
Estudio teórico conformacional del ácido …148.206.53.84/tesiuami/206320125.pdfcomplejación y en...
28
Transcript of Estudio teórico conformacional del ácido …148.206.53.84/tesiuami/206320125.pdfcomplejación y en...


2
Estudio teórico conformacional del ácido etilendiamino tetraacético
Mariano Rodríguez Bautista
Área de Fisicoquímica Teórica. Departamento de Química.
División de Ciencias Básicas e Ingeniería.
Universidad Autónoma Metropolitana-Unidad Iztapalapa
Resumen
En este trabajo se realizó un estudio conformacional del ácido etilendiamino tetraacético (EDTA)
neutro, usando la teoría de funcionales de la densidad (TFD). Para la búsqueda conformacional, se
utilizó como punto de partida la información de rayos X, obteniendo que las geometrías resultantes
difieren de manera apreciable de la geometría de partida. Por lo que se exploró la superficie de energía
potencial variando diferentes ángulos diedros, los cuales se eligieron de acuerdo a la simetría exhibida
por el sistema. Debido a la probabilidad de formación de puentes de hidrógeno se utilizó una
corrección a las interacciones de largo alcance sobre el funcional de intercambio y correlación PBE,
usando diferentes conjuntos de funciones de base (DZVP, DZVP/A1 y TZVP). De todas las estructuras
encontradas se eligieron aquellas que no diferían en más de 12 kcal/mol de la más estable, encontrando
16 conformaciones del EDTA donde ninguna de las estructuras corresponde a la de rayos X. Los
resultados obtenidos de los primeros 5 confórmeros de menor energía fueron contrastados con los
resultados generados por el método MP2/6-311G** y MP2/6-31++G**. Se encontró que la corrección
a la dispersión en el funcional de intercambio y correlación es muy importante para que exista un
comportamiento similar entre el método MP2 y TFD. La caracterización de puentes de hidrógeno en las
conformaciones del EDTA encontradas nos indica que estas interacciones inducen a que el EDTA
predomine en una forma compacta con estructuras semejantes a aquellas reportadas por rayos X donde
el EDTA forma complejos con un átomo metálico. Con el fin de conocer el efecto del solvente sobre la
estructura del EDTA, se estudiaron diferentes confórmeros del EDTA con 10 moléculas de agua usando
un método estocástico para la búsqueda conformacional. Se encontró que los puentes de hidrógeno
formados con el agua provocan una transferencia de protón intramolecular de un grupo carboxilo a un
grupo amino y una transferencia intermolecular entre el EDTA y una molécula de agua, concluyendo
que el medio ambiente provoca transferencias de protón en el EDTA, como aquella sugerida por lo
estudios de rayos X en el EDTA sin acomplejarse.

3
I. Introducción
Un grupo interesante de reacciones químicas, es la complejación de iones metálicos con agentes
específicos. Es importante mencionar que la complejación es una manera conveniente para separar,
inactivar o movilizar iones metálicos[1]. El ácido etilendiaminotetraacético (EDTA), en su forma de sal
metal alcalina, es uno de los reactivos clásicos más importantes para la determinación complejométrica
de iones metálicos en solución[2]. El EDTA y ligandos similares pueden ser usados para remover iones
metálicos tóxicos de aguas contaminadas y suelos, e incluso del cuerpo humano, es también aplicado
en terapias de quelación, por solubilización de metales, así como en numerosas aplicaciones
industriales como en el proceso de blanqueado de celulosa para fabricar papel[3]. Además, se ha
ocupado en odontología, donde se puede hacer remoción de sarro dentario o pueden realizarse
cavidades molares[4].
De acuerdo a los estudios experimentales del EDTA, se han reportado seis constantes de acidez (pka)
de las cuales cuatro de ellas pertenecen a las desprotonaciones de los grupos carboxilos (pka1=1.15,
pka2=1.15, pka3=2.12 y pka4=2.57) y dos pertenecen a las desprotonaciones de grupos amino
(pka5=6.16 y pka6=10.08)[3][5]. También se han reportado entalpías de formación y energías libres de
Gibbs entre el EDTA y algunos metales. En general, el EDTA interacciona con metales divalentes y en
algunas ocasiones los metales presentan números de oxidación mayores a dos. Es importante resaltar
que en todos los reportes se hace énfasis en el papel que juega el solvente en el proceso de
complejación y en que el EDTA se encuentra totalmente desprotonado cuando se forma el complejo
EDTA-Metal; es importante mencionar que existen estudios donde han variado el pH de la solución
donde se lleva a cabo la complejación, y de acuerdo a esta variación de pH han encontrado que el pH
debe ser básico, por lo tanto, el EDTA se encuentra desprotonado. Debido a que solamente utilizan al
EDTA en su forma desprotonada para formar el complejo EDTA-Metal, se aclara en los reportes que es
muy difícil realizar un estudio cuando el EDTA está complejando el metal y no se encuentra totalmente
desprotonado. Es importante mencionar un trabajo de resonancia magnética nuclear (RMN), donde
miden los cambios de señales de protón en el EDTA libre y en el complejo EDTA-Metal. Estas
mediciones las realizaron de acuerdo a los protones de los acetato-metileno (ubicados en los brazos del
EDTA) y a los protones del esqueleto-metileno (ubicado en la parte central del EDTA que une a los
grupos amino). Uno de los resultados importantes en ese reporte, es la estructura hexacoordinada
octahédrica presentada por el complejo EDTA-Metal que puede presentar dos diferentes
conformaciones [6].

4
Por otro lado, de acuerdo a los trabajos teóricos reportados hasta el momento podemos decir que se han
enfocado a la interacción EDTA-Metal, donde se han reportado energías libres de Gibbs, entalpías de
formación, constantes cinéticas y constantes de equilibrio. En algunos reportes se hace énfasis en la
contribución del solvente (donde se han usado los modelos continuos, explícitos y mixtos) y la
competitividad del Metal por el EDTA. En términos estructurales se ha encontrado la importancia de la
simetría que exhibe el EDTA, la forma de sus brazos (grupos carboxilos) en la interacción con el metal
para adoptar la estructura que forma el complejo EDTA-Metal, que en su mayoría es una estructura
hexacoordinada y en algunas ocasiones pentacoordinada. Dichas estructuras presentan variaciones
debido a la naturaleza del metal o a cambios de temperatura. En un reporte se utiliza la estructura del
EDTA-metal reportada por la técnica de rayos X y a partir de esa información se modela tal
interacción, usando para este fin TFD considerando solamente un conjunto de funciones de base y un
funcional de intercambio y correlación. Además, no consideran las contribuciones del solvente y la
temperatura. Es importante mencionar que en todos los trabajos utilizaron al EDTA en su forma
totalmente desprotonada.
De lo que pudimos encontrar en la literatura, es evidente que no existe un solo estudio teórico del
EDTA sin complejación y por lo tanto es necesario un trabajo teórico que reporte las posibles
conformaciones del EDTA libre con diferentes grados de protonación, puesto que se sabe
experimentalmente que el EDTA tiene seis constantes de acidez (pka) reportadas[3][5]. Esto nos lleva a
que la parte esencial de nuestro trabajo esta enfocado en el estudio de las posibles conformaciones de
EDTA libre neutro como molécula aislada y su interacción de cada una de estas conformaciones con
moléculas de agua explicitas (microsolvatación).
II. Metodología y detalles computacionales
La búsqueda conformacional del EDTA libre neutro se llevó a cabo de la siguiente manera: 1) Se partió
de una estructura de rayos X del EDTA reportada por M. F. C. Ladd y D. C. Povey en 1973[7], en esta
estructura los autores sugieren que se presenta transferencia de protones intramolecular de los grupos
carboxilos a los grupos aminos (Figura 1a). 2) Adicionalmente a esta estructura, propusimos otra a
partir de la estructura de rayos X con los protones en los grupos carboxilos como se muestra en la
Figura 1b.

