
Cromatografía en Capa Fina
65
Cromatografía en capa fina Este artículo o sección necesita una revisión de ortografía y gramática . Puedes colaborar editándolo (lee aquí sugerencias para mejorar tu ortografía ). Cuando esté corregido, borra este aviso, por favor. Puedes ayudarte del corrector ortográfico , activándolo en: Mis preferencias → Accesorios → Navegación → El corrector ortográfico resalta errores ortográficos con un fondo rojo. Tinta negra siendo separada en una placa de CCF La cromatografia en capa fina (CCF), TLC (Thin layer chromatography) es una técnica cromatográfica . La fase estacionaria es una capa uniforme de un adsorbente mantenido sobre una placa, la cual puede ser de vidrio, aluminio u otro soporte. En cromatografía en capa fina se utiliza una placa cromatográfica inmersa verticalmente en un eluyente apolar; La placa cromatográfica consiste en una fase estacionaria polar (comúnmente se utiliza sílica gel) adherida a una superficie sólida con algún agente cementante. El eluyente debe ser un compuesto líquido apolar, generalmente orgánico. Para realizar la CCF, se debe apoyar la placa cromatografica sobre algún recipiente o camara que contenga la fase líquida a aproximadamente 1 cm (la distancia entre el principio de la placa y la muestra que se desea analizar). Índice
description
instrumental
Transcript of Cromatografía en Capa Fina

Cromatografía en capa finaEste artículo o sección necesita una revisión de ortografía y gramática.Puedes colaborar editándolo (lee aquí sugerencias para mejorar tu ortografía). Cuando esté corregido, borra este aviso, por favor.Puedes ayudarte del corrector ortográfico, activándolo en: Mis preferencias → Accesorios → Navegación → El corrector ortográfico resalta errores ortográficos con un fondo rojo.
Tinta negra siendo separada en una placa de CCF
La cromatografia en capa fina (CCF), TLC (Thin layer chromatography) es una técnica cromatográfica. La fase estacionaria es una capa uniforme de un adsorbente mantenido sobre una placa, la cual puede ser de vidrio, aluminio u otro soporte.
En cromatografía en capa fina se utiliza una placa cromatográfica inmersa verticalmente en un eluyente apolar; La placa cromatográfica consiste en una fase estacionaria polar (comúnmente se utiliza sílica gel) adherida a una superficie sólida con algún agente cementante. El eluyente debe ser un compuesto líquido apolar, generalmente orgánico. Para realizar la CCF, se debe apoyar la placa cromatografica sobre algún recipiente o camara que contenga la fase líquida a aproximadamente 1 cm (la distancia entre el principio de la placa y la muestra que se desea analizar).
Índice 1 Aplicación de las muestras 2 Elección del eluyente 3 Desarrollo de la cromatografía 4 Localización de sustancias 5 Constantes Rf Y Rx
Aplicación de las muestrasLos productos a examinar se disolverán, cuando sea posible, en un disolvente orgánico que tenga un punto de ebullición lo suficientemente bajo para que se evapore después de la aplicación, lo más común es usar acetona. Frecuentemente se emplean disoluciones al 1%, de manera que al aplicar 2 µl resulta en la carga 20 µg de producto sólido. Muchos

reactivos de revelado llegan a detectar 0.1 µg de material; por esto con esta carga puede llegarse a observar un 5% de impurezas.
Existen una gran variedad de micropipetas y microjeringuillas para realizar el proceso de siembra de la muestra a analizar. También pueden usarse tubos capilares. El proceso de siembra se realiza tocando con la punta del capilar (micropipeta, etc) sobre la placa preparada. Dejando una distancia al borde inferior de un centímetro aproximadamente. El punto de aplicación de la muestra se denomina toque.
Una vez colocado el toque se deja secar para evaporar el disolvente, de forma que en la placa solo quedará la muestra a analizar. se anota en el cuaderno, no solamente en adsorbente empleado, sino la identidad del eluyente y la relación de los dos, o más que se hayan utilizado.
Elección del eluyenteLa elección del eluyente depende del componente que se va a separar y del material en que la separación se lleva a cabo. Un método que se emplea para la selección del eluyente es una cromatografía en capa fina con distintos disolventes y unas muestras patrón.
Principales eluyentes en orden creciente de polaridad:
Éter de petróleo Éter dietílico Ciclohexano Acetato de etilo Tetracloruro de carbono† Piridina Benceno† Etanol Cloroformo† Metanol Diclorometano Agua Ácido acético
† compuestos cancerígenos.
En la elección del eluyente influyen varios factores:
Precio. Pureza. No utilizar mezclas de eluyentes a menos que se conozcan las proporciones de
los disolventes que conformen la mezcla (reproducibilidad). No utilizar compuestos muy volátiles. Evitar que contengan trazas de metales (catalizadores). La elección del eluyente se realiza de forma empírica. Hay que estudiar la
polaridad del componente y probar con eluyentes cada vez menos polares.

1. Toque de la muestra sin aplicar ningún eluyente.2. Aplicando un eluyente poco polar.3. Aplicando un eluyente más polar.
Al aplicar en primer lugar eluyentes poco polares, podemos seguir utilizando la misma placa para aplicar otros eluyentes más polares, hasta dar con el más apropiado.
Otra técnica para realizar la elección del eluyente consiste en sembrar varias muestras distanciadas suficientemente, y aplicar con un tubo capilar distintos eluyentes sobre el centro de cada muestra. Esto permite desarrollar cada eluyente radialmente por capilaridad, de forma que se aprecie el eluyente con el cual la separación se realiza de una manera más eficaz.
Desarrollo de la cromatografíaEl desarrollo de los cromatogramas en capa fina se realiza normalmente por el método ascendente, esto es, al permitir que un eluyente ascienda por una placa casi en vertical, por la acción de la capilaridad. La cromatografía se realiza en una cubeta. Para conseguir la máxima saturación posible de la atmósfera de la cámara, las paredes se impregnan del eluyente.
Generalmente el eluyente se introduce en la cámara una hora antes del desarrollo, para permitir la saturación de la atmósfera. El tiempo de desarrollo, por lo general, no llega a los 30 minutos. El tiempo de una cromatografía cualitativa suele ser de un par de minutos, mientras que el tiempo de una cromatografía preparativa puede llegar a un par de horas.
Las placas pueden desarrollarse durante un tiempo prefijado, o hasta que se alcance una línea dibujada a una distancia fija desde el origen. Esto se hace para estandarizar los valores de RF. Frecuentemente esta distancia es de 10 cm, ya que parece ser la más conveniente para medir valores de RF. Después del desarrollo, las placas pueden secarse rápidamente con una corriente de aire caliente.
La mejor posición de desarrollo para un componente es el punto medio entre el origen y el frente del eluyente, ya que permite separar las impurezas que se desplazan con mayor y menor velocidad. El frente del eluyente nunca debe llegar a tocar el borde de la placa.
Localización de sustanciasSi los compuestos separados no son coloreados es necesario revelar la posición de dichos compuestos, para ello existen dos tipos de métodos:
Métodos químicos. Métodos físicos.
Métodos químicos Consisten en realizar una reacción química entre un reactivo revelador y los componentes separados, para ello se pulveriza la placa con los reactivos reveladores.

Generalmente se utiliza como reactivo revelador yodo, el cual forma complejos coloreados con los componentes orgánicos (con tonos amarillo-marrón), pero las manchas desaparecen con el tiempo con lo que es conveniente señalar las manchas aparecidas.
Otro reactivo revelador bastante utilizado es el ácido sulfúrico, que reacciona con los componentes orgánicos produciendo manchas negras.
También es utilizado el permanganato potásico, que deja unas manchas de color amarillo. El tamaño de las manchas no está relacionado con la cantidad de componente separado.
Además de estos reveladores generales, existen otros específicos:
2,4 - dinitrofenilhidracina (para aldehidos y cetonas). Verde de bromocresol (para ácidos carboxílicos). Paradimetil aminobenzaldehido (para aminas). Ninhidrina (para aminoácidos).
Métodos físicos. El más común consiste en añadir al adsorbente un indicador fluorescente. De tal forma que al colocar la placa bajo una lámpara ultravioleta, y dependiendo del indicador y de la longitud de onda, aparecen manchas fluorescentes en las zonas en las que hay componentes, o en otros casos aparece toda la placa fluorescente excepto donde hay componentes.
Algunos compuestos poseen cierta fluorescencia (aunque no es normal) con lo que pueden ser detectados directamente en una lámpara de ultravioleta.
Constantes Rf Y RxLa constante RF (Ratio of Front) es simplemente una manera de expresar la posición de un compuesto sobre una placa como una fracción decimal, mide la retención de un componente. Se define como: RF= Distancia de la muestra desde el origen/Distancia del eluyente desde el origen
La distancia recorrida por el compuesto se mide generalmente desde el centro de la mancha, los cálculos se simplifican si el denominador es 10. Para que los RF sean reproducibles deben ser fijadas una serie de condiciones (Espesor de la placa, fase móvil, fase estacionaria, cantidad de muestra). El máximo valor de RF que se puede alcanzar es de 1, lo ideal es un RF entre 0.55 y 0.7.
Para averiguar si dos compuestos son iguales, se colocan ambos sobre la misma placa y se desarrolla con varios eluyentes. Una vez desarrollados se calculan los RF y si son distintos, puede deducirse con toda seguridad que no se trataba del mismo compuesto. Por el contrario si los RF son iguales los compuestos pueden ser iguales o no serlo.
Si se sospecha que dos compuestos son muy parecidos se eluyen sobre la misma placa con el mismo eluyente u otros de menor polaridad, hasta apreciar una separación mínima. En este caso no se pueden usar reveladores químicos, ya que alterarían los compuestos, sino indicador ultravioleta.

También se puede operar de la manera siguiente: Se selecciona un compuesto (X), que tenga una posición de desarrollo conveniente; todos los demás compuestos sobre la placa se relacionan con éste.
De esta manera se tiene el RX:
Rx= Distancia recorrida por el compuesto de referencia(X)/Distancia recorrida por el eluyente
Categoría:
Cromatografía
12. cromatografÍa > 12.2 cromatografÍa EN capa FiNa Y eN columna
Índice
12.2.1 Cromatografía en capa fina (CCF)
12.2.2 Cromatografía en columna (CC)
12.2.1 Cromatografía en capa fina (CCF)
En la cromatografía en capa fina (CCF) la fase estacionaria consiste en una capa delgada de un adsorbente (como por ejemplo gel de sílice, alúmina o celulosa) depositada sobre un soporte plano como una placa de vidrio, o una lámina de aluminio o de plástico.
La CCF es una técnica analítica y tiene como objetivo el análisis de una mezcla de componentes. +
El proceso es similar a la cromatografía de papel con la ventaja de que se desarrolla más rápidamente, proporciona mejores separaciones y se puede elegir entre diferentes adsorbentes. La CCF es una técnica estándar en el laboratorio de química orgánica. Debido a su simplicidad y velocidad, la CCF se utiliza a menudo para monitorizar las reacciones químicas y también para el análisis cualitativo de los productos de una reacción, puesto que permite conocer de manera rápida y sencilla cuántos componentes hay en una mezcla.

