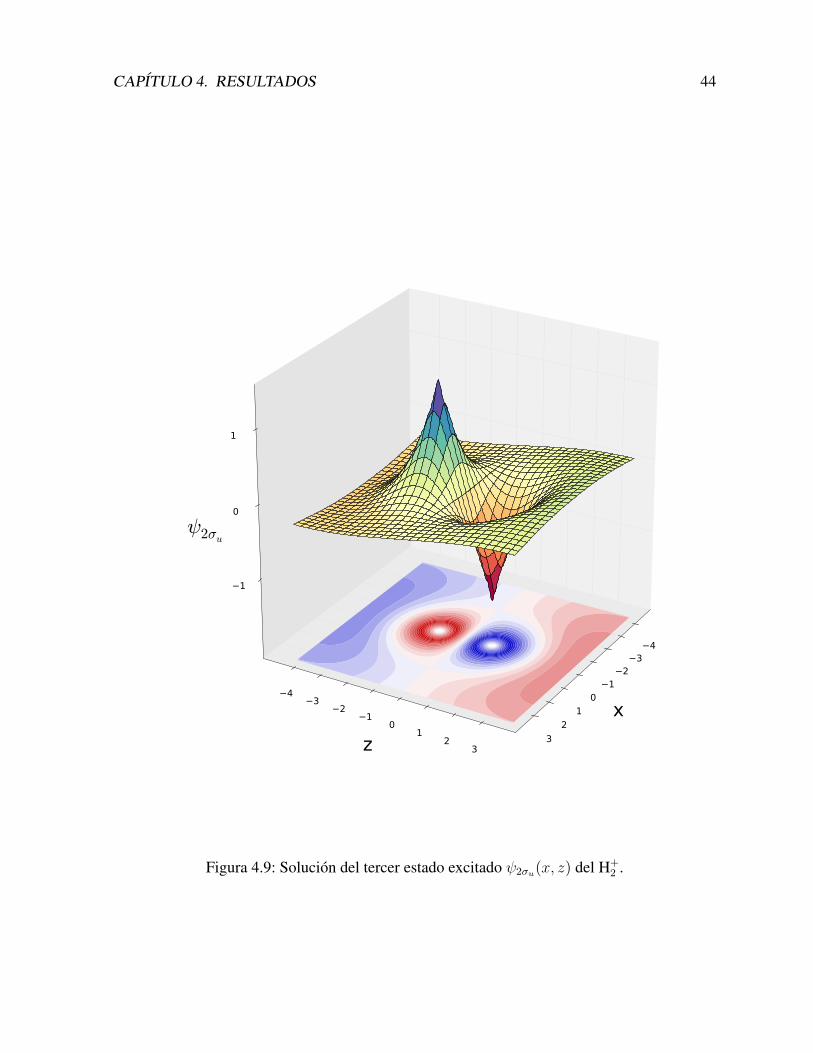
CAPITULO 4. RESULTADOS 44
x
−4−3
−2−1
01
23z
−4−3
−2−1
01
23
ψ2σu
−1
0
1
Figura 4.9: Solucion del tercer estado excitado ψ2σu(x, z) del H+2 .
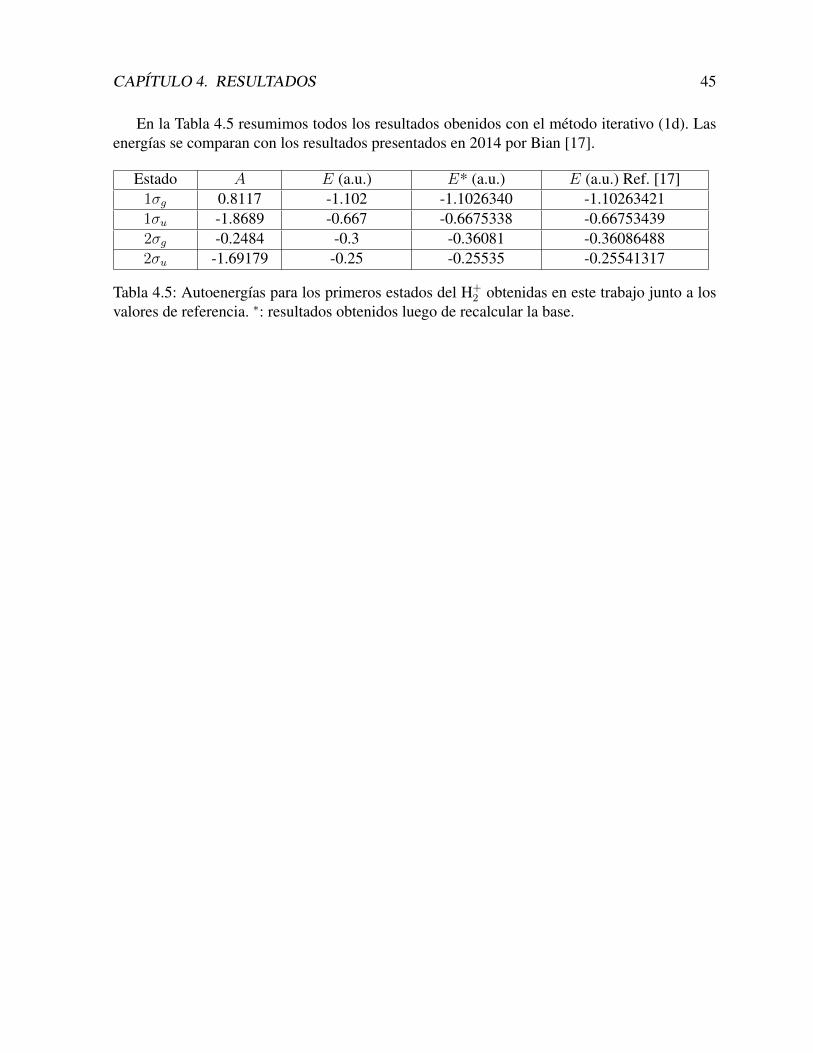
CAPITULO 4. RESULTADOS 45
En la Tabla 4.5 resumimos todos los resultados obenidos con el metodo iterativo (1d). Lasenergıas se comparan con los resultados presentados en 2014 por Bian [17].
Estado A E (a.u.) E* (a.u.) E (a.u.) Ref. [17]1σg 0.8117 -1.102 -1.1026340 -1.102634211σu -1.8689 -0.667 -0.6675338 -0.667534392σg -0.2484 -0.3 -0.36081 -0.360864882σu -1.69179 -0.25 -0.25535 -0.25541317
Tabla 4.5: Autoenergıas para los primeros estados del H+2 obtenidas en este trabajo junto a los
valores de referencia. ∗: resultados obtenidos luego de recalcular la base.

CAPITULO 4. RESULTADOS 46
4.3. Metodo Directo (2d): estados excitados del H+2
Si bien hemos obtenido excelentes resultados con el metodo iterativo (1d), y aunque esta esla forma tradicional con la que se obtienen los mejores resultados citados en nuestro resumen(Seccion 1.4), este procedimiento no aprovecha en su totalidad las ventajas de los metodos es-pectrales, como sı lo hace la resolucion completa del Hamiltoniano, sin separar las coordenadas.
Diagonalizar directamente el Hamiltoniano 2d es una tarea muy costosa desde el punto devista computacional. Teniendo en cuenta que cada coordenada debe representarse con cientosde puntos, la matriz a diagonalizar toma rangos que son intratables numericamente. Podemosmostrar como ejemplo el problema de la diagonalizacion directa del atomo de Helio. Este es unproblema de dos partıculas (asumiendo un nucleo fijo de masa infinita), y por lo tanto hay seiscoordenadas independientes. Si queremos obtener resultados razonables, debemos incluir en lagrilla numerica por lo menos 100 puntos por coordenada. Este calculo recien pudo ser realizadohace unos anos, cuando se pudo contar con supercomputadoras [43, 44]. Este procedimiento,puramente cuantico, y no perturbativo, se considera exacto, ya que las unicas aproximacionesque se introducen son numericas. Es decir, no se hace ninguna simplificacion fısica del proble-ma, y si se quiere mejorar la precision de los resultados, se deben considerar unicamente losaspectos numericos.
Logicamente, se puede utilizar un metodo espectral en lugar de diferencias finitas. Estoreduce en forma considerable el tamano de las matrices a diagonalizar. Con el metodo GSF, lasbases en las que se representan las soluciones incluyen el comportamiento fısico correcto. Deesta manera, el tratamiento numerico esta optimizado, y al mismo tiempo, se lo puede seguirconsiderando dentro de la clasificacion de puramente cuantico y no perturbativo.
Como discutimos anteriormente, introducimos el Hamiltoniano completo del ion molecu-lar H+
2 , asumiendo los nucleos estaticos, y separamos la coordenada azimutal (reiteramos queconsideramos para simplificar m = 0, pero esto no es restrictivo). Con la expansion de las so-luciones en las bases GSF, se obtiene la ecuacion (3.27), que repetimos, levemente modificadaaquı: ∑
ij
aij
{αi Vs(ξ) + p2s ξ
2 − βj}Sij(ξ, η) =
∑ij
aij p2 (ξ2 − η2)Sij(ξ, η)
, (4.10)
donde definimos la base bidimensional
Sij(ξ, η) = Si(ξ)Sj(η) . (4.11)
Esta ecuacion permite apreciar la gran ventaja de nuestro metodo GSF. En primer lugar, alhaber resuelto las ecuaciones de la base Sturmiana, todas las derivadas han sido eliminadas, yse reemplazan por terminos muy simples, que simplifican el calculo. Por otra parte, al utilizarbases optimizadas, el tamano de las bases se reduce significativamente. Tal es ası que en nuestroscalculos incluımos solo 18 funciones (3 Sj(η) y 6 Si(ξ) ). Finalmente, la diagonalizacion directade esta pequena matriz produce como resultado 18 estados, no solo el estado fundamental.