5
a) b)
Figura 1. a) Estructura reportada por M. F. C. Ladd y D. C. Povey en 1973, c) Estructura propuesta.
Ambas estructuras se optimizaron usando el funcional de intercambio y correlación PBE[8] y la base
DZVP con el conjunto de funciones auxiliar A1[9]. 3) Sobre cada una de las estructuras optimizadas se
giraron dos ángulos diedros para hacer la búsqueda conformacional. El ángulo diedro presentado en la
Figura 1a, definido por los átomos N-C-C-N el cual divide a la molécula en dos fragmentos que
contienen dos “brazos” cada uno de ellos. Sobre cada brazo se definió el ángulo N-C-C-O y
precisamente este fue el segundo ángulo diedro que se usó para la búsqueda conformacional. Cada uno
de estos ángulos se variaron en 20 grados partiendo de 0 a 180 y de 0 a -180. Para cada valor que
adquirían estos ángulos se optimizó cada estructura fijando los dos ángulos. Una vez que se tiene
construida la superficie de energía potencial, se ubicaron todas las conformaciones que presentaron un
mínimo y sobre ellas se realizó una optimización sin restricciones. Con afán de ampliar la exploración
en la búsqueda conformacional, se realizó sobre cada conformación una dinámica molecular usando el
método semiempírico PM3[10] y después de terminado el tiempo de la corrida se realizó su respectiva
optimización.
Debido a los posibles puentes de hidrógeno que se pueden formar en el EDTA se consideraron los
efectos de dispersión, o de largo alcance, a través de la corrección de Grimme (DISP)[11] teniendo así
el método PBE+DISP. Ambos funcionales de intercambio y correlación se acoplaron con las funciones
de base DZVP/A1, DZVP y TZVP[9], para tener idea del efecto de las bases auxiliares y el tamaño del
conjunto de funciones de base. Además, se utilizó la Teoría de Perturbaciones de Moller y Plesset a
segundo orden (MP2)[12] utilizando dos conjuntos de funciones base 6-311G** y 6-31++G**[13].
Este método se aplicó sólo a las estructuras de más baja energía encontradas con el método
PBE+DISP/TZVP.

6
Sobre todas las estructuras encontradas con el método PBE+DISP/DZVP/A1 se incluyeron 10
moléculas de agua, dos por cada grupo carboxilo y una por cada grupo amino del EDTA. Las
posiciones de la moléculas de agua se obtuvieron al aplicar un método estocástico, el cual se basa en
colocar dentro de una caja de tamaño predefinido y aplica el método de recocido simulado. En nuestro
caso el tamaño de la caja que se usó fue de 8.5 Angstroms. De este procedimiento se obtuvieron cientos
de estructuras y se eligieron solamente aquellas donde las moléculas de agua rodearan al máximo al
EDTA. Finalmente, las estructuras elegidas fueron optimizadas con el método PBE+DISP/DZVP/A1.
Naturalmente, se espera que aparezcan puentes de hidrógeno inter e intra molecular. Debido a los
posibles contactos de este tipo fue necesario diseñar un algoritmo para poder contabilizar y caracterizar
los puentes de hidrógeno. Así, se programó este algoritmo en el lenguaje de programación en C. Los
criterios que usa dicho algoritmo para decidir si se presenta un puente de hidrógeno son: Distancia de
2.5 Angstroms entre el hidrógeno y el átomo que lo acepta (H…A), distancia de 3.0 Angstroms entre el
átomo donador y el átomo aceptor de hidrógeno (D…H) , y el ángulo de 135.0 grados formado por el
donador - hidrógeno – aceptor (D…H…A); para que se cumpla este tipo de contacto los valores que
encuentra el programa deben ser menor o igual a los impuestos. El programa con algunas corridas se
reporta en el Anexo A.
El método estocástico empleado usa la implementación del método PM3 que se encuentra en
Gaussian09. Todas las optimizaciones de los métodos basados en TFD y en MP2 se realizaron con el
código NWChem v6.1.