Procedimiento
Una placa de CCF es una lámina de vidrio, metal o plástico recubierta con una capa delgada de un sólido adsorbente (gel de sílice o alúmina). Se deposita una pequeña cantidad de la muestra problema en disolución en un punto en la parte inferior de la placa. Entonces la placa se introduce en una cubeta cromatográfica, de forma que sólo la parte inferior de la placa queda sumergida en el líquido. Este líquido o eluyente es la fase móvil y asciende por la placa de CCF por capilaridad.
A medida que el eluyente pasa por el lugar donde está la mancha de la mezcla problema se establece un equilibrio entre las moléculas de cada uno de los componentes en la mezcla que son adsorbidas y las que se encuentran en disolución.
En principio, los componentes se diferenciarán en solubilidad y en la fuerza de su adsorción, de forma que unos componentes se desplazarán más que otros. Cuando el eluyente llega a la parte superior de la placa, esta se saca de la cubeta, se seca, y los componentes separados de la mezcla se visualizan.
Visualización de las manchas
Si los compuestos son coloreados se pueden observar las manchas a simple vista. Si no es así, hay varios métodos para visualizar las manchas correspondientes a cada componente de la mezcla.
Utilizar luz ultravioleta (UV254) para observar la placa. Normalmente se adiciona un colorante fluorescente al adsorbente, de forma que la placa sea fluorescente en todas partes excepto donde haya una mancha correspondiente a un compuesto orgánico.
Utilizar reveladores, por ejemplo, vapores de yodo que es un reactivo inespecífico.

Emplear reactivos específicos para desarrollar coloración en las manchas. Esto se puede hacer sumergiendo la placa de CCF en una disolución que los contenga o en forma de spray.
Cálculo del factor de retención Rf
Cuando son visibles, se puede determinar para cada una de las manchas el valor de Rf (factor de retención), o la distancia que cada compuesto se desplaza en la placa. Cada compuesto tiene un Rf característico que depende del disolvente empleado y del tipo de placa de CCF utilizada, pero es independiente del recorrido del disolvente. De esta manera se puede ayudar a identificar un compuesto en una mezcla al comparar su Rf con el de un compuesto conocido (preferiblemente cuando se hacen eluir en la misma placa de CCF).
12.2.2 Cromatografia en columna
Es una técnica de purificación, puesto que permite aislar los compuestos deseados de una mezcla.
Procedimiento
La cromatografía en columna utiliza una columna de vidrio vertical que se llena con un soporte sólido adsorbente (fase estacionaria: los más utilizados son gel de sílice (SiO2) y alúmina (Al2O3))+. La muestra que se quiere separar se deposita en la parte superior de este soporte. El resto de la columna se llena con el eluyente (disolvente que constituye la fase móvil) que, por efecto de la gravedad, hace mover la muestra a través de la columna. Se establece un equilibrio entre el soluto adsorbido en la fase estacionaria y el disolvente eluyente que fluye por la columna. Debido a que cada uno de los componentes de una mezcla establecerá interacciones diferentes con la fase estacionaria y la móvil, serán transportados a diferentes velocidades y se conseguirá su separación. Así, de manera similar a otros tipos de cromatografía, las diferencias en las velocidades de desplazamiento a través del medio sólido se corresponden con diferencias en los tiempos de elución por la parte inferior de la columna para cada uno de los componentes de la muestra original, que se recogerán en fracciones diferentes.

La polaridad del eluyente afecta las velocidades relativas con las que los diferentes componentes de la mezcla se mueven en la columna. Los disolventes polares compiten más eficientemente con las moléculas polares de una mezcla por los lugares polares del adsorbente. Por lo tanto, un disolvente polar desplazará las moléculas, incluyendo las más polares, rápidamente a través de la columna. Si el disolvente es muy polar la elución será muy rápida y generalmente habrá poca separación de los componentes de la mezcla. Si por el contrario el disolvente es muy apolar, no eluirán los compuestos de la columna. Por lo tanto, la elección del eluyente es crucial para el éxito de la cromatografía en columna. A menudo se utiliza un gradiente creciente de polaridad para la elución. La CCF se utiliza para determinar y elegir el sistema solvente adecuado para cada separación.
En 1978 se introdujo una versión modificada denominada cromatografía en columna rápida. La diferencia con la cromatografía en columna tradicional es que en la técnica rápida el disolvente se hace atravesar la fase estacionaria aplicando una presión positiva. Esto hace que las separaciones mejoren en resolución y se pueda disminuir el tiempo de elución, por lo cual constituye un método de elección.
Análisis de los eluatos de la columna
Si los compuestos separados en una cromatografía en columna son coloreados, el progreso de la separación se puede monitorizar visualmente. No obstante, a menudo los compuestos que deben ser aislados suelen ser incoloros. En este caso, se recogen secuencialmente pequeñas fracciones de eluatos en tubos rotulados y la composición de cada fracción se analiza por cromatografía en capa fina. Una vez identificadas las diferentes fracciones que contienen el mismo producto, se reúnen, se elimina el disolvente y se analiza la identidad de los componentes por métodos espectroscópicos.
Información previa.
Departamento al que corresponde la práctica: Química Orgánica.
Titulo de la práctica: Cromatografía en capa fina
Numero de la práctica: 8

Fecha de realización: 22 / 04 / 03
Objetivos de la práctica.
Aprendizaje de la técnica de cromatografía en capa fina
Conocimiento de conceptos cromatográficos: polaridad, fase móvil, fase estacionaria, factor de retención, etc.
Determinación de los componentes orgánicos presentes en una muestra usando sustancias patrón.
Antecedentes.
La cromatografia es una técnica de análisis químico utilizada para separar sustancias puras de mezclas complejas. Esta técnica depende del principio de adsorción selectiva (no confundir con absorción). La cromatografía fue descubierta por el botánico ruso, de origen italiano, Mikhail Tswett en 1906, pero su uso no se generalizó hasta la década de 1930. Tswett separó los pigmentos de las plantas (clorofila) vertiendo extracto de hojas verdes en éter de petróleo sobre una columna de carbonato de calcio en polvo en el interior de una probeta. A medida que la solución va filtrándose por la columna, cada componente de la mezcla precipita a diferente velocidad, quedando la columna marcada por bandas horizontales de colores, denominadas cromatogramas. Cada banda corresponde a un pigmento diferente.
La cromatografía es probablemente la más versátil de las técnicas de separación: es aplicable a cualquier mezcla soluble o volátil. De hecho las técnicas de separación suelen dividirse en dos grandes grupos: cromatográficas y no cromatográficas. La elección de una técnica cromatográfica concreta dependerá de la naturaleza y cantidad de la muestra, del objetivo de la separación y de las limitaciones del tiempo y equipo asequible.
Podemos distinguir 3 tipos de cromatografía dependiendo si la fase estacionaria/fase móvil es sólido/líquido, líquido/líquido o líquido/gas:
Si es sólido/líquido, cabe distinguir entre:
Cromatografía de adsorción: El sólido adsorbe al componente que inicialmente estaba en fase móvil (liquida o gaseosa) (Fuerzas de Van der Waals).
Cromatografia de cambio iónico: el sólido es un cambiador de iones (fuerzas electrostáticas).
Cromatografia de exclusión (o de geles): el sólido es un gel formado por polímeros no iónicos porosos que retienen a las moléculas de soluto según su tamaño.
Cromatografia de afinidad: es un tipo especial de cromatografia de adsorción, utilizada especialmente en bioquímica, en la que un sólido tiene enlazado un llamado ligando de afinidad que puede ser por ejemplo, un indicador enzimático o un anticuerpo.

Si es líquido/líquido, cabe distinguir:
Cromatografía de partición: el soluto se reparte entra la fase móvil (liquido o gas) y la fase estacionaria.
Si es líquido/gas, cabe distinguir entre:
Cromatografía gas-liquido (CGL), que es una cromatografía de partición.
Cromatografía gas-sólido (CGS), que es una cromatografía de adsorción.
Si es un fluido supercrítico, (fluido calentado a una temperatura superior a la temperatura critica, pero simultáneamente comprimido a una presión mayor que su presión critica), se trata de la cromatografía de fluidos supercríticos (CFS), que puede ser de partición o de adsorción.
Las técnicas cromatográficas, según el dispositivo utilizado para conseguir la separación entre la fase móvil y la estacionaria, pueden ser: en columna y plana.
Cromatografía en columna: Se utiliza un tubo cilíndrico, en cuyo interior se coloca la fase estacionaria y a su través se hace pasar la fase móvil. El flujo de la fase móvil (liquido o gas) a través de la estacionaria se consigue:
1)por presión,
2)por capilaridad,
3) por gravedad.
Cromatografia plana: La fase estacionaria esta colocada en una superficie plana que en realidad es tridimensional, aunque una de sus dimensiones es muy reducida, por lo que puede considerarse bidimensional. Se divide en dos tipos generales:
1. -Cromatografía en papel: en la que el papel absorbente actúa como soporte de la fase estacionaria (cromatografia de partición). Una muestra líquida fluye por una tira vertical de papel absorbente, sobre la cual se van depositando los componentes en lugares específicos.
2. -Cromatografía en capa fina: en la que un sólido actúa como fase estacionaria (cromatografia de partición), se extiende en una capa delgada sobre una placa, generalmente de vidrio.
El uso de la cromatografía está ampliamente extendido en el análisis de alimentos, medicinas, sangre, productos petrolíferos y de fisión radiactiva.
Procedimiento experimental.
Materiales:
4 placas de cromatografía

2 vasos de precipitado de 100ml
2 Placa Petri
1 probeta de 10 ml
Varillas de vidrio
Reactivos:
Acetona de lavar
Acetato de etilo
Hexano
Patrón 1: Mentol
Patrón 2: Acetoacetato de etilo
Patrón 3: Pirogalol
Patrón 4: Floroglucina
Patrón 5: Benzofenona
Muestra problema P1
Muestra problema P2
Revelador anisaldehído (225 ml etanol + 12 ml anisaldehído + 13 ml H2SO4)
- Obtención de capilares:
Para la realización de la práctica se deben de obtener 7 capilares de 10 cm aproximadamente, uno para cada muestra ya sea patrón o problema. Se podría hacer solo un capilar para las 7 muestras, pero ello conllevaría que fuese limpiado el capilar cada vez que lo usemos con una muestra distinta a la anterior, con lo que se irían arrastrando errores.
Para la realización de los capilares, se tomará una varilla de vidrio de unos 15 cm aproximadamente, esta será calentada en un mechero Bunsen hasta q se reblandezca la sección que estamos calentando. Llegado a este punto, apartamos la varilla de la llama y rápidamente estiramos axialmente.
- Realización de la cromatografía en capa fina:
En este apartado se realizaran 4 placas de cromatografía 4'5 x 5 cm, las cuales solo se podrán coger por los bordes y nunca tocar la parte de celulosa (la blanca), porque ello acarrearía errores en la medición.