CAPITULO 4. RESULTADOS 47
Con esto obtenemos el espectro correspondiente, evitando la necesidad de realizar iteracionesseparadas para cada estado.
En lo que respecta a los aspectos tecnicos de los calculos, debemos discutir acerca del va-lor del parametro libre en la generacion de las bases. Dado que queremos llegar a resultadosglobales, y no de un unico estado, elegimos como energıa Es =-0.2 a.u.. Este valor, esta lejosde la energıa del estado fundamental, pero permitira obtener con mayor precision a los estadosexcitados. En la Tabla (4.6) mostramos las energıas que obtenemos con el metodo GSF directo(2d), para los primeros 7 estados, con una unica diagonalizacion. En este caso, los resultados secomparan con los de Madsen y Peek [15], respetando la notacion de los estados.
Estado E (a.u.) E (a.u.) Ref. [15]1Sσg -1.102630 -1.102634212Pσu -0.66753431 -0.667534392Sσg -0.360863 -0.360864883Pσu -0.25541312 -0.255413173Dσg -0.2357775 -0.235777633Sσg -0.1776 -0.177681054Pσu -0.133 -0.137312924
Tabla 4.6: Energıas de los primeros estados del H+2 obtenidas mediante el metodo GSF directo
(2d).
Puede observarse que se obtienen excelentes resultados, para todos los niveles energeticos,con muy bajo costo computacional. Si se desea en algun caso particular una mejor precision,puede utilizarse el valor de la energıa que se obtiene para ese nivel particular, e introducirlocomo parametro externo Es en la generacion de la base radial, del mismo modo que lo hicimosen el metodo 1d. Dado que la generacion de la base es un calculo unidimensional, y que la matriz2d tiene tan solo unas pocas decenas de elementos, este procedimiento de ulterior optimizaciontiene muy bajo costo.
4.4. Otros iones: HHe+2 y HLi+3
Una vez desarrollado el metodo y habiendo estudiado los aspectos numericos concernientesal uso de los parametros, criterios de convergencia, etc., podemos avocarnos a resolver otrasmoleculas. En principio, estudiamos sistemas diatomicos y monoelectronicos, como por ejem-plo, los iones moleculares HHe+2 y HLi+3. En la Tabla (4.7) presentamos nuestros resultados,junto a estimaciones de otros autores. Para permitir la comparacion directa, hemos realizado loscalculos a distancias internucleares fijas (en estos iones, el valor que mayormente se encuentraen la literatura es R = 4 a.u.).
Para estimar las ventajas de nuestro metodo, debemos considerar el costo computacional delos resultados realizados con los metodos con los que estamos comparando. En los resultados deAvery et al. [27] se utilizan diez elementos de base (FSC) para cada nucleo. Kereselidze et al.
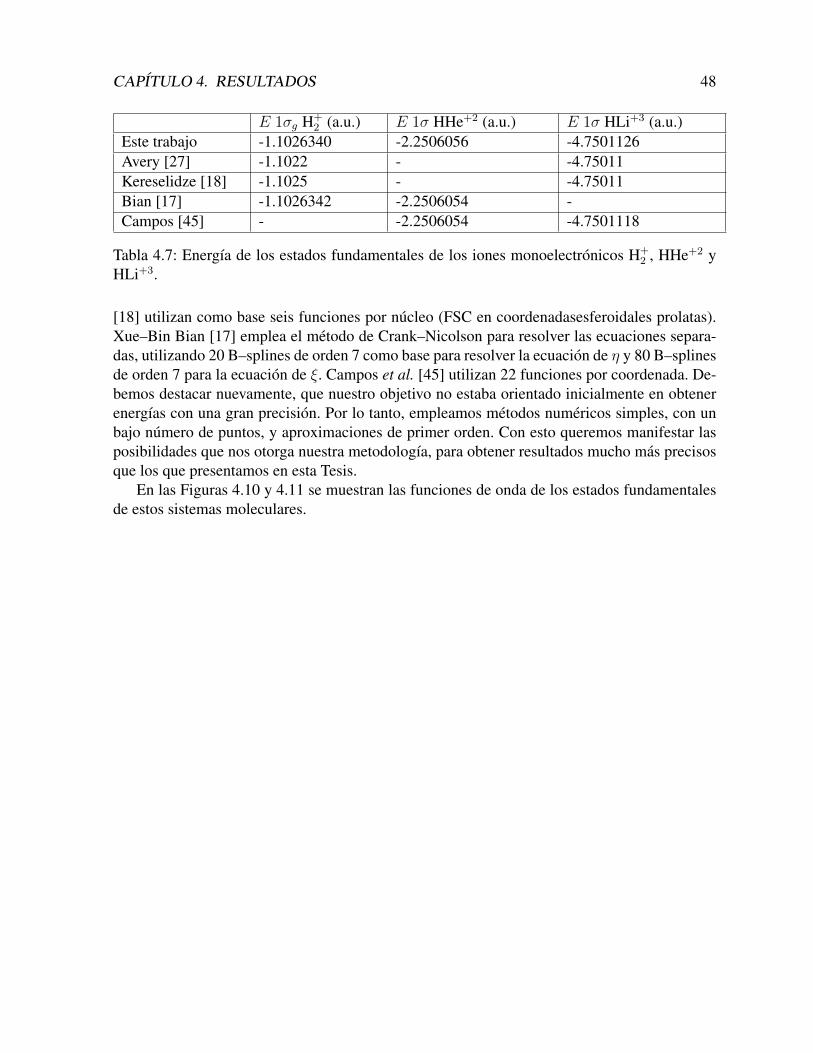
CAPITULO 4. RESULTADOS 48
E 1σg H+2 (a.u.) E 1σ HHe+2 (a.u.) E 1σ HLi+3 (a.u.)
Este trabajo -1.1026340 -2.2506056 -4.7501126Avery [27] -1.1022 - -4.75011Kereselidze [18] -1.1025 - -4.75011Bian [17] -1.1026342 -2.2506054 -Campos [45] - -2.2506054 -4.7501118
Tabla 4.7: Energıa de los estados fundamentales de los iones monoelectronicos H+2 , HHe+2 y
HLi+3.
[18] utilizan como base seis funciones por nucleo (FSC en coordenadasesferoidales prolatas).Xue–Bin Bian [17] emplea el metodo de Crank–Nicolson para resolver las ecuaciones separa-das, utilizando 20 B–splines de orden 7 como base para resolver la ecuacion de η y 80 B–splinesde orden 7 para la ecuacion de ξ. Campos et al. [45] utilizan 22 funciones por coordenada. De-bemos destacar nuevamente, que nuestro objetivo no estaba orientado inicialmente en obtenerenergıas con una gran precision. Por lo tanto, empleamos metodos numericos simples, con unbajo numero de puntos, y aproximaciones de primer orden. Con esto queremos manifestar lasposibilidades que nos otorga nuestra metodologıa, para obtener resultados mucho mas precisosque los que presentamos en esta Tesis.
En las Figuras 4.10 y 4.11 se muestran las funciones de onda de los estados fundamentalesde estos sistemas moleculares.
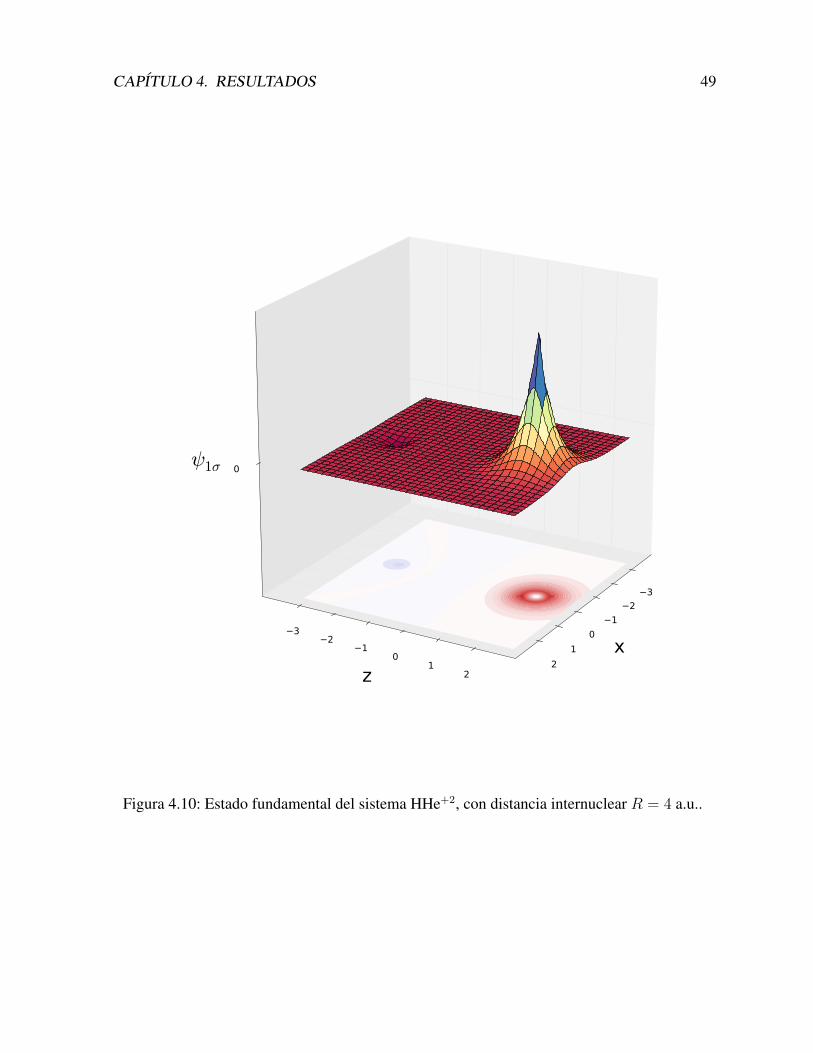
CAPITULO 4. RESULTADOS 49
x
−3−2
−10
12z
−3−2
−10
12
ψ1σ 0
Figura 4.10: Estado fundamental del sistema HHe+2, con distancia internuclear R = 4 a.u..
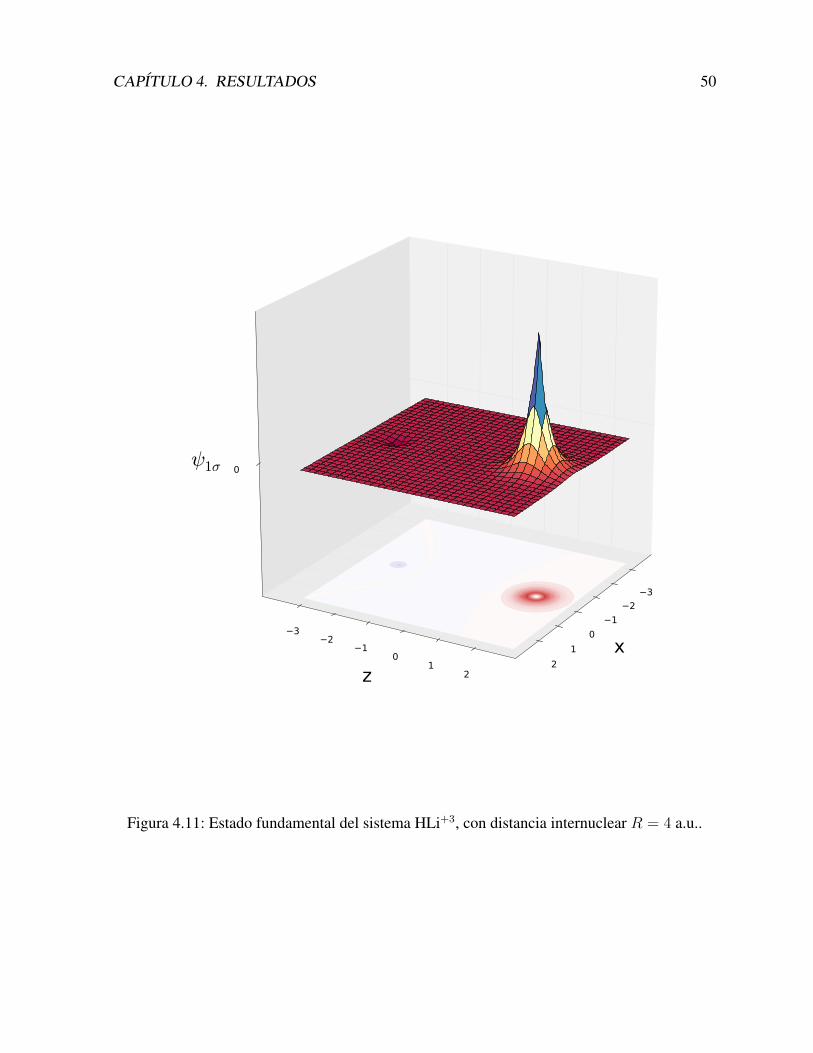
CAPITULO 4. RESULTADOS 50
x
−3−2
−10
12z
−3−2
−10
12
ψ1σ 0
Figura 4.11: Estado fundamental del sistema HLi+3, con distancia internuclear R = 4 a.u..