7
II. Resultados
IIa. Energías conformacionales
De acuerdo al desarrollo de metodología expuesta anteriormente, se obtuvieron las estructuras que se
muestran en la Figura 2. Ahora bien, cuando comparamos las estructuras de las Figuras 1 y 2,
encontramos una gran diferencia entre ellas, por lo tanto, las estructuras que se encuentran en fase
sólida no corresponden a las estructuras en forma aislada.
a) b)
Figura 2. Geometrías optimizabas con el método PBE/DZVP/A1. a) Estructura a partir de la Figura 1a.
b) Estructura a partir de la Figura 1b.
Debido a que las geometrías optimizadas difieren de las geometrías de partida se exploró la superficie
de energía potencial de acuerdo a lo señalado en la metodología, obteniendo como resultado 35
estructuras. Al fijar un intervalo de energía conformacional entre 0 y 12 kcal/mol, se obtuvieron 18
conformaciones del EDTA. De las conformaciones obtenidas, encontramos que en algunas de ellas hay
presencia de puentes de hidrógeno y por esa razón se utilizó el método PBE+DISP. Al usar este método
se encontraron 16 estructuras que se encuentran en el intervalo mencionado. Las energías relativas
encontradas por los métodos PBE y PBE+DISP, con los tres conjuntos de funciones de base DZVP/A1,
DZVP y TZVP, se reportan en la Tabla 1.
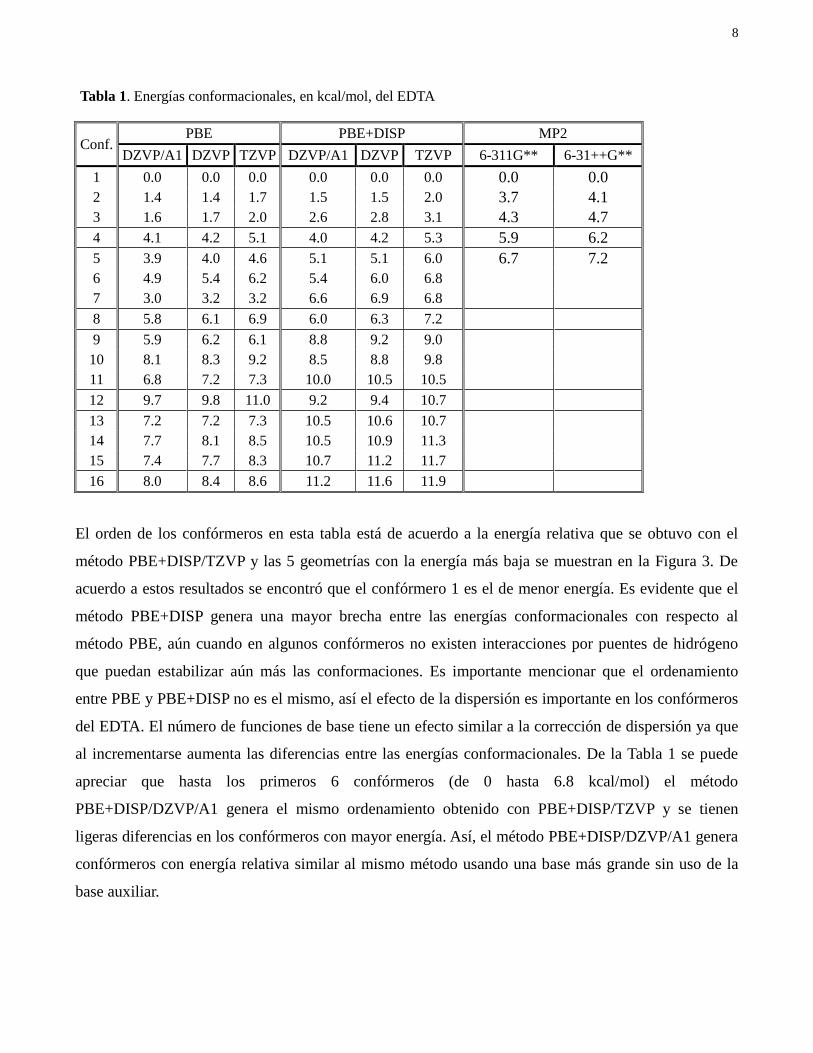
8
Tabla 1. Energías conformacionales, en kcal/mol, del EDTA
Conf. PBE PBE+DISP MP2
DZVP/A1 DZVP TZVP DZVP/A1 DZVP TZVP 6-311G** 6-31++G**
1 0.0 0.0 0.0 0.0 0.0 0.0 0.0 0.0
2 1.4 1.4 1.7 1.5 1.5 2.0 3.7 4.1
3 1.6 1.7 2.0 2.6 2.8 3.1 4.3 4.7
4 4.1 4.2 5.1 4.0 4.2 5.3 5.9 6.2
5 3.9 4.0 4.6 5.1 5.1 6.0 6.7 7.2
6 4.9 5.4 6.2 5.4 6.0 6.8
7 3.0 3.2 3.2 6.6 6.9 6.8
8 5.8 6.1 6.9 6.0 6.3 7.2
9 5.9 6.2 6.1 8.8 9.2 9.0
10 8.1 8.3 9.2 8.5 8.8 9.8
11 6.8 7.2 7.3 10.0 10.5 10.5
12 9.7 9.8 11.0 9.2 9.4 10.7
13 7.2 7.2 7.3 10.5 10.6 10.7
14 7.7 8.1 8.5 10.5 10.9 11.3
15 7.4 7.7 8.3 10.7 11.2 11.7
16 8.0 8.4 8.6 11.2 11.6 11.9
El orden de los confórmeros en esta tabla está de acuerdo a la energía relativa que se obtuvo con el
método PBE+DISP/TZVP y las 5 geometrías con la energía más baja se muestran en la Figura 3. De
acuerdo a estos resultados se encontró que el confórmero 1 es el de menor energía. Es evidente que el
método PBE+DISP genera una mayor brecha entre las energías conformacionales con respecto al
método PBE, aún cuando en algunos confórmeros no existen interacciones por puentes de hidrógeno
que puedan estabilizar aún más las conformaciones. Es importante mencionar que el ordenamiento
entre PBE y PBE+DISP no es el mismo, así el efecto de la dispersión es importante en los confórmeros
del EDTA. El número de funciones de base tiene un efecto similar a la corrección de dispersión ya que
al incrementarse aumenta las diferencias entre las energías conformacionales. De la Tabla 1 se puede
apreciar que hasta los primeros 6 confórmeros (de 0 hasta 6.8 kcal/mol) el método
PBE+DISP/DZVP/A1 genera el mismo ordenamiento obtenido con PBE+DISP/TZVP y se tienen
ligeras diferencias en los confórmeros con mayor energía. Así, el método PBE+DISP/DZVP/A1 genera
confórmeros con energía relativa similar al mismo método usando una base más grande sin uso de la
base auxiliar.