Una vez obtenidas las placas, marcamos a lápiz una línea a 1 cm de la base. Sobre esa línea marcamos 7 puntos, donde se depositaran con los capilares pequeñas cantidades de las muestras.
En los vasos de precipitado, que actúan como tanque de elución, se pondrán las disoluciones. Utilizaremos 4 disoluciones de acetato de etilo (Ae) y hexano (Hex), cuyas composiciones serán del 20, 40, 60 y 80 % de Ae/Hex. Solamente utilizaremos 5 ml de cada disolución, la cual no debe sobrepasar la línea dibujada a lápiz.
Finalmente se revelarán las placas usando dos agentes reveladores:
Revelador físico, usaremos una lámpara de rayos UVA, donde pondremos con unas pinzas las placas cromatográficas. Las marcas que aparezcan se señalarán con lápiz.
Revelador químico, una vez pasada la placa por los rayos UVA, se introducirá con una pinza en un revelador anisaldehído y posteriormente será colocada en una placa calefactora a 100º C. Finalmente anotamos los colores obtenidos.
Resultados.
Para el calculo del factor de retención, Rf, se usará la siguiente expresión:
Rf = distancia recorrida por el compuesto
distancia recorrida por el disolvente
Se tomará como distancia del compuesto la distancia comprendida entre la línea trazada donde se ha depositado el compuesto y el centro de la mancha.
Placa 1
Sistema eluyente: 20% Ae/Hex
Distancia recorrida por el disolvente: 35 mm
Patrón Recorrido (mm) Rf UV Color Nombre1 24 0.685 No Azul Mentol
2 17 0.485 Si Rojo Acetoacetato de etilo
3 2 0.057 Si Violeta Pirogalol4 0 0 Si Amarillo Floroglucina5 27 0.771 Si Incoloro benzofenona
Placa 2
Sistema eluyente: 40% Ae/Hex
Distancia recorrida por el disolvente: 36 mm

Patrón Recorrido (mm) Rf UV Color Nombre1 30 0.833 No Azul Mentol
2 26 0.722 Si Rojo Acetoacetato de etilo
3 9 0.250 Si Violeta Pirogalol4 4 0.111 Si Amarillo Floroglucina5 31 0.861 Si Incoloro benzofenona
Placa 3
Sistema eluyente: 60% Ae/Hex
Distancia recorrida por el disolvente: 37 mm
Patrón Recorrido (mm) Rf UV Color Nombre1 34 0.918 No Azul Mentol
2 30 0.810 Si Rojo Acetoacetato de etilo
3 21 0.567 Si Violeta Pirogalol4 14 0.378 Si Amarillo Floroglucina5 35 0.945 Si Incoloro benzofenona
Placa 4
Sistema eluyente: 80% Ae/Hex
Distancia recorrida por el disolvente: 37 mm
Patrón Recorrido (mm) Rf UV Color Nombre1 34 0.918 No Azul Mentol
2 32 0.864 Si Rojo Acetoacetato de etilo
3 28 0.756 Si Violeta Pirogalol4 23 0.621 Si Amarillo Floroglucina5 35 0.945 Si Incoloro benzofenona
Discusión de resultados.
En algunas placas no se ve el mentol, esto es debido a que es posible q hayan habido otras reacciones las cuales impiden poder valorarlas.
En el factor de retención, las distancias han sido medidas con una regla a simple vista, por lo que los valores de polaridad no son exactos sino una simple aproximación.
Conclusiones.

La técnica de cromatografía en capa fina (CCF) tiene numerosas ventajas, aparte de ser rápida, sencilla, barata... tiene también la ventaja de utilizar poca cantidad de muestra.
Las muestras problemas P1 y P2 son mezclas de muestras patrón, siendo P1 una mezcla de floroglucina y mentol, y P2 una mezcla de Pirogalol y Benzofenona. Esto lo hemos averiguado gracias a las muestras patrón, ya que al ser las muestras problema mezclas entre ellas, al meterlas en el tanque de elución, el disolvente subirá por la placa e irá separando la mezcla, dejando abajo el patrón más polar. Luego, gracias a los reveladores físicos y químicos, se ve que esta compuesta la mezcla problema.
A partir del factor de retención sabremos que sustancia es más polar, siendo el mas polar aquel que tenga un Rf menor.
benzofenona > mentol > acetoacetato de etilo > pirogalol > floroglucina
+ Polar - Polar
1. Resumen 2. Antecedentes 3. Investigación 4. Procedimiento de análisis 5. Determinación de la eficacia de desorción
ResumenLa cromatografía de gases es el procedimiento comúnmente utilizado en el análisis químico, en concreto cromatografía de gases consiste en una muestra que se vaporiza y se inyecta en la cabeza de la columna cromatografía. La muestra se transporta a través de la columna por el flujo de fase inerte, móvil gaseosa. La propia columna contiene una fase líquida estacionaria que se adsorbe sobre la superficie de un sólido inerte.
AntecedentesLa cromatografía de gases fue inventado por AJP Martin, quien, con RLM Synge, propusieron que la fase líquida móvil utilizada en la cromatografía líquida podría ser reemplazado por un gas adecuado. La base de esta recomendación fue que, debido a difusividad de los gases es mucho más alta de solutos en los gases en comparación con los líquidos, el equilibrio en un proceso cromatográfico sería mucho más rápido y por lo tanto, las columnas mucho más eficiente con tiempos de separación mucho más cortos. Así, el concepto de cromatografía de gases fue concebido hace más de cincuenta años.La primera separación publicado por cromatografía de gases fue la de una serie de ácidos grasos, un procedimiento de valoración se utiliza, junto con una micro bureta, como detector, tiempo después la bureta se automatizó para proporcionar una respuesta integra más eficaz. El cromatógrafo de gases fue también uno de los primeros instrumentos de análisis que se asocian con un ordenador que controlaba el análisis, procesar los datos e informó de los resultados.Una forma más sofisticada de la cromatografía de gases fue construido por James y Martin y descrito por James en 1955. El instrumento era un dispositivo algo voluminoso con una columna recta llena con una camisa de vapor. Inicialmente, el detector se encuentra en la base de la columna y consistía en el dispositivo de valoración automática, la separación se presenta como un cromatograma en forma de una serie de pasos, la altura de cada paso de ser proporcional a la masa de soluto diluida. El aparato fue utilizado con éxito para separar algunos ácidos grasos, pero la capacidad limitada del dispositivo para detectar sólo material iónico motivado Martin para desarrollar un detector más versátil, el balance densidad del gas.
InvestigaciónDiagrama de flujo funcional del Cromatografo de gases

Principio de funcionamiento del cromatografo de gases:Puesto que estoy estudiando ingeniería química petrolera, relacionare al cromatógrafo de gases es con una destilación fraccionada con millones de platos.Tenemos un gas que buscamos analizar y separar en sus componentes, este se pasa por un aparato a una temperatura fija, en este aparato, el gas pasa por una columna que contiene un sólido ,o en su defecto, un sólido embebido en líquidos; esto se llama Fase Fija y el gas, se llama fase móvil.La columna es muy larga, como de unos 2 mts y medio cm de espesor, este tubo está lleno de polvo de cerámica, el cual está en forma de espiral metido en un horno a unos 150ºCLa separación se efectúa porque los diferentes componentes de la mezcla de gases interactúan con la fase fija. Los que más sean afines a la fase fija, tardan más en salir del tubo y los otros tardan menos. Con este principio, la separación de los componentes.Luego, a la salida pones un detector que analiza los gases y te informa que componente sale primero y cuál después, a medida que sale se van quemando y por la cantidad de calor sabemos la cantidad y por el tiempo en que salen la sustancia que es y la concentración de cada componente.Aplicaciones industriales de la cromatografía de gasesLa cromatografía de gases se puede aplicar a gases y cualquier compuesto que pueda ser volatilizado o co nvertido en un derivado volátil; la cromatografía de gases tiene amplia aplicación, en las industrias se enfoca principalmente a evaluar la pureza de los reactantes y productos de reacción o bien a monitorear la secuencia de la reacción, para los fabricantes de reactivos químicos su aplicación para la determinación de la pureza es lo más importante.En la investigación es un auxiliar indispensable para diversas técnicas de evaluación, entre las principales están los estudios cinéticos, análisis de adsorción a temperatura programada, determinación de áreas específicas por adsorción de gas y determinación de isotermas de adsorción.En el campo también pueden ser aplicados, principalmente en estudios de contaminantes del agua: insecticidas en agua, pesticidas en aguas de lagos, lagunas, ríos; desechos industriales descargados en ríos o lagunas.En la industria del petróleo juega una función primordial, por medio de la cromatografía se pueden analizar los constituyentes de las gasolinas, las mezclas de gases de refinería, gases de combustión, etc.Medición de hidrocarburos mediante cromatografía de gases.Este método de análisis se ha desarrollado para determinar concentraciones medias ponderadas en el tiempo de vapores de hidrocarburos aromáticos en aire, mediante la utilización de equipos de toma de muestras de baj o caudal, tanto para muestreos personales como en lugares fijos. No puede ser utilizado para medir concentraciones instantáneas ó fluctuaciones de concentración en periodos cortos de tiempo.MetodologíaLa muestra se recoge haciendo pasar una cantidad conocida de aire a través de un tubo relleno dé carbón activo, mediante una bomba de muestreo personal, que dando los vapores orgánicos adsorbidos sobre el carbón. Posteriormente se desorben con sulfuro de carbono y se analiza la disolución resultante en un cromatógrafo de gases equipado con detector de ionización de llama.

Se obtienen las áreas de los picos de los analitos de interés y del patrón interno, determinando la cantidad' presente en la muestra.A partir de la masa de los analitos presentes en la muestra se obtienen las concentraciones ambientales.Reactivos y productosGasesNitrógeno purificadoHidrógeno purificadoAire sintético puroReactivosBencenoNOTA: SUSTANCIA TOXICA Y FÁCILMENTE INFLAMABLE.ToluenoNOTA: SUSTANCIA NOCIVA Y FÁCILMENTE INFLAMABLE.EtilbencenoNOTA: SUSTANCIA NOCIVA Y FÁCILMENTE INFLAMABLE. p-XilenoNOTA: SUSTANCIA NOCIVA.Trimetilbenceno n-PropilbencenoNOTA: SUSTANCIA IRRITANTE.Sulfuro de carbono, debe estar exento de compuestos que convivan con los analitos de interés. NOTA: SUSTANCIA MUY INFLAMABLE Y MUY TOXICA.DisolucionesDisolución desorbente: sulfuro de carbono conteniendo el patrón interno en una concentración de 1 µl/ml.Disolución patrón para la calibración a un nivel de concentración. Se prepara añadiendo mediante micro jeringas de precisión, una cantidad determinada de cada analito a un volumen de disolución desorbente a fin de obtener una disolución patrón de concentración similar a la muestra a analizar. Dicha concentración se debe expresar en términos de mg/ml de disolución desorbente.Disolución patrón para la calibración multinivel. Se preparan cinco disoluciones añadiendo mediante micro jeringas de precisión diferentes cantidades de cada analito a un volumen de disolución desorbente a fin de obtener disoluciones patrón de concentraciones que cubran el intervalo de aplicación del método. Dichas concentraciones se deben expresar en términos de mg/ml de disolución desorbente.
Procedimiento de análisisPreparación de muestras y blancosAñadir 1 ml de la disolución desorbente (4.3.1.) a un tubo roscado y cerrarlo inmediatamente. Hacer u na muesca en el tubo de carbón enfrente de la primera sección de carbón activo y romper el tubo. Se saca y se desecha la lana de vidrio. Añadir la primera sección de carbón al tubo con la disolución desorbente y volver a cerrar. Agitar el tubo ocasionalmente durante un período de 30 minutos para asegurarse que la desorción sea máxima. Repetir el mismo procedimiento para la segunda sección de carbón utilizando otro tubo roscado.CalibraciónAnálisis cromatográficoCondiciones cromatografías. Las condiciones típicas de trabajo para el cromatógrafo de gases equipado según se indica. son las siguientes:
Temperatura del inyector 230 oC-Temperatura del horno 100 oC
-Temperatura del detector 250 oC-Gas portador nitrógeno 30 ml/min
-Hidrógeno 40 ml/min-Aire sintético 300 ml/min
Determinación de la eficacia de desorciónLa eficacia de desorción de los vapores de hidrocarburos aromáticos puede variar con el tipo y lote de carbón usado, siendo necesario calcularla para cada lote de carbón y para cada analito sobre el intervalo de aplicación del método.Para calcular dicha eficacia de desorción, se inyectan diferentes cantidades de los analitos de interés en al menos tres tubos conteniendo 100 mg de carbón (primera sección de un tubo de muestreo) para cubrir el intervalo de aplicación del método. Una vez adicionados los contaminantes a los tubos de carbón, se guardan refrigeradas durante toda la noche para asegurar la completa adsorción. Estos tubos se tratan