Capıtulo 5
Conclusiones
En esta tesis se establecieron los principios teoricos del metodo de Funciones SturmianasGeneralizadas (GSF) en coordenadas esferoidales prolatas. Se elaboraron las ecuaciones quedeterminan la generacion de las funciones de base, y su posterior utilizacion en la resolucion deproblemas moleculares, tanto ligados como colisionales.
Se escribieron los codigos computacionales necesarios para la implementacion del metodo,y se realizo un estudio exhaustivo del manejo de los mismos. Esto incluye la determinacion delos valores de los parametros libres, el estudio detallado de convergencias, analisis de errores ydesempenos de ejecucion.
Dado que se trata del trabajo inicial en el area, esta Tesis se focalizo en sistemas molecularesdiatomicos, con un unico electron. Se resolvio el problema fısico de la molecula ionizadasH+
2 , y se encontraron las funciones de onda y las energıas de los primeros estados excitados,en concordancia hasta con 8 cifras significativas con los mejores resultados que existen en laactualidad. Se estudiaron los cambios que se producen al variar la distancia internuclear, y dadoque el metodo computacional continua siendo robusto, aun a distancias muy pequenas, se pudoestudiar el lımite atomico, en el cual ambos nucleos se encuentran juntos.
Se estudiaron otros iones moleculares, como el HHe+2 y HLi+3. En estos casos, los re-sultados publicados en la literatura provienen de calculos en los que se asume una distanciainternuclear fija R = 4 a.u.. Hemos reproducido esos resultados con una notable concordancia.
El metodo GSF ha sido implementado con dos metodologıas diferentes. La primera es ite-rativa, aprovechando la separabilidad de las ecuaciones en coordenadas prolatas. El segundometodo consiste en la diagonalizacion directa del Hamiltoniano. En ambos casos, se explotaronlas enormes ventajas que ofrece el metodo GSF. Basicamente, estas provienen de la correc-ta eleccion de las condiciones asintoticas de las bases, que permiten reducir notoriamente eltamano de los calculos, y tambien acelerar las convergencias.
La comparacion con las otras metodologıas muestran que nuestro metodo es mas efectivo,porque demanda menores recursos computacionales. De todos modos, estas ventajas se haranmucho mas notorias cuando abordemos problemas colisionales. A diferencia de los estados liga-dos – que pueden expandirse con pocas funciones –, los estados del continuo requieren enormescantidades de elementos de base, ya que se extienden hasta grandes distancias. Las FuncionesSturmianas Generalizadas, por su parte, al tener las condiciones asintoticas del problema, solo
51

CAPITULO 5. CONCLUSIONES 52
necesitan resolver el problema a cortas distancias. Esta caracterıstica hace que ciertos proble-mas, intratables desde el punto de vista numerico, de ahora en mas puedan ser considerados aabordar en el futuro.

Apendice A
Combinaciones Lineales de OrbitalesAtomicos
La aproximacion de Born–Oppenheimer ofrece una gran simplificacion al problema mole-cular, desacoplando los movimientos de los electrones y los nucleos. Sin embargo, la solucionexacta de la ecuacion de Schrodinger electronica aun sigue siendo irresoluble. Uno de los meto-dos utilizados para resolver este problema es el metodo variacional, que utiliza como base a losorbitales atomicos.
Las Combinaciones Lineales de Orbitales Atomicos (LCAO) se utilizan para representar alos orbitales moleculares como combinacion de los orbitales atomicos:
ψ =∑n
cnφn . (A.1)
Los valores optimos de los coeficientes cn se encuentran aplicando el principio variacional, loque implica resolver la ecuacion secular
H− ES = 0 , (A.2)
donde los elementos de matriz se calculan como
Hrs = 〈φr|H|φs〉 (A.3)
para el Hamiltoniano H, ySrs = 〈φr|φs〉 (A.4)
para la matriz de solapamiento S. Estas ecuaciones tienen soluciones no triviales solo si eldeterminante de la matriz es nulo. Escribimos esta condicion como
|H− ES| = 0 . (A.5)
Las bases φn deberıan ser orbitales atomicos cuyas coordenadas esten centradas en los di-ferentes nucleos. Sin embargo, por cuestiones practicas, se emplean normalmente funcionesGaussianas, u orbitales de Slater. Las ventajas de estos ultimos es que son funciones con la
53