9
Figura 3. Estructuras de los 5 confórmeros más estables del EDTA de acuerdo al método
PBE+DISP/TZVP.
En ocasiones, la corrección a la dispresión propuesta por Grimme ha generado cierta incertidumbre ya
que los efectos de muchos cuerpos no se consideran correctamente. Por esa razón, para los primeros 5
confórmeros, de la Tabla 1, se aplicó el método MP2 con los conjuntos de funciones de base 6-311G**
y 6-31++G**. Los resultados de este método también se reportan en la Tabla 1, donde se puede
observar que el método MP2, y ambos conjuntos de funciones base, predicen el mismo ordenamiento
que el predicho por PBE+DISP/DZVP/A1. Es importante resaltar que este método abre aún más la
brecha en las energías conformacionales. Ya se había mencionado que la corrección de Grimme
también muestra este efecto, sin embargo, no lo hace tanto como MP2.
Conf. 1 Conf. 2 Conf. 3
Conf. 4 Conf. 5
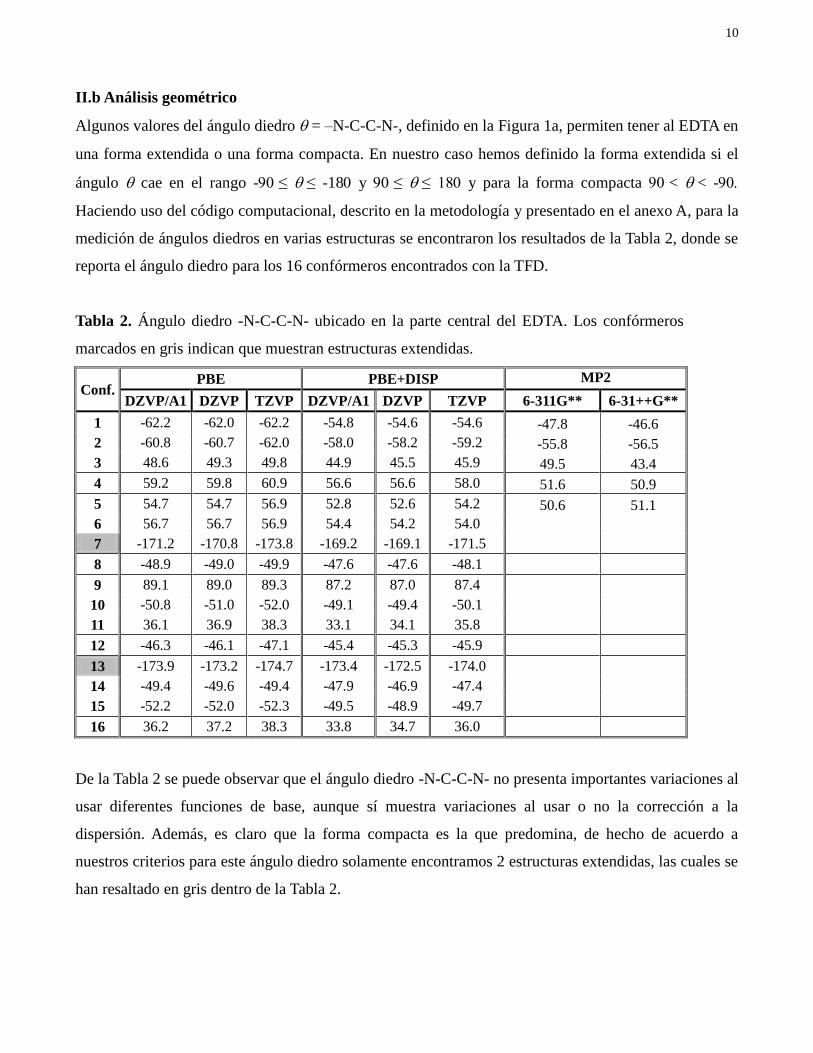
10
II.b Análisis geométrico
Algunos valores del ángulo diedro = –N-C-C-N-, definido en la Figura 1a, permiten tener al EDTA en
una forma extendida o una forma compacta. En nuestro caso hemos definido la forma extendida si el
ángulo cae en el rango -≤ ≤ -18 y ≤ ≤ 18 y para la forma compacta < < -
Haciendo uso del código computacional, descrito en la metodología y presentado en el anexo A, para la
medición de ángulos diedros en varias estructuras se encontraron los resultados de la Tabla 2, donde se
reporta el ángulo diedro para los 16 confórmeros encontrados con la TFD.
Tabla 2. Ángulo diedro -N-C-C-N- ubicado en la parte central del EDTA. Los confórmeros
marcados en gris indican que muestran estructuras extendidas.
Conf. PBE PBE+DISP MP2
DZVP/A1 DZVP TZVP DZVP/A1 DZVP TZVP 6-311G** 6-31++G**
1 -62.2 -62.0 -62.2 -54.8 -54.6 -54.6 -47.8 -46.6
2 -60.8 -60.7 -62.0 -58.0 -58.2 -59.2 -55.8 -56.5
3 48.6 49.3 49.8 44.9 45.5 45.9 49.5 43.4
4 59.2 59.8 60.9 56.6 56.6 58.0 51.6 50.9
5 54.7 54.7 56.9 52.8 52.6 54.2 50.6 51.1
6 56.7 56.7 56.9 54.4 54.2 54.0
7 -171.2 -170.8 -173.8 -169.2 -169.1 -171.5
8 -48.9 -49.0 -49.9 -47.6 -47.6 -48.1
9 89.1 89.0 89.3 87.2 87.0 87.4
10 -50.8 -51.0 -52.0 -49.1 -49.4 -50.1
11 36.1 36.9 38.3 33.1 34.1 35.8
12 -46.3 -46.1 -47.1 -45.4 -45.3 -45.9
13 -173.9 -173.2 -174.7 -173.4 -172.5 -174.0
14 -49.4 -49.6 -49.4 -47.9 -46.9 -47.4
15 -52.2 -52.0 -52.3 -49.5 -48.9 -49.7
16 36.2 37.2 38.3 33.8 34.7 36.0
De la Tabla 2 se puede observar que el ángulo diedro -N-C-C-N- no presenta importantes variaciones al
usar diferentes funciones de base, aunque sí muestra variaciones al usar o no la corrección a la
dispersión. Además, es claro que la forma compacta es la que predomina, de hecho de acuerdo a
nuestros criterios para este ángulo diedro solamente encontramos 2 estructuras extendidas, las cuales se
han resaltado en gris dentro de la Tabla 2.

11
Es importante mencionar que las estructuras que forma el EDTA en complejos con metales presenta
formas compactas ya que a partir de lo reportado por rayos X encontramos 12 estructuras [14]–[25], de
donde se observa que este ángulo presenta dos valores, uno negativo, -59.0±6.3 grados, y otro positivo,
58.1±12.6 grados. A partir de la Tabla 2 es evidente que la TFD predice que el ángulo -N-C-C-N- de los
dos confórmeros de más baja energía caen en el rango del valor negativo que presenta este ángulo en
los complejos de EDTA. Y el ángulo de los confórmeros 3 al 6 caen en el intervalo del valor positivo de
este ángulo en algunos complejos de EDTA. Podemos concluir así que las estructuras del EDTA libre
neutro están en conformaciones óptimas para formar complejos con metales. También se pueden ver
estos resultados de otra manera ya que se puede pensar que las conformaciones del EDTA predisponen
a que sus complejos formen estructuras compactas.
Como ya se mencionó anteriormente, algunas de las 16 estructuras reportadas en las Tablas 1 y 2
presentan puentes de hidrógenos. De acuerdo al código computacional descrito en la metodología se
indican en la Tabla 3 los confórmeros donde se presenta esta interacción.
Tabla 3. Las casillas en gris indican que si se presenta un puente de hidrógeno.
Conf. PBE PBE+DISP MP2
DZVP/A1 DZVP TZVP DZVP/A1 DZVP TZVP 6-311G** 6-31++G**
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16

12
La importancia de la corrección a la dispersión se pone evidente en los confórmero 4 y 10 ya que en
esos confórmeros el método PBE+DISP, con cualquier conjunto de funciones de base, indica que sí
existe al menos un puente de hidrógeno mientras que con el método PBE no presenta tal interacción
con todas las funciones de base. Es importante mencionar que el método PBE+DISP es insensible a las
funciones de base tratadas en este trabajo por que en los confórmeros donde presenta puentes de
hidrógeno no importa el conjunto de funciones de base. Esta conclusión no es aplicable al método MP2
por que en el confórmero 3 es necesario el uso de funciones difusas para obtener esta interacción.
Por supuesto que nuestro código permite caracterizar los puentes de hidrógeno al reportar distancias
A..H-D y A..D, así como el ángulo AHD. En la Tabla 4, reportamos estos parámetros geométricos para
todas las estructuras que presentan puentes de hidrógeno. En esta tabla se está reportando los valores
promedios obtenidos con cada una de las estructuras. En particular, para el método PBE+DISP/TZVP
se están reportando también las respectivas incertidumbres. Si tomamos como referencia el método
PBE+DISP/TZVP encontramos que todas las demás metodologías predicen mayores longitudes en el
puente de hidrógeno, aún el método MP2 genera este resultado aunque los resultados de este método
caen dentro del intervalo generado por nuestro método de referencia. Con respecto al ángulo AHD se
obtiene que los métodos basados en la TFD predicen menores ángulos con respecto a nuestro método
de referencia. Para el método de MP2/6-31++G** se obtienen un valor mayor para este parámetro, con
respecto al método PBE+DISP/TZVP, lo cual significa que aún cuando el método MP2/6-31++G**
predice mayores distancias para el puente de hidrógeno, genera puentes de hidrógeno más lineales. Es
muy probable que el método de Grimme solamente actúe de manera importante sobre la interacción
A..H pero no toma en cuenta de manera correcta el medio ambiente que hay a su derredor.
Tabla 4. Valores promedios de las distancias, A..H, A..D, y el ángulo AHD en los puentes de
hidrógeno reportados en la Tabla 1.
Distancia/Angstroms Ángulo/Grados
A…H A…D A…H…O
PBE
DZVP/A1 1.968 2.898 154.9
DZVP 1.975 2.900 154.2
TZVP 1.896 2.840 158.3
PBE+DISP
DZVP/A1 1.924 2.865 157.2
DZVP 1.920 2.860 156.9
TZVP 1.865 ± 0.082 2.815 ± 0.053 159.9 ± 6.0
MP2 6-311G** 1.909 2.835 160.0
6-31++G** 1.932 2.859 158.5