como muestras. Paralelamente debe prepararse un tubo blanco por cada concentración, de la misma manera que las muestras, excepto que no se le ha añadido contaminante.Asimismo, se preparan dos o tres patrones inyectando el mismo volumen de los contaminantes en 1 m l de disolución desorbente, cola misma micro jeringa utilizada en la preparación de las muestras.Tanto los tubos blancos como de muestra, se desorben con 1 ml de disolución desorbente, analizándose dichas disoluciones, así como las disoluciones patrón de la misma manera que se ha descrito.Cálculoso Cálculo de la eficacia de desorciónLa eficacia de desorción (ED) se calcula basándose en los resultados mediante la siguiente expresión:
donde:mi es la cantidad promedio (mg) de analito recuperada en la primera sección del tubo de carbón (tubo tratado como muestra).m es la cantidad promedio (mg) de analito añadida al patrón. mb es la cantidad de analito (mg) encontrada en el blancoPrecisiónEl coeficiente de variación del método, calculado a partir de los datos intra laboratorio de muestras captadas en atmósferas de hidrocarburos aromáticos de concentraciones conocidas, es inferior a 3% en todo el intervalo de aplicación del método.De esta manera, con los cálculos ya mencionados y algunos otros cálculos de calibración, junto con la comparación de anexos, que no hemos adicionado en este trabajo por cuestiones de extensión, podemos obtener las cantidades y concentraciones de los hidrocarburos que mencionamos mediante la cromatografía de los gases.
Leer más: http://www.monografias.com/trabajos94/cromatografia-gases/cromatografia-gases.shtml#ixzz2zv8zWfUU
Experimento: Cromatografía la tinta de un bolígrafo
Cromatografía la tinta de un bolígrafo
.
Áreas de conocimiento:
Química
Recomendado para (nivel estudios/edad):
General
Experimento presentado por:
Entidad:Universidad de Alcalá

Equipo:Facultad de Ciencias Químicas de la Universidad de Alcalá
Feria: IX Feria Madrid es Ciencia (2008)
Este experimento ha sido contrastado empíricamente
no
¿Has realizado un experimento similar pero no exactamente igual?
Crea una variante de este experimento.
Escribe el título a continuación y modifica la ficha para crear una nueva.
Resumen:
Práctica sobre una de las técnicas de separación y purificación más importante en el
mundo de la Química:la cromatografía
Fundamento científico:
La cromatografía se puede emplear como método de separación y análisis cualitativo y
cuantitativo de compuestos químicos. De todas las posibles variantes que se engloban dentro
de la cromatografía nos centraremos en la cromatografía en papel. La cromatografía en papel
es una separación química que se basa en los distintos tipos de afinidad de los productos
químicos por, en este caso, el papel y el disolvente. Los compuestos con más afinidad con el
papel van a ser los que menos migren, mientras que los más susceptibles de migrar serán los
compuestos menos afines con dicho soporte.
Materiales utilizados:
Papel cromatográfico (recortado en rectángulos)
Bolígrafos de colores
Etanol (eluyente)
Cubeta cromatográfica (puede ser un tarro de cristal convencional)
Conceptos relacionados con este experimento (expresiones clave):
Conceptos relacionados con este experimento (expresiones clave):
· química · cromatografía
Experimento-varia Cromatografía la Agregar o editar

Desarrollo y montaje del experimento:
Consejos y Advertencias
Se pueden hacer varias separaciones simultáneas alineando puntos sobre la misma línea. Con
bolígrafos BIC se consiguen las siguientes separaciones:
Azul: morado + azul claro
Rojo: rosa + naranja
Negro: violeta + amarillo
Verde: verde + azul claro
Aunque está demostrado con estos modelos y colores, se puede utilizar otro tipo de bolígrafo.
Los de tinta líquida proporcionan buenos resultados.
Paso a seguir:
*1.Recortar un rectángulo de papel cromatográfico. Pintar con lápiz una línea paralela a los
lados más pequeños del rectángulo y a un centímetro de distancia de uno de los bordes.
Paso a seguir:
*2.Dibujar uno o más puntos gordos con la tinta de los bolígrafos
Paso a seguir:
*3.Introducir el papel verticalmente y con el/los puntos dibujados hacia abajo en la cubeta
cromatográfica, en la que previamente hemos vertido etanol (menos de un centímetro de altura
del disolvente)
Paso a seguir:
*4.Dejar que el disolvente suba a través del papel cromatográfico por capilaridad, arrastrando
parte de la tinta según avanza.
Paso a seguir:
*5.Una vez que se haya completado la subida del etanol se habrá obtenido una separación de
los diferentes colorantes que forman parte de la tinta del bolígrafo.

Paso a seguir:
*6.Interpretar los resultados: normalmente para conseguir un determinado color es preciso
recurrir a mezclas de colorantes.
Colabora en mejorar esta fichaPara mejorar la ficha de este experimento se han hecho las peticiones siguientes.
Para hacer las mejoras solicitadas edita la ficha clicando la pestaña "editar con formulario"
en la parte superior de la página.
ComentariosVer o añadir comentarios sobre este experimento
Datos relacionados con Cromatografía la tinta de un bolígrafo — Búsqueda de páginas
similares con +.Ver como RDF
Descripción experimento Práctica sobre una de las técnicas de separación y purificación más importante en el mundo de la Química:la cromatografía
Entidad Universidad de Alcalá +Equipo Facultad de Ciencias Químicas de la Universidad de AlcaláExperimento Cromatografía la tinta de un bolígrafo +Expresión clave química + y cromatografía +Feria IX Feria Madrid es Ciencia (2008) +Hay video no +Nivel Educativo General +Pag imagen experimento Imagen:Sinimagen.png +Título experimento Cromatografía la tinta de un bolígrafo +Ultima edicion 25 abr 2014 20:20 +Área conocimiento Química +
Cromatografía: ¿Qué hay en una gota de tinta?

Introducción.
Los científicos y los analistas necesitan con frecuencia separar los componentes de una mezcla como paso previo a su identificación. La cromatografía es una técnica de separación de sustancias que se basa en las diferentes velocidades con que se mueve cada una de ellas a través de un medio poroso arrastradas por un disolvente en movimiento. Vamos a utilizar esta técnica para separar los pigmentos utilizados en una tinta comercial.
Material necesario.
Una tira de papel poroso. Se puede utilizar el papel de filtro de una cafetera o incluso recortar el extremo (sin tinta) de una hoja de periódico.
Rotuladores o bolígrafos de distintos colores.
Un vaso
Un poco de alcohol
Prodecimiento.
Recorta una tira del papel poroso que tenga unos dos o tres dedos de ancho y que sea un poco mas larga que la altura del vaso.
Enrolla un extremo en un bolígrafo (puedes ayudarte de cinta adhesiva) de tal manera que el otro extremo llegue al fondo del vaso. (ver dibujo)
Dibuja una mancha con un rotulador negro en el extremo libre de la tira, sin tocar el borde, de forma que no quede sumergida en el alcohol (ver

paso siguiente). Procura que sea intensa y que no ocupe mucho (ver dibujo)
Echa en el fondo del vaso alcohol, hasta una altura de un dedo aproximadamente.
Sitúa la tira dentro del vaso de tal manera que el extremo quede sumergido en el alcohol pero la mancha que has hecho sobre ella quede fuera de él.
Puedes tapar el vaso para evitar que el alcohol se evapore.
Observa lo qué ocurre: a medida que el alcohol va ascendiendo a lo largo de la tira, arrastra consigo los diversas pigmentos que contiene la mancha de tinta. Como no todos son arrastrados con la misma velocidad, al cabo de un rato se ven franjas de colores.
Repite la experiencia utilizando diferentes tintas.
Repite la experiencia utilizando las mismas tintas pero con otros líquidos que sirvan como disolvente.
Entrega un informe a tu profesor según sus instrucciones.

Información adicional: Fundamento teórico de la cromatografía.
En la cromatografía sobre papel se distinguen dos fases:
Fase móvil, constituida por el disolvente que asciende por el papel, al que se denomina eluyente.
Fase estacionaria, formada por el agua contenida en el papel de filtro, que retiene de forma diferente cada uno de los componentes de la sustancia a analizar.
Esa diferente retención del agua contenida en el papel sobre los componentes de la sustancia provoca que, según avanza el frente de disolvente, cada uno de ellos sea arrastrado a diferente distancia del punto de partida, pudiendo tanto observarse el número de componentes que forman la sustancia original como determinar mediante análisis posteriores de qué tipo de componentes se trata.
experimentos caseros
¿De Que esta Hecho una Tinta? - Cromatografia
Objetivo:
Utilizar la técnica de cromatografía para separar los componentes
de una tinta comercial
Fundamento Teórico:
Los biólogos, médicos y químicos necesitan con frecuencia separar
los componentes de una mezcla como paso previo a su
identificación. La cromatografía es una técnica de separación de
sustancias que se basa en las diferentes velocidades con que se
mueve cada una de ellas, a través de un medio poroso, arrastradas
por un disolvente en movimiento. Vamos a aplicar esta técnica para
separar los pigmentos utilizados en una tinta comercial.

Materiales:
• Una tira de papel de filtro o poroso (se puede usar el papel del fi
ltro de una cafetera o incluso recortar el extremo –sin tinta– de una
hoja de periódico)
• Rotuladores o bolígrafos de distintos colores
• Un vaso
• Un poco de alcohol
Procedimiento:

1. Recorta una tira del
papel poroso que tenga unos 4 cm de ancho y una altura un poco
mayor a la del vaso.
2. Enrolla un extremo en un bolígrafo (puedes fijarlo con cinta
adhesiva) de tal manera que el otro extremo llegue al fondo del vaso.
3. Pinta una mancha con un rotulador negro en el extremo libre de la
tira, a unos 2 cm del borde. Procura que sea intensa, pero que no
ocupe mucho espacio.