APENDICE A. COMBINACIONES LINEALES DE ORBITALES ATOMICOS 54
misma forma que los orbitales atomicos, pero permiten ser contraıdos ajustando los parametroscorrespondientes. Las funciones Gaussianas, por otra parte, tienen la enorme ventaja de proveerexpresiones analıticas para las integrales de todas las interacciones, incluyendo aquellas queinvolucran dos centros.
Ilustraremos el uso del metodo variacional con el sistema molecular mas simple: la moleculade Hidrogeno ionizada H+
2 .El Hamiltoniano del problema molecular esta dado por
He = − ~2
2µ∇2 − 1
r1− 1
r2+
1
R, (A.6)
donde ri son las distancias del electron a los respectivos nucleos, y µ es la masa reducida.Obviamente, cuando el electron se aproxima al nucleo 1, r1 � r2, y el Hamiltoniano es aproxi-madamente
He ≈ −~2
2µ∇2 − 1
r1+
1
R, (A.7)
que corresponde al Hamiltoniano de un atomo de hidrogeno, mas la repulsion entre los nucleos.Dada la simetrıa del problema, se puede aproximar el mismo caso, analogo, cuando el electroneste muy cerca del otro nucleo.
Esto sugiere que la aproximacion mas simple mediante LCAO, para los primeros estados(sin normalizar), resulta
ψ+ ≈ φ1 + φ2 y ψ− ≈ φ1 − φ2 , (A.8)
donde φi son orbitales atomicos del Hidrogeno sobre el nucleo i.Para un conjunto base de dos orbitales atomicos, uno sobre el atomo 1 y el otro identico
sobre el atomo 2, el determinante (A.5) tiene un rango de 2× 2. Para abreviar, denominamos alos elementos de matriz
S11 = S22 = 1 S12 = S21 = S ,
yH11 = H22 = α H12 = H21 = β ,
donde α es la integral de Coulomb y β es la integral de resonancia. El determinante resulta∣∣∣∣ α− E β − E Sβ − E S α− E
∣∣∣∣ = 0 , (A.9)
y sus raıces son
E± =α± β1± S
. (A.10)
Los valores correspondientes para los coeficientes reales de las funciones de onda normali-zadas son
c1 = c2 c1 =1√
2 + 2Spara E+ =
α + β
1 + S,

APENDICE A. COMBINACIONES LINEALES DE ORBITALES ATOMICOS 55
c1 = −c2 c1 =1√
2− 2Spara E− =
α− β1− S
. (A.11)
La integral de Coulomb α se calcula (abreviamos |φi〉 por |i〉) haciendo
α = 〈 1 | H |1 〉 = E1s + J , (A.12)
donde el primer termino se obtiene debido a que φ1 es autofuncion del Hamiltoniano atomico yresulta
E1s = 〈 1 | − ~2
2me
∇2 − 1
r1| 1 〉 (A.13)
mientras que J corresponde a la energıa de interaccion entre la densidad electronica φ21 con el
nucleo 2, mas la repulsion entre ambos nucleos
J = −〈 1 | 1
r2| 1 〉+
1
R= −
∫φ21
r2dτ +
1
R. (A.14)
La integral de resonancia β por su parte resulta
β = 〈 1 | H | 2 〉 = E1s S +K (A.15)
dondeK = −〈 1| 1
r1| 2 〉+
S
R= −
∫φ1φ2
r1dτ +
S
R. (A.16)
Para el caso de la moleculaH+2 , es de suponer que la menor energıa se obtendra en alguna de
las combinaciones que involucre a φ1 = φ2 = φ1s. En ese caso, J , S y K poseen las siguientesexpresiones analıticas:
J = e−2R(
1 +1
R
), (A.17)
S = e−R(
1 +R +1
3R2
), (A.18)
K =S
R− e−R(1 +R) = e−R
[1
R− 2
3R
]. (A.19)
En la Figura A.1 se muestran estas integrales, en funcion de R.
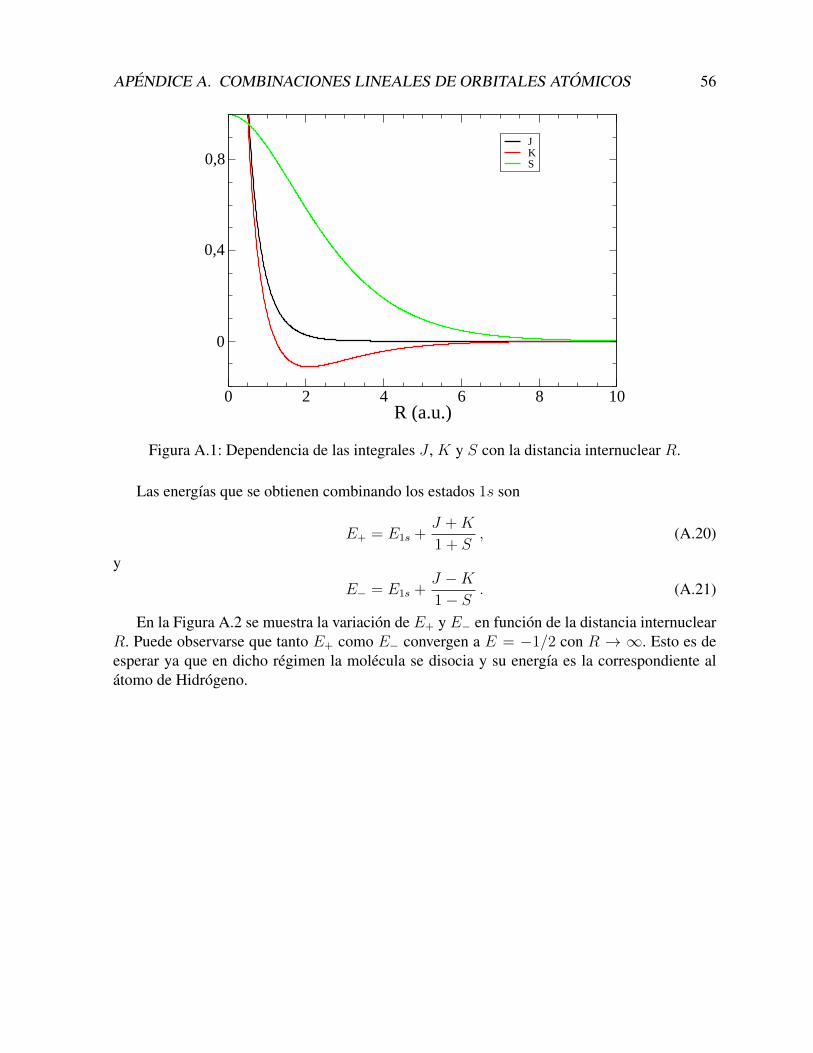
APENDICE A. COMBINACIONES LINEALES DE ORBITALES ATOMICOS 56
0 2 4 6 8 10R (a.u.)
0
0,4
0,8
JKS
Figura A.1: Dependencia de las integrales J , K y S con la distancia internuclear R.
Las energıas que se obtienen combinando los estados 1s son
E+ = E1s +J +K
1 + S, (A.20)
y
E− = E1s +J −K1− S
. (A.21)
En la Figura A.2 se muestra la variacion de E+ y E− en funcion de la distancia internuclearR. Puede observarse que tanto E+ como E− convergen a E = −1/2 con R → ∞. Esto es deesperar ya que en dicho regimen la molecula se disocia y su energıa es la correspondiente alatomo de Hidrogeno.
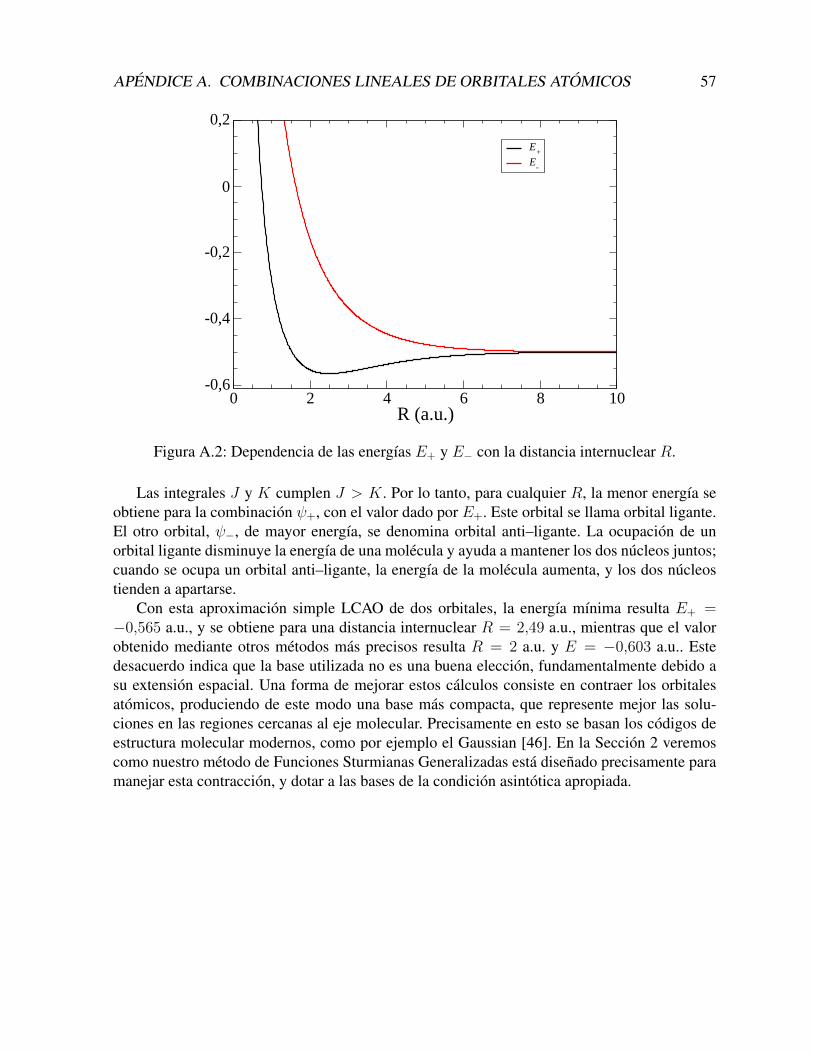
APENDICE A. COMBINACIONES LINEALES DE ORBITALES ATOMICOS 57
0 2 4 6 8 10R (a.u.)
-0,6
-0,4
-0,2
0
0,2
E+
E-
Figura A.2: Dependencia de las energıas E+ y E− con la distancia internuclear R.
Las integrales J y K cumplen J > K. Por lo tanto, para cualquier R, la menor energıa seobtiene para la combinacion ψ+, con el valor dado por E+. Este orbital se llama orbital ligante.El otro orbital, ψ−, de mayor energıa, se denomina orbital anti–ligante. La ocupacion de unorbital ligante disminuye la energıa de una molecula y ayuda a mantener los dos nucleos juntos;cuando se ocupa un orbital anti–ligante, la energıa de la molecula aumenta, y los dos nucleostienden a apartarse.
Con esta aproximacion simple LCAO de dos orbitales, la energıa mınima resulta E+ =−0,565 a.u., y se obtiene para una distancia internuclear R = 2,49 a.u., mientras que el valorobtenido mediante otros metodos mas precisos resulta R = 2 a.u. y E = −0,603 a.u.. Estedesacuerdo indica que la base utilizada no es una buena eleccion, fundamentalmente debido asu extension espacial. Una forma de mejorar estos calculos consiste en contraer los orbitalesatomicos, produciendo de este modo una base mas compacta, que represente mejor las solu-ciones en las regiones cercanas al eje molecular. Precisamente en esto se basan los codigos deestructura molecular modernos, como por ejemplo el Gaussian [46]. En la Seccion 2 veremoscomo nuestro metodo de Funciones Sturmianas Generalizadas esta disenado precisamente paramanejar esta contraccion, y dotar a las bases de la condicion asintotica apropiada.