13
II.c Microsolvatación
Se exploró el efecto del solvente sobre las conformaciones del EDTA a través de una microsolvatación
usando 10 moléculas de agua y de acuerdo a lo expuesto en la metodología. Sólo presentaremos en este
reporte tres estructuras (Figura 5) que consideramos importantes.
a) b)
c)
Figura 5. Estructuras del EDTA microsolvatadas con 10 moléculas de agua, a) y b) Presentan
transferencia de protón intra e inter molecular respectivamente y c) presenta puente de hidrógeno
intermolecular.

14
En las Figuras 5a y 5b se observan transferencias de protón, intra e inter molecular. Cabe aclarar que la
transferencia se presenta de manera natural durante el proceso de optimización. Ahora bien,
observamos que en la figura 5a, se transfiere el protón del grupo carboxilo, el cual está formando dos
puentes de hidrógeno, al nitrógeno dentro del EDTA, dicha transferencia es similar a lo sugerido por
los estudios de ratos X en el EDTA. Por otro lado, encontramos que en la figura 5b, la transferencia de
protón se da del grupo carboxilo hacia una molécula de agua, dicha transferencia es muy importante,
porque está indicando que el EDTA se desprotona fácilmente a través de los grupos carboxilos.
Finalmente, en la Figura 5c se muestra que una molécula de agua está formando tres puentes de
hidrógeno y tiene la posibilidad de transferir un protón al átomo de nitrógeno. Sin embargo, a
diferencia del carboxilo de la Figura 5a, no se presenta tal transferencia. Naturalmente, las moléculas
de agua deben de estar en otras condiciones para transferir un protón ya que es bien sabido que el
EDTA se encuentra protonado en pHs bajos. Cómo resultado final de esta discusión podemos decir que
el medio ambiente induce sobre el EDTA transferencias de protón intra e intermoleculares.

15
III. Conclusiones
En EDTA aislado puede encontrarse en muchas conformaciones, debido a los grados de libertad que exhibe.
Pero ninguna de las conformaciones que encontramos del EDTA aislado corresponde a la estructura de
rayos X. Además, puede tenerse una descripción del EDTA aislado bastante confiable usando el método
PBE+DISP/DZVP/A1. El método es relativamente más bajo en costo computacional que el método
PBE+DISP/(DZVP ó TZVP) y MP2/(6-311G** ó 6-31++G**). Una de las razones que lo hace confiable, es
que el método PBE+DISP y los tres conjuntos de funciones base (DZVP/A1, DZVP y TZVP) generan el
mismo ordenamiento conformacional que el método MP2 y los dos conjuntos de funciones base (6-311G**
y 6-31++G**) en las primeras 5 conformaciones de más baja energía.
Los puentes de hidrógeno inducen a que el EDTA predomine en una forma compacta y no extendida. Se
encontró que en las seis conformaciones de mínima energía del EDTA, asemejan a los complejos reportados
en rayos X en donde el EDTA acompleja un solo átomo metálico. Por lo tanto, la estructura del EDTA se
encuentra en conformaciones óptimas para formar complejos con un átomo metálico. También este
resultado puede pensarse de distinta manera: que las conformaciones del EDTA predisponen a que sus
complejos formen estructuras compactas.
La contabilización de puentes de hidrógeno en cada conformación del EDTA, sirvió para saber en qué
conformaciones, respecto a cada una de las metodologías ocupadas, forman al menos un puente de
hidrógeno. Obteniendo como resultado, que la corrección a la dispersión propuesta por Grimme en el
funcional de intercambio y correlación es muy importante, puesto que no importa el tamaño del conjunto de
funciones base para observar que en las mismas conformaciones se forman al menos un puente de
hidrógeno y que a su vez concuerda con las cinco conformaciones de mínima energía obtenidas con MP2/6-
31++G**.
La caracterización de puentes de hidrógeno en las conformaciones del EDTA, arrojó resultados importantes
para la elección del método con un costo computacional bajo y que además describa de manera adecuada
los puentes de hidrógeno.
En la microsolvatación encontramos que el EDTA presenta transferencia de protón inter e intra molecular.
En la transferencia inter molecular el grupo carboxilo cede el protón a una molécula de agua y en la
transferencia intra molecular el grupo carboxilo cede el protón al grupo amino. En este ultimo caso es
importante, puesto que, encontramos que la estructura del EDTA microlsolvatada transfiere un protón de
manera similar a lo reportado por los estudios de rayos X. El resultado en ambas transferencias de protón
inter e intra molecular nos indica la facilidad que tienen los grupos carboxilos de desprotornarse, por lo
tanto, el medio ambiente provoca dichas transferencias de protón en el EDTA.

16
IV. Referencias
[1] J. Sillanpää, Reijo Aksela and Kari Laasonen, Density functional complexation study of metal ions
with (amino) polycarboxyl acid ligands, Phys. Chem. Chem. Phys., 2003, 5, pp 3382–3393.
[2] Attila Kovács, Denes S. Nemcsok, Tamás Kocsis, Bonding interaction in EDTA complexes, Journal
of Molecular Structure: THEOCHEM, 2010, 950, pp 93-97.
[3] M.A. Marini, W.J. Evans and R.L. Berger, Use of the twin-cell differential titration calorimeter for
binding studies. I. EDTA and its calcium complex, Journal of Biochemical and Biophysical Methods,
1985, 10 pp 273-285.
[4] Ismael Yévenes, Juan Reyes Y. y col. Desmineralización de premolares con soluciones de EDTA a
diferentes pH de Incubación, Revista Dental de Chile, 2002, 93,1, pp 17-20.
[5] Yuri V. Griko, Energetics of Ca+2
- EDTA interactions: calorimetric study, Biophysical Chemistry,
1999, 79, pp 117 – 127.
[6] Steven Han, Young Ba, Determination of the Concentrations of Metal Cations in Aqueous Solutions
Using Pronton NMR Spectral Area Integration of the EDTA Complexes, Journal of Solution Chemistry,
Vol. 33, No.3, 2004
[7] M. F. C. Ladd and D. C. Povey, Crystallographic and spectroscopic studies on
ethylenediaminetetraacetic acid (EDTA). II. Comparison of two structure determinations of [beta]-
EDTA, Acta Cryst., (1973), B29, 2973.
[8] J.P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett. 77, 3865 (1996); 78, 1396 (1997)
[9] N. Godbout, D. R. Salahub, J. Andzelm, and E. Wimmer, Can. J. Chem. 70, 560 (1992).
[10] J.J.P. Stewart, J. Comput. Chem., 10, 221 (1989).
[11]S. Grimme J. Comp. Chem. 25 1463 (2004).
[12] Møller, C.; Plesset, M. S. Phys. ReV. 1934, 46, 618.
[13] R. Krishnan, J.S. Binkley, R. Seeger, J.A. Pople, J. Chem. Phys. 72, 650 (1980)
[14] T.N.Polynova, L.A.Zasurskaya, A.B.Ilyukhin, Potassium ethylenediaminotetra-acetato-aluminium
dihydrate, Kristallografiya(Russ.)(Crystallogr.Rep.) (1997), 42, 168.
[15] Shuang Chen, S.Hoffmann, Y.Prots, Jing-Tai Zhao, R.Kniep, catena-((m9-ethylenediamine-
N,N,N',N'-tetraacetato)-diaqua-di-barium hemihydrate), Z.Anorg.Allg.Chem. (2010), 636, 1710.
[16] X.Solans, S.Gali, M.Font-Altaba, J.Oliva, J.Herrera, Hexa-aqua-magnesium(ii) aqua-
(ethylenediaminetetra-acetato)-cadmium(ii) trihydrate, Acta Crystallogr.,Sect.C:Cryst.Struct.Commun.
(1983), 39, 438.