4. En el fondo del vaso, vierte alcohol hasta una altura de 1 cm,
aproximadamente.
5. Sitúa la tira dentro del vaso de tal manera que el extremo quede
sumergido en el alcohol, pero la mancha fuera de él.
6. Puedes tapar el vaso para evitar que el alcohol se evapore.
7. Observa lo que ocurre: a medida que el alcohol va ascendiendo a
lo largo de la tira, arrastra consigo los diversos pigmentos que
contiene la mancha de tinta. Como no todos son arrastrados con la
misma velocidad, al cabo de un rato se ven franjas de colores.
8. Repite la prueba empleando agua en lugar de alcohol.
9. Una vez más, pero ahora utiliza una tira de papel de cuaderno y
alcohol. Si lo deseas, repite la experiencia utilizando diferentes
colores de tintas; así descubrirás los pigmentos que los componen.
Explicacion
La mancha de tinta se separa en sus diferentes componentes
porque el color que observamos es el resultado de una mezcla de
diferentes pigmentos, los cuales fueron separados mediante la
técnica de cromatografía.
Debido a que el agua no es un disolvente de la tinta, no separa los
diferentes pigmentos como lo hace el alcohol. De la misma manera,
la hoja de cuaderno, por no ser un material poroso, no favorece que

el alcohol arrastre los diferentes pigmentos de la tintA
ESTE ERA UN EXPERIMENTO Y AQUI VEMOS OTRO MUY INTERESANTE Y FACIL DE HACER
Los biólogos, médicos y químicosnecesitan con frecuencia separar los componentes de una mezcla como paso previo a su identificación. La cromatografía es una técnica de separación de sustancias que se basa en las diferentes velocidades con que se mueve cada una de ellas a través de un medio poroso arrastradas por un disolvente en movimiento. Vamos a utilizar esta técnica para separar los pigmentos utilizados en una tinta comercial.
Material necesario Una tira de papel poroso. Se puede utilizar el papel de filtro de una cafetera o incluso
recortar el extremo (sin tinta) de una hoja de periódico. Rotuladores o bolígrafos de distintos colores. Un vaso Un poco de alcohol
Prodecimiento Recorta una tira del papel poroso que tenga unos 4 cm
mas larga que la altura del vaso. Enrolla un extremo en un bolígrafo (puedes ayudarte de cinta adhesiva) de tal
manera que el otro extremo llegue al fondo del vaso. (ver dibujo) Dibuja una mancha con un rotulador negro en el extremo libre de la tira, a unos
cm del borde. Procura que sea intensa y que no ocupe mucho. (ver dibujo) Echa en el fondo del vaso alcohol, hasta una altura de Sitúa la tira dentro del vaso de tal manera que el extremo quede sumergido en el
alcohol pero la mancha que has hecho sobre ella quede fuera de él. Puedes tapar el vaso para evitar que el alcohol se evapore. Observa lo que ocurre : a medida que el alcohol va ascendiendo a lo largo de la tira,

arrastra consigo los diversas pigmentos que contiene la mancha de tinta. Como no todos son arrastrados con la misma velocidad, al cabo de un rato se vencolores.
Repite la experiencia utilizando diferentes tintas.
Cromatografía
La cromatografía permite separar los componentes de la tinta
Alumnado:Carlos Flox CanoMaría García CastañedaCelia García JiménezCristina Gámez PonceDaniel León PolainaRhaisa LópezLiraProfesor: Emilio Martínez Torres.Asignatura: El medio natural I: Física, Química y su didáctica.Curso: 3º Grado de Maestro en Educación Primaria – B

ÍNDICE1. 1. ¿Qué es la cromatografía?2. 2. Fases de la cromatografía. 3. 3. Tipología.4. 4. Cromatografía en papel, ¿qué es?5. 5. ¿Cómo separar los componentes de la tinta?6. 6. ¿Para qué sirve? Aplicaciones de la cromatografía.7. 7. Conclusión.8. 8. Bibliografía.9. 9. Webgrafía.
1. 1. ¿Qué es la cromatografía?
Es la técnica más desarrollada en los últimos años, empleada en la química analítica, así como la mejor técnica para separar las sustancias en química.Su empleo presenta notables ventajas:a. Es sencilla, rápida y no requiere aparatos complicados.b. Abarca escalas microanalíticas hasta escalas industriales.c. Es una técnica poca o nada destructiva que puede aplicarse a sustancias lábiles.Constituye una metodología imprescindible en estudios bioquímicos, toxicológicos, estructurales, etc. No solo utilizando como técnica de separación e identificación, sino como método preparatorio, incluso a escalas industriales.La palabra cromatografía proviene de “kromatos” (color) y “graphos” (escrito).Una de las definiciones de la técnica más correcta seria, la dada por Keulemans:"Método físico de separación en el que los componentes a separar se distribuyen en dos fases, una de las cuales constituye un hecho estacionario de gran desarrollo superficial y la otra un fluido que pasa a través o a lo largo del lecho estacionario (fase móvil)."En todo proceso cromatográfico la fase móvil es la que provoca un movimiento de las distintas especias para que abandonen el medio soporte, y la fase estacionaria la que suministra el efecto retardador, selectivo para cada componente, que condiciona que cada uno de ellos se desplace con distinta velocidad.
1. 2. Fases de la cromatografía.
En la cromatografía sobre papel se distinguen dos fases:
Fase móvil, constituida por el disolvente que asciende por el papel, al que se denomina eluyente.
Fase estacionaria, formada por el agua contenida en el papel de filtro, que retiene de forma diferente cada uno de los componentes de la sustancia a analizar.
Esa diferente retención del agua contenida en el papel sobre los componentes de la sustancia provoca que, según avanza el frente de disolvente, cada uno de ellos sea arrastrado a diferente distancia del punto de partida, pudiendo tanto observarse el

número de componentes que forman la sustancia original como determinar mediante análisis posteriores de qué tipo de componentes se trata.
1. 3. Tipología.
Según la clasificación de los métodos cromatográficos se pueden distinguir:
Según como se coloque en contacto la fase móvil y estacionaria: cromatografía en columna, cromatografía plana, y cromatografía en papel.
Según el tipo de fase móvil utilizada: Cromatografía gaseosa y cromatografía líquida y electroforesis (parecido a las demás cromatografías pero los colores aparecen en el papel a través de corrientes eléctricas).
También destacar el uso del cromatograma como la representación gráfica de la señal en función del tiempo una vez que la muestra es inyectada a un sistema cromatográfico.
1. 4. Cromatografía en papel, ¿qué es?
La cromatografía en papel es la técnica de separación e identificación de sustancias químicas mediante un disolvente que se mueve sobre hojas o tiras de papel de filtro.Se toma una pieza de pape de filtro y cerca de uno de sus extremos se deposita una gota de la disolución que contiene una mezcla de sustancias que se quieren separar. Se dejan secar la gota, donde quedará una mancha de las sustancias mezcladas. El extremos del papel más próximo a la mancha se introduce en un disolvente apropiados, pero sin que la mancha llegue a introducirse en él. Existen muchos tipos de cromatografías sobre papel, una primera división de las variadas clases puede ser:
1. a. Cromatografía ascendente: el disolvente se encuentra en el fondo del recipiente que sostiene al papel y va subiendo a través de él por capilaridad.
2. b. Cromatografía descendente: el disolvente está en un recipiente del que cuelga un papel, fluye por él hacia abajo por una combinación de capilaridad y gravedad.
En ambos casos el disolvente avanza a lo largo del papel pasando por encima la mancha, al avanzar disuelve y arrastra las sustancias de la mancha, cada una de ellas se mueve, por lo general a distinta velocidad que las otras. Se deja actuar el disolvente un tiempo determinado, se seca el papel y se observan las sustancias separadas si tienen color o, en caso de no tener color, se procede al revelado por una reacción química apropiada. Esta técnica se conoce como cromatografía monodimensional y el papel resultante recibe el nombre de cromatograma monodimendional.

1. 5. ¿Cómo separar los componentes de la tinta?
Material necesario.
Una tira de papel poroso. Se puede utilizar el papel de filtro de una cafetera o incluso recortar el extremo (sin tinta) de una hoja de periódico.
Rotuladores o bolígrafos de distintos colores. Un vaso Un poco de alcohol.
Procedimiento.
Recorta una tira del papel poroso que tenga unos dos o tres dedos de ancho y que sea un poco más larga que la altura del vaso.
Enrolla un extremo en un bolígrafo (puedes ayudarte de cinta adhesiva) de tal manera que el otro extremo llegue al fondo del vaso. (ver dibujo).
Dibuja una mancha con un rotulador negro en el extremo libre de la tira, sin tocar el borde, de forma que no quede sumergida en el alcohol (ver paso siguiente). Procura que sea intensa y que no ocupe mucho (ver dibujo).
Echa en el fondo del vaso alcohol, hasta una altura de un dedo aproximadamente.
Sitúa la tira dentro del vaso de tal manera que el extremo quede sumergido en el alcohol pero la mancha que has hecho sobre ella quede fuera de él.
o Puedes tapar el vaso para evitar que el alcohol se evapore.o Observa lo qué ocurre: a medida que el alcohol va ascendiendo a lo
largo de la tira, arrastra consigo los diversos pigmentos que contiene la mancha de tinta. Como no todos son arrastrados con la misma velocidad, al cabo de un rato se ven franjas de colores.
Repite la experiencia utilizando diferentes tintas. Repite la experiencia utilizando las mismas tintas pero con otros
líquidos que sirvan como disolvente.
1. 6. ¿Para qué sirve? Aplicaciones de la cromatografía.
El uso de la cromatografía está ampliamente extendido en el análisis de alimentos,

medicinas, sangre, productos petrolíferos y de fisión… A continuación, se muestran algunas de sus aplicaciones más importantes:
En los problemas relacionados con la contaminación ambiental, se utiliza para la contaminación de algunos pesticidas (insecticidas, larvicidas, hervicidas, etc.). También es posible la determinación de hidrocarburos aromáticos polinucleares que son contaminantes atmosféricos muy importantes.
En cromatografía líquida, el análisis de compuestos vitamínicos es posible en corto tiempo.
Las pruebas de ADN se basan en la cromatografía denominada electroforesis, donde en el cromatograma, se muestran perfectamente los nucleótidos que hay en el ADN.
El análisis de medicamentos es también una aplicación muy interesante, la determinación de los componentes activos de una tableta analgésica. También el análisis de barbitúricos, anticonceptivos, etc.
1. 7. Conclusión.
Al observar e investigar sobre dicha información, "Cromatografía", hemos llegado a entender que para realizar cualquier separación de mezclas primero debemos saber sobre su estado físico, características y propiedades.Es interesante realizar una mezcla, pero es más importante tener claro cuales componentes se mezclan para que, a la hora de separar los componentes, usemos la técnica más adecuada.
1. 8. Bibliografía.
“Análisis Instrumental”. Rubinson y Rubinson. 2001. Editorial Pretice Hall. “Análisis Químico Cuantitativo”. Daniel Harris. 3era edición. 2007. Editorial Reverté.
“Principios de Análisis Instrumental”. 5ª Edición. Skoog Holler Nieman.“Cromatografía sobre papel y capa fina. Electroferesis”. (I. Smith y J. Feinberg).“Química Analítica Cualitativa”. (Burriel).
1. 9. Webgrafía.
https://www.itescam.edu.mx/principal/sylabus/fpdb/recursos/r49995.PDF
Métodos de separación Cromatografía36 respuestas
Métodos de separación Cromatografía.

Cromatografía:
La separación de determinados componentes de una mezcla la cual sea homogénea,
Técnica que se usa para permitir separar aquellos componentes de una mezcla, para ello se hace pasar a través de un absorbente (que se adhiere a una superficie).
Se conoce y utiliza como metodología más simple es la que usa papel como medio absorbente, el papel es el filtro en esta Cromatografía, y el solvente el liquido alcohol o agua.
Estos componentes se separan cuando estos componentes manifiestan sus diferentes afinidades por el filtro de papel o bien el disolvente que acciona.
Podemos ver que la tinta de plumón parece como totalmente homogénea, sin embargo al estar formada por distintos componentes se pueden separar con facilidad, para ello solo requerimos dejar correr en un medio que sea absorbente por acción de un disolvente.
Nombremos algunos ejemplos que se pueden usar para este método, los productos que se usan como medio de absorción pueden ser, arena, papel, tiza, filtro, etc.