Apendice B
Nomenclatura
Los diferentes estados electronicos se denominan segun diversos criterios, que pueden serlas energıas, las propiedades de simetrıa, los momentos angulares y de spin, y los estados a loscuales se disocian cuando la distancia internuclear se hace suficientemente grande. Ningunode estos criterios por sı mismo puede determinar en forma inambigua al estado de cualquiermolecula.
La primer diferencia importante entre un estado molecular y un estado atomico, radica enel rol que tiene el numero cuantico principal n. A diferencia de los atomos, donde este nume-ro determina el orden energetico, en las moleculas esto solo ocurre para estados Rydberg. Losestados inferiores con diferente momento angular pueden estar separados con mayor diferenciaenergetica que aquellos con distintos numeros cuanticos n, lo que le resta sentido a la caracte-rizacion de estados moleculares por medio de este numero.
En las moleculas en general, los nucleos producen un campo que no es invariante ante rota-ciones, por lo cual, el momento angular no se conserva. En el caso particular de las moleculasdiatomicas, la cantidad que se conserva es la proyeccion del momento angular sobre el eje nu-clear, que se denota con el sımbolo Λ. A los estados con Λ = 0, 1, 2 se los denomina con lossımbolos Σ,Π,∆, · · · etc.. Si bien existen ciertas excepciones (como por ejemplo, la moleculaNO), en general, el estado fundamental de una molecula diatomica es un estado Σ.
Ademas, en una molecula diatomica, el estado de electrones de la molecula se caracterizapor el spin total S de todos los electrones de la molecula. Si S no es cero entonces hay unadegeneracion de 2S + 1 con respecto a las direcciones del espın total (ignorando la estructurafina). El numero 2S + 1 se llama multiplicidad, como en el caso de los atomos, y se escribecomo un superındice antes de la letra griega que indica la proyeccion del momento angular a lolargo del eje nuclear. Por ejemplo, 2Πu denota un estado de electrones impares con S = 1/2 yΛ = 1. En el caso de moleculas monoelectronicas, el ındice 2S + 1 resulta igual a 2 para todoslos estado, por lo cual, se contrae.
La energıa de una molecula diatomica es invariante ante la reflexion sobre un plano queatraviese el eje nuclear. Esta simetrıa se esquematiza en la Figura B.1(a). Sin embargo, ante estaoperacion el valor de Λ cambia de signo. Esto significa que todos los estados, excepto los Σ,estan doblemente degenerados. La funcion de onda de un estado Σ solo puede ser multiplicadapor una constante como resultado de la reflexion. La doble reflexion es equivalente a la trans-
58

APENDICE B. NOMENCLATURA 59
formacion identidad, por lo tanto los autovalores del operador de reflexion (para estados Σ) son±1. Los estados que cambian de signo, y los que no cambian de signo ante una reflexion, sedenotan como Σ− y Σ+, respectivamente.
En el ion de la molecula de hidrogeno se observa otra simetrıa, que consiste en la inversionde las coordenadas electronicas, manteniendo las coordenadas de los nucleos sin cambios. Estasimetrıa se esquematiza en la Figura B.1(b). Para una molecula con dos nucleos identicos, existeun centro de simetrıa en el punto medio de la lınea de union de los nucleos. El Hamiltonianosigue invariante a la inversion de las coordenadas electronicas, y este operador de transforma-cion tambien conmuta con el operador momento angular orbital. Esto permite que los estadossean etiquetados dependiendo de la paridad de la funcion de onda. Para un Λ dado, los estadospares (g – gerade) son invariantes a tal cambio, mientras que los estados impares (u – ungerade)cambian de signo.
Esto da como resultado la descripcion completa del estado del electron dado por 2S+1Λ±g,ucon el (±) incluido solo para estados Σ.
Figura B.1: (a) Reflexion de las coordenadas electronicas sobre un plano que contiene al ejemolecular. (b) Inversion de todas las coordenadas respecto al origen (r → −r).
Otros metodos para etiquetar los estados de moleculas diatomicas, se basan en los estadosatomicos en los que esta se disocia (R → ∞), o bien en etiquetar el estado que se obtendrıa alunir los atomos (R→ 0). Estos metodos de nomenclatura se denominan de Atomos separados yAtomos unidos, respectivamente. En estas notaciones se reemplaza la letra griega mayuscula poruna letra griega minuscula, respetando el mismo orden. Para el caso de los atomos separados,si al disociarse la molecula se obtienen estados atomicos 1s, el estado molecular se nota 1sσgo 1sσu, donde tanto σ como g respetan las reglas mencionadas anteriormente. Sin embargo,el estado 1sσu, bajo la notacion de atomos unidos, resulta un estado p. Concretamente, dichoestado se nota 2pσ. Los lımites bajo los cuales se realizan estas nomenclaturas se esquematizanen la siguiente figura:

APENDICE B. NOMENCLATURA 60
Figura B.2: Esquema de las nomenclaturas utilizadas. Se muestra la notacion atomica, la nota-cion de Atomos separados y finalmente la notacion de Atomos unidos.
La correlacion entre notaciones, para los primeros estados de una molecula diatomica ho-monuclear se muestra en la siguiente Tabla, ordenada energeticamente para el lımite R→ 0.
Atomos unidos Atomos separados1sσ 1sσg2sσ 2sσg2pσ 1sσu2pπ 2pπu3sσ 2pσg3pσ 2sσu3pπ 3pπg3dσ 3sσg3dπ 2pπg4sσ 3pσg4pσ 2pσu4dπ 3pπu
Tabla B.1: Correlacion entre notaciones.
En la Tabla B.2 se muestran algunos ejemplos de los primeros estados moleculares y losestados atomicos en los que se disocian, bajo la notacion de atomos separados.
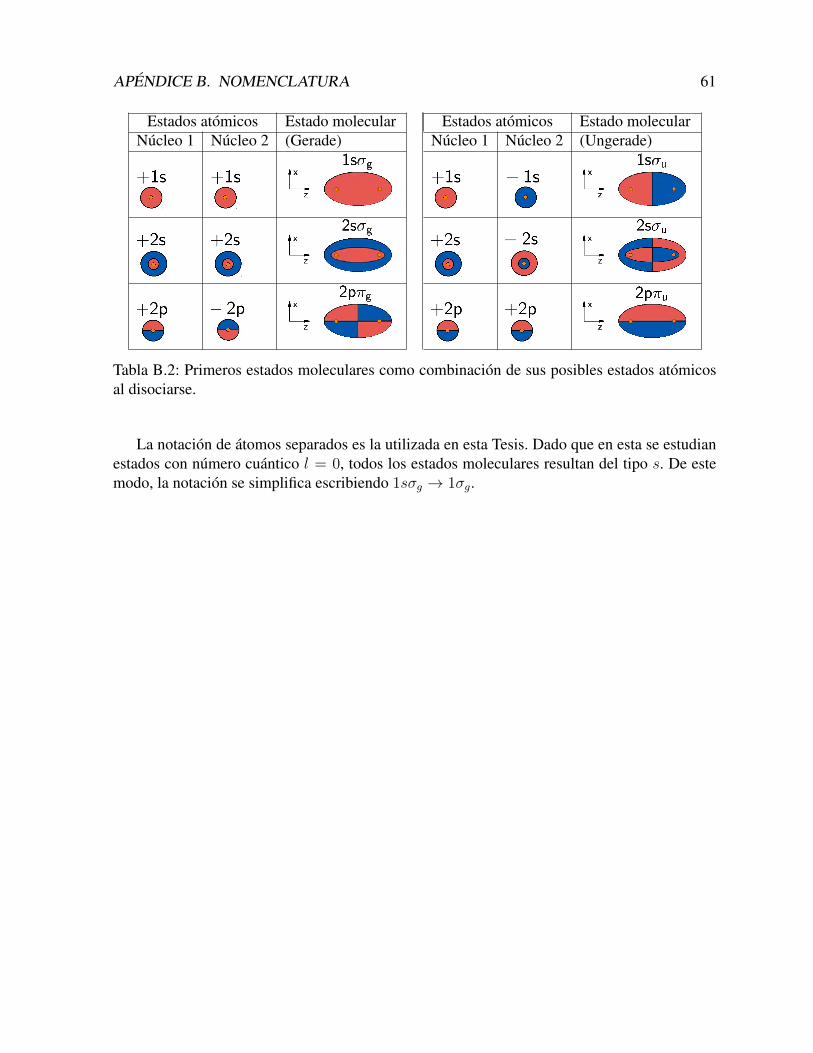
APENDICE B. NOMENCLATURA 61
Estados atomicos Estado molecular Estados atomicos Estado molecularNucleo 1 Nucleo 2 (Gerade) Nucleo 1 Nucleo 2 (Ungerade)
Tabla B.2: Primeros estados moleculares como combinacion de sus posibles estados atomicosal disociarse.
La notacion de atomos separados es la utilizada en esta Tesis. Dado que en esta se estudianestados con numero cuantico l = 0, todos los estados moleculares resultan del tipo s. De estemodo, la notacion se simplifica escribiendo 1sσg → 1σg.

Apendice C
Coordenadas esferoidales prolatas
Las coordenadas esferoidales prolatas proporcionan un sistema muy importante para proble-mas con dos centros. En particular la ecuacion de onda electronica de la molecula de hidrogenoionizada es separable en estas coordenadas. Tomando como referencia el sistema de la FiguraC.1
Figura C.1: Molecula de hidrogeno ionizada H+2 .
las coordenadas prolatas [47, 48, 49] se definen como
ξ =r1 + r2R
; η =r1 − r2R
; φ = arctan(yx
). (C.1)
donde 1 ≤ ξ <∞, −1 ≤ η < 1 y 0 ≤ φ < 2π. Con esta definicion
r1 =R
2(ξ + η) y r2 =
R
2(ξ − η) . (C.2)
62

APENDICE C. COORDENADAS ESFEROIDALES PROLATAS 63
Las relaciones de correspondencia con las coordenadas cartesianas (x, y, z) quedan definidaspor
x =R
2
√(ξ2 − 1)(1− η2) cosφ ,
y =R
2
√(ξ2 − 1)(1− η2) sinφ ,
z =R
2ξ η . (C.3)
Analizando las coordenadas en un plano con φ constante, vemos que las curvas con ξ cons-tante forman elipses, cuyos focos son los nucleos, mientras que las η constante forman hiperbo-las. En todo el espacio, las superficies con ξ constante conforman elipsoides confocales derevolucion, y las superficies η constante forman hiperboloides de revolucion alrededor del ejez. Las superficies con el angulo azimutal φ constante forman semiplanos.
Figura C.2: Coordenadas esferoidales prolatas.
La interseccion de estas tres superficies ubican unıvocamente un punto en el espacio.
C.1. LaplacianoLa formula general para el Laplaciano en un sistema de coordenadas (q1, q2, q3) ortogonales
es
∇2 =1
h1h2h3
[∂
∂q1
(h2h3h1
∂
∂q1
)+
∂
∂q2
(h1h3h2
∂
∂q2
)+
∂
∂q3
(h1h2h3
∂
∂q3
)], (C.4)

APENDICE C. COORDENADAS ESFEROIDALES PROLATAS 64
donde hn es el factor de escala de la coordenada enesima y se calcula segun
h2n =
(∂x
∂qn
)2
+
(∂y
∂qn
)2
+
(∂z
∂qn
)2
. (C.5)
Con la forma general del factor de escala para un sistema de coordenadas ortonormal, elelemento de volumen dV queda definido con
dV = h1 h2 h3 dq1 dq2 dq3 . (C.6)
Con estas definiciones estamos en condiciones de derivar el Laplaciano en coordenadasesferoidales prolatas. En primer lugar, se deben reemplazar los (x, y, z) obtenidos en (C.3),en las expresiones correspondientes para hallar los hn (Ecuacion C.5). Por ejemplo, para lacoordenada ξ se obtiene
h2ξ =
(R
2
ξ(1− η2)√(ξ2 − 1)(1− η2)
cosφ
)2
+
(R
2
ξ(1− η2)√(ξ2 − 1)(1− η2)
sinφ
)2
+
+
(R
2η
)2
, (C.7)
Simplificando la expresion, y aplicando analogamente el mismo procedimiento a las otras coor-denadas, se obtiene
hξ =R
2
√ξ2 − η2ξ2 − 1
,
hη =R
2
√ξ2 − η21− η2
,
hφ =R
2
√(ξ2 − 1)(1− η2) . (C.8)
Reemplazando los factores hn (C.8) en (C.4), el Laplaciano en esferoidales prolatas resulta
∇2 =4
R2(ξ2 − η2)
{∂
∂ξ
[(ξ2 − 1)
∂
∂ξ
]+
∂
∂η
[(1− η2) ∂
∂η
]+
+ξ2 − η2
(ξ2 − 1)(1− η2)∂2
∂φ2
}. (C.9)

Apendice D
Construccion Numerica de las GSF
En nuestro trabajo hemos calculado las funciones de base mediante diferencias finitas. Dadoque convertimos las ecuaciones a sistemas de ecuaciones unidimensionales, ha sido suficiente,en la mayorıa de los casos, la utilizacion de la aproximacion en primer orden.
Vamos a describir, en primer lugar, los metodos numericos utilizados para generar las basesGSF para estados ligados. Las soluciones Sn(r) de la ecuacion (2.11), con un potencial auxiliarUaux Coulombiano y energıa Es negativa, tienen un comportamiento asintotico decreciente (ex-ponencial). Si la carga Z ∈ R, entonces la funcion asintotica tambien es real, el flujo es nulo, ylas soluciones son estacionarias. El espectro en este caso, es discreto e infinito.
Se comienza integrando la ecuacion desde el origen, asumiendo un comportamiento Sn(r →0) = rl+1. La integracion se realiza hasta un punto de cotejo rm, ubicado a una distancialevemente mayor al ultimo punto de inflexion. A partir de este punto, se supone que la solucionya adquiere su forma asintotica, pero no se puede seguir integrando hacia afuera, ya que siemprela solucion esta contaminada por una fraccion divergente, y a grandes distancias, aun en el casoen que esta fraccion fuera muy pequena, la solucion total va a diverger. Es de suponer tambienque a esta distancia el potencial generador (que es de corto alcance) Vgen es nulo. Por lo tanto,podemos calcular el punto de inflexion rt como aquel en el cual se satisface
Uaux(rt) +l(l + 1)
2r2t+ βn Vgen(rt) = Es . (D.1)
En la region externa, mas alejada que el ultimo punto de inflexion, se integra la ecuacionhacia adentro, desde un infinito practico r∞, donde se asume que la funcion se comporta comoe−κr. El valor de r∞ se elige de manera tal que
κ r∞ =√−2(E − U(r∞)) r∞ ≈ 40 . (D.2)
El valor de la funcion en este punto Sn(r∞), es aproximadamente 10−12 veces el maximo valor.Por lo tanto, podemos considerarlo tecnicamente nulo. Se comienza desde allı la integracionhacia adentro asumiendo el comportamiento asintotico adecuado.
En ambas integraciones (tanto hacia afuera como hacia adentro), se requiere un valor inicialdel autovalor βn. Si el problema es Coulombiano, podemos utilizar las cargas Coulombianasanalıticas. Luego, se deben seguir los siguientes pasos:
65