17
[17] E.F.K.McCandlish, T.K.Michael, J.A.Neal, E.C.Lingafelter, N.J.Rose, catena((m3-
Ethylenediamine-tetra-acetato)-cobalt(ii)-tetra-aqua-cobalt dihydrate), Inorg.Chem. (1978), 17, 1383.
[18] X.Solans, M.Font-Altaba, J.Oliva, J.Herrera, (Ethylenediaminetetra-acetato)-zinc-(tetra-aqua-
magnesium) dihydrate, Acta Crystallogr.,Sect.C:Cryst.Struct.Commun. (1983), 39, 435.
[19] B.L.Barnett, V.A.Uchtman, catena-((m2-Ethylenediaminetetra-acetato)-tri-aqua-di-calcium
tetrahydrate), Inorg.Chem. (1979), 18, 2674.
[20] T.V.Filippova, T.N.Polynova, A.L.Il'inskii, M.A.Porai-Koshits, L.I.Martynenko, Ammonium
diaqua-(ethylenediamine-N,N'-tetra-acetato)-erbium(iii) trihydrate,
Zh.Strukt.Khim.(Russ.)(J.Struct.Chem.) (1977), 18, 1127.
[21] A.I.Pozhidaev, M.A.Porai-Koshits, T.N.Polynova, Diaqua-(ethylenediaminetetra-acetato-
N,N',O,O',O'',O''')-zirconium dihydrate, Zh.Strukt.Khim.(Russ.)(J.Struct.Chem.) (1974), 15, 644.
[22] R.L.Davidovich, A.B.Ilyukhin, Sh.-Zh.Hu, catena-((Hydrogen ethylenediamino-tetra-acetato)-
bismuth(iii) dihydrate), Kristallografiya(Russ.)(Crystallogr.Rep.) (1998), 43, 653.
[23] N.N.Anan'eva, T.N.Polynova, M.A.Porai-Koshits, Dilithium aqua-(1,2-ethylenediaminetetra-
acetato)-manganese(ii) tetrahydrate, Zh.Strukt.Khim.(Russ.)(J.Struct.Chem.) (1974), 15, 261.
[24] M.Shimoi, Y.Orita, T.Uehiro, I.Kita, T.Iwamoto, A.Ouchi, Y.Yoshino, (Hydrogen ethylenediamine-
tetra-acetato)-antimony(iii) dihydrate, Bull.Chem.Soc.Jpn. (1980), 53, 3189.
[25] F.P.van Remoortere, J.J.Flynn, F.P.Boer, Aqua-(ethylenediaminetetra-acetato)-tin(iv), Inorg.Chem.
(1971), 10, 2313.

18
Anexo A
#include <stdio.h>
#include <stdlib.h>
#include <string.h>
#include <math.h>
/* Programa para calcular ángulos diedros
y encontrar posibles puentes de hidrógeno.
Mariano Rodríguez
Mayo de 2012
*/
main()
{
int i, j, k, p, b, total, imprimir;
double pi, tol_distance, tol_ang, dist_rest;
double dist1, dist2, dAtom, angulo, angulo2, angulo3, prodpunt, norma,
normb, angdiedro, pructpunto, angulopH;
int *posicion;
double *x, *y, *z;
char **simbol;
double *newposicionx, *newposiciony, *newposicionz;
double *new2posicionx, *new2posiciony, *new2posicionz;
double *new3posicionx, *new3posiciony, *new3posicionz;
double *new4posicionx, *new4posiciony, *new4posicionz;
double *cordiedx, *cordiedy;
double *dH_atom;
double *vector_1, *vector_2;
char atom[3], nombre[80], respuesta[3];
char basura1[80], basura2[80];
FILE *archivo;
FILE *archivo_salida;
pi = 4.f*atan(1.f);

19
do
{
printf("Deme el nombre del archivo de coordenadas:");
scanf("%s",nombre);
archivo = fopen(nombre,"r");
fscanf(archivo, "%d", &total);
fscanf(archivo, "%5s %s", basura1, basura2);
/*Asignación de memoria dinámica */
posicion = (int *) malloc(5*sizeof(int));
if (posicion == NULL) {
free(posicion);
posicion = 0;
printf("Error en arreglo posicion, insuficiente RAM\n");
exit(1);
}
x = (double *) malloc(total*sizeof(double));
if (x == NULL) {
free(x);
x = 0;
printf("Error en arreglo x, insuficiente RAM\n");
exit(1);
}
y = (double *) malloc(total*sizeof(double));
if (y == NULL) {
free(y);
y = 0;
printf("Error en arreglo y, insuficiente RAM\n");
exit(1);
}
z = (double *) malloc(total*sizeof(double));
if (z == NULL) {
free(z);
z = 0;
printf("Error en arreglo z, insuficiente RAM\n");
exit(1);
}
simbol = (char **) malloc(total*sizeof(char *));
if (simbol == NULL) {
free(simbol);
simbol = 0;
printf("Error en arreglo simbol, insuficiente RAM\n");
exit(1);
}