Lo más utilizado es el papel de filtro, siempre en los laboratorios de estudio, se ase una demostración con el papel de filtro y tinta de plumón de agua, la razón es que para poder separar estos componentes de la mezcla, se nos torna sencillo ya que utilizamos como disolvente el agua.
Para lograr un buen resultado se debe tener en cuenta, que para lograr la separación el disolvente no puede ni debe estar en contacto con la mezcla, este debe llegar a ella por medio de la absorción.
Separación de Mezclas Cromatografía y Centrifugación
Metodos de separacion de Mezcla “Inicio”

Esta entrada se publicó en mezclas, Química, Recomendados, Separación de mezclas y está etiquetada con absorbente, agua, arena, Cromatografía, disolvente, filtro, homogenea, mescla, metodo, Metodos de separación Cromatografía, mezcla, papel, separación, superficie, tiza en septiembre 10,
High-performance liquid chromatographyFrom Wikipedia, the free encyclopedia
High-performance liquid chromatography
An HPLC. From left to right: A pumping device generating a gradient of two different solvents- a steel
enforced column and a detector for measuring the absorbance.
Acronym HPLCClassification ChromatographyAnalytes organic molecules

biomoleculesionspolymers
Other techniques
Related
ChromatographyAqueous normal-phase chromatographyHydrophilic Interaction ChromatographyIon exchange chromatographySize exclusion chromatographyMicellar liquid chromatography
Hyphenated Liquid chromatography-mass spectrometry
A modern self-contained HPLC.
Schematic representation of an HPLC unit. (1) Solvent reservoirs, (2) Solvent degasser, (3) Gradient valve, (4) Mixing vessel for delivery of the mobile phase, (5) High-pressure pump, (6) Switching valve in "inject position", (6') Switching valve in "load position", (7) Sample injection loop, (8) Pre-column (guard column), (9) Analytical column, (10) Detector (i.e. IR, UV), (11) Data acquisition, (12) Waste or fraction collector.
High-performance liquid chromatography (HPLC; formerly referred to as high-pressure liquid chromatography), is a technique in analytic chemistry used to separate the components in a mixture, to identify each component, and to quantify each component. It relies on pumps to pass a pressurized liquid solvent containing the sample

mixture through a column filled with a solid adsorbent material. Each component in the sample interacts slightly differently with the adsorbent material, causing different flow rates for the different components and leading to the separation of the components as they flow out the column.
HPLC has been used for medical (e.g. detecting vitamin D levels in blood serum), legal (e.g. detecting performance enhancement drugs in urine), research (e.g. separating the components of a complex biological sample, or of similar synthetic chemicals from each other), and manufacturing (e.g. during the production process of pharmaceutical and biological products) purposes.[1]
Chromatography can be described as a mass transfer process involving adsorption. HPLC relies on pumps to pass a pressurized liquid and a sample mixture through a column filled with a sorbent, leading to the separation of the sample components. The active component of the column, the sorbent, is typically a granular material made of solid particles (e.g. silica, polymers, etc.), 2–50 micrometers in size. The components of the sample mixture are separated from each other due to their different degrees of interaction with the sorbent particles. The pressurized liquid is typically a mixture of solvents (e.g. water, acetonitrile and/or methanol) and is referred to as a "mobile phase". Its composition and temperature play a major role in the separation process by influencing the interactions taking place between sample components and sorbent. These interactions are physical in nature, such as hydrophobic (dispersive), dipole–dipole and ionic, most often a combination thereof.
HPLC is distinguished from traditional ("low pressure") liquid chromatography because operational pressures are significantly higher (50–350 bar), while ordinary liquid chromatography typically relies on the force of gravity to pass the mobile phase through the column. Due to the small sample amount separated in analytical HPLC, typical column dimensions are 2.1–4.6 mm diameter, and 30–250 mm length. Also HPLC columns are made with smaller sorbent particles (2–50 micrometer in average particle size). This gives HPLC superior resolving power when separating mixtures, which is why it is a popular chromatographic technique.
The schematic of an HPLC instrument typically includes a sampler, pumps, and a detector. The sampler brings the sample mixture into the mobile phase stream which carries it into the column. The pumps deliver the desired flow and composition of the mobile phase through the column. The detector generates a signal proportional to the amount of sample component emerging from the column, hence allowing for quantitative analysis of the sample components. A digital microprocessor and user software control the HPLC instrument and provide data analysis. Some models of mechanical pumps in a HPLC instrument can mix multiple solvents together in ratios changing in time, generating a composition gradient in the mobile phase. Various detectors are in common use, such as UV/Vis, photodiode array (PDA) or based on mass spectrometry. Most HPLC instruments also have a column oven that allows for adjusting the temperature the separation is performed at.
Contents 1 Operation 2 Types

o 2.1 Partition chromatography o 2.2 Normal–phase chromatography o 2.3 Displacement chromatography o 2.4 Reversed-phase chromatography (RPC) o 2.5 Size-exclusion chromatography o 2.6 Ion-exchange chromatography o 2.7 Bioaffinity chromatography o 2.8 Aqueous normal-phase chromatography
3 Isocratic and gradient elution 4 Parameters
o 4.1 Theoretical o 4.2 Internal diameter o 4.3 Particle size o 4.4 Pore size o 4.5 Pump pressure
5 See also 6 References 7 Further reading 8 External links
OperationThe sample mixture to be separated and analyzed is introduced, in a discrete small volume (typically microliters), into the stream of mobile phase percolating through the column. The components of the sample move through the column at different velocities, which are function of specific physical interactions with the sorbent (also called stationary phase). The velocity of each component depends on its chemical nature, on the nature of the stationary phase (column) and on the composition of the mobile phase. The time at which a specific analyte elutes (emerges from the column) is called its retention time. The retention time measured under particular conditions is considered an identifying characteristic of a given analyte.
Many different types of columns are available, filled with sorbents varying in particle size, and in the nature of their surface ("surface chemistry"). The use of smaller particle size packing materials requires the use of higher operational pressure ("backpressure") and typically improves chromatographic resolution (i.e. the degree of separation between consecutive analytes emerging from the column). In terms of surface chemistry, sorbent particles may be hydrophobic or polar in nature.
Common mobile phases used include any miscible combination of water with various organic solvents (the most common are acetonitrile and methanol). Some HPLC techniques use water-free mobile phases (see Normal-phase chromatography below). The aqueous component of the mobile phase may contain acids (such as formic, phosphoric or trifluoroacetic acid) or salts to assist in the separation of the sample components. The composition of the mobile phase may be kept constant ("isocratic elution mode") or varied ("gradient elution mode") during the chromatographic analysis. Isocratic elution is typically effective in the separation of sample components that are not very different in their affinity for the stationary phase. In gradient elution the composition of the mobile phase is varied typically from low to high eluting strength.

The eluting strength of the mobile phase is reflected by analyte retention times with high eluting strength producing fast elution (=short retention times). A typical gradient profile in reversed phase chromatography might start at 5% acetonitrile (in water or aqueous buffer) and progress linearly to 95% acetonitrile over 5–25 minutes. Periods of constant mobile phase composition may be part of any gradient profile. For example, the mobile phase composition may be kept constant at 5% acetonitrile for 1–3 min, followed by a linear change up to 95% acetonitrile.
The chosen composition of the mobile phase (also called eluent) depends on the intensity of interactions between various sample components ("analytes") and stationary phase (e.g. hydrophobic interactions in reversed-phase HPLC). Depending on their affinity for the stationary and mobile phases analytes partition between the two during the separation process taking place in the column. This partitioning process is similar to that which occurs during a liquid–liquid extraction but is continuous, not step-wise. In this example, using a water/acetonitrile gradient, more hydrophobic components will elute (come off the column) late, once the mobile phase gets more concentrated in acetonitrile (i.e. in a mobile phase of higher eluting strength).
The choice of mobile phase components, additives (such as salts or acids) and gradient conditions depends on the nature of the column and sample components. Often a series of trial runs is performed with the sample in order to find the HPLC method which gives adequate separation.
Types
Partition chromatography
HILIC Partition Technique Useful Range
Partition chromatography was the first kind of chromatography that chemists developed. The partition coefficient principle has been applied in paper chromatography, thin layer chromatography, gas phase and liquid-liquid applications. The 1952 Nobel Prize in chemistry was earned by Archer John Porter Martin and Richard Laurence Millington Synge for their development of the technique, which was used for their separation of amino acids. Partition chromatography uses a retained solvent, on the surface or within the grains or fibers of an "inert" solid supporting matrix as with paper chromatography; or takes advantage of some coulombic and/or hydrogen donor interaction with the

stationary phase. Analyte molecules partition between a liquid stationary phase and the eluent. Just as in Hydrophilic Interaction Chromatography (HILIC; a sub-technique within HPLC), this method separates analytes based on differences in their polarity. HILIC most often uses a bonded polar stationary phase and a mobile phase made primarily of acetonitrile with water as the strong component. Partition HPLC has been used historically on unbonded silica or alumina supports. Each works effectively for separating analytes by relative polar differences. HILIC bonded phases have the advantage of separating acidic, basic and neutral solutes in a single chromatographic run.[2]
The polar analytes diffuse into a stationary water layer associated with the polar stationary phase and are thus retained. The stronger the interactions between the polar analyte and the polar stationary phase (relative to the mobile phase) the longer the elution time. The interaction strength depends on the functional groups part of the analyte molecular structure, with more polarized groups (e.g. hydroxyl-) and groups capable of hydrogen bonding inducing more retention. Coulombic (electrostatic) interactions can also increase retention. Use of more polar solvents in the mobile phase will decrease the retention time of the analytes, whereas more hydrophobic solvents tend to increase retention times.
Normal–phase chromatography
Normal–phase chromatography was one of the first kinds of HPLC that chemists developed. Also known as normal-phase HPLC (NP-HPLC) this method separates analytes based on their affinity for a polar stationary surface such as silica, hence it is based on analyte ability to engage in polar interactions (such as hydrogen-bonding or dipole-dipole type of interactions) with the sorbent surface. NP-HPLC uses a non-polar, non-aqueous mobile phase (e.g. Chloroform), and works effectively for separating analytes readily soluble in non-polar solvents. The analyte associates with and is retained by the polar stationary phase. Adsorption strengths increase with increased analyte polarity. The interaction strength depends not only on the functional groups present in the structure of the analyte molecule, but also on steric factors. The effect of steric hindrance on interaction strength allows this method to resolve (separate) structural isomers.
The use of more polar solvents in the mobile phase will decrease the retention time of analytes, whereas more hydrophobic solvents tend to induce slower elution (increased retention times). Very polar solvents such as traces of water in the mobile phase tend to adsorb to the solid surface of the stationary phase forming a stationary bound (water) layer which is considered to play an active role in retention. This behavior is somewhat peculiar to normal phase chromatograhy because it is governed almost exclusively by an adsorptive mechanism (i.e. analytes interact with a solid surface rather than with the solvated layer of a ligand attached to the sorbent surface; see also reversed-phase HPLC below). Adsorption chromatography is still widely used for structural isomer separations in both column and thin-layer chromatography formats on activated (dried) silica or alumina supports.
Partition- and NP-HPLC fell out of favor in the 1970s with the development of reversed-phase HPLC because of poor reproducibility of retention times due to the presence of a water or protic organic solvent layer on the surface of the silica or alumina