APENDICE D. CONSTRUCCION NUMERICA DE LAS GSF 66
1. Con el autovalor escogido, se calcula el ultimo punto de inflexion rt (ecuacion (D.1)).
2. Se integra hacia afuera, hasta el punto de cotejo rm, levemente mas alejado que rt. Estasolucion se denomina Sout
n , y se puede normalizar∫ rm
0
Soutn Vgen(r) Sout
n dr = 1 . (D.3)
3. Se debe controlar que el numero de nodos de la funcion sea el correcto. Este debe ser(n− l − 1). Si no lo es, se cambia el autovalor βn en un 10 % y se comienza de nuevo laiteracion.
4. Se integra hacia adentro, desde r∞ hacia rm, y se llama a esta solucion Sinwn . Se escalea
esta funcion, para obtener una solucion total continua, es decir, Sinwn (rm) = Sout
n (rm).
5. Se calcula la correccion ∆βn
∆βn =Sn(rm)(dS
inwn
dr− dSout
n
dr)|r=rm
2(∫ rm0
Soutn (r)V (r)Sout
n (r) +∫∞rmSinwn (r)V (r)Sinw
n (r)), (D.4)
determinada por la diferencia entre las derivadas de las dos funciones en el punto decotejo.
6. Se realiza el chequeo de convergencia. El proceso se repite, hasta que ambas curvas ten-gan la misma derivada en el punto de cotejo.
En general, con unas pocas iteraciones se obtiene una convergencia muy rapida. Obviamen-te, esto depende de la eleccion del autovalor inicial, y del criterio de convergencia impuesto. Sibien este algoritmo permite obtener en solo 4 o 5 pasos un resultado correcto con cerca de 10 ci-fras significativas, es importante apreciar hasta que grado de precision se requieren calcular losautovalores, para que la funcion de onda tenga un comportamiento asintotico adecuado. En laTabla D.1 se muestran los resultados parciales que se van obteniendo en los pasos sucesivos, pa-ra un problema Coulombiano, en el cual la energıa Es = −2 a.u. (Uaux(r) = 0, Vgen(r) = −1
r).
Este problema admite solucion exacta para las cargas βn = 2n. En la tabla mostramos los re-sultados para n = 5, por lo cual la carga deberıa tomar el valor β5 = 10. Para poder apreciarla convergencia, mostramos las diferencias de las derivadas logarıtmicas ∆η5 = ηinw5 − ηout5 ,donde
ηn ≡dSndr
Sn, (D.5)
y el correspondiente factor de correccion ∆βn dado en (D.4). Tambien hemos calculado lassoluciones con una grilla numerica de pocos puntos (N = 500), ya que de otra manera laconvergencia serıa inmediata.
A pesar que las iteraciones sucesivas van cambiando el autovalor en una figura significativamuy alta, este cambio tiene un efecto dramatico en la autofuncion correspondiente. Dado quela solucion numerica es una combinacion de dos soluciones exponenciales (una decreciente y
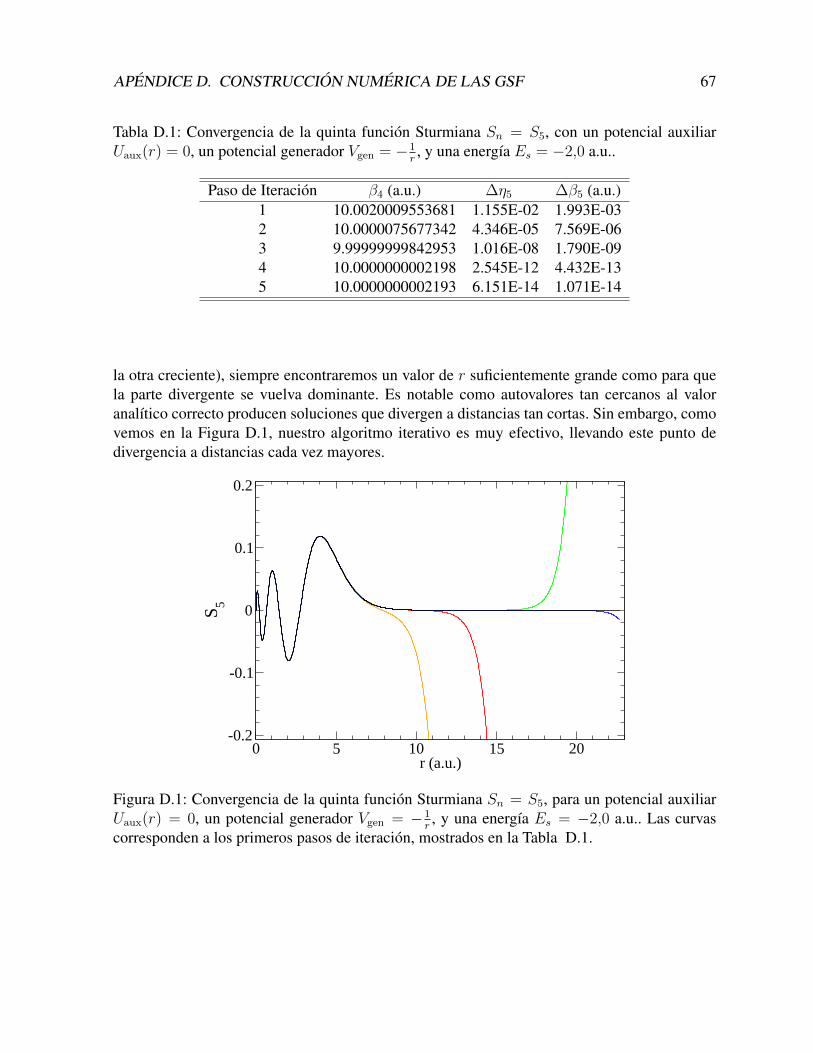
APENDICE D. CONSTRUCCION NUMERICA DE LAS GSF 67
Tabla D.1: Convergencia de la quinta funcion Sturmiana Sn = S5, con un potencial auxiliarUaux(r) = 0, un potencial generador Vgen = −1
r, y una energıa Es = −2,0 a.u..
Paso de Iteracion β4 (a.u.) ∆η5 ∆β5 (a.u.)1 10.0020009553681 1.155E-02 1.993E-032 10.0000075677342 4.346E-05 7.569E-063 9.99999999842953 1.016E-08 1.790E-094 10.0000000002198 2.545E-12 4.432E-135 10.0000000002193 6.151E-14 1.071E-14
la otra creciente), siempre encontraremos un valor de r suficientemente grande como para quela parte divergente se vuelva dominante. Es notable como autovalores tan cercanos al valoranalıtico correcto producen soluciones que divergen a distancias tan cortas. Sin embargo, comovemos en la Figura D.1, nuestro algoritmo iterativo es muy efectivo, llevando este punto dedivergencia a distancias cada vez mayores.
0 5 10 15 20r (a.u.)
-0.2
-0.1
0
0.1
0.2
S5
Figura D.1: Convergencia de la quinta funcion Sturmiana Sn = S5, para un potencial auxiliarUaux(r) = 0, un potencial generador Vgen = −1
r, y una energıa Es = −2,0 a.u.. Las curvas
corresponden a los primeros pasos de iteracion, mostrados en la Tabla D.1.

Apendice E
Polinomios de Legendre
Los polinomios de Legendre cumplen la ecuacion diferencial (E.1)
d
dx
[(1− x2
) d
dxPn (x)
]+ n (n+ 1)Pn (x) = 0 , (E.1)
esta ecuacion, llamada Ecuacion Diferencial de Legendre, puede resolverse usando el metodode serie de potencias. En general la serie de potencias obtenida converge cuando |x| < 1 y en elcaso particular de que n sea un entero no negativo (0, 1, 2, ...) las soluciones forman una familiade polinomios ortogonales llamados Polinomios de Legendre.
Una importante propiedad de los polinomios de Legendre es que estos son ortogonales conrespecto al producto escalar definido en L2 en el intervalo −1 ≤ x ≤ 1:∫ 1
−1Pm(x)Pn(x)dx =
2
2n+ 1δmn . (E.2)
Los polinomios de Legendre pueden construirse usando la relacion de recurrencia (E.3)
(n+ 1)Pn+1 = (2n+ 1)xPn − nPn−1 , (E.3)
de la cual podemos realizar el siguiente despeje:
xPn =1
2n+ 1[(n+ 1)Pn+1 + nPn−1] . (E.4)
Al multiplicar por otro polinomio de Legendre Pm e integrando en ambos miembros seobtiene ∫ 1
−1Pm xPn dx =
1
2n+ 1
∫ 1
−1Pm [(n+ 1)Pn+1 + nPn−1] dx .
Utilizando la propiedad de ortogonalidad (E.2) se obtiene∫ 1
−1Pi xPj dx =
2
2i+ 1
[j + 1
2j + 1δi,j+1 +
j
2j + 1δi,j−1
]. (E.5)
68

APENDICE E. POLINOMIOS DE LEGENDRE 69
De manera analoga se pueden resolver todas las integrales del tipo∫ 1
−1Pm x
k Pn dx ,
con k εN. Por ejemplo para k = 2 la integral se calcula utilizando:
x2Pn = x(xPn) = xn+ 1
2n+ 1Pn+1 + x
n
2n+ 1Pn−1 . (E.6)
Para ello calculamos:
xPn+1 =n+ 2
2n+ 3Pn+2 +
n+ 1
2n+ 3Pn (E.7)
y tambien
xPn−1 =n
2n− 1Pn +
n− 1
2n− 1Pn−2 . (E.8)
La integral entonces queda:∫ 1
−1Pi x
2 Pj dx =2
2i+ 1
[j + 1
2j + 1
j + 2
2j + 3δi,j+2 +
+
(j + 1
2j + 1
j + 1
2j + 3+
j
2j + 1
j
2j − 1
)δi,j +
+j
2j + 1
j − 1
2j − 1δi,j−2
]. (E.9)