20
for(i=0 ; i < total ; i++){
simbol[i] = (char *) malloc(2*sizeof(char));
if (simbol[i] == NULL) {
free(simbol[i]);
simbol[i] = 0;
printf("Error en arreglo simbol, insuficiente RAM\n");
exit(1);
}
}
//En nuevas posiciones
newposicionx = (double *) malloc(total*sizeof(double));
if (newposicionx == NULL) {
free(newposicionx);
newposicionx = 0;
printf("Error en arreglo newposicionx, insuficiente RAM\n");
exit(1);
}
newposiciony = (double *) malloc(total*sizeof(double));
if (newposiciony == NULL) {
free(newposiciony);
newposiciony = 0;
printf("Error en arreglo newposiciony, insuficiente RAM\n");
exit(1);
}
newposicionz = (double *) malloc(total*sizeof(double));
if (newposicionz == NULL) {
free(newposicionz);
newposicionz = 0;
printf("Error en arreglo newposicionz, insuficiente RAM\n");
exit(1);
}
new2posicionx = (double *) malloc(total*sizeof(double));
if (new2posicionx == NULL) {
free(new2posicionx);
new2posicionx = 0;
printf("Error en arreglo new2posicionx, insuficiente RAM\n");
exit(1);
}
new2posiciony = (double *) malloc(total*sizeof(double));
if (new2posiciony == NULL) {
free(new2posiciony);
new2posiciony = 0;
printf("Error en arreglo new2posiciony, insuficiente RAM\n");
exit(1);
}

21
new2posicionz = (double *) malloc(total*sizeof(double));
if (new2posicionz == NULL) {
free(new2posicionz);
new2posicionz = 0;
printf("Error en arreglo new2posicionz, insuficiente RAM\n");
exit(1);
}
new3posicionx = (double *) malloc(total*sizeof(double));
if (new3posicionx == NULL) {
free(new3posicionx);
new3posicionx = 0;
printf("Error en arreglo new3posicionx, insuficiente RAM\n");
exit(1);
}
new3posiciony = (double *) malloc(total*sizeof(double));
if (new3posiciony == NULL) {
free(new3posiciony);
new3posiciony = 0;
printf("Error en arreglo new3posiciony, insuficiente RAM\n");
exit(1);
}
new3posicionz = (double *) malloc(total*sizeof(double));
if (new3posicionz == NULL) {
free(new3posicionz);
new3posicionz = 0;
printf("Error en arreglo new3posicionz, insuficiente RAM\n");
exit(1);
}
new4posicionx = (double *) malloc(total*sizeof(double));
if (new4posicionx == NULL) {
free(new4posicionx);
new4posicionx = 0;
printf("Error en arreglo new4posicionx, insuficiente RAM\n");
exit(1);
}
new4posiciony = (double *) malloc(total*sizeof(double));
if (new4posiciony == NULL) {
free(new4posiciony);
new4posiciony = 0;
printf("Error en arreglo new4posiciony, insuficiente RAM\n");
exit(1);
}

22
new4posicionz = (double *) malloc(total*sizeof(double));
if (new4posicionz == NULL) {
free(new4posicionz);
new4posicionz = 0;
printf("Error en arreglo new4posicionz, insuficiente RAM\n");
exit(1);
}
cordiedx = (double *) malloc(2*sizeof(double));
if (cordiedx == NULL) {
free(cordiedx);
cordiedx = 0;
printf("Error en arreglo cordiedx, insuficiente RAM\n");
exit(1);
}
cordiedy = (double *) malloc(2*sizeof(double));
if (cordiedy == NULL) {
free(cordiedy);
cordiedy = 0;
printf("Error en arreglo cordiedy, insuficiente RAM\n");
exit(1);
}
//En Puente de hidrógeno
dH_atom = (double *) malloc(total*sizeof(double));
if (dH_atom == NULL) {
free(dH_atom);
dH_atom = 0;
printf("Error en arreglo dH_atom, insuficiente RAM\n");
exit(1);
}
vector_1 = (double *) malloc(3*sizeof(double));
if (vector_1 == NULL) {
free(vector_1);
vector_1 = 0;
printf("Error en arreglo vector_1, insuficiente RAM\n");
exit(1);
}
vector_2 = (double *) malloc(3*sizeof(double));
if (vector_2 == NULL) {
free(vector_2);
vector_2 = 0;
printf("Error en arreglo vector_2, insuficiente RAM\n");
exit(1);
}

23
/*Termina asignación de memoria dinámica */
//Comienza el programa
for(i = 0; i < total; i++){
fscanf(archivo, "%s %lf %lf %lf", atom, &x[i], &y[i], &z[i]);
strcpy(simbol[i], atom);
}
close(archivo);
printf("Deme las posiciones en orden del diedro que quiere medir:\n");
for(p = 0; p < 4; p++){
scanf("%d", &posicion[p]);
}
/* Resta de vectores para hacer una traslacián del átomo 2 al origen */
for(i = 0; i < total; i++){
newposicionx[i]=x[i]-x[posicion[1]-1];
newposiciony[i]=y[i]-y[posicion[1]-1];
newposicionz[i]=z[i]-z[posicion[1]-1];
}
/* Rotación de la molécula hacia el eje y */
angulo=atan(newposicionx[posicion[2]-1]/newposiciony[posicion[2]-1]);
if(newposicionx[posicion[2]-1]>0 && newposiciony[posicion[2]-1]>0)
angulo=angulo;
else
if(newposicionx[posicion[2]-1]<0 && newposiciony[posicion[2]-
1]>0)
angulo=angulo;
else
if(newposicionx[posicion[2]-1]<0 &&
newposiciony[posicion[2]-1]<0)
angulo=angulo+pi;
else
if(newposicionx[posicion[2]-1]>0 &&
newposiciony[posicion[2]-1]<0)
angulo=angulo-pi;

24
for(i = 0; i < total; i++){
new2posicionx[i]=-
newposiciony[i]*sin(angulo)+newposicionx[i]*cos(angulo);
new2posiciony[i]=newposiciony[i]*cos(angulo)+newposicionx[i]*sin(angulo);
new2posicionz[i]=newposicionz[i];
}
angulo2=atan(new2posicionz[posicion[2]-1]/new2posiciony[posicion[2]-
1]);
if(new2posicionz[posicion[2]-1]>0 && new2posiciony[posicion[2]-1]>0)
angulo2=angulo2;
else
if(new2posicionz[posicion[2]-1]<0 && new2posiciony[posicion[2]-
1]>0)
angulo2=angulo2;
else
if(new2posicionz[posicion[2]-1]<0 &&
new2posiciony[posicion[2]-1]<0)
angulo2=angulo2+pi;
else
if(new2posicionz[posicion[2]-1]>0 &&
new2posiciony[posicion[2]-1]<0)
angulo2=angulo2-pi;
for(i = 0; i < total; i++){
new3posicionz[i]=-
new2posiciony[i]*sin(angulo2)+new2posicionz[i]*cos(angulo2);
new3posiciony[i]=new2posiciony[i]*cos(angulo2)+new2posicionz[i]*sin(angulo2
);
new3posicionx[i]=new2posicionx[i];
}
angulo3=atan(new3posicionx[posicion[0]-1]/new3posicionz[posicion[0]-
1]);
if(new3posicionx[posicion[0]-1]>0 && new3posicionz[posicion[0]-1]>0)
angulo3=angulo3;
else
if(new3posicionx[posicion[0]-1]<0 && new3posicionz[posicion[0]-
1]>0)
angulo3=angulo3;
else
if(new3posicionx[posicion[0]-1]<0 &&
new3posicionz[posicion[0]-1]<0)
angulo3=angulo3+pi;
else
if(new3posicionx[posicion[0]-1]>0 &&
new3posicionz[posicion[0]-1]<0)
angulo3=angulo3-pi;