chromatographic media. This layer changes with any changes in the composition of the mobile phase (e.g. moisture level) causing drifting retention times.
Recently, partition chromatography has become popular again with the development of HILIC bonded phases which demonstrate improved reproducibility, and due to a better understanding of the range of usefulness of the technique.
Displacement chromatography
The basic principle of displacement chromatography is: A molecule with a high affinity for the chromatography matrix (the displacer) will compete effectively for binding sites, and thus displace all molecules with lesser affinities.[3] There are distinct differences between displacement and elution chromatography. In elution mode, substances typically emerge from a column in narrow, Gaussian peaks. Wide separation of peaks, preferably to baseline, is desired in order to achieve maximum purification. The speed at which any component of a mixture travels down the column in elution mode depends on many factors. But for two substances to travel at different speeds, and thereby be resolved, there must be substantial differences in some interaction between the biomolecules and the chromatography matrix. Operating parameters are adjusted to maximize the effect of this difference. In many cases, baseline separation of the peaks can be achieved only with gradient elution and low column loadings. Thus, two drawbacks to elution mode chromatography, especially at the preparative scale, are operational complexity, due to gradient solvent pumping, and low throughput, due to low column loadings. Displacement chromatography has advantages over elution chromatography in that components are resolved into consecutive zones of pure substances rather than “peaks”. Because the process takes advantage of the nonlinearity of the isotherms, a larger column feed can be separated on a given column with the purified components recovered at significantly higher concentration.
Reversed-phase chromatography (RPC)
A chromatogram of complex mixture (perfume water) obtained by reversed phase HPLCFor more details on this topic, see Reversed-phase chromatography.
Reversed phase HPLC (RP-HPLC) has a non-polar stationary phase and an aqueous, moderately polar mobile phase. One common stationary phase is a silica which has been surface-modified with RMe2SiCl, where R is a straight chain alkyl group such as C18H37 or C8H17. With such stationary phases, retention time is longer for molecules which are

less polar, while polar molecules elute more readily (early in the analysis). An investigator can increase retention times by adding more water to the mobile phase; thereby making the affinity of the hydrophobic analyte for the hydrophobic stationary phase stronger relative to the now more hydrophilic mobile phase. Similarly, an investigator can decrease retention time by adding more organic solvent to the eluent. RP-HPLC is so commonly used that it is often incorrectly referred to as "HPLC" without further specification. The pharmaceutical industry regularly employs RP-HPLC to qualify drugs before their release.
RP-HPLC operates on the principle of hydrophobic interactions, which originates from the high symmetry in the dipolar water structure and plays the most important role in all processes in life science. RP-HPLC allows the measurement of these interactive forces. The binding of the analyte to the stationary phase is proportional to the contact surface area around the non-polar segment of the analyte molecule upon association with the ligand on the stationary phase. This solvophobic effect is dominated by the force of water for "cavity-reduction" around the analyte and the C18-chain versus the complex of both. The energy released in this process is proportional to the surface tension of the eluent (water: 7.3×10−6 J/cm², methanol: 2.2×10−6 J/cm²) and to the hydrophobic surface of the analyte and the ligand respectively. The retention can be decreased by adding a less polar solvent (methanol, acetonitrile) into the mobile phase to reduce the surface tension of water. Gradient elution uses this effect by automatically reducing the polarity and the surface tension of the aqueous mobile phase during the course of the analysis.
Structural properties of the analyte molecule play an important role in its retention characteristics. In general, an analyte with a larger hydrophobic surface area (C-H, C-C, and generally non-polar atomic bonds, such as S-S and others) is retained longer because it is non-interacting with the water structure. On the other hand, analytes with higher polar surface area (conferred by the presence of polar groups, such as -OH, -NH2, COO– or -NH3
+ in their structure) are less retained as they are better integrated into water. Such interactions are subject to steric effects in that very large molecules may have only restricted access to the pores of the stationary phase, where the interactions with surface ligands (alkyl chains) take place. Such surface hindrance typically results in less retention.
Retention time increases with hydrophobic (non-polar) surface area. Branched chain compounds elute more rapidly than their corresponding linear isomers because the overall surface area is decreased. Similarly organic compounds with single C-C-bonds elute later than those with a C=C or C-C-triple bond, as the double or triple bond is shorter than a single C-C-bond.
Aside from mobile phase surface tension (organizational strength in eluent structure), other mobile phase modifiers can affect analyte retention. For example, the addition of inorganic salts causes a moderate linear increase in the surface tension of aqueous solutions (ca. 1.5×10−7 J/cm² per Mol for NaCl, 2.5×10−7 J/cm² per Mol for (NH4)2SO4), and because the entropy of the analyte-solvent interface is controlled by surface tension, the addition of salts tend to increase the retention time. This technique is used for mild separation and recovery of proteins and protection of their biological activity in protein analysis (hydrophobic interaction chromatography, HIC).

Another important factor is the mobile phase pH since it can change the hydrophobic character of the analyte. For this reason most methods use a buffering agent, such as sodium phosphate, to control the pH. Buffers serve multiple purposes: control of pH, neutralize the charge on the silica surface of the stationary phase and act as ion pairing agents to neutralize analyte charge. Ammonium formate is commonly added in mass spectrometry to improve detection of certain analytes by the formation of analyte-ammonium adducts. A volatile organic acid such as acetic acid, or most commonly formic acid, is often added to the mobile phase if mass spectrometry is used to analyze the column effluent. Trifluoroacetic acid is used infrequently in mass spectrometry applications due to its persistence in the detector and solvent delivery system, but can be effective in improving retention of analytes such as carboxylic acids in applications utilizing other detectors, as it is a fairly strong organic acid. The effects of acids and buffers vary by application but generally improve chromatographic resolution.
Reversed phase columns are quite difficult to damage compared with normal silica columns; however, many reversed phase columns consist of alkyl derivatized silica particles and should never be used with aqueous bases as these will destroy the underlying silica particle. They can be used with aqueous acid, but the column should not be exposed to the acid for too long, as it can corrode the metal parts of the HPLC equipment. RP-HPLC columns should be flushed with clean solvent after use to remove residual acids or buffers, and stored in an appropriate composition of solvent. The metal content of HPLC columns must be kept low if the best possible ability to separate substances is to be retained. A good test for the metal content of a column is to inject a sample which is a mixture of 2,2'- and 4,4'- bipyridine. Because the 2,2'-bipy can chelate the metal, the shape of the peak for the 2,2'-bipy will be distorted (tailed) when metal ions are present on the surface of the silica.[citation needed]..
Size-exclusion chromatography
For more details on this topic, see size-exclusion chromatography.
Size-exclusion chromatography (SEC), also known as gel permeation chromatography or gel filtration chromatography, separates particles on the basis of molecular size (actually by a particle's Stokes radius). It is generally a low resolution chromatography and thus it is often reserved for the final, "polishing" step of the purification. It is also useful for determining the tertiary structure and quaternary structure of purified proteins. SEC is used primarily for the analysis of large molecules such as proteins or polymers. SEC works by trapping these smaller molecules in the pores of a particle. The larger molecules simply pass by the pores as they are too large to enter the pores. Larger molecules therefore flow through the column quicker than smaller molecules, that is, the smaller the molecule, the longer the retention time.
This technique is widely used for the molecular weight determination of polysaccharides. SEC is the official technique (suggested by European pharmacopeia) for the molecular weight comparison of different commercially available low-molecular weight heparins.
Ion-exchange chromatography
For more details on this topic, see Ion-exchange chromatography.
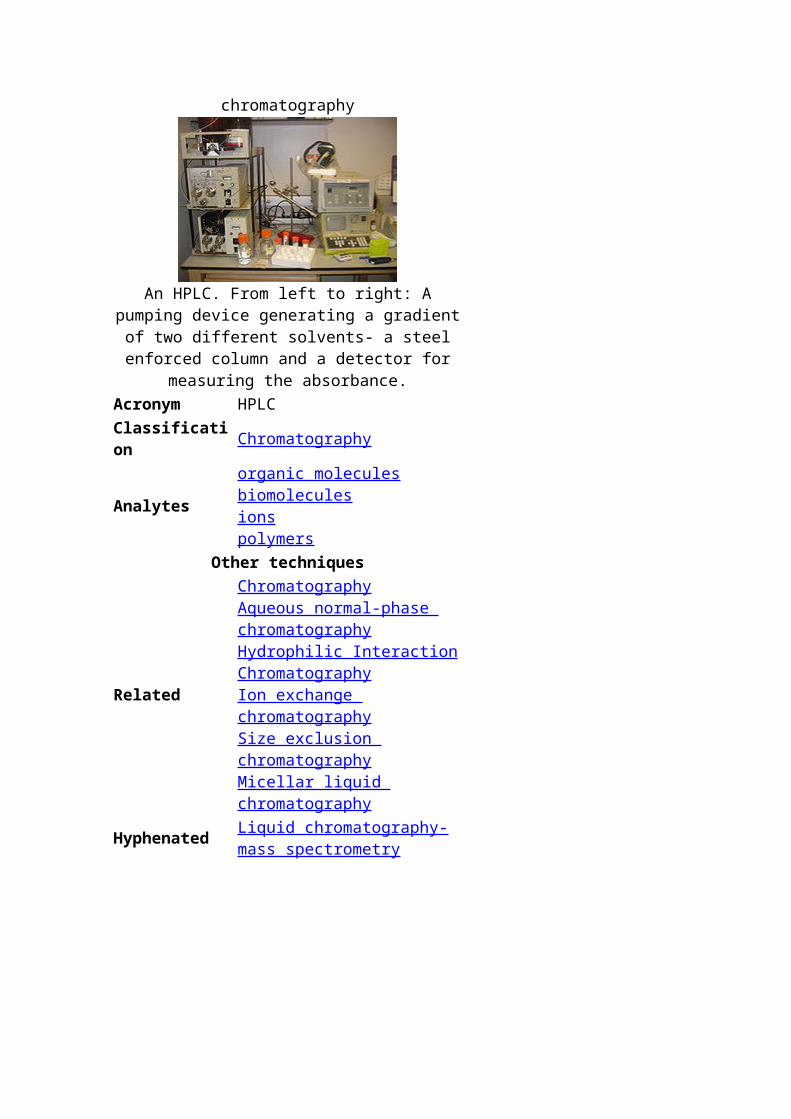
In ion-exchange chromatography (IC), retention is based on the attraction between solute ions and charged sites bound to the stationary phase. Solute ions of the same charge as the charged sites on the column are excluded from binding, while solute ions of the opposite charge of the charged sites of the column are retained on the column. Solute ions that are retained on the column can be eluted from the column by changing the solvent conditions (e.g. increasing the ion effect of the solvent system by increasing the salt concentration of the solution, increasing the column temperature, changing the pH of the solvent, etc...).
Types of ion exchangers include:
Polystyrene resins – These allow cross linkage which increases the stability of the chain. Higher cross linkage reduces swerving, which increases the equilibration time and ultimately improves selectivity.
Cellulose and dextran ion exchangers (gels) – These possess larger pore sizes and low charge densities making them suitable for protein separation.
Controlled-pore glass or porous silica
In general, ion exchangers favor the binding of ions of higher charge and smaller radius.
An increase in counter ion (with respect to the functional groups in resins) concentration reduces the retention time. A decrease in pH reduces the retention time in cation exchange while an increase in pH reduces the retention time in anion exchange. By lowering the pH of the solvent in a cation exchange column, for instance, more hydrogen ions are available to compete for positions on the anionic stationary phase, thereby eluting weakly bound cations.
This form of chromatography is widely used in the following applications: water purification, preconcentration of trace components, ligand-exchange chromatography, ion-exchange chromatography of proteins, high-pH anion-exchange chromatography of carbohydrates and oligosaccharides, and others.
Bioaffinity chromatography
For more details on this topic, see Affinity chromatography.
This chromatographic process relies on the property of biologically active substances to form stable, specific, and reversible complexes. The formation of these complexes involves the participation of common molecular forces such as the Van der Waals interaction, electrostatic interaction, dipole-dipole interaction, hydrophobic interaction, and the hydrogen bond. An efficient, biospecific bond is formed by a simultaneous and concerted action of several of these forces in the complementary binding sites.
Aqueous normal-phase chromatography
Aqueous normal-phase chromatography (ANP) is a chromatographic technique which encompasses the mobile phase region between reversed-phase chromatography (RP) and organic normal phase chromatography (ONP). This technique is used to achieve unique selectivity for hydrophilic compounds, showing normal phase elution using reversed-phase solvents.[citation needed]