Bibliografıa
[1] Thomson, J. J. On rays of positive electricity, The London, Edinburgh, and Dublin Philo-sophical Magazine and Journal of Science, 13, 561 (1907).
[2] Brasefield, C. J. “The Spectrum of the Hydrogen Molecular Ion”, Proceedings of the Na-tional Academy of Sciences, 14, 686 (1928).
[3] Pauli, W. “uber das Modell des Wasserstoffmolekulions”, Annalen der Physik, 373, 177(1922).
[4] Niessen, K. F. “Zur quantentheorie des wasserstoffmolekulions”, Annalen der Physik, 375, 129 (1923).
[5] Alexandrow, W. “Das Wasserstoffmolekulion und die Undulationsmechanik”. Annalen derPhysik, 386 , 603 (1926).
[6] Burrau, O. “Berechnung des energiewertes des wasserstoffmolekel-ions (H+2 ) im nor-
malzustand”, Naturwissenschaften, 15, 16 (1927).
[7] Wilson, A. H. “The ionised hydrogen molecule”. Proceedings of the Royal Society ofLondon, Series A, Containing Papers of a Mathematical and Physical Character, 118, 635(1928).
[8] Hylleraas, E. A. “uber die Elektronenterme des Wasserstoffmolekuls. Zeitschrift fur Phy-sik”, 71, 739 (1931).
[9] Jaffe, G. “Zur theorie des Wasserstoffmolekulions. Zeitschrift fur Physik A Hadrons andNuclei”, 87, 535 (1934).
[10] Bates, D. R., Ledsham, K., and Stewart, A. L. “Wave functions of the hydrogen molecularion”, Philosophical Transactions of the Royal Society of London A: Mathematical, Physicaland Engineering Sciences, 246, 215 (1953).
[11] Teller, E. “uber das Wasserstoffmolekulion”, Zeitschrift fur Physik A Hadrons and Nuclei,61, 458 (1930).
[12] Steensholt, G. “Zur numerischen Berechnung der Potentialkurven des Wasserstoffmo-lekulions”, Zeitschrift fur Physik A Hadrons and Nuclei , 100, 547 (1936).
70

BIBLIOGRAFIA 71
[13] Chakravarty, S. K. “Quantization under two centres of forces.—Part I. The hydrogen mo-lecular Ion”. The London, Edinburgh, and Dublin Philosophical Magazine and Journal ofScience, 28, 423 (1939).
[14] Bates, D. R., and Reid, R. H. G. “Electronic eigenenergies of the hydrogen molecular ion”,Advances in atomic and molecular physics, 4, 13 (1968).
[15] Madsen, M. M. and Peek, J.M. “Atomic Data and Nuclear Data Tables”, 2, 171 (1970).
[16] Scott, T. C., Aubert-Frecon, M., and Grotendorst, J. “New approach for the electronicenergies of the hydrogen molecular ion”, Chemical physics, 324, 323 (2006).
[17] Bian, X. B. “Photoionization of atoms and molecules studied by the Crank-Nicolson met-hod”, Physical Review A, 90, 033403 (2014).
[18] Kereselidze, T., Chkadua, G., and Defrance, P. “Coulomb Sturmians in spheroidal coor-dinates and their application for diatomic molecular calculations”, Molecular Physics, 113,3471 (2015).
[19] Kereselidze, T., Chkadua, G., Defrance, P., and Ogilvie, J. F. “Derivation, properties andapplication of Coulomb Sturmians defined in spheroidal coordinates”, Molecular Physics,114, 148 (2016).
[20] Shull, H., and and Lowdin, P. O., J. Chem. Phys. 30, 617 (1959).
[21] Avery, J., and Avery, J. “Generalized sturmians and atomic spectra”, ISBN 981-256-806-9.Published by World Scientific Publishing Co., Pte. Ltd., Singapore, (2006).
[22] Rotenberg, M. “Application of Sturmian functions to the Schrodinger three-body problem:Elastic e+-H scattering”, Annals of Physics, 19, 262 (1962).
[23] Rotenberg, M. “Theory and application of Sturmian functions”, Advances in Atomic andMolecular Physics, 6, 233 (1970).
[24] Aquilanti V., Cavalli S., Coletti C. and Grossi G., Chem. Phys. 209, 405 (1996).
[25] Aquilanti V. and Caligiana A., Chem. Phys. Lett. 366, 157 (2002).
[26] Koga T. and Matsuhashi T., J. Chem. Phys. 89, 983 (1988).
[27] Avery, J., and Avery, J. “Can Coulomb Sturmians be used as a basis for N-electron mole-cular calculations?”, The Journal of Physical Chemistry A, 113, 14565 (2009).
[28] Avery J. and Avery J., Molec. Phys. bf 110, 1593 (2012).
[29] Rawitscher, G. “Positive Energy Weinberg States for the Solution of Scattering Problems”,Phys. Rev. C , 25, 2196 (1982).

BIBLIOGRAFIA 72
[30] Rawitscher, G. “Iterative Solution of Integral Equations on a Basis of Positive EnergySturmian Functions”, Phys. Rev. E, 85, 026701 (2012).
[31] Yu Ovchinnikov, S.; Macek, J. H. “Positive Energy Sturmian States for Two-CoulombCenter Problems”, Phys. Rev. A, 55, 3605 (1997).
[32] Macek, J. H.; Yu Ovchinnikov, S.; Gasaneo, G. “Exact Solution for Three Particles Inter-acting via Zero-Range Potentials”, Phys. Rev. A, 73, 032704 (2006).
[33] Randazzo, J. M., “Metodos ab initio para el problema de tres cuerpos con interaccionesCoulombianas” , PhD thesis, Instituto Balseiro, Universidad Nacional de Cuyo Division deColisiones Atomicas, (2009).
[34] Frapiccini, A. L., “Descripcion de la dinamica de electrones en interaccion con ato-mos”, PhD thesis, Instituto Balseiro, Universidad Nacional de Cuyo Division de ColisionesAtomicas, (2009).
[35] Ambrosio, M. J., “Simple y doble ionizacion de Helio por impacto de electrones”, PhDthesis, Universidad Nacional del Sur Division de Colisiones Atomicas, (2014).
[36] Granados, C., “Application of Generalized Sturmian Basis Functions to Molecular Sys-tems”, PhD thesis, Universite de Lorraine, (2016).
[37] Gasaneo, G., Ancarani, L. U., Mitnik, D. M., Randazzo, J. M., Frapiccini, A. L., andColavecchia, F. D. “Three-Body coulomb problems with generalized sturmian functions”,Adv. Quantum Chem, 67, 153 (2013).
[38] Mitnik, D. M., Colavecchia, F. D., Gasaneo, G., and Randazzo, J. M. “Computationalmethods for Generalized Sturmians basis”, Computer Physics Communications, 182, 1145(2011).
[39] Gasaneo, G., Ancarani, L. U., Mitnik, D. M., Randazzo, J. M., Frapiccini, A. L., andColavecchia, F. D., “Three-Body coulomb problems with generalized sturmian functions”.Adv. Quantum Chem, 67, 153 (2013).
[40] https://www.wolfram.com/mathematica/ (2017).
[41] Linear Algebra PACKage, http://www.netlib.org/lapack/ (2017).
[42] Schaad, L. J., and Hicks, W. V.“Equilibrium Bond Length in H+2 ”, The Journal of Chemical
Physics, 53, 851 (1970).
[43] Mitnik, D. M. “Helium atom in a box: Doubly excited levels within the S-wave model”,Physical Review A, 70, 022703 (2004).
[44] Mitnik, D. M. “Helium atom in a box: a fully quantal solution”, Nuclear Physics A, 790,784c (2007).

BIBLIOGRAFIA 73
[45] Campos, J. A., Nascimento, D. L., Cavalcante, D. T., Fonseca, A. L. A., and Nunes, A. O.C. “Determination of electronic energy levels for the heteromolecular ions HeH2+, LiH3+,and BeH4+ from the Hamilton–Jacobi equation”, International journal of quantum che-mistry, 106, 2587 (2006).
[46] http://gaussian.com/ (2017).
[47] LEY-Koo, E. “Desarrollos armonicos del potencial de Coulomb en coordenadas cilındri-cas, parabolicas y esferoidales”, Revista Mexicana de Fısica, 5 785 (1993).
[48] Celemin, H., Machado, E. J., and Cardona-Bedoya, J. A. “Solucion de la ecuacion deSchrodinger para el atomo de hidrogeno en coordenadas esferoidales prolatas”, Respuestas,15, 43 (2015).
[49] Ley-Koo, E., and Cruz, S. A. “The hydrogen atom and the H+2 and HeH++ molecular ions
inside prolate spheroidal boxes”, The Journal of Chemical Physics, 74, 4603 (1981).


