25
for(i = 0; i < total; i++){
new4posicionx[i]=-
new3posicionz[i]*sin(angulo3)+new3posicionx[i]*cos(angulo3);
new4posicionz[i]=new3posicionz[i]*cos(angulo3)+new3posicionx[i]*sin(angulo3
);
new4posiciony[i]=new3posiciony[i];
}
cordiedx[0]=new4posicionx[posicion[0]-1];
cordiedy[0]=new4posicionz[posicion[0]-1];
cordiedx[1]=new4posicionx[posicion[3]-1];
cordiedy[1]=new4posicionz[posicion[3]-1];
/* Producto punto */
prodpunt=cordiedx[0]*cordiedx[1]+cordiedy[0]*cordiedy[1];
/* Norma de cada vector */
norma=sqrt(pow(cordiedx[0],2.f)+pow(cordiedy[0],2.f));
normb=sqrt(pow(cordiedx[1],2.f)+pow(cordiedy[1],2.f));
/* Calculo del ángulo diedro */
angdiedro=acos(prodpunt/(norma*normb))*(180.f/pi);
if (new4posicionx[posicion[3]-1] < 0.f) angdiedro = -1.f*angdiedro;
strcat(nombre,"_salida");
archivo_salida=fopen(nombre,"w");
fprintf(archivo_salida,"%s\n",nombre);
fprintf(archivo_salida,"Õngulo diedro ");
for(p = 0; p < 4; p++){
fprintf(archivo_salida,"-(%d)%s-",posicion[p],simbol[posicion[p]-1]);
};
fprintf(archivo_salida," = %6.2f\n", angdiedro);
//Criterios para puente de hidrógeno
tol_distance = 2.5f;
dist_rest = 3.f;
tol_ang = 135.0f;
fprintf(archivo_salida,"-----------------------------------Puente de
hidrógeno-------------------------------------\n");

26
fprintf(archivo_salida,"Criterios de búsqueda: A..H <= %3.1f Angstroms
A..D <= %3.1f Angstroms y A..H-D => %4.2f Grados\n", tol_distance,
dist_rest ,tol_ang);
fprintf(archivo_salida,"----------------------------------------------
---------------------------------------------\n");
// Primero buscamos cada hidrógeno y comparamos la distancia con los demás
átomos
// del sistema.
for(i=0; i < total; i++){
if(strcmp(simbol[i],"H") == 0) {
for (j = 0; j < total; j++) dH_atom[j] = 0.f;
for(j=0; j < total; j++){
if (j != i) {
dist1=pow((x[j]-x[i]),2.f)+pow((y[j]-y[i]),2.f)+pow((z[j]-
z[i]),2.f);
dist1 = sqrt(dist1);
if (dist1 <= tol_distance) dH_atom[j] = dist1;
}
}
for (j = 0; j < total; j++) {
if (dH_atom[j] != 0.f) {
for (k = j + 1; k < total; k++) {
if (dH_atom[k] != 0.f) {
// Voy a ver si el ángulo es mayor a tol_ang
vector_1[0]=x[i]-x[j];
vector_1[1]=y[i]-y[j];
vector_1[2]=z[i]-z[j];
vector_2[0]=x[i]-x[k];
vector_2[1]=y[i]-y[k];
vector_2[2]=z[i]-z[k];
pructpunto=vector_1[0]*vector_2[0]+vector_1[1]*vector_2[1]+vector_1[2]*vect
or_2[2];
angulopH=acos(pructpunto/(dH_atom[j]*dH_atom[k]))*(180.f/pi);
if (angulopH >= tol_ang) {
imprimir = 0;
if (strcmp("H",simbol[j]) != 0 && strcmp("H",simbol[k]))
{
if(dH_atom[k] < 2.f || dH_atom[j] < 2.f){
dist2 = pow((x[j]-x[k]),2.f)+pow((y[j]-
y[k]),2.f)+pow((z[j]-z[k]),2.f);
dAtom=sqrt(dist2);
if(dAtom <= dist_rest){
fprintf(archivo_salida,"d(H%d-%s%d)= %5.3f, d(H%d-
%s%d)= %5.3f, d(%s%d-%s%d)= %5.3f, ángulo(%s%d-H%d-%s%d)= %6.2f\n",i+1,
simbol[j], j+1, dH_atom[j], i+1, simbol[k], k+1, dH_atom[k],simbol[j], j+1,
simbol[k], k+1, dAtom, simbol[j], j+1, i+1, simbol[k], k+1, angulopH);

27
}//Termina comp dAtom
}//Termina if dH_atom
}//Termina if printf
} // Termina if angulopH
} // Termina if dH_atom[k]
} // Termina for sobre k
} // Termina if dH_atom[j]
} // Termina for sobre j
}
}
close(archivo_salida);
printf("\nSe creo un archivo de resultados %s\n",nombre);
printf("\n¿Quiere volver a ejecutar el programa? (si/no):");
scanf("%s", respuesta);
printf("\n");
free(vector_2);
free(vector_1);
free(dH_atom);
free(cordiedy);
free(cordiedx);
free(new4posicionz);
free(new4posiciony);
free(new4posicionx);
free(new3posicionz);
free(new3posiciony);
free(new3posicionx);
free(new2posicionz);
free(new2posiciony);
free(new2posicionx);
free(newposicionz);
free(newposiciony);
free(newposicionx);
for (i = total-1; i >= 0; i--) free(simbol[i]);
free(simbol);
free(z);
free(y);
free(x);
free(posicion);
} while(strcmp(respuesta,"si") == 0);
}

28
Ejemplo
El programa pide un archivo de entrada* y la posición de los átomos del diedro que quiera conocer y al
finalizar la ejecución genera un archivo de salida. El contenido que podrá encontrar dentro del archivo
de salida es el siguiente:
conformer_1_sem_mp2_6-311+G.xyz_salida
Ángulo diedro -(7)N--(6)C--(1)C--(2)N- = -46.63
-----------------------------------Puente de hidrógeno--------------------------------------------------------------
Criterios de búsqueda: A..H <= 2.5 Angstroms A..D <= 3.0 Angstroms y A..H-D => 135.00 Grados
--------------------------------------------------------------------------------------------------------------------------
d(H26-O13)= 1.834, d(H26-O20)= 0.984, d(O13-O20)= 2.799, ángulo(O13-H26-O20)= 166.09
d(H30-O17)= 1.834, d(H30-O19)= 0.984, d(O17-O19)= 2.800, ángulo(O17-H30-O19)= 166.08
*El archivo de entrada debe cumplir con las siguientes características:
36 Número de átomos
TE= -1099.09682048 Energía electrónica
C -0.41835199 1.94505979 1.13374489 Geometría
N -1.22628074 0.76627416 0.83164262
C -1.97099957 0.91180855 -0.42147285
C -1.21278637 0.27454481 -1.58695807
O -0.86683602 0.89994513 -2.58216410
: : : :
: : : :
: : : :
: : : : H -1.42545245 0.44821936 2.87276346
H 1.52513329 1.75749424 -2.25180399


