Isocratic and gradient elutionA separation in which the mobile phase composition remains constant throughout the procedure is termed isocratic (meaning constant composition). The word was coined by Csaba Horvath who was one of the pioneers of HPLC.[citation needed],
The mobile phase composition does not have to remain constant. A separation in which the mobile phase composition is changed during the separation process is described as a gradient elution.[4] One example is a gradient starting at 10% methanol and ending at 90% methanol after 20 minutes. The two components of the mobile phase are typically termed "A" and "B"; A is the "weak" solvent which allows the solute to elute only slowly, while B is the "strong" solvent which rapidly elutes the solutes from the column. In reversed-phase chromatography, solvent A is often water or an aqueous buffer, while B is an organic solvent miscible with water, such as acetonitrile, methanol, THF, or isopropanol.
In isocratic elution, peak width increases with retention time linearly according to the equation for N, the number of theoretical plates. This leads to the disadvantage that late-eluting peaks get very flat and broad. Their shape and width may keep them from being recognized as peaks.
Gradient elution decreases the retention of the later-eluting components so that they elute faster, giving narrower (and taller) peaks for most components. This also improves the peak shape for tailed peaks, as the increasing concentration of the organic eluent pushes the tailing part of a peak forward. This also increases the peak height (the peak looks "sharper"), which is important in trace analysis. The gradient program may include sudden "step" increases in the percentage of the organic component, or different slopes at different times – all according to the desire for optimum separation in minimum time.
In isocratic elution, the selectivity does not change if the column dimensions (length and inner diameter) change – that is, the peaks elute in the same order. In gradient elution, the elution order may change as the dimensions or flow rate change.[citation needed]
The driving force in reversed phase chromatography originates in the high order of the water structure. The role of the organic component of the mobile phase is to reduce this high order and thus reduce the retarding strength of the aqueous component.
Parameters
Theoretical
HPLC separations have theoretical parameters and equations to describe the separation of components into signal peaks when detected by instrumentation such as by a UV detector or a mass spectrometer. The parameters are largely derived from two sets of chromatagraphic theory: plate theory (as part of Partition chromatography), and the rate theory of chromatography / Van Deemter equation. Of course, they can be put in practice through analysis of HPLC chromatograms, although rate theory is considered the more accurate theory.

They are analogous to the calculation of retention factor for a paper chromatography separation, but describes how well HPLC separates a mixture into two or more components that are detected as peaks (bands) on a chromatogram. The HPLC parameters are the: efficiency factor(N), the retention factor (kappa prime), and the separation factor (alpha). Together the factors are variables in a resolution equation, which describes how well two components' peaks separated or overlapped each other. These parameters are mostly only used for describing HPLC reversed phase and HPLC normal phase separations, since those separations tend to be more subtle than other HPLC modes (e.g. ion exchange and size exclusion).
Void volume is the amount of space in a column that is occupied by solvent. It is the space within the column that is outside of the column's internal packing material. Void volume is measured on a chromatogram as the first component peak detected, which is usually the solvent that was present in the sample mixture; ideally the sample solvent flows through the column without interacting with the column, but is still detectable as distinct from the HPLC solvent. The void volume is used as a correction factor.
Efficiency factor (N) practically measures how sharp component peaks on the chromatogram are, as ratio of the component peak's area ("retention time") relative to the width of the peaks at their widest point (at the baseline). Peaks that are tall, sharp, and relatively narrow indicate that separation method efficiently removed a component from a mixture; high efficiency. Efficiency is very dependent upon the HPLC column and the HPLC method used. Efficiency factor is synonymous with plate number, and the 'number of theoretical plates'.
Retention factor (kappa prime) measures how long a component of the mixture stuck to the column, measured by the area under the curve of its peak in a chromatogram (since HPLC chromatograms are a function of time). Each chromatogram peak will have its own retention factor (e.g. kappa1 for the retention factor of the first peak). This factor may be corrected for by the void volume of the column.
Separation factor (alpha) is a relative comparison on how well two neighboring components of the mixture were separated (i.e. two neighboring bands on a chromatogram). This factor is defined in terms of a ratio of the retention factors of a pair of neighboring chromatogram peaks, and may also be corrected for by the void volume of the column. The greater the separation factor value is over 1.0, the better the separation, until about 2.0 beyond which an HPLC method is probably not needed for separation.
Resolution equations relate the three factors such that high efficiency and separation factors improve the resolution of component peaks in a HPLC separation.
Internal diameter
The internal diameter (ID) of an HPLC column is an important parameter that influences the detection sensitivity and separation selectivity in gradient elution. It also determines the quantity of analyte that can be loaded onto the column. Larger columns are usually seen in industrial applications, such as the purification of a drug product for later use. Low-ID columns have improved sensitivity and lower solvent consumption at the expense of loading capacity.

Larger ID columns (over 10 mm) are used to purify usable amounts of material because of their large loading capacity.
Analytical scale columns (4.6 mm) have been the most common type of columns, though smaller columns are rapidly gaining in popularity. They are used in traditional quantitative analysis of samples and often use a UV-Vis absorbance detector.
Narrow-bore columns (1–2 mm) are used for applications when more sensitivity is desired either with special UV-vis detectors, fluorescence detection or with other detection methods like liquid chromatography-mass spectrometry
Capillary columns (under 0.3 mm) are used almost exclusively with alternative detection means such as mass spectrometry. They are usually made from fused silica capillaries, rather than the stainless steel tubing that larger columns employ.
Particle size
Most traditional HPLC is performed with the stationary phase attached to the outside of small spherical silica particles (very small beads). These particles come in a variety of sizes with 5 µm beads being the most common. Smaller particles generally provide more surface area and better separations, but the pressure required for optimum linear velocity increases by the inverse of the particle diameter squared.[5][6][7]
This means that changing to particles that are half as big, keeping the size of the column the same, will double the performance, but increase the required pressure by a factor of four. Larger particles are used in preparative HPLC (column diameters 5 cm up to >30 cm) and for non-HPLC applications such as solid-phase extraction.
Pore size
Many stationary phases are porous to provide greater surface area. Small pores provide greater surface area while larger pore size has better kinetics, especially for larger analytes. For example, a protein which is only slightly smaller than a pore might enter the pore but does not easily leave once inside.
Pump pressure
Pumps vary in pressure capacity, but their performance is measured on their ability to yield a consistent and reproducible flow rate. Pressure may reach as high as 40 MPa (6000 lbf/in 2 ), or about 400 atmospheres. Modern HPLC systems have been improved to work at much higher pressures, and therefore are able to use much smaller particle sizes in the columns (<2 μm). These "Ultra High Performance Liquid Chromatography" systems or RSLC/UHPLCs can work at up to 100 MPa (15,000 lbf/in2), or about 1000 atmospheres. The term "UPLC" is a trademark of the Waters Corporation, but is sometimes used to refer to the more general technique.
See also History of chromatography Csaba Horváth

Ion chromatography Column chromatography Capillary electrochromatography Micellar liquid chromatography
References1. Gerber, F.; Krummen, M.; Potgeter, H.; Roth, A.; Siffrin, C.; Spoendlin, C.
(2004). "Practical aspects of fast reversed-phase high-performance liquid chromatography using 3μm particle packed columns and monolithic columns in pharmaceutical development and production working under current good manufacturing practice". Journal of Chromatography A 1036 (2): 127–133. doi:10.1016/j.chroma.2004.02.056. PMID 15146913. edit
2. Lindsay, S. ; Kealey, D. (1987). High performance liquid chromatography. Wiley. from review Hung, L. B.; Parcher, J. F.; Shores, J. C.; Ward, E. H. (1988). "Theoretical and experimental foundation for surface-coverage programming in gas-solid chromatography with an adsorbable carrier gas". J. Am. Chem. Soc. 110 (11): 1090. doi:10.1021/ac00162a003.
3. Displacement Chromatography . Sacheminc.com. Retrieved on 2011-06-07.4. Snyder, Lloyd R. and Dolan, John W. (2006). High-Performance Gradient
Elution: The Practical Application of the Linear-Solvent-Strength Model. Wiley Interscience. ISBN 0470055510.
5. Majors, Ronald E.. (2010-09-07) Fast and Ultrafast HPLC on sub-2 μm Porous Particles — Where Do We Go From Here? – LC-GC Europe. Lcgceurope.com. Retrieved on 2011-06-07.
6. Xiang, Y.; Liu Y. and Lee M.L. (2006). "Ultrahigh pressure liquid chromatography using elevated temperature". Journal of Chromatography A 1104 (1–2): 198–202. doi:10.1016/j.chroma.2005.11.118. PMID 16376355.
7. Horváth, Cs.; Preiss B.A. and Lipsky S.R. (1967). "Fast liquid chromatography. Investigation of operating parameters and the separation of nucleotides on pellicular ion exchangers". Analytical Chemistry 39 (12): 1422–1428. doi:10.1021/ac60256a003. PMID 6073805.
Further reading L. R. Snyder, J.J. Kirkland, and J. W. Dolan, Introduction to Modern Liquid
Chromatography, John Wiley & Sons, New York, 2009. M.W. Dong, Modern HPLC for practicing scientists. Wiley, 2006. L. R. Snyder, J.J. Kirkland, and J. L. Glajch, Practical HPLC Method
Development, John Wiley & Sons, New York, 1997. S. Ahuja and H. T. Rasmussen (ed), HPLC Method Development for
Pharmaceuticals, Academic Press, 2007. S. Ahuja and M.W. Dong (ed), Handbook of Pharmaceutical Analysis by HPLC,
Elsevier/Academic Press, 2005. Y. V. Kazakevich and R. LoBrutto (ed.), HPLC for Pharmaceutical Scientists,
Wiley, 2007. U. D. Neue, HPLC Columns: Theory, Technology, and Practice, Wiley-VCH,
New York, 1997. M. C. McMaster, HPLC, a practical user's guide, Wiley, 2007.

External links
Wikimedia Commons has media related to High-performance liquid chromatography.
Look up HPLC in Wiktionary, the free dictionary.
Liquid Chromatography at DMOZ














![Cromatografia Papel- Capa Fina[1]](https://static.fdocuments.ec/doc/165x107/55cf99e3550346d0339fa4a6/cromatografia-papel-capa-fina1.jpg)



